Embed Size (px)
Citation preview

ĐẠI HỌC HUẾ
TRƯỜNG ĐẠI HỌC KHOA HỌC
NGUYỄN THỊ QUỲNH TRANG
NGHIÊN CỨU PHÁT TRIỂN PHƯƠNG PHÁP
CHEMOMETRIC ĐỂ XÁC ĐỊNH ĐỒNG THỜI
CÁC CHẤT CÓ PHỔ HẤP THỤ PHÂN TỬ
XEN PHỦ NHAU VÀ ÁP DỤNG TRONG
PHÂN TÍCH DƯỢC PHẨM
LUẬN ÁN TIẾN SĨ HÓA HỌC
HUẾ - NĂM 2018

ĐẠI HỌC HUẾ
TRƯỜNG ĐẠI HỌC KHOA HỌC
NGUYỄN THỊ QUỲNH TRANG
NGHIÊN CỨU PHÁT TRIỂN PHƯƠNG PHÁP
CHEMOMETRIC ĐỂ XÁC ĐỊNH ĐỒNG THỜI
CÁC CHẤT CÓ PHỔ HẤP THỤ PHÂN TỬ
XEN PHỦ NHAU VÀ ÁP DỤNG TRONG
PHÂN TÍCH DƯỢC PHẨM
Chuyên ngành: Hóa học Phân tích
Mã số: 62440118
LUẬN ÁN TIẾN SĨ HÓA HỌC
Người hướng dẫn khoa học:
1. PGS.TS. Trần Thúc Bình
2. PGS.TS. Ngô Văn Tứ
HUẾ - NĂM 2018

ii
LỜI CAM ĐOAN
Luận án này được hoàn thành tại Trường Đại học Khoa học, Đại học Huế, dưới
sự hướng dẫn của PGS.TS. Trần Thúc Bình và PGS.TS. Ngô Văn Tứ. Tôi xin cam
đoan luận án này là công trình nghiên cứu của tôi. Các kết quả trong luận án là trung
thực, được các đồng tác giả cho phép sử dụng và chưa từng được ai công bố trước đó.
Tác giả
Nguyễn Thị Quỳnh Trang

iii
LỜI CẢM ƠN
Luận án này được hoàn thành với sự hỗ trợ, giúp đỡ, động viên của các Thầy
Cô, gia đình và bạn bè, đồng nghiệp.
Với tình cảm chân thành, tôi xin bày tỏ lòng biết ơn sâu sắc tới
PGS.TS. Trần Thúc Bình và PGS.TS. Ngô Văn Tứ là những người thầy đã tận
tâm hướng dẫn, giúp đỡ và động viên tôi trong suốt quá trình làm luận án.
Tôi xin gửi lời cảm ơn đến Ban Giám đốc Đại học Huế, Ban Giám hiệu
Trường Đại học Khoa học, Phòng Đào tạo Sau đại học, Quý Thầy Cô trong Bộ môn
Hóa học phân tích, khoa Hóa học của trường Đại học Khoa học cùng Quý Thầy Cô
giảng dạy lớp nghiên cứu sinh, các nghiên cứu sinh, học viên cao học đã tận tình
giúp đỡ và tạo mọi điều kiện thuận lợi trong suốt thời gian học tập và nghiên cứu.
Lời sau cùng, tôi xin được bày tỏ tình cảm đặc biệt và lòng biết ơn chân thành
tới những người thân trong gia đình, tới những người bạn, tới lãnh đạo và các đồng
nghiệp khoa Khoa học Môi trường, trường Đại học Sài Gòn đã luôn kịp thời động
viên, ủng hộ và tạo mọi điều kiện thuận lợi nhất trong suốt thời gian học tập và
hoàn thành luận án.
Tác giả
Nguyễn Thị Quỳnh Trang

iv
MUC LUC
LỜI CAM ĐOAN ..................................................................................................... ii
LỜI CẢM ƠN .......................................................................................................... iii
MUC LUC ................................................................................................................ iv
KY HIÊU VIÊT TĂT .............................................................................................. vi
DANH MUC CAC BIÊU, BẢNG ......................................................................... vii
DANH MUC HINH VE, BIÊU ĐÔ, SƠ ĐÔ ........................................................... x
MỞ ĐẦU .................................................................................................................... 1
CHƯƠNG 1. TỔNG QUAN ..................................................................................... 5
1.1. Định luật Bouguer Lambert Beer và tính chất cộng tính độ hấp thụ quang ..... 5
1.1.1. Định luật Bouguer Lambert Beer ............................................................. 5
1.1.2. Tính chất cộng tính độ hấp thụ quang ...................................................... 5
1.2. Một số phương pháp phân tích quang phổ UV-VIS kết hợp với chemometric
xác định đồng thời các cấu tử có phổ hấp thụ xen phủ nhau ...................................... 6
1.2.1. Phương pháp Vierordt .............................................................................. 7
1.2.2. Phương pháp quang phổ đạo hàm ........................................................... 8
1.2.3. Phương pháp bình phương tối thiểu....................................................... 11
1.2.4. Phương pháp bình phương tối thiểu từng phần (Partial Least Square -
PLS) ........................................................................................................................... 13
1.2.5. Phương pháp hồi quy cấu tử chính (Principal Component Regression -
PCR) .......................................................................................................................... 15
1.2.6. Phương pháp mạng nơron nhân tạo (Artificial Neural Networks - ANN)
................................................................................................................................... 19
1.2.7. Phương pháp lọc Kalman ....................................................................... 22
1.3. Tổng quan về dược phẩm đa thành phần và các hoạt chất nghiên cứu .......... 33
1.3.1. Giới thiệu về các dược phẩm đa thành phần ......................................... 33
1.3.2. Telmisartan (TEL), hydrochlorothiazide (HYD) .................................... 34
1.3.3. Paracetamol (PAR)và caffeine (CAF) .................................................... 37
1.3.4. Paracetamol (PAR) và ibuprofen (IB) ................................................... 41
1.3.5. Amlodipine besylat (AML), hydroclorothiazid (HYD), valsartan (VAL)
................................................................................................................................... 43
CHƯƠNG 2. NỘI DUNG VÀ PHƯƠNG PHAP NGHIÊN CỨU ...................... 48
2.1. Nội dung nghiên cứu ...................................................................................... 48
2.2. Phương pháp nghiên cứu ................................................................................ 48

v
2.2.1. Đối tượng và phạm vi nghiên cứu .......................................................... 48
2.2.2. Phương pháp lọc Kalman và chương trình tính ..................................... 49
2.2.3. Phương pháp bình phương tối thiểu sử dụng phần mềm simulan ......... 54
2.2.4. Phương pháp phổ đạo hàm .................................................................... 55
2.4.5. Phương pháp xây dựng chương trình máy tính ...................................... 55
2.2.6. Phương pháp đánh giá độ tin cậy của phương pháp phân tích ............. 55
2.2.7. Phương pháp xử lý số liệu ...................................................................... 57
2.2.8. Chuẩn bị mẫu cho phân tích và tính kết quả .......................................... 58
2.2.9. Thiết bị, dụng cụ và hóa chất. ................................................................ 62
CHƯƠNG 3. KÊT QUẢ VÀ THẢO LUẬN ......................................................... 65
3.1. Lựa chọn giá trị khởi tạo ban đầu ................................................................... 65
3.1.1. Lựa chọn giá trị khởi tạo ngẫu nhiên ..................................................... 66
3.1.2. Lựa chọn giá trị khởi tạo giả định.......................................................... 69
3.1.3. Lựa chọn giá trị khởi tạo gần đúng ........................................................ 73
3.2. Chương trình máy tính để tính toán theo thuật toán lọc Kalman ................... 77
3.3. Kiểm chứng phương pháp Kalman đối với hỗn hợp hai cấu tử ..................... 78
3.3.1. Phổ hấp thụ quang và phổ đạo hàm ....................................................... 78
3.3.2. Kiểm định phương pháp đối với dung dịch chuẩn phòng thí nghiệm .... 82
3.4. Kiểm chứng phương pháp đối với hỗn hợp ba cấu tử ..................................... 96
3.4.1. Phổ hấp thụ của hỗn hợp........................................................................ 97
3.4.2. Kiểm định phương pháp đối với dung dịch chuẩn phòng thí nghiệm .... 98
3.5. Quy trình phân tích theo phương pháp lọc Kalman ..................................... 104
3.6. Áp dụng thực tế ............................................................................................ 105
3.6.1. Kiểm soát chất lượng phương pháp phân tích ..................................... 106
3.6.2. Hàm lượng các hoạt chất trong các thuốc đa thành phần ................... 132
KÊT LUẬN ............................................................................................................ 135
DANH MUC CÁC CÔNG TRÌNH CÔNG BỐ KÊT QUẢ NGHIÊN CỨU CỦA
LUẬN ÁN ............................................................................................................... 137
TÀI LIÊU THAM KHẢO .................................................................................... 138
PHU LUC ............................................................................................................... 153

vi
KY HIÊU VIÊT TĂT
Tiếng Việt Tiếng Anh Viết tắt
Amlodipin besilat Amlodipine besylat AML
Bình phương tối thiểu nghịch đảo Inverse least square ILS
Bình phương tối thiểu riêng phần Partial least square PLS
Bình phương tối thiểu cổ điển Classical least square BPTT
Cafein Caffeine CAF
Cấu tử chính Principal Components PC
Dược điển Mỹ 38 The United States
Pharmacopoeia 38, 2015. USP38
Dược điển Việt Nam IV Pharmacopoeia vietnamica
2009-2010
DĐVN
IV
Giới hạn định lượng Limit of quantity LOQ
Giới hạn phát hiện Limit of detection LOD
Hồi quy cấu tử chính Principal component regression PCR
Hydroclorothiazid Hydrochlorothiazide HYD
Ibuprofen Ibuprofen IB
Mạng nơron nhân tạo Artificial Neural Networks ANN
Mạng nơron nhân tạo kết hợp hồi
quy thành phần chính
Principal component regression-
Artificial Neural Networks
PCR-
ANN
Paracetamol Paracetamol PAR
Phổ tử ngoại – khả kiến Ultraviolet-visible spectroscopy UV-VIS
Sắc kí lỏng hiệu năng cao High Performance Liquid
Chromatography HPLC
Valsartan Valsartan VAL
Ghi chú: Trong luận án, tên hóa chất và hoạt chất theo quy định trong Dược điển
Việt Nam IV.

vii
DANH MUC CAC BIÊU, BẢNG
Bảng 1.1. Một số nghiên cứu ở Việt Nam và thế giới sử dụng thuật toán lọc Kalman
ứng dụng vào phân tích ........................................................................... 32
Bảng 1.2. Một số nghiên cứu xác định TEL và HYD .............................................. 36
Bảng 1.3. Một số nghiên cứu xác định PAR và CAF .............................................. 40
Bảng 1.4. Một số nghiên cứu xác định PAR và IB .................................................. 43
Bảng 1.5. Một số nghiên cứu xác định AML, HYD và VAL .................................. 47
Bảng 2.1 .Kết quả đo quang phổ của hỗn hợp hai chất ............................................ 51
Bảng 2.2. Kết quả tính toán nồng độ các chất trong hỗn hợp và hiệp phương sai ở
mỗi bước sóng (theo phương pháp lọc Kalman) .................................... 53
Bảng 2.3. Kết quả tính toán nồng độ, hiệp phương sai, mức cập nhật INV ở các
bước sóng khác nhau cho trường hợp hỗn hợp ba cấu tử 1, 2 và 3 ........ 54
Bảng 3.1. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp theo phương pháp
Kalman với cách lựa chọn giá trị khởi tạo ngẫu nhiên ........................... 66
Bảng 3.2. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp theo
phương pháp Kalman với cách lựa chọn giá trị khởi tạo ngẫu nhiên ..... 68
Bảng 3.3. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng phương pháp
Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 1 ....... 70
Bảng 3.4. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng phương pháp
Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 2 ....... 70
Bảng 3.5. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định –
Phương án 1 ............................................................................................ 71
Bảng 3.6. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định –
Phương án 2 ............................................................................................ 72
Bảng 3.7. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng phương pháp
Kalman với cách lựa chọn giá trị khởi tạo gần đúng .............................. 75
Bảng 3.8. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo gần đúng ........ 76
Bảng 3.9. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng phương pháp
Kalman và BPTT .................................................................................... 83
Bảng 3.10. Kết quả xác định nồng độ TEL, HYD trong hỗn hợp và sai số tương đối
của phương pháp quang phổ đạo hàm .................................................... 84

viii
Bảng 3.11. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng phương
pháp Kalman và BPTT khi có nhiễu ...................................................... 85
Bảng 3.12. Kết quả xác định nồng độ PAR, CAF trong hỗn hợp và sai số tương đối
của ba phương pháp ................................................................................ 86
Bảng 3.13. Kết quả xác định nồng độ PAR, IB trong hỗn hợp và sai số tương đối
của các phương pháp .............................................................................. 88
Bảng 3.14. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp TEL và HYD
................................................................................................................ 90
Bảng 3.15. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm ....... 92
Bảng 3.16. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp PAR và CAF
................................................................................................................ 93
Bảng 3.17. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm ....... 94
Bảng 3.18. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp PAR và IB 95
Bảng 3.19. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm ....... 96
Bảng 3.20. Kết quả xác định nồng độ AML, HYD, VAL trong hỗn hợp và sai số
tương đối của các phương pháp .............................................................. 98
Bảng 3.21. Kết quả xác định độ lặp lại của phương pháp đối với hỗn hợp AML,
HYD và VAL ........................................................................................ 100
Bảng 3.22. So sánh kết quả của 2 phương pháp đối với hỗn hợp H4 .................... 101
Bảng 3.23. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman và BPTT khi có nhiễu ....................................... 103
Bảng 3.24. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc
quang khi xác định TEL và HYD trong thuốc Micardis(R) Plus ......... 107
Bảng 3.25. Độ lặp lại và độ đúng của các phương pháp trắc quang và chemometric-
trắc quang khi phân tích đồng thời TEL và HYD trong dược phẩm .... 108
Bảng 3.26. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc
quang khi xác định PAR và CAF trong thuốc Panadol Extra .............. 109
Bảng 3.27. Độ lặp lại và độ đúng của các phương pháp trắc quang và chemometric-
trắc quang khi phân tích đồng thời PAR và CAF trong dược phẩm ... 110
Bảng 3.28. Kết quả đánh giá độ lặp lại của các phương pháp chemmoetric-trắc
quang khi xác định PAR và IB trong thuốc Alaxan ............................. 111
Bảng 3.29. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc
quang khi xác định PAR và IB trong thuốc Lopenca ........................... 112
Bảng 3.30. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc
quang khi xác định PAR và IB trong thuốc Protamol .......................... 113

ix
Bảng 3.31. Độ lặp lại và độ đúng của các phương pháp trắc quang và chemometric-
trắc quang khi phân tích đồng thời PAR và IB trong dược phẩm ....... 114
Bảng 3.32. Kết quả đánh giá độ lặp lại của phương pháp Kalman và BPTT khi xác
định AML, HYD và VAL trong thuốc Exforge HCT .......................... 116
Bảng 3.33. Độ lặp lại và độ đúng của các phương pháp trắc quang và chemometric-
trắc quang khi phân tích đồng thời AML, HYD và VAL trong dược
phẩm ..................................................................................................... 117
Bảng 3.34. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời TEL và HYD trong thuốc Micardis(R) Plus ..... 119
Bảng 3.35. So sánh độ đúng của các phương pháp chemometric-trắc quang với
phương pháp HPLC khi xác định TEL và HYD trong thuốc Micardis(R)
Plus ....................................................................................................... 121
Bảng 3.36. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời PAR và CAF trong thuốc Panadol Extra ......... 122
Bảng 3.37. So sánh độ đúng của các phương pháp chemometric-trắc quang với
phương pháp HPLC khi xác định PAR, CAF trong thuốc Panadol Extra
.............................................................................................................. 123
Bảng 3.38. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời PAR và IB trong thuốc Alaxan, Lopenca và
Protamol ................................................................................................ 125
Bảng 3.39. So sánh các phương pháp chemometric với phương pháp HPLC khi xác
định hàm lượng PAR, IB trong thuốc Lopenca (*) ................................ 128
Bảng 3.40. Kết quả xác định độ đúng của phương pháp khi phân tích mẫu thực tế
thuốc Exforge ........................................................................................ 129
Bảng 3.41. So sánh các phương pháp chemometric với phương pháp HPLC khi xác
định hàm lượng AML, HYD và VAL trong thuốc Exforge HCT ........ 131
Bảng 3.42. Hàm lượng các hoạt chất trong một số loại thuốc đa thành phần đang lưu
hành ở thị trường Việt Nam (phân tích theo phương pháp Kalman).... 133

x
DANH MUC HINH VE, BIÊU ĐÔ, SƠ ĐÔ
Hinh 1.1. (a) Phổ hấp thụ; (b) phổ đạo hàm bậc 2; (c) phổ đạo hàm bậc 4; (d) phổ
hấp thụ của trans-stilbene trong cyclohexane; (e) phổ đạo hàm bậc 2 của
trans-stilbene trong cyclohexane; (f) phổ đạo hàm bậc 4 của trans-
stilbene trong cyclohexane. .................................................................... 10
Hinh 1.2. Mô hình hoạt động của mạng nơron được thể hiện ở hình 1.1. ............... 21
Hình 2.1. Sơ đồ các bước tính toán theo phương pháp chemometric- trắc quang sử
dụng thuật toán lọc Kalman (sử dụng phần mềm máy tính). ................. 50
Hình 2.2. Sơ đồ tiến trình phân tích mẫu dược phẩm theo phương pháp
chemometric-trắc quang. ........................................................................ 58
Hình 3.1. Sơ đồ chương trình tính toán theo thuật toán lọc Kalman với giải pháp lựa
chọn giá trị khởi tạo gần đúng (áp dụng cho hệ 2 và 3 cấu tử). ............. 77
Hình 3.2. Phổ hấp thụ quang của dung dịch chuẩn HYD, TEL, hỗn hợp TEL và
HYD trong dung môi NaOH 0.1M. ........................................................ 78
Hình 3.3. Phổ đạo hàm bậc một của các dung dịch chuẩn TEL, HYD và dung dịch
hỗn hợp TEL, HYD. ............................................................................... 79
Hình 3.4. Phổ hấp thụ quang của dung dịch chuẩn PAR, CAF, hỗn hợp PAR và
CAF. ........................................................................................................ 80
Hình 3.5. Phổ đạo hàm bậc 1 của dung dịch chuẩn PAR và CAF. .......................... 80
Hình 3.6. Phổ hấp thụ quang của dung dịch chuẩn PAR, IB, hỗn hợp PAR và IB. 81
Hình 3.7. Phổ đạo hàm bậc 1 của dung dịch chuẩn PAR và IB trong môi trường
đệm photphat pH = 7. ............................................................................. 82
Hình 3.8. Phổ hấp thụ của các dung dịch chuẩn AML, HYD, VAL và hỗn hợp
AML, HYD, VAL đo được trong dung môi methanol. .......................... 97
Hình 3.9. So sánh kết quả của 2 phương pháp đối với hỗn hợp H4 ...................... 102
Hình 3.10. Quy trình phân tích mẫu thực tế theo phương pháp Kalman. .............. 104

1
MỞ ĐẦU
Thuật ngữ chemometric được đưa ra đầu tiên vào năm 1972 bởi Svante Wold
(người Thụy Điển) và Bruce R. Kowalski (người Mỹ). Sau đó sự ra đời của Hiệp hội
Chemometric vào năm 1974 đã đưa ra định nghĩa đầu tiên của ngành chemometric, đó
là việc ứng dụng các phương pháp toán học, thống kê, đồ họa,… để quy hoạch thực
nghiệm, tối ưu hóa các thông tin hóa học trích ra từ tập số liệu phân tích và đưa ra tối
đa những thông tin hữu ích từ tập số liệu ban đầu [26], [45], [46], [88].
Chemometric được ứng dụng rất nhiều trong các lĩnh vực như hóa học môi
trường, hóa học hữu cơ, hóa sinh, hóa học lý thuyết, thống kê trong hóa học và đặc biệt
là đã xác lập được vị trí quan trọng trong ngành hóa học phân tích [45], [88].
Hóa học phân tích là công cụ phục vụ đắc lực trong các lĩnh vực khoa học và
công nghệ, như hóa học, sinh học, nông học, y học, thực phẩm…, đặc biệt là trong
ngành dược phẩm. Ở Việt Nam, dược phẩm và chất lượng dược phẩm hiện tại đang là
vấn đề được xã hội và các cơ quan quản lý nhà nước quan tâm. Trong những năm gần
đây, thị trường thuốc đang phát triển nhanh cả về sản xuất và kinh doanh. Theo thống
kê của Cục quản lý Dược Việt Nam, năm 2005 có khoảng 10.000 mặt hàng thuốc lưu
hành trên thị trường với trên 800 hoạt chất. Chỉ 10 năm sau, năm 2015, số lượng thuốc
đã tăng lên nhanh chóng với trên 25.000 mặt hàng, khoảng 1.500 hoạt chất [7]. Mỗi
hoạt chất trong dược phẩm đều có những tác dụng dược lý riêng biệt, do đó để tăng
cường tác dụng chữa bệnh, người ta đã bào chế ra các loại thuốc chứa đa thành phần.
Đối với những người lớn tuổi và người già, bệnh tăng huyết áp và tim mạch là
một trong những nguyên nhân gây tử vong hàng đầu trên thế giới nói chung và Việt
Nam nói riêng. Trên thị trường hiện nay, có khá nhiều loại thuốc được dùng để điều trị
bệnh cao huyết áp, trong đó các chế phẩm đa thành phần nhằm phối hợp tác dụng dược
lý của các dược chất vẫn được ưu tiên nghiên cứu và bào chế. Ví dụ như thuốc viên nén
Exforge, viên nén Exforge HCT, viên nén Co-Diovan… Những loại thuốc này chứa
các hoạt chất amlodipine, hydrochlorothiazide và valsartan hay telmisartan và
hydrochlothiazide [38], [87].

2
Bên cạnh các loại thuốc điều trị huyết áp, tim mạch thì tại Việt Nam, có gần
200 chế phẩm thuốc giảm đau phối hợp khác nhau cũng đang lưu hành. Trong đó
thuốc kết hợp hai thành phần là paracetamol (thuốc giảm đau, hạ sốt) và ibuprofen
(thuốc chống viêm không steroid) hay sự kết hợp của paracetamol và caffein có
tác dụng làm hạ sốt, giảm đau và chống viêm nhanh, tốt hơn, hiệu quả hơn so với
dùng paracetamol hay ibuprofen, caffein đơn độc. Chính vì vậy, việc nghiên cứu
xác định đồng thời các hoạt chất trong các dược phẩm (hay thuốc) đa thành phần,
đặc biệt là các hoạt chất có phổ hấp thụ quang xen phủ nhau là rất cần thiết và
đang là thách thức đối với các nhà hóa học phân tích.
Ngày nay, với sự tiến bộ không ngừng trong lĩnh vực phân tích hóa học trên
thế giới, hàng loạt các phương pháp phân tích công cụ hiện đại ra đời và được đưa
vào ứng dụng trong phân tích dược phẩm. Tuy nhiên, hai bài toán liên quan đặt ra là
i) xử lý bộ dữ liệu khổng lồ thu được từ các phương pháp phân tích, và ii) ở các
nước đang phát triển - như Việt Nam – thì nhu cầu xây dựng được các phương pháp
phân tích sao cho tiết kiệm được chi phí và thời gian phân tích mà vẫn bảo đảm tiêu
chuẩn về kết quả phân tích được đặt lên hàng đầu. Một lời giải chung cho cả hai bài
toán trên là ứng dụng phương pháp chemometric.
Các phương pháp chemometric đã được các nhà nghiên cứu trong và ngoài
nước quan tâm trong nhiều năm qua để phân tích đồng thời hỗn hợp các chất trong
các đối tượng khác nhau, trong đó có dược phẩm. Các công trình nghiên cứu cho
thấy, các phương pháp chemometric thường được dùng nhiều nhất là phương pháp
bình phương tối thiểu riêng phần (PLS), phương pháp hồi quy cấu tử chính (PCR),
phương pháp bình phương tối thiểu cổ điển (BPTT), phương pháp mạng nơron nhân
tạo (ANN), phương pháp phổ đạo hàm, phương pháp lọc Kalman (Kalman filter)…
Mỗi phương pháp đều có những ưu điểm và hạn chế riêng. Phương pháp BPTT có
thể sử dụng toàn bộ số liệu đo phổ để lập ra hệ m phương trình n ẩn số (m>n). Phép
biến đổi ma trận theo nguyên tắc của phương pháp bình phương tối thiểu sẽ cho ra
các kết quả mắc sai số thỏa mãn yêu cầu. Tuy nhiên nếu trong bộ số liệu đo phổ có
nhiều nhiễu (hay sai số đo phổ) và/hoặc khi các cấu tử có tương tác với nhau tạo ra
hiệu ứng quang học làm thay đổi hệ số hấp thụ của từng cấu tử, thì phương pháp

3
này không loại được nhiễu, dẫn đến kết quả phân tích mắc sai số lớn [2]; Phương
pháp ANN có nhược điểm là thời gian luyện mạng lâu và nó đòi hỏi nhiều thuật
toán khác nhau, nên khi xây dựng một mô hình phân tích, đòi hỏi phải thử nhiều mô
hình khác nhau để tìm được cấu trúc mạng tối ưu [25]. Phương pháp phổ đạo hàm
không áp dụng được khi mẫu chứa nhiều cấu tử có phổ hấp thụ quang xen phủ nhau
hoặc tương tự nhau, vì rất khó để lựa chọn được một bước sóng thích hợp để xác
định một cấu tử nào đó, hoặc phổ đạo hàm của chúng vẫn có các cực đại hấp thụ
trùng nhau [2], [27]. Phương pháp lọc Kalman có thể loại bỏ được tối đa các nhiễu
và do đó giảm tối đa sai số, nhưng hạn chế của phương pháp này là phải lựa chọn
các giá trị khởi tạo cho bộ lọc, tức là phải chọn được giá trị ban đầu phù hợp của
hàm lượng các chất phân tích trong hỗn hợp của chúng và sai số kèm theo (được thể
hiện qua phương sai). Nếu các giá trị khởi tạo (nồng độ và phương sai) không phù
hợp, kết quả cuối cùng sẽ mắc sai số lớn [49], [101], [110].
Trên thế giới đã có một số nghiên cứu áp dụng phương pháp lọc Kalman vào
chemometric – trắc quang để xác định đồng thời hỗn hợp 2 hoặc 3 chất trong dược
phẩm [50], [121], song các nghiên cứu đó hoặc không đưa ra cách lựa chọn giá trị
khởi tạo phù hợp hoặc không đề cập đến các giá trị khởi tạo và do vậy, rất khó áp
dụng cho các phòng thí nghiệm phân tích. Ở nước ta, Mai Xuân Trường [27] đã
nghiên cứu áp dụng phương pháp lọc Kalman để xác định đồng thời các vitamin
trong dược phẩm, các nguyên tố đất hiếm…nhưng do tác giả cũng không giới thiệu
về cách chọn giá trị khởi tạo và do vậy, đã hạn chế khả năng áp dụng phương pháp
đề xuất vào thực tế. Nói chung, mặc dù phương pháp lọc Kalman có nhiều ưu điểm
khi áp dụng vào phương pháp chemometric - trắc quang để xác định đồng thời các
hợp chất, đặc biệt là các chất có phổ hấp thụ phân tử xen phủ nhau, nhưng cho đến
nay, các nghiên cứu áp dụng nó vẫn rất hạn chế.
Xuất phát từ các vấn đề trên, rõ ràng những nghiên cứu phát triển phương
pháp chemometric – trắc quang kết hợp với sử dụng phương pháp lọc Kalman là rất
cần thiết, đặc biệt là trong định lượng đồng thời các hỗn hợp chất khó phân tích –
các hỗn hợp chứa các chất có phổ hấp thụ quang xen phủ nhau - trong các đối tượng

4
mẫu khác nhau, trong đó có các mẫu dược phẩm. Song, thách thức đặt ra là phải tìm
được giải pháp phù hợp để lựa chọn giá trị khởi tạo cho bộ lọc Kalman sao cho đưa
ra các kết quả phân tích chính xác (độ lặp lại và độ đúng tốt) hay mắc sai số chấp
nhận được, đồng thời cần xây dựng được quy trình phân tích theo phương pháp
chemmometric – trắc quang kết hợp với phương pháp lọc Kalman sao cho có thể áp
dụng thuận lợi trong trong lĩnh vực kiểm nghiệm dược phẩm ở nước ta. Với các lí do
đó, đề tài “Nghiên cứu phát triển phương pháp chemometric để xác định đồng thời các
chất có phổ hấp thụ phân tử xen phủ nhau và áp dụng trong phân tích dược phẩm”
được thực hiện nhằm mục đích:
i) Xây dựng được quy trình phân tích chemometric - trắc quang kết hợp với
phương pháp lọc Kalman để phân tích đồng thời hỗn hợp 2 và 3 chất có phổ hấp thụ
quang xen phủ nhau trong các mẫu dược phẩm;
ii) Áp dụng quy trình xây dựng được để phân tích đồng thời hỗn hợp 2 và 3 chất
trong một số loại dược phẩm đang lưu hành trên thị trường Việt Nam.
Để đạt được mục đích trên, các nhiệm vụ chính của luận án bao gồm:
1. Nghiên cứu nhằm tìm ra giải pháp phù hợp để lựa chọn được giá trị khởi
tạo cho bộ lọc Kalman để áp dụng trong phương pháp chemometric – trắc quang
xác định đồng thời hỗn hợp các chất có phổ hấp thụ phân tử xen phủ nhau.
2. Nghiên cứu xây dựng chương trình phần mềm cho phép xác định nhanh
và thuận lợi hỗn hợp các cấu tử có phổ hấp thụ quang phân tử xen phủ nhau theo
phương pháp chemometric – trắc quang kết hợp với phương pháp lọc Kalman.
3. Nghiên cứu xây dựng quy trình phân tích theo phương pháp
chemometric – trắc quang kết hợp với phương pháp lọc Kalman và phần mềm xây
dựng được để xác định đồng thời các cấu tử có phổ hấp thụ quang xen phủ nhau
trong các mẫu dược phẩm (hay thuốc).
4. Áp dụng quy trình xây dựng được để phân tích đồng thời các hoạt chất
trong một số loại thuốc (đang lưu hành ở nước ta) chứa 2 hoặc 3 chất có phổ hấp
thụ quang xen phủ nhau. Đồng thời, đánh giá độ tin cậy của quy trình phân tích qua
độ đúng và độ lặp lại.

5
CHƯƠNG 1. TỔNG QUAN
1.1. Định luật Bouguer Lambert Beer và tính chất cộng tính độ hấp thụ quang
Tất cả các phương pháp phân tích quang phổ đều dựa trên các định luật cơ bản
của sự hấp thụ bức xạ điện từ. Các định luật cơ bản này là: Định luật Bouguer
Lambert, định luật Beer, định luật hợp nhất Bouguer Lambert Beer. [2], [9], [15],
[22].
Trong đó, để xác định đồng thời nhiều cấu tử theo phương pháp phân tích
quang phổ, hầu hết đều dựa trên cơ sở định luật hợp nhất Bouguer Lambert Beer và
tính cộng tính độ hấp thụ quang của các cấu tử trong hỗn hợp [11].
1.1.1. Định luật Bouguer Lambert Beer
Khi chiếu một chùm tia sáng có năng lượng nhất định vào một dung dịch chứa
cấu tử hấp thụ ánh sáng thì cấu tử đó sẽ hấp thụ chọn lọc một số tia sáng. Độ hấp
thụ quang của cấu tử ty lệ thuận với nồng độ của cấu tử trong dung dịch và bề dày
lớp dung dịch mà ánh sáng truyền qua [15], [16], [22].
Định luật Bouguer Lambert Beer (từ đây về sau được gọi tắt là định luật Beer)
có thể được biểu diễn bằng phương trình toán học như sau [15], [16], [22]:
Aλ = ελ.l.C (1.1)
Trong đó: Aλ: độ hấp thụ quang của dung dịch ở bước sóng λ
ελ: hệ số hấp thụ mol phân tử của cấu tử tại bước sóng λ
l: bề dày lớp dung dịch (cm)
C: nồng độ của cấu tử trong dung dịch (mol/lít)
Điều kiện sử dụng định luật: Chùm tia sáng đơn sắc; dung dịch phải loãng;
chất phân tích phải bền và bền dưới tác dụng của tia UV-VIS.
1.1.2. Tính chất cộng tính độ hấp thụ quang
Tính chất cộng tính đã được biết từ lâu và được xác nhận bằng thực nghiệm.
Tất cả các phương pháp phân tích định lượng xác định đồng thời hàm lượng các cấu
tử hấp thụ ánh sáng có mặt trong dung dịch đều dựa trên định luật này.
Tính chất cộng tính độ hấp thụ được phát biểu: Nếu trong dung dịch chứa n cấu
tử hấp thụ ánh sáng không tương tác hóa học với nhau, ở điều kiện tuân theo định luật

6
cơ bản của sự hấp thụ ánh sáng thì mật độ quang của một dung dịch như vậy bằng
tổng mật độ quang của tất cả các cấu tử hấp thụ ánh sáng chứa trong dung dịch tại
bước sóng xác định.
Tính chất cộng tính được viết như sau:
Aλ = A1,λ + A2,λ +…+ Ai,λ +…+ An,λ = ∑ 𝐴𝑖,
𝑛𝑖=1 (1.2)
Trong đó:
Aλ: độ hấp thụ ánh sáng của dung dịch hỗn hợp chứa n cấu tử ở bước sóng λ
Ai,λ: độ hấp thụ ánh sáng của cấu tử thứ i ở bước sóng λ; n là số cấu tử hấp
thụ ánh sáng có trong hỗn hợp; với i = 1 ÷ n.
Từ (1.1) có thể viết lại phương trình (1.2) như sau:
Aλ = ε1,λ.l.C1 + ε2,λ.l.C2 +…+ εn,λ.l.Cn = ∑ 𝜀𝑖,𝜆.. 𝑙. 𝐶𝑖𝑛𝑖=1 (1.3)
Khi phân tích đồng thời nhiều cấu tử trong hỗn hợp thì tính chất cộng tính của
độ hấp thụ quang thường không thỏa mãn một cách nghiêm ngặt làm ảnh hưởng
đáng kể đến độ chính xác của kết quả phân tích. Vì vậy, việc kiểm tra tính cộng tính
độ hấp thụ quang của hỗn hợp trước khi phân tích là hết sức quan trọng.
Để kiểm tra tính cộng tính độ hấp thụ quang của hỗn hợp ở các điều kiện khác
nhau, ta phải so sánh tổng độ hấp thụ quang của các dung dịch chứa từng cấu tử
riêng lẻ với độ hấp thụ quang của dung dịch hỗn hợp chứa các cấu tử đó, được đo
trong cùng một điều kiện. Kết quả kiểm tra sẽ cho biết có sự ảnh hưởng giữa các
cấu tử hấp thụ quang trong dung dịch hỗn hợp hay không? Trong khoảng nồng độ
nào thì độ hấp thụ quang của dung dịch nghiên cứu còn tuân theo định luật Bughe-
Lămbe- Bia và thỏa mãn tính cộng tính độ hấp thụ quang để có thể áp dụng.
Nguyên nhân làm sai lệch tính cộng tính độ hấp thụ quang có thể là do tương
tác hóa học giữa các cấu tử trong hỗn hợp, cũng có thể do tương tác vật lý (lực ion,
các cấu tử có sự hút, đẩy lẫn nhau….), do hiện tượng khuyếch tán, tán xạ ánh sáng
của các cấu tử, đặc biệt là khi số cấu tử trong hỗn hợp càng lớn [15], [16], [22].
1.2. Một số phương pháp phân tích quang phổ UV-VIS kết hợp với
chemometric xác định đồng thời các cấu tử có phổ hấp thụ xen phủ nhau
Phương pháp đường chuẩn, phương pháp thêm chuẩn, phương pháp vi sai là
những phương pháp được áp dụng để xác định các cấu tử trong dung dịch có phổ

7
hấp thụ không xen phủ nhau và tuân theo định luật Bia. Tuy nhiên, đối với dung
dịch hỗn hợp mà các cấu tử có phổ hấp thụ xen phủ nhau thì việc tính toán rất
phức tạp. Vì vậy, dựa trên cơ sở định luật Beer nhiều phương pháp phân tích đã ra
đời cho phép xác định các cấu tử trong hỗn hợp có phổ hấp thụ xen phủ nhau mà
không cần che, tách. Sau đây là một số phương pháp chemometric xác định các
chất có phổ hấp thụ xen phủ nhau.
1.2.1. Phương pháp Vierordt
1.2.1.1. Giới thiệu chung
Phạm vi: Phương pháp Vierordt chủ yếu được dùng với các hệ có từ hai đến
ba cấu tử mà độ hấp thụ quang của các cấu tử đó xen phủ nhau không nhiều.
Điều kiện để áp dụng phương pháp này là các cấu tử trong hỗn hợp phải tuân
theo định luật Beer và thỏa mãn tính cộng tính của độ hấp thụ quang.
Ứng dụng: phương pháp Vierordt được dùng trong việc phân tích hỗn hợp các
chất hữu cơ, các loại dược phẩm, các hỗn hợp chất màu [43], [70], [87].
1.2.1.2. Cơ sở lý thuyết của phương pháp
Để xác định nồng độ của các cấu tử trong hỗn hợp, lần đầu tiên Vierordt đã đo
độ hấp thụ quang của dung dịch hỗn hợp ở các bước sóng khác nhau, sau đó thiết
lập hệ phương trình bậc nhất mà số phương trình bằng số ẩn số (số cấu tử trong hỗn
hợp), giải hệ phương trình này sẽ tính được nồng độ của các cấu tử.
Với hỗn hợp chứa n cấu tử ta cần phải lập hệ n phương trình n ẩn số. Hệ
phương trình này được thiết lập bằng cách đo độ hấp thụ quang của hỗn hợp ở n
bước sóng khác nhau.
A(λ1)= Ɛ11C1l + Ɛ21C2l + …+ Ɛi1Cil+… + Ɛn1Cnl
A(λ2)= Ɛ12C1l + Ɛ22C2l + …+ Ɛi2Cil+… + Ɛn2Cnl
… … … … ….
A(λn)= Ɛ1nC1l + Ɛ2nC2l + …+ ƐinCil+… + ƐnnCnl (1.4)
Trong đó:
A(λ1), A(λ2),…, A(λn): Độ hấp thụ quang của hỗn hợp ở bước sóng λ1, bước
sóng λ2, …, và bước sóng λn.

8
Ɛin: Hệ số hấp thụ phân tử của cấu tử i tại bước sóng λn (được xác định bằng
cách đo độ hấp thụ quang của dung dịch chỉ chứa cấu tử i ở bước sóng λn)
l: Bề dày lớp dung dịch (cm)
Ci: Nồng độ của cấu tử thứ i trong hỗn hợp (mol/L). Với i = 1 ÷ n
Giải hệ n phương trình với n ẩn số là C1, C2, …, Cn sẽ tìm được nồng độ của các
cấu tử. Khi số cấu tử trong hỗn hợp ít thì việc giải hệ n phương trình tuyến tính khá
đơn giản. Tuy nhiên khi số cấu tử lớn thì việc giải hệ phương trình phức tạp hơn.
Phương pháp Vierordt chủ yếu được vận dụng để tìm cách giải hệ phương
trình như sau: giải bằng đồ thị, giải bằng phép ma trận vuông, phương pháp khử
Gauss,... để xác định nồng độ của mỗi cấu tử [2], [24], [29], [30].
1.2.1.3. Ưu điểm và nhược điểm
Ưu điểm: Phương pháp Vierordt đơn giản, dễ thực hiện.
Nhược điểm: Chỉ áp dụng được khi số cấu tử trong dung dịch hỗn hợp ít, phổ
hấp thụ quang phân tử xen phủ nhau không nhiều, tính chất cộng tính độ hấp thụ
quang được thỏa mãn nghiêm ngặt, thiết bị đo quang tốt thì phương pháp cho kết
quả khá chính xác. Đối với hệ nhiều cấu tử, đặc biệt là khi phổ của các cấu tử xen
phủ nhau nhiều, tính chất cộng tính độ hấp thụ quang không được thỏa mãn nghiêm
ngặt, thiết bị đo có độ chính xác không cao thì phương pháp không chính xác và có
sai số lớn [2], [28] [55], [56]. Bởi vậy mặc dù phương pháp Vierordt tuy ra đời đã
lâu, nhưng ứng dụng trong thực tế còn rất ít. Tuy nhiên đây là cơ sở lý thuyết cơ
bản, đặt nền móng cho các nhà khoa học sau này phát triển, cải tiến để xây dựng
nên các phương pháp mới [28].
1.2.2. Phương pháp quang phổ đạo hàm
1.2.2.1. Giới thiệu chung
Phương pháp quang phổ đạo hàm là một kỹ thuật chuyển đổi phổ hấp thụ dựa
trên phép lấy đạo hàm bậc nhất, bậc hai hoặc bậc cao hơn.
Một số tác giả đã sử dụng phương pháp phổ đạo hàm xác định đồng thời các
vitamin tan trong nước [18], [21], [22], [28] cũng như xác định đồng thời các chế
phẩm dược dụng khác [13], [18], [28], [31], [108].
Các kết quả thu được có sai số trong khoảng 1,7 5% [28].

9
Trên thế giới, phương pháp phổ đạo hàm được ứng dụng để phân tích các chế
phẩm dược dụng [28], [54], [57], [92], [93], [94], [95] cũng như hỗn hợp các chất
vô cơ, hữu cơ [28], [48], [52], [53], [80], [118].
1.2.2.2. Cơ sở lý thuyết của phương pháp
Theo định luật Bia thì: A = ε.l.C
Lấy phổ đạo hàm bậc 1, bậc 2 hoặc bậc cao hơn của độ hấp thụ quang A theo
bước sóng λ, ta được:
dA dε = C.l.
dλ dλ
2 2
2 2
d A d ε = C.l.
dλ dλ
.................................. (1.5)
n n
n n
d A d ε = C.l.
dλ dλ
Vì độ dày l của một lớp dung dịch luôn không đổi và tại một bước sóng nhất
định đạo hàm của ε là một hằng số nên giá trị đạo hàm của A còn phụ thuộc tuyến
tính với nồng độ C của dung dịch.
* Phổ đạo hàm của hỗn hợp nhiều chất
Phổ đạo hàm của hỗn hợp nhiều chất có tính chất cộng tính nên:
Ahh = A1 + A2 + … + An
1 2dA dAdA dA
= ...dλ dλ dλ dλ
hh n
2 22 2
1 2
2 2 2 2
d A d Ad A d A = ...
dλ dλ dλ dλ
hh n
…………………………………
1 2d A d Ad A d A
= ...dλ dλ dλ dλ
n nn n
hh n
n n n n
(1.6)
Trong đó: 1, 2,…, n là: chất 1, chất 2,…, chất n.

10
Hinh 1.1. (a) Phổ hấp thụ; (b) phổ đạo hàm bậc 2; (c) phổ đạo hàm bậc 4;
(d) phổ hấp thụ của trans-stilbene trong cyclohexane; (e) phổ đạo hàm bậc 2
của trans-stilbene trong cyclohexane; (f) phổ đạo hàm bậc 4 của trans-stilbene
trong cyclohexane.
Hình 1.1 minh họa sự chuyển từ dạng phổ hấp thụ UV-Vis sang dạng phổ đạo
hàm, cho thấy thông tin thu được trên phổ đạo hàm nhiều hơn phổ hấp thụ ban đầu
(số cực trị tăng khi số bậc đạo hàm tăng). Trong đó:
- Các cực trị ở đạo hàm bậc n – 1 sẽ có giá trị 0 tại đạo hàm bậc n, còn các
điểm uốn ở đạo hàm bậc n – 1 lại trở thành điểm cực trị ở các ở đạo hàm bậc n. Do
đó, xuất phát từ 1 peak ban đầu sau n lần đạo hàm sẽ cho n cực trị mới được gọi là
cực ảo (virtual extrema) hay các vệ tinh (satellites).
- Sự xuất hiện của các cực trị ảo này sẽ làm tăng khả năng phân tích phổ hấp
thụ ban đầu, có thể ứng dụng được trong phân tích định tính.
Như vậy, dùng phương pháp phổ đạo hàm ta có thể tách phổ gần trùng nhau
thành những phổ mới và khi đó ta có thể chọn được những bước sóng mà tại đó chỉ
có duy nhất 1 cấu tử hấp thụ quang còn các cấu tử khác không hấp thụ, nhờ đó mà
có thể xác định được từng chất trong hỗn hợp. Bằng toán học, người ta xây dựng

11
được phần mềm khi đo phổ của dung dịch hỗn hợp có thể ghi ngay được phổ đạo
hàm các bậc của phổ đó. Căn cứ vào các giá trị phổ đạo hàm ta lựa chọn được bước
sóng xác định đối với từng cấu tử.
1.2.2.3. Ưu điểm và nhược điểm
Ưu điểm: Xác định các chất có phổ hấp thụ xen phủ nhau hoặc rất sát nhau.
Tăng độ tương phản giữa các phổ có độ bán rộng khác nhau. Hầu hết các kết quả
đều cho thấy phương pháp có độ tin cậy cao.
Nhược điểm: Khi bậc đạo hàm càng cao thì độ nhạy phép đo càng giảm. Đối
với những chất có phổ tương tự nhau hay hỗn hợp phức tạp có nhiều cấu tử có phổ
hấp thụ xen phủ nhau thì khó áp dụng phổ đạo hàm. Phương pháp phổ đạo hàm chỉ
được áp dụng khi số cấu tử trong dung dịch ít và phổ hấp thụ quang phân tử của
chúng không trùng nhau. Trường hợp dung dịch có nhiều cấu tử và phổ hấp thụ
quang phân tử tương tự nhau thì không thể áp dụng phương pháp phổ đạo hàm và
bậc đạo hàm càng cao thì độ nhạy của phép xác định càng giảm [27].
1.2.3. Phương pháp bình phương tối thiểu
1.2.3.1. Giới thiệu chung
Hạn chế của phương pháp Vierordt là sử dụng dữ liệu ít, trong khi đó mỗi giá
trị đo bao giờ cũng kèm theo sai số của thiết bị, điều đó dẫn đến kết quả tính toán
không được chính xác. Phương pháp bình phương tối thiểu cho hệ đa biến (Least
Squares, LS) được biết dưới tên gọi là ma trận K, áp dụng định luật Bia mở rộng
tính toán các hệ số hấp thụ trên phần phổ lớn hơn nhiều so với phương pháp hồi quy
bình phương tối thiểu đơn giản [2].
1.2.3.2. Cơ sở lý thuyết của phương pháp
Áp dụng định luật Bia cho hệ gồm n cấu tử tại m bước sóng (m > n) và độ hấp
thụ của hệ có tính chất cộng tính tại một bước sóng.
Với εi: độ hấp thụ phân tử của cấu tử thứ i;
Ci: nồng độ của cấu thử thứ i trong hỗn hợp.
Ta được hệ phương trình tuyến tính gồm m phương trình, n ẩn số:

12
A1 = ε11.C1 + ε12.C2+ … + ε1i.Ci + … +ε1n.Cn
A2 =ε21.C1 + ε22.C2 + … + ε2i.Ci + … + ε2n.Cn
…
Aj = ε1j.C1 + εj2.C2 + … + εji.Ci + … + εjn.Cn (1.7)
…
Am =εm1.C1 + εm2.C2 + … +εmi.Ci + … + εmn.Cn
Khi đo phổ hấp thụ quang của dung dịch ở bước sóng thứ j ta được giá trị yj.
Giá trị này thường mắc phải sai số đo nên sẽ khác với giá trị thực Aj một đại lượng
sj. Trong đó sj là sai số đo và si = yj – Aj.
Hàm biễu diễn sai số bình phương toàn phần:
22
1 1 2 2
1 1
( ) ( .C .C ... .C ... .C )m m
j j j j j ji i jn n
j j
S y A y
(1.8)
Để S đạt cực tiểu thì đạo hàm của S theo các biến Ci phải bằng 0. Nếu ta lấy
đạo hàm S theo biến x1 và cho đạo hàm bằng 0 sẽ nhận được phương trình sau:
1 1 2 2 1 1
11
2 ( .C .C ... .C ... .C ) . .( ) 0m
j j j ji i jn n j j j
j
dSy y
dC
Suy ra:
2
1 1 1 2 2 1 1 1
1 1 1 1 1
.C . .C ... . .C ... . .C . 0m m m m m
j j j j ji i j jn n j j
j j j j j
e y
(1.9)
Tương tự cũng lấy đạo hàm S theo các biến Ci còn lại và cho các giá trị đạo
hàm này bằng 0, kết hợp với phương trình (1.7) ở trên ta được hệ phương trình sau:
1 1 1 2 1 2 1 1 1
1 1 1 1 1
1 2 1 2 2 2 2 2 2
1 1 1 1 1
1 1 2 2
1 1
. . ... . ... . . 0
. . ... . ... . . 0
...
. . ...
m m m m m
j j j j i j ji n j jn j j
j j j j j
m m m m m
j j j j i j ji n j jn j j
j j j j j
m m
ji j ji j
j j
C C C C y
C C C C y
C C C
1 1 1
1 1 2 2
1 1 1 1 1
. ... . . 0
...
. . ... . ... . . 0
m m m
i ji ji n ji jn ji j
j j j
m m m m m
jn j jn j i jn ji n jn jn jn j
j j j j j
C y
C C C C y
(1.10)

13
Đặt: 1
.m
ji ji jk
j
a
, 1
.m
k jk j
j
b y
, với 1,i n , 1,k n
Ta được hệ phương trình viết gọn lại như sau:
a11.C1 + a12.C2 + … + a1i.Ci + … + a1n.Cn = b1
a21.C1 + a22.C2 + … + a2i.Ci + … + a2n.Cn = b2
…
ak1.C1 + ak2.C2 + … + aki.Ci + … + akn.Cn = bk (1.11)
…
an1.C1 + an2.C2 + … + ani.Ci + … + ann.Cn = bn
Các giá trị aki, bk trong hệ (1.11) được tính từ các giá trị đo ban đầu eji thông
qua phương pháp bình phương tối thiểu. Hệ phương trình (1.10) là một hệ phương
trình tuyến tính gồm n phương trình, n ẩn số, giải hệ này để xác định nồng độ của
các cấu tử Ci trong hệ bằng phương pháp Gauss [2].
1.2.3.3. Ưu điểm và nhược điểm
Ưu điểm: Phương pháp này có thể sử dụng toàn bộ số liệu phổ để lập ra hệ
phương trình tuyến tính có số phương trình nhiều hơn số ẩn. Quá trình biến đổi dựa
trên nguyên tắc của phép bình phương tối thiểu sẽ mắc sai số nhỏ nhất, do đó nâng
cao độ chính xác của phép xác định. Nồng độ được tính toán tương đối nhanh và có
thể dùng cho các hỗn hợp phức tạp.
Nhược điểm: Phải biết thành phần định tính của mẫu. Khi các cấu tử có tương
tác với nhau tạo ra hiệu ứng quang học làm thay đổi hệ số hấp thụ của từng cấu tử
thì kết quả phân tích cũng không chính xác.
1.2.4. Phương pháp bình phương tối thiểu từng phần (Partial Least Square -
PLS)
1.2.4.1. Giới thiệu chung
PLS là một phương pháp dùng để xây dựng mối quan hệ hồi quy giữa hai biến
số với biến số ẩn trong ma trận X và ma trận Y, trong đó X là biến độc lập và Y là
biến phụ thuộc [51],[78],[121].
Phương pháp bình phương tối thiểu riêng phần (PLS) là phương pháp đa biến
dùng để mô hình hóa mối quan hệ giữa biến độc lập X và biến phụ thuộc Y, từ đó

14
có thể đoán được thông tin trong Y khi đã biết các thông tin của X và ngược lại.
Mục đích của PLS là giảm số biến và tạo ra các phần tử không liên quan, sau đó
biểu diễn phương trình bình phương tối thiểu với những phần tử này.
Phương pháp này đã được áp dụng để xác định các hỗn hợp dược phẩm, kim
loại, thuốc trừ sâu [28], [85], [50].
1.2.4.2. Cơ sở lý thuyết của phương pháp
Thuật toán PLS được giải bằng cách tối ưu hóa giá trị đồng phương sai
(covariance) giữa ma trận X và Y. Hai ma trận X và Y được phân tích thành hai ma
trận trị số (score matrices) T và U, và ma trận trọng số (loading matrices) P và Q.
Có hai dạng khác nhau của trọng số trong PLS, trọng số bình phương tối thiểu riêng
phần W là một trong số hai dạng đó và được tính theo công thức:
W = (Y’. X). inv (X’. X)
T = Y . W (1.12)
Mỗi ma trận X và Y sẽ được chuyển thành hai ma trận nhỏ
P = (Y’. t) . inv (t’ . t)
Q = (X’ . t) . inv (t’ . t) (1.13)
Từ phương trình hồi quy tuyến tính tổng quát dạng x = a + by, hệ số hồi quy b
và a được tính theo công thức:
b= w . inv (P’ . w) . Q (1.14)
a = mean (X)- mean (Y) . b (1.15)
Với mẫu chưa biết nồng độ, từ ma trận tín hiệu đo y0 của mẫu sẽ xác định
được nồng độ của các chất dựa vào hệ số hồi quy b đã tính:
X0 = a + y0 . b (1.16)
Mục đích của PLS là giảm số biến và tạo ra các phần tử không liên quan nhau
sau đó biễu diễn phương trình bình phương tối thiểu với những phần tử này. PLS
mô hình hóa đồng thời cả 2 biến X và Y để tìm ra những biến ẩn trong X, từ đó có
thể đoán được biến ẩn trong Y [26],[121].
1.2.4.3 Ưu điểm và nhược điểm
- Ưu điểm:

15
Phương pháp PLS khác với các phương pháp hồi quy khác ở chỗ nó thích hợp
cho những tập số liệu có số thí nghiệm ít hơn số biến và sự tương quan giữa các
biến độc lập và có tính chất cộng tính cao.
Giảm số biến và tạo ra các cấu tử không liên quan sau đó biểu diễn phương
trình bình phương tối thiểu với những cấu tử này.
Phương pháp PLS cho kết quả có độ chính xác cao và tiện lợi. Từ nội dung
của thuật toán, ta có thể lập trình theo nhiều ngôn ngữ khác nhau như ngôn ngữ
Pascal, ngôn ngữ C+, C++….
Ưu thế của phương pháp PLS so với phương pháp mạng nơron là thời gian
tính toán ít và khả năng hội tụ nhanh.
Phương pháp này dùng được cho các hỗn hợp phức tạp.
- Nhược điểm:
Phương pháp PLS đòi hỏi khá phức tạp về mặt toán học trong khi đó lại không
đơn giản được nhiều số biến phân tích như phương pháp hồi quy cấu tử chính
(PCR)
Việc tính toán cho phương pháp này lại chậm hơn phương pháp bình phương
tối thiểu hệ đa biến. Việc chọn các mẫu chuẩn là tương đối khó, phải dùng nhiều
mẫu.
Phương pháp BPTT đòi hỏi những cấu tử trong hỗn hợp phải cho tín hiệu có
tính chất cộng tính. Vì vậy cần phải biết tất cả các phổ của những chất gây ảnh
hưởng đến vùng phổ được đo vì chúng đều đóng góp vào đường chuẩn. Điều này có
thể được loại trừ đáng kể bằng cách phân tích dải phổ tại một thời điểm sau khi gộp
kết quả vào phép phân tích thống kê. Nó cho phép loại bỏ dải phổ không tuân theo
định luật Bia hoặc những phổ có chứa tín hiệu của ion cản [26].
1.2.5. Phương pháp hồi quy cấu tử chính (Principal Component Regression -
PCR)
1.2.5.1. Giới thiệu chung
PCR là phương pháp mở rộng về phương trình hồi quy sử dụng phân tích đa
biến áp dụng cho tập số liệu có rất nhiều biến như PLS. Phương pháp này sử dụng
phương trình hồi quy để chuyển đổi những giá trị số cấu tử chính (PC scores) thành

16
nồng độ, quy trình này còn được gọi là phân tích thành phần. PCR là một trong
những công cụ phân tích hữu hiệu cho phép giảm sai số biến trong tập số liệu nhằm
đạt được biễu diễn hai chiều từ tập số liệu đa chiều bằng cách tìm ra giá trị phương
sai lớn nhất với số cấu tử chính ít nhất.
Do phương pháp này phát triển trên cơ sở của phương pháp ILS nên để sử
dụng được các phương pháp này trong phân tích trắc quang chúng ta cần số mẫu
chuẩn tối thiểu phải bằng số thời điểm sử dụng trong đường chuẩn mã hóa, tức là số
mẫu không nhỏ hơn số cấu tử chính (PC) lựa chọn. Thông thường, với một tập số
liệu có mức độ tập trung tốt thì chỉ có một số ít các PC đầu tiên là có nghĩa (có tổng
phương sai tích lũy đủ lớn để coi rằng chúng đã chứa toàn bộ thông tin hữu ích đặc
trưng của tập số liệu). Như vậy, sử dụng mô hình PCR có thể làm giảm kích thước
của tập số liệu mà không làm mất thông tin, đồng thời có thể loại bỏ được tín hiệu
nhiễu của dữ liệu gốc.
1.2.5.2. Cơ sở lý thuyết của phương pháp
PCR – phương pháp hồi quy cấu tử chính gồm 2 quá trình: Phân tích cấu tử
chính chuyển sang tập dữ liệu mới, chứa một số ít các yếu tố quan trọng, cần thiết;
sau đó sử dụng phương pháp bình phương tối thiểu nghịch đảo để phân tích tập dữ
liệu mới này.
Trước tiên, chiếu tập số liệu tín hiệu phân tích lên không gian có ít chiều hơn
theo PCA mà không làm mất đi các thông tin quan trọng và tiến hành phân tích hồi
quy đa biến trên không gian mới này.
Các bước chính của PCR
- Bước 1: Xử lý ban đầu (không bắt buộc)
Nội dung chính của bước này là chuẩn hóa tập số liệu.
- Bước 2: Các xử lý cần thiết
Với một tập số liệu đã chuẩn hóa hoặc chưa chuẩn hóa, trước khi sử dụng đều
cần phải bình phương toàn tập dữ liệu – đây là yêu cầu bắt buộc đối với hầu hết các
hàm vectơ riêng.
D = AT.A (1.17)
Trong đó:

17
A là ma trận số liệu biểu diễn độ hấp thụ quang theo các thời điểm đo của các
dung dịch chuẩn.
AT
là ma trận chuyển vị của ma trận A.
- Bước 3: Xác định các vectơ riêng hay các PC
Có thể tính toán các vectơ riêng của tập số liệu bằng nhiều hàm toán học khác
nhau. Có 3 hàm chính, thường sử dụng là hàm NIPALS (hàm phi tuyến lặp sử dụng
kĩ thuật bình phương tối thiểu riêng phần), hàm SVD (hàm phân tách các giá trị
riêng) và hàm Princomp (hàm tính các cấu tử chính). Cần lưu ý rằng, tất cả các hàm
này đều tính toán và đưa ra tất cả các cấu tử nhưng thường không sử dụng tất cả mà
chỉ sử dụng N cấu tử đầu đủ để xác định không gian mới.
Các hàm toán học trên đều đưa ra một ma trận cột chứa các vectơ riêng - Vc - là
ma trận trong đó mỗi cột là một vectơ hay nhân tố mới – PC – của ma trận dữ liệu và
số hàng ma trận là số thời điểm đo. Mỗi nhân tố hay vectơ này lại là tổ hợp bậc nhất
của các điểm phổ ban đầu, phần đóng góp của các điểm này vào mỗi vectơ là khác
nhau tùy thuộc vào giá trị hàm phụ thuộc tại điểm đó. Những điểm có giá trị đóng
góp lớn vào các PC chứa phương sai lớn sẽ là những điểm đo có ảnh hưởng quyết
định tới kết quả tính ma trận hệ số hồi quy và kết quả hồi quy sau đó. Ma trận kết quả
thứ hai cũng rất quan trọng là ma trận phương sai của các PC: đó là dạng ma trận
chéo đối với hàm SVD, là một vectơ cột đối với hàm NIPALS và hàm Princomp.
- Bước 4: Lựa chọn các vectơ có nghĩa
Đây là bước có ảnh hưởng đặc biệt quan trọng đến bước xử lý tiếp theo. Nếu giữ
lại nhiều vectơ hơn số cần thiết thì những vectơ đó sẽ chứa cả tín hiệu nhiễu và như vậy
kết quả hồi quy sẽ mắc sai số. Nếu giữ lại không đủ số vectơ cần thiết sẽ làm mất thông
tin có ích từ tập dữ liệu, điều này cũng sẽ gây nên sai lệch giữa mô hình hồi quy thu
được và mô hình thực. Vì vậy, việc đánh giá và lựa chọn các vectơ có nghĩa là rất quan
trọng. Một số phương pháp phổ biến để xác định số PC có nghĩa: 1. Dùng các hàm chỉ
thị như CPV (tính phần trăm phương sai tích lũy), hàm IEF…; 2. Tính toán PRESS
(tổng bình phương sai số dự đoán) để đánh giá thông tin từ dữ liệu; 3. Phương pháp
đánh giá chéo; 4. Phương pháp đánh giá Xu – Kailath; 5. Đánh giá theo tiêu chuẩn
Akaike; 6. Tính phương sai của sai số tái lập VRE.

