Embed Size (px)
Citation preview

FULL PAPER
Preparation, Characterisation and Reactivity of Neutral Mono-q4-oxadiene Complexes of Molybdenum and Tungsten Containing Bidentate Donor Ligands* Thomas Schmidt*, Frank Bienewald, and Richard Goddard
Max-Planck-lnstitut fur Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mulheini an der Ruhr, Gerniany E-mail: schmidtampi-muelheim.mpg.d400.de
Received October 20, 1995
Key Words: Oxa-1 J-diene complexes I Molybdenum oxadiene complexes / Tungsten oxadiene complexes / Electrophilic alkylation / Umpolung
Neutral molybdenum and tungsten complexes containing mined by X-ray structural analysis Spectroscopic data indi- only one q4-coordinated l-oxa-1,3-diene ligand are obtained cate that an effective transfer of electron density from the diastereospecifically from the reaction of the corresponding donor atoms occurs. This is also reflected in a decreased re- dicarbonylbis(oxadiene) complexes with bidentate donor li- activity of the carbonyl ligands in the mono(oxadiene) com- gands capable of forming five-membered ring chelates. plexes towards nucleophilic alkylation. As a result, the selec- Thus, dicarbonylbis[i14-(R)-(+)-pinocarvone]metal complexes tivity in the alkylation (a formal "Umpolung" reaction) of react with 1,2-bis(dimethylphosphanyl)ethane (dmpe) or anionic intermediates generated from 2 and methyllithium N,N,N',N'-tetramethylethylenediamine (tmeda) to give the with carbon electrophiles is greatly improved, allowing ac- mono(oxadiene) compounds 1-4 in 45-95 '%I yield. The mo- cess to alkylation products, e.g. 5, that are otherwise difficult lecular structure of the dmpe-tungsten complex 3 was deter- to obtain.
From complexation reactions of a,P-unsaturated ketones, esters, and amides with molybdenum and tungsten dicar- bony1 fragments, we have hitherto obtained dicar- bonylbis(q4-1-oxa-1 ,3-diene) complexes in a variety of cases"]. Under specific reaction conditions or when highly reactive metal sources such as bis(qG-to1uene)molybdenum are employed, homoleptic tris( 1 -oxa- 1,3-diene) complcxes of these metals could also be preparedL2]. Oxadiene com- plexes in and specifically group-6 metal com- p o u n d ~ [ ~ ~ ] have already been shown to be synthetically use- ful in nucleophilic alkylation reactions of various types.
However, to our knowledge no completely characterised, neutral complexes are known in which only one oxadiene ligand is coordinated to a group-6 metal centre in an q4- bonding mode. The few cationic mono(oxadiene) complexes already known bear a dicarbonyl(cyclopentadieny1)metal fragmentL4]. Since this fragment is chemically relatively in- ert, only limited possibilities exist for further ligand vari- ation in these complexes. In the course of our investigations directed towards the synthesis and reactivity of I -oxa- 1,3- diene complexes of molybdenum and tungsten, we became interested in the preparation of uncharged mono(oxadiene) complexes. The opportunity to alter the reactivity of the coordinated oxadiene ligands by the introduction of ad- ditional ligands to the metal centre appeared to be es- pecially promising.
In the following, we report on a simple preparative ap- proach to several of the hitherto unknown neutral mono(oxadiene) complexes by partial ligand exchange from
bis(oxadiene) complexes of molybdenum and tungsten by using phosphorus and nitrogen donor ligands that are ca- pable of forming five-membered chelate rings. Compared with the bis( oxadiene) complexes, the electronic situation within these complexes appears to be significantly altered by extensive interaction of the donor atoms with the metal. This is reflected in the spectroscopic properties as well as in the reactivity of these compounds.
Synthesis and Characterisation
Initial attempts to perform selective partial ligand substi- tution reactions on bis(oxadiene) complexes. e.g. dicar- bonylbis(q4-pulegone)tungsten[11 employing monodentate donor ligands such as alkyl- and arylphosphanes or carbon monoxide led only to exchange of both the coordinated ox- adienes. Using trialkylamines. insoluble, presumably poly- meric, metal-containing products of unknown structure were obtained[5].
When, however, ligands with two donor centres are used, specifically those which can form five-membered chelate rings on coordination to the metal, a highly selective reac- tion occurs with formation of dicarbonylmono(oxadiene) complexes containing one bidentate donor ligand (equation 1). Especially in the reaction of 1,2-bis(dimethylphospha- ny1)ethane (dmpe) with the bis(q4-pinocarvone)molyb- denum complex, the starting materials have to be used in equimolar amounts and the reaction has to be performed at room temperature or below in order to circumvent extensive formation of dicarbonylbis(dmpe)molybdenum.
Chem. Ber. 1996,129,305-311 0 VCH Verlagsgcscllschaft mbH, D-69451 Weinheim, 1996 0009-2940/96/0303-0305 S 10.00+.25/0 305

FULL PAPER T. Schmidt. F. Bienewald. R. Goddard
In this special case we have so far been unsuccessful in ob- taining an analytically pure sample of the molybdenum complex 1. However, the corresponding tungsten complex could be transformed cleanly into the mono(oxadiene) com- plex 3 in 56% yield. For bidentate nitrogen ligands such as NflN’,N’-tetramethylethylenediamine (tmeda), an excess of the donor ligand can be employed since there is clearly no tendency to form a complex of the composition (tmedajzMo(CO)z, which, to our knowledge, is unknown in the literature. Complex 2 could thus be obtained as orange solid in 95% yield.
