Embed Size (px)
Citation preview

Integrantes:
Guadalupe García González
Sharemy Gómez Dorantes
Marlene Hernández Sánchez
Adrián Ricardo Hipólito Nájera
Equipo 2.
Principios de Estructura de la Materia.
Profesor: Dr. Luis Alberto Vicente Hinestroza.
Método Extendido de Hückel

2 Método Extendido de Hückel
INTRODUCCIÓN.
El método OM semiempírico de un electrón más importante para moléculas no planares es
la teoría de Hückel extendida. Wolfsberg y Helmholz usaron una versión primitiva para
tratar complejos inorgánicos; y más tarde, el método fue desarrollado y ampliamente
aplicado por Hoffmann. Este método hace uso de un Hamiltoniano de partícula
independiente, y difiere del método de Hückel fundamentalmente, en que el segundo sólo
se aplica a sistemas con electrones , mientras que el primero incluye a todos los electrones
de valencia de la molécula. Se trata de métodos aproximados que proveen información útil
sobre diferentes aspectos de la estructura electrónica y la reactividad química.
El método de Hückel extendido (EH) ha sido ampliamente empleado con éxito a efectos de
analizar la información experimental y correlacionarla con datos de tipo atómico, por
ejemplo, disposición de los orbitales atómicos, potenciales de ionización y número de
electrones. Si bien no permite una buena predictibilidad de magnitudes absolutas, como las
energías de enlace, es una metodología que revela las interacciones básicas que son
responsables del enlace químico y posibilita ubicar estas mismas magnitudes en una escala
relativa que facilita la comparación entre situaciones con similares distribuciones
geométricas o composicionales. Otra virtud del método es que permite trabajar con
sistemas que incluyen metales de transición, con varios orbitales d (interés catalítico).
El método de Hückel extendido comienza cuando se toma la función de onda electrónica
como el producto de una función de onda de valencia y una función de onda de core y
puede escribir como Eq. 1:
Ec. 1
La función de onda total de electrones de valencia se describe como un producto funciones
de onda de un electrón.
( ) ( ) ( ) ( ) Ec. 2
Donde n es el número de electrones y j identifica el orbital molecular. Cada orbital
molecular se da como combinaciones lineales de orbitales atómicos (LCAO) de valencia.
∑

3 Método Extendido de Hückel
Donde r son los orbitales atómicos de valencia elegidos que incluyen el 2s, 2px, 2py, y 2pz
de los carbonos y heteroatomos en la molécula y los orbitales 1s de los átomos de
hidrógeno. El conjunto de orbitales definidos aquí se llama conjunto base (basis set). Ya
que este conjunto base contiene sólo orbitales atómicos similares para la capa de valencia
de los átomos en una molécula, es llamado, conjunto base mínimo (minimal basis set). Se
puede deducir una ecuación matriz para todos los orbitales moleculares como en la
siguiente ecuación:
HC=SCE Ec. 4
En la teoría extendida de Hückel, el solapamiento no se desprecia, y S es la matriz de las
integrales de solapamiento. Lo siguiente es encontrar H que es una matriz cuadrada que
contiene las Hrs, las integrales de energía de un electrón, las ideas básicas son similares en
espíritu a aquellas descritas en relación con la interpretación de α y β en el método Simple
de Hückel; y C es la matriz de los coeficientes para los orbitales atómicos. Cada columna
en C es la C’ que define un orbital molecular en términos de las funciones base. E es la
diagonal de la matriz de las energías de los orbitales. Todas estás variables son matrices
cuadradas con un tamaño que es igual al número de orbitales atómicos usados en la LCAO
para la molécula bajo consideración. Similar a la teoría de orbital molecular de Hückel, la
Ecuación 3 permanece para un problema eigenvalue o de valor propio. Para cualquier
calculo extendido de Hückel, se necesita establecer estas matrices y entonces encontrar los
valores propios y vectores propios. Los valores propios son las energías de los orbitales, y
los vectores propios son los coeficientes de orbital atómico que define el orbital molecular
en términos de las funciones base.
