Embed Size (px)
Citation preview

HIGHLIGHTS
2100 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2100 $ 17.50+.50/0 Angew. Chem. 2001, 113, Nr. 11
Tanabe und Mitarbeiter stellten eine andere Methode vor,bei der keine Dehydratisierungsmittel oder -techniken nötigsind.[5] Diphenylammoniumtriflat, das aus Diphenylamin undTrifluormethansulfonsäure erhältlich ist, katalysiert die Kon-densation äquimolarer Mengen Carbonsäure und Alkohol(Tabelle 1). Die Reaktion verläuft unter Erhitzen der Reak-tanten auf 80 8C, ohne dass auf Dehydrierungstechnikenzurückgegriffen werden muss. Die Produkte konnten nachSäulenchromatographie in beträchtlichen Ausbeuten, abernicht quantitativ isoliert werden (89 ± 96 %, in einem Fall nur78 %). Primäre und sekundäre Alkohole wurden eingesetzt,und einige Funktionalitäten wie Keto- und Cyclopropylgrup-pe wurden toleriert.
Bei den hier erwähnten Reaktionsvorschriften waren dieProdukte zwar mit geringen Mengen der Ausgangssubstanzenverunreinigt, doch die hohen Ausbeuten ermöglichten eineeinfache Isolierung der Produkte mittels Säulenchromatogra-phie.[6] Die Aufarbeitung gelingt problemlos, so lange alleinauf die Isolierung der Ester abgezielt wird. Der Fall liegtjedoch anders bei in der Praxis angewendeten Prozessen, beidenen zudem nicht umgesetzte Reaktanten zurückgewonnenwerden müssen. Eine 1:1-Stöchiometrie ist nur wirklicheffektiv bei 100 % Umsetzung, anderenfalls sollten die zweiKomponenten (zurückgewonnene Carbonsäure und Alkohol)von der Produktmischung abgetrennt werden. Im Vergleichist dies jedoch weniger vorteilhaft als eine Reaktionsführung,
bei der eine der Komponenten im Überschuss vorgelegt wird.In diesem letzteren Fall ist die Zurückgewinnung nur einesReaktanten notwendig aufgrund der leichteren Erreichbar-keit einer 100-prozentigen Umsetzung. Eine Ausbeute von100 % unter Einsatz äquimolarer Mengen der Reaktantenohne jeglichen Reinigungsschritt bleibt daher noch immereine Herausforderung für die Zukunft. Die Umsätze undAusbeuten, die in den beiden hier beschriebenen Studienerzielt werden konnten, sind in diesem strengen Sinne nichtbefriedigend. Ohne Zweifel stellen die beiden Methodenjedoch einen groûen Schritt vorwärts dar, und es istzu erwarten, dass diese Erfolge den Weg zum Ziel ebnenwerden.
[1] T. W. Greene, P. G. Wuts, Protective Groups in Organic Synthesis,3. Aufl. , Wiley, New York, 1999, S. 149, S. 373.
[2] B. M. Trost, Science 1991, 254, 1471; B. M. Trost, Angew. Chem. 1995,107, 285 ± 307; Angew. Chem. Int. Ed. Engl. 1995, 34, 259 ± 281..
[3] Repräsentative Beispiele: G. A. Olah, T. Keumi, D. Meidar, Synthesis1978, 929; A. K. Kumar, T. K. Chattopadhyay, Tetrahedron Lett. 1987,28, 3713; J. Otera, N. Dan-oh, H. Nozaki, J. Org. Chem. 1991, 56, 5307;B. Das, B. Venkataiah, P. Madhusudhan, Synlett 2000, 59; A. S.-Y. Lee,H.-C. Yang, F.-Y. Su, Tetrahedron Lett. 2001, 42, 301.
[4] K. Ishihara, S. Ohara, H. Yamamoto, Science 2000, 290, 1140.[5] K. Wakasugi, T. Misaki, K. Yamada, Y. Tanabe, Tetrahedron Lett. 2000,
41, 5249.[6] Professor Hisashi Yamamoto, persönliche Mitteilung; die Isolierungs-
methode ist nicht in Lit. [4] beschrieben.
Neues aus der hochauflösenden Festkörper-NMR-Spektroskopie**
Harald Schwalbe* und Anthony Bielecki
Es ist seit langem bekannt, dass Struktur und Dynamik vonbiologischen Makromolekülen mittels NMR-Spektroskopiegenau bestimmt werden können. Die Mehrzahl der NMR-Untersuchungen wurde und wird an Proben durchgeführt, diein flüssigen Lösungsmitteln gelöst vorliegen, da unter diesenBedingungen schmalere Linienbreiten und damit bessereAuflösungen als bei Festkörperproben beobachtet werden.Häufig ist es jedoch notwendig, Festkörperspektren aufzu-nehmen, z. B. wenn man unlösliche biologische Proben wieAggregate oder Amyloide untersuchen will. Hinzu kommen
Systeme, deren Eigenschaften zwischen denen von Festkör-pern und gelösten Systemen liegen. Membranen und mem-brangebundene Proteine sind beispielsweise im engen Sinnkeine Festkörper, lassen sich aber wegen ihres hohen Gradesan molekularer Ausrichtung als Flüssigkristalle beschreiben.Weitere Beispiele sind stark viskose Flüssigkeiten oder Gele,in denen die molekulare Beweglichkeit gering oder räumlicheingeschränkt ist. Für alle diese Systeme können Festkörper-NMR-Untersuchungen besonders wertvoll sein, vorausge-setzt man weiû, mit welchen Festkörper-NMR-Methodenman die interessantesten Informationen erhält.