18
- Bước 5: Tính toán lại
Sau khi loại bỏ các vectơ riêng không có nghĩa, chúng ta cũng loại được tín hiệu
nhiễu của dữ liệu gốc và cần tính lại dữ liệu sau khi loại bỏ sai số. Như vậy, khi tính
toán ở hệ tọa độ mới ta đã loại bỏ được tín hiệu nhiễu trong tập dữ liệu ban đầu.
- Bước 6: Xây dựng đường chuẩn
Khi xây dựng đường chuẩn PCR theo phương pháp ILS, điểm khác biệt duy
nhất là tập dữ liệu sử dụng. Các bước tiến hành bao gồm:
Xác định phép chiếu trong hệ tọa độ mới:
Aj = A.Vc (1.18)
Trong đó:
Aj: là ma trận số liệu ở hệ tọa độ mới
A: là ma trận gốc.
Vc: là ma trận các vectơ riêng có nghĩa.
- Thay thế A bằng Aj trong phương trình hồi quy:
C = Aj.F (1.19)
Trong đó F được tính theo công thức:
F = (AjT. Aj)-1.Aj
T.C (1.20)
- Nồng độ chất phân tích trong mẫu chưa biết được tính theo công thức:
Cx = Ax.Vc.F = Ax.Fcal (1.21)
Với Fcal = Vc.F đóng vai trò tương tự ma trận P trong phương trình của ILS.
1.2.5.3. Ưu điểm và nhược điểm
- Ưu điểm:
Hội tụ đầy đủ các ưu điểm của phương pháp ILS đồng thời khắc phục được
các nhược điểm của phương pháp ILS do tiến hành tính toán trên toàn phổ.
Phương pháp này cho phép loại bỏ sai số nhiễu phổ và sai số ngẫu nhiên trong
quá trình đo khi lựa chọn được số PC phù hợp.
Đối với trường hợp sử dụng phổ toàn phần, khi dùng các phương pháp khác
như BPTT, kết quả tính cuối cùng là kết quả tính trung bình trên toàn phổ nên kém
chính xác hơn trường hợp dùng phổ chọn lọc. Khi sử dụng mô hình PCR, tuy kết
quả vẫn tính trên tất cả các điểm nhưng đóng góp của các điểm đo sẽ khác nhau tùy

19
theo lượng đóng góp của từng điểm này vào các PC được chọn mà lượng đóng góp
này lại được phân tích dựa trên tín hiệu đo tại từng thời điểm của các mẫu chuẩn.
Do có sự phân biệt và chọn lọc trong đánh giá mỗi điểm đo nên kết quả thu được sẽ
chính xác hơn phương pháp tính trung bình trên toàn phổ ở các phương pháp phổ
toàn phần khác.
- Nhược điểm
Vì PCR là một phương pháp hoạch định nên đôi khi những quan sát có thể dẫn
đến những giải nghĩa sai lệch. Khi đó, sẽ gây ra hiện tượng, trong những cấu tử bị
loại bỏ khi phân tích cấu tử chính, có những thông tin hữu ích sẽ “vô tình” bị mất đi
cùng với những cấu tử đó, và lúc này chắc chắn rằng các giá trị nhiễu vẫn tồn tại ít
nhiều trong các cấu tử chính được lưu giữ lại dùng trong sự hồi quy. Như vậy, tính
hiệu lực của phương pháp PCR không còn cao [26], [121].
1.2.6. Phương pháp mạng nơron nhân tạo (Artificial Neural Networks - ANN)
1.2.6.1. Giới thiệu chung
Mạng nơron nhân tạo là một tập hợp các nơron được đặt trong những lớp cách
biệt, mỗi nơron trong một lớp được nối với tất cả các nơron khác ở lớp kế tiếp và
xác định bằng những tín hiệu chỉ được truyền theo một hướng qua mạng.
Phương pháp mạng nơron nhân tạo được ứng dụng để xác định đồng thời các
cấu tử theo phương pháp trắc quang [28], [33].
- Nguyên tắc: Đặt các nơron sao cho chúng ở trong những lớp cách biệt, mỗi
nơron trong một lớp được nối với tất cả các nơron khác ở lớp kế tiếp và xác định
bằng những tín hiệu chỉ được truyền theo một hướng qua mạng. Đó chính là mô
hình mạng nơron [11].
- Ứng dụng: ANN có rất nhiều ứng dụng trong nhiều ngành và lĩnh vực khác
nhau:
+ Giải các bài toán phân lớp: bài toán này đòi hỏi giải quyết vấn đề phân
loại các đối tượng thành các nhóm dựa trên những đặc điểm của các nhóm đối
tượng. Trên cơ sở này người ta sử dụng ANN trong nhận dạng chữ viết, tiếng nói,
phân loại gen, phân loại chất lượng sản phẩm

20
+ Bài toán dự báo: mạng ANN đã được ứng dụng trong việc xây dựng mô
hình dự báo sử dụng tập dữ liệu trong quá khứ để dự đoán số liệu cho tương lai (dự
báo thời tiết).
+ Bài toán điều khiển và tối ưu hoá: ANN được sử dụng trong hệ điều khiển
tự động cũng như trong việc giải quyết rất nhiều bài toán tối ưu trong thực tế
+ Nhìn chung, ANN là công cụ cho phép tiếp cận có hiệu quả để giải quyết
các bài toán có tính phi tuyến tính, biến động, dữ liệu có nhiễu và đặc biệt là trong
trường hợp các mối quan hệ mà bản chất vật lý của các quá trình cần nghiên cứu
không dễ dàng nhận biết và thể hiện chúng hay còn gọi là các tập mờ [20].
+ Ứng dụng trong hoá học phân tích: Việc nghiên cứu xác định đồng thời
nhiều cấu tử mà phổ của các đại lượng vật lý đo được của chúng xen phủ nhau đã
được nhiều tác giả quan tâm nghiên cứu. Để xác định đồng thời nhiều cấu tử có nhiều
phương pháp: phương pháp trắc quang đạo hàm, phương pháp chuẩn đa biến sử dụng
bình phương tối thiểu (BPTT, ILS, PLS)... nhưng phương pháp đạo hàm sẽ làm giảm
độ nhạy của phép phân tích còn trong nhiều trường hợp các phương pháp bình
phương tối thiểu không thích hợp vì tín hiệu đo không có tính cộng tính [72].
Hiện nay nhiều công trình nghiên cứu sử dụng ANN được triển khai thực hiện
ở rất nhiều phòng thí nghiêm trên thế giới. ANN cho phép mô hình hoá các mối
quan hệ phi tính phức tạp. Nó cho phép giải quyết mối quan hệ mà trong đó có
những quá trình xảy ra chưa được biết hoặc những thông tin về hệ còn chưa đầy đủ
hay hệ mờ.
1.2.6.2. Cơ sở lý thuyết của phương pháp
Quá trình vận hành mạng nơron: mỗi nơ ron nhận một tín hiệu từ nơron của
lớp trước và mỗi tín hiệu này được nhân với hệ số riêng. Những tín hiệu vào có
trọng số được gom lại và qua một hàm hạn chế dùng để căn chỉnh tín hiệu ra (kết
quả) vào một khoảng giá trị xác định. Sau đó, tín hiệu ra của hàm hạn chế được
truyền đến tất cả các nơron của lớp kế tiếp. Như thế, để sử dụng mạng giải bài toán,
chúng ta sử dụng những giá trị tín hiệu vào cho các lớp đầu. Cho phép tín hiệu lan
truyền qua mạng và đọc các giá trị kết quả sau lớp ra.

21
Hinh 1.2. Mô hình hoạt động của mạng nơron được thể hiện ở hình 1.1.
Độ chính xác của tín hiệu ra (kết quả) phụ thuộc vào trọng số của các nơron,
nên cần phải hiệu chỉnh các trọng số để giải với từng bài toán cụ thể. Để hiệu chỉnh
được trọng số cần các thông tin lan truyền ngược. Quá trình lan truyền ngược được
thực hiện với một số bước lặp. Lúc đầu, các kết quả thu được sẽ là hỗn loạn. Kết
quả này được so sánh với kết quả đã biết và tín hiệu sai số bình phương trung bình
sẽ được tính. Sau đó, giá trị sai số sẽ được lan truyền trở lại mạng và những thay đổi
nhỏ được thực hiện đối với các trọng số trong mỗi lớp. Sự thay đổi trọng số được
tính toán sao cho giảm tín hiệu sai số đối với trường hợp đang xét. Toàn bộ quá
trình được lặp lại đối với mỗi bài toán và sau đó lại quay trở về bài toán đầu tiên và
cứ thế tiếp tục. Vòng lặp được lặp lại cho đến khi sai số toàn cục rơi vào vùng xác
định bởi một ngưỡng hội tụ nào đó. Tất nhiên không bao giờ các kết quả thu được
có độ chính xác tuyệt đối [11], [74].
1.2.6.3. Ưu điểm và nhược điểm
- Ưu điểm
Phương pháp cho phép xác định đồng thời nhiều cấu tử khi phổ của chúng
trùng lấn nhau ngay cả khi độ hấp thụ quang đo được không có tính cộng tính.
Trong khi đó các phương pháp khác như trắc quang đạo hàm, Vierordt đòi hỏi các
đại lượng đó phải có tính cộng tính.
Mạng ANN cho phép xác định đồng thời nhiều cấu tử mà trong hệ có nhiều
quá trình xảy ra còn chưa biết hay còn gọi là hệ mờ, nhờ vậy mà ANN có thể xác

22
định bằng phương pháp trắc quang ngay cả khi trong dung dịch có sự tạo phức cạnh
tranh, thuốc thử tạo phức màu không đủ dư và khi nồng độ các cấu tử cần xác định
không nằm trong khoảng tuyến tính.
ANN cho phép xác định đồng thời nhiều cấu tử mà phổ của chúng trùng lấn nhau
bằng các kỹ thuật khác nhau như: điện hoá, trắc quang động học, huỳnh quang tia X...
- Nhược điểm
Thời gian luyện mạng thường khá lâu.
Chưa có phần mềm tiện ích để sử dụng ngay, đòi hỏi người thực hiện phải
nắm rõ thuật toán để viết chương trình trên các phần mềm khác (Pascal, Matlab, C+,
... ) mới sử dụng được.
ANN có rất nhiều thuật toán khác nhau, do đó khi xây dựng một mô hình phân
tích chất, đòi hỏi người sử dụng phải thử nhiều mô hình để tìm được cấu trúc mạng
tối ưu.
Thực hiện các thí nghiệm phức tạp, khó áp dụng vào thực tế. Để xây dựng
được chương trình theo phương pháp mạng nơron có kết quả cao là rất khó và đòi
hỏi người lập trình phải có kiến thức tốt về tin học.
1.2.7. Phương pháp lọc Kalman
1.2.7.1. Giới thiệu chung
Năm 1960 R.E. Kalman đã công bố một bài báo nổi tiếng giới thiệu một giải
pháp mới (áp dụng thuật toán học đại số đệ quy) để giải quyết vấn đề - lọc nhiễu
(hay sai số) của các dữ liệu tuyến tính và rời rạc nhằm đưa ra các kết quả (hay thông
tin) chính xác nhất; Giải pháp đó được gọi là bộ lọc Kalman và phương pháp đó
mang tên phương pháp lọc Kalman [121]. Kể từ đó, do có những ưu điểm lớn trong
tính toán, phương pháp lọc Kalman ngày càng được nghiên cứu và ứng dụng, nhiều
vào thực tế, đặc biệt là trong hệ thống định vị và dẫn đường.
Trong lĩnh vực hóa học, phương pháp lọc Kalman cũng đã được nghiên cứu và
ứng dụng để xác định đồng thời các cấu tử (hay các chất phân tích) trong hỗn hợp
của chúng như: xác định đồng thời các nguyên tố đất hiếm, các hoạt chất trong các
mẫu dược phẩm đa thành phần… và đặc biệt là xác định đồng thời các chất trong
các hỗn hợp khó phân tích – các hỗn hợp chứa các chất có phổ hấp thụ quang xen
phủ nhau [27], [101], [126].

23
Cần thấy rằng, trong phương pháp phân tích bất kỳ nói chung và phương pháp
phân tích trắc quang nói riêng, kết quả cuối cùng (nồng độ chất phân tích trong
mẫu) luôn mắc sai số (còn gọi là nhiễu), gây ra do nhiều nguyên nhân khác nhau,
nhưng nói chung, có thể chia thành 2 loại sai số (nhiễu) chính – nhiễu đo và nhiễu
hệ thống [8], [14, 22]:
- Nhiễu đo: Nhiễu đo là sai số của phép đo độ hấp thụ quang; Sai số này gây ra do
thiết bị đo có thể bị nhiễu nhiệt, nhiễu điện, nhiễu từ, do sai lệch về đơn sắc của bức xạ
tới, do độ sai lệch về cường độ bức xạ tới… Sai số này có thể là sai số ngẫu nhiên, hoặc
sai số hệ thống (sai số hệ thống có thể gây ra do thiết bị đo bị trôi dạt về một phía),
hoặc sai số thô (gây ra do sốc nhiệt, sốc điện, sốc từ); Sai số thô sẽ cho ra các giá trị đo
nằm ngoài phân bố chuẩn (còn được gọi là các giá trị nằm ngoài/outlier);
- Nhiếu hệ thống: Nhiễu hệ thống là nhiễu gây ra do hệ thống phân tích, dẫn
đến sai số nồng độ; Nhiễu này có thể do đóng góp của nhiều nguyên nhân khác
nhau như: sai số pha nồng độ dung dịch chuẩn (gây ra do độ tinh khiết của hóa chất,
độ chính xác của dụng cụ thủy tinh đo lường, độ chính xác của cân phân tích…);
Sai số của phép hồi quy tuyến tính (khi định lượng bằng phương pháp đường chuẩn
hoặc thêm chuẩn); Sai số do tính toán (chẳng hạn, do làm tròn sai hoặc cách lấy số
con số có nghĩa không đúng); Sai số do bản thân phương pháp trắc quang gây ra
(chẳng hạn, do tính chất cộng tính của độ hấp thụ quang khác nhau, tương tác của
các cấu tử trong hệ, ảnh hưởng của môi trường nền/matrix…) [14], [15]..
Cả 2 nguồn nhiễu (sai số) nói trên được gọi chung nhiễu (noise). Ngoài ra, khi
phân tích các mẫu thực tế, việc lấy mẫu kém đại diện cũng gây ra sai số lấy mẫu và
do vậy, cũng đóng góp vào sai số của kết quả phân tích cuối cùng. Trong thực tế,
việc lấy mẫu luôn được xem là đại diện hay nói cách khác, có thể bỏ qua sai số lấy
mẫu, tức là chấp nhận nó chỉ mắc sai số ngẫu nhiên và chấp nhận được. Như vậy,
thách thức lớn đối với các phương pháp phân tích là phải có giải pháp phù hợp để
loại bỏ tối đa sai số (nhiễu) với chấp nhận rằng, bỏ qua sai số lấy mẫu.
Giả sử dung dịch mẫu chứa một chất phân tích và tiến hành đo ở k bước sóng,
sẽ thu được bộ dữ liệu tín hiệu đo X (độ hấp thụ quang ở k bước sóng), bao gồm cả
tín hiệu đúng (hay tín hiệu sạch) S và tín hiệu nhiễu N. Nói cách khác, X bao hàm
cả S và N ở k bước sóng [27], [123], [124]:

24
X(k) = S(k) + N(k) (1.22)
Do tín hiệu nhiễu N (ở bước sóng k bất kỳ) là một đại lượng ngẫu nhiên, nên
luôn tuân theo phân bố chuẩn có giá trị thực (hay trung bình toàn bộ) là 0:
∑ 𝑁(𝑘)𝑚
𝑘=1
𝑚= 0 khi m đủ lớn (1.23)
Như vậy, để loại bỏ N, có thể lấy tổng của X trên một cửa sổ có kích thước m:
∑ 𝑋(𝑘) = ∑ 𝑆(𝑘)𝑚𝑘=1
𝑚𝑘=1 (1.24)
Các phương pháp chemometric-trắc quang nêu trên như: phương pháp phổ đạo
hàm, phương pháp bình phương tối thiểu, phương pháp phân tích cấu tử chính…
thường chỉ loại bỏ được nhiễu hệ thống (hay sai số nồng độ), mà chưa loại bỏ được
nhiễu đo (sai số đo độ hấp thụ quang), vì các phương pháp đó chấp nhận cả các độ
hấp thụ quang mắc sai số lớn để tính nồng độ chất phân tích, rồi từ đó, áp dụng các
thuật toán để tính ra nồng độ cuối cùng sao cho sai số nồng độ là nhỏ nhất.
Trong khi đó, phương pháp lọc Kalman cho phép loại bỏ cả 2 loại nhiễu nêu
trên và do vậy, nó cho ra các kết quả tính toán (nồng độ chất phân tích) tin cậy hơn
hay mắc sai số nhỏ hơn, hay nói cách khác, gần với giá trị thực của nó hơn. Do vậy,
có thể cho rằng, so với các phương pháp nêu trên, phương pháp lọc Kalman có
nhiều lợi thế hơn.
Trong phương pháp lọc Kalman, người ta dùng bộ lọc Kalman, cho phép loại
bỏ dần các giá trị mắc sai số lớn (hay nhiễu lớn) để thu được các giá trị mắc sai số
nhỏ nhất (hay nhiễu nhỏ nhất). Như vậy, bộ lọc Kalman là một tập hợp các phương
trình toán học để tính toán một cách hiệu quả nhằm ước lượng được trạng thái nồng
độ chất phân tích trong hệ nghiên cứu sao cho nồng độ đó có phương sai (hay sai số)
nhỏ nhất. Rõ ràng, nhiệm vụ của bộ lọc Kalman nhằm loại bỏ nhiễu trong phương
pháp trắc quang và ở mỗi bước sóng, bộ lọc Kalman đã thực hiện một bước lọc nhiễu.
Bộ lọc nhằm loại bỏ nhiễu trong phương pháp trắc quang theo nguyên tắc: từ trang
thái khởi tạo ban đầu (gồm nồng độ chất phân tích và phương sai kèm theo ở bước
sóng đầu tiên), sẽ ước lượng được trạng thái tiếp theo (nồng độ mới có phương sai
nhỏ hơn ở bước sóng tiếp theo) và cứ tiếp tục như vậy, cho đến bước sóng cuối cùng
của bộ dữ liệu phổ (độ hấp thụ quang ở k bước sóng), sẽ thu được nồng độ chất phân
tích có phương sai (hay sai số) nhỏ nhất.

25
1.2.7.2. Cơ sở lý thuyết của phương pháp lọc Kalman
Trong phương pháp trắc quang, tín hiệu đo A (độ hấp thụ quang) tuân theo
định luật Beer: A = ε.l.C. Giả sử hỗn hợp (dung dịch phân tích) chứa 2 chất (1 và 2)
được đo A ở bước sóng λk, sẽ có (chấp nhận chiều dài cuvet l không đổi) [14]. [15]:
Độ hấp thụ quang đối với chất 1: A1 = ε1(k)C1
Độ hấp thụ quang đối với chất 2: A2 = ε2(k)C2 (2.1)
Suy ra độ hấp thụ quang của hệ: A = A1 + A2 = ε1(k)C1 + ε2(k)C2
Hay biểu diễn ở dạng ma trận, sẽ có A = ( )1 2( )k k 1
2
C
C
(2.2)
Một cách tổng quát, đối với hệ có n cấu tử (chất phân tích), ở bước sóng k, có
thể viết: ma trận độ hấp thụ quang A(k) là tích của 2 ma trận – ma trận độ hấp thụ
phân tử (k) và ma trận nồng độ C(k):
𝐴(𝑘) = (𝑘)𝐶(𝑘) (2.3)
Trong đó,
1( )
2( )( )
( )
...
k
kk
n k
C
CC
C
æ ö÷ç ÷ç ÷ç ÷ç ÷ç ÷ç ÷= ç ÷÷ç ÷ç ÷ç ÷ç ÷ç ÷çè ø
là vector nồng độ của các cấu tử, với Ci(k) (với i = 1 – n) là
nồng độ của cấu tử thứ i tại bước sóng thứ k . Vector này được gọi là vector trạng
thái nồng độ.
11 1
( )
1
...
... ... ...
...
n
k
m mn
e e
e
e e
æ ö÷ç ÷ç ÷ç ÷ç= ÷ç ÷ç ÷ç ÷÷çè ø
là ma trận các giá trị hệ số hấp thụ quang.
Do phương pháp trắc quang luôn có nhiễu, nên một cách tổng quát, có thể viết
ma trận độ hấp thụ quang A(k) ở bước sóng k như ở (2.4):
( ) ( ) ( ) ( )k k k kA C ve= + (2.4)
Trong đó, v(k) là nhiễu của độ hấp thụ quang ở bước sóng k.

26
Khi đo độ hấp thụ quang ở số bước sóng đủ lớn (m bước sóng hay k = 1 – m),
thì nhiễu đo tuân theo phân bố chuẩn có giá trị thực (hay trung bình số học với m
lớn) là 0 [12], [25]:
( )
1 0
M
k
k
v
M= =
å
(2.5)
Như vậy, để lọc bỏ nhiễu, đơn giản nhất là lấy tổng của các độ hấp thụ quang
đo được ở số bước sóng đủ lớn (hay được gọi độ hấp thụ quang trong cửa sổ có kích
thước m). Khi đó, độ hấp thụ quang ở m bước sóng được biểu diễn ở dạng ma trận
như sau:
( ) ( ) ( )
1 1
M M
k k k
k k
A Ce
= =
»å å (2.6)
Trong phương trình (2.6), sử dụng dấu xấp xỉ với hàm ý rằng, thực tế không
thể loại bỏ hoàn toàn nhiễu, mà chỉ có thể giảm nhiễu để thu được độ hấp thụ quang
mắc ít nhiễu nhất (hay được coi là sạch nhiễu), tức là giảm tối đa ma trận nhiễu (k).
Giả sử trường hợp đơn giản nhất là mẫu chứa một chất phân tích (có nồng độ
biết trước 10 µg/L) và tiến hành xác định nồng độ đó lặp lại n lần: Lần thứ nhất, xác
định được kết quả C1 là 9,9 µg/L, lần thứ 2 được kết quả C2 là 10,3 µg/L. Vấn đề đặt
ra là lựa chọn kết quả nào cho nồng độ chất phân tích trong mẫu? Thông thường,
người ta chọn giá trị trung bình số học của 2 kết quả đó, bằng 10,1 µg/L. Song, nếu
tiếp tục xác định nồng độ chất trong mẫu thêm 2 lần nữa, có thể sẽ được kết quả
trùng hoặc khác so với 2 kết quả trên. Như vậy, nhất thiết phải xác định xem, phần
đóng góp của mỗi kết quả vào kết quả trung bình cuối cùng là bao nhiêu. Để giải
quyết điều đó, người ta đưa ra đại lượng Hệ số hiệu chỉnh a (corection factor; thể
hiện phần đóng góp của mỗi kết quả Ci với i = 1 – 2, vào kết quả cuối cùng C và
luôn ≤ 1):
( )1 21C C Ca a= + -
(2.7)

27
Rõ ràng, trong bất kỳ trường hợp nào (khi xác định nồng độ chất trong mẫu),
cũng cần tìm a sao cho thể hiện đúng đặc trưng của quá trình đo và cho phép loại
bỏ được nhiễu đo (nhiễu đo độ hấp thụ quang), để từ đó, xác định được giá trị nồng
độ gần nhất với giá trị thực của nó. Điều này sẽ được đề cập chi tiết hơn khi mô tả
các đại lượng và phương trình toán của phương pháp lọc Kalman được đề cập sau.
Nguyên tắc tính toán (hay hoạt động) của bộ lọc Kalman [101], [110],
[121], [126]:
Để giảm nhiễu trong phương pháp chemometric-trắc quang sử dụng thuật toán
lọc Kalman, người ta dùng bộ lọc Kalman và nguyên tắc tính toán của bộ lọc
Kalman được thực hiện theo trình tự sau:
- Từ dữ liệu phổ đo được đối với mỗi cấu tử (giá trị độ hấp thụ quang A ở k
bước sóng lựa chọn của các dung dịch chuẩn của các cấu tử), tính toán được hệ số
hấp thụ phân tử của mỗi cấu tử (tính theo công thức của định luật Beer);
- Tiếp theo, trước hết cần đưa ra giá trị khởi tạo ban đầu (giá trị đầu vào) của
nồng độ mỗi cấu tử (Co) trong hệ và phương sai ban đầu Po (phương sai của các
nồng độ Co) cho bộ lọc Kalman. Việc lựa chọn giá trị Co và Po ban đầu tùy thuộc
vào quan điểm của người nghiên cứu, có thể là lựa chọn giá trị ngẫu nhiên hoặc giá
trị lựa chọn theo chủ ý của người nghiên cứu. Từ Co ban đầu đó và các giá trị biết
trước của mỗi cấu tử, sẽ tính được độ hấp thụ quang lý thuyết ở bước sóng đầu tiên
(k = 1) là A1lt (tính theo công thức của định luật Beer). Từ dữ liệu phổ đo được đối
với hỗn hợp các cấu tử (độ hấp thụ quang A ở k bước sóng của dung dịch hỗn hợp
các cấu tử), sẽ xác định được độ hấp thụ quang ở bước sóng đầu tiên A1. Từ đó, tính
toán được Độ lệch (hay sai số) giữa A1lt và A1. Độ lệch này là cơ sở để tiếp tục điều
chỉnh (hay cập nhật) giá trị khởi tạo ban đầu để tính toán được giá trị mới C1 (giá trị
dự báo) của mỗi cấu tử trong hệ và phương sai mới P1 (phương sai dự báo) ở bước
sóng tiếp theo (bước sóng thứ 2).
- Tiếp theo, cập nhật giá trị đầu vào cho bộ lọc (hay giá trị cập nhật) về nồng
độ và phương sai để tính toán ở bước sóng tiếp theo (bước sóng thứ 2): Nồng độ
được chấp nhận bằng Co ước lượng ban đầu cộng với phần sai số (tính toán từ độ
lệch về độ hấp thụ quang ở trên) và phương sai bằng phương sai ước lượng ban đầu

28
Po cộng với phần nhiễu/sai số (gây ra do độ lệch trên). Như vậy, từ giá trị nồng độ
và phương sai cập nhật, theo cách tương tự trên, sẽ tính toán được giá trị mới C2
(giá trị dự báo) và phương sai P2 (phương sai dự báo). Một cách tổng quát, ở một
bước sóng k bất kỳ, giá trị đầu vào (hay cập nhật) cho bộ lọc được gọi là giá trị ước
lượng (estimated, viết tắt là est); giá trị tính toán được tiếp theo, được gọi là giá trị
dự báo (predicted, viết tắt là pri).
- Phép tính toán tương tự trên được lặp đi lặp lại cho đến bước sóng cuối cùng
và lúc này, sẽ thu được giá trị nồng độ của mỗi cấu tử trong hệ (C) gần nhất với giá
thực của nó và phương sai P nhỏ nhất. Đến đây bộ lọc dừng lại (hay phép tính theo
phương pháp lọc Kalman kết thúc) và cho ra kết quả cuối cùng là C và P ứng với
mỗi cấu tử trong hệ.
Các đại lượng và phương trình toán trong thuật toán lọc Kalman [101],
[110], [121], [126]:
Trong thuật toán của phương pháp lọc Kalmnan, người ta sử dụng một số đại
lượng và các phương trình toán học mô tả như sau:
- Giả sử 𝐶𝑝𝑟𝑖 (𝑘) và 𝐶𝑒𝑠𝑡 (𝑘) lần lượt là vector dự báo và vector ước lượng của
trạng thái nồng độ C tại bước sóng k. Giá trị ước lượng là “đầu vào” (giá trị này
được cập nhật liên tục ở từng bước sóng dựa vào Độ lệch giữa giá trị độ hấp thụ
quang tính theo lý thuyết và giá trị đo được bằng thực nghiệm), để tính toán giá trị
dự báo (tính theo mô hình toán của bộ lọc Kalman) và cứ liên tục như vậy cho đến
bước sóng cuối cùng.
- Như vậy, một cách tổng quát, bộ lọc kalman gồm 2 vector: vector ước lượng
và vector dự báo, trong đó, vector ước lượng ở bước sóng k bất kỳ (làm đầu vào cho
phép lọc tiếp theo) sẽ bằng vector dự báo ở bước sóng trước đó (đầu ra của mỗi
phép lọc). Nói cách khác, ma trận dự báo của trạng thái nồng độ ở bước sóng (k – 1)
(đầu ra của bộ lọc) là Cpri(k-1) bằng ma trận ước lượng của trạng thái nồng độ ở bước
sóng k (đầu vào cho phép lọc tiếp theo) là Cest(k):
( ) ( 1)est k pri kC C -= (2.10)

29
- Từ đó, có thể thấy rằng, ở bước sóng k bất kỳ, có 2 nguồn sai số: ma trận sai
số của giá trị dự báo epri(k) và ma trận sai số của giá trị ước lượng eest(k) (lưu ý rằng, ma
trận sai số của giá trị ước lượng đã được cập nhật, tức là đã tính đến độ lệch giữa 2
giá trị độ hấp thụ quang – giá trị lý thuyết và giá trị đo được bằng thực nghiệm):
( ) ( ) ( )pri k k pri ke C C= - và
( ) ( ) ( )est k k est ke C C= - (2.11)
Trong đó, C(k) là ma trận trạng thái nồng độ thực của các cấu tử ở bước sóng k.
Từ đó, tính toán được ma trận hiệp phương sai của 2 nguồn sai số trên:
( )( ) ( ) ( ), T
pri k pri k pri kP E e e=
( )( ) ( ) ( ), T
est k est k est kP E e e= (2.12)
Trong đó, E là ký hiệu của ma trận sai số của hiệp phương sai ; T là ký hiệu
của ma trận chuyển vị.
- Cần thấy rằng, trong phương pháp lọc Kalman, các giá trị ước lượng của
nồng độ ở bước sóng k bất kỳ đều đã được cập nhật, tức là đã tính đến phần hiệu
chỉnh sai số, gây ra do độ lệch giữa giá trị độ hấp thụ quang lý thuyết và thực
nghiệm, tức là:
( )( ) ( ) ( ) ( ) ( ) ( )est k pri k k k k pri kC C K A Ce= + -
(2.13)
Trong đó, ma trận ( )K k đóng vai trò như Hệ số hiệu chỉnh a đề cập ở trên;
Ma trận giá trị ước lượng trạng thái nồng độ Cest(k) sẽ bằng ma trận giá trị dự báo
nồng độ Cpri(k) cộng thêm phần hiệu chỉnh sai số, mà có tính đến Độ lệch giữa ma
trận độ hấp thụ quang thực nghiệm A(k) và ma trận độ hấp thụ quang lý thuyết = tích
ma trận các hệ số hấp thụ phân tử (k) và ma trận giá trị nồng độ dự báo Cpri(k).
Trong mô hình gốc của phương pháp lọc Kalman, người ta gọi độ lệch giữa
ma trận độ hấp thụ quang thực nghiệm và ma trận độ hấp thụ quang lý thuyết là
Mức cập nhật (Inovation, viết tắt là INV), tức là: ( )( ) ( ) ( ) ( )k k k pri kINV A Ce= - , còn
ma trận K(k) được gọi là Hệ số hiệu chỉnh Kalman (Kalman gain).

30
- Vấn đề đặt ra là cần tính Hệ số hiệu chỉnh Kalman ở bước sóng k bất kỳ sao
cho phù hợp. Về mặt toán học, Hệ số hiệu chinh Kanmal phù hợp (hay tối ưu) là hệ
số thỏa mãn điều kiện - hiệp phương sai của sai số ước lượng là nhỏ nhất, nghĩa là:
( )( ) ( ) ( ), minT
est k est k est kP E e e= ® (2.14)
Để tìm cực trị của ma trận hiệp phương sai Pest(k), cần thay ma trận sai số ước
lượng là ( )( ) ( ) ( ) ( ) ( )est k k k k pri ke K A Ce= - vào phương trình (2.14), rồi lấy đạo hàm
theo biến ( )K k , sẽ tìm được ma trận Hệ số hiệu chỉnh Kalman K(k), mà tại đó hàm
đạt cực tiểu:
( )
1
( ) ( ) ( ) ( ) ( ) ( ) ( )T T
k pri k k k pri k k kK P P Re e e-
= + (2.15)
Giải pháp giảm nhiễu nhờ bộ lọc Kalman [49], [101], [110], [120], [124]:
Với các đại lượng và phương trình của phương lọc Kalman nêu trên, giải pháp
giảm nhiễu nhờ bộ lọc Kalman đượcthực hiện như sau:
Giả sử mẫu chứa đồng thời nhiều cấu tử và khi đo, đã thu được độ hấp thụ
quang (các giá trị rời rạc) ở k bước sóng và độ hấp thụ quang đo được luôn tuân
theo theo định luật Beer. Lúc này, có thể viết 2 phương trình trạng thái như sau:
i) Phương trình trạng thái nồng độ thể hiện: Ma trận nồng độ ở bước sóng k
bất kỳ C(k) bằng ma trận nồng độ ở bước sóng trước đó C(k-1) cộng với ma trận
nhiễu w(k):
( ) ( 1) ( )k k kC C w (2.8)
Vector w trong phương trình (2.8) thể hiện nhiễu hệ thống (còn gọi là nhiễu
trắng) mô tả nhiễu của toàn bộ hệ thống (tức là bao hàm các loại nhiễu hay các loại
sai số gây ra do hệ thống phân tích) ở bước sóng thứ k ( k );
ii) Phương trình trạng thái độ hấp thụ quang: Ma trận độ hấp thụ quang ở bước
sóng k bất kỳ A(k) bằng tổng của ma trận tích (k)C(k) và ma trận nhiễu v(k):
( ) ( ) ( ) ( )k k k kA C ve= + (2.9)
Trong đó, vector v thể hiện nhiễu đo (nhiễu của phép đo độ hấp thụ quang).

31
Cần thấy rằng, vector v và w là các biến ngẫu nhiên, độc lập nhau và và do
vậy, chúng luôn tuân theo phân bố chuẩn (phân bố Gauss). Như thế, cả v và w đều
có trung bình toàn bộ (hay giá trị thực) là 0 và ma trận hiệp phương sai tương ứng
là Q(k) và R(k) ở bước sóng thứ k (khái niệm hiệp phương sai ở đây được hiểu là
phương sai của 2 hoặc nhiều biến đồng thời, do trong hệ chứa 2 hoặc hơn 2 chất
phân tích):
( )( ) ( )0,k kw N Q: , ( )( ) ( )0,k kv N R:
Mặt khác, do trạng thái nồng độ (nồng độ chất trong hệ) là không thay đổi ở
mọi bước sóng, nên về nguyên tắc, hiệp phương sai Q(k) ít biến động. Còn R(k) là
phương sai thể hiện nhiễu của phép đo độ hấp thụ quang ở bước sóng thứ k bất kỳ.
Như đã đề cập ở trên, nhiễu trong phương pháp phân tích bao gồm nhiễu hệ
thống, thể hiện qua hiệp phương sai Q(k) và nhiễu đo, thể hiện qua hiệp phương sai
R(k). Song, đối với phương pháp lọc Kalman, khi đã biết trước số cấu tử trong hệ và
cho rằng, trạng thái nồng độ ít thay đổi theo bước sóng, thì không cần hiệu chỉnh
khi tính hiệp phương sai Q(k).
Mặt khác, mức cập nhật ( )( ) ( ) ( ) ( )k k k pri kINV A Ce= - thể hiện độ lệch giữa độ
hấp thụ quang thực nghiệm (đo được) và độ hấp thụ quang lý thuyết (tính theo định
luật Beer) và như vậy, mức cập nhật đó nhân với hệ số hiệu chỉnh Kalman thể hiện
mức độ hiệu chỉnh sai số (hay mức lọc) của bộ lọc Kalman. Do mức cập nhật INV(k)
là một đại lượng ngẫu nhiên, nên về nguyên tắc, hiệu quả lọc sẽ tốt nhất khi mức
cập nhật ( )kINV tuân theo phân bố chuẩn có giá trị thực (hay trung bình toàn bộ)
bằng 0, tức là: ( )
1
10
n
k
k
INVn
=
®å . Khi điều này không thỏa mãn, hiệu quả của bộ
lọc sẽ giảm đi, do khi đó, mức cập nhật INV(k) là biến tương quan với các tham số
ước lượng và do vậy, dẫn đến sai số lớn hơn khi tính theo mô hình của phương pháp
lọc Kalman. Như thế, để đạt được hiệu quả lọc tốt nhất, cần tính toán hiệp phương
sai R(k) sao cho phù hợp. Trong thuật toán lọc Kalman, để thỏa mãn điều kiện
( )
1
10
n
k
k
INVn
=
®å , người ta chọn ra những vùng bước sóng thỏa mãn điều kiện đó

32
(được gọi là các cửa sổ làm mịn/smooth windows) để tính toán ma trận R(k). Ma trận
R(k) được tính toán từ mức cập nhật INV(k-j) (với j là số thứ tự bước sóng trong m
bước sóng của cửa sổ làm mịn) và được hiệu chỉnh bằng cách trừ đi tích của ma trận
hệ số hấp thụ phân tử (k) và hiệp phương sai Ppri(k) như sau [49], [101], [110]:
( ) ( ) ( ) ( ) ( ) ( )
1
1.
mT
k k j k j k pri k k
j
R INV INV Pm
e e- -
=
= -å (2.22)
1.2.7.3. Ưu điểm và nhược điểm
Tuy có nhiều ưu điểm (lọc được tối đa nhiễu hay sai số), nhưng phương pháp
lọc Kalman có nhược điểm là phải lựa chọn được giá trị khởi tạo phù hợp, tức là phải
đưa ra gia trị khởi tạo ban đầu về nồng độ mỗi chất phân tích trong hệ và phương sai
(hay sai số) kèm theo, để cung cấp dữ liệu “đầu vào” cho bộ lọc bắt đầu hoạt động (hay
tính toán). Nếu giá trị khởi tạo ban đầu (nồng độ và phương sai) không phù hợp, kết
quả cuối cùng sẽ mắc sai số lớn [49], [101], [110]. Cho đến nay, tuy đã có nhiều nghiên
cứu áp dụng phương pháp lọc Kalman, nhưng vẫn chưa có giải pháp thống nhất về việc
lựa chọn giá trị khởi tạo cho bộ lọc Kalman và do vậy, việc lựa chọn giá trị khởi tạo
cho bộ lọc Kalman vẫn là một thách thức đối với các nhà hóa học phân tích.
Bang 1.1. Một số nghiên cứu ở Việt Nam và thế giới sử dụng thuật toán lọc Kalman
ứng dụng vào phân tích
Phương pháp xác định Đối tượng phân tích Tác gia, năm, nguồn
[TLTK]
Sắc kí lớp mỏng kết hợp
thuật toán lọc Kalman
Hợp chất PAH’s
(polyaromatic hydrocarbons)
David D. Gerow và
cộng sự- 1988
[49]
Trắc quang UV-Vis kết
hợp thuật toán lọc
Kalman
Phenol, resorcinol
Xle Yu-Long và cộng
sự - 1992
[125]
Trắc quang UV- Vis kết
hợp thuật toán lọc
Kalman
Pr3+ (tạo phức với EDTA)
Rutan Sarah C và cộng
sự - 1984
[101]
Trắc quang UV- Vis kết
hợp thuật toán lọc
Kalman
Các nguyên tố đất hiếm, các
vitamin nhóm B, hỗn hợp
paracetamol và ibuprofen
Mai Xuân Trường-
2008
[27]

33
Như vậy, trên thế giới đã có một số nghiên cứu áp dụng phương pháp lọc
Kalman vào chemometric – trắc quang để xác định đồng thời hỗn hợp 2 hoặc 3 chất
trong dược phẩm [50], [121], song các nghiên cứu đó hoặc không đưa ra cách lựa
chọn giá trị khởi tạo phù hợp hoặc không đề cập đến các giá trị khởi tạo và do vậy,
rất khó áp dụng cho các phòng thí nghiệm phân tích. Ở nước ta, Mai Xuân Trường
[27] đã nghiên cứu áp dụng phương pháp lọc Kalman để xác định đồng thời các
vitamin trong dược phẩm, các nguyên tố đất hiếm…nhưng do tác giả cũng không
giới thiệu về cách chọn giá trị khởi tạo và do vậy, đã hạn chế khả năng áp dụng
phương pháp đề xuất vào thực tế. Nói chung, mặc dù phương pháp lọc Kalman có
nhiều ưu điểm khi áp dụng vào phương pháp chemometric - trắc quang để xác định
đồng thời các hợp chất, đặc biệt là các chất có phổ hấp thụ phân tử xen phủ nhau,
nhưng cho đến nay, các nghiên cứu áp dụng nó vẫn rất hạn chế.
1.3. Tổng quan về dược phẩm đa thành phần và các hoạt chất nghiên cứu
1.3.1. Giới thiệu về các dược phẩm đa thành phần
Thuốc luôn là một nhu cầu không thể thiếu được trong đời sống con người.
Cũng như mọi ngành khác, thuốc đòi hỏi một nền sản xuất ngày càng cao và phát
triển theo sự phát triển và tiến bộ của loài người.
Trải qua mấy ngàn năm lịch sử phát triển, tiến bộ của loài người, cùng với
những cuộc Cách mạng khoa học kỹ thuật cũng như nhu cầu ngày càng gia tăng của
con người về phòng và chữa bệnh, kỹ thuật sản xuất thuốc cũng ngày càng phát
triển với những dạng thuốc tinh tế hơn, phức tạp hơn. Cụ thể là các dạng thuốc viên,
viên nén, viên bao, viên nang, thuốc tác dụng kéo dài, thuốc tiêm, dịch truyền,…[9]
Ngày nay, thị trường thuốc đang phát triển nhanh cả về sản xuất và kinh
doanh. Các loại thuốc ngoại nhập được cấp số đăng ký lưu hành ngày càng nhiều.
Theo thống kê của Cục quản lý Dược Việt Nam vào năm 2005, có khoảng 6000
mặt hành thuốc trong nước và khoảng 4000 thuốc ngoại nhập (ước tính trên 800
hoạt chất). Chỉ 10 năm sau, năm 2015, số lượng thuốc đã tăng lên chóng mặt với
trên 25.000 mặt hàng (khoảng 1.500 hoạt chất). Trong số đó, các thuốc đa thành
phần đang trở nên phổ biến và chiếm một ty lệ khá cao. Các nhà sản xuất thường
phối hợp nhiều dược chất trong một công thức bào chế, đặc biệt là các thuốc hạ

34
nhiệt, giảm đau, ho, sổ mũi,… với mục đích phối hợp tác dụng dược lý của các
dược chất để tăng hiệu quả điều trị đồng thời làm giảm tác dụng phụ và tiện sử
dụng cho bệnh nhân.
Để phối hợp tác dụng dược lý trong một chế phẩm thuốc sử dụng cho bệnh
nhân, thuốc đa thành phần thường được phối hợp với các hoạt chất trong cùng một
nhóm thuốc như: kháng sinh, vitamin, thuốc hạ nhiệt giảm đau, thuốc sốt rét,
thuốc chống lao,... Cũng có thể kết hợp các hoạt chất của 2, 3 nhóm thuốc như
vitamin với thuốc hạ nhiệt, kháng sinh với thuốc chống viêm,…
Nói chung, trên thế giới và ở Việt Nam, các thuốc có nhiều thành phần đang
được sản xuất ngày càng nhiều.
1.3.2. Telmisartan (TEL), hydrochlorothiazide (HYD)
1.3.2.1. Giới thiệu về TEL [3],[6],[31], [43].
- Telmisartan có tên gọi là: 4’-[(1,4’-dimethyl-2’ propyl [2,6 -bi-1H-
benzimidazol]-1’-yl)-methyl] [1,1’-biphenyl]-2-carboxylic acid.
- Công thức hóa học: C33H30N4O2
- Công thức cấu tạo:
- Tính chất vật lý: là chất bột kết tinh màu trắng hoặc hơi vàng, thực tế không
tan trong nước, hơi tan trong metanol, ít tan trong metylen clorua. Tan tốt trong
dung dịch kiềm.
- Dược lý và cơ chế tác dụng: Telmisartan là một chất đối kháng đặc hiệu của
thụ thể angiotensin II ở cơ trơn thành mạch và tuyến thượng thận. Angiotensin II là
chất gây co mạch, kích thích vỏ thượng thận tổng hợp và giải phóng aldosteron,
kích thích tim. Aldosteron làm giảm bài tiết natri và tăng bài tiết kali ở thận.
Telmisartan ngăn cản chủ yếu angiotensin II vào thụ thể ở cơ trơn mạch máu và

35
tuyến thượng thận, gây giãn mạch và giảm tác dụng của aldosteron. Ở người, liều
80 mg telmisartan ức chế hầu như hoàn toàn tăng huyết áp do angiotensin II. Thông
thường, huyết áp động mạch giảm tối đa đạt được 4 – 8 tuần sau khi bắt đầu điều trị.
Ở người tăng huyết áp, telmisartan làm giảm huyết áp tâm thu và tâm trương mà
không thay đổi tần số tim. Tác dụng chống tăng huyết áp của telmisartan cũng
tương đương với các loại thuốc chống tăng huyết áp loại khác.
1.3.2.2. Giới thiệu về hydrochlorothiazide (HYD)[3], [4], [6], [31], [43], [63],
[104], [111].
- Hydrochlorothiazide có tên gọi là: 2H-1,2,4-benzothiadiazine-7-sulfonamide,
6-cloro-3,4-dihydro-1,1-dioxide.
- Công thức hóa học: C7H8ClN3O4S2
- Công thức cấu tạo:
- Tính chất vật lý: là chất bột kết tinh màu trắng, rất ít tan trong nước, hơi tan
trong ethanol 96%, tan trong aceton, tan trong dung dịch kiềm loãng.
- Đặc tính phổ hấp thụ UV: dung dịch HYD trong methanol và HCl có cực đại
hấp thụ tại 226 nm, 271 nm và 317 nm.
- Dược lực học
+ HYD và các thuốc lợi tiểu thiazid làm tăng bài tiết natri clorid và
nước kèm theo do cơ chế ức chế tái hấp thu các ion natri và clorid ở ống lượn xa.
+ HYD có tác dụng hạ huyết áp, trước tiên có lẽ do giảm thể tích huyết
tương và dịch ngoại bào liên quan đến sự bài niệu natri. Tác dụng hạ huyết áp của
HYD thể hiện chậm sau 1-2 tuần, còn tác dụng lợi tiểu xảy ra nhanh, có thể thấy
ngay sau vài giờ.
1.3.2.3. Một số phương pháp định lượng đồng thời TEL và HYD
Một số phương pháp định lượng đồng thời TEL và HYD được liệt kê ở bảng
1.2

36
Bang 1.2. Một số nghiên cứu xác định TEL và HYD
Phương pháp Khoảng tuyến
tính (µg/mL) R2
LOD
(µg/mL)
LOQ
(µg/mL) RSD (%)
Độ thu hồi
Rev (%)
Tác giả, năm, nguồn
[TLTK]
Vierordt 2-20 μg/ml 0,999 - 0,226 (µg/mL) (TEL); 0,125 (µg/mL) (HYD)
0,069 – 0,483 (TEL); 0,060 – 0,176 (HYD)
98,22 – 102,66 (TEL); 96,19 -101,59 (HYD)
Rekha Gangola và cộng sự-2011 [64]
Vierord, ty lệ độ hấp thụ, đạo hàm bậc nhất
4 - 24 μg/ml và 2 - 14 μg/ml cho TEL và HYD
- - - ± 0,115 đến ± 0,519
99,95% - 100,087 %.
Chinmaya C Behera, và cộng sự – 2014 [42]
Vierordt 4 - 24 µg/ml cho TELvà 2 - 8 µg/ml cho HYD
0,997 - 0,998
0,431 mg/mL (TEL); 0,130 mg/mL (HYD)
0,260 mg/mL (TEL); 0,789 mg/mL (HYD)
0,437 % (TEL); 0,123 % (HYD)
99,67 (TEL); 99,53 (HYD)
Manish Kvà cộng sự– 2014 [87]
Ty lệ độ hấp thụ (phương pháp A) và quang phổ đạo hàm (phương pháp B)
5 - 25μg/mL
0,996 (TEL) và
0,994 (HYD)
- - 0,592 (TEL) và 0,859 (HYD)
98,75 % (TEL) và 96,8 % (HYD)
Agarkar A.R và cộng sự – 2013 [30]
Phổ đạo hàm giao điểm không
2 - 40 μg/mL (TEL) và 2 - 20 μg/mL (HYD).
-
0,5945 μg/ml (TEL) và 0,6633 μg/ml (HYD)
1,801 μg/ml (TEL) và 2,010 μg/ml (HYD)
- 98,18 - 99,67 % (TEL) và 98,148 - 99,23% (HYD)
Piusha Shakya và cộng sự- 2015 [108]
PLS 1 - 5 mg/L (TEL); 0.5 – 2,5 mg/mL (HYD)
0,9987 (TEL); 0,9956 (HYD)
- - - 100,14 (TEL) ; 98,32 (HYD)
Lakshmi KS và cộng sự – 2010 [84]

37
1.3.3. Paracetamol (PAR)và caffeine (CAF)
1.3.3.1.Giới thiệu chung về paracetamol
Paracetamol hay acetaminophen (tên được chấp nhận tại Hoa Kỳ) là thuốc có
tác dụng hạ sốt và giảm đau, tuy nhiên không như aspirin nó không hoặc ít có tác
dụng chống viêm. So với các thuốc chống viêm không steroit (nonsteroidal
antiinflammatory drugs - NSAIDs), paracetamol có rất ít tác dụng phụ với liều điều
trị nên được cung cấp không cần kê đơn ở hầu hết các nước.
- Tên IUPAC hệ thống: N-(4-hydroxyphenyl) acetamide
- Công thức phân tử: C8H9NO2
- Khối lượng phân tử: 151,17 g/mol
- Công thức cấu tạo:
- Tính chất vật lý: Paracetamol là chất bột kết tinh màu trắng, không mùi, vị
đắng nhẹ. Độ tan trong nước: 0,1 ÷0 ,5 g/100ml nước tại 22 0C. Ngoài ra còn có khả
năng tan trong etanol, dung dịch kiềm, dung dịch axit...
- Chế phẩm tan ít trong nước, tan nhiều hơn trong nước sôi, khó tan trong
clorofom, ete, etanol và các dung dịch kiềm... dung dịch bão hòa trong nước có pH
khoảng 5,3 - 5,6; pKa = 9,51.
- Dược lý và cơ chế tác dụng: PAR là thuốc giảm đau hạ sốt hữu hiệu có thể
thay thế aspirin, tuy vậy khác với aspirin, PAR không có hiệu quả điều trị viêm. Với
liều ngang nhau tính theo gam, PAR có tác dụng giảm đau và hạ sốt tương tự như
aspirin. PAR làm giảm thân nhiệt ở người bệnh sốt, nhưng hiếm khi làm giảm thân
nhiệt ở người bình thường.
- Tác dụng: Paracetamol là chất chuyển hóa có hoạt tính của phenacetin, là thuốc
hữu hiệu để điều trị các chứng đau và sốt từ nhẹ đến vừa; tuy vậy, khác với aspirin,
paracetamol không có hiệu quả điều trị viêm. Thuốc có hiệu quả nhất trong việc điều trị
các chứng đau có cường độ thấp và nguồn gốc không phải nội tạng.