Los elementos de la matriz H se asignan usando valores experimentales, los cuales hacen al
método un método de orbital molecular semi-empírico. Los elementos de la matriz
Hamiltoniana fuera de la diagonal se dan por una aproximación debida a Wolfsberg y
Helmholtz que los relaciona a los elementos de la diagonal y al elemento de la matriz de
solapamiento:
Hij =
K(Hii+Hjj)Sij Ec. 5
El razonamiento para esta expresión es que la energía debe ser proporcional a la energía de
los orbitales atómicos, y debe se más grande cuando el solapamiento de los orbitales
atómicos es más grande. La contribución de estos efectos a la energía se escala por el
parámetro K. Hoffman asignó el valor de K como 1.75 después de un estudio del efecto de
este parámetro sobre las energías de orbitales ocupados para el eteno. Las Hii son elegidas
como potenciales de ionización de estado de valencia con un signo menos para indicar
unión. Es común en muchos estudios teóricos usar el orbital molecular extendido de Hückel
como un paso preliminar para determinar los orbitales moleculares por un método más

4 Método Extendido de Hückel
sofisticado, como el método CNDO/2 y métodos de química cuántica ab inicio. Un
programa reciente para el método extendido de Hückel es YaeHMOP el cual sus siglas en
inglés son "yet another extended Hückel molecular orbital package" otro paquete de orbital
molecular de Hückel extendido. El método extendido de Hückel se usa para determinar los
orbitales moleculares, pero no es muy exitoso en determinar la geometría estructural de una
molécula orgánica. Puede, sin embargo, determinar la energía relativa de diferentes
configuraciones geométricas. Esto incluye cálculos de las interacciones electrónicas de una
manera bastante sencilla, donde la repulsión electrón-electrón no está explícitamente
incluida y el total de energía es sólo una suma de términos para cada electrón en la
molécula. Se encontró que el método de Extendido de Hückel proporcionaba ángulos de
enlace más bien precisos para las moléculas cuyos enlaces no eran demasiado polares, pero
fallaba en las predicciones de los ángulos de enlace de moléculas con enlaces muy polares
(Por ejemplo, H2O, que predice que el lineal). El método no es fiable para predecir
longitudes de enlace, momentos dipolares y barreras de rotación interna; ni para predecir
conformaciones moleculares. El valor real del método de Hückel Extendido está en la
naturaleza cualitativa de los resultados y en las interpretaciones que pueden proporcionar
sus resultados.
EJEMPLOS
Molécula de Helio Protonado.
La protonación de un átomo de Helio genera He-H+, la molécula heteronuclear más simple.
Conceptualmente, también puede formarse por la unión ya sea de Helio dicatiónico y un
ion hidruro o de helio catiónico y un átomo de hidrogeno.
Su baja simetría hace que esta molécula resulte mejor que el H2 para ilustrar los cálculos de
la mecánica cuántica molecular (la mayoría de las moléculas tienen poca o nula simetría)
A continuación se ilustran los pasos del cálculo:
1) Elección de longitud de enlace
Se elige una longitud de enlace aceptable: 0.800Å (la longitud del enlace H-H es 0.742 Å y
la de H-X es 1.0 Å, donde X es puede ser Li, Be, B, C, N, O y F).
Las coordenadas cartesianas pueden escribirse H1 (0,0,0) He2 (0,0,0.800)
2) Integrales y matriz de traslape
Los orbitales base a utilizar son el 1s del Hidrógeno (φ1) y el orbital 1s del Helio (φ2) Las
integrales necesarias son y , donde ∫ .
Las funciones de Slater para φ1 y φ2 son:
( ) (
)
( ) (
)

5 Método Extendido de Hückel
Son valores razonables ξH=1.24a.u. y ξHe=2.0925a.u., mientras que las integrales de traslape
son (si los orbitales φ1 y φ2 están normalizados) y (para
las funciones ∫ ∫ )
Por lo que la matriz de traslape será:
(
)
3) Construcción de la matriz del Hamiltoniano
Se necesitan los elemento de la matriz y donde las integrales
⟨ ⟩ son estimadas a partir de las integrales de traslape y las energías de
ionización:
⟨ ⟩ ⟨ ⟩
( )
Se utilizan las energías de ionización experimentales:
( ) ( )
Para una aproximación inicial se toma K=1.75 obteniendo:
( )( )( )
La matriz resultante será:
(
)
4) Obtención de los valores propios y vectores propios
Se realiza la ortogonalización de la matriz S así como la diagonalización de la matriz H
para obtener los valores propios del producto de la matriz donde C’ son los
coeficientes de las funciones de base (orbitales atómicos).
