Embed Size (px)
Citation preview

Neuroscience Vol. 34, No. 1, pp. 225-234, 1990 Printed in Great Britain
0306-4522/90 $3.00 + 0.00 Pergamon Press plc
0 1990 IBRO
INTRASTRIATAL INJECTION OF DL-a-AMINOADIPATE REDUCES KAINATE TOXICITY IN VITRO
G. J. MCBEAN
Department of Biochemistry, Trinity College, Dublin 2, Ireland
Abstract-Intrastriatal injection of the excitatory amino acid analogue DL-a-aminoadipate (100 pg in 2 ~1 saline, pH 7.4) into anaesthetized rats caused a significant reduction in striatal glutamine synthetase activity in the ipsilateral compared to the contralateral striatum 6 h after the injection. Striatal neurons were unaffected by this treatment, and by 24 h after the injection, glutamine synthetase activity had returned to normal. In contrast to the situation in viuo, incubation of coronal slices (which included the striatum) in vitro with DL-a-aminoadipate (1-3 mM) for periods of up to I h caused no change in glutamine synthetase activity. Increased doses of DL-a-aminoadipate coupled with longer incubation times led to widespread neuronal degeneration within the striatum.
Preparation of coronal slices from striata which had been injected 6 h previously with DL-a-amino- adipate, and subsequently incubated with 300 PM kainate, showed a marked survival of some neurons particularly those ordering the injection tract. The toxicity of SOO~M N-methyl-o-aspartate in similar slices was unchanged. Conversely, co-incubation of DL-a-aminoadipate with excitotoxins in vitro provided protection of striatal cells against degeneration by N-methyl-D-aspartate, but not kainate.
These findings suggest that, in uiuo, DL-a-aminoadipate has a specific effect on glial cell metabolism which, in contrast to incubation of coronal slices with the compound in oitro, is not related to the amino acid antagonist properties associated with the u-isomer. Thus, the reduced toxicity of kainate observed in striatal slices following DL-a-aminoadipate injection in uiuo may indicate a non-neuronal site of action of kainate.
Several workers have described a specific effect of the six-carbon analogue of glutamate DL-a-aminoadipate (m-aAA) on the viability of glial cells either in uiuo or in vitro. In both the ependymal and glial cells of the arcuate nucleus of the hypothalamus,” and in retinal Muller cells,‘3~‘7~22 severe morphological changes were observed at 4-6 h after DL-c~AA injec- tion. Neurons in both these areas were unaffected by this treatment and glial cell morphology was normal by 24 h after the injection. Studies on the stereospe-
cificity of the reaction showed that the DL-racemate of AA was gliotoxic and the L-isomer both gliotoxic and neurotoxic, whereas the D-isomer was mildly gliotoxic, but not neurotoxic.‘9 Inclusion of DL-uAA in the culture medium of dissociated postnatal
cerebellar cells caused a time- and dose-dependent loss in the viability of glial cells, as indicated by immunofluorescent labelling of these cells with glial fibrillary acidic protein (GFAP) antibodies.‘,* How- ever, longer times of exposure coupled with higher concentrations of DL-uAA did cause additional loss of neurons.’ This study investigates whether injection of DL-&AA in vim is toxic to striatal cells, as previous reports on the selectivity of DL-a AA as a striatal toxin have given divergent results.6.28 An examination of the specificity of DL-QAA as a gliotoxin during incu-
Abbreviations: DL-uAA, DL-a-aminoadipate; DL-APV, DL-
aminophosphonovalerate; GFAP, glial fibrillary acidic protein; GHA, y-glutamylhydroxamate; GS, glutamine synthetase; NMDA, N-methyl-o-aspartate.
