Embed Size (px)
Citation preview

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis
OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis
Marisa Babb, Pierre Chesnot, Emily Levine, Sam Seymour, and Job Shiach
Dept. of Bioengineering, University of California Berkeley, Berkeley, CA 94720
(Dated: 11 December 2015)
Multiple Sclerosis (MS) is a devastating disease with no cure. Current treatments aim to slow progression
and manage symptoms, necessitating continuous care for the remainder of patients’ lives. The need for long
term care is a major contributor to estimated worldwide healthcare costs of up to $144 billion annually. Here,
we describe OliGro, a novel solution to restore motor skills and, consequently, MS patient autonomy through
extended release of therapeutic antibodies. Anti-LINGO-1, an FDA-approved drug to regenerate the myelin
sheath, will be housed within a PEG-PLA block co-polymer hydrogel. This scaffold will be packaged into
a native membrane-cloaked nanoparticle to limit immune response to the particle and facilitate intravenous
delivery. Receptors for myelin-specific molecules in the central nervous system will be used for targeting our
particles, with the secondary effect of competing with T-cells that act to degrade the myelin sheath.
Keywords: Hydrogel, Nanoparticle, Drug Delivery, Multiple Sclerosis
I. INTRODUCTION
Multiple Sclerosis (MS) is an autoimmune disease in
which the insulating myelin that sheathes neuronal ax-
ons in the central nervous system are damaged. This
disrupts the ability of the neurons to communicate and
consequently causes a wide range of physical and mental
symptoms. There is no cure for this debilitating dis-
ease, but current ”disease modifying” therapies aim to
speed recovery from attacks, slow progression, and man-
age symptoms.
There are four patterns of MS with differing symp-
tom progressions and histological manifestations, but in
all cases, T-cells, a type of lymphocyte that aids in the
body’s defense, play an active role. T-cells gain access to
the brain through disrupted or leaky tight junctions in
the blood brain barrier (BBB), which is normally highly
selective in the healthy central nervous system (CNS).
The T-cells recognize the myelin as foreign and attack
it, triggering an extensive inflammatory process that can
further impede axonal transmission.
The name multiple sclerosis refers to the scars that
form in the white matter of the CNS. In the healthy
brain, oligodendrocytes are responsible for creating the
myelin sheath that insulates the conductive axons. As
MS progresses, however, oligodendrocytes are less effec-
tive in rebuilding the myelin sheath, and these succes-
sively less effective attempts at remyelination cause a
scar-like plaque to build up around the damaged regions.
A. Clinical Need
The majority of the research into MS treatments fo-
cuses on a stem cell-based myelin regeneration approach.
While stem cells represent a very promising field, their
use poses potential problems that include ethical con-
cerns, potential immune reaction and tumor formation,
limited differentiation ability, and utilization unknowns
depending on the type of stem cell being used. In ad-
dition, inclusion of stem cells in any therapy introduces
more stringent regulation guidelines and greatly increases
the complexity of the FDA regulatory process. For this
reason, we have decided to take an alternative approach
to the problem.
Taking a look at the economic impact of this disease
helps put it into context on a global scale. With current

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 2
costs reaching $57,500 per patient per year in the United
States, MS is one of the most expensive diseases to treat,
second only to heart failure.1 The estimated 2.5 million
cases worldwide bring the total cost of this disease to
around $144 billion per year. Lowering this staggering
figure is part of our goal with OliGro. Rather than uti-
lizing expensive stem cell therapies, our solution reduces
the direct cost of treatment by relying on polymer syn-
thesis. Even more significantly, our therapy minimizes
assistive care costs by making patients autonomous once
again.
B. Proposed Solution
Here, we propose a non-stem cell-based approach for
regenerating the myelin sheath as well as protecting the
myelin from additional damage through systemic deliv-
ery. With systemic delivery, the biocompatibility of our
material is a significant design challenge; however, we
plan to ”cloak” our nanoparticle within a native cell
membrane that the body recognizes as ”self” and thus
evades an unwanted immune response. Specifically, we
intend to cloak our particle with the isolated membranes
of naive, patient-specific T-cells since they contain the
major histocompatibility complexes that identify cells as
”self”.2 We will also investigate the potential for ”off-the-
shelf” non-patient-derived T-cells for use in later genera-
tions of the therapy, which would be cheaper but poten-
tially less safe. Before culturing cells for membrane iso-
lation, T-cells will be transfected with a vector for mem-
brane expression of one or more molecules to facilitate
effective targeting of our nanoparticles.
In order for OliGro to be effective, the ability to mi-
grate through the BBB is essential. Our primary ap-
proach relies on the small size of the nanoparticle to fa-
cilitate diffusion through the damaged, leaky tight junc-
tions. If this approach is ineffective, we propose an ad-
ditional method for allowing the T-cells to penetrate the
BBB through signaling molecules expressed on the sur-
FIG. 1. Cascade of T-cell transmigration through theblood brain barrier by expressing activated α4β1 inte-grin. From Engelhardt 3 .
face of the cloaking lipid membrane. Early studies of
T-cell migration show that only activated T-cells are ca-
pable of bypassing the healthy BBB in MS patients.3
Further research has shown that the active component on
these infiltrating T-cells is the α4β1 integrin (also known
by antigen CD49D, or as the alpha 4 subunit of the α4β1
receptor). This mediates T-cell attachment to the signal-
ing endothelial cells at the capture phase of the cascade
leading to transmigration through the BBB (Fig. 1).3 If
necessary, we will include this α4β1 integrin in the T-
cell membrane that cloaks our nanoparticle to promote
perfusion into the CNS via the traditional T-cell route
characterized by the disease.
Another design challenge for our therapy is effective
targeting of myelin. This is complicated by the fact that
myelin is expressed in both the central and peripheral
nervous systems. Myelin proteolipid protein (PLP) is
a protein that makes up more than 50% of the CNS
myelin proteins. It is ubiquitous across myelin within
the CNS and is found in very low amounts in the periph-
eral nervous system (0.05% of PNS myelin proteins).4
We propose incorporating anti-PLP into the nanoparticle
membrane as a means of specifically targeting it to CNS
myelin. Additionally, PLP has been identified as one
of several targets of invasive T-cells, and therefore our