38
- Quá liều
+ Nhiễm độc PAR có thể do dùng một liều độc duy nhất hoặc do uống
lặp lại liều lớn hơn PAR hoặc do uống thuốc dài ngày. Hoại tử gan phụ thuộc liều
là tác dụng độc cấp tính nghiêm trọng nhất do quá liều và có thể gây tử vong.
+ Khi bị ngộ độc nặng, ban đầu có thể kích thích hệ thần kinh trung
ương, kích động và mê sảng. Tiếp theo có thể là ức chế hệ thần kinh trung ương,
sững sờ, hạ thân nhiệt, mệt lả, thở nhanh, nông, mạch nhanh, yếu, không đều,
huyết áp thấp và suy tuần hoàn. Cơn co giật ngẹt thở gây tử vong có thể xảy ra.
Thường hôn mê xảy ra trước khi chết đột ngột hoặc sau vài ngày hôn mê.
+ Khi nhiễm độc nặng cần rửa dạ dày trong mọi trường hợp, tốt nhất
trong vòng 4 giờ sau khi uống. Liệu pháp giải độc chính là dùng những hợp chất
sulfhydryl, có lẽ tác động một phần do bổ sung dự trữ glutathion ở gan. N-
acetylcystein có tác dụng khi uống hoặc tiêm tĩnh mạch. Ngoài ra có thể dùng
thuốc tẩy muối hoặc nước chè đặc để làm giảm hấp thu PAR. [5], [10], [33], [117]
1.3.3.2. Giới thiệu chung về caffeine [32]
- Tên quốc tế: Caffeine.
- Một số tên khác: Trimethylxanthine, Coffeine, Theine, Mateine, Guaranine,
Methyltheobromine hay 1,3,7-trimethylxanthine.
- Công thức phân tử: C8H10N4O2 ; - Khối lượng mol phân tử: 194,19 (g/mol).
- Công thức cấu tạo:
- Tên IUPAC: 1,3,7-trimethylxanthine
- Tính chất vật lý:
+ Caffeine ở dưới dạng tinh thể trắng, mịn hay bột kết tinh trắng, hoặc không
màu, không mùi, vị hơi đắng. Nhiệt độ nóng chảy ở khoảng 2340C - 2390C vụn nát ngoài
không khí khô. Khi đun nóng ở 1000C sẽ mất nước và thăng hoa ở khoảng 200 0C.

39
+ Caffeine tan ít trong nước, dễ tan trong nước sôi và clorofom, một
phần trong etanol (ở nhiệt độ bình thường 1 lít nước chỉ hòa tan 20g caffeine,
nhưng 1 lít nước sôi hòa tan tới 700g caffeine). Caffeine tan trong các dung dịch
axit và trong các dung dịch đậm đặc của benzoat hay salicylat kiềm.
- Tính chất hóa học: Caffeine là một chất có tính bazơ yếu, chỉ tạo muối với
các axit mạnh và các muối này không bền, dễ bị phân hủy. Trong môi trường kiềm
caffeine không bền, dễ bị phân hủy thành chất cafeidin không có tác dụng như
caffeine nữa nhưng không độc.
- Công dụng: Caffeine có tác dụng kích thích hoạt động hệ thần kinh trung
ương chọn lọc trên vỏ não, làm tăng khả năng nhận thức, tăng khả năng làm việc trí
óc, làm giảm cảm giác mệt mỏi, buồn ngủ. Thuốc có tác dụng kích thích, liều cao
làm tim đập nhanh, co bóp mạnh, tăng lưu lượng máu qua tim. Thuốc có tác dụng
lợi tiểu nhưng kém theophyllin và theobromin.
- Ảnh hưởng của caffeine: Caffeine khi dùng với liều lượng nhiều sẽ gây ra
các ảnh hưởng như căng thẳng thần kinh, hưng phấn, tăng huyết áp, giãn nở phế
quản, lợi tiểu (từ 300mg/ngày trở lên), kích thích nhu động ruột, mất ngủ. Tổ chức
Y tế Thế giới (WHO) không xếp caffeine vào nhóm các chất gây nghiện. Đến nay
vẫn không có dấu hiệu gì rõ ràng chứng minh caffeine nguy hại đến sức khỏe, ngay
cả những trường hợp sử dụng thường xuyên caffeine trong thời gian dài. Tuy nhiên
việc dùng caffeine nhiều có thể dẫn tới sự phụ thuộc về tâm lý, trong trường hợp
này mùi vị cà phê, khẩu vị người uống và truyền thống cũng đóng một vai trò quan
trọng. Nếu dùng caffeine với liều lượng cao có thể làm tăng nhịp tim và lợi tiểu.
Tuy vậy nếu uống những loại đồ uống chậm giải phóng caffeine như guarana hay
chè đen thì có thể hạn chế được các ảnh hưởng tiêu cực của caffeine cũng như tận
dụng được các tác dụng của nó.
1.3.3.3.Một số phương pháp xác định đồng thời PAR và CAF
Một số phương pháp định lượng đồng thời PAR và CAF được liệt kê ở bảng
1.3.

40
Bang 1.3. Một số nghiên cứu xác định PAR và CAF
Phương pháp Khoảng tuyến tính (µg/mL)
R2 LOD
(µg/mL)
LOQ
(µg/mL) RSD Độ thu hồi Rev (%)
Tác giả, năm, nguồn [TLTK]
Hiệu chỉnh độ hấp thu
1,5– 7,0 (PAR); 0,1 – 3,0 (CAF)
- - - < 2 99,93 – 100,41 Ahmed Ashoura và cộng sự – 2015 [31]
HPLC 10 – 60 0,999 - - - 98 – 102 Khoshayand M.R và cộng sự – 2008 [82]
Trắc quang - - - - - 99,29 – 100,19 Sena Marcelo M. và cộng sự– 2004 [105]
(PCR), (PLS) (ANN)
- - - -
Đối với PAR: 1,56 (PRC); 1,56 (PLS); 0,78 (ANN); Đối với CAF: 1,26 (PRC); 1,27(PLS); 1,15 (ANN).
Đối với PAR: lần lượt là 100.45 (PRC); 100,05 (PLS); 100,92 (ANN); Đối với CAF: 100,21 (PRC); 99,85 (PLS); 100,37 (ANN).
A. Hakan Aktaş và cộng sự– 2014 [32]
Ty số độ hấp thụ
2÷16(PAR); 2÷32(CAF)
0,990 0,998
- - - 95.46(PAR); 98.66% (CAF)
Vijaya Vichare và cộng sự – 2010 [117]
Dấu “-“ là không có thông tin

41
1.3.4. Paracetamol (PAR) và ibuprofen (IB)
1.3.4.1. Giới thiệu chung về Paracetamol (xem mục 1.3.3.1)
1.3.4.2. Giới thiệu chung về Ibuprofen
- Tên quốc tế: Axit- 2-[4-(2-metyl propyl)phenyl] propanoic
- Công thức phân tử: C13H18O2
- Công thức cấu tạo:
- Tính chất vật lý
+ Ibuprofen là chất bột kết tinh màu trắng hoặc tinh thể không màu.
Nhiệt độ nóng chảy: 760C.
+ Tan ít trong nước (<1mg/ml), dễ tan trong axeton, ete, diclometan,
tan trong các dung dịch kiềm và cacbonat kiềm (do nhóm chức axit), tan trong
axit.
- Dược động học
+ Hấp thu: Dược động học của ibuprofen có liên hệ tuyến tính với liều
dùng. Đạt được nồng độ tối đa trong huyết thanh 90 phút sau khi uống thuốc.
Thức ăn có thể làm giảm độ hấp thu của thuốc.
+ Phân bố: 99% ibuprofen gắn kết với protein huyết tương. Trong hoạt
dịch, ibuprofen đạt được nồng độ ổn định khoảng giữa giờ thứ 2 và giờ thứ 8 sau
khi uống thuốc. Nồng độ tối đa trong hoạt dịch chiếm khoảng 1/3 nồng độ tối đa
trong huyết tương. Sau khi uống 400 mg ibuprofen mỗi 6 giờ ở phụ nữ cho con
bú, lượng ibuprofen có thể tìm thấy trong sữa mẹ là 1mg/ngày.
+ Chuyển hóa: Ibuprofen không có tác dụng cảm ứng emzyme. 90%
ibuprofen được chuyển hóa dưới dạng không hoạt động.
+ Thải trừ: Thải trừ chủ yếu qua nước tiểu: Trong 24 giờ, 10% dưới
dạng không thay đổi, 90% dưới dạng không hoạt động, chủ yếu là dưới dạng liên
hợp với acid glucuronic. Thời gian bán thải của thuốc là 1-2 giờ.

42
+ Việc sử dụng ibuprofen quá liều đã trở nên phổ biến từ khi nó được
cấp phép bán không đơn. Các triệu chứng xảy ra khi sử dụng quá liều gây ra các
tác động dược lý của ibuprofen bao gồm đau bụng, buồn nôn, nôn mửa, buồn ngủ,
chóng mặt, đau đầu, ù tai. Hiếm khi có các triệu chứng nghiêm trọng hơn như
chảy máu tiêu hóa, co giật, tăng kali máu, hạ huyết áp, hôn mê, rối loạn chức năng
gan, suy thận cấp, chứng xanh tím da, rối loạn hô hấp. Không thể xác định chính
xác liều tử vong, vì liều này có thể thay đổi tùy theo tuổi, trọng lượng và các bệnh
lý đi kèm ở mỗi bệnh nhân khác nhau.
+ Liệu pháp điều trị phần lớn dựa vào triệu chứng, trong trường hợp
biết sớm thì khử nhiễm dạ dày bằng cách dùng than hoạt tính. Than thấm hút
thuốc trước khi thuốc có thể được hấp thu vào hệ tuần hoàn. Trong đa số các
trường hợp uống ibuprofen chỉ gây ra ảnh hưởng nhẹ và điều trị quá liều là đơn
giản. Các biện pháp thường dùng là thông qua theo dõi nước tiểu và tiến hành theo
dõi chức năng thận. Ibuprofen bị giữ lại bởi các protein có trong máu, nên sự bài
tiết tối thiểu qua thận của thuốc không thay đổi. Khi bệnh nhân qua được giai đoạn
ngộ độc cấp tính thì sẽ không có di chứng gì
- Tác dụng:
+ Ibuprofen là thuốc chống viêm không steroid, có tác dụng giảm đau,
hạ sốt, chống viêm, dạng bào chế phổ biến nhất là viên nén. Điều trị đau từ nhẹ đến
vừa và các tình trạng viêm, như đau bụng kinh, nhức nửa đầu, đau hậu phẫu, đau
răng, bệnh cơ xương khớp, viêm khớp, viêm đa khớp dạng thấp, bệnh quanh khớp,
viêm bao gân, mô mềm, bong gân, căng cơ.
+ Cơ chế tác dụng chống viêm của ibuprofen là ức chế tổng hợp các
chất trung gian hóa học gây viêm đặc biệt là prostaglandin bằng cách ức chế enzyl
cyclooxygennase (COX) là enzym tổng hợp prostaglandin. Ngoài ra thuốc còn đối
kháng hệ enzym phân hủy protein, ngăn cản quá trình biến đổi protein làm bền
vững màng lysosom và đối kháng tác dụng của các chất trung gian hóa học như
bradykinin, serotonin, histamin, ức chế hóa hướng động bạch cầu, ức chế sự di
chuyển của bạch cầu tới tổ chức bị viêm.

43
+ Cơ chế tác dụng giảm đau của ibuprofen cũng như các thuốc giảm
đau chống viêm không steroid khác, chúng có tác dụng giảm đau nhẹ và vừa bằng
cách làm giảm tổng hợp prostaglandin F2.
- Sự chuyển hóa của ibuprofen trong cơ thể: Ibuprofen hấp thụ tốt ở đường
tiêu hóa, nồng độ tối đa của thuốc trong huyết tương đạt được sau từ 1 đến 2 giờ
dùng thuốc. Thuốc kết hợp mạnh với protein có trong huyết tương. Thời gian bán
hủy của thuốc khoảng 2 giờ, ibuprofen đào thải nhanh qua nước tiểu (1% dưới dạng
thuốc nguyên và 14% dưới dạng liên hợp). Ibuprofen cũng bền vững ít nhất 4
ngày. [5], [4], [58] [119]
1.3.4.3.Một số phương pháp xác định đồng thời paracetamol và ibuprofen
Một số phương pháp định lượng đồng thời PAR và IB được liệt kê ở bảng 1.4.
Bang 1.4. Một số nghiên cứu xác định PAR và IB
Phương pháp
Khoảng tuyến tính (µg/mL)
R2 LOD
(µg/mL)
LOQ
(µg/mL) RSD
Độ thu hồi
Rev(%)
Tác giả, năm, nguồn [TLTK]
UPLC/
DAD
5 – 60 (PAR); 5 – 100 (IB)
>0,999 - - <2 98 - 102 Trần Thị Ngọc Vân và cộng sự - 2017
[28]
Chuyển dịch bước sóng
20 – 40 (PAR); 1 – 32
(IB) -
-
- <2
99,5 (PAR);
100,3 (IB)
Vũ Đăng Hoàng và cộng sự - 2014 [120]
Phổ hồng ngoại chuyển đổi Fourier
0.9999 (IB) and 0.9998
(PAR) - - - -
100,35 (PAR),
98,70 (IB)
Muhammad Ali Mallah - 2012 [90]
Dấu “-“ là không có thông tin
1.3.5. Amlodipine besylat (AML), hydroclorothiazid (HYD), valsartan (VAL)
1.3.5.1. Giới thiệu chung về amlodipine [4], [5], [35], [68], [114]
- Công thức phân tử: C20H25ClN2O5.C6H6O3S.
- Khối lượng phân tử: 567,05.

44
- Tên khoa học: 3-Ethyl 5-methyl(±) -2-[aminoethoxy)methyl]-4-(o-
chlorophenyl)-1,4-dihydro-6-methyl-3,5-pyridinedicarboxylate,
monobenzenesulfonate
- Công thức cấu tạo:
- Tính chất vất lý- hóa học: Bột màu trắng hoặc gần như trắng, dễ tan trong
methanol, hơi tan trong ethanol khan, khó tan trong nước và 2-propanol. Đặc tính
phổ hấp thụ UV: Dung dịch AML trong dung dịch HCl 0,1N trong methanol có cực
đại hấp thụ tại 360nm (quét phổ từ 300nm đến 400nm).
- Dược lực học
+ AML là dẫn xuất của dihydropyridin có tác dụng chẹn dòng vào
calci qua màng tế bào. AML ngăn chặn kênh calci loại L phụ thuộc điện thế, tác
động trên các cơ trơn mạch máu và tim.
+ AML có tác dụng chống tăng huyết áp bằng cách trực tiếp làm giãn
cơ trơn quanh động mạch ngoại biên và ít có tác dụng hơn trên kênh calci cơ tim.
AML cũng có tác dụng tốt là giảm sức cản mạch máu thận, do đó làm tăng lưu
lượng máu ở thận và cải thiện chức năng thận.
+ AML không có ảnh hưởng xấu đến nồng độ lipid trong huyết tương
hoặc chuyển hóa glucose, do đó có thể dùng AML để điều trị tăng huyết áp ở
người bệnh đái tháo đường.
+ Tác dụng chống đau thắt ngực.
- Dược động học
+ Sinh khả dụng của AML khi uống khoảng 60-80% và không bị ảnh
hưởng bởi thức ăn. Nồng độ đỉnh trong huyết tương đạt được sau khi uống liều
khuyến cáo 6 đến 12 giờ.

45
+ Phân bố: Thuốc liên kết với protein huyết tương cao (trên 98%).
+ Chuyển hóa trong gan thành các chất mất hoạt tính và bài tiết qua
nước tiểu.
+ Ở người suy gan, nửa đời của AML tăng, vì vậy có thể cần phải
giảm liều hoặc kéo dài thời gian giữa các liều dùng.
- Chỉ định: Điều trị tăng huyết áp, điều trị đau thắt ngực.
- Chống chỉ định: Quá mẫn cảm với dihydropyridin.
1.3.5.2. Giới thiệu chung về hydroclorothiazid (xem mục 1.3.1.2)
1.3.5.3. Giới thiệu chung về Valsartan[4], [5], [35],[68], [114]
- Công thức phân tử: C24H29N5O3
- Khối lượng phân tử 435,52
- Tên khoa học: N-[p-(o-1H-Tetrazol-5-ylphenyl)benzyl]-N-valery-L-valine.
- Công thức cấu tạo:
- Tính chất vật lý, hóa học: Bột màu trắng hoặc gần như trắng, dễ hút ẩm,
không tan trong nước, tan tự do trong ethanol khan, hơi tan trong methylene clorid
[5], [47].
- Dược lực học [5]: Valsartan là thuốc đối kháng thể typ 1 của angiotensin II
(AT1). Valsartan ức chế chọn lọc angiotensin II gắn vào thụ thể AT1 ở nhiều mô
khác nhau, trong đó có cơ trơn mạch máu và tuyến thượng thận, làm hạ huyết áp
bằng cách đối kháng các tác dụng gây ra bởi angiotensin II.
- Dược động học
+ Hấp thu: VAL hấp thu nhanh sau khi uống. Sinh khả dụng đường
uống đạt khoảng 25%. Thời gian đạt nồng độ cực đại trong huyết tương trong
khoảng 2 đến 4 giờ sau khi dùng thuốc.

46
+ Phân bố: VAL liên kết mạnh với protein huyết tương (khoảng 94-
97%), chủ yếu là albumin huyết thanh.
+ Chuyển hóa: VAL không được chuyển hóa đáng kể, chỉ có 20% liều
tìm thấy dưới dạng chất chuyển hóa.
+ Thải trừ: VAL được thải trừ chủ yếu qua đường mật vào phân
(khoảng 83%) nhưng cũng qua thận vào nước tiểu (khoảng 13% liều).
- Chỉ định: Điều trị tăng huyết áp ở người lớn và trẻ em trên 6 tuổi, có
thể dùng đơn độc hoặc phối hợp với các thuốc chống tăng huyết áp loại khác. Điều
trị bệnh thận do đái tháo đường ở người tăng huyết áp. Điều trị suy tim sung huyết,
người tăng huyết áp suy tim. Điều trị sau nhồi máu cơ tim trên bệnh nhân suy thất
trái hoặc rối loạn chức năng tâm thu thất trái nhằm giảm tỉ lệ tử vong do tim mạch.
1.3.5.4. Một số phương pháp xác định đồng thời AML, HYD và VAL
Một số phương pháp định lượng đồng thời AML, HYD và VAL được liệt kê ở
bảng 1.5.
Nhận xét chung:
Từ các vấn đề trên, nghiên cứu phát triển phương pháp chemometric – trắc
quang kết hợp với sử dụng phương pháp lọc Kalman là rất cần thiết, đặc biệt là
trong định lượng đồng thời các hỗn hợp chất khó phân tích – các hỗn hợp chứa các
chất có phổ hấp thụ quang xen phủ nhau - trong các đối tượng mẫu khác nhau, trong
đó có các mẫu dược phẩm. Thách thức đặt ra là phải tìm được giải pháp phù hợp để
lựa chọn giá trị khởi tạo cho bộ lọc Kalman sao cho đưa ra các kết quả phân tích
chính xác hay mắc sai số chấp nhận được, đồng thời xây dựng được quy trình phân
tích theo phương pháp chemmometric – trắc quang kết hợp với phương pháp lọc
Kalman sao cho có thể áp dụng thuận lợi trong trong lĩnh vực kiểm nghiệm dược
phẩm ở nước ta.
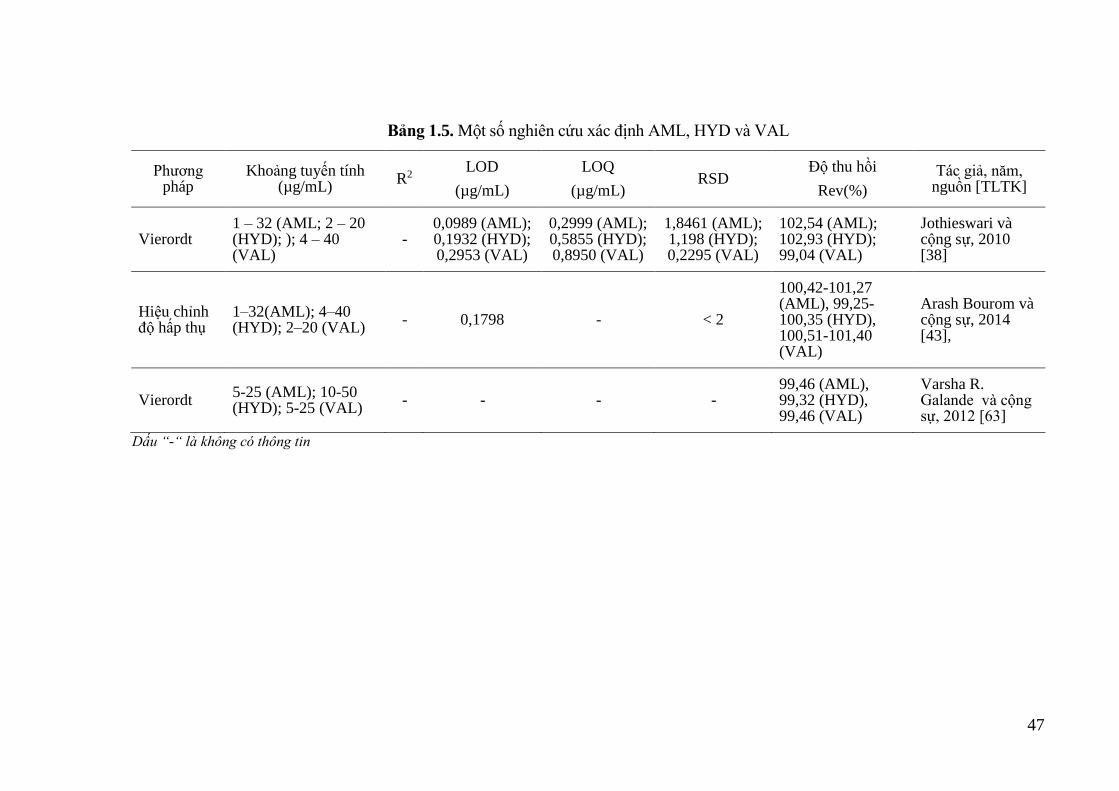
47
Bang 1.5. Một số nghiên cứu xác định AML, HYD và VAL
Phương pháp
Khoảng tuyến tính (µg/mL)
R2 LOD
(µg/mL)
LOQ
(µg/mL) RSD
Độ thu hồi
Rev(%)
Tác giả, năm, nguồn [TLTK]
Vierordt 1 – 32 (AML; 2 – 20 (HYD); ); 4 – 40 (VAL)
- 0,0989 (AML); 0,1932 (HYD); 0,2953 (VAL)
0,2999 (AML); 0,5855 (HYD); 0,8950 (VAL)
1,8461 (AML); 1,198 (HYD); 0,2295 (VAL)
102,54 (AML); 102,93 (HYD); 99,04 (VAL)
Jothieswari và cộng sự, 2010 [38]
Hiệu chỉnh độ hấp thụ
1–32(AML); 4–40 (HYD); 2–20 (VAL)
- 0,1798 - < 2
100,42-101,27 (AML), 99,25-100,35 (HYD), 100,51-101,40 (VAL)
Arash Bourom và cộng sự, 2014 [43],
Vierordt 5-25 (AML); 10-50 (HYD); 5-25 (VAL)
- - - - 99,46 (AML), 99,32 (HYD), 99,46 (VAL)
Varsha R. Galande và cộng sự, 2012 [63]
Dấu “-“ là không có thông tin

48
CHƯƠNG 2. NỘI DUNG VÀ PHƯƠNG PHAP NGHIÊN CỨU
2.1. Nội dung nghiên cứu
Để đạt được mục tiêu đề ra của luận án là góp phần phát triển phương pháp
chemometric-trắc quang sử dụng thuật toán lọc Kalman để áp dụng trong phân tích
dược phẩm, các nội dung nghiên cứu bao gồm:
1) Nghiên cứu nhằm tìm ra giải pháp phù hợp để lựa chọn được giá trị khởi
tạo (giá trị nồng độ và phương sai ban đầu) cho bộ lọc Kalman để áp dụng trong
phương pháp chemometric – trắc quang xác định đồng thời hỗn hợp các chất có phổ
hấp thụ quang xen phủ nhau (hỗn hợp chứa 2 chất và hỗn hợp chứa 3 chất).
2) Nghiên cứu xây dựng chương trình máy tính theo thuật toán lọc Kalman
trên phần mềm Microsoft-Excel 2016 với ngôn ngữ lập trình Visual Basic for
Applications, cho phép tính toán nhanh nồng độ các cấu tử có phổ hấp thụ quang
phân tử xen phủ nhau trong hệ nghiên cứu (chứa 2 hoặc 3 chất đồng thời).
3) Kiểm định độ tin cậy của phương pháp phân tích – Phương pháp
chemometric-trắc quang sử dụng thuật toán lọc Kalman (tính toán bằng chương
trình phần mềm đã xây dựng được): So sánh phương pháp phân tích với phương
pháp chemometric-trắc quang khác (phương pháp bình phương tối thiểu dùng phổ
toàn phần và phương pháp phổ đạo hàm) khi phân tích mẫu chuẩn phòng thí nghiệm
(chứa 2 hoặc 3 chất phân tích).
4) Xây dựng quy trình phân tích theo phương pháp chemometric-trắc quang sử
dụng thuật toán lọc Kalman (tính toán bằng chương trình phần mềm đã xây dựng được).
5) Áp dụng quy trình phân tích xây dựng được vào thực tế - phân tích các
mẫu dược phẩm đa thành phần (chứa 2 hoặc 3 thành phần) đang lưu hành ở thị
trường Việt Nam.
2.2. Phương pháp nghiên cứu
2.2.1. Đối tượng và phạm vi nghiên cứu
- Phương pháp chemometric-trắc quang sử dụng thuật toán lọc Kalman, phương
pháp bình phương tối thiểu dùng phổ toàn phần và phương pháp phổ đạo hàm;

49
- Các hoạt chất có phổ hấp thụ quang xen phủ nhau và thỏa mãn điều kiện:
phổ của chúng thỏa mãn định luật Beer và tính cộng tính độ hấp thụ quang, bao
gồm các dược phẩm đa thành phần thuộc các nhóm thuốc giảm đau hạ sốt, nhóm
thuốc tim mạch và điều trị huyết áp (các nhóm thuốc này đang lưu hành ở thị trường
Việt Nam):
i. Các thuốc chứa paracetamol và ibuprofen; paracetamol và caffeine
ii. Các thuốc chứa telmisartan và hydrochlorothiazide;
iii. Các thuốc chứa amlodipine, hydrochlorothiazide và valsartan.
2.2.2. Phương pháp lọc Kalman và chương trình tính
Dựa vào cơ sở lý thuyết, phương pháp lọc Kalman và chương trình tính được
thực hiện theo các bước như sau (Hình 2.1):
i) Ghi phổ của dung dịch đơn chất phân tích (dung dịch chuẩn phòng thí
nghiệm) và dung dịch hỗn hợp các chất phân tích, thu được bộ dữ liệu phổ (độ hấp
thụ quang ở k bước sóng lựa chọn) ở dạng file có đuôi txt (số bước sóng lựa chọn
tùy thuộc vào đặc điểm của các cấu tử trong hệ nghiên cứu);
ii) Nhập file dữ liệu phổ đơn chất và hỗn hợp chất vào chương trình phần mềm
máy tính (lập trình trên phần mềm Microsoft-Excel 2016) để tính các giá trị (hệ số
hấp thụ phân tử) của các đơn chất;
iii) Chạy bộ lọc Kalman:
- Đưa ra giá trị khởi tạo ban đầu, gồm: ước lượng đầu tiên của trạng thái nồng
độ Cest(0) và hiệp phương sai của sai số Pest(0) (nội dung nghiên cứu (1) sẽ đưa
ra giá trị khởi tạo ban đầu);
- Ngoại suy dự báo trạng thái nồng độ:
( ) ( 1)pri k est kC C -= (2.1)
- Ngoại suy hiệp phương sai của sai số:
( ) ( 1)pri k est kP P -= (2.2)
- Tính toán Lợi Kalman:
( )1
( ) ( ) ( ) ( ) (k) ( ) ( )T T
k pri k k k pri k kK P P Re e e-
= + (2.3)

50
- Cập nhật ước lượng trạng thái nồng độ:
( )( ) ( ) ( ) ( ) ( ) ( )est k pri k k k k pri kC C K A Ce= + -
(2.4)
- Cập nhật hiệp phương sai của sai số:
( ) ( ) ( ) ( )est k k k pri kP INV K Peé ù= -ê úë û (2.5)
- Các bước tính toán trên được thực hiện từ bước sóng thứ nhất đến bước sóng
cuối cùng. Cuối cùng, chương trình tính sẽ cho ra kết quả gồm: Nồng độ mỗi
cấu tử trong hệ và hiệp phương sai của sai số. Hiệp phương sai này thường
bé nhất ở bước sóng cuối cùng.
Hinh 2.1. Sơ đồ các bước tính toán theo phương pháp chemometric- trắc quang
sử dụng thuật toán lọc Kalman (sử dụng phần mềm máy tính).
Để minh họa cho cách tính toán theo thuật toán lọc Kalman, dưới đây đưa ra
thí dụ cho trường hợp hệ chứa 2 hoặc 3 cấu tử (chất phân tích).
2.2.2.1. Trường hợp hệ gồm 2 cấu tử:
Giả sử có hỗn hợp gồm 2 chất (chất 1 và 2) có nồng độ tương ứng là C1 = 10
µg/mL và C2 = 5 µg/mL với kết quả đo độ hấp thụ quang của dung dịch hỗn hợp đó
ở 161 bước sóng khác nhau nêu ở bảng 2.1 (ở đây không đưa ra độ hấp thụ quang ở
các bước sóng tương ứng đối với dung dịch chuẩn mỗi chất):

51
Bang 2.1 .Kết quả đo quang phổ của hỗn hợp hai chất(*)
STT Bước sóng (λ) (nm)
Độ hấp thụ quang đo được (A)
Hệ số hấp thụ phân tử của chất 1 (ε1)
Hệ số hấp thụ phân tử của chất 2 (ε2)
1 300,0 0,0226521 1983,31 702,81
2 299,5 0,0257293 2190,43 899,40
3 299,0 0,0290797 2418,52 1129,23
4 298,5 0,0326358 2644,93 1392,85
5 298,0 0,0364914 2871,66 1701,28
6 297,5 0,0406435 3114,71 2042,77
… … … … …
160 220,5 0,6043200 37667,60 46086,91
161 220,0 0,6083060 36860,40 48546,23
(*) Các giá trị độ hấp thụ quang 1 và 2 được tính toán từ bộ dữ liệu phổ độ hấp thụ quang của mỗi chất.
Từ các số liệu ở bảng 2.1, trình tự tính toán như sau:
i) Độ hấp thụ quang đo được đối với hỗn hợp chất là vector ( ) 161 1iA A´
é ù= ê úë û có
kích cỡ 161 dòng, 1 cột, thu được:
0,02265207
0,02572930
...
0,60830600
A
é ùê úê úê ú=ê úê úê úë û
ii) Ma trận ( , 161 2)i je e
´é ù= ê úë û
có kích cỡ 161 dòng, 2 cột ứng với các giá trị hệ số
hấp thụ phân tử của 2 chất ở mỗi bước sóng. Các giá trị ở đây được tính toán từ bộ
dữ liệu phổ của riêng mỗi chất và tính theo định luật Beer, thu được:
1983,31 702,812
2190, 43 899, 400
... ...
36860, 40 48546, 212
e
é ùé ùé ùê úê úê úê úê úê úê úê úê ú= ê úê úê úê úê úê úê úê úê úê úë ûë ûë û
iii) Giả sử nhiễu hệ thống (nhiễu trạng thái nồng độ) có hiệp phương sai
( ) 1kQ = và nhiễu đo có hiệp phương sai là ( ) 1kR = ; Chấp nhận hiệp phương sai của
sai số là P(0) = 1 (giá trị khởi tạo phương sai). (k ở đây là thể hiện bước sóng k bất kỳ);

52
iv) Tính toán ở bước 1:
- Chọn giá trị khởi tạo ban đầu cho nồng độ: Chọn vector ước lượng nồng độ
ban đầu là (0)
0
0C
é ùê ú=ê úë û
, tức là chấp nhận nồng độ chất 1 và chất 2 ban đầu bằng 0.
Lưu ý rằng, nồng độ ước lượng cho các bước tính toán tiếp theo Cest(k) sẽ thay đổi và
tiệm cận dần đến giá trị thực của nó trong dung dịch hỗn hợp, còn giá trị hiệp
phương sai P(k) sẽ ngày càng giảm. Từ đó suy ra:
(1) (0)
0
0pri estC C
é ùê ú= =ê úë û và
(1) (0) 1pri estP P
- Tính Hệ số hiệu chỉnh Kalman ở bước sóng thứ nhất:
1
(1) (1) (1) (1) (1) (1) (1)
1983,311
702,812
1983,31 0,000161983,31 702,81
0,00039
2 1 1702,812
T T
pri priK P H H P H R
- Tính mức cập nhật INV ở bước sóng thứ nhất:
[ ]
(1) (1) (1) (1)
0,0226520
1983,31 702,812 0,02570
0 2937
estINV A Ce= -
é ùê ú= - =ê úë û
- Tính nồng độ ước lượng cho bước sóng thứ nhất là Cest(1), mà trong đó, nó
được cập nhật phần INV(1) và hệ số hiệu chỉnh Kalman K(1):
(1) (1) (1) (1)
6
6
.
0 10,051 100,0257293
0 0,00016 4,127 10
0,00039
est priC C K INV
Rõ ràng, giá trị nồng độ ước lượng đã thay đổi theo hướng gần với thực tế hơn
(do nó khác 0 hay khác giá trị khởi tạo ban đầu Cest(0) = 0).
- Tính hiệp phương sai cho bước sóng thứ nhất Pest(1):
(1) (1) (1)
7
.
1 1983,31 702,812 1 1,783.100,00
0,00039
016
est estP INV K P
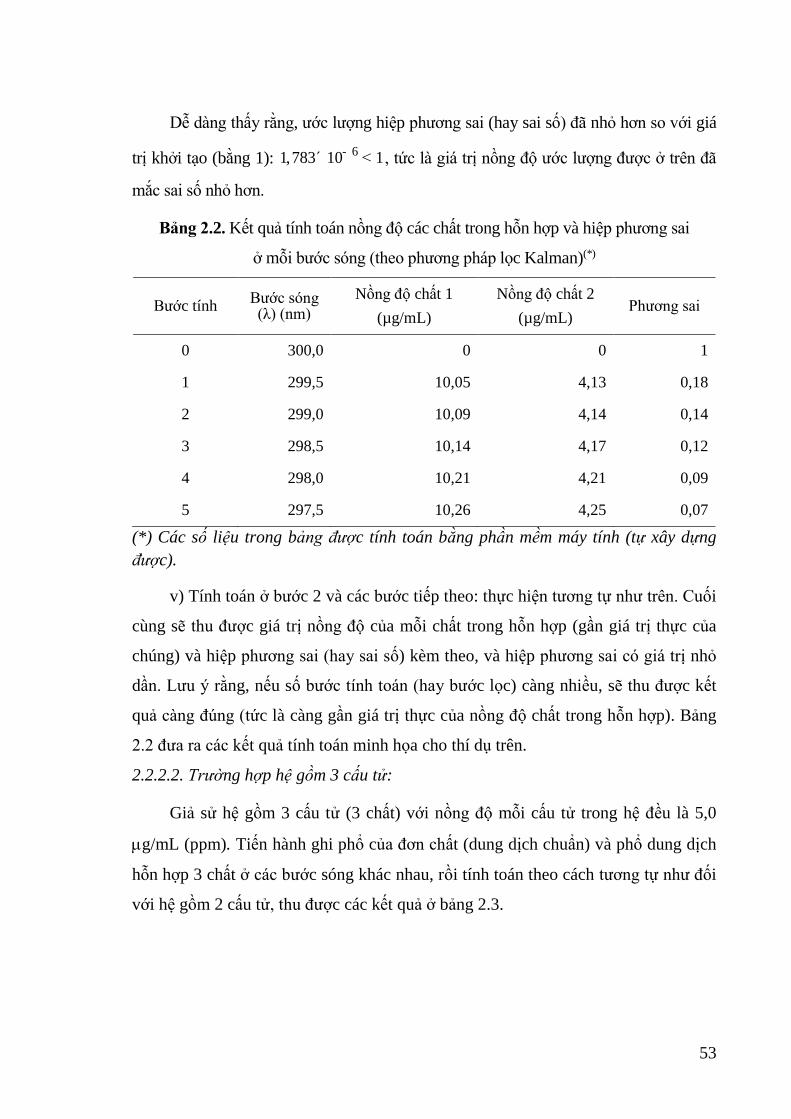
53
Dễ dàng thấy rằng, ước lượng hiệp phương sai (hay sai số) đã nhỏ hơn so với giá
trị khởi tạo (bằng 1): 61,783 10 1-´ < , tức là giá trị nồng độ ước lượng được ở trên đã
mắc sai số nhỏ hơn.
Bang 2.2. Kết quả tính toán nồng độ các chất trong hỗn hợp và hiệp phương sai
ở mỗi bước sóng (theo phương pháp lọc Kalman)(*)
Bước tính Bước sóng (λ) (nm)
Nồng độ chất 1
(µg/mL)
Nồng độ chất 2
(µg/mL) Phương sai
0 300,0 0 0 1
1 299,5 10,05 4,13 0,18
2 299,0 10,09 4,14 0,14
3 298,5 10,14 4,17 0,12
4 298,0 10,21 4,21 0,09
5 297,5 10,26 4,25 0,07
(*) Các số liệu trong bảng được tính toán bằng phần mềm máy tính (tự xây dựng
được).
v) Tính toán ở bước 2 và các bước tiếp theo: thực hiện tương tự như trên. Cuối
cùng sẽ thu được giá trị nồng độ của mỗi chất trong hỗn hợp (gần giá trị thực của
chúng) và hiệp phương sai (hay sai số) kèm theo, và hiệp phương sai có giá trị nhỏ
dần. Lưu ý rằng, nếu số bước tính toán (hay bước lọc) càng nhiều, sẽ thu được kết
quả càng đúng (tức là càng gần giá trị thực của nồng độ chất trong hỗn hợp). Bảng
2.2 đưa ra các kết quả tính toán minh họa cho thí dụ trên.
2.2.2.2. Trường hợp hệ gồm 3 cấu tử:
Giả sử hệ gồm 3 cấu tử (3 chất) với nồng độ mỗi cấu tử trong hệ đều là 5,0
g/mL (ppm). Tiến hành ghi phổ của đơn chất (dung dịch chuẩn) và phổ dung dịch
hỗn hợp 3 chất ở các bước sóng khác nhau, rồi tính toán theo cách tương tự như đối
với hệ gồm 2 cấu tử, thu được các kết quả ở bảng 2.3.

54
Bang 2.3. Kết quả tính toán nồng độ, hiệp phương sai, mức cập nhật INV
ở các bước sóng khác nhau cho trường hợp hỗn hợp ba cấu tử 1, 2 và 3(*)
Bước sóng
Cest (1) (µg/mL)
Cest (2) (µg/mL)
Cest (3) (µg/mL)
Pest (1) Pest (2) Pest (3) INV(k)
340 4,8 5,1 4,9 0,0049 0,0052 0,0050 0
330 5,0 5,0 4,8 0,0049 0,0052 0,0050 0,0608
320 5,0 5,0 4,8 0,0049 0,0052 0,0050 0,0546
310 5,0 5,0 4,8 0,0049 0,0052 0,0050 0,0512
300 5,0 5,0 4,8 0,0049 0,0052 0,0050 0,0541
290 5,0 5,0 4,8 0,0049 0,0052 0,0050 0,0659
280 5,0 5,1 4,8 0,0049 0,0052 0,0050 0,0702
270 5,0 5,1 4,8 0,0047 0,0049 0,0047 -0,0172
260 5,0 5,1 4,8 0,0043 0,0045 0,0043 0,0060
250 5,0 5,1 4,8 0,0040 0,0042 0,0040 0,0242
240 5,0 5,1 4,8 0,0038 0,0039 0,0038 -0,0013
230 5,0 5,1 4,8 0,0034 0,0036 0,0034 0,0084
225.5 5,0 5,1 4,8 0,0031 0,0033 0,0032 -0,0895
(*) - Cest (1), Cest (2) và Cest (3) là các giá trị nồng độ ước lượng của chất 1, 2 và 3; Pest (1), Pest (2),
Pest (3) là hiệp phương sai (hay sai số) ứng với chất 1, 2 và 3; INV(k) là mức cập nhật ở bước sóng
thứ k.
- Các số liệu trong bảng được tính toán bằng phần mềm máy tính (tự xây dựng
được).
Từ bảng 2.3 nhận thấy: Giá trị trung bình số học của mức cập nhật INV(k) (bằng
0,02395) đã gần tiệm cận đến giá trị thực của nó (bằng 0), tức là bộ lọc Kalman đạt
hiệu quả tốt. Mặt khác, các giá trị hiệp phương sai trong các bước tính toán có xu thế
giảm dần, nghĩa là sai số ngày càng nhỏ và nồng độ các cấu tử càng tiệm cận dần đến
giá trị thực của chúng.
2.2.3. Phương pháp bình phương tối thiểu sử dụng phần mềm simulan [2]
- Bước 1. Chuẩn bị các dung dịch chuẩn riêng từng cấu tử và hỗn hợp của
chúng.

55
- Bước 2: Ghi phổ hấp thụ quang của dung dịch chuẩn để tính ma trận hệ số
hấp thụ của các cấu tử: = (ij )mxn.
- Bước 3: Ghi phổ hấp thụ quang của dung dịch hỗn hợp, nhập ma trận độ hấp
thụ quang đo được A: A = (Ai1)mx1.
- Bước 4: Giải hệ m phương trình n ẩn số theo phương pháp bình phương tối
thiểu: A= . C để tìm ra nồng độ C.
2.2.4. Phương pháp phổ đạo hàm
Bước 1. Chuẩn bị các dung dịch chuẩn riêng từng cấu tử và hỗn hợp của
chúng.
Bước 2: Ghi phổ hấp thụ quang và phổ đạo hàm, tìm bước sóng đo thích hợp
mà tại đó giá trị phổ đạo hàm của một chất cần phân tích khác 0 hoặc cực đại, còn
giá trị phổ đạo hàm của chất kia bằng 0.
Bước 3: Sau khi xác định được bước sóng đo ở một bậc đạo hàm nhất định,
tiến hành định lượng các chất theo phương pháp đường chuẩn hoặc thêm chuẩn.
2.4.5. Phương pháp xây dựng chương trình máy tính
- Việc tính toán để xác định nồng độ các chất theo phương pháp lọc Kalman là
khá phức tạp, vì vậy cần lập trình trên máy tính để tính toán nhanh và thuận lợi cho
người sử dụng;
- Chọn phần mềm mã nguồn mở là Microsoft-Excel để không vi phạm bản
quyền;
- Chọn ngôn ngữ và công cụ visual basic for application;
- Chương trình xây dựng được phải có cấu trúc gồm: Các khối đầu vào (nhập
dữ liệu) và các khối đầu ra trung gian (theo các bước tính toán của thuật toán lọc
Kalman), và cuối cùng là khối đầu ra của chương trình tính toán (xuất kết quả ra ở
dạng bảng số liệu).
2.2.6. Phương pháp đánh giá độ tin cậy của phương pháp phân tích
2.2.6.1. Sai số tương đối [98]
Theo định nghĩa, sai số là độ lệch giữa kết quả đo và giá trị thực của nó. Trong
thực tế đo lường hóa học, khi đo hay phân tích một mẫu, thường không biết trước
giá trị thực của nó, nên về nguyên tắc, khó xác định được sai số.

56
Khi nghiên cứu cơ bản trong phòng thí nghiệm, thường người ta pha các dung
dịch chuẩn từ hóa chất tinh khiết và chấp nhận giá trị nồng độ của dung dịch chuẩn
là giá trị thực. Khi đó, sẽ xác định được sai số của phép đo/phân tích là độ lệch giữa
kết quả xác định được (C, g/mL) và nồng độ của dung dịch chuẩn (Co, g/mL). Sai
số này có thể biểu diễn ở dạng sai số tuyệt đối (có đơn vị trùng với đơn vị của nồng
độ) hoặc sai số tương đối (có đơn vị %). Trong nghiên cứu kiểm định phương pháp
chemmometric-trắc quang sử dụng thuật toán lọc Kalman, chúng tôi sử dụng sai số
tương đối (đối với dung dịch chuẩn phòng thí nghiệm) để đánh giá sai số của
phương pháp. Khi đó, sai số tương đối (RE %) được tính theo công thức:
(2.11)
2.2.6.2. Độ lặp lại [40], [116]
Khi nghiên cứu phát triển phương pháp phân tích hoặc trước khi áp dụng một
phương pháp phân tích kỳ vào thực tế, bắt buộc phải kiểm soát lượng (quality
control) hay thẩm định phương pháp qua các thông số thể hiện năng lực của phương
pháp (performance parameters). Trong đó, hai thông số quan trọng nhất là độ đúng
(truenss) và độ lặp lại (precision). Nếu phân tích trong nội bộ phòng thí nghiệm,
người ta dùng thuật ngữ độ lặp lại hay độ chụm (repeatability), nếu phân tích trong
nhiều phòng thí nghiệm khác nhau, người ta dùng thuật ngữ độ tái lặp hoặc độ hồi
phục (reproducibility). Chỉ khi một phương pháp phân tích vừa đạt được độ đúng
tốt và độ lặp lại tốt, mới được xem là đạt được độ chính xác (accurate) cao.
Trong nghiên cứu của luận án, độ lặp lại của phương pháp phân tích trong nội
bộ phòng thí nghiệm (repeatability) được đánh qua độ lệch chuẩn tương đối (RSD):
(2.12)
Trong đó, S: độ lệch chuẩn (có đơn vị trùng với đơn vị của nồng độ) với n
phép đo lặp lại; CTB: giá trị trung bình của nồng độ (µg/mL);
Người ta thừa nhận rằng, khi xác định nồng độ C trong nội bộ phòng thí
nghiệm, nếu thu được giá trị RSD (%) ≤ ½ RSDHorwitz thì phương pháp đạt được độ
lặp lại tốt (hay đạt yêu cầu) Trong đó, RSDHorwitz là RSD được tính theo phương
trình Horwitz [116]:
0
0
C CRE(%) .100
C
S.100RSD(%)
x

57
RSDHorwitz = 2(1 – 0.5*lgC) (2.13)
Trong đó, C là nồng độ chất phân tích được biểu diễn dưới dạng phân số.
2.2.6.3. Độ đúng [40]
Độ đúng của phương pháp phân tích là độ gần của kết quả đo/phân tích với giá
trị thực của nó. Để đánh giá độ đúng của phương pháp phân tích, có thể thực hiện
theo 3 cách: i) Phân tích Mẫu vật liệu so sánh được cấp chứng chỉ (certified
reference material), có giá trị nồng độ chất ghi trong chứng chỉ đi kèm mẫu được
chấp nhận là giá trị thực; ii) Phân tích mẫu thực tế được thêm chuẩn (spiked
sample) và đánh giá độ đúng qua độ thu hồi; iii) Phân tích mẫu bằng phương pháp
chuẩn khác, rồi so sánh kết quả phân tích của phương pháp đang dùng và phương
pháp chuẩn đó bằng phương pháp thống kê.
Trong nghiên cứu của luận án, hai cách đánh giá độ đúng (ii) và (iii) được sử
dụng. Phương pháp chuẩn được chọn để so sánh (khi phân tích các mẫu dược phẩm)
là phương pháp sắc ký lỏng hiệu năng cao (HPLC).
Theo cách (ii) ở trên, độ thu hồi được tính theo công thức:
𝑅𝑒𝑣(%) = 𝐶𝑥− 𝐶0
𝐶𝑡 (2.14)
Trong đó, Ct: nồng độ của chất chuẩn thêm vào mẫu (µg/mL); Cx: nồng độ
chất xác định được trong mẫu sau khi thêm chuẩn (µg/mL); C0: nồng độ chất xác
định được trong mẫu khi chưa thêm chuẩn (µg/mL).
Độ thu hồi (Rev) là đại lượng phụ thuộc vào nồng độ chất phân tích. Để đánh
giá xem, độ thu hồi là bao nhiêu (ứng với mức nồng độ đang phân tích), thì phương
pháp phân tích được xem là đạt được độ đúng tốt, cần tuân thủ các hướng dẫn (hay
quy định) của Hiệp hội các nhà hóa học phân tích, Mỹ (AOAC) [40].
2.2.7. Phương pháp xử lý số liệu
Áp dụng phần mềm Microsoft-Excel 2016 với công cụ Data Analysis để xử lý
các số liệu thí nghiệm: Tính toán các đại lượng thống kê (trung bình số học, độ lệch
chuẩn, RSD); so sánh hai độ lặp lại (hay hai phương sai), dùng kiểm định F (F-test);
So sánh hai giá trị trung bình, dùng kiểm định t (t-test); So sánh hai phương pháp,
dùng kiểm định t theo cặp (paired-t-test)...
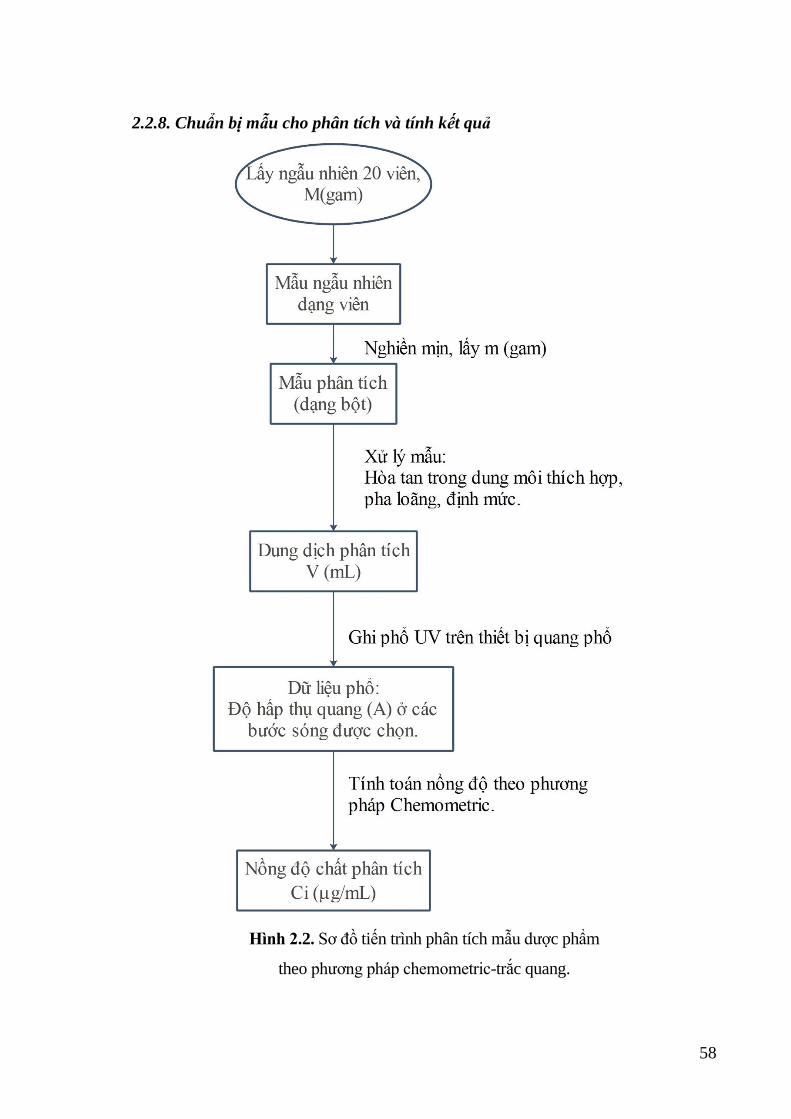
58
2.2.8. Chuẩn bị mẫu cho phân tích và tính kết quả
Hinh 2.2. Sơ đồ tiến trình phân tích mẫu dược phẩm
theo phương pháp chemometric-trắc quang.

