Embed Size (px)
Citation preview

Lezione 7
Allineamento di sequenze biologiche

Allineamento di sequenze
Determinare la similarità e dedurre l’omologia
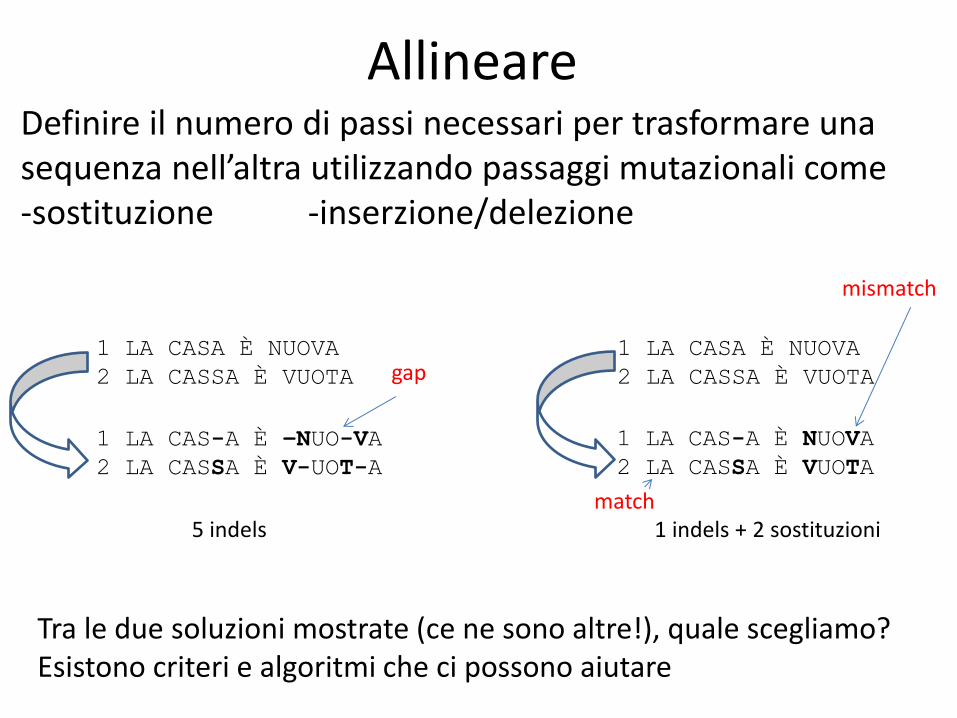
Allineare
1 LA CASA È NUOVA
2 LA CASSA È VUOTA
Definire il numero di passi necessari per trasformare una sequenza nell’altra utilizzando passaggi mutazionali come -sostituzione -inserzione/delezione
1 LA CASA È NUOVA
2 LA CASSA È VUOTA
1 LA CAS-A È NUOVA
2 LA CASSA È VUOTA
1 indels + 2 sostituzioni
Tra le due soluzioni mostrate (ce ne sono altre!), quale scegliamo? Esistono criteri e algoritmi che ci possono aiutare
match
mismatch
1 LA CAS-A È –NUO-VA
2 LA CASSA È V-UOT-A
5 indels
gap

Perchè allineare?
• Per fornire una misura di quanto sequenze nucleotidiche o aminoacidiche siano “imparentate”, abbiano in comune
• Questa parentela ci permette di fare inferenze biologiche in termini di
– relazioni strutturali
– relazioni funzionali
– relazioni evolutive
• Alignment-based database searching

Terminologia
• La misura QUANTITATIVA: Similarità
– Si esprime in genere come % di identità, quantifica i cambiamenti che sono avvenuti dal momento della divergenza tra due specie (sostituzioni, In-dels)
– Identifica i residui cruciali per mantenere la struttura o la funzione di una proteina
Alti livelli di similarità possono indicare una divergenza recente tra le sequenze, una storia evolutiva comune, simile funzione biologica