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 3
particle may have the additional benefit of competitive
displacement of the T-cells, leading to enhanced preven-
tion of additional myelin degeneration.4 Other potential
CNS myelin targets include myelin basic protein (MBP),
oligodendrocyte-specific protein (OSP), and myelin oligo-
dendrocyte glycoprotein (MOG).4
Once our nanoparticles make it past the BBB (Fig.
2 B,D.) and target the myelin sheath, they will deliver
a therapeutic antibody that can aid in the regeneration
of myelin and improvement of nerve signalling. LINGO-
1 (leucine-rich repeat and immunoglobulin-like domain-
containing Nogo receptor-interacting protein 1) is a pro-
tein expressed almost exclusively in CNS neurons and
oligodendrocytes, and acts as a negative regulator of
oligodendrocyte differentiation and myelination5. With
this in mind, we propose to deliver an anti-LINGO-1 an-
tibody into the MS-ravaged axonal landscape to aid in
the promotion of oligodendrocyte differentiation and re-
myelination.
We will consider the potential to increase the efficacy
of our particles through engineering the PLP receptors
and anti-LINGO-1 antibody to have greater affinity for
their ligand and receptor. Greater affinity for PLP may
allow greater competitive displacement of T-cells along
the myelin sheath as well as better targeting of the parti-
cles, while greater affinity of anti-LINGO-1 may improve
the efficacy of the treatment and reduce the frequency of
infusion.
II. MANUFACTURING METHODS AND
VERIFICATION OF DESIGN
While the rationale behind our approach is valid and
based on well-established science, the novelty of this
treatment demands that we optimize a number of de-
sign features. It is likely that this optimization will be a
balancing act, where desired properties of our nanopar-
ticles may be inversely dependent on a single design fea-
ture of the particle. Specifically, testing of our particle is
FIG. 2. Our proposed solution encompasses A.) intra-venous delivery of our therapeutic, cloaked nanoparticle.B.) Once in the bloodstream, the nanoparticle will reachthe leaky blood brain barrier and diffuse through dueto its small size or by the same mechanism as activatedt-cells. C.) The nanoparticle is encapsulated in a naiveT-cell membrane tagged with myelin-specific targetingPLP. Contained inside is the anti-LINGO-1 antibodywithin a PEG-PLA hydrogel scaffold. D.) This nanopar-ticle will attach to the damaged myelin sites of neuronalaxons, and, while the hydrogel scaffold degrades, the an-tibody will diffuse out and promote myelin regenerationthrough oligodendrocyte proliferation.
designed for optimizing physical features of our nanopar-
ticle, such as properties of the polymer scaffold, concen-
tration of targeting receptors, and particle size and dis-
tribution. These physical features will all be optimized
for the desired in vivo properties, most importantly to
allow for infrequent systemic delivery with limited toxic-
ity and off-target effects, with drug delivery for effective
remyelination as the ultimate goal.
It would be naive to claim that efforts to design our
particle will follow a linear trajectory. As experiments
are conducted and analyzed in light of other results, an
iterative verification and validation process will occur in
which it will be necessary to re-assess the physical prop-
erties and biological effects of our nanoparticle. As this
process occurs it will likely be necessary to make changes
to our manufacturing and testing protocols.

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 4
A. Hydrogel
Inside our nanoparticle, we expect to use a hydrogel
copolymer scaffold carrying anti-LINGO-1 antibodies in
suspension. Controlled degradation of the hydrogel scaf-
fold will allow for therapeutic release kinetics of the an-
tibody. The primary components of this hydrogel will
be poly(ethylene glycol) (PEG), which has been shown
to effectively resist protein adsorption and therefore be
relatively bioinert.6 The second functional polymer to be
used will be poly(lactic acid) (PLA). When crosslinked
with PEG, this polymer is known to degrade completely
and without toxic effects in primate brains.7 In addition,
PLA has well-documented and predictable degradation
rates on the order of a few weeks.8 Adjusting the PEG-
PLA copolymer ratio can be used to fine tune the degra-
dation rate of our own hydrogel to match maturation
time scales of oligodendrocytes in human embryos, which
is in the range of 4 weeks to 2 months.9,10
1. Hydrogel Production
This hydrogel will be created by physical gelation, or
using thermoresponsive attractive properties of the hy-
drophobic PLA domains in the polymer chains to cause
them to spontaneously fold and create a soft physical
network (Fig. 3). This technique has been explored ex-
tensively in the literature for PEG/PLGA combinations,
but is much less common for PEG/PLA and, therefore,
will require optimization.11 To start, we will photopoly-
merize triblock copolymers of PLA-mPEG-PLA in solu-
tion. We will then add the antibodies to be delivered
and form nanoparticles around this solution (see section
on ”Nanoparticle Formation Through Microfluidic Jet-
ting”). The result will be a nanoparticle solution that
is ready to be injected into the patient, with no further
polymerization required. Once in the body, the tempera-
ture will naturally increase and cause the polymer to gel
and form a scaffold for our therapy.
FIG. 3. Physical gelation of PLA-mPEG-PLA copoly-mer. The polymer chains are initially dissolved in solu-tion and solidify to form a gel at an Upper Critical Solu-tion Temperature (UCST) that is designed to be belowbody temperature.
If, using this approach, we are unable to target a gel
phase transition temperature below 37◦C or obtain a sat-
isfactory degradation rate, we will move to a chemically
bonded copolymer network. This method will involve
photopolymerization, again within nanoparticle (or after
formation of the particle). In this procedure, we will ini-
tially functionalize the PEG with methacrylate groups
to cause crosslinking and mix concentrated solutions of
both PEG and PLA components in a reaction vessel.12
The antibodies will also be added, along with an initia-
tor. We will again form the nanoparticles around this
mixture then wash the nanoparticles, and move them to
PBS. At this point, we will subject them to UV radiation
to chemically polymerize the gel network from the inside.
For initial experimentation, all the materials required
for production can be obtained from Sigma Aldrich. For
the purpose of animal studies, we can buy generic anti-
LINGO-1 solution from the chemical manufacturer for
around $360/100 mg of concentrated solution. However,
the actual anti-LINGO-1 solution that will be used for
OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 5
human trials will need to be FDA approved. Currently,
there is a therapeutic anti-LINGO-1 antibody named
BIIB033 undergoing phase II clinical trials. If these trials
prove to be successful, we may be able to license BIIB033
in our own therapy.
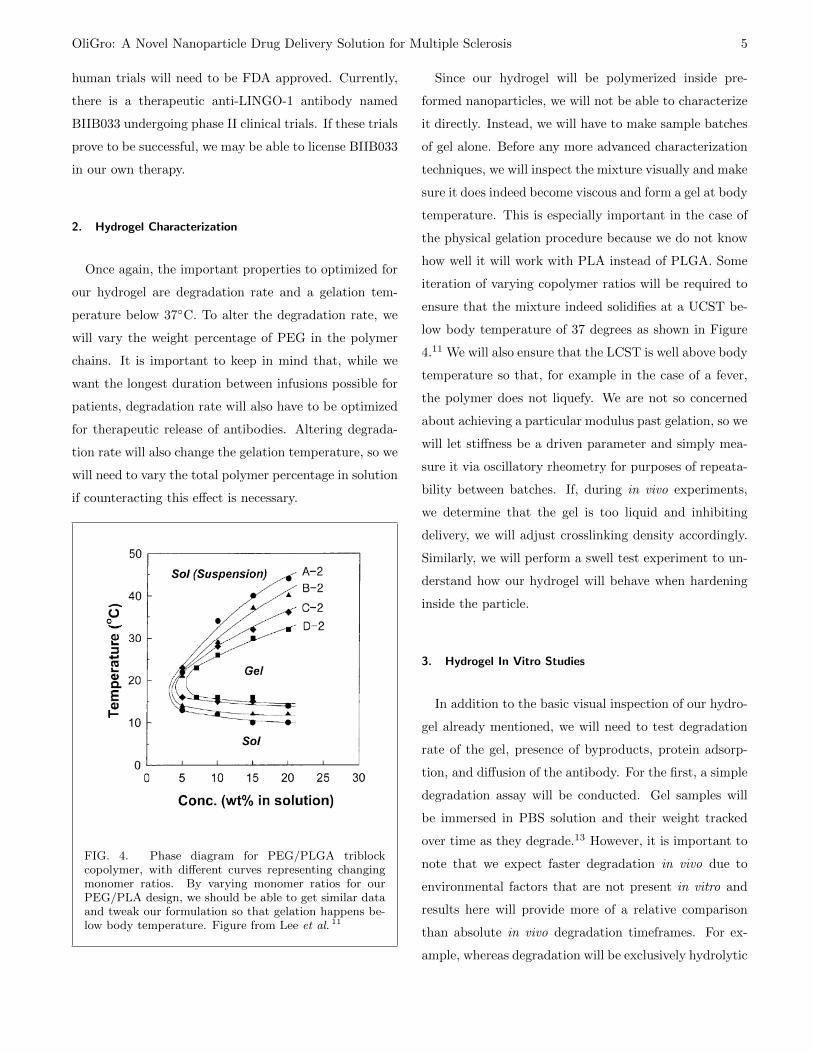
2. Hydrogel Characterization
Once again, the important properties to optimized for
our hydrogel are degradation rate and a gelation tem-
perature below 37◦C. To alter the degradation rate, we
will vary the weight percentage of PEG in the polymer
chains. It is important to keep in mind that, while we
want the longest duration between infusions possible for
patients, degradation rate will also have to be optimized
for therapeutic release of antibodies. Altering degrada-
tion rate will also change the gelation temperature, so we
will need to vary the total polymer percentage in solution
if counteracting this effect is necessary.
FIG. 4. Phase diagram for PEG/PLGA triblockcopolymer, with different curves representing changingmonomer ratios. By varying monomer ratios for ourPEG/PLA design, we should be able to get similar dataand tweak our formulation so that gelation happens be-low body temperature. Figure from Lee et al. 11
Since our hydrogel will be polymerized inside pre-
formed nanoparticles, we will not be able to characterize
it directly. Instead, we will have to make sample batches
of gel alone. Before any more advanced characterization
techniques, we will inspect the mixture visually and make
sure it does indeed become viscous and form a gel at body
temperature. This is especially important in the case of
the physical gelation procedure because we do not know
how well it will work with PLA instead of PLGA. Some
iteration of varying copolymer ratios will be required to
ensure that the mixture indeed solidifies at a UCST be-
low body temperature of 37 degrees as shown in Figure
4.11 We will also ensure that the LCST is well above body
temperature so that, for example in the case of a fever,
the polymer does not liquefy. We are not so concerned
about achieving a particular modulus past gelation, so we
will let stiffness be a driven parameter and simply mea-
sure it via oscillatory rheometry for purposes of repeata-
bility between batches. If, during in vivo experiments,
we determine that the gel is too liquid and inhibiting
delivery, we will adjust crosslinking density accordingly.
Similarly, we will perform a swell test experiment to un-
derstand how our hydrogel will behave when hardening
inside the particle.
3. Hydrogel In Vitro Studies
In addition to the basic visual inspection of our hydro-
gel already mentioned, we will need to test degradation
rate of the gel, presence of byproducts, protein adsorp-
tion, and diffusion of the antibody. For the first, a simple
degradation assay will be conducted. Gel samples will
be immersed in PBS solution and their weight tracked
over time as they degrade.13 However, it is important to
note that we expect faster degradation in vivo due to
environmental factors that are not present in vitro and
results here will provide more of a relative comparison
than absolute in vivo degradation timeframes. For ex-
ample, whereas degradation will be exclusively hydrolytic

