Embed Size (px)
Citation preview
![Page 1: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/1.jpg)
Métodos de
la mecánica cuántica
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
la mecánica cuántica
Posgrado en Ciencias Químicas
1
![Page 2: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/2.jpg)
Mecánica Cuántica
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
2
![Page 3: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/3.jpg)
Métodos mecánico-cuánticos
Basados en función de onda
- de primeros principios (ab initio)
- semiempíricos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Basados en densidad electrónica
- ab initio
- ¿semiempíricos?
3
![Page 4: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/4.jpg)
ab initio
• Métodos variacionales
• Métodos de perturbacionales
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Otros métodos
4
![Page 5: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/5.jpg)
Semiempíricos
- aquellos que desprecian algunas integrales
o bien:
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
- aquellos que reemplazan su valor basados en
valores experimentales.
5
![Page 6: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/6.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Métodos basados
en la mecánica cuántica
ab initio
Hartree-Fock (HF)
Moller-Plesset (MP2, MP3 …) *
Interacción de configuraciones (CI)
Campo autoconsistente multiconfiguracional (MCSCF)
Interacción de configuraciones multireferencial (MRCI)
Cúmulo acoplado (CC)
Monte Carlo cuántico (QMC)
Funcionales de la densidad (DFT)
-Huckel
6
en la mecánica cuántica
semiempíricos
-Huckel
-Extended Huckel (EH)
- Pariser-Parr-Pople (PPP)
- Cancelación completa del
traslape diferencial (CNDO)
- Cancelación modificada del
traslape diatómico (MNDO)
- Cancelación intermedia modificada
del traslape diferencial (MINDO)
- Cancelación intermedia del traslape
diferencial (INDO)
- Cancelación intermedia modificada
del traslape diferencial (MINDO)
- Cancelación intermedia del traslape
diferencial de Zerner (ZINDO, INDO/S)
- Austin Model 1 (AM1)
- Parametrization method 3 (PM3)
- …
![Page 7: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/7.jpg)
Aspectos a tomar en consideración
• La energía de correlación:
• La base atómica (número de funciones base)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Ecorr= Eexacta - EHF
• Esto lleva al problema del escalamiento:
N3 N10
7
DFT HF MP CI
![Page 8: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/8.jpg)
Desempeño y exactitud de los diferentes métodos
HF << MP2 < CISD ≅ MP4 ≅ CCSD < CCSD(T) < CCSD(T) < Full CI
Fuentes de error:
- Aproximación Born-Oppenheimer
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
- Aproximación Born-Oppenheimer(Ψ depende de las posiciones nucleares no de sus momenta)
- El uso de bases incompletas
- La incorporación parcial de la correlación
- La omisión de efectos relativistas.
8
![Page 9: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/9.jpg)
Nivel de teoría-base atómica
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
STO-3G 3-21-G 6-31G(d) . . . ∞∞∞∞
HF HF/STO-3G HF/3-21-G HF/3-21-Glímite
HF
correlación tamaño de la base
9
CISD
CISDT
. . .
Full CIsol.
