Embed Size (px)
Citation preview

Next Generation Sequencing for Invertebrate Virus Discovery
-a practical approach
Sijun Liu amp Bryony C Bonning
Iowa State University USA 8-14-2013 SIP Pittsburgh
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
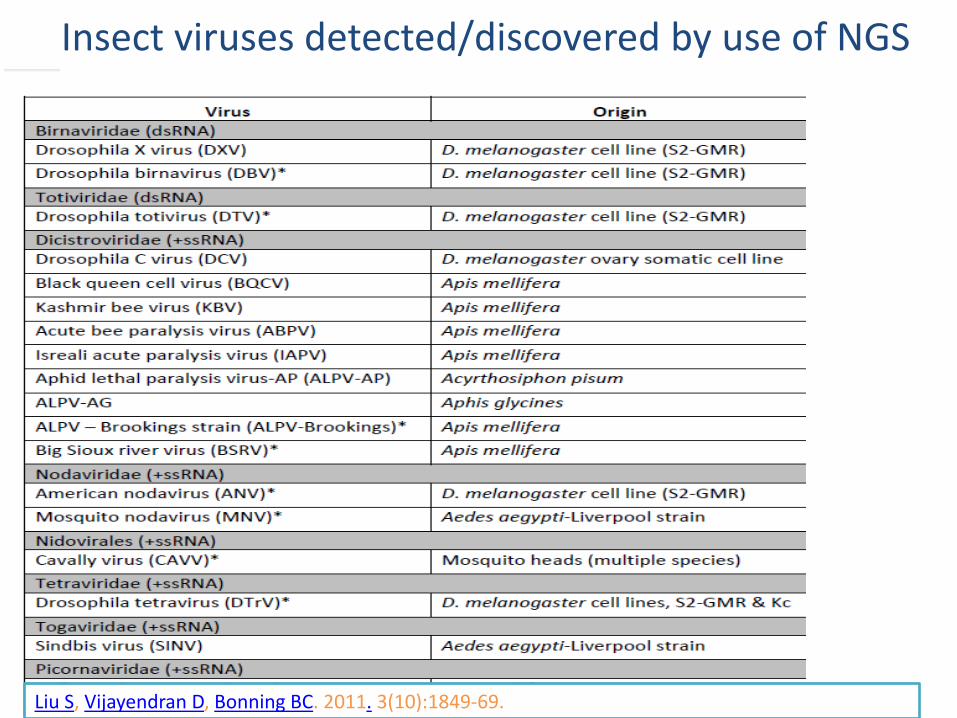
Insect viruses detecteddiscovered by use of NGS
Liu S Vijayendran D Bonning BC 2011 3(10)1849-69
Collect samples that show disease symptoms
Isolate viruses
Observe virus particles
Identify viral genomes
Clone genomic DNARNA - sequence (Sanger sequencing) - assemble viral genome
Traditional Approach for Virus Discovery
Advantages of NGS for Virus Discovery
bull Many viruses are latent or asymptomatic
bull NGS can identify viral sequences without background information on viruses
bull Viral genomes are assembled de novo without reference sequences
bull NGS has revolutionized virus discovery
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
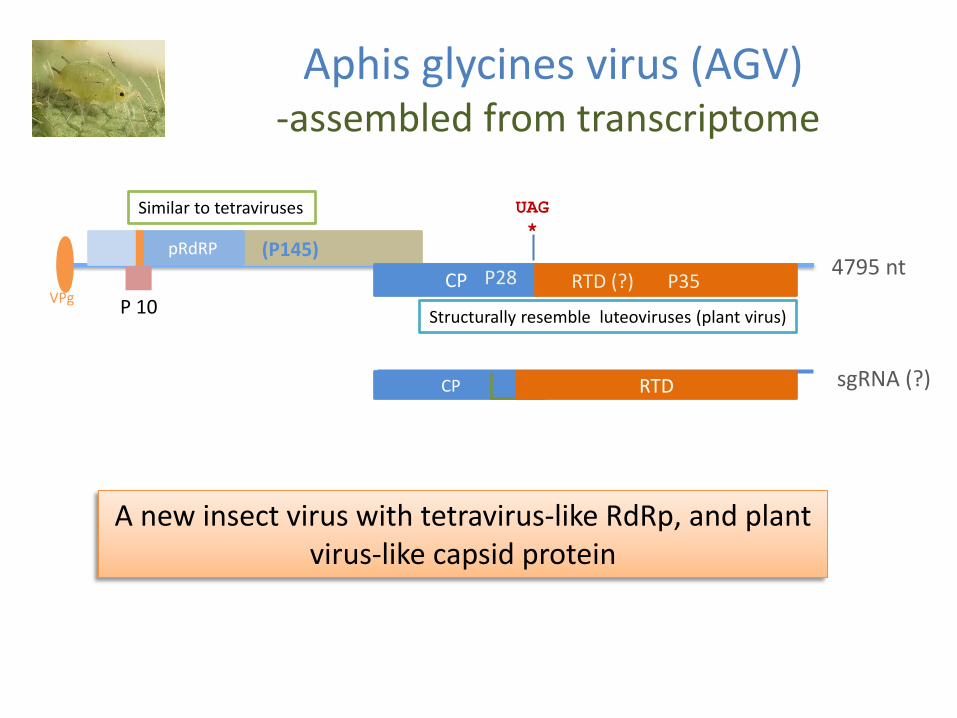
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
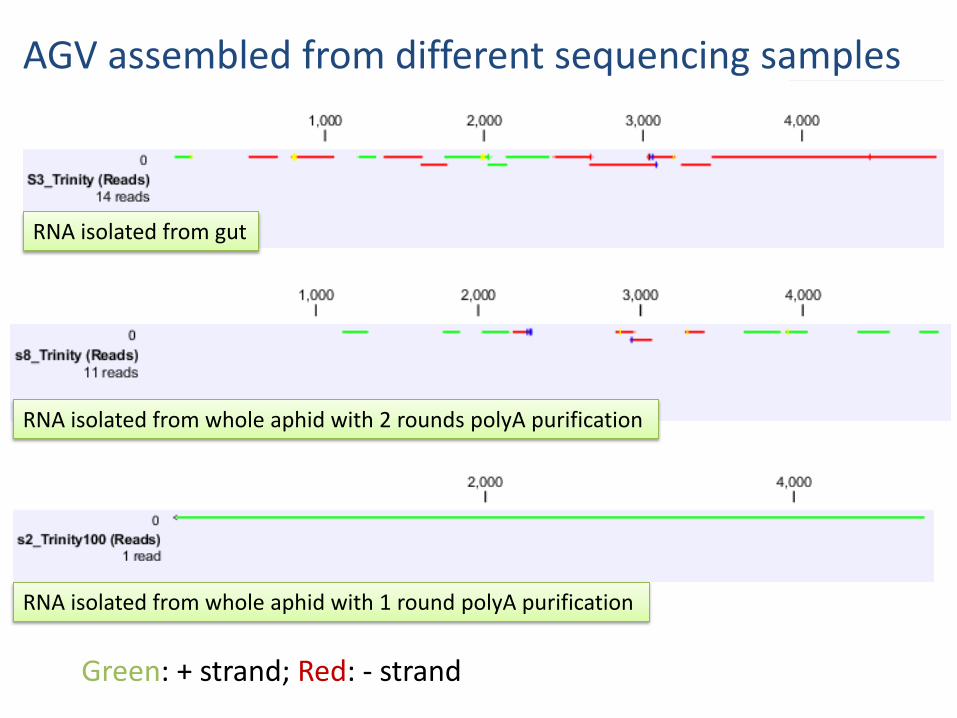
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
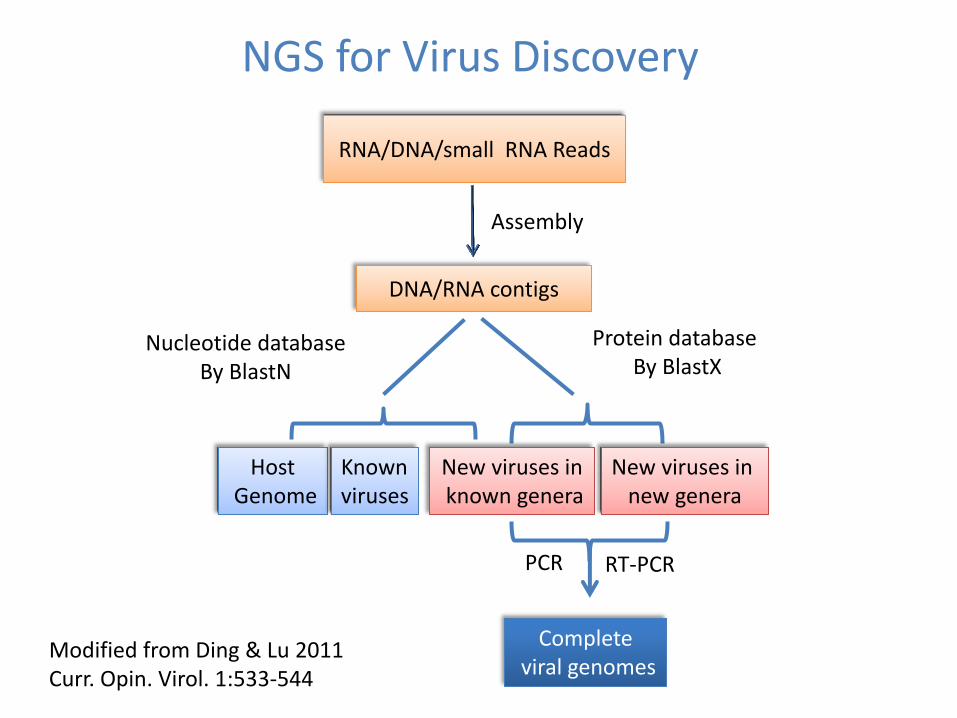
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Insect viruses detecteddiscovered by use of NGS
Liu S Vijayendran D Bonning BC 2011 3(10)1849-69
Collect samples that show disease symptoms
