Embed Size (px)
Citation preview

Research Article
Myeloid Differentiation Factor 88 Signaling inBone Marrow–Derived Cells Promotes GastricTumorigenesis by Generation of InflammatoryMicroenvironmentYusuke Maeda1,2,3, Kanae Echizen1,4, Hiroko Oshima1, Liang Yu5, Natthiya Sakulsak6,Osamu Hirose7, Yoichi Yamada7, Tadatsugu Taniguchi8, Brendan J. Jenkins5,Hideyuki Saya2, and Masanobu Oshima1,4
Abstract
It has been established that COX-2 and downstream signal-ing by prostaglandin E2 (PGE2) play a key role in tumorigenesisthrough generation of inflammatory microenvironment. Toll-like receptor (TLR) signaling through myeloid differentiationfactor 88 (MyD88) also regulates inflammatory responses intumors. However, the relationship between these distinct path-ways in tumorigenesis is not yet fully understood. We hereininvestigated the role of MyD88 in gastric tumorigenesis usingGan mice, which develop inflammation-associated gastrictumors due to the simultaneous activation of the COX-2/PGE2pathway and Wnt signaling. Notably, the disruption of Myd88in Gan mice resulted in the significant suppression of gastrictumorigenesis with the inhibition of inflammatory responses,even though COX-2/PGE2 pathway is constitutively activated.Moreover, Myd88 disruption in bone marrow–derived cells
(BMDCs) in Gan mice also suppressed inflammation andtumorigenesis, indicating that MyD88 signaling in BMDCsregulates the inflammatory microenvironment. We also foundthat expression of Tlr2 and its coreceptor Cd14 was inducedin tumor epithelial cells in Gan mice, which was suppressedby the disruption of Myd88. It has already been shown thatTLR2/CD14 signaling is important for stemness of intestinalepithelial cells. These results indicate that MyD88 in BMDCs,together with COX-2/PGE2 pathway, plays an essential role inthe generation of the inflammatory microenvironment, whichmay promote tumorigenesis through induction of TLR2/CD14pathway in tumor epithelial cells. These results suggest thatinhibition of TLR/MyD88 signaling together with COX-2/PGE2pathway will be an effective preventive strategy for gastric cancer.Cancer Prev Res; 9(3); 253–63. �2016 AACR.
IntroductionGastric cancer is the second leading cause of death from
malignancy worldwide (1).Helicobacter pylori (H. pylori) infectioninduces chronic gastritis, which is closely associated with gastriccancer (2, 3). It has been shown that COX-2 expression is induced
by H. pylori infection in the stomach (4), and that the activationof downstream signaling by prostaglandin E2 (PGE2) plays a keyrole in tumorigenesis in a variety of organs (5). We previouslyshowed that induction of COX-2/PGE2 pathway in the gastricmucosa ofK19-C2mEmice by expression of Ptgs2 and Ptges causesinflammatory responses in the glandular stomach (6). Moreover,Wnt signaling activation in the gastric mucosa of K19-Wnt1miceby expression of Wnt1 results in the development of limiteddysplastic lesions; however, the simultaneous activation of Wntsignaling and COX-2/PGE2 pathway in Gan mice leads to devel-opment of inflammation-associated gastric tumors (7, 8). Theseresults indicate the role of COX-2/PGE2 pathway-induced inflam-mation in gastric tumorigenesis. In the gastric tumor stroma,proinflammatory cytokines including TNFa, IL1b, and IL11 areexpressed and promote tumorigenesis through enhancement ofundifferentiated status, recruiting of myeloid-derived suppressorcells and increasing tumor cell survival (9–11).
On the other hand, it has been shown that innate immuneresponses through Toll-like receptors (TLRs) promote tumordevelopment through the regulation of inflammatory andimmune responses in tumor tissues (12, 13). Mouse geneticsstudies have indicated that signaling through myeloid differen-tiation factor 88 (MyD88), which is an effector molecule of theTLRs, is required for the development of sporadic intestinaltumors (14–16), colitis-associated colon cancer (17, 18), gastriccancer (19), liver cancer (20), skin tumors, and sarcomas (21).
1Division of Genetics, Cancer Research Institute, Kanazawa University,Kanazawa, Japan. 2Division of Gene Regulation, Institute forAdvancedMedical Research, Keio University,Tokyo, Japan. 3ResearchFellow of Japan Society for the Promotion of Science (JSPS), Tokyo,Japan. 4AMED-CREST, AMED, Japan Agency for Medical Researchand Development, Tokyo, Japan. 5Centre for Innate Immunity andInfectious Diseases, Hudson Institute of Medical Research, MonashUniversity, Clayton, Australia. 6Department of Anatomy, Faculty ofMedical Science, Naresuan University, Phitsanulok, Thailand. 7Facultyof Electrical and Computer Engineering, Institute of Science andEngineering, Kanazawa University, Kanazawa, Japan. 8Department ofMolecular Immunology, Institute of Industrial Science, The Universityof Tokyo, Tokyo, Japan.
Note: Supplementary data for this article are available at Cancer PreventionResearch Online (http://cancerprevres.aacrjournals.org/).
Corresponding Author: Masanobu Oshima, Division of Genetics, CancerResearch Institute, Kanazawa University, Kanazawa, Ishikawa 920-1192, Japan.Phone: 81-76-264-6760; Fax: 81-76-234-4519; E-mail:[email protected]
doi: 10.1158/1940-6207.CAPR-15-0315
�2016 American Association for Cancer Research.
CancerPreventionResearch
www.aacrjournals.org 253
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

MyD88 has also been shown to promote tumor metastasis (22).Moreover, MyD88 signaling induces the expression of COX-2 andinflammatory cytokines in tumor tissues, which contribute to theinflammatory microenvironment generation (14, 17, 18, 20).However, relationship between the COX-2/PGE2 pathway andTLR/MyD88 signaling in the inductionof inflammatory responsesand the promotion of tumorigenesis is not yet fully understood.
Here, we examined the role of MyD88 in gastric tumorigenesisusing Gan mice, in which COX-2/PGE2 pathway is constitutivelyactivated. Importantly, we show that MyD88 signaling in BMDCsis required for the generation of the inflammatory microenviron-ment in tumors even though the COX-2/PGE2 pathway is acti-vated. Moreover, inflammatory responses induce the expressionof TLR2 and CD14 in tumor epithelial cells, which have beenshown to play tumor-promoting roles through enhancement ofstem cell properties (16, 23, 24). These results indicate that boththe MyD88 signaling and the COX-2/PGE2 pathway are requiredfor generation of the inflammatory microenvironment, whichmay promote tumorigenesis through the induction of TLR2/CD14 signaling in tumor cells. These results suggest that targetingboth TLR/MyD88 signaling and the COX-2/PGE2 pathwaywill bean effective preventive strategy for gastric cancer.