exacta
![Page 10: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/10.jpg)
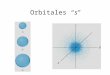
Espín orbitales
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
10
![Page 11: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/11.jpg)
Las coordenadas esféricas polares
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
11
![Page 12: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/12.jpg)
Funciones radiales del hidrógeno
1s2s2p
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
12
2p3s3p3d
![Page 13: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/13.jpg)
Funciones radiales
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
13
![Page 14: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/14.jpg)
Funciones angulares hidrogenoides
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
14
![Page 15: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/15.jpg)
La ecuación de Schrodinger
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
La ecuación dependiente del tiempo
15
La ecuación independiente del tiempo
![Page 16: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/16.jpg)
Las soluciones: orbitales hidrogenoides
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
La parte radial
Factor de normalización
16
![Page 17: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/17.jpg)
Las soluciones …cont.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
La parte angular
17
![Page 18: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/18.jpg)
Funciones ortonormalizadas
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
18
![Page 19: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/19.jpg)
Otro ejemplo: He
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
19
dos formas de decir lo mismo
![Page 20: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/20.jpg)
Orbitales moleculares: LCAO
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Funciones base
20
Un caso particular: H2
Funciones base
![Page 21: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/21.jpg)
La molécula de H2
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
21
![Page 22: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/22.jpg)
H2
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
22
![Page 23: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/23.jpg)
H2
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
23
![Page 24: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/24.jpg)
Sistemas polielectrónicos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
24
![Page 25: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/25.jpg)
Sistemas polielectrónicos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
25
![Page 26: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/26.jpg)
Sistemas polielectrónicos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
26
![Page 27: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/27.jpg)
Multiconfiguraciones
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
σ∗
27
σ
20 > 11 > 02 >
ψ σ σHF = ≡ ⟩...( ) ...( *) ...2 0 20
![Page 28: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/28.jpg)
Hartree-Fock-SCF
Ψtot= Ψel Ψnuc (Born-Oppenheimer)
Etot= Eel + Enuc
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Htot= Tn + Te + Vne + Vee + Vnn
![Page 29: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/29.jpg)
Hartree-Fock-SCF
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Hamiltoniano para un electrón en un espín-orbital χi en el campo de los núcleos y
los otros electrones en sus espín-orbitales χj .
core Coulomb (Jj (1))
29
intercambio (Jj (1))
![Page 30: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/30.jpg)
Hartree-Fock-SCF
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
30
El operador de Fock es un Hamiltoniano monoelectrónico efectivo en un sistema
polielectrónico.
![Page 31: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/31.jpg)
El método de campo autoconsistenteSCF
• Parte de una función de onda tipo determinante de Slater y busca el conjunto de orbitales χj que minimicen el valor esperado del hamiltoniano electrónico del átomo. Para esto:
1- Se selecciona un primer conjunto aproximado de funciones orbitales χj , j= 1…N.
2- Se calcula el efecto que sobre un electrón i tienen los demás
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
2- Se calcula el efecto que sobre un electrón i tienen los demás electrones. Para ello se sustituyen las funciones χj y se resuelve la ecuación de Fock, obteniéndose la función χi y las energías orbitales εi.
El nuevo conjunto de orbitales es mejor que el anterior. El segundo paso se repite cuantas veces sea necesario hasta que el conjunto de orbitales de salida y de entrada sean idénticos, es decir, cuando se alcance la autoconsistencia.
31
![Page 32: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/32.jpg)
Las ecuaciones de Roothaan-Hall
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Para sistemas moleculares, Roothaan y Hall, independiente y simultáneamente,
propusieron una estrategia para resolver un sistema molecular de N electrones con
N/2 orbitales (capa cerrada)
A partir de la expresión para la energía HF:
32
![Page 33: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/33.jpg)
Las ecuaciones de Roothaan-Hall
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Empleando la aproximación orbital para un sistema
molecular mediante:
33
Se requiere un conjunto de coeficientes Cνi que correspondan a la función de onda con la
menor energía.
![Page 34: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/34.jpg)
Las ecuaciones de Roothaan-Hall
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
La forma estándar de los elementos de la matriz de Fock en las ecuaciones de
Roothaan-Hall es:
34
Donde son los elementos de la matriz de densidad P
FC = SCE
La expresión matricial quedaría como:
![Page 35: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/35.jpg)
Secuencia del cálculo
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Calcular las integrales de la matriz de Fock (F)
• Calcular la matriz de superposición S
• Diagonalizar S
• Obtener S-1/2
• Generar una matriz de densidad inicial P
35
• Construir la matriz de Fock a partir de las integrales y de la matriz P
• Construir F’= S-1/2 F S-1/2
• Resolver la ecuación secular |F’-EI|= 0
• Calcular la matriz de coeficientes C de C= S-1/2 C´
• Calcular una nueva matriz de densidad P a partir de C
• Revisar la convergencia, Si el cálculo ha convergido detiener, si no, regresar al paso 6 empleando la nueva matriz de densidad P.
