Embed Size (px)
Citation preview

Commission océanographique intergouvernementale
Manuels et guides
LA DETERMINATION DES HYDROCARBURES PÉTROLIERS DANS LES SÉDIMENTS
I I
Unesco 1982

PREFACE
Le présent rapport a été établi par la Commission océanographique inter- gouvernementale (COI) de l'Unesco avec la collaboration d'un consultant, M. Karsten H. Palmork, de l'Institut de recherches marines de Bergen (Norvège), à la demande du Programme des Nations Unies pour l'environnement (PNUE), par l'intermédiaire du programme du Centre d'activités du Programme pour les mers régionales, le Programme coordonné pour la surveillance et l'étude de la pollution en Méditerranée (MED POL). Le PNUE est vivement remercié pour sa contribution.
Les méthodes d'analyse présentées ont été ainsi que leurs limites examinées et approuvées par le Groupe d'experts de la COI sur les méthodes, les normes et l'interétalonnage, sous réserve qu'elles soient modifiées à mesure qu'on dispose- ra de nouvelles informations concernant le choix des solvants, l'efficacité de l'extraction, le choix des normes, etc.
Les méthodes décrites dans le présent rapport ont été mises au point afin de servir de méthodes de référence pour l'analyse du pétrole contenu dans les sédiments dans le cadre de MED POL. Toutefois, étant donné que leur applicabilité dépasse de beaucoup les limites d'une seule région, elles sont publiées a l'inten- tion des océanographes intéressés du monde entier.

- 1 -
TABLE DES MATIEFES
1 .
2 .
3 .
4 .
5 .
6 .
7 .
8 .
Pages
INTRODUCTION ........................................................ 1
PORTEE ET CHAMP D'APPLICATION ....................................... 2
REFERENCES ET BIBLIOGRAPHIE ......................................... 3
PRINCIPE ............................................................ 3
FEACTIFS ............................................................ 4
5.1 Substances chimiques ............................................ 4
5.2 Normes internes ................................................ 4
APPAREILS ET MATERIEL ............................................... 5
PROCEDURE D'ECHANTILLONNAGE ......................................... 6
7.1 Planification .................................................. 6
7.2 Sélection des sites d'échantillonnage ........................... 7
7.3 Prélèvement et stockage des échantillons ....................... 8
7.4 Types d'échantillons ........................................... 9
PROCEDURE D'ANALYSE ................................................. 10
8.1 PrépaKation des échantillons ................................... 10
8.2 Détermination du poids sec ..................................... 10
8.3 Minéralisation de la matrice sédimentaire ...................... 1 1
8.4 Extraction des hydrocarbures pétroliers ........................ 11
8.5 Séparation des alcanes et des aromatiques à l'aide de colonnes remplies d'alumine (à utiliser avant l'analyse par fluorescence- W et par chromatographie en phase gazeuse avec colonnes capillaires) ................................................... 12
8.6 Purification des extraits à l'aide de colonnes remplies de silice ......................................................... 13
8.7 Méthode de dépistage par fluorescence-UV ....................... 15
8.7.1 Etalonnage .............................................. 17
8.7.2 Essais à blanc .......................................... 19

9 .
10
Pages
8.7.3 Calcul de la concentration de l'échantillon initial ..... 19
8.7.4 Essais d'inhibition de la fluorescence .................. 19
8.8 Chromatographie en phase gazeuse utilisant des colonnes garnies . 20
8.9 Chromatographie en phase gazeuse utilisant des colonnes de silice fondue .................................................. 20
8.10 Chromatographie en phase gazeuse/spectrométrie de masse &. !.& ..... dlildtisé es ................................................... 21
PRINCIPES PERMETTANT D'ASSURER LA QUALITE DES DONNEES ............... 27
9.1 Planification .................................................. 27
9.2 Assurance de qualité ........................................... 27
9.3 Définitions de quelques termes statistiques .................... 28
9.3.1 La spécificité .......................................... 28
9.3.2 La sensibilité .......................................... 28
9.3.3 La précision ............................................ 28
9.3.4 L'exactitude ............................................. 28
CONCLUSIONS ......................................................... 29
APPENDICE I ......................................................... 31

DETERMINATION DES HYDROCARBURES PETROLIERS DANS LES SEDIMENTS
1. INTRODUCTION
La nécessité de rechercher les hydrocarbures d'origine pétrolière dans les sédiments peut avoir de multiples raisons, par exemple, pour n'en citer que quelques- unes, (i) le désir d'établir des niveaux de base avant le lancement d'opérations de forage et de production pétrolière en mer, (ii) le fait que la zone en question, un terrain de pêche, a été le site d'un déversement de pétrole et (iii) le fait que la zone en question est soumise à une pollution permanente due à un trafic maritime intense ou à la proximité d'une raffinerie. Toutes ces situations peuvent avoir des incidences sur la vie marine et la pêche. 11 est donc d'une importance primordiale de connaître l'étendue de la pollution par le pétrole, son influence sur la vie marine et la durée de ses effets.
L'analyse d'échantillons d'eau de mer révélera si cette eau est encore polluée. L'analyse d'échantillons de poissons gras ou de foie de poissons maigres indiquera dans quelle mesure les poissons ont souffert de la pollution. L'analyse d'échan- tillons de sédiments peut fournir des renseignements sur les retombées atmosphéri- ques au cours du temps, sur les niveaux de base avant l'entreprise d'activités entraînant le déversement de polluants, sur les effets d'un rejet d'hydrocarbures, sur des filtrations chroniques, sur une pollution permanente due à une raffinerie et tout simplement sur le résultat de l'association d'hydrocarbures avec des parti- cules en chute vers le fond, telles que coprolithes, organismes morts ou vivants, minéraux argileux et silicates. Ces particules sont généralement à l'origine de l'élimination des hydrocarbures de la colonne d'eau. En outre, les hydrocarbures pétroliers dissous peuvent être absorbés directement par les sé,diments selon leur teneur en matière indigène, comme, par exemple, les acides humiquesi
Quelles méthodes faut-il utiliser pour évaluer les hydrocarbures pétroliers ? De nombreuses méthodes d'analyse ont déjà été décrites : l'analyse gravimétrique (substances extractibles non volatiles), méthode par absorption de l'UV (polyolé- fines conjuguées, aromatiques), par fluorescence-UV (composés non saturés, aroma- tiques, etc. , dépendant d'une excitation-h , d'une émission-A , d'une extraction et de la volatilité) , par infrarouge (IR) (fréquence d'allongement C-H2 , 2.930 cm-') , par chromatographie en phase gazeuse utilisant des colonnes garnies (total des hydrocarbures, divers n-alcanes, rapports n-paraffine/isoprénoïdes), chromatographie en phase gazeuse utilisant des colonnes capillaires de silice fondue (divers n- alcanes et hydrocarbures aromatiques polynucléaires après une préséparation sur silice ou sur silice couverte d'alumjme ou sur alumine seulement), et par chromato- graphie en phase gazeuse/spectrométrie de masse associées à un système de données (divers composants, composants présélectionnés) en utilisant le mode de surveillance d ions sélectionnés (SIM) .
Le présent document de référence portera sur les méthodes qui ont déjà été utilisées à ce jour (fluorescence-UV, chromatographie en phase gazeuse utilisant des colonnes capillaires garnies ou de silice fondue et la chromatographie en phase gazeuse/spectrométrie de masse), qui ont toutes été approuvées par plusieurs groupes, le plus récent étant le Groupe d'experts de la GIPME sur les méthodes, les normes et l'interétalonnage, relevant de la COI, à sa quatrième session à Curaçao (Antilles néerlandaises), 25-31 mars 1982.

- 2 -
Pour ces trois méthodes, il faut appliquer avec soin la même procédure d'échan- tillonnage, d'extraction et de traitement (voir sections 7.3, 7.4 et 8.1, 8.2, 8.3, 8.4 et 8.5). Les différences se manifestent au niveau de l'analyse, de l'utilisation des appareils et de l'expression du résultat. Dans la méthode par fluorescence-UV (décrite à la section 8-71, le résultat est présenté sous la forme d'un nombre d'unités de chrysène, une expression du "pétrole" présent dans 1 'échantillon. Dans la méthode de la chromatographie en phase gazeuse utilisant une colonne garnie, le résultat est exprimé en quantité totale d'hydrocarbures. Dans la chromatographie en phase gazeuse et dans la chromatographie en phase gazeuse/spectrométrie de masse utilisant des colonnes capillaires, les résultats peuvent être exprimés en quantités de tel ou tel composant : n-alcanes, hydrocarbures aromatiques polynucléaires et/ou autres composants présélectionnés en utilisant la chromatographie en phase gazeuse/ spectrométrie de masse selon le mode de surveillance d'ions sélectionnés.
Les types de pétrole qui peuvent aboutir dans le milieu marin varient sensible- ment ; ils vont du pétrole brut à différents distillats tels que le fuel lourd, l'essence ou les retombées atmosphériques. Tous ces "types" de pétrole ont quelque chose en commun, ilscontiennentdes composants qui sont dissous dans l'eau de mer et des composants qui sont dégradés par des bactéries et par le rayonnement uitra- violet. 11 est donc inutile d'essayer de recnercher le "pétrole" en tant que tel, parce qu'il n'existe plus sous sa forme initiale quand il a atteint la mer. C'est pourquoi la méthode de la fluorescence-UV peut être utilisée dans ce cas comme moyen de détecter les "points chauds".
L'analyse des hydrocarbures pétroliers devrait avoir pour but de répondre aux questions suivantes : Quels sont les composants les plus dangereux ? Quels composants dérivant du pétrole aboutissent dans les sédiments ? Quand on aura répondu à ces questions, on pourra passer à l'analyse des différents composants.
2. PORTEE ET CHAMP D'APPLICATION
Les méthodes de référence décrites sont une méthode de filtrage par fluores- cence-UV, une méthode de chromatographie en phase gazeuse (détecteur à ionisation de flamme) et une méthode de chromatographie en phase gazeuse/spectrométrie de masse pour la détermination de la quantité totale et des différents composants des hydro- carbures pétroliers contenus dans les échantillons de sédiments. Pour l'analyse chimique des échantillons, les hydrocarbures sont extraits avec du pentane après saponification du sédiment en reflux pendant une heure et demie avec de la potasse alcoolique. L'extrait est séparé en une fraction d'alcane et une fraction aromatique sur une colonne remplie d'alumine (section 8.5 pour les analyses par fluorescence-UV et par chromatographie en phase gazeuse avec colonnes capillaires) et purifié sur une colonne remplie d'un gel de silice (section 8.6 pour les analyses par chromato- graphie en phase gazeuse en colonne garnie et par chromatographie en phase gazeuse/ spectrométrie de masse). La limite de détection pour le "total" des hydrocarbures pétroliers est approximativement de 1 mg/kg de sédiment et pour les différents hydro- carbures, de 100 ng/kg.

