Embed Size (px)
Citation preview
1
Introdução às reacções enzímicas.Equilíbrio químico, catálise e classificação funcional.
Aula preparada por Rui Fontes. O autor agradece a colaboração de Maria João Martins.
Bibliografia aconselhada:Nelson DL & Cox MM. (2005) Lehninger Principles of Biochemistry. 3rd ed. Worth Publishers. New York.
Chang R. (1994) Química 5ª ed. McGrow-Hill de Portugal, Lda
Fontes R (2003) Notas de termodinâmica química e calorimetria (A energia como um conceito menos intuitivo que o que parece).
Fontes R (2005) Notas sobre Introdução às reacções enzímicas. Equilíbrio químico e classificação funcional. 2
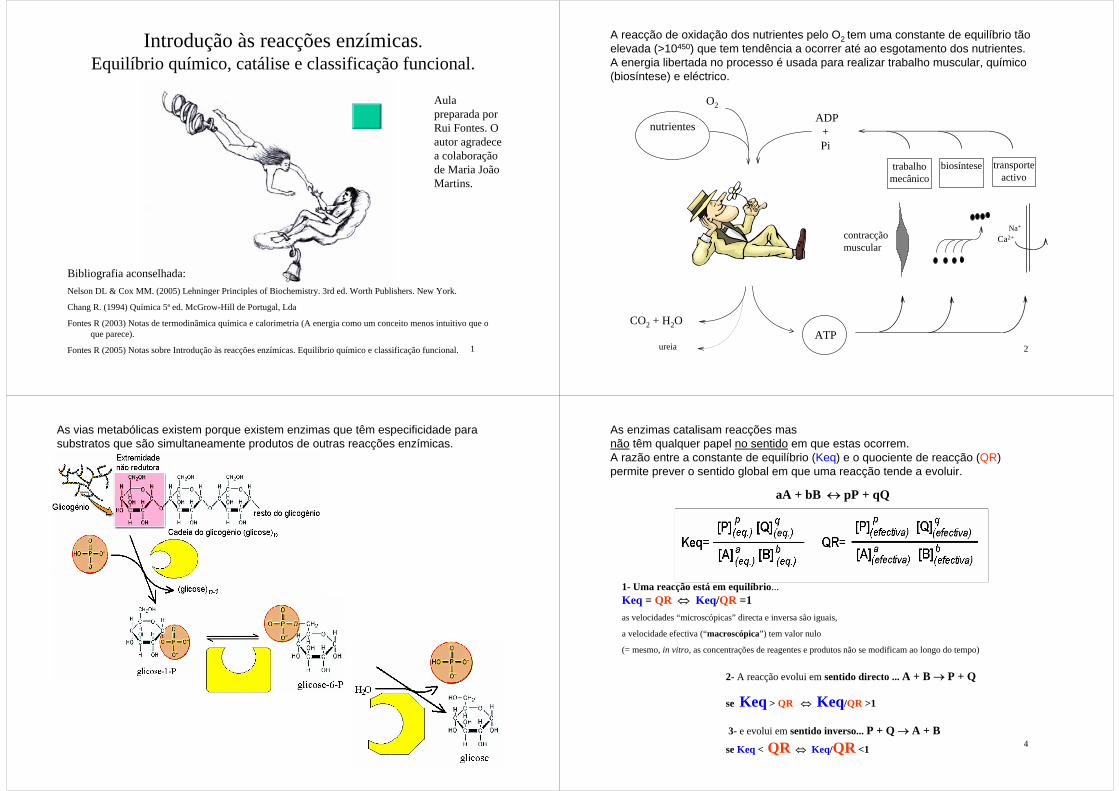
contracção muscular
nutrientes
O2
ADP+Pi
ureia
CO2 + H2OATP
transporteactivo
biosíntesetrabalhomecânico
Na+
A reacção de oxidação dos nutrientes pelo O2 tem uma constante de equilíbrio tão elevada (>10450) que tem tendência a ocorrer até ao esgotamento dos nutrientes.A energia libertada no processo é usada para realizar trabalho muscular, químico (biosíntese) e eléctrico.
Ca2+
3
As vias metabólicas existem porque existem enzimas que têm especificidade para substratos que são simultaneamente produtos de outras reacções enzímicas.