bation of brain slices in vitro has also been made, and it has been ascertained whether any disruption to glial cell metabolism by DL-U&i either in vitro or in uivo affects the ability of the neurotoxin kainic acid to cause degeneration of striatal neurons. Since most neurotoxin experiments in uivo have a time course typically between 24 h and 2 weeks,” yet the effects of DL-~LAA application in vivo are no longer apparent at
24 h after the injection, I7 the incubation of brain slices
with neurotoxins in vitro prepared 6 h after intrastri- atal injection of DL-aAA, permits an effective reduc- tion in the time-scale of a neurotoxicity experiment. In view of the reported gliotoxic and neurotoxic properties of the L-isomer of uAA,‘~ it was considered preferable to use the racemate in these studies since its toxic actions have been reported to be confined to
glial cells.‘9 The disruption to glial cell metabolism caused by
DL-uAA was estimated by measuring changes in the enzyme glutamine synthetase (GS) which is localized almost exclusively in glial cells.26
EXPERIMENTAL PROCEDURES
Preparation of brain slices and incubation in vitro All experiments were performed using 20&250 g female
Wistar rats. Animals were killed by cervical dislocation and the brains rapidly removed. Coronal slices (0.5 mm), which included the striatum, corpus callosum and part of the cerebral cortex, were prepared using a Campden Vibroslice, and placed, either singly or in pairs, in a number of open perfusion chambers (2 cm diameter). Each chamber was perfused with oxygenated Krebs Bicarbonate Medium
225

226 G. J. MCBEAN
(contents, in mM: NaCI, 109.6; KCl, 4.7; KH,PO,, 1.2; M&O,. 1.2: NaHCO,. 25: GaCl,. 2.5 and elucose. 11.5). pH 7.4, at 30” at a rate ofO.S-I.6 ml/min. Alter the initial preincubation period of 60 min, drugs under study were added, and the incubation continued for a maximum period of 60 min. The slices were then removed and processed for either biochemical or histological analysis. The activity of GS was measured in striatal homogenates (in 0.1 M sodium acetate buffer, pH 5.5) by the method of Schousboe,26 in which the product of the reaction is y-glutamylhydroxamate (GHA). The protein content of each sample was measured according to the method of Markwell ef a~‘.‘~
Histological examination of the tissue was made by fixation of each slice in 2% paraformaldehyde-1% glutar- aldehyde in 0.1 M phosphate buffer, pH 7.4, for 40 min. followed by dehydration and paraffin impregnation. Sections (6 pm) were stained in Thionin for examination by light microscopy. Photomicrographs of Nissl-stained sections were taken using a Nikon Optiphot microscope with a Nikon UFX camera attachment.
In vivo injections
Rats were anaesthetized with sodium pentobarbitone (50mg/kg, i.p.) and placed in a stereotaxic apparatus. A small burr hole was made in the skull overlying the left striatum (coordinates AP + 7.6 mm, ML + 2.2 mm from lambda according to the atlas of Konig and Klippel).’ Injections were made into the left striatum at a depth of 5.3 mm from the level of the dura using a blunt-tip Hamilton syringe. DL-aAA (1OOpg in 2 ~1 phosphate-buffered saline neutralized with NaOH) was injected over a 2min period. and the needle was left in place for a further 5 min. Animals were allowed to recover for either 6 or 24 h after the injection. Striata were dissected out for estimations of GS activity and protein content by the methods described. Histological examination was performed after transcardial perfusion of anaesthetized animals with 2% paraformaide- hyde-- I % glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Coronal slices were prepared for in t&o incubation experi- ments as described above.
~ateriuis
ok-a-A, kainic acid and quinolinic acid were purchased from Sigma Chemical Co.. Poole. Dorset. U.K. DL-2- Aminophosphonovaleric acid and N-methyl-D-aspartate (NMDA) were obtained from Cambridge Research Biochemicals, Cambridge, U.K. All other chemicals and reagents were supplied by either BDH Ltd, Poole, U.K. or Sigma.
RESULTS
Incubation of coronal slices with AL-~-aminoadipate in vitro
Inclusion of ~L-~AA (1-3 mM) in the superfusing buffer for 1 h or less caused no alteration in GS
activity compared to control slices which were incu- bated in buffer alone (Table 1). Examination of paraffin-embedded sections showed good preserva- tion of striatal neurons (karyopyknosis generally absent; Fig. IS), compared to untreated controls (Fig. IA). Increased doses of DL-GIAA coupled with longer incubation times again led to no change in GS activity, but widespread degeneration of striatal neurons was apparent, which was characterized by loss of definition of the cell membrane coupled with swollen and pyknotic nuclei (Fig. IC). Striatal astrocytes are small and well dispersed and thus not easily identified by light microscopy.
Table 1. Striatal glutamine synthetase activity in brain slices incubated with DL-a-aminoadipate
Time (min) Control 1 mM DL-C( AA
40 1.44 + 0.07 (5) 1.65 + 0.07 (4) 60 1.85 ho.17 (2) 1.60 zt 0.05 (2)
120 2.26 + 0.07 (5) 2.64 & 0. I I (6)
Coronal slices were incubated in physiological buffer at 30“, as described in Experimental Procedures. After a preincubation period of 60min, 1 mM DL-aAA was added to the superfusion medium, and the incuba- tion continued for the times indicated. Control slices were incubated throughout in buffer. Glutamine syn- thetase activity was measured in each slice by the method of Schousboe.26 Values are mean + S.E.M. Units: nmol GHA formed/mg protein per h. The figures in parentheses refer to the number of observations, assayed in duplicate.
E_xcitotoxin -induced degeneration in vitro
Incubation of coronal slices with either quinolinate
(500 PM) or kainate {3~~M) induced widspread degeneration of striatal cells within 40 min, compared to control (see Fig. 2A-C). NMDA (5OO~M) was also toxic to striatal cells under these conditions {results not shown). Co-incubation of 1 mM DL-aAA with either 500 p M quinolinate (Fig. 2D) or 300 p M kainate (Fig. 2E) provided protection of striatal neurons against the toxic effects of the former, but not the latter, compound. Preincubation of coronal slices with 1 mM DL-uAA for 40 min, followed by a 10 min washout period, before the addition of 500pM quinolinate did not lessen the ability of I mM DL-aAA to antagonize the toxic effects of quinolinate (results not shown).