- 3 -
3. REFERENCES ET BIBLIOGRAPHIE
Anon, 1980. Guidelines for Data Aquisition and Data Quality Evaluation in Environ- mental Chemistry. Analyt. Chem. 52 (1980) 2242-2249.
Farrington, J. W., 1980. An Overview of the Biogeochemistry of Fossile Fuel Hydro- carbons in the Marine Environment, in Petroleum in the Marine Environment, Advances in Chemistry Series 185, Leonid Petrakis et Fred T. Weiss (dir. publ.), American Chemical Society, Washington D.C. 1980.
Friedrich, H., 1973. Marine Biology. An introduction to its problems and results. Sidgwick & Jackson, London.
Futoma, D. J. ; Ruven Smith, S. ; Smith, T. E. ; J. Tanaka, 1981. Polycyclic Aromatic Hydrocarbons in Water Systems. CRS Press, Inc., Boca Raton, Florida, 1981.
Hites, R. A. ; R. E. Laflamme ; J. G. Windsor jr., 1980. Polycyclic Aromatic Hydro- carbons in Marine/Aquatic Sediments: Their Ubiquity, in Petroleum in the Marine Environment, Advances in Chemistry Series 185, Leonid Petrakis et Fred T. Weiss (dir. publ.) , American Chemical Society, Washington D.C. 1980.
McAuliffe, C. D., 1980. The Multiple Gas-Phase Equilibration Method and its Appli- cation to Environmental Studies, in Petroleum in the Marine Environment, Advances in Chemistry Series 185, Leonid Petrakis et Fred T. Weiss (dir. publ.), American Chemical Society, Washington D.C., 1980.
Moore, H. B. ; Neil, R. G., 1930. An Instrument for Sampling marine muds. J. mar. biol. Ass. U.K. XVI: 589-594. -
Overton, E. B. ; J. L. Laseter, 1980. Distribution of Aromatic Hydrocarbons in Sediments from Selected Atlantic, Gulf of Mexico, and Pacific Outer Conti- nental Shelf Areas, in Petroleum in the Marine Environment, Advances in Chemistry Series 185, Leonid Petrakis et Fred T. Weiss (dir. publ.), American Chemical Society, Washington D.C. 1980.
4. PRINCIPE
Après le prélèvement du sédiment à l'aide d'une benne van Veen ou d'un carot- tier à gravité, les hydrocarbures pétroliers sont isolés des échantillons de sédi- ment par saponification avec de la potasse alcoolique pendant une heure et demie suivie d'une extraction à l'aide de pentane. Un sous-échantillon est utilisé pour la détermination du poids sec. Dans les échantillons fortement pollués, la quantité "totale" d'hydrocarbures pétroliers peut être évaluée par la méthode du filtrage par fluorescence-UV (section 8.7) ainsi que par la chromatographie en phase gazeuse avec colonnes garnies et un détecteur à ionisation de flamme, intégrant à la fois le mélange complexe résolu et le mélange non résolu (section 8.8). Un pétrole aussi semblable que possible au polluant du sédiment est utilisé comme norme externe. Certains hydrocarbures aromatiques non biogenes (par exemple, les naphtalènes, les phénanthrènes et les dibenzothiophènes), qui sont les aromatiques dominant dans les échantillons pollués par des hydrocarbures pétroliers, sont analysés quantitative- ment à l'aide d'une colonne capillaire remplie de silice fondue, de trois normes internes (le diphényle-d l'anthracène-d et le pyrène d ) et d'un spectromètre de masse comme détecteur. 1 o' 1 o' 10

- 4 -
Dans le cas d'un échantillon de sédiment anoxique, le soufre élémentaire pourrait perturber la chromatographie en phase gazeuse. On peut remédier à ce pro- blème en ajoutant une goutte ou deux de mercure métallique pendant la période de reflux ou en utilisant une colonne contenant du cuivre. Toutefois, si l'échantillon est saponifié avec de la potasse alcoolique, le soufre est complètement éliminé.
5. REACTIFS
Tous les réactifs utilisés doivent être de la meilleure qualité possible : p.a. ou qualité nécessaire pour la chromatographie. Si les résultats des essais à blanc sont trop élevés, il faut redistiller les solvants utilisés pour améliorer leur qualité au maximum.
5.1 Subs tances chimiques
5.1.1 Eau distillée
5.1.2 Méthanol p.a. Merck 6009.
5.1.3 Potasse caustique (KOH) p.a. Merck 5033.
5.1.4 Pierres réfractaires, alundum (RI maille approx. 8 à 14 Cat. No 1590-D 18, Arthur H. Thomas Company, Philadelphie, Pa. 19105 (Etats-Unis d'Amérique).
5.1.5 Pentane, Uvasol Merck 7179
5.1.6 Hexane, Uvasol Merck 4369
5.1.7 Azote, qualité 3.
5.1.8 Acide chromique contenant 4 g de bichromate de potassium (K Cr O ) dans un 2 2 7 litre d'acide sulfurique concentré.
5.1.9 Gel de silice (Kieselgel 60) dimension granulométrique 0,063-0,125 mm (maille ASTM 120-230) Merck 9386.
5.1.10 Oxyde d'aluminium, activité (anionotropique) de l'acide Woelm, qualité I pour la chromatographie. M. Woelm Eschwege (Allemagne).
5.2 Normes internes :
5.2.1 Diphényle-d MD-208 (99 % atome DI Lot no B-1011. Merck et Co., Inc. Rahway, N. i? ou Merck Sharp et Dohme Canada Limited, Montréal (Canada) (MSD-ISOTOPES)
5.2.2 anthracene-d MD-46 (98 % atome D) Lot no C-570. Merck et Co., Inc. 10 Rahway, N. J.
ou Merck Sharp et Dohme Canada Limited, Montréal (Canada) (MSD-ISOTOPES)
5.2.3 pyrène-d MD-363 (98 % atome D) Lot nu C-569. Merck et Co., Inc. Rahway, 10 N. J.
ou Merck Sharps et Dohme Canada Limited, Montréal (Canada) (MSD-ISOTOPES)

- 5 -
5.2.4 Un mélange contenant approximativement 0,5kg de 5.2.1, 5.2.2 et 5.2.3 par ml dissous dansl'nexane. Le mélange peut être préparé comme suit : trans- vaser 100 mg de 5.2.1, 5.2.2 et 5.2.3 dans une fiole jaugée et diluer jus- qu'à 100 ml avec de l'hexane; solution A.
Prendre 1 ml de la solution A et diluer jusqu'à 100 ml avec de l'hexane. La solution obtenue (solution B) contient approximativement 10 A g de chacune des normes par ml.
Prendre 5 ml de la solution B et diluer jusqii'à 100 ml avec de l'hexane. La solution obtenue (solution C) contient approximativement O , 5 Ag/ml de chacune des normes deutériques. La pesée doit se faire au 0,0001 g près et être notée pour que le titre de la solution puisse être calculé exactement.
6. APPAREILS ET MATERIEL
6.1 Boites isolantes en plastique, pour les périodes chaudes réfrigérateur (+1 à +4OC) ou glace pour le refroidissement.
6.2 Congélateur (-18 à -2OoC), si les échantillons voyagent plus de 48 heures.
6.3 Feuille d'aluminium propre pour envelopper les échantillons de sédiments.
6.4 Bocaux en verre propres pour les échantillons de sédiments.
6.5 Etiquettes ou marques pour l'identification des échantillons de sédiments (voir fig. 5).
6.6 Six flacons de verre de 250 ml; Quickfit ou Jena.
6.7 +
Une balance, E Mettler type B5, no 14788 (- O,OOl+g) et une autre à charge- ment par le haut, type K71 no 88298 (100-200 g) (- O, 1 9).
6.8 Isomantel, type FSE,&/lL/6, 220/240 volts 6x280 watts, phase S/PH, circuits 1, numéro de série WB 4649, Isopad Ldt., Bosehamwood, Herts.
6.9 Isopad, F.E.R. Control, type no 7422 Bx.
6.10 Six entonnoirs de séparation de verre de 250 ml avec des Douchons de téflon; verre de borosilicate BS 2021.
6.11 Un Rotavapor ; Rotavapor Buchi, EL, Glasapparatefabrik, Flawil (Suisse) avec bain thermostatique et thermostat.
6.12 Ballons de verre 5 fond rond de 100 ml pour le Rotavapor.
6.13 Paquets de fioles (2 mi) 5080-8712, fiole-4330-0525 Pl Hewlett Packard.
6.14 Paquets de capsules, 5080-8713, capsule-1540-0132 P, Hewlett Packard.
6.15 Une pince pour le capsulage des fioles ; Wheaton 224301, Millville, N.J.
6.16 Seringue (100,;~l) pour le transvasement des solvants (Hamilton).