Complex Yield [“/.I
1 M = M o X = P 58
2 M = M o X = N 95
3 M = W X = P 56
4 M = W X = N 4s
Analogous mono(oxadiene) complexes were also ob- tained with 1,2-bis(diphenylphosphanyl)ethane as donor li- gand. The solubility of these compounds, however, is ex- tremely low in many organic solvents so that NMR charac- terisation as well as reactivity studies of these compounds are dif€icult‘61.
Interestingly, all our attempts to obtain mono(oxadiene) complexes in a one-pot-procedure starting from a reactive metal source such as (thf),Mo(CO), or (EtCN),W(CO), and equimolar amounts of the oxadiene and the chelating ligand were unsuccessf~l~~]. The reasons for this unexpected behaviour are still under investigation, but presumably the special type of ligand exchange reaction required for the selective formation of mono(oxadiene) complexes necessi- tates the bis(oxadienc) compounds as starting materials (vide infra).
The new complexes have a much lower solubility than the bis(oxadiene) complexes, especially in nonpolar solvents such as hydrocarbons, but also in diethyl ether. By judicious choice of a solvent, e.g. n-pentane, the newly formed com- plexes could be precipitated from the reaction mixture and were thus immediately obtained in analytically pure form by simple filtration and washing with pentane. A significant shift of the carbonyl stretching vibrations towards smaller wave numbers is observed in their IR spectra. This obser- vation can easily be explained in terms of increased back- donation of electron density into the x* orbitals of the car-
bony1 ligands caused by the strong donor character of the newly introduced chelating ligand. Interestingly, IR spectra recorded from solid samples, e.g. KBr pellets, usually exhi- bit an additional splitting of the CO stretching vibrations, presumably due to some kind of mechanical or electronic coupling arising from intermolecular interactions in the crystal. This phenomenon and its correlation with results from crystal structure determinations currently is being in- vestigated[’]. These splittings disappear when spectra are re- corded in THF as well as hexane solutions. at least for the tmeda-molybdenum complex 2. In the mass spectra of the tmeda complexes 2 and 4, the identity of the fragments de- noted [Mi - 2 CO - 2H] was unambiguously assigned by high-resolution MS whereas the fragment [Mi -
CH2NMe2] could not be detected[’]. All mass spectra show that the complexes have, apart from loss of carbon monox- ide, only weak tendency for successive fragmentation of the different ligands.
In the 13C-NMR spectra, the most remarkable differ- ences between mono- and bis(oxadiene) complexes are the shift values of the carbonyl ligands. In general, they are shifted downfield by 7-29 ppm for the molybdenum and 2.5-24 ppm for the tungsten complexes. The large differ- ences between minimum and maximum deviations can be understood in terms of the geometrical requirement for one of the CO ligands to be trans to one of the donor atoms while the other has to be trans to either the ketone or the alkene x moiety of the organic ligand (see Scheme 1). As a consequence, the t r i m relationship of one of the carbonyl ligands changes upon oxadiene ligand substitution, whereas the other does not. The shift values of the coordinated car- bon atoms of the organic ligand respond quite differently to the ligand exchange. Whereas the shift of the quaternary alkene carbon signals remains almost unchanged, the meth- ylene carbon signal may be shifted in either direction by 4-14 ppm, depending on the specific complex. Most re- markably, the keto carbonyl signal is significantly shifted towards higher Geld in all cases [shift differences A&: 24.7 (compound 2), 36.1 (3) and 27.4 (4)].
Even with the geometrical restrictions discussed above, there are still several remaining possibilities for the arrange- ment of the ligands in the niono(oxadiene) complexes. This is illustrated in Scheme 1 (the nomenclature has been chosen according to the IUPAC scheme for octahedral co- ordination ge~metries[~]). Nevertheless, all the ligand ex- change reactions we have investigated so far have only led to one isomer of the corresponding mono(oxadiene) com- plexes. All attempts to uncover the identity of the selectively formed isomer on the basis of analysis of steric crowding or from spectroscopic data turned out to be highly specula- tive, so a crystal structure analysis of 3 was performed.