Antagonism of agonist -induced degeneration in vitro by DL-aminophosphonoi>alerate
Figure 3A indicates that the presence of I mM DL-aminophosphonovalerate (DL-APV) in the incu- bation medium for as long as 1 h did not alter the ability of striatial cells to survive incubation in t)itro. Like T)L-@AA, 1 mM IX.-APV provided protection of striatal cells against 500 JIM quinolinate (Fig. 3B, C) but not 300pM kainate (Fig. 3D, E), when present in the incubation medium at the same time as the agonists.
intrastriata! injection of DL-a-aminoadipate in vivo
Injection of DL-c(AA (1OOyg in 2 ~1) into the left striatum induced a significant reduction in GS activity at 6, but not 24 h after the injection, compared to the right, uninjected striatum (Table 2). The morphology of striatal neurons was unaffected by this treatment, and there was no glial proliferation around the site of injection (Fig. 4A. B). There was no change seen in striatal GS activity 6 h after intrastriatal application of an equimolar concentration of VL-APV (61 pg in 2 ~1) Table 2).
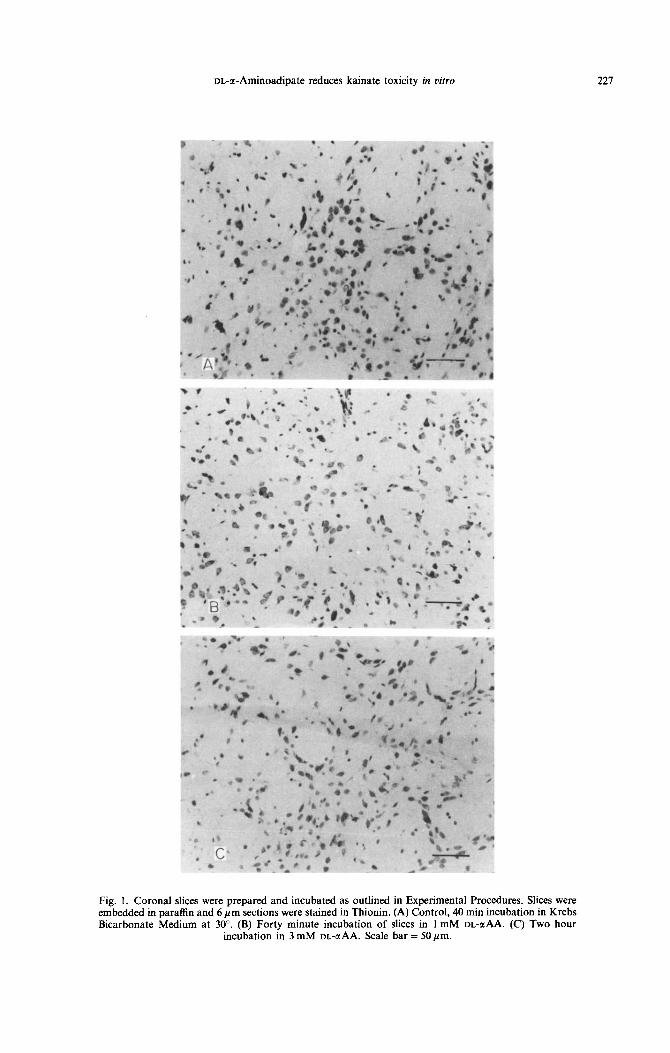
Fig.
Bicat
DL-a-Aminoadipate reduces kainate toxicity in vitro 221
1. Core ural :dded in pa: :bonate Mt
slices were prepared and incubated as outlined in Experimental Proced raffin and 6 pm sections were stained in Thionin. (A) Control, 40 min inct dium at 30”. (B) Forty minute incubation of slices in 1 mM DL-aAA
incubation in 3 mM DL-aAA. Scale bar = 50 pm.