- 6 -
6.17
6.18
6.19
6.20
6.21
6.22
NOTE
Seringues (10 et 1,ill) pour l'injection de l'échantillon dans la chromato- graphie en phase gazeuse et dans la chromatographie en phase gazeuse/spec- trométrie de masse, seringue Hamilton 7001, Supelco, 1 mc. Bellefonte, Pennsylvanie 16823.
Chromatographe en phase gazeuse pour colonne garnie et colonne capillaire de silice fondue.
Chromatographe en phase gazeuseLspectromètre de masse/système de données adaptés aux colonnes capillaires de verre ou aux colonnes de silice fondue.
Colonnes capillaires de verre ou de silice fondue, approximativement 25 m x 0,21 mm de diamètre intérieur, aux parois enduites de SE-52, SE 54 ou SP-LlOU.
Séchoir ou four (100' a 3OOOC).
Spectrophotomètre par f luorescence-W.
:,Les ustensiles de verre qui sont utilisés pour la premiere fois (section 8) doivent être parfaitement nettoyés de la manière suivante :
- laver avec de l'eau et du savon
- rincer à l'eau propre, de préférence sous le robinet et rincer enfin avec de l'eau distillée et laisser égoutter
- laisser 1'ust.ensile dans l'acide chromique (5.1.8) , de préférence la nuit
- rincer à l'eau et enfin plusieurs fois avec de l'eau distillée et égoutter.
toute
laisser
7. PROCEDURE D'ECHANTILLONNAGE
L'échantillonnage est la base des études sur la pollution par les hydro- carbures pétroliers, c'est donc une procédure très importante. Le prélèvement d'échantillons de sédiments à des fins d'analyse chimique peut être divisé en quatre phases : (1) planification, (2) sélection des sites d'échantillonnage et détermination du nombre d'échantillons nécessaires pour étudier la zone en question, (3) les techniques de collecte (benne van Veen ou carottier à gravité) et de stoc- kage d'échantillons , et (4) type d'échantillon.
7.1 P 1 an if i cat i on
Un plan d'échantillonnage doit être conqu en fonction de la nature de la situation à étudier.
I1 est peu probable que les sédiments contiennent une quantité importante d'hydrocarbures dissous ; en effet il faut que le pétrole traverse la colonne d'eau pour parvenir aux sédiments. Beaucoup plus vraisemblablement les hydrocarbures solubles ont ëté éliminés avant que les gouttelettes atteignent les sédiments (McAuliffe, 1980).

- 7 -
Avant d'entreprendre une étude ou une surveillance il est indispensable d'exé- cuter une enquête initiale : le taux de sédimentation est normalement faible et un échantillonnage annuel ou biennal devrait être suffisant pour une surveillance des tendances.
7.2 Sélection des sites d'échantillonnage
Les sites d'échantillonnage sont normalement choisis sur une vaste grille qui couvre la zone géographique touchée par un déversement de pétrole, laquelle est choisie comme zone de référence, ou susceptible d'être le théâtre d'opérations de forage 2 l'avenir (Fig. 1).
Etant donné que l'analyse des échantillons d'hydrocarbures pétroliers est couteuse, il faut déterminer avec soin le nombre des échantillons recueillis et les sites d'échantillonnage. Les échantillons doivent être choisis de manière à per- mettre d'atteindre les objectifs ; cela signifie généralement qu'on procédera à un suréchantillonnage sachant cependant qu'un certain nombre d'échantillons seulement seront choisis pour être analysés à un stade ultérieur.
Les échantillons doivent être recueillis en plusieurs exemplaires toutes les fois que cela sera possible à chaque point de la grille. L'analyse des doubles per- mettra de déterminer la variance des paramètres mesurés. Cette variance servira ensuite à indiquer le nombre de doubles nécessaires pour déceler un changement sta- tistique dans les valeurs mesurées dans certaines limites de confiance. Le nombre de doubles calculé à des fins statistiques suppose que les valeurs sont "normalement distribuées" (c'est-à-dire courbe de Gauss, fig. 2).
Fig. 1 - Réseau pour l'échantillonnage de sédiments. O-stations d'échantillon- nages, &forage et, la flèche indique la direction du courant résiduel.
- 8 -
Y
- 1
X X X
Fig. 2 - Distribution normale, courbe de Gauss. On observe généralement cette distribution si les hydrocarbures pétroliers
présents dans le sédiment représentent les retombées atmosphériques au cours du temps. si les composants présents proviennent d'un deversement de pétrole récent avec une distribution inegale de pétrole dans les sédiments, les concentrations d'hydrocarbures dans un groupe de doubles risquent de ne pas être distribuées nor- malement et, par conséquent, les tests-t de Student ne sont pas applicables. Dans de tels cas, ces échantillons doivent être classés en fonction de la source de la pollution.
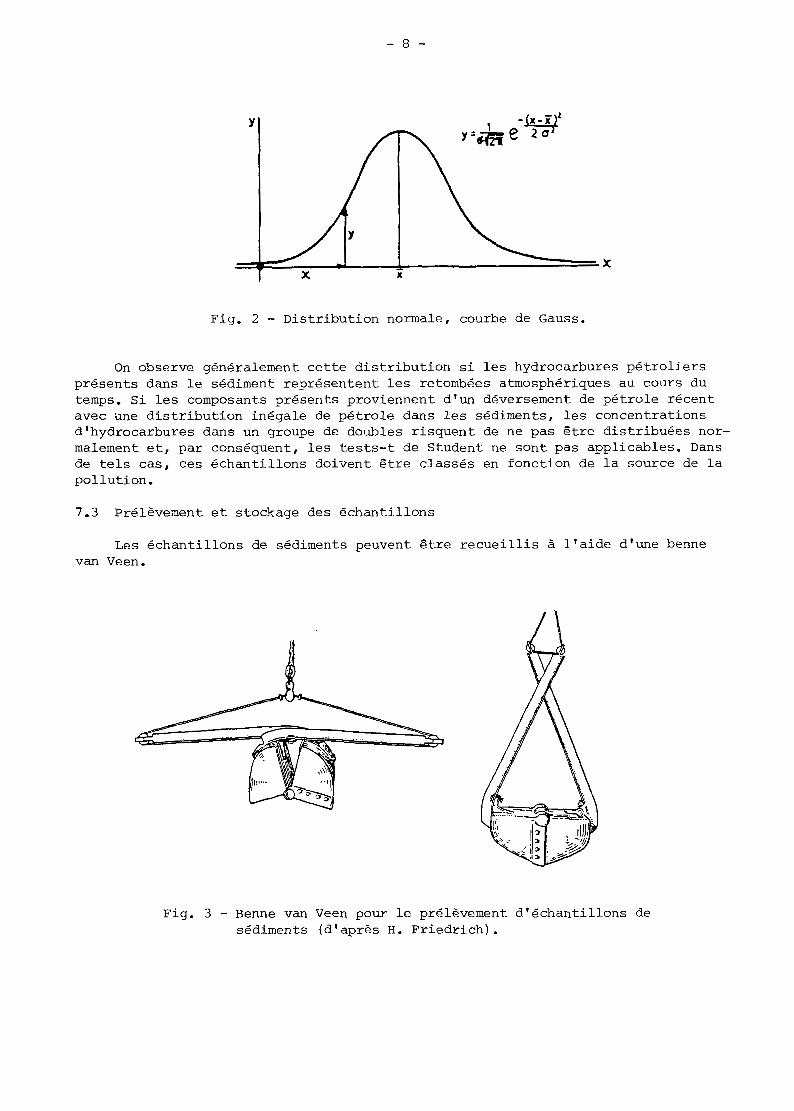
7.3 Prélèvement et stockage des échantillons
Les échantillons de sédiments peuvent être recueillis à l'aide d'une benne van Veen.
Fig. 3 - Benne van Veen pour le prélevernent d'échantillons de sédiments (d'après H. Friedrich).

- 9 -
Après récupération de l'appareil, il faut laisser l'eau s'égoutter en évitant de perturber la couche de surface des échantillons. Les doubles des échantillons, d'en- viron 500 ml, doivent être prélevés soigneusement sur les cinq premiers centimètres de la couche par l'ouverture située en haut de l'appareil (fig. 3). Dès que les doubles sont prélevés, ils doivent être enveloppés dans une feuille d'aluminium OU placés dans des bocaux de verre et immédiatement congelés à - 2OoC afin d'être ana- lysés ultérieurement.
Les échantillons de sédiments peuvent également être recueillis à l'aide d'un carottier doublé de verre (MOORE et NEIL, 1930, fig. 4).
Le manchon de verre contenant l'échantillon de sédiment peut être congelé. On peut extraire le sédiment du manchon après l'avoir décongelé a.vec une serviette chaude et le couper à la longueur désirée avec un couteau.
a.
e au
b. sédiment
Fig. 4 - a. Manchon de verre d'un carottier à gravité. Le sédiment de la partie supérieure peut servir à des analyses de tendances. b. Ensemble d'un préleveur d'échantillons du fond.
Lorsque l'échantillon de sédiment est récupéré, il est très important de le stocker dans un endroit sûr avec une étiquette indiquant son identité et tous les renseignements nécessaires (fig. 5).
L'échantillon doit &tre constamment protégé de la lumière et gardé au frais soit dans un réfrigérateur (+ 1 2 + 4OC), soit dans une boîte isolante refroidie avec de la glace jusqu'à ce qu'il puisse être congelé, de préférence à -18 ou -2OOC.
7.4 Types d'échantillons
Dans certains cas, lors du prélèvement d'échantillons (par exemple pendant les déversements de pétrole), il est recommandé de recueillir également d'autres types d'échantillons aux fins d'analyse chimique. Pour établir la source et l'identité du pétrole qui cause la pollution, il faut recueillir un échantillon de ce pétrole (1 à 5 9). Dans certains cas, il faut prélever des échantillons d'eau (3 à 5 1) pour déterminer si la pollution des masses,d'eau par le pétrole est toujours nota- ble et il.faudrait aussi recuejllir des organismes marins pour y rechercher les hydrocarbures pétroliers et étab1i.r ainsi le degré de pollution.

