Embed Size (px)
Citation preview

Praktikum der Ionenchromatographie – Version 12
khv – 1 – 22/12/2000
Praktikum der Ionenchromatographie Eine Einführung
Claudia Eith Prof. Dr. Maximilian Kolb Prof. Dr. Andreas Seubert Dr. Kai Henning Viehweger (Hrsg.)
Metrohm Monographie 8.792.2001 d
8.792.2003 e

Praktikum der Ionenchromatographie – Version 12
khv – 2 – 22/12/2000
Inhaltsverzeichnis 1 Die Autoren ...................................................................................................................................... 4 2 Einführung ........................................................................................................................................ 5 3 Theoretischer Teil............................................................................................................................. 6
3.1 Historie und Bedeutung der Ionenchromatographie ................................................................. 6 3.2 Grundlagen der Chromatographie ............................................................................................ 7
3.2.1 Einteilung und Begriffe der Chromatographie ................................................................... 7 3.2.2 Theoretische Konzepte zur Beschreibung der Chromatographie ................................... 10
3.3 Grundlagen der Ionenchromatographie (IC)........................................................................... 14 3.3.1 Terminologie und Einordnung in die LC .......................................................................... 14 3.3.2 Der Ionenaustausch......................................................................................................... 15 3.3.3 Die Ionenpaarbildung....................................................................................................... 16 3.3.4 Der Ionenausschluss ....................................................................................................... 17
3.4 Retentionsmodelle in der Ionenchromatographie ................................................................... 18 3.4.1 Retentionsmodelle in der Anionenchromatographie........................................................ 18 3.4.2 Retentionsmodelle in der Kationenchromatographie....................................................... 23
3.5 Detektionssysteme in der Ionenchromatographie .................................................................. 26 3.5.1 Elektrochemische Detektoren.......................................................................................... 27 3.5.2 Spektroskopische Detektionen ........................................................................................ 31
3.6 Stationäre Phasen in der Ionenchromatographie ................................................................... 32 3.6.1 Übersicht gebräuchlicher stationärer Phasen.................................................................. 32 3.6.2 Stationäre Phasen für die Anionenchromatographie....................................................... 33 3.6.3 Stationäre Phasen in der Kationenchromatographie....................................................... 34 3.6.4 Kationenaustauscher auf Silicagelbasis .......................................................................... 34 3.6.5 Kationenaustauscher auf Basis von organischen Polymeren ......................................... 35 3.6.6 Pellikulare Kationenaustauscher ..................................................................................... 35 3.6.7 Stationäre Phasen in der Ionenausschlusschromatographie .......................................... 35 3.6.8 Die Bedeutung der Kapazität von Ionenaustauschern .................................................... 35
3.7 Eluenten in der Ionenchromatographie................................................................................... 36 3.7.1 Anionenchromatographie................................................................................................. 37
3.8 Kationenchromatographie....................................................................................................... 39 3.8.1 Kationenchromatographie von Alkali-, Erdalkali- und Ammonium-Ionen mit Leitfähigkeitsdetektion.................................................................................................................... 39 3.8.2 Kationenchromatographie von Übergangs- und Erdalkalimetall-Ionen mit Nachsäulenderivatisierung und photometrischer Detektion........................................................... 40 3.8.3 Ionenausschlusschromatographie................................................................................... 41
4 Praktischer Teil............................................................................................................................... 42 4.1 Hinweise zum praktischen Arbeiten........................................................................................ 42 4.2 Versuche zur Theorie der Ionenchromatographie .................................................................. 45
4.2.1 Versuch 1 – Ionenchromatographie mit und ohne chemische Suppression ................... 45 4.2.2 Versuch 2 – Kapazität von Trennsäulen.......................................................................... 49 4.2.3 Versuch 3 – Selektivität von Trennsäulen ....................................................................... 52 4.2.4 Versuch 4 – Kalibration, Nachweis- und Bestimmungsgrenzen in der Ionenchromatographie ................................................................................................................... 57 4.2.5 Versuch 4a – Bestimmung von Anionen mit chemischer Suppression........................... 58 4.2.6 Versuch 5 – Veränderung der Selektivität mit Hilfe von Kronenethern (18 Crown-6) ..... 60 4.2.7 Versuch 6 – Veränderung der Selektivität mit Hilfe von Komplexbildnern ...................... 63 4.2.8 Versuch 7 – Anreicherungstechnik .................................................................................. 68
4.3 Versuche für die Bestimmung von Anionen............................................................................ 71 4.3.1 Versuch 8 – Bestimmung von Anionen in Trinkwasser ................................................... 71 4.3.2 Versuch 9 – Anionen in Ethanol ...................................................................................... 74 4.3.3 Versuch 10 – Anionen in Kopfsalat.................................................................................. 80 4.3.4 Versuch 11 – Phosphorsäure in Cola.............................................................................. 83 4.3.5 Versuch 12 – Anionen im Wein ....................................................................................... 88 4.3.6 Versuch 13 – Bestimmung von Verunreinigungen in Borat – Chlorid und Sulfat in Borax 93 4.3.7 Versuch 14 – Bestimmung von Anionen in Abwasser..................................................... 96 4.3.8 Versuch 15 – Fluorid in Zahnpaste................................................................................ 101 4.3.9 Versuch 16 – Anionen in braunem und weissem Zucker .............................................. 105 4.3.10 Versuch 17 – Verunreinigungen im Wasserstoffperoxid ............................................... 110
4.4 Versuche für die Bestimmung von Kationen......................................................................... 115

Praktikum der Ionenchromatographie – Version 12
khv – 3 – 22/12/2000
4.4.1 Versuch 18 – Alkli- und Erdalkalimetalle in Trinkwasser und Mineralwasser ............... 115 4.4.2 Versuch 19 – Bestimmung der Übergangsmetalle ........................................................ 119 4.4.3 Versuch 20 – Verunreinignungen im Silikagel – Bestimmung von Calcium- und Magnesiumionen .......................................................................................................................... 125 4.4.4 Versuch 21 – Kosmetik und Korrosionsschutz: Bestimmung von Ethanolamin und Alkalimetallen ............................................................................................................................... 128 4.4.5 Versuch 22 – Alkali- und Erdalkalimetalle im Wein ....................................................... 132
5 Literatur ........................................................................................................................................ 136 5.1 Literatur zum Praktikumsbuch .............................................................................................. 136

Praktikum der Ionenchromatographie – Version 12
khv – 4 – 22/12/2000
1 Die Autoren
Claudia Eith
Chemiestudium an der Fachhochschule Aalen, Praxissemester im Bereich der Trinkwasser und Abwasseranalytik in Adelaide (Australien), seit 2000 im Bereich Forschung & Entwicklung der Metrohm AG.
Maximilian Kolb
Chemiestudium an der Technischen Universität München, Promotion auf dem Gebiet der homogenen Katalyse, danach für 5 Jahre Leiter des Sachgebiets Gewässergüte am Wasserwirtschaftsamt Traunstein. Seit 1982 Professor an der Fachhochschule Aalen; Arbeitsgebiete: Umwelttechnik, Umweltanalytik und Chemometrie.
Andreas Seubert
Chemiestudium an der Universität Hannover, Promotion 1990: «Ultraspurenanalytik in hochreinen Refraktärmetallen mit ionenchromatographischer Spuren-Matrix-Trennung». Habilitation 1995: «Einsatzgebiete der On-line Kopplung HPLC-Atomspektrometrie in der Elementanalytik», 1998 bis 2000 Vertretungsprofessur für Analytische Chemie an der Universität Gh Kassel, seit März 2000 Professur für Analytische Chemie an der Philipps-Universität Marburg.
Kai Henning Viehweger
Studium der Chemie an der Universität Hamburg, Diplomarbeit in organischer Analytik. Promotion im Bereich der marinen und ästuarinen Ökosystemforschung, seit 1996 im Bereich Marketing der Metrohm AG – International Sales Manager Ionenchromatographie.

Praktikum der Ionenchromatographie – Version 12
khv – 5 – 22/12/2000
2 Einführung Es ist immer eine Herausforderung, Einblick in Dinge zu nehmen, die sich nicht direkt offenbaren. Die Gründe hierfür reichen von einfacher Neugier bis zur schlichten Notwendigkeit zu überleben. Es gibt viele Möglichkeiten hinter die Kulissen zu schauen. Die einfachste ist, sich der menschlichen Sinne zu bedienen: hören, fühlen, riechen, schmecken und sehen. Die Alchemie hat sich dieser menschlichen Sinne früher gerne bedient. Deshalb schmecken Säuren heute sauer und Brom hat seinen Namen von «bromos», griechisch für stinken. Chrom zeigt sich dem Auge farbig, da «chroma» sprachgeschicht-lich gleich Farbe ist. Gefühlsbetont wird die Alchemie mit Schimpfwörtern wie «Du Nickel» und Kobold bzw. Kobalt. Beide haben die Eisenherstellung unsere Vorfahren empfindlich gestört.
Viele einzelne Dinge zeigen sich nicht direkt. Sie sind gut gemischt oder die menschliche Sensorik reicht nicht aus, sie zu identifizieren. Dies ist der Moment an dem die Analytik ins Spiel kommt. Sie kann aus einer undefinierten Gemengelage heraus präzise Informationen extrahieren, die mit den menschlichen Sinnen zu verstehen sind.
Obwohl der Organismus voll davon ist, zeigen sie sich den menschlichen Sinnen nicht direkt. Gemeint sind Ionen, jene geladenen Atome oder Moleküle, die integraler Bestandteil fast aller lebender und toter Materie sind. Ionen sind dafür verantwortlich, dass die Nerven Informationen weiterleiten, dass die Verdauung funktioniert, dass der Blutdruck stimmt und genug Sauerstoff ins Blut gelangt. Ionen bringen das Salz ins Meer, sie regeln den Durst und ionische Bestandteile dienen als Nahrung für alle Lebewesen – von der Bakterie bis zum Menschen.
Das Wissen um Art und Menge von Ionen, die sich in Umwelt befinden, hilft biochemische und ökologische Zusammenhänge zu verstehen. Die Kenntnis der Ionenkonzentrationen in Lebensmitteln gibt Hinweise darauf, ob diese gesund oder vielleicht schädlich sind.
Es gibt viele Möglichkeiten, Ionen qualitativ – nach ihrer Art – und quantitativ – nach ihrer Menge – zu bestimmen. Beide Informationen sind wichtig. Ein Verfahren, um an diese Information zu gelangen ist die Ionenchromatographie. Chromatographie heisst eigentlich «Schreiben mit Farbe». In der traditionellen Analytik bedeutet dies, Substanzen nach ihrer Farbe zu trennen und nachher mit dem Auge zu bestimmen. Obwohl sich nicht alle Ionen durch sichtbare Farbe auszeichnen, ist der Begriff geblieben, nur werden heute andere Verfahren zur Bestimmung eingesetzt.
Die Ionenchromatographie ist ein Mitglied in der grossen Familie dieser chromatographischen Verfahren. Mit ihr lassen sich – sehr vereinfacht dargestellt – alle Ionen bestimmen, die ein bzw. zwei Ladungen tragen. War die Ionenchromatographie oder «IC» in der Vergangenheit ein sehr teures Verfahren, so ist sie heute wesentlich preiswerter geworden. Sie entwickelt sich deshalb zu einem universellen und leistungsfähigen analytischen Werkzeug, das leicht zu verwenden ist.
Das «Praktikum der Ionenchromatographie» zeigt, dass die IC keine abgehobene analytische Spielart ist, sondern auf ganz alltägliche Fragen schnell Antworten geben kann: Ist das Trinkwasser für die Säuglingsernährung geeignet? Wie viel Nitrat ist im Spinat? Warum verkalkt die Waschmaschine? Belastet das Abwasser die Umwelt? Da eine genaue analytische Praxis ohne theoretischen Hintergrund nur schwerlich möglich ist, enthält die vorliegende Monographie auch hierzu detaillierte Informationen in einem eigenen theoretischen Teil.
Mit dem «Praktikum der Ionenchromatographie» ist es möglich, nicht nur die Grundzüge der IC kennen zu lernen. Es dient auch dazu, einen generellen Einblick in die generellen Prinzipien der Chromatographie zu geben. Und die Chromatgraphie leistet vieles: sie stillt naturwissenschaftliche Neugier und sie sichert das gesunde Überleben in einer belasteten Umwelt.

Praktikum der Ionenchromatographie – Version 12
khv – 6 – 22/12/2000
3 Theoretischer Teil
3.1 Historie und Bedeutung der Ionenchromatographie Die Anfänge der Ionenchromatographie (IC) oder genauer gesagt der Ionenaustauschchromatographie reichen bis in die Mitte des vorigen Jahrhunderts zurück. In den Jahren von 1935 bis 1950 wurde das Wissen um Ionenaustauscher und deren Anwendung durch das «Manhattan-Project» erheblich erweitert. Die 50er und 60er Jahre dienten der Erarbeitung von theoretischen Modellen zum Verständnis des Ionenaustauschs und der darauf basierenden Ionenchromatographie. In den 70er Jahren wurden kontinuierliche Detektoren eingesetzt und damit der Schritt von der Niederdruck- zur Hochleistungschromatographie vollzogen.
Tabelle 1 Geschichte des Ionenaustausches und der darauf basierenden Ionenchromatographie
um 1850 Ackerböden als Ionenaustauscher für Mg2+, Ca2+ und NH4
+ Thomson u. Way
1935 Sulfonierter u. aminierter Kondensations-polymere (Phenol/Formaldehyd)
Adams, Holmes
1942 Sulfonierte PS/DVB-Harze als Kationenaustauscher (Manhattan-Projekt)
d’Alelio
1947 Aminierte PS/DVB-Harze als Anionenaustauscher
McBurney
1953 Ionenausschlusschromatographie Wheaton, Baumann
1957 Makroporöse Ionenaustauscher Corte, Meyer, Kunin u.a.
1959 Grundlagen für das theoretische Verständnis Helfferich
1967-70 Pellikulare Ionenaustauscher Horvath, Kirkland
1975 Ionenaustauschchromatographie mit Leitfähigkeitsdetektion mittels »Stripper»
Small, Stevens, Baumann
1979 Leitfähigkeitsdetektion ohne »Stripper» Gjerde, Fritz, Schmuckler
1976-80 Ionenpaarchromatographie Waters, Bidlingmeier, Horvath u.a.
Der Begriff der Ionenchromatographie wurde im Jahre 1975 mit der Einführung der Leitfähigkeitsdetektion kombiniert mit einer chemischen Leitfähigkeitsreduzierung durch Small, Stevens und Bauman entwickelt und danach lange Zeit als Handelsname für Vermarktungszwecke eingesetzt. Mittlerweile hat sich der abkürzende Begriff Ionenchromatographie als Oberbegriff für die Methoden Ionenaustausch- , Ionenausschluss- und Ionenpaar-Chromatographie innerhalb der Hochleistungsflüssigchromatographie (HPLC) etabliert [1]. Gerade im Bereich der Anionenbestimmung nimmt die IC heute die dominierende Stellung ein, während die zur Bestimmung von bevorzugt kationisch vorliegenden Elementen gebräuchlichen atomspektrometrischen Verfahren bei den elektronegativen Anionenbildnern der fünften bis siebten Hauptgruppe des Periodensystems nur eine eingeschränkte Leistungsfähigkeit besitzen.
Das wichtigste Einsatzgebiet der Anionenchromatographie stellt heute die routinemäßige Untersuchung wässriger Systeme dar, in der die Trinkwasseranalytik eine zentrale Bedeutung besitzt [2,3,4]. Daneben wird die IC für die Elementspeziesanalyse von anionisch auftretenden Elementen oder Komplexen eingesetzt, wobei überwiegend umweltrelevante Fragestellungen bearbeitet werden. Das dritte grosse Anwendungsgebiet der Anionenchromatographie ist die Ultraspurenanalytik in hochreinen Prozesschemikalien, wie sie vor allem in der Halbleiterindustrie benötigt werden.
Üblicherweise bestehen Anionenaustauscher für die HPLC heute aus sphärischen Polymerpartikeln mit einem Durchmesser von etwa 5 bis 15 µm. Auf die Polymeroberfläche werden mit Hilfe unterschiedlicher Verfahren sogenannte Ankergruppen aufgebracht, die als Abstandhalter (Spacer)
HPLC
LC

Praktikum der Ionenchromatographie – Version 12
khv – 7 – 22/12/2000
zwischen dem Basispolymer und den eigentlichen funktionellen Gruppen dienen. Diese bestehen im Regelfall aus quartären Ammonium-Ionen, welche an den Ankergruppen chemisch fixiert sind. Die Gesamtzahl der funktionellen Gruppen wird als Austauschkapazität bezeichnet und stellt allgemein ein zentrales Charakteristikum von Ionenaustauschern dar.
Kommerzielle Packungsmaterialien für die Anionenchromatographie sind mit Austauschkapazitäten zwischen 50 und 100 µMol pro Trennsäule von niederkapazitiver Natur. Dies ist in der dominierenden Anwendung der für Ionen universell einsetzbaren Leitfähigkeitsdetektion (LF) begründet, weil eine empfindliche Detektion der Analyten eine möglichst geringe Eigenleitfähigkeit der verwendeten Elutionssysteme erfordert. Bei niederkapazitiven Anionenaustauschern genügen dafür in der Regel sehr verdünnte wäßrige Lösungen von NaOH oder Carbonat-Puffern, deren Eigenleitfähigkeit zudem noch chemisch unterdrückt (suppressiert) werden kann [2,4].
Heute werden in der Anionenchromatographie grösstenteils funktionelle Gruppen vom Typ I (Trimethylammonium, TMA) und Typ II (Dimethylethanolammonium, DMEA) verwendet. Da die eigentliche Wechselwirkung zwischen der stationären Phase und den Analyt-Anionen an den funktionellen Gruppen stattfindet, hat deren Struktur einen entscheidenden Einfluß auf das Selektivitätsverhalten der Packungsmaterialien. Nach den bisherigen Erkenntnissen ist dabei insbesondere die Polarität der funktionellen Gruppen, die durch die Zahl der Hydroxyethylreste (-CH2CH2OH) am quartären Stickstoff gesteuert werden kann, von Bedeutung [2,4].
Der Begriff «Ionenchromatographie» umfasst alle Trennungen ionischer Spezies innerhalb der HPLC mit online-Detektion und ist somit von apparativen Limitierungen weitgehend befreit [5]. Die IC hat sich wegen der mittlerweile vielfältigen Auswahl an Trennsäulen, Elutionssystemen und Detektoren vor allem in der Anionen-Analytik zur Methode der Wahl entwickelt. Grund hierfür ist, dass für Anionen nur wenige Trennungsgänge existieren, die in der Praxis kaum sinnvoll zu verwenden sind. Gravimetrische und volumetrische Verfahren sind durch ihre Empfindlichkeit und ihre Selektivität limitiert. Auch die stürmische Entwicklung der Gaschromatographie ab 1965 brachte für Anionen keine grossen Vorteile, da die nichtflüchtigen Ionen zunächst derivatisiert werden müssen und die Empfindlichkeit nicht den heutigen Anforderungen an die Spurenanalytik gerecht wird [6]. Für die Kationenanalytik existieren leistungsfähige atomspektrometrische Alternativen zur IC, z.B. die ICP-AES/MS, so dass der Stellenwert der Kationenchromatographie verglichen zur Anionenchromatographie wesentlich geringer ist. Im Bereich der Analytik der Alkali- und Erdalkalimetalle sowie bei der Bestimmung von Ammonium-Stickstoff (Trinkwasseranalytik) hat die Kationenchromatographie aber eine gewisse Bedeutung. Bei der Speziierung von ionischen Verbindungen ist die IC in Verbindung mit elementspezifischen Detektoren unverzichtbar. Einen guten Überblick über die Anwendungen der IC in den verschiedensten Bereichen der Analytik geben die Werke von Haddad et al. und Weiss [2,4].
3.2 Grundlagen der Chromatographie
3.2.1 Einteilung und Begriffe der Chromatographie Die Chromatographie ist ein physikalisch-chemisches Verfahren zur Trennung von Substanzgemischen. Der Trenneffekt beruht auf einer wiederholten Verteilung zwischen zwei Phasen, von denen eine Phase als stationär (ruhend) betrachtet wird, während die zweite, mobile Phase sich in einer bestimmten Richtung bewegt [7,8]. Nach dem Aggregatzustand der beiden beteiligten Phasen werden chromatographische Techniken eingeteilt in:
flüssig gasförmig
fest LSC GSC
flüssig LLC GLC
mobile Phase
stat
ionä
re P
hase
Abbildung 1 Einteilung chromatographischer Methoden nach Aufbau von stationärer und mobiler Phase
Praktikum der Ionenchromatographie – Version 12
khv – 8 – 22/12/2000
Eine weitere Unterscheidung chromatographischer Verfahren kann nach den grundlegenden Vorgängen während des Trennvorganges, wie etwa Adsorption oder Verteilung, bzw. nach der Art der Ausführungstechnik (Säulen- oder Planarchromatographie) erfolgen [9].
Retentionsparameter Betrachtet man ein Stoffgemisch und unterwirft dieses einer chromatographischen Trennung, so wird sich für jede Komponente ein Verteilungsgleichgewicht zwischen mobiler und stationärer Phase ausbilden. Eine Stofftrennung ist nur dann erfolgreich, wenn sich die Verteilungskoeffizienten D der Komponenten hinreichend voneinander unterscheiden. D ist definiert als das Verhältnis der Konzentrationen eines Stoffes A in der stationären (Index S) und der mobilen Phase (Index M):
M
S A [A]
[A]D = (1)
Dementsprechend werden Stoffe mit einem hohen Verteilungskoeffizienten D stärker zurückgehalten (retardiert) als solche mit einem kleinen D. Der Vorgang der chromatographischen Trennung wird in Form eines Chromatogrammes dargestellt, welches die Aufzeichnung eines Detektorsignals als Funktion des Elutionsvolumens der mobilen Phase oder der Zeit darstellt. Somit entspricht es einem Konzentrations- oder Massenprofil als Funktion der Zeit. Das Detektorsignal soll dabei proportional zur Konzentration eines Analyten am Ende der Trennstrecke sein [8]. Die Verweil- oder Bruttoretentionszeit tR eines Stoffes auf der stationären Phase setzt sich, wie in Gleichung 2 dargestellt, additiv aus der Nettoretentionszeit tS, welche dem realen Aufenthalt auf der Trennstrecke entspricht, und der Durchflusszeit der mobilen Phase ohne Wechselwirkung, der Totzeit tM, zusammen.
MSR ttt += (2)
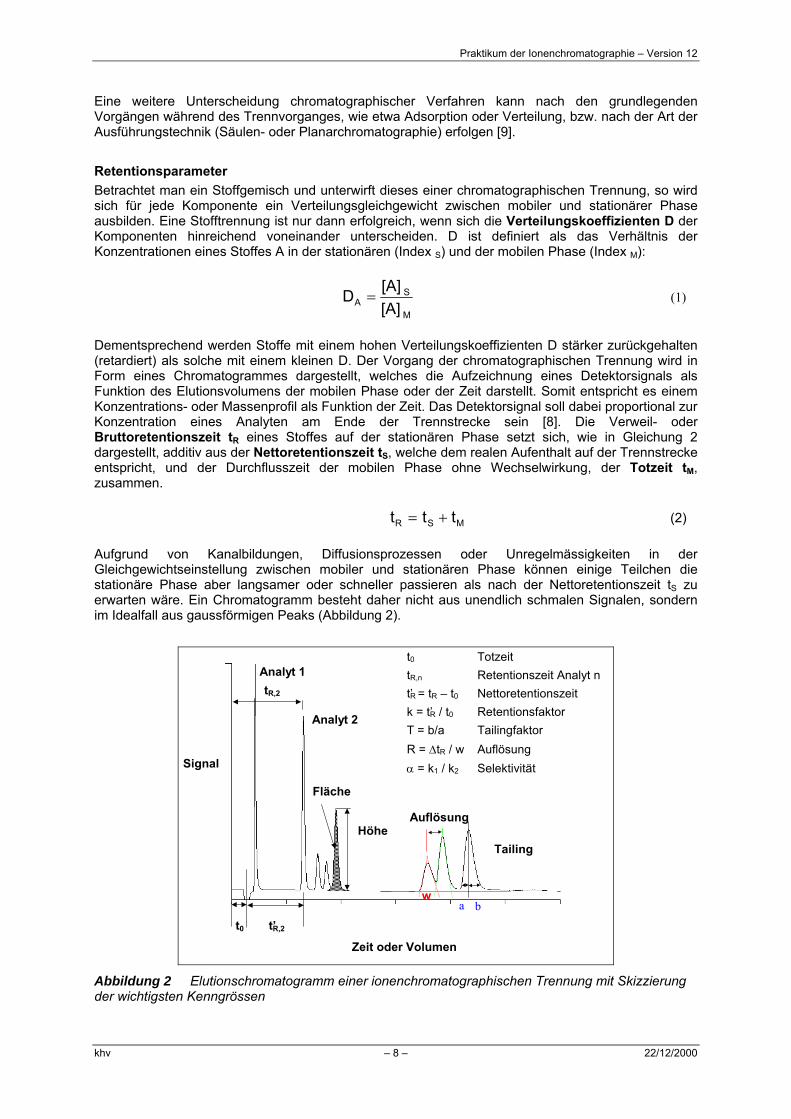
Aufgrund von Kanalbildungen, Diffusionsprozessen oder Unregelmässigkeiten in der Gleichgewichtseinstellung zwischen mobiler und stationären Phase können einige Teilchen die stationäre Phase aber langsamer oder schneller passieren als nach der Nettoretentionszeit tS zu erwarten wäre. Ein Chromatogramm besteht daher nicht aus unendlich schmalen Signalen, sondern im Idealfall aus gaussförmigen Peaks (Abbildung 2).
Signal
Zeit oder Volumen
Höhe
t0
Analyt 1
Analyt 2
tR,2
t’R,2
wa b
Tailing
Auflösung
Fläche
t0 Totzeit
tR,n Retentionszeit Analyt n
t’R = tR – t0 Nettoretentionszeit
k = t’R / t0 Retentionsfaktor
T = b/a Tailingfaktor
R = ∆tR / w Auflösung
α = k1 / k2 Selektivität
Abbildung 2 Elutionschromatogramm einer ionenchromatographischen Trennung mit Skizzierung der wichtigsten Kenngrössen

Praktikum der Ionenchromatographie – Version 12
khv – 9 – 22/12/2000
Durch Diffusionsprozesse, die mit zunehmender Verweilzeit auf der stationären Phase an Einfluss gewinnen, beobachtet man eine mit der Retentionszeit einer Substanz zunehmende Bandenverbreiterung. Dieses Phänomen ist für alle chromatographischen Verfahren charakteristisch.
Wie bereits erwähnt, stellt ein Peak in einem Chromatogramm idealerweise eine Gaussverteilung dar. Abbildung 3 zeigt schematisch eine Gaussverteilung.
Abbildung 3 Gaussverteilung mit den wichtigsten Kenngrössen
Die Breite bei halber Höhe wird als Halbwertsbreite b0,5 bezeichnet und entspricht der 2,354-fachen Varianz σ der Verteilung. Die Basisbreite w ist definiert durch die Differenz der Schnittpunkte der Wendetangenten mit der y-Achse, gleichbedeutend der 4-fachen Varianz der Gauss-Funktion. Beide Grössen sind ein Mass für die Leistungsfähigkeit einer chromatographischen Trennsäule und können bei idealer Peakform zur Berechnung der Bodenzahlen herangezogen werden.
Abweichungen von der idealen Peakform lassen sich durch einen sogenannten Asymmetriefaktor T beschreiben. Er ist definiert als das Verhältnis der Strecken A und B zwischen der Mittelsenkrechten und den Flanken der Verteilung bei 10 % ihrer Höhe (Abbildungen 2 und 3) und berechnet sich zu:
ABT = . (3)
Für Gauss-Peaks ist T = 1. Abweichungen zu grösseren T-Werten werden als Tailing, zu kleineren als Fronting bezeichnet. In der Praxis strebt man Symmetriefaktoren von T = 0,9 bis 1,1 an.
Retentionsfaktor, Selektivität und Auflösung Da die Bruttoretentionszeit tR massgeblich von den chromatographischen Bedingungen abhängt, ist sie nur unter definierten Bedingungen für eine Substanz charakteristisch und damit zur qualitativen Identifizierung geeignet. Man führt daher eine dimensionslose Grösse ein, den Retentionsfaktor k‘, welcher den Vergleich verschiedener chromatographischer Systeme erlaubt. Er gibt an, wieviel länger sich eine Substanz auf der Trennstrecke aufhält als in der mobilen Phase [8]. Mathematisch ist der Retentionsfaktor definiert als Produkt des Verteilungskoeffizienten D und dem Phasenvolumenverhältnis von mobiler und stationärer Phase bzw. als das Verhältnis von Nettoretentionszeit zur Totzeit. Auch möglich ist eine Berechnung über die Länge der Trennstrecke L und der Geschwindigkeit der mobilen Phase u (Gleichung 4).
1 - Lt u
tt =
VVD = k' R
M
S
M
S ⋅=⋅ (4)

Praktikum der Ionenchromatographie – Version 12
khv – 10 – 22/12/2000
Bei kleinen Werten von k‘ eluiert eine Substanz nahe an der Totzeit bzw. am Totvolumen des chromatographischen Systems, was gleichbedeutend mit einer schlechten Trennung ist. Ist k‘ sehr gross, bedeutet dies zwar eine gute Trennung, gleichzeitig aber auch eine hohe Verweilzeit auf der Trennstrecke und damit eine Peakverbreiterung. Im Realfall sollte der Retentionsfaktor zwischen 2 und 5 liegen.
Zwei Stoffe werden nur dann ausreichend getrennt, wenn sich ihre Retentionsfaktoren hinreichend voneinander unterscheiden. Der Selektivitätskoeffizient α, auch relativer Trennfaktor genannt, gilt als Mass für die Trennbarkeit zweier Substanzen und ist wie folgt definiert:
121
2
S1
S2
MR1
MR2 k' k' mit k'k'
= t
t =
t - t
t - t = >α (5)
Lassen sich zwei Substanzen nicht auftrennen, ist α = 1 und es kommt zur Koelution. Je grösser α ist, desto besser die Trennung. Allerdings vergrössert sich mit zunehmendem α auch der Zeitaufwand für die Trennung, so dass man in der Praxis Selektivitätskoeffizienten von α = 1,5 anstrebt [10].
Der Selektivitätskoeffizient beschreibt nicht die Güte eines Trennvorganges. Die Auflösung R (Resolution) berücksichtigt nicht nur die relativen Lagen der Peaks zueinander, sondern auch ihre Halbwertsbreiten (b0,5) bzw. die Basisbreiten (w), wie aus Gleichung 6 hervorgeht.
(0,5)21 (0,5)
R1R2
21
R
2) w- (wR1R2
b - bt-t
1,198 = wwt 2
= t - t
= R21
⋅−∆⋅
(6)
Ist die Differenz der Retentionszeiten zweier Peaks gross im Verhältnis zu ihren Basis- oder Halbwertsbreiten, erhält man eine hohe Auflösung. Unter Voraussetzung einer idealen Peaksymmetrie können zwei Stoffe bei R = 0,5 noch identifiziert werden. Für qualitative Trennungen sollte R = 1 betragen (4σ-Trennung), für eine Quantifizierung strebt man eine Auflösung von R = 1,2 bis 1,5 an [25]. Auflösungen von R ≥ 2 (8σ-Trennung) sind aufgrund der damit verbundenen langen Analysezeiten nicht erwünscht.
3.2.2 Theoretische Konzepte zur Beschreibung der Chromatographie
Das Modell der theoretische Trennstufen Das dem Destillationsprozess entstammende Modell der theoretischen Trennstufen dient der Beschreibung chromatographischer Trennungen [11]. Es unterteilt die stationäre Phase in einzelne Abschnitte, den theoretischen Trennstufen oder Böden, auf denen prinzipiell genau einmal eine völlig reversible und unendlich schnelle Gleichgewichtseinstellung zwischen mobiler und stationärer Phase erfolgt. Die Leistungsfähigkeit (Effizienz) eines chromatographischen Systems wird daher durch eine möglichst hohe Anzahl theoretischer Trennstufen charakterisiert.
Die theoretische Trennstufenzahl N kann unter Verwendung der Varianz sowie der Basis- und Halbwertsbreiten direkt aus einem Chromatogramm ermittelt werden und berechnet sich wie folgt [12]:
2R
2
0,5
R2
R t =
bt
(2) ln 8 = wt
16 = N ⎟⎠
⎞⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⋅⋅⎟
⎠
⎞⎜⎝
⎛σ
(7)
Anstelle der Trennstufenzahl kann auch die Trennstufenhöhe HETP (Height Equivalent to a Theoretical Plate) zur Beschreibung der Trennleistung herangezogen werden.
2
R2
R
0,52
wt
16L =
tb
(2) ln8
L = L
= NL HETP ⎟
⎠
⎞⎜⎝
⎛⋅⎟⎟
⎠
⎞⎜⎜⎝
⎛⋅
⋅=
σ (8)
Aus den Gleichungen 5 ... 8 lässt sich ableiten, dass eine stationäre Phase mit einer sehr grossen Zahl an theoretischen Trennstufen auch dann noch Substanzen voneinander separieren kann, wenn deren Selektivitätskoeffizienten oder Auflösung sich kaum voneinander unterscheiden. Die

Praktikum der Ionenchromatographie – Version 12
khv – 11 – 22/12/2000
Gleichungen erlauben ebenso die Berechnung der Zahl an theoretischen Böden, die zur Bewältigung eines Trennproblems zwingend notwendig ist.
Das Modell der theoretischen Trennstufen kann zur Erklärung des Auftretens gaussförmiger Signale in der Chromatographie herangezogen werden, wenn man zugrunde legt, dass es aufgrund von Strömungs- und Diffusionsprozessen nur zu einer endlich schnellen und unvollständigen Gleichgewichtseinstellung zwischen mobiler und stationärer Phase kommt. Daraus resultiert eine Bandenverbreiterung, da eine am Anfang der Trennstrecke schmale Substanzzone mit zunehmender Verweilzeit auf der stationären Phase deutlich auseinandergezogen wird.
Die Berechnung der theoretischen Trennstufenzahl gemäss Gleichung 7 setzt eine ideale Peakform voraus, welche aber im Realfall nur sehr selten gegeben ist. Bei asymmetrischen Peakformen muss die Berechnung über die Momentenmethode erfolgen [13]. Gleichung 9 ergibt unter Einbeziehung des Symmetriefaktors näherungsweise sinnvolle Werte.
1,25+T
bt
41,7 = N
2
0,5
R ⎥⎦
⎤⎢⎣
⎡
⋅ (9)
Eine effektive Trennstufenzahl n, welche der realen Trennleistung näher kommt als die theoretische Trennstufenzahl N, ist um den Retentionsfaktor k‘ korrigiert und berechnet sich zu:
2
k'+1k' N = n ⎟
⎠⎞
⎜⎝⎛⋅ (10)
Die dynamische Theorie (Van-Deemter-Theorie) Die entscheidende Schwäche des Modells der theoretischen Trennstufen besteht insbesondere darin, dass der Destillation und der Chromatographie zwei grundlegend verschiedene physikalisch-chemische Prozesse zugrunde liegen. Ausserdem werden keine Annahmen über den Einfluss wichtiger experimentell zugänglicher Parameter gemacht, die nicht die Art oder Qualität der stationären Phase selbst betreffen [14,15]. Dies können sein:
• Flussrate der mobilen Phase • Partikeldurchmesser in der stationären Phase • Schichtdicke von Oberflächenfilmen auf dem Packungsmaterial
Daneben haben auch Grössen wie die Diffusionskoeffizienten in der mobilen und stationären Phase, die Temperatur oder das Detektorvolumen in der Flüssigchromatographie eine grosse Bedeutung für die Trennleistung.
Die von van Deemter entwickelte dynamische Theorie stellt im Prinzip eine Erweiterung des theoretischen Trennstufenmodells unter Einbeziehung von nichtidealen Randbedingungen dar [16]. Folgende Annahmen werden gemacht:
• Keine spontane und ungehemmte Einstellung von Gleichgewichten • Verzögerter Massentransport in der stationären und mobilen Phase • Keine über den Säulenquerschnitt homogene Geschwindigkeit der mobilen Phase • Auftreten von Streudiffusion und Ausbildung von Kanälen in der stationären Phase • Longitudinaldiffusion unabhängig von der Geschwindigkeit der mobilen Phase und direkt
proportional zur Verweilzeit auf der Trennstrecke
Der Zusammenhang zwischen den genannten dynamischen Effekten und der theoretischen Trennstufenhöhe wird durch die Van-Deemter-Gleichung hergestellt.
u C uB A HETP ⋅++= (11)
Die drei Terme A, B und C hängen dabei in unterschiedlicher Weise von der Strömungsgeschwindigkeit u der mobilen Phase ab. Die Terme A und B beschreiben den gesamten Massentransport durch die stationäre Phase, während der Term C durch Störungen bei der Gleichgewichtseinstellung zwischen mobiler und stationärer Phase bestimmt wird.