![Page 36: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/36.jpg)
Las funciones base actuales• Los conjuntos base estándar para cálculos de estructura
electrónica emplean combinaciones lineales de funciones gausianas para formar orbitales atómicos. Los conjuntos base asignan un grupo de funciones base a cada átomo en una molécula para aproximar sus orbitales.
• A mayor tamaño de los conjuntos base mejor aproximarán a los orbitales al imponerles menos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
aproximarán a los orbitales al imponerles menos restricciones para moverse en el espacio.
• Las funciones base que son, a su vez, formadas por combinaciones lineales de funciones gausianas se denominan funciones contraídas. Y a las funciones gausianas que las componen se les llama primitivas.
• A una función base formada por una sola gausiana se le conoce como no contraída.
36
![Page 37: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/37.jpg)
Las funciones base• Orbitales hidrogenoides
• Orbitales tipo Slater (STOs)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Orbitales Gausianos (GTOs)
• Los pseudopotenciales (ECPs)
37
![Page 38: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/38.jpg)
Características
• Los hidrogenoides poseen una parte radial muy compleja.
• Los STOs son más simples y la variable ζ se puede parametrizar, aunque, a diferencia de los hidrogenoides, carecen de nodos.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
los hidrogenoides, carecen de nodos.
• Para moléculas poliatómicas, las integrales multicéntricas con STOs son difíciles de evaluar pues no tienen solución analítica.
• Los GTOs son más fáciles de evaluar.
38
![Page 39: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/39.jpg)
Características
• Una base mínima es la formada por el menor
número de funciones estrictamente
necesarias para un átomo. Por ejemplo:
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
H: 1s C: 1s , 2s , 2px , 2py , 2pz
• Las bases mínimas no son adecuadas para
describir distribuciones electrónicas no
esféricas en las moléculas.
39
![Page 40: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/40.jpg)
STOs
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
40
El exponente ζ depende de la carga nuclear efectiva, es decir.
incorpora de una manera estática el efecto pantalla.
Por ejemplo:
![Page 41: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/41.jpg)
GTOs cartesianos
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
( )( )
2 2 2
13 2
4- ( )
8 ! ! !2( , , ; , , , ) e
(2 )!(2 )!(2 )!
i j k
i j k x y zi j k
x y z i j k x y zi j k
αααϕ α
π
+ +
+ +
=
41
- α es una constante que controla la extensión radial de la función.
- la suma de los enteros i, j, k es igual al número cuántico de momento
angular de la función: 0 para una s, 1 para para una p, 2 para una
función d, etc.
Se necesitan un gran número de gausianas para representar un orbital
molecular con una precisión aceptable.
![Page 42: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/42.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
42
![Page 43: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/43.jpg)
Bases gausianas
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
43
![Page 44: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/44.jpg)
3 ejemplos de primitivas gausianas
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
44
![Page 45: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/45.jpg)
Base mínima: contiene sólo la cantidad estrictamente
necesaria de funciones para cada ubicar a los
electrones de un átomo. Emplea un tamaño fijo para
Tipos de funciones base
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
cada tipo de orbital (STO-3G)
Doble zeta: emplea dos funciones base por cada OA
Triple zeta: emplea tres funciones base por cada OA
![Page 46: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/46.jpg)
Tipos de funciones base
Funciones base de “valencia dividida”
● orbitales del core representados por una función
base.
● orbitales de valencia: se dividen, arbitrariamente, en
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
● orbitales de valencia: se dividen, arbitrariamente, en
muchas funciones.
Esto permite mayor flexibilidad para la descripción
de los orbitales de valencia.