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 6
in vitro, we would expect some enzymatic degradation in
vivo that would speed up the process. In addition, al-
though we do not expect any, we can verify that any
byproducts of the degrading hydrogel are not toxic, as
was done in Bjugstad et al. 7 with St. Kitts green mon-
keys.
In order to understand the biocompatibility of our hy-
drogel, we will also want to analyze surface protein ad-
sorption. To do this, hydrogel samples will be immersed
in blood plasma and CSF in vitro, and ellipsometry will
be conducted to measure the thickness of any result-
ing protein layer adsorbed onto their surface. While
ellipsometry is not easy to do with non-reflective sur-
faces, it can be done with hydrogels by using a metallic
substrate.14 Making use of the hydrogels transparency,
the light from the ellipsometer is able to travel through
the protein layer, the gel sample, reflect off the metallic
substrate, and refract again through both the gel and the
protein layer. In the case of our own hydrogel, we may
encounter some difficulty if the gel is too opaque. Since
transparency is a function of where in the sol-gel transi-
tion the temperature is, we could adjust it to get a clear
gel and therefore better results.
The antibodies we expect to use are in the range of
3-9 nm and the mesh size of our hydrogel will need to
be much larger than this in order to allow for diffusion
over time.15,16 Rather than trying to verify this mesh size
using Scanning Electron Microscopy or Nuclear Magnetic
Resonance Spectroscopy, which can be very expensive
techniques and may not be necessary, we believe it would
be sufficient to conduct an in vitro diffusion study. In
this experiment, we will fluorescently tag the antibodies
and form the hydrogel in their solution. We will then
track the movement of antibodies in hydrogel samples
over time.
B. Native T-Cell Membrane Cloaking
We have chosen to cloak the nanoparticles in a na-
tive cell membrane to help disguise them from immune
response.17 Specifically, we have chosen to commandeer
T-cell membranes because we know T-cells can infiltrate
the BBB in MS patients without prompting a systemic
autoimmune response.
1. T-Cell Culture
In our in vivo animal testing, we will need to determine
whether naive (non-activated) T-cell precursors ought to
be collected from spleen or lymph node samples, or if T-
cells collected from blood will suffice. Though it would ul-
timately be simpler to collect autologous T-cells from pa-
tients blood, the T-cells circulating in blood have already
been activated by their TCR-specific antigen, and this
could potentially present a problem for our use. There
are several methods and kits available for collecting naive
T-cells from murine spleen and blood, for example, from
vendors like Miltenyli Biotec or Abcam.18
T-cells will be isolated from other tissue or blood cells
using magnetic-activated cell sorting (MACS) beads from
Miltenyli Biotec. Briefly, cells are incubated with mag-
netic nanoparticles coated with either anti-CD8 or anti-
CD4 antibodies, which bind to CD8+ or CD4+ T-cells
in solution. The solution is then transferred to a column
placed in a strong magnetic field, and the T-cells attached
to magnetic nanoparticles will adhere to the column while
other cells will flow through. T-cells are then collected
in a separate vessel after removing the column from the
magnetic field.
Through our in vitro and in vivo testing, we will assess
whether there is a difference between using CD8+ (cyto-
toxic, MHC-I-selective) T-cell or CD4+ (helper, MHC-
II-selective) T-cell membranes to cloak our nanoparticle.
Both cell types play a role in the pathology of MS, but
it could be potentially more expensive to isolate two cell