Materials and MethodsAnimal model experiments
The construction ofGanmice, the gastric tumormodel that wasused in the current study, is described in Supplementary Infor-mation. Myd88-mutant mice were purchased (Oriental Bio-Service), and Irf5-mutant mice were constructed as describedpreviously (25). For the gastric tumor phenotype analyses, eachgenotype mice were euthanized and examined at 40 to 50 weeksof age. For the preneoplastic lesion analyses, K19-Wnt1mice wereexamined at 45 to 50 weeks of age. To induce gastric mucosalregeneration, tamoxifen (150 mg/kg) was administered to 10-week-old wild-type mice by oral gavage for 5 continuous days.This resulted in a loss of parietal cells, leading to mucosalregeneration (26). All of the mice used in this study were bornand raised in the same room of the Kanazawa University SPFfacility (Kanazawa, Japan). The protocols of all animal experi-ments were approved by the Committee on Animal Experimen-tation of Kanazawa University (Kanazawa, Japan).
The measurement of tumor volume and scoring ofpreneoplastic lesions
Ganmouse tumors are not polypotic, thus the numbers cannotbe counted. Thus, the tumor heightwasmeasuredusing histologicsections, and the relative tumor height was compared with thecontrol. The number of preneoplastic lesions of K19-Wnt1mousestomach was counted using six independent histologic sectionsfromeachmouse, and themeannumber of lesions per sectionwascalculated.
Histology and IHCTissues were fixed in 4% paraformaldehyde, paraffin-embed-
ded, and sectioned at 4-mm thickness. The sections were stainedwith haematoxylin and eosin (H&E) or processed for IHC. Theantibodies and visualization methods are described in Supple-mentary Information. The mean index for Ki-67 or F4/80 wascalculated by counting the labeled cells per microscopic field at200� in five fields. The immunostaining-positive area in the field
was measured using ImageJ 1.48 software program (NIH,Bethesda, MD). Apoptotic cells were detected using an ApopTagPeroxidase In Situ Apoptosis Detection Kit (Millipore).
PCR array analysesTotal RNAwas extracted from themouse gastric tumors and the
normal wild-typemouse stomach (n¼ 3 for each genotype) usingISOGEN (Nippon Gene), and purified using an RNeasy Mini Kit(Qiagen). Inflammation-related genes were analyzed using theRT2 Profiler PCR Array Mouse Innate & Adaptive ImmuneResponses (Qiagen). Thedatawere analyzedusing theRT2ProfilerPCR Array Data Analysis version 3.5 software (Qiagen) and werecompared with the wild-type mice.
Western blotting analysesTissues were homogenized in lysis buffer and the supernatant
protein sample was separated in SDS-polyacrylamide gel. Theantibodies are described in Supplementary Information. The ECLDetection System (GE Healthcare) was used to detect the signals.
Bone marrow transplantation and X-ray CTBonemarrow cells were prepared from the femurs and tibias of
donormice. Recipientmicewere irradiated (9Gy), followed by anintravenous injection of 2� 106 bone marrow cells. The x-ray CTimages of the gastric tumors were examined using a LaTheta LCT-100 instrument (Aloka) at every 4weeks until 20weeks after bonemarrow transplantation. The tumor areas of the slice images weremeasured using the ImageJ 1.48 software program (NIH) asdescribed previously (9).
Real-time RT-PCRTotal RNAs were extracted from cells or tumor tissues using
ISOGEN (Nippon Gene), reverse-transcribed using the Prime-Script RT reagent kit (Takara), and PCR-amplified by a StratageneMx3000P instrument (Agilent Technologies) using SYBR PremixExTaqII (Takara). The primers used for the real-time RT-PCRexcept for Cd14 were purchased from Takara, and the primer IDsand sequences for Cd14 are shown in Supplementary Table S1.
Cell culture experimentsThe human gastric cancer cell lines, AGS (ATCC), MKN74
(RIKEN BioResource Center, Japan), SNU484 [Korean Cell LineBank (KCLB)], and the mouse macrophage cell line, RAW264(RIKEN BioResource Center, Japan), were cultured in RPMI1640medium supplemented with 10% FBS. Cell lines were authenti-cated by isoenzyme analysis or short tandem repeat by the ATCC,RIKEN, and KCLB. All cell lines were initially expanded andcryopreserved within 1 month of receipt. Cells were used for 3months after thawing frozen vials. Construction of knockdowncell lines and the preparation of the conditioned medium (CM)are described in Supplementary Information.
Next-generation sequencing analysesTotal RNAwas extracted from themouse gastric tumors and the
normal wild-typemouse stomach (n¼ 3 for each genotype) usingISOGEN (Nippon Gene) and purified using an RNeasy Mini Kit(Qiagen). A sequencing analysis and upstream pathway analysiswere performed according to the methods described in Supple-mentary Information. The sequence results were deposited in theGene Expression Omnibus, as accession GSE70135.
Maeda et al.
Cancer Prev Res; 9(3) March 2016 Cancer Prevention Research254
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

Statistical analysisThe data were analyzed using the unpaired t test and are
presented as the mean � SE or SD. A value of P < 0.05 wasconsidered to be statistically significant.
ResultsThe suppression of gastric tumor developmentin Gan Myd88�/� mice
Innate immune signaling throughTLR/MyD88has been shownto regulate immune responses in tumors (12, 13). IFN regulatoryfactor 5 (IRF5) has also been shown to play a role in the TLR/MyD88 pathway through the induction of proinflammatorycytokines, including TNFa (25). Because TNFa plays a tumor-promoting role in Gan mice (9), we examined the roles of bothMyD88 and IRF5 in the current study. The expression of bothMYD88 and IRF5was significantly increased in human intestinal-type gastric cancer, and significant upregulation ofMyd88was alsofound in Ganmouse gastric tumors (Supplementary Fig. S1). Wethus crossedGanmicewithMyd88 and Irf5 gene knockoutmice togenerate GanMyd88�/� and Gan Irf5�/�mice, in whichMyd88 orIrf5 was disrupted in both epithelial and stromal cells, respec-tively. Although the control Gan mice developed large gastrictumors by 40 to 50 weeks of age, gastric tumorigenesis wassignificantly suppressed in the Gan Myd88�/� mice, but not inthe Gan Irf5�/� mice (Fig. 1A). The mean tumor sizes in the GanMyd88�/�micewere 49%of the controlGanmice (Fig. 1B). Theseresults indicate that MyD88, but not IRF5, plays a role in gastrictumor development.
Apoptotic cells were found on the tumor surface in both thecontrol Gan and the Gan Irf5�/� mice, and similar apoptotic celllocalization was found in theGanMyd88�/�mice, indicating thatMyD88 pathway does not affect apoptosis (Fig. 1C). In contrast,Ki-67–labeled cells were found throughout the tumor tissues ofGan andGan Irf5�/�mice,whereas theyweremostly limited to theproliferating zone at the gland neck in the Gan Myd88�/� mice.Consistently, the Ki-67–labeling index was reduced, to a signif-icant extent, in the Gan Myd88�/� mouse tumors, suggesting thatMyD88 signaling promotes tumor cell proliferation (Fig. 1D).
The suppression of the inflammatory responses inGan Myd88�/� tumors
In Gan mouse gastric tumors, macrophages infiltrated thetumor stroma and generated an inflammatorymicroenvironment(Fig. 2A). In the Gan Myd88�/� mouse tumors, the number ofmacrophages was significantly decreased in comparison to Ganand Gan Irf5�/� mouse tumors (Fig. 2A and B).