Terminologia
• Una valutazione di STATO: Omologia
– Implica l’esistenza di relazioni evolutive
– Geni omologhi: geni che si sono originati per divergenza da un antenato comune
– I geni SONO o NON SONO omologhi, non esiste una misura quantitativa dell’omologia
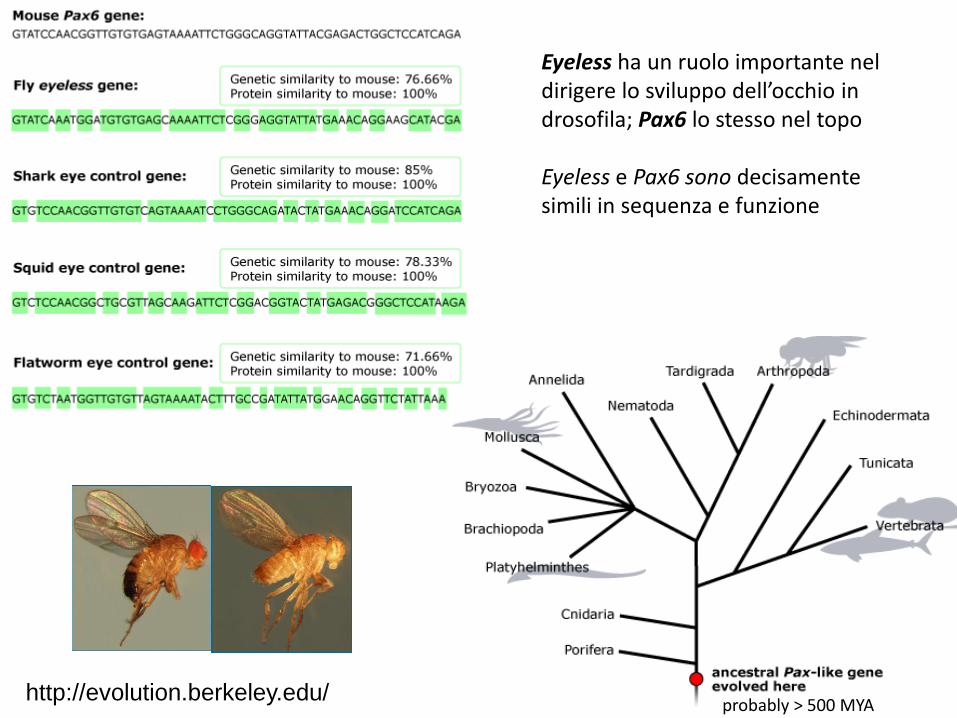
Eyeless ha un ruolo importante nel dirigere lo sviluppo dell’occhio in drosofila; Pax6 lo stesso nel topo Eyeless e Pax6 sono decisamente simili in sequenza e funzione
probably > 500 MYA http://evolution.berkeley.edu/

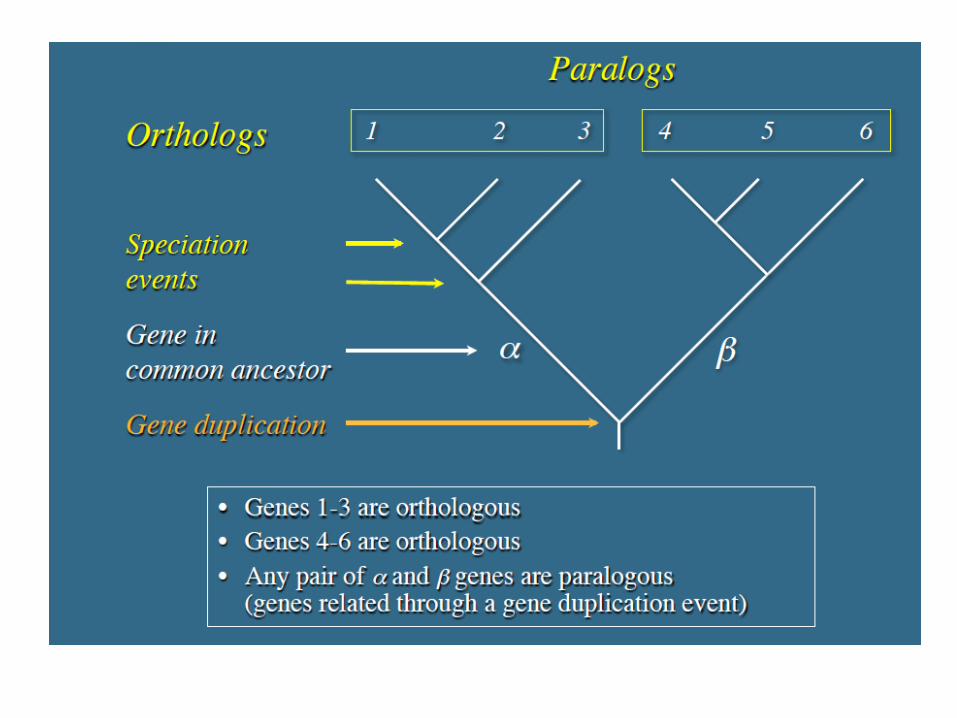
Terminologia Ortologhi: Geni che si sono separati in seguito ad un evento di speciazione
• Le sequenze discendono da un antenato comune
• Molto probabilmente codificano per proteine con domini simili e simili strutture tridimensionali
• Spesso mantengono funzioni simili
• Possono essere usati per predire funzioni geniche in genomi nuovi
Paraloghi: Geni che si sono evoluti per duplicazione in una specifica linea evolutiva
• E’ meno probabile che mantengano funzioni simili, più comunemente evolvono nuove funzioni

