Embed Size (px)
Citation preview

Introduction à la phylogénie*
* Notes basées en partie sur les notes de cours de Nadia El-Mabrouk
Tree of Life web project:http://tolweb.org/tree/phylogeny.html

* 1. Théorie de l’évolution
* 2. Arbres de phylogénie
* Arbres enracinés et non enracinés
* La donnée du problème
* 3. Construction d’arbres de phylogénie
* Présentation des méthodes
* Méthodes de distance:
* distances ultramétriques
* distances additives
* Méthodes de parcimonie:
* phylogénie parfaite
* phylogénie parfaite généralisée
* phylogénie maximale
* Méthodes de maximum de vraisemblance
PLAN
IFT6291, A2006, Sylvie HamelUniversité de Montréal 1Phylogénie

1. Théorie de l’évolutionTous les organismes vivants dérivent d’un ancêtre commun.
La diversité est due à la spéciation i.e à la séparation d’une espèce en deux espèces différentes.
Idée de base: Les caractères sont transmis d’une génération à l’autre et, au cours de l’évolution, ces caractères subissent une série de mutations
2Phylogénie
Nous allons parler d’arbres de phylogénie même si un des grands débat, en phylogénétique, est de savoir si l’évolution peut être vu comme un arbre.
Plusieurs aspects de l’évolution moléculaires ne peuvent être représentés par un arbre. Ex. Transferts horizontaux.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

2. Arbres de phylogénie
3Phylogénie
ancêtre commun
Espèces actuelles
ancêtre commun ou point dansl’histoire où les espèces ont divergé
Taille: peut représenter le nombre de mutations ou le temps d’évolution
Arbres racinés
Les noeuds internes peuvent être étiquettés par les séquences les plus probables des ancêtres
Noeud de degré > 3: Noeud non résolu. Ordre de speciation non déterminé
IFT6291, A2006, Sylvie HamelUniversité de Montréal

4Phylogénie
Noeud de degré > 3: Noeud non résolu. Ordre de speciation non déterminé
IFT6291, A2006, Sylvie HamelUniversité de Montréal

5Phylogénie
Différentes représentations d’arbres enracinés:
1) Cladogram: indique simplement les relations d’ancêtre entre les espèces
Les espèces A et B ont un ancêtre commun plus récent que les espèces A et C
IFT6291, A2006, Sylvie HamelUniversité de Montréal

2) Arbres additifs: la taille des branches indique, par exemple, le nombre de substitutions entre les deux séquences
6Phylogénie
Différentes représentations d’arbres enracinés:
IFT6291, A2006, Sylvie HamelUniversité de Montréal

7Phylogénie
3) Arbres ultramétriques: Arbres additifs où les feuilles sont équidistantes de la racine.
Différentes représentations d’arbres enracinés:
Hypothèse sous-jacente: horoge molécule i.e taux de mutation constant
IFT6291, A2006, Sylvie HamelUniversité de Montréal

8Phylogénie
Arbres enracinés versus arbres non enracinés:Arbres enracinés: Direction représentant le temps d’évolution: plus un noeud est proche de la racine, plus il est vieux.
Arbres non enracinés: Ne permet pas de déduire des relations de descendance
Plusieurs méthodes de constructions d’arbres de phylogénie génèrent des arbres non enracinés. Il faut ensuite trouver un “outgroup” pour enraciner l’arbre.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

9Phylogénie
La donnée du problème:Étant donné n espèces, calculer un arbre qui retrace l’évolution de ces espèces.
L’information sur les espèces peut prendre deux formes différentes:
E1 . . . Ej . . . En
d(Ei,Ej)
E1
Ei
En
...
...
t1 . . . t j . . . tn
e(Ei, t j)
E1
Ei
En
...
...
1) distances 2) caractères
état du caractère pour l’espècet j Ei
IFT6291, A2006, Sylvie HamelUniversité de Montréal

10Phylogénie
3. Construction d’arbres de phylogénieTrois méthodes principales:
1) Méthodes de distance: - Entrée: ensemble de distances (ex: distance d’édition)- Problème: Construire l’arbre de phylogénie en “accord” avec cet ensemble de distances
- Distances ultramétriques ou additives -> problèmes faciles à traiter
- Distances générales -> heuristiques nous donne l’arbre qui fournit la “meilleure approximation”
2) Méthodes de parsimonie: arbres qui explique l’évolution des espèces par un nombre minimal de mutations.
- Calcul du score d’un arbre donné- Recherche, parmi tous les arbres, de l’arbre de score minimal. Parcimonie maximale = nombre minimal de mutations
3) Méthodes de maximun de vraisemblance: Méthode probabiliste permettant de trouver la séquence de noeuds internes la plus probable
IFT6291, A2006, Sylvie HamelUniversité de Montréal