Isolate viruses
Observe virus particles
Identify viral genomes
Clone genomic DNARNA - sequence (Sanger sequencing) - assemble viral genome
Traditional Approach for Virus Discovery
Advantages of NGS for Virus Discovery
bull Many viruses are latent or asymptomatic
bull NGS can identify viral sequences without background information on viruses
bull Viral genomes are assembled de novo without reference sequences
bull NGS has revolutionized virus discovery
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
Insect viruses detecteddiscovered by use of NGS
Liu S Vijayendran D Bonning BC 2011 3(10)1849-69
Collect samples that show disease symptoms
Isolate viruses
Observe virus particles
Identify viral genomes
Clone genomic DNARNA - sequence (Sanger sequencing) - assemble viral genome
Traditional Approach for Virus Discovery
Advantages of NGS for Virus Discovery
bull Many viruses are latent or asymptomatic
bull NGS can identify viral sequences without background information on viruses
bull Viral genomes are assembled de novo without reference sequences
bull NGS has revolutionized virus discovery
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Collect samples that show disease symptoms
Isolate viruses
Observe virus particles
Identify viral genomes
Clone genomic DNARNA - sequence (Sanger sequencing) - assemble viral genome
Traditional Approach for Virus Discovery
Advantages of NGS for Virus Discovery
bull Many viruses are latent or asymptomatic
bull NGS can identify viral sequences without background information on viruses
bull Viral genomes are assembled de novo without reference sequences
bull NGS has revolutionized virus discovery
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Advantages of NGS for Virus Discovery
bull Many viruses are latent or asymptomatic
bull NGS can identify viral sequences without background information on viruses
bull Viral genomes are assembled de novo without reference sequences
bull NGS has revolutionized virus discovery
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
sgRNA ()
4795 nt CP
pRdRP
VPg
CP
(P145)
P 10
RTD
P28 RTD () P35
UAG
Aphis glycines virus (AGV) -assembled from transcriptome
Similar to tetraviruses
Structurally resemble luteoviruses (plant virus)
A new insect virus with tetravirus-like RdRp and plant virus-like capsid protein
Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Outline
bull Introduction Why use NGS ndash Traditional approach for virus discovery ndash Next Generation Sequencing (NGS) ndash Advantages of NGS for virus discovery
bull How itrsquos done ndash Sample selection ndash Sequencing library preparation ndash Sequencing method ndash Assembly of sequencing reads ndash Identification of viral sequence ndash Assembly of viral genome
Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Sample Selection
bull Small sample size (10 ug or less RNA adequate)
-but the more the better
bull Tissue vs whole organism
-sequencing depth
bull Virus purification
-helps to identify full-length sequence
-better approach for DNA viruses
Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Sequencing Technologies
o Short reads (35-250 nt)
1 Genome Analyzer IIx (GAIIx) HiSeq2000 HiSeq2500 MiSeq ndash Illumina
(Hiseq2000 capable of up to 600Gb per run)
1 SOLiD 5500xl System ndash Applied Biosystems
2 HeliScopetrade Single Molecule Sequencer - Helicos
o Long reads (400-20000 nt)
1 Genome Sequencer FLX System (454) ndash Roche
2 PacBio RS - Pacific Bioscience
3 Personal Genome Machine Ion Proton - Ion Torrent
4 GridION ndash Oxford Nanopore
Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Preparation of Sequencing Library
Library type Viral genomes Sequence recovery
mRNA DNARNA +++ possible full-length
Small RNA DNARNA +++
DNA DNA +++ possible full-length
DNA or RNA isolated from viruses
DNARNA +++++ full-length
mRNA purification may result in loss of sequences for viruses that lack polyA tails
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
AGV assembled from different sequencing samples
RNA isolated from gut
RNA isolated from whole aphid with 2 rounds polyA purification
RNA isolated from whole aphid with 1 round polyA purification
Green + strand Red - strand
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
Assembly
DNARNA contigs
RNADNAsmall RNA Reads
Host Genome
Known viruses
New viruses in known genera
Complete viral genomes
New viruses in new genera
Nucleotide database By BlastN
Protein database By BlastX
PCR RT-PCR
NGS for Virus Discovery
Modified from Ding amp Lu 2011 Curr Opin Virol 1533-544
Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Assembly of Sequencing Reads -pre-processing of sequence data
bull Remove potential adaptor index sequences
bull Check sequencing quality
ndash Quality score GC content
ndash Read length distribution
ndash Overrepresented sequences
ndash etc
bull If necessary trim bases with low quality
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
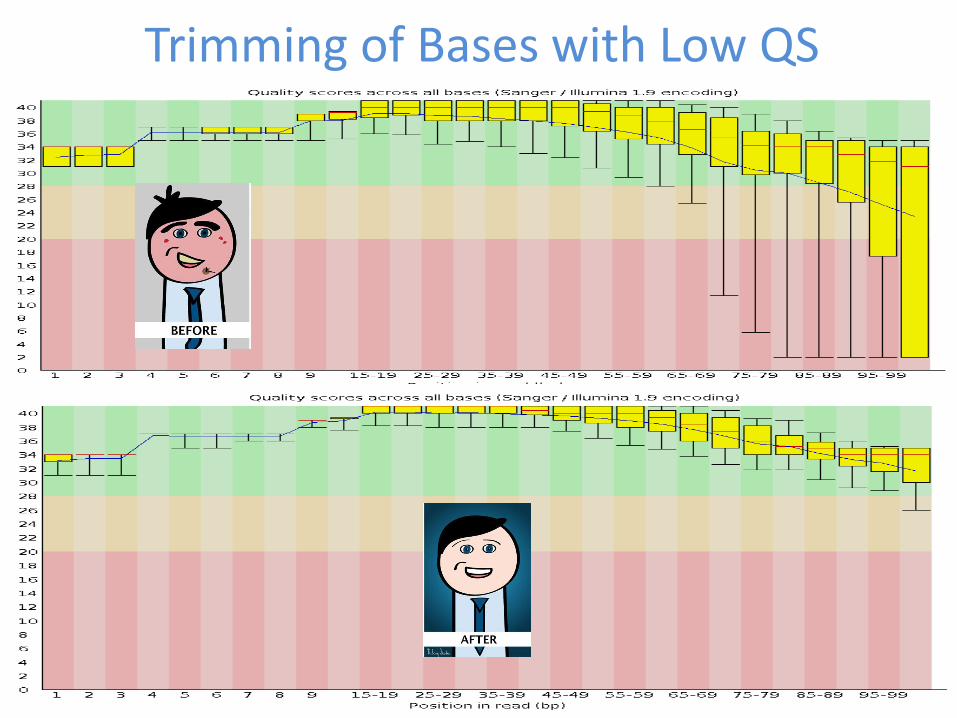
Trimming of Bases with Low QS
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
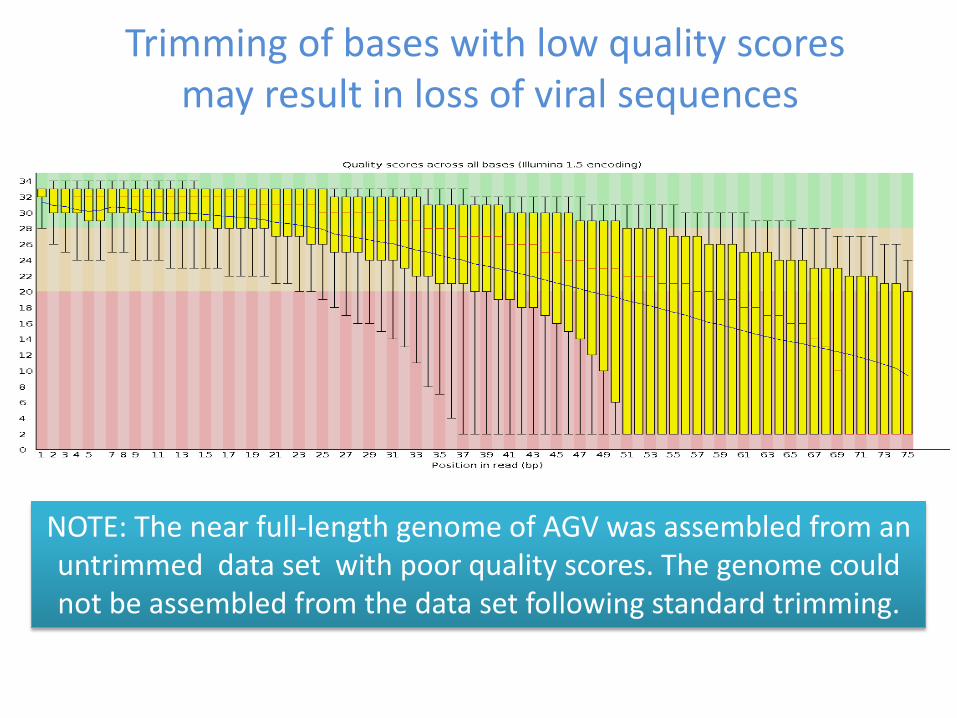
Trimming of bases with low quality scores may result in loss of viral sequences
NOTE The near full-length genome of AGV was assembled from an untrimmed data set with poor quality scores The genome could not be assembled from the data set following standard trimming
Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Software for Checking Sequence Quality-FastQC
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
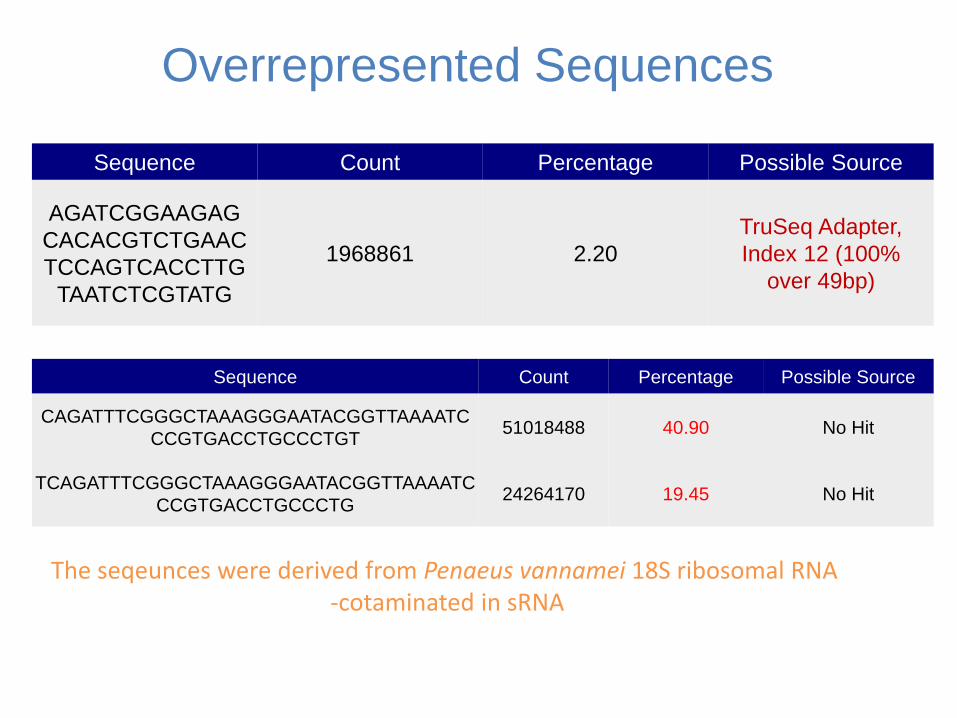
Sequence Count Percentage Possible Source
AGATCGGAAGAG
CACACGTCTGAAC
TCCAGTCACCTTG
TAATCTCGTATG
1968861 220
TruSeq Adapter
Index 12 (100
over 49bp)
Overrepresented Sequences
Sequence Count Percentage Possible Source
CAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTGT 51018488 4090 No Hit
TCAGATTTCGGGCTAAAGGGAATACGGTTAAAATC
CCGTGACCTGCCCTG 24264170 1945 No Hit
The seqeunces were derived from Penaeus vannamei 18S ribosomal RNA -cotaminated in sRNA
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
Software for manipulating sequencing data
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
CLC Genomics Workbench (US$5000 per copy gtUS$1000per year for update)
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
Assembly of Sequencing Reads
bull de novo assembly or mapping (alignment) -de novo assembly searching for new viruses no reference is needed -mapping re-sequencing SNP isolate need reference sequences (MARA GATK and other toolkits) bull de novo assembly may provide extra information about
known viral sequences Shrimp virus Infectious myonecrosis virus (IMNV a dsRNA virus) - documented seq 7560 bp (Poulos et al JGV 2006 87 987-996)