Allineamenti globali e locali
• Globale
– trova l’allineamento ottimale sul totale della lunghezza delle sequenze
– È la soluzione migliore per sequenze di lunghezza simile ed omologhe
– Al dimiuire del grado di similarità (es. aumento distanza evolutiva, alto tasso di ricombinazione) i metodi di allineamento globale tendono a peggiorare molto in efficienza

Allineamenti globali e locali • Locale
– Ha lo scopo di trovare regioni simili (es. domini) in due sequenze (“paired subsequences”)
– Le regioni fuori dalle aree di allineamento locale vengono escluse
– Può essere generato più di un allineamento locale per ogni coppia di sequenze confrontate
– Scelta indicata nel caso di due sequenze a similarità ridotta o di differenti lunghezze

Local vs. Global Alignment
• Global Alignment
• Local Alignment—migliore per trovare regioni conservate
--T—-CC-C-AGT—-TATGT-CAGGGGACACG—A-GCATGCAGA-GAC | || | || | | | ||| || | | | | |||| | AATTGCCGCC-GTCGT-T-TTCAG----CA-GTTATG—T-CAGAT--C
tccCAGTTATGTCAGgggacacgagcatgcagagac
||||||||||||
aattgccgccgtcgttttcagCAGTTATGTCAGatc

Allineamenti locali: perchè?
• Due geni in specie diverse possono essere simili in corte regioni conservate e diversi nel resto della sequenza.
• Esempio:
– I geni Homeobox (chiaramente omologhi) hanno corte regioni chiamate omeodomini altamente conservate tra specie.
– Un allineamento globale non troverebbe gli omeodomini perchè cercherebbe di allineare l’INTERA sequenza

Allineamento: ipotesi circa l’omologia
posizionale (discendenza da antenato comune)
di due residui in due (o più) sequenze
GCGGCCCATCAGGTAGTTGGTGG GCGTTCCATCCTGGTTGGTGTG
Sequenza della specie 1 sequenza della specie 2
Sequenza ancestrale
(prima della speciazione: antenato comune delle due sequenze.
Non direttamente osservabile (a meno di avere il DNA antico),
ma ricostruibile
11111111112222
base nr 12345678901234567890123
GCGGTCCATCAGCTGGTTGGTGG
presente
passato
G > T pos 4
Del AG pos 11 e 12
Ins T pos 23
T > C pos 5
C > G pos 13
G > A pos 15

Un allineamento a coppie consiste di una serie di residui o basi accoppiati, una per sequenza. Ci sono tre tipi di coppie:
(a) match = stesso nucleotide (o AA) in entrambe le sequenze (b) mismatch = diverso nucleotide (o AA) in una delle sequenze (c) gap = una base (o AA) in una sequenza e niente nell’altra
1111111111222 2
1234567890123456789012 3
Specie1 GCGGCCCATCAGGTAGTTGGTG-G
Specie2 GCGTTCCATC--CTGGTTGGTGTG
aaabbaaaaaccbabaaaaaaaca
Come si può fare in modo non manuale??

Nelle prossime diapositive cercheremo di rispondere alla domanda: su che cosa si basa un allineatore (algoritmo di allineamento) per operare un allineamento?
Come si può fare in modo non manuale??