Die Herausforderungen der Festkörper-NMR-Spektrosko-pie liegen in den starken anisotropen Wechselwirkungen derdurch den Raum vermittelten Dipol-Dipol-Kopplung, diehäufig auch als dipolare Kopplung bezeichnet wird, und derorientierungsabhängigen chemischen Verschiebung, der che-mischen Verschiebungsanisotropie (CSA). (Einige Kernehaben auûerdem ein elektrisches Quadrupolmoment, daseine zusätzliche anisotrope Wechselwirkung erzeugt, auf diehier aber nicht eingegangen wird.) In Lösung werden dieseanisotropen Wechselwirkungen durch schnelle molekulareBewegung ausgemittelt, sodass lediglich der Einfluss auf die
[*] Prof. Dr. H. Schwalbe, Dr. A. BieleckiDepartment of ChemistryandMIT/Harvard Center for Magnetic Resonanceat the Francis Bitter Magnet LaboratoryMassachusetts Institute of Technology77 Massachusetts Avenue, Cambridge, MA 02139 (USA)Fax: (�1) 617-253-5405E-mail : [email protected]
[**] Wir danken dem Massachusetts Institute of Technology, der Karl-Winnacker-Stiftung und dem NIH/NCRR-Programm (RR00995) fürfinanzielle Unterstützung sowie Elke Duchardt für Hilfe beim An-fertigen von Abbildungen.
HIGHLIGHTS
Angew. Chem. 2001, 113, Nr. 11 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2101 $ 17.50+.50/0 2101
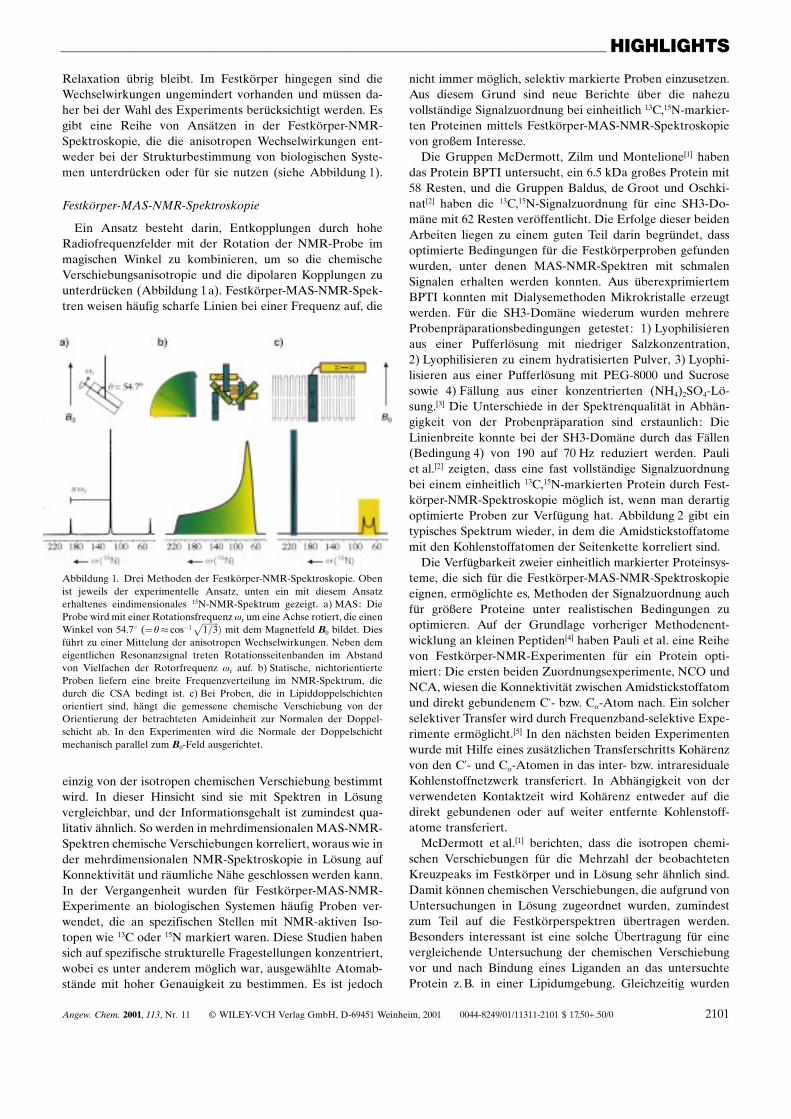
Relaxation übrig bleibt. Im Festkörper hingegen sind dieWechselwirkungen ungemindert vorhanden und müssen da-her bei der Wahl des Experiments berücksichtigt werden. Esgibt eine Reihe von Ansätzen in der Festkörper-NMR-Spektroskopie, die die anisotropen Wechselwirkungen ent-weder bei der Strukturbestimmung von biologischen Syste-men unterdrücken oder für sie nutzen (siehe Abbildung 1).
Festkörper-MAS-NMR-Spektroskopie
Ein Ansatz besteht darin, Entkopplungen durch hoheRadiofrequenzfelder mit der Rotation der NMR-Probe immagischen Winkel zu kombinieren, um so die chemischeVerschiebungsanisotropie und die dipolaren Kopplungen zuunterdrücken (Abbildung 1 a). Festkörper-MAS-NMR-Spek-tren weisen häufig scharfe Linien bei einer Frequenz auf, die
Abbildung 1. Drei Methoden der Festkörper-NMR-Spektroskopie. Obenist jeweils der experimentelle Ansatz, unten ein mit diesem Ansatzerhaltenes eindimensionales 15N-NMR-Spektrum gezeigt. a) MAS: DieProbe wird mit einer Rotationsfrequenz wr um eine Achse rotiert, die einenWinkel von 54.78 (�q� cosÿ1
��������1=3
p � mit dem Magnetfeld B0 bildet. Diesführt zu einer Mittelung der anisotropen Wechselwirkungen. Neben demeigentlichen Resonanzsignal treten Rotationsseitenbanden im Abstandvon Vielfachen der Rotorfrequenz wr auf. b) Statische, nichtorientierteProben liefern eine breite Frequenzverteilung im NMR-Spektrum, diedurch die CSA bedingt ist. c) Bei Proben, die in Lipiddoppelschichtenorientiert sind, hängt die gemessene chemische Verschiebung von derOrientierung der betrachteten Amideinheit zur Normalen der Doppel-schicht ab. In den Experimenten wird die Normale der Doppelschichtmechanisch parallel zum B0-Feld ausgerichtet.
