Embed Size (px)
Citation preview

QUÍMICA ORGÁNICA
Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 1
2019
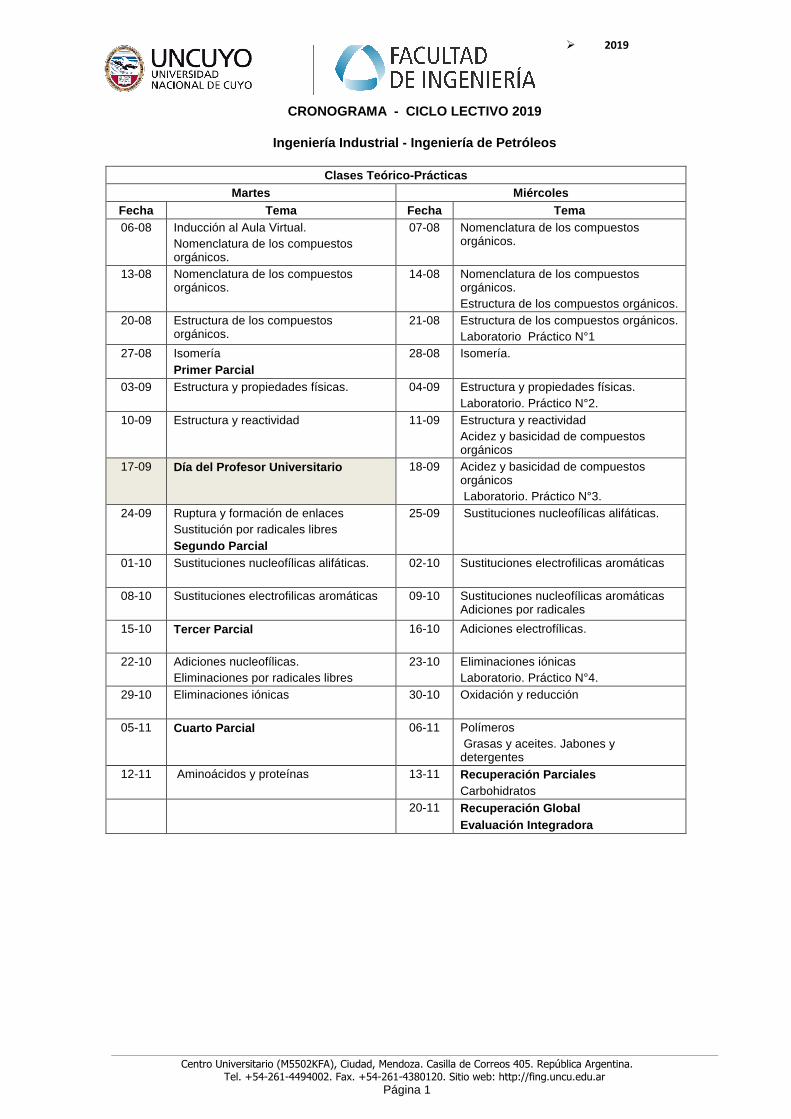
CRONOGRAMA - CICLO LECTIVO 2019
Ingeniería Industrial - Ingeniería de Petróleos
Clases Teórico-Prácticas Martes Miércoles
Fecha Tema Fecha Tema 06-08 Inducción al Aula Virtual.
Nomenclatura de los compuestos orgánicos.
07-08 Nomenclatura de los compuestos orgánicos.
13-08 Nomenclatura de los compuestos orgánicos.
14-08 Nomenclatura de los compuestos orgánicos. Estructura de los compuestos orgánicos.
20-08 Estructura de los compuestos orgánicos.
21-08 Estructura de los compuestos orgánicos. Laboratorio Práctico N°1
27-08 Isomería Primer Parcial
28-08 Isomería.
03-09 Estructura y propiedades físicas.
04-09 Estructura y propiedades físicas. Laboratorio. Práctico N°2.
10-09 Estructura y reactividad
11-09 Estructura y reactividad Acidez y basicidad de compuestos orgánicos
17-09 Día del Profesor Universitario 18-09 Acidez y basicidad de compuestos orgánicos Laboratorio. Práctico N°3.
24-09 Ruptura y formación de enlaces Sustitución por radicales libres Segundo Parcial
25-09 Sustituciones nucleofílicas alifáticas.
01-10 Sustituciones nucleofílicas alifáticas. 02-10 Sustituciones electrofilicas aromáticas
08-10 Sustituciones electrofilicas aromáticas
09-10 Sustituciones nucleofílicas aromáticas Adiciones por radicales
15-10 Tercer Parcial 16-10 Adiciones electrofílicas.
22-10 Adiciones nucleofílicas. Eliminaciones por radicales libres
23-10 Eliminaciones iónicas Laboratorio. Práctico N°4.
29-10 Eliminaciones iónicas
30-10 Oxidación y reducción
05-11 Cuarto Parcial
06-11 Polímeros Grasas y aceites. Jabones y detergentes
12-11 Aminoácidos y proteínas 13-11 Recuperación Parciales Carbohidratos
20-11 Recuperación Global Evaluación Integradora
Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 1
2019
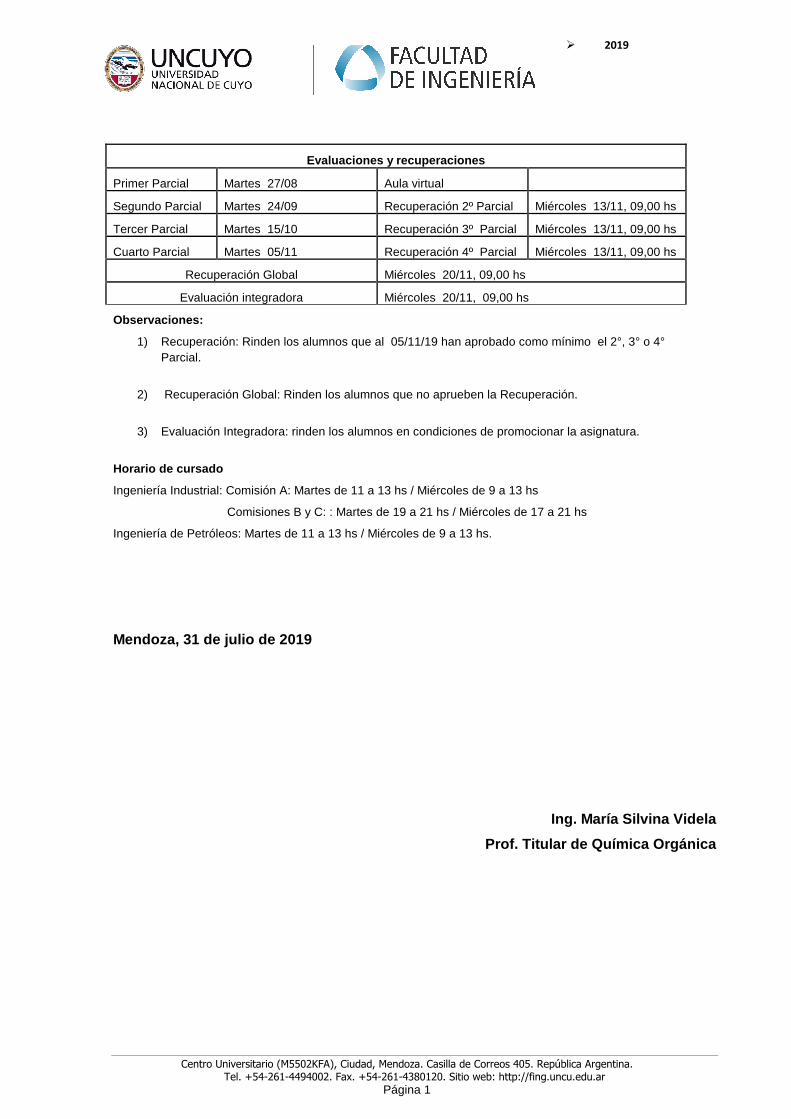
Evaluaciones y recuperaciones
Primer Parcial Martes 27/08 Aula virtual
Segundo Parcial Martes 24/09 Recuperación 2º Parcial Miércoles 13/11, 09,00 hs
Tercer Parcial Martes 15/10 Recuperación 3º Parcial Miércoles 13/11, 09,00 hs
Cuarto Parcial Martes 05/11 Recuperación 4º Parcial Miércoles 13/11, 09,00 hs
Recuperación Global Miércoles 20/11, 09,00 hs
Evaluación integradora Miércoles 20/11, 09,00 hs
Observaciones:
1) Recuperación: Rinden los alumnos que al 05/11/19 han aprobado como mínimo el 2°, 3° o 4° Parcial.
2) Recuperación Global: Rinden los alumnos que no aprueben la Recuperación.
3) Evaluación Integradora: rinden los alumnos en condiciones de promocionar la asignatura.
Horario de cursado
Ingeniería Industrial: Comisión A: Martes de 11 a 13 hs / Miércoles de 9 a 13 hs
Comisiones B y C: : Martes de 19 a 21 hs / Miércoles de 17 a 21 hs
Ingeniería de Petróleos: Martes de 11 a 13 hs / Miércoles de 9 a 13 hs.
Mendoza, 31 de julio de 2019
Ing. María Silvina Videla
Prof. Titular de Química Orgánica

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 1
2019
Facultad de Ingeniería - Universidad Nacional de Cu yo
P1- PROGRAMA DE ASIGNATURA
Asignatura: Química Orgánica
Profesor Titular: Ing. María Silvina Videla
Carrera: Ingeniería de Petróleos, Industrial
Año: 2019 Semestre: 4° Horas Semestre: 90 Horas Semana: 6
OBJETIVOS
• Adquirir los conocimientos fundamentales sobre el carbono, los compuestos que éste constituye, y sus reacciones más comunes.
• Relacionar los conocimientos de la química del carbono con diferentes procesos en el contexto industrial, económico, medioambiental y social.
• Proporcionar a los estudiantes una base de conocimientos de la química del carbono con las que pueda continuar sus estudios de Ingeniería con un alto grado de autonomía.
En términos de competencias, el alumno podrá: • Identificar los aspectos principales de la terminología química y nomenclatura de
compuestos orgánicos. • Emplear en forma sistemática y progresiva los conceptos de la teoría estructural para
estudiar las propiedades físicas y químicas de los compuestos del carbono. • Reconocer y relacionar las principales propiedades químicas de los compuestos orgánicos y
biológicos. • Distinguir los aspectos estructurales de los elementos químicos y sus compuestos,
incluyendo estereoquímica. • Adquirir destreza en el manejo del carácter tridimensional de los compuestos orgánicos y el
curso estereoquímico de las reacciones más importantes. • Identificar la naturaleza y el comportamiento de los grupos funcionales en moléculas
orgánicas. • Describir y diferenciar los tipos principales de reacciones químicas y las características
asociadas a cada una de ellas. • Describir y diferenciar mecanismos de reacción simples e interpretar su papel en el diseño
y control de las condiciones de una reacción. • Reconocer las principales rutas sintéticas en química orgánica, incluyendo la
interconversión de grupos funcionales y la formación de enlaces carbono-carbono y carbono - heteroátomo.
• Identificar la relación entre propiedades macroscópicas y propiedades de átomos y moléculas individuales, incluyendo macromoléculas (naturales y sintéticas), polímeros y otros materiales.
• Describir y relacionar la estructura y reactividad de las principales clases de biomoléculas y la química de los principales procesos biológicos.

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 2
2019
• Predecir mecanismos de reacción y condiciones para llevarlos a cabo. • Identificar sustancias químicas de importancia biológica e industrial. • Relacionar conceptos y sustancias químicas con situaciones de la vida diaria y de su futura
profesión. • Investigar e identificar los riesgos ambientales de algunas sustancias orgánicas. • Manipular material de laboratorio y desarrollar técnicas para identificar propiedades físicas y
químicas de compuestos orgánicos. • Recolectar e interpretar datos procedentes de observaciones y medidas en el laboratorio. • Resolver problemas de aplicación. • Participar y colaborar en trabajos grupales. • Discutir y argumentar resultados en grupo.
CONTENIDOS
UNIDAD 1: CLASIFICACIÓN Y NOMENCLATURA DE LOS COMPU ESTOS ORGANICOS
Conceptos generales. Alcanos, alquenos y alquinos. Hidrocarburos alicíclicos. Hidrocarburos aromáticos. Haluros de alquilo. Alcoh oles. Éteres. Aldehídos y cetonas. Ácidos carboxílicos. Aminas. Moléculas con varios g rupos funcionales.
UNIDAD 2: ESTRUCTURA DE LOS COMPUESTOS ORGÁNICOS. I SOMERÍA
2.A. Estructura de los compuestos orgánicos
Teoría estructural clásica. Fórmulas estructurales de las moléculas orgánicas. Teoría de repulsión del par electrónico en la capa de valenci a. Teoría del enlace de valencia. Ángulos de enlaces. Energías y longitudes de enlace s. Teoría de la resonancia. Formas contribuyentes. Energía de resonancia. Aromaticidad . Estructura del benceno. Criterios de aromaticidad.
2.B. Isomería
Isomería plana o estructural: Isómeros de cadena. I sómeros de posición. Isómeros de función. Propiedades de los isómeros estructurales.
Isomería espacial o estereoisomeria: Isomería confo rmacional. Isomería configuracional. Isomería Geométrica. Isomería Óptica: Centro quiral , actividad óptica, enantiómeros, diasterómeros, par racémico y forma meso. Propieda des de los estereoisómeros.
UNIDAD 3: PROPIEDADES FÍSICAS Y QUÍMICAS DE LOS CO MPUESTOS ORGÁNICOS
3.A. Estructura y propiedades físicas
Fuerzas intermoleculares: Fuerzas de Van der Waals, atracción dipolo-dipolo, puente hidrógeno. Punto de ebullición. Punto de fusión. So lubilidad. Solvatación de iones y de moléculas covalentes.
Electronegatividad. Polaridad de enlaces. Momento d ipolar. Moléculas polares y no polares.

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 3
2019
3.B. Estructura y reactividad
Efectos sobre la reactividad (Efectos inductivo, es térico y de resonancia). Análisis de efectos sobre las estructuras de compuestos orgánic os.
3.C. Acidez y basicidad de compuestos orgánicos
Ácidos y bases de Lewis. Ácidos y Bases de Brönsted Lowry. Acidez de alquinos, alcoholes, fenoles y ácidos carboxílicos. Influenci a de sustituyentes sobre la acidez.
Basicidad de alcoholes, aminas alifáticas y aromáti cas. Influencia de sustituyentes sobre la basicidad
UNIDAD 4: REACCIONES DE SUSTITUCIÓN EN COMPUESTOS ORGÁNICOS ALIFÁTICOS
4.A. Introducción a la reactividad de los compuesto s orgánicos
Ruptura y formación de enlaces. Homólisis y heteról isis. Electrófilos y nucleófilos. Mecanismos de reacción. Avance de la reacción: camb ios de energía.
4.B. Sustituciones por radicales libres
Halogenación de alcanos. Intermedios. Estados de tr ansición. Cinética. Diagramas de energía. Energía de activación. Calor de reacción. Importancia de las sustituciones por radicales libres.
4.C. Sustituciones nucleofílicas alifáticas
Sustituciones nucleofílicas en haluros de alquilo y alcoholes. Cinética. Orden de reacción y molecularidad. Diagramas de energía. Influencia d e la estructura y la solvatación sobre la reactividad. Reordenamientos o transposiciones d e iones carbonio.
UNIDAD 5: REACCIONES DE SUSTITUCIÓN EN COMPUESTOS O RGÁNICOS AROMÁTICOS
5.A. Sustituciones electrofílicas aromáticas
Mecanismo General. Diagrama de Energía. Sustitucion es electrofílicas más comunes. Efectos de los sustituyentes sobre la reactividad y la orientación. Aplicaciones de las sustituciones electrofílicas en la industria químic a orgánica. Polimerización por condensación: Resinas fenólicas.
5.B. Sustituciones nucleofílicas aromáticas
Sustituciones nucleofílicas en compuestos aromático s. Cinética. Diagramas de energía. Efecto de los sustituyentes sobre la reactividad. A plicaciones de las sustituciones nucleofílicas aromáticas.

