Embed Size (px)
Citation preview

PROTEIN METABOLISM
Nada A.Abotemin
-Amino acid pool
- Protein turnover - Nitrogen Balance
- Digestion of Dietary Proteins
- Removal of Nitrogen from Amino Acids
A. Transamination
B. oxidative deamination
- Transport of ammonia to the liver
- Urea Cycle
- Transport of ammonia in the circulation
- Hyperammonemia - Amino Acid Degradation and Synthesis
- Metabolic Defects in Amino Acid Metabolism
- Other Nitrogen-Containing Compounds

- 1 -
Protein metabolism
Amino acid catabolism is part of the larger process of the metabolism of nitrogen-containing molecules. Nitrogen enters the body in a variety of compounds present in food, the most important being amino acids contained in dietary protein. Nitrogen leaves the body as urea, ammonia, and other products derived from amino acid metabolism. The role of body proteins in these transformations involves two important concepts: the amino acid pool and protein turnover.
A. Amino acid pool
Free amino acids are present throughout the body, for example, in cells, blood, and the extracellular fluids. For the purpose of this discussion, envision all these amino acids as if they belonged to a single entity, called the amino acid pool. This pool is supplied by three sources:
1) amino acids provided by the degradation of body proteins,
2) amino acids derived from dietary protein, and
3) synthesis of nonessential amino acids from simple intermediates of metabolism (Figure 1).
Conversely, the amino pool is depleted by three routes:
1) synthesis of body protein,
2) amino acids consumed as precursors of essential nitrogen-containing small molecules, and
3) conversion of amino acids to glucose, glycogen, fatty acids or CO2 (Figure 1).
In healthy, well fed individuals, the input to the amino acid pool is balanced by the output, that is, the amount of amino acids contained in the pool is constant. The amino acid pool is said to be in a steady state.

- 2 -
Figure (1): Sources and fates of amino acids.
B. Protein turnover
Most proteins in the body are constantly being synthesized and then degraded, permitting the removal of abnormal or unneeded proteins.
Rate of turnover: In healthy adults, the total amount of protein in the body remains constant, because the rate of protein synthesis is just sufficient to replace the protein that is degraded. This process, called protein turnover, leads to the hydrolysis and resynthesis of 300–400 g of body protein each day. The rate of protein turnover varies widely for individual proteins. Short-lived proteins (for example, many regulatory proteins and misfolded proteins) are rapidly degraded, having half-lives measured in minutes or hours. Long-lived proteins, with half-lives of days to weeks, constitute the majority of proteins in the cell. Structural proteins, such as collagen, are metabolically stable, and have half-lives measured in months or years.
Protein degradation: There are two major enzyme systems responsible for degrading damaged or unneeded proteins:

- 3 -
1- the energy-dependent ubiquitin-proteasome mechanism : Proteasomes mainly degrade endogenous proteins, that is, proteins that were synthesized within the cell.
2- the nonenergy-dependent degradative enzymes (acid hydrolases) of the lysosomes: Lysosomal enzymes degrade primarily extracellular proteins, such as plasma proteins that are taken into the cell by endocytosis, and cell-surface membrane proteins that are used in receptor-mediated endocytosis.
Ubiquitin-proteasome proteolytic pathway: Proteins selected for degradation by the ubiquitin-proteasome mechanism are first covalently attached to ubiquitin, a small, globular protein. Ubiquitination of the target substrate occurs through linkage of the α-carboxyl glycine of ubiquitin to a lysine ε-amino group on the protein substrate by a three-step, enzyme-catalyzed process.
Proteins tagged with ubiquitin are then recognized by a large, barrel-shaped, macromolecular, proteolytic complex called a proteasome, (Figure 2). The proteasome cuts the target protein into fragments that are then further degraded to amino acids, which enter the amino acid pool. The degradation of proteins by the ubiquitin-proteosome complex (unlike simple hydrolysis by proteolytic enzymes) requires adenosine triphosphate (ATP)—that is, it is energy-dependent.
Figure (2): The ubiquitin-proteasome degradation pathway of proteins

- 4 -
Nitrogen Balance
Catabolism of amino acids leads to a net loss of nitrogen from the body .This loss must be compensated by the diet in order to maintain a constant amount of body protein ,Nitrogen balance studies evaluate the relationship between the nitrogen intake ( in the form of protein ) and nitrogen excretion.
Three situations are possible as follows:
1- Nitrogen equilibrium:
●In normal adults nitrogen intake = nitrogen excretion. The subject is said to be in nitrogen equilibrium or balance.
In this situation ,the rate of protein synthesis is equal to the rate of the rate of degradation. ●
2- Positive nitrogen balance :
In whish nitrogen intake ˃ nitrogen excretion●
It shows that nitrogen is retained in the body, which means that protein is laid down. ●
This occurs in growing infants and pregnant women. ●
3- Negative nitrogen balance:
In which nitrogen intake ˂ nitrogen excretion .this occurs during serious illness and major injury and trauma ,in advanced cancer and following failure to ingest adequate or sufficient high quality protein ( malnutrition).If the situation is prolonged, it will ultimately lead to death.
Digestion of Dietary Proteins
Proteins are generally too large to be absorbed by the intestine. Proteins must, therefore, be hydrolyzed to yield their constituent amino acids, which can be absorbed. Proteolytic enzymes responsible for degrading proteins are produced by three different organs: the stomach, the pancreas, and the small intestine (Figure 3).
[Note: An example of an exception to this rule is that newborns can take up maternal antibodies in breast milk ,e.g. immunoglobin IgA from colostrum of maternal milk are absorbed intact without loss of biologic activity , so that they provide passive immunity to the infant. In contrast to the situation encountered in the newborn infants , in some adult individuals ,small amount of intact proteins may be absorbed through the intestinal mucosa. These proteins often cause undesirable immunological responses (formation of antibodies against the foreign protein ) and are responsible for the symptoms of food allergies.
A. Digestion of proteins by gastric secretion
The digestion of proteins begins in the stomach, which secretes gastric juice—a unique solution containing hydrochloric acid and the proenzyme, pepsinogen.