We thus performed PCR array analyses to examine the expres-sion of inflammation-related genes in the respective genotypemouse tumors. In the control Gan mouse tumors, expressionlevels of proinflammatory cytokines Il17a, Tnf, Il1a, Il1b, Il6, andIl23a, chemokine receptors Ccr4 and Ccr6, and TLRs, Tlr1, Tlr2,and Tlr5 were significantly increased (Fig. 2C and D). Althoughthe upregulation of Il1b, Il6,Ccr4, andCcr6was suppressed inGanIrf5�/� mouse tumors, most other inflammation-related geneswere upregulated, suggesting that IRF5 signaling may be lessinvolved in the inflammatory responses in gastric tumors. Impor-tantly, in the Gan Myd88�/� mouse tumors, induction of inflam-mation-related genes was significantly suppressed, indicating thatMyD88 signaling is essential for inflammatorymicroenvironmentgeneration (Fig. 2C and D).
We also found substantially decreased levels of phosphorylatedIkB and NF-kB in Gan Myd88�/� mouse tumors (Fig. 2E). More-over,we confirmedby IHC thatNF-kB is activated inmacrophagesof Gan mouse tumors, which was suppressed in Gan Myd88�/�
mice (Fig. 2F). InGanmouse tumors, the COX-2/PGE2 pathway isconstitutively activated by transgenic expression of Ptgs2 and Ptgesencoding COX-2 and mPGES-1, respectively; we confirmedthe expression of COX-2 and mPGES-1 in Gan Myd88�/� mousetumors. In two Gan Myd88�/� mice, mPGES-1 levels weredecreased in comparison to control, possibly due to the decreasein endogenous mPGES-1 levels by suppression of inflammation(Fig. 2G). We previously showed that Il1r1 disruption did notaffect the inflammation phenotype in K19-C2mEmice, indicatingthat the IL1R/MyD88 pathway is not required for COX-2/PGE2pathway-induced inflammation (27). Accordingly, these resultsindicate that TLR/MyD88 signaling is required for the generationof the COX-2/PGE2 pathway-induced inflammatory microenvi-ronment through the activation of NF-kB. Interestingly, the levelof active b-catenin was also decreased by Myd88 disruption (Fig.2E), suggesting that MyD88 has a role in activation of Wntsignaling in tumor cells (see below).
We next examined the expression of MyD88-dependent genesin another inflammation-associated gastric tumor model,gp130F/Fmice, in which hyperactivation of Stat3 promotes gastrictumorigenesis via the TLR2/MyD88 cascade (19, 28, 29). Theexpression levels of Tnf, Cd14, Casp1, Il1a, and Tlr2 were signif-icantly increased in gp130F/F tumors; however, this upregulationwas not suppressed in the gp130F/F Myd88�/� mouse tumors, inwhich Myd88 was disrupted in both epithelial and stromalcells (Supplementary Fig. S2). It is therefore possible that thehyperactivation of Stat3 (in gp130F/F mice) can mimic the role ofCOX-2/PGE2 and MyD88 (in Gan mice) in the generation of theinflammatory microenvironment; however, this possibilityremains to be investigated.
The suppression of gastric tumorigenesis by Myd88 disruptionin BMDCs
To examine the role of MyD88 in BMDCs in gastric tumor-igenesis, we next performed bone marrow transplantation fromMyd88�/�, Irf5�/�, or GFP transgenic mice into Gan mice togenerate the respective bone marrow chimeric mice. In thesemodels, Myd88 or Irf5 was disrupted in the BMDCs but not inthe epithelial cells of the tumor tissues. Chronologic X-ray CTanalyses showed a significant increase in the gastric tumorvolume of the bone marrow–transplanted (BMT)-Gan micethat received bone marrow from GFP or Irf5�/� mice duringthe 20-week observation period (Fig. 3A and B). In contrast,gastric tumor growth was significantly suppressed in the BMT-Gan mice that received Myd88�/� mouse bone marrow. Mac-rophage infiltration into the tumor tissues was significantlydecreased by the disruption of Myd88 in BMDCs (Fig. 3C andD). We further examined the gene expression in gastric tumorsof BMT-Gan mice by next-generation sequencing. We foundthat the levels of inflammatory cytokines and chemokines weredecreased in the BMT-Gan from Myd88�/� mice in comparisonwith the control Gan mice (Fig. 3E). Consistently, NF-kBactivation was suppressed in macrophages by the disruptionof Myd88 in BMDCs (Fig. 2F). Taken together, these resultsindicate that MyD88 signaling in BMDCs is important for theinflammatory microenvironment generation, which may play atumor-promoting role.
MyD88 and Inflammation in Gastric Tumorigenesis
www.aacrjournals.org Cancer Prev Res; 9(3) March 2016 255
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

The induction of TLR2 and CD14 expression in tumor cells byactivated macrophages
Among the MyD88-dependent upregulated genes in Ganmouse tumors, we focused on TLR2 and its coreceptor CD14because we had previously demonstrated that epithelial TLR2expression is important for gastric tumorigenesis in gp130F/Fmice(28). We found that the expression levels TLR2 and CD14 wereincreased significantly in human gastric cancer and Gan mousetumor tissues which were amixture of epithelial and stromal cells(Supplementary Fig. S3). We thus performed laser microdissec-tion–based RT-PCR, and found that the expression of Tlr2 andCd14 in Gan mouse tumors was predominantly detected in E-cadherin (Cdh1)-expressing tumor epithelial cells rather thanstromal cells (Fig. 4A). These results suggest that the inflammatorymicroenvironment induces the expression of Tlr2 and Cd14 intumor epithelial cells.
To further confirm the inflammation-dependent induction ofTLR2 and CD14 in tumor cells, we treated gastric cancer cells withCM from activated RAW264 macrophages treated with LPS.Importantly, we found that the expression levels of TLR2 andCD14 were significantly increased in AGS, MKN74, and SNU484cells after treatment with LPS-stimulated RAW264 CM but notwith control CM (Fig. 4C). Moreover, the induction of TLR2 andCD14 in gastric cancer cells was significantly suppressed whenMyd88 expression was knocked down by CRISPR/Cas9 system(KD-Myd88) in RAW264 cells (Fig. 4B and C). These resultsindicate that MyD88-dependent macrophage-derived factorsupregulate TLR2 and CD14 in cancer cells. Moreover, the expres-sion of stem cell marker CD44 was significantly increased in AGScells when the cells were treated with LPS CM, whereas it was
suppressed by knockdown of Myd88 expression in RAW264macrophages (Fig. 4D). These results indicate thatMyD88-depen-dent macrophage-derived factor(s) induces the TLR2/CD14 sig-naling pathway in cancer cells, whichmaypromote tumorigenesisby enhancing the undifferentiated status of the tumor cells.
TheMyD88 pathway helpsmaintain the undifferentiated statusof tumor cells
Tamoxifen treatment induces loss of parietal cells in mousestomach, which leads to expansion of undifferentiated cell pop-ulation and regeneration of the gastric mucosa (SupplementaryFig. S4) as described previously (26). Notably, the expressionlevels of Tlr2 and Cd14 were significantly increased in theseregenerating gastric mucosa, suggesting a role for TLR2/CD14signaling in undifferentiated epithelial cells in the stomach.