The results, summarised in Figure 1, clearly illustrate that the ad isomer is actually formed, where “ad” denotes the position of the newly introduced bidentate donor ligand. This is also in accordance with several other crystal struc- ture determinations of related compounds we have carried out[’]. Although the structural formula in Scheme I sug- gests some steric crowding between the pinocarvone and
306 Chem. Ber. 1996, 129, 305-311
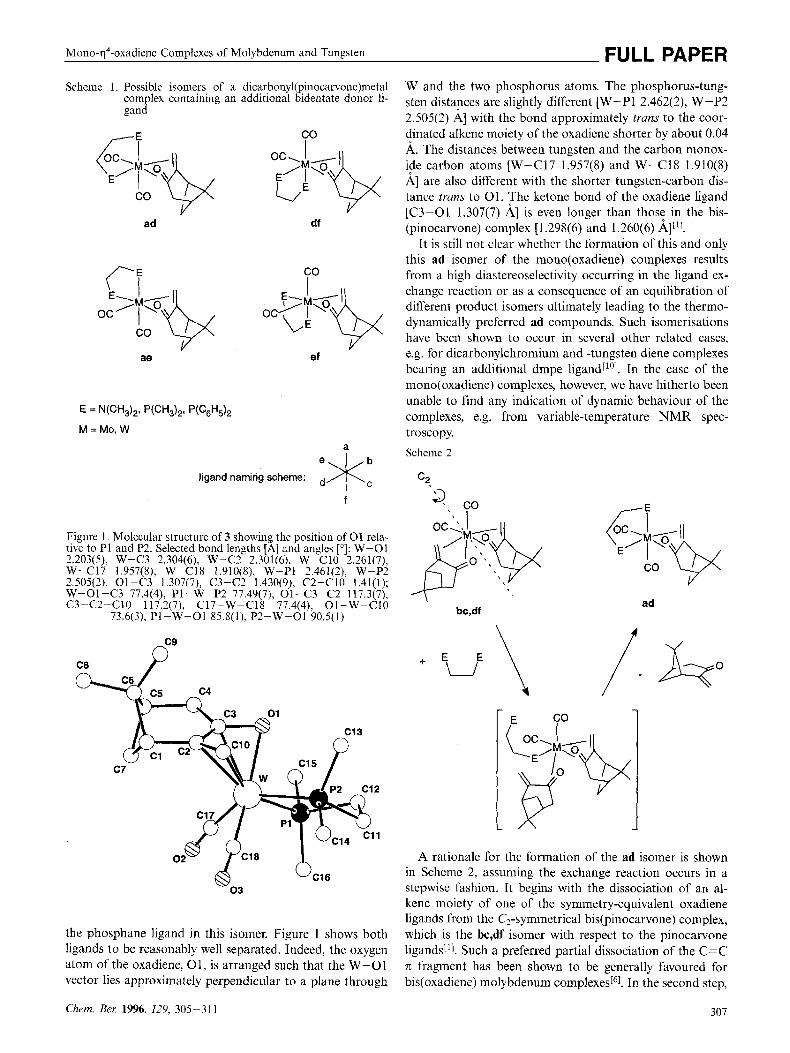
Mono-q4-oxddiene Complexes of Molybdenum and Tungbten FULL PAPER Scheme 1. Possible isomers of a dicarbonyl(piiiocarvoiic)metal
complex containing an additional bidelitate donor li- gand
ad df
co I
ae ef
ligand naming scheme: e*: ,, f
Figure 1. Molecular structure of 3 showing the position of 0 1 rela- tive to PI aiid P2. Selected bond lengths [A] and angles ["I: W-01 2.203(5). W-C3 2.304(6), W-C2 2.301(6), W-ClO 2.261(7), W--C17 1.957(8), W-CI8 1.910(8), W-PI 2.461(2), W-P2 2.505(2), Ol-C3 1.307(7), C3-C2 1.430(9), C2-CIO l.41(1); W-OI-C3 77.4(4), PI-W-P2 77.49(7), 01-C3-C2 117.3(7), C3-C2-C10 117.2(7), C17-W-Cl8 77.4(4), 01-W-C10
73.6(3), PI-W-Ol 85.8(1). P2-W-01 90.5(1)
uC16 03
the phosphane ligand in this isomer, Figure 1 shows both ligands to be reasonably well separated. Indeed, the oxygen atom of the oxadiene, 01, is arranged such that the W-01 vector lies approximately perpendicular to a plane through
Chm. BUK 1996. 129, 305-31 1
W and the two phosphorus atoms. The phosphorus-tung- sten distances arc slightly different W-Pl 2.462(2), W-P2 2.505(2) A] with the bond approximately trans to the coor- dinated alkene moiety of the oxadiene shorter by about 0.04 A. The distances between tungsten and the carbon monox- ide carbon atoms [W-C17 1.957(8) and W-C18 1.910(8) A] are also different with the shorter tungsten-carbon dis- tance trans to 0 1 . The ketone bond of the oxadiene ligand [C3-01 1.307(7) A] is even longer than those in the bis- (pinocarvone) complex [ 1.298(6) and 1.260(6) 811'1.
It is still not clear whether the formation of this and only this ad isomer of the mono(oxadiene) complexes results from a high diastereoselectivity occurring in the ligand ex- change reaction or as a consequence of an equilibration of different product isomers ultimately leading to the thermo- dynamically preferred ad compounds. Such isomerisations have been shown to occur in several other related cases, e.g. for dicarbonylchromium and -tungsten diene complexes bearing an additional dmpe ligand[l01. In the case of the mono( oxadiene) complexes, however, we have hitherto been unable to find any indication of dynamic behaviour of the complexes, e.g. from variable-temperature troscopy. Scheme 2
/E
NMR spec-
\
bc,df ad
A rationale for the formation of the ad isomer is shown in Scheme 2, assuming the exchange reaction occurs in a stepwise fashion. It begins with the dissociation of an al- kene moiety of one of the symmetry-equivalent oxadiene ligands from the C,-symmetrical bis(pinocarvone) complex, which is the bc,df isomer with respect to the pinocarvone ligandsr']. Such a preferred partial dissociation of the C=C n: fragment has been shown to be generally favoured for bis(oxadiene1 molybdenum complexed61. In the second step,
307

T. Schmidt, F. Bienewald, R. Goddard FULL PAPER one of the donor atoms of the bidentate ligand becomes coordinated, and finally the other donor atom replaces the ketone moiety of the partly coordinated oxadiene by a backside attack, which can be envisioned to be sterically favourable. The second ligand cannot be involved as its al- kene moiety is in the trans position and thus is not geo- metrically accessible to the donor ligand.