- 10 -
CHAIN OF CUSTODY RECORD
EHVIRONMEHTAL PROTECTION AGENCY
SAMPLE NO.
SOURCE OC SAMCLI
Fig. 5 - Etiquette permettant d'identifier les échantillons recueillis (utilisée par la U.S. Environmental Protection Agency).
8. PROCEDURE D'ANALYSE
8.1 Préparation des échantillons
Peser un ballon à fond rond, propre et sec, sans le bouchon à l'émeri et noter le poids. Peser dans ce ballon approximativement 150 g au O l l g près de sédiments en partie dégelés, noter le poids exact et calculer le poids humide.
8.2 Détermination du poids sec
Une bouteille propre, sans son bouchon à l'émeri, est placée dans le séchoir ou dans le four (lOO°C, 2 heures), à l'aide d'une pince propre. I1 est important d 'utiliser des pinces chaque fois qu'on touche le verre pour éviter de laisser des traces de doigts et des particules de poussière sur la bouteille. Le bouchon et la bouteille sont placés à refroidir dans un dessiccateur. -
La bouteille vide et l&.bouchon sont ensuite soigneusement pesés sur une balance de précision. Le poids obtenu est le poids de la bouteille vide et du bouchon séchés Noter le poids. Enlever le bouchon et placer 25 à 50 g de sédiments dans la bouteille et reboucher.
Déterminer soigneusement le poids de la bouteille, du bouchon et du sédiment. Le poids obtenu est le poids humide du sédiment plus le poids de la bouteille et du bouchon.
Placer la bouteille dans le séchoir ou dans le four (lOO°C), enlever le bouchon et le placer également dans le four.
Après 24 heures, remettre le bouchon sur la bouteille, retirer la bouteille avec le bouchon du four et les placer tous les deux à refroidir dans un dessiccateur, Peser la bouteille bouchée et noter le poids. Répéter le cycle de séchage jusqu'à ce que la différence entre les pesées suivantes atteigne moins de 5 % du poids total ; noter le poids humide et le poids sec et calculer leur rapport.

- 11 -
Note : Lors de l'analyse des hydrocarbures pétroliers dans les sédiments, l'alté- ration est moindre si l'on recherche le poids humide de l'échantillon au lieu de faire sécher celui-ci à 100OC. Néanmoins, si l'on calcule à la fois le poids humide et le poids sec, ces valeurs peuvent être comparées à celles qui ont été relevées précédemment.
8.3 Minéralisation de la matrice sédimentaire
Le matériel nécessaire à cette phase est présenté sur la figure 6. Placer 80 à 100 g de l'échantillon de sédiments humides dans un ballon à fond rond, ajouter 100 ml de méthanol redistillé, 3 g de KOH et de pierres réfractaires. Ajouter 1 ml d'un mélange d'une solution titrée interne contenant, à raison de 0,5 W/mi de cha- que substance, du biphényle, de l'anthracène et du pyrène deutériques dissous dans l'hexane. Chauffer le mélange au reflux pendant une heure et demie (Fig. 7).
8.4 Extraction des hydrocarbures pétroliers
Refroidir l'extrait contenant du méthanol à la température de la pièce, le transvaser dans un entonnoir de séparation (Fig. 8) et extraire deux fois avec 25 ml de pentane (Uvasol). Séparer la phase méthanol de la phase pentane et trans- férer celle-ci dans une fiole d'évaporation et réduire le volume à 0,5 ml en uti- lisant un Rotavapor (Fig. 9). Transvaser le volume réduit dans une fiole de verre en utilisant du pentane (Fig. 10). Concentrer cet extrait soigneusement à approxi- mativement 0,2 ml en utilisant du gaz N sec avant purification sur une colonne 2 remplie d' alumine (5.1.10) .
Fig. 6 - Vue d'ensemble du matériel nécessaire au traitement des échantillons de sédiments : A = N (5.1.7), B = Fiole (6.13), C = KOH (5-1.31, D = méthanol (5.1 -2): E = pierres réfractaires (5.1.4) , F = norme interne (5.2) , G = pentane (5.1.5), H = échantillon de sédiment, I = entonnoir de séparation (6.10) et J = dispositif pour le reflux des échantillons (6.8).

- 12 -
8.5 Séparation des alcanes et des aromatiques à l'aide de colonnes remplies d'alumine (à utiliser avant l'analyse par fluorescence-UV et par chromato- graphie en phase gazeuse avec colonnes capillaires)
Une pipette Pasteur équipée d'un bouchon de laine de verre est remplie d'en- viron 5 cm (1,15 g) d'oxyde d'aluminium (5.1.10). La colonne est rincée trois fois avec 2 ml de pentane (5.1.5) et l'extrait d'échantillon est ajouté en haut de la colonne. L'élution est exécutée comme le montre le tableau 1. Les différents éluats sont desséchés sous l'action d'un flux de gaz N Pour la méthode de la fluorescence- W, dissoudre les résidus contenant les aromatiques (c'est-à-dire les fractions 3 et 4) dans de l'hexane sans aromatiques et transvaser dans une fiole jaugée de 1 ou 5 ml et ajouter de l'hexane jusqu'au trait de repère. Pour l'analyse par chromato- graphie en phase gazeuse, dissoudre les résidus dans une quantité connue d'hexane (entre 40 pl et 1 ml selon la concentration des hydrocarbures).
2'
9
Fig. 7 - La fiole contenant le sédiment, le méthanol, le KOH et les fragments de pierres réfractaires est prête à être chauffée au reflux.

- 13 -
8.6 Purification des extraits à l'aide de colonnes remplies de silice
Une pipette Pasteur équipée d'un bouchon de laine de verre est remplie de 0,5 g de silice (5.1.9). Rincer la colonne tro,is fois avec du pentane (5.1.5), puis placer l'échantillon en haut de la colonne et rincer la fiole trois fois avec 1,0 ml de pentane. Eluer les hydrocarbures pétroliers totalement avec 12 ml de pentane en tout. Dessécher l'extrait au pentane sous l'action d'un flux de gaz N
Tableau 1. L'élution des différentes fractions dans la séparation des alcanes et des aromatiques (provenant d'un extrait de sédiment).
2'
Volume
en ml
Rapport Fractions du solvant résultantes No d'éluat d'éluat Solvant d'élution
1 4 Pentane Alcanes
2 4 Pen tane
3 4 Pentane : dichlorométhane 7:3
Solvants seulement
(a) Aroma tiques
4 4 Dich lorométhane Aromatiques supérieurs (b)
(Voir fig. 13 p. 17 pour l'illustration des séparations).
(a) Voir fig. 14 (a) pour l'équivalent de l'éluat 3.
(b) Voir fig. 14 (b) pour l'équivalent de l'éluat 4.
Pour les analyses par chromatographie en phase gazeuse avec une colonne garnie ainsi que par chromatographie en phase gazeuse/spectrométrie de masse selon le mode de surveillance d'ions sélectionnés, dissoudre le résidu dans une quantité connue d'hexane (entre 40 Hl et 1 ml selon la concentration d'hydrocarbures).

- 14 -
Fig. 8 - Transfert de l'extrait contenant du méthanol dans un entonnoir de séparation (6.10).
Fig. 9 - Réduction de l'extrait contenant du pentane dans des conditions de pression réduite avec le Rotavapor (6.11).

- 15 -
8.7 La méthode de dépistage par fluorescence-UV
L'analyse des hydrocarbures pétroliers par la méthode de la fluorescence-UV est présentée ci-après. Lorsqu'on procède à ces analyses, il faut tenir compte des points mentionnés dans l'Appendice I.
Un échantillon de l'extrait dissous dans du ri-hexane doit ëtre placé dans un récipient en verre de silice de 1 cm muni d'un couvercle. Mesurer l'intensité de la fluorescence à 360 nm (excitation à 310 nm). 11 faudrait, si possible, explorer les spectres d'excitation et de fluorescence pour chaque échantillon. Pour le me- lange des substances fluorescentes (c'est-à-dire essentiellement benzènes de substi- tution, naphtalènes et composés aromatiques polynucléaires) présentes dans le pé- trole brut et dans le fuel-oil résiduel l'excitation est la plus forte à 3:O nm et la fluorescence la plus intense vers 360 nm.
Fig. 10. Transfert du volume réduit de l'extrait dissous dans du pentane dans une fiole (6.13) avec une pipette Pasteur.

