Embed Size (px)
DESCRIPTION
gfhf
Citation preview

III. MATERIALES Y METODOS
III.1 MATERIALES
Plano general de la ciudad de Tingo María y aledaños.
Libreta de apuntes.
Cámara fotográfica.
cronometro
Materiales de escritorio.
Vehículos motorizados.
Material bibliográfico.
Instrumento captador de contaminante.
GPS.
Algodón.
Bolsas herméticas.
Pipetas.
Material de vidrio de boro silicato 3.3 de acuerdo con ISO 3585
Vasos de precipitado o matraces Erlenmeyer de capacidad
aproximada de 100 ml.
Vidrios de reloj.
Pipetas de un enrase, de acuerdo con ISO 648.
Probetas de 100 ml de capacidad.
Matraces aforados, en el intervalo de 10 ml a 1 l de acuerdo con
ISO 1042.
Botellas de polipropileno, de capacidad entre 100 ml y 1 litro.
Para captador del contaminante

Clavos de 1 ½ pulg.
Clavos de 3 pulg.
1 Madera de 6cmx2cmx2.5m
4 Maderas de 3cmx1cmx15cm
REACTIVOS
Agua destilada o desionizad
Ácido nítrico, HNO3
Concentrado, mínimo 65% (m/m), densidad aproximada 1,40 g/ml.
Ácido clorhídrico, HCl
Concentrado, mínimo 32%(m/m), densidad aproximada 1,16 g/ml.
Nitrato de lantano (III),La(NO3)3 • 6H2O
Nitrato de cesio, CsNO3
Disolución de ácido nítrico al 10% (V/V)
Se toman 100 ml de ácido nítrico.
Disolución de lantano de 50 mg/ml
Disolución de cesio de 50 mg/ml
EQUIPOS
Aparatos volumétricos que operan a émbolo, de acuerdo con ISO
8655
Placa calefactora, controlada termostáticamente, capaz de
mantener una temperatura de al menos 150°C.
Espectrofotómetro de absorción atómica, equipado con
quemadores para uso con llamas de aire-acetileno y monóxido de
dinitrógeno-acetileno
Balanza analítica, con una precisión de ± 1 mg como mínimo.

III.2 METODOLOGIA
III.2.1 Ubicación política
El trabajo se desarrollará en en la ciudad de Tingo María, distrito de
Rupa Rupa, provincia de Leoncio Prado, Región Huánuco.
III.2.2 Ubicación geográfica
III.2.3 Aspectos ambientales
Ecológicamente de acuerdo a la clasificación de zonas de vida o
formaciones vegetales del mundo y el diagrama bioclimático de
HOLDRIDGE (1982), Tingo María se encuentra en la formación vegetal
bosque muy húmedo Pre-montano Tropical bmh-PT, y de acuerdo a las
regiones naturales del Perú corresponde a Rupa Rupa o Selva Alta.
Hidrográficamente pertenece a la cuenca del río Huallaga; el
comportamiento climático es variable, con una precipitación anual pro-
medio de 3328.9 mm. Las mayores precipitaciones se producen entre
los meses de septiembre a abril y alcanza un máximo extremo en el mes
de febrero con un promedio mensual de 608.4 mm. En los últimos años
se han registrado los siguientes datos climatológicos relacionados con el
proyecto (Estación meteorológica José Abelardo Quiñones, 2010).
Temperatura máxima : 30,70 º C

Temperatura mínima : 18,90 º C
Temperatura promedio : 24,90 º C
Humedad relativa promedio : 86 %
Velocidad del viento máxima : 22,2 m/s
III.2.4 Reconocimiento del área de estudio
Se realizó a través del reconocimiento de campo de la ciudad de Tingo
María y y los puntos críticos con más congestión vehicular.
Una vez determinado los focos de contaminación se procede al
reconocimiento in situ, de manera explorativo y descriptiva y detallar los
puntos críticos a través coordenadas geográficas establecidos por los
grupos de práctica.
Posteriormente se elaboró un cronograma de trabajo (monitoreo) de 06
días establecidos en una sola semana donde se detalla 3 monitores
diarios, detallando las horas precisas del monitoreo, el personal
responsable de la operación y el punto preciso de monitoreo.
3.2.5 Monitoreo de muestras
Para monitorear los contaminantes del aíre se realizó a través del uso de
un captador de contaminante hecho artesanalmente con madera de 2.5
m de largo en cuyo extremo se colocara un marco de madera de 20cm x
30cm en cuya parte central se ubicara una película de algodón
extendido que servirá como captador de los diferentes tipos de
contaminantes que son emanados o se encuentran suspendidos en el
ambiente aéreo.
El tiempo de duración en el proceso de monitoreo es de 20 a 30 minutos
por cada turno y punto.