Reactivity
When dicarbonylbis(oxadiene) complexes of molyb- denum and tungsten are treated with Grignard or organoli- thium compounds, highly diastereoselective 1,2-alky- l a t i ~ n [ ~ ~ ] of one of the coordinated oxadienes (e. g. observed with the pulegone complex) and/or equally selective 1,4-re- duction of one ligand (predominant reaction for the pino- carvone complexes) takes p l a ~ e [ ~ ~ , ~ ] , depending on the ac- tual starting material. In the latter reaction, the formation of the reduction product isopinocamphone can be ex- plained by invoking an oxametallacyclic intermediate B (Scheme 3) which is then hydrolytically cleaved.
Scheme 3. Schematic reaction pathways for the subsequent trcat- ment of oxadiene complexes with nucleophiles (R’Li) and electrophiles (R2-Hal or H20)
OC\
R’, R2 = alkyl M=Mo, W
W Hal = Br. I
R1 Li I 1- 1-
\P1 ’ oc oc \ , R 1 ‘
2. H20 I H,O
Scheme 4
2
1. MeLi (1.2 eq.),THF 2. t-BuBr, -78°C + 20°C \ 3. H20,CAN
Y (42 Yo)
(4:3) (4:3)
(35%) ( 3 : l )
(24%) k o 7 ( 3 : l )
(9%) (-1
1. MeLi (1.1 eq.),THF 2. t-BuBr, -78°C + 20°C \ 3. HZO
1
The anionic intermediates A andlor B proposed for these reactions can also react with electrophiles other than water (Scheme 3). Such generation of anionic intermediates by the
R1 1 - 1 action of hard carbon nucleophiles, e.g. methyllithium, and / subsequent alkylation reaction with electrophiles such as
alkyl halides results in an overall “Umpolung” of the oxadi- ene P-carbon atom. A related nickel-mediated reaction has been recently reported to proceed with unsaturated alde- hydes to yield P-alkylated silyl enol ethers[“].
C Performed with dicarbonylbis(pinocarvone)molybdenum or -tungsten complexes as starting materials, the reactions yielded only a complex mixture of products (Scheme 4, bot- tom), as additional competition arose from an intramolecu- lar carbon monoxide insertion with formation of an acyl- enolate intermediate C (Scheme 3) , which gave rise to the formation of a 1 ,Cdicarbonyl compound after intramolecu- lar migration of R’ and subsequent hydrolysis. This latter
I H‘O
R 1 k
308 Chem. Ber. 1996, 129, 305-311

FULL PAPER Mono-q4-oxadiene Complexes of Molybdenum and Tungsten
reaction is already known to be the typical pathway fol- lowed by oxadiene carbonyliron complexes on treatment with various nucle0philes[~~1.
In contrast, when mono(oxadiene) complexes such as 2 were employed, yield and selectivity of such reactions could be significantly improved. Specifically, the acylation reac- tion arising from attack on the carbonyl ligand was effec- tively suppressed (Scheme 4, top). Additionally, the liber- ation of one unreacted ligand as in the case of the bis(oxa- diene) complexes was no longer observed, as expected.
The increased electron density in mono(oxadiene) com- plexes bearing additional donor ligands, which is evident from spectroscopic data, e.g. the IR spectra (vide supra), is likely to be responsible for the lack of acylation in these cases. However, hydrolysis of the anionic intermediates is still competing so that significant amounts of the pinocam- phones are also obtained. Due to base-catalysed isomeri- sation, a mixture of diastereomeric products was formed in these cases.
Similar reactions could also be performed with other electrophiles such as methyl iodide or isopropyl bromide. The reaction was especially effective when ethyl 2-bromo-2- methylpropionate was employed. In this case a 4: 1 mixture of the P-alkylation products 2-[2-(ethoxycarbonyl)-2- methylpropyl]-6,6-dimethylbicyclo[3.l.l]heptan-3-one and reduction products was obtained in an isolated yield of 41 %. Thus, the reaction appears to be especially suitable for tertiary halides.
In summary, the "Umpolung" methodology of the p-PO- sition of an a,P-unsaturated ketone is a useful method for the synthesis of alkylation products that are otherwise diffi- cult to obtain. However, it is essential to use electronically
no(oxadiene) complexes with additional donor li- starting materials in order to obtain acceptable
product selectivities in these reactions. Financial support from the Fonds der Chemisehen Industrie, the
Studienstiftung des Deutschen Volkes, the Fritz ter Meer-Stiftung, and the Mux-Planck-GeselI.~chaft is gratefully acknowledged.
Experimental All manipulations of organometallic compounds were carried
out under purified, dry argon by employing standard Schlenk tech- niques. Solvents were dried by using Na[AIEt4] as drying agent, subsequently distilled and saturated with argon. - Melting points: determined in capillary tubes sealed under argon, uncorrected. - The molybdenum and tungsten bis(q4-(R)-(+)-pinocarvone) com- plexes were prepared according to the published procedures[']. - IR: Nicolet 7199 FT-IR. - MS: Varian 311A; for metal-containing ions, signals corresponding to the major isotopes 98Mo and 184W are reported. - 'H, 13C and 31P NMR: Bruker AC 200. Standards: Si(CH3)4 ('H), solvent signal (I3C), H3P04, ext. (31P). Multiplicities of I3C-NMR signals obtained from DEPT experiments are given in parentheses. Additional signal splitting arising from coupling to phosphorus is denoted in brackets. - Elementary analyses: Dornis & Kolbe, Microanalytical Laboratory, Mulheim an der Ruhr.