tbati (Cl
Slices on in 1 I Two
Krebs
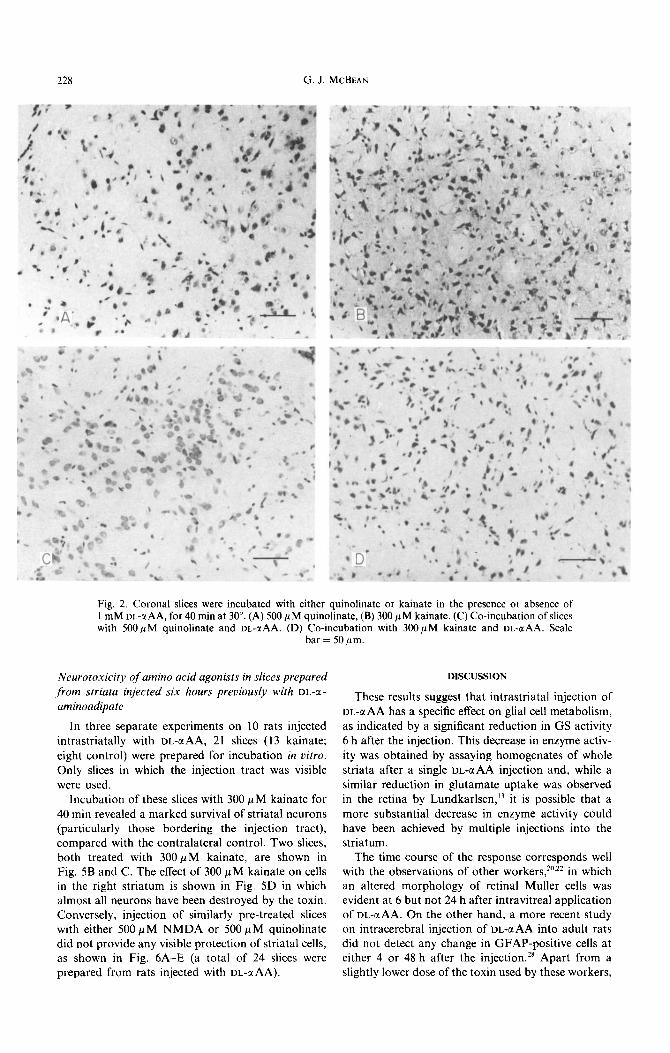
228 G. J. MCBEAN
Fig. 2. Coronal slices were incubated with either quinolinate or kainate in the presence or absence of 1 mM UL-a AA, for 40 min at 30”. (A) 500 PM quinolinate, (B) 300 PM kainate. (C) Co-incubation of slices with 500 PM quinolinate and DL-aAA. (D) Co-incubation with 300 PM kainate and DL-@AA. Scale
bar = 50 pm.
Neurotoxicity of amino acid agonists in slices prepared ,from striata injected six hours previously with DL-a- aminoadipate
In three separate experiments on 10 rats injected intrastriatally with DL-aAA, 21 slices (13 kainate;
eight control) were prepared for incubation in vitro. Only slices in which the injection tract was visible were used.
Incubation of these slices with 300 PM kainate for 40 min revealed a marked survival of striatal neurons (particularly those bordering the injection tract), compared with the contralateral control. Two slices, both treated with 300pM kainate, are shown in Fig. 5B and C. The effect of 300 PM kainate on cells in the right striatum is shown in Fig. SD in which almost all neurons have been destroyed by the toxin. Conversely, injection of similarly pre-treated slices with either 500 PM NMDA or 500 PM quinolinate did not provide any visible protection of striatal cells, as shown in Fig. 6A-E (a total of 24 slices were prepared from rats injected with DLGAA).
DISCUSSION
These results suggest that intrastriatal injection of
DL-a AA has a specific effect on glial cell metabolism, as indicated by a significant reduction in GS activity 6 h after the injection. This decrease in enzyme activ-
ity was obtained by assaying homogenates of whole striata after a single DbaAA injection and, while a similar reduction in glutamate uptake was observed in the retina by Lundkarlsen,” it is possible that a
more substantial decrease in enzyme activity could have been achieved by multiple injections into the striatum.