einzig von der isotropen chemischen Verschiebung bestimmtwird. In dieser Hinsicht sind sie mit Spektren in Lösungvergleichbar, und der Informationsgehalt ist zumindest qua-litativ ähnlich. So werden in mehrdimensionalen MAS-NMR-Spektren chemische Verschiebungen korreliert, woraus wie inder mehrdimensionalen NMR-Spektroskopie in Lösung aufKonnektivität und räumliche Nähe geschlossen werden kann.In der Vergangenheit wurden für Festkörper-MAS-NMR-Experimente an biologischen Systemen häufig Proben ver-wendet, die an spezifischen Stellen mit NMR-aktiven Iso-topen wie 13C oder 15N markiert waren. Diese Studien habensich auf spezifische strukturelle Fragestellungen konzentriert,wobei es unter anderem möglich war, ausgewählte Atomab-stände mit hoher Genauigkeit zu bestimmen. Es ist jedoch
nicht immer möglich, selektiv markierte Proben einzusetzen.Aus diesem Grund sind neue Berichte über die nahezuvollständige Signalzuordnung bei einheitlich 13C,15N-markier-ten Proteinen mittels Festkörper-MAS-NMR-Spektroskopievon groûem Interesse.
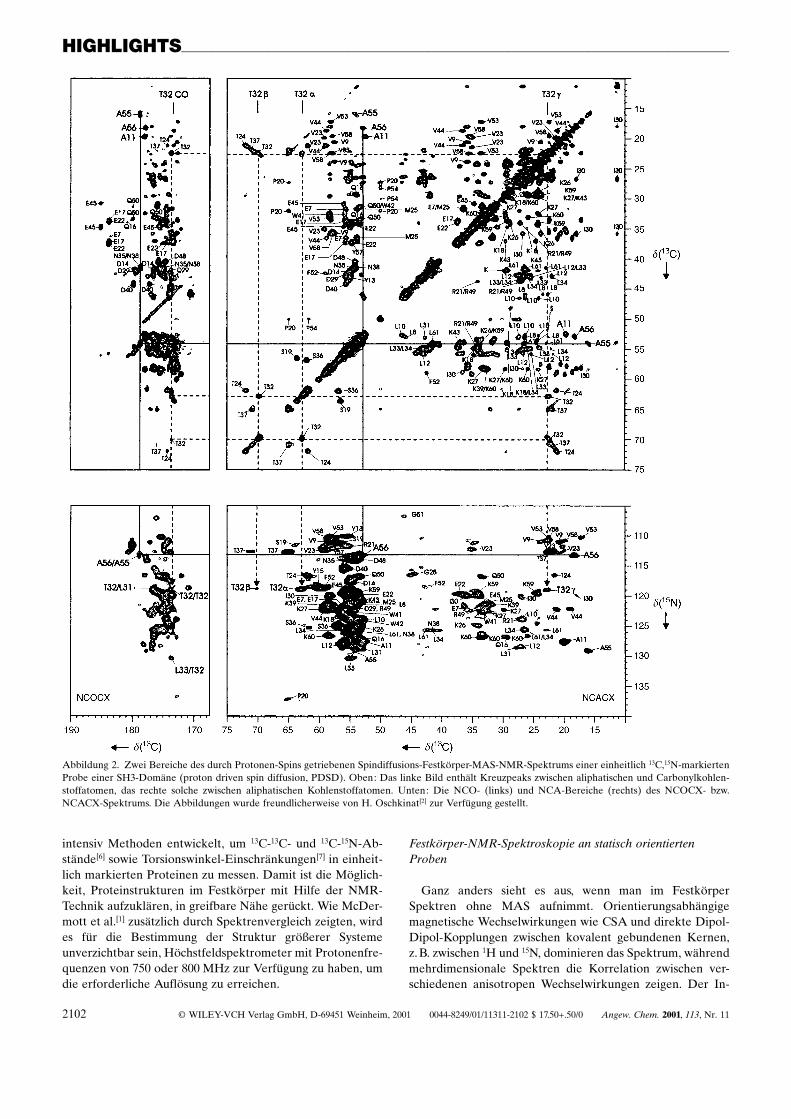
Die Gruppen McDermott, Zilm und Montelione[1] habendas Protein BPTI untersucht, ein 6.5 kDa groûes Protein mit58 Resten, und die Gruppen Baldus, de Groot und Oschki-nat[2] haben die 13C,15N-Signalzuordnung für eine SH3-Do-mäne mit 62 Resten veröffentlicht. Die Erfolge dieser beidenArbeiten liegen zu einem guten Teil darin begründet, dassoptimierte Bedingungen für die Festkörperproben gefundenwurden, unter denen MAS-NMR-Spektren mit schmalenSignalen erhalten werden konnten. Aus überexprimiertemBPTI konnten mit Dialysemethoden Mikrokristalle erzeugtwerden. Für die SH3-Domäne wiederum wurden mehrereProbenpräparationsbedingungen getestet: 1) Lyophilisierenaus einer Pufferlösung mit niedriger Salzkonzentration,2) Lyophilisieren zu einem hydratisierten Pulver, 3) Lyophi-lisieren aus einer Pufferlösung mit PEG-8000 und Sucrosesowie 4) Fällung aus einer konzentrierten (NH4)2SO4-Lö-sung.[3] Die Unterschiede in der Spektrenqualität in Abhän-gigkeit von der Probenpräparation sind erstaunlich: DieLinienbreite konnte bei der SH3-Domäne durch das Fällen(Bedingung 4) von 190 auf 70 Hz reduziert werden. Pauliet al.[2] zeigten, dass eine fast vollständige Signalzuordnungbei einem einheitlich 13C,15N-markierten Protein durch Fest-körper-NMR-Spektroskopie möglich ist, wenn man derartigoptimierte Proben zur Verfügung hat. Abbildung 2 gibt eintypisches Spektrum wieder, in dem die Amidstickstoffatomemit den Kohlenstoffatomen der Seitenkette korreliert sind.
