Embed Size (px)
Citation preview

BRAIN RESEARCH
ELSEVIER Brain Research 734 (1996) 203-212
Research report
Progressive expression of immunomolecules on activated microglia and invading leukocytes following focal cerebral ischemia in the rat
Hiroyuki Kato a, *, Kyuya Kogure b, Xiao-Hong Liu a, Tsutomu Araki a, Yasuto Itoyama a a Department of Neurology, Tohoku University School of Medicine, 1-i Seiryo-machi, Aoba-ku, Sendai 980-77, Japan
b Foundation for Brain Function and Diseases and the Institute ofNeuropathology, Fukaya 366, Japan
Accepted 15 May 1996
Abstract
In order to evaluate the involvement of inflammatory reactions following focal cerebral ischemia in the rat, we immunohistochemi- cally visualized microglial cells and blood-borne leukocytes (neutrophils and monocytes) using various antibodies directed against immunomolecules expressed on these cells. Focal cerebral ischemia was produced by intraluminal occlusion of the right middle cerebral artery for 1 h. The brains were perfusion-fixed at 4 h, 1 day, 3 days, 7 days and 14 days after ischemia. Frozen brain sections were prepared and stained with monoclonal antibodies to complement receptor type 3 (OX42), major histocompatibility complex (MHC) class I and class II antigens (OX18 and OX6, respectively), a pan-macrophage/monocyte marker (ED1), intercellular adhesion molecule-1 (ICAM-1), LFA-1 c~ chain (CDlla) and [3 chain (CD18), and T cells (CD5). In ischemic areas where infarction developed later, microglial cells were destroyed (beginning at 4 h), neutrophils migrated (1-3 days), and then monocytes/macrophages infiltrated and covered the entire lesion (3-14 days). The invading leukocytes expressed CD11 and CD18 adhesion molecules on their cell surface while ICAM-1 was expressed on endothelial cells. In surrounding areas, in contrast, there was a rapid activation of microglia showing morphological changes and upregulation of OX42 immunoreactivity (4 h-7 days), especially in the transitional rim of the infarct (7 days). ED1 and MHC antigens were expressed on both activated microglia and invading leukocytes. Thus, developing infarction was accompanied by accumulation of inflammatory cells of both intrinsic (microglia) and extrinsic (leukocytes) origins. The results suggest that the relative importance of each source is determined by the time after ischemia and the site within the lesion, and that the expression of immunological molecules plays an important role in eliciting such inflammatory reactions.
Keywords: Cerebral ischemia; Cerebral infarction; Microglia; Leukocyte; Immunohistochemistry; Inflammation; Rat
1. Introduct ion
Cytotoxic, inflammatory reactions caused by microglial activation and blood-borne neutrophils and monocytes have been implicated in the pathogenesis of ischemia/reperfu- sion brain injury [14,15,18]. Reaction of microglial cells to ischemia is very rapid and they can be activated within minutes after ischemia [23]. Activated microglia may de- velop into ameboid microglia (brain macrophages) when neurons are lethally injured [1,14,15,31,32]. On the other hand, neutrophils begin to infiltrate into an infarct area
* Corresponding author. Present address: Department of Neurology, Washington University School of Medicine, Box 8111,660 South Euclid Avenue, St, Louis, MO 63110, USA. Fax: + 1 (314) 362-9462.
0006-8993/96/$15.00 Published by Elsevier Science B.V. Pll S0006-8993(96)0063 6- 1
12-24 h after ischemia, and then monocytes begin to invade massively after 2 - 3 days [12,18,20]. It has been suggested that fully activated microglia and infiltrating neutrophils and monocytes lead to secondary injury to potentially viable tissue. Neutrophils may damage tissue by reducing blood flow as a result of capillary plugging or release of vasoconstrictive mediators, and by secreting cytotoxic substances such as proteases, oxygen free radi- cals, and lipid-derived mediators [6,18,21]. Microglia- or monocyte-derived macrophages also respond to injury by phagocytosis of damaged tissue as well as by secretion of cytotoxic agents.
It has been documented that a variety of immunological surface marker molecules are expressed or upregulated in inflammatory cells following cerebral ischemia. A number of immunological markers, such as major histocompatibil- ity complex (MHC) class I and class II antigens, are
204 H. Kato et al. / Brain Research 734 (1996) 203-212
expressed on the surface of activated microglia after is- chemia [11,16,17,24]. Furthermore, adhesion and migra- tion of blood-borne neutrophils and monocytes occur via highly specific receptor-ligand interactions. The adhesion is regulated in part by interactions between intercellular adhesion molecule-1 ( ICAM-I) on endothelial cells and a group of C D l l / C D 1 8 glycoproteins on leukocytes [21,27,30]. However, only a few papers have specifically addressed the time course and magnitude of postischemic microglial activation and leukocyte accumulation in the brain with regard to the expression of immunological molecules in focal cerebral ischemia [25]. The purpose of this study was therefore to investigate the expression of immunomolecules and adhesion molecules on microglia and blood-borne leukocytes following focal cerebral is- chemia in the rat.