(1,2-Bis(dimethylphosphunyl) ethune]dicurbonyl[q4- (R) - (+)-pinocurvone]molybdenum(O) (1): To a solution of 244 mg (0.54 mmol) of dicarbonylbis[q4-(R)-( +)-pinocarvone]molybdenum
Chem. Ber. 1996, 129, 305-311
in 15 ml of n-pentane was added 75 mg (0.5 mmol) of 1,2-bis(dime- thylphosphany1)ethane (dmpe) by means of a synnge. Withm I nun an orange-brown precipitate formed. After stirring for 30 min at room temp., the reaction mixture was filtered through a sintered glass frit, washed twice with 2-ml portions of n-pentane, and dried under high vacuum (0.1 Pa) for 2.5 h. A brown solid was obtined that could not be recrystalhsed without undergoing extensive de- composition. From the 3'P-NMR spectra, a mnor contamination of the product by cis-(dmpe)2Mo(CO)2 (A2X2 pattern centred at 6 = 41.8 and 23.3, J p p = 17 Hz) could be detected['*]. Crude yield of 1: 131 mg (58%0), dec. above 100°C. - IR (KBr): 3 = 1972 s
MHz): S = 54.46 (A part, AB pattern, 2Jpp = 30.0 Hz), 41.84 (B part, AB pattern). - MS (EI, 70 eV), m/z (%): 454 (9) [M+], 426
Dicurbonyl[q4-(R) -( + ) - p i n o c a r v o n e / ( ~ ~ N ' , N - t e t r u m e t h y l - ethylenedcumine)molybdenum(O) (2): To a solution of 1.09 g (2.4 mmol) of dicarbonylbis[q4-(R)-( +)-pin0car~0ne]mo1ybdenum(0) in 15 ml of benzene was added 291 mg (2.5 mmol) of "N',N'- tetramethylethylenediamine at room temp. After stirring for 30 min, TLC monitoring showed the starting molybdenum complex to have disappeared completely (solvent: diethyl etherlhexane, 1 : I ; Rf = 0.65). This spot was replaced by free pinocarvone (Rf = 0.63, colourless spot, UV-detected) and the product complex (remaining at the starting line). The reaction mixture was filtered through a D4 sintered glass frit, benzene and pinocarvone were removed under reduced pressure, and the orange solid residue was dried under high vacuum (0.1 Pa). Yield: 958 mg (%YO), m.p. 130°C (dec.). - IR (KBr): P = 3022 w cm-', 2977 s, 2916 s, 2842 s, 2800 m (CH), 1862 s, 1858 S, 1771 s, 1748 s [Mo(CO)~], 1469 s, 1406 s, 1385 s, 1298 m, 1019 m, 959 m, 809 s; IR (4% solution in THF): C = 1878 s cm-I,
2.63 (s, 3H, NCH,), 2.60-2.50 (m, IH), 2.47 (s, 3H, NCH3), 2.45-2.30 (m, ZH), 2.28 (s, 3H, NCH,), 2.17-2.05 (m, 3H), 2.01 (s,2H), 1.85-1.70(m, IH), 1.68(s, 3H,NCH3), 1.36(s, lH), 1.24
z): F = 259.19 (s, CO), 237.61 (s, CO), 158.59 (s,
Cm-', 1883 S, 1840 S, 1775 S [Mo(C0)2]. - 31P NMR (C&, 81.0
(31) [Mt - CO], 396 (100) [Mt - 2 CO - 2H], 41 (21).
1773 S. - 'H NMR (C6D6, 200.1 MHz): 6 = 3.02-2.80 (m, IH),
1.18 (s, lH), 1.14 (s, 3H, CCH3). - I3C NMR
5.44 (s , C=CH*), 59 76 (t), 59.48 (t), 54.41 (q, NCH3), 54.11 (4, NCH3), 53.91 (q, NCH3), 48.70 (t), 48.24 (d), 45.36 (q, NCH3), 42.24 [s, C(CH&], 41.19 (d), 36.55 (t), 32.04 (t), 27.14 (q, C-CH3), 21.72 (q, C-CH3). - MS (EI, 70 eV), m/z (YO):
72 (14), 58 (100) [Me2NCH:], 28 (16). - C18H30M~N203 (418.4): calcd. C 51.67, H 7.23, Mo 22.93, N 6.75; found C 51.49, H 7.30, Mo 22.74, N 6.75.