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 7
FIG. 5. ProImmune Protocol for Magnetic Cell Separa-tion.
types instead of one. Notably, if we are required to dis-
tinguish between naive and active T-cells, naive T-cells
are characterized by the absence of CD25, CD44, and
CD69, while these cytokines are upregulated in activated
T-cells.19 For the purposes of our experimental design
outline, we will proceed with assuming CD8+ T-cells.
Cells will be cultured as outlined in Lewis et al. 18 af-
ter being isolated. Briefly, The isolated T-cells will be
plated and coated with anti-CD28 and anti-CD3 in PBS
at for 24 hours. After 24 hours, IL-7 and IL-2 will be
added. The cells will be grown for 24 hours, harvested,
and replated for 3 days. After which time, the cells will
be transfected with a vector encoding the genes for the
protein which will be incorporated into the nanoparticle
membrane.
2. Gene Delivery
As outlined previously, we have also chosen to incor-
porate an antibody for myelin α-PLP into the isolated
membrane, in addition to potentially adding α4β1 inte-
grin for BBB penetration if necessary. In order to inte-
grate α-PLP and α4β1 into the naive CD8+ T-cells, a
transfection method will be used. Transfection works by
introducing a gene vector into the cell using a chemical,
electrical, or viral delivery method (lipofectamine, elec-
troporation, and lentivirus, respectively). All methods
will be tested to identify which is most effective for the
CD8+ T-cell populations. Once inside the cell, the trans-
fected plasmid will take advantage of the host cells cen-
tral dogma to express the protein of interest using a vec-
tor (pjP008) that has been designed to incorporate pro-
teins into the membrane as described in Protein Science.
MACS will be used to isolate the successful transfections
using antibodies for α4β1 and α-PLP. Immunoprecipi-
tation along with western blot analysis will be used to
confirm incorporation into the cell membrane.
It is possible that proteins endogenous to the naive
T-cell membrane prove to be reactive within the body.
Clustered Regularly Interspaced Short Palindromic Re-
peats (CRISPR)-Cas9 is a protein, which, along with
a guide RNA (gRNA), is capable of inducing a double
strand break in a targeted genes DNA and silencing that
gene.20 Should there be a reactive protein native to naive
T-cells on the membrane, CRISPR/Cas9 genome editing
techniques will be used to knock-out the reactive protein
using the genome editing kit from Clontech. This tech-
nique will be valuable in investigating the potential for
removing markers of ”self” and preventing recognition of
”nonself” (i.e. major histocompatibility complex) if we
are to use engineered generic T-cells for a later version of
our therapy.

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 8
C. Nanoparticle
Once we have shown we can effectively culture T-
cells and incorporate proteins of interest into their mem-
branes, we will need to isolate these membranes so we
can package and cloak our nanoparticles. Repeated rapid
freeze-thaw cycles in a low-detergent disruption buffer are
known to effectively lyse cells and begin the process of
membrane isolation. Pelleting at low speeds can remove
cellular debris while keeping lipids in solution. Following
this with washing with buffered solution (pelleting lipids
through ultracentrifugation and resuspension) and son-
ication will further contribute to the isolation of T-cell
precursor membranes and formation of vesicles. In order
to preserve functionality of surface membrane proteins,
some or all steps may require protease inhibitors. Effec-
tive protease inhibition can be assessed through western
blot if necessary. Membrane isolation and vesicle forma-
tion may be sensitive to specific parameters (i.e. number
of freeze-thaw cycles, buffer solution, sonication length
and intensity); the final procedure will need to be op-
timized for effective formation of nanoparticles. If nec-
essary, affinity purification will be possible through the
targeting of native proteins or membrane expression of a
high affinity tag such as polyhistidine in our T-cell trans-
fection process.
For storage, membrane vesicles will be frozen in
buffered solution (stability over time will need to
be assessed). Alternatively, isolated vesicles may be
lyophilized and stored in this dried form.
1. Nanoparticle Formation Through Microfluidic Jetting
For formation of our nanoparticles, suspension of the
isolated lipids will occur in an organic solvent after again
pelleting the bilayers through ultracentrifugation and re-
moving the disruption buffer. The solvent we use will
need to be optimized for the function of our nanoparti-
cles, as organic solvents can denature proteins that we
want to be functional on the surface of our particles for
effective cloaking and targeting.
Microfluidic jetting is a technique that can be fine-
tuned to produce a uniformly sized vesicle through rela-
tively straightforward means. Formation of a lipid bilayer
is accomplished by placing an organic solution contain-
ing lipids into two connected wells. The addition of small
drops of immiscible solution to each well allows for the
formation of a lipid bilayer between the wells. A narrow
inkjet tip is used to jet a second solution across the bi-
layer, causing vesicles to bud off of the bilayer and form
independent vesicles (Fig.6).21
FIG. 6. Nanovesicle formation from Coyne et al. 21
For our application, the solution jetted across the lipid
bilayer to form the nanoparticles will contain our poly-
mer components in solution. To prevent rupture of the
vesicles, it will be important for the osmolarity of the
buffer into which the nanoparticles are being formed (the
immiscible solution used to form the lipid bilayer be-
tween wells) to be similar to that of our polymer compo-
nents. Nanoparticle size can be controlled during jetting
by varying the distance between the inkjet nozzle and
the voltage applied to the inkjets piezoelectric actuator.
Researchers have found that smaller nanovesicles are gen-
erally more stable with smaller diameters.21 However, if
we are unable to form particles of our desired size, we can
consider introducing non-native lipids to the solubilized
cell membrane solution in order to alter packing param-

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 9
eters. By introducing lipids with varying chain length or
head group areas, we can change the critical packing pa-
rameter, CPP, where CPP = v/a0IC (v, hydrocarbon tail
volume; a0, optimal head group area; IC , critical chain
length). While we do not want to change the CPP such
that our particles will form micelles, altering the param-
eter slightly will influence the ability of the lipid bilayers
to form particles of various sizes.
An alternative method to achieving cloaking will in-
volve rehydration/resuspension of a mixture of polymer-
ized, dried hydrogel particles and lyophilized membrane
vesicles in the presence of buffered solution. Rehydration
and resuspension in a concentrated environment may im-
prove efficiency in coating of particles, however, this ap-
proach will likely be less precise and may result in signif-
icant waste compared to in-vesicle polymerization.
Dynamic light scattering will provide insight into par-
ticle size uniformity through measuring the scattering
or reflection of light off of the nanoparticles in solution.
We anticipate a uniform size distribution of the particles
based on previous work.21 If necessary, chromatographic
purification based purely on size exclusion may be effec-
tive for desired nanoparticle isolation, however, if this
method is not effective, an experimentally-determined
ionic gradient should prove sufficient for isolation of de-
sired nanoparticles.
III. PERFORMANCE VALIDATION OF DESIGN
Below, we outline many of the in vitro and in vivo
experiments required to assess the performance of our
nanoparticle design. A common mouse model for multi-
ple sclerosis will be used for in vivo experiments before
moving into clinical trials with patients. The experimen-
tal autoimmune encephalitis (EAE) mouse is representa-
tive of many of the disease characteristics found in multi-
ple sclerosis, and is therefore optimal for this testing. To
induce EAE, mice will be injected with myelin oligoden-
drocyte glycoprotein peptide (MOGaa35−55) followed by
immunization using nonviable Mycobacterium tuberculo-
sis, then inject pertussis toxin into the abdomen. While
several other EAE models and methods to induce en-
cephalitis, this particular model has been shown to pro-
vide leakiness in the brain in order to simulate barrier
bypass.22 A power analysis will be conducted in order to
determine the number of mice necessary for the extensive
and diverse testing described.
A. Toxicity and Off-Target Effects
As a systemically delivered antibody-based therapy,
our nanoparticle has significant potential for off-target
effects and in vivo toxicity. The complexity of these ef-
fects renders them difficult to predict, but can in part be
assessed through a variety of in vitro assays. These assays
will provide indications of upstream results that may lead
to undesirable effects when delivered in vivo. Further-
more, these in vitro assays will help indicate whether our
particle will have the opportunity to encounter and mi-
grate through the BBB, rather than total sequestration
through, for example, a thrombotic response. Once the
results of in vitro assays yield satisfactory results, we will
progress into in vivo studies that will show us the local-
ization, or distribution in the body, of our antibody and
provide a variety of downstream toxicity results. These
in vivo tests will also help identify the clearance mech-
anisms of our particle (i.e. hepatic) and whether the
particle is actually reaching the myelin target.
1. Protein Interaction
In order to understand the behavior of our nanoparticle
in the complex environment of the body it is important
to conduct in vitro assays to elucidate the way in which
our nanoparticle associates with other proteins and cells.
These assays will be conducted in conditions that reflect
both blood medium, due to the planned systemic delivery
of our therapy, and cerebrospinal fluid (CSF) medium, to