- 16 -
Fig. 11. Capsulage à l'aide d'une pince (6.15) de la fiole contenant l'extrait à injecter dans le chromatographe en phase gazeuse.
Fig. 12. Injection de l'échantillon dans le chromatographe en phase gazeuse, HP 5880, (6.18) équipé d'une colonne capillaire de silice fondue (6.20).
- 17 -
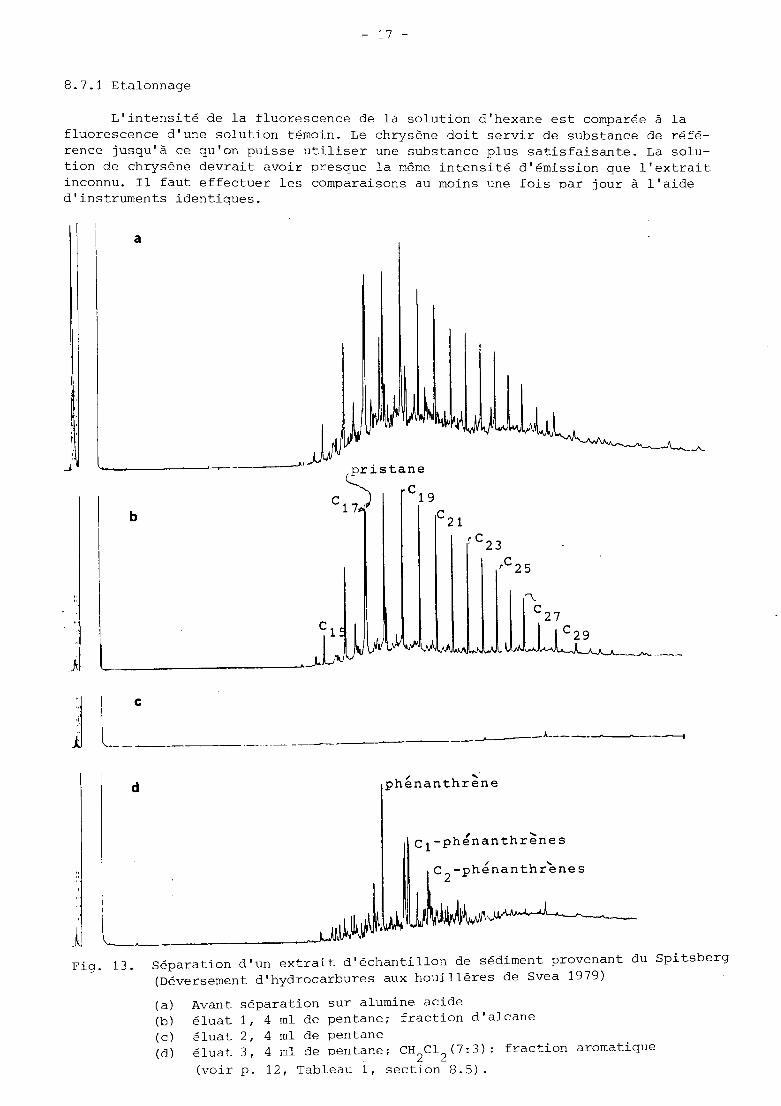
8.7.1 Etalonnage
L'intensité de la fluorescence de la solution d'hexane est comparée à la fluorescence d'une solution témoin. Le chrysène doit servir de substance de réfé- rence jusqu'à ce qu'on puisse utiliser une substance plus satisfaisante. La solu- tion de chrysène devrait avoir presque la même intensité d'émission que l'extrait inconnu. 11 faut effectuer les comparaisons au moins une fois par jour à l'aide d'instruments identiques.
a
h tane
'23
d p h 6 n an th r ;.ne I
q-phénanthrènes
C -phénanthrènes 1 2
Fig. 13. Séparation d'un extrait d'échantillon de sédiment provenant du Spitsberg (Déversement d'hydrocarbures aux houillères de Svea 1979)
(a) (b) éluat 1, 4 ml de pentane; fraction d'alcane (c) iluat 2, 4 ml de pentane (d)
Avant séparation sur alumine acide
éluat 3, 4 ml de pentane; CH C1 (7:3): fraction aromatique (voir p. 12, Tableau 1, section 8.5).
2 2
- 18 -
Fig. 14a.
4J U id Ici a, u Li ru
N
1:
a, C #a, Li c ci E: ru E:
‘al s a
1 21
d a, c a d h c 4J
’al E!
e.4
a I io
m
c id Li O 5 d U4
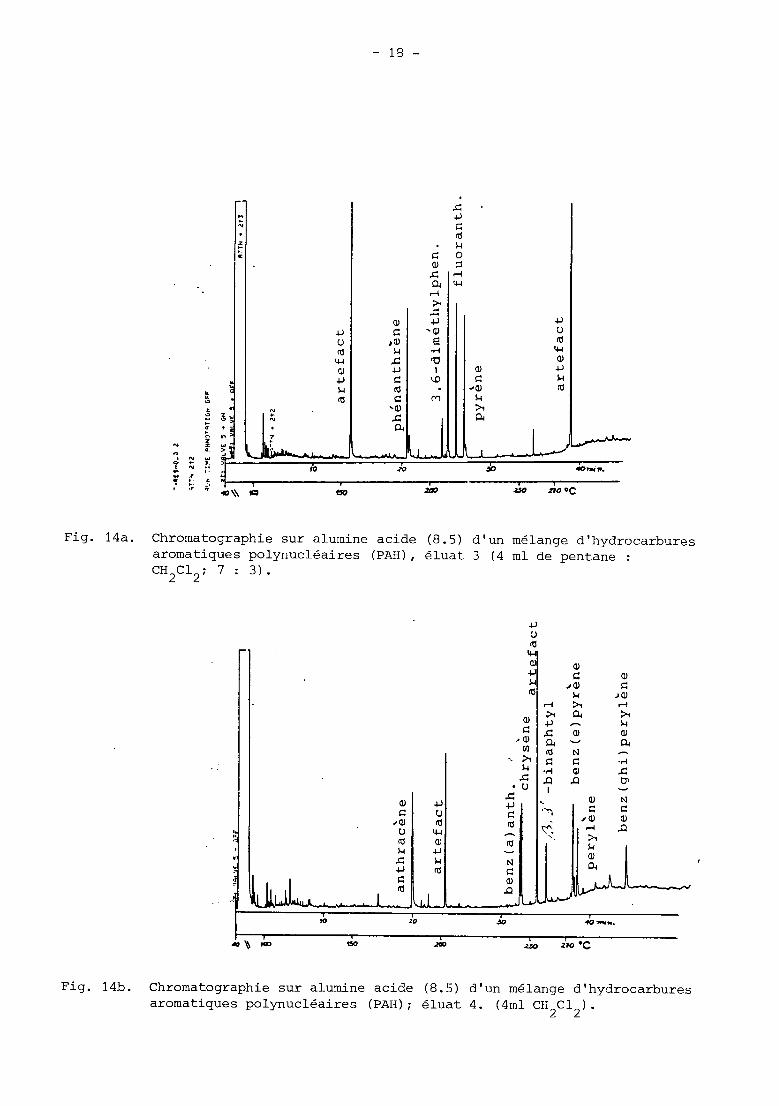
Chromatographie sur alumine acide (8.5) aromatiques polynucléaires (PAH), éluat CH2C12; 7 : 3).
. _
,!
4 ‘i
al C
.al U id LI c c,
+ c m Iu a, u Li id
d’un mélange d’hydrocarbures 3 (4 ml de pentane :
al _i Q
a, E:
Aal LI
.-ln h a u - e a , a - i d N C E : 4 a, Q Q
I al
a, C
.a, rl $i LI a, a 4 c P N E: a, Ll
- Y
Fig. 14b. Chromatographie sur alumine acide (8.5) d’un mélange d’hydrocarbures aromatiques polynucléaires (PAH); éluat 4. (4ml CH C1 ). 2 2

- 19 -
8.7.2 Essais à blanc
Pendant l'opération, on prendra bien soin de ne pas contaminer les échan- tillons ; par exemple, on évitera de laisser inutilement l'échantillon de sédiment, le n-hexane ou l'extrait final en contact avec l'atmosphère ou d'autres sources possibles de contamination. 11 faut vérifier fréquemment que les solvants et l'appa- reillage ne sont pas contaminés en pratiquant des essais à blanc. Le mieux à cette fin consiste à procéder à l'ensemble de l'opération sans l'échantillon de sédiment. Lorsque aucours des essais à blanc la fluorescence est suffisamment intense pour indiquer une contamination (c'est-à-dire que la fluorescence est supérieure à celle correspondant à 1 microgramme par litre de chrysène), il convient d'éliminer les sources de contamination au lieu de corriger les données effectivement obtenues d'après les résultats des essais à blanc.
8.7.3 Calcul de la concentration de l'échantillon initial
La concentration des hydrocarbures dans l'extrait d'échantillon, en équi- valents chrysène, s'obtient par la courbe d'étalonnage du chrysène et est corrigée en cas de fluorescence au cours des essais en blanc. La concentration de l'échan- tillon initial en équivalents chrysëne est alors calculée selon la formule suivante :
B x C D
A = - - -
A = Concentration des hydrocarbures dans les échantillons initiaux en/,L4/g
B = Concentration dans l'extrait d'échantillon eny*g/ml
C = Volume de l'extrait en ml
D = Poids de l'échantillon initial en grammes
Le résultat est la concentration en équivalents chrysène en/l.g/grammes.
8.7.4 Essai d'inhibition de la fluorescence
11 arrive des cas OU de fortes concentrations de composés autres que des hydrocarbures peuvent être présentes dans l'échantillon et inhiber la fluorescence des hydrocarbures dans celui-ci. On peut réaliser l'expérience suivante pour véri- fier si tel est le cas :
(i) on mesure la fluorescence de l'extrait d'échantillon ;
(ii) on mesure la fluorescence de la solution témoin de chrysëne ;
(iii) on combine des volumes égaux de l'extrait d'échantillon et de la solution témoin de chrysène et l'on mesure la fluorescence du mélange.
La fluorescence du mélange doit être égale à :
fluorescence de + fluorescence de la solution tSmoin de chrysène l'extrait d'échantillon
2 2