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 4
2019
UNIDAD 6: REACCIONES DE ADICION
6.A. Adiciones por radicales libres
Hidrogenación de alquenos, alquinos e hidrocarburos aromáticos. Calores de hidrogenación. Diagramas de energía. Aplicaciones d e las adiciones por radicales libres.
6.B. Adiciones electrofílicas
Adiciones electrofílicas en alquenos y alquinos. Me canismo de reacción. Orientación de la adición. Importancia industrial de las adiciones el ectrofílicas.
6.C. Adiciones nucleofílicas
Adiciones nucleofílicas en aldehídos y cetonas. Cin ética. Diagramas de energía. Reactividad de aldehidos y cetonas. Importancia de las adiciones nucleofílicas.
UNIDAD 7: REACCIONES DE ELIMINACIÓN
7.A. Eliminaciones por radicales libres
Cracking de alcanos. Cinética. Diagramas de energí a. Importancia industrial de las eliminaciones por radicales libres.
7.B. Eliminaciones iónicas
Reacciones de eliminación iónica en haluros de alqu ilo y alcoholes. Cinética. Orden de reacción y molecularidad. Diagramas de energía. Inf luencia de la estructura y la solvatación sobre la reactividad. Reordenamientos o transposiciones de iones carbonio. Competencia con las sustituciones nucleofílicas. Im portancia industrial de las eliminaciones iónicas.
UNIDAD 8: OXIDACIÓN Y REDUCCIÓN DE COMPUESTOS ORGÁ NICOS
Combustión. Calor de combustión. Uso de hidrocarbur os como combustibles. Oxidación química de hidrocarburos y compuestos orgánicos oxi genados.
Reducción de hidrocarburos alifáticos y aromáticos. Reducción de compuestos orgánicos oxigenados. Reducción de nitrocompuestos aromáticos
UNIDAD 9: MACROMOLÉCULAS
9.A. Polímeros
Generalidades. Clasificación: Polímeros naturales y sintéticos. Características estructurales. Peso molecular.

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 5
2019
Polimerización por condensación. Polamidas, poliést eres y poliuretanos. Copolimerización.
Polimerización por adición. Polimerización por radi cales libres, aniónica y catiónica. Caucho natural. Cauchos sintéticos.
Configuración de los polímeros. Propiedades.
9.B. Grasas y aceites. Jabones y detergentes.
Estructura de grasas y aceites. Ácidos grasos omega . Grasas trans. Hidrólisis.
Estructura de los jabones. Acción limpiadora. Desve ntajas. Detergentes: estructura, clasificación y ventajas respecto de los jabones.
9.C. Aminoácidos y proteínas
Estructura de aminoácidos. Propiedades ácido-base. Punto isoeléctrico. Unión peptídica. Péptidos y proteínas.
9.D. Carbohidratos
Monosacáridos: Definición. Clasificación. D-(+)-Glu cosa (Configuración. Estructura cíclica). D-(-)-Fructosa (Configuración. Estru ctura cíclica). Mutarrotación.
Disacáridos Reductores y No Reductores. Inversión de la sacarosa.
Polisacáridos: Generalidades (Almidón y Celulosa).
TRABAJOS PRÁCTICOS DE LABORATORIO
Nº1: El Petróleo crudo. Propiedades de interés para su comercialización como energía primaria.
Nº2: Solubilidad de compuestos orgánicos.
Nº3: Exploración experimental en casa. pH de compue stos orgánicos.
Nº4: Acidez y basicidad de compuestos orgánicos.
Nº5: Determinación de detergentes en aguas residual es
METODOLOGÍA DE ENSEÑANZA
La asignatura consta de clases teórico – prácticas, trabajos prácticos de laboratorio y trabajos prácticos en Aula Virtual. Se emplea una metodología que va de lo particular a lo general (método inductivo), y de lo general a lo particular (método deductivo), simulaciones y experimentaciones, para favorecer el aprendizaje.
Clases teórico – prácticas: los alumnos se distribuyen en cuatro grupos. El docente responsable del curso introduce los conceptos básicos a fin de que el alumno conozca la terminología asociada y comprenda las aplicaciones del tema.
Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 6
2019
El alumno cuenta con apuntes de la materia y bibliografía indicada por la cátedra para responder un cuestionario sobre el tema y resolver los ejercicios que le son indicados por el docente, pudiendo consultar a éste toda vez que sea necesario.
Las clases tienen carácter de obligatorias.
Trabajos prácticos de laboratorio: Cada curso se organiza en comisiones para la realización de los prácticos. El alumno cuenta con una Guía de Trabajos Prácticos de Laboratorio que incluye conceptos teóricos. Dichos prácticos son de asistencia obligatoria debiendo, además, rendir y aprobar un pre práctico para su realización.
Trabajos prácticos en aula virtual: el alumno, en forma individual o grupal, tiene actividades a desarrollar y entregar previo al parcial correspondiente a la unidad temática abordada.
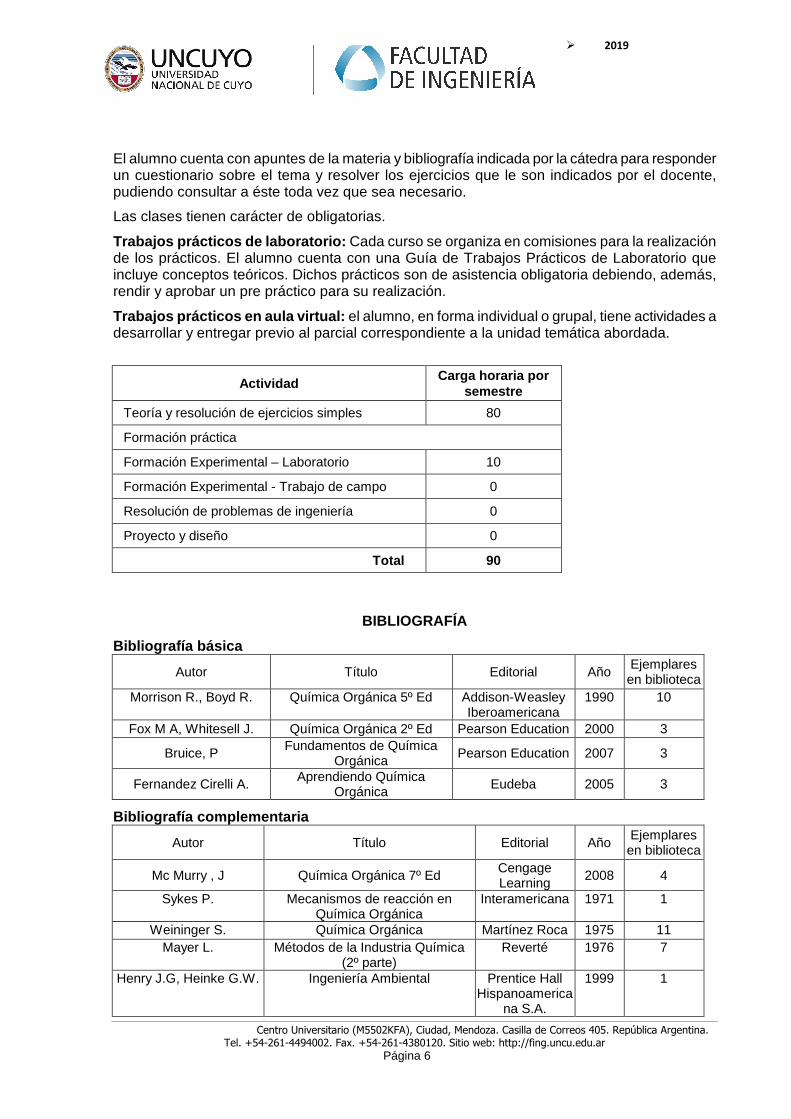
Actividad Carga horaria por
semestre
Teoría y resolución de ejercicios simples 80
Formación práctica
Formación Experimental – Laboratorio 10
Formación Experimental - Trabajo de campo 0
Resolución de problemas de ingeniería 0
Proyecto y diseño 0
Total 90
BIBLIOGRAFÍA
Bibliografía básica
Autor Título Editorial Año Ejemplares en biblioteca
Morrison R., Boyd R. Química Orgánica 5º Ed Addison-Weasley Iberoamericana
1990 10
Fox M A, Whitesell J. Química Orgánica 2º Ed Pearson Education 2000 3
Bruice, P Fundamentos de Química Orgánica
Pearson Education 2007 3
Fernandez Cirelli A. Aprendiendo Química Orgánica
Eudeba 2005 3
Bibliografía complementaria
Autor Título Editorial Año Ejemplares en biblioteca
Mc Murry , J Química Orgánica 7º Ed Cengage Learning
2008 4
Sykes P. Mecanismos de reacción en Química Orgánica
Interamericana 1971 1
Weininger S. Química Orgánica Martínez Roca 1975 11 Mayer L. Métodos de la Industria Química
(2º parte) Reverté 1976 7
Henry J.G, Heinke G.W. Ingeniería Ambiental Prentice Hall Hispanoamerica
na S.A.
1999 1

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 7
2019
EVALUACIONES (S/ Ord. 108-10_CS) 1. Evaluaciones : Durante el cursado se toman cuatro evaluaciones parciales que tienen carácter práctico (ejercicios de aplicación), las que se aprueban con un 60 % del total. El alumno que, habiendo rendido las cuatro evaluaciones parciales, no apruebe ninguna de ellas, NO regulariza la materia. Se podrán recuperar un máximo de tres evaluaciones parciales, exigiéndose como mínimo la aprobación del 2º, 3º o 4º parcial para acceder a las instancias recuperatorias. Las notas de los recuperatorios serán de carácter sustitutivo. El 1º parcial y su respectiva recuperación se realizarán a través del aula virtual de la cátedra. El alumno que no apruebe la recuperación, deberá rendir la Recuperación Global, que se aprueba con un 60 % del total. Los alumnos que estén en condiciones de acceder a la promoción de la materia, deben rendir una evaluación integradora, para acreditar el espacio curricular como promoción sin examen final. La misma se aprobará alcanzando un porcentaje igual o superior al 70 %. Tanto los parciales como las recuperaciones y la evaluación integradora, son escritos. 2. Promoción: Para alcanzar la condición de promoción de la asignatura, el alumno deberá cumplir con los siguientes requisitos: - Asistencia obligatoria a clases teórico-prácticas, debiendo registrar el 75 % de asistencia a la fecha de cada parcial. - Asistencia obligatoria a prácticos de laboratorio, los cuales se aprobarán rindiendo un pre práctico individual. - Aprobación de las cuatro evaluaciones parciales. Para promocionar la materia, no se puede desaprobar ninguna de las evaluaciones parciales. - Aprobación de la evaluación integradora. La calificación definitiva de la materia promovida por el estudiante, se obtiene del siguiente promedio:
( )2
IntegradorNotaparcialespromedioNotafinalNota
+=
Cuando la fracción sea de 50 centésimos se coloca el numero entero inmediato superior 3. Regularidad: Para obtener la regularidad el alumno debe cumplir con las siguientes obligaciones: - Asistencia obligatoria a clases teórico-prácticas, debiendo registrar el 75 % de asistencia a la fecha de cada parcial. - Asistencia obligatoria a prácticos de laboratorio, los cuales se aprobarán rindiendo un pre práctico individual. - Aprobación de las cuatro evaluaciones parciales o de sus respectivas recuperaciones. Se podrán recuperar como máximo tres evaluaciones parciales bajo las condiciones consignadas en el inciso 1 (evaluaciones parciales). El alumno que no apruebe la recuperación, deberá rendir la Recuperación Global. 4. Examen final: Alumnos regulares: obtenida la regularidad, el alumno queda habilitado para rendir el examen final oral. Cuando el alumno ingresa a la mesa extrae dos bolillas de examen y dispone de quince minutos para hacer una revisión rápida. Los profesores integrantes de la mesa eligen los temas a evaluar de las bolillas sorteadas para dar orden al examen, pudiendo incluir durante el

Centro Universitario (M5502KFA), Ciudad, Mendoza. Casilla de Correos 405. República Argentina. Tel. +54-261-4494002. Fax. +54-261-4380120. Sitio web: http://fing.uncu.edu.ar
Página 8
2019
examen otros temas pertenecientes al programa. Dichos temas pueden ser de teoría o prácticas de laboratorio. Alumnos libres: el alumno en condición de libre debe rendir un examen escrito de carácter práctico (ejercicios de aplicación similares a los dictados en la práctica de la materia) el mismo día del examen final, previo al oral, debiendo aprobar el primero para pasar al segundo. El examen oral seguirá la misma metodología de los alumnos regulares. La nota final estará determinada por las dos instancias evaluativas. Ambos exámenes versarán sobre la totalidad del programa vigente de la asignatura.
Criterios de evaluación: Se tendrán en cuenta los siguientes aspectos:
- La apropiación de los contenidos de la asignatura. - La resolución correcta de situaciones problemáticas. - La coherencia en la expresión oral y escrita. - La precisión en el empleo del vocabulario específico de la disciplina. - La relación pertinente entre conceptos. - La consistencia en el tratamiento o análisis de los temas. - La organización lógica de los contenidos desarrollados. - La pertinencia y suficiencia en los argumentos que se aportan.
Programa de examen
Bolilla 1: Temas: 2B – 4B – 5A – 9D Bolilla 2: Temas: 2A – 4C – 3C – 9C Bolilla 3: Temas: 3A – 5B – 7B – 9B Bolilla 4: Temas: 2B – 5A – 8 – 9D Bolilla 5: Temas: 3B – 6A – 7B – 9A Bolilla 6: Temas: 3C – 4B – 6C – 9A Bolilla 7: Temas: 3B – 4C – 6B – 9C Bolilla 8: Temas: 3C – 5B – 7A – 9B Bolilla 9: Temas: 5A – 6C – 7B – 8
Mendoza, 31 de julio de 2019
Ing. María Silvina Videla
Titular Química Orgánica

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción 1
INTRODUCCIÓN
La química orgánica es la química de los compuestos del carbono. El nombre engañoso “orgánico” es una reliquia de los tiempos en que los compuestos químicos se dividían en dos clases: inorgánicos y orgánicos, según de dónde provenían. Los compuestos inorgánicos eran aquellos que provenían de los minerales y los orgánicos los que se obtenían de fuentes vegetales y animales, o sea: de materiales producidos por organismos vivos. Hace años, los químicos pasaban mucho tiempo haciendo extracciones, purificando y analizando las sustancias provenientes de animales y plantas. Se sentían motivados por su curiosidad natural con respecto a la materia viva, así como por el deseo de obtener, a partir de la naturaleza, ingredientes para las medicinas, colorantes y otros productos de utilidad. Los científicos pensaban que los compuestos que se encuentran en la materia viva eran diferentes de las otras sustancias y que contenían una fuerza vital intangible que les daba vida. Esta idea hizo que los químicos no trataran de obtener compuestos orgánicos en el laboratorio. Sin embargo, en 1828 el químico alemán Friedrich Wöhler, que tenía entonces 28 años, accidentalmente preparó urea (que, como es sabido, es un constituyente de la orina) al calentar cianato de amonio, una sustancia inorgánica (o mineral). Este experimento y otros parecidos desacreditaron poco a poco la teoría de la fuerza vital y abrieron el camino para la moderna química orgánica sintética. Los compuestos de fuentes orgánicas tenían esto en común: todos contenían el elemento carbono. Aún después de que quedó establecido que estos compuestos no necesariamente debían provenir de fuentes vivas, ya que podían hacerse en el laboratorio, resultó conveniente mantener el nombre orgánico para describirlos, así como también compuestos similares, reteniéndose hasta la fecha esta división entre compuestos inorgánicos y orgánicos. Aunque aún hoy muchos compuestos del carbono se aíslan más convenientemente de fuentes vegetales y animales, la mayoría de ellos se obtienen por síntesis (la síntesis consiste en ir uniendo moléculas pequeñas y relativamente sencillas, para formar otras más grandes y complejas). A veces, se sintetizan de sustancias inorgánicas, tales como carbonatos y cianuros, pero más a menudo se parte de otros compuestos orgánicos. Hay dos grandes fuentes de las que se pueden obtener sustancias orgánicas simples: el petróleo y el carbón (ambas son orgánicas en el sentido tradicional, puesto que son producto de la descomposición de plantas y animales). Estas sustancias simples se emplean como elementos constructivos, a partir de los cuales se pueden hacer compuestos más complicados. Algunos ejemplos de sustancias que inicialmente se aislaron de la naturaleza pero que actualmente se producen sintéticamente para su uso comercial son las vitaminas, los aminoácidos, el colorante índigo, el alcanfor, que es un repelente de polillas y el antibiótico penicilina. La mayoría de los fármacos que se utilizan en medicina son sintéticos (incluyendo la aspirina, el éter, la novocaína y los barbituratos). A pesar de que algunas veces el término sintético se ve con recelo, como si conllevara algo artificial o no natural, los productos naturales sintéticos antes mencionados son, de hecho, idénticos a los compuestos que se extraen de fuentes naturales. ¿Qué tienen en especial los compuestos del carbono que justifiquen su separación de los de todos los demás cientos y tantos elementos del Sistema Periódico? Parcialmente, al menos, la respuesta parece ser esta: hay muchísimos compuestos del carbono y sus moléculas pueden ser muy grandes y complejas. El número de compuestos que contienen carbono es varias veces mayor que el número de sustancias que no lo contienen. Se conocen moléculas orgánicas que contienen miles de átomos, cuyo arreglo puede ser muy complicado, aún en moléculas relativamente pequeñas. ¿Qué propiedad tan especial posee el carbono como para permitirle formar tantos compuestos? Los átomos de carbono pueden unirse entre sí hasta un grado que es imposible para átomos de cualquier otro elemento. Pueden formar cadenas de miles de átomos o anillos de todos los tamaños: estas cadenas y anillos pueden tener ramificaciones y uniones cruzadas- A los carbonos de estas cadenas y anillos se unen otros átomos: principalmente hidrógeno y además también flúor, cloro, bromo, yodo, oxígeno, nitrógeno, azufre, fósforo y muchos otros. Cada arreglo atómico diferente corresponde a un compuesto distinto y cada compuesto tiene su conjunto de características químicas y físicas. No es sorprendente que hoy se conozcan 16 millones de compuestos del carbono y que se conozcan miles más cada año. No es sorprendente que el estudio de su química sea un campo especializado. La química orgánica está presente en la vida diaria. Estamos formados y rodeados por compuestos orgánicos. Casi todas las reacciones de la materia viva involucran compuestos orgánicos: es imposible comprender la