2. Materials and Methods
2.1. Induction o f focal cerebral ischemia
Male adult Wistar rats (Funabashi Farm, Shizuoka, Japan) weighing 250-260 g were used. Focal cerebral ischemia was induced by transient occlusion of the right middle cerebral artery (MCA) for 1 h, followed by reperfu- sion for 4 h, 1 day, 3 days, 7 days and 14 days. Each group consisted of five rats. The method of occlusion has been described by Nagasawa and Kogure [26]. Briefly, the animals were anesthetized with 2% halothane in a mixture of 30% oxygen and 70% nitrous oxide. The right external carotid artery was exposed and an 18 mm long 4-0 nylon thread which had been coated with silicon to thicken the distal two-thirds was inserted from the proximal portion of the artery into the internal carotid artery. Then, the origin of the fight MCA and anterior and posterior communicat- ing arteries was occluded intraluminally by the silicon- coated embolus. After 1 h of MCA occlusion, the embolus was removed to allow reperfusion of the ischemic area via the right internal carotid artery and anterior and posterior communicating arteries. When the wound was closed, anesthesia was discontinued and the development of left hemiparesis with upper limb dominance was observed. Body temperature during surgery was maintained at 37 .0- 37.5°C using a heating pad and a lamp. Sham-operated animals were also prepared in the same way except for insertion of an embolus.
After survival for the periods indicated above, the ani- mals were anesthetized with pentobarbital (50 m g / k g , i.p.) and the brains were perfusion-fixed with 4% paraformal- dehyde in 0.1 M phosphate buffer (pH 7.4). The brains were then removed and postfixed in the same fixative for 6 h at 4°C and then immersed in 30% sucrose solution in phosphate buffer at 4°C. The brains were frozen and coronal sections with a thickness of 18 p~m were cut at the
level of the striatum on a cryostat at -20°C . Part of the sections were stained with Cresyl violet for histopathologic observations.
2.2. Immunohistochemistry
The sections were immunohistochemically stained with monoclonal antibodies raised against complement receptor type 3 (CR3) or CD1 lb (OX42, Serotec), MHC class I and class II (Ia) antigens (OX18 and OX6, respectively, Serotec), and a pan-macrophage/monocyte marker ED1 (Serotec). We also used monoclonal antibodies to rat ICAM-I (CD54, Serotec), and its ligand rat LFA-I c~ chain (CD1 la, Serotec) and 13 chain (CDIS, Serotec), and rat T cells (CD5, Serotec). The sections were preincubated with 10% normal serum in phosphate buffered saline (PBS) for 30 rain. This was followed by an overnight incubation at 4°C with one of the antibodies (diluted 1:500 except for CD1 la, CD18, and ICAM-1 which were diluted l:100). The sections were then incubated with a biotinyl- ated second antibody (preadsorbed with rat IgG) diluted 1:200 for 1 h and then placed in avidin-biotin-horseradish peroxidase solution for 30 rain (Vectastain elite ABC kit, Vector Laboratories). Finally, the sections were reacted with diaminobenzidine and H 2 0 2 for color development. Negative control studies were performed by replacing the primary antibodies with PBS.
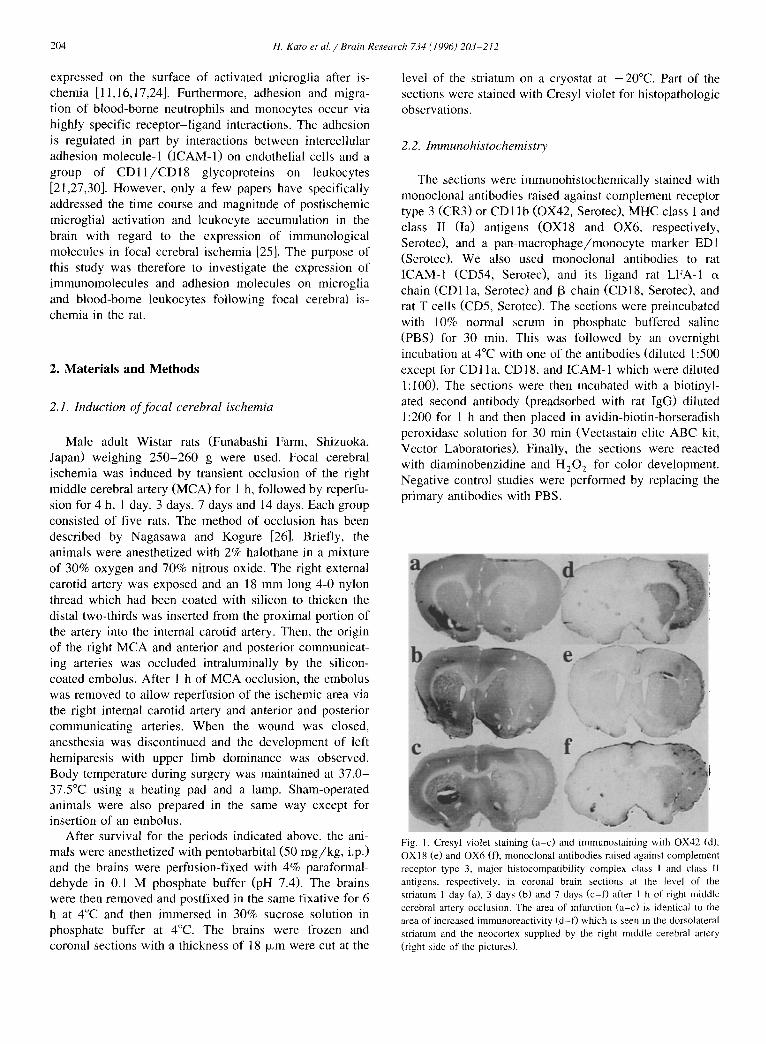
Fig. 1. Cresyl violet staining (a-c) and immunostaining with OX42 (d), OXI 8 (e) and OX6 (f), monoclonal antibodies raised against complement receptor type 3, major histocompatibility complex class I and class II antigens, respectively, in coronal brain sections at the level of the striatum 1 day (a), 3 days (b) and 7 days (c-D after 1 h of right middle cerebral artery occlusion. The area of infarction (a-c) is identical to the area of increased immunoreactivity (d-f) which is seen in the dorsolateral striatum and the neocortex supplied by the right middle cerebral artery (right side of the pictures).