59
(*) Kỹ thuật xử lý mẫu tùy thuộc vào đặc tính của hỗn hợp chất phân tích trong mẫu
Chuẩn bị mẫu cho phân tích và tính kết quả:
2.2.8.1. Mẫu thuốc Micardis(R) Plus chứa hỗn hợp TEL và HYD
- Xử lý mẫu [42], [85]:
+ Cân 20 viên thuốc; Tính khối lượng trung bình của một viên ( M ); Nghiền
thuốc thành bột mịn (dùng cối và chày mã não) và trộn đều;
+ Lấy m (g) bột thuốc vào bình thủy tinh 250 mL; Thêm vào bình 100 mL
dung dịch NaOH 0,10 M; Lắc đều và đem siêu âm trong 30 phút; Kết thúc siêu âm,
định mức bằng dung dịch NaOH 0,10 M; Đem lọc dung dịch qua màng lọc
xenluoaxetate 20-25 μm (Whatman 41); Đây là dung dịch đầu cho phân tích (dung
dịch A).
+ Lấy 2,5 mL dung dịch A vào bình định mức 50 mL; Định mức bằng dung
dịch NaOH 0,10 M. Đây là dung dịch mẫu (dung dịch B) được đem đo phổ hấp thụ
quang.
- Tính hàm lượng chất phân tích trong mẫu:
Hàm lượng x của chất phân tích trong một viên (x, mg/viên) được tính theo
công thức:
m
10.M.Cx a
(2.15)
Trong đó, M : khối lượng trung bình của một viên thuốc (g); Thuốc
Micardis(R) Plus có M = 0,4382 g; [Ca] : nồng độ của chất phân tích xác định
được trong dung dịch B (g/mL); m: khối lượng mẫu bột thuốc (g);.
Từ quy trình xử lí mẫu, ta có công thức tính hàm lượng chất rút gọn là:
x = C.5 (mg/viên) (2.16)
2.2.8.2. Mẫu thuốc Panadol Extra chứa hỗn hợp PAR và CAF
- Xử lý mẫu [42], [85]:
+) Cân 20 viên thuốc; Tính khối lượng trung bình của một viên ( M ); Nghiền
thuốc thành bột mịn (dùng cối và chày mã não) và trộn đều;

60
+) Cân gam mẫu cho vào bình định mức 250 mL, hòa tan trong khoảng
200 mL nước cất, siêu âm 30 phút, định mức đến vạch. Đem lọc dung dịch qua
màng lọc xenluoaxetate 20-25 μm (Whatman 41). Lấy 10 mL dung dịch sau khi lọc
pha loãng trong bình định mức 100 mL. Lấy 5 mL dung dịch trong bình 100 mL
pha loãng trong bình 25 mL thu được dung dịch mẫu để đo quang.
- Tính hàm lượng chất phân tích trong mẫu:
Hàm lượng PAR và CAF trong 1 viên được tính theo công thức:
(mg/viên) (2.17)
Trong đó:
M: Khối lượng trung bình 1 viên (gam)
m: Khối lượng mẫu cân để phân tích (gam)
C: Nồng độ chất đo được trong dung dịch mẫu (µg/mL)
V: Thể tích định mức ban đầu (250 ml)
K: Hệ số pha loãng (K = 50)
Khối lượng trung bình mỗi viên = 0,6965 g.
Từ quy trình xử lí mẫu, ta có công thức tính hàm lượng chất rút gọn là:
x = C.63 (mg/viên) (2.18)
2.2.8.3. Đối với hỗn hợp PAR và IB
- Xử lý mẫu [91], [119].
+ Chọn ngẫu nhiên 20 viên thuốc, cân và tính khối lượng trung bình mỗi viên
M, nghiền toàn bộ 20 viên đó thành bột mịn, trộn đều.
+ Cân chính xác m (g) lượng bột mẫu tương đương 2M/5 lượng viên
thuốc, cho vào cốc thủy tinh, thêm khoảng 30 mL dung môi đệm photphat pH =
7, khuấy đều cho mẫu tan hết. Cho dung dịch mẫu theo đũa thủy tinh vào bình
định mức 100 mL. Dùng dung môi tráng cốc thủy tinh nhiều lần rồi cho dung
dịch vào bình định mức; định mức bằng dung môi đến vạch 100 mL; lắc để trộn
đều dung dịch, lọc dung dịch bằng màng lọc xenluoaxetate 20-25 μm (Whatman
41). Ta thu được dung dịch (1).
M
5
C.V.K.Mx
m.1000
M

61
+ Dùng pipet lấy chính xác 10 mL dung dịch (1) vừa thu được đem pha loãng
bằng dung môi, định mức thành 100 mL, trộn đều thu được dung dịch (2).
+ Dùng pipet lấy chính xác 10 mL dung dịch (2) vừa thu được đem pha loãng
bằng dung môi, định mức thành 100 mL, trộn đều ta thu được dung dịch mẫu (3).
Đây là dung dịch mẫu sử dụng để đo quang.
- Tính hàm lượng chất phân tích trong mẫu:
Hàm lượng PA và IB trong 1 viên được tính theo công thức:
(mg/viên) (2.19)
Trong đó: M: Khối lượng trung bình 1 viên (gam)
m: Khối lượng mẫu cân để phân tích (gam)
C: Nồng độ chất đo được trong dung dịch mẫu (µg/mL)
V: Thể tích định mức ban đầu (100 ml)
K: Hệ số pha loãng (K = 100)
Thay các giá trị đã biết vào phương trình, ta có:
Từ quy trình xử lí mẫu, ta có công thức tính hàm lượng chất rút gọn là:
x = C.25 (mg/viên) (2.20)
2.2.8.4. Đối với hỗn hợp AML, HYD và VAL
- Xử lý mẫu [64].
Cân 20 viên thuốc, tính khối lượng trung bình của một viên ( ). Nghiền
thuốc thành bột mịn và trộn đều. Cân chính xác m (g) bột thuốc tương đương với 1
viên thuốc (ứng với 10 mg AML; 12,5 mg HYD và 160 mg VAL) cho vào bình
thủy tinh có nút nhám 250 mL. Thêm vào bình 50 mL dung môi methanol nguyên
chất lắc đều. Siêu âm dung dịch trong 30 phút. Sau khi siêu âm, lọc dung dịch qua
giấy lọc băng xanh vào bình định mức 100 mL và định mức đến vạch bằng dung
môi. Lấy chính xác 10 mL dung dịch này cho vào bình định mức 100 mL thứ 2,
định mức đến vạch bằng dung môi, lắc đều. Lại lấy chính xác 10 mL dung dịch ở
bình định mức 2 cho vào bình định mức 100 mL thứ 3, định mức đến vạch bằng
dung môi, lắc đều ta được dung dịch mẫu cần đo.
Khối lượng trung bình của một viên: = 4,1252 g.
C.V.K.Mx
m.1000
)/(..10
viênmgm
MCx
M
M

62
** Công thức tính hàm lượng chất
. . .
.1000
CV K Mx
m (mg/viên) (2.21)
Trong đó: M : khối lượng trung bình 1 viên (g);
m: khối lượng mẫu cân để phân tích (g);
C: nồng độ chất đo được trong dung dịch mẫu (g/mL);
V: thể tích định mức ban đầu (100 mL);
K: hệ số pha loãng (K=100).
Từ quy trình xử lí mẫu, với m = M ta có công thức tính hàm lượng chất rút
gọn là:
x = C.10 (mg/viên) (2.22)
2.2.9. Thiết bị, dụng cụ và hóa chất.
2.2.9.1. Thiết bị và dụng cụ
- Máy quang phổ UV – Vis (hiệu Jasco V630 -Nhật)
- Cân phân tích hiệu Precisa XB 220A độ chính xác 0,0001 g, Thụy Sĩ.
- Máy cất nước hai lần hiệu Aquatron, Anh.
- Máy lắc hiệu IKA KS 130 basic, Đức.
- Các dụng cụ khác: Micropipet của hãng HTL- Đức, pipet, bình định mức,
cốc thủy tinh, bình tam giác, đũa thuy tinh, giấy lọc, phễu, các lọ đựng hoá chất và
mẫu, bếp điện, ...
2.2.9.2. Hóa chất
- Chất chuẩn Telmisartan: Bộ Y tế - Viện kiểm nghiệm thành phố Hồ Chí
Minh. Hàm lượng TEL 98,97%.
- Chất chuẩn Hydrochlorothiazide: Bộ Y tế - Viện kiểm nghiệm thành phố Hồ
Chí Minh. Hàm lượng HYD 98,10 %.
- Chất chuẩn Ibuprofen: Bộ Y tế - Viện tiêu chuẩn dược dụng Hà Nội. Hàm
lượng IB 100%.
- Chất chuẩn Paracetamol: Trung tâm kiểm nghiệm thuốc TW, chuẩn dược
điển Việt Nam: Hàm lượng PAR 98,86%.

63
- Chất chuẩn Cafein: Trung tâm kiểm nghiệm thuốc TW, chuẩn dược điển
Việt Nam. Hàm lượng CAF 100,26 %.
- - Chất chuẩn AML: Trung tâm kiểm nghiệm thuốc TW. Hàm lượng AML
100,43%.
- Chất chuẩn HYD: Trung tâm kiểm nghiệm thuốc TW. Hàm lượng HYD
HYD 99,55%.
- Chất chuẩn VAL: Trung tâm kiểm nghiệm thuốc TW. Hàm lượng VAL
98,38%.
- Thuốc Panadol Extra: Sản xuất tại công ty cổ phần dược phẩm Sanofi-
Synthelabo- Việt Nam, số lô/ngày sản xuất: 15123, HDS: 13/10/2017, hàm lượng
ghi trên nhãn 500 mg PAR và 65 mg CAF/viên.
- Thuốc Colocol Extra: Sản xuất tại công ty CP dược phẩm Sao Kim, số
lô/ngày sản xuất: 040715, HSD: 16/07/2018, hàm lượng ghi trên nhãn 500 mg PAR
và 65 mg CAF/viên
- Thuốc viên nén Micardis(R) Plus:Sản xuất tại Boehringer Ingelheim Pharma
GmbH & Co. KG Binger Str. 173 55216 Ingelheim am Rhein, Germany, số lô:
505392, ngày sản xuất: 26/06/2015, hạn dùng: 26/06/2018, SĐK: VN-16587-13,
hàm lượng ghi trên nhãn của TEL là 40,0 mg và HYD là 12,5 mg/viên
- Thuốc Alaxan: Sản xuất tại công ty United International Pharma, số lô/ngày
sản xuất 407251, HSD: 04/05/2017, hàm lượng ghi trên nhãn 325 mg PA và 200 mg
IB/viên.
- Thuốc Di-afasawic: Sản xuất tại công ty Dược phẩm Quang Minh – Wa
Pharma USA, số lô/ngày sản xuất 0020215, HSD: 07/02/2017, hàm lượng ghi trên
nhãn 300 mg PA và 200 mg IB/viên.
- Thuốc Protamol: Sản xuất tại công ty Hóa – Dược phẩm Mekophar, số
lô/ngày sản xuất 14001NN, HSD: 04/02/2017, hàm lượng ghi trên nhãn 325 mg PA
và 200 mg IB/viên.
- Thuốc Lopenca: Sản xuất tại công ty Dược Hậu Giang, số lô/ngày sản xuất
020214, HSD: 25/02/2017, hàm lượng ghi trên nhãn 325 mg PA và 200 mg IB/viên.

64
- Thuốc viên nén Exforge HCT: Sản xuất tại Novartis Farmaceutica S.A.
Ronda Santa Maria 158, 08210 Barberà del Vallès, Barcelona, Tây Ban Nha, số lô:
BK917, ngày sản xuất: 09/2016, hạn dùng: 08/2018, SĐK: VN-19287-15, hàm
lượng ghi trên nhãn 10 mg AML; 12,5 mg HYD và 160 mg VAL.

65
CHƯƠNG 3. KÊT QUẢ VÀ THẢO LUẬN
3.1. Lựa chọn giá trị khởi tạo ban đầu
Khi đề cập đến cơ sở lý thuyết của phương pháp lọc Kalman, đã cho rằng,
phương pháp này có hạn chế là phải tìm được cách phù hợp để lựa chọn giá trị khởi
tạo ban đầu cho thuật toán lọc Kalman. Nếu lựa chọn sai, sẽ đưa đến kết quả tính
toán sai. Đối với dung dịch chứa hỗn hợp 2 hoặc 3 chất, giá trị khởi tạo ban đầu là
các giá trị ước lượng nồng độ và hiệp phương sai kèm theo (từ đây để cho gọn, gọi
tắt là phương sai) của mỗi chất trong hỗn hợp. Nhiều nghiên cứu trước đây đã đưa
ra các cách lựa chọn các giá trị khởi tạo khác nhau, song nói chung, có thể phân
chia thành 2 nhóm giải pháp lựa chọn giá trị khởi tạo ban đầu:
Nhóm 1: Giá trị khởi tạo được lựa chọn ngẫu nhiên (hay gọi tắt là lựa chọn giá
trị khởi tạo ngẫu nhiên), tức là có thể chọn một giá trị bất kỳ cho nồng độ (C),
chẳng hạn bằng 0 hoặc 0,5…, và giá trị bất kỳ của phương sai (P), chẳng hạn bằng 1
[27], [112];
Nhóm 2: Giá trị khởi tạo được chọn theo cách giả định (hay gọi tắt là giá trị
khởi tạo giả định), tức là (i) dựa vào hiểu biết của người nghiên cứu đối với mẫu
đang khảo sát, để lựa chọn giá trị C và phương sai P theo kinh nghiệm cá nhân;
hoặc (ii) qua khảo sát trên một số thí nghiệm ban đầu để đưa ra giá trị C và P khởi
tạo; (iii) hoặc dựa vào nguyên tắc của phương pháp trắc quang, sơ bộ tính toán giá
trị C (đối với mỗi chất trong hỗn hợp) qua giải hệ phương trình tuyến tính (theo
nguyên tắc của định luật Beer) ở một số bước sóng lựa chọn và chọn giá trị khởi tạo
P dựa vào một hướng dẫn nào đó về sai số chấp nhận được (chẳng hạn, dựa vào
phương trình Horwitz để tính RSD, rồi từ đó tính được độ lệch chuẩn và phương sai
ứng với một giá trị C xác định)… [49], [101], [110].
Nói chung, có thể cho rằng, cho đến nay, chưa có một giải pháp thống nhất để
lựa chọn giá trị khởi tạo (C và P) phù hợp cho thuật toán lọc Kalman. Do vậy, điều
này vẫn là một thách thức đối với các nhà hóa học phân tích khi nghiên cứu áp dụng
thuật toán lọc Kalman. Dưới đây, chúng tôi sẽ khảo sát cả 3 cách để lựa chọn giá trị
khởi tạo phù hợp: theo cách của nhóm 1, nhóm 2 và cách mới.

66
3.1.1. Lựa chọn giá trị khởi tạo ngẫu nhiên
Theo cách này, chọn giá trị khởi tạo ngẫu nhiên là có thể chọn một giá trị bất
kì cho nồng độ (0)estC và phương sai (0)estP [27], [112].
Đối với hỗn hợp chứa 2 chất hoặc 3 chất (là hỗn hợp các chất chuẩn trong
phòng thí nghiệm), trong nghiên cứu này đều lựa chọn ngẫu nhiên giá trị khởi tạo
ban đầu đối với mỗi chất đều là nồng độ (0)estC = 0,3 µg/mL và phương sai (0)estP
= 1. Việc lựa chọn (0)estC = 0,3 µg/mL, tuy là ngẫu nhiên, nhưng có chủ ý nhất
định, vì dựa vào ước lượng thô về giới hạn phát hiện (LOD) của phương pháp trắc
quang khoảng 0,1 µg/mL, ứng với giới hạn định lượng (LOQ) khoảng 0,3 µg/mL
(nhiều công bố đã xác nhận điều này).
3.1.1.1. Đối với hệ chứa hai chất telmisartan (TEL) và hydrochloruathiazide (HYD)
Áp dụng phương pháp chemometric-trắc quang sử dụng thuật toán lọc Kalman
(từ đây, để cho gọn, gọi tắt là phương pháp Kalman) để xác định nồng độ mỗi chất
trong hỗn hợp của chúng.
Từ bộ dữ liệu phổ của dung dịch chuẩn mỗi chất (TEL, HYD) và dung dịch
hỗn hợp của chúng (trong khoảng bước sóng 220 nm – 340 nm), sử dụng thuật toán
lọc Kalman và chương trình máy tính (tự xây dựng) để tính toán, thu được các kết
quả ở bảng 3.1. Tiêu chí để đánh giá độ tin cậy của phương pháp đối với mỗi chất ở
đây là dựa vào sai số tương đối RE (%).
Bang 3.1. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp
theo phương pháp Kalman với cách lựa chọn giá trị khởi tạo ngẫu nhiên(*)
Hỗn hợp H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
C (µg/mL) 0,30 0,30 0,30 0,30 0,30 0,30 0,30 0,30 0,30
RE (%) -70 -85 -90 -93 -94 -95 -96 -96 -97
HYD
Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
C (µg/mL) 0,30 0,30 0,30 0,30 0,30 0,30 0,30 0,30 0,30
RE(%) -97 -96 -96 -95 -94 -93 -90 -85 -70

67
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả ở bảng 3.1 cho thấy, ở các nồng độ khác nhau của 2 chất (TEL và
HYD) trong hỗn hợp: Nồng độ mỗi chất dao động trong khoảng 1,00 µg/mL – 9,00
µg/mL và tỉ lệ nồng độ giữa chúng (ppm/ppm) dao động trong khoảng 1/9 – 9/1,
các kết quả xác định được đều mắc sai số tương đối (RE) rất lớn: đối với cả 2 chất
RE khoảng 70 % – 97 % và đều mắc sai số hệ thống âm. Như vậy, giá trị khởi tạo
lựa chọn ( (0)estC = 0,3 µg/mL và phương sai (0)estP = 1) là chưa phù hợp, dẫn đến
kết quả mắc sai số không chấp nhận được. Theo chúng tôi, có thể phương pháp
Kalman đang dùng đã hiểu rằng, giá trị nồng độ 0,3 µg/mL đó là thuộc một phân bố
chuẩn khác với giá trị thực nào đó, chứ không thuộc phân bố chuẩn ứng với các
nồng độ thực của chất phân tích từ 1,00 đến 9,00 µg/mL. Hay nói cách khác, khi
việc lựa chọn giá trị khởi tạo nồng độ quá xa so với giá trị thực của nó, phương
pháp Kalman sẽ không giải “hội tụ”, mà bị “phân kỳ”, dẫn đến sai số không chấp
nhận được.
3.1.1.2. Đối với hỗn hợp ba chất amlodipine (AML), valsartan (VAL) và HYD,
Từ bộ dữ liệu phổ của dung dịch chuẩn mỗi chất (AML, HYD và VAL) và
dung dịch hỗn hợp của chúng (trong khoảng bước sóng 230 – 340 nm), áp dụng
phương pháp Kalman để xác định nồng độ mỗi chất trong hỗn hợp của chúng theo
cách tương tự như đối với hỗn hợp 2 chất, thu được các kết quả ở bảng 3.2.
Kết quả ở trên cũng cho thấy rằng, với cách lựa chọn giá trị khởi tạo ngẫu
nhiên, trừ AML và HYD trong hỗn hợp H1, phương pháp Kalman cho ra các kết
quả mắc sai số khá lớn với RE khoảng 40 % – 98 % đối với cả 3 chất trong các hỗn
hợp khảo sát H1 – H4 (nồng độ của mỗi chất trong hỗn hợp dao động trong khoảng
0,250 µg/mL – 16 µg/mL) (bảng 3.2). Điều này có thể được giải thích theo cách
tương tự như đối với hỗn hợp chứa 2 chất ở trên: giá trị nồng độ khởi tạo quá xa so
với giá trị thực của nó, nên phương pháp giải bị “phân kỳ”. Riêng đối với AML và
HYD trong hỗn hợp H1, do giá trị nồng độ khởi tạo (0,3 µg/mL) khá gần với giá trị
thực của nồng độ AML (0,250 µg/mL) và nồng độ HYD (0,325 µg/mL), nên kết

68
quả mắc sai số nhỏ hơn: Nồng độ AML mắc sai số 20 %, còn nồng độ HYD mắc sai
số 6 % (bảng 3.2).
Bang 3.2. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp
theo phương pháp Kalman với cách lựa chọn giá trị khởi tạo ngẫu nhiên(*)
AML
Hỗn hợp H1 H2 H3 H4
Co (µg/mL) 0,250 0,50 1,00 5,00
C (µg/mL) 0,300 0,300 0,300 0,304
RE (%) 20 -40 -70 -94
HYD
Co (µg/mL) 0,325 0,65 1,30 5,00
C (µg/mL) 0,307 0,304 0,302 0,299
RE (%) -6 -53 -77 -94
VAL
Co (µg/mL) 4,00 8,00 16,00 5,00
C (µg/mL) 0,301 0,300 0,300 0,299
RE (%) -93 -97 -98 -94
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Từ các kết quả khảo sát với hỗn hợp chứa 2 chất và 3 chất ở trên, có thể cho
rằng, cách lựa chọn giá trị khởi tạo ngẫu nhiên sẽ không cho ra các kết quả tin cậy,
vì nếu giá trị nồng độ khởi tạo ngẫu nhiên khác nhiều so với giá trị thực của nó,
phương pháp Kalman sẽ bị mắc sai số lớn hay bị “phân kỳ”. Ở đây, cũng lưu ý rằng,
các chất trong các hỗn hợp khảo sát có phổ hấp thụ quang xen phủ nhau, nên ảnh
hưởng qua lại giữa chúng trong phương pháp Kalman là rất lớn và do vậy, để
phương pháp cho ra kết quả tin cậy (hay mắc sai số nhỏ), cần có cách lựa chọn giá
trị khởi tạo phù hợp hơn. Cũng cần thấy rằng, các nhận xét trên chỉ đúng cho trường
hợp các chất trong hỗn hợp khảo sát là các hoạt chất trong các mẫu dược phẩm và
chúng có phổ hấp thụ quang xen phủ nhau. Trong các trường hợp cụ thể khác,
chẳng hạn, hỗn hợp các kim loại có phổ hấp thụ quang xen phủ nhau, phương pháp
Kalman có thể vẫn cho kết quả tin cậy. Điều này đã được tác giả ở [27] công bố khi
nghiên cứu phân tích đồng thời hỗn hợp các nguyên tố đất hiếm.

69
3.1.2. Lựa chọn giá trị khởi tạo giả định
Kết quả khảo sát với cách lựa chọn giá trị khởi tạo ngẫu nhiên ở trên (đối với
hỗn hợp các hoạt chất trong dược phẩm) đã cho rằng, phương pháp Kalman cho ra
các kết quả mắc sai số lớn (hay không tin cậy), trừ khi giá trị khởi tạo ngẫu nhiên
(giá trị nồng độ) gần với giá trị thực của nồng độ chất trong hỗn hợp. Để khắc phục
hạn chế đó, ở đây, tiến hành lựa chọn giá trị khởi tạo theo cách khác – lựa chọn giá
trị khởi tạo giả định.
Theo cách này, một số nghiên cứu đã cho ra các kết quả chấp nhận được
[49], [101], [110]. Theo các nghiên cứu đó, cần khảo sát sơ bộ nhiều lần với các giá
trị khởi tạo ban đầu khác nhau cho Cest(0) và Pest(0) để từ đó, chọn ra giá trị khởi tạo
cho nồng độ và phương sai. Một số nghiên cứu khác cho rằng, có thể thực hiện các
khảo sát sơ bộ đó một cách tự động (nhờ các chương trình máy tính tự xây dựng),
song, nói chung tốc độ tính toán tự động chưa tốt (hay còn chậm và mất thời gian)
và phải trải qua nhiều lần tính toán lặp lại mới có thể lựa chọn được giá trị khởi tạo
phù hợp [50], [99], [124].
Trong nghiên cứu này, chúng tôi khảo sát một cách lựa chọn giá trị khởi tạo
giả định khác so với các nghiên cứu trước đây (đối với hệ 2 hoặc 3 chất):
- Phương án 1: Giải hệ 2 (hoặc 3) phương trình với 2 (hoặc 3) ẩn số là nồng
độ chất) ở 2 (hoặc 3) bước sóng gần nhau (phương trình phụ thuộc giữa độ hấp thụ
quang và nồng độ chất trong hỗn hợp với các hệ số hấp thụ phân tử biết trước, tính
toán từ phổ của dung dịch chuẩn đơn cấu tử/hay đơn chất), sẽ xác định được nồng
độ các chất trong hỗn hợp, và lấy chúng làm các giá trị khởi tạo nồng độ. Còn giá trị
khởi tạo phương sai được lựa chọn ngẫu nhiên, chẳng hạn bằng 1.
- Phương án 2: Lựa chọn giá trị nồng độ khởi tạo ngẫu nhiên (nhưng có chủ
ý) là 0,3 µg/mL (cho mỗi chất bất kỳ trong hỗn hợp 2 hoặc 3 chất). Nhưng đối với
phương sai, giá trị khởi tạo cho nó không chọn ngẫu nhiên, mà được tính toán theo
phương trình Horwitz: Với nồng độ C = 0,3 µg/mL = 3.10-7, tính toán được phương
sai bằng 0,003 và lựa chọn giá trị này làm giá trị khởi tạo.
3.1.2.1. Đối với hệ hai cấu tử TEL và HYD

70
Áp dụng phương pháp Kalman cho bộ dữ liệu phổ đơn chất và hỗn hợp 2
chất (trong khoảng bước sóng 220 nm – 340 nm) với cách lựa chọn giá trị khởi tạo
giả định (theo phương án 1 và phương án 2), thu được các kết quả ở bảng 3.3 và
3.4. Ở đây, đối với phương án 1, để tính toán các giá trị nồng độ khởi tạo, chúng tôi
lựa chọn 2 bước sóng 220 nm và 220,5 nm để giải 2 phương trình tương ứng và tính
toán được C1(0) và C2(0) đối với mỗi hỗn hợp (H1 – H9) (các kết quả chi tiết về C1(0)
và C2(0) đối với các hỗn hợp khảo sát được nêu ở Phụ lục 6)
Bang 3.3. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 1(*)
Hỗn hợp H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
C (µg/mL) 0,99 1,99 2,95 3,88 5,03 6,07 7,18 7,99 9,00
RE (%) -0,9 -0,6 -2 -3 -0,6 1 3 -0,1 0
HYD Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
C (µg/mL) 8,91 7,84 6,86 6,02 5,06 3,95 3,01 1,98 1,03
RE (%) -1,1 -2,0 -2,0 0,4 1,3 -1,2 0,3 -0,8 3
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Bang 3.4. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 2(*)
Hỗn hợp H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
C (µg/mL) 0,30 0,30 0,31 0,31 0,32 0,35 0,38 0,42 0,48
RE (%) -70,0 -84,9 -89,8 -92,3 -93,5 -94,2 -94,6 -94,7 -94,6
HYD Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
C (µg/mL) 0,30 0,31 0,31 0,31 0,31 0,31 0,31 0,31 0,30
RE (%) -96,6 -96,2 -95,6 -94,8 -93,7 -92,2 -89,7 -84,7 -69,7
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả ở bảng 3.3 và 3.4 cho thấy:

71
- Theo phương án 1, phương pháp Kalman cho kết quả tin cậy về nồng độ
các chất trong hỗn hợp với các sai số RE < 3 % (đối với cả TEL và HYD). Tuy
vậy, theo phương án này, cách thực hiện khá phức tạp và phụ thuộc vào 2 bước
sóng lựa chọn để giải phương trình xác định các giá trị nồng độ khởi tạo. Mặt khác,
khi áp dụng vào thực tế, do ảnh hưởng của pha nền (matrix), phép đo phổ có thể
mắc sai số lớn hơn, nên phương án này có thể mắc sai số lớn hơn;
- Theo phương án 2, phương pháp Kalman cho kết quả mắc sai số lớn, dù
rằng giá trị phương sai khởi tạo đã được giả định phù hợp hơn so với cách chọn giá
trị phương sai ngẫu nhiên (bằng 1) như ở trường hợp trước (mục 3.1.1);
- Các kết quả trên cho phép nhận xét rằng, giữa nồng độ và phương sai, giá
trị khởi tạo nồng độ đóng vai trò quan trọng hơn (hay quyết định hơn) đến sai số
của kết quả cuối cùng (khi xác định theo phương pháp Kalman). Rõ ràng, cần phải
có cách phù hợp hơn để lựa chọn giá trị khởi tạo nồng độ.
3.1.2.2. Đối với hệ 3 cấu tử AML, HYD và VAL
Bang 3.5. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 1(*)
Kí hiệu H1 H2 H3 H4
AML
Co (µg/mL) 0,250 0,50 1,00 5,00
C (µg/mL) 1,731 0,478 0,530 5,032
RE (%) -30,8 -4,5 -47 0,6
HYD
Co (µg/mL) 0,325 0,65 1,30 5,00
C (µg/mL) 2,794 0,495 1,610 5,910
RE (%) -14,0 -23,8 23,85 18,2
VAL
Co (µg/mL) 4,00 8,00 16,00 5,00
C (µg/mL) 4,796 11,053 29,067 3,949
RE (%) 19,9 38,2 81,7 -21,03
(*) Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được

72
Từ bộ dữ liệu phổ của dung dịch chuẩn mỗi chất (AML, HYD và VAL) và
dung dịch hỗn hợp của chúng (trong khoảng bước sóng 230 nm – 340 nm), áp dụng
phương pháp Kalman để xác định nồng độ mỗi chất trong hỗn hợp của chúng theo
cách tương tự như đối với hỗn hợp 2 chất, thu được các kết quả ở bảng 3.5 và 3.6. Ở
đây, đối với phương án 1, để tính toán các giá trị nồng độ khởi tạo, chúng tôi lựa
chọn 3 bước sóng 230 nm, 230, 5 nm và 231 nm để giải 3 phương trình tương ứng
và tính toán được C1(0), C2(0) và C3(0) đối với mỗi hỗn hợp (H1 – H4) (các kết quả chi
tiết về C1(0), C2(0) và C3(0) đối với các hỗn hợp khảo sát được nêu ở Phụ lục 6).
Bang 3.6. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo giả định – Phương án 2(*)
Hỗn hợp H1 H2 H3 H4
AML
Co (µg/mL) 0,250 0,50 1,00 5,00
C (µg/mL) 0,300 0,300 0,282 0,477
RE (%) 20,0 -40,0 -71,8 -90,5
HYD
Co (µg/mL) 0,325 0,65 1,30 5,00
C (µg/mL) 0,301 0,304 0,368 0,443
RE (%) -7,4 -53,2 -71,7 -91,1
VAL
Co (µg/mL) 4,00 8,00 16,00 5,00
C (µg/mL) 0,319 0,454 0,542 0,289
RE (%) -92,0 -94,3 -96,6 -94,2
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả ở bảng 3.5 và 3.6 cho thấy:
- Theo phương án 1, ngoại trừ trường hợp đối với AML trong hỗn hợp H2 và
H4 (sai số RE < 4,5 %), các trường hợp còn lại đều có sai số lớn với RE khoảng 14
% – 82 %. Như vậy, khác với hệ 2 cấu tử (nồng độ của chúng chỉ mắc sai số với RE
< 3%), đối với hệ 3 cấu tử, phương pháp mắc sai số lớn hơn nhiều. Rõ ràng, khi số
cấu tử trong hệ tăng lên, ảnh hưởng qua lại của chúng sẽ lớn hơn, dẫn đến việc giải
hệ 3 phương trình với 3 ẩn số (nồng độ chất trong hệ) sẽ mắc sai số lớn hơn. Rõ

73
ràng, phương án 1 chỉ áp dụng được cho hệ 2 cấu tử. Mặt khác, phương án cũng khá
phức tạp, vì sai số của phương pháp phụ thuộc vào các bước sóng được lựa chọn để
thiết lập và giải phương trình.
- Theo phương án 2, cũng tương tự như trường hợp hệ 2 cấu tử, mặc dù việc đưa
ra giá trị khởi tạo cho phương sai tiếp cận với thực tế hơn (do được ước lượng từ phương
trình Horwitz), song phương pháp vẫn mắc sai rất lớn với RE khoảng 7% – 97 %).
Đến đây, có thể thấy rằng, cả 2 cách lựa chọn giá trị khởi tạo cho nồng độ và
phương sai – lựa chọn giá trị khởi tạo ngẫu nhiên và lựa chọn giá trị khởi tạo giả
định – đều chưa cho kết quả tốt (hay mắc sai số lớn), trừ khi giá trị khởi tạo nồng độ
được chọn ngẫu nhiên, hoặc được tính toán như phương án 1 (thuộc cách lựa chọn
gái trị khởi tạo giả định), gần với giá trị thực của nồng độ chất trong hệ. Rõ ràng,
cần phải có một cách lựa chọn giá trị khởi tạo khác, sao cho giá trị nồng độ khởi tạo
của chất trong hệ càng gần với giá trị thực của nó càng tốt.
Xuất phát từ những lí do trên, cần phải đề xuất một giải pháp lựa chọn giá trị
khởi tạo mới nhằm đáp ứng 3 yêu cầu:
- Giá trị nồng độ khởi tạo càng gần với giá trị thực của chất trong hệ càng tốt;
- Phương sai (hay sai số) của nồng độ không nên lựa chọn ngẫu nhiên, mà
nên lựa chọn sao cho phù hợp với các hướng dẫn của quốc tế khi xác định một nồng
độ C bất kỳ, chẳng hạn, dựa vào phương trình Horwitz để ước lượng giá trị phương
sai khởi tạo;
- Giải pháp khởi tạo đưa ra phải sao cho dễ dàng áp dụng vào thực tế khi
phân tích một hỗn hợp chất bất kỳ, mà chưa biết trước nồng độ của chúng.
3.1.3. Lựa chọn giá trị khởi tạo gần đúng
Qua nghiên cứu lý thuyết và thực nghiệm đối với các hệ chứa 2 – 3 cấu tử có
phổ hấp thụ quang xen phủ nhau và để đáp ứng các yêu cầu nêu trên, chúng tôi lựa
chọn giải pháp khởi tạo mới (được gọi là lựa chọn giá trị khởi tạo gần đúng). Giải
pháp như sau:
- Áp dụng phương pháp bình phương tối thiểu thông thường (viết tắt là
BPTT) để giải hệ m phương trình với n ẩn số (m là số bước sóng được lựa chọn để
quét phổ hấp thụ quang của dung dịch hỗn hợp các cấu tử, n là số cấu tử trong hệ),
sử dụng phương pháp khử Gauss để đưa hệ phương trình về dạng n phương trình
với n ẩn số; Các phương trình của hệ có dạng bội tuyến tính và thỏa mãn tính cộng

74
tính của độ hấp thụ quang [2]; Nồng độ các cấu tử thu được từ việc giải hệ phương
trình đó được chọn làm giá trị khởi tạo nồng độ Cest(0); Theo cách này, các giá trị
nồng độ ước lượng ban đầu tương đối gần với giá trị thực của nồng độ cấu tử trong
hệ đang nghiên cứu, bất kể là hệ đã biết trước nồng độ thực (chẳng hạn, dung dịch
chuẩn của hỗn hợp các cấu tử) hoặc chưa biết trước nồng độ thực của các cấu tử
trong hệ (chẳng hạn, mẫu thực tế);
- Áp dụng phương trình Horwitz để ước lượng giá trị phương sai ứng với
nồng độ C của mỗi cấu tử trong hệ và chấp nhận giá trị thu được là giá trị khởi tạo
cho phương sai đối với mỗi cấu tử Pest(0); Theo cách này, sẽ khắc phục được hạn chế
của 2 giải pháp lựa chọn giá trị khởi tạo trước (lựa chọn giá trị khởi tạo ngẫu nhiên
và giả định) – đưa ra giá trị khởi tạo cho phương sai một cách ngẫu nhiên và thiếu
thực tế, chẳng hạn, khi nồng độ cấu tử trong hệ là 0,3 µg/mL, mà lại chọn giá trị
phương sai bằng 1 là không phù hợp, mà nên phải cỡ 0,01 – 0,02 (µg/mL)2. Giá trị
phương sai Pest(0) ứng với nồng độ Cest(0) đối với mỗi cấu tử trong hệ được tính từ
phương trình Horwitz như sau:
- Từ công thức (3.1),
RSD (%) = ×100or(0)
H witzest
S
C (3.1)
Tính được độ lệch chuẩn S [RSDHorwitz*Cest(0)]/100;
Trong đó, RSDHorwitz được tính theo công thức (3.2), mà trong đó Cest(0) được
biểu diễn bằng phân số.
( ) (0)
1 0.5lgC% 2 est
HorwitzRSD-
= (3.2)
- Từ S, tính được phương sai S2 Pest(0).
- Khi phân tích lặp lại trong nội bộ phòng thí nghiệm, người ta chấp nhận
rằng, nếu đạt được độ lặp lại với RSD ½ RSDHorwitz, thì phương pháp có độ lặp lại
thỏa mãn yêu cầu. Như vậy, độ lệch chuẩn S tính được giảm một nửa và từ đó, Pest(0)
ở trên phải giảm đi ¼. Nghĩa là, khi chọn giá trị phương sai khởi tạo ứng với nồng
độ Cest(0), phải lấy giá trị Pest(0) ở trên chia cho 4.
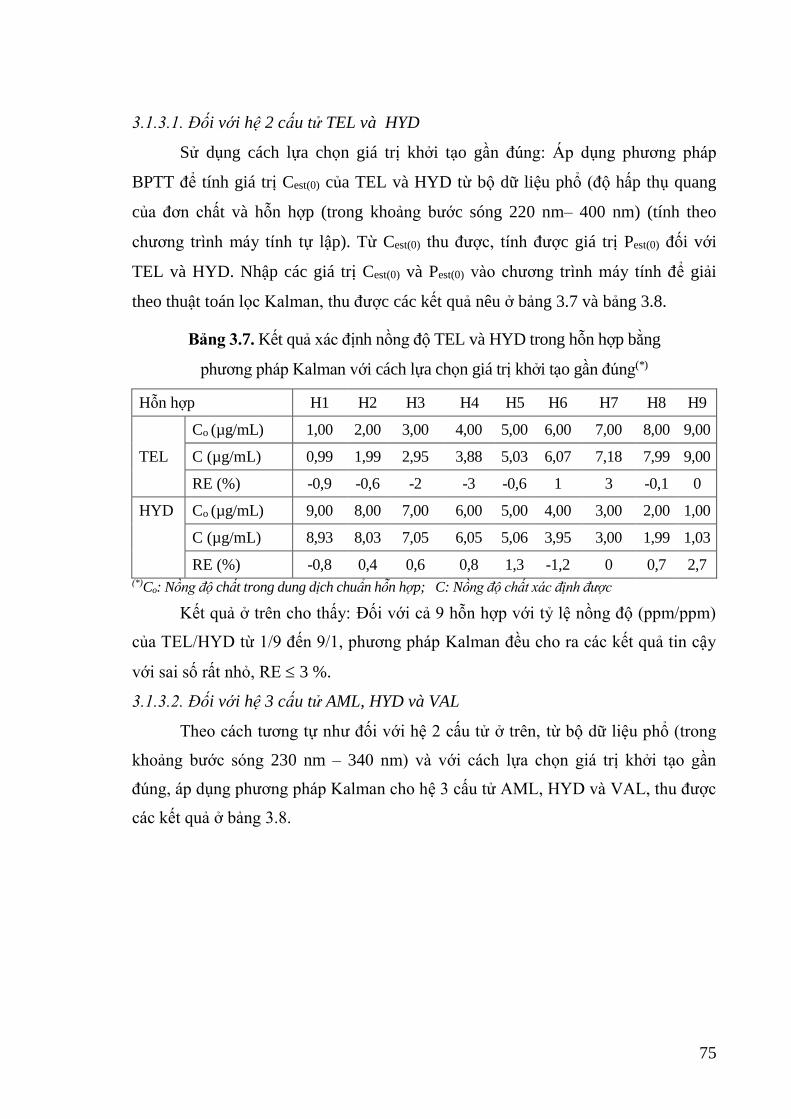
75
3.1.3.1. Đối với hệ 2 cấu tử TEL và HYD
Sử dụng cách lựa chọn giá trị khởi tạo gần đúng: Áp dụng phương pháp
BPTT để tính giá trị Cest(0) của TEL và HYD từ bộ dữ liệu phổ (độ hấp thụ quang
của đơn chất và hỗn hợp (trong khoảng bước sóng 220 nm– 400 nm) (tính theo
chương trình máy tính tự lập). Từ Cest(0) thu được, tính được giá trị Pest(0) đối với
TEL và HYD. Nhập các giá trị Cest(0) và Pest(0) vào chương trình máy tính để giải
theo thuật toán lọc Kalman, thu được các kết quả nêu ở bảng 3.7 và bảng 3.8.
Bang 3.7. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp bằng
phương pháp Kalman với cách lựa chọn giá trị khởi tạo gần đúng(*)
Hỗn hợp H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
C (µg/mL) 0,99 1,99 2,95 3,88 5,03 6,07 7,18 7,99 9,00
RE (%) -0,9 -0,6 -2 -3 -0,6 1 3 -0,1 0
HYD Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
C (µg/mL) 8,93 8,03 7,05 6,05 5,06 3,95 3,00 1,99 1,03
RE (%) -0,8 0,4 0,6 0,8 1,3 -1,2 0 0,7 2,7 (*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả ở trên cho thấy: Đối với cả 9 hỗn hợp với ty lệ nồng độ (ppm/ppm)
của TEL/HYD từ 1/9 đến 9/1, phương pháp Kalman đều cho ra các kết quả tin cậy
với sai số rất nhỏ, RE 3 %.
3.1.3.2. Đối với hệ 3 cấu tử AML, HYD và VAL
Theo cách tương tự như đối với hệ 2 cấu tử ở trên, từ bộ dữ liệu phổ (trong
khoảng bước sóng 230 nm – 340 nm) và với cách lựa chọn giá trị khởi tạo gần
đúng, áp dụng phương pháp Kalman cho hệ 3 cấu tử AML, HYD và VAL, thu được
các kết quả ở bảng 3.8.

76
Bang 3.8. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp
bằng phương pháp Kalman với cách lựa chọn giá trị khởi tạo gần đúng (*)
Hỗn hợp H1 H2 H3 H4
AML
Co (µg/mL) 0,250 0,50 1,00 5,00
C (µg/mL) 0,253 0,511 1,016 4,981
RE (%) 1,2 2,2 1,6 0,4
HYD
Co (µg/mL) 0,325 0,65 1,30 5,00
C (µg/mL) 0,320 0,646 1,290 5,064
RE (%) -1,5 -0,6 -0,8 1,3
VAL
Co (µg/mL) 4,00 8,00 16,00 5,00
C (µg/mL) 3,99 8,06 16,05 4,821
RE (%) -0,2 0,8 0,3 -3,6
*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được .
Các kết quả cho thấy, phương pháp cho ra các kết quả tin cậy về nồng độ của
3 cấu tử trong hệ với sai số nhỏ, RE 4 %.
Như vậy, đối với cả hệ 2 và 3 cấu tử, giải pháp lựa chọn giá trị khởi tạo gần
đúng đều cho kết quả tin cậy hơn 2 giải pháp lựa chọn giá trị khởi tạo ngẫu nhiên và
giả định. Song, để khẳng định chắc chắn hơn về giải pháp lựa chọn giá trị khởi tạo
gần đúng cũng như lợi thế của phương pháp Kalman (với giải pháp lựa chọn đó),
cần có những nghiên cứu so sánh phương pháp Kalman với một số phương pháp
truyền thống khác như: Phương pháp chemometric-trắc quang sử dụng thuật toán
bình phương tối thiểu (viết tắt là BPTT), phương pháp phổ đạo hàm (viết tắt là
PĐH) khi xác định nồng độ các cấu tử trong hỗn hợp của chúng cả trong dung dịch
chuẩn và mẫu thực tế (mẫu dược phẩm).

77
3.2. Chương trình máy tính để tính toán theo thuật toán lọc Kalman
Hinh 3.1. Sơ đồ chương trình tính toán theo thuật toán lọc Kalman với
giải pháp lựa chọn giá trị khởi tạo gần đúng (áp dụng cho hệ 2 và 3 cấu tử).
Tiến trinh tính toán như sau: Pha dung dịch chuẩn của các đơn chất, hỗn
hợp và ghi phổ UV (Đo độ hấp thụ quang (hay mật độ quang) của các dung dịch
chuẩn đơn chất và hỗn hợp trong vùng bước sóng khảo sát; Lưu các file dữ liệu phổ
ở dạng đuôi txt; Nhập file dữ liệu phổ (có đuôi txt) vào vùng nhập dữ liệu của
chương trình tính; Chạy modul phương pháp BPTT để xác định các giá trị hệ số hấp
thụ …. .Cuối cùng cho ra các giá trị nồng độ các cấu tử trong hệ. Đây cũng chính
là các giá trị nồng độ khởi tạo Cest(0); Các giá trị Cest(0) được tự động đưa vào modul
tính phương sai và cho ra kết quả các giá trị Pest(0) và đây là các giá trị phương sai
khởi tạo;
Các giá trị khởi tạo Cest(0), Pest(0) và bộ dữ liệu phổ đơn chất và hỗn hợp của
chúng (đã lưu sẵn trong chương trình tính từ khi nhập dữ liệu đầu vào) được tự
động chuyển vào modul tính toán theo thuật toán lọc Kalman và cuối cùng xuất kết

78
quả ra ở dạng bảng dữ liệu (trên excel). Chương trình cho phép in ra các kết quả về
nồng độ của mỗi cấu tử trong hỗn hợp và sai số tương đối RE tương ứng.
3.3. Kiểm chứng phương pháp Kalman đối với hỗn hợp hai cấu tử
Để kiểm chứng phương pháp Kalman với giải pháp lựa chọn giá trị khởi tạo
mới – lựa chọn giá trị khởi tạo gần đúng (và áp dụng chương trình máy tính đã lập
để tính toán nhanh), tiến hành xác định nồng độ các cấu tử (chất phân tích) trong
hỗn hợp của chúng bằng 3 phương pháp để so sánh: i) phương pháp Kalman,; ii)
phương pháp chemometric-trắc quang, sử dụng thuật toán bình phương tối thiểu
dùng phổ toàn phần (viết tắt là BPTT) và phương pháp chemometric-trắc quang
dùng phổ đạo hàm (viết tắt là PĐH). Các hỗn hợp (chứa 2 cấu tử) khảo sát gồm:
Telmisartan (TEL) và Hydrochlothiazide (HYD); Paracetamol (PAR) và
Cafein (CAF); Paracetamol (PAR) và Ibuprofen (IB).
3.3.1. Phổ hấp thụ quang và phổ đạo hàm
3.3.1.1. Đối với TEL và HYD
Theo [30], [42], [65] cả hai hoạt chất TEL và HYD đều tan tốt trong dung môi
NaOH 0,1 M. Vì vậy chọn dung môi NaOH 0,1 M làm dung môi hòa tan trong quá
trình xác định đồng thời TEL và HYD bằng phương pháp quang phổ – chemometric
dùng phổ toàn phần.
Phổ hấp thụ của các dung dịch chuẩn TEL, HYD (10,0 g/mL) trong môi
trường NaOH 0,1M và hỗn hợp của chúng ở khoảng bước sóng 220 nm – 340 nm
được biểu diễn ở hình 3.2.
240 260 280 300 320 3400.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
(4)(3)
(2)
(1)
A
(nm)
(1) HYD 10
(2) TEL 10
(3) HH 10-10 theo LT
(4) HH 10-10 theo TN
Hinh 3.2. Phổ hấp thụ quang của dung dịch chuẩn HYD, TEL, hỗn hợp TEL và HYD
trong dung môi NaOH 0.1M.

79
Từ hình 3.2 cho thấy: Trong khoảng bước sóng 240 nm – 335 nm thì phổ hấp thụ
của các dung dịch chuẩn của TEL và HYD xen phủ nhau và có tính chất cộng tính.
Dựa trên phổ hấp thụ của dung dịch chuẩn TEL 10 µg/mL và HYD 10 µg/mL
và dung dịch hỗn hợp TEL 16 µg/mL, HYD 5 µg/mL (lấy tỉ lệ giống với tỉ lệ trong
thuốc). Tiến hành ghi phổ đạo hàm bậc một của các dung dịch trong khoảng bước
sóng 220 nm - 340 nm, khoảng cách bước sóng ghi giá trị phổ là 0,2 nm. Kết quả
được thể hiện ở hình 3.3
220 240 260 280 300 320 340-0.04
-0.02
0.00
0.02
0.04
0.06
0.08
A'
(nm)
TEL
HYD
HH1
HH2
HH3
Hinh 3.3. Phổ đạo hàm bậc một của các dung dịch chuẩn TEL, HYD
và dung dịch hỗn hợp TEL, HYD.
Từ hình 3.3. cho thấy: Trong khoảng bước sóng 220 nm - 340 nm phổ đạo hàm bậc
một của hai chất xen phủ nhau. Tại bước sóng 322,8 nm giá trị phổ đạo hàm bậc một của
HYD bằng 0, còn giá trị phổ đạo hàm bậc một của TEL khác không. Do đó có thể chọn
bước sóng 1 = 322,8 nm để định lượng TEL. Còn tại bước sóng 282,4 nm, giá trị phổ đạo
hàm bậc một của TEL và HYD đạt cực trị. Do đó có thể chọn bước sóng 2 = 282,4 nm để
định lượng HYD sau khi đã định lượng TEL trước đó và trừ sự đóng góp của giá trị phổ đạo
hàm bậc một của TEL vào phổ đạo hàm bậc một của hỗn hợp hai chất tại bước sóng này.
3.3.1.2. Đối với PAR và CAR
Pha các dung dịch chuẩn PAR 5 µg/mL, CAF 5 µg/mL. Quét phổ của các
dung dịch PAR 5 µg/mL và CAF 5 µg/mLvà hỗn hợp của chúng từ 200 nm - 300
nm. Phổ hấp thụ phân tử của các dung dịch được thể hiện ở hình 3.4.