D(i,j)
11Phylogénie
Méthodes de distance- n séquences; D(i,j) est la distance entre les deux séquences i et j
- Algorithme de clustering UPGMA (Unweight Pair Group Method with Arithmetic Means):
Procéder par regroupement des séquences les plus proches. À chaque étape, les deux regroupements les plus “proches” sont fusionnés.
- : Distance entre deux regroupements et = moyenne des distances entre les paires de séquences entre les deux regroupementsdi, j Ci Cj
IFT6291, A2006, Sylvie HamelUniversité de Montréal

12PhylogénieIFT6291, A2006, Sylvie Hamel
Université de Montréal

13Phylogénie
Arbres et distances ultramétriquesLa construction d’un arbre par UPGMA sous-entend un modèle d’évolution faisant intervenir l’hypothèse de l’horloge moléculaire: taux de mutation constant
UPGMA trouve LE bon arbre ssi il existe un arbre ultramétrique pour D
Définition: Soit D une matrice symétrique n X n. Un arbre ultramétrique associé à D est un arbre A tel que:
1) L’arbre A a n feuilles étiquettées par les lignes de la matrice D (les espèces)
2) Chaque noeud de A est étiquetté par une valeur D(i,j) et a au moins deux enfants
3) Sur tout chemin de la racine à une feuille, les étiquettes sont strictement décroissantes
4) L’étiquette du plus petit ancêtre commun de i et j est D(i,j)
IFT6291, A2006, Sylvie HamelUniversité de Montréal

14Phylogénie
Arbres et distances ultramétriques (suite)
Remarque: A a au plus n-1 noeuds internes. Donc, si la matrice D à plus de n-1 valeurs, il n’existe pas d’arbres ultramétriques pour D
IFT6291, A2006, Sylvie HamelUniversité de Montréal

15Phylogénie
Détection et construction d’arbres ultramétriques
Idée:
i j k
d(i,j)
d(i,k) = d(j,k)
i j k
d(j,k)
d(i,j) = d(i,k)
i j k
d(i,j) = d(i,k) = d(j,k)
Définition: Une matrice symétrique D est ultramétrique si pour tout i,j et k max{ d(i,j), d(i,k), d(j,k) }
n’est pas unique.
Théorème: Une matrice symétrique admet un arbre ultramétrique ssi elle est ultramétrique.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

16Phylogénie
Théorème 2: Si D est une matrice ultramétrique, alors l’arbre ultramétrique est unique
Théorème 3: Si D est ultramétrique, alors l’arbre ultramétrique peut être construit en O(n2)
Conséquence: Si D reflète la distance d’évolution entre les espèces, alors on trouve nécessairement le vrai arbre
Détection et construction d’arbres ultramétriques
IFT6291, A2006, Sylvie HamelUniversité de Montréal

17Phylogénie
Comment obtenir des données ultramétriques
- Distances étiquettant les noeuds des arbres ultramétriques supposés refléter le temps qui s’est écoulé depuis la séparation des deux espèces
- Théorie de l’horloge moléculaire (1960): Pour une protéine donnée, le taux de mutations acceptées par intervalle de temps est constant. Ce taux de mutations varie selon les protéines
- Avec cette théorie, si k mutations acceptés entre une protéine chez l’espèce A et cette même protéine chez l’espèce B, alors on peut estimer à k/2 le nombre de mutations survenues pour chaque espèces depuis la divergence. Cela permet d’obtenir des données ultramétriques
- Si nos données réelles ne sont pas ultramétriques une possibilité est de les modifier de façon minimale pour quelles le deviennent et construire l’arbre par la suite
IFT6291, A2006, Sylvie HamelUniversité de Montréal