H. Kato et a l . /Brain Research 734 (1996) 203-212 205
3. Resul ts
3.1. Histopathology
The histopathologic changes were confined to the terri- tory of the occluded fight MCA, i.e., the right frontopari- etal cortex and the dorsolateral part of the striatum. The lesion in these areas evolved into extensive neuronal necrosis over a period of 4 h-1 day followed by infarct by 7 days. The distribution of brain damage detected by Cresyl violet staining is shown in Fig. 1, Fig. and Fig. , and was mostly identical to the areas with increased immunoreactivities.
Four hours after MCA occlusion, many neurons in the ischemic areas, more strikingly in the striatum than in the cortex, lost normal neuronal morphology and showed shrunken cell bodies and perineuronal vacuolations (Fig. 2b) in comparison with sham-operated brains (Fig. 2a). The neuronal changes in the neocortex were prominent throughout layers 2-6, and the brain regions involved in the ischemic lesion at this time point were not identical in all animals. After 1 day and 3 days, these changes became more evident (Fig. 2c,d) and the ischemic areas were well demarcated (Fig. 2g). In most rats, the lesion involved the entire territory supplied by the MCA, suggesting that penumbra areas were incorporated into infarction. Edema-
O ~
G dtr !
%-" ,~
m
~Q
|
- ID
X
I~ 1 1 1
$,
~t
!
Fig. 2. Histopathological observations of the neocortex (layers 3 -5 ) following 1 h of middle cerebral artery occlusion, a-f : center of ischemia, g,h: transitional zone between non-infarcted (left half of the panels) and infarcted (right half) areas, a: sham-operation. Normal appearance, b: 4 h after ischemia, many cortical neurons are shrunken, c: after 1 day, the neurons are disappearing, d: after 3 days, microvessels are newly formed, e: after 7 days, infiltration of mononuclear macrophage-like cells is evident, f: after 14 days, macrophage-like cells decrease in number, g: after 3 days, the infarcted area is clearly demarcated, h: after 7 days, there is a marked accumulation of microglia-like cells in the transitional rim of the infarct. Cresyl violet stain. Bar = 0.05 mm (a) and 0.1 mm (h).

206 H. Kato et al./Brain Research 734 (1996) 203-212
Fig. 3. Immunostaining with OX42 (a-O and OX6 (g-j), monoclonal antibodies raised against complement receptor type 3 and major histocompatibility complex class II antigen, respectively, in the neocortex (layers 3-5) following I h of middle cerebral artery occlusion, a: sham-operation. Resting microglia are immunoreactive to OX42. b: after 4 h, microglia have fewer processes and stained slightly stronger, but they appear fragmented, c: after I day, the fragmentation is more evident, and OX42-positive round cells (leukocytes) begin to appear, d: after 3 days, many OX42-positive leukocytes can be seen. e: after 7 days, there is a dense accumulation of larger round cells (monocytes/macrophages) whose surface is OX42 positive, f: after 1 day, in the surrounding area (cingulate cortex), many microglial cells are activated and present larger cell bodies and stouter processes, g: after 1 day, a few leukocytes are OX6 positive, h,i: after 3 days (h), many leukocytes are OX6-positive but the number of OX6-positive cells does not increase after 7 days (i). j: after 14 days, OX6-positive macrophages are still observed. Scale bar = 0.05 mm.
tous c h a n g e s were obv ious , n e u r o n s were d i sappear ing ,
and sca t t e red neu t roph i l s were o b s e r v e d in the i s c h e m i c
areas. Af t e r 3 days, and more s t r ik ingly af ter 7 days, we
o b s e r v e d m i c r o v e s s e l p ro l i fe ra t ion and a m a s s i v e infi l t ra-
t ion o f m o n o c y t e s / m a c r o p h a g e s in the infarc t areas (Fig.