The time course of the response corresponds well with the observations of other workers,*‘,** in which an altered morphology of retinal Muller cells was evident at 6 but not 24 h after intravitreal application of DL-aAA. On the other hand, a more recent study on intracerebral injection of DL-aAA into adult rats did not detect any change in GFAP-positive cells at either 4 or 48 h after the injection.*’ Apart from a slightly lower dose of the toxin used by these workers,
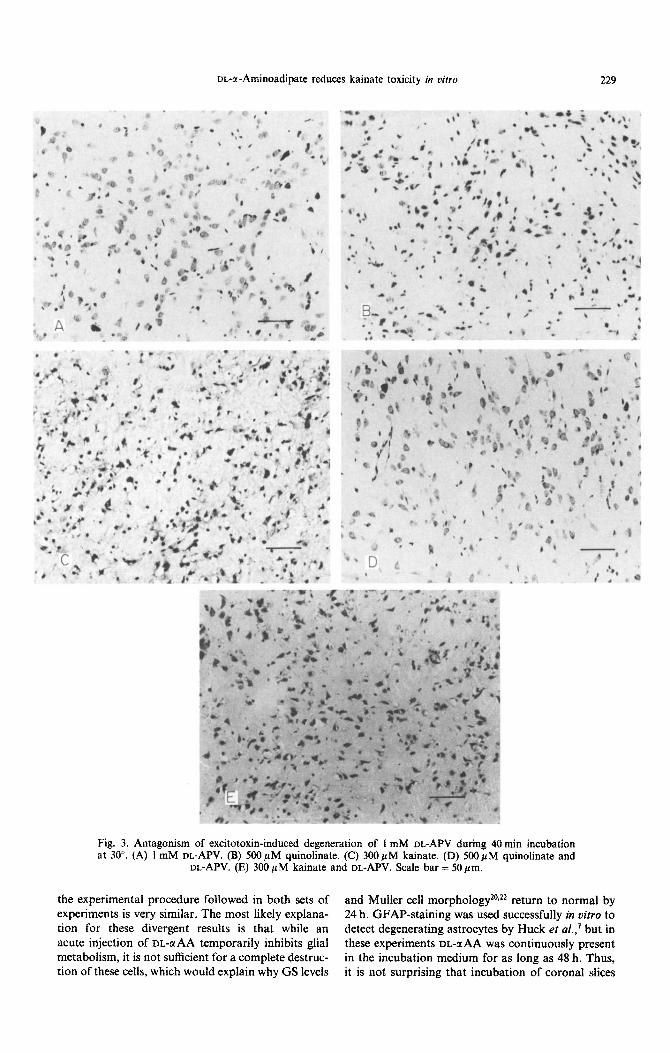
DL-a-Aminoadipate reduces kainate toxicity in vitro 229
Fig. 3. Antagonism of excitotoxin-induce degeneration of I mM DL-APV during 40min incubation at 30”. (A) 1 mM DL-APV. (B) 500 PM quinolinate. (C) 300 PM kainate. (D) 500 PM quinolinate and
DL-APV. (E) 300 PM kainate and DL-APV. Scale bar = 50 pm.
the experimental procedure followed in both sets of and Muller cell morphology20~22 return to normal by experiments is very similar. The most likely explana- 24 h. GFAP-staining was used successfully in vitro to tion for these divergent results is that while an detect degenerating astrocytes by Huck et uf.,’ but in acute injection of DL-GINA temporarily inhibits glial these experiments DL-a A.4 was continuously present metabolism, it is not sufficient for a complete destruc- in the incubation medium for as long as 48 h. Thus, tion of these cells, which would explain why GS levels it is not surprising that incubation of coronal slices
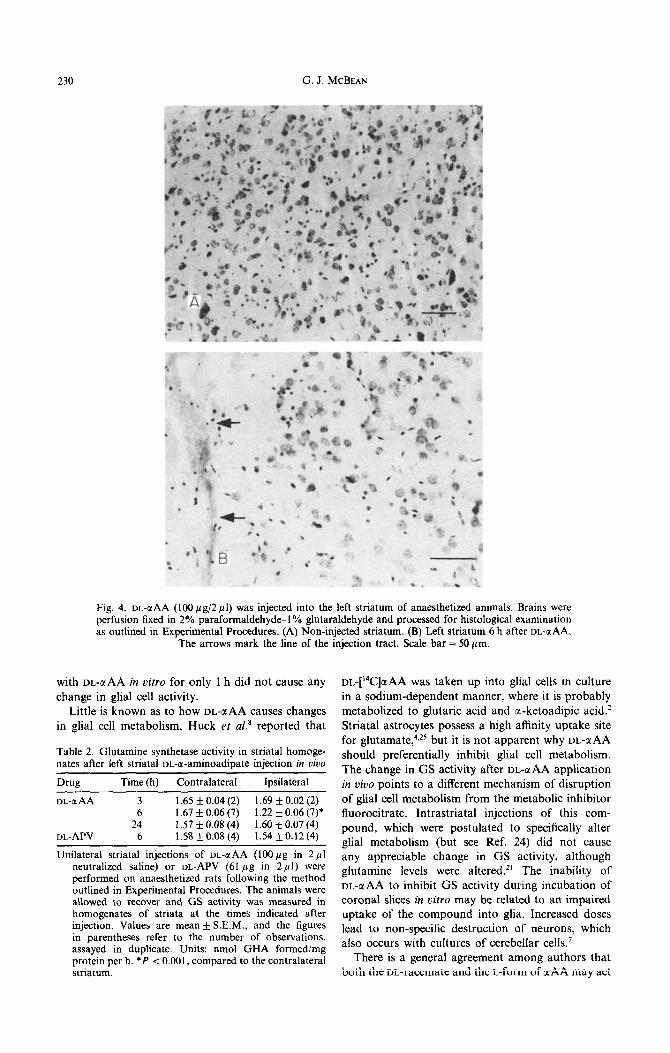
230 G. J. MCBEAN
Fig. 4. or-aAA (100 pg/2 ~1) was injected into the left striatum of anaesthetized animals. perfusion fixed in 2% paraformaldehyde-1% glutaraldehyde and processed for histological as outlined in Experimental Procedures. (A) Non-injected striatum. (B) Left striatum 6 h al
The arrows mark the line of the injection tract. Scale bar = 50 pm.