(
) (
) (
) (
)
Obteniendo así los valores energéticos de -25.5eV y -5.95eV

6 Método Extendido de Hückel
Molécula de Etileno
Otro ejemplo al que puede aplicarse el método extendido de Hückel es la molécula de
etileno, para la cual se siguen los mismos pasos de calculo que los ilustrados para el Helio
protonado, pero considerando los orbitales π.
Para este cálculo se tomaran en cuenta los siguientes datos:
Los orbitales atómicos a considerar serán el 1s para el Hidrogeno y el 2s, 2px, 2py y 2pz
para el Carbono, cuyos potenciales de ionización corresponden a -13.60eV para el 1s del
Hidrógeno, -21.40eV para el 2s del Carbono, -11.40eV para los 2p del Carbono.
El sistema de coordenadas se plantea situando a los átomos de Carbono sobre el eje x
Todos los orbitales moleculares deben ser simétricos o anti-simétricos al elemento de
simetría de la molécula.
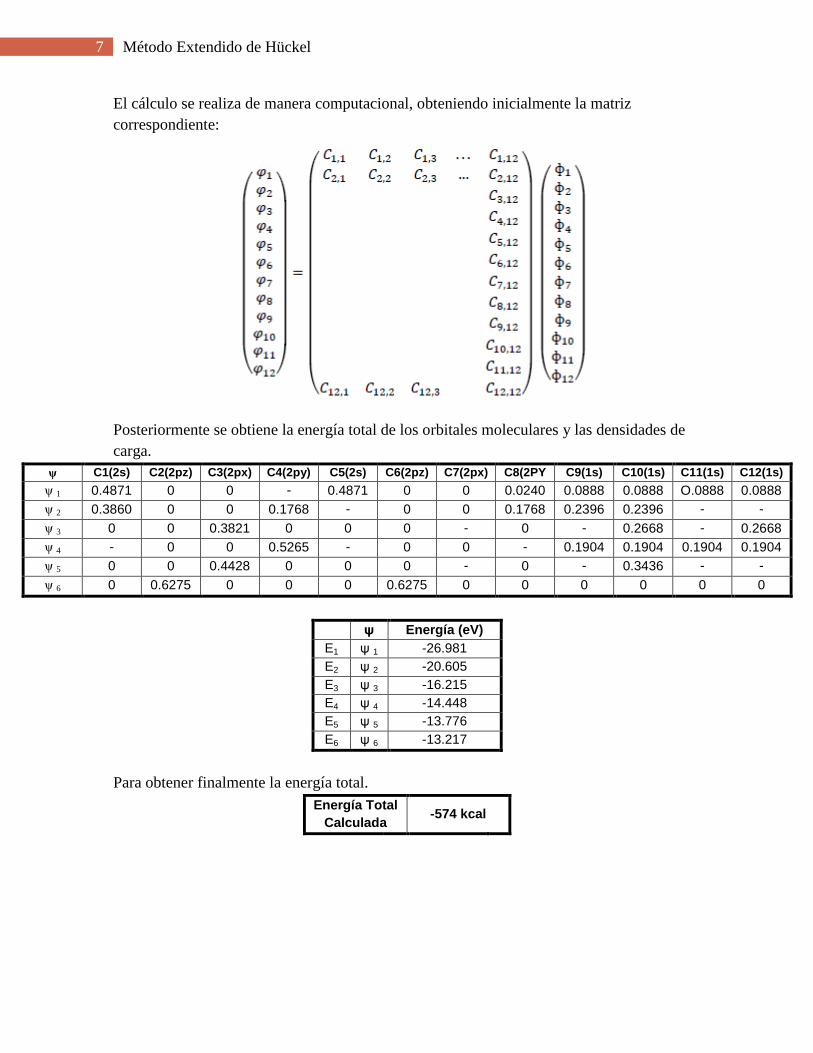
7 Método Extendido de Hückel
El cálculo se realiza de manera computacional, obteniendo inicialmente la matriz
correspondiente:
Posteriormente se obtiene la energía total de los orbitales moleculares y las densidades de
carga.