- 5 -
1- Hydrochloric acid: Stomach acid is too dilute (pH 2–3) to hydrolyze proteins. The acid functions instead to kill some bacteria and to denature proteins, thus making them more susceptible to subsequent hydrolysis by proteases.
2- Pepsin: This acid-stable endopeptidase is secreted by the serous cells of the stomach as an inactive zymogen (or proenzyme), pepsinogen. In general, zymogens contain extra amino acids in their sequences, which prevent them from being catalytically active. [Note: Removal of these amino acids permits the proper folding required for an active enzyme.] Pepsinogen is activated to pepsin, either by HCl, or autocatalytically by other pepsin molecules that have already been activated. Pepsin releases peptides and a few free amino acids from dietary proteins.
Figure (3): Digestion of dietary proteins by the proteolytic enzymes of the gastrointestinal tract.
B. Digestion of proteins by pancreatic enzymes
On entering the small intestine, large polypeptides produced in the stomach by the action of pepsin are further cleaved to oligopeptides and amino acids by a group of pancreatic proteases
1- Specificity: Each of these enzymes has a different specificity for the amino acid R-groups adjacent to the susceptible peptide bond. For example, trypsin cleaves only when the carbonyl group of the peptide bond is contributed by arginine or lysine. These enzymes, like pepsin described above, are synthesized and secreted as inactive zymogens.
2- Release of zymogens: The release and activation of the pancreatic zymogens is mediated by the secretion of cholecystokinin and secretin, two polypeptide hormones of the digestive tract.
3- Activation of zymogens: (Enteropeptidase) an enzyme synthesized by and present on the luminal surface of intestinal mucosal cells of the brush border membrane—converts the pancreatic zymogen

- 6 -
trypsinogen to trypsin by removal of a hexapeptide from the NH2-terminus of trypsinogen. Trypsin subsequently converts other trypsinogen molecules to trypsin by cleaving a limited number of specific peptide bonds in the zymogen. Enteropeptidase thus unleashes a cascade of proteolytic activity, because trypsin is the common activator of all the pancreatic zymogens.
4-Abnormalities in protein digestion: In individuals with a deficiency in pancreatic secretion (for example, due to chronic pancreatitis, cystic fibrosis, or surgical removal of the pancreas), the digestion and absorption of fat and protein is incomplete. This results in the abnormal appearance of lipids (called steatorrhea,) and undigested protein in the feces.
C. Digestion of oligopeptides by enzymes of the small intestine
The luminal surface of the intestine contains aminopeptidase—an exopeptidase that repeatedly cleaves the N-terminal residue from oligopeptides to produce free amino acids and smaller peptides.
D. Absorption of amino acids and dipeptides
Free amino acids are taken into the enterocytes up by a Na+-linked secondary transport system. Di- and tripeptides, however, are taken up by a H+-linked transport system. There, the peptides are hydrolyzed in the cytosol to amino acids before being released into the portal system. Thus, only free amino acids are found in the portal vein after a meal containing protein. These amino acids are either metabolized by the liver or released into the general circulation.
Transport of Amino Acids into Cells
The concentration of free amino acids in the extracellular fluids is significantly lower than that within the cells of the body. This concentration gradient is maintained because active transport systems, driven by the hydrolysis of ATP, are required for movement of amino acids from the extracellular space into cells. At least seven different transport systems are known that have overlapping specificities for different amino acids.
Removal of Nitrogen from Amino Acids
Removing the α-amino group is essential for producing energy from any amino acid, and is an obligatory step in the catabolism of all amino acids. Once removed, this nitrogen can be incorporated into other compounds or excreted, with the carbon skeletons being metabolized.
A. Transamination: the funneling of amino groups to glutamate
1-The first step in the catabolism of most amino acids is the transfer of their α-amino group to α-ketoglutarate (Figure 4).
2- The products are an α-keto acid (derived from the original amino acid) and glutamate.
α-Ketoglutarate plays a pivotal role in amino acid metabolism by accepting the amino groups from other amino acids, thus becoming glutamate.
Glutamate produced by transamination can be oxidatively deaminated (see below), or used as an amino group donor in the synthesis of nonessential amino acids.

- 7 -
This transfer of amino groups from one carbon skeleton to another is catalyzed by a family of enzymes called aminotransferases (formerly called transaminases). These enzymes are found in the cytosol and mitochondria of cells throughout the body—especially those of the liver, kidney, intestine, and muscle. All amino acids, with the exception of lysine and threonine, participate in transamination at some point in their catabolism. [Note: These two amino acids lose their α-amino groups by deamination].
Figure (4): Aminotransferase reaction using α-ketoglutarate as the amino-group acceptor
1- Substrate specificity of aminotransferases: Each aminotransferase is specific for one or, at most, a few amino group donors. Aminotransferases are named after the specific amino group donor, because the acceptor of the amino group is almost always α-ketoglutarate. The two most important aminotransferase reactions are catalyzed by alanine aminotransferase (ALT) and aspartate aminotransferase (AST), Figure 5.
Figure (5): Reactions catalyzed during amino acid catabolism. A. Alanine aminotransferase (ALT). B. Aspartate aminotransferase (AST).

- 8 -
a-Alanine aminotransferase (ALT): Formerly called glutamate-pyruvate transaminase, ALT is present in many tissues. The enzyme catalyzes the transfer of the amino group of alanine to α-ketoglutarate, resulting in the formation of pyruvate and glutamate.
b-Aspartate aminotransferase (AST): AST formerly called glutamate-oxaloacetate transaminase, AST is an exception to the rule that aminotransferases funnel amino groups to form glutamate. During amino acid catabolism, AST transfers amino groups from glutamate to oxaloacetate, forming aspartate, which is used as a source of nitrogen in the urea cycle.
2-Mechanism of action of aminotransferases: All aminotransferases require the coenzyme pyridoxal phosphate (a derivative of vitamin B6), which is covalently linked to the ε-amino group of a specific lysine residue at the active site of the enzyme
3-Diagnostic value of plasma aminotransferases:
Aminotransferases are normally intracellular enzymes, with the low levels found in the plasma representing the release of cellular contents during normal cell turnover. The presence of elevated plasma levels of aminotransferases indicates damage to cells rich in these enzymes. For example, physical trauma or a disease process can cause cell lysis, resulting in release of intracellular enzymes into the blood. Two aminotransferases—AST and ALT—are of particular diagnostic value when they are found in the plasma.
a- Liver disease: Plasma AST and ALT are elevated in nearly all liver diseases, but are particularly high in conditions that cause extensive cell necrosis, such as severe viral hepatitis, toxic injury, and prolonged circulatory collapse. ALT is more specific than AST for liver disease, but the latter is more sensitive because the liver contains larger amounts of AST. Serial enzyme measurements are often useful in determining the course of liver damage.
b-Nonhepatic disease: Aminotransferases may be elevated in nonhepatic disease, such as myocardial infarction and muscle disorders. However, these disorders can usually be distinguished clinically from liver disease.
B. Glutamate dehydrogenase: the oxidative deamination of amino acids
In contrast to transamination reactions that transfer amino groups, oxidative deamination by glutamate dehydrogenase results in the liberation of the amino group as free ammonia (Figure 6). These reactions occur primarily in the liver and kidney. They provide α-keto acids that can enter the central pathway of energy metabolism, and ammonia, which is a source of nitrogen in urea synthesis.
1- Glutamate dehydrogenase: As described above, the amino groups of most amino acids are ultimately funneled to glutamate by means of transamination with α-ketoglutarate. Glutamate is unique in that it is the only amino acid that undergoes rapid oxidative deamination—a reaction catalyzed by glutamate dehydrogenase. Therefore, the sequential action of transamination (resulting in the collection of amino groups from other amino acids onto α-ketoglutarate to produce glutamate) and the oxidative deamination of that glutamate (regenerating α-ketoglutarate) provide a pathway whereby the amino groups of most amino acids can be released as ammonia.