80
200 220 240 260 280 300
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
(4)
(3)
(2)
(1) CAF 5 g/mL
(2) PAR 5 g/mL
(3) HH 5 - 5 TN
(4) HH 5 - 5 LT
(1)
A
nm)
Hinh 3.4. Phổ hấp thụ quang của dung dịch chuẩn PAR, CAF, hỗn hợp PAR và CAF.
Từ hình 3.4 cho thấy: Trong khoảng bước sóng từ 215 nm – 300 nm thì phổ
hấp thụ của các dung dịch chuẩn của PAR và CAF xen phủ nhau và có tính chất
cộng tính.
Lấy phổ đạo hàm của các dung dịch chuẩn PAR 5 µg/mL và CAF 5 µg/mL từ
phổ hấp thụ phân tử của chúng. Phổ đạo hàm bậc 1 của dung dịch PAR 5 µg/mL và
CAF 5 µg/mL được biểu diễn ở hình 3.5.
200 220 240 260 280 300
-0,04
-0,02
0,00
0,02
0,04
0,06
0,08
216,4205,4
(2)
(1) PAR 5 g/mL
(2) CAF 5 g/mL
A'
nm)
(1)
Hinh 3.5. Phổ đạo hàm bậc 1 của dung dịch chuẩn PAR và CAF.
Từ hình 3.5 cho thấy: Tại bước sóng 205,4 nm giá trị phổ đạo hàm bậc 1 A’CAF =
0, A’PAR ≠ 0. Có thể sử dụng bước sóng này để xác định giá trị phổ đạo hàm riêng phần
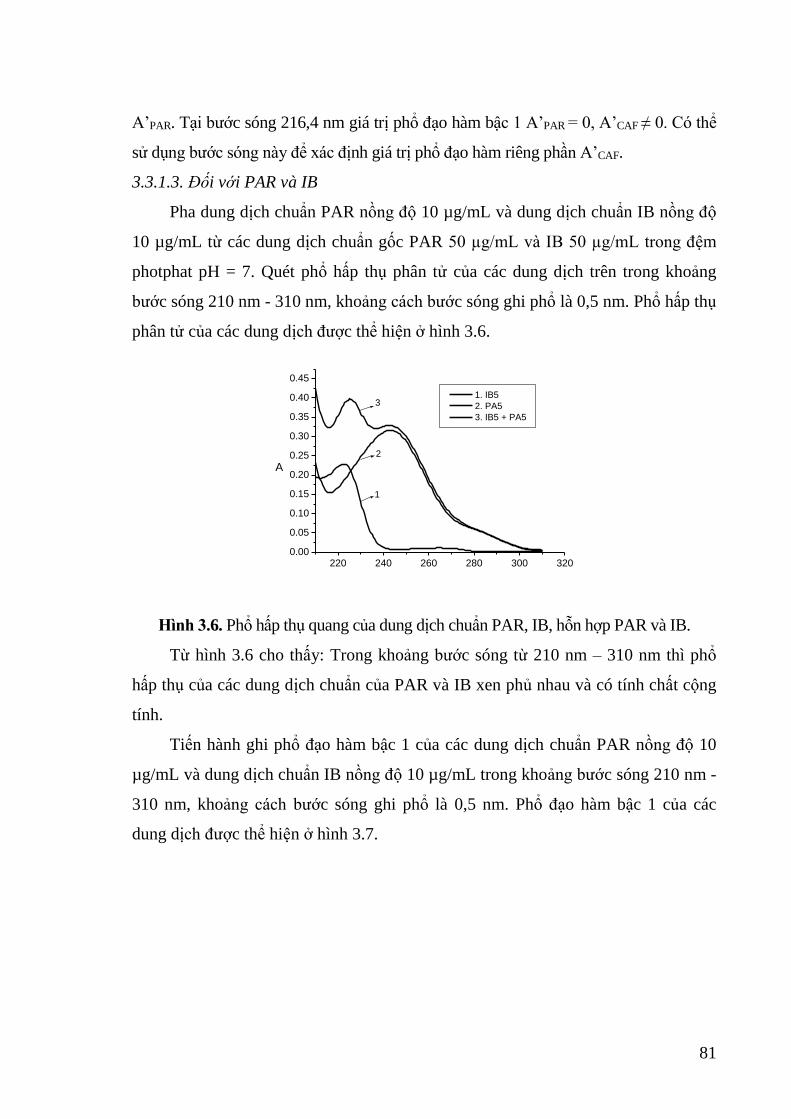
81
A’PAR. Tại bước sóng 216,4 nm giá trị phổ đạo hàm bậc 1 A’PAR = 0, A’CAF ≠ 0. Có thể
sử dụng bước sóng này để xác định giá trị phổ đạo hàm riêng phần A’CAF.
3.3.1.3. Đối với PAR và IB
Pha dung dịch chuẩn PAR nồng độ 10 µg/mL và dung dịch chuẩn IB nồng độ
10 µg/mL từ các dung dịch chuẩn gốc PAR 50 µg/mL và IB 50 µg/mL trong đệm
photphat pH = 7. Quét phổ hấp thụ phân tử của các dung dịch trên trong khoảng
bước sóng 210 nm - 310 nm, khoảng cách bước sóng ghi phổ là 0,5 nm. Phổ hấp thụ
phân tử của các dung dịch được thể hiện ở hình 3.6.
Hinh 3.6. Phổ hấp thụ quang của dung dịch chuẩn PAR, IB, hỗn hợp PAR và IB.
Từ hình 3.6 cho thấy: Trong khoảng bước sóng từ 210 nm – 310 nm thì phổ
hấp thụ của các dung dịch chuẩn của PAR và IB xen phủ nhau và có tính chất cộng
tính.
Tiến hành ghi phổ đạo hàm bậc 1 của các dung dịch chuẩn PAR nồng độ 10
µg/mL và dung dịch chuẩn IB nồng độ 10 µg/mL trong khoảng bước sóng 210 nm -
310 nm, khoảng cách bước sóng ghi phổ là 0,5 nm. Phổ đạo hàm bậc 1 của các
dung dịch được thể hiện ở hình 3.7.
220 240 260 280 300 3200.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
3
2
1
A
1. IB5
2. PA5
3. IB5 + PA5
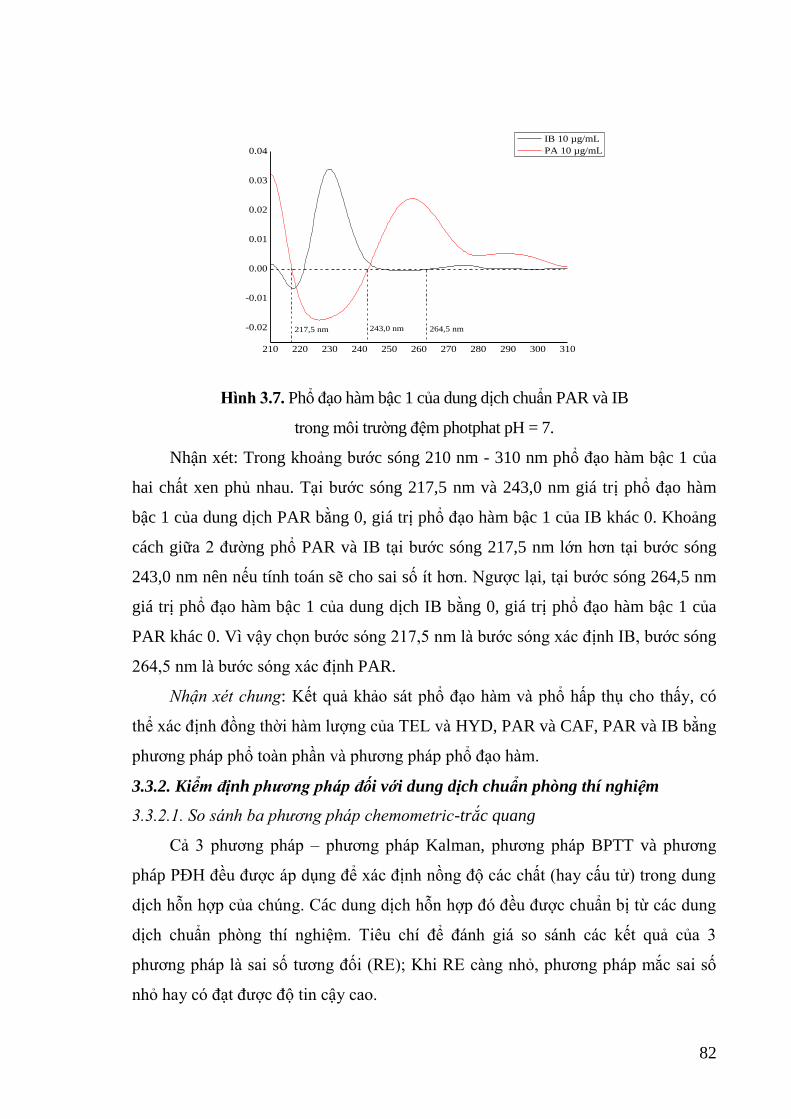
82
210 220 230 240 250 260 270 280 290 300 310
-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
217,5 nm 243,0 nm 264,5 nm
IB 10 µg/mL
PA 10 µg/mL
Hinh 3.7. Phổ đạo hàm bậc 1 của dung dịch chuẩn PAR và IB
trong môi trường đệm photphat pH = 7.
Nhận xét: Trong khoảng bước sóng 210 nm - 310 nm phổ đạo hàm bậc 1 của
hai chất xen phủ nhau. Tại bước sóng 217,5 nm và 243,0 nm giá trị phổ đạo hàm
bậc 1 của dung dịch PAR bằng 0, giá trị phổ đạo hàm bậc 1 của IB khác 0. Khoảng
cách giữa 2 đường phổ PAR và IB tại bước sóng 217,5 nm lớn hơn tại bước sóng
243,0 nm nên nếu tính toán sẽ cho sai số ít hơn. Ngược lại, tại bước sóng 264,5 nm
giá trị phổ đạo hàm bậc 1 của dung dịch IB bằng 0, giá trị phổ đạo hàm bậc 1 của
PAR khác 0. Vì vậy chọn bước sóng 217,5 nm là bước sóng xác định IB, bước sóng
264,5 nm là bước sóng xác định PAR.
Nhận xét chung: Kết quả khảo sát phổ đạo hàm và phổ hấp thụ cho thấy, có
thể xác định đồng thời hàm lượng của TEL và HYD, PAR và CAF, PAR và IB bằng
phương pháp phổ toàn phần và phương pháp phổ đạo hàm.
3.3.2. Kiểm định phương pháp đối với dung dịch chuẩn phòng thí nghiệm
3.3.2.1. So sánh ba phương pháp chemometric-trắc quang
Cả 3 phương pháp – phương pháp Kalman, phương pháp BPTT và phương
pháp PĐH đều được áp dụng để xác định nồng độ các chất (hay cấu tử) trong dung
dịch hỗn hợp của chúng. Các dung dịch hỗn hợp đó đều được chuẩn bị từ các dung
dịch chuẩn phòng thí nghiệm. Tiêu chí để đánh giá so sánh các kết quả của 3
phương pháp là sai số tương đối (RE); Khi RE càng nhỏ, phương pháp mắc sai số
nhỏ hay có đạt được độ tin cậy cao.
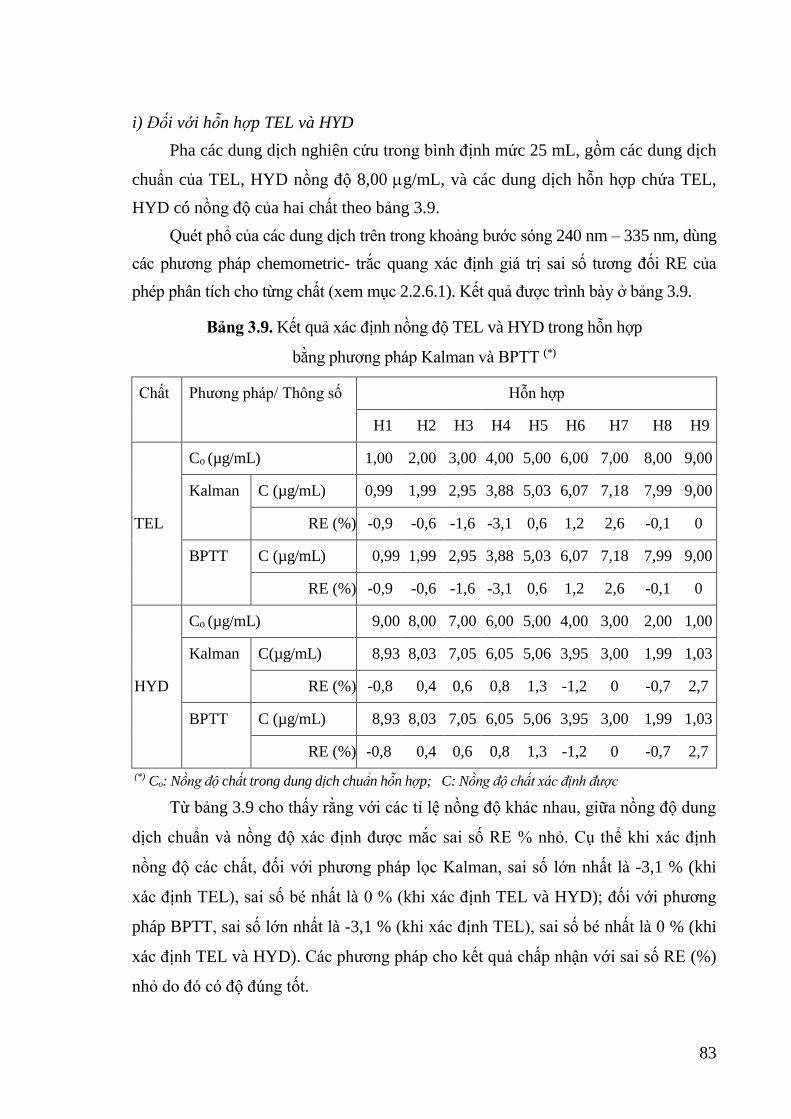
83
i) Đối với hỗn hợp TEL và HYD
Pha các dung dịch nghiên cứu trong bình định mức 25 mL, gồm các dung dịch
chuẩn của TEL, HYD nồng độ 8,00 g/mL, và các dung dịch hỗn hợp chứa TEL,
HYD có nồng độ của hai chất theo bảng 3.9.
Quét phổ của các dung dịch trên trong khoảng bước sóng 240 nm – 335 nm, dùng
các phương pháp chemometric- trắc quang xác định giá trị sai số tương đối RE của
phép phân tích cho từng chất (xem mục 2.2.6.1). Kết quả được trình bày ở bảng 3.9.
Bang 3.9. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp
bằng phương pháp Kalman và BPTT (*)
Chất Phương pháp/ Thông số Hỗn hợp
H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
Kalman C (µg/mL) 0,99 1,99 2,95 3,88 5,03 6,07 7,18 7,99 9,00
RE (%) -0,9 -0,6 -1,6 -3,1 0,6 1,2 2,6 -0,1 0
BPTT C (µg/mL) 0,99 1,99 2,95 3,88 5,03 6,07 7,18 7,99 9,00
RE (%) -0,9 -0,6 -1,6 -3,1 0,6 1,2 2,6 -0,1 0
HYD
Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
Kalman C(µg/mL) 8,93 8,03 7,05 6,05 5,06 3,95 3,00 1,99 1,03
RE (%) -0,8 0,4 0,6 0,8 1,3 -1,2 0 -0,7 2,7
BPTT C (µg/mL) 8,93 8,03 7,05 6,05 5,06 3,95 3,00 1,99 1,03
RE (%) -0,8 0,4 0,6 0,8 1,3 -1,2 0 -0,7 2,7
(*) Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Từ bảng 3.9 cho thấy rằng với các tỉ lệ nồng độ khác nhau, giữa nồng độ dung
dịch chuẩn và nồng độ xác định được mắc sai số RE % nhỏ. Cụ thể khi xác định
nồng độ các chất, đối với phương pháp lọc Kalman, sai số lớn nhất là -3,1 % (khi
xác định TEL), sai số bé nhất là 0 % (khi xác định TEL và HYD); đối với phương
pháp BPTT, sai số lớn nhất là -3,1 % (khi xác định TEL), sai số bé nhất là 0 % (khi
xác định TEL và HYD). Các phương pháp cho kết quả chấp nhận với sai số RE (%)
nhỏ do đó có độ đúng tốt.
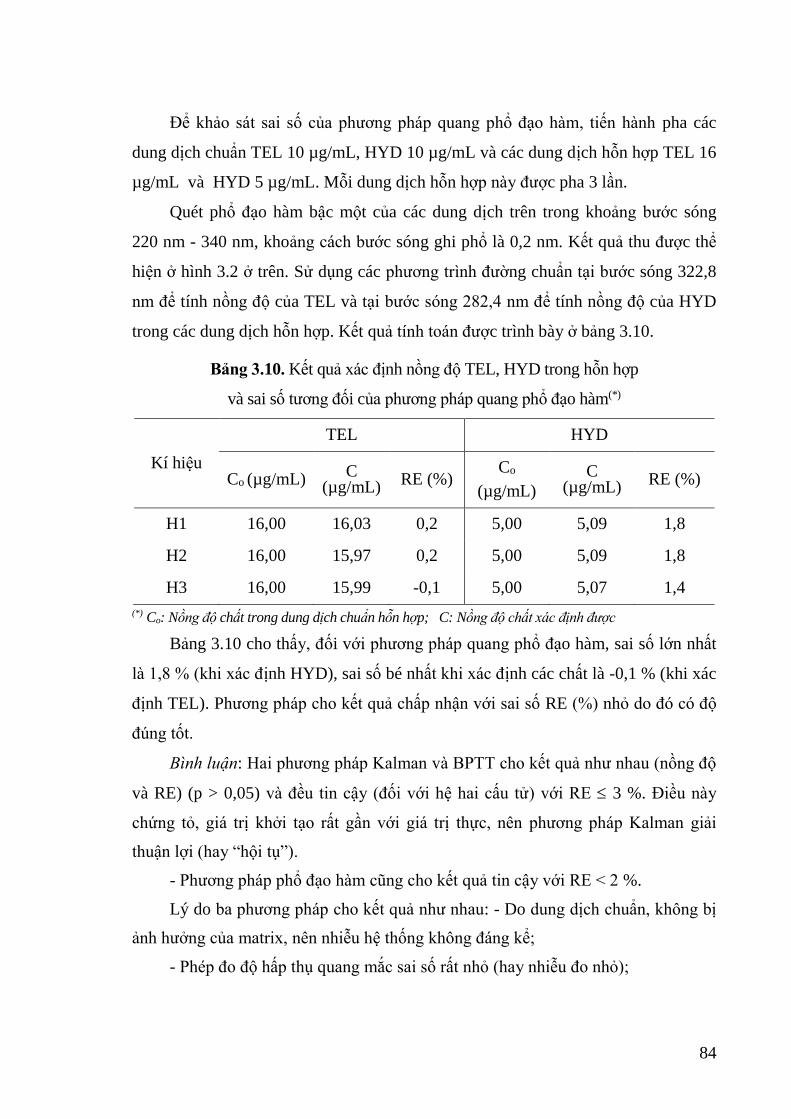
84
Để khảo sát sai số của phương pháp quang phổ đạo hàm, tiến hành pha các
dung dịch chuẩn TEL 10 µg/mL, HYD 10 µg/mL và các dung dịch hỗn hợp TEL 16
µg/mL và HYD 5 µg/mL. Mỗi dung dịch hỗn hợp này được pha 3 lần.
Quét phổ đạo hàm bậc một của các dung dịch trên trong khoảng bước sóng
220 nm - 340 nm, khoảng cách bước sóng ghi phổ là 0,2 nm. Kết quả thu được thể
hiện ở hình 3.2 ở trên. Sử dụng các phương trình đường chuẩn tại bước sóng 322,8
nm để tính nồng độ của TEL và tại bước sóng 282,4 nm để tính nồng độ của HYD
trong các dung dịch hỗn hợp. Kết quả tính toán được trình bày ở bảng 3.10.
Bang 3.10. Kết quả xác định nồng độ TEL, HYD trong hỗn hợp
và sai số tương đối của phương pháp quang phổ đạo hàm(*)
Kí hiệu
TEL HYD
Co (µg/mL) C
(µg/mL) RE (%)
Co
(µg/mL)
C (µg/mL)
RE (%)
H1 16,00 16,03 0,2 5,00 5,09 1,8
H2 16,00 15,97 0,2 5,00 5,09 1,8
H3 16,00 15,99 -0,1 5,00 5,07 1,4
(*) Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Bảng 3.10 cho thấy, đối với phương pháp quang phổ đạo hàm, sai số lớn nhất
là 1,8 % (khi xác định HYD), sai số bé nhất khi xác định các chất là -0,1 % (khi xác
định TEL). Phương pháp cho kết quả chấp nhận với sai số RE (%) nhỏ do đó có độ
đúng tốt.
Bình luận: Hai phương pháp Kalman và BPTT cho kết quả như nhau (nồng độ
và RE) (p > 0,05) và đều tin cậy (đối với hệ hai cấu tử) với RE 3 %. Điều này
chứng tỏ, giá trị khởi tạo rất gần với giá trị thực, nên phương pháp Kalman giải
thuận lợi (hay “hội tụ”).
- Phương pháp phổ đạo hàm cũng cho kết quả tin cậy với RE < 2 %.
Lý do ba phương pháp cho kết quả như nhau: - Do dung dịch chuẩn, không bị
ảnh hưởng của matrix, nên nhiễu hệ thống không đáng kể;
- Phép đo độ hấp thụ quang mắc sai số rất nhỏ (hay nhiễu đo nhỏ);
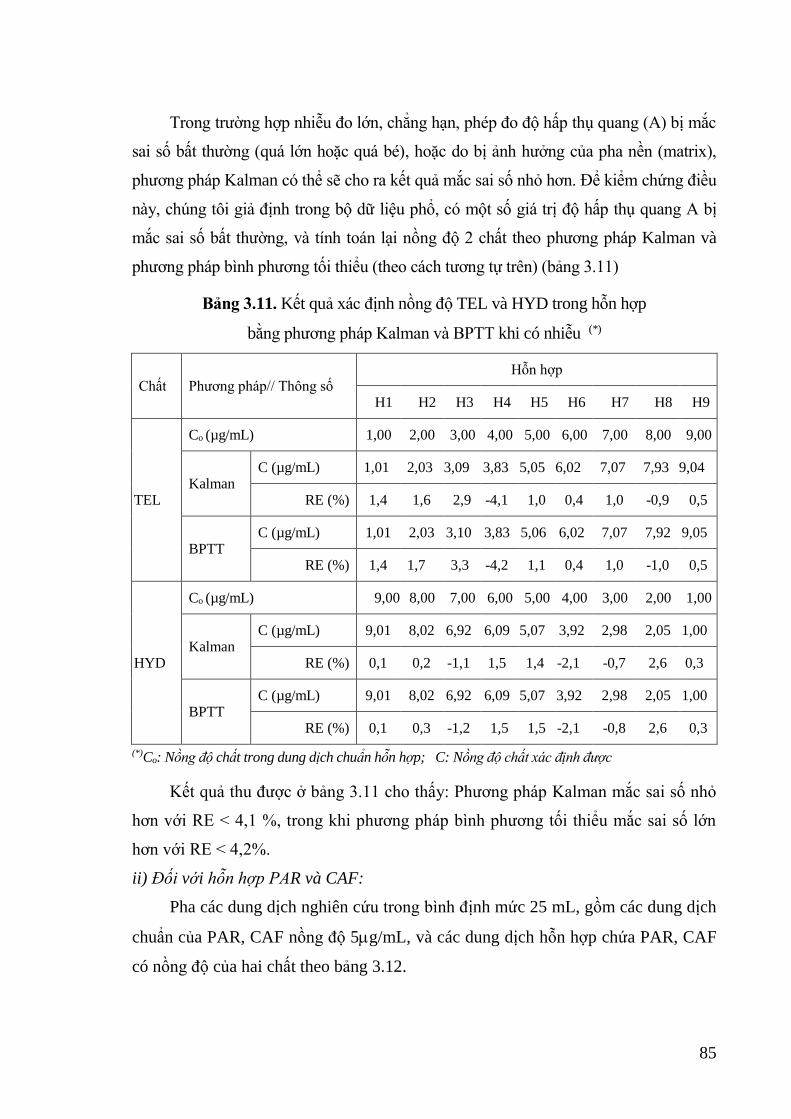
85
Trong trường hợp nhiễu đo lớn, chẳng hạn, phép đo độ hấp thụ quang (A) bị mắc
sai số bất thường (quá lớn hoặc quá bé), hoặc do bị ảnh hưởng của pha nền (matrix),
phương pháp Kalman có thể sẽ cho ra kết quả mắc sai số nhỏ hơn. Để kiểm chứng điều
này, chúng tôi giả định trong bộ dữ liệu phổ, có một số giá trị độ hấp thụ quang A bị
mắc sai số bất thường, và tính toán lại nồng độ 2 chất theo phương pháp Kalman và
phương pháp bình phương tối thiểu (theo cách tương tự trên) (bảng 3.11)
Bang 3.11. Kết quả xác định nồng độ TEL và HYD trong hỗn hợp
bằng phương pháp Kalman và BPTT khi có nhiễu (*)
Chất Phương pháp// Thông số Hỗn hợp
H1 H2 H3 H4 H5 H6 H7 H8 H9
TEL
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
Kalman C (µg/mL) 1,01 2,03 3,09 3,83 5,05 6,02 7,07 7,93 9,04
RE (%) 1,4 1,6 2,9 -4,1 1,0 0,4 1,0 -0,9 0,5
BPTT C (µg/mL) 1,01 2,03 3,10 3,83 5,06 6,02 7,07 7,92 9,05
RE (%) 1,4 1,7 3,3 -4,2 1,1 0,4 1,0 -1,0 0,5
HYD
Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
Kalman C (µg/mL) 9,01 8,02 6,92 6,09 5,07 3,92 2,98 2,05 1,00
RE (%) 0,1 0,2 -1,1 1,5 1,4 -2,1 -0,7 2,6 0,3
BPTT C (µg/mL) 9,01 8,02 6,92 6,09 5,07 3,92 2,98 2,05 1,00
RE (%) 0,1 0,3 -1,2 1,5 1,5 -2,1 -0,8 2,6 0,3
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả thu được ở bảng 3.11 cho thấy: Phương pháp Kalman mắc sai số nhỏ
hơn với RE < 4,1 %, trong khi phương pháp bình phương tối thiểu mắc sai số lớn
hơn với RE < 4,2%.
ii) Đối với hỗn hợp PAR và CAF:
Pha các dung dịch nghiên cứu trong bình định mức 25 mL, gồm các dung dịch
chuẩn của PAR, CAF nồng độ 5g/mL, và các dung dịch hỗn hợp chứa PAR, CAF
có nồng độ của hai chất theo bảng 3.12.

86
Quét phổ của các dung dịch trên trong khoảng bước sóng từ 215 nm – 300 nm,
dùng các phương pháp chemometric để xác định nồng độ của từng cấu tử trong các
dung dịch hỗn hợp, sau đó xác định giá trị sai số tương đối RE của phép phân tích
cho từng chất (xem mục 2.2.6.1). Kết quả được trình bày ở bảng 3.12.
Bang 3.12. Kết quả xác định nồng độ PAR, CAF trong hỗn hợp và
sai số tương đối của ba phương pháp (*)
Kí hiệu H1 H2 H3 H4 H5 H6 H7 H8 H9
PAR
Co (µg/mL) 1,00 2,00 3,00 4,00 5,00 6,00 7,00 8,00 9,00
Kalman C (µg/mL) 1,00 1,94 3,02 4,05 5,01 6,03 7,01 8,18 9,05
RE (%) 0,0 -2,9 0,7 1,3 0,3 0,4 0,1 2,3 0,5
BPTT C (µg/mL) 1,00 1,94 3,02 4,05 5,01 6,03 7,01 8,18 9,05
RE (%) 0 -2,9 0,7 1,3 0,3 0,4 0,1 2,3 0,5
PĐH C (µg/mL) 0,99 1,92 3,06 3,98 4,97 5,94 6,94 8,18 8,95
RE (%) -0,04 -4,0 2,0 -0,4 -0,6 -1,1 -0,9 2,3 -0,5
CAF
Co (µg/mL) 9,00 8,00 7,00 6,00 5,00 4,00 3,00 2,00 1,00
Kalman C (µg/mL) 9,04 8,06 7,05 6,10 5,05 4,05 3,00 2,00 1,00
RE (%) 0,5 0,8 0,8 1,7 0,9 1,2 0,0 0,0 0,0
BPTT C (µg/mL) 9,04 8,06 7,05 6,10 5,05 4,05 3,00 2,00 1,00
RE (%) 0,5 0,8 0,8 1,7 0,9 1,2 0,0 0,0 0,0
PĐH
C (µg/mL) 8,97 7,98 6,96 5,98 4,95 3,93 2,92 1,98 0,98
RE (%) -0,4 -0,3 -0,6 -0,3 -1 -1,7 -2,4 -0,9 -2,3
*) Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả cho thấy, cả 3 phương pháp đều mắc sai số nhỏ (hay có độ tin cậy
cao) khi xác định đồng thời PAR và CAF trong dung dịch hỗn hợp của chúng với
RE -4,0 %. Phương pháp Kalman cho kết quả hoàn toàn tương tự như phương
pháp bình phương tối thiểu. Lý do giải thích cho điều này cũng tương tự như đối
với trường hơp hỗn hợp TEL và HYD (mục 3.3.2.1 (i)). Song, áp dụng phương
pháp kiểm định t theo cặp (paired-t-test) cho thấy:

87
- Đối với PAR trong cả 9 hỗn hợp (H1 – H9), phương pháp Kalman cho kết quả
khác với phương pháp phổ đạo hàm với p 0,03: ttính 2,58 > t(0,05;f n-1 8) 2,3;
- Tương tự, đối với CAF trong cả 9 hỗn hợp (H1 – H9), phương pháp Kalman
cũng cho kết quả khác với phương pháp phổ đạo hàm với p 0,0002: ttính 6,31 >
t(0,05;f n-1 8) 2,3;
iii) Đối với hỗn hợp PAR và IB:
Pha các dung dịch nghiên cứu trong bình định mức 25 mL, gồm các dung dịch
chuẩn của PAR, IB nồng độ 10 g/mL, và các dung dịch hỗn hợp chứa PAR, IB có
nồng độ của hai chất theo bảng 3.13.
Quét phổ của các dung dịch trên trong khoảng bước sóng từ 210 nm – 300 nm,
dùng các phương pháp chemometric để xác định nồng độ của từng cấu tử trong các
dung dịch hỗn hợp, sau đó xác định giá trị sai số tương đối RE của phép phân tích
cho từng chất (xem mục 2.2.6.1). Kết quả được trình bày ở bảng 3.13.

88
Bang 3.13. Kết quả xác định nồng độ PAR, IB trong hỗn hợp và
sai số tương đối của các phương pháp (*)
Kí hiệu H1 H2 H3 H4 H5
PAR
Co (µg/mL) 5,00 5,00 5,00 5,00 5,00
Kalman C (µg/mL) 4,98 5,00 4,87 4,87 4.87
RE (%) -0,3 0,0 -2,6 -2,6 -2,6
BPTT C (µg/mL) 4,98 5,00 4,87 4,87 4.87
RE (%) -0,3 0,0 -2,6 -2,6 -2,6
PĐH C (µg/mL) 5,02 5,02 5,03 5,13 5,20
RE (%) 0,4 0,4 0,6 2,6 4,0
IB
Co (µg/mL) 1,00 2,50 5,00 10,00 15,00
Kalman C (µg/mL) 0,96 2,44 5,00 9,91 15,19
RE (%) -3,7 -2,4 0,0 -0,9 1,3
BPTT C (µg/mL) 0,96 2,44 5,00 9,91 15,19
RE (%) -3,7 -2,4 0,0 -0,9 1,3
PĐH C (µg/mL) 1,03 2,40 4,86 9,97 14,97
RE (%) 3,0 -4,0 -2,8 -0,3 -0,2
*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Kết quả cho thấy, cả 3 phương pháp đều mắc sai số nhỏ (hay có độ tin cậy
cao) khi xác định đồng thời PAR và IB trong dung dịch hỗn hợp của chúng với RE
-4,0%. Phương pháp Kalman cho kết quả hoàn toàn tương tự như phương pháp
bình phương tối thiểu. Lý do giải thích cho điều này cũng tương tự như đối với
trường hợp hỗn hợp TEL và HYD (mục 3.3.2.1 (i)).
Như vậy, từ các bảng 3.9 đến 3.13 cho thấy rằng với các tỉ lệ nồng độ khác
nhau, giữa nồng độ dung dịch chuẩn và nồng độ xác định được mắc sai số RE %
nhỏ. Cụ thể khi xác định nồng độ các chất, đối với phương pháp lọc Kalman, sai số
lớn nhất là -3,7 % (khi xác định IB trong hỗn hợp PAR và IB), sai số bé nhất là 0 %
(khi xác định HYD trong hỗn hợp TEL và HYD); đối với phương pháp BPTT, sai

89
số lớn nhất là -3,7 % (khi xác định IB trong hỗn hợp PAR và IB), sai số bé nhất là 0
% (khi xác định TEL trong hỗn hợp TEL và HYD); đối với phương pháp quang phổ
đạo hàm, sai số lớn nhất là 4,0 % (khi xác định IB trong hỗn hợp PAR và IB), sai số
bé nhất là 0,0 % (khi xác định IB trong hỗn hợp PAR và IB). Các phương pháp cho
kết quả chấp nhận với sai số RE (%) nhỏ do đó có độ đúng tốt.
3.3.2.2. Độ lặp lại của phương pháp khi phân tích dung dịch chuẩn phòng thí
nghiệm
Để tiếp tục kiểm định phương pháp phân tích, ngoài đánh giá qua sai số tương
đối (RE) như ở trên, ở đây sẽ tiến hành đánh giá độ lặp lại của phương pháp phân
tích. Độ lặp lại là một trong những thông số quan trọng thể hiện năng lực của
phương pháp phân tích.
Cả 3 phương pháp – phương pháp Kalman, phương pháp bình phương tối
thiểu và phương pháp phổ đạo hàm đều được áp dụng để xác định nồng độ các chất
(hay cấu tử) trong dung dịch hỗn hợp của chúng. Các dung dịch hỗn hợp đó đều
được chuẩn bị từ các dung dịch chuẩn phòng thí nghiệm. Tiêu chí để đánh giá các
kết quả của 3 phương pháp là độ lệch chuẩn tương đối (RSD) được so sánh với ½
RSDHorwitz. Khi RSD xác định được nhỏ hơn ½ RSDHorwitz, phương pháp được xem
là đạt được độ lặp lại thỏa mãn yêu cầu và khi RSD càng nhỏ nghĩa là độ lặp lại của
phương pháp càng tốt.
i) Đối với hỗn hợp TEL và HYD
Tiến hành xác thí nghiệm như mục 3.3.2.1 với mỗi dung dịch hỗn hợp được
chuẩn bị và phân tích lặp lại 3 lần. Sử dụng các phương pháp chemometric để xác
định nồng độ của TEL và HYD trong các dung dịch hỗn hợp và từ đó độ lặp lại của
phương pháp được đánh giá thông qua giá trị RSD tương ứng khi so sánh với ½
RSDHorwitz (xem mục 2.2.6.2). Kết quả tính toán được trình bày ở bảng 3.14.

90
Bang 3.14. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp TEL và HYD(*)
Kí hiệu mẫu
Thông số TEL HYD
Kalman BPTT Kalman BPTT
H1
C (µg/mL) 0,99/1,02/1,00 0,99/1,02/1,00 8,93/8,99/9,01 8,93/8,99/9,01
TB (µg/mL) 1,00 1,00 8,98 8,98
RSD (%) 2 2 0,4 0,4
½ RSDH (%) 8,0 5,8
H2
C (µg/mL) 1,99/1,96/1,98 1,99/1,96/1,98 8,03/8,05/8,05 8,03/8,05/8,05
TB (µg/mL) 1,98 1,98 8,04 8,04
RSD (%) 1,01 1,01 0,1 0,1
½ RSDH (%) 7,2 5,9
H3
C (µg/mL) 2,95/2,95/2,95 2,95/2,95/2,95 7,05/7,05/7,06 7,05/7,05/7,06
TB (µg/mL) 2,95 2,95 7,05 7,05
RSD (%) 0 0 0,1 0,1
½ RSDH (%) 6,8 6,0
H4
C (µg/mL) 3,88/3,88/3,89 3,88/3,88/3,89 6,05/6,05/6,05 6,05/6,05/6,05
TB (µg/mL) 3,88 3,88 6,05 6,05
RSD (%) 0,3 0,3 0 0
½ RSDH (%) 6,5 6,1
H5
C (µg/mL) 5,03/5,01/5,08 5,03/5,01/5,08 5,06/5,05/5,08 5,06/5,05/5,08
TB (µg/mL) 5,04 5,04 5,06 5,06
RSD (%) 0,8 0,8 0,4 0,4
½ RSDH (%) 6,3 6,3
H6
C (µg/mL) 6,07/6,03/6,01 6,07/6,03/6,01 3,95/3,91/3,92 3,95/3,91/3,92
TB (µg/mL) 6,04 6,04 3,93 3,93
RSD (%) 0,5 0,5 0,5 0,5
½ RSDH (%) 6,1 6,5

91
Kí hiệu mẫu
Thông số TEL HYD
Kalman BPTT Kalman BPTT
H7
C (µg/mL) 7,18/7,00/7,02 7,18/7,00/7,02 3,00/2,94/2,97 3,00/2,94/2,97
TB (µg/mL) 7,07 7,07 2,97 2,97
RSD (%) 1,4 1,4 1,0 1,0
½ RSDH (%) 6,0 6,8
H8
C (µg/mL) 7,99/7,96/8,03 7,99/7,96/8,03 1,99/1,96/2,01 1,99/1,96/2,01
TB (µg/mL) 7,99 7,99 1,99 1,99
RSD (%) 0,5 0,5 1,5 1,5
½ RSDH (%) 5,9 7,2
H9
C (µg/mL 9,01/8,99/9,02 9,01/8,99/9,02 1,03/1,01/1,02 1,03/1,01/1,02
TB (µg/mL) 9,01 9,01 1,02 1,02
RSD (%) 0,2 0,2 1,0 1,0
½ RSDH (%) 5,8 8,0
(*) Đối với nồng độ C, kết quả đo trong bảng là kết quả đo lần 1/lần 2/lần 3; RSDH tính theo
hàm Horwitz [116]; TB: Trung bình số học của nồng độ
Từ bảng 3.14 cho thấy: Đối với phương pháp Kalman, giá trị RSD của TEL cả 3
lần đo lặp lại cho các mẫu từ H1 đến H9 là 2 %, của HYD là < 2 %; Đối với phương
pháp BPTT, giá trị RSD của TEL cả 3 lần đo lặp lại cho các mẫu từ H1 đến H9 là 2 %,
của HYD là < 2 %. Khi phân tích trong nội bộ PTN, nếu các giá trị RSD thu được nhỏ
hơn ½ RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40], [116]. Như vậy, phương
pháp này có độ lặp lại tốt với cả hai cấu tử.
** Phương pháp quang phổ đạo hàm:
Pha các dung dịch chuẩn TEL 10 µg/mL, HYD 10 µg/mL và các dung dịch hỗn
hợp TEL 16 µg/mL và HYD 5 µg/mL. Mỗi dung dịch hỗn hợp này được pha 3 lần.
Quét phổ đạo hàm bậc một của các dung dịch trên trong khoảng bước sóng
220 – 340 nm, khoảng cách bước sóng ghi phổ là 0,2 nm. Kết quả thu được được
thể hiện ở hình 3.3 ở trên.

92
Sử dụng các phương trình đường chuẩn tại bước sóng 322,8 nm để tính nồng
độ của TEL và tại bước sóng 282,4 nm để tính nồng độ của HYD trong các dung
dịch hỗn hợp. Kết quả tính toán được trình bày ở bảng 3.15.
Bang 3.15. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm(*)
Kí hiệu TEL HYD
C0 (µg/mL) C(µg/mL) C0 (µg/mL) C (µg/mL)
HH1 16,00 16,03 5,00 5,09
HH2 16,00 15,97 5,00 5,09
HH3 16,00 15,99 5,00 5,07
RSD (%) 0,2 0,2
½ RSDH (%) 5,3 6,3
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Từ bảng 3.15 cho thấy: Giá trị RSD của TEL cả 3 lần đo lặp lại là 0,2 %, của
HYD là 0,2 %. Khi phân tích trong nội bộ PTN, nếu các giá trị RSD thu được nhỏ
hơn ½RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40], [116].
Như vậy, các phương pháp chemometric này có độ lặp lại tốt với cả hai cấu tử.
ii) Đối với hỗn hợp PAR và CAF:
Tiến hành thí nghiệm như mục 3.3.2.1, với mỗi dung dịch hỗn hợp được chuẩn
bị và phân tích lặp lại 3 lần. Sử dụng các phương pháp chemometric để xác định
nồng độ của PAR và CAF trong các dung dịch hỗn hợp và từ đó độ lặp lại của
phương pháp được đánh giá thông quá giá trị RSD tương ứng khi so sánh với ½
RSDHorwitz (xem mục 2.2.6.2). Kết quả tính toán được trình bày ở bảng 3.16.

93
Bang 3.16. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp PAR và CAF (*)
Thông số PAR CAF
Kalman BPTT Kalman BPTT
H1
C (µg/mL) 1,00/0,99/1,00 1,00/0,99/1,00 9,04/9,00/9,08 9,04/9,00/9,08
CTB (µg/mL) 1,00 1,00 9,04 9,04
RSD (%) 1,0 1,0 0,4 0,4
½ RSDH (%) 8,0 5,8
H2
C (µg/mL) 1,94/1,95/1,93 1,94/1,95/1,93 8,06/8,09/8,03 8,06/8,09/8,03
CTB (µg/mL) 1,94 1,94 8,06 8,06
RSD (%) 0,5 0,5 0,4 0,4
½ RSDH (%) 7,2 5,9
H3
C (µg/mL) 3,02/3,01/3,03 3,02/3,01/3,03 7,05/7,03/7,09 7,05/7,03/7,09
CTB (µg/mL) 3,02 3,02 7,06 7,06
RSD (%) 0,3 0,3 0,4 0,4
½ RSDH (%) 6,8 6,0
H4
C (µg/mL) 4,05/4,07/4,03 4,05/4,07/4,03 6,10/6,13/6,07 6,10/6,13/6,07
CTB (µg/mL) 4,05 6,1 6,1
RSD (%) 0,4 0,5 0,5
½ RSDH (%) 6,5 6,1
H5
C (µg/mL) 5,01/5,00/5,03 5,01/5,00/5,03 5,05/5,04/5,07 5,05/5,04/5,07
CTB (µg/mL) 5,01 5,01 5,05 5,05
RSD (%) 0,4 0,4 0,4 0,4
½ RSDH (%) 6,3 6,3
H6
C (µg/mL) 6,03/6,05/6,01 6,03/6,05/6,01 4,05/4,06/4,04 4,05/4,06/4,04
CTB (µg/mL) 6,03 6,03 4,05 4,05
RSD (%) 0,3 0,3 0,2 0,2
½ RSDH (%) 6,1 6,5
H7
C (µg/mL) 7,01/7,04/7,01 7,01/7,04/7,01 3,00/3,02/3,00 3,00/3,02/3,00
CTB (µg/mL) 7,02 7,02 3,01 3,01
RSD (%) 0,3 0,3 0,3 0,3
½ RSDH(%) 6,0 6,8
H8
C (µg/mL 8,18/8,15/8,22 8,18/8,15/8,22 2,00/1,97/1,99 2,00/1,97/1,99
CTB (µg/mL) 8,18 8,18 1,99 1,99
RSD (%) 0,5 0,5 1,0 1,0
½ RSDH (%) 5,9 7,2
H9
C (µg/mL 9,0/9,08/9,00 9,05/9,08/9,00 1,00/1,00/0,99 1,00/1,00/0,99
CTB (µg/mL) 9,04 9,04 1,00 1,00
RSD (%) 0,4 0,4 1,0 1,0
½ RSDH (%) 5,8 8,0
(*) Đối với nồng độ C, kết quả đo trong bảng là kết quả đo lần 1/lần 2/lần 3; RSDH tính theo
hàm Horwitz [116]; TB: Trung bình số học của nồng độ

94
Từ bảng 3.16 cho thấy: Đối với phương pháp Kalman, giá trị RSD của PAR cả
3 lần đo lặp lại cho các mẫu từ H1 đến H9 là 1 %, của CAF là 1 %, đối với
phương pháp BPTT, giá trị RSD của PAR cả 3 lần đo lặp lại cho các mẫu từ H1 đến
H9 là 1 %,, của CAF là 1 %. Khi phân tích trong nội bộ PTN, nếu các giá trị
RSD thu được nhỏ hơn ½ RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40],
[116]. Như vậy, phương pháp này có độ lặp lại tốt với cả hai cấu tử.
Đánh giá độ lặp lại bằng phương pháp quang phổ đạo hàm cho hỗn hợp PAR
và CAF thì tiến hành như sau:
Pha 3 dung dịch hỗn hợp chuẩn gồm 5 µg/mL PAR và 5 µg/mL CAF (nồng độ
C0). Quét phổ hấp thụ các dung dịch trong khoảng bước sóng từ 215 nm– 300 nm. Lấy
giá trị phổ đạo hàm của hỗn hợp tại bước sóng 216,4 nm và 260 nm, xác định nồng độ
PAR và CAF theo phương pháp đường chuẩn. Kết quả được trình bày ở bảng 3.17.
Bang 3.17. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm(*)
Lần PAR CAF
C (µg/mL) RE (%) C (µg/mL) RE (%)
1 5,03 0,52 4,97 -0,69
2 5,04 0,88 4,96 -0,90
3 5,03 0,61 4,95 -0,98
TB 5,03 4,96
RSD (%) 0,2 0,2
½ RSDH(%) 6,3 6,3
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Từ bảng 3.17 cho thấy: Giá trị RSD của PAR cả 3 lần đo lặp lại là 0,2 % , của
CAF là 0,2 %. Khi phân tích trong nội bộ PTN, nếu các giá trị RSD thu được nhỏ
hơn ½ RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40], [116].
Như vậy, các phương pháp chemometric này có độ lặp lại tốt với cả hai cấu tử.
iii) Đối với hỗn hợp PAR và IB:

95
Tiến hành thí nghiệm như mục 3.3.2.1, với mỗi dung dịch hỗn hợp được chuẩn
bị và phân tích lặp lại 3 lần. Sử dụng các phương pháp chemometric để xác định
nồng độ của PAR và IB trong các dung dịch hỗn hợp và từ đó độ lặp lại của phương
pháp được đánh giá thông qua giá trị RSD tương ứng khi so sánh với ½ RSDHorwitz
(xem mục 2.2.6.2). Kết quả tính toán được trình bày ở bảng 3.18.
Bang 3.18. Kết quả xác định độ lặp của phương pháp đối với hỗn hợp PAR và IB(*)
Ký
hiệu
mẫu
Thông số PAR IB
Lần
1
Lần
2
Lần
3
CTB Lần 1 Lần 2 Lần 3 CTB
H3
CK (µg/mL)- 5,03 5,04 5,02 5,03 2,41 2,42 2,41 2,41
RSDK (%) 0,3 0,2
CB (µg/mL) 5,03 5,04 5,02 5,03 2,41 2,42 2,41 2,41
RSDB (%) 0,3 0,2
½ RSDH (%) 6,3 7,0
H4
CK (µg/mL) 4,92 4,95 4,87 4,91 9,88 9,93 9,71 9,84
RSDK (%) 0,8 1,2
CB (µg/mL) 4,92 4,95 4,87 4,91 9,88 9,93 9,71 9,84
RSDB (%) 0,8 1,2
½ RSDH (%) 6,3 5,7
H5
CK (µg/mL) 4,91 4,90 4,93 4,91 15,20 15,17 15,26 15,21
RSDK (%) 0,3 0,3
CB (µg/mL) 4,91 4,90 4,93 4,91 15,20 15,17 15,26 15,21
RSDB (%) 0,3 0,3
½ RSDH (%) 6,3 5,3
(*) RSDH tính theo hàm Horwitz [116];CK, RSDK là nồng độ, độ lặp lại tính theo phương pháp
Kalman; CB, RSDB là nồng độ, độ lặp lại tính theo phương pháp BPCT.
Bảng 3.18 cho thấy: Giá trị RSD đối với PAR là 1 %; của IB là < 1,5 %, đều
nhỏ hơn ½ RSDH. Khi phân tích trong nội bộ PTN, nếu các giá trị RSD thu được nhỏ
hơn ½ RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40], [116]. Như vậy, các
phương pháp chemometric này có độ lặp lại tốt với cả hai cấu tử.
Để đánh giá độ lặp lại của phương pháp quang phổ đạo hàm đối với hỗn hợp
PAR và IB, tiến hành phân tích hỗn hợp có nồng độ PAR 5 µg/mL và nồng độ IB 5
µg/mL. Các thí nghiệm được thực hiện trong 5 ngày liên tiếp, mỗi thí nghiệm phân

96
tích lặp lại 3 lần/ngày. Kết quả xác định độ lặp lại của phương pháp phân tích xác
định PAR và IB thu được ở bảng 3.19.
Bang 3.19. Kết quả xác định độ lặp lại của phương pháp quang phổ đạo hàm(*)
Chất Kí
hiệu
C (µg/mL) RSD (%)
TB ½ RSDH
(%) Lần1 Lần 2 Lần 3 TB
PAR
TN1 4,90 4,85 0,85 4,87 0,6
0,6 6,0
TN2 4,90 4,9 4,95 4,92 0,6
TN3 4,80 4,85 4,90 4,85 1,03
TN4 4,85 4,85 4,90 4,87 0,59
TN5 4,90 4,85 4,95 4,90 1,02
IB
TN1 5,0 4,86 5,14 5,0 2,70
2,2 6,0
TN2 5,0 4,86 4,73 4,86 2,78
TN3 4,73 4,86 4,86 4,82 1,62
TN4 5,27 5,27 5,13 5,22 1,48
TN5 5,0 5,13 5,27 5,13 2,63
(*)RSDH tính theo hàm Horwitz [116]; TB là trung bình số học của độ lặp lại
Từ bảng 3.19 cho thấy: Giá trị RSD của PAR cả 3 lần đo lặp lại từ TN1 đến TN5
là 0,6 % , của IB là 2,2 %. Khi phân tích trong nội bộ PTN, nếu các giá trị RSD thu
được nhỏ hơn ½ RSDH thì độ lặp lại của phương pháp đạt yêu cầu [40], [116].
Như vậy, các phương pháp chemometric này có độ lặp lại tốt với cả hai cấu tử.
Nhận xét chung: Kết quả từ bảng 3.14 đến bảng 3.19 cho thấy giá trị RSD của
tất cả các chất trong hỗn hợp đều nhỏ hơn giá trị ½ RSDH. Khi phân tích trong nội
bộ PTN, nếu các giá trị RSD thu được nhỏ hơn ½ RSDH thì độ lặp lại của phương
pháp đạt yêu cầu [40], [116]. Như vậy, phương pháp này có độ lặp lại tốt với tất cả
các cấu tử đã phân tích ở trên.
3.4. Kiểm chứng phương pháp đối với hỗn hợp ba cấu tử
Do đối với hỗn hợp 3 cấu tử, trên phổ đạo hàm rất khó tìm được bước sóng mà
tại đó phổ đạo hàm của một cấu tử khác 0, còn phổ đạo hàm của 2 cấu tử còn lại
bằng 0. Nói cách khác, phương pháp phổ đạo hàm chỉ áp dụng phù hợp cho hỗn hợp

97
hai cấu tử. Do vậy, ở đây phương pháp phổ đạo hàm không được khảo sát cho
trường hợp dung dịch hỗn hợp chứa 3 cấu tử. Hỗn hợp 3 cấu tử AML, HYD và
VAL được lựa chọn để kiểm chứng phương pháp Kalman và phương pháp bình
phương tối thiểu.
3.4.1. Phổ hấp thụ của hỗn hợp
Quét phổ của 4 dung dịch AML 5 g/mL, HYD 5 g/mL, VAL 5 μg/mL và
dung dịch hỗn hợp chứa AML 5 g/mL, HYD 5 g/mL và VAL 5 μg/mL trong
dung môi metanol, ở khoảng bước sóng 230 nm – 340 nm, phổ hấp thụ của các
dung dịch được biểu diễn ở hình 3.8.
(5) Đường tính theo lý thuyết của tính chất cộng tính
Hinh 3.8. Phổ hấp thụ của các dung dịch chuẩn AML, HYD, VAL và
hỗn hợp AML, HYD, VAL đo được trong dung môi methanol.
Từ hình 3.8 cho thấy phổ hấp thụ của các dung dịch chuẩn của AML, HYD và VAL
trong dung môi methanol xen phủ nhau ở khoảng bước sóng từ 230 nm đến 340 nm. Sự xen
phủ này khiến cho việc xác định đồng thời ba chất trong hỗn hợp gặp khó khăn. Vì vậy để giải
quyết vấn đề này, chúng tôi đã sử dụng phương pháp trắc quang - chemometric.
Trong khoảng bước sóng 230 nm – 340 nm phổ của hỗn hợp theo thực tế đo
và theo lý thuyết (tính theo định luật cộng tính) gần trùng khít lên nhau nên có thể
xem độ hấp thụ của dung dịch hỗn hợp chứa ba cấu tử AML, HYD và VAL có tính
chất cộng tính. Vì vậy, chúng tôi có thể xác định đồng thời hàm lượng của AML,
HYD và VAL bằng phương pháp trắc quang – chemometric dùng phổ toàn phần.
240 260 280 300 320 3400.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
4,5
3
21
A
Wavelength (nm)
1. AM5
2. HY5
3. VA5
4. AM5 + HY5 +VA5 (LT)
5. AM5 + HY5 + VA5 (TT)

98
3.4.2. Kiểm định phương pháp đối với dung dịch chuẩn phòng thí nghiệm
3.4.2.1. So sánh hai phương pháp chemometric-trắc quang
Khảo sát các ty lệ khác nhau của các hoạt chất nghiên cứu nhằm đánh giá khả
năng đáp ứng của phương pháp.
Pha các dung dịch nghiên cứu trong bình định mức 25 mL, gồm các dung dịch chuẩn
của AML, HYD, VAL có nồng độ lần lượt là 5 g/mL , 5 g/mL và 10 g/mL; Các dung
dịch hỗn hợp chứa AML, HYD và VAL có nồng độ của ba chất có ty lệ khác nhau theo
bảng 3.20.
Quét phổ của các dung dịch trên trong khoảng bước sóng 230 nm – 340 nm,
dùng các phương pháp chemometric để xác định nồng độ của từng cấu tử trong các
dung dịch hỗn hợp, sau đó xác định giá trị sai số tương đối RE của phép phân tích
cho từng chất (xem mục 2.2.6.1). Kết quả được trình bày ở bảng 3.20.
Bang 3.20. Kết quả xác định nồng độ AML, HYD, VAL trong hỗn hợp và
sai số tương đối của các phương pháp (*)
Kí hiệu H1 H2 H3 H4
AML
C0 (µg/mL) 0,250 0,50 1,00 5,00
Kalman C (µg/mL) 0,253 0,511 1,016 4,981
RE (%) 1,2 2,2 1,6 0,4
BPTT C (µg/mL) 0,253 0,511 1,017 4,841
RE (%) 1,2 2,2 1,7 -3,2
HYD
C0 (µg/mL) 0,325 0,65 1,30 5,00
Kalman C (µg/mL) 0,320 0,646 1,290 5,064
RE (%) -1,5 -0,6 -0,8 1,3
BPTT C (µg/mL) 0,320 0,645 1,284 5,109
RE (%) -1,5 -0,7 -1,2 2,2
VAL
C0 (µg/mL) 4,00 8,00 16,00 5,00
Kalman C (µg/mL) 3,99 8,06 16,05 4,821
RE (%) -0,2 0,8 0,3 -3,6
BPTT C (µg/mL) 3,99 8,06 16,04 4,870
RE (%) -0,2 0,8 0,2 -2,5
(*)Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Từ bảng 3.20 cho thấy rằng với các tỉ lệ nồng độ khác nhau, giữa nồng độ
dung dịch chuẩn và nồng độ xác định được mắc sai số RE (%) nhỏ. Cụ thể khi xác

99
định nồng độ các chất, đối với phương pháp lọc Kalman, sai số lớn nhất là -3,6 %,
sai số lớn nhất là -0,2 %; đối với phương pháp BPTT, sai số lớn nhất là -3,2 %, sai
số bé nhất là -0,2 %. Các phương pháp cho kết quả chấp nhận với sai số RE (%) nhỏ
do đó có độ đúng tốt.
3.4.2.2. Độ lặp lại của phương pháp khi phân tích dung dịch chuẩn phòng thí
nghiệm
Tiến hành thí nghiệm như mục 3.4.2.1với mỗi dung dịch hỗn hợp được chuẩn
bị và phân tích lặp lại 3 lần. Sử dụng các phương pháp chemometric để xác định
nồng độ của các chất trong các dung dịch hỗn hợp và từ đó độ lặp lại của phương
pháp được đánh giá thông qua giá trị RSD tương ứng khi so sánh với ½ RSDHorwitz
(xem mục 2.2.6.2). Kết quả tính toán được trình bày ở bảng 3.21.

