Embed Size (px)
Citation preview

Promotor: Prof. Dr. Y. Van de Peer
Begeleidster: Tineke Casneuf
Faculteit WetenschappenVakgroep Moleculaire Genetica
Departement Plant Systems Biology – VIB
Invloed van de genomische context op genexpressie vangedupliceerde genen.
James Cauwelier
Scriptie voorgelegd tot het behalen van de graad van licentiaat/master in de biotechnologie
Academiejaar 2005 - 2006


Promoter: Prof. Dr. Y. Van de Peer
Begeleidster: Tineke Casneuf
Faculteit wetenschappenVakgroep Moleculaire Genetica
Departement Plant Systems Biology – VIB
Invloed van de genomische context op genexpressie vangedupliceerde genen.
James Cauwelier
Scriptie voorgelegd tot het behalen van de graad van licentiaat/master in de biotechnologie
Academiejaar 2005 - 2006

Dankwoord
Bij het begin van deze thesis wil ik graag alle personen bedanken die me hebben geholpen
met de realisatie ervan.
Eerst wil ik Prof. Dr. Y. Van de Peer bedanken voor het opnemen van het promotorschap.
Mijn begeleidster, Tineke Casneuf, wil ik bedanken voor de begeleiding en het beantwoorden
van al mijn vragen gedurende het afgelopen jaar. Maar ook Stefanie De Bodt heeft me goed
geholpen in het promoteronderzoek, waarvoor ik haar wil bedanken.
Twee mensen wil ik hier speciaal vermelden. Mijn vriendin, die mij, ondanks de vele
eenzame uren die ik samen met de computer doorbracht, nog steeds even graag ziet en
steeds klaar staat bij problemen. Johan Vandekerkhove, mijn mentor en vriend, verdient hier
een speciale vermelding. Zonder hem zou mijn leven een héél andere wending genomen
hebben en ik bedank hem voor de begeleiding die ik van hem ontving.
Maar ook mijn grootouders wil ik hier niet vergeten. Zij hebben de rol van mijn ouders graag
op zich genomen en ik ben hen daar dankbaar voor. Een speciale plaats in mijn hart is voor
hen gereserveerd.
En last but not least, wil ik al mijn vrienden bedanken voor het tonen van interesse in mijn
werk, zelfs al meenden ze het niet.

Inhoudstafel
Dankwoord
Inhoudstafel
1 Doelstelling van de thesis ............................................................................................1
2 Literatuurstudie.............................................................................................................3
2.1 Alignering van sequenties .......................................................................................3
2.1.1 Inleiding...............................................................................................................................3
2.1.2 Substitutiematrices .............................................................................................................3
2.1.3 Sequentie-alignering...........................................................................................................4
2.2 Genduplicatie..........................................................................................................5
2.2.1 Mechanismen......................................................................................................................5
2.2.2 Identificatie van grootschalige genduplicaties ....................................................................8
2.2.3 Datering van genduplicatie met Ks....................................................................................11
2.3 Evolutie na duplicatie ............................................................................................12
2.3.1 Behoud van het gedupliceerde genetisch materiaal.........................................................13
2.3.2 Divergentie van gedupliceerde genen ..............................................................................13
2.4 Modelorganisme: Arabidopsis thaliana..................................................................15
2.5 Microarrays ...........................................................................................................17
2.5.1 Inleiding.............................................................................................................................17
2.5.2 Types en productie ...........................................................................................................18
2.5.3 Het meten van de genexpressie .......................................................................................20
2.5.4 Normalisatie van microarray data .....................................................................................21
3 Materiaal en methode..................................................................................................24

3.1 Algemeen overzicht ..............................................................................................24
3.2 Algemene technieken ...........................................................................................25
3.2.1 Perl....................................................................................................................................25
3.2.2 R en Bioconductor ............................................................................................................25
3.3 Detectie en klassificatie van ankerpunten .............................................................25
3.3.1 BLAST...............................................................................................................................25
3.3.2 Methode van Rost.............................................................................................................26
3.3.3 i-ADHoRe..........................................................................................................................26
3.3.4 i-ADHoRe2genedraw_real_TE.pl .....................................................................................28
3.3.5 rearrangement_search.pl..................................................................................................29
3.3.6 Onderverdelen in type herschikkingen .............................................................................32
3.3.7 Berekenen van de correlatie van genexpressie ...............................................................34
3.4 Promoteranalyse van gedupliceerde genen ..........................................................36
4 Resultaten....................................................................................................................38
4.1 Grootte van de ankerpunt groepen........................................................................38
4.1.1 Inleiding.............................................................................................................................38
4.1.2 Overzicht van de ankerpunt groepen ...............................................................................39
4.2 Correlatie van genexpressies................................................................................41
4.2.1 Analyse van alle herschikte en alle niet herschikte ankerpuntgenen...............................41
4.2.2 Analyse van herschikte en niet herschikte ankerpuntgenen ............................................42
4.2.3 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en herschikte
ankerpunten (eiwitcoderende herschikking)...................................................................................43
4.2.4 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en herschikte
ankerpunten (RNA coderende herschikking). ................................................................................44
4.2.5 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en herschikte
ankerpunten (transposon coderende herschikking). ......................................................................45
4.2.6 Analyse van herschikte ankerpunten (RNA coderende herschikking) en herschikte
ankerpunten (eiwitcoderende herschikking)...................................................................................45

4.2.7 Analyse van herschikte ankerpunten (TP coderende herschikking) en niet herschikte
ankerpunten (zonder pseudogenen). .............................................................................................46
4.2.8 Analyse van herschikte ankerpunten (TP coderende herschikking) en herschikte
ankerpunten (RNA coderende herschikking). ................................................................................47
4.2.9 Analyse van herschikte ankerpunten die herschikt werden door deletie in vergelijking tot
die die herschikt werden door insertie............................................................................................48
4.3 Promoter-onderzoek. ............................................................................................50
4.3.1 Inleiding.............................................................................................................................50
4.3.2 Vergelijken van de aligneerbaarheid van het upstream gebied. ......................................50
4.3.3 Verband tussen de aligneerbaarheid van de promoterregio’s en de leeftijd van duplicatie52
4.3.4 De aligneerbaarheid van de promoterregio’s in functie van de correlatie van
genexpressie. .................................................................................................................................53
5 Discussie .....................................................................................................................56
5.1 Correlatie van genexpressie..................................................................................56
5.2 Promoter onderzoek .............................................................................................57
5.3 Besluit ...................................................................................................................58
6 Bijlagen ........................................................................................................................59
6.1 Bijlage A: microarray dataset ................................................................................59
6.2 Bijlage B: Lijst met afkortingen ..............................................................................63
6.3 Bijlage C: CD-ROM...............................................................................................64
7 Referenties...................................................................................................................65

1
1 Doelstelling van de thesis
Het erfelijk materiaal van ieder organisme is vervat in zijn DNA. Dit DNA bestaat uit
coderende regio’s (de genen) en niet coderende regio’s die een structurele of
regulerende rol kunnen vervullen. Ieder gen codeert over het algemeen voor een eiwit
en komt slechts éénmaal voor in een haploïd genoom, maar dit betekent niet dat ieder
gen uniek is. In het verleden zijn sommige genen meerdere malen gekopieerd geweest,
waardoor genfamilies onderscheiden kunnen worden die bestaan uit sterk op elkaar
lijkende gensequenties. Na duplicatie van één of meerdere genen kan mutatie ervoor
zorgen dat de sequenties van de genen gewijzigd worden, waardoor ze na verloop van
tijd minder op elkaar lijken en zelfs verschillende functies kunnen gaan uitvoeren. Enkel
die wijzigingen in sequentie die een voordeel betekenen voor de overleving en
algemene fitness van het organisme worden behouden in een populatie van dat
organisme.
Onderzoek heeft aangetoond dat gen- en zelfs grootschalige genoomduplicaties
frequent voorkomen, in het bijzonder bij planten (Adams KL and Wendel JF, 2005).
Deze verdubbeling van genetisch materiaal is een belangrijke factor in het genereren
van nieuwe functies door het leveren van extra ruw genetisch materiaal waarop selectie
kan inwerken (Ohno S., 1970). Het bij een duplicatie gevormd genetisch materiaal kan
ook voordelen bieden aan een organisme doordat deze zich beter kan aanpassen aan
veranderende omgevingsfactoren. Verder zijn gedupliceerde genen ook beter bestand
tegen mutatie (Gu Z. et al., 2003). Duplicatie, gevolgd door divergentie, kan
bijvoorbeeld aanleiding geven tot het ontstaan van nieuwe soorten. Hoe dit alles in zijn
werk gaat wordt momenteel nog onvoldoende begrepen. Divergentie van genexpressie
na herschikkingen1 van het gedupliceerde materiaal kan hier een rol in spelen maar of
dit zo is, werd nog niet onderzocht. Recent werd aangetoond dat de wijze van duplicatie
(groot- of kleinschalig) een rol speelt bij de divergentie van genexpressie (Casneuf et al.,
2006). Grootschalig gedupliceerde gensegmenten vertonen een hogere correlatie van
genexpressie dan genen die op kleinere schaal gedupliceerd werden en verspreid
voorkomen in het genoom (Casneuf et al., 2006). In deze thesis wordt verder ingegaan
op deze resultaten en nagegaan of herschikkingen van grootschalig gedupliceerde
1 Met herschikkingen bedoelt men het verwijderen (deletie)van DNA, invoegen van DNA (insertie),
omdraaien van DNA (inversie) waarbij de oriëntatie van de genen in dat stuk gewijzigd worden of
translocatie waarbij DNA verplaatst wordt naar een ander gebied van het genoom.

2
Fig. 1.1: Verstoring vaneen promoterregio doordeletie van hetstroomopwaarts gelegengen. Ieder zwart blokstelt een gen voor, terwijlde promoterregio schuingearceerd is.
regio’s in het DNA van de modelplant Arabidopsis thaliana verantwoordelijk zijn voor de
divergentie van genexpressie van homologe genen. Initieel worden gedupliceerde
regio’s opgespoord en onderverdeeld in lijsten van herschikte en niet herschikte
gedupliceerde genpaartjes. Vervolgens wordt de genexpressie nagegaan. Aan de hand
van deze sets van genpaartjes en hun genexpressie kan nagegaan worden of
herschikkingen in het genoom verantwoordelijk zijn voor de divergentie van
genexpressie van homologe genen. Figuur 1.1 toont twee homologe gebieden die
ontstaan zijn door een duplicatie en die oorspronkelijk elk twee genen bevatten (zwarte
balkjes). Voor elk gen ligt een promoterregio die instaat voor de controle van initiatie van
transcriptie (grijze balkjes). Deletie (segment B, figuur 1.1) of insertie (niet getoond) van
een stroomopwaarts gelegen gen kan de promoter verstoren. Aangezien dit gebied de
initiatie van transcriptie controleert, kunnen wijzigingen in dit gebied eventueel de
oorzaak zijn van divergentie van genexpressie.
Een tweede luik van deze thesis is het nader onderzoeken van de promoterregio’s van
enkele gedupliceerde genen om meer inzicht te verkrijgen in de wijze waarop
divergentie optreedt als gevolg van de verstoring van de promoterregio.

3
2 Literatuurstudie
2.1 Alignering van sequenties
2.1.1 Inleiding
Het aligneren van sequenties is het proces waarin twee sequenties tegenover elkaar
geplaatst worden, waarbij de overeenkomsten alsook de verschillen tussen beide
opgespoord worden. Dit alignement vertelt in welke mate twee sequenties gelijkaardig zijn.
Dit gebeurt door het paren van individuele karakters van de te aligneren sequenties, waarbij
het aantal onderbrekingen (“gaps”) en niet gealigneerde karakters zo klein mogelijk
gehouden wordt, zoals in het voorbeeld hieronder (Van De Peer Y., 2005).
Sequentie 1 A G C T T G - - C C T C G C A …Sequentie 2 A G – T T G T T C C T G G C A …
Omdat het praktisch niet haalbaar is om alle mogelijke alignementen van twee of meer
sequenties te overlopen, worden algoritmen ingeschakeld om een “beste alignement” te
selecteren. Voor een alignement van twee sequenties met een lengte van 300 nucleotiden,
zouden anders 10179 mogelijkheden overlopen moeten worden wat teveel computerkracht
vereist (Van De Peer Y., 2005).
In bovenstaand voorbeeld worden nucleotidensequenties vergeleken met een alfabet met 4
karakters (A, T, G en C), waardoor ieder karakter een kans van ¼ heeft om op gelijk welke
positie enkel op basis van toeval voor te komen. Daarom kan beter van een rijker alfabet
gebruik gemaakt worden omdat de kans dat een karakter per toeval voorkomt kleiner is
naarmate het alfabet meer uitgebreid is (Van De Peer Y., 2005). Bij het aligneren van
proteïne coderende sequenties wordt beter gebruik gemaakt van de AZ-sequentie dat een
alfabet gebruikt met 20 karakters, waardoor ieder karakter per toeval gemiddeld slechts 1/20
maal voorkomt. Voor het vergelijken van niet coderende sequenties, zoals promoterregio’s,
moet steeds de nucleotide-sequentie gebruikt worden, aangezien een dergelijke sequentie
niet tot een aminozuursequentie vertaald wordt.
2.1.2 Substitutiematrices
Twee sequenties die afkomstig zijn van dezelfde gemeenschappelijke voorouder divergeren
door het optreden van mutaties in de vorm van inserties, deleties en substituties. Voor
aminozuursequenties worden bepaalde substituties beter getolereerd dan andere omdat de

4
eigenschappen van het nieuwe aminozuur (bv. grootte, lading, hydrofobiciteit) gelijkaardig
zijn aan dat van het oude aminozuur . De functie van het genproduct wordt op die manier
minder gemakkelijk gewijzigd. Een substitutiematrix wordt samengesteld op basis van een
alignement van gekende aligneerbare sequenties waarin het geobserveerde aantal van een
bepaald aminozuurpaar vergeleken wordt met het aantal dat men op basis van toeval zou
verwachten (Henikoff and Henikoff, 1992). Men bekomt een matrix die waarden bevat voor
de aligneerbaarheid van aminozuurpaartjes in aminozuursequenties met een gelijkaardige
evolutionaire afstand en context. De bekomen aligneerbaarheid van aminozuurpaartjes
wordt dan verder gebruikt voor het opstellen van een alignement van sequentie (Henikoff
and Henikoff, 1992), waardoor eerder de verandering in eigenschappen van twee sequenties
vergeleken wordt in plaats van hun sequentie.
2.1.3 Sequentie-alignering
Alignementen kunnen berekend worden via verschillende algoritmen, zoals het algoritme van
(Needleman and Wunsch, 1970) en dat van (Smith and Waterman, 1981). Beide zijn
gebaseerd op het bouwen van een score-matrix, waarin sequentie 1 de X-as en sequentie 2
de Y-as voorstelt en de vakjes van de matrix een score bevatten voor elke aminozuurparing
in de matrix. De scores zelf worden berekend aan de hand van toegekende strafpunten bij
het openen van een leegte (“gap”) in het alignement of het verlengen van zo’n “gap”, samen
met punten voor het aligneren van een aminozuurpaar, berekend aan de hand van een
substitutiematrix. In de loop van het aligneringsproces worden op die manier leegtes
geïntroduceerd, om de uiteindelijk bekomen score zo hoog mogelijk te houden.