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 10
assess behavior once the particle has crossed the BBB.
These two environments can be simulated in vitro pri-
marily through proper buffer components and protein
concentration. In healthy patients, protein concentra-
tions in CSF compared to blood are very low (about
100-fold lower). However, in MS patients, increases in
CSF concentrations of IgG and albumin are often seen.
Healthy levels of CSF protein have been established by
the Mayo Clinic (Test ID: SFIN), but MS-related in-
creases of IgG vary generally within <10-fold. For our
assays, concentrations of protein at physiological blood
and CSF levels will be conducted, with also a series of
concentrations of up to 100-fold increase of IgG in CSF-
like buffer.
Specifically, to assess protein-nanoparticle interac-
tions, we will incubate our particles with native proteins
in blood-like and CSF-like buffer for a range of times (be-
ginning with 1 minute and potentially up to 24 hours).
Gel electrophoresis under reducing and non-reducing con-
ditions will provide information about the interaction of
our particles with proteins through variation in band mi-
gration through the gel. Under non-reducing conditions,
nanoparticle-protein complexes should represent a higher
MW, slower-migrating band. The necessary controls will
include protein samples incubated without nanoparticles,
and nanoparticles incubated alone without protein. Par-
ticles will likely be selected that have minimal interaction
with proteins.
2. Thrombosis
As we will be treating patients with a potentially
thrombogenic compound, assessing thrombosis in vitro
will be an important first step. Quantification of platelet
aggregation is commonly done in both basic research
and clinical settings through spectrophotometric mea-
surements of turbidity.23,24 Turbidity measurements are
admittedly low-throughput. If we find a need for a higher
throughput assay, there are plate-based colorimetric as-
say kits and protocols available that measure ADP re-
leased from platelets. In general, these assays rely on
conversion of ADP to ATP, after which ATP-dependent
lucerifase activity affects a colorimetric change. For ease
of use and in the interest of time, we will opt for a
kit based assay. The Abcam kit improves upon the
lucerifase-based assay and provides a fluorescent alterna-
tive: For both turbidity and colorimetric assays, throm-
bin should serve as a positive control and the addition of
buffer (PBS) would serve as a sufficient negative control.
We do not expect our nanoparticles to induce rapid clot-
ting and, as such, conducting these assays over a range
of incubation times should provide a sense of the kinetics
of thrombosis.25
3. Immune Response
Although we are cloaking our nanoparticle in a native
membrane, it is still possible the host immune system
may recognize components of our nanoparticle as for-
eign and initiate an immune response.26 To ensure our
nanoparticles truly evade the hosts immune system, we
want to compare the state of the immune system be-
tween both treated and sham-injected EAE and control
animals. The complement cascade is an early indication
of immune response, and we can survey this potential
activation with an ELISA assay for complement compo-
nents (kits for which are commercially available through
Abcam) in mouse plasma or serum. We can also mea-
sure cytokine blood levels with a similar cytokine array
kit (R&D Systems) to determine the relative levels of se-
lected mouse cytokines. We expect that if our nanopar-
ticle does not evoke an immune response, cytokine lev-
els between sham and treated WT mice, and sham and
treated EAE mice should be comparable, respectively.
If we do detect complement activation or a significant
change in cytokine levels in either or both comparisons,
we can follow up with histology to determine the extent
of inflammation. Briefly, animals would be sacrificed in

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 11
compliance with approved IACUC protocols and brains
would be dissected out, fixed with 4% paraformaldehyde,
and sectioned for histology according to the protocol out-
lined for the method of choice. Sections would then be
processed through immunohistochemistry with immune-
cell specific antibodies like Iba1 for microglia and GFAP
for astrocyte activation before imaging to visualize the
cellular landscape.
4. In Vivo Localization
Visualizing the localization of our nanoparticle in vivo
will help us to determine if the particle is reaching the
CNS and how the body sequesters and clears the parti-
cles. In order to model the location of our nanoparticle
noninvasively and continuously in vivo, we intend to use
near-infrared imaging techniques to detect its concentra-
tion and location dispersion. The dosing scheme will be
similar to the above immune response study, and may in-
clude the same animals. We will use a fiber optic device
that measures the signal intensities to determine kinet-
ics of our antibodies conjugated with NIR fluorospheres
or lipophilic tracers (available from Thermo Fisher and
other suppliers).27 This imaging technique will allow us
at least a 48 hour window, evaluated every 4 hours. Given
the fluorescent intensities, we would be able to determine
the localization of the antibodies systemically and at the
target CNS.28 We expect that for the first few hours post
intravenous injection, the drug will travel through the
body systemically before reaching the BBB and entering
the CNS, targeting the myelin PLP. As seen in the Figure
7, we similarly expect our nanoparticle to cluster in the
CNS, and specifically at the damaged axons. At set time
periods, animals will be sacrificed and various samples of
blood and tissue will be obtained for fluorescent quan-
tification. Blood samples may be taken at more frequent
intervals. Tissue samples will include at minimum brain,
liver, kidney and major blood vessels.
FIG. 7. In vivo NIR optical imaging of HER2-positivetumor xenograft mice post treatment with Alexa Fluor750-labeled conjugates. From Lee et al. 28
5. In Vivo Toxicity Monitoring
We plan for OliGro to deliver therapeutic effects over
the course of 1-2 months. As a result, we need to conduct
studies in the long term to determine if the drug contin-
ues to be viable and remains in large enough concentra-
tions in order to extend therapeutic effects. In order to
conduct these long term studies, we will inject labeled an-
tibodies with a different signal intensity than originally
used that targets our specific therapeutic antibody for up
to 2 months to verify drug concentration remains viable
for treatment.
In vivo pharmacokinetic and pharmacodynamic
(PKPD) assays will be conducted to assure that our
nanoparticle, delivered in a specified threshold above
expected concentrations, does not have toxic effects in
model organisms. Early studies will use rodents, while
more complex later-stage trials will be conducted in
larger animals. Generally, blood and urine samples ob-
tained at set intervals can be monitored for changes in
levels of biomarkers, such as liver transaminases AST
and ALT for hepatoxocity (increased levels indicate liver
toxicity), and creatinine for kidney function (generally