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción 2
vida, al menos desde el punto de vista físico, sin saber algo sobre química orgánica. Los constituyentes principales de la materia viva –proteínas, carbohidratos, lípidos (grasas), ácidos nucleicos (DNA, RNA), las membranas celulares, enzimas, hormonas- son orgánicos. Otras sustancias orgánicas incluyen la gasolina, aceites, las llantas de los automóviles, la ropa que utilizamos, la madera de nuestros muebles, el papel de nuestros libros, las medicinas que tomamos, los recipientes de plástico, las películas para fotografía, los perfumes, las alfombras y las telas. Diariamente, se hace referencia al polietileno, a resinas epóxicas, Estirofoam, nicotina, grasas poliinsaturadas y colesterol: todos estos términos se refieren a sustancias orgánicas. En pocas palabras, la química orgánica es mucho más que una rama de la ciencia: es parte de nuestra cultura tecnológica.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 3
Antes de comenzar el desarrollo del tema es conveniente ver algunos conceptos:
1. Los compuestos orgánicos son aquellos en cuya composición interviene el carbono.
2. La composición de un compuesto está dada por la fórmula molecular.
3. La constitución de un compuesto, es decir, su fórmula estructural o estructura, da el orden en que los átomos presentes en la molécula están unidos entre sí.
Ejemplo:
CH3- CH2- CH3
C3H8
Fórmula molecular Fórmula estructural Fórmula estructural Fórmula estructural
(desarrollada) (condensada) (de esqueleto)
H C C C H
H H H
HHH
La fórmula estructural desarrollada indica todos los enlaces presentes en la molécula.
En la fórmula estructural condensada no se desarrollan los enlaces entre atomos de carbono y de hidrógeno.
En la fórmula de esqueleto, cada línea indica los átomos de carbono enlazados, dándose por sobreentendido que los átomos de carbono están unidos a todos los átomos de hdrógeno correspondientes.
4. El átomo de carbono puede formar hasta cuatro enclaces covalentes.
5. El átomo de carbono puede constituir cadenas, que pueden ser:
- Cadenas abiertas (acíclicas) o cerradas (cíclicas). - Cadenas normales (lineales) o ramificadas.
C C C C C
CC
C C C
C
Cadena abierta normal Cadena abierta ramificada Cadena cerrada
6. Los átomos de carbono pueden unirse entre sí, formando enlaces (uniones o ligaduras), que pueden ser:
Enlaces simples Enlaces dobles Enlaces triples
Clasificación y nomenclatura de los compuestos orgánicos Conceptos generales. Alcanos, alquenos y alquinos. Hidrocarburos alicíclicos. Hidrocarburos aromáticos. Haluros de alquilo. Alcoholes. Éteres. Aldehídos y cetonas. Ácidos carboxílicos. Aminas. Mpléculas con varios grupos funcionales.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 4
C C
Enlace simple Enlace doble Enlace triple
C C C C
7. El átomo de carbono de una cadena puede ser primario, secundario, terciario o cuaternario según el número de carbonos a los que se une.
Carbono primario
CH3CH3
Carbono cuaternario
CH3CCH3
CH3
CH3
Carbono secundario
CH3
CH3CH2CHCH3
Carbono terciario
8. Se define grupo funcional como un grupo de átomos o conjunto de átomos que le otorgan a la molécula propiedades y reactividad característica.
En una molécula orgánica se puede distinguir una parte hidrocarbonada (formada por los enlaces C-H y C-C) que puede influir en las características físicas y químicas pero que, finalmente, quedan fuertemente determinadas por la otra parte de la molécula que comprende los grupos funcionales.
Se indican a continuación los principales grupos funcionales.
Serie homóloga Grupo funcional
Alquenos C = C
Alquinos
– C ≡ C –
Haluros de alquilo - X
X (halógeno) unido a carbono alifático
Haluros de arilo – X
X (halógeno) unido a carbono aromático
Alcoholes – OH
OH unido a carbono alifático
Fenoles – OH
OH unido a carbono aromático

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 5
Éteres – O –
O unido a carbonos alifáticos y/o aromáticos
Aldehídos C
O
H
Cetonas C
O
Ácidos carboxílicos C
O
OH
Aminas
– NH2 (primaria)
– NH – (secundaria)
NH
H
H
(terciaria)
Ésteres C
O
O
Haluros de acilo C
O
X
Anhídridos de ácidos C C
O
O
O
Amidas C
O
NH2
1. Clasificación de compuestos orgánicos En esta unidad estudiaremos la nomenclatura de los siguientes compuestos orgánicos:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 6
2. Nomenclatura de compuestos orgánicos A medida que se fueron descubriendo los compuestos orgánicos, se les asignaron nombres comunes, basados frecuentemente en su fuente de origen.
Dada la cantidad creciente y la complejidad de los compuestos, fue necesario desarrollar un sistema de nomenclatura, que fue denominado Sistema Derivado, comprobándose que no era aplicable en forma simple a todas las series homólogas, por lo que se planteó la necesidad de sistematizar la nomenclatura, lo cual llevó a cabo la International Union of Pure and Applied Chemistry (IUPAC) a partir de 1957, continuando hasta la fecha la revisión y actualización de las reglas de nomenclatura.
2.1. Hidrocarburos Los hidrocarburos son aquellos compuestos constituidos por carbono e hidrógeno. Pueden ser alifáticos o aromáticos.
Los hidrocarburos alifáticos se clasifican en alcanos, alquenos, alquinos y sus análogos cíclicos, llamados aliciclícos (cicloalcanos, cicloalquenos y cicloalquinos).
Los hidrocarburos aromáticos pueden contener uno o más núcleos en sus estructuras, por lo que se los clasifica como mononucleares o polinucleares y, a la vez, estos últimos pueden presentar núcleos unidos directamente (no hay carbonos comunes para distintos ciclos) o núcleos condensados (hay átomos de carbono que son compartidos o que pertenecen a más de un ciclo).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 7
2.1.1. Alcanos Uniendo tres, cuatro, cinco o más átomos de carbono se genera un gran número de compuestos con una longitud de cadena creciente.
H
CC
CC
CC
CH
HH H H
H HH
H H
H H H
H H
heptano (C7H16)
Los compuestos que surgen de esta manera se conocen como: alcanos de cadena abierta o alcanos normales y tienen la fórmula general CnH2n+2 donde n es un número entero.
A veces se hace referencia a estos compuestos como “hidrocarburos saturados”: hidrocarburos porque están formados solamente por carbono e hidrógeno y saturados porque la molécula tiene tantos hidrógenos como puede tener.
Los primeros cuatro hidrocarburos tienen nombres comunes. Los hidrocarburos saturados lineales de mayor número de átomos de carbono tienen nombres sistemáticos compuestos por un prefijo indicativo del número de átomos de carbono y la terminación ano.
Fórmula Nombre Fórmula Nombre
CH4 metano C8H18 octano
C2H6 etano C9H20 nonano
C3H8 propano C10H22 decano

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 8
C4H10 butano C11H24 undecano
C5H12 pentano C12H26 dodecano
C6H14 hexano C15H32 pentadecano
C7H16 heptano C20H42 eicosano
Aprendiendo a escribir fórmulas
¿Cómo se puede escribir butano?
Mediante su fórmula molecular C4H10.
Mediante su fórmula desarrollada, indicando todos los enlaces presentes en la molécula.
C C C CH H
H H H H
H H H H
Mediante su fórmula condensada, donde los enlaces entre átomos de carbono y de hidrógeno no se desarrollan.
CH3CH2CH2CH3
Mediante su fórmula de esqueleto, donde cada línea indica los átomos de carbono enlazados, dándose por sobreentendido que los átomos de carbono están unidos a todos los átomos de hidrógeno correspondientes.
Esta representación parece la más difícil. Sin embargo, una vez que se aprende, es la más rápida y empleada, sobre todo en moléculas de mayor tamaño cuya formulación resultaría lenta y engorrosa.
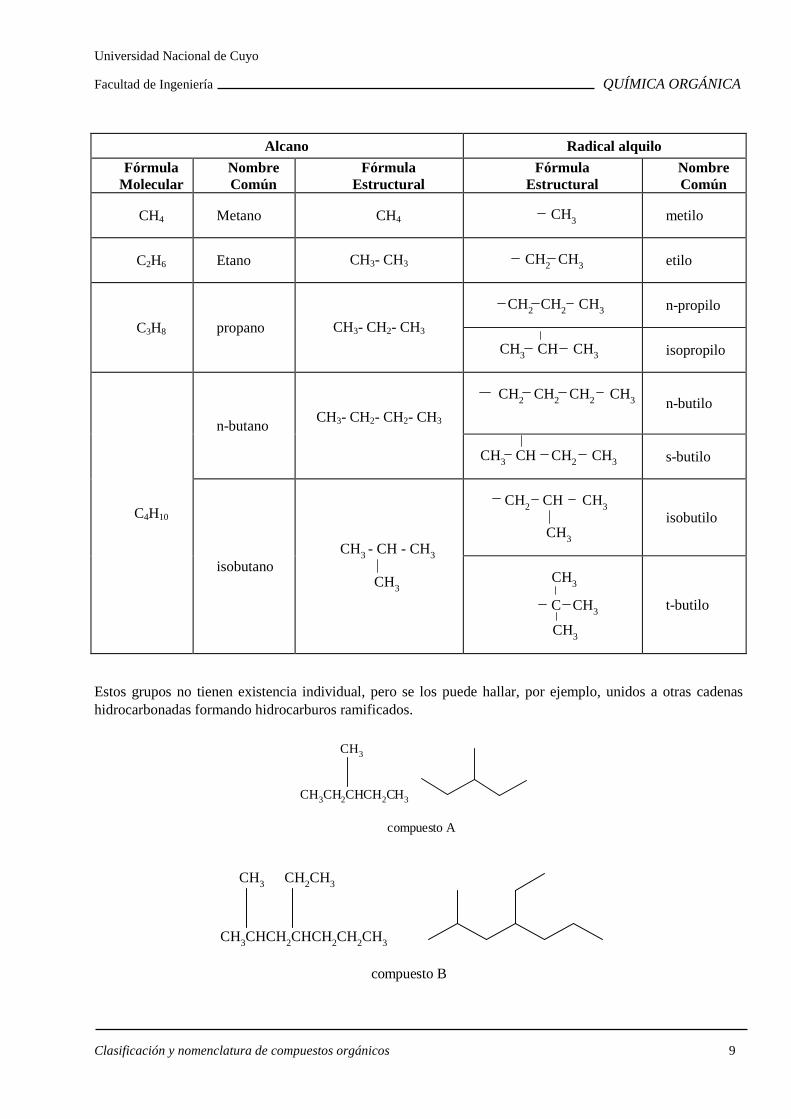
Los átomos de carbono son asombrosos por su habilidad para unirse entre sí generando una gran variedad de compuestos orgánicos. Eliminando un átomo de hidrógeno en un hidrocarburo saturado se obtiene un grupo alquilo, que se nombra cambiando la terminación ano del alcano de origen por ilo.
metilo: -CH3 etilo: -CH2CH3
A continuación se da un resumen con los alcanos y los grupos alquilo de hasta cuatro átomos de carbono.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 9
Alcano Radical alquilo
Fórmula Molecular
Nombre Común
Fórmula Estructural
Fórmula Estructural
Nombre Común
CH4 Metano CH4 CH3
metilo
C2H6 Etano CH3- CH3 CH2 CH3
etilo
C3H8 propano CH3- CH2- CH3
CH2 CH2 CH3
n-propilo
CH3 CH CH3
isopropilo
C4H10
n-butano CH3- CH2- CH2- CH3
CH2 CH2 CH2 CH3
n-butilo
CH3 CH CH2 CH3
s-butilo
isobutano CH3
- CH - CH3
CH3
CH2 CH CH3
CH3
isobutilo
C CH3
CH3
CH3
t-butilo
Estos grupos no tienen existencia individual, pero se los puede hallar, por ejemplo, unidos a otras cadenas hidrocarbonadas formando hidrocarburos ramificados.
CH3CH2CHCH2CH3
CH3
compuesto A
CH3CHCH2CHCH2CH2CH3
CH3 CH2CH3
compuesto B

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 10
¿Cómo se nombran estos compuestos?
Para llegar al nombre final se siguen sistemáticamente los siguientes pasos.
1 Nombre base
Se elige la cadena de átomos de carbono más larga, cuyo número dará origen al nombre base del compuesto.
De acuerdo a ello el compuesto A es un derivado del pentano y el compuesto B del heptano.
2 Nombre de los sustituyentes.
2a. Se ubican los sustituyentes de la cadena principal que, en los ejemplos anteriores, son un metilo en el compuesto A y un metilo y un etilo en el compuesto B.
2b. Se numera la cadena principal a fin de ubicar los átomos de carbono donde se unen los sustituyentes. Siguiendo con los ejemplos anteriores, el compuesto A se ramifica en el C-3, pero para el compuesto B existen dos posibilidades. Comenzando la numeración por un extremo se ramifica en los C-2 y 4, pero comenzando por el otro, se ramifica en los C-4 y 6.
CH3CH2CHCH2CH3
CH3
1 2 3 4 5 CH3CHCH2CHCH3CH2CH3
CH3 CH2CH3
1 2 3 4 5 6 7 CH3CHCH2CHCH3CH2CH3
CH3 CH2CH3
7 6 5 4 3 2 1
Por convención, se elige la numeración que asigna al primer carbono sustituido el menor número posible. O sea, la primera.
Si comenzando por ambos extremos al primer carbono sustituido le corresponde el mismo número, se busca la diferencia en el segundo carbono sustituido comenzando la numeración por aquel extremo que asigne a este último el número menor. Si la diferencia no estuviese en el segundo, se sigue con el tercero y así sucesivamente.
CH3CHCH2CHCH2CH2CHCH3
CH3 CH3 CH3
1 2 3 4 5 6 7 8
123
4567
8
9101112
2c. Los sustituyentes de la cadena principal se nombran precedidos por un número que indica el carbono al que está unido. Cuando se nombra un grupo alquilo se quita del nombre la o final. Los nombres y números se separan con guiones: 3-metil; 4-etil; etc.
3 Composición del nombre
Se nombran primero los sustituyentes, por orden alfabético y ligándose el último de ellos al nombre base.
Compuesto A: 3-metilpentano Compuesto B: 4-etil-2-metilheptano
Consideraciones generales
Cuando un sustituyente se repite más de una vez:
i) Se agrega un prefijo multiplicativo que indica cuantas veces se repite (di, tri, tetra, etc.).
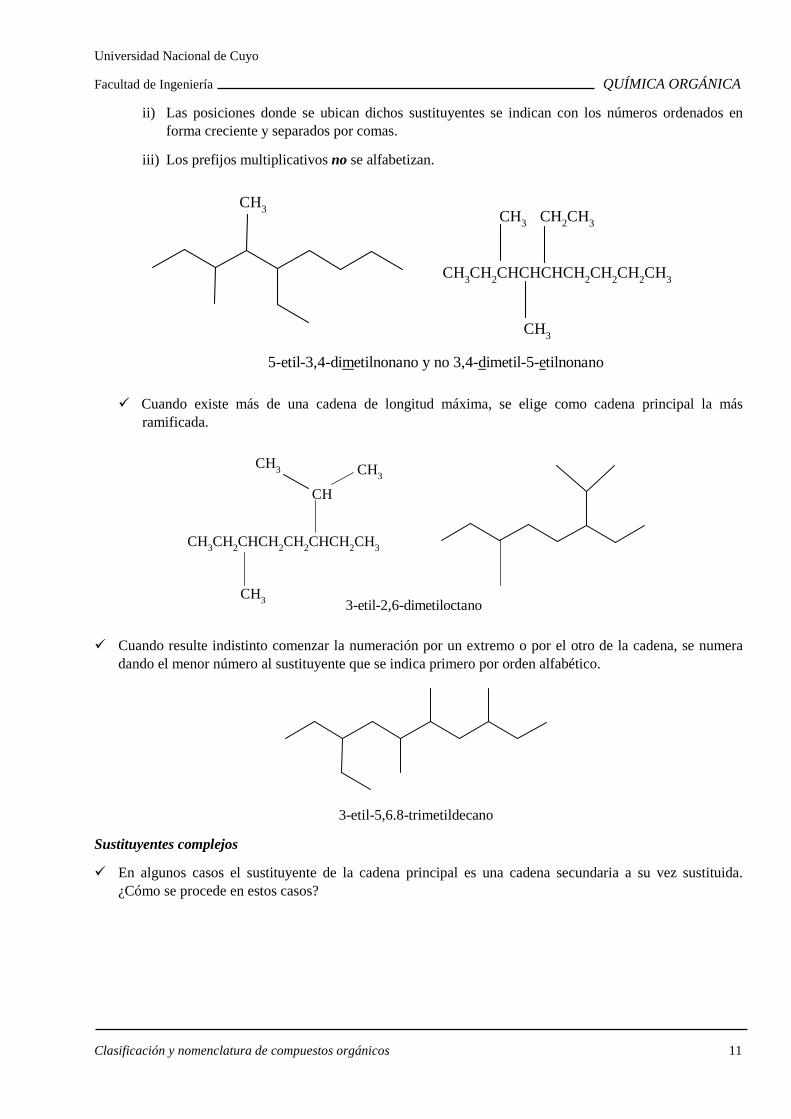
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 11
ii) Las posiciones donde se ubican dichos sustituyentes se indican con los números ordenados en forma creciente y separados por comas.
iii) Los prefijos multiplicativos no se alfabetizan.
CH3CH2CHCHCHCH2CH2CH2CH3
CH3
CH3CH2CH3CH3
5-etil-3,4-dimetilnonano y no 3,4-dimetil-5-etilnonano
Cuando existe más de una cadena de longitud máxima, se elige como cadena principal la más ramificada.
CH3CH2CHCH2CH2CHCH2CH3
CH3
CH
CH3CH3
3-etil-2,6-dimetiloctano
Cuando resulte indistinto comenzar la numeración por un extremo o por el otro de la cadena, se numera dando el menor número al sustituyente que se indica primero por orden alfabético.
3-etil-5,6.8-trimetildecano
Sustituyentes complejos
En algunos casos el sustituyente de la cadena principal es una cadena secundaria a su vez sustituida. ¿Cómo se procede en estos casos?