5
2.2 Genduplicatie
2.2.1 Mechanismen
Genduplicatie is het verdubbelen van een gen en kan op verschillende manieren tot stand
komen. Het volgende overzicht gaat in op enkele van die mechanismen.
Autopolyploïdie
Autopolyploïdie is de vorming van een
verdubbeld genoom als het gevolg van een fout
in de ontwikkeling van de gameten (Van de Peer
Y. and Meyer A., 2005). Dergelijke gameten met
een dubbel genoom bezitten steeds een even
aantal homologe chromosomen die bivalenten
kunnen vormen tijdens de meiose. Zo kunnen
nog steeds fertiele gameten gevormd worden na
een autopolyploïdie. Het organisme kan nog
steeds reproduceren, maar is niet meer in staat
om te kruisen met de oorspronkelijke diploïde
organismen. In het geval dat een dergelijke
kruissing zou optreden, worden gameten
gevormd met een oneven aantal homologe
chromosomen die niet in staat zijn om bivalenten
te vormen in de meiose en het organisme zou
zich niet kunnen reproduceren.
Figuur 2.1: In de profase I van de meiosewordt het aantal homologe chromosomengehalveerd. Deze homologen worden in deprofase II verdeeld over de gevormdegameten (links helft van de figuur). Als dooreen fout in de meiose de homologen nietgehalveerd worden in profase I, dan bekomtmen gameten met een dubbel aantalhomologen dan normaal (rechtse helft vande figuur).

6
Figuur 2.1 illustreert autopolyploïdie. Het toont een onderdeel van de meiose waarbij de
gepaarde homologe chromosomen (rood en zwart) na profase I in afzonderlijke nucleï
terecht komen (linker helft van de figuur) om daarna verdeeld te worden in 2 nieuwe nucleï
na profase II. Het resultaat is dat 4 nieuwe haploïde cellen gevormd worden. Wanneer de
meiose foutief verloopt (rechter helft van de figuur) en het genetisch materiaal na profase I
niet verdeeld wordt over 2 nucleï, zal de meiose 2 diploïde cellen produceren in plaats van 4
haploïde. De bevruchting met 2 diploïde geslachtscellen zal aanleiding geven tot een fertiel
tetraploïd organisme met verdubbeld genomisch materiaal.
Allopolyploïdie
Allopolyploïdie treedt op bij bevruchting tussen 2 organismen van een verschillende soort,
maar enkel wanneer na die bevruchting een verdubbeling optreedt van het genetisch
materiaal. In dat geval kunnen de homologe chromosomen nog correct verdeeld worden
over de gameten en aanleiding geven tot een nageslacht (Van de Peer Y. and Meyer A.,
2005).
Polyploidie is een belangrijk fenomeen bij de evolutie van planten (Adams and Wendel,
2005). Men neemt aan dat de meeste oude polyploïden op deze manier ontstaan zijn in
plaats van door autopolyploïdie (Spring, 2003).
Aneuploïdie
Aneuploïdie is een toestand waarbij meer of minder chromosomen aanwezig zijn dan de
normale set van chromosomen (Van de Peer Y. and Meyer A., 2005). Het chromosoom-
aantal is dan niet langer een exact meervoud van de haploïde set chromosomen, in
tegenstelling tot bij allopolyploïdie en autopolyploïdie. Een voorbeeld hiervan is het
“syndroom van Down”, dat veroorzaakt wordt door een trisomie van chromosoom 21 bij de
mens.
Segmentale duplicatie
Een segmentale duplicatie is de duplicatie van grote stukken DNA en is het gevolg van een
fout in het replicatieproces (Koszul et al., 2004).

7
Tandem duplicatie
Tandem duplicatie ontstaat door ongelijke crossing-over tijdens de meiose waarbij een stuk
DNA uitgewisseld wordt tussen twee homologe chromosomen (Van de Peer Y. and Meyer
A., 2005). Eén van de homologen zal een extra DNA segment bijkrijgen en het andere
homoloog zal datzelfde DNA segment verliezen. Omdat deze fout in overkruissing (“crossing
over”) locaal gebeurdt, blijven de duplicaten naast elkaar gelocaliseerd op het chromosoom
(Van de Peer Y. and Meyer A., 2005).
Retropositie
Bij retropositie wordt een genduplicaat gevormd op een nieuwe positie in het genoom.
Hiertoe wordt het gen op zijn originele plaats overgeschreven naar RNA, dat met behulp van
een reverse transcriptase aanleiding kan geven tot een DNA kopij. Een reverse
transcriptase katalyseert de polymerisatie van DNA vertrekkende van een RNA , dus tegen
de normale informatiestroom in (DNA RNA) . Omdat de RNA template enkel de
coderende sequentie van het gen bevat zonder de regulerende sequenties van de promoter,
wordt enkel het coderend gebied van het gen gedupliceerd. Het duplicaat wordt dus
afhankelijk van reeds aanwezige regulerende sequenties op zijn nieuwe positie in het
genoom (Long et al., 2003). Indien het duplicaat geen promoter meer heeft, kan het niet
worden afgeschreven en zal het eiwitproduct niet meer gevormd worden. In het geval een
gen niet langer aanleiding geeft tot een functioneel product, noemt men dit een pseudogen
en kan het verloren gaan. Wanneer een pseudogen noodzakelijk is voor de structuur van
het DNA in die regio kan de aanwezigheid ervan toch getolereerd worden. Een pseudogen
wordt dus niet zomaar verwijderd op basis van de afwezigheid van zijn product.
Laterale gen-transfer
Bij prokaryoten worden genen getransfereerd tussen organismen van dezelfde generatie
(laterale of horizontale gentransfer). Veelal leidt dit tot de uitwisseling van homologe genen
zonder dat die daarbij gedupliceerd worden, maar soms kan de transfer van nieuwe genen
zorgen voor het ontstaan van nieuwe fenotypes (Long et al., 2003). Ook bij eukaryoten doet
zich laterale gentransfer voor, maar in dat geval gaat het om organelgenen die volgens de
endosymbiont-hypothese afkomstig zijn van prokaryoten en doet de gentransfer zich voor
binnenin de eukaryote cel en niet tussen de cellen onderling.

8
2.2.2 Identificatie van grootschalige genduplicaties
Grootschalige gen- en volledige genoomduplicaties kunnen gedetecteerd worden
door het opsporen van gedupliceerde gebieden met geconserveerde gen-inhoud en
volgorde (= “colineariteit”) (Van de Peer Y. and Meyer A., 2005).
Eerst worden met BLASTp en de methode van Rost de gedupliceerde genen
opgespoord in het genoom en vervolgens wordt nagegaan welke gedupliceerde
genpaartjes samen gedupliceerd werden. Hiervoor wordt een “gene homology
matrix” (GHM) opgesteld waarbij twee segmenten ten opzichte van elkaar uitgezet
worden en gezocht wordt naar diagonale elementen die de samen gedupliceerde
genen aanduiden (figuur 2.2). Verder wordt onderzocht of de geobserveerde
diagonaliteit het gevolg kan zijn van louter toeval door het uitvoeren van een
permutatietest.
Wanneer een gedupliceerd blok geïdentificeerd wordt dat ontstaan is door eenzelfde
duplicatiegebeurtenis, worden de homologe genen van beide sequenties
“ankerpunten” genoemd.
2.2.2.1 i-ADHoRe
i-ADHoRe (Automatic Detection of Homologous Regions) is een tool om in groep
gedupliceerde genen op te sporen aan de hand van een paarsgewijze vergelijking
van genomische segmenten (Simillion et al., 2004). Hiervoor worden twee lijsten met
alle proteïne-coderende genen vergeleken en ze worden gerangschikt in de volgorde
die ze innemen op de te onderzoeken segmenten. Met BLASTp en de methode van
Rost worden eerst de homologe genparen geïdentificeerd en het resultaat hiervan
wordt opgeslagen in een (m x n)-matrix, waarbij m en n de lengte geven van de
gebruikte genlijsten. De bekomen matrix wordt de “gene homology matrix” (GHM)
genoemd omdat het alle gevonden homologen bevat (figuur 2.2). Ieder element in de
matrix stelt een gedetecteerd homoloog genpaar voor en kan positief of negatief zijn
afhankelijk van het feit of beide genen van het paar wel of niet dezelfde oriëntatie
bezitten op het genoom. Eenmaal deze matrix is samengesteld, kunnen
blokduplicaties geïdentificeerd worden als diagonale reeksen van ankerpunten, terwijl
de tandem repeats aanwezig zijn in horizontale of verticale reeksen. De tandem
repeats worden eerst herschikt tot één enkel gen, waarna clusters van diagonale
series van ankerpunten kunnen gedetecteerd worden die de paraloge gebieden
aangeven. Bij deze detectie van paraloge gebieden wordt gebruikt gemaakt van een
“maximum gap size” (G) en een “quality parameter” (Q) om te beslissen of clusters

van ankerpunten inderdaad een blokduplicatie vormen. Met een permutatietest wordt
nagegaan of de gevonden diagonaliteit significant en dus niet door toeval ontstaan is.
2.2.2
tandem duplicatie
blok duplicatie
inversie
Figuur 2.2: Een hypothetisch voorbeeld van een “Gene Homology Matrix” (GHM), waarbijde homologie wordt nagegaan tussen het genomisch segment op de x-as (segment nr. 1)en dat op de y-as (segment nr. 2). De grijze cellen stellen de ankerpunten voor, met anderewoorden de plaatsen waar het gen op de x-as het homoloog is van het gen op de y-as. Dediagonale gebieden van de matrix, inversies en tandem duplicaties zijn duidelijk zichtbaar.
(A) De originele organisatie van alle genen in hun genomische context, waarbij tandemduplicaties en inversies nog duidelijk zichtbaar zijn.
(B) Dezelfde GHM, maar na “tandem remapping” en het verwijderen van niet relevante datapunten (diegene die niet het gevolg zijn van een grootschalige duplicatie) mbv het ADHoRealgoritme. Ook geïnverteerde gebieden worden hermapt zodat diagonaliteit beterdetecteerbaar wordt.
(Gregory T., 2005; Van de Peer Y. and Meyer A., 2005) (bewerkt)
9
.2 Hidden en ghost duplications
Met de hierboven beschreven aanpak kunnen al heel wat duplicaties gevonden
worden, maar toch kan men de gevoeligheid van bovenstaande benadering nog
verbeteren (Simillion et al., 2002).
Wanneer onvoldoende gedupliceerde genen geclusterd kunnen worden over een
bepaald gebied, worden beide genomische regio’s niet als duplicaten beschouwd. Dit

betekent niet noodzakelijk dat deze geen duplicaat zijn van elkaar, want misschien
zijn ze zodanig herschikt dat de duplicatiegebeurtenis niet meer duidelijk
waarneembaar is. Om dergelijke duplicaties toch nog te herkennen kan het gebruik
van een derde segment uitkomst bieden.
Dit wordt voorgesteld in figuur 2.3 waarbij in het eerste voorbeeld een gewone
duplicatie voorgesteld wordt. In figuur 2.3.B kan de homologie tussen twee
segmenten (1 en 3) gedetecteerd worden met de hulp van een derde segment
(middelste segment) in hetzelfde organisme (organisme 1) en men noemt dit een
verborgen duplicatie (“hidden duplication”). In figuur 2.3.C wordt de homologie
tussen het 1ste en 3de segment op een gelijkaardige manier gedetecteerd, met dat
verschil dat het bijkomende segment (middelste segment) afkomstig is van een ander
organisme (organisme 2).
2.2.2
D
g
a
t
d
b
Figuur 2.3: Schematisch voorstelling van niet verborgen, verborgen (“hiddenduplication”) en spook duplicaties (“ghost duplication”)
(Van de Peer Y. and Meyer A., 2005) (bewerkt)
10
.3 Genomische profielen
oor rekening te houden met verborgen en spook duplicaties kan men de
evoeligheid voor de detectie van gedupliceerde segmenten verhogen (Simillion et
l., 2004). In gevallen van extreem verlies en/of herschikking van genen kan het
oevoegen van verborgen en spook duplicaties onvoldoende blijken om bepaalde
uplicaties te detecteren, maar kan het gebruik van een genomisch profiel uitkomst
ieden (Simillion et al., 2004).

11
Men begint, zoals voordien, met het opstellen van een GHM, waarbij een eerste
genomisch segment vergeleken wordt met een tweede segment. Als men besluit dat
beide segmenten homoloog zijn, dan worden deze samengevoegd tot een groep van
segmenten, een profiel. Voor het onderzoeken van een extra genomisch segment op
homologie met de segmenten in het profiel, worden de segmenten van het profiel
samen uitgezet op de x-as van de GHM en wordt het nieuwe segment uitgezet op de
y-as. De gevoeligheid van de detectie kan zo merkbaar verbeterd worden, want in
het profiel worden meer homologe genen gevonden dan met de standaard
benadering op basis van slechts één genomisch segment in de x-as (zie figuur 2.4).
Deze aanpak zorgt ervoor dat uitvoerig herschikte genomische segmenten toch nog
als homoloog gebied kunnen herkend worden. De bekomen groep van homologe,
gedupliceerde segmenten (de segmenten van het profiel) die ontstaan zijn door één
of meerdere duplicaties noemt men een multiplicon. Het multiplicatie niveau duidt op
het aantal colineaire genomische segmenten die in het multiplicon aanwezig zijn.
2.2.3 Datering van genduplicatie met Ks
De genetische code is degeneratief, wat wil zeggen dat verschillende codons voor hetzelfde
aminozuur coderen. Substituties van een nucleotide op de derde positie van een codon
resulteren veelal niet in een aminozuurverandering en worden synonieme substituties
Figuur 2.4: Verduidelijking vanhet gebruik van een profiel bijdetectie van gedupliceerdegensegmenten.
Het profiel bestaat hier uitgenomische segmenten A en B.Deze worden getest opcolineariteit met segment C.
Vergelijken van A met C levert 3homologe genparen (blauw).Vergelijken van B met C levert 2homologe genparen (rood).Vergelijken van C met het profiel(= A + B) levert 4 homologegenparen.
Vergelijken met een profiel levertsteeds minimaal even veelgenparen als in gelijk welk andergenomisch segment in het alsprofiel gebruikte multiplicon.
(Van de Peer and Meyer,2005)(herwerkt)