Ejemplos: 3-21G, 6-21G, 4-31G, 6-31G, y 6-311G
![Page 47: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/47.jpg)
Bases con polarización
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
47
![Page 48: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/48.jpg)
Funciones de Polarización
Representación que considera la deformación de los
orbitales atómicos.
orbital s → polarizado en combinación con orbitales p
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
orbital p → polarizado en combinación con orbitales d
La polarización de una función base con momento angular l,
se obtiene al combinar con funciones base de momento
angular l+1.
![Page 49: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/49.jpg)
Notación de Pople en GTOs
STO-3G STO-6G … STO-nG
3-21G 4-31G 6-31G
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Tipo Salter de por medio de n funciones gausianas
Bases mínimas:
Valencia dividida:
49
6-31G* 6-31G**
3-21+G 6-311++G(3df,3pd)
6 gausianas para el core 4 gausianas para la valencia: 3 contraídas y
una difusa.
Bases con polarización:
Bases con difusas y polarización: (triple zeta en la valencia)
Hay variaciones, por ejemplo: 6-31G* es equivalente a 6-31G(d).
Existen otras notaciones. Ver notación de Huzinaga para bases atómicas.
![Page 50: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/50.jpg)
Notación de Huzinaga en GTOs
STO-3G STO-6G … STO-nG
3-21G 4-31G 6-31G
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Tipo Salter de por medio de n funciones gausianas
Bases mínimas:
Valencia dividida:
50
6-31G* 6-31G**
3-21+G 6-311++G(3df,3pd)
6 gausianas para el core 4 gausianas para la valencia: 3 contraídas y
una difusa.
Bases con polarización:
Bases con difusas y polarización: (triple zeta en la valencia)
Hay variaciones, por ejemplo: 6-31G* es equivalente a 6-31G(d).
Existen otras notaciones. Ver notación de Huzinaga para bases atómicas.
![Page 51: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/51.jpg)
Bases
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
51
![Page 52: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/52.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Las bases enGaussian
52
![Page 53: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/53.jpg)
La matriz Z
• Permite expresar las posiciones atómicas de
manera que el cálculo pueda optimizar la
geometría de manera eficiente.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
53
![Page 54: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/54.jpg)
HF como punto de partida
Ecuaciones de HF
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
54
Ecuaciones de HF
Adición de más aproximaciones Adición de más determinantes
Soluciones aproximadas(métodos semiempíricos)
Convergencia haciala solución exacta
![Page 55: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/55.jpg)
H2O: densidad electrónica
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
55
![Page 56: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/56.jpg)
DFT: Hohenberg y Kohn
[ ] [ ]( ) ( ) ( ) ( )E r v r r dr F rρ ρ ρ= +∫
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
La energía del estado basal está definida unívocamente por la densidad electrónica
del estado basal. Por lo tanto, la energía es un funcional de la densidad electrónica.
La densidad electrónica debe integrar al número total de electrones N del sistema.
56
( )r dr Nρ =∫
[ ]( ) ( ) 0( )
E r r drr
δρ µ ρ
δρ − = ∫
La densidad electrónica debe integrar al número total de electrones N del sistema.
Para minimizar la energía se introduce esta restricción por medio del multiplicador
de Lagrange (-µ).
![Page 57: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/57.jpg)
DFT: Kohn-Sham
[ ]2
( ) ( ) ( )2
N
KE i iE r r r drρ ϕ ϕ ∇
= − ∑∫
[ ] [ ] [ ] [ ]( ) ( ) ( ) ( )KE H XCF r E r E r E rρ ρ ρ ρ= + +
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Funcional de intercambio y correlación
57
[ ]1
( ) ( ) ( )2
KE i i
i
E r r r drρ ϕ ϕ=
= −
∑∫
[ ]( ) ?XC
E rρ =
[ ] 1 2
1 2
1 2
( ) ( )1( )
2H
r rE r dr dr
r r
ρ ρρ =
−∫ ∫
![Page 58: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/58.jpg)
DFT funcionales de intercambio y correlación
• Funcionales locales, aprox. LDA, LSDA de Vosko-Wilk-Nussair (VWN).