Brains were examination ?ter DL-a AA.
with DL-uAA in vitro for only 1 h did not cause any
change in glial cell activity. Little is known as to how DL-uAA causes changes
in glial cell metabolism. Huck et al8 reported that
Table 2. Glutamine synthetase activity in striatal homoge- nates after left striataf DL-a-aminoadipate injection in vivo
Drug Time (h) Contralateral Ipsilateral
m-a AA 3 1.65 + 0.04 (2) 1.69 k 0.02 (2) 6 1.67 + 0.06 (7) 1.22 f 0.06 (7)*
24 1.57+0.08(4) 1.60~0.07(4) DL-APV 6 l.SSkO.O8(4) 1.54*0.12(4)
Unilateral striatal injections of DL-aAA (1OOpg in 2~1 neutralized saline) or DL-APV (61 pg in 2.~1) were performed on anaesthetized rats following the method outlined in Experimental Procedures. The animals were allowed to recover and G‘S activity was measured in homogenates of striata at the times indicated after injection. Values are mean + S.E.M., and the figures in parentheses refer to the number of observations, assayed in duplicate. Units: nmol GHA formed/mg protein per h. *P < 0.001, compared to the contralateral striatum.
Dt,-[‘4C]aAA was taken up into glial cells in culture in a sodium-dependent manner, where it is probably metabolized to glutaric acid and a-ketoadipic acid.’ Striatal astrocytes possess a high affinity uptake site for glutamate, 4,25 but it is not apparent why DL-c(AA
should preferentially inhibit glial cell metabolism. The change in GS activity after DL-CL AA application in vivo points to a different mechanism of disruption of glial cell metabolism from the metabolic inhibitor fluorocitrate. Intrastriatal injections of this com- pound, which were postulated to specifically alter glial metabolism (but see Ref. 24) did not cause any appreciable change in GS activity, although glutamine levels were altered.*’ The inability of DL-@AA to inhibit GS activity during incubation of coronal slices in vitro may be related to an impaired uptake of the compound into glia. Increased doses lead to non-specific destruction of neurons, which also occurs with cultures of cerebellar cells.’
There is a general agreement among authors that both the DL-racemate and the L-form of u AA may act
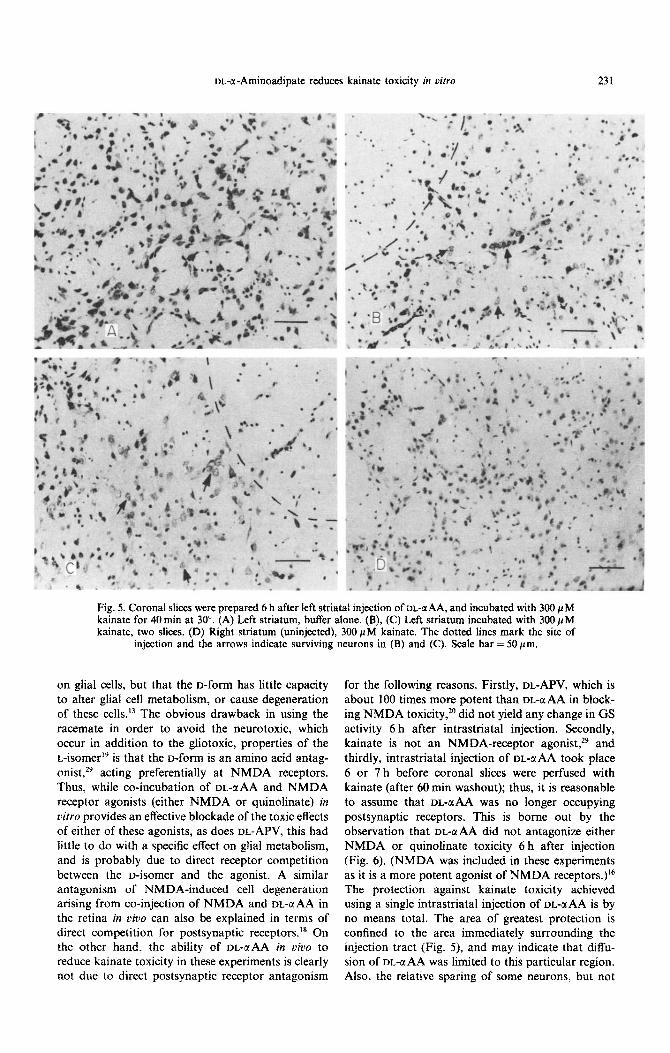
m-cr-Aminoadipate reduces kainate toxicity in vitro 231
Fig. 5. Coronal slices were prepared 6 h after left striatal injection of m-BAA, and incubated with 300 PM kainate for 40 tin at 30”. (A) Left striatum, buffer alone. (B), (C) Left striatum incubated with 300 PM kainate, two slices. (D) Right striatum (uninjected), 300 PM kainate. The dotted lines mark the site of
injection and the arrows indicate surviving neurons in (B) and (C). Scale bar = 50pm.