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 12
compuesto C
CH3CH2CH2CH2CHCHCH2CH2CH2CH2CH3
CH3
CH2CCH2CH3
CH3CH3
Para numerar la cadena secundaria siempre se considera C-1 el que está directamente unido a la cadena principal.
Comenzando por este carbono se elige la cadena más larga que servirá de base al nombre de la cadena secundaria. En este caso es de cuatro átomos de carbono.
Para nombrar los sustituyentes de la cadena secundaria se siguen las mismas reglas que para los de la cadena principal.
El nombre de la cadena secundaria tiene la terminación de un alquilo sustituyente: il .
Se encierra el nombre entre paréntesis y se precede por el número que indica el carbono de la cadena principal al que está unido, separando el número del paréntesis con un guión.
Se alfabetiza con la primera letra que queda dentro del paréntesis.
Compuesto C: 6-(2,2-dimetilbutil)-5-metilundecano.
Nombres comunes
Las reglas IUPAC permiten el empleo de algunos nombres de alcanos o grupos alquilo de uso común: Es conveniente tenerlos presentes.
Se llama iso al siguiente arreglo de átomos de carbono:
CH3 CH
CH3
En los siguientes alcanos o grupos alquilo se ejemplifica su aplicación en el nombre.
CH3 CH CH3 =
CH3
isobutano
CH3
isoheptano
CH3CH2CH2CH2CHCH3 =