Die Verfügbarkeit zweier einheitlich markierter Proteinsys-teme, die sich für die Festkörper-MAS-NMR-Spektroskopieeignen, ermöglichte es, Methoden der Signalzuordnung auchfür gröûere Proteine unter realistischen Bedingungen zuoptimieren. Auf der Grundlage vorheriger Methodenent-wicklung an kleinen Peptiden[4] haben Pauli et al. eine Reihevon Festkörper-NMR-Experimenten für ein Protein opti-miert: Die ersten beiden Zuordnungsexperimente, NCO undNCA, wiesen die Konnektivität zwischen Amidstickstoffatomund direkt gebundenem C'- bzw. Ca-Atom nach. Ein solcherselektiver Transfer wird durch Frequenzband-selektive Expe-rimente ermöglicht.[5] In den nächsten beiden Experimentenwurde mit Hilfe eines zusätzlichen Transferschritts Kohärenzvon den C'- und Ca-Atomen in das inter- bzw. intraresidualeKohlenstoffnetzwerk transferiert. In Abhängigkeit von derverwendeten Kontaktzeit wird Kohärenz entweder auf diedirekt gebundenen oder auf weiter entfernte Kohlenstoff-atome transferiert.
McDermott et al.[1] berichten, dass die isotropen chemi-schen Verschiebungen für die Mehrzahl der beobachtetenKreuzpeaks im Festkörper und in Lösung sehr ähnlich sind.Damit können chemischen Verschiebungen, die aufgrund vonUntersuchungen in Lösung zugeordnet wurden, zumindestzum Teil auf die Festkörperspektren übertragen werden.Besonders interessant ist eine solche Übertragung für einevergleichende Untersuchung der chemischen Verschiebungvor und nach Bindung eines Liganden an das untersuchteProtein z. B. in einer Lipidumgebung. Gleichzeitig wurden
HIGHLIGHTS
2102 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2102 $ 17.50+.50/0 Angew. Chem. 2001, 113, Nr. 11
intensiv Methoden entwickelt, um 13C-13C- und 13C-15N-Ab-stände[6] sowie Torsionswinkel-Einschränkungen[7] in einheit-lich markierten Proteinen zu messen. Damit ist die Möglich-keit, Proteinstrukturen im Festkörper mit Hilfe der NMR-Technik aufzuklären, in greifbare Nähe gerückt. Wie McDer-mott et al.[1] zusätzlich durch Spektrenvergleich zeigten, wirdes für die Bestimmung der Struktur gröûerer Systemeunverzichtbar sein, Höchstfeldspektrometer mit Protonenfre-quenzen von 750 oder 800 MHz zur Verfügung zu haben, umdie erforderliche Auflösung zu erreichen.
Festkörper-NMR-Spektroskopie an statisch orientiertenProben
Ganz anders sieht es aus, wenn man im FestkörperSpektren ohne MAS aufnimmt. Orientierungsabhängigemagnetische Wechselwirkungen wie CSA und direkte Dipol-Dipol-Kopplungen zwischen kovalent gebundenen Kernen,z. B. zwischen 1H und 15N, dominieren das Spektrum, währendmehrdimensionale Spektren die Korrelation zwischen ver-schiedenen anisotropen Wechselwirkungen zeigen. Der In-
Abbildung 2. Zwei Bereiche des durch Protonen-Spins getriebenen Spindiffusions-Festkörper-MAS-NMR-Spektrums einer einheitlich 13C,15N-markiertenProbe einer SH3-Domäne (proton driven spin diffusion, PDSD). Oben: Das linke Bild enthält Kreuzpeaks zwischen aliphatischen und Carbonylkohlen-stoffatomen, das rechte solche zwischen aliphatischen Kohlenstoffatomen. Unten: Die NCO- (links) und NCA-Bereiche (rechts) des NCOCX- bzw.NCACX-Spektrums. Die Abbildungen wurde freundlicherweise von H. Oschkinat[2] zur Verfügung gestellt.

HIGHLIGHTS
Angew. Chem. 2001, 113, Nr. 11 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2103 $ 17.50+.50/0 2103
formationsgehalt der Spektren hängt von der Orientierungder statischen Probe im Magnetfeld ab. Ist die Probe starkorientiert (Abbildung 1 c), wie dies bei einem in einerLipiddoppelschicht rekonstituierten Protein möglich ist, dannbesteht das Spektrum aus diskreten Signalen. Auf den erstenBlick ähnelt es somit einem in Lösung oder mit der MAS-Technik aufgenommenen Spektrum, doch gibt es einenbedeutenden Unterschied: Die Frequenzen der Kreuzpeakshängen in jeder Frequenzdimension von der Orientierung derMoleküle relativ zum magnetischen Feld ab. Diese Eigen-schaft kann dazu genutzt werden, die Orientierung einzelnerStrukturelemente eines Moleküls zu bestimmen, sodassweitere Untersuchungen die gesamte Molekülstruktur zu-gänglich machen sollten.