ψ C1(2s) C2(2pz) C3(2px) C4(2py) C5(2s) C6(2pz) C7(2px) C8(2PY
)
C9(1s) C10(1s) C11(1s) C12(1s)
ψ 1 0.4871 0 0 -
0.0240
0.4871 0 0 0.0240 0.0888 0.0888 O.0888 0.0888
ψ 2 0.3860 0 0 0.1768 -
0.3860
0 0 0.1768 0.2396 0.2396 -
0.2396
-
0.2396 ψ 3 0 0 0.3821 0 0 0 -
0.3821
0 -
0.2668
0.2668 -
0.2668
0.2668
ψ 4 -
0.0741
0 0 0.5265 -
0.0741
0 0 -
0.5265
0.1904 0.1904 0.1904 0.1904
ψ 5 0 0 0.4428 0 0 0 -
0.4428
0 -
0.3436
0.3436 -
0.3436
-
0.3436 ψ 6 0 0.6275 0 0 0 0.6275 0 0 0 0 0 0
ψ Energía (eV)
E1 ψ 1 -26.981
E2 ψ 2 -20.605
E3 ψ 3 -16.215
E4 ψ 4 -14.448
E5 ψ 5 -13.776
E6 ψ 6 -13.217
Para obtener finalmente la energía total.
Energía Total
Calculada -574 kcal

8 Método Extendido de Hückel
Molécula: Metano
1. Elección de las coordenadas:
Estructura molecular: Tetraédrica. Distancia de enlaces C-H: 1.1 Å, Angulo: 109 °
Coordenadas cartesianas ( en Å):
Coordenadas cartesianas de átomos de metano.
Átomo x y z
C 0.0 0.0 0.0
Ha 0.0 0.0 1.1
Hb 1.03709 0.0 -0.366667
Hc -0.518545 0.8998146 -0.366667
Hd -0.518545 -0.8998146 -0.366667
2. Elección de los orbitales atómicos
Para el CH4, los orbitales atómicos 1s para cada hidrógeno y para el carbono los orbitales
atómicos 2s, 2px, 2py y 2pz. Los orbitales atómicos son representados como orbitales tipo
slater (STOs). Excepto para del orbital 1s del hidrógeno, el parámetro exponencial de los
STOs están determinados por las reglas de Slater. Varios valores de orbital 1s han sido
sugeridos, estos tienen rangos que van de 1 a 1.41. Usando el valor de 1.2 que corresponde
al óptimo para el H2.
Elección de orbitales atómicos para metano
OA Átomo Tipo n l m exp
1 C 2s 2 0 0 1.625
2 C 2pz 2 1 0 1.625
3 C 2px 2 1 1 1.625
4 C 2py 2 1 1 1.625
5 Ha 1s 1 0 0 1.200
6 Hb 1s 1 0 0 1.200
7 Hc 1s 1 0 0 1.200
8 Hd 1s 1 0 0 1.200

9 Método Extendido de Hückel
3. Matriz de traslape
Esta matriz es simétrica (debido a que el traslape entre dos orbitales atómicos es
independiente a su orden numérico) y tienen elementos diagonales de unidad desde que el
orbital atómico esta normalizado. El valor cero en las primeras cuatro filas reflejan la
ortogonalidad entre los orbitales atómicos s y todos los p del carbono. Otros valores cero
resultan cuando el orbital atómico 1s del hidrógeno está centrado en un plano nodal del
orbital atómico p del carbono. La geometría del sistema está claramente reflejada en la
matriz de traslape.
El traslape de los orbitales atómicos 2pz del carbono y lo 1s del hidrogeno Ha es grande y
positivo, mientras que el traslape con el orbital atómicos Hb, Hc y Hd son negativos o de
magnitudes más pequeñas. Por otro lado los orbitales atómicos 2s del carbono con el orbital
1s con todos iguales.