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 13
CH3 CH
CH3
isopropilo
Los prefijos sec y ter no se alfabetizan.
2.2. Alquenos Los alquenos también son hidrocarburos, ya que sólo están formados por átomos de carbono e hidrógeno, pero se diferencian de los alcanos por la presencia de dobles enlaces.
Debido a que estos compuestos son capaces de tomar H2 para transformarse en hidrocarburos saturados (alcanos) se los conoce con el nombre general de hidrocarburos insaturados.
Para nombrarlos se siguen las siguientes reglas:
Se determina la cadena de átomos de carbono más larga que contiene el doble enlace y que dará origen al nombre base, cambiando la terminación ano por eno.
Se numera esa cadena de forma de asignar al doble enlace el menor número posible, independientemente de los grupos alquilo ramificantes.
La posición a la que se une el grupo funcional se indica con un número adelante del nombre y separado del mismo con un guión. El número que indica la posición del grupo funcional es el que corresponde al primero de los dos carbonos de la doble ligadura.
CH3CH C CH2CH2CH3
CH2CH2CH3
3-propil-2-hexeno
Observe que la cadena más larga es de siete átomos de carbono, pero no contiene el grupo principal. Por eso, el compuesto se nombra como un derivado del hexeno. Si la molécula presenta más de una doble ligadura, se indica el prefijo multiplicativo inmediatamente delante de la terminación “-eno” (observe que se deja la a de la terminación ano del alcano para evitar que se junten consonantes).
2,6-nonadieno
CH2 C CH CHCH2CH CH2
CH2CH3
2-etil-1,3,6-heptatrieno
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 14
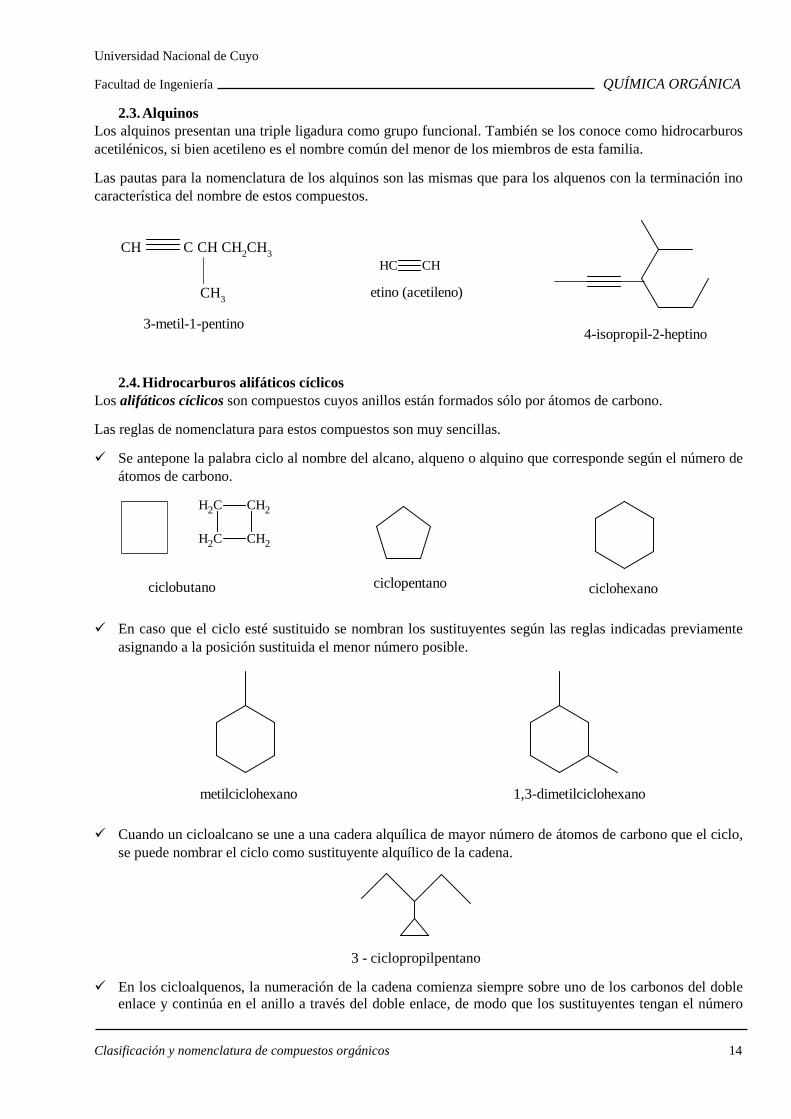
2.3. Alquinos Los alquinos presentan una triple ligadura como grupo funcional. También se los conoce como hidrocarburos acetilénicos, si bien acetileno es el nombre común del menor de los miembros de esta familia.
Las pautas para la nomenclatura de los alquinos son las mismas que para los alquenos con la terminación ino característica del nombre de estos compuestos.
CH C CH CH2CH3
CH3
3-metil-1-pentino
CH CH
etino (acetileno)
4-isopropil-2-heptino
2.4. Hidrocarburos alifáticos cíclicos Los alifáticos cíclicos son compuestos cuyos anillos están formados sólo por átomos de carbono.
Las reglas de nomenclatura para estos compuestos son muy sencillas.
Se antepone la palabra ciclo al nombre del alcano, alqueno o alquino que corresponde según el número de átomos de carbono.
CH2
CH2
CH2
CH2
ciclobutano
ciclopentano
ciclohexano
En caso que el ciclo esté sustituido se nombran los sustituyentes según las reglas indicadas previamente asignando a la posición sustituida el menor número posible.
metilciclohexano
1,3-dimetilciclohexano
Cuando un cicloalcano se une a una cadera alquílica de mayor número de átomos de carbono que el ciclo, se puede nombrar el ciclo como sustituyente alquílico de la cadena.
3 - ciclopropilpentano
En los cicloalquenos, la numeración de la cadena comienza siempre sobre uno de los carbonos del doble enlace y continúa en el anillo a través del doble enlace, de modo que los sustituyentes tengan el número
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 15
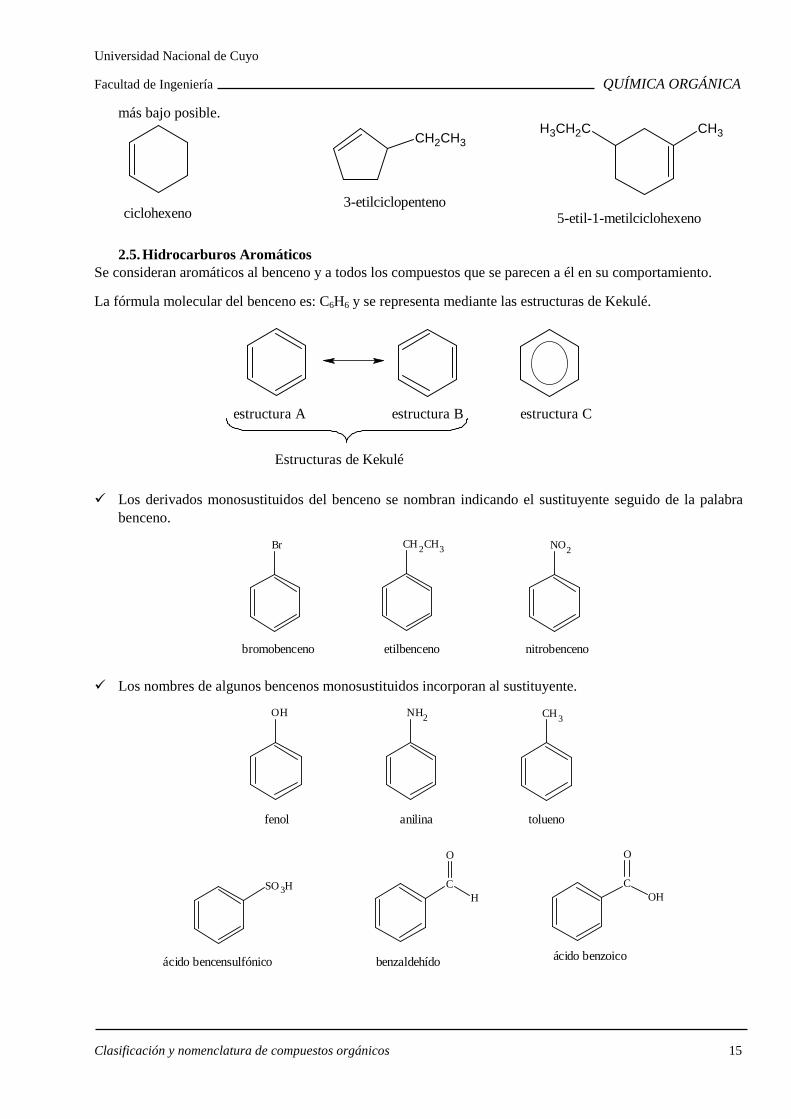
más bajo posible.
ciclohexeno
3-etilciclopenteno
CH2CH3
5-etil-1-metilciclohexeno
CH3H3CH2C
2.5. Hidrocarburos Aromáticos Se consideran aromáticos al benceno y a todos los compuestos que se parecen a él en su comportamiento.
La fórmula molecular del benceno es: C6H6 y se representa mediante las estructuras de Kekulé.
estructura A estructura B
Estructuras de Kekulé
estructura C
Los derivados monosustituidos del benceno se nombran indicando el sustituyente seguido de la palabra benceno.
Br
bromobenceno
CH2CH3
etilbenceno
NO2
nitrobenceno
Los nombres de algunos bencenos monosustituidos incorporan al sustituyente.
OH
fenol
NH2
anilina
CH3
tolueno
ácido bencensulfónico
SO3H CH
O
benzaldehído
COH
O
ácido benzoico
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 16
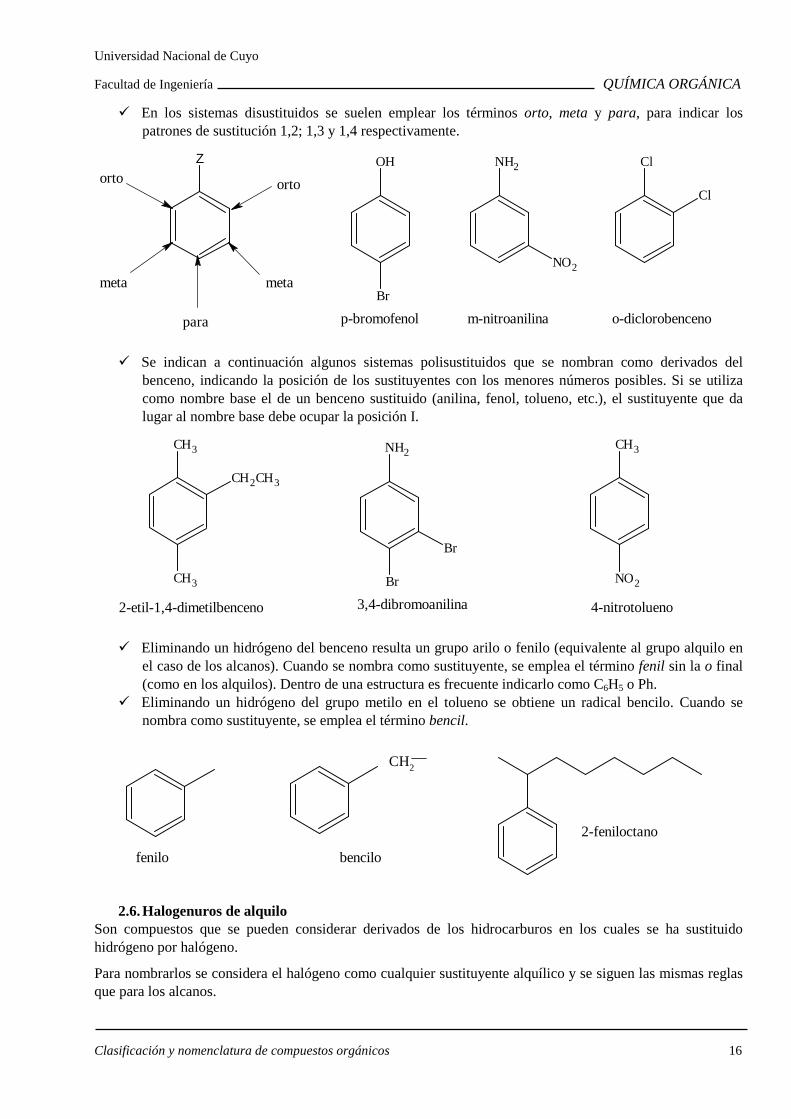
En los sistemas disustituidos se suelen emplear los términos orto, meta y para, para indicar los patrones de sustitución 1,2; 1,3 y 1,4 respectivamente.
Z
ortoorto
meta
para
meta
OH
Br
p-bromofenol
NH2
NO2
m-nitroanilina
o-diclorobenceno
Cl
Cl
Se indican a continuación algunos sistemas polisustituidos que se nombran como derivados del benceno, indicando la posición de los sustituyentes con los menores números posibles. Si se utiliza como nombre base el de un benceno sustituido (anilina, fenol, tolueno, etc.), el sustituyente que da lugar al nombre base debe ocupar la posición I.
CH3
CH3
CH2CH3
2-etil-1,4-dimetilbenceno
NH2
Br
Br
3,4-dibromoanilina
4-nitrotolueno
CH3
NO2
Eliminando un hidrógeno del benceno resulta un grupo arilo o fenilo (equivalente al grupo alquilo en el caso de los alcanos). Cuando se nombra como sustituyente, se emplea el término fenil sin la o final (como en los alquilos). Dentro de una estructura es frecuente indicarlo como C6H5 o Ph.
Eliminando un hidrógeno del grupo metilo en el tolueno se obtiene un radical bencilo. Cuando se nombra como sustituyente, se emplea el término bencil.
fenilo bencilo
2-feniloctano
CH2
2.6. Halogenuros de alquilo Son compuestos que se pueden considerar derivados de los hidrocarburos en los cuales se ha sustituido hidrógeno por halógeno.
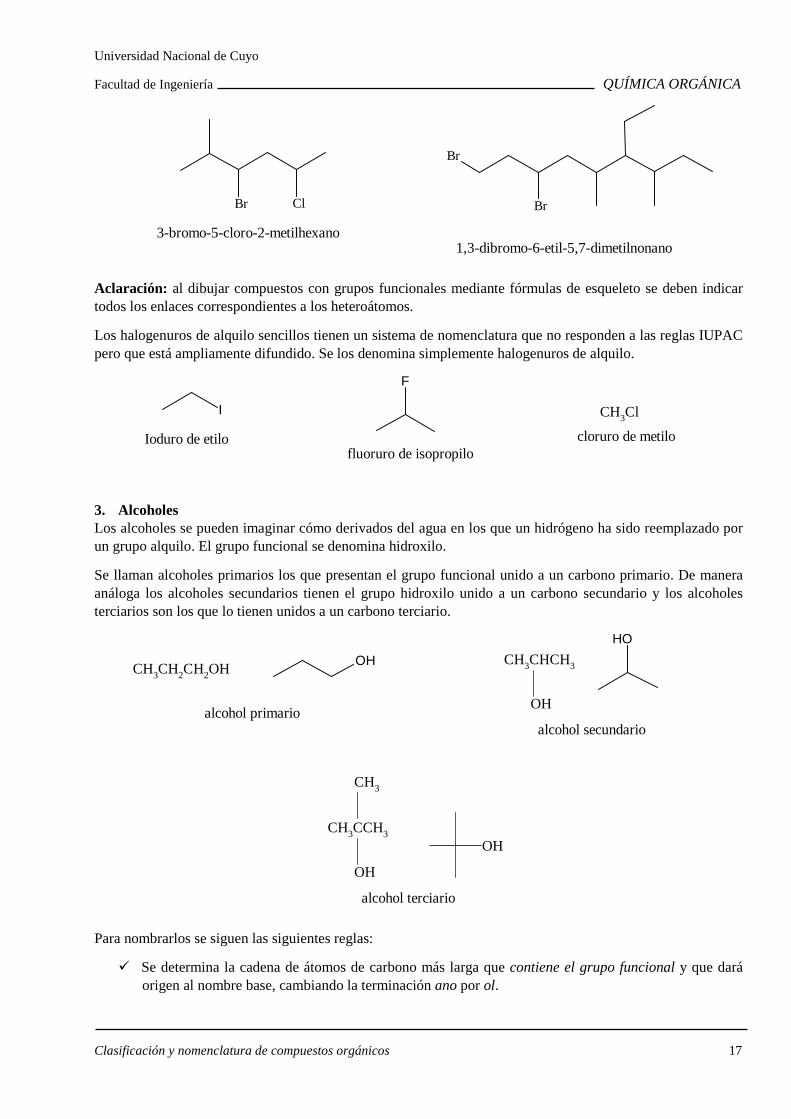
Para nombrarlos se considera el halógeno como cualquier sustituyente alquílico y se siguen las mismas reglas que para los alcanos.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 17
3-bromo-5-cloro-2-metilhexano
Br Cl
1,3-dibromo-6-etil-5,7-dimetilnonano
Br
Br
Aclaración: al dibujar compuestos con grupos funcionales mediante fórmulas de esqueleto se deben indicar todos los enlaces correspondientes a los heteroátomos.
Los halogenuros de alquilo sencillos tienen un sistema de nomenclatura que no responden a las reglas IUPAC pero que está ampliamente difundido. Se los denomina simplemente halogenuros de alquilo.
I
Ioduro de etilo
F
fluoruro de isopropilo
CH3Cl
cloruro de metilo
3. Alcoholes Los alcoholes se pueden imaginar cómo derivados del agua en los que un hidrógeno ha sido reemplazado por un grupo alquilo. El grupo funcional se denomina hidroxilo.
Se llaman alcoholes primarios los que presentan el grupo funcional unido a un carbono primario. De manera análoga los alcoholes secundarios tienen el grupo hidroxilo unido a un carbono secundario y los alcoholes terciarios son los que lo tienen unidos a un carbono terciario.
CH3CH2CH2OHOH
alcohol primario alcohol secundario
OH
CH3CHCH3
OH
alcohol terciario
CH3CCH3
OH
CH3
OH
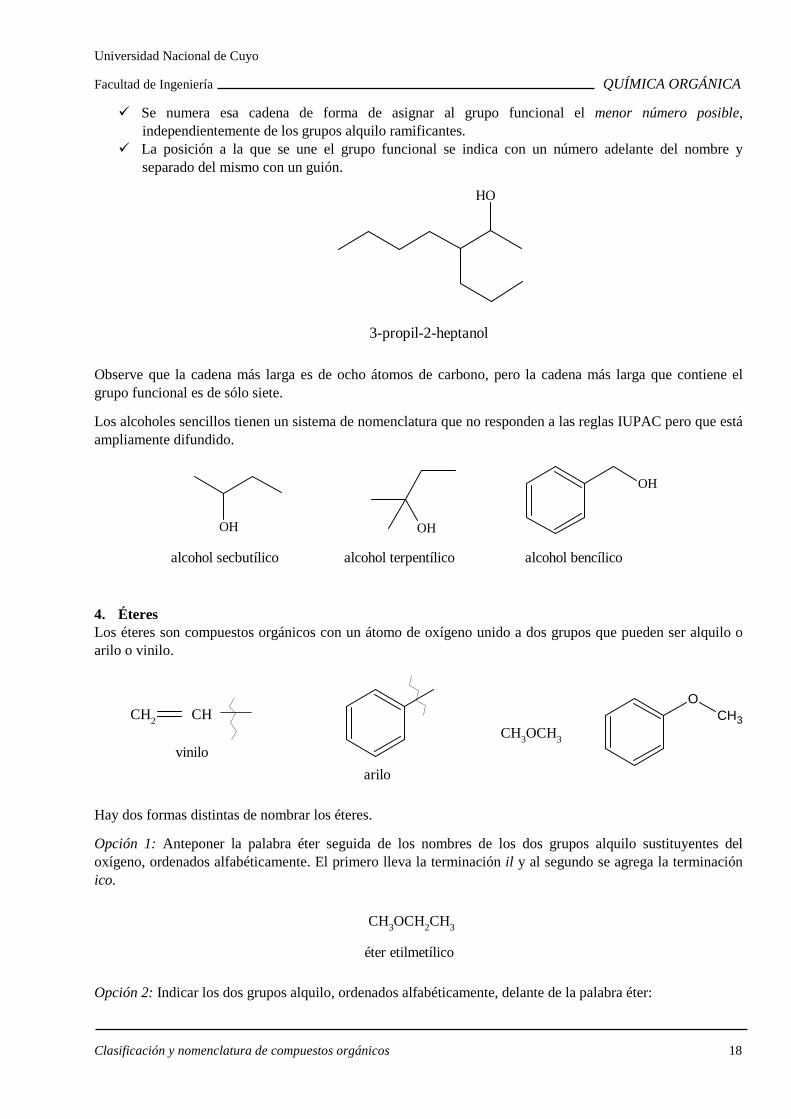
Para nombrarlos se siguen las siguientes reglas:
Se determina la cadena de átomos de carbono más larga que contiene el grupo funcional y que dará origen al nombre base, cambiando la terminación ano por ol.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 18
Se numera esa cadena de forma de asignar al grupo funcional el menor número posible, independientemente de los grupos alquilo ramificantes.
La posición a la que se une el grupo funcional se indica con un número adelante del nombre y separado del mismo con un guión.
OH
3-propil-2-heptanol
Observe que la cadena más larga es de ocho átomos de carbono, pero la cadena más larga que contiene el grupo funcional es de sólo siete.
Los alcoholes sencillos tienen un sistema de nomenclatura que no responden a las reglas IUPAC pero que está ampliamente difundido.
alcohol secbutílico alcohol terpentílico alcohol bencílico
OH OH
OH
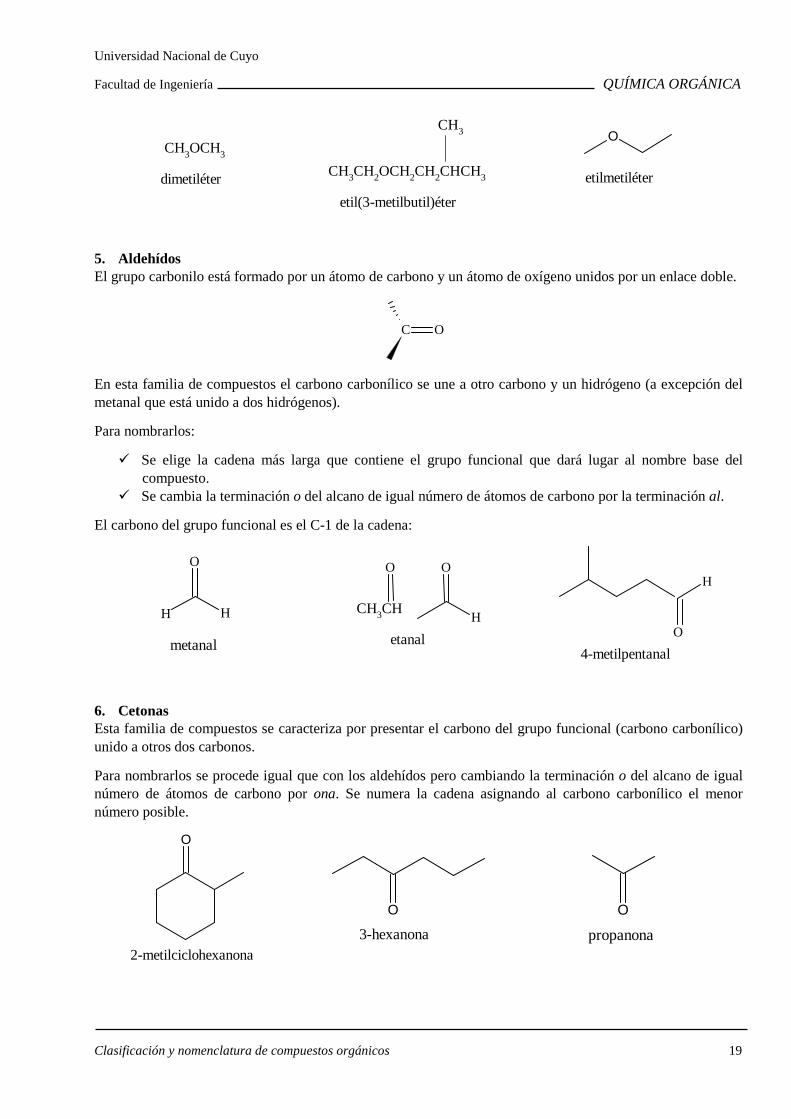
4. Éteres Los éteres son compuestos orgánicos con un átomo de oxígeno unido a dos grupos que pueden ser alquilo o arilo o vinilo.
vinilo
CH2 CH
arilo
OCH3
CH3OCH3
Hay dos formas distintas de nombrar los éteres.
Opción 1: Anteponer la palabra éter seguida de los nombres de los dos grupos alquilo sustituyentes del oxígeno, ordenados alfabéticamente. El primero lleva la terminación il y al segundo se agrega la terminación ico.
CH3OCH2CH3
éter etilmetílico
Opción 2: Indicar los dos grupos alquilo, ordenados alfabéticamente, delante de la palabra éter:
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 19
CH3OCH3
dimetiléter
CH3CH2OCH2CH2CHCH3
CH3
etil(3-metilbutil)éter
O
etilmetiléter
5. Aldehídos El grupo carbonilo está formado por un átomo de carbono y un átomo de oxígeno unidos por un enlace doble.
C O
En esta familia de compuestos el carbono carbonílico se une a otro carbono y un hidrógeno (a excepción del metanal que está unido a dos hidrógenos).
Para nombrarlos:
Se elige la cadena más larga que contiene el grupo funcional que dará lugar al nombre base del compuesto.
Se cambia la terminación o del alcano de igual número de átomos de carbono por la terminación al.
El carbono del grupo funcional es el C-1 de la cadena:
O
H H
metanal
O
H
etanal
CH3CH
O
O
H
4-metilpentanal
6. Cetonas Esta familia de compuestos se caracteriza por presentar el carbono del grupo funcional (carbono carbonílico) unido a otros dos carbonos.
Para nombrarlos se procede igual que con los aldehídos pero cambiando la terminación o del alcano de igual número de átomos de carbono por ona. Se numera la cadena asignando al carbono carbonílico el menor número posible.
O
2-metilciclohexanona
3-hexanona
O
propanona
O

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 20
7. Ácidos carboxílicos El grupo funcional que caracteriza a los ácidos carboxílicos surge de unir un grupo hidroxilo a un grupo carbonilo. Para nombrarlo se cambia la terminación o, del alcano que dará lugar al nombre base, por oico. Además se antepone la palabra ácido. El carbono del carboxilo es el C-1 de la cadena.
Ácido propanoico
CH3CH2C
O
OH
Algunos aldehídos, cetonas y ácidos carboxílicos tienen nombres comunes.
NOMBRE SISTEMÁTICO NOMBRE COMUN
HCHO Metanal Formaldehído
CH3CHO Etanal Acetaldehído
CH3COCH3 Propanona Acetona
HCOOH Acido metanoico Acido fórmico
CH3COOH Acido etanoico Acido acético
CH3CH2COOH Acido propanoico Acido propiónico
8. Aminas Las aminas son compuestos orgánicos relacionados con el amoníaco, que se forman al reemplazar uno, dos o los tres hidrógenos del amoníaco por grupos orgánicos (grupos alquilo, arilo o vinilo). El grupo funcional se denomina amino.
Dependiendo de la cantidad de grupos orgánicos unidos al nitrógeno se pueden clasificar como aminas primarias, secundarias o terciarias.
CH3CH2NH2
amina primaria
CH3CH2CH2NHCH3
amina secundaria
N
H
amina terciaria
(CH3)3N
Las aminas pueden nombrarse de diferentes formas y no hay una clara preferencia de alguna de ellas por sobre las demás.
El sistema común para las aminas simples nombra cada sustituyente alquilo en el nitrógeno en orden alfabético, seguido por el sufijo -amina. Muchas aminas aromáticas y heterocíclicas son conocidas por los nombres comunes singulares, cuyos orígenes son a menudo desconocidos para los químicos que los utilizan con frecuencia. Puesto que estos nombres no se basan en un sistema racional, es necesario memorizarlos.
Según el sistema IUPAC, el grupo amino, -NH2, se nombra como un sustituyente en el mayor grupo alquilo, en aminas primarias. En este sistema, las aminas secundarias y terciarias se nombran utilizando prefijos que incluyen a todos los carbonos, menos a los de la cadena más larga.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 21
Recientemente el Chemical Abstract Service (CA) introdujo un sistema por el cual las aminas se nombran como alcanaminas. En aminas 2º y 3º, los sustituyentes adicionales del nitrógeno son designados por el prefijo N- antes del grupo alquilo más largo, que se utiliza como la raíz del nombre.
A continuación se muestran los diferentes nombres de las cuatro aminas isómeras de fórmula molecular C4H11N:
Nombre IUPAC 1-aminobutano 2-amino-2-metilpropano 1-metilaminopropano dimetilaminoetano
Nombre CA butanamina 2-metil-2-propanamina N-metilpropanamina N,N-dimetiletanamina
Nombre común n-butilamina terbutilamina metilpropilamina etildimetilamina
NH2
NH2
NH N
NH2
N
NH
anilina piridina pirrol
9. Moléculas con varios grupos funcionales Dentro del área de la química orgánica, es frecuente encontrar moléculas que presentan varios grupos funcionales.
¿Cómo se procede en estos casos? En general, los grupos funcionales imponen al nombre un sufijo característico (ol para los alcoholes, oico para los ácidos, eno para los alquenos, etc). Pero dado que el nombre sólo puede tener un sufijo es necesario elegir, dentro del conjunto, aquél grupo funcional considerado principal.
Para regular esta selección se estableció, por convención, un orden de prioridades que se indica en la siguiente tabla en forma decreciente. Los grupos funcionales de menor prioridad se nombran, salvo excepciones, como prefijos del nombre base del compuesto.
GRUPO FUNCIONAL PREFIJO SUFIJO
acido carboxilico carboxi- -oico (ácido)
acido sulfónico sulfa- -sulfónico (ácido)
Ester alcoxicarbonil- -oato
Amida carbamoil- -amida
Nitrilo ciano- -nitrilo
Aldehído formal- -al
Cetona oxo- -ona
Alcohol hidroxi- -ol
Fenol
Amina amino- -amina