2d,e), and a m a s s i v e a c c u m u l a t i o n o f glial cel ls in a
n a r r o w t rans i t iona l zone b e t w e e n the infarc t and su r round-
ing areas (Fig. 2h). Th i s b o u n d a r y c h a n g e was m o r e
r e m a r k a b l e in the n e o c o r t e x than in the s t r ia tum. Af t e r 14
days, in fa rc t ion ful ly d e v e l o p e d and there cou ld be a cavi ty
f o r m a t i o n in the cen te r o f the infarct , but the reac t ive
ce l lu lar c h a n g e s were subs id ing (Fig. 2f). The h i s topa tho-
logic f ind ings o f this e x p e r i m e n t a l ser ies have also been
desc r ibed e l s e w h e r e [19].
3.2. Immunohistochemistry
3.2.1. 0X42 (CR3 receptor or C D l l b ) O X 4 2 i m m u n o r e a c t i v i t y was o b s e r v e d bo th in mic rog l i a
and in i n v a d i n g leukocytes . In s h a m - o p e r a t e d an imals , we
Fig. 4. lmmunostaining with OX42 (a-d) and EDI (e,O, monoclonal antibodies raised against complement receptor type 3 and a monocyte/macrophage antigen, respectively, in the neocortex (layers 3 5) following 1 h of middle cerebral artery occlusion. The transitional zone between non-infarcted area (left half of the panels) and the infarcted area (right halo are shown, a: after 1 day, there is no transitional zone althougb the morphology of microglial cells is different between the two areas, b-d: after 3 days (b), there is a formation of a transitional rim consisted of activated microglia between the infarcted and non-infarcted areas, and the cellularity of the rim becomes strikingly high after 7 days (c) and decreases after 14 days (d). e,f: the rim is less remarkably defined by ED1 immunostaining after 3 days (e) and 7 days (f). Scale bar - 0.1 mm.

H. Kato et al. / Brain Research 734 (1996) 203-212 207
observed numerous OX42-positive microglia distributed processes (Fig. 3a). After 4 h, OX42 immunoreactivity throughout the brain. The microglia had a morphology of slightly increased in microglial cells in the ischemic areas resting microglia, i.e., small cell body and ramified, thin but the morphology of the cells was irregular with fewer

208 H. Kato et al./Brain Research 734 (1996) 203 212
processes and appeared fragmented (Fig. 3b). In the sur- rounding areas such as the ipsilateral cortex supplied by the anterior cerebral artery, microglial cells had a morphol- ogy of activated microglia, i.e., contraction of their highly ramified processes thereby appeared stouter in shape and larger cell bodies. After 1 day, we observed fragmented OX42-positive substances (destroyed microglial cells) and scattered OX42-positive small, round cells (leukocytes, most likely neutrophils) in the ischemic areas (Fig. 3c and Fig. 4a), but no resting and activated microglia could be seen there. In areas surrounding the infarct, and even in part of the contralateral cingulate cortex, numerous acti- vated microglia were seen (Fig. 3f). After 3 days, we observed accumulation of OX42-positive leukocytes within the infarct (Fig. 3d) but the activated microglia in the surrounding areas, except for the transitional zone between the infarct and the surrounding area, tended to decrease in number. After 7 days, the infarct core was covered by
OX42-positive larger round cells (monocytes/macro- phages) (Fig. 3e). There was also a narrow rim of high cellularity of OX42-positive activated microglia in the transitional zone (Fig. 4c). After 14 days, the number of OX42-positive cells in the infarct decreased (Fig. 4d). The most intense OX42 immunoreactivity was seen in the infarct areas, i.e., the right frontoparietal cortex and the dorsolateral part of the striatum (Fig. 1).
3.2.2. OX6 (MHC class II antigen) Resting microglia were only rarely positive for OX6 in
sham-operated rat brain parenchyma and no changes were seen after 4 h. After 1 day, a small number of small, round cells (leukocytes) were OX6 positive in the ischemic areas (Fig. 3g) and many activated microglia in the surrounding areas were also OX6 positive. After 3 days, the number of OX6-positive leukocytes increased in the ischemic center (Fig. 3h) and the activated microglia were also OX6
!
Fig. 5. lmmunostaining with monoclonal antibodies directed to a monocyte/macrophage marker EDI (a-d), major histocompatibility complex class 1 antigen OXI8 (e), a T cell marker CD5 (f), intercellular adhesion molecule-1 (g), LFA-I c~ chain CDI la (h,i) and [3 chain CD18 (j) in the neocortex (layers 3 -5 ) following I h of right middle cerebral artery occlusion, a,b: EDl-positive leukocytes begin to appear after 1 day (a) and increase after 3 days (b). c,d: after 7 days (c), EDl-positive monocytes/macrophages cover the lesion but decrease in number after 14 days (d). e: after 3 days, both endothelial cells and leukocytes are positive for OX 18. f: a small number of leukocytes are CD5 positive after 7 days. g: endothelial cells are stained for ICAM-1 after I day. h,i: CDI l a-positive leukocytes can be seen after 3 days (h) and remarkable after 7 days (i). j: CDI 8-positive leukocytes are seen after 7 days. Scale bar = 0.05 mm.