- 20 -
Si la fluorescence est infêrieure de 20 % ou plus à l'intensité prévue, l'inhi- bition est importante et il faut s'efforcer de purifier l'échantillon (8.6).
8.8 Chromatographie en phase gazeuse utilisant des colonnes garnies
Pour évaluer la "teneur totale en pétrole" des sédiments, on peut utiliser la chromatographie en phase gazeuse avec des colonnes garnies, quoique l'on perde beaucoup d'informations. On injecte deux microlitres de l'extrait d'échantillon, qui a été soumis à une procédure de purification (8.6), dans le chromatographe en phase gazeuse (voir Tableau 2 pour les conditions chromatographiques).
La solution ci-apres pourra servir d'échantillon témoin : 2 ,ql d'une solution contenant 150 mg de pétrole brut ou du petrole en question dans 25 mL d'hexane : 12 ag sont injectés. Les chromatogrammes peuvent être intégrés à l'aide d'un inté- grateur ou d'un planimètre. I1 s'est révélé utile de n'intégrer qu'une "fenêtre" du chromatogramme, par exempln, entre n-C et n-C - il faut alors intégrer et déter- 15 26 ' miner cette aire par )~4 de pétrole de reference pour calculer la "quantité totale de pétrole" eii utilisant la zone entre n-C et n-C 26' 15
I1 convient d'analyser le pétrole de référence en appliquant des conditions identiques à celles qui sont appliquées pour les échantillons.
Tableau 2. Conditions de la chromatographie en phase gazeuse avec colonne garnie.
Un chromatographe en phase gazeuse équipé d'un détecteur à ionisation de flamme
Gaz porteur : Azote -17 mi/min
Co 1 on ne : 200 cm x 4 mm de diamètre intérieur 3 % SP 2100 sur Chromosorb W HMDSO maille 80-100
Température d'injection : 3OOOC
Température du collecteur : 350°C
Programmation de la température : Valeur initiale à 80°C Temps initial 3 min Vitesse de programmation 8OC/min Temps finai 15 min
Volume injecté : 2 M 1
8.9 Chromatographie en phase gazeuse utilisant des colonnes capillaires de silice fondue
Un volume de 1 à 2 A l d'extrait d'échantillon soumis à la procédure de sépa- ration ci-dessus (section 8.5) est injecté par la technique de l'injection non fractionnée. Les conditions chromatographiques sont indiquées dans le tableau 3.

- 21 -
Tableau 3. Conditions de la chromatographie en phase gazeuse avec colonnes capillaires
Chromatographe en phase gazeuse HP 5880A équipé d'un détecteur à ionisation de flamme.
Gaz porteur : Azote 8 psi (pression en livres par pouce carré) I 1,5 mY/min
Gaz d'appoint : Azote 40 fi/min
Colonne : 25 m x 0133 de diamètre intérieur. Silice fondue SE-54
Température d'injection : 280°C
Température du collecteur : 280°C
Programmation de la température : Valeur initiale 4OoC Temps initial 4 min Vitesse de programmation 10"C/min Temps final 15 min
Volume injecté : 1,5 ,ul non fractionné
On mesure les aires des pics en utilisant un système d'intégration électronique et on les identifie en comparant les temps de rétention de la chromatographie en phase gazeuse avec ceux des solutions témoins connues et avec les précédentes ana- lyses par chromatographie en phase gazeuse/spectrométrie de masse d'échantillons analogues dans 'des conditions identiques et avec la même colonne.
La quantification des pics identifiés et intégrés s'effectue par comparaison avec la norme interne la plus proche.
8.10 Chromatographie en phase gazeuse/spectrométrie de masse automatisées
Un volume de 0,3 ml de l'extrait d'échantillon- soumis à la procédure de puri- fication ci-dessus (8.6) est injecté par la technique de l'injection non fraction- née. Les conditions chromatographiques sont décrites dans le tableau 4.

- 22 -
Tableau 4. Conditions applicables au chromatographe en phase gazeuse/ spectromètre de masse automatisés (GC/MS/Comp.)
GC/MS/Comp. Finnigan, Modèle 9000/3200~'/6100
Gaz porteur : Hélium 1,5 d/min
Colonne : 25 m x 0,2 mm diamètre intérieur. Silice fondue Sp 2100
Température d'injection : 280°C
Programmation de la température : Valeur initiale 2OoC, puis 20-100°C en 1 min
Temps initial 1 min
: Vitesse de programmation 6"C/min à partir de 100-230°C
: Temps final 5 min isotherme
Volume injecté : 0,3 fiL non fractionné
La colonne est prolongée jusqu'à la source des ions sur le spectromètre de masse 3200 Finnigan. Les hydrocarbures aromatiques ci-après se sont révélés utilisables pour les analyses en utilisant le mode de surveillance d'ions sélectionnés, tableau 5.
Les figures 15a et 15b des pages 25 et 26 sont des exemples de chromatogrammes de certains hydrocarbures aromatiques obtenus selon le mode de surveillance d'ions sélectionnés.
Tableau 5. Hydrocarbures aromatiques sélectionnés pour les analyses
Compos ants ~~ ~~ ~~
Ions moléculaires
Naphta 1 ène m/z 128
Méthylnaphtalènes (C1)
C -naphtalènes
C ~ - n aph ta lèn e s Phénanthrène
Méthylphénanthrènes (C
C -phénanthr@nes
Méthyldibenzothiophènes (C
2
3
1
2
1
m/z 142
m/z 156
m/z 170
m/z 178
m/z 192
m/z 206
m/z 198
C -dibenzothiophènes m/z 212
C -dibenzothiophënes m/z 226 2
3

- 23 -
La quantification des naphtalènes est fondée sur la norme interne, le biphé- nyle deutérique (ion-164). L'anthracène deutérique (ion-188) est utilisé pour la quantification du phénanthrène, du méthylphénantrène, du méthyldibenzothiophène ; et le pyrène deutérique (ion-212) est utilisé pour la quantification du diméthyl- phénanthrène, du diméthyldibenzothiophène et du triméthyldibenzothiophène.
Les aires des différents pics sont calculées à l'aide d'un ordinateur 6100 Finnigan.
Les facteurs de réponse des différents hydrocarbures aromatiques corres- pondant aux solutions témoins deutériques - le biphényle, l'anthracène et le pyrène - sont déterminés en injectant des quantités connues des composants aroma- tiques choisis dans un mélange de pétrole brut Ekofisk (riche en naphtalènes et en phénanthrènes) et de brut léger d'Arabie (riche en dibenzothiophènes).
La chromatographie du brut Ekofisk (20 mg) se fait sur une courte colonne contenant de la silice (2,5 cm de silice dans une pipette Pasteur). Legel de silice (maille 120-230) est activé pendant la nuit 5 105OC availt d'être utilisé. Les al- canes (et quelques naphtalènes inférieurs) sont élués avec 7 ml de pentane. La fraction aromatique est ensuite éluée avec 5 ml de pentane. L'éluat est desséché et ensuite dissous dans 100 microlitres d'hexane.
Le pétrole brut léger d'Arabie est traité de la même manière pour obtenir un mélange aromatique riche en dibenzothiophènes.
Ces deux mélanges sont analysés par la chromatographie en phase gazeuse avec un détecteur à ionisation de flamme, une colonne capillaire et dans les mêmes con- ditions que celles qui concernent les analyses par chromatographie en phase gazeuse/ spectrométrie de masse. On part du principe que les composants qui se ressemblent donnent la même réponse par poids unitaire. La composition des mélanges aromatiques (en pourcentage) est calculée d'après les aires des chromatogrammes (phase gazeuse). Lorsque cette composition est connue, des quantités exactes de naphtalène, 2,6-di- méthylnaphtalène et 2,3,6-triméthylnaphtalène sont ajoutées pour permettre la détermination des naphtalènes. Du phénanthrène est ajouté pour déterminer le méthyl- phénanthrène et le méthyldibenzothiophène, et du fluoranthène est ajouté pour la détermination du diméthylphénanthrène, du diméthyldibenzothiophène et du triméthyl- dibenzothiophène. Les nouveaux mélanges sont alors analysés par chromatographie en phase gazeuse et les aires des pics intégrées. L'information est alors suffisante pour déterminer les naphtalènes, les phénanthrènes et les dibenzothiophènes dans le mélange aromatique.
Pour pouvoir calculer les facteurs de réponse des différents hydrocarbures aromatiques Dar la méthode de la chromatographie en phase gazeuse/spectrométrie de masse, des quantités connues des trois normes deutériques sont ajoutées aux mélanges aromatiques et les échantillons sontensuite analysés par chromatographie en phase gazeuse/spectrométrie de masse selon le mode de la surveillance d'ions sélectionnés. Puisqu'on connalt alors à la fois la quantité d'hydrocarbures aromatiques et les normes deutériques, on peut calculer les facteurs de réponse.
Exemple :
Le facteur de réponse (fr) du naphtalène par rapport au biphényle-d est le nombre qui est multiplié par l'aire du fragment 128 (le naphtalène est dékgcté par l'ion-128) pour obtenir la quantité exacte.