4
As enzimas catalisam reacções mas não têm qualquer papel no sentido em que estas ocorrem.A razão entre a constante de equilíbrio (Keq) e o quociente de reacção (QR) permite prever o sentido global em que uma reacção tende a evoluir.
aA + bB ↔ pP + qQ
2- A reacção evolui em sentido directo ... A + B → P + Q
se Keq > QR ⇔ Keq/QR >1
3- e evolui em sentido inverso... P + Q → A + Bse Keq < QR ⇔ Keq/QR <1
1- Uma reacção está em equilíbrio...Keq = QR ⇔ Keq/QR =1as velocidades “microscópicas” directa e inversa são iguais,
a velocidade efectiva (“macroscópica”) tem valor nulo
(= mesmo, in vitro, as concentrações de reagentes e produtos não se modificam ao longo do tempo)
5
Numa via metabólica existem enzimas com baixa actividade catalítica e que,consequentemente, catalisam reacções em que Keq»QR.
Outras enzimas têm uma actividade catalítica elevada e, nestes casos, Keq≈QR.
6
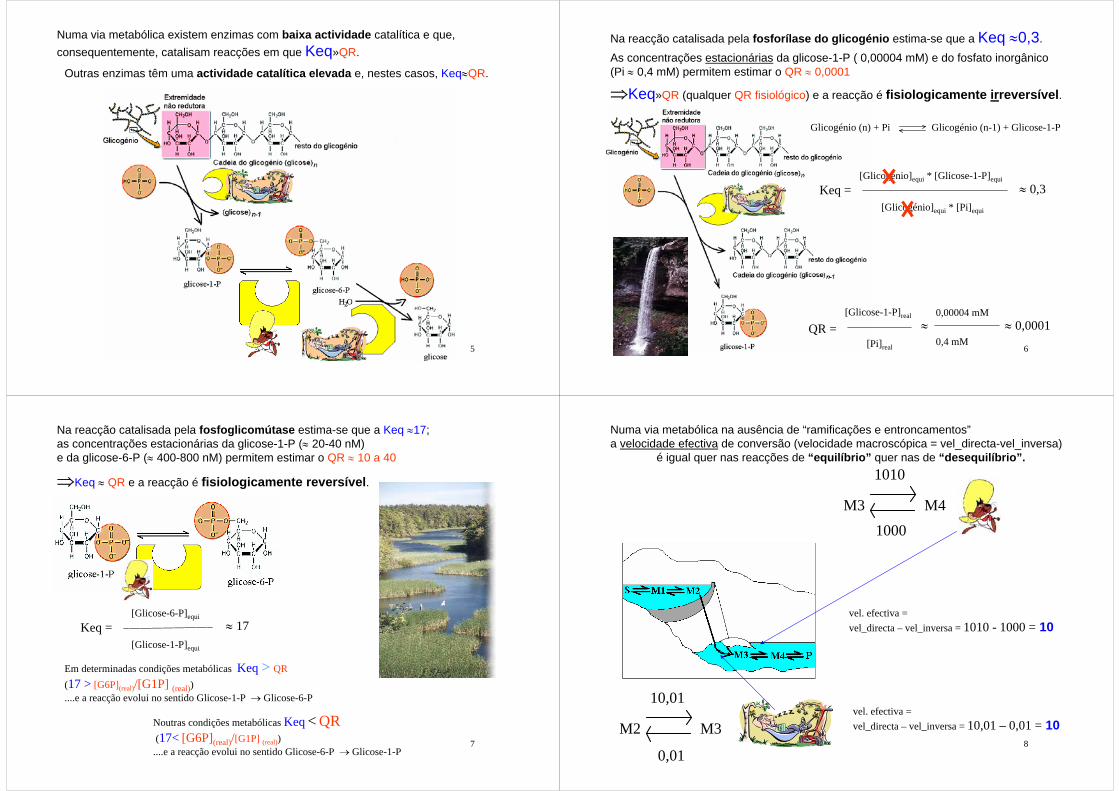
Na reacção catalisada pela fosforílase do glicogénio estima-se que a Keq ≈0,3.
Glicogénio (n) + Pi Glicogénio (n-1) + Glicose-1-P
Keq = ≈ 0,3[Glicogénio]equi * [Glicose-1-P]equi
[Glicogénio]equi * [Pi]equi
QR = ≈ 0,0001[Glicose-1-P]real
[Pi]real
≈0,00004 mM
0,4 mM
As concentrações estacionárias da glicose-1-P ( 0,00004 mM) e do fosfato inorgânico (Pi ≈ 0,4 mM) permitem estimar o QR ≈ 0,0001
⇒Keq»QR (qualquer QR fisiológico) e a reacção é fisiologicamente irreversível.