1. Matrici di punteggio e 2. Penalità per i gap
• Il vero allineamento tra due sequenze è quello che riflette in modo accurato le loro relazioni evolutive (vedi i numerini nell’esempio precedente: omologia posizionale).
• Poichè il vero allineamento non è conosciuto in pratica si cerca l’allineamento ottimale: minimizza i mismatches e i gaps secondo certi criteri….purtroppo ↓ mms ↑ gaps
↓ gaps ↑ mms
Sostituzioni o mismatch
In/del o gap
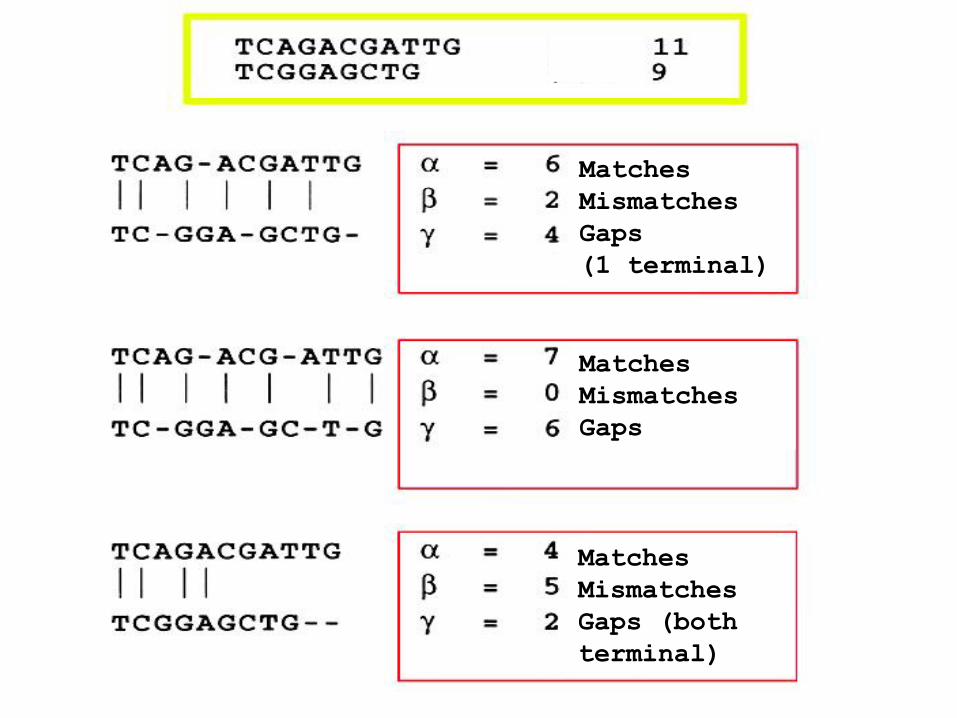
( (
(
Matches
Mismatches
Gaps
(1 terminal)
Matches
Mismatches
Gaps
Matches
Mismatches
Gaps (both
terminal)

Lo schema di punteggio include una penalizzazione per le in-del (gap penalty) e una matrice di punteggio (scoring matrix) M(a,b), che specifica ogni tipo di match (a = b) o di mismatch (a b). Le unità nella matrice di punteggio possono essere nucleotidi nelle sequenze di DNA o RNA, i codoni nelle regioni codificanti, o gli aminoacidi nelle sequenze proteiche.
Matrici di punteggio e penalità per i gap

Cos’è una matrice di punteggio?
• Matrice che associa un punteggio ad ogni coppia di entità che troviamo in un allineamento
• Ogni linea e ogni colonna rappresentano un residuo (4 nucleotidi o 20 aminoacidi)
• La diagonale è l’identità
• Il triangolo inferiore corrisponde alle sostituzioni e il superiore è simmetrico (non necessario)
• I valori negativi indicano penalità per certe sostituzioni, l’algoritmo di allineamento cercherà di evitarle
• I valori positivi indicano sostituzioni ‘accettate’ in termini evoolutivi, strutturali o funzionali
Sostituzioni : mismatches

Perché è importante capire le matrici di punteggio?
• Compaiono in ogni analisi che implichi un confronto tra sequenze
• Implicano un determinato percorso evolutivo
• Possono influenzare fortemente il risultato delle analisi
Sostituzioni : mismatches