Matriz de traslape de molécula de metano
1 2 3 4 5 6 7 8
1 1.0000 0.0 0.0 0.0 0.5133 0.5133 0.5133 0.5133
2 0.0 1.0000 0.0 0.0 -0.1618 0.4855 -0.1618 -0.1618
3 0.0 0.0 1.0000 0.0 0.4577 0.0 -0.2289 -0.2289
4 0.0 0.0 0.0 1.0000 0.0 0.0 0.3964 -0.3964
5 0.5133 0.4855 0.0 0.1805 0.1805 1.0000 0.1805 0.1805
6 0.5133 -0.1618 0.4577 1.000 1.0000 0.1805 0.1805 0.1805
7 0.5133 -0.1618 -0.2289 0.1805 0.1805 0.1805 1.0000 0.1805
8 0.5133 -0.1618 -0.2289 0.1805 0.1805 0.1805 0.1805 1.0000
4. Matriz del Hamiltoniano
Con la matriz de traslape (S), se debe encontrar la matriz del hamiltoniano (H)
HC=SCE
La energía integral Hii en el método de Huckel extendido es igual a la energía de un
electrón en determinado orbital atómico aislado en el apropiado estado. Sin embargo en un
átomo de carbono aislado, los más bajos estados de energía están asociados con la
configuración 1s22s
22p
2. En una molécula saturada con el metano, sin embargo los
electrones compartidos del carbono con cuatro hidrógenos indican que los orbitales 2s y 2p
están igualmente involucrados en la formación de los orbitales moleculares. La
configuración sp3 se refiere a los estados de valencia del carbono en esta molécula.
Para el metano
(C2s): H11 = −19.44eV= −0.7144 a.u
(C2p): H22 = H33 =H44= −10.67eV= −0.3921 a.u
(H1s): H55 = H66 =H77 =H88= −13.60eV= −0.50000 a.u
Los elementos no diagonalizados son evaluados de acuerdo a:
(
)
Donde K es un parámetro ajustable, el valor ajustado de Hoffman es 1.75.
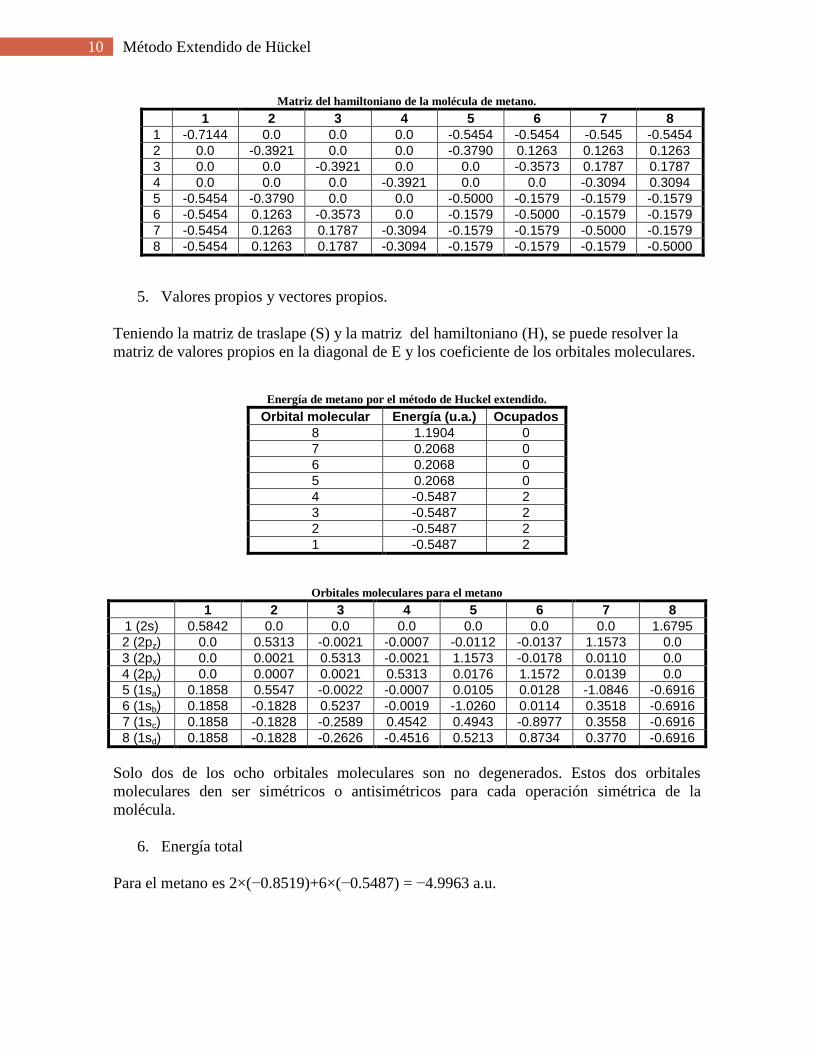
10 Método Extendido de Hückel
Matriz del hamiltoniano de la molécula de metano.