7
Noutras condições metabólicas Keq < QR(17< [G6P](real)/[G1P] (real))
....e a reacção evolui no sentido Glicose-6-P → Glicose-1-P
Na reacção catalisada pela fosfoglicomútase estima-se que a Keq ≈17; as concentrações estacionárias da glicose-1-P (≈ 20-40 nM) e da glicose-6-P (≈ 400-800 nM) permitem estimar o QR ≈ 10 a 40
Em determinadas condições metabólicas Keq > QR(17 > [G6P](real)/[G1P] (real)) ....e a reacção evolui no sentido Glicose-1-P → Glicose-6-P
Keq = ≈ 17[Glicose-6-P]equi
[Glicose-1-P]equi
⇒Keq ≈ QR e a reacção é fisiologicamente reversível.
8
Numa via metabólica na ausência de “ramificações e entroncamentos”a velocidade efectiva de conversão (velocidade macroscópica = vel_directa-vel_inversa)
é igual quer nas reacções de “equilíbrio” quer nas de “desequilíbrio”.
vel. efectiva = vel_directa – vel_inversa = 1010 - 1000 = 10
M3 M41000
1010
M2 M3
0,01
10,01vel. efectiva = vel_directa – vel_inversa = 10,01 – 0,01 = 10

9
O valor da razão Keq/QR de um sistema reactivo A → P é uma medida do “grau de desequilíbrio” da reacção;quanto maior é o valor da razão Keq/QR maior a quantidade de energia livre (ou de Gibbs) libertada no processo reactivo.
10
A equação de Gibbs relaciona a energia de Gibbs (ou função de Gibbs) de um sistema reactivo (∆G) com a razão Keq/QR
3610-6
tende a prosse-guir no sentidoinverso
Posit.<11810-3
Aparen-temente
parada
Nula= 101
-18103
tende a prosse-guir no sentido
directo
Negat.>1-36106
e a reacção
A→P
...então a energia de
Gibbs
Se a razão Keq/QR
∆GKJ/mol≈
Keq/QR
∆G (KJ/mol) ≈
- log decimal (Keq/QR) * 6
11
As reacções com ∆G negativo dizem-se exergónicas= expontâneas (na terminologia dos químicos)
Em sentido estrito as reacções endergónicas (∆G positivo Keq < QR) são apenas uma abstracção...... uma reacção não evolui em sentido inverso ao que é ditado pela razão Keq/QR
Exergónica não é sinónimo de exotérmica... 12
O ∆Gº e o ∆G’º são, simplesmente, os ∆G de sistemas reactivos que se convencionaram como sistemas padrão em química e em bioquímica, respectivamente.
Os químicos convencionaram condições padrão que lhes permitiram construir tabelas que comparam a energia livre padrão (∆Gº) de muitas reacções.
As condições padrão dos químicos são:
1- 25ºC (298 Kelvin)
2- Para definir o QR: quer os reagentes quer os produtos estão em concentração unitária
(1 atm se são gazes e 1M se são solutos não gasosos; Nota: [H+ ] = 1 ⇔ pH zero)
... excepto (se reacção em solução aquosa) a água que está na concentração 55,5 M
Consideremos a reacção de hidrólise do composto AB: AB (+ H2O) → A + B
Teríamos condições padrão e o ∆G = ∆Gº
se temperatura = 298 Kelvin
se para definir o QR:
[AB]=1 M; [A]=1M; [B]=1M; [H2O]=55,5 M
KeqRTG ln−=∆ o

13
Porque a esmagadora maioria das reacções estudadas pelos bioquímicos envolvem enzimas que, na sua maioria, não funcionam a pH 0 ([H+]=1M), os bioquímicosconvencionaram definir condições padrão transformadas.