18Phylogénie
Arbres et distances additives
- Matrice des distances D (n X n) additive:
- symétrique- la diagonale ne contient que des 0- toutes les autres entrées sont des nombres réels strictement positifs
- Arbre additif :
- contenant au moins n noeuds, en comptant les feuilles
- chaque ligne de D (espèces) correspond à une feuille
- les arcs sont étiquettés de sorte que pour chaque paire de feuille (i,j), le poids total du chemin de i à j est D(i,j)
IFT6291, A2006, Sylvie HamelUniversité de Montréal

19Phylogénie
Arbres et distances additives
Matrice additive Arbre additive pour la matrice (a)
Problème: Étant donnée une matrice additive D, trouver un arbre additif pour cette matrice ou déterminer qu’un tel arbre n’existe pas
Distances additives: contrainte moins forte que les distances ultramétriques. Par contre, les données réelles sont très rarement additive.
Un vaste domaine de recherche: comment effectuer la plus petite “déviation” possible pour que des distances deviennent additives.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

20Phylogénie
Test d’additivité des 4 pointsArbre additif pour D: Distance entre deux feuilles quelconques de l’arbre = somme des poids des arcs du chemin joignant ces deux feuilles
Pour tout ensemble de 4 espèces i,j,k et l, deux des distances D(i,j)+D(k,l), D(i,k)+D(j,l) et D(i,l)+D(j,k) sont égales et supérieures à la troisième
x
y
s
t
sxz
y tz y
sz
x
tz
IFT6291, A2006, Sylvie HamelUniversité de Montréal

21Phylogénie
Méthode de construction d’un arbre additif
Paire de feuilles voisines: Deux feuilles de l’arbre ayant le même père
- Choisir deux objets (espèces) garantis d’être voisins dans un arbre additif
- Supprimer i et j de la liste des objets et rajouter le noeud k correspondant au père commun de i et j. Distance de k à un autre objet quelconque (feuille) m: D(k,m) = 1/2 (D(i,m)+D(j,m)-D(i,j))
- De cette façon, le nombre d’objets à placer est réduit de 1 à chaque étape.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

22Phylogénie
Méthode de construction d’un arbre additif (suite)Comment déterminer, à partir de D, deux feuilles qui sont nécessairement voisines dans l’arbre additif pour D? Il ne suffit pas de choisir une paire d’objets pour lesquels la distance est minimale.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

23PhylogénieIFT6291, A2006, Sylvie HamelUniversité de Montréal

24Phylogénie
Méthode de construction d’un arbre additif
Remarque: Pour une distance additive, il n’existe pas un seul arbre additif
Heuristique: Appliquer l’algorithme plusieurs fois en modifiant l’ordre des objets dans L, ce qui entrainera des choix différents de voisins. Trouver un consensus pour l’ensemble des arbres obtenus
Lorsque la distance n’est pas additive, on peut quand même employer l’algorithme de Neighbour-Joining, mais on a pas de garantie sur la qualité del’arbre obtenu.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

25Phylogénie
Enraciner les arbres
Contrairement à l’algorithme UPGMA, Neighbour-Joining construit un arbre non enraciné. Pour raciner l’arbre, il suffit d’ajouter une espèce très éloignée des autres espèces considérées (outgroup)
Une autre statégie est de considérer comme racine le milieu d’un plus long chemin dans l’arbre (Hypothèse de l’horloge moléculaire).
IFT6291, A2006, Sylvie HamelUniversité de Montréal

Méthodes de parcimonie
26Phylogénie
Entrée: Ensemble de traits (caractères, attributs) qu’un objet peut posséder
La distribution des traits dans les objets permet de déduire des relations d’évolution
Version simplifiée du problème: Phylogénie parfaite avec caractères binaires (présents ou absents)
Soit M une matrice n X m de 0 et de 1, où n est le nombre d’espèces et m le nombre de traits. Cette matrice possède un arbre phylogénétique si
1) T a n feuilles correspondant à chacune des espèces
2) Chaque caractère ou trait est l’étiquette d’une arête
3) Les étiquettes de la racine à la feuille i énumèrent tous les caractères présents dans l’espèce i
IFT6291, A2006, Sylvie HamelUniversité de Montréal

27Phylogénie
Phylogénie parfaiteIci un arbre de phylogénie détermine des relations d’évolution entre les espèces, en terme de branchement et non de temps. Ces relations sont basées sur les hypothèses suivantes:
1) La racine représente un ancêtre commun ne présentant aucun des m traits2) Un caractère acquis n’est jamais perdu. (C’est pourquoi un trait étiquette un seul arc de l’arbre)
Exemple: Évolution des quadrilatère
fermé aligné convexe symétrie 2D
1 0 1 0
1 0 1 1
0 1 0 0
convexe symétrie
fermé aligné
IFT6291, A2006, Sylvie HamelUniversité de Montréal