[1,2-B1s(dimethylphosphanyl)ethane jdic~rbonyl[~~-(R)-(+)- pinocarvone]tungsten(O) (3): 82 mg (0.55 mmol) of 1,2-bis(dime- thylphosphany1)ethane was added to a stirred solution of 297 mg (0.55 mmol) of dicarbonylbis[q4-(R)-(+)-pinocarvone]tungsten(O) in 10 ml of n-pentane. After stirring for an hour, the orange-red supernatant solution was filtered off and the prccipitate washed twice with 2 ml of n-pentane. The filtrates as well as the washing fractions were discarded, and the precipitate was dried for 2 h un- der high vacuum (0.1 Pa) to yield analytically pure complex 3 as an orange solid. Yield: 180 mg (56%0), m.p. 195-197°C (dec.) - IR (KBr): 3 = 2975 w cm-l, 2929 m, 2911 m (CH), 1180 s, 1795 s [W(CO),], 1410 m, 1280 w, 1129 w, 931 m, 894 w, 837 w. - 'H NMR (C6D6, 200.1 MHz): 6 = 3.40-2.62 (AB pattern, J H H = 17.1 Hz, 2H, C=CH2), 2.57-2.17 (m, ZH), 2.17-2.00 (m, IH), 2.00-1.85 (m, lH), 1.31 (d, 2JpH = 8.8 Hz, 3H, PCH,), 1.22 (d, 'JPH = 8.9 Hz, 3H, PCH,), 1.17-0.25 (m, 6E9, 1.13 (s, 3H,
420 (8) [M+], 392 (0.7) [M+ - CO], 362 (32) [M+ - 2 CO - 2H],
CCH3), 1.04 (d, ' J ~ H = 9.1 Hz, 3H, PCH3), 0.93 (s, 3H, CCH3), 0.63 (d, 'JpH = 8.1 Hz, 3H, PCH3). - I3C NMR (C6D6, 50.3
309

FULL PAPER T. Schmidt, F. Bicnewald. R. Goddard
MHz): 6 = 244.70 (s [dd], CO, X part of ABX pattern, 12Jpc.l = 19.0/8.6 Hz), 222.47 (s [dd], CO, X part of ABX pattern, (2Jpc( = 6.611.9 Hz), 141.08 (s, C=O ketone), 101.11 (s, C=CH2), 46.95 (d), 42.69 (d), 42.22 [s, C(CH&], 36.59 (t [dd], X part of ABX pattern,
part of ABX pattern, /‘JP,-I = 28.8128.4 Hz, PCH2). 29.59 (t idd], X part of ABX pattern, lJpcl = 28N16.6 Hz, PCHJ, 27.39 (q,
PCH3), 16.95 (q [dd], 1’’4Jp,-( = 1933.5 Hz. PCH3), 16.23 (q [dd],
I2JpcI = 6.916.1 Hz, C=CH2), 34.86 (t), 33.50 (t), 30.75 (t [dd], X
HIC-C), 21.91 (4, H3C-C), 17.54 (9 [dd], I”4JpcI == 29.6/3.5 Hz,
1”4Jpc1 = 29.613.5 Hz, PCH3), 10.6 (9 [d], I’Jpcl = 20.1 Hz, PCHI). - 31P NMR (C6D6, 81.0 MHz): 6 = 32.42 (AB pattern [d], ‘Jwp =
262.0, 2Jpp = 13.6 Hz), 22.40 (AB pattern [d], lJWp = 207.6 Hz). - MS (EI, 70 eV), m/z (%): 540 (32) [M’], 512 (93) [M+ - CO],
C18H3003P2W (540.2): calcd. C 40.02, H 5.60. P 11.47; found C 39.84, H 5.72, P 11.55.
X-Ray Analysis of3 [ I3 ] : CI8H3,,O3P2W, M = 540.2 g . mol-’, crystal colour: red, crystal size 0.20 0.21 x 0.2$ mm, a = 12.114(1), h = 12.588(1), c = 13.651(1) A, V = 2081.7 A3, T = 293 K, dCdc. = 1.72 g . cm-j, p = 57.15 cm-I, absorption correction: psi-scan, min. trans.: 0.820, max. trans.: 0.983, F(000) = 1064 e, 2 = 4, orthorhombic, space group P2,2,2, mo. 191, Enraf-Nonius CAD4 diffractometcr, h = 0.71069 A, index range 0 6 h G 17, 0 6 k c 17, 0 c 1 19, measuring methodam-20, 3440 measured reflections [+h, +k, + I ] , [sinWh],, = 0.70 A-’, 3408 independent and 3193 observed reflections [I > 2~(1)], 217 refined parameters, structure refinement: full-matrix least-squares on e, riding hydro- gen atoms included in least-squares refinement, R = 0.042, R,, = 0.102, ]w = l/{02(e) + (0.033 E”, + 0.067 e l 2 ] , final difference Fourier p = 1.65 eA-3 in the vicinity of the tungsten atom.