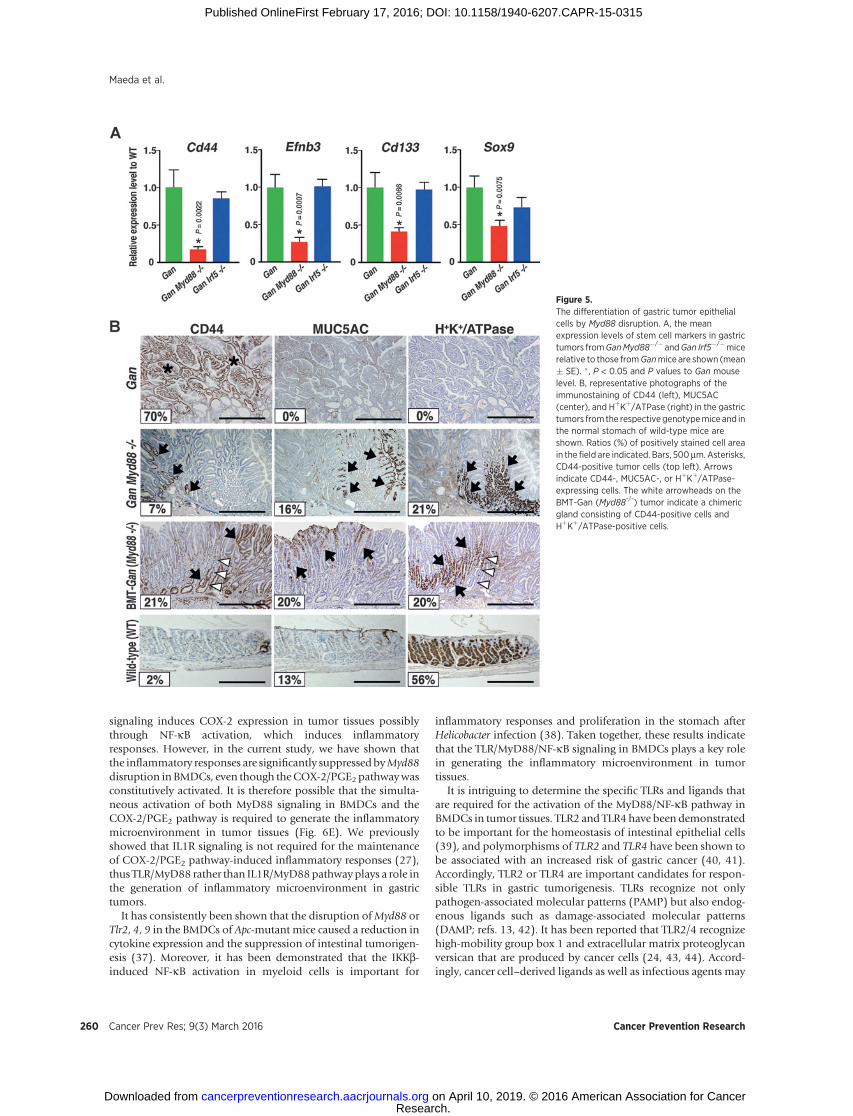
We next examined the differentiation status of the gastrictumors of Gan Myd88�/� mice, in which Myd88 was disruptedin both epithelial and stromal cells. Importantly, the expressionlevels of intestinal stem cell markers, Cd44, Ephb3, Cd133, andSox9 (30), were significantly decreased in the Gan Myd88�/�
mouse tumors but not in Gan Irf5�/� mice compared with thecontrol Ganmice (Fig. 5A). CD44 expression was not detected inthe normal glandular stomach, however, CD44-expressing cellswere found throughout the tumor tissues ofGanmice (Fig. 5B). Incontrast, there were substantial decreases in the numbers ofCD44-positive cells in Gan Myd88�/� mice and BMT-Gan fromMyd88�/� mice (Fig. 5B). We also found that the expression ofMUC5AC and HþKþ/ATPase (differentiation markers) was notexpressed in control Gan mouse tumors, but induced in thetumors of Gan Myd88�/� and BMT-Gan from Myd88�/� mice
Figure 1.The suppression of gastric tumordevelopment by Myd88 disruption.A, representative macroscopicphotographs (top) and histologicphotographs of whole views (bottom)of the indicated genotype mousegastric tumors. Arrow on theGan Myd88�/� tumor indicates asuppressed tumor lesion. Bars, 5 mm.B, the gastric tumor size of the GanMyd88�/� and Gan Irf5�/� micerelative to the mean level of controlGan mouse tumors is shown (mean%� SE). �, P < 0.05. C, representativephotographs of ApopTag (top) andanti-Ki-67 immunostaining (bottom)of the indicated genotype mousetumors. The insets are enlargedimages. Asterisk indicates aproliferating zone in a Gan Myd88�/�
mouse tumor. Bars, 200 mm. D, themean Ki-67 labeling indices of GanMyd88�/� and Gan Irf5�/� tumorsrelative to that of Gan mouse tumorsare shown (mean% � SE). � , P < 0.05.
Maeda et al.
Cancer Prev Res; 9(3) March 2016 Cancer Prevention Research256
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315
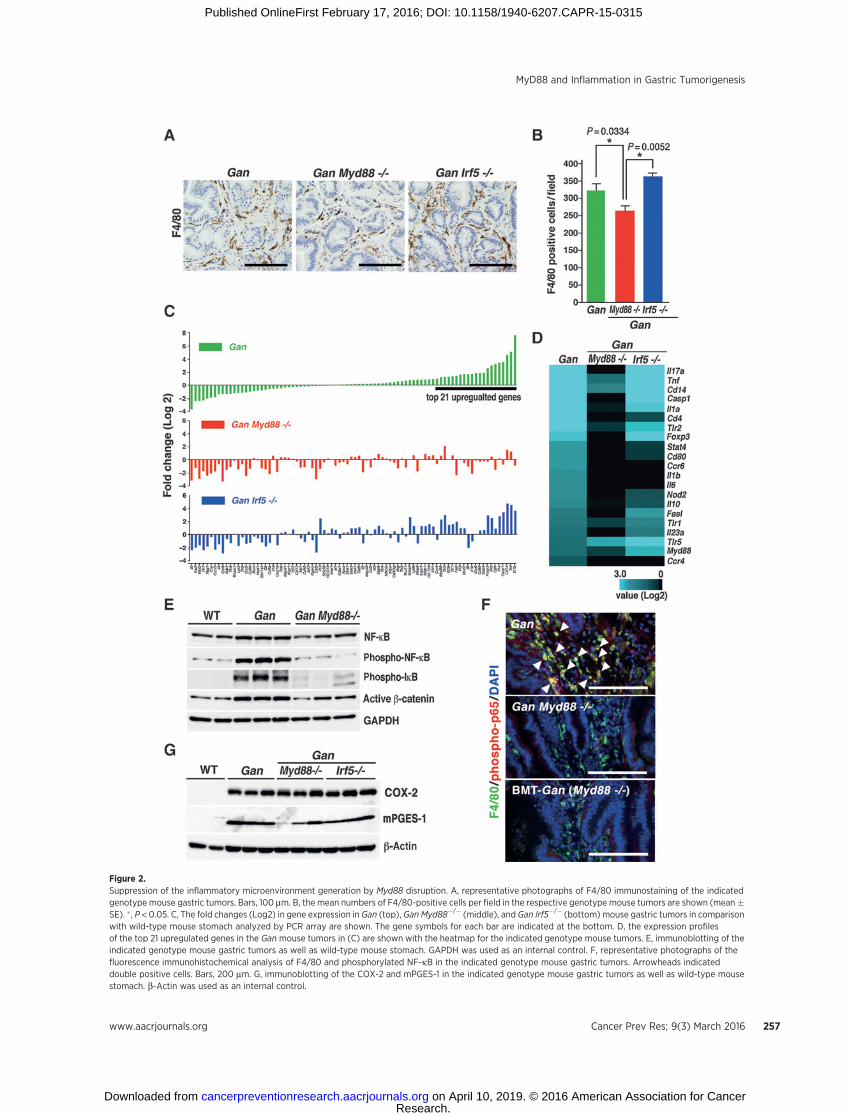
Figure 2.Suppression of the inflammatory microenvironment generation by Myd88 disruption. A, representative photographs of F4/80 immunostaining of the indicatedgenotype mouse gastric tumors. Bars, 100 mm. B, the mean numbers of F4/80-positive cells per field in the respective genotype mouse tumors are shown (mean�SE). � , P < 0.05. C, The fold changes (Log2) in gene expression in Gan (top), Gan Myd88�/� (middle), and Gan Irf5�/� (bottom) mouse gastric tumors in comparisonwith wild-type mouse stomach analyzed by PCR array are shown. The gene symbols for each bar are indicated at the bottom. D, the expression profilesof the top 21 upregulated genes in the Gan mouse tumors in (C) are shown with the heatmap for the indicated genotype mouse tumors. E, immunoblotting of theindicated genotype mouse gastric tumors as well as wild-type mouse stomach. GAPDH was used as an internal control. F, representative photographs of thefluorescence immunohistochemical analysis of F4/80 and phosphorylated NF-kB in the indicated genotype mouse gastric tumors. Arrowheads indicateddouble positive cells. Bars, 200 mm. G, immunoblotting of the COX-2 and mPGES-1 in the indicated genotype mouse gastric tumors as well as wild-type mousestomach. b-Actin was used as an internal control.