1 2 3 4 5 6 7 8
1 -0.7144 0.0 0.0 0.0 -0.5454 -0.5454 -0.545 -0.5454
2 0.0 -0.3921 0.0 0.0 -0.3790 0.1263 0.1263 0.1263
3 0.0 0.0 -0.3921 0.0 0.0 -0.3573 0.1787 0.1787
4 0.0 0.0 0.0 -0.3921 0.0 0.0 -0.3094 0.3094
5 -0.5454 -0.3790 0.0 0.0 -0.5000 -0.1579 -0.1579 -0.1579
6 -0.5454 0.1263 -0.3573 0.0 -0.1579 -0.5000 -0.1579 -0.1579
7 -0.5454 0.1263 0.1787 -0.3094 -0.1579 -0.1579 -0.5000 -0.1579
8 -0.5454 0.1263 0.1787 -0.3094 -0.1579 -0.1579 -0.1579 -0.5000
5. Valores propios y vectores propios.
Teniendo la matriz de traslape (S) y la matriz del hamiltoniano (H), se puede resolver la
matriz de valores propios en la diagonal de E y los coeficiente de los orbitales moleculares.
Energía de metano por el método de Huckel extendido.
Orbital molecular Energía (u.a.) Ocupados
8 1.1904 0
7 0.2068 0
6 0.2068 0
5 0.2068 0
4 -0.5487 2
3 -0.5487 2
2 -0.5487 2
1 -0.5487 2
Orbitales moleculares para el metano
1 2 3 4 5 6 7 8
1 (2s) 0.5842 0.0 0.0 0.0 0.0 0.0 0.0 1.6795
2 (2pz) 0.0 0.5313 -0.0021 -0.0007 -0.0112 -0.0137 1.1573 0.0
3 (2px) 0.0 0.0021 0.5313 -0.0021 1.1573 -0.0178 0.0110 0.0 4 (2py) 0.0 0.0007 0.0021 0.5313 0.0176 1.1572 0.0139 0.0 5 (1sa) 0.1858 0.5547 -0.0022 -0.0007 0.0105 0.0128 -1.0846 -0.6916
6 (1sb) 0.1858 -0.1828 0.5237 -0.0019 -1.0260 0.0114 0.3518 -0.6916 7 (1sc) 0.1858 -0.1828 -0.2589 0.4542 0.4943 -0.8977 0.3558 -0.6916 8 (1sd) 0.1858 -0.1828 -0.2626 -0.4516 0.5213 0.8734 0.3770 -0.6916
Solo dos de los ocho orbitales moleculares son no degenerados. Estos dos orbitales
moleculares den ser simétricos o antisimétricos para cada operación simétrica de la
molécula.
6. Energía total
Para el metano es 2×(−0.8519)+6×(−0.5487) = −4.9963 a.u.

11 Método Extendido de Hückel
Molécula: Agua
1. Elección de las coordenadas:
Estructura molecular: Tetraédrica. Distancia de enlaces (O-H): 0.958 Å (=1.810 u.a.),
Angulo: 104.5 °
Coordenadas cartesianas ( en u.a.):
Se situa la molécula en el plano YZ con el átomo de oxígeno en el origen y su eje de
simetría coincidiendo con el eje OZ. Coordenadas cartesianas de átomos de agua.
Átomo x y z
O 0.0 0.0 0.0
Ha 0.0 1.432 1.109
Hb 0.0 1.432 1.109
2. Elección de los orbitales atómicos
Para el H2O, los orbitales atómicos 1s para cada hidrógeno y para el oxígeno los orbitales
atómicos 2s, 2px, 2py y 2pz.