482 (100) [Mt - 2 CO - 2H], 108 (23), 53 (47), 41 (93). -
Dicurbonyl(q4- (R) - (+ ) -pinocurvone] (nl;nl; N , IT-tetramethyl- ethylenediamine~tungsten(0) (4): 69 mg (0.60 mmol) of N N N ’ , N ‘ - tetramethylethylenediamine was added to a solution of 322 mg (0.60 mmol) of dicarbonylbis[~~~-(R)-(+)-piiiocarvone]tungsten(O) in 10 ml of n-pentane. The rcsulting mixture was stirred at room temp. for 24 h and then heated at 40°C for 6 h. The suspension was then filtered through a sintered glass frit and the residue washed with two 2-ml portions of a-pentane. The combined f i l - trates were discarded, and the residue was dried under high vacuum (0.1 Pa) for 2 h. Complex 4 was obtained as orange solid. Yield: 137 mg (45%;), dec. above 180°C. - IR (KBr): P = 3020 w cm-’, 2978 m, 2922 s, 2918 & 2848 m, 2800 w (CH). 1859 s. 1850 s, 1759 s, 1737 s pN(CO),], 1469 m, 1450 m, 1408 m, 1291 w, 1262 w, 1012 w, 956 w, 810 m. - ’H NMR ([D,]THF, 200.1 MHz): 6 = 3.34 ( 5 ,
3H, NCH,), 3.05 (s, 3H, NCH3), 3.02-2.71 (m, 3H), 2.66 (s, 3H, NCH3), 2.57-2.12 (m, 6H), 2.28 (s, 3H, NCHI), 2.17 (s, IH), 1.76-1.62 (m, 2H), 1.42 (s, 3H. CCH3), 1.13 (s, 3h, CCH,). - I3C
NMR ([D,]THF; 50.3 MHz): 6 = 244.20 (s, CO), 237.71 (s, CO), 149.85 (s, C=O ketone), 94.85 (s, C=CH2), 62.66 (t), 62.39 (t), 57.71 (q, NCH3), 56.82 (q, NCHj), 55.44 (q, NCHI), 49.46 (d), 47.10 (q, NCHI), 43.67 [s, C(CH,)2], 42.65 (d), 41.78 (t), 36.34 (t), 32.86 (t), 28.11 (q, H3C-C), 22.56 (q, H,C-C). - MS (EI, 70 eV),
CO - 2H], 72 (16), 58 (100) [Me2NCH;]. - C I ~ H ~ ~ N ~ O ~ W (506.3): calcd. C 42.70, H 5.97, N 5.53; found C 42.56, H 5.92, N 5.58.
m 1 ~ (Yo): 506 ( 5 ) [M+], 478 (0.4) [M+ - CO], 448 (18) [M+ - 2
Methjdlithiurn AdditionlElectrophilic Alkylation Sequence with tert-Butyl Bromide
a) With Dicarbonylbis(r14-(R)- (+)-pinocar~~one]molybdenmi(O): A solution of 226 mg (1 .O mmol) of dicarbonylbis[q4-(R)-(+)-pino- carvone]molybdenum in 5 ml of THF was cooled to -78 “C. After the addition of 1.0 mmol of an ethereal solution of methyllithium,
the reaction mixture was allowed to warm up slowly to room temp. and stirred at that temp. for another 15 min. After cooling again to -78”C, 690 mg (5.0 mmol) of tert-butyl bromide was added by means of a syringe, the mixture was warmed up and stirred at 20°C for 24 h. THF was then removed from the reaction mixture under reduced pressure, the residual material was taken up in a mixture of 30 ml of diethyl ether and 10 ml of water. After stirring under normal atmospheric conditions for an additional hour, the aqucous layer was removed and the organic layer washed successively with 10-ml portions of water and asaturdtcd NaCl solution. After dry- ing with MgS04, the filtered solution was concentrated in vacuo to yield an oily residue which was analysed by NMR spectroscopy and GLC. Combined yield: 68%: 93.2 mg (21%) of 2-(2,2- dimethylpropyl) -6,6-dimethylbic~~clo0/3.1.1 lheptnn-3-one (5 ) (4: 3 mixture of diastereomers, product characterisation included in fol- lowing procedure). - 54.3 mg (14%) of 6,h-dimelhyl-2-(2- oxopropyl) hicyclo[3.1.I]heptcin-.7-one (6) (4: 1 mixture of di- asteromcrs); spcctroscopic data identical with those reported in ref.[I4]. - 73.0 mg (24%) ofpinocamphonelisopinocainphone (7); di- astereomeric mixture (1 :3); spectroscopic data identical with those reported in ref.[15]. - 27.0 mg (9%) of iR)-(+)-pinocarvone (8).
b) With Dirarbonyl(q4- ( R ) - (+) -pinocarvone] (NN N , “-tetra- meth~~lethylenediamine]molyhdt.num(O) ( Typical Procedure): 41 8 mg (1.0 mmol) of the mono-l-oxa-l,3-diene complex 2 was dis- solved in 5- 10 ml of THE After cooling of the solution to -78”C, 1.2 equivalents of a solution of inethyllithium in diethyl ether was added. The mixture was allowed to warm up to room temp., and after it had been stirred at that temp. for 30 min. 685 mg (5.0 mmol) of tert-butyl bromide was introduced. The reaction mixture was then stirred for 5 h at room temp. The solvents were subsequently removed under reduced pressure, the residue was taken up in 40 ml of diethyl ether, and 20 ml of water was added to the solution. Solid cerium ammonium nitrate (CAN) was slowly added with vigorous stirring until the aqueous phase turned permanently yellow. The clear, colourless ethereal layer was separated, washed with a satu- rated NaHC03 solution, a saturated NaCl solution and dried with MgS04. The colourless oily residue remaining after removal of the solvent in vacuo was analysed by NMR spectroscopy and GLC. Pure products could be obtained by chromatography on silica using diethyl etherln-hexane (1 : 1) as eluent. Combined yield: 78%: 106.4 mg (35%) of a 1 :3 mixture of pinue~phune/iuopii~o~~u~zi~hone (7); 174.8 mg (43%) of 5 (4:3 mixture of diastereomers). - Analytical data for the major diastereomer of 5: IR (neat): P = 298 s cm-’ (CH), 2871 m (CH), 1716 s (C=O), 1472 m, 1410 w, 1395 w, 1385 w, 1368 m, 1156 m. - ‘H NMR (CDC13, 200.1 MHz): 6 = 2.71-2.23 (m, 4H), 2.09-2.03 (m, 2H), 1.78 (dd, IH, JHH = 13.91 2.4 Hz), 1.33 (s, 3H, H3CC), 1.25-1.10 (m, 2H), 0.91 [s, 9H, (H?C),C], 0.89 (s. 3H, H3CC). - NMR (CDC13, 50.3 MHz): 6 = 216.00 (s, C=O), 49.22 (d), 44.65 (d), 44.46 (t), 44.01 (t), 39.03 [s, C(CH&], 37.79 (d), 31.05 [s, C(CH&I. 29.75 [q, 3 C, (H,C)3C],
mlz (%): 208 (10) [Ml]. 151 (4) [M+ - C4H9], 139 (21), 111 (26), 69 (71), 57 (100) [C4H$], 41 (44). - C14H240: calcd. 208.182715;
29.31 (t), 26.53 (9, H3C-C), 19.94 (q, H3C-C). - MS (EI, 70 eV),
found 208.181445 (high-resolution MS).