- 24 -
Le facteur de réponse pour le naphtalène est calculé comme suit :
quantité connue de napht/aire du napht
fr = quantité connue de la norme interne (biphényle-d )/ aire de la norme interne 10
18,5/733351* = o,66 37,3/980978 fr =
On calcule le facteur de réponse pour le phénanthrène en utilisant comme
10' norme interne 1 ' anthracene-d
Le mélange aromatique normalisé est composé de mélanges de brut Ekofisk et de brut léger d'Arabie dans lesquels sont ajoutés des volumes connus de solutions témoins deutériques. On détermine les quantités d'hydrocarbures aromatiques dans les mélanges de pétroles bruts par la méthode de la chromatographie en phase gazeuse/spectrométrie de masse/surveillance d'ions sélectionnés et les facteurs de réponse sont ensuite calculés (voir ci-dessus). Le mélange qui en résulte est alors utilisé comme solution témoin pour le calcul des facteurs de réponse utilisés pour l'évaluation des naphtalènes, des phénanthrènes et des dibenzothiophènes.
Tableau 6. Quantité en microgramme/millilitre des différents hydrocarbures aromatiques dans le pétrole de référence déterminée par la chromatographie en phase gazeuse (détecteur à ionisation de flamme)
Composé microgramme/ Composé microgramme/ Composé microgramme/ millilitre mi Ilil i tre mi Ili li tre
Naph t al ène 18,5 Phénanthrène 6,2 C -phénanthrène 20,5
méth. napht. 81,6 ANTHRACENE-~10 33,O C -dibenzo-
2
thiophene 47,3
C2- napht. 115,6 méth. -phérian- 20,7 C -dibenzo- 29,8 thrène thiophene
C3- napht. 75,7 méth. -dibenzo- 29,3 PYRENE-D1 O 33,2 th iophène
BIPHENYLE-~10 37,3
* La quantité de naphtalène, de phénanthrène, etc., provient du mélange normalisé.
- 25 -
99
o 1 ûQ 200 300 400
RROllFITSTRNDRRD.20-2-81
170
I TMN
L
! 1 DMN
164
156
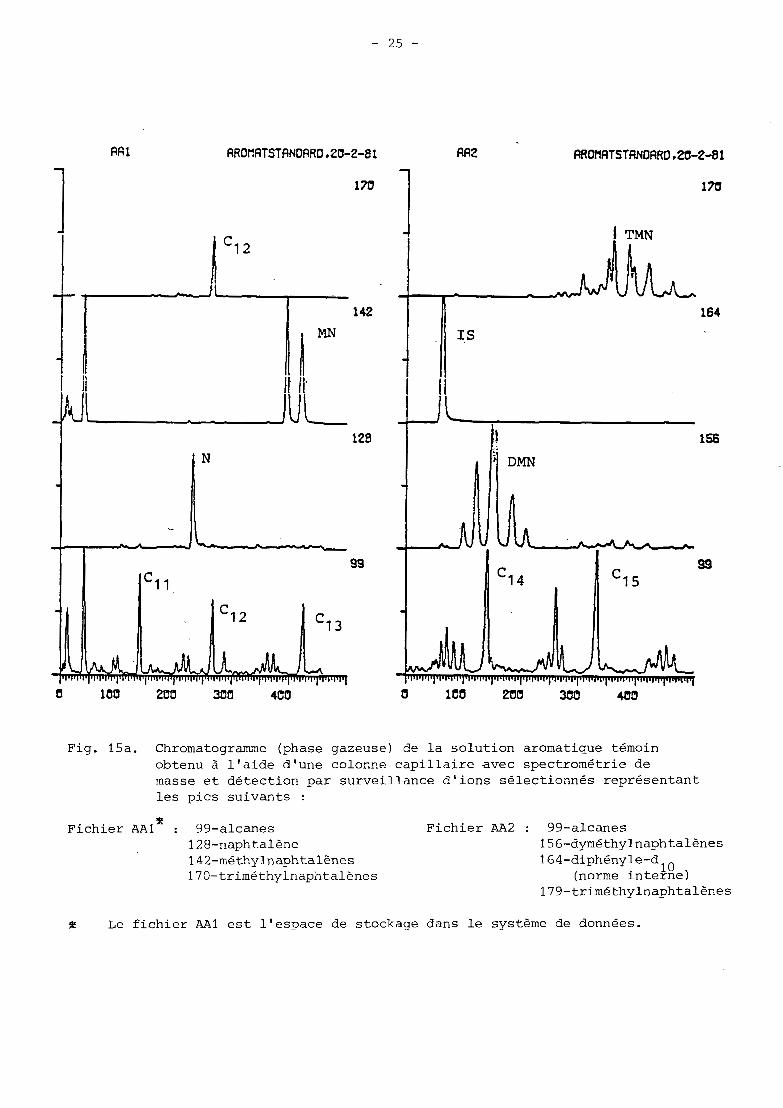
Fig. 15a. Chromatogramme (phase gazeuse) de la solution aromatique témoin obtenu à l'aide d'une colonne capillaire avec spectrométrie de masse et détection par surveillance d'ions sélectionnés représentant les pics suivants :
* Fichier AA2 : 99-alcanes Fichier AA1 : 99-alcanes 128-naphtalène 156-dyméthylnaphtalènes
170-triméthylnaphtalènes (norme interne) 1 4 2-mé thy Inaph t a lènes 1 6 4- diphény le-d 10
179-triméthylnaphtalènes
f Le fichier AA1 est l'espace de stockage dans le système de données.

- 26 -
- 3
1 198 226 MD
I TMD
Fig. 15b. Chromatogramme (phase gazeuse) de la solution aromatique témoin obtenu à l'aide d'une colonne capillaire avec spectrométrie de masse et détection par surveillance d'ions sélectionnés repré- sentant les pics suivants :
Fichier AA4 : 178-phénanthrène Fichier AA5 : 202-fluoranthène, pyrène 206-diméthylphénanthrènes 212-diméthyldibenzothio- 10 188-anthracène-d
(norme interne) 192-méthylphPnanthrènes phène, pyrène-d 10 198-méthyldibenzothiophène (norme interne)
226-triméthyldibenzothio- phène
-
- - -
192 212
r 188
IS DMP
- I 178
P -

- 27 -
9. PRINCIPES PERMETTANT D'ASSURER LA QUALITE DES DONNEES
Afin d'assurer la meilleure qualité possible aux analyses d'échantillons de l'environnement, le Comité sur l'amélioration de l'environnement de 1'American Chemical Society (ACS) a demandé à son Sous-Comité sur la chimie analytique de l'environnement d'élaborer une série de "Directives pour l'acquisition des données et l'évaluation de leur qualité dans le domaine de la chimie de l'environnement''. Ces directives visent à assurer les conditions d'une analyse chimique fiable d'échantillons de l'environnement et devraient donc permettre aux laboratoires d'échanger et d'utiliser avec plus de confiance les données obtenues. Le présent chapitre est fondé essentiellement sur les idées qui inspirent ces directives.
On effectue des analyses de l'environnement à des fins multiples (voir intro- duction). Cette large gamme d'applications des données d'analyse et les nombreuses manières d'utiliser l'information pourront nécessiter différents niveaux de certi- tude. I1 est donc important d'établir le degré d'exactitude nécessaire dans chaque situation. Les analyses de l'environnement se heurtent à de nombreuses difficultés : un grand nombre de composés organiques, une grande diversité de paramètres, l'ab- sence ou l'insuffisance de connaissances techniques, la déformation inhérente à l'échantillonnage, les erreurs systématiques et humaines figurent parmi celles qui sont mentionnées dans les "Directives" .
9.1 Planification. En raison de tous ces problèmes, il est très difficile de par- venir à l'exactitude. Il. est donc particulièrement important de bien planifier
chaque étape de l'entreprise. Un tel plan ou modèle intervient implicitement ou explicitement dans tout processus de mesure. Les données sont produites pour servir à répondre à des questions qui permettront de tirer des conclusions. Si le modèle, c'est-à-dire l'ensemble de relations entre les données et le problème, est défec- tueux, les conclusions tirées seront également défectueuses, même si les mesures sont de bonne qualité.
Le plan devrait s'appuyer sur les efforts conjugués de l'analyste, qui connaît les techniques de mesure, du scientifique qui utilisera les données et du statis- ticien qui les'évaluera en définitive. En outre, aucun programme de mesure ne devrait être entrepris avant l'établissement d'un tel plan. Celui-ci doit contenir les aspects généraux du problème et le système d'analyse à appliquer afin de les résoudre, de préférence en se référant à des détnils écrits des plans et procédures (protocoles) .
9.2 Assurance de qualité. Afin de définir les problèmes et de les résoudre, il faudrait concevoir un programme facteur de qualité qui comprendrait les aspects
suivants :
1.
2.
3.
4.
5.
S'assurer un personnel qualifié, appliquer des méthodes prévues par écrit et validées et disposer de laboratoires bien construits, bien équipés et bien entretenus.
Fournir des échantillons représentatifs et des échantillons témoins.
Utiliser des objets en verre, des solvants et tout matériel d'essai de haute qualité.
Etalonner, ajuster et entretenir le matériel.
Utiliser des échantillons témoins et des échantillons normalisés avec les observations utiles correspondantes.