Elección de orbitales atómicos para agua.
OA Átomo Tipo exp
1 O 2s 4.55
2 O 2pz 4.55 3 O 2px 4.55 4 O 2py 4.55 5 Ha 1s1 1
6 Hb 1s2 1
3. Matriz de traslape
Matriz de traslape de molécula de agua.
2s 2px 2py 2pz 1s1 1s2
2s 1.0000 0.0 0.0 0.0 0.509 0.509
2px 0.0 1.0000 0.0 0.0 0 0
2py 0.0 0.0 1.0000 0.0 0.257 -0.257
2pz 0.0 0.0 0.0 1.0000 0.213 0.213
1s1 0.509 0.0 0.275 0.213 1 0.377
1s2 0.509 0.0 -0.275 0.213 0.377 1
4. Matriz del Hamiltoniano
Con la matriz de traslape (S), se debe encontrar la matriz del hamiltoniano (H)
HC=SCE
Para el agua
(O2s): H11 = −32.20 eV
12 Método Extendido de Hückel
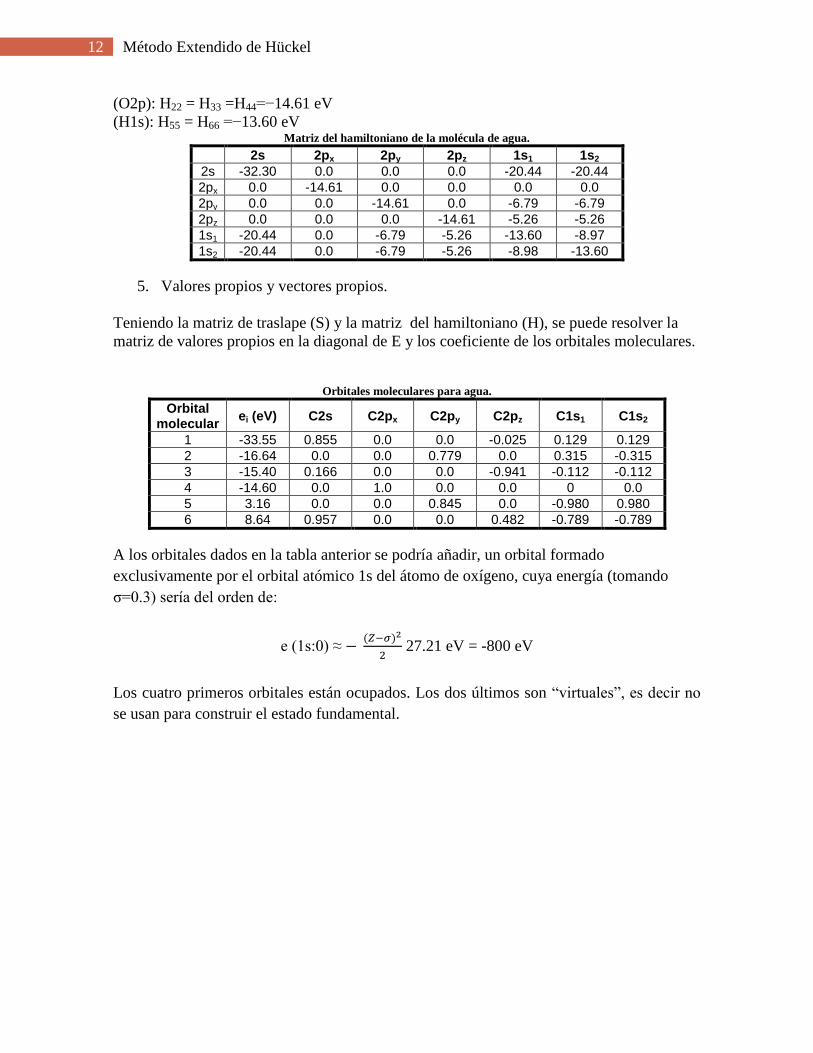
(O2p): H22 = H33 =H44=−14.61 eV
(H1s): H55 = H66 =−13.60 eV Matriz del hamiltoniano de la molécula de agua.