- 9 -
. Figure (6): Oxidative deamination by glutamate dehydrogenase
2- D-Amino acid oxidase: D-Amino acids are found in plants and in the cell walls of microorganisms, but are not used in the synthesis of mammalian proteins. D-Amino acids are, however, present in the diet, and are efficiently metabolized by the kidney and liver. D-Amino acid oxidase is an FAD-dependent peroxisomal enzyme that catalyzes the oxidative deamination of these amino acid isomers. The resulting α-keto acids can enter the general pathways of amino acid metabolism, and be re-aminated to L-isomers, or catabolized for energy.
Transport of ammonia to the liver
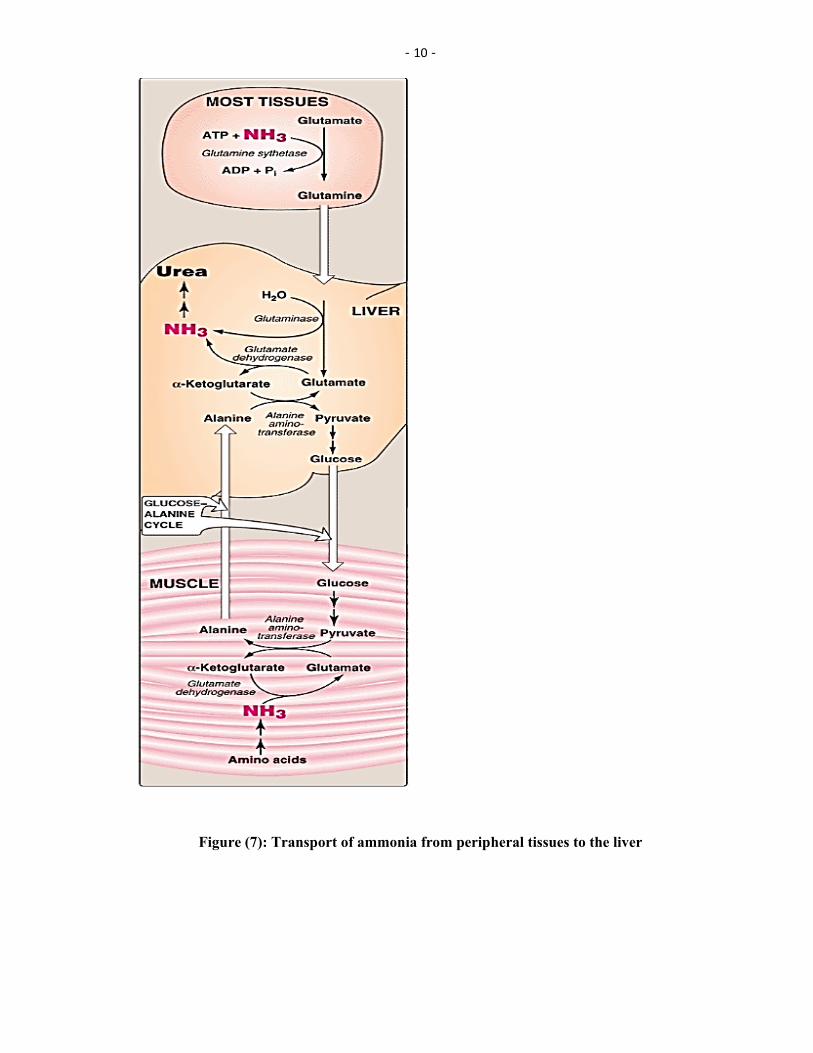
Two mechanisms are available in humans for the transport of ammonia from the peripheral tissues to the liver for its ultimate conversion to urea.
The first, found in most tissues, uses glutamine synthetase to combine ammonia with glutamate to form glutamine—a nontoxic transport form of ammonia (Figure 7). The glutamine is transported in the blood to the liver where it is cleaved by glutaminase to produce glutamate and free ammonia.
The second transport mechanism, used primarily by muscle, involves transamination of pyruvate (the end product of aerobic glycolysis) to form alanine (see Figure 7). Alanine is transported by the blood to the liver, where it is converted to pyruvate, again by transamination. In the liver, the pathway of gluconeogenesis can use the pyruvate to synthesize glucose, which can enter the blood and be used by muscle—a pathway called the glucose-alanine cycle.
- 10 -
Figure (7): Transport of ammonia from peripheral tissues to the liver

- 11 -
Urea Cycle
Urea is the major disposal form of amino groups derived from amino acids, and accounts for about 90% of the nitrogen-containing components of urine. One nitrogen of the urea molecule is supplied by free NH3, and the other nitrogen by aspartate. [Note: Glutamate is the immediate precursor of both ammonia (through oxidative deamination by glutamate dehydrogenase) and aspartate nitrogen (through transamination of oxaloacetate by AST).] The carbon and oxygen of urea are derived from CO2. Urea is produced by the liver, and then is transported in the blood to the kidneys for excretion in the urine.
A. Sequence of reactions of the cycle
The sequence of reactions involved in the biosynthesis of urea , summarized in five steps as follows:
1- Formation of carbamoyl phosphate
2-Formation of citrulline
3- Formation of argininosuccinate
4- Formation of arginine and fumarate
5-Formation of urea and ornithine
The first two reactions leading to the synthesis of urea occur in the mitochondria, whereas the remaining cycle enzymes are located in the cytosol (Figure 8).
Formation of carbamoyl phosphate: Formation of carbamoyl phosphate by carbamoyl phosphate synthetase I is driven by cleavage of two molecules of ATP. Ammonia incorporated into carbamoyl phosphate is provided primarily by the oxidative deamination of glutamate by mitochondrial glutamate dehydrogenase (see Figure 8). Ultimately, the nitrogen atom derived from this ammonia becomes one of the nitrogens of urea. Carbamoyl phosphate synthetase I requires N-acetylglutamate as a positive allosteric activator (see Figure 9).
2-Formation of citrulline: Ornithine and citrulline are basic amino acids that participate in the urea cycle. [Note: They are not incorporated into cellular proteins, because there are no codons for these amino acids Ornithine is regenerated with each turn of the urea cycle, much in the same way that oxaloacetate is regenerated by the reactions of the citric acid cycle. The release of the high-energy phosphate of carbamoyl phosphate as inorganic phosphate drives the reaction in the forward direction. The reaction product, citrulline, is transported to the cytosol.
3-Synthesis of argininosuccinate: Citrulline condenses with aspartate to form argininosuccinate. The α-amino group of aspartate provides the second nitrogen that is ultimately incorporated into urea. The formation of argininosuccinate is driven by the cleavage of ATP to adenosine monophosphate (AMP) and pyrophosphate. This is the third and final molecule of ATP consumed in the formation of urea.