100
Bang 3.21. Kết quả xác định độ lặp lại của phương pháp đối với
hỗn hợp AML, HYD và VAL(*)
Mẫu Thông số AML HYD VAL
Lần 1 Lần 2 Lần 3 TB Lần 1 Lần 2 Lần 3 TB Lần 1 Lần 2 Lần 3 TB
H1
CK (µg/mL) 0,253 0,252 0,254 0,253 0,320 0,320 0,321 0,320 3,990 3,980 4,010 3,993
RSDK (%) 0,4 0,3 0,4
CB (µg/mL) 0,253 0,253 0,254 0,253 0,319 0,319 0,321 0,319 3,993 3,981 4,009 3,994
RSDB (%) 0,4 0,4 0,4
½ RSDH 9,9 9,5 6,5
H2
CK (µg/mL) 0,511 0,510 0,514 0,512 0,646 0,645 0,650 0,647 8,060 8,044 8,109 8,071
RSDK (%) 0,4 0,5 0,4
CB (µg/mL) 0,511 0,510 0,514 0,512 0,645 0,644 0,649 0,646 8,059 8,043 8,107 8,070
RSDB (%) 0,4 0,5 0,4
½ RSDH 8,9 8,6 5,9
H3
CK (µg/mL) 1,016 1,013 1,020 1,016 1,290 1,286 1,296 1,291 16,050 15,994 16,114 16,053
RSDK (%) 0,4 0,4 0,4
CB (µg/mL) 1,017 1,013 1,021 1,017 1,284 1,279 1,290 1,284 16,037 15,980 16,101 16,040
RSDB (%) 0,4 0,5 0,4
½ RSDH 8,0 7,9 5,5
H4
CK (µg/mL) 4,981 4,971 5,008 4,987 5,064 5,054 5,089 5,069 4,821 4,811 4,844 4,825
RSDK (%) 0,4 0,4 0,4
CB (µg/mL) 4,841 4,831 4,865 4,846 5,109 5,099 5,135 5,114 4,873 4,864 4,898 4,878
RSDB (%) 0,4 0,4 0,4
½ RSDH 6,3 6,3 6,3
(*)RSDH tính theo hàm Horwitz [116]. TB: Trung bình số học của nồng độ; CK, RSDK: Nồng
độ, độ lặp lại tính được theo phương pháp Kalman; CB, RSDB: Nồng độ, độ lặp lại tính được theo
phương pháp BPCT; Nồng độ chuẩn của các dung dịch AML/HYD/VAL lần lượt trong các hỗn hợp
là H1 (0,25/0,325/4,00), H2 (0.5/0,65/8,00), H3 (1,00/1,30/16,00), H4 (5,00/5,00/5,00).
Bảng 3.21 cho thấy: Giá trị RSD của AML và VAL cả 3 lần đo lặp lại cho các
mẫu từ H1 đến H4 là 0,4 % , của HYD từ 0,4 % đến 0,5 %. Khi phân tích trong nội
bộ PTN, nếu các giá trị RSD thu được nhỏ hơn ½ RSDH thì độ lặp lại của phương

101
pháp đạt yêu cầu [116]. Như vậy, phương pháp này có độ lặp lại tốt với cả ba cấu
tử.
Từ bảng 3.21 nhận thấy: Nồng độ trung bình của ba chất AML, HYD và VAL
trong các mẫu H1, H2, H3 được tính theo hai phương pháp là như nhau (p>0,05).
Trong khi đó đối với mẫu H4 nồng độ trung bình xác định được theo hai phương
pháp khác nhau (p <0,05). Để đánh giá sự khác biệt này có ý nghĩa về mặt thống kê
hay không, sử dụng kiểm định t (t-test) để so sánh giá trị trung bình của hai phương
pháp, kết quả thu được ở bảng 3.22 và hình 3.9.
Bang 3.22. So sánh kết quả của 2 phương pháp đối với hỗn hợp H4
Chất Phương pháp
TB±S
(µg/mL) Ftính
Flt
(α= 0,05; 2; 2)
Sp ttính
tlt
(0,05; 4)
AML Kalman 4,987 ± 0,019
1,200 39,0 0,018 9,42 2,78 BPTT 4,846 ± 0,017
HYD Kalman 5,069 ± 0,018
1,063 39,0 0,018 3,03 2,78 BPTT 5,114 ±0,019
VAL Kalman 4,825 ± 0,017
1,083 39,0 0,017 3,76 2,78 BPTT 4,878 ± 0,018
(*)𝑥𝑖: Kết quả phân tích lặp lại (i = 1-3); Ftính = phương sai của mỗi phương pháp chemometric /
phương sai của phương pháp HPLC; F(0,05;2;2): Giá trị F tới hạn ở mức ý nghĩa thống kê 0,05 và
2 bậc tự do của 2 phương sai tử số và mẫu số; Sp: Phương sai chung, được tính từ 2 phương sai
của 2 phương pháp khi 2 phương sai của 2 phương pháp như nhau (Ftính< F(0,05;2;2)); f: Bậc tự
do được tính từ 2 tập số liệu của 2 phương pháp khi 2 phương pháp có phương sai khác nhau
(Ftính> F(0,05;2;2); t(0,05; f): Giá trị tới hạn của t ở mức ý nghĩa thống kê p và bậc tự do f.
TB và S: Trung bình số học và độ lệch chuẩn; CTN: Nồng độ dung dịch chuẩn phòng thí nghiệm, ở
đây các chất AML, HYD, VAL trong dung dịch đều có nồng độ 5 (µg/mL).

102
(CPTN: Nồng độ dung dịch chuẩn của mỗi chất;).
Hinh 3.9. So sánh kết quả của 2 phương pháp đối với hỗn hợp H4
Từ bảng 3.22 và hình 3.9 thấy rằng: Khi sử dụng hai phương pháp Kalman và
BPTT để tính toán nồng độ AML, HYD và VAL ở mẫu H4 đều thu được ttính> tlt. Vì
vậy có thể kết luận nồng độ trung bình tính được từ hai phương pháp là khác nhau
có ý nghĩa về mặt thống kê (với p < 0,05)
Khi đối chiếu giữa nồng độ tính được theo hai phương pháp Kalman và BPTT
với nồng độ pha chế của dung dịch chuẩn phòng thí nghiệm thì thấy rằng phương
pháp lọc Kalman cho kết quả gần với nồng độ dung dịch chuẩn phòng thí nghiệm
hơn phương pháp BPTT. Điều này được giải thích là do bản chất của phương pháp
BPTT là dựa trên nguyên lý của phương pháp bình phương cực tiểu. Phương pháp
bình phương cực tiểu hướng đến giá trị trung bình của bộ số liệu, nghĩa là nồng độ
tính được từ phương pháp bình phương cực tiểu gần với giá trị trung bình nhất chứ
chưa hẳn gần với giá trị thật nhất.
ttính = 9,42
tlt(0,05; 3) = 2,78 ttính = 3,03
tlt(0,05; 3) = 2,78 ttính = 3,76
tlt(0,05; 3) = 2,78

103
Còn phương pháp lọc Kalman, với cách khởi tạo là lấy nồng độ tìm được từ
phương pháp bình phương cực tiểu làm điểm bắt đầu cho bộ lọc và sẽ dò lại với dải
sóng trong bộ số liệu để tìm ra giá trị nồng độ gần với giá trị thật nhất bằng phương
pháp lọc, trong đó đặc trưng của bộ lọc là dãy đổi mới INV được bổ sung vào từng
bước tính để bù lại phần sai lệch giữa giá trị dự báo và giá trị đo đạc. Giá trị INV
được xác định bởi việc tối ưu sai số của mô hình dựa theo giá trị lợi Kalman (được
trình bày ở công thức 2.7 và 2.15).
Bang 3.23. Kết quả xác định nồng độ AML, HYD và VAL trong hỗn hợp
bằng phương pháp Kalman và BPTT (*) khi có nhiễu
Kí hiệu H1 H2 H3 H4
AML
C0 (µg/mL) 0,250 0,50 1,00 5,00
Kalman C (µg/mL) 0,261 0,516 1,044 5,001
RE (%) 4,40 3,13 4,40 0,02
BPTT C (µg/mL) 0,261 0,517 1,045 4,818
RE (%) 4,40 3,33 4,50 -3,65
HYD
C0 (µg/mL) 0,325 0,65 1,30 5,00
Kalman C (µg/mL) 0,322 0,643 1,283 5,049
RE (%) -0,82 -1,08 -1,28 0,99
BPTT C (µg/mL) 0,321 0,642 1,277 5,122
RE (%) -1,13 -1,23 -1,77 2,44
VAL
C0 (µg/mL) 4,00 8,00 16,00 5,00
Kalman C (µg/mL) 3,982 8,071 16,019 4,764
RE (%) -0,46 0,89 0,12 -4,72
BPTT C (µg/mL) 3,981 8,069 16,007 4,830
RE (%) -0,48 0,86 0,05 -3,39
(*) Co: Nồng độ chất trong dung dịch chuẩn hỗn hợp; C: Nồng độ chất xác định được
Chính vì bản chất của phương pháp BPTT và phương pháp Kalman như vậy
nên trong trường hợp nhiễu đo lớn, chẳng hạn, phép đo độ hấp thụ quang (A) bị
mắc sai số bất thường (quá lớn hoặc quá bé), hoặc do bị ảnh hưởng của pha nền
(matrix), phương pháp Kalman có thể sẽ cho ra kết quả mắc sai số nhỏ hơn. Để
kiểm chứng điều này, chúng tôi giả định trong bộ dữ liệu phổ, có một số giá trị độ

104
hấp thụ quang A bị mắc sai số bất thường, và tính toán lại nồng độ 3 chất theo
phương pháp Kalman và phương pháp bình phương tối thiểu (theo cách tương tự
trên). Kết quả thu được ở bảng 3.23 cho thấy: Phương pháp Kalman mắc sai số nhỏ
hơn với RE < 4,4 %, trong khi phương pháp bình phương tối thiểu mắc sai số lớn
hơn với RE < 4,5 %.
Kết quả cho thấy, đối với cả hệ 2 hoặc 3 cấu tử, phương pháp Kalman cho ra
kết quả mắc sai số nhỏ hơn phương pháp bình phương tối thiểu ( nồng độ xác định
được gần với nồng độ thật hơn). Tuy nhiên để đánh giá được độ tin cậy của phương
pháp Kalman cần phải kiểm chứng phương pháp đối với mẫu thực tế.
3.5. Quy trình phân tích theo phương pháp lọc Kalman
Từ các kết quả thu được ở trên, quy trình phân tích theo phương pháp
Kalman được đề xuất ở hình 3.10.
Hinh 3.10. Quy trình phân tích mẫu thực tế theo phương pháp Kalman.
Quy trình phân tích mẫu thực tế theo phương pháp Kalman được thực hiện
như sau:
Bước 1: Việc chuẩn bị mẫu cho các thuốc nghiên cứu được trình bày rõ ở mục
2.2.8 (trong đó giai đoạn hòa tan mẫu với sự hỗ trợ của siêu âm khoảng 30 phút).

105
Đồng thời pha các dung dịch chuẩn của các hoạt chất có mặt trong các mẫu thuốc
nghiên cứu.
Bước 2: Ghi phổ các dung dịch chuẩn và các dung dịch mẫu thuốc; Độ hấp thụ
quang A ở m bước sóng trong khoảng xác định (thường từ 230 nm – 340 nm,
khoảng cách giữa các bước sóng là 2 nm hoặc 5 nm). Dữ liệu phổ được lưu ở dạng
txt. Các file này là đầu vào cho phương pháp Kalman.
Bước 3: Nhập dữ liệu này vào ô nhập dữ liệu của chương trình tính (chương
trình viết bằng ngôn ngữ Visual basic for Applications trên phần mềm Microsoft –
Excel 2016 (chương trình chi tiết được nêu ở phụ lục 1).
Từ dữ liệu phổ của dung dịch chuẩn và dung dịch hỗn hợp, máy tính sẽ tính
được các giá trị khởi tạo:
+) Nồng độ khởi tạo Cest (0) được tính theo phương pháp bình phương tối
thiểu
+) Phương sai khởi tạo Pest (0) được tính theo phương trình Horwitz (tính từ
các giá trị Cest (0) theo cách đã mô tả ở mục 2.2.2).
Các giá trị Cest (0) và Pest (0) là các giá trị đầu vào cho thuật toán lọc Kalman.
Bước 4: Tính theo thuật toán lọc Kalman cho m bước sóng (từ bước sóng đầu
đến bước sóng cuối) như đã mô tả ở mục 2.2.2. Kết quả đầu ra của chương trình
tính là nồng độ các chất trong mẫu và sai số tương đối (RE %) kèm theo; và chương
trình tính kết thúc ở đây.
Toàn bộ quá trình chuẩn bị mẫu cho đến khi thu được kết quả cuối cùng khoảng
60 phút. Trong khoảng thời gian đó, giao đoạn chuẩn bị mẫu cho phân tích chiếm là
chiếm phần lớn thời gian, còn giai đoạn tính toán và hiện kết quả dưới 2 phút.
3.6. Ap dụng thực tế
Khi nghiên cứu phát triển phương pháp phân tích hoặc trước khi áp dụng một
phương pháp phân tích kỳ vào thực tế, bắt buộc phải kiểm soát chất lượng hay thẩm
định phương pháp (quality control) qua các thông số thể hiện năng lực của phương
pháp (performance parameters). Trong đó, hai thông số quan trọng nhất là độ đúng
(trueness) và độ lặp lại (precision). Đối với độ lặp lại, nếu phân tích trong nội bộ
phòng thí nghiệm, người ta dùng thuật ngữ độ lặp lại hay độ chụm (repeatability),

106
nếu phân tích trong nhiều phòng thí nghiệm khác nhau, người ta dùng thuật ngữ độ
tái lặp hoặc độ hồi phục (reproducibility). Chỉ khi một phương pháp phân tích vừa
đạt được độ đúng tốt và độ lặp lại tốt, mới được xem là đạt được độ chính xác
(accurate) cao.
3.6.1. Kiểm soát chất lượng phương pháp phân tích
3.6.1.1. Độ lặp lại
i) Đối với TEL và HYD trong thuốc Micardis(R) Plus
Thông tin về thuốc nghiên cứu: Thuốc viên nén Micardis(R) Plus với hàm lượng
ghi trên nhãn của TEL là 40 mg và HYD là 12,5 mg, số lô: 505392, ngày sản xuất:
26/06/2015, hạn dùng: 26/06/2018, SĐK: VN-16587-13; thuốc được sản xuất tại
Boehringer Ingelheim Pharma GmbH & Co. KG Binger Str. 173 55216 Ingelheim
am Rhein, Germany.
Áp dụng quy trình phân tích) đã được trình bày ở hình 3.10 (gọi chung là
phương pháp Kalman) để xác định đồng thời hàm lượng TEL và HYD trong mẫu
thuốc Micardis(R) Plus (thuốc điều trị huyết áp). Mặt khác, để so sánh với phương
pháp Kalman, phương pháp bình phương tối thiểu dùng phổ toàn phần (BPTT) và
phương pháp phổ đạo hàm (PĐH) cũng được áp dụng để phân tích mẫu thuốc (cả 3
phương pháp – Kalman, BPTT và PĐH được gọi chung là phương pháp
chemometric-trắc quang). Chi tiết về chuẩn bị mẫu cho phân tích theo các phương
pháp trên được nêu ở mục 2.2.8.1. Các kết quả phân tích được tính theo nồng độ chất
trong dung dịch mẫu (g/mL) và hàm lượng chất trong viên thuốc (mg/viên) (bảng
3.24). Để so sánh với các kết quả đó, nồng độ các chất (TEL và HYD) trong dung
dịch mẫu cũng được tính toán từ hàm lượng các chất trong một viên thuốc (được ghi
trên nhãn thuốc), tương ứng đối với TEL và HYD là 8,00 µg/mL và 2,5 µg/mL.
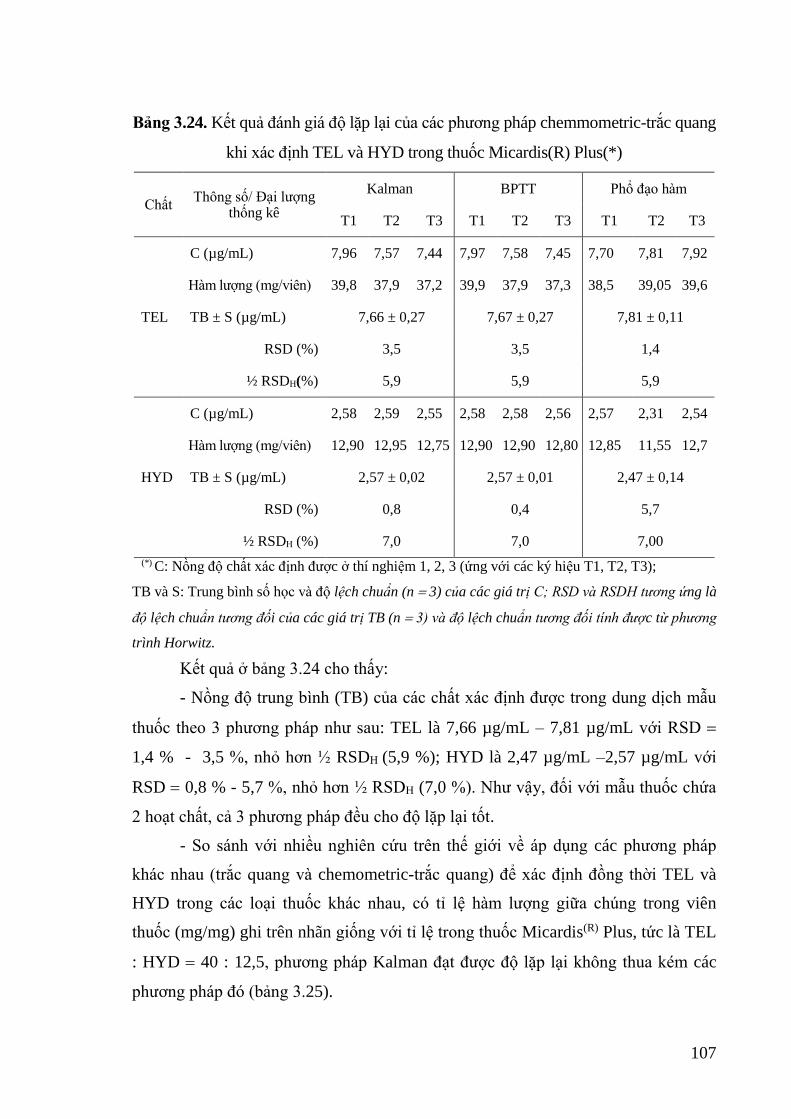
107
Bang 3.24. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc quang
khi xác định TEL và HYD trong thuốc Micardis(R) Plus(*)
Chất Thông số/ Đại lượng
thống kê
Kalman BPTT Phổ đạo hàm
T1 T2 T3 T1 T2 T3 T1 T2 T3
TEL
C (µg/mL) 7,96 7,57 7,44 7,97 7,58 7,45 7,70 7,81 7,92
Hàm lượng (mg/viên) 39,8 37,9 37,2 39,9 37,9 37,3 38,5 39,05 39,6
TB ± S (µg/mL) 7,66 ± 0,27 7,67 ± 0,27 7,81 ± 0,11
RSD (%) 3,5 3,5 1,4
½ RSDH(%) 5,9 5,9 5,9
HYD
C (µg/mL) 2,58 2,59 2,55 2,58 2,58 2,56 2,57 2,31 2,54
Hàm lượng (mg/viên) 12,90 12,95 12,75 12,90 12,90 12,80 12,85 11,55 12,7
TB ± S (µg/mL) 2,57 ± 0,02 2,57 ± 0,01 2,47 ± 0,14
RSD (%) 0,8 0,4 5,7
½ RSDH (%) 7,0 7,0 7,00
(*) C: Nồng độ chất xác định được ở thí nghiệm 1, 2, 3 (ứng với các ký hiệu T1, T2, T3);
TB và S: Trung bình số học và độ lệch chuẩn (n 3) của các giá trị C; RSD và RSDH tương ứng là
độ lệch chuẩn tương đối của các giá trị TB (n 3) và độ lệch chuẩn tương đối tính được từ phương
trình Horwitz.
Kết quả ở bảng 3.24 cho thấy:
- Nồng độ trung bình (TB) của các chất xác định được trong dung dịch mẫu
thuốc theo 3 phương pháp như sau: TEL là 7,66 µg/mL – 7,81 µg/mL với RSD
1,4 % - 3,5 %, nhỏ hơn ½ RSDH (5,9 %); HYD là 2,47 µg/mL –2,57 µg/mL với
RSD 0,8 % - 5,7 %, nhỏ hơn ½ RSDH (7,0 %). Như vậy, đối với mẫu thuốc chứa
2 hoạt chất, cả 3 phương pháp đều cho độ lặp lại tốt.
- So sánh với nhiều nghiên cứu trên thế giới về áp dụng các phương pháp
khác nhau (trắc quang và chemometric-trắc quang) để xác định đồng thời TEL và
HYD trong các loại thuốc khác nhau, có tỉ lệ hàm lượng giữa chúng trong viên
thuốc (mg/mg) ghi trên nhãn giống với tỉ lệ trong thuốc Micardis(R) Plus, tức là TEL
: HYD 40 : 12,5, phương pháp Kalman đạt được độ lặp lại không thua kém các
phương pháp đó (bảng 3.25).

108
- Mặt khác, các kết quả xác định được (C) cũng rất gần với các kết quả tính
toán từ các giá trị ghi trên nhãn viên thuốc, hay nói cách khác, cả 3 phương pháp
cũng có độ đúng tốt. Song, độ đúng của các phương pháp sẽ được khẳng định thêm
khi khảo sát độ đúng của các phương pháp nêu ở mục 3.6.1.2 dưới đây).
Bang 3.25. Độ lặp lại và độ đúng của các phương pháp trắc quang và
chemometric-trắc quang khi phân tích đồng thời TEL và HYD trong dược phẩm (*)
Phương pháp
TEL HYD
TLTK Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Kalman 38,3 3,5 101 12,9 0,8 104 Luận án
này
Trắc quang 39,7 0,33 99,6 12,2 0,51 100 [85]
Ty lệ độ hấp thụ
39,5 - 98 – 99 12,1 - 98 [30]
Chuyển dịch bước sóng
39,9 - 98 -103 12,2 - 96 – 102 [100]
PLS 40,1 0,91 101 12,3 1,04 102 [84]
PCR 41,3 0,92 99,9 12,6 0,76 97 [84]
(*) TLTK: Tài liệu tham khảo; PLS: Phương pháp bình phương tối thiểu riêng phần (Partial Least
Square); PCR: Phương pháp hồi quy cấu tử chính (Principal Component Regression). Rev(%): Độ
thu hồi. Dấu (-) là không xác định.
ii) Đối với PAR và CAF trong thuốc Panadol Extra
Thông tin về thuốc nghiên cứu: Thuốc Panadol Extra sản xuất tại công ty cổ
phần dược phẩm Sanofi –Synthelabo Việt Nam, số lô/ngày sản xuất: 15123, hạn
dùng: 13/10/2017, hàm lượng ghi trên nhãn 500 mg PAR và 65 mg CAF. Khối
lượng trung bình mỗi viên M = 0,6965g
Tương tự như đối với phân tích thuốc Micardis(R) Plus (chứa TEL và HYD),
tiến hành xác định đồng thời PAR và CAF trong mẫu thuốc Panadol Extra (thuốc
giảm đau, hạ sốt) theo 3 phương pháp, thu được các kết quả ở bảng 3.26. Chi tiết về
xử lý mẫu cho phân tích được nêu ở mục 2.2.8.2. Để so sánh, nồng độ các chất

109
trong dung dịch mẫu thuốc cũng được tính toán từ hàm lượng các chất trong viên
thuốc (ghi trên nhãn thuốc): tương ứng đối với PAR và CAF là 8,00 µg/mL và 1,0
µg/mL. Kết quả ở bảng 3.26 cho thấy:
Bang 3.26. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc quang
khi xác định PAR và CAF trong thuốc Panadol Extra(*)
Chất Thông số/ Đại lượng thống kê
Kalman BPTT Phổ đạo hàm
T1 T2 T3 T1 T2 T3 T1 T2 T3
PAR
C (µg/mL) 7,88 7,84 7,85 7,88 7,84 7,85 7,93 7,88 7,88
Hàm lượng (mg/viên)
496,2 493,8 494,7 496,2 493,8 494,7 498,0 494,9 494,9
TB ± S (µg /mL) 7,86 ± 0,02 7,86 ± 0,02 7,90 ± 0,03
RSD (%) 0,3 0,3 0,4
½ RSDH (%) 5,9 5,9 5,9
CAF
C (µg/mL) 1,07 1,08 1,06 1,07 1,08 1,06 1,07 1,07 1,06
Hàm lượng (mg/viên)
67,1 67,8 66,5 67,1 67,8 66,5 67,2 67,2 66,6
TB ± S (µg /mL) 1,07 ± 0,01 1,07 ± 0,01 1,07 ± 0,01
RSD (%) 0,9 0,9 0,9
½ RSDH (%) 8,0 8,0 8,0
(*) Các ký hiệu và các đại lượng nêu trong bảng cũng như ở bảng 3.24; các giá trị TB, S và RSD đều
ứng với n 3.
- Cả 3 phương pháp đều đạt được độ lặp lại tốt: Đối với PAR, nồng độ trung
bình (TB) trong dung dịch mẫu thuốc là 7,84 µg/mL - 7,93 µg/mL với RSD 0,3 %
- 0,4 %, nhỏ hơn ½ RSDH (5,9 %); Đối với CAF, nồng độ trung bình là 1,06 µg/mL
– 1,08 µg/mL với RSD 0,9 %, nhỏ hơn ½ RSDH (8,0 %);
- So sánh với nhiều nghiên cứu trên thế giới về áp dụng các phương pháp
khác nhau (trắc quang và chemometric-trắc quang) để xác định đồng thời PAR và
CAF trong các loại thuốc khác nhau, có tỉ lệ hàm lượng giữa chúng trong viên thuốc
(mg/mg) ghi trên nhãn giống với tỉ lệ trong thuốc Panadol Extra, tức là PAR : CAF

110
500 : 65, phương pháp Kalman đạt được độ lặp lại không thua kém các phương
pháp đó (bảng 3.27).
- Cả 3 phương pháp đều cho kết quả (về nồng độ chất trong dung dịch mẫu)
gần với kết quả được tính toán từ các giá trị ghi trên nhãn thuốc và như vậy, có thể
cho rằng, cả 3 phương pháp đều đạt được độ đúng tốt (chi tiết về đánh giá độ đúng
của 3 phương pháp được nêu ở mục 3.6.1.2 dưới đây).
Bang 3.27. Độ lặp lại và độ đúng của các phương pháp trắc quang và
chemometric-trắc quang khi phân tích đồng thời PAR và CAF trong dược phẩm (*)
Phương pháp
PAR CAF
Nguồn TLTK
Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Kalman 494,9 0,3 100 - 103 67,0 0,9 97 - 104 Luận
án này
Trắc quang 487,3 0,4 98 - 99 64,4 0,5 98 - 99 [118]
Ty lệ độ hấp thụ 477,3 0,07 98 - 99 64,1 1,2 98 - 99 [118]
PCR 499,9 0,4 97 64,0 1,2 99 [32]
PLS 499,5 1,4 97 64,9 1,7 97 [32]
ANN 502,5 2,29 99 65,1 0,7 99 [33]
(*) TLTK: Tài liệu tham khảo; PLS và PCR: Như ở bảng 3.25; ANN: Phương pháp mạng nơron
nhân tạo (Artificial Neutron Network); Rev(%): Độ thu hồi. Dấu (-) là không xác định.
iii) Đối với hỗn hợp PAR và IB trong 3 loại thuốc (Alaxan, Lopenca, Protamol)
Thông tin về thuốc nghiên cứu: Thuốc viên nén Lopenca với hàm lượng ghi
trên nhãn của PAR là 325 mg, IB là 200 mg/viên, số lô: 020214, ngày sản xuất:
02/2014, hạn dùng: 25/02/2017, thuốc được sản xuất tại CPCP Dược Hậu Giang
Cũng theo cách tương tự như đối với phân tích mẫu thuốc Micardis(R) Plus và
Panadol Extra ở trên, kết quả phân tích PAR và IB trong 3 loại thuốc (Alaxan –
thuốc giảm đau xương khớp; Lopenca – điều trị viêm khớp, giảm đau, hạ sốt;
Protamol – thuốc giảm đau, kháng viêm) bằng 3 phương pháp nêu ở bảng 3.28 –
3.30. Chi tiết về xử lý mẫu cho phân tích được nêu ở mục 2.2.8.3. Để so sánh, nồng
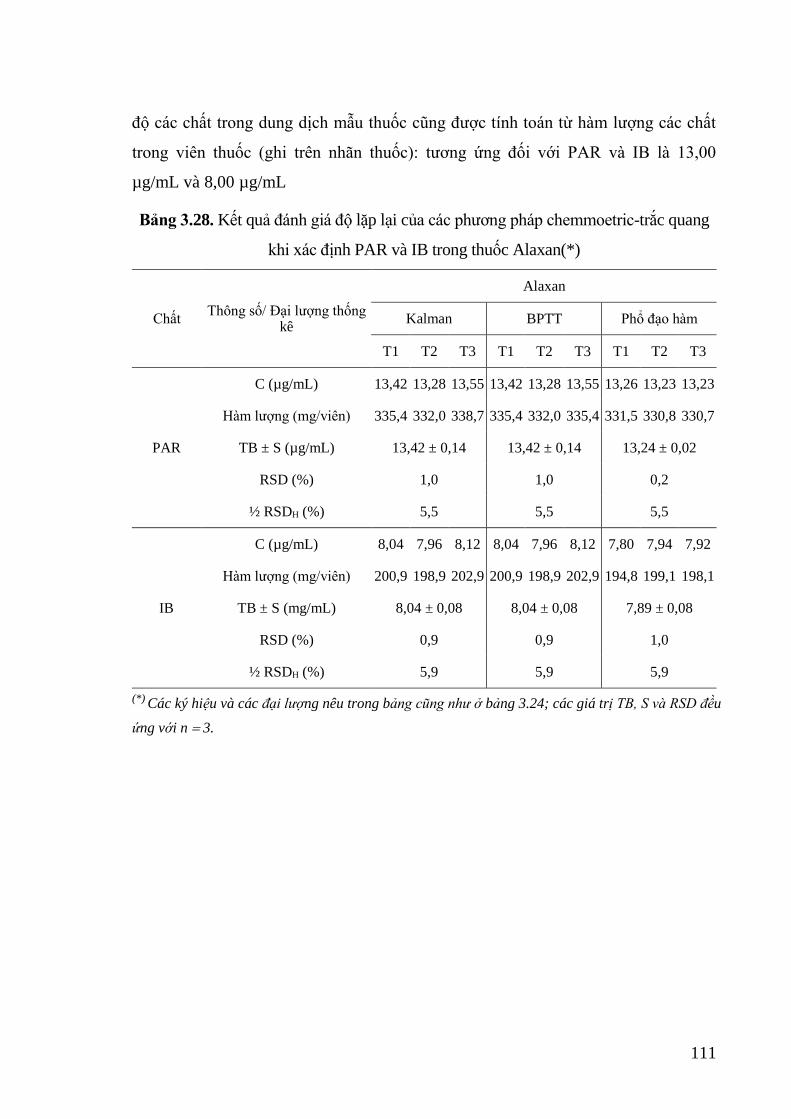
111
độ các chất trong dung dịch mẫu thuốc cũng được tính toán từ hàm lượng các chất
trong viên thuốc (ghi trên nhãn thuốc): tương ứng đối với PAR và IB là 13,00
µg/mL và 8,00 µg/mL
Bang 3.28. Kết quả đánh giá độ lặp lại của các phương pháp chemmoetric-trắc quang
khi xác định PAR và IB trong thuốc Alaxan(*)
Chất Thông số/ Đại lượng thống
kê
Alaxan
Kalman BPTT Phổ đạo hàm
T1 T2 T3 T1 T2 T3 T1 T2 T3
PAR
C (µg/mL) 13,42 13,28 13,55 13,42 13,28 13,55 13,26 13,23 13,23
Hàm lượng (mg/viên) 335,4 332,0 338,7 335,4 332,0 335,4 331,5 330,8 330,7
TB ± S (µg/mL) 13,42 ± 0,14 13,42 ± 0,14 13,24 ± 0,02
RSD (%) 1,0 1,0 0,2
½ RSDH (%) 5,5 5,5 5,5
IB
C (µg/mL) 8,04 7,96 8,12 8,04 7,96 8,12 7,80 7,94 7,92
Hàm lượng (mg/viên) 200,9 198,9 202,9 200,9 198,9 202,9 194,8 199,1 198,1
TB ± S (mg/mL) 8,04 ± 0,08 8,04 ± 0,08 7,89 ± 0,08
RSD (%) 0,9 0,9 1,0
½ RSDH (%) 5,9 5,9 5,9
(*) Các ký hiệu và các đại lượng nêu trong bảng cũng như ở bảng 3.24; các giá trị TB, S và RSD đều
ứng với n 3.

112
Bang 3.29. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc quang
khi xác định PAR và IB trong thuốc Lopenca(*)
Chất Thông số/ Đại lượng
thống kê
Lopenca
Kalman BPTT Phổ đạo hàm
T1 T2 T3 T1 T2 T3 T1 T2 T3
PAR
C (µg/mL) 12,95 12,91 13,04 12,95 12,91 13,04 12,91 12,80 12,95
Hàm lượng (mg/viên) 323,8 322,8 326,0 323,8 322,8 326,0 322,8 320,0 323,9
TB ± S (µg/mL) 12,97 ± 0,07 12,97 ± 0,07 12,89 ± 0,08
RSD (%) 0,5 0,5 0,6
½ RSDH (%) 5,5 5,5 5,5
IB
C (µg/mL) 8,50 8,48 8,56 8,50 8,48 8,56 8,59 8,65 8,67
Hàm lượng (mg/viên) 212,5 212,0 214,0 212,5 212,0 214,0 214,9 216,3 216,7
TB ± S (µg/mL) 8,51 ± 0,04 8,51 ± 0,04 8,64 ± 0,04
RSD (%) 0,5 0,5 0,5
½ RSDH (%) 5,9 5,9 5,9
(*) Các ký hiệu và các đại lượng nêu trong bảng cũng như ở bảng 3.24; các giá trị TB, S và RSD đều
ứng với n 3.
- Cả 3 phương pháp đều đạt được độ lặp lại tốt: Đối với PAR, nồng độ trung
bình (TB) là 12,97 µg/mL – 13,42 µg/mL với RSD 0,2 % - 1,0 %, nhỏ hơn ½
RSDH (5,5 %); Đối với IB là 7,89 µg/mL – 8,64 µg/mL với RSD 0,5 % - 1,2 %,
nhỏ hơn ½ RSDH (5,9 %);
- Đối với cả 3 phương pháp, các kết quả xác định được (TB) đều gần với kết
quả tính toán được từ các giá trị ghi trên nhãn và như vậy, có thể cho rằng, cả 3
phương pháp đều đạt được độ đúng tốt. Về độ đúng của 3 phương pháp, sẽ đề cập
chi tiết hơn ở mục 3.6.1.2 dưới đây).

113
Bang 3.30. Kết quả đánh giá độ lặp lại của các phương pháp chemmometric-trắc quang
khi xác định PAR và IB trong thuốc Protamol (*)
Chất Thông số/ Đại lượng
thống kê
Protamol
Kalman BPTT Phổ đạo hàm
T1 T2 T3 T1 T2 T3 T1 T2 T3
PAR
C (µg/mL) 13,28 13,15 13,29 13,28 13,15 13,29 13,17 13,16 13,19
Hàm lượng (mg/viên) 332,0 328,7 332,4 332,0 328,7 332,4 329,4 329,1 329,7
TB ± S (µg/mL) 13,24 ± 0,08 13,24 ± 0,08 13,17 ± 0,02
RSD (%) 0,6 0,6 0,2
½ RSDH (%) 5,5 5,5 5,5
IB
C (µg/mL) 7,97 7,89 7,98 7,97 7,89 7,98 7,94 8,08 8,11
Hàm lượng (mg/viên) 199,2 197,2 199,3 199,2 197,2 199,3 198,5 202,1 202,8
TB ± S (µg/mL) 7,94 ± 0,05 7,94 ± 0,05 8,04 ± 0,09
RSD (%) 0,6 0,6 1,1
½ RSDH (%) 5,9 5,9 5,9
(*) Các ký hiệu và các đại lượng nêu trong bảng cũng như ở bảng 3.24; các giá trị TB, S và RSD đều
ứng với n 3.
Do PAR và IB có mặt trong nhiều loại thuốc khác nhau đang được tiêu thụ
trên thị trường thế giới và do vậy, cũng đã có nhiều nghiên cứu áp dụng các phương
pháp khác nhau (trắc quang và chemometric-trắc quang) để xác định đồng thời
chúng. So sánh với các phương pháp đó, phương pháp Kalman cũng đạt được độ
lặp lại tương đương khi xác định PAR và IB (trong các loại thuốc khác nhau đã
nghiên cứu, tỉ lệ hàm lượng giữa PAR và IB trong viên thuốc, mg/mg, ghi trên nhãn
giống với tỉ lệ giữa chúng trong cả 3 loại thuốc Alaxan, Lopenca và Protamol, tức là
PAR : IB 325 : 200 (bảng 3.31).

114
Bang 3.31. Độ lặp lại và độ đúng của các phương pháp trắc quang và
chemometric-trắc quang khi phân tích đồng thời PAR và IB trong dược phẩm (*)
Phương pháp
PAR IB
Nguồn TLTK Hàm lượng
𝑥𝑖(mg/viên)
RSD (%)
Rev (%) Hàm lượng
𝑥𝑖(mg/viên)
RSD (%)
Rev (%)
Kalman 324,2 0,5 97,5 – 103 212,8 0,5 97,5 – 101,5 Luận án
này
Phổ đạo hàm
99,1 – 100,1 99,5 – 100,3 [120]
Chuyển dịch bước sóng
99,1 – 100,7 99,2 – 100,6 [120]
Ty lệ độ hấp thụ
99,1 – 99,2 101,5 – 101,8
[122]
BPTT 325 100,2 199,8 99,9 [122]
PCR 324,4 99,90 199,8 100,0 [122]
(*) Hàm lượng PAR: IB công bố trên mẫu thuốc là 325:200 (mg/viên); Dấu (-) là không xác định.
Cuối cùng, có thể thấy rằng, các kết quả phân tích đồng thời 2 chất có phổ
hấp thụ quang xen phủ nhau trong các thuốc chứa 2 thành phần theo cả 3 phương
pháp – Kalman, BPTT và phổ đạo hàm đều cho độ lặp lại tốt. Như vậy, đối với các
thuốc chứa 2 thành phần đã khảo sát, do ảnh hưởng qua lại của chúng trong dung
dịch phân tích không đáng kể và do độ hấp thụ quang ghi được bị nhiễu đo không
đáng kể, nên có thể khẳng định rằng, trong những trường hợp đó, đều có thể áp
dụng cả 3 phương pháp để phân tích các chất trong thuốc. Song, trong trường hợp
nhiễu đo lớn hoặc ảnh hưởng qua lại của các chất trong dung dịch mẫu lớn, việc áp
dụng phương pháp Kalman sẽ cho kết quả tốt hơn. Điều này sẽ được khẳng định
trong các nghiên cứu với thuốc chứa 3 thành phần dưới đây.
iv) Đối với hỗn hợp AML, HYD và VAL trong thuốc Exforge HCT
Khi khảo sát các dung dịch chuẩn hỗn hợp 3 chất (mục 3.4) đã cho thấy, đối với
trường hợp dung dịch chứa 3 chất có phổ hấp thụ quang xen phủ nhau, không thể hoặc
rất khó áp dụng phương pháp phổ đạo hàm. Do vậy, ở đây, để so sánh, chỉ 2 phương
pháp – Kalman và BPTT được áp dụng để phân tích đồng thời 3 chất (AML, HYD và
VAL) trong mẫu thuốc Exforge HCT (thuốc điều trị bệnh tim mạch).

115
Mẫu thuốc nghiên cứu: Thuốc viên nén Exforge HCT với hàm lượng ghi trên
nhãn của AML là 10 mg, VAL là 160 mg và HYD là 12,5 mg, số lô: BK917, ngày
sản xuất: 09/2016, hạn dùng: 08/2018, SĐK: VN-19287-15, thuốc được sản xuất tại
Novartis Farmaceutica S.A. Ronda Santa Maria 158, 08210 Barberà del Vallès,
Barcelona, Tây Ban Nha.
Tiến hành xử lý mẫu thuốc cho phân tích theo cách tương tự như nêu ở mục
2.2.8.4 và tiến hành phân tích theo 2 phương pháp – Kalman (quy trình phân tích
như ở hình 3.10) và phương pháp bình phương tối thiểu dùng phổ toàn phần
(BPTT), thu được các kết quả ở bảng 3.32. Từ các giá trị về hàm lượng các chất
trong viên thuốc (được ghi trên nhãn thuốc), cũng tính được nồng độ 3 chất AML,
HYD và VAL trong dung dịch mẫu thuốc tương ứng là 1,00 µg/mL, 1,25 µg/mL và
16,00 µg/mL.
Kết quả ở bảng 3.32 cho thấy:
- Mặc dù ty lệ các chất trong thuốc Exforge HCT rất khác nhau (AML :
HYD : VAL 1 : 1,25 : 16), nhưng cả 2 phương pháp vẫn cho độ lặp lại tốt đối với
cả 3 chất: Đối với AML, nồng độ trung bình (TB) trong dung dịch mẫu thuốc là
0,961 µg/mL – 0,962 µg/mL với RSD là 2,2 % – 2,3 %, nhỏ hơn ½ RSDH (8 %);
Đối với HYD, nồng độ trung bình là 1,163 µg/mL – 1,166 µg/mL với RSD 2,2 %,
nhỏ hơn ½ RSDH (5,5 %); Đối với VAL, nồng độ trung bình là 16,506 µg/mL –
17,340 µg/mL với RSD 2,2 % - 2,3 %, nhỏ hơn ½ RSDH (5,3 %).
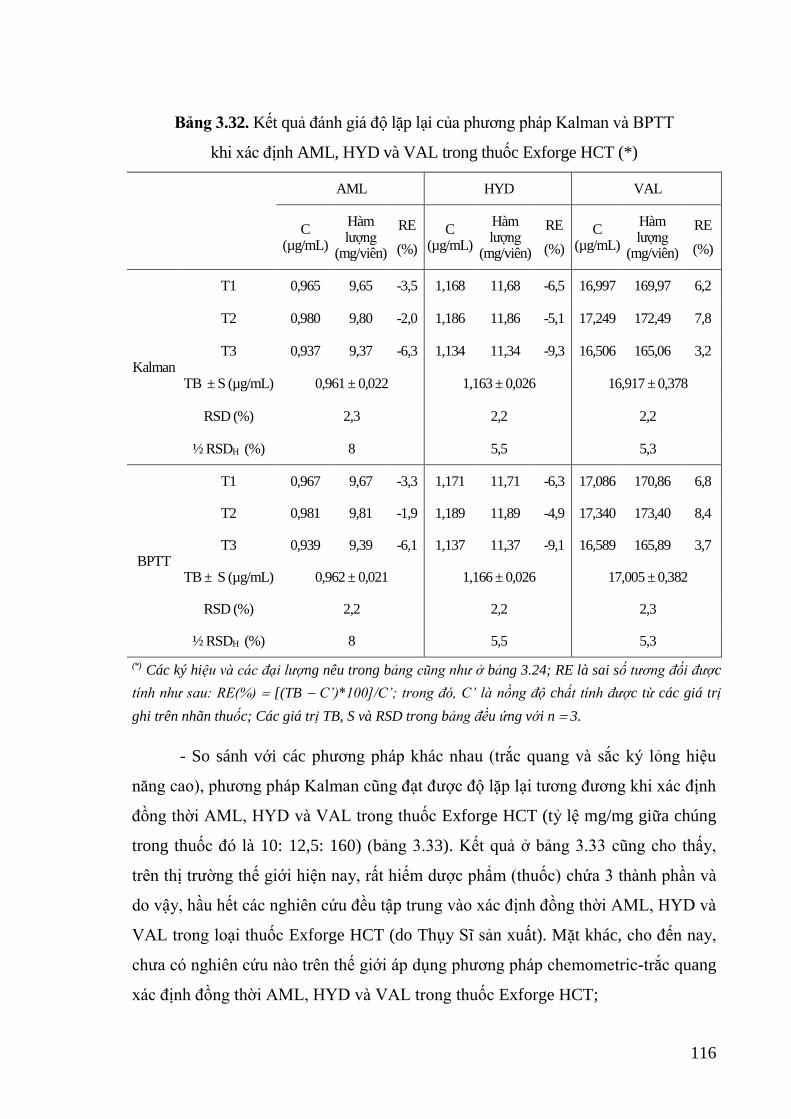
116
Bang 3.32. Kết quả đánh giá độ lặp lại của phương pháp Kalman và BPTT
khi xác định AML, HYD và VAL trong thuốc Exforge HCT (*)
AML HYD VAL
C (µg/mL)
Hàm lượng
(mg/viên)
RE
(%)
C
(µg/mL)
Hàm lượng
(mg/viên)
RE
(%)
C (µg/mL)
Hàm lượng
(mg/viên)
RE
(%)
Kalman
T1 0,965 9,65 -3,5 1,168 11,68 -6,5 16,997 169,97 6,2
T2 0,980 9,80 -2,0 1,186 11,86 -5,1 17,249 172,49 7,8
T3 0,937 9,37 -6,3 1,134 11,34 -9,3 16,506 165,06 3,2
TB ± S (µg/mL) 0,961 ± 0,022 1,163 ± 0,026 16,917 ± 0,378
RSD (%) 2,3 2,2 2,2
½ RSDH (%) 8 5,5 5,3
BPTT
T1 0,967 9,67 -3,3 1,171 11,71 -6,3 17,086 170,86 6,8
T2 0,981 9,81 -1,9 1,189 11,89 -4,9 17,340 173,40 8,4
T3 0,939 9,39 -6,1 1,137 11,37 -9,1 16,589 165,89 3,7
TB ± S (µg/mL) 0,962 ± 0,021 1,166 ± 0,026 17,005 ± 0,382
RSD (%) 2,2 2,2 2,3
½ RSDH (%) 8 5,5 5,3
(*) Các ký hiệu và các đại lượng nêu trong bảng cũng như ở bảng 3.24; RE là sai số tương đối được
tính như sau: RE(%) [(TB C’)*100]/C’; trong đó, C’ là nồng độ chất tính được từ các giá trị
ghi trên nhãn thuốc; Các giá trị TB, S và RSD trong bảng đều ứng với n 3.
- So sánh với các phương pháp khác nhau (trắc quang và sắc ký lỏng hiệu
năng cao), phương pháp Kalman cũng đạt được độ lặp lại tương đương khi xác định
đồng thời AML, HYD và VAL trong thuốc Exforge HCT (ty lệ mg/mg giữa chúng
trong thuốc đó là 10: 12,5: 160) (bảng 3.33). Kết quả ở bảng 3.33 cũng cho thấy,
trên thị trường thế giới hiện nay, rất hiếm dược phẩm (thuốc) chứa 3 thành phần và
do vậy, hầu hết các nghiên cứu đều tập trung vào xác định đồng thời AML, HYD và
VAL trong loại thuốc Exforge HCT (do Thụy Sĩ sản xuất). Mặt khác, cho đến nay,
chưa có nghiên cứu nào trên thế giới áp dụng phương pháp chemometric-trắc quang
xác định đồng thời AML, HYD và VAL trong thuốc Exforge HCT;

117
- Đối với cả 3 chất (AML, HYD và VAL), các kết quả xác định được (các
giá trị TB) đều gần với các kết quả tính toán được từ các gái trị ghi trên nhãn thuốc
hay nói cách khác, cả 2 phương pháp đều cho độ đúng tốt (hay sai số nhỏ). Như vậy,
trong trường hợp nhiễu đo độ hấp thụ quang ở các bước sóng khác nhau nhỏ, cả 2
phương pháp đều có thể áp dụng để xác định đồng thời cả 3 chất trong dung dịch
hỗn hợp của chúng.
Song cũng cần thấy rằng, trong trường hợp nhiễu đo lớn (chẳng hạn, vì một
lý do bất thường nào đó như nhiễu nhiệt, nhiễu từ, điện không ổn định...), phương
pháp Kalman cho kết quả mắc sai số nhỏ hơn so với phương pháp BPTT. Điều này
đã được khẳng định khi khảo sát các dung dịch chuẩn hỗn hợp chứa 3 chất (AML,
HYD và VAL) với giả định, phép đo độ hấp thụ quang bị mắc sai số lớn (hay nhiễu
đo lớn) ở một số bước sóng nào đó (xem mục 3.4.2.2 và xem bảng 3.23).
Bang 3.33. Độ lặp lại và độ đúng của các phương pháp trắc quang và chemometric-
trắc quang khi phân tích đồng thời AML, HYD và VAL trong dược phẩm (*)
Phương pháp
AML HYD VAL
TLTK Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Hàm lượng
(mg/viên)
RSD (%)
Rev (%)
Kalman 9,61 2,3 90-94 11,66 2,2 90-96 169,17 2,2 99-104 Luận
án này
Hiệu chỉnh độ hấp thụ
10,11 1,9 100- 101
12,73 1.3 99–100 159,64 0,4 100-101
[39]
Trắc quang
1,0 99 – 101
1,3 99-102 0,7 99-101 [62]
RP-HPLC
10,03 0,5 –1,4
100- 101
12,49 0.24 -0,36
98-99 160,05 0,8 – 1,0
99-100 [86]
HPTLC 10,03 2,0 99 - 100
1,9 99 -100 0,8 99-101 [86]
(*) RP-HPLC: Sắc ký lỏng hiệu năng cao pha đảo (Reverse Phase High Performance Liquid
Chromatography); HPTLC: Sắc ký lớp mỏng hiệu năng cao (High Performance Thin Layer
Chromatography); Rev(%): Độ thu hồi; Dấu (-) là không xác định.

118
3.6.1.2. Độ đúng
Độ đúng của phương pháp phân tích là độ gần của kết quả đo/phân tích với giá
trị thực của nó. Để đánh giá độ đúng của phương pháp phân tích, có thể thực hiện
theo 3 cách: (1) Phân tích Mẫu vật liệu so sánh được cấp chứng chỉ (certified
reference material), có giá trị nồng độ chất ghi trong chứng chỉ đi kèm mẫu được
chấp nhận là giá trị thực; (2) Phân tích mẫu thực tế được thêm chuẩn (spiked
sample) và đánh giá độ đúng qua độ thu hồi; (3) Phân tích mẫu bằng phương pháp
chuẩn khác, rồi so sánh kết quả phân tích của phương pháp đang dùng và phương
pháp chuẩn đó bằng phương pháp thống kê.
Trong nghiên cứu của luận án, hai cách đánh giá độ đúng (2) và (3) được sử
dụng. Phương pháp chuẩn được chọn để so sánh (khi phân tích các mẫu dược phẩm)
là phương pháp sắc ký lỏng hiệu năng cao (HPLC).
i) Đối với TEL và HYD trong thuốc Micardis(R) Plus
Phân tích mẫu thêm chuẩn: Tiến hành xác định độ đúng của các phương pháp
chemometric-trắc quang (phương pháp Kalman, bình phương tối thiểu và phổ đạo
hàm) khi xác định đồng thời TEL và HYD trong thuốc Micardis(R) Plus bằng cách
phân tích mẫu thuốc được thêm chuẩn (thêm chuẩn vào dung dịch mẫu thuốc), thu
được các kết quả ở bảng 3.34. Đối với mỗi phép xác định độ đúng, thực hiện lặp lại
3 lần (No1, No2, No3 ứng với các mức thêm chuẩn khác nhau: 1,00; 2,00 và 3,00
g/mL) để từ đó, tính độ thu hồi trung bình. Kỹ thuật xử lý mẫu cho phân tích được
thực hiện như ở mục 2.2.8. Dữ liệu phổ của các dung dịch chuẩn và các dung dịch
mẫu được ghi trong khoảng bước sóng 240 – 335 nm với khoảng cách giữa các
bước sóng là 0,2 nm. Dữ liệu đó là dữ liệu đầu vào để tính toán nồng độ mỗi chất
trong mẫu theo phương pháp bình phương tối thiểu (BPTT) và phương pháp
Kalman.
Đối với phương pháp phổ đạo hàm, quét phổ đạo hàm bậc 1 của dung dịch
mẫu trong khoảng bước sóng 240 - 335 nm với khoảng cách giữa các bước sóng là
0,2 nm. Sử dụng giá trị phổ đạo hàm tại 322,8 nm để định lượng TEL và tại 282,4
nm để định lượng HYD.