2s 2px 2py 2pz 1s1 1s2
2s -32.30 0.0 0.0 0.0 -20.44 -20.44
2px 0.0 -14.61 0.0 0.0 0.0 0.0
2py 0.0 0.0 -14.61 0.0 -6.79 -6.79
2pz 0.0 0.0 0.0 -14.61 -5.26 -5.26
1s1 -20.44 0.0 -6.79 -5.26 -13.60 -8.97
1s2 -20.44 0.0 -6.79 -5.26 -8.98 -13.60
5. Valores propios y vectores propios.
Teniendo la matriz de traslape (S) y la matriz del hamiltoniano (H), se puede resolver la
matriz de valores propios en la diagonal de E y los coeficiente de los orbitales moleculares.
Orbitales moleculares para agua.
Orbital molecular
ei (eV) C2s C2px C2py C2pz C1s1 C1s2
1 -33.55 0.855 0.0 0.0 -0.025 0.129 0.129
2 -16.64 0.0 0.0 0.779 0.0 0.315 -0.315
3 -15.40 0.166 0.0 0.0 -0.941 -0.112 -0.112
4 -14.60 0.0 1.0 0.0 0.0 0 0.0
5 3.16 0.0 0.0 0.845 0.0 -0.980 0.980
6 8.64 0.957 0.0 0.0 0.482 -0.789 -0.789
A los orbitales dados en la tabla anterior se podría añadir, un orbital formado
exclusivamente por el orbital atómico 1s del átomo de oxígeno, cuya energía (tomando
σ=0.3) sería del orden de:
e (1s:0) ≈ ( )
27.21 eV = -800 eV
Los cuatro primeros orbitales están ocupados. Los dos últimos son “virtuales”, es decir no
se usan para construir el estado fundamental.

13 Método Extendido de Hückel
CONCLUSIONES
Una de las grandes ventajas del Método extendido de Hückel es que puede aplicarse a una
gran cantidad de sistemas y que puede tratar a casi cualquier elemento ya que el único
parámetro especifico requerido es la energía de ionización, la cual suele encontrarse
reportada en tablas.
La capacidad de aplicar este método a sistemas grandes y a una gran variedad de elementos
es una de las razones por las que ha sido ampliamente utilizado para el cálculo de
estructuras de estado sólido y poliméricas.
Otra de sus ventajas es que el cálculo resulta más rápido que muchos otros métodos debido
a que la obtención de los elementos de la matriz del Hamiltoniano es simple, además de que
dicha matriz solo requiere ser diagonalizada una vez, es decir no requiere refinamiento,
para dar los valores y vectores propios.
De la misma manera, la debilidad del método radica en que no considera la repulsión
electrón spin o electrón-electrón, a su vez ignora el hecho de que la geometría molecular se
encuentra parcialmente determinada por la repulsión internuclear, de forma tal que este
método no suele ser utilizado para optimización de geometría sino que utiliza geometrías
experimentales.

14 Método Extendido de Hückel
BIBLIOGRAFÍA.
a) Levine, N. Ira, Química Cuántica, 5ª edición, Prentice Hall, Madrid, 2001,
p. 627-629.
b) Métodos computaciones, Cap. II,
http://www.jorgemarchetti.com.ar/Tesis/Fisica/Capitulo%20II.pdf , consultado el 21
de septiembre de 2013.
c) Hoffmann, R. J. Chem. Phys. 1963, 39, 1397-1412.
d) Ramachandran, K. I., Deepa, G. 2008. Computational Chemistry and Molecular
Modeling. Springer-Verlag Berlín Heidelberg. India. Pp. 86-88.
e) Lowe, John P. 2005. Quantum Chemistry. 3a. Elsevier. Londres. Pp. 324-335.
f) Gimarc, B. M, Molecular Structure and Bonding, Academic Press, New York,
1979, página 216.
g) Lewars, E.G. COMPUTATIONAL CHEMISTRY Introduction to the Theory and
Applications of Molecular and Quantum Mechanics, 1a. Ed, Kluwer Academic
Publishers, 2004.
h) http://idv.sinica.edu.tw/ytt/Ch4.pdf
i) Fernández, Manuel. Elementos de mecánica cuántica molecular. Universidad de
Cadiz, 1998. p. 348-354