28Phylogénie
Phylogénie parfaite (suite)Définition: Si M est une matrice booléenne n X m, pour toute colonne j,
Θ j ! {1, . . . ,n}
est défini comme i.e l’ensemble des espèces ayantle caractère j
{i |M(i, j) = 1}
Théorème: M a un arbre de phylogénie parfaite ssi pour tout j, k on a
Θ j!Θk = /0
Θ j !Θk
Θk !Θ j
ouou
IFT6291, A2006, Sylvie HamelUniversité de Montréal

29Phylogénie
Phylogénie parfaite (suite)- Traits considérés
- Les traits comportementaux sont également problématiques. Par exemple, “marcher sur les articulations” est un trait commun aux chimpanzés et aux gorilles mais pas à l’homme. Pourtant, l’arbre maintenant admis pour l’homme, le chimpanzé et le gorilleest:
- Par contre, la considération de traits morphologiques peut être problématique. Sous certaines conditions des traits similaires peuvent apparaître indépendamment.
1) morphologiques (colonne vertébrale, aile...)2) liés aux séquences d’AA ou de nucléotides (présence ou non d’un motif particulier)3) comportementaux (marcher sur les articulations, ...)
http://www.nature.com/embor/journal/v3/n4/fig_tab/embor181_f3.html
IFT6291, A2006, Sylvie HamelUniversité de Montréal

30Phylogénie
Phylogénie parfaite généralisée- Le type de nucléotide ou d’AA à une position donnée d’un alignement constitue également un trait mais ce trait n’est pas binaire. Dans le cas des nucléotides, 4 étatspossibles, dans le cas des acides aminés, 20. (Par contre, on peut se ramener à desétats binaires dans le cas des nucléotides si on considère les purines et pyrimidines)
- Ici, une phylogénie parfaite pour M est un arbre tel que
- Chaque feuille représente une espèce- Chaque arc est étiquetté par une transition particulière de l’état d’un trait i.e par une triplet (t,x,y) indiquant que le caractère t change de l’état x à y.
- Tout chemin de la racine à une feuille p décrit exactement les états des traits pour p- Chaque transition (t,x,y) n’apparaît qu’une fois
- Le problème: Étant donné une matrice M telle que chaque caractère peut avoir au plus r états, déterminer s’il existe une phylogénie parfaite pour M et si oui, en construire une.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

31Phylogénie
Phylogénie maximale
Parcimonie maximale = nombre minimal de mutations
Considérer l’ensemble T de toutes les topologies d’arbres possibles ayant les séquences comme étiquette des feuilles.
Calculer un poids pour chaque arbre T de T
Sélectionner un arbre de T de poids minimal
IFT6291, A2006, Sylvie HamelUniversité de Montréal

32Phylogénie
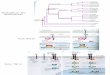
Algorithme de FitchÉtant donné un ensemble de séquences alignées, une topologie d’arbre et une colonne j de l’alignement, on veut trouver le nombre minimal de substitutions associées à cet arbre:
1) Ajouter une racine sur n’importe quel arête
IFT6291, A2006, Sylvie HamelUniversité de Montréal

33Phylogénie
Algorithme de Fitch (suite)2) Passage de bas en haut:
On traverse l’arbre des feuilles à la racine et on assigne à chaque noeud interne n, un ensemble de nucléotides possibles N de la façon suivante:Soit u et v les fils de n et U, V les ensembles de nucléotides correpondant à ces noeuds alors
N =!U !V siU "V = /0U "V sinon
{C,G}{A,C}
A
{C,G,A}
2) Passage de haut en bas:L’arbre est ensuite traversé de haut en bas et on assigne des nucléotides aux noeuds internes selonces règles:
- on assigne à la racine, un nucléotides x de son ensemble (n’importe lequel)- On assigne à un enfant v de parent u le nucléotide
!x si x !Un’importe quel nuclotides deV sinon
C
CA
A
IFT6291, A2006, Sylvie HamelUniversité de Montréal