Praktikum der Ionenchromatographie – Version 12
khv – 12 – 22/12/2000
Durch den Term A wird die Streudiffusion (Eddy-Diffusion) beschrieben, welche aufgrund eines Mehrwege-Effektes als eine Ursache für die Bandenverbreiterung anzusehen ist. Dieser auch Packungsfaktor genannte Term ist von der linearen Geschwindigkeit u der mobilen Phase zumindest in erster Näherung unabhängig. Für den Term A gilt folgende Beziehung:
pd 2 A ⋅⋅= λ (12)
In Gleichung (12) ist dp der mittlere Teilchendurchmesser in der stationären Phase, λ kennzeichnet die statistische Unregelmässigkeit der Packung, welche möglichst homogen sein und aus uniformen Teilchen bestehen sollte.
Der Term B beschreibt die Longitudinaldiffusion in oder entgegen der Strömungsrichtung der mobilen Phase. Er ist insbesondere bei Verwendung von Kapillarsäulen in der Gaschromatographie (GC) von Bedeutung, da die Diffusionskoeffizienten in Gasen um 4 bis 5 Dekaden grösser sind als in Flüssigkeiten. B berechnet sich als Produkt aus dem Diffusionskoeffizienten in der mobilen Phase DM und dem Labyrinthfaktor γ , welcher die Porosität der stationären Phase beschreibt.
MD 2 B ⋅⋅= γ (13)
Da die Bedeutung der Diffusion mit zunehmender Strömungsgeschwindigkeit der mobilen Phase abnimmt, ist B umgekehrt proportional zu u.
Der Term C wird als Massenübergangsterm bezeichnet. Der verzögerte Massentransfer zwischen mobiler und stationärer Phase hat zumeist den grössten Anteil an der Bandenverbreiterung. Die Störungen in der Gleichgewichtseinstellung zwischen mobiler und stationärer Phase werden mit zunehmendem u grösser, weshalb sich eine direkte Proportionalität zur linearen Fliessgeschwindigkeit ergibt. Die Verzögerungen im Massentransfer resultieren aus den im Vergleich zur mobilen Phase sehr kleinen Diffusionskoeffizienten DS in der stationären Phase, weshalb Teilchen, die sich gerade in den Poren der stationären Phase aufhalten, hinter dem Peakmaximum, welches sich mit der mobilen Phase weiterbewegt, zurückbleiben. Durch kurze Diffusionswege und schnelle Austauschvorgänge lässt sich der Term C deutlich verringern. Dies kann über eine Lokalisierung der Poren vor allem an der Oberfläche erfolgen, die so nur wenig in das Innere der stationären Phase hineinreichen. Der Massenübergangsterm C berechnet sich wie folgt:
( ) S
2
Ddp
k'+1k'16 = C ⋅
⋅⋅
π (14)
Die graphische Darstellung der Van-Deemter-Gleichung ergibt eine hyperbelartige Kurve, in deren Minimum sich die Fliessgeschwindigkeit u für die minimale Bodenhöhe (maximale Trennstufenzahl) bestimmen lässt (Abbildung 4).
Abbildung 4 Darstellung der Einzelterme aus der van-Deemter Theorie mit der resultierenden Van-Deemter-Kurve mit Optimum der Fliessgeschwindigkeit
Auch die dynamische Theorie geht letztendlich von idealisierten Voraussetzungen aus. Im Realfall sind die drei Terme A, B und C nur in erster Näherung unabhängig voneinander, wobei es zusätzlich einen Einfluss der Fliessgeschwindigkeit u auf die Streudiffusion (Term A) gibt. Der Term C lässt sich

Praktikum der Ionenchromatographie – Version 12
khv – 13 – 22/12/2000
in die Terme CM und CS differenzieren, die den Massentransfer in der mobilen Phase (CM) bzw. zur stationären Phase und zurück beschreiben (CS). Daher ist die ursprüngliche Van-Deemter-Gleichung in zahlreichen Anwendungsfällen für die HPLC, GC und die TLC modifiziert worden [17,18].
Moderne Flüssigchromatographie (LC) Die Flüssigkeitschromatographie LC (Liquid Chromatography) ist als Oberbegriff für zahlreiche moderne flüssigchromatographische Trennverfahren zu betrachten. Sie lässt sich auf die verschiedensten Substanzklassen anwenden und zeichnet sich durch eine exzellente analytische Leistungsfähigkeit aus. Die Ionenchromatographie (IC) ist ein Bestandteil der LC, welche die wohl zur Zeit wichtigste Trennmethode in der modernen Analytischen Chemie darstellt [3].
Die HPLC stellt eine konsequente Weiterentwicklung der klassischen Flüssigkeitschromatographie (Liquid Chromatography, LC) dar. In der klassischen LC, von Tswett im Jahre 1906 eingeführt, wurden Glasssäulen mit 1 bis 5 cm Durchmesser und einer Länge von bis zu 500 cm verwendet, die mit Trennphasen von 150 bis 200 µm Partikelgrösse gefüllt waren. Trennungen auch einfacher Stoffgemische dauerten bei mässiger Trennleistung oft mehrere Stunden. Aufgrund des in der Folgezeit entwickelten Verständnisses der Chromatographie (Gleichung 11) wurde deutlich, dass eine Leistungssteigerung nur durch eine drastische Verringerung des Partikeldurchmessers in der stationären Phase möglich war, was allerdings völlig neue Anforderungen an das chromatographische Equipment stellte.
Seit etwa 1970 steht eine spezielle und leistungsfähige Gerätetechnik zur Verfügung, die in der Lage ist, die hohen Gegendrücke von 10 bis 50 MPa zu bewältigen, welche bei Verwendung von Packungsmaterialien mit einem Durchmesser von 3 bis 10 µm und Trennsäulen von 125 bis 250 x 4 mm ID auftreten können.
Die HPLC hat sich aufgrund der drastischen Miniaturisierung zu einer rein analytischen Trennmethode entwickelt, wohingegen die klassische LC heute praktisch nur noch für präparative Zwecke angewendet wird. Die Vorteile der HPLC gegenüber der klassische LC sind dabei vor allem:
• Exzellente chromatographische Effizienz • Kontinuierliche Arbeitsweise • Online-Detektion der getrennten Substanzen • Hohe Empfindlichkeit und Reproduzierbarkeit • Nutzung der Retentionszeit zur qualitativen Identifizierung von Stoffen • Kurze Analysenzeiten
Ein HPLC-System besteht unabhängig vom Einsatzgebiet im wesentlichen aus den in der Abbildung 5 dargestellten Komponenten der Hochleistungspumpe mit Vorrat für die mobile Phase (Eluent), Injektor (Probenaufgabe), Trennsäule und Detektionssystem (inklusive Derivatisierung, Datenaufnahme und -verarbeitung):
Abbildung 5 Aufbau einer HPLC- bzw. IC-Anlage mit den wichtigsten Komponenten
Die Pumpe ist neben der Trennsäule das Kernstück eines jeden HPLC-Systems. Sie muss in der Lage sein, den Eluenten möglichst konstant und pulsationsfrei auch gegen hohe Staudrücke zu fördern. Diese machen auch die Verwendung eines speziellen Schleifen-Injektors zur Probenaufgabe notwendig. Üblich ist ein 6-Wege-Ventil, das in der Lage ist, die Probe unter Normaldruck in einer Schleife mit definiertem Volumen aufzunehmen und in das unter hohem Druck stehende HPLC-

Praktikum der Ionenchromatographie – Version 12
khv – 14 – 22/12/2000
System zu überführen. Die Zusammensetzung der mobilen Phase muss wie die Art der Trennsäule an die analytische Fragestellung angepasst werden. Dies trifft ebenso auf die Auswahl des Detektionssystems zu. Die Datenaufnahme und -verarbeitung erfolgt heute ausschliesslich computergesteuert. Je nach Problemstellung lässt sich dieser grundlegende Aufbau einer HPLC-Apparatur nahezu beliebig erweitern.
Trennprinzipien in der LC Die HPLC lässt sich anhand der verschiedenen physikalisch-chemischen Wechselwirkungen zwischen den Substanzen in einer Probe und der stationären Phase differenzieren. Obwohl im Realfall meist mehrere Mechanismen für eine erfolgreiche Trennung verantwortlich sind [9], ist eine grobe Klassifizierung nach den folgenden Trennmechanismen möglich:
• Adsorption • Verteilung • Grössenausschluss • Affinität • Ionenaustausch • Ionenpaarbildung • Ionenausschluss
Die Adsorptionschromatographie ist definiert durch Grenzflächenreaktionen, bei denen flüssige oder gasförmige Stoffe an einer festen Phase angereichert werden. Zur qualitativen und quantitativen Beschreibung von Adsorptionsprozessen existieren verschieden Modelle, wobei an dieser Stelle auf die einschlägige Literatur der Physikalischen Chemie verwiesen sei [19]. In der Anwendung lassen sich zwei Ausführungstechniken unterscheiden. Bei der Normalphasen-Chromatographie ist die stationäre Phase meist ein Silicagel und damit wesentlich polarer als das Laufmittel (Kohlenwasserstoffe). In der Umkehrphasenchromatographie (Reversed-Phase) sind die Verhältnisse genau entgegengesetzt. Aus praktischen Gründen, welche vor allem die Handhabung der Eluenten betreffen, wird heute fast nur noch die RPC eingesetzt [3,9].
Bei der Verteilungschromatographie ist die stationäre Phase eine mit der mobilen Phase nicht mischbare Flüssigkeit. Die Trennung beruht auf den unterschiedlichen Löslichkeiten der Analyten in beiden Phasen. Dabei gilt im Idealfall das Nernst’sche Verteilungsgesetz. Dieser Trennmechanismus spielt vor allem in der Gaschromatographie eine bedeutende Rolle, wenn mit Trennflüssigkeiten beschichtete Kapillaren als stationäre Phasen verwendet werden. Verteilungschromatographie kann auch in der HPLC vorliegen, wenn mit unpolaren Kohlenwasserstoffen modifizierte Silicagele, z.B. sogenannte Octadecyl-Phasen, als Trennmaterialien zum Einsatz kommen.
Die Grössenausschlusschromatographie, auch Size-Exclusion-Chromatography (SEC) genannt, ermöglicht eine Trennung nach der Molekülgrösse aufgrund von Siebeffekten. Als stationäre Phasen werden Silicagele oder organische Polymerharze mit definierter Porenstruktur verwendet. Kleinere Analyten können in die Poren diffundieren und werden retardiert. Mit zunehmender Molekülgrösse wird eine Wechselwirkung mit den Poren immer unwahrscheinlicher, bis ab einer bestimmten Grösse Moleküle ganz ausgeschlossen werden und praktisch im Totvolumen eluieren. Die SEC findet vor allem in der Polymer- und Bioanalytik breite Anwendung.
Die Affinitätschromatographie ermöglicht die Trennung von Stoffgemischen durch selektive oder spezifische Wechselwirkungskräfte. Vor allem bei Enzymen und ihren Substraten beobachtet man hochspezifische Wechselwirkungen, ebenso zwischen Antikörpern und Antigenen (Schlüssel-Schloss-Prinzip). In der Praxis werden Enzyme oder Antikörper auf einer stationären Phase chemisch immobilisiert. Befindet sich nun ein entsprechendes Substrat oder Antigen in der Probe, so wird dieses mit extremer Selektivität retardiert. Daher ist die Bioaffinitätschromatographie im Bereich der Wirkstoffanalytik (Pharmakologie) ein unverzichtbares Verfahren.
Die Ionenaustauschchromatographie (IC) wird ebenso wie die Ionenpaar- und Ionenausschlusschromatographie im folgenden Kapitel ausführlich behandelt.
3.3 Grundlagen der Ionenchromatographie (IC)
3.3.1 Terminologie und Einordnung in die LC Die Ionenaustausch- oder Ionenchromatographie (IC) stellt eine Untergruppe der HPLC dar. Laut IUPAC ist die Ionenaustauschchromatographie wie folgt definiert [7,8]:

Praktikum der Ionenchromatographie – Version 12
khv – 15 – 22/12/2000
»Bei der Ionenaustauschchromatographie beruht die Trennung auf Unterschieden in den Ionenaustauschaffinitäten der einzelnen Analyten. Wenn anorganische Ionen getrennt und mit Leitfähigkeitsdetektoren oder durch indirekte UV-Detektion nachgewiesen werden, bezeichnet man dies auch als Ionenchromatographie».
Diese Definition ist aus verschiedenen Gründen unglücklich gewählt. Die Detektionstechnik sollte unabhängig vom vorliegenden Trennmechanismus betrachtet werden. Ausserdem ist eine Beschränkung des Begriffes ‚Ionenchromatographie‘ auf anorganische Ionen nicht nachvollziehbar, da in der Praxis mit einem gegebenen System oft sowohl organische wie auch anorganische Ionen gleichzeitig getrennt und identifiziert werden können.
Besser geeignet zur Definition der Ionenchromatographie ist eine ältere, allgemeinere Definition [20]:
«Die Ionenchromatographie umfasst alle schnellen flüssigkeitschromatographischen Trennungen von Ionen in Säulen in Online Kopplung mit Detektion und Quantifizierung in einem Durchflussdetektor.»
Diese Definition charakterisiert die Ionenchromatographie unabhängig von Trennmechanismus und Detektion, grenzt sie aber gleichzeitig vom klassischen Ionenaustausch ab. Als Trennprinzipien wirken in der Ionenchromatographie:
• Ionenaustausch • Ionenpaarbildung • Ionenausschluss
Chromatographische Methoden werden durch den überwiegend vorliegenden Trennmechanismus definiert. Die Ionenaustauschchromatographie wird dabei heute vereinfacht Ionenchromatographie (IC) genannt, wobei die Ionenpaarchromatographie (IPC) und Ionenausschlusschromatographie (IEC, Ion Exclusion Chromatography) als speziellere Anwendungen gelten.
3.3.2 Der Ionenaustausch Die Ionenaustauschchromatographie (IC) beruht auf einer stöchiometrisch verlaufenden chemischen Reaktion zwischen Ionen in einer Lösung und einem üblicherweise festen Stoff, der funktionelle Gruppen trägt und Ionen aufgrund elektrostatischer Kräfte fixieren kann. In der Kationenchromatographie sind dies im einfachsten Fall Sulfonsäuregruppen, in der Anionenchromatographie quarternäre Ammoniumgruppen. Zwischen beiden Phasen können Ionen gleichsinniger Ladung theoretisch völlig reversibel ausgetauscht werden. Der Prozess des Ionenaustausches führt zu einem Gleichgewichtszustand. Auf welcher Seite dieses Gleichgewicht liegt, hängt von der Affinität der beteiligten Ionen zu den funktionellen Gruppen der stationären Phase ab. Die Abbildung 6 zeigt schematisch die Austauschvorgänge für Kationen und Anionen. Dabei werden die Analyt-Ionen mit A bezeichnet, die mit diesen um die Austauschplätze konkurrierenden Eluent-Ionen mit E.
Abbildung 6 Schematische Darstellung des Ionenaustauschprozesses in der Ionenchromatogra-phie. Links: Kationenaustausch, rechts: Anionenaustausch
Thermodynamische Aspekte des Ionenaustauschprozesses Ionenaustauscher bestehen im Normalfall aus festen Phasen, an deren Oberfläche ionische Gruppen fixiert sind. Aufgrund der Elektroneutralitätsbedingung befindet sich immer ein entgegengesetzt

Praktikum der Ionenchromatographie – Version 12
khv – 16 – 22/12/2000
geladenes Gegen-Ion in der Nähe der funktionellen Gruppe. Das Gegen-Ion stammt in der Regel aus dem Laufmittel und wird deshalb auch als Eluent-Ion bezeichnet.
Wird eine Probe aufgegeben, die zwei Analyt-Ionen A- und B- enthalten, so verdrängen diese kurzzeitig die Eluent-Ionen E- und werden an der fixierten Ladung zurückgehalten, bevor sie ihrerseits wieder durch das Eluent-Ion ausgetauscht werden. Für die Anionenchromatographie ergeben sich damit folgende reversiblen Gleichgewichte:
Harz - N+R3 E- + A- Harz - N+R3 A- + E- (15)
Harz - N+R3 E- + B- Harz - N+R3 B- + E- (16)
Durch die unterschiedlichen Affinitäten von A- und B- zu den funktionellen Gruppen ist eine Trennung der Komponenten möglich. Die Gleichgewichtskonstante K wird auch Selektivitätskoeffizient genannt und berechnet sich für das Anion A- wie folgt:
M S
M S
3
3A ][A][E
][E][A
][A]E RN-[Harz][E] ARN-[Harz
= K−−
−−
−−+
−−+
⋅⋅
=⋅⋅
(17)
Unter der Voraussetzung, dass die Konzentration der Eluent-Ionen normalerweise um mehrere Grössenordnungen höher ist als die der Analyt-Ionen, kann [E-] in mobiler und stationärer Phase als konstant betrachtet werden. Damit lassen sich der Verteilungskoeffizient DA (Gleichung 1) und der Retentionsfaktor k‘A (Gleichung 4) berechnen. Derartige Berechnungen sind aber strenggenommen nur dann zulässig, wenn die Konzentrationen in Gleichung 17 den Aktivitäten entsprechen, was nur bei unendlicher Verdünnung der Fall ist [19]. Die Aktivitäten der Ionen in der stationären Phase sind prinzipiell nicht zugänglich [4]. Für die am häufigsten eingesetzten Ionenaustauscher geringer Kapazität, die nur mit sehr verdünnten Elektrolyten als Laufmittel verwendet werden können, behilft man sich mit der Vernachlässigung der Aktivitäten. Diese sehr grobe Näherung trifft für Packungsmaterialien mit höherer Kapazität (> 200 mMol/g) und konzentriertere Eluenten nicht mehr zu, bei denen sich deutliche Abweichungen vom ‚idealen‘ Verhalten zeigen.
3.3.3 Die Ionenpaarbildung Mit Hilfe der Ionenpaarchromatographie können die gleichen Analyten getrennt werden wie mit der Ionenaustauschchromatographie, der Trennmechanismus ist jedoch ein völlig anderer. Als stationäre Phasen werden die aus der Verteilungschromatographie bekannten, völlig unpolaren Reversed-Phase-Materialien verwendet. Dem Eluenten wird ein sogenanntes Ionenpaarreagenz zugefügt, dass aus anionischen oder kationischen Tenside wie Tetraalkylammonium-Salzen bzw. n-Alkylsulfonsäuren besteht. Die Ionenpaarreagenzien bilden mit Analyt-Ionen entgegengesetzter Ladung ein ungeladenes Ionenpaar, welches an der stationären Phase durch hydrophobe Wechselwirkungen retardiert werden kann. Aufgrund der Bildungskonstanten der Ionenpaare und ihrer unterschiedlich starken Adsorption ist eine Stofftrennung möglich. Abbildung 7 zeigt vereinfacht das statische Ionenaustausch-Modell, bei dem angenommen wird, dass es erst nach Adsorption des Ionenpaarreagenzes an der stationären Phase zu Wechselwirkungen mit den Analyten kommt.

Praktikum der Ionenchromatographie – Version 12
khv – 17 – 22/12/2000
Abbildung 7 Schematische Darstellung des statischen Ionenaustausch-Modells in der Ionenpaarchromatographie (IPC). Das Trennprinzip gilt sowohl für Anionen als auch für Kationen.
3.3.4 Der Ionenausschluss Die Ionenausschlusschromatographie (IEC) dient vor allem zur Trennung von schwachen Säuren oder Basen [2,4]. Den grössten Stellenwert besitzt die IEC bei der Analytik schwacher Säuren wie Carbonsäuren, Kohlenhydraten, Phenolen oder Aminosäuren. Die Abbildung 8 zeigt das Trennprinzip der IEC am Beispiel einer Carbonsäure R – COOH.
Abbildung 8 Der Donnan-Ausschluss als Trennprinzip in der Ionenausschlusschromatographie (IEC)
Als Packungsmaterial wird in der IEC häufig ein vollständig sulfonierter Kationenaustauscher verwendet, dessen Sulfonsäuregruppen mit Protonen als Gegen-Ionen elektrisch neutral sind. Bei wässrigen Eluenten sind die funktionellen Gruppen hydratisiert. Die Hydrathülle wird durch eine (gedachte) negativ geladene Membran (Donnan-Membran) begrenzt. Sie ist nur für ungeladene, nicht dissoziierte Moleküle wie Wasser passierbar. Organische Carbonsäuren können getrennt werden, wenn starke Mineralsäuren wie Salzsäure als Laufmittel verwendet werden. Aufgrund der niedrigen Säurekonstanten (pkS-Werte) der Carbonsäuren liegen diese im stark sauren Eluenten nahezu vollständig undissoziiert vor. Sie können die Donnan-Membran durchqueren und an der stationären Phase adsorbieren, während die Chlorid-Ionen der vollständig dissoziierten Salzsäure ausgeschlossen werden.
Die Abbildung 9 zeigt eine typische Abhängigkeit des Elutionsvolumens einer Säure von deren pKs-Wert für die Trennung durch Ionenausschluss. Deutlich sind überlagerte Adsorption (längerkettige Carbonsäuren, H2S) und die Grenzen des praktischen Arbeitsbereiches zu erkennen. Die Trennung von Carbonsäuren erfolgt letztlich aufgrund ihrer unterschiedlichen pkS-Werte.

Praktikum der Ionenchromatographie – Version 12
khv – 18 – 22/12/2000
Abbildung 9 Abhängikeit des Elutionsvolumens in der Ionenausschlusschromatographie vom jeweiligen pKs-Wert der Säure
3.4 Retentionsmodelle in der Ionenchromatographie Im Idealfall wird die Retention eines Analyten in der Ionenchromatographie nur durch dessen Affinität zu den funktionellen Gruppen des Ionenaustauschers bestimmt. Diese Affinität lässt sich durch die Formulierung einer chemischen Reaktion, der Ionenaustauschreaktion, beschreiben und mit den Mitteln des Massenwirkungsgesetzes erklären.
Die im folgenden vorgestellten Retentionsmodelle versuchen, auf Basis des Massenwirkungsgesetzes Voraussagen über das Retentionsverhalten der beteiligten Analyten unter bestimmten chromatographischen Bedingungen zu treffen. Sind die resultierenden Modelle geeignet, die makroskopischen Beobachtungen zu erklären, kann mit ihrer Hilfe z.B. ein Elutionssystem auf das jeweilige Trennproblem hin optimiert werden.
3.4.1 Retentionsmodelle in der Anionenchromatographie Die folgenden Betrachtungen befassen sich zunächst mit der Elution durch gleichionische Verdrängung als einfachstem Elutionsmechanismus in der Ionenchromatographie. Das konkrete Beispiel ist auf die Anionenchromatographie bezogen, die gleichen Überlegungen gelten aber analog auch für die Kationenchromatographie. Erst bei Ergänzung des Eluenten mit Komplexbildnern muss das Retentionsmodell erweitert werden, was im Kapitel: «Retentionsmodell für die Elution in Gegenwart von Komplexbildnern» (Seite 24) passieren wird.
Retentionsmodell für Eluenten mit einem Anion Unter der Voraussetzung der Elektroneutralität ist der einfachste Ansatz für ein Retentionsmodell die gleichionische Verdrängung, bei der nur ein einziges Eluent-Ion Ey- mit einem Analyt-Anion Ax- um die funktionellen Gruppen der stationären Phase konkurriert [4]. Die Konzentration des Eluent-Anions Ey- ist dabei zeitlich konstant (isokratische Elution).
Die Austauschplätze der Trennsäule mit der Kapazität Q sind zu Beginn des chromatographischen Prozesses mit den Eluent-Anionen Ey- belegt. Wird eine Probe mit dem Analyt-Anion Ax- aufgegeben, so kommt es zwischen stationärer Phase (Index S) und mobiler Phase (Index M) zur Einstellung des folgenden Gleichgewichtes:
y · AMx- + x · ES
y- y · ASx- + x · EM
y- (18)
Das Gleichgewicht lässt sich nach dem Massenwirkungsgesetz durch eine thermodynamische Gleichgewichtskonstante beschreiben. Man erhält bei Berücksichtigung der Aktivitäten der beteiligten Ionen die folgende thermodynamische Gleichgewichtskonstante:

Praktikum der Ionenchromatographie – Version 12
khv – 19 – 22/12/2000
xE
yA
xE
yA
xyS
yxM
xyM
yxS
EA,yS
xM
yM
xS
γ γ
γ γ
][E ][A][E ][A
K−−
−−
⋅
⋅⋅
⋅⋅
=−−
−−
(19)
Die Aktivitäten der beteiligten Ionen in stationärer und mobiler Phase werden mangels Bestimmbarkeit in der stationären Phase im allgemeinen vernachlässigt und gleich Eins gesetzt.
Führt man nun für das Analyt-Anion Ax- zwei aus Kapitel 3.2.1 bekannte Grössen ein, den Verteilungskoeffizienten DA und den Retentionsfaktor k‘A .
M
S A [A]
[A] D = mit
M
S AA V
V D k' ⋅= (20)
so lässt sich Gleichung 19 durch diese Beziehungen und unter Vernachlässigung der Aktivitäten umformen zu:
x
yS
yM
y
S
MAEA, ][E
][E VV k' K ⎟⎟
⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛⋅=
−
−
(21)
Da die Konzentration der Eluent-Ionen E im Regelfall um mehrere Zehnerpotenzen grösser ist als die der Analyt-Anionen Ax-, kann man in guter Näherung annehmen, dass alle funktionelle Gruppen mit Ey- belegt sind. Die nicht bestimmbare Konzentration von Ey- in der stationären Phase lässt sich unter dieser Annahme durch die leichter zugänglichen Parameter Austauschkapazität Q und Ladung des Eluent-Anions y ersetzen:
yQ ][Ey
S =− (22)
Damit geht Gleichung 21 über in:
xyM
xy
S
M'AEA, ][E
yQ
VVk K −
⎟⎟⎠
⎞⎜⎜⎝
⎛⋅⎟⎟
⎠
⎞⎜⎜⎝
⎛⋅= (23)
Der Retentionsfaktor k‘A des Analyt-Anions Ax- ist aus einem Chromatogramm leicht zugänglich. Gleichung 23 wird daher nach dieser Grösse aufgelöst.
yx
yM
yx
y1
EA,M
S'A ][E
yQ )(K
VV
k−
−⎟⎟⎠
⎞⎜⎜⎝
⎛= (24)
Diese Gleichung ist von entscheidender Bedeutung für die Anionenchromatographie, da durch sie ein quantitativer Zusammenhang zwischen dem Retentionsfaktor k’A und einigen experimentell zugänglichen Parametern wie der Konzentration des Eluenten und der Austauschkapaziät hergestellt wird. In der Praxis arbeitet man aus Gründen der Übersicht mit der logarithmierten Form der Gleichung 24.
][E log yx log
yQlog
yx K log
y1 k' log y
MEA,A−−Φ++= mit
M
S
VV
=Φ (25)
Aus Gleichung 25 ergeben sich zunächst folgende Konsequenzen:
• Eine Erhöhung der Eluentenkonzentration [E y-] beschleunigt die Elution

Praktikum der Ionenchromatographie – Version 12
khv – 20 – 22/12/2000
o Grössere Retentionsfaktoren werden verursacht durch grössere Gleichgewichtskonstanten KA,E, höhere Austauschkapazitäten Q und ein grösseres Phasenvolumenverhältnis Φ.
• Multivalente Analyten Anx- werden stärker retardiert als monovalente Ax-, o zumindest solange die Eluentkonzentration [E y-] realtiv niedrig ist. Dies wird auch als
Elektroselektivität bezeichnet. • Multivalente Eluenten E ny- haben eine höhere Elutionskraft als monovalente E y-
o Die Elution multivalenter Analyten Anx- wird durch gesteigerte Konzentrationen monovalenter Eluent-Ionen E y- stärker beeinflusst als die monovalenter Analyten Ax-.
In erster Näherung kann angenommen werden, dass die Selektivitätskoeffizienten bei konstantem Φ unabhängig von Q sind, womit sich folgende Proportionalität ergibt:
][E
Q k' -yM
A ∝ (26)
Aus Gleichung 26 ist ersichtlich, dass bei einer Steigerung der Austauschkapazität Q die Konzentration des Eluenten [Ey-] proportional erhöht werden muss, damit konstante Retentionsfaktoren erreicht werden. Dies ist der Grund, warum in der Ionenchromatographie üblicherweise niederkapazitive Trennphasen verwendet werden, da hohe Elektrolytkonzentrationen die wichtigste Detektionsmethode in der Ionenchromatographie, die Leitfähigkeitsdetektion, praktisch unmöglich machen.
Zur Optimierung von Trennproblemen wird oft die Eluentenkonzentration [E y-] variiert. Werden alle anderen in Gleichung 25 auftretenden Parameter konstant gehalten, vereinfacht sich diese zu:
][E log yx C k' log y
M1A−−= (27)
Die graphische Auftragung der Gleichung 27 ergibt eine Gerade mit der Steigung m = - x/y und dem Achsenabschnitt C, der die Grössen Q, Φ und KA,E enthält. Bei Verwendung monoanionischer Eluenten wird m auch als effektive Ladung bezeichnet. Abbildung 10 stellt schematisch das Ergebnis der Gleichung 27 für verschiedene Kombinationen unterschiedlich geladener Eluent- und Analyt-Anionen dar.
Abbildung 10 Graphische Darstellung von Gleichung 28 für verschiedene Kombinationen unterschiedlich geladener Eluent- und Analyt-Anionen [4].
Gleichung 27 wurde bisher in einer Vielzahl von Publikationen bestätigt, allerdings unter der Voraussetzung, dass niederkapazitive Trennmaterialien und verdünnte Eluenten verwendet wurden.
Variiert man dagegen unter konstanten Bedingungen die Austauschkapazität Q, vereinfacht sich Gleichung 25 zu:

Praktikum der Ionenchromatographie – Version 12
khv – 21 – 22/12/2000
yQ log
yx C k' log A += (28)
Die graphische Darstellung dieser Gleichung ähnelt Abbildung 10, allerdings mit positiven Geradensteigungen. Chromatographische Untersuchungen zur Variation von Q wurden bisher nur einmal bei der Trennung von divalenten Kationen durchgeführt. Dabei zeigte sich, dass im Gegensatz zu den bisherigen Annahmen der Retentionsfaktor und auch die Selektivitätskoeffizienten nicht unabhängig von der Austauschkapazität betrachtet werden können. Es wird deutlich, dass zur Optimierung von Trennproblemen neben der Konzentration des Eluenten [E y-] auch die Austauschkapazität Q eine wichtige Grösse darstellt.
Die bisherigen Betrachtungen gelten nur für ein Analyt-Anion. Konkurrieren dagegen zwei unterschiedliche Anionen Ax– und Bz– um die funktionellen Gruppen, so gilt für den Selektivitätskoeffizienten KA,B:
xzS
zxM
xzM
zxS
BA, ][A ][A][B ][A
K−−
−−
⋅⋅
= (29)
Unter Berücksichtigung von Gleichung 20 erhält man zunächst die Selektivität α,
][B ][A][B ][A
k'k'
α zS
xM
zM
xS
B
ABA, −−
−−
⋅⋅
== (30)
und dann nach Umformen die Gleichungen 31 a und b,
⎟⎟⎠
⎞⎜⎜⎝
⎛⋅
−+⋅=
S
MBBA,A.B V
Vk' log
zzx K log
z1 log α (31a)
⎟⎟⎠
⎞⎜⎜⎝
⎛⋅
−+⋅=
S
MABA,A.B V
Vk' log
zzx K log
x1 log α (31 b)
die sich für gleichgeladene Analyten (x = z) vereinfachen zu:
BA,BA, K log z1 α log = (32) bzw. A,BA,B K log
x1 α log = (33)
Für die Selektivität zwischen zwei gleichgeladenen Analyt-Anionen bedeutet dies:
• sie ist nur eine Funktion des Selektivitätskoeffizienten KA,B und der Ladungen z bzw. x, • sie hängt bei konstantem KA,B weder von der Konzentration [E y-] noch von der chemischen
Beschaffenheit des Eluent-Anions ab (!)
Bei unterschiedlichen Ladungen von A und B ergibt sich:
• α hängt vom Retentionsfaktor einer der beiden Analyten ab, • die beiden Retentionsfaktoren k‘A und k’B sind nicht unabhängig voneinander (!)
An den Gleichungen 31 bis 33 ist besonders bemerkenswert, dass die Selektivitäten zweier Anionen zunächst weder von der chemischen Beschaffenheit noch von der Ladung des Eluent-Anions abhängt, solange das Phasenvolumenverhältnis und der Selektivitätskoeffizient konstant sind. In der Praxis lässt sich aber dennoch eine Veränderung von α durch die Variation von [E y-] erreichen, da sich zwei Analyten trotz gleicher Ladung deutlich in ihren chemischen Eigenschaften, z.B. Polarisierbarkeit und Hydratation, unterscheiden können, was unterschiedliche Affinitäten zur stationären Phase zur Folge hat. Diese Wechselwirkungen werden aber in der klassischen Ableitung nicht berücksichtigt.