• Funcionales corregidos por gradientes. GGAs
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Funcionales corregidos por gradientes. GGAs
(BLYP, PBE).
• Funcionales híbridos. (B3LYP)
• Meta GGAs (TPSS)
58
![Page 59: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/59.jpg)
Funcionales locales
• En los inicios de TFD se derivaron modelos a partir del gas de electrones, usando en el punto r las propiedades del gas homogéneo de electrones con densidad uniforme ρ igual a la ρ(r) que el sistema finito tiene localmente. Esta es la aproximación de densidad local (LDA). En ésta se aprovechan los resultados de las expresiones de Thomas-Fermi-Dirac para la energía cinética y el funcional de intercambio y correlación el cual se particiona en dos componentes:
E = E + E
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Exc= Ex + Ec
• para el intercambio se aprovecha el llamado funcional de intercambio de Dirac. Para la correlación Vosko, Wilk y Nussair realizaron interpolaciones de Padé a partir de cálculos Monte Carlo muy precisos en el gas de electrones realizados por Ceperley y Alder. Resulta algo sorprendente que la aproximación LDA dé tan buenos resultados para muchos sistemas atómicos y moleculares. La aproximación de densidad local se denomina LSDA.
• Vosko, S. H., Wilk, N., Nusair, M. Can. J. Phys. 1980, 58, 1200.
59
![Page 60: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/60.jpg)
Funcionales corregidos por gradientes
• Posteriormente se propuso la expansión por gradientes de la densidad para sistemas de densidad electrónica variable la cual tiene algunas dificultades, no obstante se ha desarrollado la expansión por gradientes generalizados (GGA) tanto para la parte del intercambio como para la parte de la correlación que expresa εxcen términos de las propiedades locales de la densidad, es decir, su valor y su gradiente o Laplaciano en el punto r lo cual es una
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
valor y su gradiente o Laplaciano en el punto r lo cual es una medida de las inhomogeneidades de la densidad. La inclusión de estas correcciones semilocales tiene ventajas como la de representar adecuadamente el comportamiento asintótico de la densidad. Dentro de la aproximación GGA, el funcional propuesto por Becke para el intercambio y de Lee, Yang y Parr para la correlación se denomina BLYP.
• Becke, A. D. Phys. Rev. A. 1988, 38, 3098.
• Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B. 1988, 37, 785.
60
![Page 61: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/61.jpg)
Funcionales híbridos• Existen otras aproximaciones como los tratamientos
híbridos en los que se define el funcional de intercambio como una combinación lineal de los términos de Hartree-Fock, los locales y los corregidos por gradientes. Este funcional de intercambio se combina después con uno de correlación ya sea local o también corregido por gradientes. Para este nivel de aproximación se tiene la formulación híbrida de tres parámetros sugerida también por Becke,
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
híbrida de tres parámetros sugerida también por Becke, B3LYP.
• Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
• Recientemente otras familias de funcionales han surgido, se les llama meta-GGAs.
61
![Page 62: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/62.jpg)
Desempeño de DFT
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
62
![Page 63: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/63.jpg)
OMs del aguaFacultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
63
![Page 64: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/64.jpg)
La densidad electrónica ρρρρ
tomando en cuenta que:
La densidad electrónica puede expresarse a partir de la matriz de densidad como:
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
elemento de la matriz de densidad
64
( )r dr Nρ =∫La densidad electrónica puede expresarse a partir de la matriz de densidad como:
la cual debe cumplir con:
Por lo tanto:
![Page 65: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/65.jpg)
La partición de la carga
• Existen diferentes esquemas de partición de la
carga eléctrica en una molécula.
• A estos esquemas se les conoce como análisis
de población.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
de población.
• Uno de los más conocidos y antiguos es el
análisis de población de Mülliken.