Di solito sono semplici. La più semplice: M(a,b) assegna valori positivi se a = b (match), altrimenti negativi (mismatch)
M(a,b) 0 if a b
0 if a b
DNA scoring matrices Sostituzioni : mismatches
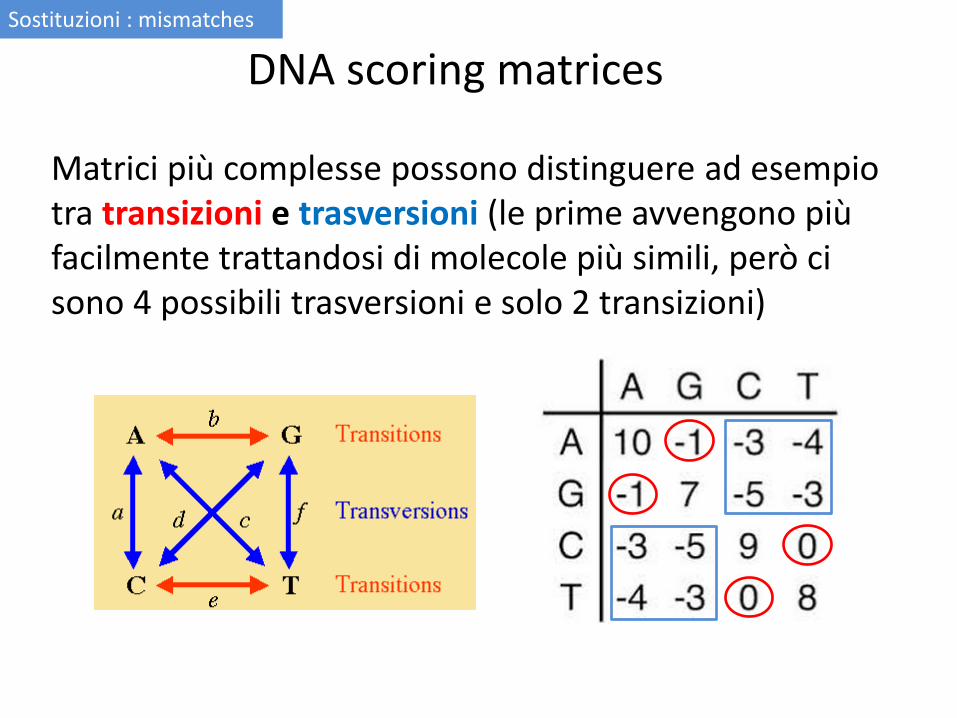
Matrici più complesse possono distinguere ad esempio tra transizioni e trasversioni (le prime avvengono più facilmente trattandosi di molecole più simili, però ci sono 4 possibili trasversioni e solo 2 transizioni)
DNA scoring matrices Sostituzioni : mismatches

Margareth Dayhoff 1965: “Atlas of potein sequences” contenente le sequenze aminoacidiche di 65 proteine Inizio delle collezioni di dati da cui avranno origine le banche dati elettroniche Dayhoff et al. nel decennio 1970-1980 hanno proposto una procedura per il calcolo di matrici di punteggio per quantificare la propensione di AA a mutare l’uno nell’altro durante l’evoluzione (matrici 20 x 20). Alla base c’è l’osservazione delle proteine note: MATRICI DI SOSTITUZIONI EMPIRICHE
Amino acid/protein scoring matrices
Sostituzioni : mismatches

Empirical substitution matrices
PAM matrix (Percent/Point Accepted Mutation Matrix) BLOSUM (BLOcks SUbstitution Matrix)
Amino acid/protein scoring matrices
Sostituzioni : mismatches
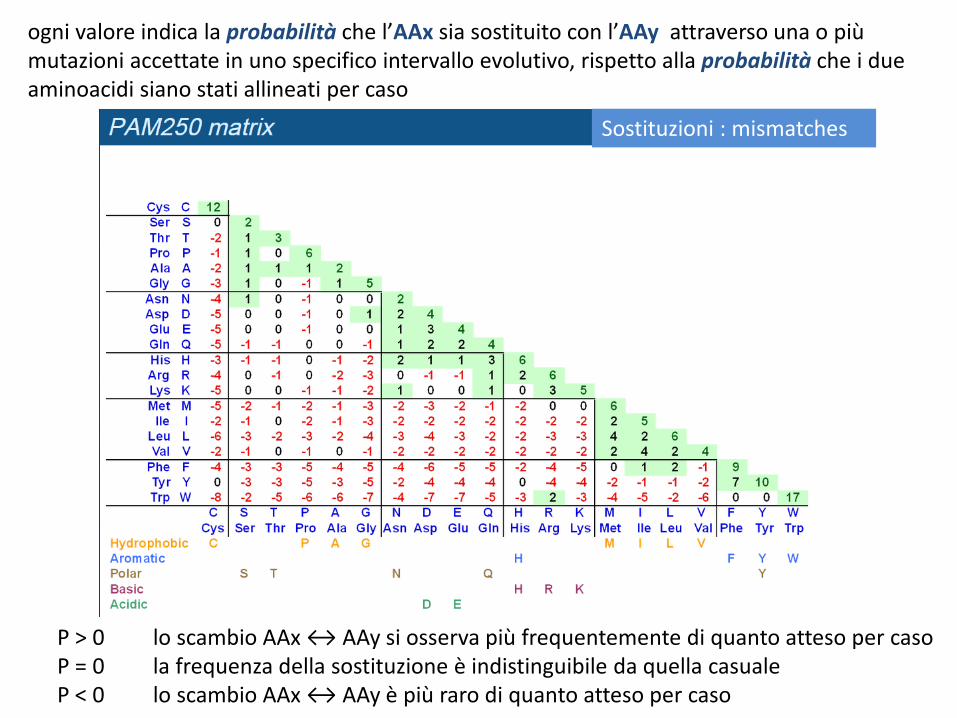
P > 0 lo scambio AAx ↔ AAy si osserva più frequentemente di quanto atteso per caso P = 0 la frequenza della sostituzione è indistinguibile da quella casuale P < 0 lo scambio AAx ↔ AAy è più raro di quanto atteso per caso
ogni valore indica la probabilità che l’AAx sia sostituito con l’AAy attraverso una o più mutazioni accettate in uno specifico intervallo evolutivo, rispetto alla probabilità che i due aminoacidi siano stati allineati per caso
Sostituzioni : mismatches