- 12 -
Figure (8): Reactions of the urea cycle
4-Cleavage of argininosuccinate: Argininosuccinate is cleaved to yield arginine and fumarate. The arginine formed by this reaction serves as the immediate precursor of urea. Fumarate produced in the urea cycle is hydrated to malate, providing a link with several metabolic pathways. For example, the malate can be transported into the mitochondria via the malate shuttle and reenter the tricarboxylic acid cycle. Alternatively, cytosolic malate can be oxidized to oxaloacetate, which can be converted to aspartate or glucose.
5-Cleavage of arginine to ornithine and urea: Arginase cleaves arginine to ornithine and urea, and occurs almost exclusively in the liver. Thus, whereas other tissues, such as the kidney, can synthesize arginine by these reactions, only the liver can cleave arginine and, thereby, synthesize urea.
6-Fate of urea: Urea diffuses from the liver, and is transported in the blood to the kidneys, where it is filtered and excreted in the urine. A portion of the urea diffuses from the blood into the intestine, and is cleaved to CO2 and NH3 by bacterial urease. This ammonia is partly lost in the feces, and is partly
- 13 -
reabsorbed into the blood. In patients with kidney failure, plasma urea levels are elevated, promoting a greater transfer of urea from blood into the gut. The intestinal action of urease on this urea becomes a clinically important source of ammonia, contributing to the hyperammonemia often seen in these patients.
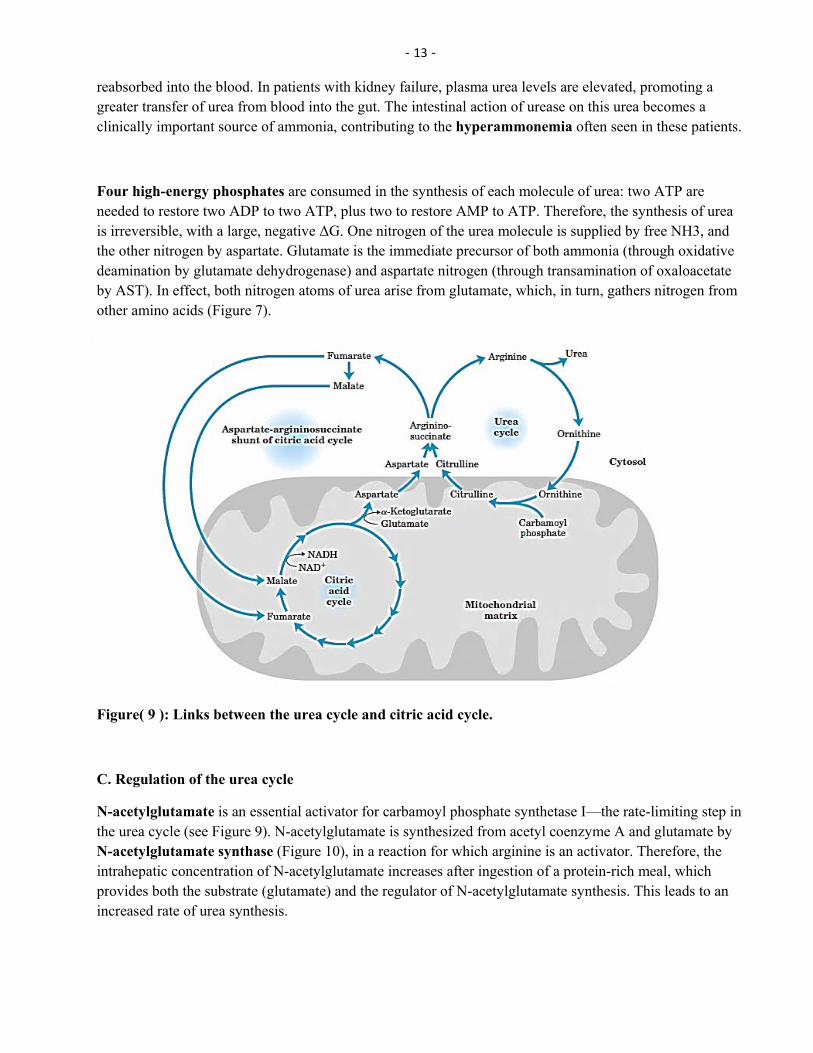
Four high-energy phosphates are consumed in the synthesis of each molecule of urea: two ATP are needed to restore two ADP to two ATP, plus two to restore AMP to ATP. Therefore, the synthesis of urea is irreversible, with a large, negative ΔG. One nitrogen of the urea molecule is supplied by free NH3, and the other nitrogen by aspartate. Glutamate is the immediate precursor of both ammonia (through oxidative deamination by glutamate dehydrogenase) and aspartate nitrogen (through transamination of oxaloacetate by AST). In effect, both nitrogen atoms of urea arise from glutamate, which, in turn, gathers nitrogen from other amino acids (Figure 7).
Figure( 9 ): Links between the urea cycle and citric acid cycle.
C. Regulation of the urea cycle
N-acetylglutamate is an essential activator for carbamoyl phosphate synthetase I—the rate-limiting step in the urea cycle (see Figure 9). N-acetylglutamate is synthesized from acetyl coenzyme A and glutamate by N-acetylglutamate synthase (Figure 10), in a reaction for which arginine is an activator. Therefore, the intrahepatic concentration of N-acetylglutamate increases after ingestion of a protein-rich meal, which provides both the substrate (glutamate) and the regulator of N-acetylglutamate synthesis. This leads to an increased rate of urea synthesis.