Praktikum der Ionenchromatographie – Version 12
khv – 22 – 22/12/2000
Retentionsmodelle für Eluenten mit mehreren Anionen Die bisherigen Betrachtungen bezogen sich auf Elutionssysteme mit nur einem Eluent-Anion. In der Praxis liegen aber meist mehrere eluierende Spezies vor, etwa bei Carbonat/Hydrogencarbonat-Puffern oder bei mehrbasigen Säuren wie Phosphorsäure, deren Dissoziation und damit Speziesverteilung stark vom pH-Wert abhängt.
Selbst in einfachen Fällen, wenn keines der beteiligten Eluent-Anionen an Säure-Base-Gleich-gewichten beteiligt ist, kann der Zusammenhang zwischen dem Retentionsfaktor k‘ und der Eluentkonzentration [E-] nicht in Form einer einfachen log-log-Beziehung nach Gleichung 28 dargestellt werden. Dies wäre nur dann möglich, wenn die Konzentration oder die Elutionskraft der übrigen Eluent-Anionen zu vernachlässigen ist, was dem Retentionsmodell für monoanionische Eluenten entsprechen würde.
In der Literatur werden mehrere Modelle beschrieben, die sich mit polyanionischen Eluenten befassen und im folgenden kurz diskutiert werden:
• Modell des dominanten Gleichgewichtes [21] • Modell der effektiven Ladung [22-24] • Modell der vielfachen Eluentspezies [25,26]
Bei Betrachtung eines auf Phosphat basierenden Eluenten H2PO4-, HPO4
2- und PO43-, (im weiteren
H2P-, HP2- und P3-) und dem monovalenten Analyt-Ion A- bilden sich folgende Gleichgewichte aus:
−− + S2M PH A −− + M2S PH A ; x1 (34)
−− + 2S2
1M HP A
−− + 2M2
1S HP A ; x2 (35)
−− + 3S3
1M P A −− + 3
M31
S P A ; x3 (36)
Die Grössen x1..3 entsprechen dabei den Anteilen der jeweiligen Reaktionen an der Retention, weshalb gilt:
x1 + x2 + x3 = 1 (37)
Sowohl das Modell des dominanten Gleichgewichtes als auch das der effektiven Ladung postulieren, obwohl mehrere Spezies vorhanden sind, eine bestimmte Ladung für das Eluent-Anion, so dass das in Kapitel 4.1.1 für monoanionische Eluenten abgeleitete Retentionsmodell verwendet werden kann.
Das Modell des dominanten Gleichgewichtes nimmt an, dass Gleichung 36 ganz auf der rechten Seite liegt, da P3- aufgrund der höheren Ladung wesentlich stärker als H2P- und HP2- an der stationären Phase gebunden wird. Damit ist P3- allein für die Elution massgeblich, so dass sich die Ladung des Eluent-Anions zu -3 ergibt. Dieses Modell erzielt allerdings nur bei multivalenten Analyten eine gute Übereinstimmung mit der Praxis [4].
Beim Modell der effektiven Ladung wird unter Berücksichtigung des pH-Wertes aus den Molenbrüchen der möglichen Spezies H2P-, HP2- und P3- eine effektive Ladung berechnet [22]. Mit dieser und den vorliegenden Konzentrationen der Eluentspezies lässt sich eine Beziehung analog Gleichung 27 aufstellen. Voraussetzung für derartige Berechnungen ist aber, dass sich die Selektivitäten der Eluent-Spezies bezüglich des Analyt-Ions A- nicht wesentlich unterscheiden. Das Modell der effektiven Ladung lässt sich vor allem bei monovalenten Analyten sinnvoll anwenden [4].
In der Realität ist das Modell der vielfachen Eluentspezies zur Beschreibung von Eluenten, deren Komponenten sich chemisch voneinander ableiten, am besten geeignet. Die folgenden Betrachtungen basieren auf dem Modell von Mongay et al. [27], welches eine Weiterentwicklung der Arbeiten von Jenke und Pagenkopf darstellt [25].
Aus den Gleichungen 34 bis 36 lässt sich das globale Gleichgewicht auf der Trennsäule darstellen (Gleichung 38). Unter Berücksichtigung von Gleichung 37 lässt sich bei Vernachlässigung der Aktivitäten die Gleichgewichtskonstante KA,P für den Austauschprozess definieren (Gleichung 39).

Praktikum der Ionenchromatographie – Version 12
khv – 23 – 22/12/2000
−−−− ⋅+⋅+⋅+⋅++ 3S
32
2S2
1M321 P
3x
HP2x
PH1x
A)xx(x (38)
−−−− ⋅+⋅+⋅+⋅++ 3M
3M
2M2
1S321 P
3x
HP2x
PH1x
A)xx(x
][HP ][HP ]P[H ][A][HP ][HP ]P[H ][A K /3X3
S/2X2
S/1X
S2M
/3X3M
/2X2M
/1XM2S
PA, 321
321
−−−−
−−−−
⋅⋅⋅⋅⋅⋅
= (39)
Die weitere mathematische Behandlung erfolgt wie bei der Ableitung des Retentionsmodells für monoanionische Eluenten. Berücksichtigt werden müssen vor allem:
• Die (mögliche) Dissoziation des Analyt-Anions A– • Die Gesamtkonzentration der Eluentspezies: cP = [H3P] + [H2P-] + [HP2–] + [P3–] • Das Ausmass der Wechselwirkungen der Eluentspezies mit den funktionellen Gruppen
Die Einführung des Retentionsfaktors k’A (Gleichung 20) und der Kapazität Q (Gleichung 22) liefert nach weiterer mathematischer Umformung einen komplizierten Ausdruck für k’A [28], der hier nur in seiner logarithmierten und weiter vereinfachten Form dargestellt wird:
P321
3A c log 3x
2x
1x
C k' log ⋅⎜⎜⎝
⎛⎟⎠
⎞++−= (40)
C3 ist eine Konstante, die analog Gleichung 27 Grössen wie das Phasenvolumenverhältnis, die Kapazität und die Gleichgewichtskonstante enthält, cP die Summe der Konzentrationen der Eluentspezies. Aus Gleichung 40 lässt sich folgern, dass die Steigungen der Geraden bei einer doppelt logarithmischen Auftragung immer kleiner sein müssen als nach dem einfachen Retentionsmodell für monoanionische Eluenten (Gleichung 27), da die Summe in der Klammer stets kleiner als Eins ist. Weiterhin wird deutlich, dass der pH-Wert einen entscheidenden Einfluss auf das Ausmass der Beeinflussung der log-log-Beziehung hat.
Für Eluentspezies, die sich chemisch nicht voneinander ableiten, existiert ein Modell von Janoš et al., welches zur Beschreibung von Eluenten entwickelt wurde, die einen Phosphatpuffer und zusätzlich Perchlorat enthalten [29]. Die Ableitung dieses Modells erfolgt analog den obigen Überlegungen, allerdings muss zusätzlich ein Austauschgleichgewicht für ein weiteres, monovalentes Eluent-Ion betrachtet werden. Die Berechnungen liefern sehr komplizierte Ausdrücke für den Retentionsfaktor, die sich aber für neutrale oder saure Eluenten drastisch vereinfachen lassen. Für den Fall, dass neben dem Perchlorat nur eine weitere monovalente Eluent-Spezies vorliegt, erhält man Gleichung 41, in der x und y die Beiträge der entsprechenden Gleichgewichtsreaktionen (x: Phosphatpuffer, y: Perchlorat) an der Retention repräsentieren. C ist wie in den übrigen Modellen eine Konstante, während der nicht näher bestimmte Faktor a berücksichtigen soll, um wieviel stärker das Perchlorat-Ion an der stationären Phase gebunden wird als die betreffende Phosphat-Spezies.
][E log i
x y a log
ix
y C k' log -ii1A ⋅⎜⎜
⎝
⎛⎟⎠
⎞+−⋅⎜⎜⎝
⎛⎟⎠
⎞+−= ∑∑ (41)
Da die Klammerterme in Gleichung 41 stets kleiner als Eins sind, ist die Steigung in der log-log-Auftragung immer geringer als nach dem einfachen Retentionsmodell zu erwarten. Das Modell liefert im konkreten Anwendungsfall gute Übereinstimmungen mit den experimentellen Daten. Es kann allerdings in der beschriebenen Form nicht auf alkalische Elutionssysteme angewendet werden.
3.4.2 Retentionsmodelle in der Kationenchromatographie Die Kationenchromatographie muss für die Betrachtung der Retentionsmodelle in zwei Untergruppen geteilt werden. Die eine Gruppe befasst sich mit Alkali und Erdalkalimetallkationen als Analyten und benötigt nur ein Elutionsystem auf Basis der gleichionischen Verdrängung. Dabei trägt die stationäre
KA,P

Praktikum der Ionenchromatographie – Version 12
khv – 24 – 22/12/2000
Phase Carbonsäure-Gruppen als Funktionalität. Bei der Trennung von zwei- und höhergeladenen Metallionen kommt man um den Einsatz eines Komplexbildners nicht herum, dessen Einfluss auf die Retention im Kapitel «Retentionsmodell für die Elution in Gegenwart von Komplexbildnern» auf Seite 24 beschrieben wird.
Retentionsmodell für Eluenten mit einem Kation Die Erläuterungen des Abschnitts «Retentionsmodell für Eluenten mit einem Anion» gelten in analoger Weise für die Kationenchromatographie mit Elution durch gleichionische Verdrängung. In der Praxis relevant ist dies für die Bestimmung von Alkali- und Erdalkalimetalle, Ammonium und kurzkettigen Aminen. Als Eluentkationen werden neben H+ auch organische Kationen wie die 2,3-Diaminopropionsäure (DAP) in Verbindung mit verdünnter Salzsäure eingesetzt. Je nach eingestelltem pH-Wert des Eluenten liegt die DAP in den ionischen Formen (1) und (2) vor (Abbildung 11). Nach einer Suppression erhält man die zwitterionische Form (3), die keine Eigenleitfähigkeit besitzt.
Abbildung 11 Ionische Spezies der Diaminopropionsäure
Retentionsmodell für die Elution in Gegenwart von Komplexbildnern In der Kationenchromatographie werden zur Trennung von Erdalkali-, Übergangs- und Schwermetall-Ionen Eluenten eingesetzt, die zusätzlich zum Eluentkation En+ noch einen Komplexbildner enthalten. Als Komplexbildner werden hauptsächlich Dicarbonsäuren H2L wie Weinsäure, Oxalsäure, Citronensäure und auch Pyridindicarbonsäure, eingesetzt. Die Analyten bilden mit den Anionen der Komplexbildner HL– sowie L2– unterschiedlich stabile Komplexe mit verschiedenen Stöchiometrien. Durch die Komplexierung wird die effektive, d.h. im zeitlichen Mittel vorliegende Ladung der Analyten herabgesetzt. Da dies entsprechend der Kinetik der Komplexbildung und der Stabilitätskonstanten der Komplexe erfolgt, nehmen die Selektivitätsunterschiede zu und eine Trennung auch ähnlicher Analyten wird möglich. Neben dem Ionenaustausch ist daher die Komplexbildung entscheidend für die Trennung der höhergeladenen Metall-Ionen.
Mex+ + L2– MeLx-2 [ ]
[ ][ ]−+
−
= 2x
2x
MeL LMeMeLK mit (42)
MeLx-2 + L2– MeL2x-4 [ ]
[ ][ ]−
−
= 22-x
4x
MeL LMeLMeLK mit
2 (43)
MeLx-2 + L2– MeL2x-4
[ ][ ][ ]−−
−
= 22x
4x
MeL LMeLMeLK mit
2 (44)
Mex+ + HL– MeHLx-1 [ ][ ][ ]−+
−
=HLMe
MeHLK mit x
1x
HL (45)
Um den Einfluss des Komplexbildners auf die ionenchromatographische Trennung zu berücksichtigen, wird das Retentionsmodell der gleichionischen Verdrängung (siehe Kapitel «Retentionsmodell für Eluenten mit einem Anion» auf Seite 18) erweitert. Als Einflussgrösse, die das Ausmass der

Praktikum der Ionenchromatographie – Version 12
khv – 25 – 22/12/2000
Komplexierung des Analyten beschreibt, wird der αM-Wert eingeführt. Der Bruchteil αM der freien Analyt-Ionen in der mobilen Phase ist gegeben als
[ ]
[ ] [ ] [ ] [ ][ ][ ]Me'Me
MeLMeLMeHLMeMeα
x
4x2
2x1xx
x
M
+
−−−+
+
=+++
= (45)
mit der Gesamtkonzentration [Me’] der Metall-Ionen. Der αM-Wert lässt sich aus den Komplexbildungskonstanten, den Säuredissoziationskonstanten der Carbonsäure und dem pH-Wert des Eluenten berechnen. Für den Verteilungskoeffizienten DMe erhält man unter Berücksichtigung der Komplexbildung:
[ ]
[ ][ ][ ]+
== xx
Mx
Me MeMeRα
Me'MeRD (46)
Setzt man voraus, dass nur freie Analyt-Ionen Mex+ mit den Carbon- oder Sulfonsäure-Gruppen wechselwirken und c(Ez+) >> c(H+), so ergibt sich für Gleichung 21:
[ ]xx
+
−
⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎟⎠
⎞⎜⎜⎝
⎛= y
y
M
MeE Me, E
yQ
Φ αk' K (47)
In der logarithmierten Form erhält man für Gleichung 47 analog zu Gleichung 25:
][E log yx log
yQlog
yx K log
y1 α log k' log y
MEMe,MMe+−Φ+++= (48)
Liegen nebeneinander mehrere kationische Metall-Spezies vor, z. B. Mex+ und MeHL(x-1)+, resultiert daraus im Chromatogramm in der Regel jedoch nur ein Peak für den betreffenden Analyten. Die Anzahl der auftretenden Peaks ist abhängig von der Kinetik der Komplexierungs- und Dekomplexierungs-Gleichgewichte in der mobilen Phase. Man erhält nur einen Peak für den Fall, dass sich die Komplexgleichgewichte in der mobilen Phase schnell einstellen im Vergleich zur Aufenthaltszeit des Komplexes in der stationären Phase. Verläuft die Umkomplexierung hingegen nur langsam, können asymmetrische oder multiple Peaks auftreten.
Unter der Annahme, dass alle in der mobilen Phase vorliegenden Metall-Spezies mit der stationären Phase wechselwirken können, ergibt sich für den experimentell bestimmten Kapazitätsfaktor k’exp des Analyten:
2-x2x1-x1xxx MeLMeLMeHLMeHLMe Mexp' α k'α k'αk'k −−++ ++= (49)
Bei der Betrachtung der Abhängigkeit des Kapazitätsfaktors von den Einflussgrössen Q, [Ey+] sowie αM wird jedoch der in Gleichung 48 dargestellte Zusammenhang zugrunde gelegt, da die divalenten Analyten mit starken Komplexbildnern überwiegend neutrale oder anionische Komplexe bilden.
Berechnung von αM-Werten Der αM-Wert ist nach Gleichung 45 definiert als das Verhältnis der Konzentration der freien Metall-Ionen zur Gesamtkonzentration der Metall-Ionen. Die Konzentrationen der in der mobilen Phase vorliegenden Metall-Spezies lassen sich mit den jeweiligen Komplexbildungskonstanten und den Säuredissoziationskonstanten der verwendeten Carbonsäure berechnen.
Bei der Verwendung von Weinsäure als Komplexbildner im Eluenten werden von den Erdalkali-, Übergangs- und Schwermetallen vornehmlich die neutralen 1:1-Komplexe MeL sowie in geringerem Masse die Hydrogentartrat-Komplexe MHL+ gebildet. Für die Berechnung von αM ergibt sich für Weinsäure-Eluenten:

Praktikum der Ionenchromatographie – Version 12
khv – 26 – 22/12/2000
[ ]
[ ] [ ] [ ] LHLMeHLLLMeL2
2
M cαKcαK11
MeHLMeLMeMeα
++=
++=
++
+
(50)
wobei cL die Gesamtkonzentration der Weinsäure und αHL sowie αL die Molenbrüche der Säure-Anionen HL- und L2- sind.
Mit Oxalsäure und/oder Pyridindicarbonsäure bilden einige Metall-Ionen neben den 1:1-Komplexen auch stabile MeL2
2--Komplexe, so dass sich αM wie folgt berechnet:
[ ]
[ ] [ ] [ ] 2L
2LMeLMeLLLMeL
22
2
2
M cαKKcαK11
MeLMeLMeMeα
2++
=++
=−+
+
(51)
Berechnung der Säuredissoziation Der pH-Wert und die Konzentration des Komplexbildners in der mobilen Phase bestimmen die Konzentration an Ligand und damit das Ausmass der Komplexierung der Analyten. Eine zweiprotonige Säure dissoziiert in zwei Stufen:
H2L HL- + H+ [ ][ ][ ]LH
HHLK mit2
S1
+−
= (52)
HL- L2- + H+ [ ][ ][ ]−
+−
=HL
HLK mit2
S2 (53)
mit den Säurekonstanten KS1 und KS2
. Die zur Berechnung des αM-Wertes verwendeten Molenbrüche αH L2
, αHL sowie αL ergeben sich aus den Massenwirkungsgesetzen der einzelnen Deprotonierungsschritte:
[ ]
[ ] [ ] [ ][ ]
[ ] [ ]211
2
SSS2
2
22
2LH
K KH KHH
LHLLHLHα
++=
++=
++
+
−− (54)
[ ]
[ ] [ ] [ ][ ]
[ ] [ ]211
1
SSS2
S2
2HL
K KH KH
H KLHLLH
HLα++
=++
=++
+
−−
−
(55)
[ ]
[ ] [ ] [ ] [ ] [ ]211
21
SSS2
SS2
2
2
LK KH KH
K KLHLLH
Lα++
=++
=++−−
−
(56)
3.5 Detektionssysteme in der Ionenchromatographie Zur Detektion von Substanzen stehen im Bereich der HPLC eine Reihe von unterschiedlichen Verfahren zur Verfügung, wobei die Wahl eines geeigneten Detektors im Hinblick auf die analytische Fragestellung erfolgen muss. Die Anforderungen an einen Detektor lassen sich wie folgt zusammenfassen:
• Hohe Messempfindlichkeit und kurze Ansprechzeiten • Proportionalität des Messsignals zur Analytkonzentration (grosser linearer Bereich) • Geringe Veränderung der Basislinie (Drift) • Geringes Eigenrauschen • Möglichst kleines Eigenvolumen zur Verminderung der Bandenverbreiterung

Praktikum der Ionenchromatographie – Version 12
khv – 27 – 22/12/2000
Allgemein werden selektive und nicht selektive Detektoren unterschieden. Während ein selektiver Detektor direkt auf eine Eigenschaft des Analyten anspricht, reagieren unselektive Detektoren auf eine durch den Analyten hervorgerufene Änderung einer physikalischen Eigenschaft des gesamten Elutionssystems. Da sich die Detektoren zur Verwendung mit der Ionenchromatographie im Prinzip nicht von denen für die ‚konventionelle‘ HPLC unterscheiden, werden die wichtigsten Detektionssysteme in diesem Abschnitt zumindest angesprochen. Der universellste und gebräuchlichste Detektor in der IC ist der Leitfähigkeitsdetektor.
3.5.1 Elektrochemische Detektoren
Leitfähigkeitsdetektion Die Leitfähigkeitsdetektion oder auch konduktometrische Detektion hat in der Ionenchromatographie einen Anteil von etwa 55 % [4]. Aus Sicht der verkauften Ionenchromatographen dürfte dieser Anteil heute noch wesentlich höher liegen. Die Leitfähigkeitsdetektion ist ein unselektives Detektionsprinzip, wobei auch in diesem Fall direkte und indirekte Bestimmungen möglich sind. Da in der Ionenchromatographie wässrige Elektrolyte als Laufmittel verwendet werden, muss der Detektor auf die relativ geringe, durch die Analyt-Ionen verursachte Veränderung der Gesamtleitfähigkeit des Eluenten ansprechen. Durch die Verwendung sogenannter Suppressionstechniken kann die Eigenleitfähigkeit bestimmter Eluenten drastisch reduziert werden, wodurch im Falle der starken Säureanionen eine deutliche Empfindlichkeitssteigerung erreicht wird.
Die Leitfähigkeit κ einer Lösung wird messtechnisch als Kehrwert des Widerstandes R bestimmt, den eine Lösung zwischen zwei Elektroden der Fläche A und des Abstandes L verursacht.
κ = L / A R (57)
Die Äquivalentleitfähigkeit Λ einer Lösung ermittelt sich zu:
Λ = κ / c (58)
Die Grenzleitfähigkeit Λ∞ und die Abhängigkeit der Leitfähigkeit von der Konzentration kann anhand der Gleichung 59 ermittelt werden. Die Konstanten A und B sind empirische Stoffkonstanten.
Λ = Λ∞ - (A + B Λ∞) c (59)
Die Leitfähigkeit eines Elektrolyten setzt sich additiv aus den Ionenleitfähigkeiten Λ-Anion und Λ
+Kation
zusammen
κ = c (Λ-Anion- + Λ
+Kation+) (60)
Abbildung 12 Aufbau einer Leitfähigkeitsmesszelle.
Nach dem Kohlrausch’schen Gesetz gilt, dass die Leitfähigkeit einer verdünnten Lösung proportional zur Summe der Leitfähigkeiten aller Ionen multipliziert mit deren Konzentrationen ist.

Praktikum der Ionenchromatographie – Version 12
khv – 28 – 22/12/2000
1000
i∑Λ=
ciκ (61)
wobei κ die Leitfähigkeit in S cm-1, Λ die Grenzleitfähigkeit in S cm2 (z mol L-1)-1 und c die Konzentration in z mol L-1 (z entspricht der Ladung des Ions) ist. Der Faktor 1000 resultiert daraus, dass 1 Liter 1000 cm3 entspricht.
Die Leitfähigkeitsänderung durch den Analyten ist proportional zu dessen Konzentration im Effluat.
( )
1000c ΛΛ
∆ SES −=κ (62)
wobei S und E für die Analyt- bzw. Eluent-Ion steht. Da bei der Leitfähigkeitsdetektion die Änderung der Leitfähigkeit gemessen wird, treten in der Anionenchromatographie bei hohen Grundleitfähigkeiten nur geringe Leitfähigkeitsänderungen auf. Es ist daher meist günstig, die Grundleitfähigkeit möglichst gering zu halten.
Abbildung 13 Verlauf der Leitfähigkeit des Eluates einer ionenchromatographischen Trennung eines Mehrstoffgemisches. Gezeigt sind die Verläufe für einen Eluenten mit hoher (rot) bzw. niedriger (blau) Leitfähigkeit
Die Leitfähigkeit eines Eluates in der Ionenchromatographie kann entweder direkt oder nach Durchlaufen eines Suppressors bestimmt werden. Diese Varianten werden auch als Ein- bzw. Suppressortechnik bezeichnet. Welche der beiden Ausführungsformen zu bevorzugen ist, kann anhand von Überschlagsrechnungen ermittelt werden.
Verwendet man im Bereich der Anionenchromatographie die direkte Leitfähigkeitsdetektion, so erhält man für Empfindlichkeit der Bestimmung κPeak ausgedrückt als Differenz zwischen der Leitfähigkeit von Analyt und Eluentanion für den Fall Chlorid als Analyt und Carbonat als Eluent-Anion folgende Gleichungen:
κPeak ≈ cAnalyt (Λ-Cl- - Λ
-CO
32- ) ⇒ κPeak ≈ cAnalyt (76 - 72)
κPeak ≈ cAnalyt • 4
Wird der Eluent an die Bedürfnisse der direkten Leitfähigkeitsdetektion angepasst, so erhält man für das obige Beispiel durch Ersatz des Carbonat-Eluenten durch einen Phthalat-Eluenten folgende Empfindlichkeit:
κPeak ≈ cAnalyt (Λ-Cl- - Λ
-Phthalat2- ) ⇒ κPeak ≈ cAnalyt (76 - 38)
κPeak ≈ cAnalyt • 38

Praktikum der Ionenchromatographie – Version 12
khv – 29 – 22/12/2000
Im Fall der chemischen Suppression der Leitfähigkeit des Eluenten ist die Empfindlichkeit des Verfahrens nur noch durch die Äquivalentleitfähigkeit der zu detektierenden Substanz bestimmt. Im Fall von Cl- gilt dann:
κPeak ≈ cAnalyt (Λ-Cl- + Λ
+H+ ) ⇒ κPeak ≈ cAnalyt (76 + 350)
κPeak ≈ cAnalyt • 426
Aus obigen Überschlagsrechnung folgt für Anionen, dass die direkte Leitfähigkeitsdetektion um den Faktor 10 unempfindlicher ist als die Leitfähigkeitsdetektion nach chemischer Suppression.
Für die Kationenchromatographie ergibt sich analog die folgende Überschlagsrechnung, wobei als Analyt Na+ und als Eluent H+ eingesetzt wird. Im Fall der direkten Leitfähigkeitsdetektion (HCl/NaCl) gilt
κPeak ≈ cAnalyt (Λ+Na+ - Λ
+H+ ) ⇒ κPeak ≈ cAnalyt (50 – 350)
κPeak ≈ cAnalyt • – 300
Bei chemischer Suppression der Leitfähigkeit des H+-Ions durch Umsatz zu Wasser gilt:
κPeak ≈ cAnalyt (Λ+Na+ + Λ
-OH-) ⇒ κPeak ≈ cAnalyt (76 + 198)
κPeak ≈ cAnalyt • 248
Damit ergibt sich für die direkte Leitfähigkeitsdetektion von Kationen eine höhere Empfindlichkeit als für die Leitfähigkeitsdetektion nach chemischer Suppression.
Tabelle 2 Äquivalentleitfähigkeit Λ∞ einiger Ionen
Kationen Λ+ (S cm2 eq-1) Anionen Λ− (S cm2 eq-1) H+ 350 OH– 198 Li+ 39 F– 54 Na+ 50 Cl– 76 K+ 74 Br– 78 NH4
+ 73 I– 77 ½ Mg2+ 53 NO2
– 72 ½ Ca2+ 60 NO3
– 71 ½ Sr2+ 59 HCO3
– 45 ½ Ba2+ 64 ½ CO3
2– 72 ½ Zn 2+ 52 H2PO4
– 33 ½ Hg 2+ 53 ½ HPO4
2– 57 ½ Cu 2+ 55 1/3 PO4
3– 69 ½ Pb 2+ 71 ½ SO4
2– 80 ½ Co 2+ 53 CN– 82 1/3 Fe3+ 70 SCN– 66 N(Et)4
+ 33 Acetat 41 ½ Phthalat 38 Propionat 36 Benzoat 32 Salicylat 30 ½ Oxalat 74
Die Suppressoren für die Ionenchromatographie können entweder als diskontinuierlich arbeitetende Packed-Bed-Suppressoren gemäss Abbildung 14 oder als kontinuierlich arbeitende Membransuppres-soren ausgelegt sein. Der Packed-Bed-Suppressor in der von Metrohm verwendeten Revolveraus-führung, besitzt drei gleichartige Suppressoreinheiten, von denen jeweils eine Einheit als Suppressor

Praktikum der Ionenchromatographie – Version 12
khv – 30 – 22/12/2000
dient, die zweite regeneriert und die dritte mit Wasser gespült wird. Nach Ablauf einer Analyse wird der Revolver um 120o gedreht und die zuvor gespülte Säule wird als Suppressor benutzt. Auf diese Weise wird eine quasi-kontinuierliche Arbeitsweise ermöglicht.
Detektor Waste Waste
Eluat H2SO4 Wasser
3 2 1
Abbildung 14 Schematischer Aufbau eines Packed-Bed-Suppressors für eine quasi-kontinuierliche Arbeitsweise
Der in Abbildung 15 skizzierte Membransuppressor ermöglicht eine kontinuierliche Arbeitsweise, ist aber durch den Einsatz von Ionenaustauschermembranen anfällig gegenüber Belegungen der Membranoberfläche, die dann eine reduzierte Suppressionskapazität und schliesslich ein Versagen des Suppressors zur Folge haben.
Eluat DetektorH2SO4
H2SO4
Abfall
Abfall
Ionenaustausch- Membran
Abbildung 15 Schematischer Aufbau eines kontinuierlich arbeitenden Membransuppressors
Amperometrische Detektion Voltammetrische Detektoren können im Prinzip auf alle Verbindungen angewendet werden, die leicht reduzierbare oder oxidierbare funktionelle Gruppen tragen. Der amperometrische Detektor ist die wichtigste Variante. Bei ihm wird zwischen einer Arbeits- und einer Referenzelektrode eine bestimmte Spannung angelegt. Passiert nun ein elektrochemisch aktiver Analyt die Messstrecke, dessen Halbstufenpotential so liegt, dass bei der angelegten Spannung eine Reduktion oder Oxidation abläuft, fliesst ein Strom, der das Messsignal darstellt. Die Amperometrie ist sehr empfindlich, wobei der Stoffumsatz nur etwa 10 % beträgt. Im Bereich der Ionenanalytik lassen sich neben einigen Kationen (Fe3+, Co2+) vor allem Anionen wie Nitrit, Nitrat, Thiosulfat sowie Halogene und Pseudohalogene bestimmen. Die wichtigsten Anwendungsfälle liegen jedoch in der Analytik von Zuckern mittels Anionenchromatographie und in der klinischen Analytik. Der coulometrische Detektor liefert aufgrund einer anderen Arbeitsweise einen quantitativen Stoffumsatz, woraus allerdings keine gesteigerte Empfindlichkeit resultiert.
Potentiometrische Detektion Bei der potentiometrischen Detektion wird mit ionensensitiven Elektroden gearbeitet, die teilweise eine sehr hohe Selektivität besitzen. Die Sensoren müssen trotz ihrer notwendigen starken Miniaturisierung zuverlässig arbeiten können, was in der Praxis immer noch Probleme bereitet. Daher bleibt die Anwendung der potentiometrischen Detektion, insbesondere in der Ionenchromatographie, bisher auf wenige Spezialfälle beschränkt.

Praktikum der Ionenchromatographie – Version 12
khv – 31 – 22/12/2000
3.5.2 Spektroskopische Detektionen
Photometrische Detektion Die photometrische oder UV/VIS-Detektion ist aufgrund ihres sehr grossen Anwendungsbereiches in der HPLC die wichtigste Detektionsvariante, da nahezu alle organischer Moleküle über chromophore Gruppen verfügen, die im UV- oder im VIS-Bereich zu absorbieren vermögen. Voraussetzung ist, dass der verwendetet Eluent im betreffenden Wellenlängenbereich keine Absorption zeigt. Bei einer direkten Detektion im Absorptionsmaximum eines Analyten ist die UV/VIS-Detektion praktisch selektiv. Substanzen, die im betrachteten Wellenlängenbereich nur eine geringe oder gar keine Absorption zeigen, können indirekt bestimmt werden, indem man im Absorptionsmaximum des Elutionssystems misst. Im Bereich der Analytik anorganischer Ionen spielt die UV/VIS-Detektion eine geringere Rolle. Während von den einfachen Anionen nur Analyten wie Nitrat, Bromid oder Iodid absorbieren, sind wichtige Analyten wie Fluorid, Sulfat oder Phosphat nur indirekt zugänglich [4]. Viele Kationen absorbieren überhaupt nicht, aber insbesondere multivalente und Übergangsmetalle können in einer Nachsäulenderivatisierung mit Chelatbildnern wie 4-(2-Pyridylazo)-Resorcinol (PAR) oder Tiron umgesetzt werden, wodurch farbige Komplexe entstehen. Redoxaktive Analyten wie Bromat und andere Oxohalogenid-Ionen können durch eine Nachsäulenreaktion mit einem elektrochemisch aktiven Indikator der UV/VIS-Detektion zugänglich gemacht werden.
Fluoreszenzdetektion Die Fluoreszenzdetektion ist sehr empfindlich und immer dann möglich, wenn Analyten zur Fluoreszenz angeregt werden können, was vor allem bei organischen Verbindungen mit ausgedehnten π-Elektronensystemen der Fall ist. Typische Anwendungen liegen daher im Bereich der medizinischen bzw. organischen Analytik. Im Zusammenhang mit der Ionenchromatographie wird die Fluoreszenzdetektion in einigen Spezialfällen angewandt, da nur besondere Ionen wie Ce3+ direkt zugänglich sind und nicht fluoreszierende Ionen erst nach einer Derivatisierung detektiert werden können. Die Entwicklung von Elutionssystemen für diese Detektionsmethode ist wegen ihrer starken Anfälligkeit gegen Störungen durch Verunreinigungen sehr schwierig. Weiterhin ist der lineare Bereich des Verfahrens aufgrund von Selbstabsorptionseffekten mit oft weniger als 2 Grössenordnungen recht gering.
Kopplungstechniken Die sogenannten Kopplungstechniken stellen die Verbindung eines chromatographischen Systems mit einer eigenständigen Analysenmethode, zumeist spektrometrische Verfahren, dar [3]. Diese Verfahren haben in den letzten Jahren deutlich an Bedeutung gewonnen. Während im Bereich der Gaschromatographie Kopplungen mit der Massenspektrometrie (GC-MS) wohl etabliert sind, bereitet die Kopplung der HPLC mit spektrometrischen Verfahren grössere technische Probleme. Für die klassische HPLC, also der Analytik von organischen Verbindungen, stehen heute Kopplungen mit der Massenspektrometrie (LC-MS), der IR-Spektroskopie (LC-FTIR) und der Kernresonanzspektroskopie (LC-NMR) zur Verfügung [3]. Für die Ionenchromatographie (IC) finden insbesondere leistungsfähige atomspektrometrische Detektoren Verwendung. Beispiele sind die Atomemission und die Massenspektrometrie mit dem induktiv gekoppelten Plasma (IC-ICP-AES, MS), welche aufgrund ihrer Elementspezifität und Empfindlichkeit ausgezeichnete Leistungsdaten liefern. Daher werden solche Kopplungen trotz der vergleichsweise hohen Anschaffungskosten im Bereich der Spezies- und Ultraspurenanalytik von Elementen angewendet.
Refraktometrie Die Differentialrefraktometrie ist eine weitere auf einem optischen Verfahren beruhende Detektionsmethode. Dieser Detektor wird auch RI-Detektor genannt (engl. Refractive Index). Er ist völlig unspezifisch und kann universell eingesetzt werden, da die Messgrösse eine vom Analyten bewirkte Änderung des Brechungsindex des reinen Eluenten ist. Allerdings resultiert daraus auch aufgrund der starken Temperaturabhängigkeit des Brechungsindex eine grosse Anfälligkeit gegenüber Störungen. Das Verfahren ist, absolute Temperaturkonstanz vorausgesetzt, über drei Grössenordnungen linear. Da einfache anorganische Ionen einen extrem niedrigen Brechungsindex besitzen, können diese nur indirekt unter Verwendung eines mit stark brechenden Verbindungen versetzten Eluenten bestimmt werden.