Las muestras fueron recolectadas en bolsas herméticas y rotuladas con
sus respectivas fechas y hora de recolección, que son utilizadas para el
traslado del material que será analizado.
3.2.6 Manipulación de muestras previa al análisis
Las muestras, una vez recepcionadas y aceptadas, han sido
manipuladas de manera que se asegure su no contaminación y su
perfecta identificación. En el caso de que las muestras hayan de ser
analizadas por diferentes unidades analíticas, es recomendable disponer
de un registro de distribución de muestra y establecer un orden de
reparto o de prioridades.
3.2.7 Conservación de muestras previa al análisis
El almacenamiento de muestras se realizó en las condiciones adecuadas
en lugares frescos de preferencia.
3.2.8 Análisis de muestras.
Las muestras recolectadas fueron llevadas al laboratorio de suelos de la
Universidad nacional agraria de la selva.
El análisis de las muestras es el método por el cual se determinan los
componentes de una muestra, las concentraciones, de cada uno de ellos.
Los métodos de medición que utilizan muestreadores, requieren por lo
general que una vez que se ha muestreado el contaminante sea
necesario analizarlo por alguno del siguiente método.

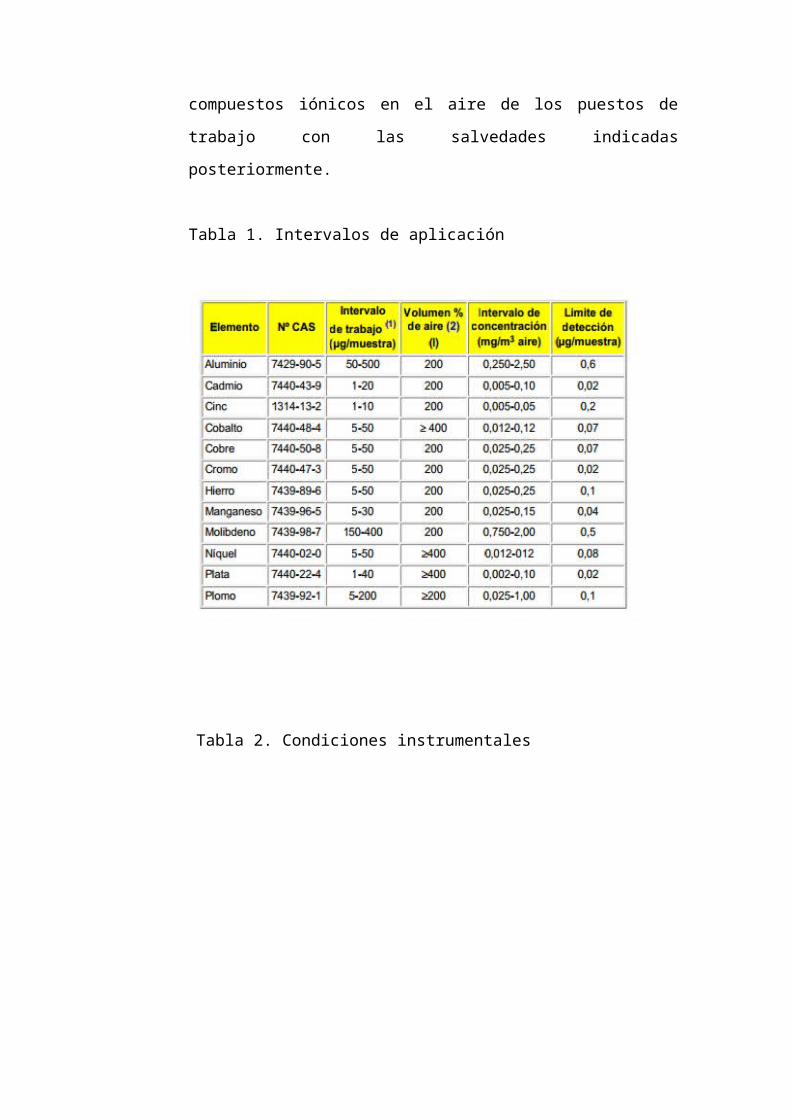
3.2.8.1 Espectrofotometría de Absorción Atómica
Se describe en este método el procedimiento a seguir y el equipo
necesario para la determinación de la concentración promedio de
aluminio, cadmio, cinc, cobalto, cobre, cromo, hierro, manganeso,
molibdeno, níquel, plata y plomo (Tabla 1) y sus compuestos iónicos
en el aire de los puestos de trabajo con las salvedades indicadas
posteriormente.
Tabla 1. Intervalos de aplicación
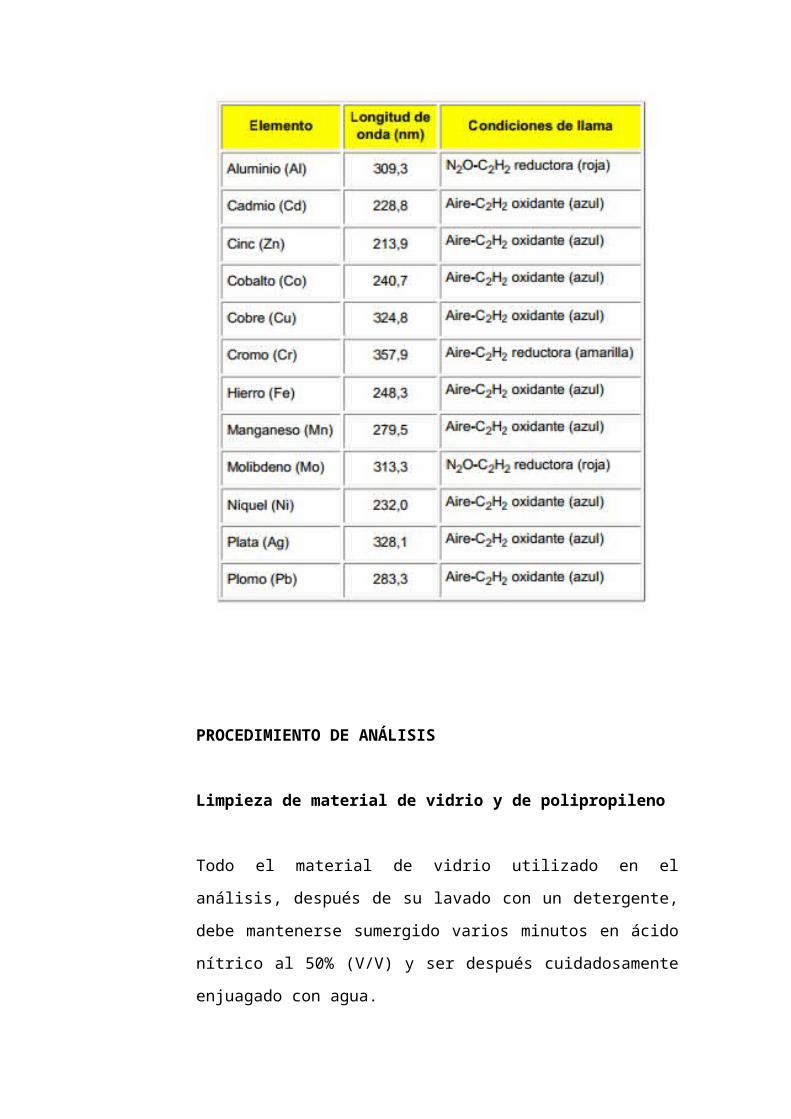
Tabla 2. Condiciones instrumentales