MyD88 and Inflammation in Gastric Tumorigenesis
www.aacrjournals.org Cancer Prev Res; 9(3) March 2016 257
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315
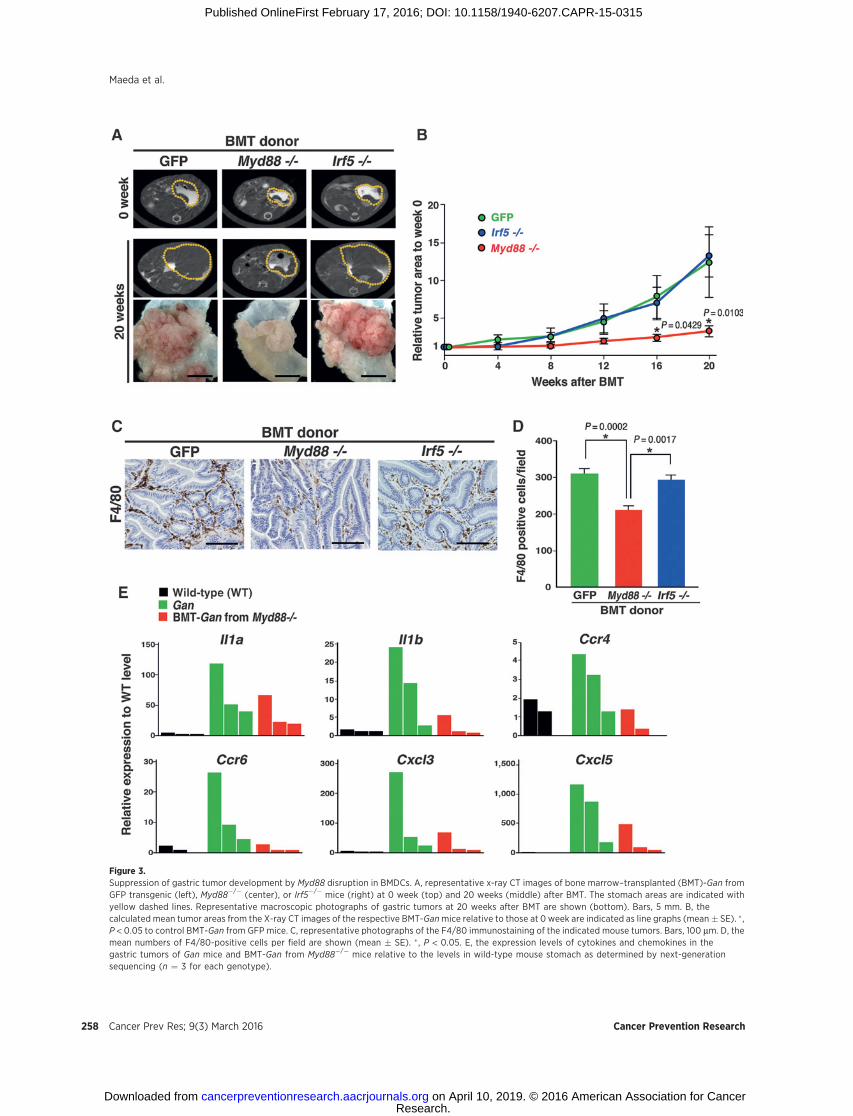
Figure 3.Suppression of gastric tumor development by Myd88 disruption in BMDCs. A, representative x-ray CT images of bone marrow–transplanted (BMT)-Gan fromGFP transgenic (left), Myd88�/� (center), or Irf5�/� mice (right) at 0 week (top) and 20 weeks (middle) after BMT. The stomach areas are indicated withyellow dashed lines. Representative macroscopic photographs of gastric tumors at 20 weeks after BMT are shown (bottom). Bars, 5 mm. B, thecalculated mean tumor areas from the X-ray CT images of the respective BMT-Ganmice relative to those at 0 week are indicated as line graphs (mean� SE). � ,P < 0.05 to control BMT-Gan from GFP mice. C, representative photographs of the F4/80 immunostaining of the indicated mouse tumors. Bars, 100 mm. D, themean numbers of F4/80-positive cells per field are shown (mean � SE). � , P < 0.05. E, the expression levels of cytokines and chemokines in thegastric tumors of Gan mice and BMT-Gan from Myd88�/� mice relative to the levels in wild-type mouse stomach as determined by next-generationsequencing (n ¼ 3 for each genotype).
Maeda et al.
Cancer Prev Res; 9(3) March 2016 Cancer Prevention Research258
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

(Fig. 5B). Notably, there are chimeric glands consisting of bothCD44-positive cells and HþKþ/ATPase-positive cells in BMT-Ganfrom Myd88�/� mice, suggesting that tumor epithelial cells aredifferentiated by Myd88 disruption in BMDCs (Fig. 5B, whitearrowheads).