Praktikum der Ionenchromatographie – Version 12
khv – 32 – 22/12/2000
3.6 Stationäre Phasen in der Ionenchromatographie Eine effiziente Ionenchromatographie verlangt Packungsmaterialien, die aus sehr kleinen, möglichst sphärischen Teilchen mit enger Teilchengrössenverteilung bestehen. Verwendet werden Partikeldurchmesser im Bereich von 2 bis 10 µm. Weiterhin soll das Packungsmaterial eine möglichst schnelle Kinetik des Ionenaustausches zeigen. Dies bestimmt neben der Teilchengrösse die Leistungsfähigkeit von Ionenaustauschern.
3.6.1 Übersicht gebräuchlicher stationärer Phasen Für die Ionenchromatographie eignen sich die unterschiedlichsten Materialien anorganischer und organischer Natur. Allen ist gemeinsam, dass sie an ihrer Oberfläche funktionelle Gruppen tragen, die Ionen auszutauschen vermögen. Folgende Stoffklassen können verwendet werden [4]:
• Modifizierte organische Polymerharze • Modifizierte Silicagele • Anorganische Salze (z.B. Polyphosphate) • Gläser • Zeolithe • Metalloxide (z.B. Al2O3) • Cellulosederivate
Daneben sind auch sehr komplex zusammengesetzte Systeme möglich, etwa Anionenaustauscher, deren funktionelle Gruppen aus Alkalimetall-Ionen bestehen, die durch Kronenether an eine stationäre Phase gebunden sind. Die wichtigsten Vertreter in der Praxis basieren auf modifizierten organischen Polymer-Harzen und Silicagelen. Abbildung 16 gibt eine Übersicht über die in der IC eingesetzten Trennmaterialien:
Polymerharze
makroporös mikroporös
oberflächenfunktionalisierte Materialien Agglomerierte Materialien
elektrostatischeBindung
hydrophobe Bindung
mechanische
PS/DVB- Copolymere
PS/MA- Copolymere
Ionenaustauscher
Silicagele
polymer- beschichtet
oberflächen - funktionalisiert
Abbildung 16 Gebräuchliche stationäre Phasen in der Ionenchromatographie [4]
Alle stationären Phasen lassen sich je nach Anwendungsbereich (Anionen- oder Kationenchromatographie) oder der Struktur der funktionellen Gruppe noch weiter differenzieren. Die Packungsmaterialien auf der Basis von Silicagel wurden ursprünglich für die Ionenchromatographie verwendet. Sie besitzen zwar eine sehr gute Trennleistung und sind mechanisch extrem stabil, können aber aufgrund ihrer chemischen Labilität nur im pH-Bereich zwischen 2 und 7 eingesetzt werden.
Etwa ab 1980 standen erstmals Ionenaustauscher auf Basis organischer Polymere zur Verfügung, die in der Ionenchromatographie verwendet werden konnten und durch Modifikation handelsüblicher Adsorberharze hergestellt wurden. Heute werden üblicherweise Packungsmaterialien eingesetzt, die entweder auf Polystyrol-Divinylbenzol-Copolymeren (PS-DVB) oder Methacrylat-Polymeren (MMA) beruhen.
Die beiden Grundtypen der Copolymere unterscheiden sich vor allem in ihrer Polarität. Während die PS-DVB-Copolymere völlig unpolar sind und RP-Phasen darstellen, sind die MMA-Polymere recht

Praktikum der Ionenchromatographie – Version 12
khv – 33 – 22/12/2000
polar. Dieser Umstand ist in der IC von Vorteil, da polarere Trennphasen weniger zu sekundären Wechselwirkungen wie Adsorption neigen.
Der grösste Vorteil der organischen Polymerharze liegt in ihrer grossen chemischen Stabilität über den gesamten pH-Bereich. Ihre chromatographische Effizienz ist nach anfänglichen Problemen im Bereich der Silicagele einzuordnen. Bei MMA-Phasen muss allerdings mit einer eingeschränkten mechanischen Stabilität gerechnet werden, die die Länge der zu verwendenden Trennsäule oder die maximal mögliche Flussrate des Eluenten begrenzt.
In der Ionenchromatographie werden heute zwei prinzipiell unterschiedlich aufgebaute Arten von stationären Phasen verwendet, die oberflächenfunktionalisierten und die pellikularen Ionenaustauscher. Bei den ersteren sind die funktionellen Gruppen direkt auf der Polymeroberfläche oder in den Poren lokalisiert, während bei den pellikularen Materialien sehr kleine, ebenfalls oberflächenfunktionalisierte Partikel an grössere Kernteilchen gebunden sind [4]. Die Bindung kann mechanisch oder durch hydrophobe bzw. elektrostatische Wechselwirkungen erfolgen. Abbildung 17 zeigt schematisch den Aufbau beider Typen von Packungsmaterialien am Beispiel von Anionenaustauschern.
N+R3
R3+N
R3+N
R3+N
N+R3
N+R3
N+R3
N+R3
Polymerpartikel
a)
Polymerpartikel
N+R3
R3+N
R3+N
R3+N
N+R3
N+R3
N+R3
N+R3
aminiertes Latexparikel
Bindungs- material
b)
Abbildung 17 Prinzipieller Aufbau von oberflächenfunktionalisierten (a) und pellikularen Anionenaustauschern mit mechanischer Bindung (b)
Die pellikularen Packungsmaterialien besitzen eine höhere chromatographische Effizienz, da durch die grössere Entfernung der funktionellen Gruppen vom Basismaterial die Diffusionswege sehr kurz gehalten werden, woraus ein exzellenter Massentransfer resultiert. Die chemische Stabilität dieser Trennphasen ist aber deutlich geringer als die der oberflächenfunktionalisierten Materialien.
3.6.2 Stationäre Phasen für die Anionenchromatographie Die eigentliche funktionelle Gruppe im Bereich der Anionenchromatographie wird üblicherweise durch die Umsetzung einer Ankergruppe mit einem geeigneten Amin erzeugt. Dadurch entstehen auf der Polymeroberfläche fixierte Ammonium-Ionen. Zur Trennung von Anionen mittels IC werden fast ausschliesslich auf Stickstoff basierende funktionelle Gruppen verwendet. Dies ist vor allem in ihrer ausserordentlichen chemischen Stabilität und der fast unbegrenzten Zahl der möglichen Substituenten am Stickstoffatom begründet.
Die Generierung von Ammoniumgruppen auf Polymeroberflächen erfolgt durch Umsetzung einer Ankergruppe mit einem Amin.
Die Alkylreste am positiv geladenen Stickstoff können in weiten Grenzen variiert werden. Im einfachsten Fall ist R = H und es entsteht ein primäres (1°) Ammonium-Ion. Dieses kann aber bei höheren pH-Werten deprotoniert werden und damit seine Ladung verlieren. Bei Packungsmaterialien dieser Art ist die Austauschkapazität vom pH-Wert des Eluenten abhängig, weshalb sie auch als schwach basisch bezeichnet werden. Substituiert man nun sukzessive die Wasserstoffatome durch Alkylgruppen, entstehen zunächst sekundäre (2°) und tertiäre (3°) Ammoniumgruppen, die ebenfalls noch deprotoniert werden können. Erst wenn alle Reste R aus Alkylgruppen bestehen, ist die Kapazität oder Ladung vom pH-Wert des Eluenten unabhängig und man erhält stark basische, quartäre (4°) Anionenaustauscher. Für die Chromatographie strebt man üblicherweise eine pH-unabhängige Kapazität an, so dass nur vollständig alkylierte Materialien verwendet werden. Bei

Praktikum der Ionenchromatographie – Version 12
khv – 34 – 22/12/2000
speziellen Anwendungen wie der Protein-Analytik oder Anreicherungstechniken kommen aber auch schwach basische Materialien zum Einsatz.
Die beiden wichtigsten funktionellen Gruppen in der Anionenchromatographie leiten sich vom Trimethylamin (TMA) und Dimethylethanolamin (2-Dimethylaminoethanol, DMEA) ab. Praktisch alle kommerziell verfügbaren Trennmaterialien verwenden eine dieser Gruppen. TMA-Gruppen werden in der Literatur auch als Typ I, DMEA-Gruppen als Typ II bezeichnet. Weitere Funktionalitäten, die mit den beiden erstgenannten in enger Beziehung stehen, sind in der Abbildung 18 gezeigt:
Abbildung 18 Übersicht über die wichtigen bzw. in Rahmen dieser Arbeit verwendeten funktionellen Gruppen
TMA: Trimethylamin (Typ I) DEMA: Diethanolmethylamin EDMA: Ethyldimethylamin TEA: Triethanolamin DMEA: Dimethylethanolamin (Typ II)
Bei kommerziellen Materialien, die sich ohnehin meist vom Typ I oder II ableiten, ist der genaue Aufbau der funktionellen Gruppe ein gut gehütetes Geheimnis [4].
3.6.3 Stationäre Phasen in der Kationenchromatographie Im der Kationenchromatographie sind sowohl Materialien auf Basis Silicagel als auch auf Basis von Polymeren im Gebrauch. Im Gegensatz zur Anionenchromatographie mit ihren meist alkalischen Eluenten sind die Bedingungen der Kationenchromartographie auch für Silicagele geeignet.
3.6.4 Kationenaustauscher auf Silicagelbasis Bei Kationenaustauschern auf Silicagelbasis wird unterschieden zwischen direkt funktionalisierten Materialien und solchen, die mit einem Polymer beschichtet sind.
Bei den direkt funktionalisierten Materialien wird praktisch nur von stark sauren Austauschern mit der Sulfonsäuregruppe berichtet [a2,a4]. Diese zeigen eine gute chromatographische Effizienz, sind aber für eine simultane Bestimmung von Alkali- und Erdalkalimetallen aufgrund der grossen Affinitätsunterschiede nicht geeignet.
Bei polymerbeschichteten Silicagelen, sog. Schomburg-Phasen, wird die Silikat-Oberfläche mit einem »Prepolymer» belegt, welches anschliessend durch Quervernetzung immobilisiert wird. Durch nachträgliche Funktionalisierung sind verschiedene Austauschertypen darstellbar. Zur Darstellung von schwach sauren Kationenaustauschern wird Polybutadienmaleinsäure (PBDMA) verwendet, die dann in-situ radikalisch quervernetzt wird [30]. Aufgrund der dünnen Polymerschicht von etwa 1 bis 5 nm [31] sind die Diffusionswege der Analyten kurz, dies führt zu einer hohen chromatographischen Effizienz.
Es gibt eine grosse Anzahl von Applikationen für die Ionenaustauscher auf Silicagelbasis vorhanden. Die simultane Trennung von Alkali- und Erdalkalimetallen gehört zu der häufigsten Anwendung, die Trennung von Übergangs- und Schwermetall-Ionen ist ebenfalls möglich. Der universellen Anwendung der Ionenaustauscher auf Silicagelbasis stehen jedoch einige Nachteile entgegen:
• Bei pH-Werten <2 wird die Bindung zwischen Silizium-Grundgerüst und funktioneller Gruppe zunehmend geschwächt, was zu einer allmählichen Auswaschung der Funktionalitäten und damit zu einem Kapazitätsverlust führt.
• Bei pH-Werten >7 nimmt die Löslichkeit des Silicagels deutlich zu, was die mechanische Stabilität der Packung herabsetzt, zu einem Brechen der Partikel führt und ein Totvolumen am Säulenanfang verursacht.

Praktikum der Ionenchromatographie – Version 12
khv – 35 – 22/12/2000
3.6.5 Kationenaustauscher auf Basis von organischen Polymeren Es werden überwiegend Harze aus Styrol-Divinylbenzol-Copolymerisaten als Grundgerüst verwendet. Die für Silicagel-Austauscher angeführten Einschränkungen bestehen hier nicht, sie sind über den gesamten pH-Bereich von 0-14 einsetzbar und auch inert gegenüber Fluorid. Die Druckbeständigkeit ist zwar geringer als bei den Silicagelen, aber ausser bei einigen Methacrylat-Harzen im allgemeinen ausreichend.
Als funktionelle Gruppen werden in kommerziell erhältlichen Kationenaustauschern überwiegend Sulfonsäuregruppen verwendet. Diese können direkt oder über einen Spacer variabler Länge an das aromatische System gebunden sein. Die Sulfonsäureaustauscher mit Spacer zeigen eine deutlich höhere chromatographische Effizienz, die Länge des Spacers hat aber eher untergeordnete Bedeutung. Für die simultane Bestimmung von Alkali- und Erdalkalimetallen sind sie aufgrund der grossen Affinitätsunterschiede nicht geeignet.
3.6.6 Pellikulare Kationenaustauscher Neben den direkt funktionalisierten Harzen gibt es noch pellikulare Austauscher. Sie haben einen doppelten Schichtaufbau, da es nicht möglich ist, einen vollständig aminierten Substratpartikel darzustellen. Darum wird ein totalsulfonierter Substratpartikel erst mit einer Schicht aus aminierten und dann mit einer Schicht aus sulfonierten Latexteilchen umgeben. Die Fixierung erfolgt über elektrostatische und van-der-Waals-Wechselwirkungen. Der relativ grosse Durchmesser des Substratpartikels (10-30µm) bewirkt einen vergleichsweise geringen Staudruck. Der kleine Durchmesser (20-250 nm) der Latexteilchen ermöglicht kurze Diffusionswege mit entsprechend schnellen Austauschprozessen und damit eine hohe Effizienz der Trennsäule. Nachteilig bei den pellikularen Materialien ist ihre Empfindlichkeit gegenüber organischen Lösungsmitteln [32] und gegenüber mobilen Phasen mit hoher Ionenstärke, da hier allmählich die Latexteilchen verdrängt werden.
Die simultane Bestimmung von Alkali- und Erdalkalimetallen ist mit einer abgewandelten Variante möglich, hier ist die aminierte Schicht kovalent gebunden. Jedoch sind die Selektivitätsunterschiede zwischen Kalium, Magnesium und Calcium unnötig gross, so das relativ lange Analysenzeiten benötigt werden. Dies ist auf die verwendete, stark saure Sulfonsäuregruppe zurückzuführen.
3.6.7 Stationäre Phasen in der Ionenausschlusschromatographie Die Auswahl stationärer Phasen für die IEC ist recht begrenzt. Wichtig für den Trennprozess ist die Ausbildung der Donnan-Membran durch dissozierte funktionelle Gruppen. Deren Anzahl sollte recht hoch sein. Weiterhin soll das Packungsmaterial eine Adsorption der Analyten auf der stationären Phase möglichst vermeiden. Da sich die Analyten im wesentlichen aus den Gruppen der Carbonsäuren und Zucker gruppieren, ist hier eine möglichst polare Oberfläche wünschenswert. An die Kinetik der Ionenaustauschreaktion wird praktisch keine Anforderung gestellt, da die Trennung auf einem Ausschlussmechanismus basiert. Eingesetzt werden praktisch ausschliesslich niedrig quervernetze PS/DVB Polymere, die vollständig sulfoniert werden.
3.6.8 Die Bedeutung der Kapazität von Ionenaustauschern Neben der Natur des Grundgerüstes und der Art der funktionellen Gruppe ist die Austauschkapazität Q die entscheidende Grösse zur Charakterisierung von Ionenaustauschern. Durch sie lässt sich angeben, wie viele Plätze für einen Ionenaustausch zur Verfügung stehen.
Die Austauschkapazität wird üblicherweise pro Gramm trockenes Packungsmaterial angegeben, entweder in Mikroequivalenten (µeq/g) oder Mikromolen (µMol/g) [2]. Daneben sind auch eine Reihe weiterer Angaben gebräuchlich [33]. Für analytische Zwecke lassen sich Ionenaustauscher grob nach ihrer Kapazität einstufen in:
• niederkapazitive Materialien: Q < 100 µMol/g • mittelkapazitive Materialien: 100 < Q < 200 µMol/g • hochkapazitive Materialien: Q > 100 µMol/g
Die im klassischen Ionenaustausch verwendeten Trennphasen sind mit 3 bis 5 mMol/g durchweg hochkapazitiver Natur [4].
Die obige Definition der Kapazität beruht auf einer vollständigen Gleichgewichtseinstellung zwischen stationärer und mobiler Phase, weshalb sie auch als statische Kapazität bezeichnet wird. Unter der

Praktikum der Ionenchromatographie – Version 12
khv – 36 – 22/12/2000
dynamischen (effektiven) Kapazität versteht man dagegen die Zahl der funktionellen Gruppen, die tatsächlich während eines chromatographischen Prozesses zur Verfügung steht. Sie wird immer kleiner sein als die statische Kapazität [2,4].
Die Austauschkapazität lässt sich auf verschiedene Arten bestimmen [33]:
• volumetrisch bzw. titrimetrisch • durch Elementaranalyse • über die Bestimmung von Retentionszeiten
Alle Verfahren werden bei demselben Material unterschiedliche Werte für Q liefern. In der Praxis werden volumetrische Verfahren am häufigsten angewendet. Bei Anionenaustauschern wird dabei eine gepackte Trennsäule oder eine definierte Menge an Harz z.B. mit einer Chlorid-Lösung beladen. Nach Auswaschen der Chlorid-Reste kann mit Nitrat eluiert werden. Die eluierte Menge an Chlorid, welche der statischen Austauschkapazität unter Gleichgewichtsbedingungen entspricht, wird dann mit einer AgNO3-Lösung titriert und quantifiziert.
Die pH-Abhängigkeit von Anionenaustauschern ist meist zu vernachlässigen. Bei Kationenaustauschern ist die Kapazität der schwach sauren Carbonsäure-Gruppen (R-COOH) nur bei höheren pH-Werten gegeben (Deprotonierung), während die stark sauren Sulfonsäure-Gruppen (R-SO3H) stets vollständig deprotoniert vorliegen und eine pH-unabhängige Kapazität liefern.
Im Zusammenhang mit den Retentionsmodellen (Kapitel 3.4) ist die Bedeutung von Q für die Auswahl von Elutions- und Detektionssystemen deutlich geworden. Die universelle Leitfähigkeitsdetektion war mit Eluenten hoher Ionenstärke praktisch nicht zu realisieren, gleichgültig welche speziellere Technik angewendet wurde. Dies ist der Grund, warum seit Einführung der Ionenchromatographie 1975 fast nur niederkapazitive Trennsäulen verwendet werden. Hochkapazitive Ionenaustauscher sind in der IC bisher nur in Verbindung mit Detektionstechniken beschrieben worden, die nicht so stark vom Elutionssystem limitiert werden, wie etwa die UV/VIS-Detektion [2,4].
Während niederkapazitive Trennmaterialien recht gut zur Analytik von Proben geringer Ionenstärke, also geringer Matrixbelastung, geeignet sind, erreichen sie bei hohen Ionenstärken schnell ihre Grenzen. Aufgrund ihrer geringen Anzahl an funktionellen Gruppen kommt es zu Überladungseffekten und damit zur Peakdeformation und einem drastischen Einbruch der Effizienz. Auch treten Probleme auf, wenn die Analyten in sehr unterschiedlichen Konzentrationsverhältnissen vorliegen, was im Bereich der Ultraspurenanalytik nahezu immer der Fall ist.
3.7 Eluenten in der Ionenchromatographie Wie bei allen flüssigchromatographischen Trennungen stellt auch in der Ionenchromatographie die mobile Phase denjenigen Parameter dar, der am leichtesten variiert werden kann, um eine Trennung zu beeinflussen. Die Trennsäule oder das Detektionssystem sind dagegen in den meisten Fällen vorgegeben.
Die Auswahl eines geeigneten Elutionssystems kann anhand der verschiedensten Kriterien erfolgen. Zu beachten sind für die Anionenchromatographie unter anderem folgende Parameter [4]:
• Kompatibilität mit der Detektionsmethode • Chemische Natur und Konzentration des Eluent-Ions • pH-Wert • Pufferkapazität • Gehalt an organischen Lösemitteln (Modifier)
In der gängigen Literatur [2,4,5] erfolgt die Diskussion der mobilen Phasen im wesentlichen unter dem Aspekt, ob sie für die sogenannte Einsäulen- oder Suppressortechnik geeignet sind. Dies soll auch im folgenden durchgeführt werden, wobei der Schwerpunkt aber im Hinblick auf die Thematik dieser Arbeit auf die Austauschkapazität Q der Trennsäulen gelegt wird. Zunächst erfolgt eine kurze Erläuterung der Begriffe «Einsäulentechnik» und «Suppressortechnik». Die Betrachtungen beschrän-ken sich wie in den vorausgehenden Kapiteln hauptsächlich auf die Anionenchromatographie.

Praktikum der Ionenchromatographie – Version 12
khv – 37 – 22/12/2000
3.7.1 Anionenchromatographie
Einsäulentechnik Bei der Einsäulentechnik, welche durch Gjerde und Mitarbeiter 1979 in der Ionenchromatographie etabliert wurde [33], ist der Ausgang einer Trennsäule direkt mit einem Detektor verbunden, was dem klassischen Aufbau einer HPLC-Apparatur entspricht. In Abgrenzung zur zweiten prinzipiellen Variante der IC wird diese Technik auch als ‚Nicht-Suppressierte-Ionenchromatographie‘ bezeichnet. Der die Trennsäule verlassende Eluent und die darin enthaltenen Analyten werden chemisch nicht verändert. Die Anzahl möglicher Elutionssysteme der unterschiedlichsten chemischen Natur ist praktisch unbegrenzt. Zu berücksichtigen ist, vom eigentlichen Trennproblem abgesehen, lediglich die Kompatibilität des Eluenten mit dem Detektor. Wenn z.B. die direkte UV-Detektion angewendet werden soll, darf der Eluent nicht im betreffenden Spektralbereich absorbieren. Bezüglich der Detektionsmethoden gibt es bei der Einsäulentechnik keine Beschränkungen, so dass im Prinzip alle gängigen HPLC-Detektoren Verwendung finden können.
Für niederkapazitive Anionenaustauscher (Q < 100 µMol/Trennsäule) stehen für die Einsäulentechnik eine ganze Reihe von Eluenten unterschiedlicher chemischer Natur zur Verfügung. Die Konzentration der Eluenten liegt dabei üblicherweise im unteren mMol/kg-Bereich oder noch darunter. Verwendet werden können unter anderem die unten genannten Substanzklassen [4]. Daneben können in spezielleren Fällen auch Komplexbildner wie EDTA oder Borat-Mannitol-Komplexe zum Einsatz kommen.
• Aromatische Carbonsäuren • Aliphatische Carbonsäuren • Sulfonsäuren • Alkalihydroxide • Anorganische Säuren wie H2SO4, HCl oder H3PO4
Die wichtigste Verbindungsklasse stellen die aromatischen Carbonsäuren und ihre Salze dar. Die am häufigsten verwendeten Vertreter sind in Abbildung 19 dargestellt.
Abbildung 19 Strukturen der wichtigsten aromatischen Carbonsäuren zur Verwendung mit der Einsäulentechnik.
Diese Substanzklasse findet deshalb so starke Verwendung, da die Lösungen der Säuren und deren Salze eine sehr hohe Elutionskraft bei relativ geringer Eigenleitfähigkeit besitzen, so dass sie zur direkten LF-Detektion eingesetzt werden können. Bei den mehrbasigen Säuren kann die Ladung und damit die Elutionskraft über den pH-Wert gesteuert werden. Dieser muss allerdings sehr exakt eingehalten werden. Aromatische Carbonsäuren besitzen eine hohe Absorption im UV-Bereich, so dass sie auch vorteilhaft mit der indirekten UV/VIS-Detektion angewendet werden können.
Für aromatische Sulfonsäuren wie p-Toluolsulfonsäure gelten im Prinzip die gleichen Aussagen, wobei diese aber immer deprotoniert vorliegen und daher keine Pufferkapazität besitzen. Ihre Elutionskraft lässt sich nur über die Konzentration steuern.
Aliphatische Carbonsäuren wie Oxalsäure und Citronensäure zeigen hohe Eigenleitfähigkeiten, sind aber UV-transparent, so dass sie mit der direkten UV/VIS-Detektion eingesetzt werden. Dies gilt ebenso für aliphatische Sulfonsäuren, von denen Methansulfonsäure am häufigsten verwendet wird. Die höheren Homologen beider Verbindungsklassen bieten aufgrund ihrer langen Kohlenwasserstoffketten und der damit verbundenen geringen Equivalentleitfähigkeiten die Möglichkeit der Anwendung der direkten LF-Detektion [2,4].
Alkalihydroxide lassen sich nur sehr begrenzt in der Einsäulentechnik einsetzen, da das OH–-Ion von allen möglichen Eluent-Anionen die geringste Affinität zu quartären Ammonium-Gruppen besitzt.

Praktikum der Ionenchromatographie – Version 12
khv – 38 – 22/12/2000
Daher sind selbst bei niedrigen Kapazitäten hohe Eluentkonzentrationen notwendig, so dass allenfalls die indirekte LF-Detektion verwendet werden kann. Gut möglich ist dagegen die direkte UV/VIS-Detektion, wobei aber auch das eigentlich UV-transparente Hydroxid-Ion bei höheren Konzentrationen eine deutliche Absorption unterhalb 220 nm zeigt.
Bei Verwendung von anorganischen Säuren oder deren Salzen ist man aufgrund ihrer nahezu vollständigen Dissoziation und damit hohen Leitfähigkeit auf photometrische Detektoren angewiesen. Beim Einsatz von Phosphorsäure oder Phosphaten lässt sich aber die Pufferkapazität und die Elutionskraft über den pH-Wert des Eluenten steuern.
Die meisten der hier genannten Eluenten lassen auch andere Detektionssysteme wie Amperometrie, Fluorimetrie oder Kopplungstechniken zu. Zur Verwendung der genannten Elutionssysteme mit diesen Detektoren sei aber an dieser Stelle auf die einschlägige Literatur verwiesen [2,4,5].
Suppressortechnik Die Suppressortechnik ist die bei der Einführung der IC ursprünglich verwendete Detektionstechnik [1]. Im Gegensatz zur Einsäulentechnik verwendet diese Methode ausschliesslich die Leitfähigkeitsdetektion (LF-Detektion). Bei der Suppressortechnik wird zwischen Trennsäule und Detektor ein sogenannter Suppressor geschaltet, weshalb diese auch als ‚Suppressierte-Ionenchromatographie‘ bezeichnet wird [2,4]. Im Suppressor werden sowohl der Eluent als auch die Analyten chemisch modifiziert, was im Hinblick auf die nachfolgende Leitfähigkeitsdetektion von besonderer Bedeutung ist. Der Suppressor hat die Aufgabe, die Eigenleitfähigkeit des Eluenten zu reduzieren und, wenn möglich, die Detektierbarkeit der Analyten zu erhöhen.
Das Prinzip der chemischen Suppression ist in den Gleichungen 63 und 64 für einen Anwendungsfall aus der Anionenchromatographie dargestellt. Verwendet wird ein NaHCO3-Eluent, als Analyt fungiert das Chlorid-Ion. Die Suppression erfolgt mit einem stark sauren Kationenaustauscher in der H+-Form.
R-SO3-H+ + Na+ + HCO3
- R-SO3-Na+ + H2O + CO2 (63)
R-SO3-H+ + Na+ + Cl- R-SO3
-Na+ + H+ + Cl- (64)
Die Suppressoreinheit besteht im einfachsten Fall aus einer der Trennsäule nachgeschalteten Säule, woraus sich der alte Begriff ‚Zweisäulentechnik‘ erklärt. Der Eluent Natriumbicarbonat wird gemäss Gleichung 63 neutralisiert, da die Natrium-Ionen durch Protonen ersetzt werden. Dadurch wird die Eigenleitfähigkeit des Eluenten drastisch herabgesetzt. Der Analyt Cl- wird selbst nicht verändert (Gleichung 64), allerdings wird sein Gegen-Ion Na+ durch H+ ausgetauscht, welches eine deutlich grössere Equivalentleitfähigkeit besitzt [19]. Da der Detektor die Summe der Leitfähigkeiten von Analyt- und Gegen-Ion als Signal registriert, ergibt sich also aus beiden ablaufenden Reaktionen ein deutlicher Empfindlichkeitsgewinn.
Die ursprünglich als Suppressoren verwendeten Säulen mit Kationenaustauschern in der H+-Form tragen deutlich zur Bandenverbreiterung bei. Ausserdem können sie nur diskontinuierlich betrieben werden, da der Kationenaustauscher regeneriert werden muss. Deshalb werden heute überwiegend kontinuierlich arbeitende Membransuppressoren verwendet, bei denen der Regenerant, normalerweise verdünnte Schwefelsäure, und der Eluent im Gegenstromprinzip aneinander vorbei geführt werden [6].
Die Suppressortechnik hat trotz ihrer Vorzüge aber auch einige entscheidende Nachteile. In der Praxis lassen sich in der Anionenchromatographie nur solche Eluenten erfolgreich suppressieren, die auf Alkalihydroxiden oder -carbonaten basieren. Anionen schwacher Säuren, z.B. Acetat oder Fluorid, liegen nach der Suppressionsreaktion praktisch vollständig protoniert vor, so dass ihre Detektierbarkeit im Vergleich zur Einsäulentechnik wesentlich geringer ist. Höherwertige Kationen müssen vor der Analyse entfernt werden, da sie schwer lösliche Hydroxide bilden, die auf der Trennsäule ausfallen und zur Verstopfung führen können.
Wie bereits deutlich wurde, ist die Suppressortechnik gleichbedeutend mit der chemischen Suppression des Eluenten und der Anwendung der direkten LF-Detektion [2,4]. Diese Technik ist in der Anionenchromatographie ausserordentlich weit verbreitet, vor allem weil sie in vielen Fällen empfindlicher als die direkte LF-Detektion in der Einsäulentechnik ist. Die Eigenleitfähigkeit von klassischen Eluenten (z.B. 2 mMol/kg Phthalat, pH = 8) in der Einsäulentechnik liegt bei etwa 200 µS/cm. Bei suppressierbaren Eluenten ist sie mindestens um eine Grössenordnung niedriger.

Praktikum der Ionenchromatographie – Version 12
khv – 39 – 22/12/2000
Die chemische Unterdrückung der Eigenleitfähigkeit ist nur bei sehr wenigen Eluenten möglich. Dies sind Lösungen von [4]:
• Alkalihydroxiden • Alkalicarbonaten und -hydrogencarbonaten • Boraten (z.B. B4O7
2–) • Aminosäuren
In der Praxis haben von den genannten Eluenten aber nur die Alkalihydroxide und die Carbonat-Puffer eine grössere Bedeutung, womit die Auswahl an potentiellen mobilen Phasen sehr beschränkt ist.
Das Hydroxid-Ion ist ein extrem schwaches Eluent-Ion, so dass auch bei niederkapazitiven Trennmaterialien bereits mit hohen Konzentrationen oberhalb von 50 mMol/kg gearbeitet werden muss. Bei Anwendung sehr polarer funktioneller Gruppen kann die relative Retentionskraft des OH–-Ions unter Ausnutzung der Hydroxid-Selektivität deutlich erhöht werden. Bei OH–-Eluenten ist eine Manipulation von Retentionszeiten oder Selektivitäten nur über die Konzentration möglich.
Der Einsatz von Alkalicarbonaten und- Alkaliydrogencarbonaten ermöglicht wesentlich flexiblerer Elutionssysteme. Beide Spezies, HCO3
– und CO32–, liegen nach der Suppression als Kohlensäure
H2CO3 vor, die nur zu einem sehr geringen Anteil dissoziiert ist. Hydrogencarbonat ist in seiner Elutionskraft nur geringfügig stärker als Hydroxid, während Carbonat einen relativ starken Eluenten darstellt. Beide Anionen werden üblicherweise zusammen verwendet, was zu einem Eluenten mit Pufferwirkung führt, dessen Elutionskraft sehr leicht über die Konzentrationen der beiden Komponenten und deren Konzentrationsverhältnis gesteuert werden kann. Aufgrund der Ladungen der Eluent-Spezies lassen sich die Selektivitäten für mono- und multivalente Analyten sehr gezielt beeinflussen. Das Konzentrationsverhältnis beider Eluent-Ionen lässt sich zudem sehr genau über den pH-Wert einstellen, weshalb der sinnvolle pH-Bereich für HCO3
–/CO32–-Eluenten zwischen 8 und 11
liegt. Wie auch bei den OH--Eluenten kann die Elution durch Verwendung stationärer Phasen mit polaren funktionellen Gruppen beschleunigt werden.
Beide Elutionssysteme lassen sich bei oberflächenfunktionalisierten Anionenaustauschern mit Erfolg nur dann anwenden, wenn entweder das Grundgerüst (Methacrylat-Copolymere) oder aber die funktionellen Gruppen eine hohe Polarität besitzen. Bei auf PS-DVB basierenden Trennsäulen werden für weiche Analyten (Nitrat, Bromid) auch bei Anwendung polarer Funktionalitäten sehr schlechte Peaksymmetrien und lange Retentionszeiten beobachtet. Bei pellikularen Materialien treten diese Effekte nicht so stark hervor, was ihre ausserordentlich grosse Verbreitung in der Suppressortechnik erklärt [2,4].
3.8 Kationenchromatographie
3.8.1 Kationenchromatographie von Alkali-, Erdalkali- und Ammonium-Ionen mit Leitfähig-keitsdetektion
Für die ionenchromatographische Trennung von Alkalimetall- und Ammonium-Ionen sowie kurzkettigen aliphatischen Aminen auf sulfonierten Trennphasen verwendet man vorwiegend Mineralsäuren wie HCl oder HNO3 als Eluent [4]. Die Säurekonzentration des Eluenten richtet sich dabei nach Art und Kapazität des eingesetzten Kationenaustauschers und liegt bei einigen mMol/l. Divalente Kationen wie die Erdalkalimetalle können mit Mineralsäuren nicht eluiert werden, da sie eine deutlich höhere Affinität zur stationären Phase aufweisen und eine drastische Erhöhung der Säurekonzentration die Detektion zu unempfindlich machen würde, bzw. die Suppression nicht mehr gewährleistet wäre. Alternativ können zur Trennung der Erdalkalimetall-Ionen auch organische Basen wie das Ethylendiamin verwendet werden. Dies liegt bei niedrigen pH-Werten protoniert als zweiwertiges Kationen vor.
Die simultane Analyse von Alkali- und Erdalkalimetall-Kationen an stark sauren Kationenaustauschern erfolgt hauptsächlich mit Eluenten, die Salzsäure und 2,3-Diaminopropionsäure enthalten [2]. Durch Variation des pH-Wertes ist es möglich, den Protonierungsgrad der Amino-Gruppen und damit die Elutionskraft der 2,3-Diaminopropionsäure zu verändern (siehe Kapitel: Retentionsmodell für Eluenten mit einem Kation).
In ionenchromatographischen Systemen ohne Suppression können neben den Mineralsäuren schwache organische Säuren als Eluenten eingesetzt werden. Anwendung finden z.B. Oxalsäure, Citronensäure, Weinsäure. Komplexbildner wie 2,6-Pyridindicarbonsäure (PDCA) und 18-Krone-6, um die Analysenzeiten einzelner Kationen selektiv zu beeinflussen.