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 12
a ratio of blood creatinine to urine creatinine, with an
increase in blood indicating decreased golumerular fil-
tration). Timepoints and duration will need to be set
depending on initial PKPD assays. Biomarker data will
be normalized to a baseline sample obtained before ad-
ministration of drug, and data will be compared to ani-
mals dosed with saline (sham), vesicles alone (no hydro-
gel contents), and nanoparticles without the therapeutic
antibody. Histology will be conducted on animals after
sacrifice to identify any toxic effects, especially in the
primary organs such as the liver, kidneys and brain.
B. Drug Delivery and Release Kinetics
A critical aspect of OliGro will be controlled release of
the anti-LINGO-1 antibody. To avoid injection fatigue,
which can lead to low patient compliance, we want our
drug to have a therapeutic effect as long as possible, ide-
ally over the course of at least 1-2 months. First, we
want to ensure proper drug delivery to the myelin sheath
by confirming permeability of OliGro through the BBB.
Then, we want to optimize a robust drug release profile.
1. Blood Brain Barrier Permeability
For in vitro simulation of our device across the BBB,
we intend to use microscopy of primary mouse brain
microvascular endothelial cells (pMBMECs) harvested
from a mouse whose BBB has been previously compro-
mised following the EAE model. The monolayer of pMB-
MECs will be placed in a commercially available two-
chamber assay where live action T-cell extravasation can
be imaged during flow. Fluorescently tagged intracel-
lular adhesion molecules (I-CAM) will allow us to de-
termine whether our α4β1 tagged naive T-cell is capa-
ble of rolling, attaching and bypassing the blood brain
barrier.22
Following this experiment, we can confirm BBB pen-
etration in vivo. This will be achieved using two-
FIG. 8. Experimental setup of two-photon intravital flu-orescence videomicroscopy (IVM) live-imaging EAE andcontrol mice spinal cord window for T cell transmigra-tion to CSF and spinal cord. From Coisne, Lyck, andEngelhardt 22 .
photon Intravital fluorescence videomicroscopy (IVM).
Our empty, naive T-cells tagged with α4β1 integrin will
be fluorescently tagged and washed will a buffer prior
to injection into the carotid artery of EAE and control
mice. The surgical window of the spinal cord will im-
aged in real-time, recording the attachment of T-cells to
the endothelial layer and quantifying T-cell infiltration
into the spinal cord.22 We will also conduct this study
using the tagged nanoparticle without the α4β1, as the
size may allow simple diffusion through the leaky bar-
rier. This will allow us to determine which nanoparticle
version is optimal for entry and targeting and bypassing
the BBB. We primarily expect that the nanoparticle can
enter through this simple diffusion method.
2. Antibody Release Kinetics
An anti-LINGO-1 antibody called BIIB033 was discov-
ered and has begun to be commercialized by the company
Biogen. Randomized phase I trials verified the safety and
tolerability of anti-LINGO-1 in healthy volunteers and
MS patients,29 and a phase II clinical trial is currently
underway. Initial reports suggest that intravenous doses

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 13
above 100 mg/kg in humans could lead to serum con-
centrations that are predicted to have pharmacological
activity; however, we will have to follow the phase II and
III clinical trials to determine the actual efficacious con-
centration for clinical use. We can use 100 mg/kg as a
baseline concentration goal for initial studies.
Commercial vendors of Anti-LINGO-1 antibodies, such
as Sigma Aldrich, ThermoFisher Scientific and Millipore,
cite the antibodys size to be about 83 kDa, which means
we would need to deliver at least 7.26x1017 antibodies per
kg to have an effect. Assuming a hydrodynamic radius of
about 9 nm for anti-LINGO-1 (Pepinsky et al. 30 report a
hydrodynamic radius of 15 nm for the antibody-antigen
complex and Mosyak et al. 31 report a hydrodynamic ra-
dius of 6.2 nm for the LINGO-1 protein), the spherical
volume of a single antibody is 3.05x103 nm3. If we esti-
mate the total volume available within each nanoparticle
to be between 6.55x104-5.24x105 nm3 (based on a 50-
100 nm diameter range for each nanoparticle), we could
fix a maximum of 171 antibodies in each nanoparticle.
With this calculation, we would need to deliver 4.24x1015
nanoparticles per kg to have an effect. Of course, this
does not take into the account the amount of volume
taken up by our hydrogel scaffold within the nanopar-
ticle, and we ought to assume that the majority of the
internal nanoparticle volume will indeed be occupied by
hydrogel. In our experimental designs, we will try to fit
as many antibodies into our hydrogel as possible. We will
have to deliver a greater concentration of nanoparticles
than estimated above, which will highly depend on this
final distribution of antibodies within our hydrogel.
Another crucial consideration in assessing drug deliv-
ery will be the actual rate of drug released from the hy-
drogel over time as it degrades. We do not have to take
the T-cell membrane degradation rate into account be-
cause its half life is extremely small and T-cells generally
have rapid turnover. The majority of drug is released
during the second diffusion phase of a triphasic release
process, and we want to design our hydrogel as such so
this phase lasts at least a month in vivo. Besides con-
trolling the degradation rate of the hydrogel, the rate of
drug release depends on other factors such as: the size
and shape of the scaffold (here, we will test various sizes
of spherical gels) and the porosity, or, degree of cross-
linking within our hydrogel.
To assess drug release kinetics from the hydrogel in
vitro, we can start by measuring drug concentration in
a solution surrounding our drug-studded hydrogel over
time. Gels would be placed in 1 ml of PBS in a multi-
well plate and shaken in a room temperature incubator.
PBS would be collected and replaced, and anti-LINGO-
1 concentration would be measured various time point
over the course of minutes, hours, and days in subsequent
rounds of experiments to complete our concentration over
time curve. We could use ELISA or ultraviolet-visible
spectroscopy (UV-Vis, antibodies generally have a UV
absorption peak near 280 nm) to assess anti-LINGO-1
concentration in collected PBS. It is likely we would be
able to run these tests with a standard IgG, which would
be cheaper than using our specific anti-LINGO-1 anti-
body, and still measure similar release kinetics although
the comparison test would have to be run to ensure this.
We will also want to test drug release kinetics in cell
culture once our baseline is established in well-plates to
ensure the rate of drug release is consistent in a more
physiologically relevant environment.
C. Therapeutic Performance
Finally, it will be important to ensure that our
nanoparticle actually achieves the desired therapeutic ef-
fect that we are seeking. We will start with an in vitro
approach in which we assess axonal recovery and re-
myelination in a co-cell culture of axons and oligoden-
drocytes. Moving on to animal tests, we will want to
survey our mice throughout treatment for regained neu-
rological function alongside looking for oligodendrocyte
proliferation and myelin regeneration. A 0-5 functional