• Existen otros como el de Löwdin, el de
Wiberg, el NBO, el de Bader, etc.
65
![Page 66: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/66.jpg)
Análisis de población de Mülliken
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
µ está situado en el átomo A
Limitaciones:- No existe un operador mecánico cuántico para la carga atómica, por lo tanto, la carga
66
- No existe un operador mecánico cuántico para la carga atómica, por lo tanto, la carga
no es una variable observable.
- Mülliken supone que cada átomo tiene sus funciones base centradas en ese átomo
lo cual no siempre es cierto.
- Mülliken reparte la densidad electrónica asociada al traslape por igual entre los
átomos involucrados, lo cual no es realista y puede llevar a contradicciones.
- Las cargas de Mülliken dependen de la base empleada.
- Se dan casos extremos: Para el carbón central del isobutano se tiene que la carga da:
¡ 0.1 con 6-31G* y 1.0 con 6-311++G** !
![Page 67: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/67.jpg)
Análisis de población de Löwdin
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Trabaja sobre un conjunto base ortogonalizado del tipo:
67
Ventajas:- Evita el problema de poblaciones negativas o mayores que 2.
- Es menos dependiente de la base empleada.
![Page 68: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/68.jpg)
Potencial electrostático molecular
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
68
![Page 69: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/69.jpg)
Átomos en moléculas
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
contornos de
densidad electrónica
líneas de gradiente de
densidad electrónica
69
HF
densidad electrónica
Punto crítico
![Page 70: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/70.jpg)
Líneas de gradiente en formamida
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
H H
70
C N
O H
![Page 71: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/71.jpg)
Topología del MEP
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
71
![Page 72: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/72.jpg)
Los protocolos Gn de Pople
• Pretenden alcanzar resultados con una exactitud
alta a un costo computacional razonable.
(±2 kcal mol-1, ±0.1 eV, ±0.003 H, ±8 kJ mol-1)
• Se apoyan en resultados para un gran número de
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Se apoyan en resultados para un gran número de
compuestos diversos con elementos de hasta el
tercer período.
• La idea central llegar a un resultado combinando
cálculos de diferentes niveles de teoría.
72
![Page 73: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/73.jpg)
Los protocolos Gn … cont.
• La secuencia sugerida es:
1- Optimizar la geometría con HF/6-31G*
2- Reoptimizar geometría con MP2/6-31G*
3- Calcular un solo punto con MP4/6-31G*
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
3- Calcular un solo punto con MP4/6-31G*
4- Refinar la energía calculando con QCISD(T)
y bases con polarización.
5- Corregir por ZPE a partir del análisis escalado
de frecuencias armónicas del primer cálculo.
73
![Page 74: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/74.jpg)
Los protocolos Gn … cont.
• G2 Conjunto de referencia: 125 energías experimentales.
Los datos experimentales de referencia son energías de atomización, de ionización y afinidades electrónicas y protónicas.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
protónicas.
• G3 conjunto de referencia: 299 energías experimentales.
Los datos experimentales de referencia son energías de atomización, de ionización, afinidades electrónicas y protónicas y entalpías de formación.
74
![Page 75: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/75.jpg)
Energía de enlace
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Energía depunto cero
![Page 76: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/76.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
X2(g) 2X(g) ∆He(T) = ∆Uel +3/2RT
Energía y entalpía
H (g) 2H(g)
Ejemplo:
H2(g) 2H(g)
∆Uel = 458.1 (kJmol-1)
∆U0 = 432.0
∆U298 = 433.21
∆H298 = 435.93
![Page 77: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/77.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Energía entalpía y entropía
• E0 = Eelec + ZPE
• E= E0 + Evib + Erot + Etransl
• H= E + RT
• G= H -TS
![Page 78: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/78.jpg)
Entalpías de reacción
H2O + H+ H3O+
∆H298 = ∆E + ∆(PV)
∆E298= ∆Ee0 + ∆(∆Ee)298 + ∆Ev
0 + ∆(∆Ev)298 + ∆Er
298 + ∆E 298
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
∆ ∆ e ∆ ∆ e ∆ v ∆ ∆ v ∆ r
∆Et298
∆Ee0 = diferencia de energía entre productos y reactivos a 0 K.