Die PISEMA-Methode[8] ist die Methode der Wahl zurUntersuchung orientierter Systeme. Bei dieser zweidimen-sionalen NMR-Methode wird die dipolare Kopplung zwi-schen 1H und 15N in der Frequenzdomäne w1 mit derzugehörigen 15N-CSA in der Frequenzdomäne w2 korreliert.Bei einheitlich 15N-markierten Membranproteinen in orien-tierten Doppelschichten liefern PISEMA-Experimente Aus-sagen über die Winkel zwischen Amideinheiten und Doppel-schicht-Normaler. Die Doppelschicht ist typischerweise me-chanisch parallel zum B0-Feld ausgerichtet.[9] Der traditionelleAnsatz zur Analyse von PISEMA-Spektren erfordert einesehr genaue Zuordnung der Kreuzpeaks, um Aussagen überdie Proteinsekundärstruktur treffen zu können. Bei gröûerenProteinen beobachtet man in zunehmendem Maûe Signal-überlagerungen, was eine vollständige Zuordnung erschwert.Diese Überlagerungen können jedoch mit einem neuendreidimensionalen PISEMA-Experiment zumindest teilweiseaufgelöst werden, indem das zweidimensionale PISEMA-Experiment zusätzlich mit der CSA des Amidprotons korre-liert wird.[10]
Marassi und Opella[11] sowie die Gruppe um Cross[12] habenkürzlich festgestellt, dass Sekundärstrukturinformation auchdurch einfache Analyse der 2D-PISEMA-Spektren erhaltenwerden kann. Erstaunlicherweise kann damit das Vorliegeneiner Helix nachgewiesen werden. Zudem lassen sich ihrNeigungswinkel relativ zur Doppelschicht und auf der Grund-lage vorläufiger Signalzuordnung auch die Orientierung einerWiederholungseinheit innerhalb der Helix in Bezug auf dieRotation um die Helixachse sowie die Helixhändigkeitbestimmen. Die dipolare Kopplung zwischen 1H und 15Nsowie die 15N-CSA können als Tensoren geschrieben werden.Auch wenn die Unterschiede in Gröûe und Orientierung derCSA der 15N-Kerne durchaus signifikant sein und interessanteInformationen über Struktur und Dynamik enthalten können,wurde in der hier besprochenen Analyse von konstanten undeinheitlichen Werten für die CSA-Tensoren in der Peptid-ebene[13] ausgegangen. Unter diesen Annahmen beschreibtGleichung (1) das zweidimensionale Signal F(w2 ,w1) desPISEMA-Experiments in guter Näherung. In dieser Glei-
F(w2,w1) � F(CSA(1,t), Dipol-Dipol-Kopplung(1,t))
� (s11(ÿ0.828 cos 1 sin tÿ 0.558 sin 1 sin tÿ 0.047 cos t)2
� s22(0.554 cos 1 sin tÿ 0.803 sin 1 sin tÿ 0.220 cos t)2 (1)
� s33(ÿ0.088 cos 1 sin t� 0.206 sin 1 sin tÿ 0.975 cos t)2),
nk/2((ÿ0.326 cos 1 sin t� 0.034 sin 1 sin tÿ 0.946 cos t)2ÿ 1))
chung sind s11� 64, s22� 77 und s33� 217 die Hauptkompo-nenten des 15N-CSA-Tensors; der Dipoltensor und die s33-Komponente des CSA-Tensors stehen in einem Winkel vonq� 178 zueinander (siehe Abbildung 3 b). nk �ÿ10.4 kHz istdie dipolare Kopplung für eine Amideinheit, die parallel zu B0
liegt, t beschreibt die Neigung der Helixachse bezogen auf dasMagnetfeld B0 (und die Normale der Lipiddoppelschicht) und1 die Orientierung einer Amideinheit bei Rotation um dieHelixachse.
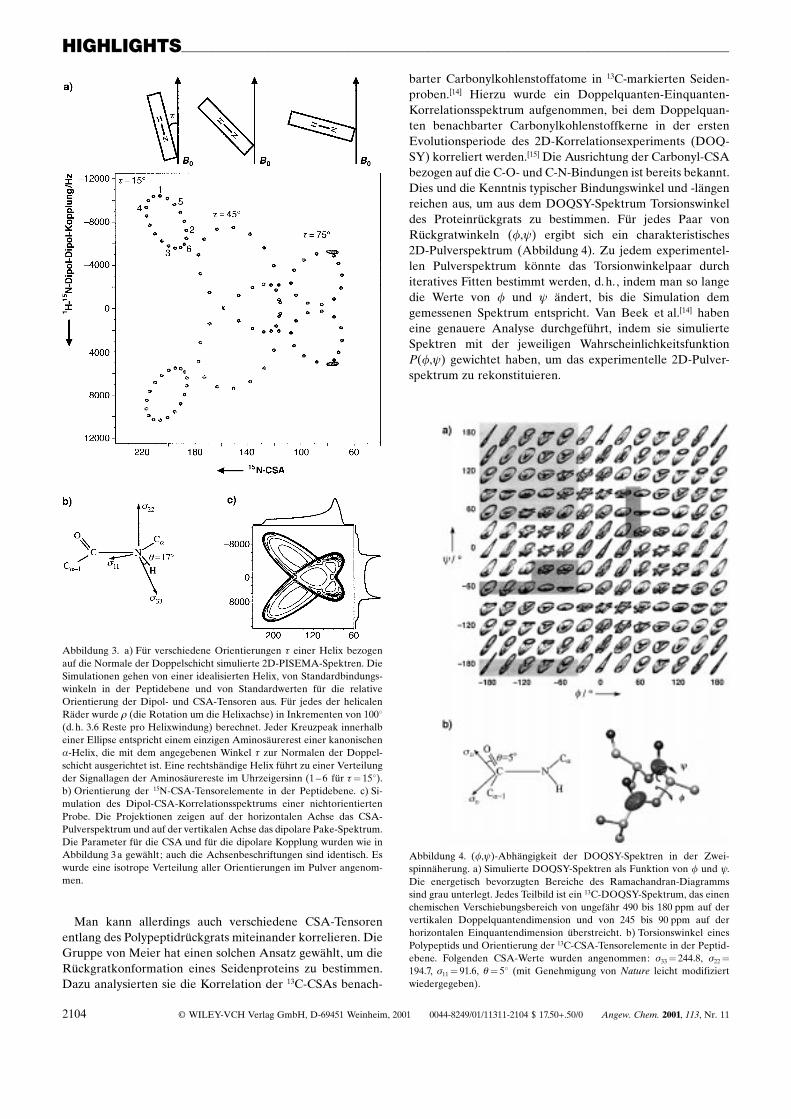