... a única diferença relativamente às condições padrão dos químicos é que em vez de convencionar pH 0 convencionou-se que: [H+] = 10-7 ⇔ pH = 7
∆Gº, Keq, ∆G’º e Keq’ são medidas da diferença entre a estabilidade termodinâmica dos produtos e dos reagentes nas condições convencionadas como padrão
Consideremos uma reacção de isomerização AH↔BH mas em que, num sistema tamponado a pH 7,B se ioniza libertando um protão: AH → B- + H+
A Keq dos bioquímicos (para além da água) ignora também a [H+] considerando-a fixada a 10-7 M, ...e quando queremos dar ênfase a essa ideia, podemos escrever Keq’
Falar de ∆G’º duma reacção é apenas uma outra maneira de falar da Keq’
14
Contudo, por razões de tradição, nas equações em que intervém o NAD+ ou o NADP+ éfrequente escrever-se:
Lactato + NAD+ ↔ Piruvato + NADH + H+
glicose-6-P + NADP+ ↔ 6-fosfogliconolactona + NADPH + H+
Em bioquímica, as concentrações da água e dos protões são frequentemente ignoradas nas equações que exprimem Keq e QR; a água e os protões também são frequentemente ignorados nas equações das reacções escritas pelos bioquímicos.
Porque na razão que exprime o “grau de desequilíbrio” de uma reacção (Keq/QR) em sistema aquoso e a pH constante as concentrações da água e dos protões são iguais no numerador e no denominador é frequente os protões e a água serem ignorados nas equações dos bioquímicos.
É normal escrever-seATP → ADP + Pi ouATP + H2O → ADP + Pi
mas, por exemplo, a pH 7, seria mais correcto: ATP4- + H2O → ADP3- + Pi2- + H+
15
O valor de ∆Gº’ é apenas um equivalente da Keq’ e, em geral, não nos diz nada acerca do sentido em que a reacção tende a evoluir nem da reversibilidade ou irreversibilidade do processo em sistemas biológicos.
molKJRTG /33,0lnº' +≈−=∆
molKJRTG /200001,0
3,0ln' −≈−=∆
A reacção de fosforólisedo glicogénio éexergónica porque o ∆G’ é negativo
16
As tabelas com potenciais de oxi-redução padrão permitem prever o sentido em que uma determinada reacção de oxi-redução tende a evoluir quando as condições são padrão. O ∆Eº e o ∆E’º são equivalentes à Keq.
O potencial de oxiredução padrão de um determinado par oxidante/redutor (exemplos: O2/H2O, piruvato/lactato, NAD+/NADH) é uma medida da estabilidade relativa das formas oxidada e reduzida:
quanto maior o valor do potencial de oxi-redução padrão maior a estabilidade da forma reduzida relativamente à forma oxidada desse par.
Par redox E’º (volt)
½ O2 / H2O + 0,815
piruvato / lactato -0,185
NAD+ / NADH -0,315
Da tabela podemos deduzir que, em condições padrão (pH 7, 25ºC, concentrações 1M ou 1 atm para reagentes e produtos),
as reacções em que o oxigénio oxida o NADH
ou o piruvato oxida o NADH
⇔ têm Keq’ > 1 e, portanto, se QR = 1 evoluem no sentido indicado.
2e-
∆E’º = 0,059n
log Keqlog Keq =
n ∆E’º
0,059

17
Numa reacção de oxi-redução, para além das equivalências que também se aplicam às outras reacções (∆G’ Keq’/QR’ e ∆G´º Keq’), existem outras equivalências que relacionam
∆E’ ∆G’ Keq’/QR’ e ∆E’º ∆G’º Keq’.
18
O potencial de oxi-redução padrão positivo (∆E’º > 0) indica-nos que Keq’> 1. O potencial oxi-redução positivo (∆E’ > 0) indica-nos que a razão Keq’ / QR’ > 1.
Se numa reacção de oxiredução: ∆E´º > 0 Keq’>1 ∆G’º< 0
As expressões equivalentes para condições diferentes das condições padrão são:∆E´ > 0 Keq’/QR’>1 ∆G < 0
19
Conhecer o ∆G (⇔ razão Keq/QR) de um sistema reactivo indica-nos o sentido em que a reacção pode evoluir ... mas não nos diz nada acerca da velocidade em que ela ocorre.
1- Algumas reacções são muito lentas:
A Keq da reacção de oxidação da glicose (glicose + 6 O2 → 6 CO2 + 6 H2O)
é cerca de 10500 M-1 ∆G’º = - 2840 KJ/mol
a reacção tem tendência a evoluir até ao consumo total do reagente limitante (em geral a glicose)
...mas, à temperatura ambiente e na ausência de enzimas, posso ter
glicose em contacto com O2 durante milhares de anos que não acontece nada.