∆ (∆Ee)298 = diferencia en el cambio de la energía electrónica entre 0 y 298 K.
∆Ev0 = diferencia entre la energía de punto cero de los productos y los reactivos.
∆(∆Ev)298 = diferencia en el cambio de la energía vibracional entre 0 y 298 K.
∆Er298 = diferencia entre la energía rotacional de productos y reactivos.
∆Et298 = diferencia entre la energía translacional de productos y reactivos.
∆(PV)= término de trabajo por presión volumen. (-RT en este caso)
78
![Page 79: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/79.jpg)
Los modos normales de vibración
La energía interna a una tempertatura es:
U(T)= Utrans(T) + Urot(T)+ Uvib(T) + Uvib(0)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
79
![Page 80: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/80.jpg)
Efectos del disolvente
• Los efectos del disolvente son importantes en
muchos casos.
• La forma de incluirlos depende de los recursos
disponibles y de lo que se pretenda calcular.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
disponibles y de lo que se pretenda calcular.
• La forma más simple es modificar la
permitividad del medio reemplazando la ε0
por otro valor.
• Hay otras opciones…
80
![Page 81: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/81.jpg)
Aplicaciones
A pesar de las aproximaciones, los métodos de estructura
electrónica son de enorme utilidad para:
• Estudios de estructura y energética de moléculas.
• Calcular diversas propiedades moleculares y espectrales
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
• Calcular diversas propiedades moleculares y espectrales
• Para estudiar intermediarios de reacciones.
• Para estudiar reactividad y selectividad.
81
![Page 82: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/82.jpg)
La reacción química
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
E Energía de activación
estado de transición
82
∆E = E(final) - E(inicial) < 0
t
edo. inicial
edo. final
![Page 83: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/83.jpg)
Desempeño de los modelos teóricos en la termoquímica de las reacciones
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
83
![Page 84: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/84.jpg)
Superficies de energía potencial
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
84
2 grados de libertad
![Page 85: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/85.jpg)
La geometría
La determinación eficiente de geometrías de equilibrio y de estados de transición es importante para comparaciones termoquímicas y cinéticas ya que la estructura electrónica es muy sensible a pequeños cambios en la geometría.
El procedimiento para aceptar una geometría como optimizada comprende satisfacer varios criterios, por ejemplo:
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
comprende satisfacer varios criterios, por ejemplo:
1- Que los cambios en la geometría entre un ciclo y el siguiente no afecten la energía más de un cierto valor arbitrario pequeño.
2- Que el gradiente de la energía de la estructura optimizada sea cercano a cero.
3- Que los parámetros geométricos (distancias, ángulos, etc.) entre un ciclo y el siguiente no cambien más allá de ciertos valores específicos pequeños.
85
![Page 86: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/86.jpg)
Geometrías del estado de transición
• Las geometrías del estado de transición entre
los reactivos y los productos de una reacción
no pueden predecirse fácilmente. Algunas
razones de ello es que no se tiene suficiente
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
razones de ello es que no se tiene suficiente
evidencia experimental de éstos
86
![Page 87: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/87.jpg)
Búsqueda de estados de transición
Presentan algunas particularidades
- El problema matemático de encontrar un estado de transición (ET) es más complicado que el de un mínimo.
- La hipersuperficie de energía potencial en la vecindad de un ET es más plana que en la vecindad de un mínimo.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
ET es más plana que en la vecindad de un mínimo.
- Debido a que un ET involucra enlaces parciales o casi nulos, deben aplicarse métodos que incorporen adecuadamente el tratamiento de la correlación electrónica.