PROCEDIMIENTO DE ANÁLISIS
Limpieza de material de vidrio y de polipropileno
Todo el material de vidrio utilizado en el análisis, después de su
lavado con un detergente, debe mantenerse sumergido varios
minutos en ácido nítrico al 50% (V/V) y ser después
cuidadosamente enjuagado con agua.
Las botellas de polipropileno se limpiarán sumergiéndolas en ácido
nítrico diluido al 10% (V/V) durante varias horas y posteriormente
enjuagándolas con agua.
Preparación de la muestra
Se abren los portafiltros de muestras y blancos y se transfiere cada
filtro a un vaso o matraz Erlenmeyer.
Digestión por vía húmeda
Se añaden aproximadamente 5 ml de ácido nítrico concentrado ,
se cubre cada vaso con un vidrio de reloj y se calientan en placa
calefactora a 140oC en el interior de una vitrina extractora de
gases. Si las cantidades de materia orgánica fueran elevadas,
serían necesarias nuevas adiciones de ácido nítrico concentrado
hasta completar la digestión de la muestra, lo que se aprecia por el
aspecto claro y transparente de la misma. Una ligera ebullición
deberá permitir evaporar la mayor parte del líquido sin llegar ase
quedad.
Se retira el vidrio de reloj y se lava con 2 ó 3 ml de ácido nítrico
diluido al 10% (V/V) recogiendo el residuo de lavado en el. Vaso
que contiene la muestra. Se calienta de nuevo esta disolución a
140°C para reducir el volumen hasta aproximadamente 0,5 ó 1ml.
Se retira la muestra y se deja enfriar.
Las disoluciones se transfieren cuantitativamente a matraces
aforados de 10 ml de capacidad, enjuagando cuidadosamente los
vasos o erlenmeyers que contienen la muestra y completando hasta
dicho volumen con ácido nítrico diluido. Si se prevén interferencias
(no espectrales) se añaden los reactivos adecuados para minimizar
su efecto.
Preparación de las disoluciones de calibración.
El blanco de disolvente ISO 6955 es ácido nítrico diluido al 10%
(V/V).
Se prepara una disolución de compensación del cero de ISO
6955)y al menos tres patrones que cubran el intervalo de
concentraciones de las muestras a analizar. Es aconsejable trabajar
dentro del intervalo lineal del metal en cuestión. Si hay presentes
interferencias (no espectrales) los patrones se compensan de la
misma forma que las muestras.
Las disoluciones de compensación de cero y las disoluciones de
calibración preparadas, pueden ser almacenadas en botellas de
polipropileno por un tiempo máximo de una semana.
Calibración
Selección de la longitud de onda de análisis.
Se selecciona la longitud de onda recomendada en la Tabla 2 para
cada metal, o bien otra línea alternativa teniendo en cuenta la
concentración de las muestras, la sensibilidad de cada línea y la
relación señal-ruido que puede presentar cada una de ellas. No es
aconsejable el uso de lámparas multielementales que puedan dar
lugar a interferencias espectrales. Si a la longitud de onda escogida
para el análisis puede producirse absorción inespecífica 5.1.5 de
ISO 6955 debe disponerse de algún dispositivo corrector de dicha
absorción.

Ajuste del espectrofotómetro
Se seguirán las recomendaciones del fabricante relativas a los
parámetros operativos del instrumento: intensidad de lámpara,
anchura de rendija, etc. y se utilizará para cada metal la llama y
características indicadas en la Tabla 2.
Curva de calibración
Se ajusta el cero del espectrofotómetro mientras se aspira blanco
de disolvente. Se aspiran a la llama la disolución de compensación
del cero y las disoluciones de calibración, y se mide la absorbancia
registrada a la longitud de onda elegida, utilizando corrección de la
absorción inespecífica si fuera necesario.
Se prepara un gráfico de calibración representando la absorbancia
de las disoluciones de calibración frente a la concentración del
metal de interés en µg/ml en las respectivas disoluciones. La
absorbancia es proporcional a la concentración de metal en
disolución en el intervalo lineal de la curva de calibración. Es
aconsejable trabajar en dicho margen lineal.
Determinación
Se ajusta el cero del espectrofotómetro mientras se aspira el blanco
de disolvente. Si se produce deriva de la línea de base mientras se
aspira el blanco de disolvente, el cero del espectrofotómetro debe
reajustarse.

Las disoluciones obtenidas, de la preparación de las muestras y de
los blancos de muestra se aspiran a la llama del espectrofotómetro
de absorción atómica y se miden las absorbancias a la longitud de
onda elegida, utilizando corrección para la absorción inespecífica si
fuera necesario. Cada cinco o diez muestras se aspirará un patrón
de concentración intermedia.
Si la lectura del patrón de concentración intermedia indica que se
ha producido un cambio inaceptable en la sensibilidad del sistema,
la secuencia analítica debe interrumpirse y recalibrarse el
espectrofotómetro. La concentración del metal de interés en las
disoluciones de muestras se determina a partir de la curva de
calibración.
Cuando se encuentren altas concentraciones de metal, se diluye
una alícuota de la disolución de muestra hasta que la
concentración esté dentro del intervalo de calibración. Se harán
todas las diluciones con disolución de compensación de cero y
Se anotará el factor de dilución F. Alternativamente, puede usarse
una línea analítica menos sensible, o bien puede girarse la cabeza
del quemador para reducir la respuesta del espectrofotómetro.
CÁLCULOS
Determinación de la concentración de analito en la curva de
calibración. La concentración de metal presente en la muestra,
expresada en microgramos por mililitro de disolución, se determina
por interpolación de la lectura obtenida, en la correspondiente curva
de calibración.