H. Kato et al. / Brain Research 734 (1996) 203-212 209
positive in the transitional rim of the infarct. After 7 days, OX6-positive leukocytes were seen in the infarct area but did not increase in number (Fig. 3i). After 14 days, OX6-positive macrophages in the infarct were both round and ameboid in shape (Fig. 3j).
3.2.3. ED1 (a macrophage / monocyte marker) No EDl-positive cells were seen in the brain
parenchyma of sham-operated animals and 4 h after MCA occlusion. After 1 day, a small number of ED-1 positive small, round cells (leukocytes) could be seen in the is- chemic areas (Fig. 5a). After 3 days, EDl-positive leuko- cytes distributed throughout the infarct areas (Fig. 5b). Activated microglia in the transitional rim and in the surrounding areas were also ED1 positive. After 7 days, EDl-positive larger round cells (monocytes/macrophages) covered the entire infarct (Fig. 5c) and EDl-positive acti- vated microglia accumulated in the transitional zone (Fig. 4f). After 14 days, EDl-positive cells decreased in number in the infarct (Fig. 5d).
abundant in the periphery of the infarct (Fig. 5h). After 7 days, many monocytes/macrophages were immunoreac- tire for CD1 la (Fig. 5i) but they decreased in number after 14 days.
3.2.8. CD18 (LFA-I[3) No immunoreactivity was seen in sham-operated rat
brains and after 4 h. A few (1 day) and a small number of (3 days) small, round cells in the ischemic areas were CD18-positive. After 7 days, larger round cells (mono- cytes/macrophages) in the infarct expressed CD18 (Fig. 5j) and activated microglia in the transitional and sur- rounding areas were also CD18 positive. After 14 days, activated microglia in the transitional zone were CD18 positive but the number of positive leukocytes decreased strikingly in the central area of the infarct.
4. Discussion
3.2.4. OX18 (MHC class I antigen) No OX18-positive cells, except for faint staining in
endothelial cells, were seen in sham-operated rat brain parenchyma and after 4 h. After 1 day, endothelial cells in the ischemic areas and activated microglia in the transi- tional rim were OX18 positive. After 3 days, many OX18-positive small, round cells (leukocytes) were seen in the ischemic center in addition to the endothelial staining (Fig. 5e). After 7 days, OX18-positive monocytes/macro- phages were additionally seen within the infarct but de- creased by 14 days. In general, there was much more MHC class I than class II expression.
3.2.5. CD5 (a T cell marker) No CD5-positive cells were seen in sham-operated ani-
mals and until 3 days after ischemia. After 7 days, a small number of leukocytes within the infarct were positive for CD5 (Fig. 5f), but not after 14 days.
3.2.6. CD54 (ICAM-1) No remarkable immunoreactivity was seen in sham-op-
erated rats and after 4 h. After 1 day, endothelial cells within the ischemic areas were positive for ICAM-1 (Fig. 5g). After 3 days, smaller microvessels (neovasculariza- tion) also expressed ICAM-I and this vessel staining was stronger in the periphery of the infarct. After 7 days, infiltrating leukocytes were slightly immunoreactive for ICAM-1. The vascular ICAM-1 immunoreactivity de- creased after 14 days.
3.2.7. CDl la (LFA-lc~) No CDlla-posit ive cells could be seen in sham-oper-
ated animals and after 4 h. A few (1 day) and many (3 days) small, round cells (most likely neutrophils) in the infarct areas expressed CDl la , and the cells were more
The present study showed that: (1) various im- munomolecules were expressed on activated microglia in response to focal cerebral ischemia, most strikingly in the transitional zone, and less remarkably in the surrounding areas, but not in the center of ischemia where infarction developed; and (2) blood-borne leukocytes migrated into the center of ischemia, expressing a variety of surface immunomolecules on them, while adhesion molecules were upregulated on endothelial cells; those molecules are con- sidered essential for the adhesion and migration of leuko- cytes into damaged brain tissue. Thus, both intrinsic and extrinsic inflammatory reactions could be seen following focal ischemia in and around the developing cerebral infarction.
The adult mammalian brain contains two major classes of mononuclear phagocytes in response to brain injury: resident microglia and blood-borne monocytes [14,15]. Resting microglia are distributed throughout the normal adult brain in significant numbers and currently not clearly understood how these cells contribute to brain function [31 ]. These microglia undergo marked changes in morphol- ogy to activated forms in response to ischemia [11,16,17,23]. Microglial cells can be activated within minutes after ischemia and change their morphology into that of activated microglia with stouter processes and larger cell bodies [23]. In this study, such activated mi- croglia could be seen in the surrounding areas as early as 4 h after ischemia. In the center of ischemia, however, this type of microglial activation was not seen after ischemia. Because brain cells of any type are difficult to survive in the center of ischemia where infarction develops, we pre- sume that microglia also die there. In contrast, microglial activation and accumulation were striking in the transi- tional zone. The shortening of microglial processes may represent the further conversion of activated microglia to

210 H. Kato et al. / Brain Research 734 (1996) 203 212
phagocytic ameboid forms in this transitional rim of the infarct. It is well documented that fully activated ameboid microglia are phagocytes (brain macrophages) [14,15].