119
Bang 3.34. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời TEL và HYD trong thuốc Micardis(R) Plus (*)
Chất Cthêm (µg/mL) 1,00 2,00 3,00 RevTB
(%) Đại lương\Thí nghiệm No1 No2 No3 No1 No2 No3 No1 No2 No3
TEL
PP
Kalman
Co (µg/mL) 7,96 7,57 7,44 7,96 7,57 7,44 7,96 7,57 7,44
C (µg/mL) 8,94 8,59 8,47 10,00 9,56 9,47 11,06 10,59 10,45
Rev(%) 98 102 103 102 100 102 103 101 100 101
PP
BPTT
Co (µg/mL) 7,97 7,58 7,45 7,97 7,58 7,45 7,97 7.58 7,45
C (µg/mL) 8,96 8,60 8,48 10,01 9,57 9,48 11,07 10,60 10,46
Rev(%) 99 102 103 102 100 102 103 101 100 101
PP Phổ
đạo
hàm
Co (µg/mL) 7,70 7,81 7,92 7,70 7,81 7,92 7,70 7,81 7,92
C (µg/mL) 8,83 8,73 8,90 9,87 9,77 9,77 10,88 10,69 10,89
Rev (%) 113 92 98 109 98 93 106 96 99 100
HYD
PP
Kalman
Co (µg/mL) 2,58 2.59 2,56 2,58 2,59 2,56 2,58 2,59 2,56
C (µg/mL) 3,63 3,64 3,62 4,65 4,64 4,62 5,67 5,66 5,63
Rev(%) 105 105 106 104 103 103 103 102 102 104
PP
BPTT
Co (µg/mL 2,58 2,59 2,56 2,58 2,59 2,56 2,58 2,59 2,56
C (µg/mL 3,63 3,64 3,62 4,66 4,64 4,62 5,67 5,66 5,62
Rev(%) 105 105 106 104 103 103 103 102 102 104
PP Phổ
đạo
hàm
Co (µg/mL) 2,57 2,31 2,54 2,57 2,31 2,54 2,57 2,31 2,54
C (µg/mL) 3,52 3,29 3,61 4,49 4,31 4,33 5,55 5,52 5,60
Rev(%) 95 98 97 96 100 90 99 107 103 99 (*) Cthêm: Nồng độ chất chuẩn thêm vào mẫu; Co: Nồng độ chất trong mẫu; C: Nồng độ chất
trong mẫu đã được thêm chuẩn; Rev: Độ thu hồi; RevTB: Độ thu hồi trung bình (n 9).
Kết quả ở bảng 3.34 cho thấy:
- Cả 3 phương pháp đều đạt được độ đúng tốt với độ thu hồi thỏa mãn yêu cầu:
Theo AOAC (Hiệp hội các nhà hóa học phân tích Mỹ), khi phân tích những nồng độ
cỡ 1 ppm - 10 ppm (ppm ≈ g/mL), nếu đạt được độ thu hồi trong khoảng 80 - 110
%, là đạt yêu cầu [40].
- Phương pháp phổ đạo hàm tuy đạt được độ thu hồi khá tốt (trung bình
khoảng 99 – 100 %), nhưng lại dao động trong khoảng rộng hơn (95 – 107 %).
Phương pháp Kalman và phương pháp BPTT cũng đạt được độ thu hồi khá tốt
(trung bình là 101 % đối với TEL và 104 % đối với HYD), nhưng lại dao động
trong khoảng hẹp hơn (98 – 103 % đối với TEL và 102 – 106 % đối với HYD) so

120
với phương pháp phổ đạo hàm. Nói cách khác, phương pháp Kalman và phương
pháp BPTT cho độ đúng tốt hơn so với phương pháp phổ đạo hàm (đối với cả TEL
và HYD). Các kết quả trên cũng cho thấy, ảnh hưởng qua lại giữa 2 chất trong dung
dịch và ảnh hưởng của các thành phần tá dược (có mặt trong thuốc) đối với cả 2
phương pháp phân tích – Kalman và BPTT, là không đáng kể.
So sánh với phương pháp HPLC: Để khẳng định chắc chắn hơn, độ đúng của
các phương pháp chemometric-trắc quang cũng được đánh giá qua so sánh với
phương pháp HPLC (được xem là phương pháp chuẩn) khi phân tích thuốc
Micardis(R) Plus (phương pháp HPLC xác định TEL – Dược điển Việt Nam/DĐVN
IV và xác định HYD – Dược điển Mỹ/USP 38).
Kết quả ở bảng 3.35 cho thấy:
- Đối với TEL, phương pháp Kalman và BPTT có độ lặp lại kém hơn so với
phương pháp HPLC (do các giá trị Ftính lớn hơn giá trị tới hạn F(0,05;2,2), nhưng vẫn
cho kết quả không khác so với phương pháp HPLC với p > 0,05 (các giá trị ttính đều
nhỏ hơn nhiều so với giá trị tới hạn t(0,05; f)). Phương pháp phổ đạo hàm tuy có độ lặp
lại tương đương với phương pháp HPLC, nhưng lại có giá trị ttính nhỏ hơn không
nhiều so với giá trị tới hạn t(0,05; f)), tức là cho kết quả sai khác nhiều hơn so với
phương pháp HPLC;
- Đối với HYD, phương pháp Kalman và BPTT có độ lặp lại tương đương với
phương pháp HPLC (do các giá trị Ftính nhỏ hơn nhiều giá trị tới hạn F(0,05;2,2), và
cho kết quả không khác so với phương pháp HPLC với p > 0,05 (các giá trị ttính đều
nhỏ hơn nhiều so với giá trị tới hạn t(0,05; f)). Trong khi đó, phương pháp phổ đạo
hàm có độ lặp lại kém hơn so với phương pháp HPLC, nhưng vẫn cho kết quả
không sai khác so với phương pháp HPLC (do có giá trị ttính nhỏ hơn giá trị tới hạn
t(0,05; f)), nhưng nhỏ hơn không nhiều;
- Tuy độ lặp lại của 3 phương pháp (đánh giá qua S hoặc S2) có khác nhau,
nhưng chúng đều đạt được độ đúng tốt (đối với cả TEL và HYD) khi so sánh với
phương pháp HPLC với p > 0,05.
So sánh với các kết quả nghiên cứu liên quan: So sánh với kết quả công bố của
một số tác giả khác trên thế giới, độ đúng của phương pháp Kalman không thua
kém so với các phương pháp phân tích khác (trắc quang và chemometric-trắc

121
quang) khi xác định đồng thời TEL và HYD trong các loại thuốc khác nhau: Đối
với TEL và HYD, các phương pháp khác đạt được độ thu hồi tương ứng là 98 – 103
% và 96 – 102 % (các số liệu chi tiết về độ thu hồi được nêu ở bảng 3.25).
Bang 3.35. So sánh độ đúng của các phương pháp chemometric-trắc quang với
phương pháp HPLC khi xác định TEL và HYD trong thuốc Micardis(R) Plus (*)
Đại lượng thống kê
TEL HYD
Kalman BPTT Phổ đạo
hàm HPLC Kalman BPTT
Phổ đạo hàm
HPLC
xi (mg/viên)
39,8
37,9
37,2
39,9
37,9
37,3
38,5
39,1
39,6
38,4
38,2
38,4
12,9
13,0
12,8
12,9
12,9
12,8
12,9
12,5
12,7
12,9
12,8
13,0
TB (mg/viên) 38,3 38,4 39,1 38,3 12,9 12,9 12,4 12,9
S (mg/viên) 1,4 1,4 0,6 0,1 0,1 0,1 0,7 0,1
Ftính 136 139 23 1,1 3,4 44
F(0,05;2;2) 39 39
Sp 0,4 0,1 0,1
F 2 2 4 4 4 2
ttính 0,04 0,04 2,21 0,28 0,14 1,23
t(0,05; f) 4,30 4,30 2,78 2,78 2,78 4,30
(*) xi: Các kết quả phân tích lặp lại (i = 1-3), được trích từ các bảng kết quả đánh giá độ lặp lại của
phương pháp Kalman, BPTT và phổ đạo hàm khi phân tích TEL và HYD; TB: trung bình số học (n
= 3); S: độ lệch chuản; Ftính = phương sai (S2) của mỗi phương pháp / phương sai của phương
pháp HPLC; F(0,05;2;2): Giá trị F tới hạn ở mức ý nghĩa thống kê 0,05 và 2 bậc tự do của 2 phương
sai tử số và mẫu số; Sp: Phương sai chung, được tính từ 2 phương sai của 2 phương pháp khi 2
phương sai của 2 phương pháp như nhau (Ftính< F(0,05;2;2)); f: Bậc tự do được tính từ 2 tập số liệu
của 2 phương pháp khi 2 phương pháp có phương sai khác nhau (Ftính> F(0,05;2;2); t(0,05; f): Giá trị tới
hạn của t ở mức ý nghĩa thống kê 0,05 và bậc tự do f.
ii) Đối với PAR và CAF trong thuốc Panadol Extra
Phân tích mẫu thêm chuẩn: Theo cách tương tự như đối với phân tích thuốc
Micardis(R) Plus, tiến hành xác định độ đúng của các phương pháp chemometric-trắc
quang khi xác định đồng thời PAR và CAF trong thuốc Panadol Extra bằng cách

122
phân tích mẫu thuốc được thêm chuẩn (thêm chuẩn vào dung dịch mẫu thuốc, thêm
4 và 8 g/mL mỗi chất), thu được các kết quả ở bảng 3.36. Đối với mỗi phép xác
định độ đúng, thực hiện lặp lại 3 lần. Dữ liệu phổ của các dung dịch chuẩn và các
dung dịch mẫu được ghi trong khoảng bước sóng 215 nm – 300 nm với khoảng
cách giữa các bước sóng là 0,2 nm.
Đối với phương pháp phổ đạo hàm, quét phổ đạo hàm bậc 1 của dung dịch
mẫu trong khoảng bước sóng 215 nm - 300 nm với khoảng cách giữa các bước sóng
là 0,2 nm. Sử dụng giá trị phổ đạo hàm tại 260 nm để định lượng PAR và tại 216
nm để định lượng CAF.
Bang 3.36. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời PAR và CAF trong thuốc Panadol Extra (*)
Phương pháp
PAR CAF
C0
(μg / mL) Rev (%)
C0
(μg / mL) Rev (%)
Kalman 7,88 101 - 103 1,05 96 – 105
BPTT 7,88 101 – 103 1,04 96 -103
Phổ đạo hàm 7,88 103 - 105 1,06 101 - 107
(*) Cthêm: Nồng độ chất chuẩn thêm vào mẫu; Co: Nồng độ chất trong mẫu; C: Nồng độ chất trong
mẫu đã được thêm chuẩn; Rev: Độ thu hồi; RevTB: Độ thu hồi trung bình (n 6).
Kết quả ở bảng 3.36 cho thấy:
- Cả 3 phương pháp đều đạt được độ đúng tốt với độ thu hồi thỏa mãn yêu cầu:
Theo AOAC (Hiệp hội các nhà hóa học phân tích Mỹ), khi phân tích những nồng độ
cỡ 1 ppm - 10 ppm (ppm ≈ g/mL), nếu đạt được độ thu hồi trong khoảng 80 - 110
%, là đạt yêu cầu [40].
- Đối với phương pháp Kalman, độ thu hồi trung bình của PAR là 102 %, của
CAF là 99 %; đối với phương pháp BPTT, độ thu hồi trung bình của PAR là 102 %,
của CAF là 98 %; đối với phương pháp quang phổ đạo hàm, độ thu hồi trung bình
của PAR là 104 %, của IB là 103 %. Nói cách khác, phương pháp Kalman và
phương pháp BPTT cho độ đúng tốt hơn so với phương pháp phổ đạo hàm (đối với
cả PAR và CAF). . Các kết quả trên cũng cho thấy, ảnh hưởng qua lại giữa 2 chất

123
trong dung dịch và ảnh hưởng của các thành phần tá dược (có mặt trong thuốc) đối
với cả 2 phương pháp phân tích – Kalman và BPTT, là không đáng kể.
So sánh với phương pháp HPLC: Để khẳng định chắc chắn hơn, độ đúng của
các phương pháp chemometric-trắc quang cũng được đánh giá qua so sánh với
phương pháp HPLC (được xem là phương pháp chuẩn) khi phân tích thuốc
Micardis(R) Plus (phương pháp HPLC xác định PAR – Dược điển Việt Nam/DĐVN
IV và xác định CAF – Dược điển Mỹ/USP 38).
Bang 3.37. So sánh độ đúng của các phương pháp chemometric-trắc quang với
phương pháp HPLC khi xác định PAR, CAF trong thuốc Panadol Extra (*)
Các đại lượng thống kê
Hàm lượng PAR (mg/viên) Hàm lượng CAF (mg/viên)
Kalman BPTT Phổ đạo
hàm HPLC Kalman BPTT
Phổ đạo hàm
HPLC
xi (mg/viên)
496,2
483,8
494,7
496,2
493,8
494,7
498,0
494,8
494,8
497,8
495,3
496,9
67,1
67,8
66,2
67,1
67,8
66,2
67,3
67,9
66,5
67,5
68,2
67,9
TB (mg/viên)
494,9 494,9 495,9 496,7 67,0 67,0 67,2 67,9
S 1,2 1,2 1,8 1,3 0,8 0,8 0,70 0,35
Ftính 1,09 1,09 2,13 5,22 5,22 4,0
F(0,05;2;2) 39,0 39,0 39,0 39,0 39,0 39,0
Sp 1,24 1,24 1,58 0,62 0,62 0,56
ttính 1,75 1,75 0,62 1,65 1,65 1,40
t(0,05; f) 2,78 2,78 2,78 2,78 2,78 2,78
(*) xi: Các kết quả phân tích lặp lại (i = 1-3), được trích từ các bảng kết quả đánh giá độ lặp lại của phương pháp Kalman, BPTT và phổ đạo hàm khi phân tích TEL và HYD; TB: trung bình số học (n = 3); S: độ lệch chuản; Ftính = phương sai (S2) của mỗi phương pháp / phương sai của phương pháp HPLC; F(0,05;2;2): Giá trị F tới hạn ở mức ý nghĩa thống kê 0,05 và 2 bậc tự do của 2 phương sai tử số và mẫu số; Sp: Phương sai chung, được tính từ 2 phương sai của 2 phương pháp khi 2 phương sai của 2 phương pháp như nhau (Ftính< F(0,05;2;2)); f: Bậc tự do được tính từ 2 tập số liệu của 2 phương pháp khi 2 phương pháp có phương sai khác nhau (Ftính> F(0,05;2;2); t(0,05; f): Giá trị tới hạn của t ở mức ý nghĩa thống kê 0,05 và bậc tự do f.
Kết quả ở bảng 3.35 cho thấy: Tuy độ lặp lại của 3 phương pháp (đánh giá qua
S hoặc S2) có khác nhau, nhưng chúng đều đạt được độ đúng tốt (đối với cả PAR và
CAF) khi so sánh với phương pháp HPLC với p > 0,05.
So sánh với các kết quả nghiên cứu liên quan: So sánh với kết quả công bố
của một số tác giả khác trên thế giới, độ đúng của phương pháp Kalman không thua
kém so với các phương pháp phân tích khác (trắc quang và chemometric-trắc

124
quang) khi xác định đồng thời PAR và CAF trong các loại thuốc khác nhau: Đối với
cả TEL và HYD, các phương pháp khác đạt được độ thu hồi là 97 - 99 % (các số
liệu chi tiết về độ thu hồi được nêu ở bảng 3.27).
iii) Đối với hỗn hợp PAR và IB trong thuốc Alaxan, Lopenca và Protamol
Phân tích mẫu thêm chuẩn: Theo cách tương tự như đối với phân tích thuốc
Micardis(R) Plus và thuốc Panadol Extra, tiến hành xác định độ đúng của các
phương pháp chemometric-trắc quang khi xác định đồng thời PAR và IB trong các
mẫu thuốc Alaxan, Lopenca và Protamol bằng cách phân tích mẫu thuốc được thêm
chuẩn (thêm chuẩn vào dung dịch mẫu thuốc, thêm 2 và 4 g/mL mỗi chất), thu
được các kết quả ở bảng 3.38. Đối với mỗi phép xác định độ đúng, thực hiện lặp lại
3 lần. Dữ liệu phổ của các dung dịch chuẩn và các dung dịch mẫu được ghi trong
khoảng bước sóng 230 nm – 340 nm với khoảng cách giữa các bước sóng là 0,5 nm.
Đối với phương pháp phổ đạo hàm, quét phổ đạo hàm bậc 1 của dung dịch
mẫu trong khoảng bước sóng 230 nm - 340 nm với khoảng cách giữa các bước sóng
là 0,5 nm. Sử dụng giá trị phổ đạo hàm tại 26,5 nm để định lượng PAR và tại 217,5
nm để định lượng IB.
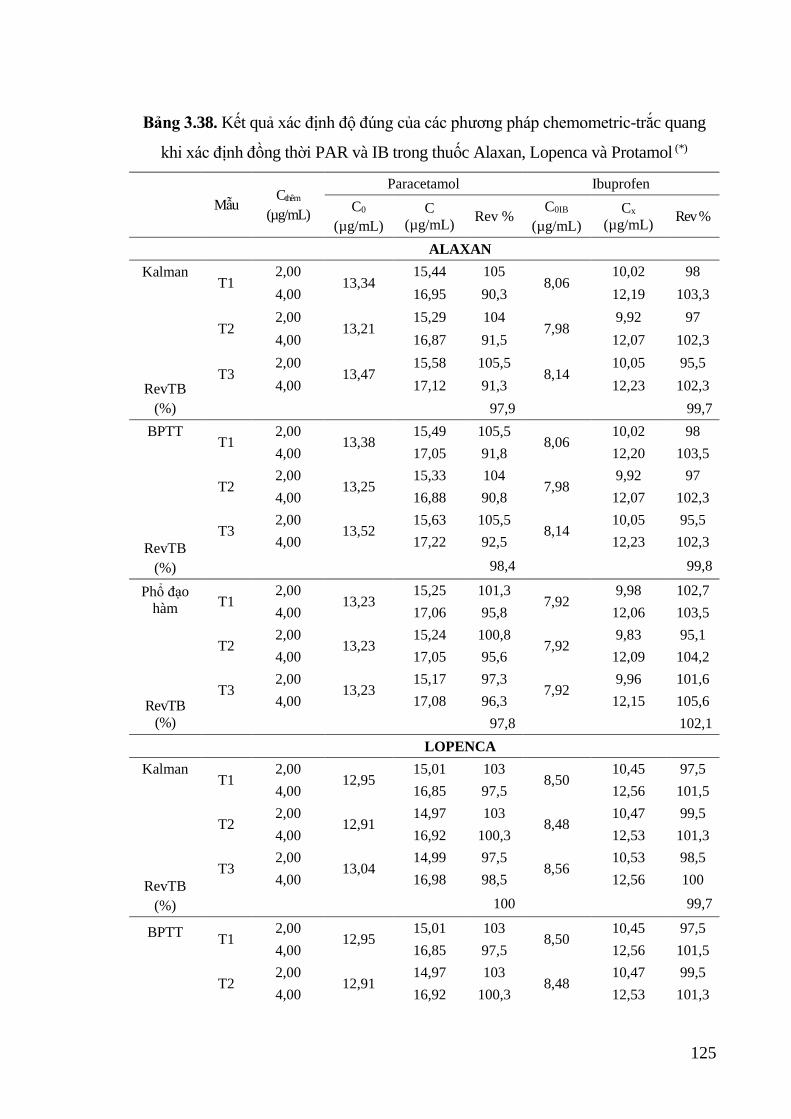
125
Bang 3.38. Kết quả xác định độ đúng của các phương pháp chemometric-trắc quang
khi xác định đồng thời PAR và IB trong thuốc Alaxan, Lopenca và Protamol (*)
Mẫu Cthêm
(µg/mL)
Paracetamol Ibuprofen
C0
(µg/mL)
C
(µg/mL) Rev %
C0IB
(µg/mL)
Cx
(µg/mL) Rev %
ALAXAN
Kalman
RevTB
(%)
T1 2,00
13,34 15,44 105
8,06 10,02 98
4,00 16,95 90,3 12,19 103,3
T2 2,00
13,21 15,29 104
7,98 9,92 97
4,00 16,87 91,5 12,07 102,3
T3 2,00
13,47 15,58 105,5
8,14 10,05 95,5
4,00 17,12 91,3 12,23 102,3
97,9 99,7
BPTT
RevTB
(%)
T1 2,00
13,38 15,49 105,5
8,06 10,02 98
4,00 17,05 91,8 12,20 103,5
T2 2,00
13,25 15,33 104
7,98 9,92 97
4,00 16,88 90,8 12,07 102,3
T3 2,00
13,52 15,63 105,5
8,14 10,05 95,5
4,00 17,22 92,5 12,23 102,3
98,4 99,8
Phổ đạo
hàm
RevTB
(%)
T1 2,00
13,23 15,25 101,3
7,92 9,98 102,7
4,00 17,06 95,8 12,06 103,5
T2 2,00
13,23 15,24 100,8
7,92 9,83 95,1
4,00 17,05 95,6 12,09 104,2
T3 2,00
13,23 15,17 97,3
7,92 9,96 101,6
4,00 17,08 96,3 12,15 105,6
97,8 102,1
LOPENCA
Kalman
RevTB
(%)
T1 2,00
12,95 15,01 103
8,50 10,45 97,5
4,00 16,85 97,5 12,56 101,5
T2 2,00
12,91 14,97 103
8,48 10,47 99,5
4,00 16,92 100,3 12,53 101,3
T3 2,00
13,04 14,99 97,5
8,56 10,53 98,5
4,00 16,98 98,5 12,56 100
100 99,7
BPTT
T1 2,00
12,95 15,01 103
8,50 10,45 97,5
4,00 16,85 97,5 12,56 101,5
T2 2,00
12,91 14,97 103
8,48 10,47 99,5
4,00 16,92 100,3 12,53 101,3

126
Mẫu Cthêm
(µg/mL)
Paracetamol Ibuprofen
C0
(µg/mL)
C
(µg/mL) Rev %
C0IB
(µg/mL)
Cx
(µg/mL) Rev %
RevTB
(%)
T3 2,00
13,04 14,99 97,5
8,56 10,53 98,5
4,00 16,98 98,5 12,56 100
100 99,7
Phổ đạo
hàm
RevTB
(%)
T1 2,00
4,00 12,79
14,83 101,8 8,71
10,74 101,8
16,61 95,6 12,73 100,5
T2 2,00
4,00 12,79
14,89 105,1 8,71
10,63 96,6
16,61 95,6 12,81 102,7
T3 2,00
4,00 12,79
14,88 104,4 8,71
10,78 103,8
16,64 96,2 12,82 102,9
99,8 101,4
PROTAMOL
Kalman
RevTB
(%)
T1
2,00 13,20
15,32 106 8,00
10,12 106
4,00 17,22 100,5 12,10 102,5
T2 2,00
13,07 15,17 105
7,92 10,06 107
4,00 17,05 99,5 11,98 101,5
T3 2,00
13,21 15,34 106,5
8,00 10,12 106
4,00 17,26 101,3 12,12 103
103,1 104,3
BPTT
RevTB
(%)
T1 2,00
13,24 15,37 106,5
8,00 10,12 106
4,00 17,28 101 12,10 102.5
T2 2,00
13,11 15,21 105
7,92 10,06 107
4,00 17,10 99,8 11,98 101,5
T3 2,00
13,26 15,38 106
8,01 10,12 106
4,00 17,32 101,5 12,12 103
103,3 104,3
Phổ đạo
hàm
RevTB
(%)
T1 2,00
13,17 15,24 103,4
7,92 9,98 103
4,00 16,98 95,2 11,98 101,6
T2 2,00
13,17 15,14 98,5
7,92 9,83 95,8
4,00 16,96 95,8 12,06 103,6
T3 2,00
13,178 15,17 100
7,92 9,96 101,8
4,00 17,01 95,6 11,79 96,8
98,1 100,4
(*)Co: Nồng độ chất trong mẫu; Ct: Nồng độ chất chuẩn thêm vào mẫu; Cx: Nồng độ chất xác định
được trong mẫu đã thêm chuẩn.
Kết quả ở bảng 3.38 cho thấy:

127
- Cả 3 phương pháp đều đạt được độ đúng tốt với độ thu hồi thỏa mãn yêu cầu:
Theo AOAC (Hiệp hội các nhà hóa học phân tích Mỹ), khi phân tích những nồng độ
cỡ 1 ppm - 10 ppm (ppm ≈ g/mL), nếu đạt được độ thu hồi trong khoảng 80 - 110
%, là đạt yêu cầu [40].
- Đối với phương pháp Kalman, độ thu hồi trung bình của PAR trong ba loại
thuốc nghiên cứu là 98 - 103 %, của IB là 100 – 104 %; đối với phương pháp
BPTT, độ thu hồi trung bình của PAR là 98 – 103 %, của IB là 100 - 104 %; đối với
phương pháp quang phổ đạo hàm, độ thu hồi trung bình của PAR là 98 - 100 %, của
IB là 100 - 102 %. Các kết quả trên cũng cho thấy, ảnh hưởng qua lại giữa 2 chất
trong dung dịch và ảnh hưởng của các thành phần tá dược (có mặt trong thuốc) đối
với cả 2 phương pháp phân tích – Kalman và BPTT, là không đáng kể.
So sánh với phương pháp HPLC: Để khẳng định chắc chắn hơn, độ đúng của
các phương pháp chemometric-trắc quang cũng được đánh giá qua so sánh với
phương pháp HPLC (được xem là phương pháp chuẩn) khi phân tích thuốc
Micardis(R) Plus (phương pháp HPLC xác định PAR – Dược điển Việt Nam/DĐVN
IV và xác định IB – Dược điển Mỹ/USP 38).
Kết quả ở bảng 3.39 cho thấy: Tuy độ lặp lại của 3 phương pháp (đánh giá
qua S hoặc S2) có khác nhau, nhưng chúng đều đạt được độ đúng tốt (đối với cả
PAR và IB) khi so sánh với phương pháp HPLC với p > 0,05.
So sánh với các kết quả nghiên cứu liên quan: So sánh với kết quả công bố
của một số tác giả khác trên thế giới, độ đúng của phương pháp Kalman không thua
kém so với các phương pháp phân tích khác (trắc quang và chemometric-trắc
quang) khi xác định đồng thời PAR và IB trong các loại thuốc khác nhau: Đối với
PAR và IB, các phương pháp khác đạt được độ thu hồi tương ứng là 99 – 101 % và
98 – 102 % (các số liệu chi tiết về độ thu hồi được nêu ở bảng 3.31).

128
Bang 3.39. So sánh các phương pháp chemometric với phương pháp HPLC
khi xác định hàm lượng PAR, IB trong thuốc Lopenca (*)
Các đại lượng
thống kê
Hàm lượng PAR (mg/viên) Hàm lượng IB (mg/viên)
Kalman BPTT Phổ đạo
hàm HPLC Kalman BPTT
Phổ đạo hàm
HPLC
xi (mg/viên) 323,8
/322,8
/326,0
323,8
/322,8
/326,0
322,8
/320,0
/323,9
324,4
/325,7
/325,4
212,5
/212,0
/214,0
212,5
/212,0
/214,0
214,9/
216,3
/216,7
214,0
/213,6
/214,9
TB (mg/viên) 324,2 324,2 322,2 325,2 212,8 212,8 216,0 214,2
S (mg/ viên) 1,6 1,6 2,0 0,7 1,0 1,0 0,9 0,6
Ftính 5,78 5,78 8,73 2,44 2,44 2,02
F(0,05;2;2) 39,0 39,0 39,0 39,0 39,0 39,0
Sp 1,25 1,25 1,50 0,87 0,87 0,82
ttính 0,94 0,94 2,39 1,87 1,87 2,70
t(0,05; f) 2,78 2,78 2,78 2,78 2,78 2,78
(*) xi: Các kết quả phân tích lặp lại (i = 1-3), được trích từ các bảng kết quả đánh giá độ lặp lại của
phương pháp Kalman, BPTT và phổ đạo hàm khi phân tích TEL và HYD; TB: trung bình số học (n
= 3); S: độ lệch chuản; Ftính = phương sai (S2) của mỗi phương pháp / phương sai của phương
pháp HPLC; F(0,05;2;2): Giá trị F tới hạn ở mức ý nghĩa thống kê 0,05 và 2 bậc tự do của 2 phương
sai tử số và mẫu số; Sp: Phương sai chung, được tính từ 2 phương sai của 2 phương pháp khi 2
phương sai của 2 phương pháp như nhau (Ftính< F(0,05;2;2)); f: Bậc tự do được tính từ 2 tập số liệu
của 2 phương pháp khi 2 phương pháp có phương sai khác nhau (Ftính> F(0,05;2;2); t(0,05; f): Giá trị tới
hạn của t ở mức ý nghĩa thống kê 0,05 và bậc tự do f.
iv) Đối với hỗn hợp AML, HYD và VAL trong thuốc Exforge HCT
Phân tích mẫu thêm chuẩn: Theo cách tương tự như đối với các mẫu thuốc
chứa 2 thành phần, tiến hành xác định độ đúng của các phương pháp chemometric-
trắc quang khi xác định đồng thời AML, HYD và VAL trong mẫu thuốc Exforge
HCT bằng cách phân tích mẫu thuốc được thêm chuẩn (thêm chuẩn vào dung dịch
mẫu thuốc, thêm 0,25 g/mL và 0,5 g/mL đối với AML; thêm 0,3g/mL và 0,6
g/mL đối với HYD; thêm 4,0 g/mL và 8,0 g/mL đối với VAL), thu được các kết
quả ở bảng 3.40. Đối với mỗi phép xác định độ đúng, thực hiện lặp lại 3 lần. Dữ
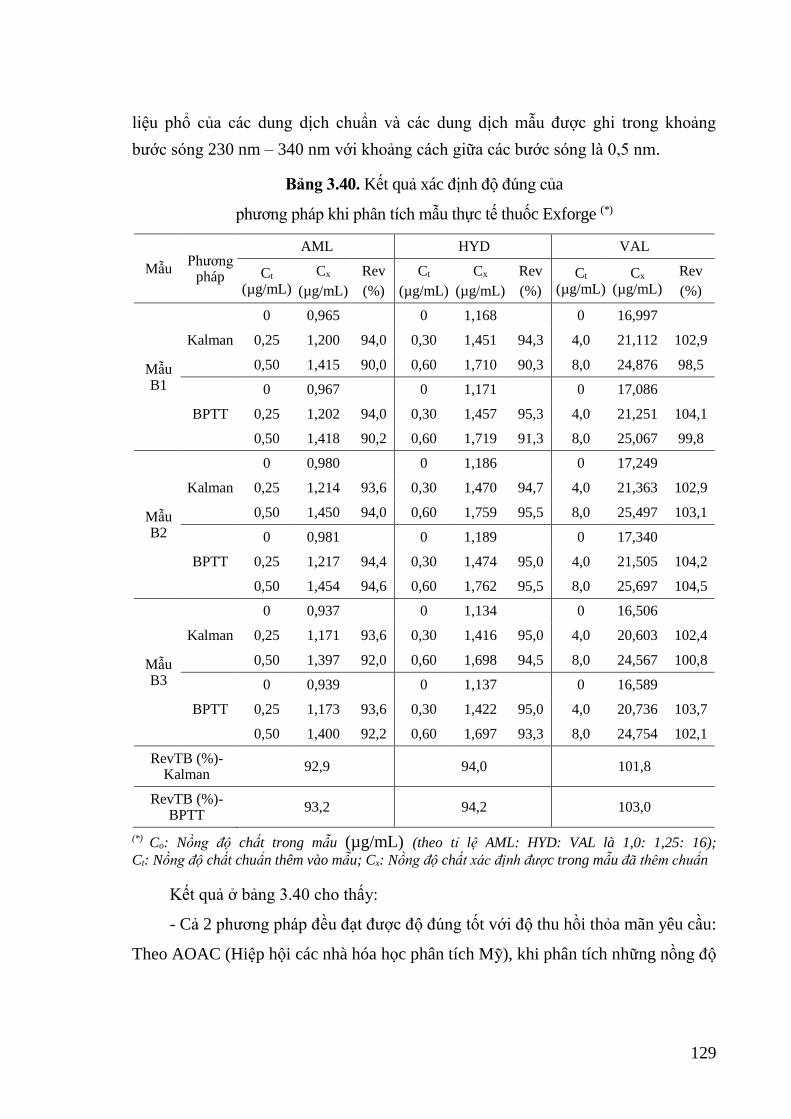
129
liệu phổ của các dung dịch chuẩn và các dung dịch mẫu được ghi trong khoảng
bước sóng 230 nm – 340 nm với khoảng cách giữa các bước sóng là 0,5 nm.
Bang 3.40. Kết quả xác định độ đúng của
phương pháp khi phân tích mẫu thực tế thuốc Exforge (*)
Mẫu Phương
pháp
AML HYD VAL
Ct
(µg/mL)
Cx
(µg/mL)
Rev
(%)
Ct
(µg/mL)
Cx
(µg/mL)
Rev
(%)
Ct (µg/mL)
Cx
(µg/mL)
Rev
(%)
Mẫu B1
Kalman
0 0,965 0 1,168 0 16,997
0,25 1,200 94,0 0,30 1,451 94,3 4,0 21,112 102,9
0,50 1,415 90,0 0,60 1,710 90,3 8,0 24,876 98,5
BPTT
0 0,967 0 1,171 0 17,086
0,25 1,202 94,0 0,30 1,457 95,3 4,0 21,251 104,1
0,50 1,418 90,2 0,60 1,719 91,3 8,0 25,067 99,8
Mẫu B2
Kalman
0 0,980 0 1,186 0 17,249
0,25 1,214 93,6 0,30 1,470 94,7 4,0 21,363 102,9
0,50 1,450 94,0 0,60 1,759 95,5 8,0 25,497 103,1
BPTT
0 0,981 0 1,189 0 17,340
0,25 1,217 94,4 0,30 1,474 95,0 4,0 21,505 104,2
0,50 1,454 94,6 0,60 1,762 95,5 8,0 25,697 104,5
Mẫu B3
Kalman
0 0,937 0 1,134 0 16,506
0,25 1,171 93,6 0,30 1,416 95,0 4,0 20,603 102,4
0,50 1,397 92,0 0,60 1,698 94,5 8,0 24,567 100,8
BPTT
0 0,939 0 1,137 0 16,589
0,25 1,173 93,6 0,30 1,422 95,0 4,0 20,736 103,7
0,50 1,400 92,2 0,60 1,697 93,3 8,0 24,754 102,1
RevTB (%)-Kalman
92,9 94,0 101,8
RevTB (%)-BPTT
93,2 94,2 103,0
(*) Co: Nồng độ chất trong mẫu (µg/mL) (theo tỉ lệ AML: HYD: VAL là 1,0: 1,25: 16);
Ct: Nồng độ chất chuẩn thêm vào mẫu; Cx: Nồng độ chất xác định được trong mẫu đã thêm chuẩn
Kết quả ở bảng 3.40 cho thấy:
- Cả 2 phương pháp đều đạt được độ đúng tốt với độ thu hồi thỏa mãn yêu cầu:
Theo AOAC (Hiệp hội các nhà hóa học phân tích Mỹ), khi phân tích những nồng độ

130
cỡ 1 ppm - 10 ppm (ppm ≈ g/mL), nếu đạt được độ thu hồi trong khoảng 80 - 110
%, là đạt yêu cầu [40].
- Độ thu hồi trung bình của các chất theo phương pháp Kalman và BPTT lần
lượt là: của AML đều là 93 %, của HYD là 97 % và 94 %, của VAL là 102 % và
103 %. Nói cách khác, phương pháp Kalman và phương pháp BPTT cho độ đúng
tốt hơn so với và phương pháp BPTT (đối với cả AML, HYD và VAL).
So sánh với phương pháp HPLC: Để khẳng định chắc chắn hơn, độ đúng của
các phương pháp chemometric-trắc quang cũng được đánh giá qua so sánh với
phương pháp HPLC (được xem là phương pháp chuẩn) khi phân tích thuốc
Micardis(R) Plus ((AML theo DĐVN IV, VAL theo USP 38, HYD theo USP 38).
Kết quả ở bảng 3.41 cho thấy, đối với cả 3 chất, kết quả của phương pháp
Kalman và phương pháp BPTT đều cho kết quả không sai khác có ý nghĩa thống kê
so với phương pháp HPLC (do các giá trị ttính đều nhỏ hơn giá trị ttới hạn với p >
0,30). Tuy vậy, dựa vào các giá trị p (mức ý nghĩa thống kê), có thể nhận xét rằng,
phương pháp Kalman cho kết quả gần với kết quả của phương pháp HPLC hơn (p
0,55 – 0,96) so với phương pháp BPTT (p 0,38 – 0,66) hay nói cách khác, phương
pháp Kalman đạt được độ đúng tốt hơn so với phương pháp BPTT (khi so sánh với
phương pháp HPLC).
So sánh với các kết quả nghiên cứu liên quan: So sánh với kết quả công bố
của một số tác giả khác trên thế giới, độ đúng của phương pháp Kalman không thua
kém so với các phương pháp phân tích khác (trắc quang và chemometric-trắc
quang) khi xác định đồng thời AML, HYD và VAL trong các loại thuốc khác nhau:
Đối với AML, HYD và VAL, các phương pháp khác đạt được độ thu hồi tương ứng
là 99 – 101 % ; 99 – 102 % và 99 – 101 % (các số liệu chi tiết về độ thu hồi được
nêu ở bảng 3.33).

131
Bang 3.41. So sánh các phương pháp chemometric với phương pháp HPLC
khi xác định hàm lượng AML, HYD và VAL trong thuốc Exforge HCT(*)
Chất
PT
Đại lượng
thống kê
Phương pháp phân tích
Kalman BPTT HPLC
AML
𝑥𝑖(mg/viên) 9,65/9,80/9,37 9,67/9.81/9,39 9,54/9,41/9,59
x (mg/viên) 9,61 9,62 9,51
S (mg/viên) 0,22 0,21 0,09
Ftính/
F(0,05;2;2) 5,30/19 5,30/19
Sp 0,16 0,16
ttính/ t(0,05; f) 0,53/4,3 0,63/4,3
P 0,65 0,59
HYD
𝑥𝑖(mg/viên) 11,68/11,86/11,34 11,71/11,89/11,37 11,72/11,76/11,41
x (mg/viên) 11,66 11,66 11,63
S (mg/viên) 0,26 0,26 0,19
Ftính/
F(0,05;2;2) 1,9/19 1,9/19
Sp 0,34 0,34
ttính/ t(0,05; f) -0,06/4,3 0,51/4,3
P 0,96 0,66
VAL
𝑥𝑖(mg/viên) 169,97/172,49/165,0
6
170,86/173,40/165,8
9
166,35/168,81/167,8
2
x (mg/viên) 169,17 167,66
S (mg/viên) 3,78 3,82 1,24
Ftính/
F(0,05;2;2) 9,32/19 9,5/9
Sp 0,10 0,10
ttính/ t(0,05; f) 0,71/4,30 1,11/4,30
P 0,55 0,38 (*) 𝑥𝑖: Kết quả phân tích lặp lại (i = 1-3); Ftính = phương sai của phương pháp Kalman (hoặc
BPTT) / phương sai của phương pháp HPLC; F(0,05;2;2): Giá trị F tới hạn ở mức ý nghĩa thống
kê 0,05 và 2 bậc tự do của 2 phương sai tử số và mẫu số; Sp: Phương sai chung, được tính từ 2
phương sai của 2 phương pháp khi 2 phương sai của 2 phương pháp như nhau (tức là khi Ftính<
F(0,05;2;2)); t(0,05;f=4): Giá trị tới hạn của t ở mức ý nghĩa thống kê p=0,05 và bậc tự do f=4.

132
3.6.2. Hàm lượng các hoạt chất trong các thuốc đa thành phần
Các kết quả nghiên cứu ở trên đã cho phép khẳng định rằng, phương pháp
Kalman đạt được độ chính xác cao (độ đúng tốt và độ lặp lại tốt) khi phân tích các
mẫu dược phẩm (thuốc) hay nói cách khác, hoàn toàn có thể áp dụng phương pháp
Kalman để phân tích các loại thuốc đa thành phần (chứa 2 hoặc 3 hoạt chất).
Trên thị trường Việt Nam hiện nay, nhiều loại thuốc chứa 2 thành phần (hay
2 hoạt chất) và chỉ một loại thuốc 3 thành phần đang được tiêu thụ. Các loại thuốc
đó chủ yếu thuộc các nhóm thuốc điều trị các loại bệnh huyết áp (thuốc Micardis(R)
Plus ), hạ sốt và giảm đau (thuốc Panadol Extra, Alaxan, Lopenca, Protamol ), tim
mạch (thuốc Exforge HCT). Phương pháp tiêu chuẩn hiện nay (theo quy định của
ngành dược Việt Nam) là phương pháp HPLC. Song, quy trình phân tích theo
phương pháp HPLC đòi hỏi nhiều kỹ thuật phức tạp, chi phí phân tích cao (do đòi
hỏi thiết bị và hóa chất đắt tiền).
Nhằm góp phần đưa ra một phương pháp phân tích đơn giản hơn, chi phí
phân tích rẻ hơn (do thiết bị trắc quang không đắt và hóa chất cần cho phân tích đơn
giản) để phân tích các loại thuốc đa thành phần đang lưu hành ở nước ta, quy trình
phân tích xây dựng được – phương pháp chemometric-trắc quang sử dụng thuật
toán lọc Kalman với chương trình phần mềm cho phép tính toán nhanh (gọi tắt là
phương pháp Kalman, như nêu ở hình 3.10) được áp dụng. Mục đích ở đây là nhằm
kiểm nghiệm xem hàm lượng các hoạt chất ghi trên nhãn thuốc có đúng không?
Theo quy định của ISO [40], khi đánh giá hàm lượng của hoạt chất ghi trên nhãn,
cần phải so sánh khoảng tin cậy 95 % của kết quả xác định được (trung bình số học
biên giới tin cậy 95 %) với giá trị ghi trên nhãn.
Các kết quả phân tích một số loại thuốc đa thành phần theo phương pháp
Kalman được nêu ở bảng 3.42. Thông tin về các loại thuốc, kỹ thuật lấy mẫu và
chuẩn bị mẫu cho phân tích, ghi dữ liệu phổ (độ hấp thụ quang trong khoảng bước
sóng xác định tùy thuộc vào dung dịch mẫu) tương tự như đã nêu ở mục 3.6.1 và
3.6.2 (khi đề cập về độ lặp lại và độ đúng của phương pháp phân tích).

133
Bang 3.42. Hàm lượng các hoạt chất trong một số loại thuốc đa thành phần đang
lưu hành ở thị trường Việt Nam (phân tích theo phương pháp Kalman) (*)
Tên thuốc Hoạt chất
Hàm lượng xác định được (mg/viên)
TB S (n 3)
(mg/viên)
95%
(mg/viên)
Hàm lượng ghi trên nhãn thuốc
(mg/viên)
Micardis(R) Plus
TEL 39,8 / 37,9 / 37,2 38,3 1,3 3,34 40,0
HYD 12,90 / 12,95 / 12,75 12,87 0,1 0,26 12,5
Panadol Extra
PAR 496,2 / 493,8 / 494,7 494,9 1,2 3,01 500,0
CAF 67,1 / 67,8 / 66,5 67,1 0,7 1,62 65,0
Alaxan PAR 335,4 / 332,0 / 338,7 335,4 3,4 8,32 325,0
IB 200,9 / 198,9 / 202,9 200,9 2,0 4,97 200,0
Protamol PAR 332,0 / 328,7 / 332,4 331,0 2,0 5,04 325,0
IB 199,2 / 197,2 / 199,3 198,6 1,2 2,94 200,0
Lopenca PAR 323,8 / 322,8 / 326,0 324,2 1,6 4,07 325,0
IB 212,5 / 212,0 / 214,0 212,8 1,0 2,59 200,0
Exforge HCT
AML 9,65 / 9,80 / 9,37 9,61 0,2 0,54 10,0
HYD 11,68 / 11,86 / 11,34 11,63 0,3 0,66 12,5
VAL 169,97 / 172,49 / 165,06 169,17 3,8 9,39 160
(*) Các số liệu về hàm lượng xác định được ghi trong cột thứ 3 ở bảng được trích từ các bảng
tương ứng khi đánh giá độ lặp lại và độ đúng của phương pháp phân tích (mục 3.6.1 và 3.6.2);
TB S: Trung bình số học và độ lệch chuẩn; 95% : Biên giới tin cậy 95%; 95%
t(0,95;2)*S/(n)1/2 , với n 3.
Các kết quả ở bảng 3.42 cho thấy:
- Hầu hết các giá trị về hàm lượng hoạt chất ghi trên nhãn đều đạt yêu cầu, do
các giá trị đó đều nằm trong khoảng tin cậy 95 % của các kết quả xác định được, tức
là có thể xem giá trị ghi trên nhãn và giá trị trung bình số học (TB) là như nhau, vì
chúng đều thuộc cùng một phân bố chuẩn;
- Có 3 trường hợp cần lưu ý:
+ Hàm lượng HYD trong thuốc Micardis(R) Plus xác định được lớn hơn so với
giá trị ghi trên nhãn (p < 0,05), do giá trị ghi trên nhãn thấp hơn khoảng tin cậy 95 %;

134
+ Hàm lượng IB trong thuốc Lopenca xác định được lớn hơn so với giá trị ghi
trên nhãn (p < 0,05), do giá trị ghi trên nhãn thấp hơn khoảng tin cậy 95 %;
+ Hàm lượng HYD trong thuốc Exforge HCT xác định được nhỏ hơn so với
giá trị ghi trên nhãn (p < 0,05), do giá trị ghi trên nhãn cao hơn khoảng tin cậy 95 %.
Tuy vậy, cũng cần thấy rằng, các kết quả thu được ở trên chỉ là những kết quả
mang tính nghiên cứu, vì chỉ lấy mẫu ngẫu nhiên ở một nơi xác định và trong một
thời điểm xác định. Song, các kết quả trên cũng một lần nữa cho phép khẳng định
rằng, hoàn toàn có thể áp dụng phương pháp Kalman để phân tích các mẫu thuốc đa
thành phần (nêu ở bảng) đang lưu hành ở thị trường nước ta.

135
KÊT LUẬN
Từ các kết quả nghiên cứu lý thuyết và thực nghiệm, đề tài luận án đi đến các
kết luận chính như sau:
1) Trên cơ sở khảo sát các giải pháp lựa chọn giá trị khởi tạo cho thuật toán lọc
Kalman, lần đầu tiên đã tìm được giải pháp mới - lựa chọn giá trị khởi tạo gần đúng
về nồng độ (bằng cách tính toán theo phương pháp bình phương tối thiểu dùng phổ
toàn phần) và phương sai (tính toán theo phương trình Horwitz). Giải pháp khởi tạo
mới này cho phép áp dụng thuận lợi phương pháp chemmometric-trắc quang sử dụng
thuật toán lọc Kalman (phương pháp Kalman) để xác định đồng thời hai hoặc ba chất
có phổ hấp thụ quang xen phủ nhau trong hỗn hợp của chúng.
2) Kết quả kiểm định phương pháp Kalman đối với ba dung dịch chuẩn hỗn
hợp (mỗi dung dịch chứa hai chất) và một dung dịch chuẩn hỗn hợp chứa ba chất
(các chất có phổ hấp thụ quang xen phủ nhau) đã cho thấy, khi phép đo độ hấp thụ
quang mắc sai số đáng kể (hay nhiễu đo lớn), đặc biệt là đối với hỗn hợp chứa ba
chất, phương pháp Kalman mắc sai số nhỏ và có độ lặp lại tốt hơn so với phương
pháp bình phương tối thiểu dùng phổ toàn phần.
3) Lần đầu tiên đã xây dựng được quy trình phân tích đồng thời các chất có
phổ hấp thụ quang xen phủ nhau trong các mẫu dược phẩm (thuốc) đa thành phần
chứa hai hoặc ba hoạt chất bằng phương pháp Kalman. Mặt khác, đã thiết lập được
chương trình máy tính sử dụng ngôn ngữ lập trình Visual basic for Applications trên
phần mềm Microsoft – Excel 2016 đi kèm quy trình phân tích và do vậy, cho phép
tính toán nhanh và thuận lợi khi áp dụng vào thực tế kiểm nghiệm dược phẩm ở các
phòng thí nghiệm của nước ta. Quy trình xây dựng được không chỉ có thao tác đơn
giản hơn, mà còn cho phép giảm chi phí phân tích so với phương pháp chuẩn là
phương pháp sắc ký lỏng hiệu năng cao (HPLC).
4) Độ đúng và độ lặp lại của quy trình phân tích (hay phương pháp) xây dựng
được đã được kiểm tra khi phân tích các mẫu thuốc chứa 2 hoặc 3 hoạt chất (các
hoạt chất có phổ hấp thụ quang xen phủ nhau): Đối với thuốc chứa 2 hoạt chất,
phương pháp được đạt độ đúng tốt với độ thu hồi 93 % – 102 % và độ lặp lại tốt với

136
RSD < 2,5 % (n 3); Đối với thuốc chứa 3 hoạt chất, phương pháp cũng đạt được
độ đúng tốt với độ thu hồi 90 % – 107% và độ lặp lại tốt với RSD < 3,5 % (n 3).
So sánh với phương pháp HPLC - phương pháp chuẩn, phương pháp Kalman cũng
đạt được độ đúng tốt (với p < 0,05) khi phân tích các thuốc chứa 2 hoặc 3 thành
phần.
5) Đã áp dụng quy trình phân tích xây dựng được vào thực tế để xác định đồng
thời hỗn hợp 2 hoặc 3 hoạt chất có phổ hấp thụ quang xen phủ nhau trong một số
loại thuốc đa thành phần đang lưu hành trên thị trường, gồm các nhóm thuốc khác
nhau: thuốc điều trị huyết áp, hạ sốt và giảm đau, tim mạch. Đặc biệt, lần đầu tiên
đã áp dụng phương pháp Kalman xác định đồng thời ba hoạt chất (AML, HYD và
VAL) trong thuốc Exforge HTC và đạt được độ đúng và độ lặp lại tốt, không thua
kém các phương pháp khác đã và đang sử dụng hiện nay. Điều này sẽ đóng góp tích
cực vào lĩnh vực kiểm nghiệm dược phẩm ở nước ta.