Praktikum der Ionenchromatographie – Version 12
khv – 40 – 22/12/2000
Bei der Durchführung der Leitfähigkeitsdetektion unterscheidet man zwei Arten. Ist die Grundleitfähig-keit des Eluenten hoch, z. B. bei verdünnten Mineralsäuren aufgrund der hohen Leitfähigkeit der H+-Ionen, bietet sich die Möglichkeit der direkten Leitfähigkeitsdetektion an, wobei die Analyten eine deutlich geringere Leitfähigkeit als der Eluent aufweisen. Es werden daher negative Peaks erzeugt, die aber durch Umpolung des Detektors oder durch eine einfache Invertierung in übliche Chromato-gramme verwandelt werden können. Lässt die Qualität des Leitfähigkeitsdetektors die empfindliche Leitfähigkeitsmessung auf hohem Niveau nicht zu, so wird auch in der Kationenchromatographie mit Suppression gearbeitet. Die Konstruktion von Supressoren für die Kationenchromatographie ist weit-aus schwieriger als in der Anionenchromatographie und auch ihre Lebensdauer ist deutlich geringer.
3.8.2 Kationenchromatographie von Übergangs- und Erdalkalimetall-Ionen mit Nachsäulen-derivatisierung und photometrischer Detektion
Für die Trennung von Übergangs- und Schwermetall-Ionen sind monovalente Kationen wie H+ oder Na+ als Eluent ungeeignet, da sich die Selektivitätskoeffizienten der Analyten bei gleicher Ladungszahl kaum unterscheiden. Die Trennung lässt sich jedoch durch Einführung eines Sekundärgleichgewichts realisieren. Hierzu werden als Eluent komplexierende Carbonsäuren (siehe Abbildung 20) wie Citro-nensäure, Oxalsäure und Weinsäure eingesetzt. Diese bilden mit den Metall-Ionen neutrale oder anio-nische Komplexe (siehe Kapitel Retentionsmodelle in der Ionenchromatographie).
OHO
OH
OH
OHO O
HO
OH
O
OH O
O
OH
O OH
HO
Weinsäure Oxalsäure Citronensäure Abbildung 20 Verschiedene Carbonsäuren als Eluenten in der Kationenchromatographie.
Durch die Komplexierung der Metall-Kationen wird die effektive Ladungsdichte der Analyten verringert. Zudem erhöht man durch die unterschiedlichen Komplexbildungskonstanten für die einzelnen Metall-Ionen die Selektivität der Trennung. Der Elutionsmechanismus resultiert aus der gleichionischen Verdrängung durch das Gegenion (»pushing»-Effekt) und aus der Komplexbildung (»pulling»-Effekt) durch den komplexierenden Liganden [4].
In Abbildung 21 werden die am Elutionsmechanismus beteiligten Gleichgewichte zwischen dem Analyten M2+, dem Komplexbildner L2- und den Gegenionen E+ schematisch dargestellt.
S O 3 - SO3
-
Me 2 + 2 E++
L 2 - + 2 H+H 2 L
Me L
"pulling"-Effekt des Komp l e x b i l d n e r s
"pushing"-Effekt des G e g e n i o n s
Abbildung 21 Schematische Darstellung der am Austauschprozess beteiligten Gleichgewichte [4].
Das Ausmass der gleichionischen Verdrängung durch die Gegenionen E+ wird durch die Affinität des Kations zur stationären Phase bestimmt. Bei monovalenten Gegenionen wirkt das Kation mit der grösseren Affinität zur stationären Phase stärker eluierend. Den Einfluss des Komplexbildners kann

Praktikum der Ionenchromatographie – Version 12
khv – 41 – 22/12/2000
man durch pH-Wert und Konzentration des Eluenten variieren. Zudem kann man das Elutionsvermögen durch den Einsatz mehrerer Komplexbildner sowie durch Verwendung eines zweiwertigen Kations als Gegenion beeinflussen.
Während für die Detektion von Alkali- und Erdalkalimetallen beide Anwendungsformen der Leitfähigkeitsdetektion geeignet sind, kann man Übergangs- und Schwermetall-Ionen nur durch direkte Leitfähigkeitsmessung ohne Suppressor bestimmen. Bei der Anwendung der Suppressor-Technik werden diese Metalle durch die Suppressorreaktion in meist unlösliche Hydroxide überführt. Die direkte Leitfähigkeitsdetektion ist nur möglich, wenn man beim Einsatz von niedrigkapazitiven Kationenaustauschern Eluenten mit geringer Grundleitfähigkeit verwendet.
Zur Detektion von Übergangs- und Schwermetall-Ionen wird daher hauptsächlich die Nachsäulenderivatisierung zu photometrisch detektierbaren Metall-Komplexen eingesetzt. Dabei wird dem Effluat nach der analytischen Trennsäule in einem Nachreaktor ein metallochromes Reagenz beigemischt, das sich mit den Analyten zu farbigen Metall-Komplexen umsetzt, die photometrisch detektiert werden. Als Farbreagenz lassen sich eine Vielzahl von Azofarbstoffen einsetzen [2,4], die mit einer grossen Anzahl an Metall-Kationen reagieren. Übergangsmetalle und die Lanthanide reagieren mit 4-(2-Pyridylazo)resorcinol (PAR) (Abbildung 22) zu farbigen Komplexen. Lanthanide und Actinide lassen sich mit 2,7-bis(2-Arsenophenylazo)-1,8-dihydroxynaphthalin-3,6-disulfonsäure (Arsenazo III) detektieren. Brenzkatechin-3,5-disulfonsäure (Tiron) ist zur Nachsäulenderivatisierung von Aluminium geeignet.
NN N OH
HO
4-(2-Pyridylazo)resorcin (PAR) Abbildung 22 Struktur des Metallindikators PAR
Zur Detektion der Übergangs- und Schwermetall-Ionen wird hauptsächlich PAR verwendet. PAR bildet mit Fe, Co, Ni, Cu, Zn, Mn, Pb und Cd farbige Komplexe, die bei Wellenlängen von 490 bis 520 nm mit Absorptionskoeffizienten bis zu 104 l mol-1 cm-1 absorbieren und photometrisch detektiert werden können. Die Empfindlichkeit des Verfahrens beruht darauf, dass die Extinktionskoeffizienten der Metall-PAR-Komplexe gross sind gegenüber dem Absorptionskoeffizienten des Reagenzes bei der verwendeten Wellenlänge.
3.8.3 Ionenausschlusschromatographie Die Auswahl des Elutionsmittels in der IEC ist wie die Wahl des Packungsmaterials wenig aufregend. Angefangen von reinem Wasser bis hin zu verdünnten Mineralsäuren reicht die Palette. Für die Auswahl des jeweils am besten geeigneten Systems muss die Detektion beachtet werden. Die häufigsten Detektionen sind die photometrische und die Leitfähigkeitsdetektion. Im Fall der Photometrie wird häufig verdünnte Schwefel- oder Perchlorsäure aufgrund ihrer absoluten UV-Transparenz eingesetzt. Soll die Leitfähigkeitsdetektion verwendet werden, so findet meist verdünnte Salzsäure in Verbindung mit einem Kationenchromatographie-Suppressor Anwendung.
Die Konzentration der Säure bestimmt die Dissoziation der Analyten und damit auch deren Retention. Allgemein verringert sich die Retention mit steigender Säurekonzentration. Die Peakform wird ebenso von der Säurekonzentration beeinflusst. Für organische Säuren ist reines Wasser aufgrund einer steigenden Adsorption meist nicht geeignet, für Carbonat und Borsäure liefert es dagegen hervorragende Resultate.

Praktikum der Ionenchromatographie – Version 12
khv – 42 – 22/12/2000
4 Praktischer Teil Im praktischen Teil des Lehrbuches werden Versuche vorgestellt, die einen ausführlichen Streifzug durch die Welt der Ionenchromatographie ermöglichen. Begonnen wird mit den Versuchen zur Theorie der Ionenchromatographie (Seite 45), es geht weiter im zweiten Teil auf Seite 71 mit der Analyse von Anionen und im dritten und letzten Teil ab Seite115 werden organische und anorganische Kationen analysiert.
Die Versuche können prinzipiell auf jedem Ionenchromatographen durchgeführt werden. Einige Anforderungen an die Ausstattung des Gerätes sollten trotzdem erfüllt sein:
• pulsationsarme Doppelkolbenpumpe, die möglichst ohne externe Stickstoff-/oder Heliumver-sorgung auskommt
• Flussregelung entsprechend den Säulenanforderungen • elektrisches Injektionsventil • Anschlussmöglichkeiten für verschiedene Probenschleifen und Anreicherungssäulen • elektronische Suppression für Anionen und Kationen • chemische Suppression für Anionen • temperaturstabilisierter Leitfähigkeitsdetektor, möglichst besser ± 0.01 °C • thermisch und elektronisch abgeschirmtes Gehäuse • Anionen- und Kationenbetrieb • PC-Kontrolle ist empfehlenswert
Alle aus dem Hause Metrohm stammenden kompakten oder modularen C-Systeme erfüllen selbstverständlich diese Anforderungen. Der Basic IC 792 ist allerdings speziell für Ausbildung und Lehre konzipiert und deshalb für die nachfolgenden Versuch besonders zu empfehlen.
Abbildung 23 Der Metrohm Basic IC 792: speziell für Forschung und Lehre entwickelt
4.1 Hinweise zum praktischen Arbeiten
Bakterielles Wachstum
Um bakterielles Wachstum zu verhindern sollten Eluenten, Spül- und Regenerationslösungen immer frisch angesetzt und nicht nach längerer Zeit wiederverwendet werden. Sollten sich trotzdem Bakterien oder Algen bilden, kann dem Eluenten 5 % Methanol oder Aceton zugesetzt werden. Dies ist bei der Verwendung von Membransuppressoren nicht möglich, da diese durch organische Lösungsmittel zerstört werden können. Das Metrohm Suppressor Modul «MSM» aber ist 100 % lösungsmittelbeständig.
Chemikalienqualität
Sämtliche Chemikalien sollten mindestens die Qualität p.a. oder puriss. besitzen. Die Standards müssen speziell für die Ionenchromatographie geeignet sein.

Praktikum der Ionenchromatographie – Version 12
khv – 43 – 22/12/2000
Entgasen des Eluenten Um Blasenbildung zu verhindern, wird empfohlen, das für die Eluentenherstellung verwendete Wasser vor Zugabe der Chemikalien zu entgasen. Hierfür wird für circa 10 Minuten ein Wasserstrahl- oder Ölpumpenvakuum oder ein Ultraschallbad verwendet
Kationenanalysen
Bei allen durchgeführten Analysen müssen die Proben mit Salpetersäure (circa 100 µL einer 2 mol/L HNO3 auf 100 mL Probe) angesäuert werden (pH 2.5 – 3.5), da ansonsten für die zweiwertigen Kationen keine reproduizierbaren Resultate erhalten werden.
Lagerung der Trennsäulen
• METROSEP IC Anionensäule Anion Dual 1 (6.1006.020)
Die Säulenkartusche wird kurzfristig (Tage) im verschlossenen Halter gelagert. Bei langfristiger Lagerung (Wochen) wird die Säule im mitgelieferten Aufbewahrungsgefäss in Aceton/Wasser (1:9) im Dunkeln gelagert.
Abbildung 24 METROSEP IC Anionensäule Anion Dual 1 (6.1006.020) mit Säulenhalter
• METROSEP IC Anionensäule A SUPP 5 (6.1006.530)
Die Säule wird im Eluenten gelagert.
• METROSEP IC Kationensäule C2 (6.1010.210)
Die Säule wird im Eluent im Kühlschrank (4 °C) gelagert. Es ist unbedingt darauf zu achten, dass die Säule nicht austrocknet.
Abbildung 25 METROSEP IC Kationensäule C2 (6.1010.210

Praktikum der Ionenchromatographie – Version 12
khv – 44 – 22/12/2000
• METROSEP IC Kationensäule Nucleosil S5A (6.1007.000)
Die Säule wird kurzfristig (Tage) im Eluenten gelagert, langfristig (Wochen) in Methanol/Wasser (1:4).
• METROSEP Organic Acids (6.1005.200)
Die Säule wird für kurze Zeit (Tage) im Eluenten und für längere Zeit (Wochen) in aq. demin gelagert.
• Trennsäulen anderer Hersteller
Bitte beachten Sie die Angaben der Hersteller.
Säulenauswahl
Die meisten der vorgestellten Versuche, werden auf sehr kostengünstigen Säulen ausgeführt – METROSEP Dual 1 für die Anionen und METROSEP C2 für die Kationen. Diese Säulen liefern eine ausreichend gute Trennung für alle beschriebenen Experimente. Selbstverständlich gibt es aus der METROSEP-Produktlinie Trennsäulen, die wesentlich leistungsfähiger, deshalb aber auch teurer sind.
Sicherheitshinweise
Bei der Durchführung aller Versuche sind Schutzbrille, Schutzkittel und ggf. Schutzhandschuhe zu tragen. Die Sicherheitshinweise für die Chemikalien (R/S-Sätze) sind unbedingt zu beachten.
Stillegen des Gerätes
Wird über längere Zeit (> 1 Woche) mit dem Ionenchromatographen nicht gearbeitet, so sollte die Trennsäule ausgebaut und der Ionenchromatograph mit Methanol/Wasser (1:4) gespült werden. Dabei ist darauf zu achten, dass auch alle drei Kammern des Suppressors gespült sind.
Umweltschutz
Ein grosser Vorteil der Ionenchromatographie ist, das meistens mit wässrigen Medien gearbeitet wird. Die in der Ionenchromatographie verwendeten Chemikalien sind deshalb weitestgehend ungiftig und belasten die Umwelt nicht. Trotzdem muss darauf geachtet werden, wenn mit Säuren, Basen, organischen Lösungsmitteln oder Schwermetallstandards gearbeitet wird, dass diese nach Gebrauch ordnungsgemäss entsorgt werden.
Wasserqualität
In der Ionenchromatographie wird vorwiegend mit wässrigen Medien gearbeitet. Die Wasserqualität ist deshalb ganz entscheidend für eine gute Chromatographie. Ist die Qualität ungenügend, so sind es die Ergebnisse definitiv auch. Zusätzlich besteht die Gefahr, Geräte und Trennsäulen zu beschädigen. Das verwendete demineralisierte Wasser «aq. demin.» sollte deshalb eine Widerstand grösser 18 MΩ aufweisen und partikelfrei sein. Es ist deshalb zu empfehlen, das Wasser über einen 0.45 µm Filter zu filtrieren.

Praktikum der Ionenchromatographie – Version 12
khv – 45 – 22/12/2000
4.2 Versuche zur Theorie der Ionenchromatographie
4.2.1 Versuch 1 – Ionenchromatographie mit und ohne chemische Suppression Die Äquivalentleitfähigkeit Λ∞ setzt sich immer additiv aus den Äquivalentleitfähigkeiten aller Anionen Λ∞
– und Kationen Λ∞+ in der Lösung zusammen:
Λ∞ = Λ∞+ + Λ∞
–
Generell gilt, dass die Leitfähigkeit mit der Elektrolyt- bzw. Ionenkonzentration steigt. Ein linearer Zusammenhang ergibt sich nur für verdünnte Lösungen, da Λ konzentrationsabhängig ist (Kohlrausches Gesetz). Die tabellierten Werte (vergl. Kapitel «Leitfähigkeitsdetektion») gelten für Λ∞ – Äquivalentleitfähigkeit in unendlich verdünnten Lösungen.
Die Grösse der Äquivalentleitfähigkeit ist stark temperaturabhängig (± 2 % / °C). Vor allem bei Messungen ohne chemische Suppression, das heisst auf einem hohen Hintergrund, machen sich Fehler aus einer Temperaturänderung besonders stark bemerkbar. Die Temperaturschwankungen sollten deshalb unbedingt kleiner 0.01 °C sein.
In der Ionenchromatographie ohne chemische Suppression, bei der die Hintergrundleitfähigleit nur elektronisch unterdrückt wird, sollten die Eluenten eine möglichst geringe Leitfähigkeit aufweisen. Hierfür kommen die Salze schwacher Säuren wie der Phthalsäure, Salicylsäure und Benzoesäure in Frage. Der ohne Suppression bestimmte Messwert ist von Differenz der Äquivalentleitfähigkeiten zwischen Probeion und Eluentuion abhängig.
∆Λ ~ (ΛP – ΛE)
Negative Peaks treten immer dann auf, wenn die Leitfähigkeit des Probeions niedriger als die des Eluentions ist. Ein Beispiel dafür ist das Phosphatanion. Bei einem pH = 5 liegt es überwiegend als H2PO4
– vor. Da die Äquivalentleitfähigkeit Λ∞ des H2PO4– mit 33 S*cm2/mol niedriger ist als die
Äquivalentleitfähigkeit Λ∞ des Phthalats mit 38 S*cm2/mol ist, erhält man einen kleinen, negativen Peak. Abhilfe schafft ein höherer pH-Wert, bei dem das Phosphat als HPO4
2– vorliegt, dessen Äquivalentleitfähigkeit Λ∞ 57 S*cm2/mol beträgt
Ionenchromatographie mit chemischer Suppression bedeutet, dass die Hintergrundleitfähigkeit chemisch herabgesetzt wird. Ein Eluent mit hoher Leitfähigkeit wird in einer Nachsäulenreaktion – der Suppressionsreaktion – zu einem Eluenten mit geringer Leitfähigkeit umgesetzt.
In der Anionenanalytik werden hierfür im Eluenten die Salze schwacher Säuren eingesetzt z. B. HCO3
–, CO3
2– und BO33–. Im Suppressor werden alle Kationen im Eluenten und in der Probe gegen H+
ausgetauscht. Gemäss nachfolgender Reaktion entstehen aus dem Eluenten schwach dissoziierte Säuren:
Na+ + HCO3- ⎯⎯⎯ →⎯
++−+ NaH H2CO3
Die entstehende Kohlensäure liegt überwiegend als CO2 + H2O vor. Daher ist die Restleitfähigkeit sehr gering. Da auch die Gegenionen der zu bestimmenden Anionen im Suppressor gegen H+ ausgetauscht werden lässt sich folgende Gleichung formulieren:
Na+ + Cl- ⎯⎯⎯ →⎯++−+ NaH
H+ + Cl-
Statt des ursprünglich in der Probe vorhandenen Na+ und Cl– wird nun die wesentlich höhere Äquivalentleitfähigkeit von H+ und Cl– gemessen und dies zusätzlich noch auf einer geringen Hintergrundleitfähigkeit. Theoretisch lässt sich ein Signal erwarten, dass um den Faktor 10 höher liegt als bei einer Messung ohne chemische Suppression. In der Praxis gefunden wird hingegen nur ein Empfindlichkeitszuwachs im Bereich eines Faktors von 2 bis 4.
Im Gegensatz zur linearen Kalibration beim Arbeiten ohne chemische Suppression, erhält man beim Arbeiten mit chemischer Suppression eine quadratische Kalibrationsfunktion, deren Berechnung die Kalibration mehrerer Punkte erforderlich macht. Der Konzentrationsbereich, in dem man mit linear

Praktikum der Ionenchromatographie – Version 12
khv – 46 – 22/12/2000
arbeiten kann ist wesentlich geringer (ca. 1/20 bis 1/50) als beim Arbeiten ohne chemische Suppression.
Lerninhalte
• Prinzipieller Aufbau eines IC-Systems
• Unterschiede zwischen dem Arbeiten mit chemischer und ohne chemische Suppression
• Bestimmung der unterschiedlichen Empfindlichkeiten der beiden Methoden
Versuch 1a – Messung ohne chemische Suppression
Tabelle 3 Parameter – Versuch 1a
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 8 mmol/L Phthalsäure, 2% Acetonitril; pH = 4.1 (TRIS)
Leitfähigkeit ca. 400 µS/cm Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat) Methode exp_01_n.mtw System anonsupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 20 min Loop 100 µL Polarität +
Herstellung des Eluenten
1.33 g Phthalsäure in 20 ml Acetonitril und etwas Wasser lösen, danach auf 1 L auffüllen. Durch Zugabe von TRIS (fest) (ca. 1 g) den pH-Wert auf 4.1 einstellen. Das verwendete Wasser vor der Zugabe der Chemikalien für 10 min am Wasserstrahlpumpenvakuum oder im Ultraschallbad entgasen.
Chromatogramm 1 Standardlösung (versuch1a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 47 – 22/12/2000
Tabelle 4 Komponenten – Versuch 1a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.6 5 2310 51 2 Chlorid 4.9 5 3680 71 3 Nitrit 5.7 5 3670 36 4 Bromid 6.7 10 3960 63 5 Nitrat 7.8 10 3890 74 6 Sulfat 10.9 10 3430 88 7 Systempeak 17
Versuch 1b – Messung mit chemischer Suppression
Tabelle 5 Parameter – Versuch 1b
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat) Methode exp_01_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 16 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Chromatogramm 2 Standardlösung (versuch1b.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 48 – 22/12/2000
Tabelle 6 Komponenten – Versuch 1b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.8 2 2780 48 2 Chlorid 5.7 5 4100 81 Systempeak 6.8 3 Nitrit 7.3 5 3900 46 4 Bromid 8.6 10 4510 69 5 Nitrat 10.4 10 4480 87 6 Phosphat 11.7 10 3220 43 7 Sulfat 14.4 10 3660 113

Praktikum der Ionenchromatographie – Version 12
khv – 49 – 22/12/2000
4.2.2 Versuch 2 – Kapazität von Trennsäulen Abgeschnittene Peaks, Peaks in der Form von Haifischflossen, veränderte Retentionszeiten für gleiche Ionen, Peaks die Tailing oder Fronting zeigen – all dies kann eine gemeinsame Ursache haben: Überladung der Säule.
Jede Säule hat eine endliche Zahl von Ionentauscherplätzen. Bevor die Probe injiziert wird, sind diese vollständig mit Eluentionen belegt. Wird die Probe aufgegeben beginnt der Ionenaustausch: Eluentionen werden gegen Probeionen und Probeionen gegen Eluentionen ausgetauscht. Da sich die Ionenspezies in ihren Bindungskonstanten unterscheiden, eluieren sie unterschiedlich schnell von der Säule. Das gewünschte Ergebnis ist die Trennung des Substanzgemisches und damit das Chromatogramm.
Dieser Prozess funktioniert nur dann einwandfrei, wenn die Zahl der Austauschplätze höher als die Zahl der von der Probe benötigten Bindungsplätze ist. Als Beispiel: eine Säule mit einer Kapazität von 1 mEq (Milli-Equivalent) kann maximal 1 mmol einwertiger Ionen binden und trennen. Ist die Ionenkonzentration der Probe höher, ist die Interaktion zwischen Probeionen und den Austauscherplätzen der stationären Phase nicht mehr für alle Ionen möglich und die Ionen werden nicht mehr ausreichend getrennt In der Praxis verschlechtert sich die Chromatographie bereits bei einem Belegungsgrad grösser drei Viertel der theoretischen Säulenkapazität.
Ein zweiter Effekt, der die Trennung negativ beeinflusst, ist der Umstand, das jedes Ion prinzipiell auch als Eluention agieren kann. Mit der Überladung der Säule werden zu viele Eluentionen durch die Probeionen ersetzt. Damit kommt es zu unkalkulierbaren Veränderungen der Säulengleichgewichte und die Trennung verschlechtert sich.
Die Kapazität einer Säule kann bestimmt werden, in dem sie vollständig mit Chloridionen belegt wird. Nach einem Spülschritt mit demineralisiertem Wasser werden die Chloridionen mit einem Carbonateluenten eluiert und dann mittels Ionenchromatographie oder argentometrischer Titration quantifiziert.
Im nachfolgenden Experiment wird die Chloridkonzentration bis zur Überladung der Säule gesteigert. Einige Effekte, die mit der Überladung der Säule einhergehen werden im folgenden beschrieben:
→ die Retentionszeiten der nachfolgenden Ionen verkürzen sich,
→ der Peak des dominaten Ione wird abgeschnitten,
→ der Peak des dominanten Ions tailt,
→ die Anzahl der theoretischen Bodenzahl «theoretical plate counts» (TP) wird geringer,
→ das Area/Hight- Verhältnis wird schlechter und
→ die Peaksymmetrie verschlechtert sich.
Die Peaksysmmetrie verschlechtert sich auch bei verstopfter Säulenfritte, erhöhtem Totvolumen und bedingt durch Absorptionserscheinungen auf dem Säulenmaterial.
Lerninhalte
• Erklären der chromatographischen Parameter: Retentionszeit, Auflösung, Fläche, Bodenzahlen, Symmetrie und «Area-Hight»-Verhältnis
• Einfluss einer dominanten Hauptkomponente auf Komponenten wesentlich geringerer Konzentration: Veränderung der o.a. Parameter

Praktikum der Ionenchromatographie – Version 12
khv – 50 – 22/12/2000
Tabelle 7 Parameter – Versuch 2
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat)
+ NaCl (ca. 1 g eingewogen) Methode exp_02_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 16 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und danach 20 mL Aceton zugeben.
Chromatogramm 3 Standardlösung mit sehr hoher Chloridkonzentration (versuch2.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 51 – 22/12/2000
Tabelle 8 Komponenten – Versuch 2
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl 4.2.2.1.1 Area [µS/cm*sec
] 1 Chlorid 7.4 n.b. n.b. n.b. 2 Nitrit 9.4 5 35540 16 3 Bromid 9.7 10 15710 52 4 Nitrat 10.9 10 7350 87 5 Phosphat 11.8 10 4540 19 6 Sulfat 13.9 10 1360 109
Bemerkungen
Natriumchlorid wird eingewogen. Die Retentionszeiten und die Werte für Bodenzahl und Fläche (Area) können je nach eingewogener NaCl-Konzentration variieren.

Praktikum der Ionenchromatographie – Version 12
khv – 52 – 22/12/2000
4.2.3 Versuch 3 – Selektivität von Trennsäulen Wird die Stärke des Eluenten gegen den Logarithmus der Retentionszeit von ein- und zweiwertigen Ionen aufgetragen, so erhält man eine Gerade. Die Steilheit dieser Geraden ist grösser für zweiwertige als für einwertige Ionen. Die Steigerung der Elutionsstärke des Eluenten hat damit einen grösseren Einfluss auf die Retentionszeit der zweiwertigen als auf die Retentionszeit der einwertigen Ionen.
Dieser Einfluss ist am Sulfat-Ion zu beobachten. Sulfat wird mit gesteigerter Eluentkonzentration verhältnismässig stärker beschleunigt, als die einwertigen Ionen.
Generell eluieren zweiwertige Ionen im Eluenten stärker als einwertige Ionen, da sie zwei Bindungen mit der stationäre Phase eingehen können und so stärker mit dieser in Wechselwirkung treten können.
Natronlauge hat bei gleicher Molarität einen grösseren Einfluss auf den pH-Wert des Eluenten als Carbonat/Bicarbonat. Die Elutionskraft des OH–-Ions ist aber verglichen mit der des Hydrogencarbonats niedriger, da das Hydroxyl-Ion eine geringere Wechselwirkung mit der stationären Phase aufweist.
Die Retentionszeit des Phosphats ist stark pH-abhängig. Bei hohem pH-Wert verschiebt sich das Gleichgewicht von PO4
2– in Richtung PO43–. Damit verlängert sich die Retentionszeit des Phosphats
während sich die Retentionszeiten aller anderen Ionen weiter verkürzen.
Lerninhalte
• Vergleich von Eluenten mit einwertigen und zweiwertigen Anionen
• Vergleich von Natronlauge- und Carbonat/Hydrogencarbonateluenten
• Einfluss des Eluent-pH-Wertes auf die Retention des Phosphats
Tabelle 9 Parameter – Versuch 3a bis 3d
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluenten a) 1 mmol/L Na2CO3 / 4 mmol/L NaHCO3
b) 4 mmol/L Na2CO3 / 1 mmol/L NaHCO3 c) 4 mmol/L Na2CO3 / 1 mmol/L NaOH (pH=11-12) d) 5 mmol/L Na2CO3 / 5 mmol/L NaOH (pH=11-12) Leitfähigkeit nach chemischer Suppression ca. 17 µS/cm
Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat) Methode exp_03_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer a) 30 min
b) 12 min c) 10 min d) 8 min
Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +

Praktikum der Ionenchromatographie – Version 12
khv – 53 – 22/12/2000
Versuch 3a – 1 mmol/L Na2CO3 / 4 mmol/L NaHCO3
Herstellung des Eluenten
106 mg Natriumcarbonat (wasserfrei) und 336 mg Natriumhydrogencarbonat in 1 L Reinstwasser lösen.
Chromatogramm 4 Standardlösung – 1 mmol/L Na2CO3 / 4 mmol/L NaHCO3 (versuch3a.gif)
Tabelle 10 Komponenten – Versuch 3a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 4.5 2 3340 49 2 Chlorid 7.2 5 4730 83 3 Nitrit 9.5 5 4360 47 4 Bromid 11.4 10 1830 69 5 Nitrat 14.0 10 4660 92 6 Phosphat 20.4 10 3530 46 7 Sulfat 26.5 10 4120 118

Praktikum der Ionenchromatographie – Version 12
khv – 54 – 22/12/2000
Versuch 3b – 4 mmol/L Na2CO3 / 1 mmol/L NaHCO3
Herstellung des Eluenten
424 mg Natriumcarbonat (wasserfrei) und 84 mg Natriumhydrogencarbonat in 1 L Reinstwasser lösen.
Chromatogramm 5 Standardlösung – 4 mmol/L Na2CO3 / 1 mmol/L NaHCO3 (versuch3b.gif)
Tabelle 11 Komponenten – Versuch 3b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.4 2 2820 50 2 Chlorid 5.1 5 4350 84 3 Nitrit 6.5 5 3980 47 4 Bromid 7.5 10 4690 71 5 Nitrat + Phosphat 9.0 10 2840 139 6 Sulfat 10.1 10 3750 117

Praktikum der Ionenchromatographie – Version 12
khv – 55 – 22/12/2000
Versuch 3c – 4 mmol/L Na2CO3 / 1 mmol/L NaOH
Herstellung des Eluenten
424 mg Natriumcarbonat (wasserfrei), 40 mg NaOH in 1 L Reinstwasser lösen.
Chromatogramm 6 Standardlösung – 4 mmol/L Na2CO3 / 1 mmol/L NaOH (versuch3c.gif)
Tabelle 12 Komponenetentabelle – Versuch 3c
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.3 2 2390 51 2 Chlorid 4.5 5 3010 86 3 Nitrit 5.6 5 3650 49 4 Bromid 6.3 10 4220 74 5 Nitrat 7.5 10 3790 96 6 Phosphat + Sulfat 8.3 10 / 10 2510 171

Praktikum der Ionenchromatographie – Version 12
khv – 56 – 22/12/2000
Versuch 3d – 5 mmol/L Na2CO3 / 5 mmol/L NaOH
Eluentherstellung
424 mg Natriumcarbonat (wasserfrei), 200 mg NaOH in 1 L Reinstwasser lösen und entgasen.
Chromatogramm 7 Standardlösung – 5 mmol/L Na2CO3 / 5 mmol/L NaOH (versuch3d.gif)
Tabelle 13 Komponenten – Versuch 3d
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.0 2 2470 49 2 Chlorid 4.1 5 3850 86 3 Nitrit 5.0 5 3600 48 4 Bromid 5.6 10 4200 71 5 Sulfat 6.2 10 2990 120 6 Nitrat 6.6 10 5130 103 7 Phosphat 7.7 10 2570 46

Praktikum der Ionenchromatographie – Version 12
khv – 57 – 22/12/2000
4.2.4 Versuch 4 – Kalibration, Nachweis- und Bestimmungsgrenzen in der Ionenchromatographie
Wesentliche Parameter für analytische Bestimmungsmethoden sind Linearitätsbereich, Nachweis-grenze, Bestimmungsgrenze. Die mathematischen Verfahren hierzu sind z.B. in der DIN-Norm 32645 festgelegt.
Werden Chromatogramme mit einem Leitfähigkeitsdetektor aufgenommen, so wird meist die Peakfläche zur Auswertung herangezogen. Die Peakfläche ist proportional der Substanzmenge. Trägt man die Peakfläche gegen die Konzentration auf, so erhält man die Kalibrationsfunktion. Sie ist linear für Messungen ohne chemische Suppression. Sie folgt in erster Näherung einer quadratischen Funktion bei Messungen mit chemischer Suppression. Auswertungsprogramme berechnen die Kalibrationsfunktionen automatisch.
Mit der Nachweisgrenze ist die minimale Konzentration eines Analyten gemeint, bei der dieser mit bekannter statistischer Sicherheit noch nachgewiesen werden kann. So berechnet man die theoretisch niedrigste Konzentration, die gerade noch von einem Blindwert zu unterscheiden ist.
Es gibt zwei Methoden, die Nachweisgrenze (NG) zu bestimmen:
- die Leerwert- oder Blindwertmethode.
Die Blindprobe sollte eine Probe sein, die das zu bestimmende Ion nicht enthält. Die wiederholte Messung einer Blindprobe ergibt zu einer Konzentration «0» (X-Wert) Messwerte (Y-Werte), deren Dichtemittel als Leer- oder Blindwert bezeichnet wird. Dem maximalen Y-Wert wird mittels der Kalibriergerade auf der X-Achse ein Konzentrationswert zugeordnet, der gleich der Nachweisgrenze ist.
- Kalibrierkurvenmethode
Diese Methode kommt zur Anwendung, wenn effektiv kein Blindwert bestimmt werden kann, da das zu untersuchende Ion in der Probe nicht nachweisbar ist. Für die Kalibrierkurvenmethode werden unterschiedliche Konzentrationen eine Ions mehrfach vermessen. Aus der Standardabweichung ergibt sich ein Vertrauensbereich. Damit entspricht der Konzentration «0» ein bestimmtes Y-Intervall. Über die Kalibrierfunktion wird dem Y-Intervall eine Konzentrationsintervall gleichgesetzt, dessen Maximum die Nachweisgrenze ist.
Oft wird zur Ermittlung der Nachweisgrenze auch das Signal-/Rauschverhältnis herangezogen. Zum Beispiel wird als Nachweisgrenze die Analytkonzentration bezeichnet, bei der das Messsignal 2x oder 3x grösser als das Rauschen der Basislinie ist.
Die Bestimmungsgrenze (BG) ist erst dann erreicht, wenn der Messfehler im Vergleich zum Analysenwert einen bestimmten Wert unterschreitet, z. B. 1/3. Erst dann wird ein Zahlenwert im Analysenbefund angegeben, da andernfalls der Messfehler als zu gross im Vergleich zum Analysenwert betrachtet wird. Als Abschätzung gilt, dass die Bestimmungsgrenze um den Faktor drei höher als die Nachweisgrenze ist
Lerninhalte
• Was bedeutet Kalibration
• Vergleich einer Einpunkt- mit einer Mehrpunktkalibration – Fehlerabschätzung
• Bestimmung des Systemrauschens
• Abschätzung der Nachweisgrenzen