- 14 -
Figure (10): Formation and degradation of Nacetylglutamate, an allosteric activator of carbamoyl phosphate synthetase I
.
Ammonia is produced by all tissues during the metabolism of a variety of compounds, and it is disposed of primarily by formation of urea in the liver. However, the level of ammonia in the blood must be kept very low, because even slightly elevated concentrations (hyperammonemia) are toxic to the central nervous system (CNS). There must, therefore, be a metabolic mechanism by which nitrogen is moved from peripheral tissues to the liver for ultimate disposal as urea, while at the same time low levels of circulating ammonia must be maintained.
B. Transport of ammonia in the circulation
Although ammonia is constantly produced in the tissues, it is present at very low levels in blood. This is due both to the rapid removal of blood ammonia by the liver, and the fact that many tissues, particularly muscle, release amino acid nitrogen in the form of glutamine or alanine, rather than as free ammonia.
Urea: Formation of urea in the liver is quantitatively the most important disposal route for ammonia. Urea travels in the blood from the liver to the kidneys, where it passes into the glomerular filtrate.
Glutamine: This amide of glutamic acid provides a nontoxic storage and transport form of ammonia .The ATP-requiring formation of glutamine from glutamate and ammonia by glutamine synthetase occurs primarily in the muscle and liver, but is also important in the nervous system, where it is the major mechanism for the removal of ammonia in the brain. Glutamine is found in plasma at concentrations higher than other amino acids—a finding consistent with its transport function. Circulating glutamine is removed by the liver and the kidneys and deaminated by glutaminase.
C. Hyperammonemia
The capacity of the hepatic urea cycle exceeds the normal rates of ammonia generation, and the levels of serum ammonia are normally low (5–50 µmol/L). However, when liver function is compromised, due either to genetic defects of the urea cycle, or liver disease, blood levels can rise above 1,000 µmol/L. Such hyperammonemia is a medical emergency, because ammonia has a direct neurotoxic effect on the CNS. For example, elevated concentrations of ammonia in the blood cause the symptoms of ammonia

- 15 -
intoxication, which include tremors, slurring of speech, somnolence, vomiting, cerebral edema, and blurring of vision. At high concentrations, ammonia can cause coma and death .
Why are high levels of ammonia toxic?
The toxic effect of ammonia may be due to :
●A decrease in the formation of ATP by citric acid cycle because of diversion of excessive amounts of
-ketoglutarate from citric acid cycle intermediates to form glutamate and glutamine in the brain and thus lowering the rate of oxidation of glucose , the major fuel of the brain or
●By increased formation of GABA(gamma amino butyric acid) from glutamate ,figure (10) leads to impaired neural transmission process .
and glutamate which +4,formed from NH elevated levels of glutaminehat One more possibility is t●
produce osmotic effect that lead directly to swelling of brain .
Decarboxylase
Glutamate---------------------------------→ Gamma aminobutyric acid ( GABA)
2PLP CO
Figure (11): Decarboxylation of glutamate
Amino Acid Degradation and Synthesis
The catabolism of the amino acids found in proteins involves the removal of α-amino groups, followed by the breakdown of the resulting carbon skeletons. These pathways converge to form seven intermediate products: oxaloacetate, α-ketoglutarate, pyruvate, fumarate, succinyl coenzyme A (CoA), acetyl CoA, and acetoacetate.
These products directly enter the pathways of intermediary metabolism, resulting either in the synthesis of glucose or lipid or in the production of energy through their oxidation to CO2 and water by the citric acid cycle.
Glucogenic and Ketogenic Amino Acids
Amino acids can be classified as glucogenic, ketogenic, or both based on which of the seven intermediates are produced during their catabolism (see Figure 12).
A. Glucogenic amino acids
Amino acids whose catabolism yields pyruvate or one of the intermediates of the citric acid cycle are termed glucogenic or glycogenic. These intermediates are substrates for gluconeogenesis and, therefore, can give rise to the net formation of glucose or glycogen in the liver and glycogen in the muscle.

- 16 -
B. Ketogenic amino acids
Amino acids whose catabolism yields either acetoacetate or one of its precursors (acetyl CoA or acetoacetyl CoA) are termed ketogenic (see Figure 20.2). Acetoacetate is one of the “ketone bodies,” which also include 3-hydroxybutyrate and acetone. Leucine and lysine are the only exclusively ketogenic amino acids found in proteins. Their carbon skeletons are not substrates for gluconeogenesis and, therefore, cannot give rise to the net formation of glucose or glycogen in the liver, or glycogen in the muscle.
Figure (12):Classification of amino acids
Catabolism of the Carbon Skeletons of Amino Acids
The pathways by which amino acids are catabolized are conveniently organized according to which one (or more) of the seven intermediates listed above is produced from a particular amino acid.
A. Amino acids that form oxaloacetate
Figure (12): Metabolism of asparagine and aspartate

- 17 -
Asparagine is hydrolyzed by asparaginase, liberating ammonia and aspartate Figure (13). Aspartate loses its amino group by transamination to form oxaloacetate .
B. Amino acids that form α-ketoglutarate
Glutamine is converted to glutamate and ammonia by the enzyme glutaminase. Glutamate is converted to α-ketoglutarate by transamination, or through oxidative deamination by glutamate dehydrogenase. Figure(14)
- Ketoglutarate
Transamination
+4NH Glutamate-L +
4HN
Mg-ATP
Glutamine synthetase Glutaminase
Mg-ADP + Pi
Glutamine
Figure (14): Synthesis and degradation of glutamine
Proline: This amino acid is oxidized to glutamate. Glutamate is transaminated or oxidatively deaminated to form α-ketoglutarate.
Arginine: This amino acid is cleaved by arginase to produce ornithine and urea . [Note: This reaction occurs primarily in the liver as part of the urea cycle. Ornithine is subsequently converted to α-ketoglutarate.
C. Amino acids that form pyruvate
Alanine: This amino acid loses its amino group by transamination to form pyruvate (Figure 4)
methylenetetrahydrofolate. Serine can also -10,N5This amino acid can be converted to glycine and N Serine:be converted to pyruvate by serine dehydratase (15).
-10,N5This amino acid can either be converted to serine by addition of a methylene group from N Glycine:methylenetetrahydrofolic acid (see Figure 15), or oxidized to CO2 and NH3.
Cystine: This amino acid is reduced to cysteine, using NADH + H+ as a reductant. Cysteine undergoes desulfuration to yield pyruvate.
Threonine: This amino acid is converted to pyruvate or to α-ketobutyrate, which forms succinyl CoA.