on glial cells, but that the o-form has little capacity to aiter glial cell metabolism, or cause degeneration of these cells.13 The obvious drawback in using the racemate in order to avoid the neurotoxic, which occur in addition to the gliotoxic, properties of the L-isomerI is that the D-form is an amino acid antag- onist,29 acting preferentially at NMDA receptors. Thus, while co-incubation of DL-uAA and NMDA receptor agonists (either NMDA or quinolinate) in oitro provides an effective blockade of the toxic effects of either of these agonists, as does DL-APV, this had little to do with a specific effect on glial metabolism, and is probably due to direct receptor competition between the D-isomer and the agonist. A similar antagonism of NMDA-induced cell degeneration arising from co-injection of NMDA and DL-IYAA in the retina in uivo can also be explained in terms of direct competition for postsynaptic receptors.” On the other hand, the ability of DL-UAA in vivo to reduce kainate toxicity in these experiments is clearly not due to direct postsynaptic receptor antagonism
for the following reasons. Firstly, DL-APV, which is about 100 times more potent than DL-aAA in block- ing NMDA toxicity, 2o did not yield any change in GS activity 6 h after intrastriatal injection. Secondly, kainate is not an NMDA-receptor agonist,29 and thirdly, intrastriatal injection of DL-@AA took place 6 or 7 h before coronal slices were perfused with kainate (after 60 min washout); thus, it is reasonable to assume that DL-ati was no longer occupying postsynaptic receptors. This is borne out by the observation that DL-UAA did not antagonize either NMDA or quinolinate toxicity 6 h after injection (Fig. 6). (NMDA was included in these experiments as it is a more potent agonist of NMDA receptors.)16 The protection against kainate toxicity achieved using a single intrastriatal injection of DL-QAA is by no means total. The area of greatest protection is confined to the area immediately surrounding the injection tract (Fig. 5), and may indicate that diffu- sion of DL-C(AA was limited to this particular region. Also, the relative sparing of some neurons, but not

232 G. J. MCBEAN
Fig. 6. Coronal slices were prepared 6 h after left striatal injection of m-a AA and incubated with 500 PM NMDA for 40 min. (A) Left striatum, buffer alone. (B), (C) Left striatum, incubated with 500 PM NMDA, two views. (D) Right striatum (uninjected), 5OOpM NMDA. (E) Left striatum, incubated with 500 @M
quinolinate. The dotted lines mark the site of injection. Scale bar = SO pm.
others in this area, points to a differential susceptibil- The mechanism by which kainate causes ity of striatal neurons to kainate. This has particular degeneraation of neurons has been the subject relevance to the recent report3 on the selective sparing of much speculation. Both kainate’.15 and quino- of cholinergic neurons after intrastriatal injection of linate2’ require the presence of an intact excitatory quinolinate, and future experiments will be aimed at afferent pathway before toxicity can be observed, addressing this question. which implies some interaction with presynaptic

DL-a-Aminoadipate reduces kainate toxicity in vitro 233
elements. However, there are differences in the mode of action of these two classes of neuro- toxins, and while quinolinate toxicity can be ex- plained in terms of an interaction with pre- and/or postsynaptic receptors, 27 the effects of kainate may be more complex. It has been suggested that the kainate-evoked release of glutamate observed in cerebellar slicesr” comes from a non-synaptic pool, and recent evidence has shown that kainate (but not NMDA) alters glutamate efflux through an inhibition of the normal uptake carrier for gluta- mate,23 which could equally well be located on astrocytes as on neurons.4 Glial cells have been shown to contain a “neurotoxic activity” which
can be released from these cells by potassium-ion depolarization.‘2
While it is not possible to conclude at present whether DL-uAA reduces kainate toxicity by a block- ade of the uptake carrier or by altered levels of glutamate/glutamine caused by an inhibition of glial cell metabolism, the model presented here provides scope for studying the involvement of glial cells in the onset of neurodegeneration by kainate. There are several aspects of the interaction which require further study; for instance, whether inhibitors of glutamine synthetase, such as methionine sulphoxi- mine,” also protect cells against the neurotoxicity of kainate.
Acknowledgements-l thank Prof. K. F. Tipton for many useful discussions and advice, and Dr M. Rowan for the use of the stereotaxic equipment. This work was supported by the Health Research Board of Ireland.
1.
2.
3.
4.
5.
6.
7.
8.
9. 10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20. 21.
22.
23.
24.
25.
26.