137
DANH MUC CÁC CÔNG TRÌNH CÔNG BỐ
KÊT QUẢ NGHIÊN CỨU CỦA LUẬN ÁN
[1] Nguyễn Thị Quỳnh Trang, Trần Thúc Bình, Châu Viết Thạch (2017). Xác
định đồng thời Paracetamol và Cafein trong hỗn hợp bằng phương pháp trắc
quang kết hợp thuật toán lọc Kalman, Tạp chí phân tích hóa, lý và sinh học,T-
22, tr.14-21.
[2] Nguyen Thi Quynh Trang, Tran Thuc Binh, Vo Thi Kim Truc, Ngo Van Tu
(2017). Simultaneous determination of telmiasartan and hydrochlorothiazide
in pharamacy by full spectrum spectrophometric method using Kalman filter
algorithm, Conference proceeding, The 5th Analytical Vietnam Conference
2017, pp.22-29.
[3] Tran Thuc Binh, Nguyen Thi Quynh Trang, Vo Thi Kim Truc, Ngo Van Tu
(2017). Simultaneous spectrophotometric determination of telmiasartan and
hydrochlorothiazide in pharamaceutical product by least-square method using
full spectra, Conference proceeding, The 5th Analytica Vietnam Conference
2017, pp.14-21.
[4] Nguyễn Thị Quỳnh Trang, Trần Thúc Bình, Ngô Văn Tứ (2017). Xác định
đồng thời amlodipine, hydrochlorothiazide và valsartan trong dược phẩm bằng
phương pháp trắc quang- chemometric dùng phổ toàn phần.Tạp chí Khoa học
- Khoa học Tự nhiên, Đại học Huế, 126(1D), tr.125-137.
[5] Trần Thúc Bình, Nguyễn Thị Quỳnh Trang, Nguyễn Thị Hồng Vân (2017).
Xác định đồng thời Paracetamol và Ibuprofen trong dược phẩm bằng phương
pháp quang phổ đạo hàm, Tạp chí phân tích hóa, lý và sinh học, T-22, tr.8-16.
[6] Nguyễn Thị Quỳnh Trang, Trần Thúc Bình, Nguyễn Thị Hồng Vân (2017).
Xác định đồng thời paracetamol và ibuprofen trong dược phẩm bằng phương
pháp trắc quang phổ toàn phần dùng thuật toán lọc Kalman. Tạp chí Khoa học
và Công nghệ, Chuyên san Khoa học Tự nhiên, Kỹ thuật và Công nghệ,
Trường Đại học Khoa học, Đại học Huế, 11(1), tr.93-104

138
TÀI LIÊU THAM KHẢO
TIẾNG VIÊT
[1] Ngọc Bích, Tường Thụy, Quỳnh Nga (2012), C# dành cho người tự học -
Tập 1, NXB Thông Tin & Truyền Thông.
[2] Trần Thúc Bình (2002), Nghiên cứu phương pháp xác định đồng thời các
chất có phổ hấp phụ xen phủ nhau sử dụng vi tính, Luận án tiến sĩ hóa học,
Đại học Quốc gia Hà Nội, Hà Nội.
[3] Bộ Y tế (2002), Dược điển Việt Nam III, Nhà xuất bản Y học, Hà Nội.
[4] Bộ Y tế (2009), Dược điển Việt Nam IV, NXB Y học, Hà Nội.
[5] Bộ Y tế (2015), Dược thư quốc gia Việt Nam, NXB Y học, Hà Nội, tr.187-
188, tr. 777-779, tr.1453-1455.
[6] Bộ Y tế (2007), Kiểm nghiệm dược phẩm, NXB Y học Hà Nội.
[7] Bộ Y tế – Cục quản lý dược (2016), Báo cáo thống kê ngành Dược.
[8] Hoàng Minh Châu, Từ Văn Mạc, Từ Vọng Nghi (2002), Cơ sở hóa học phân
tích, NXB Khoa học và kỹ thuật Hà Nội, Hà Nội.
[9] Hoàng Minh Châu (2017), Công nghệ bào chế dược phẩm, chương trình đào
tạo dược sĩ đại học, NXB Giáo dục.
[10] Huỳnh Minh Châu, Lý Dự Thư, Hồ Hồng Khánh, Nguyễn Hồng Thanh Vi,
Nguyễn Ánh Mai (2010), Phát triển phương pháp xác định đồng thời
paracetamol, ibuprofen và caffeine bằng phương pháp trắc quang kết hợp
bình phương tối thiểu từng phần, Hội nghị khoa học lần thứ IX Trường Đại
học Khoa học tự nhiên - Đại học Quốc gia Tp.Hồ Chí Minh, Tp.Hồ Chí
Minh.
[11] Bùi Công Cường, Nguyễn Doãn Phước (2001), Hệ mờ, mạng nơron và ứng
dụng, NXB Khoa học và Kỹ thuật.
[12] Tô Anh Dũng, Huỳnh Minh Trí (2013), Giáo trình xác suất thống kê, NXB
Đại học Quốc gia Tp. Hồ Chí Minh, Tp. Hồ Chí Minh.
[13] Trần Đức Thục Đoan, Vĩnh Định (2002), Áp dụng quang phổ đạo hàm phân
tích thuốc đa thành phần: các hỗn hợp speudoephedrine triprolidine;

139
betamethasone-chlorpheniramin; metronidazola spiramycine, Tạp chí Y học
Tp. Hồ Chí Minh, Tập 6 (1), tr. 263-265
[14] Trần Tứ Hiếu (2003), Phân tích trắc quang, NXB Đại học Quốc gia Hà Nội,
Hà Nội.
[15] Trần Tứ Hiếu (2004), Hóa học phân tích, NXB Đại học Quốc gia Hà Nội, Hà
Nội.
[16] Trần Tứ Hiếu, Đặng ứng Vận (2007), Phương pháp trắc quang định lượng
đồng thời các vitamin B1, B2, B3, B6, B12 và vitamin PP trong hỗn hợp theo
phương pháp lọc kalman, Tạp chí phân tích Hóa, Lý và Sinh học - Tập 12,
Số 2/2007, tr. 21- 30.
[17] Đặng Trần Phương Hồng, Trịnh Văn Quỳ (1997), Định lượng đồng thời
sulfadoxin và pyrimethamin trong viên nén fassnida bằng phương pháp
HPLC và phương pháp quang phổ đạo hàm, Thông báo kiểm nghiệm, Viện
kiểm nghiệm – Bộ y tế, tr. 1-11.
[18] Đặng Trần Phương Hồng, Trịnh Văn Quỳ (1998), Định lượng đồng thời
vitamin B1 và vitamin B6 bằng phương pháp quang phổ đạo hàm, Thông báo
kiểm nghiệm, Viện kiểm nghiệm – Bộ y tế, tr. 176 - 180.
[19] Phan Văn Khoa, Lưu Trường Văn, Lê Kiều (2006), Mạng nơron nhân tạo
(ANN) và giới thiệu một số nghiên cứu, ứng dụng trong dự án đầu tư, Tạp
chí Kinh tế, www.tnu.edu.vn/sites/duclv/.../Mang_no_ron_nhan_tao.doc.
[20] Nguyễn Văn Ly, Nguyễn Tẫn Sĩ (2005), Xác định đồng thời các vitamin B1,
B6 và B12 trong hỗn hợp bằng phương pháp trắc quang dùng phổ đạo hàm,
Tuyển tập công trình khoa học – Hội nghị khoa học phân tích hóa, lý và sinh
học Việt Nam lần thứ hai, tr. 86-89.
[21] Phạm Việt Nga (1996), Phân tích các vitamin tan trong nước của một số chế
phẩm polyvitamin bằng quang phổ đạo hàm bậc nhất, Thông báo kiểm
nghiệm, Viện kiểm nghiệm – Bộ y tế, tr. 21-27.
[22] Từ Vọng Nghi (2001), Hóa học phân tích, NXB Đại học Quốc gia Hà Nội,
Hà Nội.

140
[23] Quách Tuấn Ngọc (2002), Ngôn ngữ lập trình Pascal, NXB Thống kê, Hà
Nội.
[24] Tống Đình Quỳ, Giáo trình xác suất thống kê, NXB ĐH Bách Khoa, Hà Nội.
[25] Tạ Thị Thảo (2005), Giáo trình chemometrics, Bộ môn hóa học phân tích,
Khoa Hóa học, trường Đại học Khoa Học Tự Nhiên - Đại học Quốc Gia Hà
Nội, Hà Nội.
[26] Trần Tường Thụy, Phạm Phương Hoa (2013), Tự học C# bằng hình ảnh,
NXB Từ Điển Bách Khoa.
[27] Mai Xuân Trường (2008), Nghiên cứu phương pháp hấp thụ quang phân tử
xác định đồng thời các chất có phổ hấp thụ xen phủ nhau dựa trên thuật toán
lọc Kalman, Luận án tiến sĩ hóa học, Đại học Quốc gia Hà Nội, Hà Nội.
[28] Trần Thị Ngọc Vân, Trần Thị Tiết Nghĩa (2017), Xây dựng qui trình định
lượng đồng thời paracetamol, ibuprofen và tạp C của ibuprofen bằng phương
pháp UPLC/DAD, Tạp chí Nghiên cứu dược và Thông tin thuốc, tập 8, tr.
15– 20.
[29] Đặng Ứng Vận (2006), Giáo trình Hóa tin cơ sở, NXB Đại học Quốc gia Hà
Nội, Hà Nội.
TIẾNG ANH
[30] Agarkar A.R., Bafana S.R., Mundhe D.B., Suruse S.D., Jadhav K.B. (2013),
Spectrophotometric determination and validation of telmisartan and
hydrochlorthiazide in pure andtablet dosage form, Current Pharma Research,
Vol. 3(2), pp. 777-783.
[31] Ahmed Ashoura, Maha A. Hegazyb, Mohamed Abdel-Kawyb, Mohammad
B. ElZeinyc (2015), Simultaneous spectrophotometric determination of
overlapping spectra of paracetamol and caffeine in laboratory prepared
mixtures and pharmaceutical preparations using continuous wavelet and
derivative transform, Journal of Saudi Chemical Society, Vol. 19, Issue 2,
pp. 186-192.

141
[32] Aktas A. Hakan, Kitis Filiz (2014), Spectrophotometric simultaneous
determination of caffeine and paracetamol in commercial pharmaceutical by
principal componentregression, partial least squares and artificial neural
networks chemometric methods, doi: 10.5562/cca2214, Croat. Chem. ACTA
CCACAA, 87(1), pp. 69–74.
[33] Aktas A. Hakan, Pekcan Hulya (2013). Chemometric Methods for the
Simultaneous Spectrophotometric Determination of Caffeine, Theobromine
and Theophylline in Tea, Asian Journal of Chemistry, Vol. 25, No. 15
(2013), 8333-8338.
[34] Alnajjar A.O. (2011), Validation of a capillary electrophoresis method for
the simultaneous determination of amlodipine besylate and valsartan in
pharmaceuticals and human plasma, Journal of AOAC International, 94 (2),
pp.498-502.
[35] Altinoz Sacide, Toptan Suna (2002), Determination of tartrazine and
Ponceau-4R in various food samples by Vierordt’s method and ratio spectra
first-order derivative UV spectrophotometry, Journal of Food compostion
and Analysis, 15(6), pp. 667 – 683.
[36] Altinoz Sacide, Toptan Suna (2003), Simultaneous determination of
Indigotin and Ponceau-4R in food samples by using Vierordt’s method, ratio
spectra first order derivative and derivative UV spectrophotometry, Journal
of Food compostion and Analysis, 4 (4), pp. 517 – 530.
[37] Alula Melisew Tadele, Mohamed Abdel-Maaboud I., BekhiAdnan A. t
(2010), Simultaneous spectrophotometric determination of iron (II) and
copper (II) in tablets by chemometric methods, Thai J. Pharm, Sci, 34, pp.
93-106.
[38] Anandakumar Jothieswari K., SanthVijaya I D., Vijayakumar B., Priya D.,
Rathinaraj Stephen B. (2010), A Validated UV Spectrophotometric Method
for the Simultaneous Estimation of Amlodipine Besylate, Valsartan and
Hydrochlorothiazide in Combined Tablet Dosage Form, Journal of

142
Pharmaceutical and Biomedical Sciences (JPBMS), Vol. 05, Issue 05, pp.5-
13.
[39] Anandakumar K., Jayamariappan M. (2011), Absorption Correction Method
for the Simultaneous Estimation of Amlodipine Besylate, Valsartan and
Hydrochlorothiazide in Bulk and in Combined Tablet Dosage Form,
International Journal of Pharmacy and Pharmaceutical Sciences, Vol 3, Issue
1, pp.23-27.
[40] AOAC International (2012), AOAC® Guidelines for Single Laboratory
Validation of Chemical Methods for Dietary Supplements and Botanicals.
[41] Battu Prasanna Reddy, Reddy MS (2009), RP-HPLC Method for
Simultaneous Estimation of Paracetamol and Ibuprofen inTablets, Asian J.
Research Chem, 2 (1): Jan.-March, pp.70-72.
[42] Behera Chinmaya C., Joshi Vishal, Pillai Sujit, Validated Gopkumar P.
(April - June, 2014), Spectrophotometric methods for estimation of
telmisartan and hydrochlorothiazide in combined tablet dosage form,
RRJPA, Volume 3, Issue 2.
[43] Bouromandnasr Arash, Naik L.Saida (2014), Absorption Correction Method
for the Simultaneous Estimation of Amlodipine Besylate, Valsartan and
Hydrochlorothiazide in Bulk and in Combined Tablet Dosage Form,
International Journal of Scientific Engineering and Technology Research,
Vol.03, Issue.34, pp.6821-6825.
[44] Brereton Richard G. (2003), Chemometrics: Data Analysis for the laboratory
and Chemical plant, John Wiley and Sons Ltd, England, ISBNs: 0-471-
48977-8 (HB), 0-471-48978-6 (PB).
[45] British Pharmacopocia (2014), Amlodipine Besilate, Hydrochlorothiazide,
Hydrochlorothiazide Tablets, Liquid Chromatography, Valsartan, vol. I, II,
III, V, pp. 21, 83,604.
[46] Burn D.T., Tungkananuruk N. and other (2005), Derivative
spectrophotometric determination of acetaminophen by formation of [Fe(II)-
(2,2’-bipyridyl)3]2+, Che Anal, Vol 50, pp. 475.

143
[47] Chui C.K., Chen G. (1999), Kalman Filtering with Real-Time Applications,
Springer-Verlag Berlin Heidelberg, NewYork.
[48] Chusnul Chotimah, Sudjadi, Sugeng Riyanto, Abdul Rohman (2015),
Simultaneous determination of metamizole, thiamin and pyridoxin using
UV-spectroscopy in combination with multivariate calibration, Adv. Pharm.
Bull, 5(4), pp. 593-598.
[49] David D. Gerow and Sarah C. Rutan (1988), Background Correction for
Fluorescence Detection in Thin- Layer Chromatography Using Factor
Analysis and the Adaptive Kalman Filter, Anal. Chem, pp. 847-852.
[50] David Harvey (2000), Modern Analytical Chemistry, Mc Graw Hill.
[51] Delaye F., Gaye M.D., Aaron J.J. (1989), Evaluation of computer-assisted
second-derivative ultraviolet spectrophotometry for analysis of binary
mixtures, Analytica chimica acta, Vol 223(2), pp.395-402.
[52] Diaz T.G., Valenzuela I.A., Salina F. (1994), Multicomponent determination
of the pesticide naptalam and its metabolites in river water, by applying
partial least squarses calibration to the derivative spectrophotometric signals,
J.Anal Chem., Vol 350, pp 692-701.
[53] Dinç Erdal (1999), A comparative study of the ratio spectra derivative
spectrophotometry, Vierordt’s method and high-performance liquid
chromatography applied to the simultaneous analysis of caffeine and
paracetamol in tablets, Journal of Pharmaceutical and Biomedical Analysis,
Vol 21(4), pp. 723 – 730.
[54] Dinc E., Onur F. (1998), Comarison of ratio spectra derivative
spectrophotometry and Vierordt’s methol applied to quantitative analysis of
pseudoepedrine hydrochloride and triprolidine hydrochloride in table, STP
pharma Sci, Vol 8(3), pp. 203-208.
[55] Dinc E., Onur F. (1999), Application of ratio spectra derivative
spectrophotometry and Vierordt’s methol to quantitative analysis of
paraxetamol and metamizod in a pharmaceutical formulation, Sci pharma,
Vol 67, pp. 57-68.

144
[56] Dinc E., Yucesoy C., Onur F. (2002), Simultaneous Spectrophotometri
determination of mefenanamic acid and paraxetamol in ta pharmaceutical
preparation using ratio spetra derivative spectrophotometry and chemometric
methods, Journal of pharmaceutical and biomedical analysis, Vol 28(6), pp.
1091-1100.
[57] Dipankar Basu, Mahalanabis Kumar K., Bimal Roy (1998), Application of
least squares method in matrix form: simultaneous determination of
ibuprofen and paracatemol in tablets, Journal of Pharmaceutical and
Biomedical Analysis, 16, pp. 809-812.
[58] Dogan Kul D., Topal B, Kutucu T., Uslu B, Ozkan S.A. (2010), High-
Performance Liquid Chromatographic and First Derivative of the Ratio
Spectrophotometric Determination of Amlodipine and Valsartan in Their
Binary Mixtures, Journal of AOAC International, 93 (3), pp.882-890.
[59] Dumancas Gerard G., Muriuki Mary, Purdie Neil, Reilly Lisa (July 6-8,
2011), Simultaneous Spectrophotometric and Chemometric Determination of
Oleic, Linoleic, and Linolenic Fatty Acids in Vegetable Oils, Proceedings of
the World Congress on Engineering, Vol III, London, U.K.
[60] Dunkerley S., Adams M.J. (1997), The simultaneous determination of
caffeine, aspirin and paracetamol by principal components regression using
automatic dilution and calibration, Laboratory Automation and Information
Management, 33, pp. 107-117.
[61] Erk N., Özkan Y., Banoğlu E., Özkan S.A., Şentürk Z. (2001), Simultaneuos
determination of paracetamol and methocarbamol in tablets by ratio spectra
derivative spectrophotometry and LC, Journal of Pharmaceutical and
Biomedical Analysis, 24, pp. 469 – 475.
[62] Gadepalli Shankar Ganesh, Deme Pragney, Kuncha Madhusudana, Sistla
Ramakrishna (2014), Simultaneous determination of amlodipine. Valsartan
ang hydrochlorothiazide by LC-ESI-MS/MS and its application to
pharmacokinetics in rats, Journal of Pharmaceutical Analysis, 4 (6), pp.399-
406.

145
[63] Galande Varsha R., Baheti K. G., Indraksha S., Dehghan M.H. (2012),
Estimation of Amlodipine Besylat, Valsartan and Hydrochlorothiazide in
Bulk Mixture and Tablet by UV Spectrophotometry, Indian Journal of
Pharmaceutical Sciences, pp.18-23.
[64] Gangola Rekha, Kaushik Sunil, Sharma Paras (2011), Spectrophotometric
Simultaneous Determination of Hydrocholorothiazide and Telmisartan in
Combined Dosage Form, African Journal of Pure and Applied Chemistry, 2
(4), pp. 46-49.
[65] Gangola Rekha, Singh Narendra, Gaurav Anand, Maithani Mukesh, Singh
Ranjit (2011), Spectrophotometric simultaneous determination of
Hydrochlothiazide and telmisartan in combined dosage form by dual
wavelength method, International journal of comprehensive pharmacy, 2(04),
pp. 36 – 40.
[66] Gohel Nikunj Rameshbhai, Patel Bhavin Kiritbhai, Parmar Vijaykumar
Kunvarji (2013), Chemometrics-Assisted UV Spectrophotometric and RP-
HPLC Methods for the Simultaneous Determination of Tolperisone
Hydrochloride and Diclofenac Sodium in their Combined Pharmaceutical
Formulation, Sci Pharm, 81: 983–1001.
[67] Gupta K.R., Mahapatra A.D., Wadodkar A.R., Wadodkar S.G. (2010),
Simultaneous UV Spectrophotometric Determination of Valsartan and
Amlodipine in Tablet, International Journal of ChemTech Research, 2 (1),
pp.551-556.
[68] Gupta K.R., Taine M.R., Wadodkar S.G., Wankhede S.B. (2007), RP –
HPLC method for simultaneous estimmation of telmisartan and
Hydrochlothiazide in tablet dosage form, Indian Journal of Pharmacy
Science, 6 (2), pp. 298 – 300.
[69] Gupta N.K., Peepliwal A., Rathore D.S, Gupta P. (2015), Simultaneous
spectrophotometric estimation of telmisartan and amlodipine besylate in
tablet dosage form, Indian, J. Pharm. Biol. Res., 3(3), pp. 50-54.

146
[70] Hargis Larry G (1988), Analytical chemistry, Printon press, Philippines,
pp.50-53.
[71] He CY, Sun YM, Wu GH, Chen R (2001), Application of artifical neural
network to simultaneous spectrophotometric determination of Cu, Co and Ni,
Guang Pu Xue Yu Guang Pu Fen Xi, 21(5), pp,719-722.
[72] H.M. Stionary Office (2001), British Pharmacopeia, London.
[73] Howard Demuth Mark Beale (1998), Neural Network Toolbox For Use with
MATLAB.
[74] Isha1Azizul, Yusof Nor Azah, Malik Mazura Abdul (2007), Simultaneous
Spectrophotometric Determination of Pb(II) AND Cd(II) usng artificial
neural networks, Journal of Physical Science, Vol. 18(1), 1–10.
[75] Ivanovic D., Medenica M., Markovic G. (2000), Second – derivative
spectrophotometric assay of pseudoephedrine, ibuprofen and loratadine in
pharmaceuticals, Arzneimittel-Forschung/Drug Research, 50 (11), pp. 1004 –
1008.
[76] Jahanbakhsh (2005), Simultaneous Spectrophotometric Determination of
Group B Vitamins Using Parallel Factor Analysis: PARAFAC, Journal of the
Chinese Chemical Society, 52, 1123-1129.
[77] Jürgen W. Einax, Heinz W. Zwanziger, Sabine Geiß (1997), Chemometrics
in Environmental Analysis, Analytical Chemistry, Wiley-VCH Verlag
GmbH.
[78] Khajehsharifi Habiboallah, Pourbasheer Eslam, Tavallali Hossein, Sarvi
Solmaz, Sadeghi Maasumeh (16 February 2014), The comparison of partial
least squares and principal component regression in simultaneous
spectrophotometric determination of ascorbic acid, dopamine and uric acid in
real samples, Arabian Journal of Chemistry.
[79] Kevser S., Esma T. (2004) Second derivative spectrophotometric method for
simultaneous determination of cobalt, nickel and iron using 2-(5-bromo-2-
pyridylazo)-5-diethylaminophenol, Talanta, Vol 62(5), pp. 971-976.

147
[80] Kim H. Esbensen (2004), Multivariate Data Analysis– In Practice 5th Edition
CAMO, Ålborg University, Esbjerg.
[81] Khoshayand M.R., Abdollahi H., Ghaffari A., Shariatpanahi M., Farzanegan
H (2010), Simultaneous spectrophotometric determination of paracetamol,
phenylephrine and chlropheniramine in pharmaceuticals using chemometric
approaches, DARU, Vol. 18, No. 3, Correspondence.
[82] Khoshayand M.R., Abdollahi H., Shariatpanahi M., Saadatfard A.,
Mohammadi A. (2008), Simultaneous spectrophotometric determination of
paracetamol, ibuprofen and caffeine in pharmaceuticals by chemometric
methods, Spectrochimica Acta Part A, 70, pp. 491 – 499.
[83] Lakshmi Karunanidhi Santhana, Lakshmi Sivasubramanian (2012),
Simultaneous Analysis of Losartan Potassium, Amlodipine Besylate ang
Hydrochlorothiazide in Bulk and in Tablets by High-Performance Thin
Layer Chromatography with UV Absorption Densitometry, Journal of
Analytical Methods in Chemistry.
[84] Lakshmi KS, Lakshmi S. (2010), Design and optimization of a chemometric-
assisted spectrophotometric determination of telmisartan and
hydrochlorothiazide in pharmaceutical dosage form, Lakshmi and
Sivasubramanian. J Young Pharm.;2(1), pp. 85-89.
[85] Manish K., Ajay G., Singh M.P. (2014), Development and validation of UV-
spectrophotometric method for the simultaneous estimation of telmisartan
HCL and hydrochlorothiazide as API and in combination in tablet dosage
form, Manish K, IJPRBS, Volume 3(3), pp. 73-86.
[86] Manish Sharma, Charmy Kothari, Omkar Sherikar and Priti Mehta (2014),
Concurrent Estimation of Amlodipine Besylate, Hydrochlorothiazide and
Valsartan by RP-HPLC, HPTLC and UV–Spectrophotometry, Journal of
Chromatographic Science;52, pp. 27–35.
[87] Miller James N., Miller Jane C. (2005), Statistics and chemometrics for
analytical chemistry, 5th ed, Pearson Education Limited, England.

148
[88] Mosnica Ferraro C. F. et al. (2004), Chemometric determination of amiloride
hydrochloride, Hydrochlothiazide and timolol maleate in synthetic mixtures
and pharmaceutical formulations, Journal of Pharmaceutical and Biomedical
Analysis, 34, pp. 305 – 314.
[89] Moussa B.A., El-Zaher A.A, Mahrouse M.A., Ahmed M.S. (2013),
Simultaneous determination of amlodipine besylate and atorvastatin calcium
in binary mixture by spectrofluorimetry and HPLC coupled with
fluorescence detection, Analytical Chemistry Insights, 8, pp.107-115.
[90] Muhammad Ali Mallah, Sherazi Syed Tufail Hussain, Ahmed Sarfaraz
Mahesar, Khaskheli Abdul Rauf (2012), Simultaneous Quantification of
Ibuprofen and Paracetamol in Tablet Formulations Using Transmission
Fourier Transform Infrared Spectroscopy, American Journal of Analytical
Chemistry, 3, pp. 503-511.http://dx.doi.org/10.4236/ajac.2012.38067
Published Online August 2012 (http://www.SciRP.org/journal/ajac)
[91] Nagaraj, Vipul K., Rajshree M. (2007), Simultaneous quantitative resolution
of atorvastatin calcium and fenofibrate in pharmaceutical preparation by
using derivative calcium and fenofibrate in pharmaceutical preparation by
using derivative ratio spectrophotometry and chemometric calibrations,
Analytical sciences, Vol 23(4), pp. 445.
[92] Nevado J.J.B., Cabanillas C.G., Salcedo A.M.C. (1994), Simultaneous
determination of furaltadone and chloramphenicol in pharmaceuticals by
derivative spectrophotometry and the derivative of the radio spectra, Journal
of Analytical Chemistry, Vol 349 (10-11), pp. 756-760.
[93] Nevado J.J.B., Flores J.R., Pardo M.L.M. (1994), Simultaneous
determination of furaltadone and chloramphenicol in pharmaceuticals by
derivative spectrophotometry and the derivative of the radio spectra,
Fresenius’J.Ana.Chem, Vol 349 (10-11), pp. 756-760.
[94] Palabiyik I.M., Dinc E., Onur O. (2004), Simultaneous Spectrophotometric
determination of pseudoephedrine hydochloride and ibuprofen in a
pharmaceutical preparation using ratio spectra drivative spectrophotometry

149
and multivariate calibration techniques, Journal of pharmaceutical and
biomedical analysis, Vol 34 (3), pp. 473-483.
[95] Palabiyik I.Murat, Simultaneous Spectrophotometric determination of
paracetamol and methocarbamol in a pharmaceutical preparation using
chemometric techniques, Journal.teb.org. pp.111-115.
[96] Palled M.S., Rajesh P.M.N., Chatter M., Bhatt A.R. (2005), RP – HPLC
determination of Telmisartan in tablet dosage forms, Indian Journal of
pharmaceutical sciences, vol 67 (1), pp. 108 – 110.
[97] Palled M.S., Chatter M., Rajesh P.M.N., Bhatt A.R. (2006), Difference
spectrophotometric determination of Telmisartan in tablet dosage forms,
Indian Journal of pharmaceutical sciences, 68(50), pp. 685 – 686.
[98] Paul Mac Berthouex, Linfield C. Brown (2002), Statistics for Environmental
Engineers, Lewis Publishers.
[99] Rajni Rohilla, Usha Gupta (Oct. 2012), Simultaneous Spectrophotometric
Determination of Copper and Lead in Natural water samples and industrial
effluents in Micellar Media by the H-Point Standard Addition Method
(HPSAM), Int. J. Res. Chem. Environ, Vol.2, Issue 4, pp. 93-100.
[100] Rekha Gangola, Narendra Singh, Anand Gaurav, Mukesh Maithani and
Ranjit Singh (2011), Spectrophotometric simultaneous determination of
Hydrochlorothiazide and Telmisartan in Combined dosage form by dual
wavelength method, Gangola R et al. / Pharmacie Globale (IJCP), 2 (04)
[101] Rutan Sarah C., Brown Steven D. (1984), Adaptive Kalman Filtering Used to
compensate for model errors in multicomponent methods, Analytica Chimica
Acta, 160, pp. 99-119.
[102] Säbel Crystal E., Neureuther Joseph M. (2010), A spectrophotometric
method for the determination of zinc, copper, and cobalt ions in
metalloproteins using Zincon, Analytical Biochemistry, 397, 218–226.
[103] Salameh Azimi, Mohammad Rofouei Kazem, Sharifkhani M. Samira (2011),
Simultaneous Spectrophotometric Determination of Ag+ and Cu2+ by Partial

150
Least Square Regression, Journal of Materials Science and Engineering B 1,
895-900
[104] Satana E., Altinay S., Göğer N.G., Ozkan S.A., Sentürk Z. (2001),
Simultaneous Determination of Valsartan and Hydrochlorothiazide in
Tablets by First-Derivative Ultra-violet Spectrophotometry and LC, Journal
of Pharmaceutical and Biomedical Analysis, 25 (5-6), pp.1009-1013.
[105] Sena Marcelo M., Poppi Ronei J. (2004), N-way PLS applied to
simultaneous spectrophotometric determination of acetylsalicylic acid,
paracetamol and caffeine, Journal of Pharmaceutical and Biomedical
Analysis, 34, pp. 27 – 34.
[106] Seyed Karim Hassaninejad-Darzia, Abdolraouf Samadi-Maybodib, Seyed
Mohsen Nikou (2016), UV-Vis Spectrophotometry and Multivariate
Calibration Method for Simultaneous Determination of Theophylline,
Montelukast and Loratadine in Tablet Preparations and Spiked Human
Plasma. Iranian Journal of Pharmaceutical Research, 15 (3), pp. 379-391.
[107] Shaalan R.A., Belal T.S. (2010), Simultaneous Spectro-fluorimetric
Determination of Amlodipine besylate and Valsartan in Their
CombinedmTablets, Drug Testing and Analysis, 2 (10), pp.489-493.
[108] Shakya Piusha, Jain Pushpendra Kumar, Gajbhiye Asmita, Shrivastava S.P
(2015), Simultaneous estimation of telmisartan and hydrochlorothiazide by
derivative spectroscopy, Int J Pharm Pharm Sci, Vol 7, Issue 6, pp. 386-388.
[109] Shah Umang, Patel Sharddha, Raval Manan, Desai Pankti (Jul-Sep 2015),
Chemometric assisted spectrophotometric methods for the simultaneous
determination of Rifampicin and Piperine in bulk and capsule, Indian Journal
of Pharmaceutical Education and Research, Vol 49, Issue 3.
[110] Smit H. C. (1985), The Use of Kalman Filtering and Correlation Techniques
in Analytical Calibration Procedures, Journal of Research of the National
Bureau of Standards, pp.441-450.
[111] Stolarczyk Mariusz, Maslanka Anna, Krzek Jan, Milczarek Joanna (2008),
Application of Derivative Spectrophotometryfor Determination of Enalapril,

151
Hydrocholorothiazide and Valsartan in complex Pharmaceutical
Preparations, Acta Poloniae Pharmaceutica, 65 (3), pp.275-281.
[112] Tanase Gh. Dobre, Jose G. Sanchez Marcano (2007), Chemical Engineering:
Modelling, Simulation and Similitude, John Wiley and Sons Ltd, England.
[113] Tarighat Maryam Abbasi, Afkhamib Abbas (2012), Simultaneous
Spectrophotometric Determination of Cu(II), Co(II) and Ni(II) using Ratio
Spectra-Continuous Wavelet Transformation in some Food and
Environmental Samples. J. Braz. Chem. Soc., Vol. 23, No. 7, 1312-1319.
[114] Taverniers I., Loose De M., Bockstaele Van E. (2004), Trends in quality in
the analytical laboratory, II, Analytical method validation and quality
assurance, TrAC Trends in Analytical Chemistry, 23 (8), pp. 535-552.
[115] The United States Pharmacopoeia 37 (2014), Chromatography, Amlodipine
Besylate, Amlodipine Besylate Tablets, Hydrochlorothiazide Capsule,
Hydrochlorothiazide Tablets, Valsartan, Valsartan Tablets, Vol I, II, III,
pp.301-308, 1760-1762, 3246-3248, 5115-5117.
[116] Thompson M., Lowthian P. J. (1995), A Horwitz-like function describes
precision in a proficiency test, Analyst, 120 (2), pp. 271-272.
[117] Veeranna V., Suryanarayana V.Rao, Balanraju J.N. (2012), Simultaneous
Fourth Order Derivative Spectrophotometric Determination of Cobalt, Nickel
and Copper Using Anthrone Phenylhydrazone (APH), Chem. Sci. Trans., 1
(2), pp. 321-328.
[118] Vijaya Vichare, Preeti Mujgond, Vrushali Tambe, Dhole S.N (2010),
Simultaneous Spectrophotometric determination of Paracetamol and Caffeine
in Tablet Formulation, Int.J. PharmTech Res., 2(4).
[119] Vijaya Vichare, Vrushali Tambe, Vrushali Kashikar, Dhole S.N. (2011),
Spectrophotometric simultaneous determination of amlodipine besylate and
hydrochlorothiazide in combined tablet dosage form by simultaneous
equation, absorption ratio and first order derivative spectroscopy methods,
Int. J. Chem. Res., Vol 2, Issue 1, pp. 710.

152
[120] Vu Dang Hoang, Dong Thi Ha Ly, Nguyen Huu Tho, Nguyen Thi Minh Hue
(2014), UV Spectrophotometric Simultaneous Determination of Paracetamol
and Ibuprofen in Combined Tablets by Derivative and Wavelet
Transforms,The Scientific World Journal, Volume 2014, Article ID 313609,
13 pages, http://dx.doi.org/10.1155/2014/313609
[121] Wan E.A., Merwe Van der R. (October 2000), The unscented Kalman filter
for nonlinear estimation, in Proceedings of Symposium 2000 on Adaptive
Systems for Signal Processing, Communication and Control (AS-SPCC),
IEEE, Lake Louise, Alberta, Canada.
[122] Wafaa S. Hassan (2008),Determination of Ibuprofen and Paracetamol in
Binary Mixture UsingChemometric-Assisted Spectrophotometric
Methods,American Journal of Applied Sciences 5 (8), pp.1005-1012
[123] Wiley-VCH Verlag GmbH, Kim H. Esbensen (2004), Multivariate Data
Analysis– In Practice 5th Edition CAMO, Ålborg University, Esbjerg.
[124] www.cs.unc.edu/~welch/kalman.
[125] www.cs.unc.edu/~welch/kalman./kalmanIntro.htm.
[126] Xle Yu-Long, Wang Ji-Hong, Liang Yl-Zeng, Yu Ru-Qium (1992), Robust
Kalman filter as a chemometric method for analytical data processing,
Analytica Chimica Acta, 269, pp. 307-316.
[127] Zareena Yasmeen, T.Mamatha, Heena Farheen, Husna Kanwal Qureshi
(2013) , Dissolution method development and validation for combination of
Ibuprofen and Paracetamol tablets, Asian J Pharm Clin Res, Vol 6, Suppl 2,
pp.164-168.

153
PHU LUC
Phụ lục 1. Chương trình tính toán của thuật toán lọc Kalman
Option Explicit
Dim x_est, x, v, w, Q, r, B, x_pre, P_est, P_pre, KG, H, z, INV, GG, A_est As
Variant
'Khoi tao cac ma tran va cac bien so dung chung trong chuong trinh
'Khoi tao cac ma tran va cac bien so dung trong chuong trinh
ReDim z(1 To n)
ReDim x_est(1 To n, 1 To p)
ReDim P_est(1 To n, 1 To p)
ReDim x_pre(1 To n, 1 To p)
ReDim P_pre(1 To n, 1 To p)
ReDim r(1 To n)
ReDim H(1 To n, 1 To p)
ReDim KG(1 To n, 1 To p) 'Kalman Gain
ReDim INV(1 To n)
ReDim B(1 To p)
ReDim x(1 To p, 1 To p + 1)
ReDim GG(1 To p)
Sub MainProgram_Click()
'Chuong trinh chinh
Dim k, i, j, p, n, m, vk2 As Integer
Dim rsd, var, sd As Double
'Nhap ma tran do do hap thu quang z
For k = 1 To n
input z(k)
Next
'Nhap ma tran he so hap thu quang
For k = 1 To n

154
For i = 1 To p
input H(k, i)
Next
Next
'Thu gon he tinh toan
Call LMS(p, H, z, x)
'Giai he theo Gauss - Jordan, ket qua tinh luu tai cot p+1 cua ma tran x
Call GJ(p, x)
'Gan gia tri cho uoc luong dau tien x_est(1) la ket qua cua bpct
For i = 1 To p
x_est(1, i) = x(i, p + 1)
Next
'Tinh phuong sai va do lech chuan cua cac gia tri do dac
var = Application.WorksheetFunction.VarP(z)
sd = Application.WorksheetFunction.Sqrt(var)
'Tinh rsd theo Horwitz va khoi tao phuong sai nhieu do dau tien P_est(1) theo cong
thuc(2.10)
For i = 1 To p
rsd = Application.WorksheetFunction.Power(2, (1 - 0.5 * Log(x_est(1, i)) /
Log(10)))
P_est(1, i) = x_est(1, i) * rsd * sd / 2
Next
'Tinh toan hoi quy theo Adaptive Kalman Filter
Call AKF(H, z)
'
End Sub
Sub AKF(ByVal H As Variant, ByVal z As Variant)
'AKF = Adaptive Kalman Filter
Dim k, i, j, p, n, m, vk2 As Integer

155
Dim x_est, x, v, w, Q, r, B, x_pre, P_est, P_pre, KG, H, z, INV, GG, A_est As
Variant
Dim Rkm, Rks, a, d, G As Double
'Tinh toan hoi quy tu buoc thu 2 den buoc thu n
For k = 2 To n
' Ngoai suy trang thai nong do
Call Veql(p, k, k - 1, x_pre, x_est)
' Ngoai suy phuong sai nhieu do dac
Call Veql(p, k, k - 1, P_pre, P_est)
' Tinh toan thich nghi he so do dac R(k) theo cong thuc(2.9)
m = k - 1
Rkm = 0
Rks = 0
r(k) = r(1)
INV(k) = z(k) - Vmult(p, k, k, H, x_pre)
For i = 1 To p
Rks = Rks + (H(k, i) * P_pre(k, i)) * (H(k, i) * P_pre(k, i))
Next
' Lay so buoc thich nghi > 30 de thoa man so mau thong ke toi thieu theo phan
phoi Gauss
If m > 30 Then
m = 30
End If
If k > m Then
For j = 1 To m
Rkm = Rkm + INV(k - j) * INV(k - j)
Next
r(k) = Rkm / m - Rks
End If
'Tinh toan cho Kalman gain va cap nhat uoc luong trang thai nong do sau do dac

156
G = Vmult(p, k, k, P_pre, H)
For i = 1 To p
GG(i) = P_pre(k, i) * H(k, i)
Next
a = (G + r(k))
If a <> 0 Then
a = 1 / a
End If
d = 1 / (1 + (a * r(k)) ^ 1 / 2)
For i = 1 To p
KG(k, i) = a * P_pre(k, i) * GG(i)
x_est(k, i) = x_pre(k, i) + KG(k, i) * INV(k)
Next
'Cap nhat uoc luong phuong sai nhieu do dac
For i = 1 To p
P_est(k, i) = P_pre(k, i) - a * d * P_pre(k, i) * G * G
Next
Next
' Ket qua cuoi cua module AKF() duoc doc tai buoc tinh cuoi cung - buoc thu n
End Sub
Sub LMS(ByVal p As Integer, ByVal H As Variant, ByVal z As Variant, ByVal x
As Variant)
'LMS=Least Mean square
'Dua he n phuong trinh, p an so thanh he vuong p phuong trinh p an so dua theo
'phuong phap binh phuong cuc tieu.
'Ket qua luu vao ma tran x la ma tran mo rong cua he p phuong trinh p an so
Dim i, j, k As Integer
For i = 1 To p
x(i, p + 1) = 0
For k = 1 To n

157
x(i, p + 1) = x(i, p + 1) + H(k, i) * z(k)
Next
Next
For i = 1 To p
For j = 1 To p
x(i, j) = 0
For k = 1 To n
x(i, j) = x(i, j) + H(k, i) * H(k, j)
Next
Next
Next
End Sub
Sub GJ(ByVal p As Integer, ByVal x As Variant)
'GJ=Gauss-Jordan
'Giai he p phuong trinh p an so theo phuong phap bien doi ma tran Gauss Jordan
'x la ma tran mo rong cua he n phuong trinh - n an so
Dim num_rows, num_cols, r1, r2, c, i As Integer
Dim tmp, factor, tiny As Double
tiny = 0.000001
num_rows = p
num_cols = p + 1
ReDim GJ(1 To num_rows)
For r1 = 1 To p
If Abs(x(r1, r1)) < tiny Then
For r2 = r1 + 1 To num_rows
If Abs(x(r1, r1)) > tiny Then
For c = 1 To num_cols
tmp = x(r1, c)
x(r1, c) = x(r2, c)
x(r2, c) = tmp

158
Next
Exit For
End If
Next
End If
If Abs(x(r1, r1)) > tiny Then
For r2 = r1 + 1 To num_rows
factor = -x(r2, r1) / x(r1, r1)
For c = r1 To num_cols
x(r2, c) = x(r2, c) + factor * x(r1, c)
Next
Next
End If
Next
Dim rcnt As Integer
For rcnt = 1 To num_rows
r1 = num_rows - rcnt + 1
tmp = x(r1, num_cols)
For r2 = r1 + 1 To num_rows
tmp = tmp - x(r1, r2) * x(r2, num_cols)
Next
x(r1, num_cols) = tmp / x(r1, r1)
Next
End Sub
Public Static Function Vmult(ByVal p As Integer, ByVal rx As Integer, ByVal ry
As Integer, x As Variant, y As Variant) As Double
' Ham nhan 2 ma tran
Dim result As Double
Dim i As Integer
result = 0

159
For i = 1 To p
result = result + x(rx, i) * y(ry, i)
Next
Vmult = result
End Function
Sub Veql(ByVal p As Integer, ByVal rx As Integer, ByVal ry As Integer, x As
Variant, y As Variant)
' Thu tuc gan 2 ma tran bang nhau
Dim i As Integer
For i = 1 To p
x(rx, i) = y(ry, i)
Next
End Sub

160
Phụ lục 2. Kết quả phân tích mẫu thuốc Micardis Plus để xác định TEL
và HYD

161
162

163
164
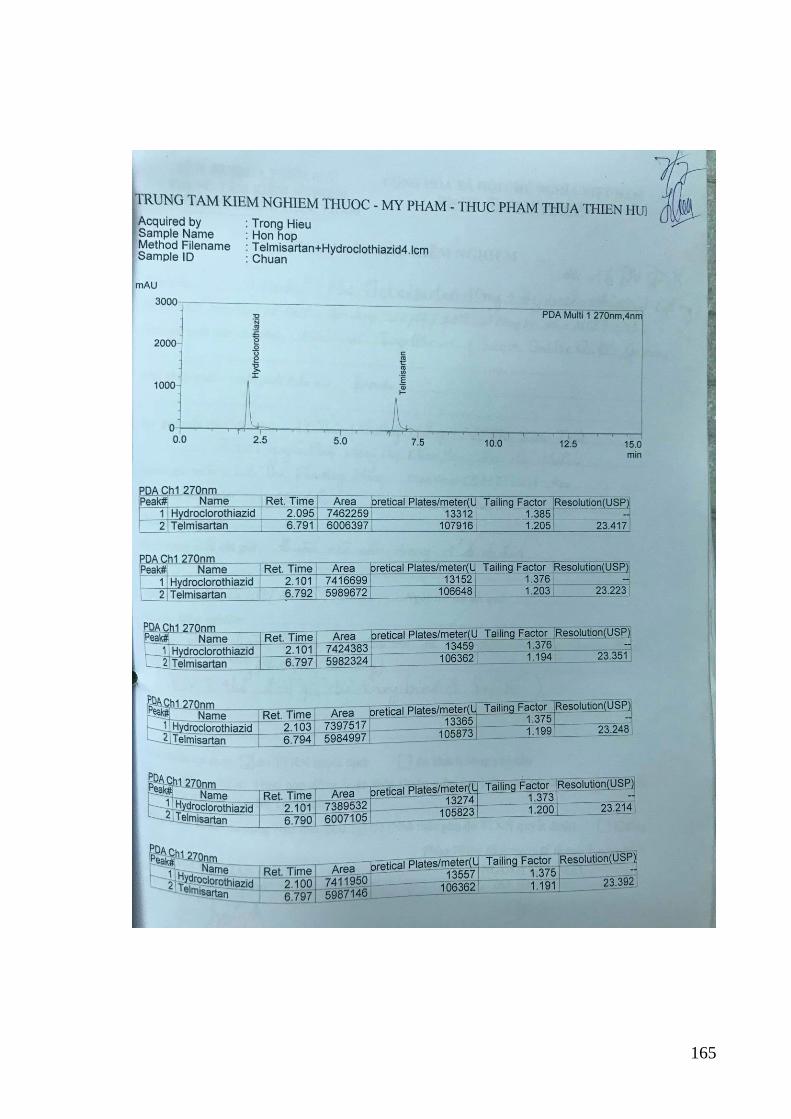
165

166
Phụ lục 3. Kết quả phân tích mẫu thuốc Panadol Extra để xác định PAR
và CAF

167

168

169

170

171
Phụ lục 4. Kết quả phân tích mẫu thuốc Exforge HCT để xác định AML,
HYD và VAL.

172

173

174
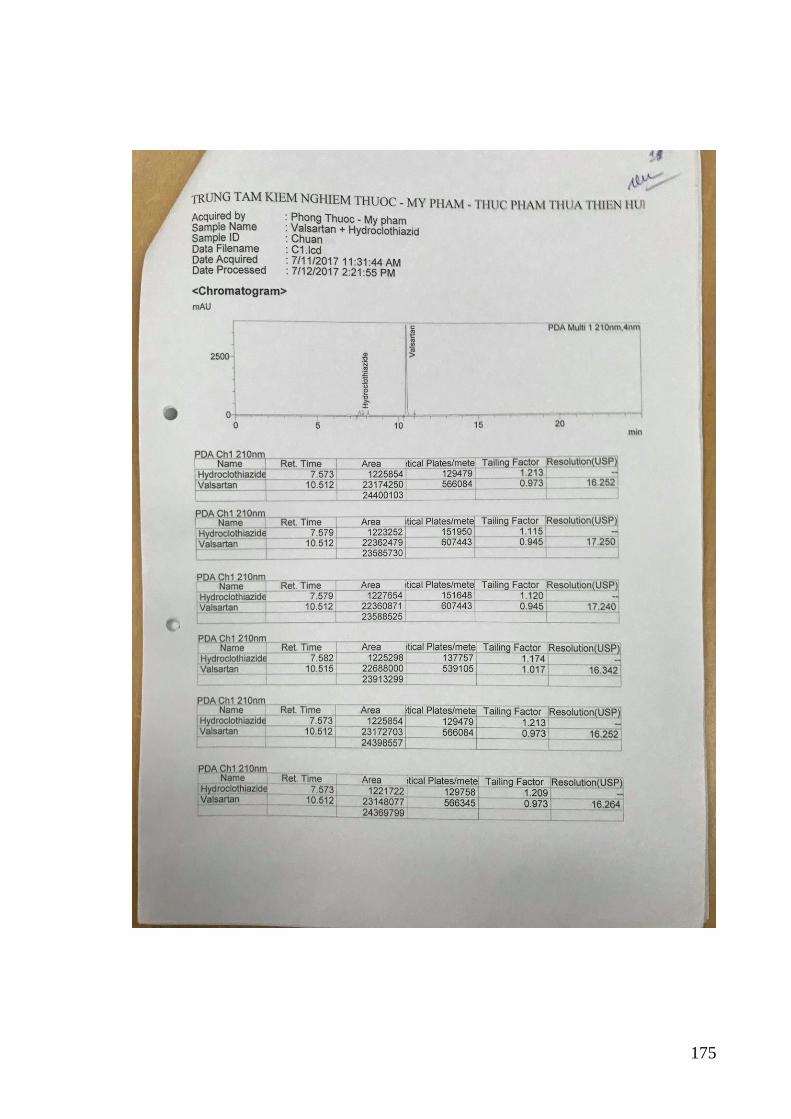
175

176

177

178
Phụ lục 5. Kết quả phân tích mẫu thuốc Lopenca để xác định PAR và IB
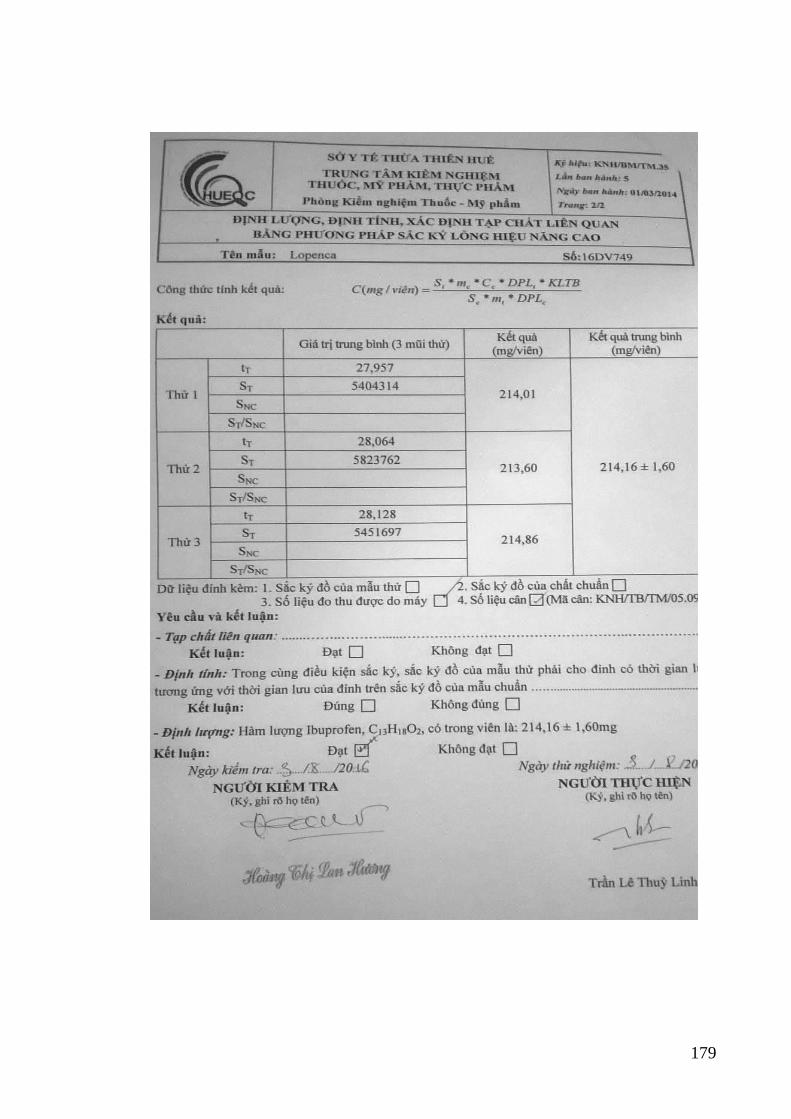
179
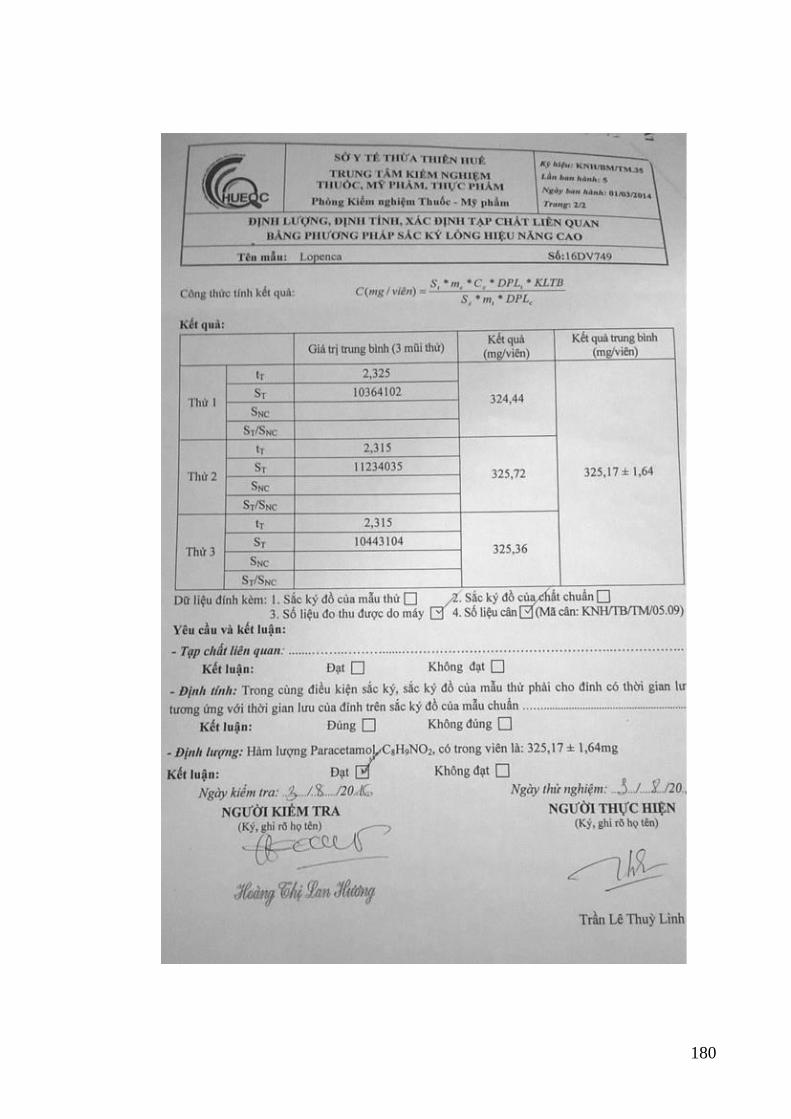
180
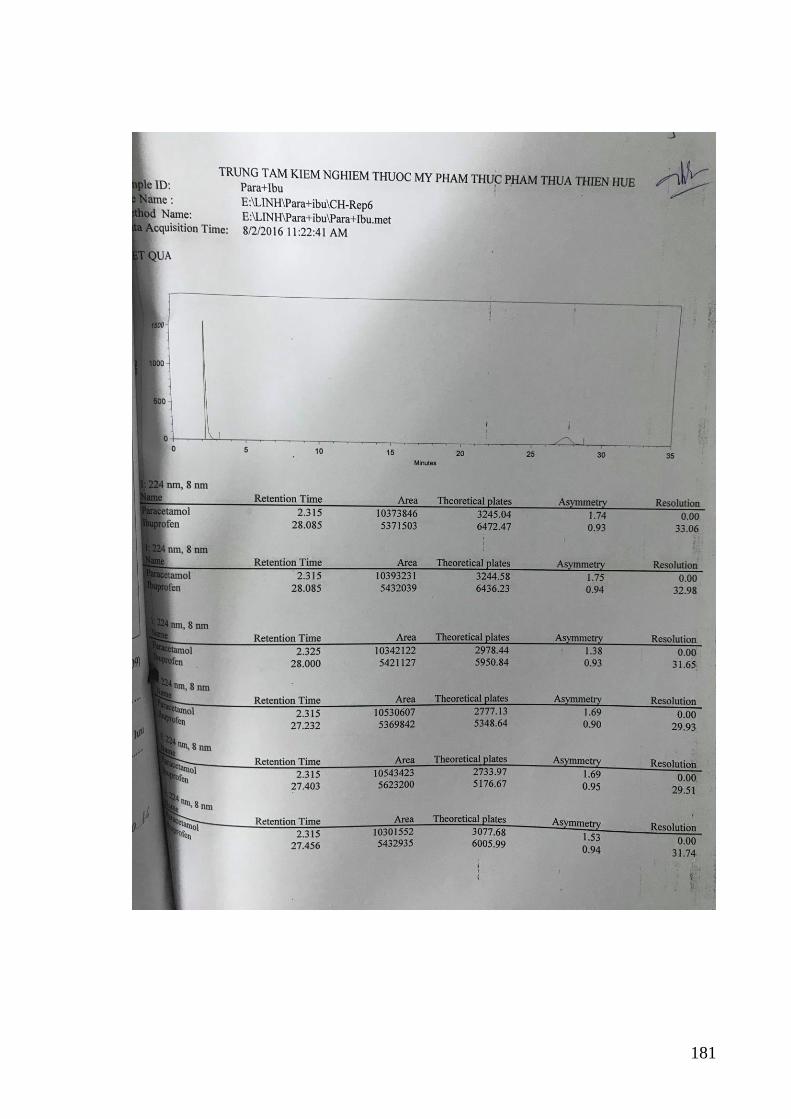
181

182

183

184





