* Dedicated to Professor Dr. Marianne Baudler on the occasion of her 75th birthday.
[‘I Th. Schmidt, P. Betz, C. Kriiger, .I Orgunornet. Chem. 1991, 402, 196-206; Th. Schmidt, F. Bienewald, R . Goddard, .I Chem. Soc., Chem. Commun. 1994, 1857-1858.
1’1 Th. Schmidt, S. Neis, J Organomet. Chem. 1992, 430, C5-C9; Th. Schmidt, S. Neis, R. Goddard. Z. Naturforsch., Part B, 1995, 50. 315-325; Th. Schmidt, R. Goddard, 1 Chem. Soc,, Dalton Trans. 1995, 1563 - 1568.
310 Chem. Ber. 1996, 129, 305-311

Mono-q4-oxadiene Complexes of Molybdenum and Tungsten
K31 For example: [3aJ S. E. Thomas, T. N. Danks, D. Rakshit, Phil. Truns. R. Soc. London A 1988, 326, 611-617; T. N. Danks, D. Rakshit, S. E. Thomas, 1 Chem. Soc., Perkin Trans. I , 1988, 2091-2093. - [3bl Th. Schmidt. F. Bicnewald. GIT Fuchz. Lab. 1993,37, 761-762.
[41 E.e.: P. L. Watson. R. G. Bergman. J Am. Chem. Soc. 1979, ZO?, 2055-2062; H. G. Alt, HrE. Engelhardt, B. Wrackmeyer, R. D. Rogers, J Organomet. Chem. 1989,379, 289-301; €1. G. Alt, G. S. Herrmann, U. Thewalt, ibid. 1987, 327, 237-246; M. Green, R. J. Mercer, A. G. Orpen, C. J. Schaverien, I. D. Willi- ams, 1 Chem. Sor., Dalton Trans. 1986, 1971-1982.
['I Th. Schmidt, R. Goddard, unpublished results. Th. Schmidt, Habilitation Thesis, Universitat Koln, 1992.
['I F. Bienewald. Th. Schmidt, unpublished results. [*I We thank M. MaJau for performing the HRMS determi-
nations. [''I B. P. Block, W. H. Powell, W. C. Fernelius, Inorganic Chemical
Nomenclature - Principles and Practice, ACS Professional Ref- erence Book, ACS, Washington, DC, 1990, p. 141.
FULL PAPER [lo] C. G. Kreiter, Adv. Organomet. Chem. 1986, 26, 297-375, and
references therein. [ ' ' I J. R. Johnson, P. S. Tully, P. B. Mackenzie, M. Sabat, J Am.
Chem. Soc. 1991, 113, 6172-6177; B. A. Grisso, J. R . Johnson, P. B. Mackenzie, ibid. 1992: 114, 5160-5165.
[12] J. A. Connor, G. K. McEwen, C. J. Rix, J Chem. Soc., Dalton Trans. 1974, 589-595; S. Datta, T. J. McNeese, S. S. Wreford, Inorg. Chem. 1977, 16, 2661-2663.
[I3] Additional details of the crystal structure determination may be obtained from the Fachinformationszentrum Karlsruhe, Ge- sellschaft fur wissenschaftlich-tcchnische Information, D-76344 Eggenstein-Leopoldshafen, on quoting the depository number CSD-404597, the names of the authors, and the journal ci- tation.
[I4] A. Kover, H. M. R. Hoffmann, Tetrahedron 1988, 44, 6831-6840.
[''I J. M. Coxon. G. J. Hvdes. P. J. Steel. J Chem. SOC.. Perkin Trons. 2. 1984, 1351-1355:
[95165]
Chmr. Ber: 1996, 129, 305-311 31 1


















![[Chem 211] Synthesis and reactivity of sterically encumbered diazaferrocenes.pptx](https://img.pdfslide.tips/doc/110x75/563dbba6550346aa9aaf0e3b/chem-211-synthesis-and-reactivity-of-sterically-encumbered-diazaferrocenespptx.jpg)