Praktikum der Ionenchromatographie – Version 12
khv – 58 – 22/12/2000
4.2.5 Versuch 4a – Bestimmung von Anionen mit chemischer Suppression
Tabelle 14 Parameter – Versuch 4a
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat) Methode exp_04_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 16 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen. Danach 20 ml Aceton zugeben.
Chromatogramm 8 Merge – Standardlösungen unterschiedlicher Konzentration (versuch4a.gif)
Tabelle 15 Komponenten Versuch 4a
Konz. [mg/L] Peak Komponente tR [min] Level 1 Level 2 Level 3 Level 4 1 Fluorid 3.9 1 5 25 50 2 Chlorid 5.6 1 5 25 50 3 Systempeak 6.8 4 Nitrit 7.5 1 5 25 50 5 Bromid 8.6 1 5 25 50 6 Nitrat 10.3 1 5 25 50 7 Phosphat 11.7 1 5 25 50 8 Sulfat 14.3 1 5 25 50

Praktikum der Ionenchromatographie – Version 12
khv – 59 – 22/12/2000
Versuch 4b – Bestimmung von Anionen ohne Suppression
Tabelle 16 Parameter – Versuch 4b
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 8 mmol/L Phthalsäure, 2% Acetonitril; pH = 4.1
Leitfähigkeit ca. 400 µS/cm Probe Standard (Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat, Sulfat) Methode exp_04_n.mtw System anonsupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 20 min Loop 100 µL Polarität +
Herstellung des Eluenten
1.33 g Phthalsäure in 20 ml Acetonitril und etwas Wasser lösen, danach auf 1 L auffüllen. Durch Zugabe von TRIS (fest) den pH-Wert auf 4.1 einstellen.
Chromatogramm 9 Merge – Standardlösungen unterschiedlicher Konzentration (versuch4b.gif)
Tabelle 17 Komponenetentabelle – Versuch 4b
Konz. [mg/L] Peak Komponente tR [min] Level 1 Level 2 Level 3 Level 4
1 Fluorid 3.6 1 5 25 50 2 Chlorid 4.9 1 5 25 50 3 Nitrit 5.7 1 5 25 50 4 Bromid 6.6 1 5 25 50 5 Nitrat 7.7 1 5 25 50 6 Sulfat 11.0 1 5 25 50 7 Systempeak 17.0

Praktikum der Ionenchromatographie – Version 12
khv – 60 – 22/12/2000
4.2.6 Versuch 5 – Veränderung der Selektivität mit Hilfe von Kronenethern (18 Crown-6) Mit der Zugabe von Komplexbildnern können die Retentionszeiten der Kationen verändert werden. Der Komplexbildner fungiert als Ligand, das Analytkation wird als Zentralmetallion eingebaut. Je selektiver ein Ligand bezüglich eines Zentralmetallions ist, desto stärker wird seine Retentionszeit beeinflusst. Die Retentionszeiten der übrigen Kationen werden im Idealfall nur geringfügig verändert.
Komplexbildner werden zur besseren Trennung von Alkalimetallionen eingesetzt. Der Zusatz des Kronenethers 18 Crown–6 zum Eluenten führt zu einer besseren Trennung von Na+, Ammonium und K+. Besonders drastisch ist die Erhöhung der Retentionszeit von K+. Dies lässt sich durch die Bildung des Komplexes aus K+ und Dibenzo-18-Krone-6-ether (18 Crown-6) erklären. K+ passt genau in den «Käfig» des Ethers. Es wird über die Elektronenpaare der Sauerstoffatome komplexiert. Bei gleicher Ladung wird nach der Komplexierung ein wesentlich grösseres Molekül getrennt. Bedingt durch die sterische Hinderung, verlängert sich so die Retentionszeit des Kaliums.
K+
O
O O
O
O
O
Abbildung 26 Kalium – 18 Crown – 6
Die Bezeichnung 18 Crown-6 zeigt an, dass das Ringsystem aus 18 Atomen besteht, von denen 6 Sauerstoffatome sind. Kronenether spielen aber nicht nur in der Ionenchromatographie eine wichtige Rolle, sie werden auch als ionenselektive Phase in Kaliumelektroden verwendet. Des weiteren sind sie für den Transport von Kaliumatomen durch Zellmembranen verantwortlich.
Lerninhalte
• Auswirkung eines sehr selektiven Komplexbildners auf die Retentionszeiten
• Erklärung des Effekts im Vergleich mit Versuch 6
Versuch 5a – Eluent ohne Kronenether
Tabelle 18 Parameter – Versuch 5a und 5b
Säule 6.1010.210 IC-Kationensäule Metrosep C2 100/4.0 - 7 µm Eluent a) 4 mmol/L Weinsäure/ 0.75 mmol/L Dipicolinsäure
b) 4 mmol/L Weinsäure/ 0.75 mmol/L Dipicolinsäure + 0.25 mmol/L Kronenäther Leitfähigkeit ca. 590 µS/cm
Probe Standard (Lithium, Natrium, Ammonium, Kalium, Calcium, Magnesium + 2 mmol/L HNO3)
Methode exp_05_c.mtw System cation.smt Fluss 1 mL/min Druck 8 MPa Analysendauer 12 min
17 min Loop 10 µL Polarität –
Herstellung des Eluenten
600 mg Weinsäure und 125 mg Dipicolinsäure in 100 mL Reinstwasser unter Erwärmen lösen, danach mit Reinstwasser auf 1 L auffüllen.

Praktikum der Ionenchromatographie – Version 12
khv – 61 – 22/12/2000
Chromatogramm 10 Standardlösung – Eluent ohne Kronenether (versuch5a.gif)
Tabelle 19 Komponenten – Versuch 5a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Lithium 2.7 1 4270 14 2 Natrium 3.6 5 4640 18 3 Ammonium 4.2 5 3640 19 4 Kalium 6.1 10 2890 17 5 Calcium 8.1 10 2950 33 6 Magnesium 9.9 10 2310 65

Praktikum der Ionenchromatographie – Version 12
khv – 62 – 22/12/2000
Versuch 5b – Eluent mit Kronenether
Herstellung des Eluenten
600 mg Weinsäure und 125 mg Dipicolinsäure in 100 mL Reinstwasser unter Erwärmen lösen, danach 132 mg Kronenether zugeben und mit Reinstwasser auf 1 L auffüllen.
Chromatogramm 11 Standardlösung – Eluent mit Kronenether (versuch5b.gif)
Tabelle 20 Komponenten Versuch 5b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Lithium 2.8 1 4250 14 2 Natrium 4.1 5 4550 18 3 Ammonium 5.1 5 3260 19 4 Calcium 9.3 10 2170 33 5 Magnesium 10.6 10 2200 66 6 Kalium 14.9 10 1910 17

Praktikum der Ionenchromatographie – Version 12
khv – 63 – 22/12/2000
4.2.7 Versuch 6 – Veränderung der Selektivität mit Hilfe von Komplexbildnern Bei der Analyse von Magnesium-, Natrium- und Kaliumionen neben Zink- und Calciumionen macht man sich die Eigenschaft von Zink- und Calciumionen zu nutze, mit Dipicolinsäure (DPA, 2,6-Pyridindicarbonsäure) Komplexe zu bilden.
Me2+
HH
OO
O OCC N
Me 2+ - Dipicolinkomplex
Die aus der Komplexbildung
Me2+ + (DPA) ↔ [MeDPA]2+
resultierende Konstante ist für jedes Metall unterschiedlich.
In Abhängigkeit vom pH-Wert, aber unabhängig von der Komplexbildungskonstante, können sich die folgenden Komplexe bilden: (zunehmende Dissoziation von H+ mit steigendem pH-Wert):
[Me2+(DPA)]2+ [Me2+(DPA)]+
+H+H+ H+
+H+ [Me2+(DPA)]0
saurer Bereich schwach saurer Bereich alkalischer Bereich
Je nach pH-Wert liegt also ein 2-fach positiver, 1-fach positiver oder ungeladener Komplex vor.
Primäres Trennkriterium auf einer Kationentauschersäule ist die Ladung der zu trennenden Ionen. Ungeladene Komplexe werden nicht zurückgehalten. Komplexe mit dreifach positiver Ladung würden nahezu irreversibel gebunden. Bedingt durch die Komplexbildungskonstante und den eingestellten pH-Wert liegt eine bestimmte mittlere Gleichgewichtsladung des Komplexes vor. Diese Gleichgewichtsladung bestimmt die Retentionszeit. Deshalb können die zweiwertigen Metallionen durch die Zugabe von Dipicolinsäure und einen bestimmten pH-Wert beschleunigt werden.
Einwertige Metallionen, die keinen Komplex mit der Dipicolinsäure bilden können, werden in ihrer Retentionszeit nicht beeinflusst.
Lerninhalte
• Einfluss der Komplexbildungskonstante auf die Retentionszeit – Vergleich Zink und Calcium
• Einfluss des pH-Wertes auf die Gesamtladung des Komplexes
• Erklärung des Effekts im Vergleich mit Versuch 5

Praktikum der Ionenchromatographie – Version 12
khv – 64 – 22/12/2000
Tabelle 21 Parameter – Versuch 6a bis 6d
Säule 6.1010.210 IC-Kationensäule Metrosep C2 100/4.0 – 7 µm Eluent a) 4 mmol/L Weinsäure
Leitfähigkeit ca. 500 µS/cm b) 4 mmol/L Weinsäure + 0.1 mmol/L Dipicolinsäure Leitfähigkeit ca. 500 µS/cm c) 4 mmol/L Weinsäure + 0.25 mmol/L Dipicolinsäure Leitfähigkeit ca. 520 µS/cm d) 4 mmol/L Weinsäure + 0.75 mmol/L Dipicolinsäure Leitfähigkeit ca. 590 µS/cm
Probe Standard (Natrium, Zink, Kalium, Calcium, Magnesium + 2 mmol/L HNO3) Methode exp_06_c.mtw System cation.smt Fluss 1 mL/min Druck 7 MPa Analysendauer a) 23 min
b) 20 min c) 16 min d) 13 min
Loop 10 µL Polarität -
Versuch 6a – 4 mmol/L Weinsäure
Herstellung des Eluenten
600 mg Weinsäure in 1L Reinstwasser lösen.
Chromatogramm 12 Standardlösung – Eluent 4 mmol/L Weinsäure (versuch6a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 65 – 22/12/2000
Tabelle 22 Komponenten – Versuch 6a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 4.3 5 4450 17 2 Kalium 7.8 10 2330 16 3 Zink 11.2 5 2330 11 4 Magnesium 14.4 10 2010 63 5 Calcium 19.5 10 2640 34
Versuch 6b – 4 mmol/L Weinsäure + 0.1 mmol/L Dipicolinsäure
Herstellung des Eluenten
600 mg Weinsäure und 17 mg Dipicolinsäure unter Erwärmen in 100 mL Reinstwasser lösen, danach auf 1L auffüllen.
Chromatogramm 13 Standardlösung – Eluent 4 mmol/L Weinsäure + 0.1 mmol/L Dipicolinsäure (versuch6b.gif)
Tabelle 23 Komponenten – Versuch 6b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Zink 1.9 5 554 6.7 2 Natrium 4.4 5 4390 17 3 Kalium 7.6 10 2750 16 4 Magnesium 14.8 10 2100 64 5 Calcium 17.9 10 2720 33

Praktikum der Ionenchromatographie – Version 12
khv – 66 – 22/12/2000
Versuch 6c – 4 mmol/L Weinsäure + 0.25 mmol/L Dipicolinsäure
Herstellung des Eluenten
600 mg Weinsäure und 42 mg Dipicolinsäure unter Erwärmen in 100 mL Reinstwasser lösen, danach auf 1L auffüllen.
Chromatogramm 14 Standardlösung – Eluent 4 mmol/L Weinsäure + 0.25 mmol/L Dipicolinsäure (versuch6c.gif)
Tabelle 24 Komponenten – Versuch 6c
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]– Zink ∼1.1 5 1 Natrium 4.3 5 4450 18 2 Kalium 7.8 10 2330 17 3 Calcium + Magnesium 14.4 10 + 10 2010 98

Praktikum der Ionenchromatographie – Version 12
khv – 67 – 22/12/2000
Versuch 6d – 4 mmol/L Weinsäure + 0.75 mmol/L Dipicolinsäure
Herstellung des Eluenten
600 mg Weinsäure und 125 mg Dipicolinsäure unter Erwärmen in 100 mL Reinstwasser lösen, danach auf 1L auffüllen.
Chromatogramm 15 Standardlösung – Eluent 4 mmol/L Weinsäure + 0.75 mmol/L Dipicolinsäure (versuch6d.gif)
Tabelle 25 Komponenten – Versuch 6d
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]– Zink ∼1.1 5 1 Natrium 3.8 5 4540 17 2 Kalium 6.4 10 2900 17 3 Calcium 8.5 10 2630 33 4 Magnesium 10.5 10 2190 67
Bemerkungen
Sämtliche Lösungen müssen in Kunststoffbehältern aufbewahrt werden. Für eine korrekte Natrium-Bestimmung muss jeglicher Kontakt mit Glas vermieden werden. Der pH-Wert der Standard- und Probelösungen muss zwischen 2.5 und 3.5 liegen.
Bei Wechsel des Eluenten das System einige Zeit laufen lassen bis Grundlinie konstant ist.
Zink wird durch die Dipicolinsäure komplexiert und eluiert im Frontpeak.

Praktikum der Ionenchromatographie – Version 12
khv – 68 – 22/12/2000
4.2.8 Versuch 7 – Anreicherungstechnik Die Probenaufgabe in der Ionenchromatographie erfolgt mit einer Probenschleife bzw. einem Sample loop, der auf dem Injektionsventil angebracht ist. Das Volumen der Probenschleife beträgt unter Standardbedingungen für Anionen 20 µL und für Kationen 10 µL. Mit Probenschleifen dieser Grösse erschliessen sich auf einem einfachen IC-System problemlos Nachweissgrenzen von 100 µg/L bzw 100 ppb.
Durch Vergrösserung des Sample loops z.B. auf 100 µL lassen sich die Nachweisgrenzen nur teilweises verringern: Wird ein grösseres Probenvolumen auf die Säule gebracht wird, so verschlechtert sich die Peakform und damit wiederum die Nachweisgrenze.
Eine einfache Methode zur Verringerung der Nachweisgrenzen ist dagegen die Probenanreicherung. Statt des Sample loops wird eine Anreicherungssäule auf das Injektionsventil montiert. Ein gesteigertes Probenvolumen – in unserem Beispiel 5 mL – wird auf die Anreicherungssäule gegeben. Diese Säule ist prinzipiell mit dem gleichen Material versehen, dass sich auch in der Trennsäule befindet. Dadurch wird gewährleistet, dass alle Anionen bzw. Kationen auf ihr vollständig zurückgehalten werden. Es wird wie üblich mit einem NaHCO3/Na2CO3-Eluenten gearbeitet.
Ist die Anreicherung erfolgt – Ventilposition «fill» – wird im Gegenstrom injiziert – Ventilposition «inject». Das Gegenstromverfahren wird angewendet, um den Einfluss der Anreicherungssäule auf die chromatographische Trennung zu minimieren.
Abbildung 27 Anreicherungstechnik: Abbildung links Position «fill», Abbildung rechts: Position «inject»
Mit der Anreicherungstechnik erschliessen sich selbst auf einem einfachen chromatographischen System Konzentrationen im unteren ppb-Bereich
Lerninhalte
• Was ist Probenvorbereitung?
• Wo ist die Schnittstelle zwischen Ionenchromatographie und Probenvorbereitung?
• Was ist der Vorteil von Probenanreicherung?
• Was sind die Grenzen des Verfahrens?

Praktikum der Ionenchromatographie – Version 12
khv – 69 – 22/12/2000
Tabelle 26 Parameter – Versuch 7
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Reinstwasser Methode exp_07_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 4 MPa Analysendauer 16 min Loop 5 mL auf METROSEP IC Anion Anreicherungssäule 6.1006.300 Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und 20 ml Aceton zugeben.
Chromatogramm 16 Standardlösung (1 ppb) (versuch7a.gif)
Tabelle 27 Komponenten – Versuch 7a
Peak Komponente tR [min] Konz. [µg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid + Acetat 3.8 1 + 1 1620 5.4 2 Chlorid 5.7 1 3090 5.2 3 Systempeak 6.8 4 Nitrit 7.4 1 3270 2.3 5 Bromid 8.6 1 3970 1.3 6 Nitrat 10.3 1 3600 1.9 7 Phosphat 11.6 1 3290 0.7 8 Sulfat 14.1 1 2980 1.1

Praktikum der Ionenchromatographie – Version 12
khv – 70 – 22/12/2000
Chromatogramm 17 Reinstwasser (versuch7b.gif)
Tabelle 28 Komponenten – Versuch 7b
Peak Komponente tR [min] Konz. [µg/L] Bodenzahl Area [µS/cm*sec]1 Chlorid 5.7 0.030 2240 0.17 2 Systempeak 6.8
Bemerkungen
Gefässe, Spritze und System mehrmals gründlich spülen, Plastikgefässe verwenden.

Praktikum der Ionenchromatographie – Version 12
khv – 71 – 22/12/2000
4.3 Versuche für die Bestimmung von Anionen
4.3.1 Versuch 8 – Bestimmung von Anionen in Trinkwasser Trinkwasser ist unser wichtigstes Lebensmittel. Es wird vor allem aus Grundwasser und Oberflächenwasser gewonnen. Als Oberflächenwasser wird See- und Talsperrenwasser, Uferfiltrat, mit Oberflächenwasser angereichertes Grundwasser sowie Flusswasser verwendet.
Nach DIN 2000 muss Trinkwasser folgende Grundanforderungen erfüllen:
Es soll farblos, klar, kühl und frei von fremdartigem Geruch und Geschmack sein. Es soll möglichst von Natur aus frei von Krankheitserregern und gesundheitsschädlichen Stoffen sein. Es soll nicht zu viele Salze, namentlich Härtebildner, Eisen, Mangan, sowie organische Stoffe (Moor- und Huminstoffe) enthalten. Es soll tunlichst keine Korrosion hervorrufen. Es soll stets auch der Menge nach allen Bedürfnissen der zu versorgenden Bevölkerung gerecht werden.
Je nach Verschmutzungsgrad werden verschiedene Verfahren zur Trinkwasseraufbereitung eingesetzt.
Rechen, entfernt grobe Verschmutzungen und grössere Partikel Sandfilter, dient der Filtration ausserdem finden in Sand biologische Abbauprozesse statt, die der Reinigung dienen Aktivkohlefilter adsorbieren gelöste organischen Substanzen, z. B. Pestizide Enteisenung, Entmanganung durch Oxidation von Fe(II) und Mn(II); dieser Prozess würde andernfalls in der Trinkwasserleitung stattfinden und zu braunen Trübungen oder Flocken im Trinkwasser führen. Desinfektion ist immer dann nötig, wenn das Wasser nicht frei von Krankheitserregern ist. Verwendet werden Chlor, Ozon, Chlordioxid, UV-Licht. Sicherheitschlorung vor Abgabe in die Trinkwasserleitung zur Vermeidung einer Wiederverkeimung auf dem Weg zum Verbraucher
Abbildung 28 Schema der Trinkwasseraufbereitung – Graphik von Thomas Seilnacht – Tuttling (wasserwerk.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 72 – 22/12/2000
Lerninhalte
• Untersuchung des Lebensmittels Nummer «1»
• Überprüfung der Angaben auf Mineralwasserflaschen
Tabelle 29 Grenzwerte für Trinkwasser (D)
Fluorid 1,5 mg/l als F- Nitrat 50 mg/l als NO3
-
Nitrit 0,1 mg/l als NO2-
Chlorid 250 mg/l Sulfat 250 mg/l
Tabelle 30 Parameter – Versuch 8
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Trinkwasser Methode exp_08_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 16 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und 20 mL Aceton zugeben.
Chromatogramm 18 Trinkwasser aus Herisau (Schweiz) (versuch8a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 73 – 22/12/2000
Tabelle 31 Komponenten – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Fluorid 3.7 0.05 2680 1.2 2 Chlorid 5.8 6.5 2500 104 3 Systempeak 6.8 4 Nitrat 10.3 8.4 4560 74 5 Sulfat 14.4 14.4 3670 68
Abbildung 29 Chromatogramm – Mineralwasser ohne Kohlensäure (versuch8b.gif)
Tabelle 32 Komponenten – Mineralwasser ohne Kohlensäure
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Fluorid 3.7 1.5 2560 35 2 Chlorid 5.7 14.1 2980 226 3 Systempeak 6.8 4 Bromid 8.5 0.075 4810 0.5 5 Nitrat 10.3 1.4 4400 12 6 Sulfat 14.3 300 3830 3400
Bemerkungen
Mineralwasser mit Kohlensäure muss vor der Messung entgast werden.

Praktikum der Ionenchromatographie – Version 12
khv – 74 – 22/12/2000
4.3.2 Versuch 9 – Anionen in Ethanol Die Bestimmung von Anionen in organischen Lösungsmitteln ist möglich, obwohl zu Beginn des Chromatogramms oft ausgeprägte Systempeaks auftreten. Als Beispiele soll die Analyse in folgenden Matrices dienen:
Anionen in Methanol können im ppb-Bereich bestimmt werden nach direkter Injektion (ohne Anreicherungssäule) und chemischer Suppression.
Ebenfalls direkt können Anionen im sub-ppb-Bereich in Butyrolacton bestimmt werden, und zwar ohne chemische Suppression. Butyrolacton ist ein wichtiges Lösungsmittel und Ausgangssubstanz für z. B. N-Methyl-pyrolidon, das für grosstechnisch wichtige Extraktionen (z. B. Butadien) verwendet wird.
Auch in unpolaren Lösungsmitteln, wie z. B. Toluol können Anionen bestimmt werden. Hier muss allerdings eine Extraktion mit ultrareinem Wasser durchgeführt werden. Die Analyse ist ohne chemische Suppression im ppm-Bereich möglich.
Enthält die wässrige Analysenlösung Spuren von organischen Lösungsmitteln, können diese mittels einer RP-Säule entfernt werden.
Lerninhalte
• Lebensmittelanalytik
• Einfluss der Matrix auf die Chromatographie
• Matrixeliminierung
Tabelle 33 Parameter – Versuch 9a bis 9c
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe a) Gin
b) Whisky c) Ethanol
Methode exp_09_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 5 MPa Analysendauer a) 17 min
b) 33 min c) 17 min
Loop a) 100 µL b) 100 µL c) 500 µL auf METROSEP IC Anion Anreicherungssäule 6.1006.300
Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser Autostep mit Fill
Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.

Praktikum der Ionenchromatographie – Version 12
khv – 75 – 22/12/2000
Versuch 9a – Bestimmung von Anionen in Gin
Chromatogramm 19 Standardlösung für die Bestimmung von Gin (versuch9a_std.gif)
Tabelle 34 Komponenten – Versuch 9a – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.5 0.1 4240 7.8 2 Systempeak 7.0 3 Nitrat 9.6 0.02 2980 0.9 4 Sulfat 13.0 0.4 3320 22 5 Oxalat 14.9 0.02 3710 0.7
Chromatogramm 20 Bestimmung von Gin (versuch9a_gin.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 76 – 22/12/2000
Tabelle 35 Komponenten – Versuch 9a – Gin
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.6 0.11 2230 8.8 2 Systempeak 7.0 3 Nitrat 9.7 0.014 3360 0.6 4 Sulfat 13.0 0.43 3260 23.5 5 Oxalat 14.9 0.013 3920 0.5
Versuch 9b – Bestimmung von Anionen in Whisky
Chromatogramm 21 Standardlösung für die Bestimmung von Whisky (versuch9b_std.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 77 – 22/12/2000
Tabelle 36 Komponenten – Versuch 9b – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.5 1 4700 84 2 Systempeak 7.0 3 Nitrat 9.6 0.5 3640 20 4 Phosphat 10.8 0.5 2960 11 5 Malat 12.1 0.5 3730 306 6 Sulfat 13.0 1 3350 56 7 Oxalat 14.9 1 3250 41
Chromatogramm 22 Bestimmung von Whisky (versuch9b_whisky.gif)
Tabelle 37 Komponenten – Versuch 9b – Whisky
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.6 1.3 3300 167 2 Systempeak 7.3 5 Nitrat 9.8 0.22 4000 8.7 6 Phosphat 10.9 0.43 3510 9.6 7 Malat 12.4 0.81 4580 5.8 8 Sulfat 13.1 0.88 3040 49 9 Oxalat 15.1 1.91 3540 79
Bemerkungen
Peaks 3, 4 und 10 sind unbekannt

Praktikum der Ionenchromatographie – Version 12
khv – 78 – 22/12/2000
Versuch 9c – Bestimmung von Anionen in Ethanol mit Anreicherung und Matrixeliminierung
Chromatogramm 23 Bestimmung von Anionen in Ethanol mit Matrixeliminierung (versuch9c_elim.gif)
Tabelle 38 Komponenten – Versuch 9c – mit Matrixeliminierung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 0.021 3570 9.5 2 Systempeak 6.8 3 Nitrat 9.3 0.013 3940 2.8 4 Sulfat 12.3 0.046 2130 14 5 Oxalat 14.1 0.018 3250 3.8

Praktikum der Ionenchromatographie – Version 12
khv – 79 – 22/12/2000
Chromatogramm 24 Bestimmung von Anionen in Ethanol ohne Matrixeliminierung (versuch9c.gif)
Tabelle 39 Komponenten – Versuch 9d – ohne Matrixeliminierung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] keine Peaks
Bemerkungen
Gefässe, Spritze und System mehrmals gründlich spülen. Plastikgefässe verwenden.

Praktikum der Ionenchromatographie – Version 12
khv – 80 – 22/12/2000
4.3.3 Versuch 10 – Anionen in Kopfsalat Nitrat gelangt über das Trinkwasser, aber auch durch Aufnahme pflanzlicher Nahrungsmittel (Gemüse) in den menschlichen Körper. Von der WHO (World Health Organization – Weltgesundheitsorganisation) wird empfohlen, dass die tägliche Nitrataufnahme 220 mg nicht überschreiten soll. Zur Zeit beträgt diese in der BRD etwa 130 mg. Ca. 5 % davon werden mit Fleisch- und Wurstwaren, der Rest etwa je zur Hälfte mit Trinkwasser und Gemüse aufgenommen.
Nitrat kann sich auf verschiedene Arten auf die Gesundheit des Menschen negativ auswirken. Es ist selbst relativ ungiftig. Erst bei Aufnahme von grösseren Mengen kann es Entzündungen des Magen-Darm-Trakts bewirken. Unter bestimmten Voraussetzungen aber, nämlich dem Vorliegen von Bakterien, wie sie beim Menschen im Mund vorhanden sind, wird durch das Enzym Nitratreduktase Nitrat zu Nitrit reduziert:
NO3–
EnzymNO2
–
Nitrit ist in der Lage, Hämoglobin, den roten Blutfarbstoff, zu Methämoglobin umzuwandeln (Fe2+ wird zu Fe3+ oxidiert). Methämoglobin kann im Gegensatz zu Hämoglobin keinen Sauerstoff im Blut transportieren. Bei erwachsenen Menschen kann dieser Schaden durch Stoffwechselreaktionen wieder behoben werden. Der Stoffwechsel von Säuglingen bis zum fünften Lebensmonat ist dazu jedoch nicht fähig. Durch den entstandenen Sauerstoffmangel im Blut verfärbt sich die Haut des Säuglings bläulich. Diese Blausucht oder auch Methämoglobinämie kann mitunter tödlich sein.
Aus Nitrit können durch Reaktion mit verschiedenen sekundären Aminen (zwei H-Atome des Ammoniakmoleküls sind durch Alkylreste ersetzt), welche durch Medikamente und Nahrung in den Körper des Menschen gelangen, im sauren Milieu des Magens Nitrosamine entstehen.
Dimethylnitrosamin
NR
RH N
R
RN O
Bei R = CH3:
NO2–
Nitrosamine gehören zu den am stärksten krebserzeugenden Stoffen. Sie werden im Organismus aus nicht kanzerogenen Verbindungen hergestellt.
Lerninhalte
• Lebensmittelanalytik
• Überprüfung von Lebensmitteln unter besonderer Beachtung von Nitrit und Nitrat
Tabelle 40 Parameter – Versuch 10
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Kopfsalat Methode exp_10_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 5 MPa Analysendauer 20 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.

Praktikum der Ionenchromatographie – Version 12
khv – 81 – 22/12/2000
Probenvorbereitung
Salat zerkleinern, dispergieren, Reinstwasser (Verdünnung 1:100) zugeben und filtrieren.
Chromatogramm 25 Standardlösung für die Bestimmung von Kopfsalat (versuch10a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 82 – 22/12/2000
Tabelle 41 Komponenten – Versuch 10 – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 5 4480 80 2 Systempeak 6.7 3 Nitrat 9.3 5 4320 42 4 Phosphat 10.4 5 2990 24 5 Malat 11.8 10 3140 26 6 Sulfat 12.7 2 3330 22 7 Oxalat 14.6 0.1 3500 0.9
Chromatogramm 26 Bestimmung von Kopfsalat (Verdünnung 1:100) (versuch10b.gif)
Tabelle 42 Komponenten – versuch 10 – Kopfsalat
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 540 4260 86 2 Systempeak 6.6 4 Nitrat 9.4 410 4240 34 5 Phosphat 10.4 540 2960 25 6 Malat 11.8 1160 2180 30 7 Sulfat 12.6 206 2800 23 8 Oxalat 14.5 10 2300 1
Bemerkungen
Peaks 3, 9 und 10 sind unbekannt

Praktikum der Ionenchromatographie – Version 12
khv – 83 – 22/12/2000
4.3.4 Versuch 11 – Phosphorsäure in Cola Coca-Cola ist ein alkoholfreies, kohlensäure- und coffeinhaltiges Getränk. Es wurde 1885 von dem amerikanischen Apotheker Pemberton hergestellt. Zu den Bestandteilen gehören Extrakte von Kolanüssen, bitteren Pomeranzen, Johannisbrotbaum, Ingweressenz, 12 % Zucker, 0.28 % Phosphorsäure (pH 2.7), Caramelfarbe und Kohlendioxid. Der Coffeingehalt beträgt 16 mg/100 ml.
Es ist eine Vielzahl von Colagetränken auf dem Markt (Pepsi Cola, Club Cola, River Cola, Afri Cola etc.), die sich in ihren Zusammensetzungen von Coca Cola unterscheiden können.
Die Phosphorsäure ist eine 3-basige Säure ( pKS1= 2.161; pKS2= 7.207; pKS3= 12.325). Der Anteil der verschiedenen Spezies in Abhängigkeit vom pH-Wert ergibt sich aus folgender Abbildung:
Abbildung 30 Pufferwirkung der Phosphorsäure
Häufig wird ein Gemisch aus primären und sekundärem Phosphat verwendet, dass im pH-Bereich 6 ... 8 (90 % H2PO4
– + 10 % HPO42– bis 10 % H2PO4
– + 90 % HPO42–) puffert.
Vergleiche auch Henderson-Hasselbach-Gleichung (Puffergleichung)
)()(lg
HAcAcpKpH S
−
+=
Lerninhalte
• Lebensmittelanalytik
• Chemie der Phosphorsäure
• Einfluss des pH-Wertes auf die chromatographische Trennung
• Statistik: Reproduzierbarkeit
Tabelle 43 Parameter – Versuch 11
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Cola Methode exp_11_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa

Praktikum der Ionenchromatographie – Version 12
khv – 84 – 22/12/2000
Analysendauer 18 min für Coca-Cola, 30 min für Coca-Cola light Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Eluentherstellung
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Chromatogramm 27 Chromatogramm – Standardlösung für die Bestimmung von Cola (versuch11.gif)
Tabelle 44 Komponenten – Versuch 11 – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 1 4140 15 2 Systempeak 6.7 3 Nitrat 9.4 1 4220 8.0 4 Phosphat 10.5 50 3250 261 5 Sulfat 12.7 1 2950 13

Praktikum der Ionenchromatographie – Version 12
khv – 85 – 22/12/2000
Versuch 11a – Bestimmung von Cola (1:10 Verdünnung)
Chromatogramm 28 Bestimmung von Cola (Verdünnung 1:10) (versuch11a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 86 – 22/12/2000
Tabelle 45 Komponenten – Versuch 11a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 5.9 3100 8.9 2 Systempeak 6.8 3 Nitrat 9.5 5.9 5000 4.7 4 Phosphat 10.5 510 3370 266 5 Sulfat 12.8 11.0 3520 14
Versuch 11b – Bestimmung von Cola-Light (1:10 Verdünnung)
Abbildung 31 Chromatogramm – Bestimmung von Cola light (Verdünnung 1:10) (versuch11b.gif)
Tabelle 46 Komponenetentabelle – Versuch 11b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 6.0 2990 9.2 2 Systempeak 6.7 3 Nitrat 9.4 7.3 4290 5.8 4 Phosphat 10.5 663 1850 346 5 Sulfat 12.8 14.4 1630 19
Bemerkung
Peaks 6, 7 und 8 unbekannt

Praktikum der Ionenchromatographie – Version 12
khv – 87 – 22/12/2000
Versuch 11c – Reproduzierbarkeit der Phosphatmessungen in Cola
Chromatogramm 29 Merge von 8 Messungen (unverdünnt) (versuch11_merge.gif)
Tabelle 47 Reproduzierbarkeit der Messungen
Chromatogramm Area Phosphat [µS/cm*sec]
Konz. Phosphat [mg/L]
1 3627 519 2 3629 520 3 3626 519 4 3626 519 5 3636 521 6 3638 521 7 3639 521 8 3638 521
Standardabweichung = kleiner 0.2 %
Bemerkung Die Cola muss entgast werden. Bei Cola light liegt unter dem Phosphatpeak der Süsstoff Cyclamat.