Determinación de la cantidad de analito presente en la muestra
La cantidad de metal presente en la muestra, expresada en
microgramos de metal, se calcula según la siguiente expresión:
M = (C1 x V1 x F) - (C2 x V2)………ecua. 1
Dónde:
M es la cantidad de analito, en microgramos, presente en la
muestra.
C1 es la concentración de metal en microgramos por mililitro de
disolución de muestra.
V1 es el volumen, en mililitros, hasta el cual ha sido diluida la
muestra (ej. 10 ml).
C2 es la concentración de metal, en microgramos por mililitro de
disolución del blanco de muestra.
V2 es el volumen, en mililitros, hasta el cual ha sido diluido el
blanco de muestra (ej. 10 ml);F es el factor de dilución empleado en
(si se aplica).

Determinación de la concentración de analito en aire
La concentración de metal en aire, en miligramos por metro cúbico,
se calcula según la siguiente expresión:
C = M/V
Dónde:
C es la concentración de metal en aire expresada en miligramos de
metal por metro cúbico de aire muestreado.
M es la cantidad de metal en microgramos, presente en la muestra.
V es el volumen, expresado en litros, de aire muestreado.
Cuando las condiciones de presión y temperatura durante el
muestreo varíen significativamente respecto a las de calibración
REFERENCIA BIBLIOGRAFÍA
NIOSH. General procedure for metals. Manual of Analytical Methods.
Method P & CAM 173 (Revised 3.11.77).
OSHA. Method for the analysis of metal particulates in workplace
atmospheres by atomic spectrophotometry. OSHA Manual of analytical
methods no ID-121.
NSHT. Método general para la determinación de metales en aire. HA-
2122. 1982.

Slavin, W., Atomic absorption spectroscopy. Interscience Publishers.
New York, 1968.
Analytical methods for atomic absorption spectrophotometry. The Perkin
Elmer Corporation, Norwalk, Connecticut 1982.
Analytical data for elements determined by atomic absorption
spectroscopy, Varian techtron, Walnut Creek. California,1971.
Air sampling instruments for evaluation of atmospheric contaminants,
American Conferene of Governmental Industria Hygienists, 6th ed. 1983.
Collaborative testing of NIOSH atomic absorption methods, Technical
report DHEW (NIOSH) publication n4 79-144.1979.
Verstuyft, A., W., Atomic absorption spectroscopy in the occupational
health laboratory, Occupational health chemistry,American Chemical
Society, 1980.
Real Decreto 2216/1985(1). de 23.10 (Presid., BB.OO.E. 27.11.1985,
rect 9.5.1986) "Reglamento sobre declaración desustancias nuevas y
clasificación, envasado y etiquetado de sustancias peligrosas".
Actualizado por Orden de 9-12-92(M. Relac. Cortes B.O.E. 1712-92).
ISO 3696, Agua para uso en laboratorio-especificaciones.
ISO 8655, Partes 1 a 4. Aparatos volumétricos de pistón y/o émbolo
(POVA).
ISO 3585, Instalaciones de vidrio, tuberías y ajustes. Propiedades del
vidrio borosilicatado 3.3

ISO 6955, Métodos analíticos espectroscópicos. Emisión de llama,
absorción atómica y fluorescencia atómica Vocabulario.
ISO 648 Material de vidrio de laboratorio, pipetas de un enrase.
ISO 1042, Material de vidrio de laboratorio, matraces aforados.
International Union of Pure and Applied Chemistry (I.U.P.A.C).
Compendium of analytical nomenclature Pergamon Press.1978.
ISO 5725. Precision of test methods. Determination of repeatability and
reproducibility for a standard test method byinter-laboratory tests.
ISO/TR 7708:1983 Air quality-particles size fraction subclauses for
health-related sampling





