34Phylogénie
Algorithme de Fitch (suite)C
CA
A
Donc, ici étant donné cette topologie d’arbres et ces données d’alignement, l’algorithme de Fitch nous donne 3 mutations.
Le nombre de mutations ne dépend pas du choix du nucléotide que l’on met à la racine dans la phase de haut en bas:
L’algorithme a une complexité linéaire en la taille de l’arbre.
A G
CA
AG
AA
IFT6291, A2006, Sylvie HamelUniversité de Montréal

35Phylogénie
Énumération de tous les arbres possiblesL’identification de l’arbre de parcimonie maximale requière le calcul du nombre minimal de mutations pour chaque topologie possible d’arbres.
Arbres binaires enracinés de n feuilles:
n feuilles => n-1 noeuds internes => nombre total de noeuds et de feuilles = 2n - 1 => 2n - 2 arcs
Arbres sans racines: 2n -2 noeuds + feuilles et 2n - 3 arcs.
Étant donné un arbre sans racines pour n espèces, on obtient un arbre enraciné en ajoutant une racine au milieu d’un des 2n - 3 arcs
=> Pour chaque arbre non enraciné, il y a 2n-3 arbres racinés.
IFT6291, A2006, Sylvie HamelUniversité de Montréal

36Phylogénie
Énumération de tous les arbres possiblesGénérer les 3 arbres non enracinés pour 4 espèces:
A
Pour chacun de ces arbres, on a 5 arbres avec racines donc: 3 * 5 = 15 arbres racinés pour n= 4 espèces
IFT6291, A2006, Sylvie HamelUniversité de Montréal

37Phylogénie
Énumération de tous les arbres possiblesGénérer les 15 arbres non enracinés pour 5 espèces:
Pour chacun de ces arbres, on a 7 arbres avec racines donc: (3*5)*7= 105 arbres racinés pour n= 5 espèces
On fait la même chose pour les 2 autres arbres non enracinés pour 4 espèces
IFT6291, A2006, Sylvie HamelUniversité de Montréal

8.2!1021
2.2!1020
38Phylogénie
Énumération de tous les arbres possiblesPar récurrence, on a 3*5*...*(2n-5) arbres sans racine de n feuilles
Donc, 3*5*...*(2n-5)*(2n-3) arbres enracinésde n feuilles
34 495 425 arbres racinésn = 10 => 2 027 025 arbres non enracinés
n = 20 => environ arbres non enracinés et
arbres racinés
IFT6291, A2006, Sylvie HamelUniversité de Montréal

39Phylogénie
Stratégie “branch and bound” *
*Hendy, M.D. et Peeny, D., Branch an bound algorithms to determine minimal evolutionary trees, Mathematical Biosciences, 60, pp.133-142, 1982.
Comme on vient de la voir, le nombre de topologies d’arbres croît très rapidementpar rapport au nombre d’espèces considérés.
“Branch and bound” est une statégie exacte permettant de trouver l’arbre de phylogénie maximal pour 20 espèces ou plus.
Méthode:
1) Obtenir une borne supérieure du nombres de mutations (par Neighbor Joingning, par exemple)
2) Construire toutes les topologies d’arbres en ajoutant les espèces une à une
3) Si, pour une topologie donnée, le nombre de mutations est plus grand que la borne supérieure, alors arrêter d’ajouter des espèces à cette topologie
IFT6291, A2006, Sylvie HamelUniversité de Montréal

40Phylogénie
Stratégie “branch and bound”
IFT6291, A2006, Sylvie HamelUniversité de Montréal

41Phylogénie
Inconsistance du modèle de parcimonieConsistance d’une méthode d’estimation: Capacité de converger vers une bonne valeur (ici un vrai arbre de phylogénie) avec l’augmentation des données
Supposons que nous savons que l’arbre de phylogénie de 4 séquences a,b,c et d est lesuivant:
Le taux d’évolution de c et d est beaucoup plus élevé que le taux d’évolution de a et b
Ici, les espèces a et c sont d’une côté et les espèces b et d de l’autre pourtant le nombre de mutations entre a et b est beaucoup moins élevé que le nombre entre a et c ....
Ce phénomène est appelé l’attraction des longues banches
IFT6291, A2006, Sylvie HamelUniversité de Montréal

42Phylogénie
Maximum de vraisemblance
IFT6291, A2006, Sylvie HamelUniversité de Montréal