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 22
Alqueno en- -eno
Alquino in- -ino
Halógeno halógeno-
Fenilo fenil-
Alquilo alquil-
Los halógenos, fenilos y alquilos sólo se nombran como prefijos según las reglas indicadas previamente.
Para nombrar los compuestos en forma sistemática se siguen los pasos que se detallan a continuación:
Se elige el grupo prioritario Se elige la cadena más larga que contiene al grupo prioritario. Se numera esta cadena asignando a dicho grupo el menor número posible. Se compone el nombre base con el prefijo indicativo del número de átomos de carbono de la cadena y
el sufijo indicativo del grupo prioritario. Los sustituyentes se anteponen al nombre base.
ácido 4-hidroxi-3-propilheptanoico
OH
OH
O
3-amino-4-etilciclohexanona
NH2
O
5-hidroxi-3-metil-2-pentanona
OH
O
En el caso particular de los alquenos y alquinos no existe un prefijo indicativo del grupo funcional. Cuando están presentes en la molécula como grupos no prioritarios se nombran siguiendo las pautas indicadas a continuación:
Se elige el grupo prioritario. Se elige la cadena más larga que contiene al grupo prioritario. Se numera esta cadena asignando a dicho grupo el menor número posible. Se compone el nombre base con el prefijo indicativo del número de átomos de carbono de la cadena
seguido de la sigla en o in (según contenga un grupo alqueno o alquino) y finalmente del sufijo indicativo del grupo prioritario.
Se antepone al nombre base el número que indica la posición del enlace doble o triple.
3-pentenal
H
O
O
Br
4-bromo-2-ciclohexenona
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Clasificación y nomenclatura de compuestos orgánicos 23
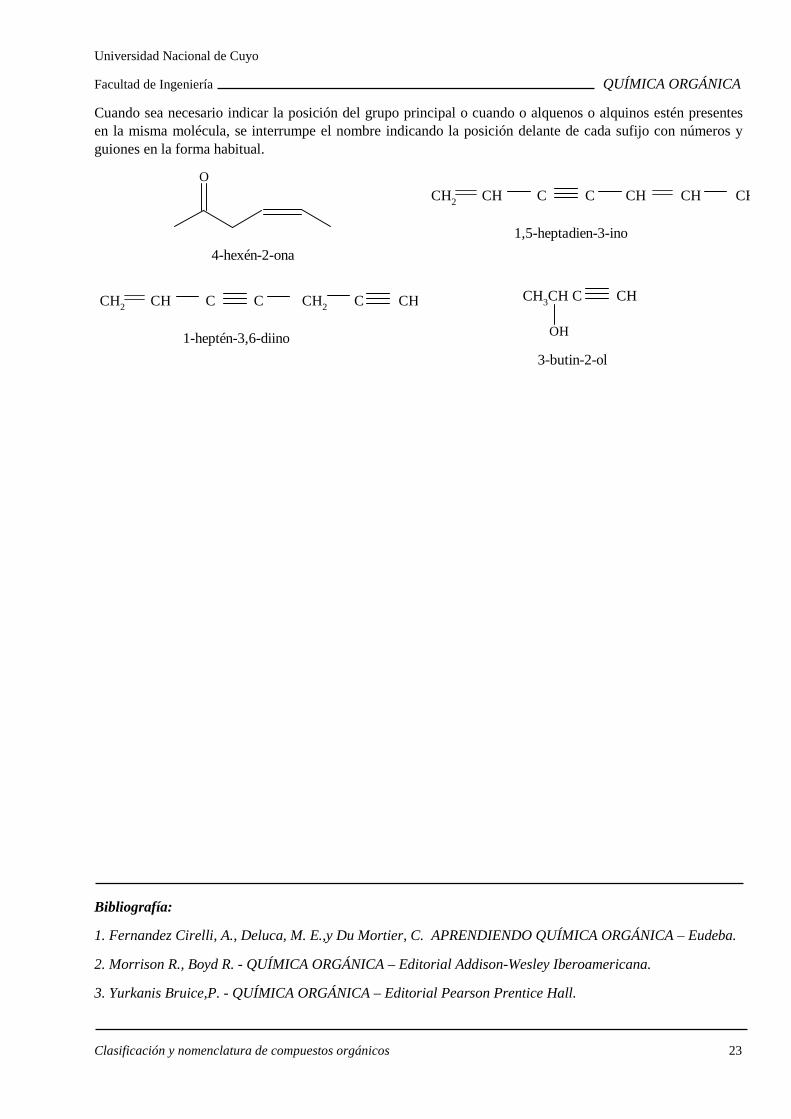
Cuando sea necesario indicar la posición del grupo principal o cuando o alquenos o alquinos estén presentes en la misma molécula, se interrumpe el nombre indicando la posición delante de cada sufijo con números y guiones en la forma habitual.
4-hexén-2-ona
O
1,5-heptadien-3-ino
CH2 CH C C CH CH CH
1-heptén-3,6-diino
CH2 CH C C CH2 C CH
3-butin-2-ol
CH3CH C CH
OH
Bibliografía:
1. Fernandez Cirelli, A., Deluca, M. E.,y Du Mortier, C. APRENDIENDO QUÍMICA ORGÁNICA – Eudeba.
2. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
3. Yurkanis Bruice,P. - QUÍMICA ORGÁNICA – Editorial Pearson Prentice Hall.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 24
La mayoría de las veces, para comprender el comportamiento de un compuesto orgánico no basta con conocer la composición del mismo, expresada a través de la fórmula molecular, sino que es necesario saber cómo están unidos los átomos en la molécula, es decir, cómo es la estructura. También la forma o configuración de la molécula es una herramienta útil cuando se estudian las propiedades de los compuestos orgánicos.
Ejemplo:
Compuesto: Metano
Fórmula Molecular: CH4
Fórmula Estructural: C
H
H H
H
Configuración: Tetraédrica regular
HC
H
HH
1. Teoría estructural clásica. La fórmula estructural y la configuración no son representaciones sin sentido, sino que si se las analiza sobre la base de la Teoría Estructural se puede arribar a información acerca de las propiedades físicas y químicas de los compuestos. Dicha Teoría explica cuál es el orden de unión de los átomos en las moléculas, qué electrones los mantienen unidos y cuáles son las formas y tamaños de las moléculas.
En toda fórmula estructural hay enlaces químicos entre los átomos, que son las fuerzas que los mantienen unidos. La Teoría Estructural Clásica (antes de 1926) menciona dos tipos de enlaces:
Enlace iónico, donde hay transferencia de electrones Enlace covalente, donde se comparten los electrones.
Tanto en el enlace iónico como en el covalente, cada átomo tiende a tomar la configuración electrónica del gas noble más próximo. A continuación se dan dos ejemplos a fin de interpretar ambos tipos de enlaces:
Estructura de los compuestos orgánicos
Teoría estructural clásica. Fórmulas estructurales de las moléculas orgánicas. Teoría de repulsión del par electrónico en la capa de valencia. Teoría del enlace de valencia. Ángulos de enlaces. Energías y longitudes de enlaces. Teoría de la resonancia. Formas contribuyentes. Energía de resonancia. Aromaticidad. Estructura del benceno. Criterios de aromaticidad.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 25
En los átomos que forman enlaces se puede observar lo siguiente:
El átomo de litio, al perder un electrón, queda con dos electrones, tomando la configuración del helio, mientras que el flúor, al ganar un electrón, queda con ocho electrones en su capa de valencia, tomando la configuración del neón. En el caso del hidrógeno, que tiene un electrón, al formar enlace presenta dos electrones como el helio.
Así, en la mayoría de los casos, para tomar la configuración del gas noble más próximo, se perderán, o ganarán o compartirán tantos electrones hasta quedar con dos (hidrógeno, litio) u ocho electrones en la capa de valencia. En este último caso, que es el más común, se dice que el átomo completa el octeto.
Por otro lado, es importante indicar que el enlace covalente puede ser, además de simple como en la molécula de hidrógeno, doble o triple. Por otro lado, la covalencia dativa es otro tipo de enlace covalente, que se presenta cuando uno de los átomos es el que aporta el par de electrones y el otro es el receptor.
Ejemplos:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 26
H C C H
H H
HH
enlace covalente simple Se suele escribir:
H C C H
H H
H H
H C C H
HH
enlace covalente doble Se suele escribir:
H C C H
H H
enlace covalente simple
H C C H
enlace covalente triple Se suele escribir:
H C C H
enlace covalente simple
A continuación se da un ejemplo donde hay covalencia dativa en la molécula de un compuesto inorgánico (ácido sulfúrico):
H O S O
O
O
enlace covalente simple H
enlace covalente dativo
electrones sin compartir
2. Estructura molecular y teorías del enlace covalente En 1926 se conoció la Mecánica Cuántica. El científico Erwin Schrödinger desarrolló expresiones matemáticas (ecuaciones de onda) para describir el movimiento de un electrón en función de su energía. Una ecuación de onda no permite dibujar una órbita alrededor del núcleo, pero da la probabilidad de encontrar el electrón en cualquier lugar particular.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 27
Orbital es la región del espacio donde es probable encontrar el electrón. El tipo de orbital que ocupa un electrón depende de su energía y se dice que es una nube que se forma como consecuencia de la difusión del electrón, cuya forma es la forma del orbital.
2.1. Orbitales atómicos Los siguientes son los orbitales atómicos que constituyen los átomos de los elementos más comunes en los compuestos orgánicos:
orbital 1s orbital 2s orbital px orbital py orbital pz
Es un orbital adireccional, cuyo centro coincide con el núcleo del átomo
Es un orbital adireccional, cuyo centro coincide con el núcleo del átomo.
Tiene mayor energía que el 1s
por la mayor distancia al núcleo, que es la causa de la menor atracción
electrostática.
Los orbitales p son de mayor energía que el 2s. Cada uno está constituido por dos lóbulos, entre los que se encuentra el núcleo.
Cada uno está orientado según un eje (x, y, z). Cada eje es perpendicular a los otros dos. Los tres orbitales p son de igual
energía.
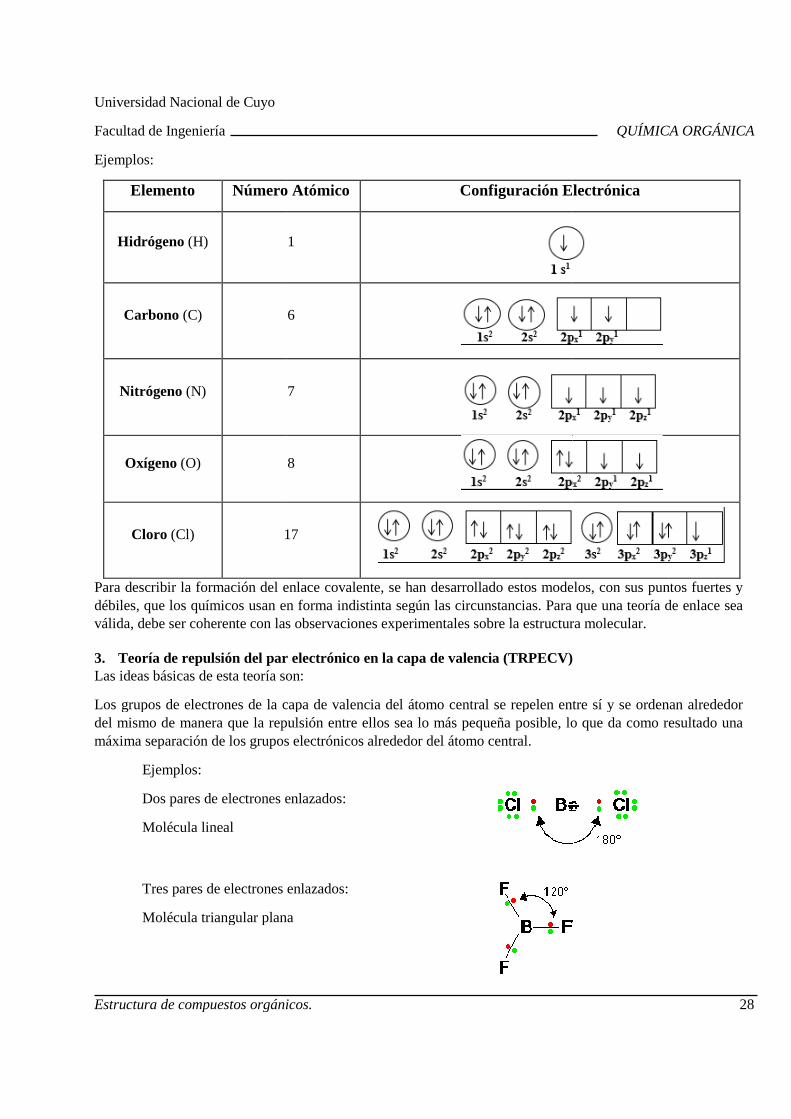
2.2. Configuración Electrónica Para establecer la configuración electrónica se debe considerar lo siguiente:
Un orbital no puede ser ocupado por más de dos electrones, los que deben ser de spines opuestos (Principio de exclusión de Pauli).
Un orbital no es ocupado mientras los orbitales de menor energía no están llenos (cada uno con dos electrones).
En un orbital no tiene lugar el apareo de electrones hasta que todos los orbitales de igual energía no contengan por lo menos un electrón (Regla de Hund).
y x
z
Universidad Nacional de Cuyo
Facultad de Ingeniería
Estructura de compuestos orgánicos.
Ejemplos:
Elemento Número Atómico
Hidrógeno (H)
Carbono (C)
Nitrógeno (N)
Oxígeno (O)
Cloro (Cl) 17
Para describir la formación del enlace covalente, se han desarrollado estos modelos, condébiles, que los químicos usan en forma indistinta según las circunstancias. Para que una teoría de enlace sea válida, debe ser coherente con las observaciones experimentales sobre la estructura molecular.
3. Teoría de repulsión del par electrónico en la capa de valencia (TRPECV)Las ideas básicas de esta teoría son:
Los grupos de electrones de la capa de valencia del átomo central se repelen entre sí y se ordenan alrededor del mismo de manera que la repulsión entre ellos sea lo más pequeña posible, lo que da como resultado unamáxima separación de los grupos electrónicos alrededor del átomo central.
Ejemplos:
Dos pares de electrones enlazados:
Molécula lineal
Tres pares de electrones
Molécula triangular plan
Estructura de compuestos orgánicos.
Número Atómico Configuración Electrónica
1
6
7
8
17
Para describir la formación del enlace covalente, se han desarrollado estos modelos, conusan en forma indistinta según las circunstancias. Para que una teoría de enlace sea
válida, debe ser coherente con las observaciones experimentales sobre la estructura molecular.
Teoría de repulsión del par electrónico en la capa de valencia (TRPECV)ideas básicas de esta teoría son:
Los grupos de electrones de la capa de valencia del átomo central se repelen entre sí y se ordenan alrededor del mismo de manera que la repulsión entre ellos sea lo más pequeña posible, lo que da como resultado una
separación de los grupos electrónicos alrededor del átomo central.
enlazados:
enlazados:
Molécula triangular plana
QUÍMICA ORGÁNICA
28
Configuración Electrónica
Para describir la formación del enlace covalente, se han desarrollado estos modelos, con sus puntos fuertes y usan en forma indistinta según las circunstancias. Para que una teoría de enlace sea
válida, debe ser coherente con las observaciones experimentales sobre la estructura molecular.
Teoría de repulsión del par electrónico en la capa de valencia (TRPECV)
Los grupos de electrones de la capa de valencia del átomo central se repelen entre sí y se ordenan alrededor del mismo de manera que la repulsión entre ellos sea lo más pequeña posible, lo que da como resultado una