Activation of Wnt signaling by MyD88 pathway ingastric tumors
We performed next-generation sequencing using the tumortissues ofGanMyd88�/�mice and BMT-Gan fromMyd88�/�miceto examine the gene expression profiles of the respective mousetumors. In the Gan Myd88�/� mouse tumors, Myd88 was dis-rupted in both the epithelial and stromal cells, whereas Myd88was disrupted only in the BMDCs in the BMT-Gan fromMyd88�/�
mice. We found that the numbers of genes that were down-regulated below 0.5-fold or upregulated above 2.0-fold in tumorsin both Gan Myd88�/� mice and BMT-Gan from Myd88�/� micecompared with control Gan mouse levels were 908 and 318,respectively (Fig. 6A). Using the sequencing results, we performedan Ingenuity Pathway Analysis (IPA) to examine the pathwaysthat were inhibited by disruption ofMyd88 in BMDCs. We foundthat NF-kB pathway was themost inhibited byMyd88 disruption,and inflammatory cytokine pathways were also significantlyinhibited (Fig. 6B and Supplementary Fig. S5). Notably, the IPAanalysis showed that the Wnt/b-catenin (CTNNB1) signaling wasalso significantly suppressed by Myd88 disruption, which wasconsistent with the immunoblotting results (Fig. 2E). These
results suggest that MyD88 signaling in BMDCs promotes tumor-igenesis through activation of Wnt signaling in tumor cells.
We previously showed that macrophages were infiltrated in thestroma of preneoplastic lesions of the K19-Wnt1mouse stomach,and thatWnt signaling activity was promoted in epithelial cells ofthese lesions, suggesting that macrophage-derived factors activateWnt signaling (31).Notably, thenumbers of preneoplastic lesionsin K19-Wnt1 Myd88�/� mice were significantly decreased incomparison with control K19-Wnt1 mice (Fig. 6C and D). It istherefore possible that the MyD88 signaling in BMDCs contri-butes to promotion of Wnt signaling activity in the tumor epi-thelial cells, which may contribute to the acceleration of theundifferentiated status and tumorigenesis.
Taken together, the current study strongly suggests that MyD88signaling inBMDCs togetherwithCOX-2/PGE2pathwaypromotetumorigenesis through the induction of the TLR2/CD14 pathwayas well as activation of Wnt signaling in tumor cells.
DiscussionIt has been established that chronic inflammation plays an
important role in cancer development through a variety ofmechanisms (32, 33), and that the COX-2/PGE2 pathway playsa key role in the promotion of tumorigenesis through the gener-ation of the inflammatory microenvironment (5, 34–36). Nota-bly, the induction of COX-2 expression in intestinal tumors wassuppressed inApcMinMyd88�/�mice (14), suggesting thatMyD88
Figure 4.The induction of TLR2 and CD14 intumor epithelial cells by activatedmacrophage-derived factors. A, themean expression levels of Cdh1, Tlr2,and Cd14 in tumor epithelial cells (GanEpi) and tumor stromal cells (Gan St)from Gan mice relative to the meanlevel of wild-type mouse epithelialcells (WTEpi) are shown (mean� SE).� , P < 0.05. B, immunoblotting ofMyD88 in the CRISPR/Cas9-mediatedMyd88-knockdown RAW264 cells.b-Actin was used as an internalcontrol. C, the mean expression levelsof CD14 and TLR2 in the indicated celllines treated with CM of parental orMyd88-knockdown (KD-Myd88)RAW264 cells with or without LPSstimulation relative to those ofuntreated control (NT) cells (shown asthe mean � SD). �, P < 0.05. D, theexpression levels of CD44 in AGS cellstreated with each RAW264 CMrelative to that treated with thecontrol CM are shown. Real-timeRT-PCR (mean � SD; left) and flowcytometry analysis (right). � , P < 0.05.CD44high population ratios areindicated in the flow cytometry graph.
MyD88 and Inflammation in Gastric Tumorigenesis
www.aacrjournals.org Cancer Prev Res; 9(3) March 2016 259
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315
signaling induces COX-2 expression in tumor tissues possiblythrough NF-kB activation, which induces inflammatoryresponses. However, in the current study, we have shown thatthe inflammatory responses are significantly suppressedbyMyd88disruption in BMDCs, even though theCOX-2/PGE2 pathwaywasconstitutively activated. It is therefore possible that the simulta-neous activation of both MyD88 signaling in BMDCs and theCOX-2/PGE2 pathway is required to generate the inflammatorymicroenvironment in tumor tissues (Fig. 6E). We previouslyshowed that IL1R signaling is not required for the maintenanceof COX-2/PGE2 pathway-induced inflammatory responses (27),thus TLR/MyD88 rather than IL1R/MyD88pathway plays a role inthe generation of inflammatory microenvironment in gastrictumors.
It has consistently been shown that the disruption ofMyd88 orTlr2, 4, 9 in the BMDCs of Apc-mutant mice caused a reduction incytokine expression and the suppression of intestinal tumorigen-esis (37). Moreover, it has been demonstrated that the IKKb-induced NF-kB activation in myeloid cells is important for
inflammatory responses and proliferation in the stomach afterHelicobacter infection (38). Taken together, these results indicatethat the TLR/MyD88/NF-kB signaling in BMDCs plays a key rolein generating the inflammatory microenvironment in tumortissues.
It is intriguing to determine the specific TLRs and ligands thatare required for the activation of the MyD88/NF-kB pathway inBMDCs in tumor tissues. TLR2 and TLR4 have been demonstratedto be important for the homeostasis of intestinal epithelial cells(39), and polymorphisms of TLR2 and TLR4 have been shown tobe associated with an increased risk of gastric cancer (40, 41).Accordingly, TLR2 or TLR4 are important candidates for respon-sible TLRs in gastric tumorigenesis. TLRs recognize not onlypathogen-associated molecular patterns (PAMP) but also endog-enous ligands such as damage-associated molecular patterns(DAMP; refs. 13, 42). It has been reported that TLR2/4 recognizehigh-mobility group box 1 and extracellular matrix proteoglycanversican that are produced by cancer cells (24, 43, 44). Accord-ingly, cancer cell–derived ligands as well as infectious agents may
Figure 5.The differentiation of gastric tumor epithelialcells by Myd88 disruption. A, the meanexpression levels of stem cell markers in gastrictumors fromGanMyd88�/� andGan Irf5�/�micerelative to those fromGanmice are shown (mean� SE). �, P < 0.05 and P values to Gan mouselevel. B, representative photographs of theimmunostaining of CD44 (left), MUC5AC(center), and HþKþ/ATPase (right) in the gastrictumors from the respective genotypemice and inthe normal stomach of wild-type mice areshown. Ratios (%) of positively stained cell areain the field are indicated. Bars, 500mm.Asterisks,CD44-positive tumor cells (top left). Arrowsindicate CD44-, MUC5AC-, or HþKþ/ATPase-expressing cells. The white arrowheads on theBMT-Gan (Myd88-/-) tumor indicate a chimericgland consisting of CD44-positive cells andHþKþ/ATPase-positive cells.
Maeda et al.
Cancer Prev Res; 9(3) March 2016 Cancer Prevention Research260
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

activate TLR2 and/or TLR4 in tumor tissues, thereby generatingthe inflammatory microenvironment.