- de novo assembled from RNA-seq 8233 bp RT-PCR proved IMNV should have at least 8233 bp
Thursday 945 am 168 Virus 4 Duan Loy
Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Trinity for Assembly
OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

OasesVelvet for Assembly
Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Running the Assembly Program
bull Two most important parameters for assembly ndash K-mers (word length) length of sequence
fragments used for joining
ndash C - coverage cut-off
bull Different combinations of K and C will result in assembly of different contigs
bull Multiple K and C should be tested for best results (Liu et al PLoS One 20127(9)e45161 doi
101371journalpone)
Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Multiple K Test for Assembly of AGV using OasesVelvet
(Here) read = contig Green + strand Red - strand
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
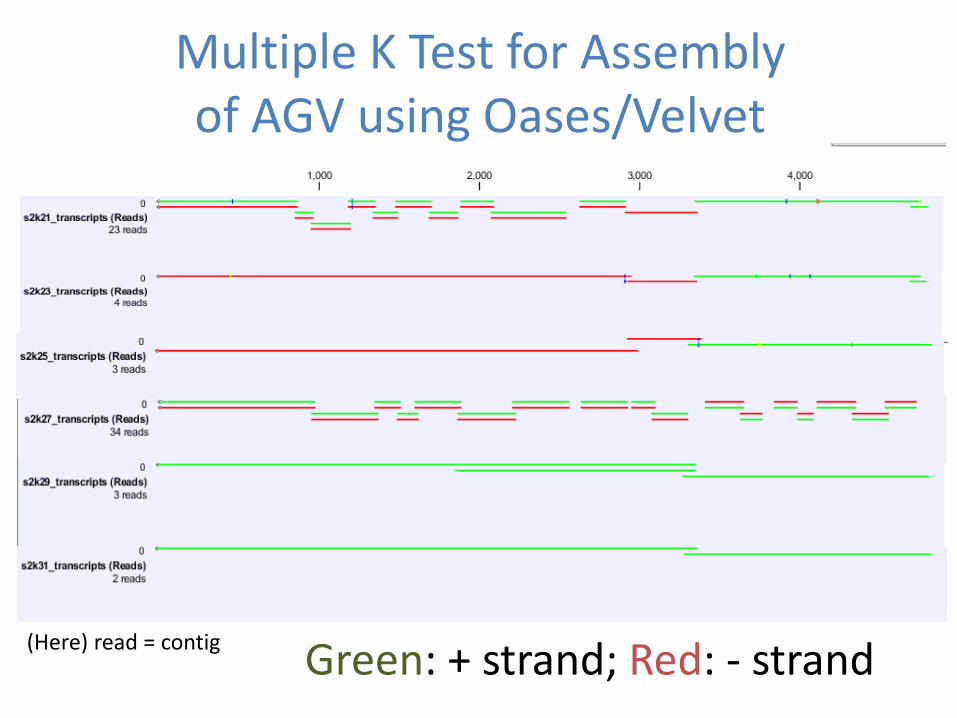
bull Annotation of contigs
-search for viral genes using BLASTx or BLASTn
bull BLAST against NCBI database
bull BLAST using your own databases
bull Blast2GO platform
-annotation of contigs
-motif search
-analysis of annotation data
Data Analysis How do we find viral sequences
Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Data Analysis Analyzing virus-derived contigs
bull Extract BLAST data (sequences with virus as top hit)