Universidad Nacional de Cuyo
Facultad de Ingeniería
Estructura de compuestos orgánicos.
Cuatro pares de electrones
Molécula tetraédrica
Cuatro pares de electrones
tres pares de electrones enlazados:
Molécula piramidal
Cuatro pares de electrones
dos pares de electrones enlazados:
Molécula angular
4. Teoría del enlace de valencia (TEV)Las ideas clave de esta teoría son las siguientes:
Los enlaces covalentes se forman por traslape (solapamiento) de dos orbitales atómicos (puros o híbridos), cada uno de los cuales tiene un electrón con espines opuestos.
Cada átomo enlazado retiene sus orbitales atómicos, y el par de ecomparte el espacio común entre ambos núcleos. Los electrones de la región de solapamiento, son atraídos simultáneamente por ambos núcleos, lo que mantiene unidos a los átomos.
Mientras mayor es el traslape de orbitales, el e Los enlaces que se forman por el traslape de frente de dos orbitales atómicos a lo largo de una línea
que une ambos núcleos, se llaman enlaces sigma ( Los enlaces tipo pi (π) se forman por solapamiento lateral de dos orbitales atómi
Ejemplos:
Estructura de compuestos orgánicos.
pares de electrones enlazados:
Cuatro pares de electrones y
res pares de electrones enlazados:
Cuatro pares de electrones y
os pares de electrones enlazados:
Teoría del enlace de valencia (TEV) son las siguientes:
Los enlaces covalentes se forman por traslape (solapamiento) de dos orbitales atómicos (puros o híbridos), cada uno de los cuales tiene un electrón con espines opuestos. Cada átomo enlazado retiene sus orbitales atómicos, y el par de electrones con espines opuestos comparte el espacio común entre ambos núcleos. Los electrones de la región de solapamiento, son atraídos simultáneamente por ambos núcleos, lo que mantiene unidos a los átomos.Mientras mayor es el traslape de orbitales, el enlace es más fuerte. Los enlaces que se forman por el traslape de frente de dos orbitales atómicos a lo largo de una línea que une ambos núcleos, se llaman enlaces sigma (σ).
se forman por solapamiento lateral de dos orbitales atómi
QUÍMICA ORGÁNICA
29
Los enlaces covalentes se forman por traslape (solapamiento) de dos orbitales atómicos (puros o
lectrones con espines opuestos comparte el espacio común entre ambos núcleos. Los electrones de la región de solapamiento, son atraídos simultáneamente por ambos núcleos, lo que mantiene unidos a los átomos.
Los enlaces que se forman por el traslape de frente de dos orbitales atómicos a lo largo de una línea
se forman por solapamiento lateral de dos orbitales atómicos.
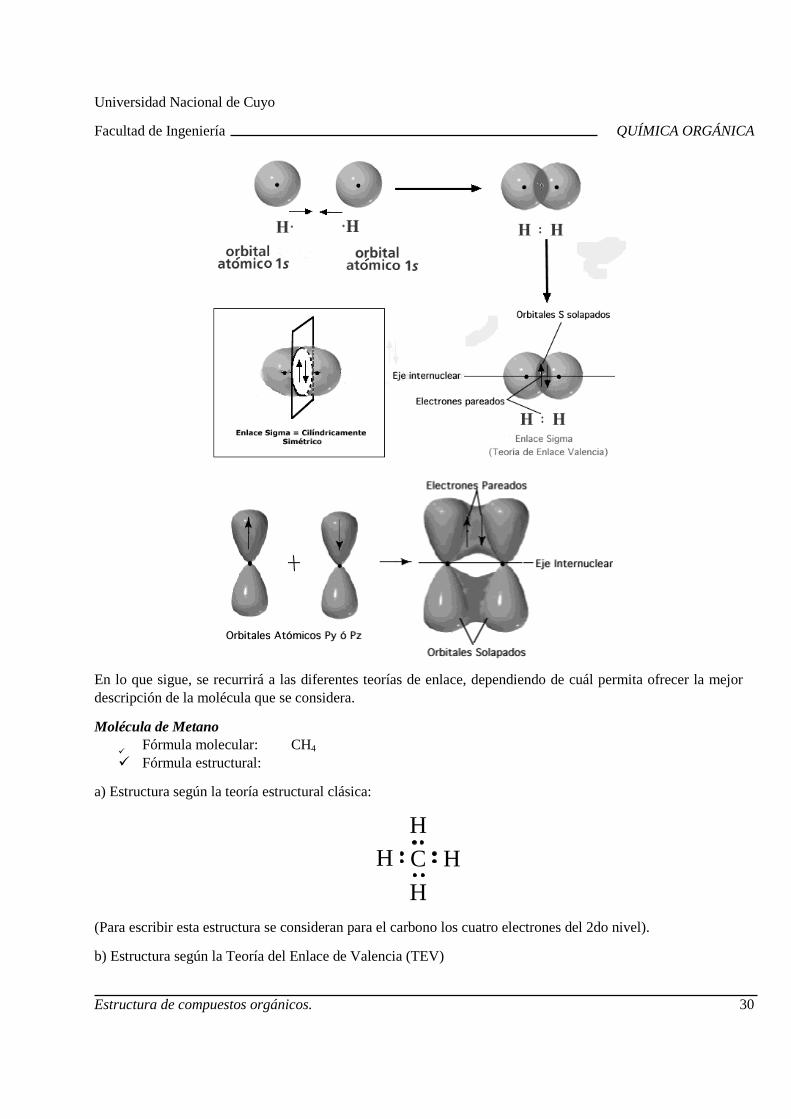
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 30
En lo que sigue, se recurrirá a las diferentes teorías de enlace, dependiendo de cuál permita ofrecer la mejor descripción de la molécula que se considera.
Molécula de Metano
Fórmula molecular: CH4
Fórmula estructural:
a) Estructura según la teoría estructural clásica:
H
H
H
H C
(Para escribir esta estructura se consideran para el carbono los cuatro electrones del 2do nivel).
b) Estructura según la Teoría del Enlace de Valencia (TEV)

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 31
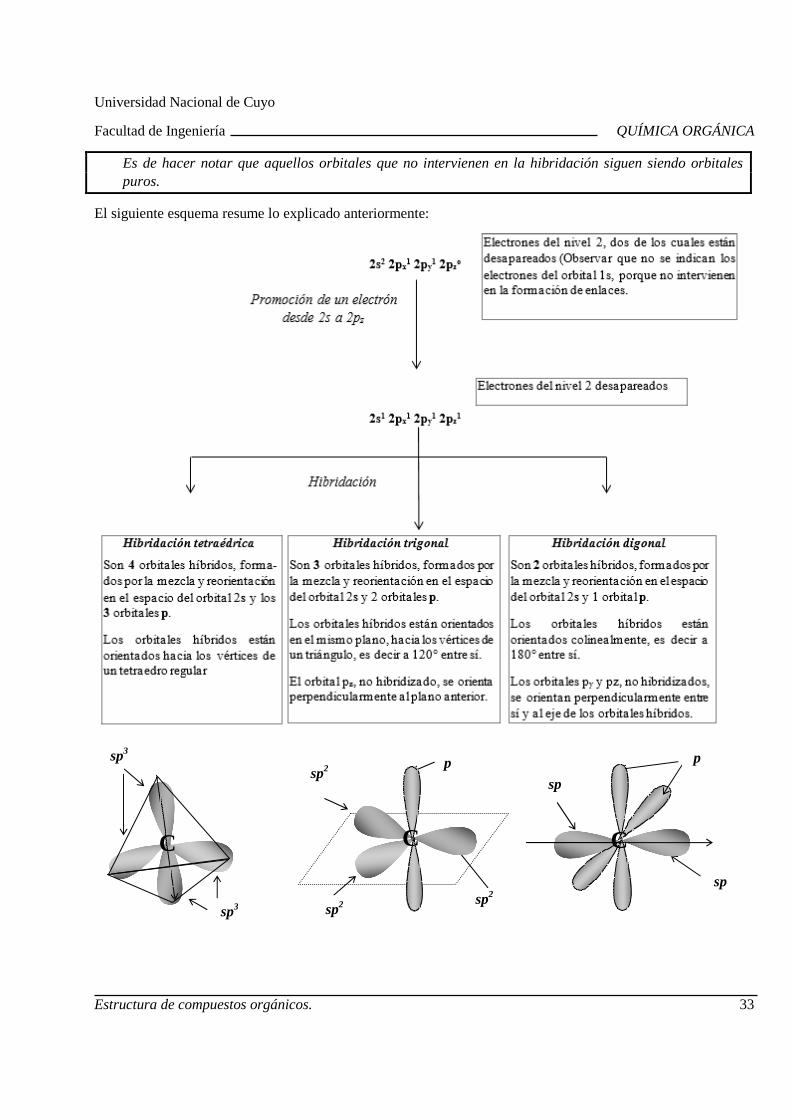
Para establecer la estructura según la Teoría Estructural posterior a 1926, se debe considerar la hibridación del carbono, entendiéndose por hibridación la mezcla y reorientación en el espacio de orbitales atómicos.
Es importante aclarar que la hibridación de orbitales atómicos puros da por resultado orbitales atómicos no puros o híbridos.
Si bien la hibridación es el resultado de cálculos complejos realizados por la Mecánica Cuántica, en este curso sólo veremos las conclusiones del análisis.
En lo que sigue, se presenta la hibridación del carbono en el metano:
Cada orbital híbrido está formado por dos lóbulos, uno de mayor tamaño y otro más pequeño, orientados según un eje. Generalmente, se representa el lóbulo mayor.
Un orbital híbrido sp3
o bien
Los cuatro orbitales híbridos se distribuyen espacialmente, lo más alejados posible entre ellos, orientados hacia los vértices de un tetraedro regular, formando sus ejes ángulos de 109º 28’.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 32
Cuatro orbitales híbridos sp3: 4sp3
* sp3 es el nombre de cada orbital híbrido e indica que proviene de la mezcla de un orbital s y 3
orbitales p.
En la molécula de metano, cada orbital híbrido está solapado con un orbital 1s del hidrógeno, resultando cuatro enlaces sigma. Los ángulos de enlace en esta molécula, como consecuencia de la orientación de los orbitales sp3 hacia los vértices de un tetraedro regular, son de 109º 28’.
− 4 enlaces σ :
∗ superposición de sp3 de C y 1s de H
− Configuración o forma de la molécula: molécula tetraédrica.
Siempre que un átomo de carbono está unido a cuatro átomos, su hibridación es sp3 o tetraédrica (ej: carbono que forma enlaces C-H y C-C).
Sin embargo, en muchos casos el átomo de carbono se encuentra unido a tres átomos (ej: carbono del enlace C=C) presentando hibridación sp2 (trigonal), o bien, a dos átomos (ej.: carbono del enlace C≡C), siendo la hibridación sp (digonal).
Hibridación del carbono
Tetragonal (Ej.: alcanos)
Trigonal (Ej.: alquenos)
Digonal (Ej.: alquinos)
Resumiendo:
1°) La configuración electrónica del carbono (1s2 2s2 2px1 2py
1 ) no es la que presenta este elemento cuando forma parte de compuestos.
2°) Si un electrón del orbital 2s, al ser excitado, pasa al orbital 2pz, la nueva configuración electrónica (1s2 2s1 2px
1 2py1 2pz
1), con cuatro electrones desapareados, no se corresponde con la del carbono que forma parte de compuestos orgánicos, ya que poseería un electrón en un orbital de menor energía y adireccional (2s) y tres electrones en orbitales p, de mayor energía y perpendiculares entre sí.
3°) Si cuatro, o tres, o dos de los orbitales atómicos puros (2s1 2px1 2py
1 2pz1) se mezclan y reorientan en el
espacio (hibridación de orbitales atómicos), de acuerdo a la necesidad del carbono de formar enlaces, se obtendrán nuevos orbitales atómicos no puros, denominados orbitales híbridos sp3, sp2o sp, según la cantidad de orbitales puros que hayan intervenido en la hibridación.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 33
Es de hacer notar que aquellos orbitales que no intervienen en la hibridación siguen siendo orbitales puros.
El siguiente esquema resume lo explicado anteriormente:
C
p
sp2 sp2
sp2
C
sp3
sp3
p
sp
sp
C

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 34
A continuación se describen algunas moléculas según la Teoría del Enlace de Valencia:
a) Compuesto: etano
Fórmula molecular: C2H6 Fórmula estructural (Teoría estructural clásica):
H C C H
H H
H H
Fórmula estructural (Teoría del Enlace de Valencia):
o Hibridación del carbono: sp3
Configuración electrónica del carbono: 1s2 4 sp3 Configuración electrónica del hidrógeno: 1s1
Nota: no se representa el orbital 1s.
Molécula de etano:
− 7 enlaces σ:
∗ superposición de sp3 de C y sp3 de C (1enlace σ)
∗ superposición de sp3 de C y 1s de H (6 enlaces σ)
Configuración o forma de la molécula: Molécula tetraédrica en cada carbono
b) Compuesto: eteno
Fórmula molecular: C2H4 Fórmula estructural (Teoría estructural clásica):
H C C H
H H

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 35
Fórmula estructural (Teoría del Enlace de Valencia): o Hibridación del C: sp2
Configuración electrónica del C: 1s2 3sp2 2pz1
Observaciones:
3sp2 significa 3 orbitales híbridos, cada uno de los cuales posee un electrón y es denominado sp2, es decir, cada uno formado por hibridación de un orbital s y dos orbitales p (ej.: px y py), quedando sin hibridizar el otro orbital p (ej.: pz), con un electrón.
Los orbitales sp2, al igual que los sp3, también están formados por dos lóbulos, pero sólo se representa uno. En este caso, el lóbulo que no se representa es mayor que el correspondiente al del sp3 y el que se representa es menor que el de sp3. Esto se debe al aumento de carácter s y disminución del carácter p de los orbitales híbridos sp2.
Un orbital híbrido sp2:
o bien
Orbital híbrido sp2
Molécula de eteno:
− 5 enlacesσ: ∗ superposición de sp2 de C y 1s de H (4 enlaces
σ)
∗ superposición de sp2 de un C y sp2 del otro C (1 enlace σ)
− 1 enlaceπ: ∗ superposición de orbitales p paralelos: pz de un
C y pz del otro C (1 enlace π)
− Configuración o forma de la molécula: molécula planar
c) Compuesto: etino
Fórmula molecular: C2H2
Configuración electrónica del H: 1s1

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 36
Fórmula estructural (Teoría estructural clásica):
H C C H
Fórmula estructural (Teoría del Enlace de Valencia): o Hibridación del C: sp
Configuración electrónica del C: 1s2 2sp 2py1 2pz
1
Observaciones:
2sp significa 2 orbitales híbridos, cada uno de los cuales posee un electrón y es denominado sp, es decir, cada uno formado por hibridación de un orbital s y un orbital p (ej.: px), quedando sin hibridizar los otros dos orbitales p (ej.: py y pz), cada uno con un electrón.
Los orbitales sp, al igual que los sp3 y sp2, también están formados por dos lóbulos, pero sólo se representa uno. En este caso, el lóbulo que no se representa es mayor que el de sp2 y el que se representa es menor que el de sp2. Esto se debe al aumento de carácter s y disminución del carácter p de los orbitales híbridos sp2.
Un orbital híbrido sp :
orbital híbrido sp
Configuración electrónica del H: 1s1

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 37
Molécula de etino:
− 3 enlaces σ: ∗ superposición de sp de C y 1s de H (2enlaces) ∗ superposición de sp de un C y sp de otro C
(1enlace) − 2 enlacesπ: ∗ superposición de orbitales p paralelos: pz - pz y
py - py. − Configuración o forma de la molécula:
molécula lineal
Observaciones:
Podemos observar en los ejemplos que, además de los enlaces σ (superposición o solapamiento “s-s”, “híbrido-híbrido” e “híbrido-s”), se presentan enlaces π, que se forman por solapamiento de orbitales atómicos p paralelos. Estos enlaces, más débiles que los σ, constan de dos nubes electrónicas, una por encima y otra por debajo del plano nodal (plano perpendicular al eje del orbital p). En el plano nodal no hay probabilidad de encontrar el electrón.
En la TEV el enlace π se puede representar como sigue:
Como el enlace π es más débil que el σ, para diferenciar estos tipos de enlaces en la Teoría Estructural Clásica se suelen representar como sigue:
π
C C
σ
C CDoble enlace (un σ y un π): Triple enlace (un σ y dos π)
5. Ángulos de enlaces. Energías y longitudes de enlaces. Se han determinado las siguientes longitudes de enlaces entre carbonos:
Compuesto Longitud de enlace carbono/carbono etano 1,53 Aº eteno 1,34 Aº etino 1,21 Aº