We also showed that inflammatory responses induced theexpression of TLR2 and CD14 in tumor epithelial cells (Fig.6E). We previously showed that TLR2/MyD88 signaling in tumorcells promotes gastric tumorigenesis in gp130F/F mice (19, 28).Consistent with these findings, it has been shown that TLR2signaling in cancer cells plays a tumor-promoting role throughthe enhancement of stemness (23, 24). Moreover, the epithelialcell–specific deletion ofMyd88was observed to suppress intestinal
tumorigenesis in ApcMin mice (16). These results suggest that theTLR2/CD14 signaling in "epithelial cells" promotes tumor devel-opment through the maintenance of the undifferentiated status oftumor cells. Notably, it has been reported that H. pylori infectionactivates TLR2 but not TLR4 (45). Accordingly, it is possible thatH. pylori infection can promote gastric tumorigenesis through theactivationof TLR2on tumorepithelial cells.On theotherhand, theNF-kBpathway is activated inbothdifferentiated (tubular typeandpapillary type) and undifferentiated (mucinous type) humangastric cancers, which suggests that the MyD88/NF-kB pathway is
Figure 6.The suppression of Wnt signaling byMyd88 disruption. A, a Venn diagram of the genes downregulated below <0.5-fold (top) or upregulated >2.0-fold (bottom) inbothGanMyd88�/� and BMT-Gan fromMyd88�/�mice are shown. B, the list of upstream regulators inhibited in the gastric tumors in bothGanMyd88�/� and BMT-Gan from Myd88�/� mice as determined by the IPA (z-score < �2.9). The orange-colored lines indicate cytokine pathways. The red-colored lines indicatetheNF-kB,MyD88, andWnt/b-catenin (CTNNB1) pathways. C, representative photographs of H&E (top) and immunostaining for Ki-67 (bottom) using serial sectionsof the gastricmucosa of the indicated genotypemice. Arrows indicate preneoplastic lesions ofK19-Wnt1mice. Bars, 200 mm. D, the numbers of preneoplastic lesionsin control K19-Wnt1 and K19-Wnt1 Myd88�/� mice are shown (mean � SE). Asterisks indicate P < 0.05. E, a schematic drawing showing the role of MyD88 ingastric tumorigenesis. TLR/MyD88 signaling and COX-2/PGE2 pathways in BMDCs cooperatively generate inflammatory microenvironment, which inducesexpression of TLR2 and CD14 in tumor epithelial cells. The promotion of Wnt signaling through activation of NF-kB is a possible mechanism underlying TLR2/CD14-dependent tumorigenesis in tumor epithelial cells.
www.aacrjournals.org Cancer Prev Res; 9(3) March 2016 261
MyD88 and Inflammation in Gastric Tumorigenesis
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

less involved in the regulation of the histologic subtypes ofestablished gastric cancer (Supplementary Table S2).
The expression of Tlr2 and Cd14 in intestinal and mammaryepithelial cells is associated with the activation of Wnt signal-ing, which may explain the increased stemness and tumorige-nicity (16). We herein showed that Wnt/b-catenin signaling issignificantly suppressed and that differentiation is induced ingastric tumors by Myd88 disruption, supporting the idea thatTLR2/CD14 signaling enhances the stemness of tumor cellsthrough activation of Wnt signaling. It has been reported thatNF-kB activation promotes Wnt signaling in the intestinalepithelial cells, which causes dedifferentiation and the acqui-sition of stem cell properties (46). It is therefore possible thatthe activation of NF-kB by TLR2/CD14/MyD88 signalingincreases the Wnt signaling activity in tumor epithelial cells,thereby contributing to the maintenance of undifferentiatedstatus of tumor cells (Fig. 6E).
In the current study, we showed that TLR/MyD88 signaling inBMDCs is required for the generation of the inflammatory micro-environment in cooperation with the activation of the COX-2/PGE2pathway.MyD88-dependentmacrophage-derived factors inthe inflammatory microenvironment induce the expression ofTLR2 and CD14 in tumor cells. Wnt signaling activation is apossible mechanism underlying the TLR2/CD14/MyD88 signal-ing–induced tumor promotion. On the basis of these findings, wepropose that the targeting of TLR/MyD88 signaling together withthe COX-2/PGE2 pathway will be an effective preventive strategyagainst gastric cancer.
Disclosure of Potential Conflicts of InterestH. Saya reports receiving commercial research grants from Daiichi Sankyo
Co., Ltd. and Eisai Co., Ltd. No potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: M. OshimaDevelopment of methodology: Y. MaedaAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Maeda, K. Echizen, H. Oshima, L. Yu, N. Sakulsak,B.J. JenkinsAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Y. Maeda, K. Echizen, H. Oshima, O. Hirose,Y. Yamada, T. Taniguchi, B.J. JenkinsWriting, review, and/or revision of the manuscript: B.J. Jenkins, M. OshimaAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): T. Taniguchi, H. SayaStudy supervision: H. Saya, M. Oshima
AcknowledgmentsThe authors thank Manami Watanabe and Ayako Tsuda for technical
assistance. The computations were partially performed on the NIG supercom-puter at the ROIS National Institute of Genetics, Japan.
Grant SupportThisworkwas supported byAMED-CREST, AMED, JapanAgency forMedical
Research and Development, Japan (to M. Oshima), and Grants-in-Aid forScientific Research on Innovative Areas from theMinistry of Education, Culture,Sports, Science and Technology of Japan (#22114005 and #15H02362; toM. Oshima). This work was also supported in part by the National Health andMedical Research Council of Australia (to B. Jenkins) as well as the OperationalInfrastructure Support Program by the Victorian Government of Australia (to B.Jenkins).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received August 14, 2015; revised November 19, 2015; accepted December11, 2015; published OnlineFirst February 17, 2016.
References1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA
Cancer J Clin 2005;55:74–108.2. Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adeno-
carcinomas. Nat Rev Cancer 2002;2:28–37.3. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest
2007;117:60–9.4. McCarthy CJ, Crofford JL, Greenson J, Scheiman JM. Cyclooxygenase-2
expression in gastric antral mucosa before and after eradication of Helico-bacter pylori infection. Am J Gastroenterol 1999;94:1218–23.
5. Wang D, DuBois RN. Eicosanoids and cancer. Nat Rev Cancer 2010;10:181–93.
6. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic gastric tumorsinduced by activated macrophages in COX-2/mPGES-1 transgenic mice.EMBO J 2004;23:1669–78.
7. Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM, OshimaM. Carcinogenesis in mouse stomach by simultaneous activation of theWnt signaling and prostaglandin E2 pathway. Gastroenterology 2006;13:1086–95.
8. Oshima H, Oguma K, Du YC, OshimaM. Prostaglandin E2, Wnt, and BMPin gastric tumor mouse models. Cancer Sci 2009;100:1779–85.
9. Oshima H, Ishikawa T, Yoshida GJ, Naoi K, Maeda Y, Naka K, et al. TNF-a/TNFR1 signaling promotes gastric tumorigenesis through induction ofNoxo1 and Gna14 in tumor cells. Oncogene 2014;33:3820–9.
10. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Over-expression of Interleukin-1b induces gastric inflammation and cancermobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408–19.
11. Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, et al.Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinaltumorigenesis and can be targeted therapeutically. Cancer Cell 2013;24:257–71.
12. Salcedo R, Cataisson C, Hasan U, Yuspa SH, Trincheri G. MyD88and its divergent toll in carcinogenesis. Trends Immunol 2013;34:379–89.
13. Pradere JP,DapitoDH, Schwabe RF. The Yin and Yangof Toll-like receptorsin cancer. Oncogene 2014;33:3485–95.
14. Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinaltumorigenesis through the adaptor protein MyD88. Science 2007;317:124–7.
15. Lee SH, Ju LL, Ganzales-Navajas J, Seo GS, Shen C, Brick J, et al. ERKactivation drives intestinal tumorigenesis in Apcmin/þ mice. Nat Med2010;16:665–70.