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 14
scoring scale is widely used to measure neurological func-
tion of the EAE animal model, with a higher score indi-
cating a more severe disease state. Mice are scored daily
for clinical symptoms of EAE, as follows: 0, healthy; 1,
loss of tail tone; 2, ataxia and/or paresis of hindlimbs;
3, paralysis of hindlimbs and/or paresis of forelimbs; 4,
tetraparalysis; 5, moribund or dead.32
FIG. 9. Multi-compartment neuron-glia co-culture mi-crosystem as outlined in Park et al. 33
1. Remyelination Assessment in Cell Culture
To assess remyelination in vitro, we will start using a
physiologically relevant, multi-compartmental microflu-
idic neuron/glia co-culture as done in Park et al. 33 (Fig.
9). This cell culture will serve as our control, and
we will add active T-cells to a separate co-culture to
model MS. In order to test if our drug can effectively
cause remyelination, either complete (PLP/α4β1 and
anti-LINGO-1), empty (PLP/α4β1), negative (neither
PLP/α4β1 nor anti-LINGO1), or naked (without anti-
LINGO-1) nanoparticles will be added at logarithmic
doses to the two cultures. Testing of the nerve function
and myelination will be carried out using immunohisto-
chemistry, transmission electron microscopy, and whole
cell patch clamp to measure the amount of myelination,
myelin morphology, and conductance of the neuron (G),
respectively.34
We hypothesize the following results:
Healthy MS
CompleteSlightincrease inmyelination
Protection and increasedmyelination to physiologicalnumbers, healthy G
Empty No changeInhibition of myelindegradation until the NPdegrades, decreased G
NakedSlightincrease inmyelination
Increased oligodendrocytenumber, same degradationrate, poor morphology,decreased G
Negative No changeTotal degradation of myelin,very poor G
2. Oligodendrocyte Proliferation and Remyelination In
Vivo
Another pivotal performance experiment is to mea-
sure the extent of oligodendrocyte and anticipated sub-
sequent remyelination in vivo. Does our nanoparticle ac-
tually deliver the anti-LINGO-1 drug effectively enough
to stimulate oligodendrocyte differentiation and remyeli-
nation? Do we also see increased myelination in our
control animals, and does this possibly impede with
healthy neuronal function? We could perform immuno-
histochemistry on sections from animals (both control
and EAE, with nanoparticle and sham treatment) in-
jected intraperitoneally once a day for two weeks with
bromodeoxyuridine (BrdU, 100 mg/kg bw), the thymi-
dine analog incorporated into DNA of dividing cells dur-
ing S-phase and used for mitotic labeling. We would
stain fixed brain sections for oligodendrocyte progenitor
cells with an NG2 antibody, young and mature oligoden-
drocytes with an RIP antibody, and a BrdU antibody.35
If our therapy stimulated oligodendrocyte proliferation,

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 15
we would expect to see an increase in BrDU+ cells that
also express NG2+ or RIP+ immunoreactivity in treated
animals.
To complement this experiment, we would also want to
measure a reduction in demyelinated regions in treated
animals. Luxol blue and Bielshowsky are commonly used
to visualize myelin and axons, respectively, under light
microscopy. We would follow the protocol outlined in
Zhang et al. 35 to measure the extent of myelinated re-
gions in sham and treated animals compared to baseline.
We could also assess remyelination post mortem via elec-
tron microscopy. Alternatively, if we have access to an
MRI that could image mice, we could assess and quantify
myelinated regions post treatment in vivo, as is done to
measure clinical diagnosis and prognosis in human MS
patients.36
IV. CONCLUSION
As an entirely novel treatment for MS, our proposed
therapy presents significant hurdles that must be over-
come for success. However, current treatments for MS
are clearly inadequate due to their preventative nature, a
lack of efficacy in some patients and their significant side
effects. As a lifelong disease with expensive pharmaceu-
tical therapies and the need for occupational assistance,
MS also represent a significant financial burden in a world
where healthcare is shifting ever more towards a discus-
sion of quality- and value-based decision making. Our
nanoparticles will not only be cheaper than the current
standard of care, but they will also improve quality of
life and reduce costs by allowing patients to return to au-
tonomy. While challenging, the novelty of our proposed
treatment presents a potential groundbreaking change for
the patients themselves as well as a cost-effective option
for the healthcare system.
1A. Pietrangelo and V. Higuera, “Multiple sclerosis by the num-
bers: facts, statistics, and you,” (2015).2B. Alberts, A. Johnson, and J. Lewis, “Molecular biology of the
cell,” (Garland Science, New York, 2002) Chap. T Cells and
MHC Proteins, 4th ed.3B. Engelhardt, “Molecular mechanisms involved in T cell mi-
gration across the blood-brain barrier,” J Neural Transm 133,
477–485 (2006).4J. M. Greer, “Autoimmune T-cell reactivity to myelin prote-
olipids and glycolipids in Multiple Sclerosis,” Mult Scler Int 2013
(2013).5S. Mi, A. Sandrock, and R. Miller, “LINGO-1 and its role in
CNS repair,” Int J Biochem Cell Biol 40, 1971–1978 (2008).6R. Michel, S. Pasche, M. Textor, and D. Castner, “Influence
of PEG architecture on protein adsorption and conformation,”
Langmuir 21, 12327–12332 (2005).7K. Bjugstad, J. D.E. Redmond, K. Lampe, D. Kern, J. Sladek,
and M. Mahoney, “Biocompatability of PEG-based hydrogels in
primate brain,” Cell Transplant 17, 409–415 (2008).8K. Kim, M. Yu, X. Zong, J. Chiu, D. Fang, Y.-S. Seo, B. Hsiao,
B. Chu, and M. Hadjiargyrou, “Control of degradation rate
and hydrophilicity in electrospun non-woven poly(D,L-lactide)
nanofiber scaffolds for biomedical applications,” Biomaterials 24,
4977–4985 (2003).9C. Hiemstra, W. Zhou, Z. Zhong, M. Wouters, and J. Fei-
jen, “Rapidly in situ forming biodegradable robust hydrogels by
combining stereocomplexation and photopolymerization,” J Am
Chem Soc 129, 9918–9926 (2007).10I. Jakovcevski, R. Filipovic, Z. Mo, S. Rakic, and N. Zecevic,
“Oligodendrocyte development and the onset of myelination in
the human fetal brain,” Front Neuroanat 3, 1–15 (2009).11D. Lee, M. S. Shim, S. Kim, H. Lee, I. Park, and T. Chang,
“Novel thermoreversible gelation of biodegradable PLGA-block-
PEO-block-PLGA triblock copolymers in aqueous solution,”
Macromol Rapid Commun 22, 587–592 (2001).12A. Sawhney, C. Pathak, and J. Hubbell, “Bioerodible hydrogels
based on photopolymerized poly(ethylene glycol)-co-poly(alpha-
hydroxy acid) diacrylate macromers,” Macromolecules 26, 581–
587 (1993).13S. Hahn, J. Park, T. Tomimatsu, and T. Shimoboji, “Synthesis
and degradation test of hyaluronic acid hydrogels,” Int J Biol
Macromol 40, 374–380 (2007).14D. Miller and N. Peppas, “The use of ellipsometry to study ad-
sorption on hydrogels,” Biomaterials 6, 33–40 (1985).15J. Armstrong, R. Wenby, H. Meiselman, and T. Fisher, “The
hydrodynamic radii of macromolecules and their effect on red
blood cell aggregation,” Biophys J 87, 4259–4270 (2004).16T. Jssang, J. Feder, and E. Rosenqvist, “Photon correlation
spectroscopy of human IgG,” J Protein Chem 7, 165–171 (1988).17C.-M. Hu, R. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana,
D. Dehaini, P. Nguyen, P. Angsantikul, C. Wen, A. V. Kroll,
C. Carpenter, M. Ramesh, V. Qu, S. Patel, J. Zhu, W. Shi,
F. Hofman, T. Chen, W. Gao, K. Zhang, S. Chien, and L. Zhang,
“Nanoparticle biointerfacing by platelet membrane cloaking,”
Nature 526, 118–121 (2015).