BLOSUM (BLOcks SUbstitution Matrix)
• Henikoff and Henikoff (1992): matrice basata su molte più osservazioni della PAM: scambi aminoacidici calcolati su circa 2000 «blocchi»
• Blocco: regione conservata di una famiglia di proteine senza indels
• Direttamente calcolate sulla base di allineamenti locali – Probabilità di sostituzione (conservazione)
– Frequenza degli aminoacidi
Sostituzioni : mismatches
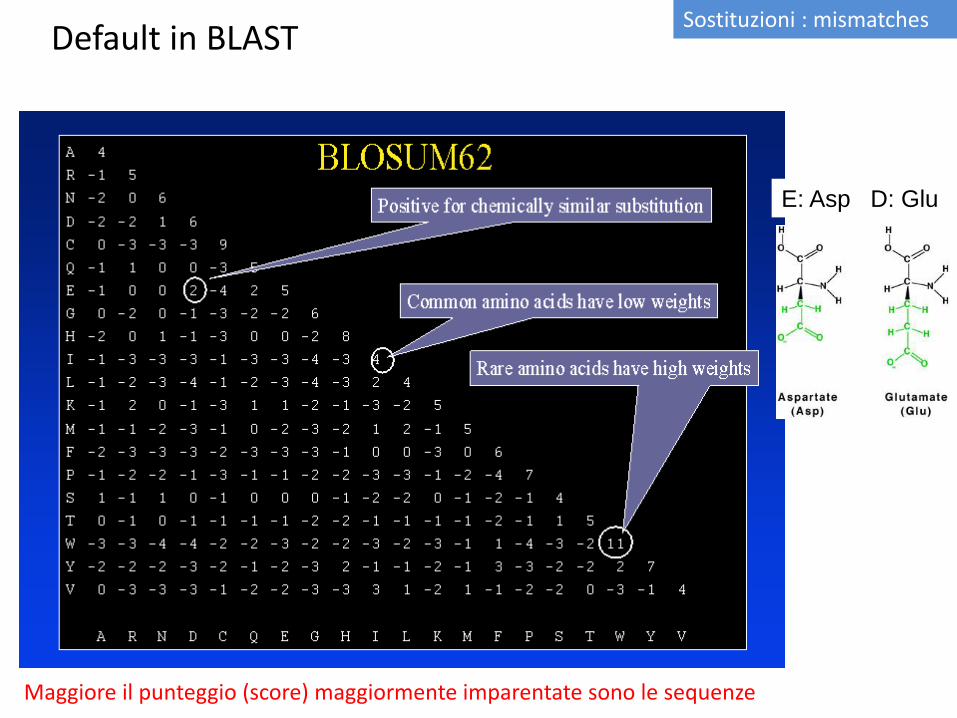
E: Asp D: Glu
Default in BLAST Sostituzioni : mismatches
Maggiore il punteggio (score) maggiormente imparentate sono le sequenze