Praktikum der Ionenchromatographie – Version 12
khv – 88 – 22/12/2000
4.3.5 Versuch 12 – Anionen im Wein Wein ist das Erzeugnis, das ausschließlich durch vollständige oder teilweise alkoholische Gärung der frischen, auch eingemaischten Weintrauben oder des Traubenmostes gewonnen wird, heisst es im Deutschen Weingesetz. Most enthält u. a. 12 – 25 % Kohlenhydrate (Glucose, Fructose) und 0.9 – 1.5 % Säuren, die wichtigsten sind L-(+) Weinsäure (2R, 3R) und Äpfelsäure, daneben Citronensäure, Ketoglutarsäure, Bernsteinsäure und Milchsäure.
Wichtig für die Beurteilung des Mosts ist der Oechsle-Grad (Oe°); er ist umso höher, je grösser der Zuckergehalt ist und zeigt an, wie viel Gramm 1 L Most bei 20 °C mehr wiegt als 1 L destilliertes Wasser. Zum Beispiel besitzt Most mit dem spezifischen Gewicht 1,115 kg/l (115 g mehr als 1 L Wasser) 115 °Oechsle. Aus dem Oechsle-Grad lässt sich durch einfache Rechnung der Zucker- und auch der Alkoholgehalt ermitteln. Aus 1,7 g Zucker entsteht 1 mL (0,794 g) Ethanol. Bei 12 – 15 Vol% Alkohol kommt die Gärung zum Stillstand, da sich die Hefen mit Alkohol selbst vergiften und absterben.
Das Aroma des Weins besteht aus 600 – 800 Komponenten: Kohlenwasserstoffe, Alkohole, Aldehyde, Ketone, Säuren, Ester, Lactone, Ether, Phenole und vieles mehr.
Analytisch von Interesse sind 2R-, 3R-Weinsäure, Äpfelsäure, Citronensäure, Milchsäure und Bernsteinsäure. Der Gesamtsäuregehalt (berechnet als Weinsäure) beträgt ca. zwischen 5,5 und 8,5 g/l. Essigsäure, Propionsäure, höhere Fettsäuren und anomale Mengen an Milchsäuren kommen im «kranken» Wein vor und werden vornehmlich durch Mikroorganismen gebildet.
Von «Weinfehler» spricht man bei einem unausgeglichenem Zucker-/Säureverhältnis. Zum Beispiel muss bei trockenen Weinen der Zuckergehalt unter 9 g/L liegen; der Säuregehalt darf nicht zu niedrig, nämlich höchstens 2 g unter dem Zuckergehalt sein.
COOH
C OHH
C HHO
COOH
2
3
L-(+) Weinsäure
(2R, 3R)-Form
Die alkoholische Gärung läuft nach folgender Gleichung ab:
C6H12O6 + 2 ADP + 2 P ⎯⎯ →⎯Hefe 2 C2H5OH + 2 CO2 + 2ATP
Abbildung 32 Abkürzungen: ADP = Adenosindiphosphat, ATP = Adenosintriphosphat, P = Phosphat

Praktikum der Ionenchromatographie – Version 12
khv – 89 – 22/12/2000
Tabelle 48 Säuregehalt (mg/l) deutscher Weine
Qualitätswein Riesling Müller-Thurgau bestimmter Anbaugebiete Portugieser
Rotwein Kabinett Spätlese Auslese Beerenaus-
lese, Eiswein Silvaner Kabinett
Spätlese Auslese Beeren- auslese
M M M M M M M M M M Oxalsäure 15,7 13,0 11,0 45,8 16,0 21,7 16,7 40,9 15,4 19,5 Bernsteinsäure 194,0 260,1 205,8 282,1 230,2 149,2 243,8 215,1 264,9 269,2 Fumarsäure 44,2 31,9 24,6 28,3 20,8 36,0 20,9 51,8 27,0 40,6 Glutarsäure 3,7 1,9 4,5 2,6 2,3 7,0 2,6 2,2 1,9 6,2 c- bzw. t- Aconit-säure 2,1 1,9 + 5,5 4,9 6,4 1,4 2,5 1,6 4,5 Glycolsäure 1,8 1,3 + + 1,4 6,9 3,7 3,4 1,7 5,8 D- + L-Milchsäure 1789 2364 319,8 159,8 151,0 2141 793 221,0 208,1 2096 Anglycerinsäure 43,5 62,6 50,1 39,5 59,0 53,0 49,6 87,0 68,8 49,9 Triglycerinsäure ± ± ± ± ± ± ± ± ± ± 3-M-2,3-DHBS ± ± ± ± ± ± ± ± ± ± Äpfelsäure 3971 5080 3850 3871 4235 2251 2893 3163 3410 2897 Weinsäure 1586 994 1428 1431 817 1251 1280 1014 805 887 Citramalsäure 94,5 53,7 63,0 18,6 46,8 48,2 31,3 33,8 24,2 41,0 α-Hydroxyglutarsäure 24,9 27,5 18,2 24,5 28,4 20,6 18,8 15,3 26,9 22,2 Citronensäure 138,9 150,2 241,1 222,8 298,6 287,3 200,7 221,7 240,2 335,1 Glyoxylsäure + 1,0 + + 4,0 6,1 2,9 1,8 1,1 3,1 Brenztraubensäure 20,6 29,8 17,8 12,4 16,8 8,6 17,6 22,7 18,6 33,0 α-Ketoglutarsäure 41,1 45,7 38,6 36,8 55,7 35,6 41,0 33,7 40,3 27,5 Gluconsäure 59,0 357,2 54,5 95,9 452,1 337,2 53,9 131,1 373,9 540 Schleimsäure – + – – 4,1 20,4 – + 4,5 11,2 Glucuronsäure 2,1 3,4 3,8 5,8 7,9 14,5 1,7 6,0 7,6 10,2 Galacturonsäure 11,1 11,7 14,1 19,7 22,0 43,4 10,0 17,3 23,2 34,3 L-Ascorbinsäure 12,9 9,0 13,7 10,1 9,8 9,6 12,0 10,4 9,7 8,5 Gesamt-Säuren 7977 9491 6349 6302 6474 6744 5685 5284 5565 7333
Abbildung 33 Anmerkungen
- = nicht nachweisbar M = Mittelwert (mg/l) aus 4 bzw. 2 Einzelanalysen 3-M-2,3-DHBS = 3-Methyl-2,3-dihydroxybuttersäure + = in Spuren ± = in Spuren (ca. 5 mg/l) vorhanden, nicht ausgewertet

Praktikum der Ionenchromatographie – Version 12
khv – 90 – 22/12/2000
Lerninhalte
• Lebensmittelanalytik
• Ionenausschlusschromatographie
• Welche Ionen können mittels Ionenaustauschchromatographie bestimmt werden?
Tabelle 49 Parameter – Versuche 12a bis 12c
Säule 6.1005.200 Metrosep Organic Acids Eluent 0.5 mmol/L H2SO4 / Aceton (85:15) Probe Wein Methode exp_12_o.mtw System orgacids.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 20 min Loop 20 µL Polarität +
Eluentherstellung
0.5 mmol H2SO4 (850 mL) und Aceton (150 mL) mischen.
Probenvorbereitung
Probe 1:100 verdünnen, Wein filtrieren durch 45 µm Filter.
Versuch 12a – Bestimmung von Anionen in Weisswein
Chromatogramm 30 Standard für die Bestimmung von organischen Säuren in Wein (versuch12.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 91 – 22/12/2000
Tabelle 50 Komponenten – Versuch 12a – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Citrat 7.2 5 26200 1.1 2 Tartrat 7.5 20 7990 31 3 Malat 8.3 5 9210 4.5 4 Succinat 9.9 5 7140 1.2 5 Lactat 10.8 20 8830 11 6 Acetat 13.1 10 11500 1.2 7 Systempeak 15.9
Chromatogramm 31 Bestimmung organischer Säuren in Weisswein (Verdünnung 1:100) (versuch12a.gif)
Tabelle 51 Komponenten – Versuch 12a – Weisswein
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Citrat 7.2 2 Tartrat 7.5 1210 8430 19 4 Malat 8.2 7 Succinat 9.8 350 5660 0.8 8 Lactat 10.7 2190 8820 13 9 Acetat 13.0 710 5200 0.8
10 Systempeak 15.8
Bemerkung
Peaks 3, 5, 6 nicht identifiziert

Praktikum der Ionenchromatographie – Version 12
khv – 92 – 22/12/2000
Versuch 12b – Bestimmung von Anionen in Rotwein
Abbildung 34 Chromatogramm – Bestimmung organischer Säuren in Rotwein (Verdünnung 1:100) (versuch12b.gif)
Tabelle 52 Komponenten – Versuich 12b – Rotwein
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Citrat 7.2 2 Tartrat 7.5 2230 8300 34 4 Malat 8.2 7 Succinat 9.9 410 10500 1.0 8 Lactat 10.8 1560 9570 8.9 9 Acetat 13.1 1010 7950 1.1
10 Systempeak 15.9
Bemerkung
Peaks 3, 5, 6 nicht identifiziert; Citrat und Malat können nicht korrekt quantifiziert werden.

Praktikum der Ionenchromatographie – Version 12
khv – 93 – 22/12/2000
4.3.6 Versuch 13 – Bestimmung von Verunreinigungen in Borat – Chlorid und Sulfat in Borax Borax (Dinatriumtetraborat, Na2B4O7·10 H2O, Na2[B4O5(OH)4]·8H2O) hat folgende Struktur:
BHOO
O
B
B
OH
OH
O
OB OHO 2 Na 8 H2O
Abbildung 35 «Borax» (Dinatriumtetraborat, Na2B4O7·10 H2O, Na2[B4O5(OH)4]·8H2O)
Borax löst beim Schmelzen zahlreiche Metalloxide unter Bildung charakteristischer Färbungen. Die «Boraxperle» ist aus dem anorganischen Praktikum bekannt. Borax wird u. a. für die Herstellung von Gläsern, Glasuren für Steingut, Porzellan und als Flussmittel beim Hartlöten verwendet. Bei 100°C verliert Borax 5 Wassermoleküle und geht in das Pentahydrat «Juwelierborax» über. Durch die zugesetzten Lötflussmittel werden Oberflächenverunreinigungen – überwiegend Oxidschichten – zerstört. Sie würden die oberflächliche Legierungsbildung zwischen Lot, dies sind Legierungen aus Silber, Kupfer und Zinn, und dem Grundwerkstoff beeinträchtigen.
Für die Bestimmung von Cl– und SO42– wird die Boraxlösung auf einen pH-Wert < 4 eingestellt. Bei
diesem pH-Wert liegt praktisch ausschliesslich die Borsäure H3BO3 oder (B(OH)3) vor. Die Borsäure ist eine sehr schwache, einbasige Säure, deren pKS-Wert mit 9.25 etwa dem von HCN entspricht. Borsäure ist kein H+-Donor (Brönsted-Säuredefinition), sondern ein OH–-Akzeptor (Lewis-Säuredefinition).
B(OH)3 + HOH H+ + B(OH)4-
Die Ionenchromatographie ohne chemische Suppression verwendet für die Bestimmung der Verunreinigungen im Borat einen saure Eluenten mit einem pH = 4.1. Bei diesem pH liegt das Borat als Borsäure B(OH)3 vor. Die Borsäure zeigt im Gegensatz zu den negativ geladenen Anionen keine Interaktion mit der stationären Phase der Trennsäule. Sie wird daher im Totvolumen eluiert.
Wird hingegen mit chemischer Suppression gearbeitet, so kommt der schwach alkalische Carbonat-/Bicarbonateluent zum Einsatz. So ist es zwar möglich Borat als Anion zu trennen, jedoch schlägt die Detektion fehl. Der Grund hierfür liegt im Suppressor. Dieser tauscht alle Na+-Ionen gegen H+-Ionen aus. Es bildet sich die nur sehr schwach dissoziierte Borsäure, die mit einem Leitfähigkeitsdetektor nicht reproduzierbar bestimmt werden kann.
Lerninhalte
• Starke und schwache Säuren
• Vergleich zwischen Messungen mit elektronischer und chemischer Suppression

Praktikum der Ionenchromatographie – Version 12
khv – 94 – 22/12/2000
Versuch 13a – Messung ohne chemische Suppression (mit elektronischer Suppression)
Tabelle 53 Parameter – Versuch 13a
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 8 mmol/L Phthalsäure, 2% Acetonitril; pH = 4.1 (TRIS)
Leitfähigkeit ca. 400 µS/cm Probe 1 g/L Borax in Reinstwasser aufgestockt mit 1 ppm Chlorid und Sulfat Methode exp_13_n.mtw System anonsupp.smt Fluss 0.5 mL/min Druck 2 MPa Analysendauer 23 min Loop 100 µL Polarität +
Eluentherstellung
1.33 g Phthalsäure in 20 ml Acetonitril und etwas Wasser lösen, dann in entgastes Wasser geben und auf 1 L auffüllen. Durch Zugabe von TRIS (fest) (ca. 1 g) den pH-Wert auf 4.1 einstellen.
Chromatogramm 32 Borat aufgestockt mit 1 ppm Chlorid und 1 ppm Sulfat – ohne chemische Suppression (versuch13a.gif)
Tabelle 54 Komponenten – Versuch 13a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Chlorid 4.4 1 2790 12 2 Sulfat 9.0 1 3270 8.4 3 Systempeak 19
Versuch 13b – Messung mit chemischer Suppression Tabelle 55 Parameter – Versuch 13b

Praktikum der Ionenchromatographie – Version 12
khv – 95 – 22/12/2000
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe 1 g/L Borax aufgestockt mit 1 ppm Chlorid, Sulfat Methode exp_13_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 2 MPa Analysendauer 16 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Eluentherstellung
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Chromatogramm 33 Borat aufgestockt mit 1 ppm Chlorid und 1 ppm Sulfat – mit chemischer Suppression
Tabelle 56 Komponenten – Versuch 13b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Chlorid 5.4 1 4170 15 2 Sulfat 13.7 1 3290 11

Praktikum der Ionenchromatographie – Version 12
khv – 96 – 22/12/2000
4.3.7 Versuch 14 – Bestimmung von Anionen in Abwasser Abwasser wird überwiegend in biologischen Kläranlagen mit Hilfe von Bakterien gereinigt. Organische Substanzen werden dabei unter Sauerstoffverbrauch weitgehend oxidiert.
Org. Substanzen + O2 ⎯⎯⎯⎯ →⎯Bakterien CO2 + H2O + Zellmasse
Neben der Oxidation von organischen Substanzen kann bei entsprechender Auslegung der Anlage auch Ammonium zu Nitrat oxidiert werden (Nitrifikation).
NH4+ + 2 O2 ⎯⎯⎯⎯ →⎯Bakterien NO3
- + H2O + 2 H+
In sauerstofffreien Becken benutzen Bakterien den Nitratsauerstoff zur Oxidation von organischen Substanzen (Denitrifikation).
Org. Substanzen + 2 NO3- ⎯⎯⎯⎯ →⎯Bakterien 2 CO2 + 2 OH- + 2 H2O + N2↑
Phosphat – wie NH4+ und NO3
– ein Pflanzennährstoff – kann z. B. durch Zusatz von Fe3+-Salzlösungen ausgefällt werden. Neben den sogenannten Summenparametern – Biochemischer Sauerstoffbedarf (BSB), Chemischer Sauerstoffbedarf (CSB), Total Organic Carbon (TOC) – die ein Maß für die organische Belastung des Abwassers bzw. auch Gewässers darstellen, ist also vor allem die Analyse von NH4
+, NO3-, PO4
3- wichtig. Cl–, SO42– werden nur in Sonderfällen analysiert.
Für kommunale Kläranlagen von mehr als 100 000 sogenannten Einwohnergleichwerten (1 Einwohnergleichwert entspricht der Schmutzfracht eines Einwohners) sind die folgenden Grenzwerte (D) festgelegt:
BSB 15 mg/l CSB 75 mg/l NH4-N 10 mg/l* NGes 18 mg/l* (Σ aus NH4-N, NO2-N, NO3-N) PO4-PGes 1 mg/l
* Gilt nur für Abwassertemperaturen ≥ 12 °C, da die Nitrifikation stark durch die Temperatur beeinflusst wird.
NH4-N, NO2-N, NO3-N bedeutet, dass sich die Werte auf den in den Ionen enthaltenen Stickstoff beziehen, analoges gilt für PO4-P.
Lerninhalte
• Umweltanalytik
• Probenvorbereitungstechnik
• Analytik von Substanzgemischen mit grossen Konzentrationsunterschieden – dynamischer Bereich

Praktikum der Ionenchromatographie – Version 12
khv – 97 – 22/12/2000
Tabelle 57 Parameter – Versuch 14a und 14b
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Herisau Abwasser Methode exp_14_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 2 MPa Analysendauer 15 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Eluentherstellung
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und 20 mL Aceton zugeben.
Probenvorbereitung
Probe filtrieren
Versuch 14a – Zulauf der Kläranlage
Chromatogramm 34 Standardchromatogramm für die Bestimmung des Kläranlagenzulaufs (versuch14a1.gif)
Tabelle 58 Komponenten – Versuch 14a – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Chlorid 5.2 100 3470 2270 2 Nitrat 9.1 2 4330 16 3 Phosphat 10.3 5 3040 21 4 Sulfat 12.5 100 3820 1470

Praktikum der Ionenchromatographie – Version 12
khv – 98 – 22/12/2000
Chromatogramm 35 Bestimmung des Kläranlagenzulaufs (versuch14a2.gif)
Tabelle 59 Komponenten – Versuch 14a – Kläranlagenzulauf
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Chlorid 5.2 128 2980 2910 5 Nitrat 9.2 2.1 4620 16 6 Phosphat 10.3 7.0 3040 30 7 Sulfat 12.5 132 3720 1940
Bemerkungen
Peaks 2, 3, 4 und 8 nicht bestimmt

Praktikum der Ionenchromatographie – Version 12
khv – 99 – 22/12/2000
Versuch 14b – Ablauf der Kläranlage
Chromatogramm 36 Standardchromatogramm für die Bestimmung des Kläranlagenablaufs (versuch14b1.gif)
Tabelle 60 Komponenten – Versuch 14b – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.6 0.1 2900 2.1 2 Chlorid 5.2 5 4140 78 3 Nitrat 9.1 10 4210 87 4 Sulfat 12.5 5 3420 53

Praktikum der Ionenchromatographie – Version 12
khv – 100 – 22/12/2000
Chromatogramm 37 Bestimmung des Kläranlagenablaufs (versuch14b2.gif)
Tabelle 61 Komponenten – Versuch 14b – Kläranlagenablauf
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Fluorid 3.6 0.05 2900 1.1 2 Chlorid 5.2 7.3 3870 114 4 Nitrat 9.2 8.5 4390 73 5 Sulfat 12.5 6.8 3460 71
Bemerkungen
Peak 3 nicht bestimmt

Praktikum der Ionenchromatographie – Version 12
khv – 101 – 22/12/2000
4.3.8 Versuch 15 – Fluorid in Zahnpaste Zahnpasten sind aus unterschiedlichen Bestandteilen zusammengesetzt:
Wasser – 30-40 %
Putzkörper – sind unlösliche, anorganische Substanzen, die die Reinigungswirkung der Zahnbürste verstärken. Die Partikelgrösse ist < 15 µm. Verbreitete Putzkörper sind Al2O3, CaCO3, CaHPO4·2H2O, SiO2·H2O, Natriumaluminiumsilikat.
Fluorid – verstärkt die Resistenz der Zahnschmelze gegen den Angriff von Säuren, die von bakteriellem Plaque (Zahnbelag) stammen. Ausserdem ermöglichen Fluoride die Mineralisierung des Zahnschmelzes (der anorganische Anteil des Zahnschmelzes besteht im wesentlichen aus Calciumphosphat, Hydroxyapatit, Calciumcarbonat, Magnesiumphosphat, Calciumfluorid und Calciumchlorid). Fluorid ist daher wichtig für die Kariesprophylaxe. Die am meisten verwendeten Fluor-Verbindungen sind Natriumfluorid, Natriummonofluorphosphat und quartäre Ammoniumfluoride, die ebenfalls freie Fluoridionen enthalten. In einigen Ländern wird Fluorid dem Trinkwasser zugesetzt.
P
OH
OFO- Na+
Natriummonofluorphosphat
Tenside (z. B. Natriumlaurylsulfat) – erniedrigen die Oberflächenspannung und tragen zur gleichförmigen Verteilung der Zahnpasta bei. Sie verstärken die Reingungswirkung, besonders an Stellen, die mit der Zahnbürste schwierig zu erreichen sind. Weitere Bestandteile von Zahnpasten sind z. B.:
Feuchtemittel (z. B. Glycerin, Sorbit, Polyethylenglycol) – verbessern die Stabilität in der Kälte und verhindern das Austrocknen.
Binde- und Verdickungsmittel (z. B. Natriumcarboxymethylcellulose) – verhindern die Trennung in Feststoff und flüssige Phase.
Süßstoffe (z. B. Natriumsaccharin) – verbessern den Geschmack der Zahnpasta.
Konservierungsmittel (z. B. 4-Hydroxybenzoesäure) – werden eingesetzt, um die Zahnpasta vor mikrobieller Zersetzung zu schützen.
Aromatisierungsmittel – werden zugesetzt, um die Akzeptanz der Zahnpasta und das Hygienebewusstsein zu erhöhen. Verwendet werden u. a. Pfefferminzöle, Menthol, Anisöl, Eucalyptusöl, Gewürzöle, Zitronenöl.
Mit der ionenchromatographischen Analytik wird sowohl Fluorid aus Natriumfluorid wie auch Natriummonofluorphosphat erfasst.
Bemerkung
Citrat eluiert auf der Metrosep Anion Dual 1 (3 x 150) erst nach ca. 90 min.
Zur Überprüfung ob Citrat in der Zahnpasta vorhanden ist, wird in Versuch 15 b anstelle der Anion Dual 1 die Vorsäule Metrosep Anion Precolumn cartridge (6.1006.030) verwendet.
Lerninhalte
• Qualitätskontrolle
• Probenvorbereitung

Praktikum der Ionenchromatographie – Version 12
khv – 102 – 22/12/2000
Versuch 15a – Zahnpasta (1) ohne Citrat
Tabelle 62 Parameter – Versuch 15a
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Standard (Fluorid, Chlorid, Phosphat, Sulfat, Saccharin)
Zahnpasta Methode exp_15_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 50 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Probenvorbereitung
1 g Zahnpasta wird in 19 mL Reinstwasser gelöst, danach durch einen 45 µL Filter filtriert.
Chromatogramm 38 Zahnpaste (1) ohne Citrat (Verdünnung 1:20)(versuch15a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 103 – 22/12/2000
Tabelle 63 Komponenten – Versuch 15a
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Fluorid 3.9 1304 1357 1663 2 Formiat 4.5 52 1643 19.8 3 Chlorid 5.6 66 4325 55.3 4 Systempeak 6.7 5 Monofluorphosphat 10.2 4527 3.1 6 Phosphat 11.5 260 3047 55.4 7 Sulfat 14.1 1346 3831 880 8 Saccharin 42.6 160 1917 179
Versuch 15b – Zahnpasta (2) mit Citrat
Tabelle 64 Parameter – Versuch 15b
Säule 6.1006.030 Metrosep Anion Precolumn cartridge Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Zahnpasta Methode exp_15b_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 3 MPa Analysendauer 25 min Loop 20 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Eluentherstellung
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Probenvorbereitung
1 g Zahnpasta wird in Reinstwasser gelöst (Verdünnung 1:20), danach durch einen 45 µL Filter filtriert.

Praktikum der Ionenchromatographie – Version 12
khv – 104 – 22/12/2000
Chromatogramm 39 Zahnpaste (2) mit Citrat (Verdünnung 1:20)(versuch15b.gif)
Tabelle 65 Komponenten – Versuch 15b
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Saccharin 8.2 160 510 177 2 Citrat 16.2 2600 267 360

Praktikum der Ionenchromatographie – Version 12
khv – 105 – 22/12/2000
4.3.9 Versuch 16 – Anionen in braunem und weissem Zucker Zucker sind organische Verbindungen mit einer Halbacetal-bildenden Carbonylgruppe und mehreren Hydroxygruppen. Im allgemeinen Sprachgebrauch versteht man unter Zucker das Disaccharid Saccharose.
OH
OH
H
H
OHH
OH
CH2OH
H
O
CH2OH
OCH2OH
H
H
OH H
OH
Abbildung 36 Disaccharid Saccharose
Für die Herstellung von Zucker aus Zuckerrüben werden diese zunächst gewaschen und geschnitzelt und dann in sogenannten Diffusionsanlagen im Gegenstrom mit Wasser ausgelaugt. Dabei gehen die löslichen Bestandteile – Zucker, Salze, Säuren, Proteine, Pektine – in Lösung. Durch Zugabe von gebranntem Kalk (CaO) wird der größte Teil der Nichtzuckerstoffe ausgefällt. Im Anschluss wird Kohlendioxid CO2 zur Fällung des überschüssigen Calciumhydroxids als CaCO3 verwendet. Nach einer Filtration wird die Zuckerlösung in mehrstufigen Verdampfern zu «Dicksaft» aufkonzentriert, filtriert und noch weiter eingedickt bis sich der Zucker als «Weisszuckerfüllmasse» ausscheidet. Dieser Zucker wird in Zentrifugen abgetrennt und nach Reinigung (Umkristallistaion) als schneeweißer Kristallzucker mit 99,95 % Reinheit erhalten. Als Zentrifugenablauf der letzten Stufe fällt ein braungefärbter Sirup – die Melasse – an.
Brauner Zucker wird weniger intensiv gereinigt und u. U. durch Zusatz von Melasse eingefärbt. Er enthält neben Verunreinigungen Alkali- und Erdalkaliionen sowie Anionen und weist anderen Geschmack als weißer Kristallzucker auf.
Vor der Bestimmung von Anionen können diese Verunreinigungen mittels RP-Säule abgetrennt werden.
Lerninhalte
• Lebensmittelanalytik
• Prozesskontrolle
• Überprüfung stark zuckerhaltiger Lebensmittel z.B. Honig

Praktikum der Ionenchromatographie – Version 12
khv – 106 – 22/12/2000
Tabelle 66 Parameter – Versuch 15a und 15b
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe a) Brauner Zucker
b) Weisser Zucker Methode exp_16_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 4 MPa Analysendauer 17 min Loop 100 µL Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Herstellung des Eluenten
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 mL Aceton zugeben.
Probenvorbereitung
Zucker in Reinstwasser (Verdünnung 1:10) lösen und über 0.45 µm filtrieren.
Versuch 16a – Brauner Zucker
Chromatogramm 40 Standardlösung für die Bestimmung von braunem Zucker (versuch16a1.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 107 – 22/12/2000
Tabelle 67 Komponenten – Versuch 16a – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.3 20 3640 2200 2 Systempeak 6.9 3 Nitrat 9.6 0.1 3700 3.9 4 Phosphat 10.7 10 3330 245 5 Malat 12.1 5 3430 62 6 Sulfat 13.0 5 3600 294 7 Oxalat 14.9 1 3220 40
Chromatogramm 41 Bestimmung von braunem Zucker (Verdünnung 1:10) (versuch16a2.gif)
Tabelle 68 Komponenten – Versuch 16a – brauner Zucker
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.4 217 3480 2380 2 Systempeak 7.2 3 Nitrat 9.7 1.8 2870 7.0 4 Phosphat 10.8 84 3320 205 5 Malat 12.2 55 3840 68 6 Sulfat 13.0 74 3980 435 7 Oxalat 14.9 6.9 3530 28
Bemerkungen
Bei braunem Zucker können zu späteren Retentionszeiten Verunreinigungen eluieren, welche die nachfolgenden Chromatogramme unter Umständen stören könnten. Es ist deshalb ratsam erst den weissen und dann den braunen Zucker zu vermessen.

Praktikum der Ionenchromatographie – Version 12
khv – 108 – 22/12/2000
Versuch 16b – Weisser Zucker
Chromatogramm 42 Standardlösung für die Bestimmung von weissem Zucker (versuch16b1.gif)
Tabelle 69 Komponenten – Versuch 16b – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.5 0.1 4408 8.1 2 Systempeak 7.0 3 Nitrat 9.6 0.1 3682 4.0 4 Malat 12.0 0.1 3587 1.2 5 Sulfat 12.9 0.1 2995 5.9 6 Oxalat 14.8 0.1 3260 3.7

Praktikum der Ionenchromatographie – Version 12
khv – 109 – 22/12/2000
Chromatogramm 43 Bestimmung von weissem Zucker (Verdünnung 1:10) (versuch16b2.gif)
Tabelle 70 Komponenten – Versuch 16b – weisser Zucker
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec] 1 Chlorid 5.5 1.3 4183 10.3 2 Systempeak 7.2 3 Nitrat 9.8 0.8 4157 3.2 4 Malat 12.2 1.5 2833 1.8 5 Sulfat 13.1 0.9 3404 5.2 6 Oxalat 15.0 1.4 3432 5.3

Praktikum der Ionenchromatographie – Version 12
khv – 110 – 22/12/2000
4.3.10 Versuch 17 – Verunreinigungen im Wasserstoffperoxid Reines Wasserstoffperoxid (H2O2) schmilzt bei 0.4 °C und siedet bei 150 °C. Es kann im Labor nur unter hohem Sicherheitsaufwand durch fraktionierte Kristallisation rein hergestellt werden. Als Industriechemikalie steht H2O2 als wässrige Lösung in Konzentrationen von 35, 50, 60 und 70 % zur Verfügung. Die stark exotherme Zersetzung von H2O2 in H2O und O2 erfolgt nicht spontan, sie wird jedoch durch Schwermetalle katalysiert.
2 H2O + O2 + 196,2 kJ2 H2O2
Durch den Zusatz von Stabilisatoren werden die Schwermetallionen komplexiert. Die Zersetzung wird so gehemmt. Die charakteristische Eigenschaft von H2O2 ist seine oxidierende Wirkung. Das Normalpotential (E0) beträgt + 1.8 V bei pH = 0 und + 0,78 V bei einem pH = 14.
Wasserstoffperoxid zählt zu den wichtigen Produkten der Grundstoffchemie mit einer Jahresproduktion von 2,7 Millionen Tonnen. Es wird grosstechnisch durch Umsetzung von Anthrahydrochinon mit O2 aus der Luft hergestellt. Dabei entsteht das entsprechende Anthrachinon und H2O2. Anthrachinon wird durch elementaren Wasserstoff am Palladium-Katalysator wieder zu Anthrahydrochinon reduziert und zurück in den Kreislauf geführt.
Ein Grossteil des H2O2 wird für die Papier- und Zellstoffbleiche verwendet. In vielen Ländern ist die Chlorbleiche (mit Cl2 oder ClO2), bei der biologisch schwer abbaubare chlorierte organische Verbindungen ins Abwasser gelangen, ganz oder teilweise durch andere Verfahren ersetzt. Dabei hat H2O2 eine grosse Bedeutung.
Weitere Anwendungsgebiete sind:
Enteisenung und Entmanganung von Trinkwasser Entgiftung cyanidhaltiger Abwässer (CN– → CNO–) z. B. Galvanikabwasser mit H2O2 oder H2O2/H2SO4 Die Kombination H2O2 und UV liefert HO-Radikale (Hydroxylradikale), die äusserst starke Oxidationsmittel sind (E0 = + 2,8 V). Sie sind daher für die Reinigung spezieller Abwässer von Interesse Vielversprechend ist die Entwicklung eines Verfahrens zur Herstellung von Propylenoxid aus Propen und H2O2 (Titanschichtkatalysatoren) anstelle des abwasserbelastenden Chlorhydrinverfahrens. Unter den H2O2-Folgeprodukten sind Natriumperborat und Natriumpercarbonat am wichtigsten; sie werden als Bleichmittel in Pulverwaschmittel eingesetzt. H2O2 wird in der Elektronikindustrie bei der Herstellung gedruckter Schaltungen und zur Reinigung von Silizium-Wafern verwendet.
Die Bestimmung von Verunreinigungen im H2O2 hat vor allem in der zuletzt angeführten Elektronikindustrie grosse Bedeutung. Schon kleinste Mengen (ng/L oder ppt) von Fremdionen verschlechtern die Ausbeute an Wafern signifikant, da zum Beispiel Phosphat und Nitrat als n-Fehlstellen im Siliziumkristall wirken.
Lerninhalte
• Prozesskontrolle
• Matrixeliminierung
• Probenvorbereitung
• Selektivität unterschiedlicher Säulen

Praktikum der Ionenchromatographie – Version 12
khv – 111 – 22/12/2000
Tabelle 71 Parameter – Versuch 17a und 17b
Säule 6.1006.020 Metrosep Anion Dual 1 (3 x 150 mm) Eluent 2.4 mmol/L NaHCO3/ 2.5 mmol/L Na2CO3 + 2% Aceton
Leitfähigkeit nach chemischer Suppression ca. 16 µS/cm Probe Wasserstoffperoxid 30 %, suprapur Methode exp_17_s.mtw System asupp.smt Fluss 0.5 mL/min Druck 2 MPa Analysendauer 17 min Loop 1 mL auf METROSEP IC Anion Anreicherungssäule 6.1006.300 Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +
Eluentherstellung
265 mg Natriumcarbonat (wasserfrei) und 201.5 mg Natriumhydrogencarbonat in 980 mL Reinstwasser lösen und entgasen. Danach 20 ml Aceton zugeben.
Probenaufgabe
System mit Reinstwasser gut spülen, dann 1 mL Probe aufgeben und mit 1 mL Reinstwasser spülen.
Chromatogramm 44 Standardlösung für die Bestimmung von Anionen in Wasserstoffperoxid (versuch_17.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 112 – 22/12/2000
Tabelle 72 Komponenten – Versuch 17 – Standardlösung
Peak Komponente tR [min] Konz. [µg/L] Bodenzahl Area [µS/cm*sec]1 Acetat 4.2 5 2620 108 2 Formiat 4.4 1 2940 222 3 Chlorid 5.3 1 3300 587 4 Systempeak 6.9 5 Nitrat 9.2 0.1 3730 31 5 Sulfat 12.4 0.5 3160 163 6 Oxalat 14.2 0.1 3010 21
Versuch 17a – Wasserstoffperoxid mit Matrixeliminierung
Chromatogramm 45 Bestimmung von Wasserstoffperoxid (30 %) mit Matrixeliminierung (versuch_17_a.gif)
Tabelle 73 Komponenten – Versuch 17a
Peak Komponente tR [min] Konz. [µg/L] Bodenzahl Area [µS/cm*sec]2 Acetat 4.2 8.8 1780 3 Formiat 4.4 0.83 2170 5 Chlorid 5.3 0.86 2960 507 6 Systempeak 7.2 7 Nitrat 9.3 0.15 4560 47 8 Sulfat 12.5 0.44 2520 142
10 Oxalat 14.1 0.05 2480 10
Bemerkung
Gefässe, Spritze und System mehrmals gründlich spülen. Plastikgefässe verwenden. Unbedingt Schutzbrille tragen! Peaks 1, 4 und 9 nicht quantifiziert.