- Se sabe relativamente poco respecto de las geometrías experimentales de los ET respecto de las geometrías de equilibrio.
87
![Page 88: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/88.jpg)
Puntos de silla
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
88
Una estructura de transición no es lo mismo que un estado de transición.
La primera es un máximo en la PES a lo largo de la ruta de reacción, la segunda
es un pico en el perfil de energía libre de la reacción.
La primera es independiente de la temperatura, la segunda no.
![Page 89: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/89.jpg)
Cl- + CH3-Cl
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
89
![Page 90: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/90.jpg)
Estados de transición
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
90
![Page 91: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/91.jpg)
Superficies
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
91
![Page 92: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/92.jpg)
Algoritmos de búsqueda de TSs
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
92
![Page 93: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/93.jpg)
Búsqueda de estados de transición
¿Cómo proponer un ET?
- Base su propuesta en una estructura relacionada con un problema similar conocido.
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
- Base su propuesta en un “promedio” entre la estructura del reactivo y el producto.
- Base su propuesta en su “intuición química” al especificar distancias y ángulos críticos de acuerdo con un mecanismo de reacción preconcebido.
- Verifique sus resultados (Hessiano, coordenadas, modos vibracionales)
93
![Page 94: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/94.jpg)
Aplicaciones
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
94
![Page 95: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/95.jpg)
Métodos de la mecánica molecular
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
Asisted Model Building with Energy Refinement (AMBER)
Chemistry at Harvard Macromolecular Mechanics (CHARMM)
Consistent Force Field (CFF)
Carbohidrate Hidroxyls represented by External Atoms (CHEAT)
DREIDING
Empirical Conformational Energy Program for Peptides (ECEPP)
95
Campos de fuerzas
Empirical Conformational Energy Program for Peptides (ECEPP)
Empirical Force Field (EFF)
Gronigen Molecular Simulation (GROMOS)
Molecular Mechanics (MM1, MM2, MM3, MM4)
Universal Force Field (UFF)
Optimized Potentials for Liquid Simulation (OPLS)
Tripos
…
![Page 96: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/96.jpg)
continuará…
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
96ψMC = ⟩ + ⟩ + ⟩a a a1 2 320 11 02[ ] [ ] [ ] [ ] ( ) ( )
s xcE T J E r v r drρ ρ ρ ρ ρ= + + + ∫
![Page 97: Métodos de la mecánica cuántica - [DePa] Departamento ...depa.fquim.unam.mx/amyd/archivero/MecanicaCuantica22-nov-08_5823.pdf · de orbitales de salida y de entrada sean idénticos,](https://reader030.pdfslide.tips/reader030/viewer/2022022705/5bd8a59309d3f22b078c3ee3/html5/page/97.jpg)
Facultad de Química
Departamento de Química Inorgánica y Nuclear
Dr. Sigfrido Escalante Tovar
97

![TRPECV y OM - [DePa] Departamento de Programas …depa.fquim.unam.mx/amyd/archivero/seminario1_23605.pdf · los orbitales de simetría adecuada. Los orbitales moleculares de la misma](https://img.pdfslide.tips/doc/110x75/5ba4a06c09d3f257608b6243/trpecv-y-om-depa-departamento-de-programas-depafquimunammxamydarchiveroseminario123605pdf.jpg)



![Teoría de Orbitales Moleculares - [DePa] Departamento de ...depa.fquim.unam.mx/amyd/archivero/TOM_27453.pdf · En general, un orbital molecular de enlace tiene una energía menor](https://img.pdfslide.tips/doc/110x75/5aded1c27f8b9af05b8b9bda/teora-de-orbitales-moleculares-depa-departamento-de-depafquimunammxamydarchiverotom27453pdfen.jpg)





![2do Depa Pedagogia[1]](https://img.pdfslide.tips/doc/110x75/56d6bea11a28ab301692efd5/2do-depa-pedagogia1.jpg)