Wegen der Regelmäûigkeit einer helicalen Struktur sinddie Amidbindungen gleichförmig entlang der Helix orientiert.Dies führt zu einer grob elliptischen Anordnung der Kreuz-peaks in einem 2D-PISEMA-Spektrum. Abbildung 3 a zeigtsimulierte PISEMA-Spektren als Funktion des Neigungswin-kels t der Helixachse bezogen auf die Normale der Lipiddop-pelschicht und der Orientierung 1 spezifischer Aminosäure-reste. Position und Form der im PISEMA-Spektrum beob-achteten Ellipsen zeigen den Neigungswinkel t der Helix an.Für einen Winkel t� 158 werden 2 Ellipsen aus jeweils18 Peaks an den beiden Rändern des 15N-Spektrums beob-achtet. Die Feinstruktur jeder Ellipse, 18 Peaks für einekanonische a-Helix, ist auf die verschiedenen Aminosäure-reste innerhalb der Helix zurückzuführen. Die Signallagezeigt die Position der Amideinheiten nach Rotation um dieHelixachse an. Die Zahlen für die einzelnen Aminosäurerestein Abbildung 3 a belegen, dass die Abfolge der Kreuzpeaksinnerhalb einer Ellipse durch die helicale Händigkeit be-stimmt wird. Durch Analyse der Spektren können alsoHändigkeit, Neigung und Rotationsverschiebung eines jedenRestes einer Helix (idealerweise 3.6 Reste pro Helixwindung)bestimmt werden und die Signale allein aufgrund der Anord-nung ihrer 2D-Korrelationspeaks im PISEMA-Spektrumzugeordnet werden.
Festkörper-NMR-Spektroskopie an statischen,nichtorientierten Proben
Spektren von statischen, nichtorientierten Festkörperpro-ben (Abbildung 1 b) weisen groûe qualitative Unterschiede zuden Spektren orientierter Proben auf. Durch die unterschied-liche Ausrichtung der Moleküle im Magnetfeld ergibt sicheine Frequenzverteilung, d. h., das Signal jeder molekularenEinheit liefert ein sogenanntes Pulverspektrum. Dies führthäufig zu unerwünschter spektraler Überlagerung, wenn dieProbe nicht spezifisch markiert ist. In mehrdimensionalenSpektren von statischen Proben beobachtet man in jederDimension breite Signale, was typischerweise das Bild einerhügeligen Landschaft ergibt, aus dem sich die anisotropenWechselwirkungen und die Art ihrer Korrelation ablesenlassen. Das Spektrum einer statischen, nichtorientierten Pro-be, in dem 1H-15N-Dipol-Dipol-Kopplungen mit den 15N-CSAs korreliert sind, ist in Abbildung 3 c wiedergegeben; eskann als gewichtete Summe über alle Helixorientierungen t
und 1 aus Abbildung 3 a interpretiert werden. Solche 2D-Pulverspektren werden gewöhnlich durch Simulation unditeratives Anpassen analysiert und letztlich aufgrund derrelativen Orientierung der Dipol- und CSA-Tensoren be-stimmt.
HIGHLIGHTS
2104 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2104 $ 17.50+.50/0 Angew. Chem. 2001, 113, Nr. 11
Abbildung 3. a) Für verschiedene Orientierungen t einer Helix bezogenauf die Normale der Doppelschicht simulierte 2D-PISEMA-Spektren. DieSimulationen gehen von einer idealisierten Helix, von Standardbindungs-winkeln in der Peptidebene und von Standardwerten für die relativeOrientierung der Dipol- und CSA-Tensoren aus. Für jedes der helicalenRäder wurde 1 (die Rotation um die Helixachse) in Inkrementen von 1008(d. h. 3.6 Reste pro Helixwindung) berechnet. Jeder Kreuzpeak innerhalbeiner Ellipse entspricht einem einzigen Aminosäurerest einer kanonischena-Helix, die mit dem angegebenen Winkel t zur Normalen der Doppel-schicht ausgerichtet ist. Eine rechtshändige Helix führt zu einer Verteilungder Signallagen der Aminosäurereste im Uhrzeigersinn (1 ± 6 für t� 158).b) Orientierung der 15N-CSA-Tensorelemente in der Peptidebene. c) Si-mulation des Dipol-CSA-Korrelationsspektrums einer nichtorientiertenProbe. Die Projektionen zeigen auf der horizontalen Achse das CSA-Pulverspektrum und auf der vertikalen Achse das dipolare Pake-Spektrum.Die Parameter für die CSA und für die dipolare Kopplung wurden wie inAbbildung 3 a gewählt; auch die Achsenbeschriftungen sind identisch. Eswurde eine isotrope Verteilung aller Orientierungen im Pulver angenom-men.