- 18 -
Figure (15): A. Interconversion of serine and glycine, and oxidation of glycine.
B. Dehydration of serine to form pyruvate
D. Amino acids that form fumarate
Phenylalanine and tyrosine: Hydroxylation of phenylalanine leads to the formation of tyrosine (Figure 16). This reaction, catalyzed by phenylalanine hydroxylase, is the first reaction in the catabolism of phenylalanine. Thus, the metabolism of phenylalanine and tyrosine merge, leading ultimately to the formation of fumarate and acetoacetate. Phenylalanine and tyrosine are, therefore, both glucogenic and ketogenic.
Inherited deficiencies: Inherited deficiencies in the enzymes of phenylalanine and tyrosine metabolism lead to the diseases phenylketonuria, and alkaptonuria, and the condition of albinism.
Figure (16):Degradation of phenylalanine
E. Amino acids that form succinyl CoA:
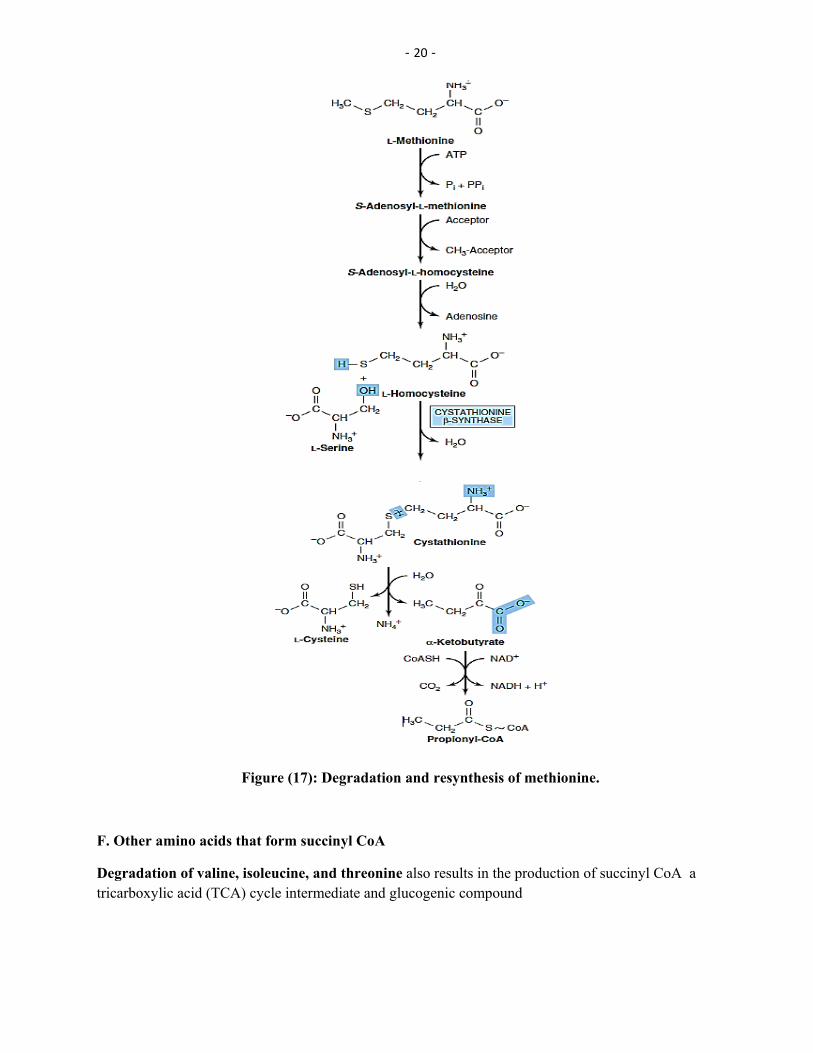
Methionine : Methionine is one of four amino acids that form succinyl CoA. This sulfur-containing amino acid deserves special attention because it is converted to S-adenosylmethionine (SAM), the major methyl-group donor in one-carbon metabolism (Figure 17).

- 19 -
Synthesis of SAM: Methionine condenses with adenosine triphosphate (ATP), forming SAM—a high-energy compound that is unusual in that it contains no phosphate. The formation of SAM is driven, in effect, by hydrolysis of all three phosphate bonds in ATP
Activated methyl group: The methyl group attached to the tertiary sulfur in SAM is “activated,” and can be transferred to a variety of acceptor molecules, such as norepinephrine in the synthesis of epinephrine. The methyl group is usually transferred to oxygen or nitrogen atoms, but sometimes to carbon atoms.
Hydrolysis of SAM: After donation of the methyl group, S-adenosylhomocysteine is hydrolyzed to homocysteine and adenosine. Homocysteine has two fates: If there is a deficiency of methionine, homocysteine may be remethylated to methionine (17). If methionine stores are adequate, homocysteine may enter the transsulfuration pathway, where it is converted to cysteine
-5methyltetrahydrofolate (N-5Homocysteine accepts a methyl group from N Resynthesis of methionine:. The methyl l2in a reaction requiring methylcobalamin, a coenzyme derived from vitamin BTHF) -methyl
-methyl-5derivative to homocysteine, and cobalamin is recharged from N 12group is transferred from the BTHF.
Synthesis of cysteine: Homocysteine condenses with serine, forming cystathionine, which is hydrolyzed requiring sequence has the net effect of –6). This vitamin B71ketobutyrate and cysteine (see Figure -to α
converting serine to cysteine, and homocysteine to α-ketobutyrate, which is oxidatively decarboxylated to form propionyl CoA. Propionyl CoA is converted to succinyl CoA. Because homocysteine is synthesized from the essential amino acid methionine, cysteine is not an essential amino acid as long as sufficient methionine is available.
Figure( 16): Formation of S-adenosylmethionine. ~CH3 represents the high group transfer potential of “active methionine.”
- 20 -
Figure (17): Degradation and resynthesis of methionine.
F. Other amino acids that form succinyl CoA
Degradation of valine, isoleucine, and threonine also results in the production of succinyl CoA a tricarboxylic acid (TCA) cycle intermediate and glucogenic compound