This study showed that the activated microglia in the transitional and surrounding areas expressed many im- munomolecules that were not detected on resting microglia as has been documented in transient global ischemia [ 11,13,16,17,23,24]. The monoclonal antibody OX42 rec- ognizes an antigen associated with CR3 which binds the intermediate C3 cleavage product C3bi [28]. Complement receptors are present on cells that participate in immune and inflammatory reactions, such as neutrophils, eosinophils, monocytes/macrophages, and lymphocytes [10], OX42 immunoreactivity was present on both resting, activated, and phagocytic microglia. These findings raise the possibility of a nonimmunological involvement of this antigen. CR3 may function as a kind of adhesion molecule maintaining the close apposition of microglial and neu- ronal membranes because the CR3 molecule or CD1 lb has been shown to mediate adhesion of myelomonocytic cells [29,30,33].
One aspect of the microglial activation was the elevated expression of MHC antigens [11,13,16,17,24]. In the nor- mal brain parenchyma, no activated microglia bearing MHC antigens were observed. However, the brain mi- croglia were an inducible site for the expression of MHC molecules especially in the transitional rim of the infarct. MHC class I and class II antigens are essential for the interaction between T cells and macrophages in the induc- tion phase of an immune response [33]. Expression of MHC antigens by microglia may indicate an elevated state of immune responsiveness in the brain. However, the upregulation of MHC antigens by activated microglia may also be involved in processes other than antigen presenta- tion to T lymphocytes because T cell infiltration was seen only within the infarct but not in the surrounding area where microglia expressed these antigens. If the peripheral immune system is not involved, one may postulate a non-immunological function of MHC antigens during mi- croglial reactions. The presence of these antigens on mi- croglia might facilitate interactions among this cell type as a cell adhesion molecule [8].
In the normal brain parenchyma, no macrophages bear- ing ED1 antigen could be detected. The morphological changes of microglial cells and the expression of a macrophage marker EDI in the transitional rim suggest that microglia were activated into brain macrophages. ED1 is found in most rat macrophage subpopulations as well as in monocytes in the peripheral blood [7]. In vitro studies have shown that microglial cells, when stimulated, release a variety of cytotoxic agents that may be important media- tors of neuronal injury, such as certain kinds of cytokines, reactive oxygen radicals, proteases, and glutamate [1,14,15]. Microglial ability to serve as effector cells that secrete those substances, as well as their potential to become antigen-presenting cells and phagocytes, could
contribute to the pathogenesis of brain damage after is- chemia.
In the center of ischemia where infarction develops and blood-brain barrier is broken down, neutrophils and monocytes progressively migrate into the ischemic tissue [12,18,20]. The leukocytes expressed various im- munomolecules on them including MHC and ED1 antigens like activated microglia. Neutrophil and monocyte adhe- sion has been shown to occur via highly specific receptor- ligand interactions with endothelium [30]. Key determi- nants for leukocyte adhesion to endothelium are glyco- proteins on the leukocyte cell surface known as integrins, which contain CDl l and CD18 noncovalently associated subunits. The most important integrins are LFA-1 (CD 11 a / C D 18) and Mac- 1 (CD 11 b / C D 18), both of which contains a common CD18 protein as their [3 subunit. The receptor for their integrins is the ICAM-I, which is ex- pressed on endothelial cells after ischemia and its expres- sion is modulated by a wide spectrum of inflammatory mediators such as interleukin-I [21,27]. Adhesion to en- dothelial cells and migration by chemotactic factors are critical stages in the infiltration of them into injured tissue. In accordance with this notion, we observed the expression of ICAM-I on the endothelial cells in the center of is- chemia and the presence of CD1 la, CDI lb and CD18 on the invading leukocytes. The leukocyte invasion occurred only in the area that developed infarction, suggesting that the leukocytes responded to a biological signal produced within the infarct.
In the MCA occlusion/reperfusion model in rats which we employed in this study, neutrophil accumulation in ischemic areas has been documented [12,20,21]. The num- ber of neutrophils in the ischemic areas begin to increase after 12 h, reaches a peak at 1-3 days, and disappears after 7 days. Many monocytes, on the other hand, are observed in the infarct areas after 3 and 7 days. However, it is unclear whether neutrophils arrive in the infarct area be- fore or after tissue necrosis develops and whether preced- ing neutrophil infiltration is a prerequisite for the later monocyte infiltration. Postischemic influx of leukocytes may contribute to ischemia/reperfusion injury by plugging the microvasculature [6] and by releasing cytotoxic prod- ucts [18,21]. Actually, neutrophil depletion or treatments with monoclonal antibodies that block specific adhesion molecules (LFA-1, Mac-1, and ICAM-1) have been shown to improve electrical activity and blood flow after ischemia or to reduce post-ischemic edema formation, neutrophil infiltration, and infarct volume [2-5,9,22,34].