16. Sheeren FA, Kuo AH, vanWeele LJ, Cai S, Glykofridis I, Sikandar SS, et al. Acell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia andoncogenesis. Nat Cell Biol 2014;16:1238–48.
17. Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C.Modulation of the intestinal microbiota alters colitis-associated colorectalcancer susceptibility. PLoS One 2009;4:e6026.
18. Schiechl G, Bauer B, Fuss I, Lang SA, Moser C, Ruemmele P, et al. Tumordevelopment in murine ulcerative colitis depends on MyD88 signaling ofcolonic F4/80þCD11bhighGr1low macrophages. J Clin Invest 2011;121:1692–708.
19. Kennedy CL, Najdovska M, Tye H, McLeod L, Yu L, Jarnicki A, et al.Differential role of MyD88 and Mal/TIRAP in TLR2-mediated gastrictumorigenesis. Oncogene 2014;33:2540–6.
20. Naugler WE, Sakurai T, Kim S, Maeda S, Kim KH, Elsharkawy AM, et al.Gender disparity in liver cancer due to sex differences inMyD88-dependentIL-6 production. Science 2007;317:121–4.
21. Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schriber RD, et al.Demonstration of inflammation-induced cancer and cancer immunoe-diting during primary tumorigenesis. Proc Natl Acad Sci U S A 2008;105:652–6.
Cancer Prev Res; 9(3) March 2016 Cancer Prevention Research262
Maeda et al.
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

22. Jia RJ, Cao L, Zhang L, Jing W, Chen R, Zhu MH, et al. Enhance myeloiddifferentiation factor 88 promotes tumor metastasis via induction ofepithelial-mesenchymal transition in human hepatocellular carcinoma.Cell Death Dis 2014;5:e1103.
23. Chefetz I, Alvero AB,Holmberg JC, Lebowitz N, Craveiro V, Yang-HartwichY, et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotestumor repair and recurrence. Cell Cycle 2013;12:511–21.
24. Conti L, Lanzardo S, ArigoniM,AntonazzoR, Radaelli E, CantarellaD, et al.The noninflammatroy role of high mobility group box 1/toll-like receptor2 axis in the self-renewal of mammary cancer stem cells. FASEB J2013;27:4731–44.
25. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al.Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005;434:243–9.
26. HuhWJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, Mills JC. Tamoxifeninduces rapid, reversible atrophy, and metaplasia in mouse stomach.Gastroenterology 2012;142:21–4.
27. Oshima M, Oshima H, Matsunaga A, Taketo MM. Hyperplastic gastrictumors with spasmolytic polypeptide-expressing metaplasia caused bytumor necrosis factor-a-dependent inflammation in cyclooxygenase-2/microsomal prostaglandin E synthase-1 transgenic mice. Cancer Res2005;65:9147–51.
28. Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N,et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesisindependent of tumor inflammation. Cancer Cell 2012;22:466–78.
29. McCormack W, Oshima M, Tan P, Jenkins BJ. Toll-like receptor 2: thera-peutic target for gastric carcinogenesis. Oncotarget 2012;3:1260–1.
30. Itkovitz S, Lyubimova A, Blat IC, Maynard M, van Es J, Lees J, et al. Single-molecule transcript counting of stem-cell markers in the mouse intestine.Nat Cell Biol 2012;14:106–14.
31. Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, et al.Activated macrophages promote Wnt signalling through tumour necrosisfactor-a in gastric tumour cells. EMBO J 2008;27:1671–81.
32. Coussens LM,Werb Z. Inflammation andCancer. Nature 2002;420:860–7.33. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-
induced cancer: crosstalk between tumours, immune cells and microor-ganisms. Nat Rev Cancer 2013;13:759–71.
34. OshimaM, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kong E, et al.Suppression of intestinal polyposis inApcD716 knockoutmice by inhibitionof cyclooxygenase 2 (COX-2). Cell 1996;87:803–9.
35. Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S,et al. Acceleration of intestinal polyposis through prostaglandin receptorE2 in ApcD716 knockout mice. Nat Med 2001;7:1045–51.
36. Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT,et al. Selective PGE2 suppression inhibits colon carcinogensis andmodifieslocal mucosal immunity. Cancer Prev Res 2011;4:1198–208.
37. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al.Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012;491:254–8.
38. Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, RogersAB, et al. Conditional deletion of IkB-kinase-b accelerates Helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenter-ology 2010;138:1022–34.
39. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R.Recognition of commensal microflora by Toll-like receptors is required forintestinal homeostasis. Cell 2004;118:229–41.
40. Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA,et al. A functional polymorphism of Toll-like receptor 4 gene increasesrisk of gastric carcinoma and its precursors. Gastroenterology 2007;132:905–12.
41. Castano-Rodriguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM.The role of TLR2, TLR4 and CD14 genetic polymorphisms in gastriccarcinogenesis: a case-control study and meta-analysis. PLoS One 2013;8:e60327.
42. Kuraishy A, Karin M, Grivennikov S. Tumor promotion via injury- anddeath-induced inflammation. Immunity 2011;35:467–77.
43. Abe A, Kuwata T, Yamauchi C, Higuchi Y, Ochiai A. High mobilitygroup box1 (HMGB1) released from cancer cells induces the expressionof pro-inflammatory cytokines in peritoneal fibroblasts. Pathol Int2014;64:267–75.
44. Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al.Carcinoma-produced factors activate myeloid cells through TLR2 to stim-ulate metastasis. Nature 2009;457:102–6.
45. Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, et al.Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacterhepaticus bacteria activate innate immunity via Toll-like receptor 2 but notToll-like receptor 4. Infect Immun 2004;72:6446–54.
46. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, ZieglerPK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acqui-sition of stem-cell-like properties. Cell 2013;152:25–38.
www.aacrjournals.org Cancer Prev Res; 9(3) March 2016 263
MyD88 and Inflammation in Gastric Tumorigenesis
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315

2016;9:253-263. Published OnlineFirst February 17, 2016.Cancer Prev Res Yusuke Maeda, Kanae Echizen, Hiroko Oshima, et al. Inflammatory MicroenvironmentDerived Cells Promotes Gastric Tumorigenesis by Generation of
−Myeloid Differentiation Factor 88 Signaling in Bone Marrow
Updated version
10.1158/1940-6207.CAPR-15-0315doi:
Access the most recent version of this article at:
Material
Supplementary
1
http://cancerpreventionresearch.aacrjournals.org/content/suppl/2015/12/29/1940-6207.CAPR-15-0315.DCAccess the most recent supplemental material at:
Cited articles
http://cancerpreventionresearch.aacrjournals.org/content/9/3/253.full#ref-list-1
This article cites 46 articles, 8 of which you can access for free at:
Citing articles
http://cancerpreventionresearch.aacrjournals.org/content/9/3/253.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerpreventionresearch.aacrjournals.org/content/9/3/253To request permission to re-use all or part of this article, use this link
Research. on April 10, 2019. © 2016 American Association for Cancercancerpreventionresearch.aacrjournals.org Downloaded from
Published OnlineFirst February 17, 2016; DOI: 10.1158/1940-6207.CAPR-15-0315





