12
genoemd. Verondersteld wordt dat dergelijke substituties continu gebeuren en hun aantal
wordt als maat gebruikt voor de ouderdom van duplicatiegebeurtenissen (Hurst, 2002).
Hierbij moet opgemerkt worden dat in de realiteit toch selectie kan optreden op synonieme
substituties. Een voorbeeld hiervan volgt uit het feit dat codongebruik specifiek is voor een
organisme en het organisme hieraan aangepast is met een eigen specifieke concentratie van
tRNA’s. Daardoor kan de concentratie van die tRNA’s limiterend werken als het gebruik van
het corresponderend codon door synonieme substitutie verhoogd wordt.
De verstreken tijd sinds de duplicatiegebeurtenis, de “tijd van divergentie” (T) kan berekend
worden door T = Ks /2λ waarbij λ de gemiddelde snelheid van synonieme substitutie is en T
uitgedrukt wordt in “miljoen jaar geleden” (Van de Peer Y. and Meyer A., 2005).
2.3 Evolutie na duplicatie
Duplicatie van een gen resulteert in twee kopieën van datzelfde gen, waardoor de informatie-
inhoud van het gen nu dubbel aanwezig is (“redundantie”). Duplicatie zorgt op die manier
voor een toename van het ruw genetisch materiaal, dat na duplicatie gemakkelijker mutaties
accumuleert door het bufferende effect van de extra genkopij op eventuele nadelige
mutaties. Beide gensequenties evolueren na duplicatie waardoor ze divergeren in zowel
genexpressie als functie (Taylor and Raes, 2005). Het ontstaan van mutaties speelt een
grote rol in deze divergentie (Haldane, 1933).
Door mutatie ontstaan voordelige en nadelige allelen. Evolutie selecteert de voordelige
allelen waardoor deze verspreid worden in de populatie. Een nadelig allel wordt niet
verspreid in de populatie, maar ge-non-functionaliseerd (het verliest zijn functie) en verandert
in een pseudogen (Taylor J. S. and Raes J., 2005). Dit pseudogen kan vervolgens verwijderd
worden op voorwaarde dat het geen andere functie vervult (bv. een structurele functie). In
zeldzame gevallen kan het door mutatie ontstane allel ook voordelig zijn en behouden
worden. Het belang van grootschalige duplicaties voor evolutie werd in 1970 opgemerkt
door Ohno in zijn boek “Evolution by Gene Duplication”2. Volgens Ohno zou het niet
mogelijk geweest zijn om enkel met natuurlijk selectie de huidige diversiteit van organismen
te creëren vertrekkende van een bacterie. Uit die bacterie zouden dan enkel verscheidene
vormen van andere bacteriën kunnen ontstaan, terwijl voor de overstap naar meercelligen
meer ingrijpende veranderingen noodzakelijk zijn geweest, zoals duplicatie.
2 Ohno was niet de eerste om dit op te merken, zie Taylor & Raes (2005)

13
2.3.1 Behoud van het gedupliceerde genetisch materiaal
Het eventueel ontstaan van nieuwe voordelige genfuncties uit gedupliceerde genen is een
effect op lange termijn, maar een allel moet ook op korte termijn voordelig zijn voor het
organisme want anders kan het verloren gaan voordat voldoende mutaties kunnen
accumuleren (Van de Peer and Meyer, 2005).
Ten eerste kan het extra genetisch materiaal optreden als buffer tegen het nadelig
schommelen van omgevingsfactoren. Het organisme kan door zijn groter aantal
gedupliceerde genen beter omgaan met stress en veranderingen in zijn milieu, maar de
bijkomende gedupliceerde genen kunnen ook de nadelige effecten van mutaties teniet doen.
Wanneer zich een mutatie in een gen voordoet, is door duplicatie nog een andere correcte
kopij aanwezig die de functie kan uitvoeren waardoor het effect van de mutatie minder
ingrijpend is. Null mutaties zijn hiervan een voorbeeld waarbij het duplicaat de functie kan
overnemen (Gu et al., 2003).
2.3.2 Divergentie van gedupliceerde genen
Gedupliceerde genen accumuleren mutaties waardoor hun sequenties divergeren(Taylor J.
S. and Raes J., 2005). Bij divergentie zal de functie van het genproduct en/of het
expressiepatroon van het gen wijzigingen ondergaan. Bij divergentie van genexpressie zal
het genproduct onder andere omstandigheden of hoeveelheden geëxpresseerd worden,
zoals in een ander weefsel, of enkel onder stress.
2.3.2.1 Non-functionalisatie
De functie van het overgrote deel van gedupliceerde genen gaat verloren door het
proces van non-functionalisatie (Taylor J. S. and Raes J., 2005). Hierbij wordt het
functionele genproduct niet langer gevormd, bijvoorbeeld omdat door mutatie een
nieuw stopcodon is ontstaan in de coderende sequentie (figuur 2.7).

14
2.3.2.2 Neo-functionalisatie
In zeldzame gevallen kunnen door mutaties ook nieuwe functies ontstaan (Prince and
Pickett, 2002; Taylor and Raes, 2005), die voordien niet aanwezig waren, zoals een
nieuwe transcriptie-factor bindingsplaats in de regulatorische sequentie of een
wijziging in de actieve plaats van het genproduct waardoor dit bijvoorbeeld een ander
substraat zal accepteren (figuur 2.7).
2.3.2.3 Sub-functionalisatie
Als genen enkel geselecteerd worden op basis van aanwezigheid van voordelige of
nadelige mutaties, dan verwacht men dat de meeste gedupliceerde genen snel
verdwijnen als er niet snel nieuwe voordelige functies gevormd worden. Toch ligt het
aantal niet verdwenen gedupliceerde genen nog vrij hoog (Prince and Pickett, 2002),
wat betekent dat bepaalde mechanismen zorgen voor het behoud van gedupliceerde
genen zodat deze niet door non-functionalisatie verloren gaan.
Een mogelijke verklaring wordt geboden door het sub-functionalisatiemodel waarbij
aparte onderdelen van de gedupliceerde genen afzonderlijk mutaties accumuleren
terwijl hun functies elkaar aanvullen en samen de functies van het ancestrale gen
uitvoeren (Force et al., 1999; Taylor J. S. and Raes J., 2005). Dit mechanisme
Figuur 2.7: Na verloop van tijd accumuleren gedupliceerde genen mutaties waardoorde expressie en functie van de gedupliceerde genen divergeren. (“R” duidt op eenwijziging in een regulatorische sequentie, terwijl een wijziging van de coderendesequentie aangeduid wordt met een “C”)
(Taylor J. S. and Raes J., 2005)

15
baseert zich op het feit dat genen modulair kunnen opgebouwd zijn (zowel in
regulatorische als coderende regio’s), waarbij die verschillende modules instaan voor
onafhankelijke subfuncties. Een module in het ene gen kan dan als buffer optreden
tegen mutaties in de overeenkomstige subfunctie van zijn homoloog gen. Omdat
dergelijke gebufferde mutaties terzelfdertijd in beide kopijen van een gen optreden, is
de aanwezigheid van beide genen vereist (Force et al., 1999). Volgens dit model
zorgen mutaties eerder voor het behoud van gedupliceerde genen, dan de
verwijdering ervan zoals het geval is bij nonfunctionalisatie.
Een voorbeeld van subfunctionalisatie zijn transcriptiefactor bindingsplaatsen op de
promoter (Taylor and Raes, 2005). In de promoter van een gen kunnen verschillende
dergelijke bindingsplaatsen aanwezig zijn die de expressie van het gen onder
verschillende condities reguleren. Mutatie van één van die TF-bindingsplaatsen kan
voor een differentiële genexpressie zorgen.
Een tweede voorbeeld zijn transmembraanreceptoren die uit 3 domeinen bestaan,
een extracellulair receptor domein, transmembraan domein en intracellulair domein
met effector functie. Een mutatie in het receptor domein kan een gewijzigde
substraatsspecificiteit veroorzaken, terwijl de effector functie ongewijzigd blijft. Als na
duplicatie een dergelijke mutatie optreedt, dan zullen in het vervolg twee substraten
dezelfde intracellulaire actie uitlokken, één substraat voor elk van de duplicaten.
2.4 Modelorganisme: Arabidopsis thaliana
Arabidopsis thaliana, de zandraket, is een veelvoorkomende plant die een
gemeenschappelijke voorouder heeft met het herderstasje (Capsella bursa-pastoris). Het
behoort tot de Brassicaceae en is een angiosperm. Het volledig gesequeneerde genoom is
slechts ongeveer 125Mb groot (Arabidopsis Genome Initiative, 2000). De kleine
genoomsgrootte is een nuttige eigenschap voor onderzoeksdoeleinden, evenals de snelle
groei en beperkte omvang waardoor ze gemakkelijk in een labo te kweken zijn. Andere
voordelen voor het gebruik van A. thaliana in een labo zijn de efficiëntie van transformatie en
de overvloedige en snelle productie van zaden waardoor het eenvoudig en snel gecultiveerd
kan worden. Verder is het plantje op het gebied van ontwikkeling, reproductie en reactie op
stress analoog aan belangrijke voedingsgewassen, zoals soja, rijst, tarwe, rogge, maïs,
tomaat, katoen, aardappel en sorgum. Hierdoor is het reeds intens bestudeerd en is heel
wat informatie over A. thaliana publiek beschikbaar (The Institute for Genomic Research,
ZD).

16
Bij de analyse van de genomische sequentie van Arabidopsis thaliana is gebleken dat dit
genoom grootschalige genduplicaties of zelfs volledige genoomduplicaties heeft ondergaan
(Arabidopsis Genome Initiative, 2000). Door genverlies na duplicatie-gebeurtenissen gaat
veel colineariteit tussen de gedupliceerde regio’s verloren en wordt het moeilijker om
gedupliceerde gebieden als dusdanig te herkennen. Eerder werden reeds technieken
besproken om de gevoeligheid te verhogen in het zoeken naar in groep gedupliceerde
genen. Gebruik makend van deze technieken werden in A. thaliana homologe genomische
gebieden vaak in 5 tot 8 kopijen teruggevonden (Simillion et al., 2002). Dit impliceert drie
genoomduplicaties in de evolutionaire geschiedenis van A. thaliana. In deze thesis wordt
gezocht naar grootschalige duplicatiegebeurtenissen om onderzoek te doen naar de
divergentie van genexpressie na duplicatie. Arabidopsis thaliana, met zijn drie volledige
genoomduplicaties, vormt daarom een goede keuze voor dit onderzoek.

17
2.5 Microarrays
2.5.1 Inleiding
Microarrays zijn chips waarop enkelstrengig DNA van verschillende sequenties zijn gehecht
(Draghici, 2003). Door hybridisatie van de microarray met een doelwit-oplossing (cRNA of
cDNA), kan de expressie van duizenden genen in één experiment worden nagegaan waarbij
als het ware een snapshot van de mRNA inhoud van het organisme wordt gemaakt
(Draghici, 2003). Hierdoor zijn microarrays zéér waardevol in vele soorten onderzoek, zoals
bijvoorbeeld het onderzoek naar kanker. Bij kanker worden vele genen differentieel
geëxpresseerd in vergelijking met gezonde weefsels en met microarrays wordt het mogelijk
om kanker meer in detail te onderzoeken op expressieniveau van die differentieel
geëxpresseerde genen. Microarrays openen ook nieuwe deuren voor het ontwikkelen van
geneesmiddelen omdat het effect van een product op genexpressie van vele genen
terzelfdertijd kan worden nagegaan.
In deze thesis wordt microarray data gebruikt voor het berekenen van correlaties van
genexpressie van verschillende soorten gedupliceerde genen.
Figuur 2.8: Werking vaneen cDNA microarray.
Vertrekkende van eendoelwitweefsel wordteen mRNA extractieuitgevoerd om daarmeecDNA te bereiden datdan kan hybridiserenmet de probes van demicroarray.
(Draghici, 2003)

18
2.5.2 Types en productie
Het maken van een microarray is gesteund op één van twee principes waarbij ofwel de DNA
probes eerst worden aangemaakt en nadien op de microarray worden gehecht ofwel worden
de probes in situ gesynthetiseerd (Draghici, 2003). Bij de eerste aanpak kan ofwel met PCR
amplificatie het gewenste cDNA aangemaakt worden of kunnen oligonucleotiden synthetisch
aangemaakt worden. Het cDNA wordt nadien met een robot opgenomen en verdeeld op de
microarray.
De tweede aanpak, in situ synthese van de probes, wordt ondermeer toegepast bij Affymetrix
microarrays (GeneChip) en aangezien in deze thesis de data van dergelijke chips wordt
gebruikt, wordt het productieproces nader toegelicht in figuur 2.10 en figuur 2.11.
Figuur 2.9: Na het scannenvan de microarray wordt eenfiguur metintensiteitswaarden bekomendie later nog verwerkt wordt.Deze figuur toont hiervaneen voorbeeld van eenAffymetrix chip.
(Draghici, 2003)

19
De sequentie van iedere probe op dergelijke chips is volledig bekend, in tegenstelling tot
microarrays waar de probes niet in situ gesynthetiseerd worden. Belangrijke voordelen zijn
dat veel ruis vermeden wordt door het elimineren van verschillende stappen in het
productieproces (bv. clonering en spotten) en dat een onderscheid tussen nauw verwante
genen ook mogelijk is aangezien de probe-sequentie zelf gekozen wordt (Draghici, 2003).
De eerste stap is het aanhechten van synthetische linkers aan het glasoppervlak met
daarbovenop beschermende groepen die door belichting kunnen verwijderd worden. In de
daaropvolgende stap wordt met een lichtstraal een specifiek gebied op de array beschenen,
waardoor de bescherming van de fotogevoelige laag in dat gebied doorbroken wordt.
Vervolgens worden deoxynucleosides toegevoegd die zich op de onbeschermde regio’s
kunnen aanhechten. Het hele proces wordt herhaald, totdat op iedere spot het gewenste
oligonucleotide gesynthetiseerd is (Affymetrix, ZD; Draghici, 2003). De oligonucleotiden op
de array worden probes genoemd en zullen later hybridiseren met het doelwit (“target”).
Figuur 2.10: Affymetrix microarrays worden fotolithografisch aangemaakt waarbij met eenfotogevoelige maskerende laag gebieden worden afgeschermd, waarna basen op specifiekeplaatsen worden toegevoegd. Het herhalen van het proces van aanbrengen van bescherming,vernietigen van bescherming op specifieke plaatsen door belichting en aanhechten vandeoxynucleosiden op onbeschermde gebieden zorgt ervoor dat op iedere plaats van demicroarray het gewenste oligonucleotide bekomen wordt.
(Draghici, 2003)

20
Speciaal voor de Affymetrix technologie is een match/mismatch strategie waarbij
gecorrigeerd wordt voor achtergrond(Affymetrix, ZD; Draghici, 2003). Figuur 2.11 stelt deze
strategie schematisch voor. De match probe (PM of “perfect match”) is een sequentie
bestaande uit 25 nucleotiden die volledig complementair is met het gen dat door deze probe
moet gedetecteerd worden. De mismatch (MM) probe telt ook 25 nucleotiden en verschilt
van de match probe in slechts één nucleotide (de middelste), maar de hybridisatiecondities
worden zo gekozen dat zelfs bij een dergelijk klein verschil de target niet meer kan binden op
de mismatch probe. Beide probes liggen naast elkaar, want enkel dichtbij gelegen probes
geven een correcte schatting van het achtergrondsignaal. Een set van 16 tot 20 probe paren
(PM + MM), vormt een probeset die gebruikt wordt voor detectie van een gen. Eén gen
wordt door de Affymtrix technologie dus vertegenwoordigd door een set van match en
mismatch probes.
2.5.3 Het meten van de genexpressie
Bij het meten van genexpressie met behulp van microarrays vertrekt men van een weefsel
waarvan een mRNA extract bereid wordt. Men veronderstelt dat de concentratie van
aanwezige mRNA speciës een correcte maat vormt voor de activiteit van een gen en men wil
Figuur 2.11: Principe van de Affymetrix technologie. Wanneer het target-DNA volledigcomplementaire gebieden bevat ten opzichte van een probe, zal het daarop binden.Reactieomstandigheden worden zo gekozen, dat zelfs met één verschillend nucleotide in demismatch probe, hybridisatie niet meer mogelijk is. Verschillende match/mismatch paartjes (10)per gen zorgen ervoor dat zwakke signalen eenvoudiger te onderscheiden zijn ten opzichte vanachtergrond.
(Draghici, 2003)