- 28 -
6.
7.
8.
9.
10.
11.
12.
13.
Observer directement l'exécution de certains essais critiques.
Examiner et critiquer les résultats.
Evaluer l'efficacité interne et externe.
Utiliser des doubles des échantillons.
Comparer les résultats avec ceux d'autres laboratoires (interétalonnage)
Répondre aux plaintes des utilisateurs.
Surveiller les résultats.
Corriger les écarts par rapport aux normes de qualité.
Ces éléments de base facteurs de qualité définissent les conditions que les protocoles écrits et, notamment, toutes les procédures d'analyse doivent prévoir pour obtenir des résultats fiables.
On a beaucoup plus de chances d'obtenir des données exactes lorsqu'on se ré- fère efficacement à des solutions de référence internes et externes et lorsque le laboratoire a prouvé qu'il se conformait étroitement aux normes d'exactitude accep- tables en participant à une opération d'interétalonnage.
9.3 Définitions de quelques termes statistiques
9.3.1 d'une méthode d'analyse chimique définit jusqu'à quel degré la valeur moyenne des mesures est due à la substance à déterminer et non à
d'autres substances qui pourraient être présentes dans l'échantillon analysé.
9.3.2 La sensibilité d'une méthode d'analyse chimique est fonction de la plus petite modification de la quantité à mesurer qui produise un changement
décelable dans les résultats. Dans ce cas, ce terme est synonyme de décelabilité minimale.
9.3.3
mesure qui approche la mesure moyenne d'un nombre infini de détermination du même échantillon. (En d'autres termes, la précision implique la reproductibilité des résultats de l'analyse).
La précision d'une méthode d'analyse chimique définit jusqu'à quel degré une détermination représentative d'une substance dans un échantillon donnera une
9.3.4 L'exactitude d'une méthode d'analyse chimique définit jusqu'à quel degré la valeur moyenne des mesures obtenues par la méthode approche de la valeur
véritable de la substance mesurée (les effets perturbateurs d'autres substances étant éliminés matériellement ou mathématiquement).
- 29 -
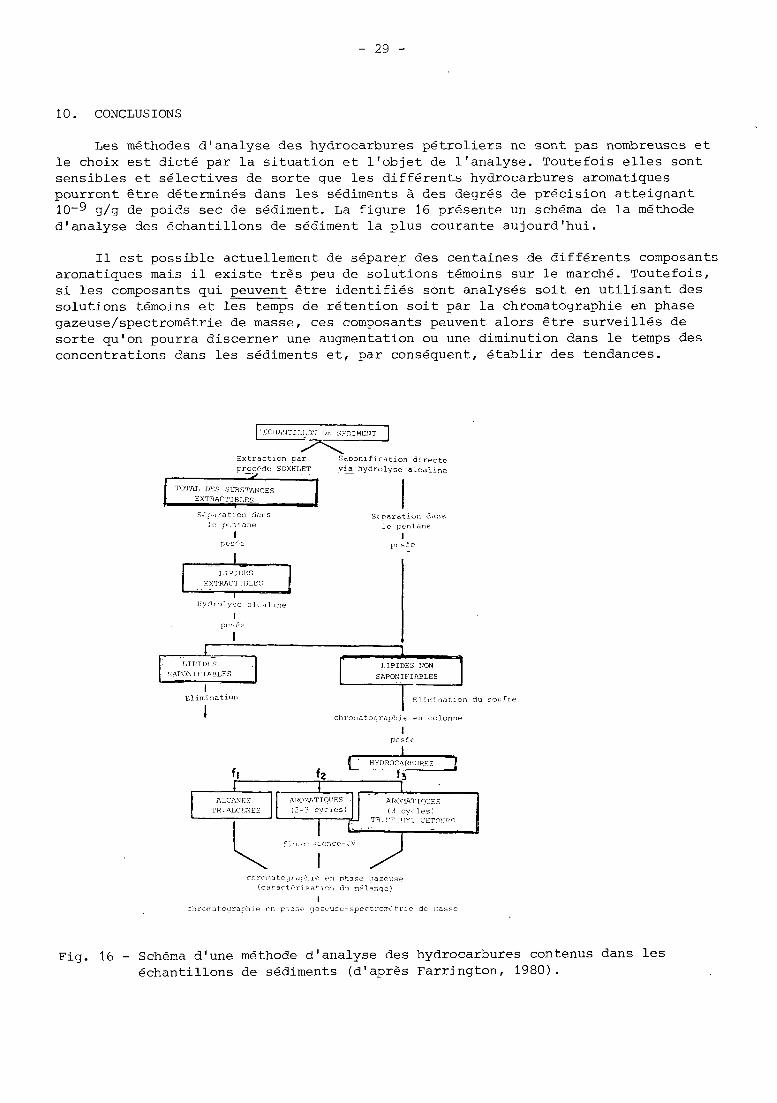
10. CONCLUSIONS
Les méthodes d'analyse des hydrocarbures pétroliers ne sont pas nombreuses et le choix est dicté par la situation et l'objet de l'analyse. Toutefois elles sont sensibles et sélectives de sorte que les différents hydrocarbures aromatiques pourrcnt être déterminés dans les sidiments à des degrés de précision atteignant 10-9 g/g de poids sec de sédiment. La figure 16 présente un schéma de la méthode d'analyse des échantillons de sédiment la plus courante aujourd'hui.
I1 est possible actuellement de séparer des centaines de différents composants aromatiques mais il existe très peu de solutions témoins sur le marché. Toutefois, si les composants qui peuvent être identifiés sont analysés soit en utilisant des solutions témoins et les temps de rétention soit par la chromatographie en phase gazeuse/spectrométrie de masse, ces composants peuvent alors être surveillés de sorte qu'on pourra discerner une augmentation ou une diminution dans le temps des concentrations dans les sédiments et, par conséquent, établir des tendances.
Extraction par Saponification directe hydrolyse alcaline
I TOTAL DES SUBSTANCES
siparation dans CiFaratiGn dans IC r,c:ntane le pentane
pcs<;t DcsGe I I
LIPIDES !ION
Elinination du soiilre
XAPONIFIARLFS
I Elimination
I cnrimatoqraphie en colonne
I pe*i.e
AROIlATICUEs
TR. !:TTiIYL CLTOirifs TR.ALCENES (2-3 cyclesi (3 cyclesi
fl "i~rc5lFn'c -uv
Fig. 16 - Schéma d'une méthode d'analyse des hydrocarbures contenus dans les échantillons de sédiments (d'apres Farrington, 1980).

- 30 -
Au cours des années à venir, l'accroissement de l'activité en mer demandera un surcroît d'efforts de la part des chimistes à la fois pour analyser les échan- tillons recueillis in situ et pour effectuer des recherches en laboratoire afin de résoudre des questions telles que la solubilité des hydrocarbures aromatiques, en général, et les variations dans le temps de la solubilité des différents composants aromatiques des sédiments.

- 31 -
APPENDICE I
GrouDe de travail ad hoc du GEMS1 sur l'analvse des hydrocarbures pétroliers dissous/dispersés dans l'eau de mer
Woods Hole, Massachusetts, Etats-Unis d'Amérique 16-18 mars 1981, pages 4-5 de l'original
Utilisation de la fluorescence-UV dans un programme de surveillance de la pollution par le pétrole
Les techniques de mesure ne constituent qu'une partie de l'ensemble du système de surveillance, qui prévoit des opérations de planification, d'échantillonnage, d'extraction, de mesure, de relevé et d'interprétation des données. La plupart de ces phases sont identiques quelle que soit la technique de mesure utilisée dans le programme de surveillance. Une évaluation complète de la simplicité, de la sensibi- lité et du coût doit tenir compte de toutes ces phases.
Le Groupe ad hoc a estimé qu'il fallait prendre en considération les réserves ci-après lorsqu'on décide d'utiliser la fluorescence-UV pour la surveillance des hydrocarbures pétroliers dissous ou dispersés :
í i)
(ii)
La technique de la fluorescence-UV donne une mesure relativement gros- sière des hydrocarbures pétroliers dissous/dispersés et il faut dis- poser d'un nombre suffisant de données (par exemple, des centaines d'échantillons pour le golfe du Mexique) pour pouvoir déterminer avec une certitude satisfaisante les "points chauds de la pollution" et un certain nombre de tendances dans la concentration des hydrocarbures pétroliers dissous/dispersés.
C'est une technique qui est facilement introduite dans les pays dont le potentiel scientifique est en cours de constitution ; c'est pourquoi son application a été recommandée pour le MAFQOLMON pour les hydrocarbures pétroliers dissous/dispersés dans l'eau de mer. Toutefois, les pays développés maîtrisant bien d'autres méthodes que la fluorescence-UV, telles que la chromatographie en phase gazeuse/spectrométrie de masse, qui permettent des analyses plus détaillées, doivent être encouragés à appliquer ces méthodes aux problèmes de la pollution en vue de mieux comprendre non seulement ce que mesure la fluorescence-W mais aussi l'ensemble de la répartition des différents types de composés des hydro- carbures pétroliers dissous/dispersés. En' outre, le Groupe ad hoc a estimé que les pays développés devaient aider les pays en développement à acquérir ces appareils plus perfectionnés et à se former aux techniques d'analyse dans le cadre des activités TEMA de la COI.
(iii). La collecte des données n'est que la première étape d'un tel programme de surveillance et il convient de prévoir une évaluation périodique des données et l'élaboration de recommandations s'inspirant de celles-ci. Cela est vrai quelle que soit la méthode emFloyée.

- 32 -
Des limitations importantes sont inhérentes à la technique de la fluo- rescence-UV. C'est pourquoi cette technique doit être considérée comme un point de départ pour ce type de programme de surveillance dont la complexité et la portée s'accroîtront à mesure que les connaissances techniques et le matériel disponible atteindront des niveaux permettant d'appliquer des techniques plus perfectionnées.
Le programme doit prévoir un effort soutenu d'expérimentation en ce qui concerne les effets de facteurs tels que l'altération de divers pétroles bruts sur les caractéristiques de leur fluorescence.
Le programme devrait prévoir que des échantillons représentatifs pro- venant de différentes régions seront soumis en laboratoire à une analyse plus détaillée par chromatographie en phase gazeuse avec colonne capil- laire de verre (GCGC) et par GCGC/spectrométrie de masse, afin de mieux évaluer les types et, si possible, les sources des composés qui provo- quent la fluorescence mesurée.
I1 a été signalé que les autres méthodes telles que la chromatographie en phase gazeuse avec colonne capillaire de verre et la chromatographie en phase gazeuse/spectrométrie de masse ne comportent pas les problèmes de normalisation inhérents à la méthode par fluorescence-UV. Le principal point à considérer dans l'application de ces méthodes à une activité mondiale telle que le MAWOLMON est le caractère limité des appareils disponibles et des compétences techniques. Toute- fois, étant donné que ces appareils et que ces compétences sont disponibles dans quelques pays développés et le seront peut-etre dans un proche avenir dans un grand nombre de pays en développement, le MARPOLMON devrait encourager toutes les fois que cela sera possible l'application de ces techniques en liaison avec la fluores- cence-UV, ce qui permettra de mieux comprendre la répartition et l'évolution des hydrocarbures pétroliers dans le milieu marin.





