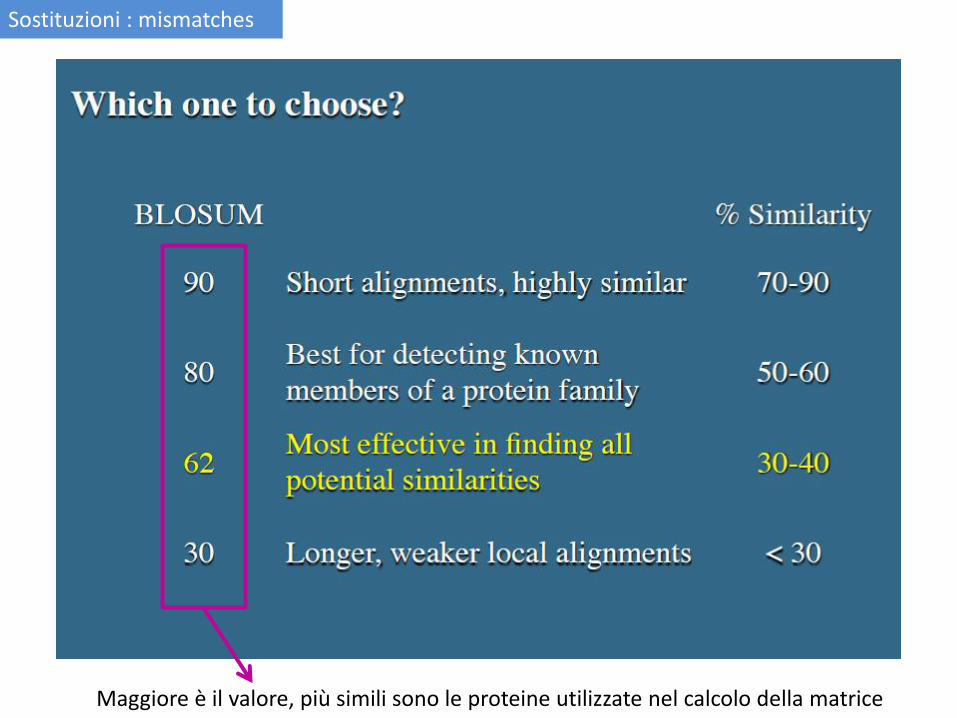
Maggiore è il valore, più simili sono le proteine utilizzate nel calcolo della matrice
Sostituzioni : mismatches
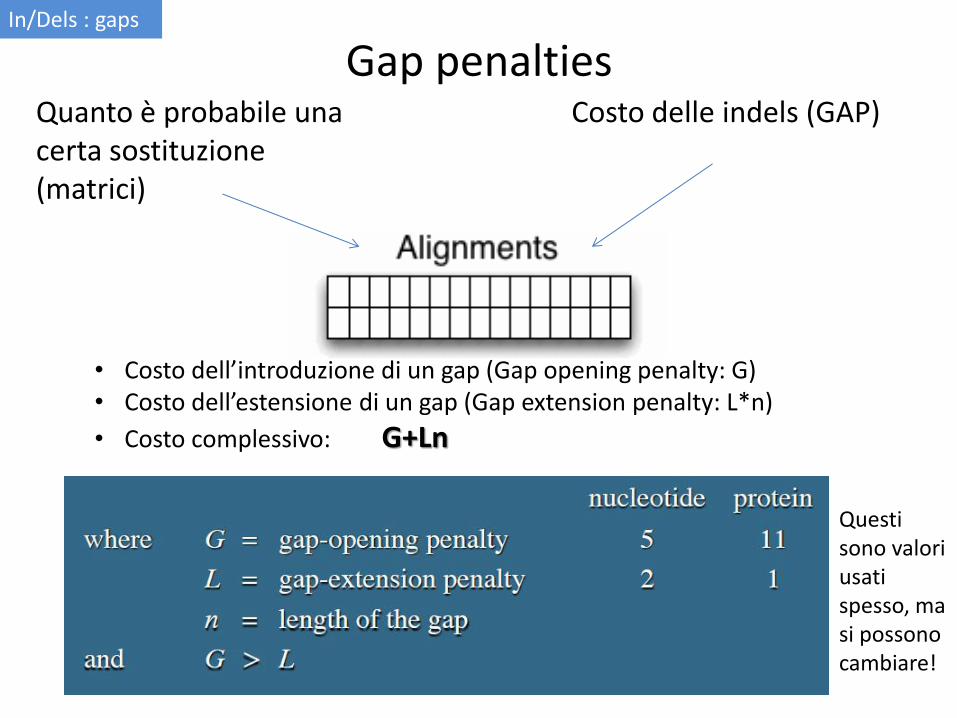
Gap penalties Costo delle indels (GAP) Quanto è probabile una
certa sostituzione (matrici)
• Costo dell’introduzione di un gap (Gap opening penalty: G) • Costo dell’estensione di un gap (Gap extension penalty: L*n)
• Costo complessivo: G+Ln
Questi sono valori usati spesso, ma si possono cambiare!
In/Dels : gaps