- 21 -
Valine and isoleucine: These amino acids are branched-chain amino acids that generate propionyl CoA, which is converted to succinyl CoA by biotin- and vitamin B12–requiring reactions [Note: Propionyl CoA, then, is generated by the catabolism of certain amino acids and odd-numbered fatty acids].
Threonine: This amino acid is dehydrated to α-ketobutyrate, which is converted to propionyl CoA and then to succinyl CoA. [Note: Threonine can also be converted to pyruvate].
Threonine
PLP Threonine dehydratase
+4NH
-Ketobutyrate
Oxidativedecarboxylation
Prpionyl-CoA
Biotin
Methyl malonyl –CoA
Vitamin B
Succinyl –CoA
TCA cycle
Figure (18):Degradation of Thrionine
G. Amino acids that form acetyl CoA or acetoacetyl CoA
Leucine, isoleucine, lysine, and tryptophan form acetyl CoA or acetoacetyl CoA directly
Leucine: This amino acid is exclusively ketogenic in its catabolism, forming acetyl CoA and acetoacetate.
Isoleucine: This amino acid is both ketogenic and glucogenic, because its metabolism yields acetyl CoA and propionyl CoA.
Lysine: An exclusively ketogenic amino acid, this amino acid is unusual in that neither of its amino groups undergoes transamination as the first step in catabolism. Lysine is ultimately converted to acetoacetyl CoA.
Tryptophan: This amino acid is both glucogenic and ketogenic because its metabolism yields alanine and acetoacetyl CoA.
Catabolism of the branched-chain amino acids
The branched-chain amino acids, isoleucine, leucine, and valine, are essential amino acids. In contrast to other amino acids, they are metabolized primarily by the peripheral tissues (particularly muscle), rather

- 22 -
than by the liver. Because these three amino acids have a similar route of catabolism, it is convenient to describe them as a group (see Figure 19).
Figure(19): Degrada on of branched chain amino acids
Role of Folic Acid in Amino Acid Metabolism
Some synthetic pathways require the addition of single carbon groups. These “one-carbon units” can exist in a variety of oxidation states. These include methane, methanol, formaldehyde, formic acid, and carbonic acid. It is possible to incorporate carbon units at each of these oxidation states, except methane, into other organic compounds. These single carbon units can be transferred from carrier compounds such as tetrahydrofolic( THF ) acid and S-adenosylmethionine to specific structures that are being synthesized or modified.
Biosynthesis of Nonessential Amino Acids
Nonessential amino acids are synthesized from intermediates of metabolism or, as in the case of tyrosine and cysteine, from the essential amino acids phenylalanine and methionine, respectively.
A. Synthesis from α-keto acids
Alanine, aspartate, and glutamate are synthesized by transfer of an amino group to the α-keto acids pyruvate, oxaloacetate, and α-ketoglutarate, respectively. These transamination reactions (Figure 5) are the

- 23 -
most direct of the biosynthetic pathways. Glutamate is unusual in that it can also be synthesized by the reverse of oxidative deamination, catalyzed by glutamate dehydrogenase.
Figure( 20): Formation of alanine, aspartate, and glutamate from the corresponding α-keto acids.
B. Synthesis by amidation
Glutamine: This amino acid, which contains an amide linkage with ammonia at the γ-carboxyl, is formed from glutamate by glutamine synthetase (see Figure 14). The reaction is driven by the hydrolysis of ATP. In addition to producing glutamine for protein synthesis, the reaction also serves as a major mechanism for the detoxification of ammonia in brain and liver.
Asparagine: This amino acid, which contains an amide linkage with ammonia at the β-carboxyl, is formed from aspartate by asparagine synthetase, using glutamine as the amide donor. The reaction requires ATP, and, like the synthesis of glutamine, has an equilibrium far in the direction of asparagine synthesis.
D. Serine, glycine, and cysteine
Serine: This amino acid arises from 3-phosphoglycerate, an intermediate in glycolysis , which is first oxidized to 3-phosphopyruvate, and then transaminated to 3-phosphoserine. Serine is formed by hydrolysis of the phosphate ester (figure 21). Serine can also be formed from glycine through transfer of a hydroxymethyl group by serine hydroxymethyl transferase (see Figure 15).

- 24 -
Glucose
3-Phosphoglycerate
+NAD
+NADH+H
3- Phosphpo-hydroxy-pyruvate
L-Glutamate
-Ketoglutarate
3-Phospho-L-serine
O2H
Pi
L-Serine
Figure(21): Synthesis of serine from a glucose molecule
Glycine: This amino acid is synthesized from serine by removal of a hydroxymethyl group, also by serine hydroxymethyl transferase (see Figure 15).
Cysteine: This amino acid is synthesized by two consecutive reactions in which homocysteine combines with serine, forming cystathionine, which, in turn, is hydrolyzed to α-ketobutyrate and cysteine (see Figure 17). Homocysteine is derived from methionine. Because methionine is an essential amino acid, cysteine synthesis can be sustained only if the dietary intake of methionine is adequate.
E. Tyrosine
Tyrosine is formed from phenylalanine by phenylalanine hydroxylase. The reaction requires molecular oxygen and the coenzyme tetrahydrobiopterin (BH4), which can be synthesized from guanosine triphosphate (GTP) by the body. One atom of molecular oxygen becomes the hydroxyl group of tyrosine, and the other atom is reduced to water. During the reaction, tetrahydrobiopterin is oxidized to dihydrobiopterin. Tetrahydrobiopterin is regenerated from dihydrobiopterin in a separate reaction requiring NADH. Tyrosine, like cysteine, is formed from an essential amino acid and is, therefore, nonessential only in the presence of adequate dietary phenylalanine (figure 16).

- 25 -
VI. Metabolic Defects in Amino Acid Metabolism
Inborn errors of metabolism are commonly caused by mutant genes that generally result in abnormal proteins, most often enzymes. The inherited defects may be expressed as a total loss of enzyme activity or, more frequently, as a partial deficiency in catalytic activity. Without treatment, the inherited defects of amino acid metabolism almost invariably result in mental retardation or other developmental abnormalities as a result of harmful accumulation of metabolites. Although more than 50 of these disorders have been described, many are rare, occurring in less than 1 per 250,000 in most populations. Phenylketonuria is the most important disease of amino acid metabolism because it is relatively common and responds to dietary treatment.
Figure (22): A deficiency in phenylalanine hydroxylase results in the disease phenylketonuria (PKU).
Phenylketonuria
Phenylketonuria (PKU), caused by a deficiency of phenylalanine hydroxylase (Figure 20.15), PKU is the most common clinically encountered inborn error of amino acid metabolism (prevalence 1:15,000). Biochemically, it is characterized by accumulation of phenylalanine (and a deficiency of tyrosine). Hyperphenylalaninemia may also be caused by deficiencies in any of the several enzymes required to synthesize BH4, or in dihydropteridine (BH2) reductase, which regenerates BH4 from BH2 (Figure 20.16). Such deficiencies indirectly raise phenylalanine concentrations, because phenylalanine hydroxylase requires BH4 as a coenzyme. BH4 is also required for tyrosine hydroxylase and tryptophan hydroxylase, which catalyze reactions leading to the synthesis of neurotransmitters, such as serotonin and catecholamines. Simply restricting dietary phenylalanine does not reverse the central nervous system (CNS) effects due to deficiencies in neurotransmitters. Replacement therapy with BH4 or L-DOPA and 5-hydroxytryptophan (products of the affected tyrosine hydroxylase– and tryptophan hydroxylase–catalyzed reactions) improves the clinical outcome in these variant forms of hyperphenylalaninemia, although the response is unpredictable.
Characteristics of classic PKU