21
met een microarray de aanwezigheid en hoeveelheid van verschillende mRNA speciës
terzelfdertijd bepalen. Twee algemene methodes worden hiervoor onderscheiden. Bij de
eerste wordt gebruik gemaakt van één (oligonucleotide microarray) en bij de tweede van
twee weefselextracten (cDNA microarray).
In het geval van de cDNA microarray wordt vertrekkende van beide weefselextracten eerst
een cDNA kopij gemaakt met reverse transcriptase (RT) en het mRNA weefselextract als
template (figuur 2.8). Bij de reverse transcriptie wordt gebruik gemaakt van fluorescent
gelabelde nucleotiden, waardoor het cDNA visualiseerbaar wordt door excitatie met licht van
de gepaste golflengte (Butte, 2002; Quackenbush, 2001). Voor beide mRNA extracten wordt
een afzonderlijk label gekozen, bijvoorbeeld cy3 en cy5. De cDNA targets worden
tegelijkertijd op dezelfde microarray chip gehybridiseert en kunnen afzonderlijk
gevisualiseerd worden door de golflengte van het gebruikt excitatielicht aan te passen.
In het geval van de oligonucleotide microarray wordt vertrekkende van één enkel
weefselextract een cDNA kopij gemaakt met reverse transcriptase, met dat verschil dat het
nu niet gemerkt wordt (Coe and Antler, ZD). Een merker wordt pas toegevoegd in de
daaropvolgende stap, waarbij door in vitro transcriptie een cRNA kopij gevormd wordt. Een
voorbeeld van dergelijke oligonucleotide microarrays is de GeneChip (Affymetrix, ZD). Bij
GeneChips wordt het target cRNA gemerkt met biotine en het is de bedoeling om via de
visualisatie van deze biotine-tags een absolute waarde te bekomen voor de genexpressie
van de geëxpresseerde genen.
Na detectie wordt door het verwerken van de foto’s (figuur 2.9) en preprocessing van de data
(achtergrond correctie en normalisatie) een absolute waarde bepaald voor de genexpressie
van de genen die corresponderen met de probes op de chip (Draghici, 2003). Een voorbeeld
van een ruwe ingescande foto’s waarop nog geen bewerkingen werden uitgevoerd, is
weergegeven in figuur 2.9 en dergelijke foto’s worden bij affymetrix genechips CEL-files
genoemd.
2.5.4 Normalisatie van microarray data
Een microarray experiment wordt uitgevoerd met als doel om biologisch significante variatie
in genexpressie te detecteren. De waargenomen variatie wordt hiertoe ingedeeld in twee
types, namelijk de interessante (biologische) variatie en de obscure variatie die het gevolg is
van ruis en systematische verschillen (Irizarry et al., 2003). De obscure variatie wil men
uiteraard vermijden of elimineren door gebruik te maken van replicatie en normalisatie.

22
Ruis treedt op bij alle microarray experimenten en is niet te vermijden. We kunnen de
effecten ervan alleen verminderen door eenzelfde experiment meerdere malen te herhalen
(replicatie) om zo een onderscheid te maken tussen biologisch significante variantie en
variantie ten gevolge van ruis (Draghici, 2003). Vele factoren geven ontstaan aan dit
fenomeen, zoals bv. mRNA bereiding, labeltype, vochtigheid, hybridisatie-omstandigheden,
…
Systematische verschillen tussen meerdere datasets zijn die verschillen die een bepaalde
wetmatigheid volgen en kunnen gecorrigeerd worden door middel van normalisatie. Die
normalisatie zorgt ervoor dat microarray data betrouwbaar kan vergeleken worden (Irizarry et
al., 2003). Een dergelijk systematisch verschil kan zich voordoen in de mRNA concentratie,
wanneer voor het ene experiment 5% meer RNA gebruikt werd voor de cDNA bereiding.
Een voorbeeld waar normalisatie vereist is, specifiek voor cDNA microarrays, is bij het
gebruik van verschillende fluorescente labels, waarbij de gemeten intensiteit van het eerste
label (bv. cy3), niet vergelijkbaar is met de gemeten intensiteit van het tweede label (bv. cy5)
(figuur 2.12) ten gevolge van een verschil in eigenschappen van de labels (Draghici, 2003).
In beide gevallen wordt nochtans hetzelfde weefselextract gebruikt bij de bereiding van de
target. Om te normaliseren voor dit verschil in intensiteit wordt de data in groepjes verdeeld
en worden een centraliteitsmaat (bv. gewogen gemiddelde) voor elk van de groepjes
berekend zoals voorgesteld in figuur 2.12. Aan de hand van de exponentiële curve van deze
gemiddelden wordt een afwijking van de ratio ten opzichte van 0 berekend om vervolgens te
corrigeren voor die afwijking. Deze normalisatie wordt LOWESS of LOESS normalisatie
(LOcally WEighted polynomial regreSSion) genoemd.
Als laatste moet ook opgemerkt worden dat het meten van expressiewaarden met behulp
van microarrays bestaande meer tijdsrovende analyses, zoals opzuivering van een eiwit en
spectroscopische concentratiebepaling, niet volledig kan vervangen want niet enkel de
transcriptie en translatie zijn van belang voor de goede werking van een genproduct
(Draghici, 2003). Vaak zijn post-translationele modificaties noodzakelijk voor het uitvoeren
van een functie en deze processen kunnen afhankelijk zijn van een groot aantal factoren die
niet in een microarray experiment in rekening kunnen gebracht worden (bv. correcte
opvouwing van het eiwit). Verder wordt bij het werken met microarrays verondersteld dat de
hoeveelheid mRNA direct proportioneel is met de hoeveelheid functioneel eiwit, maar ook dit
is niet altijd het geval, bijvoorbeeld wanneer de translatie vroegtijdig onderbroken wordt.

23
Figuur 2.12: Op de verticale as staat het logaritme van de verhouding van de intensiteiten,gemeten met twee verschillende fluorescente labels (log cy3/cy5) op hetzelfde mRNAweefselextract. Indien met beide labels dezelfde intensiteit gemeten wordt, zou men een ratio van0 bekomen, maar dit is niet het geval (linksboven). Normalisatie van deze afwijkende waardenvoor verschillende labels gebeurdt door het indelen van de data in groepen die gekenmerktworden door hun eigen gemiddeldes en varianties (rechtsboven). Vervolgens wordt eenexponentiële curve gepast door de gemiddeldes van de groepen (linksonder) om de afwijking vande ratio ten opzichte van 0 te bepalen. Op basis van die gevonden afwijking wordt een correctieuitgevoerd (rechtsonder).
(Draghici, 2003)

24
3 Materiaal en methode
3.1 Algemeen overzicht
Figuur 3.1: Deze figuur geeft een overzicht van de gebruikte technieken indeze thesis. De thesis is opgedeeld in drie onderdelen: a) het vinden van ingroep gedupliceerde genen en opdelen in soort herschikkingen; b) hetbekomen van expressie-waarden voor de te onderzoeken ankerpuntgenenen c) het onderzoeken van de aligneerbaarheid van de promoterregio’s vande gevonden ankerpuntgenen.

25
3.2 Algemene technieken
3.2.1 Perl
Perl (http://www.r-project.org) is een scripttaal die het gemakkelijk maakt om taken te
automatiseren, zo kunnen bijvoorbeeld grote hoeveelheden tekst gemanipuleerd worden
zoals kolommen verwijderen en verwisselen. Twee voorbeelden waarvoor perl in deze
thesis gebruikt wordt zijn het zoeken naar genpaartjes die aan specifieke voorwaarden
voldoen en het berekenen van correlatie coëfficiënten. Welke andere taken door middel van
perl-scripts geautomatiseerd werden, wordt duidelijk naarmate de bespreking van het
materiaal en methode vordert.
3.2.2 R en Bioconductor
Link: http://www.r-project.org
R is een gratis en open-bron statistisch software pakket met een modulaire opbouw.
Hiermee bedoelt men dat het programma uit een basis bestaat die verder aangevuld kan
worden met uitbreidingen, namelijk de modules, die specifieke functies verzorgen. De
gewenste modules kunnen eenvoudig geladen worden naargelang de behoeftes van de uit
te voeren analyse. Bioconductor (Gentleman et al., 2004) is eveneens een gratis en open-
bron software pakket, bestaande uit R-modules voor de analyse van genomische data. Voor
de analyse van Affymetrix CEL-files wordt in deze thesis een beroep gedaan op het “affy”
pakket van bioconductor.
3.3 Detectie en klassificatie van ankerpunten
3.3.1 BLAST
Link: http://www.ncbi.nlm.nih.gov/BLAST/
BLAST (Basic Local Alignment Search Tool) is een tool dat op zoek gaat naar aligneerbare
sequenties door het paarsgewijs aligneren van de sequenties (Altschul et al., 1990). Deze
alignering wordt gestart met het zoeken naar korte gelijkaardige woordjes in beide
sequenties en het blast algoritme probeert de alignering van dit woord te verlengen in beide
richtingen (5’ en 3’) totdat beide sequenties niet meer als voldoende gelijkaardig herkend
worden omdat teveel “gaps” in het alignement geïntroduceerd worden. Het zoeken naar
gelijkaardige sequenties steunt op het feit dat bepaalde mutaties in aminozuursequentie
beter geaccepteerd worden dan andere (Van de Peer, 2005). Dit is bijvoorbeeld het geval
als het aminozuur vervangen wordt door een ander, maar met gelijkaardige eigenschappen

26
(zoals grootte, lading) zodat die mutatie waarschijnlijk geen functiewijziging van het
genproduct teweeg brengt.
De kans dat een specifieke mutatie zich voordoet, kan met verschillende methodes berekend
worden en wordt daarna in een substitutiematrix opgeslaan. Deze substitutie-matrix wordt
door BLAST gebruikt om de similariteit van twee sequenties te bepalen en deze
gelijkaardigheid zal gebruikt worden om het alignement zo ver mogelijk te verlengen.
Verschillende BLAST algoritmes zijn beschikbaar:
- BLASTp voor het vergelijken van een proteïnesequentie met een proteïnedatabank,
- BLASTn voor het vergelijken van een nucleotidesequentie met een
nucleotidedatabank,
- BLASTx voor het vergelijken van een 6 leesraam-vertaling van een
nucleotidesequentie met een proteïnedatabank,
- tBLASTn voor het vergelijken van een proteïnesequentie met de 6 leesraam-vertaling
van een nucleotidedatabank en
- tBLASTx voor het vergelijken van een 6 leesraam-vertaling van een nucleotide-
sequentie met de 6 leesraam-vertaling van een nucleotidedatabank.
In deze thesis wordt enkel het BLASTp algoritme gebruikt. Hiermee wordt een proteïne
sequentie (query) vergeleken met andere proteïne sequenties, waarbij BLASTp eerst een
woord bestaande uit 3 aminozuren tracht te aligneren. Voor deze thesis wordt met BLASTp
gezocht naar alle homologe sequenties binnen het genoom van Arabidopsis thaliana om zo
groepen van genen te identificeren die samen gedupliceerd werden.
3.3.2 Methode van Rost
De methode van Rost is een methode die vertrekkende van een lijst van aligneerbare
sequenties (BLASTp output) homologe genen identificeert. Genen worden genen als
homoloog beschouwd indien ze meer dan 30% sequentie identiteit bezitten over een
aligneerbare regio van tenminste 150 aminozuren (Rost, 1999).
3.3.3 i-ADHoRe
De lijst van homologe genen is de input voor i-ADHoRe, samen met de chromosoomlijsten
van A. thaliana en andere parameters die in de athlevel2R.ini file terug te vinden zijn en
reeds in de literatuurstudie besproken werden (Simillion, 2005). i-ADHoRe geeft als output
een lijst met ankerpunten en hun beschrijving en maakt hierbij gebruik van de “map based
approach” en genomische profielen.

27
De door i-ADHoRe gebruikte parameters zijn (Simillion C., 2005):
- gap size: Geeft de maximum afstand die kan bestaan tussen de ankerpunten in een
cluster.
- cluster gap: Geeft de maximum afstand die kan bestaan tussen basisclusters van
ankerpunten. Deze basisclusters kunnen nadien samengevoegd worden indien ze
voldoende dicht bij elkaar voorkomen.
- Q value: Geeft de vereiste diagonale kwaliteit voor een gevonden ankerpuntcluster.
- ankerpunten: Geeft het minimaal aantal ankerpunten waaruit een cluster van
ankerpunten moet bestaan.
- waarschijnlijkheid cutoff: Geeft een maximum limiet voor de kans dat een gevonden
cluster door toeval ontstaan is en niet door een duplicatiegebeurtenis.
- enkel multiplicatieniveau 2: Bij het zoeken naar multiplicons van maximum niveau 2
worden geen profielen opgebouwd zoals in de literatuurstudie beschreven staat.
De in deze thesis gebruikte i-ADHoRe parameters3 zijn:
Gap size 25
Cluster gap 25
Q waarde 0,90
Ankerpunten 3
Waarschijnlijkheids cutoff 0,01
Enkel multiplicatieniveau 2 TRUE
»echo “./i-ADHoRe athlevel2R.ini” | cluster_job.pl i-ADHoRe
Dit commando start i-ADHoRe op een cluster, dit is een verzameling van computers die
samen werken alsof het één computer zou zijn, waarbij de taken over de verschillende
cluster-nodes (dit zijn de afzonderlijke computers van de cluster) verdeeld worden. De
output van i-ADHoRe wordt opgeslaan in de volgende tabellen4 (Simillion, 2005):
3 zie bijlagen op CD-ROM, map i-ADHoRe >> athlevel2R.ini
4 zie bijlagen op CD-ROM, map i-ADHoRe >> athlevel2R >> multiplicon_linair_plots >> output

28
– Multiplicons tabel: Beschrijft alle multiplicons5 voor ieder multiplicatieniveau.
Deze tabel wordt opgeslaan in de tekstfile multiplicons.txt.
– Ankerpunten tabel: Een opsomming van homologe genpaartjes met vermelding
van de vergeleken genomische segmenten voor multiplicons met
multiplicatieniveau 2. (anchorpoints.txt)
– Segmenten tabel: Geeft een overzicht van de segmenten die met elkaar
vergeleken worden in elk multiplicon. (segments.txt)
– Genen tabel: Bevat de positie van alle genen uit het configuratiebestand
(athlevel2R.ini) en info over de plaats van tandem repeats. (genes.txt)
– Lijst elementen: Een lijst van alle genen die voorkomen in de segmenten van de
multiplicons, samen met hun orientatie en positie. (list_elements.txt)
3.3.4 i-ADHoRe2genedraw_real_TE.pl
i-ADHoRe2genedraw_real_TE.pl is een perl script dat de output van i-ADHoRe neemt en
gebruikt om tekstfiles (zie bijlagen op cd-rom)6 te genereren voor ieder multiplicon met
multiplicatieniveau 2 met daarin:
– Een lijst met de elementen van het eerste segment met daarbij hun relatieve positie
op het segment, hun orientatie en hun naamcode (bv. At2g032570).
– Diezelfde lijst voor de genen van het tweede segment.
– Een lijst met ankerpunten die beide segmenten met elkaar verbindt via hun homologe
genpaartjes.
Vervolgens worden deze tekstfiles door hetzelfde script gebruikt om figuren te genereren (zie
bijlagen op cd-rom)7, waarin de genrelaties tussen beide genomische segmenten visueel
voorgesteld worden. Het volgende commando illustreert hoe dit script gebruikt kan worden:
5 Een groep van segmenten die homoloog zijn met elkaar, gevonden via de “map based approach”
met profiel (zoals besproken in de literatuurstudie).
6 Zie bijlagen op CD-ROM, map i-ADHoRe >> athlevel2R >> multiplicon_linair_plots
7 Zie bijlagen op CD-ROM, map i-ADHoRe >> athlevel2R >> multiplicon_linair_plots