bull Organize contigs that hit the same or similar viruses
bull Join contigs into viral genome
bull Design primers for PCRRT-PCR to fill sequence gaps
bull Sequence to confirm in silico cloning result
bull 5rsquo and 3rsquo RACE to identify end sequences
Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

Working with Viral Contigs
viral gene == virus
7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy

7815 8536
8874 9193
9476 9737
8523 8730
9495 9307
501 2315
2656 4084
2295 2679
1 244
522 340
6337 5056
5079 4486
4508 4062
6377 6759
6839 7221
7539
7840 7538 7310
24
4
34
0
63
37
6
37
7
67
59
6
83
9
72
21
7
31
0
97
37
75
38
Trinity Assembly of APV2 (gt9800 nt) Assembled using sRNA isolated from pea aphid
7539
APV2-Acyrthosiphon pisum virus 2 (dicistrovrius)
+ strand
- strand
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
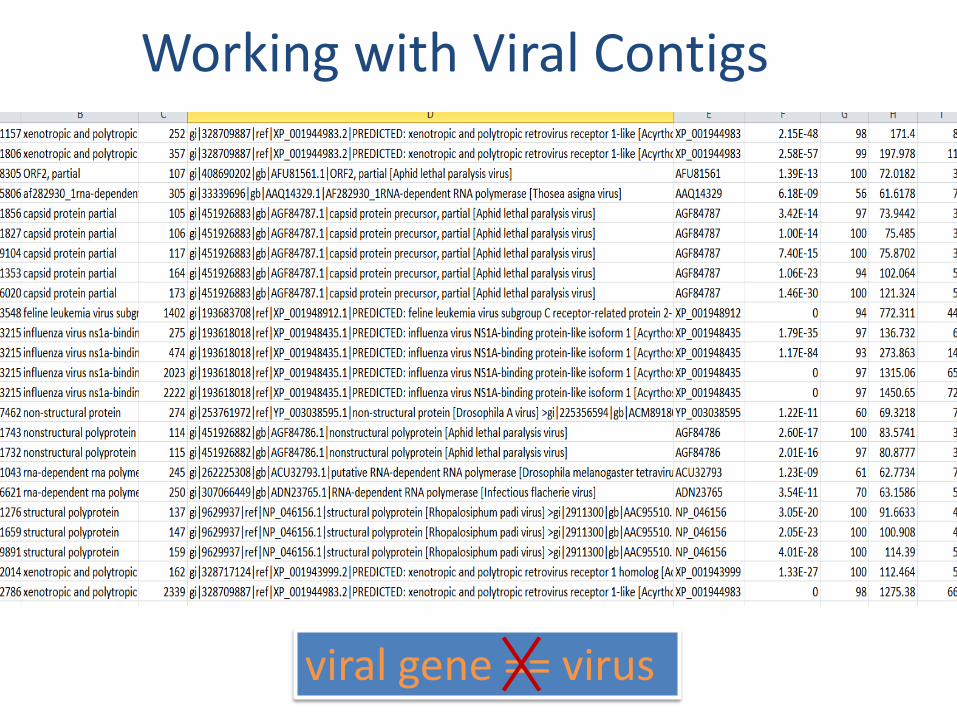
Summary
bull No single rule can be used to find a virus by NGS
bull Knowledge of virology can greatly help for analyzing NGS data
bull Manual alignment of virus derived sequences may be needed
bull Biological evidence is required for verifying true nature of viral sequences discovered by NGS
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy
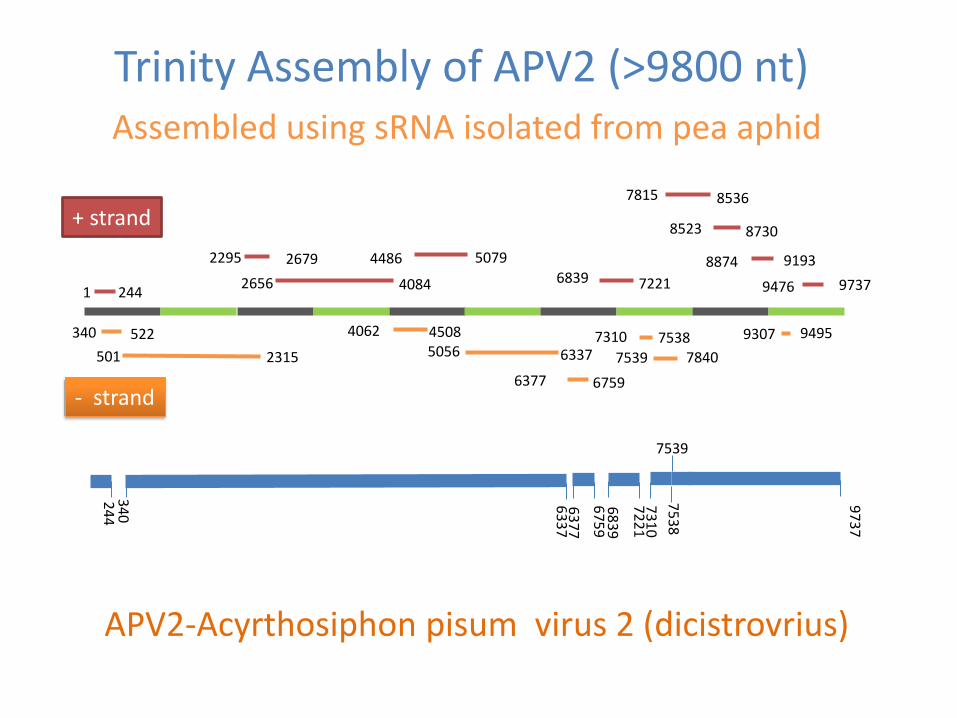
Acknowledgements
John K VanDyk Lyric Bartholomay Duan Loy





