Algoritmi di allineamento
• Obiettivo: trovare il miglior allineamento, cioè il massimo numero di simboli identici e il minor numero di gap (=minor numero di mutazioni = più breve percorso evolutivo)
• Per due sequenze di DNA di 200 basi ci sono 10153 possibili allineamenti….meglio non farli a mano!

Dynamic programming = tecnica computazionale. Si usa per effettuare ricerche complesse dividendole in una successione di piccoli passaggi, inizialmente semplici e poi più complessi. L’ultimo passaggio contiene la soluzione complessiva

41
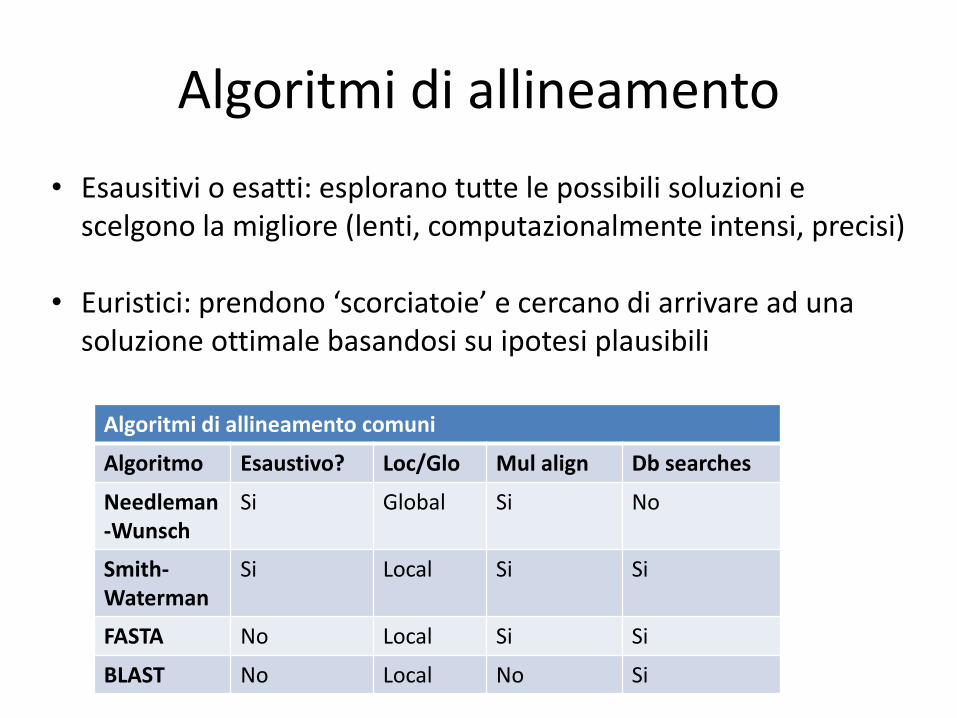
Algoritmi di allineamento
• Esausitivi o esatti: esplorano tutte le possibili soluzioni e scelgono la migliore (lenti, computazionalmente intensi, precisi)
• Euristici: prendono ‘scorciatoie’ e cercano di arrivare ad una soluzione ottimale basandosi su ipotesi plausibili
Algoritmi di allineamento comuni
Algoritmo Esaustivo? Loc/Glo Mul align Db searches
Needleman-Wunsch
Si Global Si No
Smith-Waterman
Si Local Si Si
FASTA No Local Si Si
BLAST No Local No Si

• Exact global alignment method
– Non adatto in molti casi (es. db searches, ricerca di piccole regioni di similarità, allinemanti tra sequenze con grosse differenze di lunghezza)
– Il più rigoroso e completo se lo scopo è di allineare sequenze che non si sono evolute per exon shuffling, inserzione/delezione di domini, etc.
– Il metodo migliore se le sequenze sono di lunghezze simili e si sono evolute da un antenato comune attraverso mutazioni di punto, piccole ind/dels
Needleman-Wunsch

• Exact local alignment method
– Modifica del N-W che permette di allineare in locale (non serve allineare tutta la seq)
– Allineamento molto buono per db searching, allineamento multiplo e a coppie
– Esaustivo, quindi può essere molto lento. A differenza del N-W considera qualunque allineamento che parta da qualunque posizione della sequenza, non solo quelli che cominciano all’inizio e terminano alla fine
Smith-Waterman

• Euristico locale
– Prima identifica regioni di identità tra la sequenza sonda (‘query’) e le sequenze in db. (KTUP)
– I geni o proteine con la densità maggiore di segnale vengono riesaminati
– L’allineamento viene esteso ad entrambi i lati delle regioni di match aggiungendo gaps e mismatches sulla base di matrici di punteggio
– L’allineamento ottiene un punteggio
FASTA: http://www.ebi.ac.uk/Tools/sss/fasta/ Pearson WR (1996) Effective protein sequence comparison. Academic Press Inc 227-258 Pearson WR and Lipman DJ (1998) Improved tools for biological sequence comparison. PNAS 85:2444
NB: leggere l’HELP del programma





