29
»./i-ADHoRe2genedraw_real_Te.pl athlevel2R.iniTIGRv5_lists/coding_list.txtTIGRv5_lists/non_coding_list.txt
De “coding_list.txt” en “non_coding_list.txt” bestanden zijn lijsten met daarin de genen die
wel coderen voor een functioneel polyproteïne en die genen die dit niet doen (bv.
pseudogenen). Voor deze thesis wordt hiervoor de annotatie van TIGR5 gebruikt (The
Institute for Genomic Research, ZD).
i-ADHoRe2genedraw werd gebruikt omdat de gegenereerde tekstfiles een handig overzicht
geven van alle i-ADHoRe output die voor dit onderzoek gebruikt wordt. De output van i-
ADHoRe2genedraw wordt hiertoe ingelezen met behulp van een perl-script dat de data
zodanig formateerd om de manipulatie ervan in een MySQL database te vereenvoudigen.
Hiervoor werd fill_database.pl8 gebruikt die als output een aantal tabellen genereert die dan
geïmporteerd worden in de database.
»./fill_database.pl
Er worden vier tekstfiles gegenereerd:
– elements_info: Bevat informatie over alle genen van de homologe segmenten
in de gevonden multiplicons (hun start- en stopposities, oriëntatie en naam).
– segment_pairs: Bevat informatie over start- en stopposities van de
genomische segmenten die vergeleken werden en samen in een multiplicon
geplaatst werden op basis van gevonden homologie.
– elements_info_2_segment_pairs: Verbindt elements_info aan segment_pairs
met een gemeenschappelijke kolom (element_id) in de tabel. Aan de hand
van deze tabel kan opgezocht worden welke genen tot een bepaald
genomisch segment behoren, samen met hun oriëntatie en volgorde.
– gene_relations: Geeft de genrelaties weer voor een bepaald “segment paar”
(multiplicon van niveau 2).
3.3.5 rearrangement_search.pl
Eén van de doelstellingen van deze thesis is om de correlatie van genexpressie van groepen
van ankerpuntgenen te vergelijken. Hiertoe moet de lijst met ankerpuntgenen eerst
opgesplitst worden in subgroepen, bijvoorbeeld herschikte en niet herschikte
8 zie bijlagen op CD-ROM, map i-ADHoRe >> athlevel2R >> multiplicon_lineair_plots

30
ankerpuntgenen. Hiervoor werd een script geschreven dat aan de hand van de informatie in
de MySQL database op zoek gaat naar eventuele9 herschikkingen in de promoterregio van
de ankerpunten.
»./rearrangement_search.pl pseudogenes_list.txt
Om de herschikte ankerpunten te onderscheiden van de niet herschikte overloopt het script
eerst alle multiplicons (multiplicatieniveau 2) en in ieder multiplicon overloopt het script alle
elementen van het eerste genomische segment (segment A), zoals in figuur 3.1 aangeduid
wordt met een nummering van die elementen. Het script onderzoekt daarbij of een element
een ankerpunt is (groen of rood in figuur 3.1) of niet (zwart in figuur 3.1) en indien dit zo is,
controleert het dat ankerpunt op herschikkingen. Bij deze controle op herschikkingen van
een ankerpunt (fig. 3.2), wordt eerst de orientatie opgevraagd zodat de positie van de
promoterregio gekend is. De volgende stap is een controle van de eerstvolgende elementen
op segmenten A en B vertrekkende vanaf de promoterregio’s van de te onderzoeken
ankerpunt-genen. De gevonden genen moeten een ankerpunt zijn van elkaar en indien dit
niet het geval is, zijn de bestudeerde ankerpunten herschikt geweest sinds hun duplicatie.
9 In deze thesis wordt onderzocht of deze herschikkingen zich voordoen of niet en welke invloed zeuitoefenen op de genexpressie. We splitsen de lijst met ankerpunten dus in aparte lijsten waarvan wedenken dat deze wel of niet herschikkingen bevatten in de promoterregio. De benamingen “herschikt”en “niet herschikt” moeten eerder geïnterpreteerd worden als “eventueel herschikt” en “eventueel nietherschikt”.
Figuur 3.1: Om de correlatie van genexpressie te vergelijken tussen herschikte en niet herschikteankerpunten, moet eerst een onderscheid gemaakt worden tussen de verschillende groepen vanankerpuntgenen. Herschikte ankerpunten worden aangeduid in het rood en de niet herschikte inhet groen.

31
Maar bovenstaande test geeft geen sluitend bewijs voor het al dan niet voorkomen van een
herschikking. Want indien een genpaar geïnverteerd werd, moet nog gecontroleerd worden
indien de nabije genen ook nog steeds elkaars ankerpunt zijn en of deze genen geïnverteerd
werden in dezelfde inversiegebeurtenis (figuur 3.3). En alleen als blijkt dat deze door
dezelfde inversie geïnverteerd werden, wordt besloten dat de promoterregio’s niet verstoord
werden door herschikkingen.
1
2
Figuur 3.2: Illustreert hoe gecontroleerd wordt op herschikkingen in twee gedupliceerdesegmenten. (1) Wanneer beide genen upstream van het bestudeerde ankerpunt genpaar ookankerpunten zijn en bovendien elkaars ankerpunt zijn, kan nog geen herschikking vastgesteldworden. (2) In het andere geval, wanneer de genen direct stroomopwaarts van de ankerpuntenniet elkaars ankerpunt zijn, stelt men wel een herschikking vast.

32
De inversie-controle van het ankerpunt paar verloopt
3.3.6 Onderverdelen in type herschikkingen
Herschikkingen spelen een belangrijke rol in de evolutie (Sankoff, 2003) en in deze thesis
wordt de invloed van herschikkingen op divergentie van genexpressie nagegaan. Bij het
onderverdelen van de ankerpuntgenen in een lijst van herschikte en niet herschikte
ankerpunten, stellen zich echter een aantal problemen. Wanneer stroomopwaarts van een
ankerpuntgen een pseudogen gevonden wordt, is het niet langer mogelijk om dit ankerpunt
te klassificeren als wel of niet herschikt omdat niet geweten is of het gen in de promoterregio
van het gen van het andere segment hier het ankerpunt van was of niet. Wanneer deze
ankerpunten waren, zou het ankerpunt als niet herschikt moeten worden geïdentificeerd
aangezien de promoter waarschijnlijk niet verstoord zal worden door het niet functioneel
worden van het stroomopwaartse gen. Want alhoewel selectie op het pseudogen zal
wegvallen, blijft de promoterregio van het ankerpuntgen aanwezig en onverstoord. Om
rekening te kunnen houden met de aanwezigheid van dergelijke speciale gevallen, worden
de genen die deel uitmaken van de herschikking ingedeeld in de groepen “pseudogen”, “TP”
(transposon), “RNA” (RNA coderend) en “eiwitcoderende herschikkingen”. De mogelijkheid
om deze gevallen te onderscheiden wordt toegevoegd aan het rearrangement_search.pl
script.
Een tweede probleem stelt zich in het begin en einde van een gedupliceerd segment. Als
bijvoorbeeld het eerste gen een positieve oriëntatie bezit en ook een ankerpunt is, dan wordt
deze als herschikt geïdentificeerd omdat het upstream gen geen ankerpunt vormt. Omdat de
Figuur 3.3: Een inversiecontrole neemt het gen in de promoterregio van het ankerpunt paar op heteerste segment (hier A) en zoekt het homologe gen op het tweede segment om de absolutepositie van dat gen (positie 1) te kunnen vergelijken met de absolute positie (positie 2) van hetgen van het bestudeerde ankerpunt paar op dat tweede genomisch segment (B). Enkel waneerpositie 1 kleiner is dan positie 2 bij een negatieve orientatie van het eerste ankerpuntgen (groenA), zijn de genen in de promoterregio van het bestudeerde ankerpuntpaar door dezelfde inversiegeïnverteerd, op voorwaarde dat ook de orientatie van de genen in de promoterregio’stegengesteld is.

33
informatie van het upstream gen niet in de gebruikte database aanwezig is, kan geen
verdere indeling gebeuren op de type genen die deel uitmaken van de herschikking en de
ankerpunten met + oriëntatie in het begin en – oriëntatie op het einde van de gedupliceerde
segmenten worden daarom ingedeeld in de groep “RAND” en worden bij verdere
berekeningen buiten beschouwing gelaten.
Figuur 3.4: Een insertie kan opgespoord worden wanneer slechts één van beidegenen stroomopwaarts van het ankerpunt zelf geen ankerpunt is. A) Als het gen inde promoterregio van een ankerpuntgen zelf een ankerpunt is in A. thaliana endaarbij een homoloog heeft in populier, wordt aanvaard dat het homoloog metpopulier oorspronkelijk aanwezig was en dat het niet ankerpuntgen op het anderesegment (hier segment 1 van A) na duplicatie geïnsereerd werd.
B) Als één van de genen in de promoterregio van het bestudeerdeankerpuntgenpaar homoloog is met een gen van populier, maar zelf geen homoloogheeft in A. thaliana, wordt besloten dat het oorspronkelijke duplicaat verdwenen is(deletie).

34
Als laatste kan het type herschikking in eenvoudige gevallen verder onderzocht en
geklassificeerd worden op basis van deleties of inserties in de promoterregio. Deze
klassificatie wordt uitgevoerd met een perl script dat een lijst van “eenvoudige
herschikkingen” selecteert en in die lijst zoekt naar deleties en inserties door een vergelijking
te maken met gevonden homologen in populier (Populus trichocarpa). Het script is
gebaseerd op het feit dat Arabidopsis en populier een gemeenschappelijk voorouder
hebben, waarna ze apart zijn geëvolueerd. Pas na het ontstaan van beide organismen,
heeft Arabidopsis thaliana zijn laatste genoomduplicatie ondergaan (3R) en als een gen uit
Arabidopsis een homoloog bezit in populier, maar niet in Arabidopsis, wordt dit verklaard
door een deletie van het duplicaat in Arabidopsis.
Figuur 3.4 illustreert wat bedoeld wordt met deletie en insertie. Het zoeken naar deleties en
inversies gebeurt enkel bij eenvoudige herschikkingen, waarbij slechts één van de
promoterregio’s van de ankerpuntgenen eventueel herschikt is door slechts één insertie of
één deletie. Bij meer complexe herschikkingen is het niet meer mogelijk te bepalen welke
herschikking(en) zich hebben voorgedaan. Een stroomopwaarts gen is geïnsereerd,
wanneer het geen homoloog heeft in populier én Arabidopsis, terwijl het stroomopwaartse
gen op het andere segment wel een homoloog bezit in populier én Arabidopsis. Een
stroomopwaarts gen is gedeleteerd, wanneer het stroomopwaartse gen op het andere
segment wel een homoloog bezit in populier, maar niet in Arabidopsis.
3.3.7 Berekenen van de correlatie van genexpressie
3.3.7.1 Overzicht
Voor het berekenen van de correlaties van genexpressie, wordt microarray-data gebruikt.
Een overzicht van de gebruikte microarray dataset is beschikbaar in bijlage A. De volledige
dataset bestaat uit 153 Affymetrix GeneChip slides die tot 16 experimentreeksen behoren.
Iedere reeks bestaat uit een aantal experimentele condities (aangeduid met “e”) en
tenminste één controle-slide (aangeduid met “c”) die de wild type conditie (WT) voorstelt. De
microarray data is publiek beschikbaar vanaf het “Nottingham Arabidopsis Stock Centre”
(NASC, ZD).
Volgende stappen worden ondernomen voor het vergelijken van de correlaties van
genexpressie:
– Normalisatie van de microarray dataset dient om te corrigeren voor de systematische
verschillen, zoals reeds werd toegelicht in de literatuurstudie. Bij het uitvoeren van
de normalisatie wordt RMA gebruikt en deze wordt toegepast met R en bioconductor.

35
– Unieke probe-ID’s voor de microarray data worden geselecteerd. Aan de hand van
de gemeten intensiteitswaarden in de CEL-files wordt een waarde voor de expressie
van de genen in de lijsten van herschikte en niet herschikte ankerpuntgenen
bekomen. Het selecteren van probesets die uniek zijn voor één gen is noodzakelijk
om cross-hybridisatie te vermijden.
– Per experiment zijn een aantal slides aanwezig overeenkomstig met gekozen
experimentele condities, waarbij elke slide tenminste éénmaal gerepliceerd is.
Replicatie corrigeert voor de experimentele fout bij het uitvoeren van de hybridisatie
(Draghici, 2003). Van deze gerepliceerde experimenten wordt een gemiddeld
expressiesignaal berekend. Om te corrigeren voor effecten die het gevolg zijn van
een variatie in technologie in plaats van een biologisch verschil tussen planten, wordt
voor ieder gen bovendien de intensiteitswaarde van de wild type (controle-slide)
afgetrokken van dat van de behandelde plant. De gebruikte dataset bestaat hierna
uit 49 expressiewaarden per gen, terwijl de originele dataset met replicaten en
controles 153 microarrays bevat verdeeld over 16 experimenten10.
– Een perl script11 overloopt de lijst van herschikte en niet herschikte ankerpunten en
leest de genormaliseerde microarray data in om daarmee de spearman
correlatiecoëfficiënt te berekenen. De correlatie geeft dan aan in welke mate de
expressie van de ankerpuntgenen eenzelfde patroon volgen.
– In de laatste stap worden de correlatie coëfficiënten vergeleken met behulp van R om
te controleren of deze coëfficiënten significant verschillend zijn tussen de
afzonderlijke lijsten met ankerpuntgenen.
3.3.7.2 RMA
RMA staat voor “robust multi-array average” en is een verkennende data analyse van de
ruwe microarray data op het probe-niveau (Irizarry et al., 2003). Volgende bewerkingen
worden door RMA analyse uitgevoerd:
- achtergrond correctie
- normalisatie
- log-transformatie van de PM waarden
10 Zie bijlagen op CD-ROM: dataset.pdf
11 zie bijlagen op CD-ROM: rearrangement_search >> rearrangement_search.pl

36
De door RMA gebruikte normalisatie, is “quantile normalisation” en het doel hiervan is het
verwijderen van systematische verschillen tussen afzonderlijke microarray slides. Daartoe
probeert men om de distributie van probe intensiteiten voor iedere array in een set van
arrays identiek te maken (Bolstad et al., 2003; Irizarry et al., 2003). Het wordt dan mogelijk
om de gen expressie waarden van die slides met elkaar te vergelijken (Irizarry et al., 2003).
Log-getransformeerde waarden worden gebruikt voor de genexpressie door het variantie
stabiliserende effect van deze transformatie.
3.4 Promoteranalyse van gedupliceerde genen
In deze thesis wordt onderzocht of herschikkingen van gedupliceerde genen een invloed
kunnen hebben op divergentie van genexpressie. Een dergelijke herschikking kan de
promoterregio van ankerpuntgenen namelijk verstoren. Deze verstoring kan onderzocht
worden door het vergelijken van de promoterregio’s van wel en niet herschikte
ankerpuntgenen waarbij verwacht wordt dat de promoterregio’s van herschikte
ankerpuntgenen in mindere mate aligneerbaar zullen zijn dan die van niet herschikte
ankerpuntgenen.
De alignering van een ankerpunt genpaar gebeurt als volgt:
– 2000 bp van de promoterregio van beide ankerpuntgenen worden geselecteerd, te
beginnen vanaf het startcodon (startcodon zelf wordt niet geselecteerd). Wanneer
het stroomopwaartse gen zich dichterbij bevindt dan 2000 bp, wordt enkel de
sequentie tussen beide genen geselecteerd.
– De geselecteerde promoterregio’s van beide ankerpuntgenen worden gealigneerd
met de aligneringsmethode avid (Bray et al., 2003).
– “Vista” neemt de output van avid en selecteert die regio’s die minimaal 70% identiek
zijn aan elkaar over een minimale lengte van 10 bp en zet deze uit op een plot.
– Aangezien sommige promoterregio’s geen 2000 bp lang zijn, wordt het aantal
aligneerbare basenparen gedeeld door de lengte van de kleinste promoter om zo de
aligneerbaarheid van de promoterregio’s vergelijkbaar te maken. De berekende
waarde wordt het “% alignement” genoemd.
– Verdere analyse zoals het vergelijken van het % alignement van de verschillende
lijsten met ankerpuntgenen wordt in R uitgevoerd:
Histogrammen worden getekend die het % alignement van de promoterregio’s
vergelijkt voor herschikte en niet herschikte genparen om te controleren of

37
promoterregio’s van herschikte genparen minder geconserveerd zijn dan niet
herschikte.
Het aantal gealigneerde basen voorstellen in functie van de Ks om de relatie
tussen de leeftijd van de gedupliceerde genen en de
aligneerbaarheid/conservatie van de promoters na te gaan.
Het % alignement uitzetten in een densiteitsplot in functie van de correlatie
van genexpressie om te onderzoeken of de genexpressies van ankerpunten
genen met beter aligneerbare promoters meer gecorreleerd zijn.