Praktikum der Ionenchromatographie – Version 12
khv – 113 – 22/12/2000
Versuch 17b – Wasserstoffperoxid ohne Matrixeliminierung
Chromatogramm 46 Bestimmung von Wasserstoffperoxid (30 %) ohne Matrixeliminierung (versuch_17_b.gif)
Versuch 17c – Bestimmung von Wasserstoffperoxid mit Matrixeliminierung auf einer Hochleistungssäule
Tabelle 74 Parameter – Versuch 17c
Säule METROSEP A SUPP 5-250; 6.1006.530 Eluent 1 mmol/L NaHCO3/ 3.2 mmol/L Na2CO3
Leitfähigkeit nach chemischer Suppression ca. 15 µS/cm Probe Wasserstoffperoxid 30 %, suprapur Methode exp_17c_s.mtw System asupp5.smt Fluss 1.0 mL/min Druck 11 MPa Analysendauer 30 min Loop 6.1006.300 Metrosep Anreicherungssäule Suppressor Regeneriermittel: 50 mmol/L H2SO4, Reinstwasser
Autostep mit Fill Polarität +

Praktikum der Ionenchromatographie – Version 12
khv – 114 – 22/12/2000
Chromatogramm 47 Bestimmung von Wasserstoffperoxid (30 %) mit Matrixeliminierung auf der METROSEP A SUPP 5 Säule (versuch_17_c.gif)
Tabelle 75 Komponenten – Versuch 17c
Peak Komponente tR [min] Konz. [µg/L] Bodenzahl Area [µS/cm*sec]3 Acetat 6.3 6.8 2850 219 4 Formiat 6.7 0.77 8740 123 6 Chlorid 8.9 0.78 17810 380 7 Systempeak 13.0 8 Nitrat 16.4 0.15 19100 37
10 Sulfat 23.3 0.35 16640 86 13 Oxalat 27.0 0.04 12100 7.7
Bemerkung
Peaks 1, 2, 5. 9, 10, 11 und 12 nicht identifiziert

Praktikum der Ionenchromatographie – Version 12
khv – 115 – 22/12/2000
4.4 Versuche für die Bestimmung von Kationen
4.4.1 Versuch 18 – Alkli- und Erdalkalimetalle in Trinkwasser und Mineralwasser Natürliches Wasser enthält neben den gelösten Gasen O2, N2 und CO2 auch Salze, die primär aus Böden und Gesteinen herausgelöst werden und so ins Trinkwasser gelangen. Verunreinigungen durch Abwässer kommen ebenso als Quelle für unterschiedliche Salze in Frage. Die wichtigsten Salze sind Chloride, Sulfate und Hydrogencarbonate des Calciums und Magnesiums.
Ein wichtiger Parameter des Trinkwassers sowohl was den Gebrauch als Lebensmittel wie auch seine Verwendung für Waschprozesse oder industrielle Anwendungen betrifft, ist die sogenannte Wasserhärte.
Unter Gesamthärte des Wassers versteht man die Summe der Stoffmengenkonzentration (mmol/l) von Calcium- und Magnesiumionen. Den Teil der Härte, der durch Kochen entfernt werden kann, bezeichnet man als Carbonathärte (früher temporäre Härte). Die bleibende Härte wird durch Sulfat- und Chloridionen verursacht, deren Ca- und Mg-Salze nicht durch Kochen ausgefällt werden können.
Calcium- und Magnesiumionen bilden mit den klassischen Seifen – Natriumsalze der Fettsäuren – schwerlösliche Calcium- bzw. Magnesiumseifen. Diese sind zum einen wasch-unwirksam zum anderen setzen sie sich als «Grauschleier» auf Textilien ab. Moderne Waschmittel enthalten Alkylsukfate, die keine schwerlöslichen Calcium- und Magnesiumsalze mehr bilden können. Diesen Waschmitteln ausserdem noch zugesetzte Zeolithe fungieren als Ionentauscher und entfernen Ca2+ und Mg2+-Ionen. Die Fällung von schwerlöslichem CaCO3 und basischem Magnesiumcarbonat beim Erwärmen wird so wirkungsvoll verhindert.
Erhöhte Zufuhr von Natriumionen bewirkt u. a. beim Menschen Bluthochdruck. Der tägliche Na+-Bedarf von 1 g wird z. Zt. deutlich überschritten (3 –7 g).
In der Trinkwasserverordnung (TVO), die eine Umsetzung der EG-Trinkwasserrichtlinie (80/778/EWG) ist, sind u. a. folgende Werte für die Kationen enthalten:
NH4+ 0,5 mg/l
Na+ 150 mg/l K+ 12 mg/l Mg2+ 50 mg/l Ca2+ 400 mg/l
Bei allen durchgeführten Analysen müssen die Proben mit Salpetersäure angesäuert werden (pH 2.5 – 3.5), da ansonsten für die zweiwertigen Kationen keine reproduizierbaren Resultate erhalten werden.
Lerninhalte
• Untersuchung des Lebensmittels Nummer «1»
• Überprüfung der Angaben auf Mineralwasserflaschen

Praktikum der Ionenchromatographie – Version 12
khv – 116 – 22/12/2000
Tabelle 76 Parameter – Versuche 18
Säule 6.1010.210 Metrosep C2 100 x 4.0 mm – 7 µm Eluent 4 mmol/L Weinsäure/ 0.75 mmol/L Dipicolinsäure
Leitfähigkeit ca. 590 µS/cm Probe Standard (Natrium, Ammonium, Kalium, Calcium, Magnesium +
2 mmol/L HNO3) – Leitungswasser (+ ca. 100 µL 2 mol/L HNO3 auf 100 mL Probe) bis pH = 2.5 ... 3.5) – Mineralwasser (+ ca. 100 µL 2 mol/L HNO3 auf 100 mL Probe) bis pH = 2.5 ... 3.5)
Methode exp_18_c.mtw System cation.smt Fluss 1 mL/min Druck 7 MPa Analysendauer 12 min Loop 10 µL Polarität -
Herstellung des Eluenten
600 mg Weinsäure und 125 mg Dipicolinsäure in 100 mL Reinstwasser unter Erwärmen lösen, danach mit Reinstwasser auf 1 L auffüllen.
Chromatogramm 48 Standardlösung (versuch_18.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 117 – 22/12/2000
Tabelle 77 Komponenten – Versuch 18 – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.8 0.5 4200 1.5 2 Kalium 6.5 0.2 3090 0.9 3 Calcium 8.4 20 1960 66 4 Magnesium 11.0 5 2700 34
Chromatogramm 49 Leitungswasser (Verdünnung 1:10) (versuch_18_a.gif)
Tabelle 78 Komponenten – Versuch 18 – Leitungswasser
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.8 5 4840 1.4 2 Calcium 8.7 85 2640 29 3 Magnesium 11.2 19 3520 13

Praktikum der Ionenchromatographie – Version 12
khv – 118 – 22/12/2000
Chromatogramm 50 Mineralwasser (Verdünnung 1:10) (versuch_18_b.gif)
Tabelle 79 Komponenten – Versuch 18 – Mineralwasser
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.7 4.3 4190 1.3 2 Kalium 6.4 0.8 6350 0.3 3 Calcium 8.3 344 1520 113 4 Magnesium 10.9 62 2550 43
Bemerkungen
Sämtliche Lösungen müssen in Kunststoffbehältern aufbewahrt werden. Für eine korrekte Natrium-Bestimmung muss jeglicher Kontakt mit Glas vermieden werden. Der pH-Wert der Standard- und Probelösungen muss zwischen 2.5 und 3.5 liegen.

Praktikum der Ionenchromatographie – Version 12
khv – 119 – 22/12/2000
4.4.2 Versuch 19 – Bestimmung der Übergangsmetalle Zur Trennung von Übergangsmetallionen werden der mobilen Phase Komplexbildner zugesetzt. Sie vermindern die Ladungsdichte und bewirken eine Verbesserung der Selektivität der Trennung, die für Übergangsmetallionen gleicher Ladung gering ist. Neben dem Gleichgewicht Austauscherharz / Analytion liegt bei Zusatz von Komplexbildnern ein weiteres Gleichgewicht, nämlich Metallion / Komplexbildner vor. Dieses Gleichgewicht wird ausserdem vom pH-Wert der Lösung beeinflusst.
Als Komplexbildner werden häufig schwache organische Säuren verwendet, z. B. Weinsäure, Zitronensäure, Oxalsäure, Pyridin-2,6-dicarbonsäure (Dipicolinsäure – vgl. Versuch 6 – Veränderung der Selektivität mit Hilfe von Komplexbildnern auf Seite 63). Die Zahl der Liganden L um das Zentralmetallion (Koordinationszahl) kann variieren, z. B. MeL, MeL2, MeL3. Auch die resultierende Ladung kann unterschiedlich sein. Je nach Ladung des Metallions, der Liganden und deren Zahl können kationische, neutrale und anionische Komplexe entstehen. Die Komplexe können daher an Kationenaustauschern oder Anionenaustauschern getrennt werden. Theoretisch ist auch die Verwendung von Austauschern mit Kationen- und Anionenaustauschergruppen möglich.
Durch Variation der Komplexbildner – auch 2 verschiedene Komplexbildner können der mobilen Phase zugesetzt werden – und des pH-Wertes kann die Trennung beeinflusst und optimiert werden. Die Einflüsse sind zum Teil gegenläufig und schwer vorauszusagen. Im folgenden werden einige allgemeine Hinweise gegeben:
• Unterschiedliche Stabilitäten (Komplexbildungskonstanten) beeinflussen die Trennung.
• Der Zusatz eines organischen Lösungsmittels, z. B. Aceton zum Eluenten beeinflusst ebenfalls die Trennung. Lipophile Ionen eluieren nach der Zugabe schneller.
• In der Kationenaustauschchromatographie hat sich Ethylendiamin als eluierendes Agens bewährt. Es liegt bei den üblichen Bedingungen als 2-fach protoniertes Kation vor.
• In der Kationenaustauschchromatographie bewirkt die Erhöhung des pH-Werts über die Abnahme der Gesamtladung eine Abnahme der Retentionszeit.
• In der Kationenaustauschchromatographie wird durch Erhöhung der Ligandenkonzentration in der mobilen Phase neben der Verschiebung des Komplexgleichgewichts auch die Konzentration des Gegenions H+ oder Na+ erhöht und die Verdrängung des Komplexes von den negativ geladenen Austauscherplätzen beschleunigt, d. h. die Trennleistung wird durch diesen Effekt geringer.
• In der Anionenaustauschchromatographie bewirkt die Erhöhung der Ligandenkonzentration einerseits eine Zunahme der Retentionszeit, da sich die Koordinationszahl der Komplexe erhöht. Andererseits fördern die freien anionischen Liganden die Elution der anionischen Metallkomplexe, so dass 2 gegenläufige Effekte bezüglich der Retentionszeit vorhanden sind.
Die Detektion der Komplexe kann sowohl über die Leitfähigkeit als auch photometrisch erfolgen.
Bei der Leitfähigkeitsdetektion kommt nur die Detektion ohne chemische Suppression in Frage, da bei der Suppressorreaktion zum Teil schwer lösliche Hydroxide entstehen würden.
Die photometrische Detektion wird i. a. nach einer Nachsäulenderivatisierung durchgeführt, bei der ein zugesetzter Komplexbildner den ursprünglich vorhandenen (z. B. Weinsäure, Oxalsäure...) verdrängt. Der neu gebildete und UV-oder VIS-absorbierende Komplex wird detektiert.
Lerninhalte
• Einfluss des Eluenten auf die Selektivität
• Komplexierung von Übergangsmetallionen

Praktikum der Ionenchromatographie – Version 12
khv – 120 – 22/12/2000
Tabelle 80 Parameter – Versuche 19a und 19b
Säule 6.1007.000 IC Kationensäule Nucleosil 5SA (4 x 125 mm) Eluent a) 4 mmol/L Weinsäure, 0.5 mmol/L Zitronensäure,
3 mmol/L Ethylendiamin, 5 % Aceton Leitfähigkeit ca. 500 µS/cm b) 3.5 mmol/L Oxalsäure 5 % Aceton pH = 4 (ca. 120 µL Ethylendiamin) Leitfähigkeit ca. 350 µS/cm
Probe Standard Leitungswasser
Methode exp_19_c.mtw System nucleosil.smt Fluss 1.5 mL/min Druck 13 MPa Analysendauer a) 12 min
b) 13 min Loop 100 µL Polarität -
Versuch 19a – Bestimmung der Übergangsmetalle mit einem Weinsäure-, Zitronensäure-, Ethylendiamin- und Acetoneluenten
Eluentherstellung
600 mg Weinsäure und 105 mg Zitronensäure in Reinstwasser lösen. 200 µL Ethylendiamin und 50 mL Aceton zugeben und auf 1 L auffüllen.
Chromatogramm 51 Chromatogramm – Standardlösung 1 für die Bestimmung der Übergangsmetalle (Eluent a) (versuch_19_a_std_1.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 121 – 22/12/2000
Tabelle 81 Komponenten – Versuch 19a – Standardlösung 1
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Nickel 3.4 5 1020 40 2 Zink 4.4 5 5640 36 3 Kobalt 5.6 5 4370 34 4 Eisen (II) 8.1 10 6150 30 5 Calcium 9.8 5 6340 29 6 Magnesium 10.8 5 6390 39
Chromatogramm 52 Standardlösung 2 für die Bestimmung der Übergangsmetalle (Eluent a) (versuch_19_a_std_2.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 122 – 22/12/2000
Tabelle 82 Komponenten – Versuch 19a – Standardlösung 2
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Blei 2.9 5 2680 8.6 2 Nickel 3.4 5 930 43 3 Zink 4.3 5 5510 37 4 Kobalt 5.6 5 4310 34 5 Cadmium 8.6 5 6600 15 6 Mangan 9.9 10 6270 45
Abbildung 37 Chromatogramm – Bestimmung von Leitungswasser (Eluent a – Verdünnung 1:10) (versuch_19_a_probe.gif)
Tabelle 83 Komponenten – Versuch 19a – Leitungswasser
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Zink 4.2 0.12 6360 0.9 2 Calcium 9.8 9.0 5750 53 3 Magnesium 10.6 1.9 7460 17
Versuch 19b – Bestimmung der Übergangsmetalle mit einem Oxalsäure-, Ethylendiamin-, Acetoneluenten
Eluentherstellung
315 mg Oxalsäure in Reinstwasser lösen, 50 mL Aceton zugeben und mit Reinstwasser auf 1 L auffüllen. Mit Ethylendiamin (ca. 120 µL) auf pH = 4 einstellen.

Praktikum der Ionenchromatographie – Version 12
khv – 123 – 22/12/2000
Chromatogramm 53 Standardlösung für die Bestimmung der Übergangsmetalle (Eluent b) (versuch_19_b_std.gif)
Tabelle 84 Komponenten – Versuch 19b – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Eisen 2.2 10 2110 50 2 Mangan 4.0 10 1830 110 3 Magnesium 7.5 5 3110 98 4 Calcium 12.3 5 6000 45
Chromatogramm 54 Bestimmung von Leitungswasser (Eluent b, Verdünnung 1:10) (versuch_19_b_probe.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 124 – 22/12/2000
Tabelle 85 Komponenten – Versuch 19b – Leitungswasser
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Magnesium 7.3 41 4690 2.1 2 Calcium 12.4 83 4780 9.1
Bemerkung
Calcium und Mangan coeluieren bei der Verwendung von Eluent a. Mit Eluent b sind Calcium und Mangan getrennt, Nickel, Zink und Kobalt werden stark komplexiert und eluieren im Wasserpeak.

Praktikum der Ionenchromatographie – Version 12
khv – 125 – 22/12/2000
4.4.3 Versuch 20 – Verunreinignungen im Silikagel – Bestimmung von Calcium- und Magnesiumionen
Silicagel (Kieselgel) wird z. B. durch Hydrolyse von Natriumsilikat und dessen weitgehende Entwässerung hergestellt und weist die folgende Struktur auf:
Si
Si
SiSi
Si
O
O
O
OO
O
O
OHOH
OH
Neben seiner Verwendung als technisches Adsorptionsmittel wird Silicagel sowohl in der Gaschromatographie wie auch in der Hochdruckflüssigchromatographie eingesetzt.
In der Gaschromatographie wurden zunächst – in geringem Ausmass auch noch heute – ausschliesslich gepackte Säulen eingesetzt. Dabei wird eine flüssige Phase, z. B. Siliconöl auf einen körnigen Träger, z. B. Silicagel aufgebracht. Die Trennstufenzahl dieser Säulen ist im Vergleich zur Trennstufenzahl der heute überwiegend verwendeten Kapillarsäulen gering.
Im Bereich der Hochdruckflüssigchromatographie (HPLC) wird Silicagel als stationäre Phase für Adsorptions-, Umkehr- (Reversed Phase, RP), Normalphasen- und Ionenchromatographie eingesetzt.
Auch in der Adsorptionschromatographie dient Silicagel neben Al2O3 als stationäre Phase. Die chromatographische Trennung beruht auf Dipol-induzierten Dipolbindungen, Dipol-Dipolbindungen, Wasserstoffbrückenbindungen und π-Komplexbindungen. Die Adsorptionschromatographie wird zur Trennung unpolarer Substanzen, die sich in Wasser schlecht lösen, eingesetzt.
Für die RP-Chromatographie werden die freien Silanolgruppen des Silicagels durch chemische Bindung von Octyl- oder Octadecylgruppen hydrophobiert, z. B.
Si OH + Cl Si
CH3
CH3
R Si O Si
CH3
CH3
R
Abbildung 38 Silcagel + Chlorsilan → R = (C8H17 bis C18H37)
Bei dieser Umsetzung verbleiben freie Restsilanolgruppen, die polare Verbindungen adsorbieren und zu Peak-tailing führen können. Diese Restsilanolgruppen lassen sich mit Trimethylchlorsilan deaktivieren «End capping».
In der Normalphasenchromatographie werden polare Reste an die Silanolgruppen chemisch gebunden, z. B:
(CH2)n C N oder (CH2)n NH2 (für die Zuckeranlaytik).
Mit dieser polaren stationären Phase wird eine wenig polare oder unpolare mobile Phase für die chromatographische Trennung eingesetzt. In der RP-Chromatographie, die ca. 75 % aller HPLC-Anwendungen ausmacht, sind die Polaritäten vertauscht und eine unpolare stationäre Phase wird mit einer polaren mobilen Phase verwendet. Der Name Umkehrphase bzw. Reversed Phase ist somit rein historisch begründet, da diese Methode erst später entwickelt wurde.
In der Ionenchromatographie wird Kieselgel, an das Sulfonsäuregruppen gebunden sind, als Kationenaustauscher für die Analyse divalenter Kationen eingesetzt.
Für den Einsatz von Silicagel in der Chromatographie ist die Verunreinigung mit Metallionen von Bedeutung, da diese zu unspezifischen Wechselwirkungen bzw. Blindwerten führen. Zur Analyse dieser Verunreinigungen wird ein stark saurer Kationenaustauscher verwendet, bei dem die

Praktikum der Ionenchromatographie – Version 12
khv – 126 – 22/12/2000
monovalenten Kationen im Totvolumen eluieren und nur die divalenten Kationen getrennt werden können. Fe(III) muss vor der Analyse durch Ascorbinsäure zu Fe(II) reduziert werden.
Lerninhalte
• Qualitätskontrolle
• Zusatzversuch: Spiken des Extraktes mit Fe(III);
Bestimmung mit und ohne Zugabe von Ascorbinsäure (Vitamin C)
Tabelle 86 Parameter – Versuch 20
Säule 6.1007.000 IC Kationensäule Nucleosil 5SA (4 x 125 mm) Eluent 4 mmol/L Weinsäure, 0.5 mmol/L Zitronensäure,
3 mmol/L Ethylendiamin, 5 % Aceton Leitfähigkeit ca. 500 µS/cm
Probe Silikagel (Lösung 5 %) Methode exp_20_c.mtw System nucleosil.smt Fluss 1.5 mL/min Druck 13 MPa Analysendauer 12 min Loop 100 µL Polarität -
Eluentherstellung
600 mg Weinsäure und 105 mg Zitronensäure in Reinstwasser lösen. 200 µL Ethylendiamin und 50 mL Aceton zugeben und auf 1 L auffüllen.
Chromatogramm 55 Standardlösung für die Bestimmung von Kationen in Kieselgel (versuch 20a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 127 – 22/12/2000
Tabelle 87 Komponenten – Versuch 20 – Standardlösung
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Calcium 9.6 0.5 6990 3.1 2 Magnesium 10.5 0.1 7260 0.7
Chromatogramm 56 Bestimmung von Kationen in Kieselgel (versuch20b.gif)
Tabelle 88 Komponenten – Versuch 20 – Kieselgel
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Calcium 9.7 9.4 6850 2.9 2 Magnesium 10.6 1.4 10100 0.4

Praktikum der Ionenchromatographie – Version 12
khv – 128 – 22/12/2000
4.4.4 Versuch 21 – Kosmetik und Korrosionsschutz: Bestimmung von Ethanolamin und Alkalimetallen
Die nachfolgenden drei Ethanolamine, deren systematische Namen Aminoethanol, Iminodiethanol und Nitrilotriethanol sind, werden durch Umsetzung von Ammoniak und Ethylenoxid hergestellt.
H2N CH2 CH2 OH
Abbildung 39 Monoethanolamin
Abbildung 40 Diethanolamin Abbildung 41 Triethanolamin
Alle 3 Substanzen werden u. a. zur Entfernung von CO2 und H2S aus Gasgemischen verwendet, z. B. zur Kohlendioxidentfernung aus Synthesegas und bei der Ammoniaksynthese oder aber für die Entfernung von Schwefelwasserstoff aus Raffineriegasen.
NR3 + H2O + CO2 HNR3+ + HCO3
-T
Bei höherer Temperatur wird CO2 wieder freigesetzt und das Ethanolamin NR3 in den Absorptionsprozess zurückgeführt.
2NR3 + H2S T
(HNR3)2S
Das bei höherer Temperatur wieder freigesetzte H2S wird in «Claus-Anlagen» zu flüssigem Schwefel oxidiert, der über SO2 und SO3 zur Schwefelsäure umgesetzt wird.
Monoethanolamin (MAK-Wert 3 ppm) wird als Fettsäureester in Tensiden verwendet. Diethanolamin kommt in Möbel- und Bodenpflegemitteln, Schuhcremes und Schmiermittel zum Einsatz.
Triethanolamin bildet mit Fettsäuren Triethanolaminseifen, z. B. mit Stearinsäure C18H35COOH. Diese sind in Wasser und Mineralölen leicht löslich und werden deshalb als Emulgatoren verwendet. Des weiteren können sie in kosmetischen Zubereitungen (z. B. Rasierschaum) enthalten sein. In Kraftwerken werden sie als Korrosionsinhibitoren eingesetzt, da sie einen schwach alkalischen pH-Wert stabilisieren und überschüssige H+-Ionen wegpuffern.
Für die ionenchromatographisch Bestimmung werden die Ethanolamine durch Säurezusatz protoniert und können so als Ammoniumderivate nachgewiesen werden.
Lerninhalte
• Bestimmung von anorganischen und organischen Kationen
• Selektivitätsänderung durch Zugabe von Komplexbildnern
H NCH2
CH2
CH2
CH2
OH
OHHO CH2 CH2 N
CH2
CH2
CH2
CH2
OH
OH

Praktikum der Ionenchromatographie – Version 12
khv – 129 – 22/12/2000
Tabelle 89 Parameter – Versuch 21a bis 21c
Säule 6.1010.210 Metrosep C2 100 x 4.0 mm – 7 µm Eluent a ) 4 mmol/L Weinsäure/ 0.75 mmol/L Dipicolinsäure Leitfähigkeit ca.
600 µS/cm b) 4 mmol/L Weinsäure/ 0.5 mmol/L Dipicolinsäure Leitfähigkeit ca. 570 µS/cm
Probe Ethanolamin, Triethanolamin + Standardkationen Methode exp_21_c.mtw System cation.smt Fluss 1 mL/min Druck 6 MPa Analysendauer a) 10 min
b) 13 min c) 14 min
Loop 10 µL Polarität –
Eluentherstellung
a) 600 mg Weinsäure und 125 mg Dipicolinsäure in 100 mL Reinstwasser unter Erwärmen lösen, danach mit Reinstwasser auf 1 L auffüllen.
b) 600 mg Weinsäure und 84 mg Dipicolinsäure in 100 mL Reinstwasser lösen, danach mit Reinstwasser auf 1 L auffüllen.
Chromatogramm 57 Standard 1 – Eluent a (versuch_21_a.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 130 – 22/12/2000
Tabelle 90 Komponenten – Versuch 21a – Standard 1 (Eluent a)
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.8 2 4520 6.8 2 Ammonium 4.3 2 4340 6.8 3 Monoethanolamin 4.9 7 4120 10.8 4 Kalium 6.4 5 3070 7.9 5 Triethanolamin 9.0 20 2230 6.9
Chromatogramm 58 Standard 2 – Eluent a (versuch_21_b.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 131 – 22/12/2000
Tabelle 91 Komponenten – Versuch 21 – Standard 2 (Eluent a)
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.7 2 4500 7.2 2 Ammonium 4.3 2 4190 7.0 3 Monoethanolamin 4.9 7 4380 9.8 4 Kalium 6.4 5 2930 8.6 5 Calcium + Triethanolamin 8.7 5 + 20 2070 24 6 Magnesium 11.0 5 2970 32
Chromatogramm 59 Standard 2 – Eluent b (versuch_21_c.gif)
Tabelle 92 Komponenten – Versuch 21 – Standard 2
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.9 2 4700 7.2 2 Ammonium 4.5 2 4110 7.0 3 Monoethanolamin 5.1 7 4400 9.6 4 Kalium 6.6 5 2920 8.4 5 Triethanolamin 9.3 20 2400 7.3 6 Calcium 10.8 5 3240 16 7 Magnesium 12.2 5 2820 32
Bemerkungen
Bei Verwendung von Eluent a koeluieren Calcium und Monoethanolamin. Sämtliche Lösungen müssen in Kunststoffbehältern aufbewahrt werden. Für eine korrekte Natrium-Bestimmung muss jeglicher Kontakt mit Glas vermieden werden. Der pH-Wert der Lösungen muss zwischen 2.5 und 3.5 liegen.

Praktikum der Ionenchromatographie – Version 12
khv – 132 – 22/12/2000
4.4.5 Versuch 22 – Alkali- und Erdalkalimetalle im Wein Die Mineralstoffe des Mostes sind vor allem Phosphate des Calciums, Magnesiums und Kaliums. Die Konzentrationen dieser Verbindungen betragen 3 – 5 g/L. Der Mineralstoffgehalt, auch als «Asche» bezeichnet, des Weins ist niedriger als der des Mostes, da ein Teil der Mineralstoffe im Stoffwechsel der Hefe aufgenommen wird. Ausserdem wird der Mineralstoffgehalt durch die Abscheidung weinsaurer Salze vermindert. Weine enthalten daher nur etwa 1.8 – 2.5 g/L «Asche». Wichtige kationische Bestandteile werden als Oxide angegeben und sind K2O (ca. 40 %), MgO (ca. 6 %), CaO (ca. 4 %) und Na2O (ca. 2 %).
(Vgl. Versuch 12 – Anionen im Wein, Seite 88)
Lerninhalte
• Lebensmittelanalytik
• Prozessanalytik – Kontrolle der Weinsäurefällung
Tabelle 93 Parameter – Versuch 22
Säule 6.1010.210 IC-Kationensäule Metrosep C2 100/4.0 - 7 µm Eluent 4 mmol/L Weinsäure/ 0.75 mmol/L Dipicolinsäure
Leitfähigkeit ca. 600 µS/cm Probe Standard (Natrium, Kalium, Calcium, Magnesium + 2 mmol/L HNO3)
Rotwein (Pinot Noir, Schweiz), Weisswein (Fechy, Schweiz) Methode exp_22_c.mtw System cation.smt Fluss 1 mL/min Druck 6 MPa Analysendauer 20 min Loop 10 µL Polarität -
Eluentherstellung:
600 mg Weinsäure und 125 mg Dipicolinsäure in 100 mL Reinstwasser unter Erwärmen lösen, danach mit Reinstwasser auf 1 L auffüllen.
Probenvorbereitung:
Proben filtrieren und verdünnen (1:10).

Praktikum der Ionenchromatographie – Version 12
khv – 133 – 22/12/2000
Chromatogramm 60 Standardlösung für die Bestimmung von Kationen in Wein (versuch22.gif)
Tabelle 94 Komponenten – Versuch 22 – Standard
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.7 1 4360 3.5 2 Kalium 5.9 100 1480 180 3 Calcium 8.6 5 2950 16 4 Magnesium 10.8 10 2150 66
Bemerkung
Sämtliche Lösungen müssen in Kunststoffbehältern aufbewahrt werden. Für eine korrekte Natrium-Bestimmung muss jeglicher Kontakt mit Glas vermieden werden. Der pH-Wert der Standard- und Probelösungen muss zwischen 2.5 und 3.5 liegen.

Praktikum der Ionenchromatographie – Version 12
khv – 134 – 22/12/2000
Chromatogramm 61 Bestimmung von Kationen in Weisswein (Verdünnung 1:10) (versuch22a.gif)
Tabelle 95 Komponenten – Versuch 22 – Weisswein
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.7 24 4220 8.4 5 Kalium 6.0 590 1860 106 6 Calcium 8.6 72 2920 23 8 Magnesium 11.0 59 2880 39
Bemerkung
Peaks Nr. 2, 3, 4 und 7 sind nicht bestimmt.
Chromatogramm 62 Bestimmung von Kationen in Rotwein (Verdünnung 1:10) (versuch22b.gif)

Praktikum der Ionenchromatographie – Version 12
khv – 135 – 22/12/2000
Tabelle 96 Komponenten – Versuch 22 – Rotwein
Peak Komponente tR [min] Konz. [mg/L] Bodenzahl Area [µS/cm*sec]1 Natrium 3.7 10 2000 3.6 6 Kalium 5.8 1380 1300 248 8 Calcium 8.6 50 3030 16
10 Magnesium 10.8 80 2520 53
Bemerkung
Peaks Nr. 2, 3, 4, 5, 7 und 9 sind nicht bestimmt.

Praktikum der Ionenchromatographie – Version 12
khv – 136 – 22/12/2000
5 Literatur zum Praktikumsbuch [1] H. Small, T.S. Stevens, W.C. Bauman, Anal. Chem. 47 (1975) 1801 [2] J. Weiß »Ionenchromatographie», 2. Aufl. (1991), Verlag Chemie, Weinheim [3] J.M. Mermet, M. Otto, H.M. Widmer »Analytical Chemistry», 1. Aufl. (1998), Wiley-VCH
Verlag, Weinheim-New York [4] P.R. Haddad, P.E. Jackson, »Ion Chromatography: Principles and Applications», 1. Aufl.
(1990), J. Chromatogr. Library Vol. 46, Elsevier Verlag, Amsterdam [5] J.S. Fritz, D.T. Gjerde, »Ion Chromatography», 3. Aufl., Wiley-VCH, Weinheim, 2000 [6] G. Schwedt, »Chromatographische Methoden in der Anorganischen Analytik», 1. Aufl. (1980),
Dr. Alfred Hüthig Verlag, Heidelberg [7] International Union of Pure and Applied Chemistry (IUPAC), Pure & Appl. Chem. 65 (1993),
819 [8] H. Engelhardt, L. Rohrschneider »Deutsche chromatograpische Grundbegriffe zur IUPAC-
Nomenklatur» (1998), Universität Saarbrücken [9] G. Schwedt »Chromatographische Trennmethoden», 3. Aufl. (1994), Thieme Verlag, Stuttgart [10] V.R. Meyer »Praxis der Hochleistungsflüssigchromatographie», 5. Aufl. 1988, Diesterweg
Verlag, Frankfurt/Main [11] A.J.P Martin, R.L.M. Synge, Biochem. J. 35 (1941) 1358 [12] B.A. Bindlingmeyer, F.V. Warren, Anal. Chem. 56 (1984) 1583 [13] J.P. Foley, J.G. Dorsey, Anal. Chem. 55 (1983) 730 [14] E. Grushka, L.R. Snyder, J.H. Knox, J. Chrom. Sci. 13 (1975) 25 [15] E. Katz, K.L. Ogan, R.P.W. Scott, J. Chromatogr. A 270 (1983) 51 [16] J.J Van Deemter, F.J. Zuiderweg, A.J. Klinkenberg, Chem. Eng. Sci. 5 (1956) 271 [17] J.H. Knox, H.P. Scott, J. Chromatogr. A 282 (1983) 297 [18] A. Berthold, F. Chartier, J.L. Rocca, J. Chromatogr. A 469 (1989) 53 [19] G. Wedler »Lehrbuch der physikalischen Chemie», 3. Aufl. (1987), Verlag Chemie, Weinheim [20] G. Schwedt, Fresenius Z. Anal. Chem. 320 (1985) 423 [21] M.J. van Os, J. Slania, C.L. de Ligny, W.E. Hammers, J. Agterdendos, Anal. Chim. Acta 144
(1982) 73 [22] P.R. Haddad, C.E. Cowie, J. Chromatogr. A 303 (1984) 321 [23] A. Diop, A. Jardy, M. Caude, R. Roset, Analusis 14 (1986) 67 [24] A. Jardy, M. Caude, A. Diop, C. Curvale, R. Roset, J. Chromatogr. A 439 (1988) 137 [25] D.R. Jenke, G.K. Pagenkopf, Anal. Chem. 56 (1984) 85 und 88 [26] T.B. Hoover, Sep. Sci. Tech. 17 (1982) 295 [27] P.R. Haddad, P.E. Jackson, A.L. Heckenberg, J. Chromatogr. A 346 (1985) 139 [28] P. Janoš, J. Chromatogr. A 789 (1/2) (1997) 3 [29] P. Janoš, P. Aczel, J. Chromatogr. A 749 (1996) 115 [30] P. Kolla, J. Köhler, G. Schomburg, Chromatographia 23 465 (1987) [31] C.A. Pohl, J. R. Stillian, P. E. Jackson, J. Chrom. A 789 29 (1997) [32] K. Dorfner (Hrsg.), »Ion Exchangers», 4. Aufl. (1991), Walter de Gruyter Verlag, Berlin [33] D.T. Gjerde, J.S. Fritz, G. Schmuckler, J. Chromatogr. A 186 (1979) 509











![Ionenchromatographie (IC) - Bestimmung von Anionen in ... · Abbildung 1 Chromatogramm einer Trinkwasserprobe aus der Schweiz [4] Ionenchromatographie (IC) - Bestimmung von Anionen](https://img.pdfslide.tips/doc/110x75/5e22d9f2b16c366bba257847/ionenchromatographie-ic-bestimmung-von-anionen-in-abbildung-1-chromatogramm.jpg)
![Praktikumsskript - Ionenchromatogprahie · 2016. 6. 15. · Die Messresultate werden mit der MagIC Net Software ausgewertet. Literatur [1] Praktikumsskript: Ionenchromatographie (IC)](https://img.pdfslide.tips/doc/110x75/6121c4f6ac4a9135eb3bce4a/praktikumsskript-ionenchromatogprahie-2016-6-15-die-messresultate-werden.jpg)