REFERENCES
Biziere K. and Coyle J. T. (1979) Effects of cortical ablation on the neurotoxicity and receptor binding of kainic acid in striatum. J. Neurosci. Res. 4, 383-398. Chang Y.-F. (1979) Lysine metabolism in the rat brain: the pipecolic acid-forming pathway. J. Neurochem. 30, 3477354. Davies S. W. and Roberts P. J. (1988) Sparing of cholinergic neurons following quinolinic acid lesions of the rat striatum. Neuroscience 26, 387-394. Drejer J., Larsson 0. M. and Schousboe A. (1982) Characterisation of L-glutamate and L-glutamine uptake into and release from astrocytes and neurones cultured from different brain regions. Expl Brain Res. 47, 259-269. Garthwaite J. and Regan C. M. (1980) Toxic effects of a-aminoadipate on cultured cerebellar cells. Brain Res. 194, 6033607. Hattori T. and Takeda M. (1985) Gliotoxin, a-aminoadipic acid, protects against kainic acid neurotoxicity in the striatum. Sot. Neurosci. Abstr. 11, 1195. Huck S., Grass F. and Hatten M. E. (1984) Gliotoxic effects of a-aminoadipic acid on monolayer cultures of dissociated postnatal mouse cerebellum. Neuroscience 12, 783-79 1. Huck S., Grass F. and Hortangl H. (1984) The glutamate analogue a-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effects in vitro. J. Neurosci. 4, 2650-2657. Konig J. F. R. and Klippel R. A. (1963) The Rut Bruin. Williams & Wilkins, Baltimore. Krespan B., Berl S. and Nicklas W. J. (1982) Alteration in neuronal-glial metabolism of glutamate by the neurotoxin kainic acid. J. Neurochem. 38, 509-518. Lamar C. and Sellinger 0. Z. (1965) The inhibition in oivo of cerebral glutamine synthetase and glutamine transferase by the convulsant methionine sulphoximine. Biochem. Pharmac. 14, 489-506. Lefebrve P. P., Rogister B., Delree P., Leprince P., Selak I. and Moonen G. (1987) Potassium-induced release of neuronotoxic activity by astrocytes. Bruin Res. 413, 120-128. Lunkarlsen R. (1979) The toxic effect of sodium glutamate and DL-a-aminoadipic acid on rat retina: changes in high affinity uptake of putative transmitters. J. Neurochem. 31, 1055-1061. Markwell M., Hans S. M., Biecker L. and Tolbert N. (1978) A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Analyt. Biochem. 87, 206210. McBean G. J. and Roberts P. J. (1985) Neurotoxicity of L-glutamate and m-threo-3-hydroxyaspartate in the rat striatum. J. Neurochem. 44, 247-254. McLennan H. (1984) A comparison of the effects of N-methyl-o-aspartate and quinolinate on central neurones of the rat. Neurosci. Lett. 46, 157-160. Olney J. W., Ho 0. L. and Rhee V. (1971) Cytotoxic effects of acidic and sulphur-containing amino acids on the infant mouse central nervous system. Expl Brain Res. 14, 61-76. Olney J. W., de Gubareff T. and Labruyere J. (1979) cc-Aminoadipate blocks the neurotoxic action of N-methyl- aspartate. Life Sci. 5, 537-540. Olney J. W., de Gubareff T. and Collins J. F. (1980) Stereospecificity of the gliotoxic and anti-neurotoxic actions of alpha-aminoadipate. Neurosci. L&t. 19, 227-282. Olney J. W. (1982) The toxic effects of glutamate and related compounds in the retina and the brain. Retina 2,341-359. Paulsen R. A., Contestabile A., Villani L. and Fonnum F. (1987) An in viuo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J. Neurochem. 48, 1377-1385. Pedersen 0. 0. and Karlsen R. L. (1979) Destruction of Muller cells in the adult rat by intravitreal injection of or-a-aminoadipic acid. An electron microscopic study. Expl Eye Res. 28, 569-575. Pocock J. M., Murphie H. M. and Nicholls D. G. (1988) Kainic acid inhibits the synaptosomal plasma membrane glutamate carrier and allows glutamate leakage from the cytoplasm. J. Neurochem. SO, 745-751. Sanchez-Prieto J. and Gonzales M. P. (1988) Anoxia induces a large Ca*+ independent release of glutamate in isolated nerve terminals. J. Neurochem. 50, 1322-1324. Schousboe A., Svenneby G. and Hertz L. (1979) Uptake and metabolism of glutamate in astrocytes cultured from dissociated mouse brain hemispheres. J. Neurochem. 29, 99991005. Schousboe A. (1982) Glial marker enzymes. &and. J. Immunol. 15, 339-356.

234 G. J. MCBEAN
27. Schwartz R., Foster A. C., French E. D., Whetsell W. 0. and Kohler C. (1984) Excitotoxic models for neurodegen- erative disorders. Life Sci. 35, 19-32.
28. Straffan B. N. and Crutcher K. A. (1987) Putative gliotoxin a-aminoadipate, fails to kill hippocampal astroytes in viva. Neurosci. Lett. 81, 215-220.
29. Watkins J. C. and Olverman H. J. (1987) Agonists and antagonists for excitatory amino acid receptors. Trends Neurosci. 10, 265-212.
(Accepted 10 July 1989)





