38
4 Resultaten
4.1 Grootte van de ankerpunt groepen
4.1.1 Inleiding
In deze thesis wordt gezocht naar groepen van Arabidopsis thaliana genen die samen
gedupliceerd werden. Vervolgens worden deze ankerpuntgenen opgedeeld naargelang de
eventuele aanwezigheid van structurele herschikkingen in hun upstream gebied. Een
dergelijke herschikking kan de promoterregio wijzigen, en het is de invloed van dergelijke
herschikkingen dat in dit onderdeel onderzocht wordt.
Een verdere onderverdeling gebeurt naargelang het type gen dat aanwezig is in de directe
stroomopwaartse omgeving van een ankerpuntgenpaar:
- Herschikte ankerpuntgenen:
o RNA: Eén van de directe stroomopwaartse genen codeert voor een
functioneel RNA, zoals een tRNA.
o TP: Eén van de directe stroomopwaartse genen codeert voor een
transposeerbaar element.
o Eiwitcoderend: Beide directe stroomopwaartse genen coderen voor een eiwit
dat geen transposon activiteit vertoond.
- Niet herschikte ankerpuntgenen:
o Pseudogen: Eén van de directe stroomopwaartse genen codeert voor een
pseudogen.
o Zonder pseudogen: Beide directe stroomopwaartse genen coderen voor een
eiwit.
De groep met pseudogenen in de stroomopwaartse regio vormt een speciaal geval.
Eénmaal een gen gepseudogeniseerd wordt, kan zijn sequentie snel divergeren wat de
herkenning van een ankerpunt waartoe het pseudogen behoort moeilijker kan maken. Het is
dan niet langer mogelijk om het bestudeerde ankerpunt te herkennen als wel of niet
herschikt. Verwacht wordt dat bij niet herschikte ankerpunten met een pseudogen in hun
upstream regio, de promotorregio niet aangetast wordt en de pseudogenisatie geen invloed
heeft op divergentie van genexpressie. De onderverdeling van de groep met pseudogenen
is een arbitraire keuze omdat niet geweten is of dit pseudogen deel heeft uitgemaakt van een

39
Ankerpuntgroepen voor 3R
Herschikt (1) Niet herschikt (2)
met pseudogen (2.1)
zonder pseudogen (2.2)
RNA coderend (1.1)
TP coderend (1.2)
eiwit coderend (1.3)
niet herschikt ankerpunt. Bij de verdere analyses die uitgevoerd worden op de
bovenstaande groepen wordt hier rekening mee gehouden.
Enkel ankerpuntgenen van de 3R duplicatiegebeurtenis in A. thaliana worden in aanmerking
genomen. Voor andere duplicatiegebeurtenissen is volgens de door ons gekozen methode
onvoldoende data beschikbaar.
4.1.2 Overzicht van de ankerpunt groepen
De gevonden groepen van ankerpuntgenen zijn niet allemaal even groot. Omdat het verschil
in populatiegrootte belangrijk is voor de statistische analyse van de resultaten, worden de
populatiegroottes in tabel 4.1 weergegeven.
Type niet herschikte ankerpuntgenen Aantal paartjes van ankerpunten
Totaal 256
Zonder pseudogenen in de promoterregio 225
Enkel met pseudogenen in de promoterregio 31
Type wel herschikte ankerpuntgenen Aantal paartjes van ankerpunten
Totaal 1184
Figuur 4.1: Dit stelt de onderverdeling van ankerpuntgroepen voor zoals die vergeleken worden inonderstaande analyses. Enkel ankerpunten van de 3R duplicatiegebeurtenis behoren tot dezegroepen.

40
Zonder transposon of RNA gen in de
promoterregio
1058
Met transposon in de promoterregio 61
Met RNA gen in de promoterregio 65
Herschikking door deletie in de promoterregio 6
Herschikking door insertie in de promoterregio 6
Hierbij moet nog opgemerkt worden dat het aantal herschikkingen door deletie en insertie
enkel die situaties voorstellen, waarin een deletie of insertie nog eenvoudig herkend kan
worden. Met andere inserties en deleties wordt geen rekening gehouden.
Tabel 4.1: In de rechterkolom zijn de populatiegroottes van de bekomen datasets weergegeven.

41
4.2 Correlatie van genexpressies
Doel: Wat is de invloed van herschikkingen op de divergentie van genexpressie van
ankerpuntgenen?
Expressiedata van ankerpuntgenen wordt op grote schaal vergeleken, waarbij de nadruk
gelegd wordt op het zoeken van verschillen in genexpressie tussen groepen van herschikte
en niet herschikte ankerpuntgenen. Het vergelijken van de expressiedata gebeurt via de
correlatiecoëfficiënt van de genexpressie van ankerpuntgenen. Deze coëfficiënt geeft weer
in welke mate de expressie van beide ankerpuntgenen nog dezelfde is over alle bestudeerde
microarray experimenten.
De distributie van de correlatie coëfficiënten wordt vergeleken met behulp van ANOVA
(“analysis of variance”) voor de verschillende groepen in figuur 4.1. De analyse wordt
uitgevoerd voor ankerpunten met een Ks waarde die overeenstemt met de laatste
duplicatieronde in Arabidopsis thaliana (3R) omdat enkel voor die ronde voldoende data
bekomen werd. De Ks-waarde van de geanalyseerde ankerpuntgenen voor 3R ligt tussen
0,4 en 1,0.
De anova-test gaat na of de nulhypothese kan verworpen worden (H0 = de distributie van de
correlatie coëfficiënten is gelijk). Deze test wordt niet 1 maal, maar 10 000 maal uitgevoerd
op gesampelde populaties. De nulhypothese voor de totale populatie wordt verworpen
indien de nulhypothese in meer dan 95 % van het aantal sample testen verworpen kan
worden.
Het grote verschil in populatiegrootte van de verschillende datasets (zie tabel 4.1) vraagt om
een speciale aanpak. Populaties met een te groot verschil in populatiegrootte kunnen niet
meer op een statistisch correcte manier vergeleken worden. De oplossing is het gebruik van
“sampling” (staalname), waarbij de kleinere dataset vergeleken wordt met een even grote
dataset die bekomen werd door het random samplen van de grotere dataset. De grootte van
de gebruikte datasets zijn dan gelijk en de analyse leidt tot een betrouwbaar besluit.
Bij het vergelijken van de groepen ankerpunten, worden deze telkens met een nummer zoals
in figuur 4.1 weergegeven wordt.
4.2.1 Analyse van alle herschikte en alle niet herschikte ankerpuntgenen
In deze analyse worden alle herschikte (groep 1) en alle niet herschikte (groep 2)
ankerpuntgenen vergeleken. De lijst met herschikte ankerpunten bevat genpaartjes met een

42
eiwitcoderend, RNA coderend of TP (transposon) coderend gen in de upstream regio van het
ankerpunt genpaar.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1 en 2 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1 en 2 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
5433 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Herschikte ankerpunten (groep 1) Niet herschikte ankerpunten (groep 2)
0,306 ± 0,273 0,255 ± 0,287
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
54,33% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100005433
4.2.2 Analyse van herschikte en niet herschikte ankerpuntgenen
Bij deze analyse wordt enkel rekening gehouden met de ankerpunten die herschikt werden
met een eiwitcoderend gen (groep 1.3) en met de niet herschikte ankerpunten zonder
pseudogen (groep 2.2) in de promoterregio.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.3 en 2.2 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.3 en 2.2 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
2995 / 10000 testen

43
Gemiddelde correlatie coëfficiënt van één sample test
Herschikte ankerpunten (eiwitcoderende
herschikking) (groep 1.3)
Niet herschikte (zonder pseudogenen) (groep
2.2)
0,303 ± 0,271 0,259 ± 0,295
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
29,95% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100002995
4.2.3 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en
herschikte ankerpunten (eiwitcoderende herschikking).
Enkel de niet herschikte ankerpuntgenen met een pseudogen (groep 2.1) in het upstream
gebied worden vergeleken met de ankerpunten die herschikt zijn en geen transposon of RNA
coderend gen bevatten in hun upstream gebied (groep 1.3).
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 2.1 en 1.3 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 2.1 en 1.3 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
1244 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Niet herschikte ankerpunten (enkel met
pseudogen in promoterregio) (groep 2.1)
Herschikte ankerpunten (herschikking enkel
door eiwitcoderende genen) (groep 1.3)
0,231 ± 0,227 0,303 ± 0,271

44
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
12,44% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100001244
4.2.4 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en
herschikte ankerpunten (RNA coderende herschikking).
Enkel die herschikte ankerpunt genpaartjes waarbij de upstream gebied een RNA coderend
gen bevat (groep 1.1), worden vergeleken met de ankerpunt genpaartjes die een pseudogen
bezitten in hun upstream gebied (groep 2.1).
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.1 en 2.1 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.1 en 2.1 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
2660 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Niet herschikte ankerpunten (enkel met
pseudogen in promoterregio) (groep 2.1)
Herschikte ankerpunten (herschikking enkel
door RNA-coderende genen) (groep 1.1)
0,231 ± 0,227 0,306 ± 0,273
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
26,60% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100002660

45
4.2.5 Analyse van niet herschikte ankerpunten (enkel pseudogenen) en
herschikte ankerpunten (transposon coderende herschikking).
Enkel die herschikte ankerpunt genpaartjes waarbij de upstream gebied een transposon
coderend gen bevat (groep 1.2), worden vergeleken met de ankerpunt genpaartjes die een
pseudogen bezitten (groep 2.1) in hun upstream gebied.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.2 en 2.1 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.2 en 2.1 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
1218 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Niet herschikte ankerpunten (enkel met
pseudogen in promoterregio) (groep 2.1)
Herschikte ankerpunten (herschikking enkel
door transposon-coderende genen) (groep
1.2)
0,231 ± 0,227 0,315 ± 0,248
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
12,18% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100001218
4.2.6 Analyse van herschikte ankerpunten (RNA coderende
herschikking) en herschikte ankerpunten (eiwitcoderende
herschikking).
Enkel die herschikte ankerpunt genpaartjes waarbij de upstream gebied een RNA coderend
gen bevat (groep 1.1), worden vergeleken met de ankerpunt genpaartjes die enkel
eiwitcoderende genen bevat (groep 1.3) in hun upstream gebied.

46
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.1 en 1.3 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.1 en 1.3 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
3293 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Herschikte ankerpunten (enkel met RNA-
coderende gen(en) in promoterregio; groep
1.1)
Herschikte ankerpunten (herschikking enkel
door eiwitcoderende genen; groep 1.3)
0,306 ± 0,273 0,303 ± 0,271
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
32,93% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100003293
4.2.7 Analyse van herschikte ankerpunten (TP coderende herschikking)
en niet herschikte ankerpunten (zonder pseudogenen).
Deze analyse vergelijkt de herschikte ankerpunten met een transposon (groep 1.2) in hun
upstream gebied met de niet herschikte ankerpunten zonder pseudogenen (groep 2.2) in hun
upstream gebied.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.2 en 2.2 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.2 en 2.2 is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten

47
1017 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Herschikte ankerpunten (enkel met TP-
coderende gen(en) in promoterregio; groep
1.2)
Niet herschikte ankerpunten (zonder
pseudogenen; groep 2.2)
0,315 ± 0,248 0,259 ± 0,295
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
10,17% van de sample testen een significant verschil in correlatie coëfficiënt gevonden
wordt. De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,0100001017
4.2.8 Analyse van herschikte ankerpunten (TP coderende herschikking)
en herschikte ankerpunten (RNA coderende herschikking).
Deze analyse vergelijkt de herschikte ankerpunten met een transposon in hun upstream
gebied (groep 1.2) met de herschikte ankerpunten die een RNA coderend gen in hun
upstream regio (groep 1.1) bezitten.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen 1.2 en 1.1 is gelijk
H1 = de correlatiecoëfficiënt voor groepen 1.2 en 1.1 is niet gelijk

48
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
0 / 10000 testen
Gemiddelde correlatie coëfficiënt van één sample test
Herschikte ankerpunten (met TP-coderende
gen(en) in promoterregio; groep 1.2)
Herschikte ankerpunten (met RNA-coderende
gen(en) in promoterregio; groep 1.1)
0,315 ± 0,248 0,306 ± 0,273
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
0% van de sample testen een significant verschil in correlatie coëfficiënt gevonden wordt.
De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,010000
0
4.2.9 Analyse van herschikte ankerpunten die herschikt werden door
deletie in vergelijking tot die die herschikt werden door insertie.
Deze analyse vergelijkt herschikte ankerpunt genpaartjes naargelang de wijze van
herschikking, namelijk insertie of deletie.
De onderstaande resultaten zijn weinig betrouwbaar, aangezien beide datasets slechts 6
ankerpunt genpaartjes groot zijn. Voor de volledigheid worden de bekomen resultaten toch
vermeld, alhoewel men moet opletten met het interpreteren van deze resultaten.
ANOVA: hypothesen
H0 = de correlatiecoëfficiënt voor groepen met deleties en groepen met inserties is gelijk
H1 = de correlatiecoëfficiënt voor groepen met deleties en groepen met inserties is niet gelijk
Aantal testen die duiden op significant verschil tussen de correlatie coëfficiënten
0 / 10000 testen

49
Gemiddelde correlatie coëfficiënt van één sample test
Herschikt door deletie Herschikt door insertie
0,274 ± 0,360 0,518 ± 0,300
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
0% van de sample testen een significant verschil in correlatie coëfficiënt gevonden wordt.
De correlatiecoëfficiënt van beide groepen ankerpuntgenen is gelijk.
95,010000
0