- 26 -
Elevated phenylalanine: Phenylalanine is present in elevated concentrations in tissues, plasma, and urine. Since patients can not convert phenylalanine to tyrosine by normal pathway ,some minor pathway of phenylalanine becomes prominent in phenylketonurics and accumulation of toxic derivatives of phenylalanine such as Phenyl lactate, phenyl acetate, and phenyl pyruvate, which are not normally produced in significant amounts in the presence of functional phenylalanine hydroxylase (Figure 20.17). The disease acquired its name (PKU) from the high levels of the keto acid , phenyl pyruvate in urine .
Figure (23): Pathways of phenylalanine metabolism in normal individuals and in patients with phenylketonuria.
CNS symptoms: Mental retardation, failure to walk or talk, seizures, hyperactivity, tremor, microcephaly, and failure to grow are characteristic findings in PKU. The patient with untreated PKU typically shows symptoms of mental retardation by the age of one year.
B. Maple syrup urine disease
Maple syrup urine disease (MSUD) is a rare (1:185,000), autosomal recessive disorder in which there is a partial or complete deficiency in branched-chain α-keto acid dehydrogenase, an enzyme complex that decarboxylates leucine, isoleucine, and valine (see Figure 20.10). These amino acids and their corresponding α-keto acids accumulate in the blood, causing a toxic effect that interferes with brain functions. The disease is characterized by feeding problems, vomiting, dehydration, severe metabolic

- 27 -
acidosis, and a characteristic maple syrup odor to the urine. If untreated, the disease leads to mental retardation, physical disabilities, and even death.
C. Albinism
Albinism refers to a group of conditions in which a defect in tyrosine metabolism results in a deficiency in the production of melanin. These defects result in the partial or full absence of pigment from the skin, hair, and eyes. In addition to hypopigmentation, affected individuals have vision defects and photophobia (sunlight hurts their eyes). They are at increased risk for skin cancer.
Hypopigmentation: Patients with phenylketonuria often show a deficiency of pigmentation (fair hair, light skin color, and blue eyes). The hydroxylation of tyrosine by tyrosinase, which is the first step in the formation of the pigment melanin, is competitively inhibited by the high levels of phenylalanine present in PKU.
tyrosinase
Dopa Tyrosine → Dopa
Dopamine Tyroxine Dopaquinone
Neurotransmitter
Norepinphrine Chaticholamines
Epinpherine Melanine pigmint
Figure(24): : Biosynthesis of biologically important compounds from tyrosine
E. Alkaptonuria
Alkaptonuria is a rare metabolic disease involving a deficiency in homogentisic acid oxidase, resulting in the accumulation of homogentisic acid. [Note: This reaction occurs in the degradative pathway of tyrosine] , The illness has three characteristic symptoms: homogentisic aciduria (the patient's urine contains elevated levels of homogentisic acid, which is oxidized to a dark pigment on standing), large joint arthritis, and black ochronotic pigmentation of cartilage and collagenous tissue. Patients with alkaptonuria are usually asymptomatic until about age 40. Diets low in protein—especially in phenylalanine and tyrosine—help reduce the levels of homogentisic acid, and decrease the amount of pigment deposited in body tissues. Although alkaptonuria is not life-threatening, the associated arthritis may be severely crippling.
Biologically important compounds derived from Tryptophan
)3Vitamin niacin ( vitamin B -1
2- Neurotransmitter serotonin

- 28 -
which has effects on the hypothalamic is a hormone produced by the pineal gland : Hormone melatonin -3pituitary system .Synthesis of melatonin is regulated by light –dark cycle and blood levels of melatonin rise at night.
: s disease ,Parkinson
in Parkinson, s disease , dopamine levels in the CNS are decreased because of a deficiency of cells that produce dopamine and depression is associated with low levels of serotonin.
Other Nitrogen-Containing Compounds
A. Histamine
Histamine is a chemical messenger that mediates a wide range of cellular responses, including allergic and inflammatory reactions, gastric acid secretion, and possibly neurotransmission in parts of the brain. A powerful vasodilator, histamine is formed by decarboxylation of histidine in a reaction requiring pyridoxal phosphate. It is secreted by mast cells as a result of allergic reactions or trauma. Histamine has no clinical applications, but agents that interfere with the action of histamine have important therapeutic applications.
Figure (25): Biosynthesis of histamine
C. Serotonin
Serotonin, also called 5-hydroxytryptamine, is synthesized and stored at several sites in the body (Figure 21.18). By far the largest amount of serotonin is found in cells of the intestinal mucosa. Smaller amounts occur in the central nervous system, where it functions as a neurotransmitter, and in platelets. Serotonin is synthesized from tryptophan, which is hydroxylated in a reaction analogous to that catalyzed by phenylalanine hydroxylase. The product, 5-hydroxytryptophan, is decarboxylated to serotonin. Serotonin has multiple physiologic roles, including pain perception, affective disorders, and regulation of sleep, temperature, and blood pressure.

- 29 -
Figure (26):Synthesis of serotonin.
Serotinin
Acetyl –CoA Acetyl transferase
CoA-SH
N-Acetyl serotonin
S-Adenosylmethionine Methyl transferase
N-Acetyl -5-methoxyserotonin(Melatonin)
Figure(27) : Formation of melatonin from serotonin
D. Creatine
Creatine phosphate (also called phosphocreatine), the phosphorylated derivative of creatine found in muscle, is a high-energy compound that can reversibly donate a phosphate group to adenosine diphosphate to form ATP (Figure 21.19). Creatine phosphate provides a small but rapidly mobilized reserve of high-energy phosphates that can be used to maintain the intracellular level of adenosine triphosphate (ATP) during the first few minutes of intense muscular contraction. [Note: The amount of creatine phosphate in the body is proportional to the muscle mass.

- 30 -
Figure (28):Synthesis of creatine