Man kann allerdings auch verschiedene CSA-Tensorenentlang des Polypeptidrückgrats miteinander korrelieren. DieGruppe von Meier hat einen solchen Ansatz gewählt, um dieRückgratkonformation eines Seidenproteins zu bestimmen.Dazu analysierten sie die Korrelation der 13C-CSAs benach-
barter Carbonylkohlenstoffatome in 13C-markierten Seiden-proben.[14] Hierzu wurde ein Doppelquanten-Einquanten-Korrelationsspektrum aufgenommen, bei dem Doppelquan-ten benachbarter Carbonylkohlenstoffkerne in der erstenEvolutionsperiode des 2D-Korrelationsexperiments (DOQ-SY) korreliert werden.[15] Die Ausrichtung der Carbonyl-CSAbezogen auf die C-O- und C-N-Bindungen ist bereits bekannt.Dies und die Kenntnis typischer Bindungswinkel und -längenreichen aus, um aus dem DOQSY-Spektrum Torsionswinkeldes Proteinrückgrats zu bestimmen. Für jedes Paar vonRückgratwinkeln (f,y) ergibt sich ein charakteristisches2D-Pulverspektrum (Abbildung 4). Zu jedem experimentel-len Pulverspektrum könnte das Torsionwinkelpaar durchiteratives Fitten bestimmt werden, d.h., indem man so langedie Werte von f und y ändert, bis die Simulation demgemessenen Spektrum entspricht. Van Beek et al.[14] habeneine genauere Analyse durchgeführt, indem sie simulierteSpektren mit der jeweiligen WahrscheinlichkeitsfunktionP(f,y) gewichtet haben, um das experimentelle 2D-Pulver-spektrum zu rekonstituieren.
Abbildung 4. (f,y)-Abhängigkeit der DOQSY-Spektren in der Zwei-spinnäherung. a) Simulierte DOQSY-Spektren als Funktion von f und y.Die energetisch bevorzugten Bereiche des Ramachandran-Diagrammssind grau unterlegt. Jedes Teilbild ist ein 13C-DOQSY-Spektrum, das einenchemischen Verschiebungsbereich von ungefähr 490 bis 180 ppm auf dervertikalen Doppelquantendimension und von 245 bis 90 ppm auf derhorizontalen Einquantendimension überstreicht. b) Torsionswinkel einesPolypeptids und Orientierung der 13C-CSA-Tensorelemente in der Peptid-ebene. Folgenden CSA-Werte wurden angenommen: s33� 244.8, s22�194.7, s11� 91.6, q� 58 (mit Genehmigung von Nature leicht modifiziertwiedergegeben).

HIGHLIGHTS
Angew. Chem. 2001, 113, Nr. 11 � WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 0044-8249/01/11311-2105 $ 17.50+.50/0 2105
Symmetriebeziehungen sind in Abbildung 4 deutlich zuerkennen: Man erhält identische Spektren für (f,y) und(ÿf,ÿy). Dies hat zur Folge, dass man links- und rechts-händige Helices im DOQSY-Experiment nicht unterscheidenkann. Zudem sind die Spektren der Torsionswinkelpaare(f,y) und (y,f) sehr ähnlich. Wie van Beek et al.[14] berichten,haben die beiden Konformationen sehr ähnliche CSA-CSA-Korrelationen, woraus sich ähnliche DOQSY-Spektren erge-ben. Dagegen bestehen Unterschiede in der Dipol-CSA-Korrelation, was zu einer unterschiedlichen Intensitätsvertei-lung über jedes der in Abbildung 4 a dargestellten Spektrenführt. Dadurch wird bei ausreichendem Signal-Rausch-Ver-hältnis der Experimente die strikte Symmetrie durchbrochenund eine Unterscheidung der beiden Torsionswinkelpaaremöglich.
Das DOQSY-Experiment liefert sehr eindeutig interpre-tierbare Ergebnisse, wenn das Protein an spezifischen Stellenmarkiert ist. In der Studie an Seide wurde dies durchFütterung der Seidenraupen mit Carbonyl-13C-markiertemAlanin erreicht; hiermit erhoffte man sich vor allem eineAnreicherung in Polyalaninbereichen. Die Anwendung desDOQSY-Experiments auf andere Proteine wird ähnlichspezifische Markierungsschemata erfordern. Die mechani-schen Eigenschaften von Seide hängen vom Zustand der alskleine Kristallite in die Faser eingebetteten Polyalanindomä-nen ab.[16] Für natürliche Seidenfasern zeigt das DOQSY-Spektrum, dass die Polyalanindomänen hauptsächlich in einerb-Faltblattkonformation vorliegen. In der gleichen Studie[14]
wurde ein ¹Seidenfilmª aus der von der Seidendrüse abgege-benen Flüssigkeit präpariert, dessen Polyalanindomänen nachdem DOQSY-Spektrum eine a-helicale Konformation ein-nehmen. Diese experimentellen Befunde machen deutlich,dass die Konformation des Seidenproteins kritisch von derPräparationsmethode abhängt.
Die wenigen hier vorgestellten Beispiele heben einige neueHöhepunkte der Festkörper-NMR-Spektroskopie hervor: Esscheint möglich zu sein, die NMR-Signale einheitlich mar-kierter Proteine im Festkörper zuzuordnen. Tensorkorrela-tionen in orientierten und nichtorientierten Proben ermögli-chen eine sehr genaue Bestimmung von Sekundärstruktur-elementen. Zusätzlich haben Tensorkorrelationsexperi-mente[15] eine groûe Bedeutung für die Entwicklung vonMethoden der NMR-Spektrosokopie in Lösung.[17±19] Damitkommen sich zwei in der Vergangenheit getrennte Bereicheder NMR-Spektroskopie näher.