50
4.3 Promoter-onderzoek.
4.3.1 Inleiding
Met dit promotoronderzoek wordt nagegaan of de aligneerbaarheid van de promotorregio’s
een rol speelt in de correlatie van genexpressie van een ankerpunt genpaar. Verwacht wordt
dat bij herschikkingen van gedupliceerde genen de promotorregio verstoord wordt en
daardoor divergentie van genexpressie optreedt. In dat geval verwacht men dat een
verstoring van de promotorregio herkend kan worden door een verlaging van de
aligneerbaarheid van de upstream regio. Ver worden bij oudere duplicatiegebeurtenissen
meer herschikkingen en dus een lagere aligneerbaarheid van de promotorregio’s verwacht.
4.3.2 Vergelijken van de aligneerbaarheid van het upstream gebied.
Doel: Zorgen herschikkingen voor een verlaging van de aligneerbaarheid van de upstream
regio van ankerpunten?
Door het verschil is populatiegrootte van de datasets, wordt opnieuw gebruik gemaakt van
sampling.
ANOVA: hypothesen
H0 = de % aligneerbaarheid voor groepen met herschikkingen en groepen zonder
herschikkingen is gelijk
H1 = de % aligneerbaarheid voor groepen met herschikkingen en groepen zonder
herschikkingen is niet gelijk
Aantal testen die duiden op significant verschil tussen de aligneerbaarheid van de
promoterregio’s
340 / 10000
Gemiddelde % aligneerbaarheid
Herschikt (groep 1) Niet herschikt (groep 2)
0,456 ± 0,090 % 0,451 ± 0,090 %

51
Grafiek 4.1 toont de aligneerbaarheid van de promoterregio’s voor herschikte en niet
herschikte ankerpunt genpaartjes, waarbij de % aligneerbaarheid weergeeft in welke mate
de promoterregio’s van beide ankerpuntgenen aligneerbaar zijn en de frequentie de
hoeveelheid ankerpunt genpaartjes weergeeft met die aligneerbaarheid (uitgedruk in
procenten omdat de populatiegroottes verschillend zijn).
0
10
20
30
40
50
60
10 20 30 40 50 60 70 80 90 100
%aligneerbaarheid
freq
uen
tiei
e
niet herschikte ankerpunten
herschikte ankerpunten
Besluit: De nulhypothese kan niet verworpen worden omdat bij minder dan 95%, namelijk
3,40% van de sample testen een significant verschil in % aligneerbaarheid gevonden wordt.
De % aligneerbaarheid van de promotorregio’s van beide groepen ankerpuntgenen zijn
gelijk.
95,010000
340
Dit resultaat wordt grafisch voorgesteld in grafiek 4.1 waar beide curves eenzelfde verloop
volgen.
Grafiek 4.1: De grafiek stelt de aligneerbaarheid van de promoterregio’s van herschikte en nietherschikte ankerpunten voor.

4.3.3 Verband tussen de aligneerbaarheid van de promoterregio’s en de
leeftijd van duplicatie
Doel: Is er een correlatie tusen de leeftijd van duplicatie en de aligneerbaarheid van de
promotorregio’s van ankerpunten?
Verwacht wordt dat de aligneerbaarheid van een gedupliceerd genpaar afneemt naarmate
de duplicatie zich langer geleden voordeed en meer herschikkingen zijn opgetreden. Om dit
te onderzoeken wordt in de volgende grafieken de % aligneerbaarheid uitgezet in functie van
de Ks (synonieme substituties per synonieme site), die een maat is voor de leeftijd van de
duplicatiegebeurtenis.
In tegenstelling tot de vorige analyses, die zich beperkten tot 3R ankerpunten, wordt voor
deze analyse gebruik gemaakt van de volledige groep ankerpuntgenen. Een Ks cutoff van 5
wordt gebruikt om verstoring van de grafieken door outliers tegen te gaan.
Dealigneerbaarheid van depromotorregio'svangedupliceerde herschiktegenpaartjes in functievan de
leeftijd van deduplicatie (Ks) voor Ks< 5.
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
5
0 10 20 30 40 50 60 70 80% aligneerbaarheidvandepromotorregio's
Ks
Grafiek 4.2: In de x-as staat de relatieve aligneerbaarheid van de promoterregio’s van deankerpuntgenen, met andere woorden het aantal gealigneerde basen gedeeld door de lengte vande sequenties die vergeleken werden. In de y-as staat de overeenkomstige leeftijd van duplicatievan het ankerpunt genpaar, uitgedrukt in Ks.
Deze grafiek bestudeerdt enkel de herschikte gedupliceerde genpaartjes.
52

53
Dealigneerbaarheidvandepromotorregio'svangedupliceerdeniet herschiktegenpaartjes infunctievandeleeftijdvande
duplicatie (Ks) voor Ks<5.
0
0,5
1
1,5
2
2,5
3
3,5
4
0 10 20 30 40 50 60 70 80
% aligneerbaarheidvandepromotorregio's
Ks
Besluit: Grafieken 4.2 en 4.3 tonen dat de aligneerbaarheid van de promotorregio’s van
ankerpuntgenen niet wijzigt in functie van de leeftijd van hun duplicatie.
4.3.4 De aligneerbaarheid van de promoterregio’s in functie van de
correlatie van genexpressie.
Doel: Is de correlatie van genexpressie van ankerpuntgenen hoger voor ankerpunten met
beter aligneerbare promotorregio’s?
Verwacht wordt dat een hogere correlatie van genexpressie gecorreleerd is met beter
aligneerbare promoterregio’s van ankerpuntgenen. Dit wordt onderzocht in de
densiteitsplots van grafiek 4.4 waarbij donkerder kleuren een hogere densiteit van
datapunten voorstellen. Op de X-as staat de % aligneerbaarheid. De correlatie van
genexpressie op de Y-as is een maat voor de divergentie van genexpressie van
ankerpuntgenen nadat ze ontstaan zijn door duplicatie.
Grafiek 4.3: In de x-as staat de relatieve aligneerbaarheid van de promoterregio’s van deankerpuntgenen, met andere woorden het aantal gealigneerde basen gedeeld door de lengte vande sequenties die vergeleken werden. In de y-as staat de overeenkomstige leeftijd van duplicatievan het ankerpunt genpaar, uitgedrukt in Ks.
Deze grafiek bestudeerdt enkel niet herschikte gedupliceerde genpaartjes.

54

55
Besluit: Uit grafiek 4.4 blijkt, in tegenstelling tot wat verwacht wordt, dat de correlatie van
genexpressie van ankerpuntgenen niet gecorreleerd is met de aligneerbaarheid van hun
promotorregio’s.
Grafiek 4.4: De densiteitsplots vergelijken de aligneerbaarheid van de promoterregio’s (x-as)van de ankerpuntgenen met de correlatie van genexpressie van de ankerpuntgenen (y-as).

56
5 Discussie
5.1 Correlatie van genexpressie
De belangrijkste groepen van ankerpuntgenen die bestudeerd werden, zijn de groep met
herschikte en de groep met niet herschikte ankerpuntgenen. Bij de sample testen met
ANOVA kan de nulhypothese niet verworpen worden en moet men dus besluiten dat
verstoringen in de stroomopwaartse regio geen invloed hebben op de correlatie van
genexpressie. Indien bij herschikkingen de promoterregio van de ankerpunten aangetast
zou worden, verwacht men voor gewijzigde (herschikte) promoterregio’s van ankerpunten
een lagere aligneerbaarheid van de promoters evenals gedaalde correlatie van
genexpressie. Het feit dat het belang van een eventuele verstoring van de promotorregio
voor divergentie van genexpressie niet kan aangetoond worden in dit onderzoek, betekent
dat de globale genomische context van een genpaar waarschijnlijk belangrijker is voor de
expressie van hun genen dan een eventuele verstoring van de promoterregio. Hierbij wordt
bijvoorbeeld gedacht aan het verschil in expressie bij eu- versus heterochromatine, aan de
invloed van genomische locaties zoals telomeren, centromeren en aan histon versus inter-
histon gelocaliseerde genen. Een verschil in correlatie van genexpressie tussen in groep
gedupliceerde genen (ankerpunten) en op kleine schaal gedupliceerde genen werd reeds
aangetoond (Casneuf et al., 2006).
Bij ANOVA testen tussen andere groepen van ankerpuntgenen wordt hetzelfde resultaat
gevonden. Te weinig ANOVA testen12 duiden op een verschil in correlatie van genexpressie
en de nulhypothese (correlatie coëfficiënten zijn gelijk) kan niet verworpen worden.
De correlatie van genexpressie Bij het vergelijken van herschikkingen door insertie met
herschikkingen door deletie wordt gevonden dat de correlatie coëfficiënt tussen beide
groepen opnieuw gelijk is (0 / 10000 ANOVA testen duiden op een verschil). Ook in dit geval
kan de nulhypothese niet verworpen worden en moet ze aanvaard worden. Voorzichtigheid
is hier geboden gezien de beperkte populatiegrootte van beide groepen ankerpuntgenen.
12 De precieze hoeveelheden staan vermeld in het onderdeel “resultaten”. Omdat het om
verschillende groepen gaat, wordt naar het onderdeel “resultaten” verwezen in plaats van het aantal
hier expliciet te vermelden..

57
5.2 Promoter onderzoek
De aligneerbaarheid van de promoterregio’s van ankerpuntgenen werd met ANOVA en
sampling vergeleken voor herschikte en niet herschikte ankerpuntgenen. De resultaten
tonen dat de nulhypothese niet kan verworpen worden, de aligneerbaarheid van beide
groepen is dus gelijk. Slechts 3,4 % van de sample testen toont een verschil in
aligneerbaarheid, wat onvoldoende is om te besluiten dat de aligneerbaarheid verschillend
is.
De plots met de % aligneerbaarheid in functie van de Ks tonen, tegen de verwachtingen in,
aan dat de leeftijd van een ankerpunt (Ks) geen invloed heeft op de aligneerbaarheid van zijn
promoterregio’s. Verwacht wordt dat naarmate de duplicatiegebeurtenissen langer geleden
hebben plaatsgevonden, de aligneerbaarheid afneemt. Dit is niet af te leiden uit figuren 4.2
en 4.3. De mogelijkheid bestaat dat de duplicatiegebeurtenissen te lang geleden hebben
plaatsgevonden om een vergelijking van de aligneerbaarheid van de promoterregio’s toe te
laten aangezien de promoters reeds te veel gedivergeerd zijn.
De densiteitplots met de “% aligneerbaarheid” in functie van de correlatie van genexpressie
tonen dat de correlatie van genexpressie van een ankerpunt genpaar niet gecorreleerd is
met de aligneerbaarheid van de promoterregio’s van dat genpaar.
De methode die gebruikt wordt voor het onderzoeken van de aligneerbaarheid van de
promotorregio’s is avid. Het onderzoeken van de promotorregio’s met avid geeft
onverwachte resultaten en alternatieve methoden voor promotoranalyse kunnen gebruikt
worden om de resultaten te verifiëren. Alternatieve methoden kunnen andere zaken in
rekening brengen. Als voorbeeld hierbij kan men aanhalen dat avid veronderstelt dat de
aligneerbare sequenties in dezelfde volgorde en oriëntatie voorkomen en de methode legt zo
een beperking op zijn praktische toepassing (Bray et al., 2003). Aligneerbare, maar
getransloceerde of geïnverteerde sequenties worden gewoon genegeerd.
Een mogelijke verklaring voor de gevonden resultaten is de leeftijd van de
duplicatiegebeurtenissen. Indien de promotors van de ankerpuntgenen reeds sterk
gedivergeerd zijn, zal hun aligneerbaarheid misschien niet langer correleren met de leeftijd
van duplicatie. Wanneer ankerpuntgenen van een zéér recente grootschalige
duplicatiegebeurtenis van een andere plant13 bestudeerd wordt, kan een relatie tussen de
13 De in deze thesis bestudeerde grootschalige duplicatiegebeurtenis 3R in Arabidopsis thaliana is de
meest recente in deze plant. Voor meer recente grootschalige duplicaties kan dus enkel een beroep
gedaan worden op andere planten.

58
leeftijd van duplicatie en de aligneerbaarheid van promotorregio’s eventueel wel gevonden
worden.
5.3 Besluit
Dit thesisonderzoek toont aan dat herschikkingen van gedupliceerde genen na een
grootschalige duplicatiegebeurtenis niet verantwoordelijk zijn voor een verlaging van
correlatie van genexpressie. Bovendien is de ouderdom van de duplicatiegebeurtenis (Ks)
niet gecorreleerd met de aligneerbaarheid van de promoterregio’s van de ankerpuntgenen
voor die ankerpuntgenen van de laatste duplicatieronde (3R) in A. thaliana of is dit niet meer
zichtbaar door divergentie van de promoterregio’s. Ook is voor de ankerpunten geen
correlatie tussen de aligneerbaarheid van de promoterregio’s en de correlatie van
genexpressie vastgesteld.
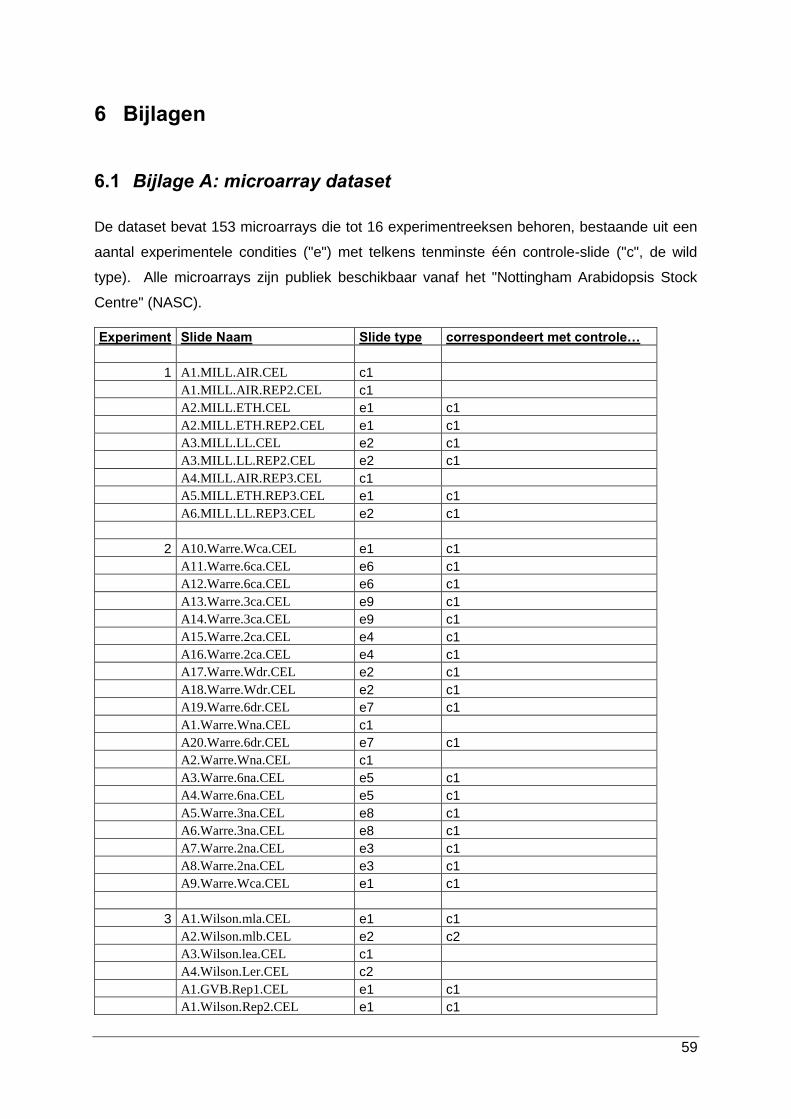
59
6 Bijlagen
6.1 Bijlage A: microarray dataset
De dataset bevat 153 microarrays die tot 16 experimentreeksen behoren, bestaande uit een
aantal experimentele condities ("e") met telkens tenminste één controle-slide ("c", de wild
type). Alle microarrays zijn publiek beschikbaar vanaf het "Nottingham Arabidopsis Stock
Centre" (NASC).
Experiment Slide Naam Slide type correspondeert met controle…
1 A1.MILL.AIR.CEL c1A1.MILL.AIR.REP2.CEL c1A2.MILL.ETH.CEL e1 c1A2.MILL.ETH.REP2.CEL e1 c1A3.MILL.LL.CEL e2 c1A3.MILL.LL.REP2.CEL e2 c1A4.MILL.AIR.REP3.CEL c1A5.MILL.ETH.REP3.CEL e1 c1A6.MILL.LL.REP3.CEL e2 c1
2 A10.Warre.Wca.CEL e1 c1A11.Warre.6ca.CEL e6 c1A12.Warre.6ca.CEL e6 c1A13.Warre.3ca.CEL e9 c1A14.Warre.3ca.CEL e9 c1A15.Warre.2ca.CEL e4 c1A16.Warre.2ca.CEL e4 c1A17.Warre.Wdr.CEL e2 c1A18.Warre.Wdr.CEL e2 c1A19.Warre.6dr.CEL e7 c1A1.Warre.Wna.CEL c1A20.Warre.6dr.CEL e7 c1A2.Warre.Wna.CEL c1A3.Warre.6na.CEL e5 c1A4.Warre.6na.CEL e5 c1A5.Warre.3na.CEL e8 c1A6.Warre.3na.CEL e8 c1A7.Warre.2na.CEL e3 c1A8.Warre.2na.CEL e3 c1A9.Warre.Wca.CEL e1 c1
3 A1.Wilson.mla.CEL e1 c1A2.Wilson.mlb.CEL e2 c2A3.Wilson.lea.CEL c1A4.Wilson.Ler.CEL c2A1.GVB.Rep1.CEL e1 c1A1.Wilson.Rep2.CEL e1 c1