OliGro: A Novel Nanoparticle Drug Delivery Solution for Multiple Sclerosis 16
18M. Lewis, E. de Leenheer, S. Fishman, L. Siew, G. Gross,
and F. Wong, “A reproducible method for the expansion of
mouse CD8+ T lymphocytes,” J Immunol Methods 417, 134–
138 (2015).19S. D. Rosa, L. Herzenberg, L. Herzenberg, and M. Roederer,
“11-color, 13-parameter flow cytometry: identification of human
naive T cells by phenotype, function and T-cell receptor diver-
sity,” Nat Med 7, 245–248 (2001).20R. Barrangou, A. Birmingham, S. Wiemann, R. Beijersbergen,
V. Hornung, and A. van Brabant Smith, “Advances in CRISPR-
Cas9 genome engineering: lessons learned from RNA interfer-
ence,” Nucleic Acids Res 43, 3407–3419 (2015).21C. Coyne, K. Patel, J. Heureaux, J. Stachowiak, D. Fletcher,
and A. Liu, “Lipid bilayer vesicle generation using microfluidic
jetting,” J Vis Exp 84, e51510 (2014).22C. Coisne, R. Lyck, and B. Engelhardt, “Live cell imaging tech-
niques to study T cell trafficking across the blood-brain barrier
in vitro and in vivo,” Fluids Barriers CNS 10 (2013).23M. Jamaluddin and L. Krishnan, “A spectrophotometric method
for following initial rate kinetics of blood platelet aggregation,”
J Biochem Biophys Methods 14, 191–200 (1987).24J. Walder and I. Klotz, “Design and operation of an adapter for
the measurement of platelet aggregation with a spectrophotome-
ter,” Anal Biochem 84, 628–632 (1978).25B. Sun, N. Tandon, N. Yamamoto, M. Yoshitake, and J. i. Kam-
bayashi, “Luminometric assay of platelet activation in 96-well
microplate,” BioTechniques 31, 1174–1181 (2001).26M. Elsabahy and K. Wooley, “Cytokines as biomarkers of
nanoparticle immunotoxicity,” Chem Soc Rev 42, 5552–5576
(2013).27V. Kalchenko, S. Shivtiel, V. Malina, K. Lapid, S. Haramati,
and T. Lapidot, “Use of lipophilic near-infrared dye in whole-
body optical imaging of hematopietic cell homing,” J Biomed
Opt 11, 050507 (2006).
28S. Lee, M. Hassan, R. Fisher, O. Chertov, V. Chernomordik,
G. Kramer-Marek, A. Gandjbakhche, and J. Capala, “Affi-
body molecules for in vivo characterization of HER2-positive tu-
mors by near infrared imaging,” Clin Cancer Res 14, 3840–3849
(2008).29J. Tran, J. Rana, F. Barkhof, I. Melamed, H. Gevorkyan,
M. Wattjes, R. de Jong, K. Brosofsky, S. Ray, L. Xu, J. Zhao,
E. Parr, and D. Cadavid, “Randomized phase I trials of
the safety/tolerability of anti-LINGO-1 monoclonal antibody
BIIB033,” Neurol Neuroimmunol Neuroinflamm 1, e18 (2014).30R. Pepinsky, J. Arndt, C. Quan, Y. Gao, O. Quintero-Monzon,
X. Lee, and S. Mi, “Structure of the LINGO-1-Anti-LINGO-
1 Li81 antibody complex provides insights into the biology of
LINGO-1 and the mechanism of action of the antibody therapy,”
J Pharacol Exp Ther 350, 110–123 (2014).31L. Mosyak, W. Wood, B. Dwyer, M. Buddha, M. Johnson,
A. Aulabaugh, X. Zhong, E. Presman, S. Benard, K. Kelleher,
J. Wilhelm, M. Stahl, R. Kriz, Y. Gao, Z. Cao, H.-P. Ling,
M. Pangalos, F. Walsh, and W. S. Somers, “The structure of
the lingo-1 ectodomain, a module implcation in central nervous
system repair inhibition,” J Biol Chem 281, 36378–36390 (2006).
32J. Zhang, Z. Zhang, D. Morris, Y. Li, C. Roberts, S. Elias,
and M. Chopp, “Neurological functional recovery after thymosin
beta4 treatment in mice with experimental auto encephalomyeli-
tis,” Neuroscience 164, 1887–1893 (2009).33J. Park, H. Koito, J. Li, and A. Han, “Multi-compartment
neuron-glia co-culture platform for localized CNS axon-glia in-
teraction study,” Lab Chip 12, 3296–3304 (2012).34R. Huval, O. Miller, J. Curley, Y. Fan, B. Hall, and M. Moore,
“Microengineered peripheral nerve-on-a-chip for preclinical phys-
iological testing,” Lab Chip 15, 2221–2232 (2015).35J. Zhang, Y. Li, J. Chen, Y. Cui, M. Lu, S. Elias, J. Mitchell,
L. Hammill, P. Vanguri, and M. Chopp, “Human bone marrow
stromal cell treatment improves neurological functional recovery
in EAE mice,” Exp Neurol 195, 16–26 (2005).36J.-M. Tillema and I. Pirko, “Neuroradiological evaluation of de-
myelinating disease,” Ther Adv Neurol Disord 6, 249–268 (2013).


![印刷用 KNET寺田121115.ppt [互換モード]...CTLを誘導し、皮内注射。生存期間24カ月。 Title Microsoft PowerPoint - 印刷用 KNET寺田121115.ppt [互換モード]](https://img.pdfslide.tips/doc/110x75/5f07bd2c7e708231d41e7f20/c-knetc-fff-ctleccoee24oe.jpg)





















![印刷用 KNET寺田121115.ppt [互換モード]](https://img.pdfslide.tips/doc/110x75/61e0955bdbb8dc1c58437ae8/-knet-.jpg)