[1] A. McDermott, T. Polenova, A. Bockmann, K. W. Zilm, E. K.Paulsen, R. W. Martin, G. T. Montelione, J. Biomol. NMR 2000, 16,209 ± 219.
[2] J. Pauli, M. Baldus, B. van Rossum, H. de Groot, H. Oschkinat,ChemBioChem, 2001, 2, 272 ± 281.
[3] J. Pauli, B. van Rossum, H. Förster, H. J. M. de Groot, H. Oschkinat, J.Magn. Reson. 2000, 143, 411 ± 416.
[4] R. G. Griffin, Nat. Struct. Biol. 1998, 5, 508 ± 512 (Supplement), zit.Lit.
[5] M. Baldus, A. T. Petkova, J. Herzfeld, R. G. Griffin, Mol. Phys. 1998,95, 1197 ± 1207.
[6] K. Nomura, K. Takegoshi, T. Terao, K. Uchida, M. Kainosho, J.Biomol. NMR 2000, 17, 111 ± 123; S. Dusold, A. Sebald, Annu. Rep.NMR Spectrosc. 2000, 41, 185 ± 264.
[7] K. Schmidt-Rohr, J. Am. Chem. Soc. 1996, 118, 7601 ± 7603; X. Feng,Y. K. Lee, D. Sandström, M. EdeÂn, H. Maisel, A. Sebald, M. H. Levitt,Chem. Phys. Lett. 1996, 257, 314 ± 320; X. Feng, P. J. E. Verdegem,Y. K. Lee, D. Sandström, M. EdeÂn, P. Bovee-Geurts, J. W. de Grip, J.Lugtenburg, H. J. M. de Groot, M. H. Levitt, J. Am. Chem. Soc. 1997,119, 6853 ± 6857; P. R. Costa, J. D. Gross, M. Hong, R. G. Griffin,Chem. Phys. Lett. 1997, 280, 95 ± 103; M. Hohwy, C. P. Jaroniec, B.Reif, C. M. Rienstra, R. G. Griffin, J. Am. Chem. Soc. 2000, 122,3219 ± 3219.
[8] H. Wu, A. Ramamoorthy, S. J. Opella, J. Magn. Reson. A 1994, 109,270 ± 272.
[9] S. J. Opella, Y. Kim, P. McDonnell, Methods Enzymol. 1994, 239, 536 ±560.
[10] F. M. Marassi, C. Ma, J. J. Gesell, S. J. Opella, J. Magn. Reson. 2000,144, 156 ± 161.
[11] F. M. Marassi, S. J. Opella, J. Magn. Reson. 2000, 144, 150 ± 155.[12] J. Wang, J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K.
Nishimura, Z. Gan, R. Fu, J. R. Quine, T. A. Cross, J. Magn. Reson.2000, 144, 162 ± 167.
[13] G. S. Harbison, L. W. Jelinski, R. E. Stark, D. A. Torchia, J. Herzfeld,R. G. Griffin, J. Magn. Reson. 1984, 60, 79 ± 82; C. J. Hartzell,M. Whitfield, T. G. Oas, G. P. Drobny, J. Am. Chem. Soc. 1987, 109,5862 ± 5866; Q. Teng, T. A. Cross, J. Magn. Reson. 1989, 85, 439 ±447.
[14] J. D. van Beek, L. Beaulieu, H. Schäfer, M. Demura, T. Asakura, B. H.Meier, Nature 2000, 405, 1077 ± 1079.
[15] M. Linder, A. Hoehner, R. R. Ernst, J. Chem. Phys. 1980, 73, 4959 ±4970; P. M. Henrichs, M. Linder, J. Magn. Reson. 1984, 58, 458 ± 461;K. Schmidt-Rohr, Macromolecules 1996, 29, 3975 ± 3981.
[16] Z. Yang, P. T. Grubb, L. W. Jelinski, Macromolecules 1997, 30, 8254 ±8261.
[17] K. Pervushin, R. Riek, G. Wider, K. Wüthrich, Proc. Natl. Acad. SciUSA 1997, 94, 12366 ± 12371.
[18] B. Reif, M. Hennig, C. Griesinger, Science 1997, 276, 1230 ± 1233; D.Yang, L. E. Kay, J. Am. Chem. Soc. 1998, 120, 9880 ± 9887; I. C. Felli,C. Richter, C. Griesinger, H. Schwalbe, J. Am. Chem. Soc. 1999, 121,1956 ± 1957; P. Pelupessy, E. Chiarparin, R. Ghose, G. Bodenhausen, J.Biomol. NMR 1999, 13, 375 ± 380.
[19] J. Boyd, C. Redfield, J. Am. Chem. Soc. 1999, 121, 7441 ± 7442; J. Boyd,C. Redfield, J. Am. Chem. Soc. 1999, 121, 9692 ± 9693.











![fgNMR 2014.ppt [Kompatibilitätsmodus] · Festkörper NMR und Spektroskopie Weicher MaterieFestkörper NMR und Spektroskopie Weicher Materie Kay Saalwächter, Günter Hempel, (Detlef](https://img.pdfslide.tips/doc/110x75/605ed99edab9e611e31707bd/fgnmr-2014ppt-kompatibilittsmodus-festkrper-nmr-und-spektroskopie-weicher.jpg)






![Prof. Dr. Paul Seidel VL Festkörper MaWi WS 2013/141 15. Magnetische Eigenschaften der Festkörper Magnetit [ mineralienatlas.de ]](https://img.pdfslide.tips/doc/110x75/55204d8449795902118d8cb1/prof-dr-paul-seidel-vl-festkoerper-mawi-ws-2013141-15-magnetische-eigenschaften-der-festkoerper-magnetit-mineralienatlasde-.jpg)