2- Outras reacções são muito rápidas
As reacções de dissociação de protões ou ligação de protões (ácido-base)aproximam-se rapidamente do equilíbrio e não necessitam de catalisadores.
20
Numa sequência de reacções (A → B → C)os ∆ G são aditivos e os Keq/QR multiplicativos
A razão Keq/QR relativamente ao processo global de transformação A → C
Consideremos uma sequência de reacções ⎯→⎯1 B ⎯→⎯2 CA
...mas no caso do ∆G pode deduzir-se que... 21)2,1( GGG ∆+∆=∆
Quando queremos exprimir quantitativamenteo estado de equilíbrio ou desequilíbrio de um sistema reactivo
as expressões ∆G e Keq/QR são equivalentes mas...
é, em geral, muito mais fácil e intuitivo usar ∆G
Keq(1,2)
QR(1,2)=
[C]eq
[A]eq
[C]real
[A]real
=Keq(1)
QR(1)x
Keq(2)
QR(2)

21
Quando A se transforma em P formam-se intermediários muito instáveis (estados de transição) A*: a Keq [A*](eq) / [A](eq) tem frequentemente um valor muito baixo⇔ ∆Gº do processo de formação do intermediário A* é elevado e positivo.
...e admitamos que na reacção A → P: Keq (1,2) = 10 ∆Gº (1,2) ≈ - 6 KJ/mol...e que na reacção A → A*: Keq (1) = 10-100 ∆Gº (1) ≈ + 600 KJ/mol
A* PConsideremos uma sequência de reacções: A1 2
No exemplo em análiseembora na reacção global A→ P
o ∆Gº (1,2) favoreça a formação de P
([P](eq)/[A](eq) = 10)
...a formação do estado de transição A* necessário para que a reacção ocorra está prejudicada por um ∆Gº1 positivo e de valor elevado.
22
Nas reacções enzímicas o estado de transição éde natureza diferente (o complexo E•A*)e termodinamicamente muito mais estável do que o que pode eventualmente existir na ausência da enzima.
Keq1A presença da enzima faz com que passe a existir um estado de transição em concentração maior ⇒ velocidade de reacção maior
Nas reacções catalisadas por enzimas forma-se umintermediário E•A*A afinidade da enzimapara o estado de transição A* estabiliza o intermediário que neste caso é o complexo E•A*
o valor da Keq correspondente à formação do intermediário aumenta
23
Afirmar que a presença de enzima aumenta a Keq relativa ao processo de formação do estado de transição é o mesmo que dizer a presença da enzimadiminui o ∆Gº relativo a esse processo.∆Gº da formação do estado de transição = energia livre de activação
As enzimas aceleram as reacções porque baixam a energia livre de de activação.
Keq1
24
As enzimas aceleram as reacções mas não alteram o sentido em que estas ocorrem: o sentido das reacções é determinado pela razão Keq/QR. A Keq e o QR (e também o ∆G’º e o ∆G) de uma reacção é independente do caminhocom que essa reacção se processa.Consideremos a transformação não catalisada enzimicamente:
Quando uma determinada enzima catalisa uma determina reacção no sentido directo
podemos estar seguros que também catalisa a reacção inversa.
Admitamos agora a mesma reacção catalisada enzimicamente pela enzima E tem os seguintes passos:
A B
Keq(1,2) =[E•A*][E] [A] [E•A*]
[E] [B]XE+A E•A* E+B
1 2
Keq = [A][B]
= [A][B]










![orgaos e tecidos03 - FMUP - Faculdade de Medicina da ...ruifonte/PDFs/PDFs_arquivados_anos... · a velocidade de entrada de glicose ↓⇒ [glicose] intracelular ↓ ⇒velocidade](https://img.pdfslide.tips/doc/110x75/5c441d0193f3c34c5a365c83/orgaos-e-tecidos03-fmup-faculdade-de-medicina-da-ruifontepdfspdfsarquivadosanos.jpg)











![Reacções de Oxidação-Redução [Modo de Compatibilidade]](https://img.pdfslide.tips/doc/110x75/5571fed549795991699c278f/reaccoes-de-oxidacao-reducao-modo-de-compatibilidade.jpg)