60
A2.GVB.Rep1.CEL e2 c2A2.Wilson.Rep2.CEL e2 c2A3.GVB.Rep1.CEL c1A3.Wilson.Rep2.CEL c1A4.GVB.Rep1.CEL c2A4.Wilson.Rep2.CEL c2
4 A1.WARRE.WTC.2..CEL c1A2.WARRE.WTW.CEL c1A3.WARRE.S6C.CEL e1 c1A4.WARRE.S6W.2..CEL e1 c1A5.WARRE.S2C.new..CEL e2 c1A6.WARRE.S2W.CEL e2 c1
5 Control.3.new.CEL c2Control.4..CEL c2Heat.3.new.CEL e3 c2Heat.4..CEL e3 c2Sen.3.new.CEL e4 c2Sen.4..CEL e4 c2
6 A1.1.cornah.icl.CEL e1 c1A1.2.cornah.icl.CEL e1 c1A1.3.cornah.icl.CEL e1 c1A2.1.cornah.irv.CEL e2 c1A2.2.cornah.irv.CEL e2 c1A2.3.cornah.irv.CEL e2 c1A3.1.cornah.msx.CEL e3 c1A3.2.cornah.msx.CEL e3 c1A3.3.cornah.msx.CEL e3 c1A4.1.cornah.wsx.CEL c1A4.2.cornah.wsx.CEL c1A4.3.cornah.wsx.CEL c1
7 A1.LLOYD.POH.CEL e1 c1A2.LLOYD.POH.CEL e1 c1A3.LLOYD.POH.CEL e1 c1A4.LLOYD.CON.CEL c1A5.LLOYD.CON.CEL c1A6.LLOYD.CON.CEL c1
8 A10.grevi.AT1.CEL e3 c1A11.grevi.AT2.CEL e3 c1A12.grevi.AT3.CEL e3 c1A1.grevi.CC1.CEL c1A2.grevi.CC2.CEL c1A3.grevi.CC3.CEL c1A4.grevi.AC1.CEL e1 c1A5.grevi.AC2.CEL e1 c1A6.grevi.AC3.CEL e1 c1A7.grevi.CT1.CEL e2 c1A8.grevi.CT2.CEL e2 c1

61
A9.grevi.CT3.CEL e2 c1
9 A1.Heggi.CAG.CEL c1A2.Heggi.CEG.CEL e3 c1A3.Heggi.HAG.CEL e2 c1A4.Heggi.HEG.CEL e1 c1A5.Heggi.CAW.CEL c1A6.Heggi.CEW.CEL e3 c1A7.Heggi.HAW.CEL e2 c1A8.Heggi.HEW.CEL e1 c1
10 A1.jones.WT1.CEL c1A2.jones.WT2.CEL c1A3.jones.rh1.CEL e1 c1A4.jones.rh2.CEL e1 c1
11 A1.deeke.tum.CEL e1 c1A2.deeke.Inf.CEL c1A3.deeke.tum.CEL e1 c1A4.deeke.Inf.CEL c1
12 A1.MUT.Top1.CEL e1 c1A2.MUT.Top2.CEL e1 c1A3.MUT.Base1.CEL e2 c2A4.MUT.Base2.CEL e2 c2A5.Turner.WT.Top1.CEL c1A6.WT.Top2.CEL c1A7.WT.Base1.CEL c2A8.WT.Base2.CEL c2
13 A1.Fille.WT.nodex.CEL c1A2.Fille.WT..dex.CEL e1 c1A3.Fille.ANGR4.12.CEL e2 c1A4.Fille.ANGR4.12.dex.CEL e3 c1A5.Fille.WTnodex.CEL c1A6.Fille.WT.dex.CEL e1 c1A7.Fille.ANGR4.12nodex.CEL e2 c1A8.Fille.ANGR4.12.dex.CEL e3 c1
14 A10.Smith.17.CEL c10 c9A11.Smith.21B.CEL c1 c10A1.Smith.21A.CEL c1 c10A2.Smith.22.CEL c2 c1A3.Smith.23.CEL c3 c2A4.Smith.1.CEL c4 c3A5.Smith.5.CEL c5 c4A6.Smith.8.45.CEL c6 c5A7.Smith.10.CEL c7 c6A8.Smith.11.CEL c8 c7A9.Smith.13.CEL c9 c8A10.smith.20h.CEL c10 c9A11.smith.24h.CEL c1 c10

62
A1.smith.00h.CEL c1 c10A2.smith.01h.CEL c2 c1A3.smith.02h.CEL c3 c2A4.smith.04h.CEL c4 c3A5.smith.08h.CEL c5 c4A6.smith.12h.CEL c6 c5A7.smith.13h.CEL c7 c6A8.smith.14h.CEL c8 c7A9.smith.16h.CEL c9 c8
15 A10.Bwoll.Col2.CEL e4 c1A1.Bwoll.COG.CEL c1A2.Bwoll.C5G.CEL e1 c1A3.Bwoll.COS.CEL c1A4.Bwoll.CSS.CEL e1 c1A5.BwolINGI.CEL e2 c1A6.Bwoll.NG2.CEL e2 c1A7.Bwoll.E11.CEL e3 c1A8.Bwoll.E12.CEL e3 c1A9.Bwoll.Col1.CEL e4 c1
16 A1.WILLA.CON.CEL c1A2.WILLA.ISOX.CEL e1 c1A1.willa.CON.REP2.CEL c1A1.willa.CON.REP3.CEL c1A2.willa.ISOX.REP2.CEL e1 c1A2.willa.ISOX.REP3.CEL e1 c1

63
6.2 Bijlage B: Lijst met afkortingen
– ANOVA
Analysis Of Variance:
– BLAST
Basic Local Alignment Search Tool
– GHM
Gene Homology Matrix: De homologie matrix die door i-ADHoRe gebruikt worden
voor het opsporen van in groep gedupliceerde genen.
– Ks
Het aantal synonieme substituties per synonieme site. Een synonieme substitutie is
hierbij een mutatie op veelal de derde positie van een codon die niet voor een
gewijzigde aminozuursequentie zorgt.
– MM
Mismatch: een probe bij de Affymetrix genechips die enkel in het middelste
nucleotide niet complementair is met de sequentie van het overeenkomstige gen.
– PCR
Polymerase Chain Reaction
– PM
Perfect Match: een probe bij de Affymetrix genechips die volledig complementair is
met de sequentie van het overeenkomstige gen.
– TIGR
The Institute of Genomic Research: Een “non-profit” centrum voor de ontcijfering en
analyse van genomische data.
– TP
Transposon: Een element die zichzelf kan kopiëren naar een andere positie in een
genoom.
– ZD
Zonder Datum: aanduiding bij referenties zonder datum

64
6.3 Bijlage C: CD-ROM
De CD-ROM met bijlagen bevindt zich in een hoesje dat op de achterkaft van deze bundel is
gekleefd. De CD-ROM bevat de files die vermeld worden in de tekst, alsook een pdf-versie
van deze thesis.

65
7 Referenties
1. Adams, K.L., and J.F. Wendel. 2005. Polyploidy and genome evolution in plants.Current Opinion Plant Biology 8:135-41.
2. Affymetrix. ZD. Affymetrix GeneChip array technology [Online]. Available byAffymetrix Inc. http://www.affymetrix.com/technology/index.affx.
3. Altschul, S.F., W. Gish, W. Miller, E.W. Myers, and D.J. Lipman. 1990. Basic localalignment search tool. J Mol Biol 215:403-10.
4. Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of theflowering plant Arabidopsis thaliana. Nature 408:796-815.
5. Bolstad, B.M., R.A. Irizarry, M. Astrand, and T.P. Speed. 2003. A comparison ofnormalization methods for high density oligonucleotide array data based on varianceand bias. Bioinformatics 19:185-93.
6. Bray, N., I. Dubchak, and L. Pachter. 2003. AVID: A global alignment program.Genome Res 13:97-102.
7. Butte, A. 2002. The use and analysis of microarray data. Nat Rev Drug Discov1:951-60.
8. Casneuf, T., S. De Bodt, J. Raes, S. Maere, and Y. Van de Peer. 2006. Nonrandomdivergence of gene expression following gene and genome duplications in theflowering plant Arabidopsis thaliana. Genome Biol 7:R13.
9. Coe, B., and C. Antler. ZD. Spot your Genes - An Overview of the MicroArray[Online] http://bioteach.ubc.ca/MolecularBiology/microarray/index.htm.
10. Draghici, S. 2003. Data analysis tools for DNA microarrays Chapman & Hall/CRC,Boca Raton.
11. Force, A., M. Lynch, F.B. Pickett, A. Amores, Y.L. Yan, and J. Postlethwait. 1999.Preservation of duplicate genes by complementary, degenerative mutations.Genetics 151:1531-45.
12. Gentleman, R.C., V.J. Carey, D.M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis,L. Gautier, Y. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R. Irizarry, F.Leisch, C. Li, M. Maechler, A.J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney,J.Y. Yang, and J. Zhang. 2004. Bioconductor: open software development forcomputational biology and bioinformatics. Genome Biol 5:R80.
13. Gregory T. 2005. The evolution of the genome Elsevier Inc.
14. Gu, Z., L. Steinmetz, X. Gu, C. Scharfe, R. Davis, and W. Li. 2003. Role of duplicategenes in genetic robustness against null mutations. Nature 421:63-66.
15. Gu Z., Steinmetz LM, Gu X, Scharfe C., Davis RW, and Li WH. 2003. Role ofduplicate genes in genetic robustness against null mutations. Nature 421:63-66.
16. Haldane, J.B.S. 1933. The Part Played by Recurrent Mutation in Evolution. TheAmerican Naturalist 67:5-19.

66
17. Henikoff, S., and J.G. Henikoff. 1992. Amino acid substitution matrices from proteinblocks. Proc Natl Acad Sci U S A 89:10915-9.
18. Hurst, D.L. 2002. The Ka/Ks ratio: diagnosing the form of sequence evolution.TRENDS in Genetics 18:486-487.
19. Irizarry, R.A., B. Hobbs, F. Collin, Y.D. Beazer-Barclay, K.J. Antonellis, U. Scherf,and T.P. Speed. 2003. Exploration, normalization, and summaries of high densityoligonucleotide array probe level data. Biostatistics 4:249-64.
20. Koszul, R., S. Caburet, B. Dujon, and G. Fischer. 2004. Eucaryotic genomeevolution through the spontaneous duplication of large chromosomal segments.Embo Journal 23:234-43.
21. Long, M., E. Betrán, K. Thornton, and W. Wang. 2003. The origin of new genes:glimpses from the young and old. Nature Reviews Genetics 4:865-875.
22. NASC. ZD. Nottingham Arabidopsis Stock Centre [Online]http://www.arabidopsis.info/.
23. Needleman, S.B., and C.D. Wunsch. 1970. A general method applicable to thesearch for similarities in the amino acid sequence of two proteins. J Mol Biol 48:443-53.
24. Ohno S. 1970. Evolution by Gene Duplication Springer Verlag., New York.
25. Prince, V.E., and F.B. Pickett. 2002. Splitting pairs: the diverging fates of duplicatedgenes. Nat Rev Genet 3:827-37.
26. Quackenbush, J. 2001. Computational analysis of microarray data. Nature ReviewsGenetics 2:418-27.
27. Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Engineering12:85-94.
28. Sankoff, D. 2003. Rearrangements and chromosomal evolution. Current Opinion inGenetics & Development 13:583-7.
29. Simillion, C. 2005. Documentation for i-ADHoRe v2.0. VIB - Ghent University.
30. Simillion, C., K. Vandepoele, Y. Saeys, and Y. Van de Peer. 2004. Building genomicprofiles for uncovering segmental homology in the twilight zone. Genome Research14:1095-106.
31. Simillion, C., K. Vandepoele, M.C. Van Montagu, M. Zabeau, and Y. Van de Peer.2002. The hidden duplication past of Arabidopsis thaliana. Proceedings of theNational Academy of Sciences 99:13627-32.
32. Smith, T.F., and M.S. Waterman. 1981. Identification of common molecularsubsequences. Journal of Molecular Biology 147:195-7.
33. Spring, J. 2003. Major transitions in evolution by genome fusions: from prokaryotesto eukaryotes, metazoans, bilaterians and vertebrates. Journal of Structural andFunctional Genomics 3:19-25.

67
34. Taylor, J.S., and Raes, J. 2005. Small-Scale Gene Duplications, p. 289-327, In G. T.R., ed. The evolution of the genome. Elsevier Inc.
35. The Institute for Genomic Research. ZD. The Institute for Genomic Research[Online] http://www.tigr.org.
36. Van de Peer, Y. 2005. GGS Course in Bioinformatics [Online]. Available by UGenthttp://bioinformatics.psb.ugent.be/intranet.php.
37. Van de Peer Y., and Meyer A. 2005. Large-Scale Gene and Ancient GenomeDuplications, p. 329-368, In G. T., ed. The evolution of the genome. Elsevier Inc.





