The origin of phagocytic cells at sites of brain injury have remained uncertain. Both ameboid microglia and blood-borne monocytes have been considered. However, when microglia are fully transformed into ameboid form, they would be indistinguishable from invading monocyte- derived macrophages. Giulian and colleagues [14,15] have shown that both intrinsic and extrinsic inflammatory re- sponses of brain occur in response to a stab wound. They

H. Kato et al. / Brain Research 734 (1996) 203-212 211
showed a rapid appearance o f a cluster of round cells
(b lood-borne monocy tes ) at the site o f the lesion, where
b l o o d - b r a i n barr ier is disrupted, and act ivated microg l ia at
regions distant to the immedia t e injury site. The type of
brain damage may de te rmine the source o f phagocytes
be ing intrinsic and extr insic to the brain, wi th the relat ive
impor tance o f each source be ing de te rmined by the charac-
teristics of the specif ic les ion [14,15]. In our study, s imilar
recru i tment o f macrophages was suggested. In the center
o f i schemia , the major i ty of mic rog l i a may die. It cannot
be d e n i e d that m i c r o g l i a w e r e t r a n s f o r m e d into
macrophages in the area o f infarction. Howeve r , this is
unl ikely because, in the infarct, we observed no act ivated
microgl ia , which t ransform into phagocyt ic amebo id mi-
c rogl ia in a s tepwise fashion [1,31]. On the other hand, we
observed a number o f act ivated microg l ia in the surround-
ing areas, par t icular ly in the transit ional r im of the infarct.
Probably, both intrinsic (microgl ia) and extr insic (mono-
cytes) macrophages invade the infarct area depending on
their locat ion and route of entry; monocy tes f rom disrupted
b lood brain barr ier into the center o f the infarct and
mic rog l i a f rom non- infarc ted surrounding areas into the
per iphery o f the infarct.
In conclus ion , the f indings of this study show that focal
cerebral i schemia leads to a local immune , in f lammatory
reaction, and suggest that this react ion is under strict
control by express ing and upregula t ing a number of im-
munolog ica l and adhesion molecules . Both intrinsic (mi-
crogl ia) and extr insic ( leukocytes) in f lammatory responses
may play roles in the d e v e l o p m e n t o f infarct ion because
those cells are known to have potent ial cy to toxic actions.
The in f lammatory react ions seen in the infarct area, transi-
t ional area, and the surrounding area were quite different.
It is of impor tance to e lucidate whether these in f lammatory
react ions only expand the i schemic damage or these reac-
tions are necessary for proper wound healing.
Acknowledgements
This study was supported in part by Grant - in -Aid for
Scient i f ic Research 07670692 f rom the Minis t ry o f Educa-
tion, Science , and Cul ture o f Japan.
References
[1] Banati, R.B., Gehrmann, J., Schubert, P. and Kreutzberg, G.W., Cytotoxicity of microglia, Glia, 7 (1993) 111-118.
[2] Bowes, M.P., Zivin, J.A. and Rothlein, R., Monoclonal antibody to ICAM-1 adhesion site reduces neurological damage in a rabbit cerebral embolism stroke model, Exp. Neurol., 119 (1993) 215-219.
[3] Chen, H., Chopp, M. and Bodzin, G., Neutropenia reduces the volume of cerebral infarct after transient middle cerebral artery occlusion in the rat, Neurosci. Res. Commun., 11 (1992) 93-99.
[4] Chopp, M., Zhang, R.L., Chen, H., Li, Y., Jiang, N. and Rusche,
J.R., Postischemic administration of an anti-Mac-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in rats, Stroke, 25 (1994) 869-876.
[5] Clark, W.M., Madden, K.P., Rothlein, R. and Zivin, J.A., Reduction of central nervous system ischemic injury in rabbits using leukocyte adhesion antibody treatment, Stroke, 22 (1991) 877-883.
[6] Del Zoppo, G.J., Schmid-SchiSnbein, G.W., Mori, E., Copeland, B.R. and Chang, C.-M., Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfu- sion in baboons, Stroke, 22 (1991) 1276-1283.
[7] Dijkstra, C.D., DiSpp, E.A., Joling, P. and Kraal, G., The heterogene- ity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies EDI, ED2 and ED3, Immunology, 52 (1985) 589-599.
[8] Doyle, C. and Strominger, J.L., Interaction between CD4 and class II MHC molecules mediates cell adhesion, Nature, 330 (1987) 256-259.
[9] Dutka, A.J., Kochanek, P.M. and Hallenbeck, J.M., Influence of granulocytopenia on canine cerebral ischemia induced by air em- bolism, Stroke, 20 (1989) 390-395.
[ 10] Fearon, D.T. and Wong, W.W., Complement ligand-receptor interac- tions that mediate biological responses, Annu. Rev. lmmuno[., 1 (1983) 243-271.
[11] Finsen, B.R., JCrgensen M.B., Diemer N.H. and Jimmer, J., Mi- croglial MHC antigen expression after ischemic and kainic acid lesions of the adult rat hippocampus, Glia, 7 (1993) 41-49.
[12] Garcia, J.H., Liu, K.F., Yoshida, Y., Lian, J., Chen, S. and Del Zoppo, G.J., Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat), Am. J. Pathol., 144 (1994) 188-199.
[13] Gehrmann, J., Bonnekoh, P., Miyazawa, T., Hossmann, K.-A. and Kreutzberg, G.W,, Immunocytochemical study of an early microglial activation in ischemia, J. Cereb. Blood Flow Metab., 12 (1992) 257-269.
[14] Giulian, D., Ameboid microglia as effectors of inflammation in the central nervous system, .L Neurosci. Res., 18 (1987) 155-171.
[15] Giulian, D., Chen, J., lngeman, LE., George, J.K, and Noponen, M., The role of mononuclear phagocytes in wound healing after trau- matic injury to adult mammalian brain, J. Neurosci., 9 (1989) 4416-4429.
[16] Jcrgensen, M.B., Finsen, B.R., Jensen, M.B., Castellano, B, Diemer, N.H. and Zimmer, J., Microglial and astroglial reactions to ischemic and kainic acid-induced lesions of the adult rat hippocampus, Exp. Neurol., 120 (1993) 70-88,
[17] Kato, H., Kogure, K., Araki, T. and Itoyama, Y., Graded expression of immunomolecules on activated microglia in the hippocampus following ischemia in a rat model of ischemic tolerance, Brain Res., 694 (1995) 85-93.
[18] Kochanek, P.M. and Hallenbeck, J.M., Polymorphonuclear leuko- cytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke, Stroke, 23 (1992) 1367-1379.
[19] Liu, X.-H., Kato, H., Araki, T., Itoyama, Y., Kato, K. and Kogure, K., An immunohistochemical study of copper/zinc superoxide dis- mutase and manganese dismutase following focal cerebral ischemia in the rat, Brain Res., 644 (1994) 257-266.
[20] Matsuo, Y,, Onodera, H., Shiga, Y., Nakamura, M., Kihara, T. and Kogure, K., Correlation between myeloperoxidase-quantified neu- trophil accumulation and ischemic brain injury in the rat. Effect of neutrophil depletion, Stroke, 25 (1994) 1469-1475.
[21] Matsuo, Y., Kihara, T., Ikeda, M., Ninomiya, M., Onodera, H. and Kogure, K., Role of neutrophils in radical production during is- chemia and reperfusion of the rat brain: effect of neutrophil deple- tion on extracellular ascorbyl radical formation, J. Cereb. Blood Flow Metab., 15 (1995) 941-947.
[22] Mori, E., Del Zoppo, G.L., Chambers, D., Copeland, B.R. and Arfors, K.E., Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons, Stroke, 23 (1992) 712-718.

212 H. Kato et al. /Brain Research 734 (1996) 203-212
[23] Morioka, T., Kalebua, A.N. and Streit, W.J., The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia, J. Cereb. Blood Flow Metab., 11 (1991) 966-973.
[24] Morioka, T., Kalebua, A.N. and Streit, W.J., Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia, Acta Neuropathol., 83 (1992) 149-157.
[25] Morioka, T., Kalehua, A.N. and Streit, W.J., Characterization of microglial reaction after middle cerebral artery occlusion in rat brain, J. Comp. Neurol., 327 (1993) 123-132.
[26] Nagasawa, H. and Kogure, K., Correlation between cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion, Stroke, 20 (1989) 1037-1043.
[27] Okada, Y., Copeland, B.R., Mori, E., Tung, M.M., Thomas, W.S. and Del Zoppo, G.J., P-selectin and intercellular adhesion molecule-1 expression after focal brain isclaemia and reperfusion, Stroke, 25 (1994) 202-21 l.
[28] Robinson, A.P., White, T.M. and Mason, D.W., Macropbage hetero- geneity in the rat as delineated by two monoclonal antibodies MRC
Ox-41 and MRC Ox-42, the latter recognizing complement receptor type 3, Immunology, 57 (1986) 239-247.
[29] Rosen, H. and Gordon, S., Monoclonal antibody to the murine type 3 complement receptor inhibits adhesion of myelomonocytic cells in vitro and inflammatory cell recruitment in vivo, J. Exp. Med., 166 (1987) 1685-1701.
[30] Springer, T.A., Adhesion receptors of the immune system, Nature, 346 (1990) 425-434.
[31] Streit, W.J., Graeber, M.B. and Kreutzberg, G.W., Functional plas- ticity of microglia: a review, Glia, 1 (1988) 301-307.
[32] Streit, W.J., Microglial-neuronal interactions, J. Chem. Neuroanat., 6 (1993) 261-266.
[33] Uranue, E.R. and Allen, P.M., The basis for the immunoregulatory role of macrophages and other accessory cells, Science, 236 (1987) 551-557.
[34] Vasthare, V.S., Heinel, L.A., Rosenwasser, R.H. and Tuma R.F., Leukocyte involvement in cerebral ischemia and reperfusion injury, Surg. Neurol., 33 (1990) 261-265.





