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 38
Considerando que en cada enlace están involucrados dos electrones, se podría afirmar que seis electrones (C≡C) acercan o unen con más fuerza que cuatro (C=C) y que cuatro electrones unen o acercan con más fuerza que dos (C-C). Sin embargo, se han observado las siguientes longitudes para enlaces C-H, donde siempre se comparten dos electrones:
Compuesto Longitud de enlace C-H etano 1,112 Aº eteno 1,103 Aº etino 1,079 Aº
Los valores anteriores nos permiten afirmar que en las longitudes de enlaces juegan un rol muy importante los tamaños de los orbitales atómicos involucrados. Un orbital s está más cerca del núcleo que un orbital p, el cual se extiende hasta cierta distancia del núcleo. En el caso de los orbitales híbridos, el sp3 tiene más carácter p y menos carácter s que un sp2, mientras que este último tiene más carácter p y menos carácter s que un sp, por lo que al aumentar el carácter s de un orbital híbrido, decrece su tamaño, con lo que también decrece la longitud del enlace con otro átomo.
Por otra parte, los enlaces más cortos son más fuertes y esto se puede comprobar con los valores de las energías de disociación homolíticas de enlaces (D) (Morrison Boyd - Química Orgánica -5a. edición- Ed. Addison Wesley Longman).
Ejemplos:
Enlace D (kcal/mol Enlace D (kcal/mol HH 104 H3C CH3 88
C2H5 H 98 H2C = CH2 163 H2C=CH H 108 HC ≡ CH 198
D, que es la Energía de Disociación de enlace, es característica de un enlace específico y es la cantidad de energía que se consume o se libera cuando se rompe o se forma dicho enlace. El siguiente ejemplo sirve para demostrar que la energía de disociación de enlace no es lo mismo que la Energía de Enlace (E):
CH3 + H CH4
D (CH3 - H) = 104 kcal/mol
CH2 + H CH3
D (CH2 - H) = 106 kcal/mol
CH + H CH2
D (CH - H) = 106 kcal/mol
C + H CH
D (C - H) = 81 kcal/mol
C + 4 H CH4
∆H = 397 kcal/mol
E (C - H)
= (397 kcal/mol) / 4 = 99 kcal/mol

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 39
La Tabla 1-2 de Morrison-Boyd da valores de energías de disociación de enlace homolítica (1) , mientras que en la tabla siguiente se muestran valores de energías de disociación de enlace heterolítica (2) , las cuales son mucho mayores que las anteriores debido a que la ruptura heterólítica de un enlace implica la formación de iones de cargas opuestas, cuya separación requiere más energía que la separación de partículas neutras.
(1) Disociación de enlace homolítica: Cuando se rompe un enlace, cada una de las partes se separa con un electrón.
(A : B A . + B .)
(2) Disociación de enlace heterolítica: Cuando se rompe un enlace, una de las partes se separa con los dos electrones (anión) y la otra queda como catión.
(A:B A : - + B +)
Ejemplos:
∗ Ruptura homolítica: H3C : Cl
H3C . + . Cl D (C-Cl)
= 84 kcal/mol
∗ Ruptura heterolítica: H3C : Cl H3C+ + : Cl - D (C-Cl) = 227 kcal/mol
6. Teoría de la Resonancia Resonancia es, básicamente, deslocalización de electrones π. La resonancia o deslocalización de electrones se presenta en moléculas con dobles o triples enlaces
conjugados (ej.: 1,3-butadieno), pero también en otras especies químicas como radicales libres, aniones y cationes:
CH2 = CH - CH = CH2 CH2 = CH - CH2° CH2 = CH - CH2
+ CH2 = CH - CH2: -
1,3-butadieno radical alilo catión alilo anión alilo
Hay pruebas (ej.: longitudes de enlaces) que demuestran, que por sí mismas cada una de ellas no representa a la especie química correspondiente. No se observa que en cada una hay deslocalización de electrones π.
Por ejemplo, si tuviéramos que escribir la estructura del 1,3-butadieno, pondríamos:
CH2 = CH - CH = CH2
Sin embargo esta no es la verdadera estructura del compuesto, ya que al poseer dobles enlaces conjugados, presentaría deslocalización de electrones π. Por lo tanto la estructura dada, por sí misma, no representaría al compuesto.
Podríamos haber escrito distintas estructuras con distintas posiciones de los electrones móviles (π ), pero ninguna en forma aislada representaría al compuesto:
-:CH2 - CH = CH - CH2+ +CH2 - CH = CH - CH2:
-
Entonces, la Teoría de la Resonancia postula que “si pueden escribirse dos o más estructuras correspondientes a un mismo compuesto, que difieran únicamente en las posiciones de los electrones, ninguna de las estructuras estará en concordancia con las propiedades del compuesto y, por lo tanto, no lo representa”.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 40
Cuando hay conjugación una única estructura no representa al compuesto porque en los dobles enlaces, además de los electrones σ que están más retenidos compartidos entre dos núcleos, los π están menos retenidos. Entonces si esos electrones pertenecen a un doble enlace aislado, es compartido por dos núcleos, estando menos retenidos que los σ (ej.: eteno). Pero si hay conjugación, en realidad esos electrones móviles pasan a moverse por toda la zona donde hay conjugación (ej.: 1,3-butadieno).
La Teoría de la Resonancia propone que esas estructuras sean utilizadas como "contribuyentes" a la estructura real, y las llama estructuras contribuyentes, o canónicas o de resonancia. No son estructuras reales.
¿Cómo representamos al compuesto?
Escribiendo las estructuras contribuyentes, que pueden ser dos o más, y entre dos estructuras se coloca una flecha de dos puntas ( ) que es el símbolo de la resonancia. Por ejemplo, al 1,3-butadieno lo representaremos como sigue:
Es decir, cuando por Teoría de la Resonancia escribimos estructuras contribuyentes estamos indicando que hay electrones móviles.
Tratemos de comprender mediante un ejemplo a escala mayor, como es el caso de un objeto que se mueve (ej.: una pelota de fútbol que cae desde un muro). Si nosotros quisiéramos dibujar en un papel a la pelota cayendo desde el muro, no podríamos. Pero sí podemos hacer una secuencia con distintas posiciones de la pelota y el conjunto nos dará la idea de la caída:
Ninguna de las pelotas en forma aislada representa la caída, pero el conjunto da idea del movimiento.
Así, como no se puede representar en el plano el movimiento de los electrones en el 1,3-butadieno, se escriben distintas estructuras, cada una de las cuales por sí misma no indica el movimiento de los electrones, pero en conjunto dan idea del mismo.
La TEV, establecida con posterioridad a la Teoría de la Resonancia, permite comprender mejor la deslocalización de electrones.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 41
Por ejemplo, en el 1,3-butadieno todos los carbonos tienen hibridación sp2. Se forman los enlaces σ orientados en un mismo plano, por solapamiento de orbitales híbridos entre sí (enlaces C1 - C2 , C2 - C3 , C3 - C4 ) y de orbitales híbridos con 1s de hidrógeno (enlaces C-H).
A cada carbono le queda un orbital pz con un electrón y, además, todos estos orbitales p están paralelos, lo que permite la formación de una nube π extendida (cada orbital p se puede solapar con los dos orbitales p vecinos), ocupada por los electrones que originalmente eran pz (cada electrón se mueve en un espacio mayor, asociado a cuatro núcleos, que el que correspondería a un π aislado):
Por la TEV se observa la deslocalización de los electrones π. Pero por la Teoría Estructural Clásica es imposible representar esa deslocalización por una única estructura. Entonces la Teoría de la Resonancia lo que hace es escribir varias estructuras que en su conjunto infieran que hay movimiento o deslocalización de electrones.
7. Aromaticidad. Si bien antes el término aromático se derivaba de la característica de poseer aroma, actualmente el significado es meramente químico, ya que se consideran compuestos aromáticos al benceno y a todas aquellas sustancias parecidas al benceno en su comportamiento químico. Por lo expuesto, resulta conveniente conocer acabadamente este compuesto, ya que si se comprende la estructura de éste, se podrán obtener criterios que permitirán determinar la aromaticidad o no de los compuestos orgánicos.
7.1. Benceno: Estructura y características
1º) La fórmula molecular del benceno es: C6 H6
2º) El benceno no reacciona como alqueno, a pesar que de acuerdo a su fórmula molecular es un compuesto muy insaturado.
Se ha comprobado que, a diferencia de los alquenos, no adiciona hidrógeno fácilmente, no reacciona con bromo en tetracloruro de carbono, no adiciona agua en presencia de ácidos, etc. El benceno adiciona hidrógeno, en presencia de catalizador, a muy altas temperaturas y grandes presiones y, lo que más llama la atención, es que con Cl2/ FeCl3 no da un producto de adición como cualquier alqueno, sino que se produce sustitución.
7.2. Estructura del benceno según la Teoría Estructural Clásica Kekulé en 1865 propuso una estructura, que satisfizo los requerimientos que los átomos de carbono formasen cuatro enlaces y que todos los hidrógenos fueran equivalentes. Pero, frente a una serie de objeciones, Kekulé

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 42
fue haciendo modificaciones para, finalmente, hablar de dos estructuras en equilibrio dinámico, fácilmente interconvertibles:
CH
CH
CH
CH
CH
CH
CH
CH
CH
CH
CH
CH
Esto también tuvo una serie de objeciones. No obstante, se considera que la propuesta de Kekulé fue un paso importante, aplicándose actualmente con significado distinto al que tuvo originalmente, lo cual veremos más adelante.
7.3. Estructura del benceno según la Teoría de la Resonancia El benceno, por tener dobles enlaces conjugados según la Teoría Estructural Clásica, es un híbrido de resonancia:
Las dos estructuras de Kekulé, que no representan a dos moléculas separadas en equilibrio, son la representación más aproximada que puede hacerse de la molécula de benceno. (I) y (II) son dos estructuras contribuyentes.
(I) (II)
El símbolo ( ), que reemplaza al de equilibrio ( ), es el que interrelaciona las estructuras contribuyentes.
En cada vértice se sobreentiende que hay un carbono, unido a tantos hidrógenos como sean necesarios para completar los cuatro enlaces del carbono (en este caso es un Hidrógeno).
Se puede observar que los simples enlaces en (I) son dobles en (II) y los dobles en (I) son simples en (II). Si se mezclan (I) y (II), o más correctamente, si se construye un híbrido de (I) y (II) los enlaces de ese híbrido (benceno) no son dobles ni simples, sino que son intermedios.
Esto se ha comprobado al determinar las longitudes de enlaces. Todos los enlaces carbono-carbono son iguales y de longitud 1,39 A°, valor intermedio entre los de los enlaces C-C (1,54 A°) y C=C (1,34 A°).
Por eso muchas veces se encontrará al benceno representado de la siguiente forma:
Con el análisis realizado se aclaran, además de lo referido a longitudes de enlaces, otros aspectos particulares del benceno:
El benceno da sólo un producto 1,2-disustituido (sustituyentes en dos carbonos adyacentes):
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 43
Correcto
X
X
Incorrecto
X
X
X
X
Único producto Dos productos que serían distintos (ver enlace entre carbonos en
cada caso)
El benceno contiene menos energía que la que correspondería a cualquiera de las estructuras de Kekulé, lo cual explica su gran estabilidad. Esto es consecuencia de la deslocalización de electrones. Por dicha razón a la energía de resonancia se llama también "de deslocalización o de estabilización".
Los siguientes estudios termoquímicos permitieron explicar la estabilidad del benceno, lo cual no había podido dilucidarse con las estructuras propuestas hasta el momento.
El ciclohexeno, que posee un doble enlace, es fácilmente hidrogenado:
CH
CH
CH
CH
CH
CH
CH2
CH2
CH2
CH2
CH2
CH2
H2/Pt
∆H = -28,6 kcal/mol
ciclohexeno ciclohexano
Por lo tanto, se podría esperar que la hidrogenación de benceno, considerado como 1,3,5-ciclohexatrieno (hipotético), liberara tres veces 28,6 kcal/mol, es decir, 85,8 kcal/mol. Sin embargo, el dato experimental es de sólo 49,8 kcal/mol. Hay una diferencia entre el valor teórico y el experimental de 36 kcal/mol.
CH
CH
CH
CH
CH
CH
CH2
CH2
CH2
CH2
CH2
CH2
3H2/Pt
∆Hteórico = -85,8 kcal/mol ∆Hexperim. = -49,8 kcal/mol
Estructura hipotética (no existe) ciclohexano
Si hacemos para la hidrogenación un diagrama de barras con estos resultados, observamos que el benceno, con deslocalización de electrones, es más estable (tiene menos energía potencial) en 36 kcal/mol que el 1,3,5-ciclohexatrieno hipotético, sin deslocalización de electrones.
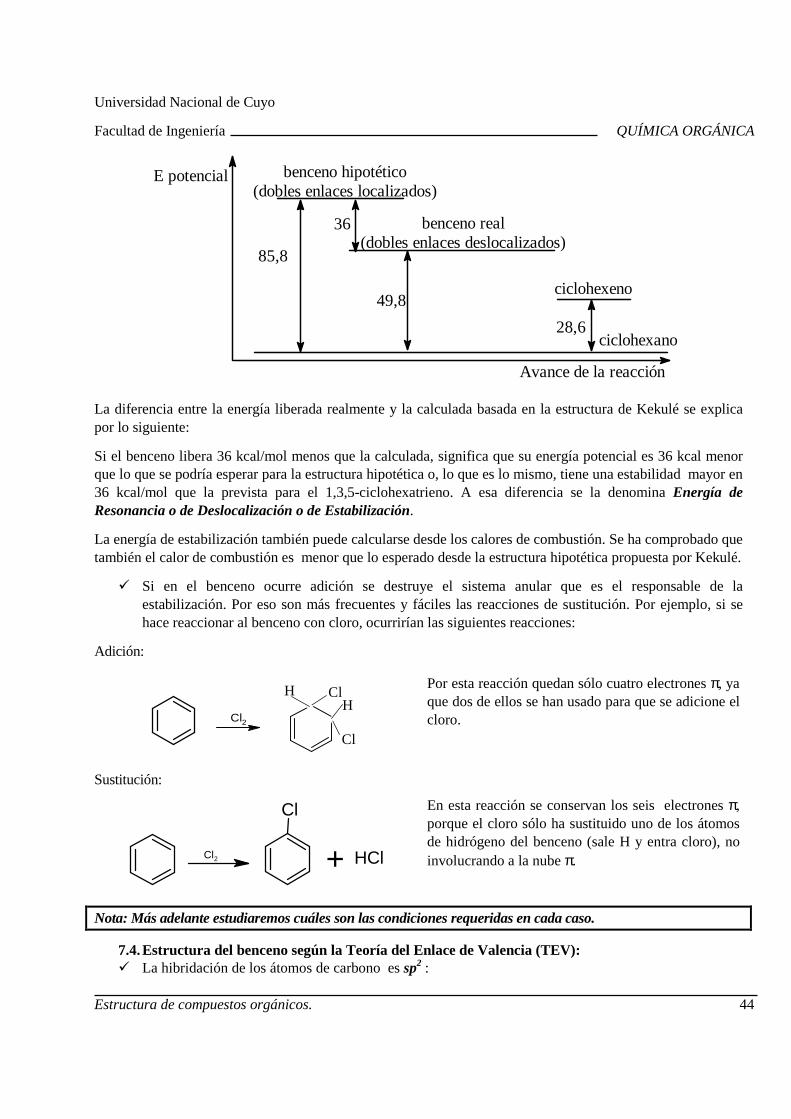
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 44
E potencial
Avance de la reacción
benceno hipotético(dobles enlaces localizados)
85,8
benceno real(dobles enlaces deslocalizados)
ciclohexano
ciclohexeno
36
49,8
28,6
La diferencia entre la energía liberada realmente y la calculada basada en la estructura de Kekulé se explica por lo siguiente:
Si el benceno libera 36 kcal/mol menos que la calculada, significa que su energía potencial es 36 kcal menor que lo que se podría esperar para la estructura hipotética o, lo que es lo mismo, tiene una estabilidad mayor en 36 kcal/mol que la prevista para el 1,3,5-ciclohexatrieno. A esa diferencia se la denomina Energía de Resonancia o de Deslocalización o de Estabilización.
La energía de estabilización también puede calcularse desde los calores de combustión. Se ha comprobado que también el calor de combustión es menor que lo esperado desde la estructura hipotética propuesta por Kekulé.
Si en el benceno ocurre adición se destruye el sistema anular que es el responsable de la estabilización. Por eso son más frecuentes y fáciles las reacciones de sustitución. Por ejemplo, si se hace reaccionar al benceno con cloro, ocurrirían las siguientes reacciones:
Adición:
Por esta reacción quedan sólo cuatro electrones π, ya que dos de ellos se han usado para que se adicione el cloro. Cl2
H
Cl
H Cl
Sustitución:
En esta reacción se conservan los seis electrones π, porque el cloro sólo ha sustituido uno de los átomos de hidrógeno del benceno (sale H y entra cloro), no involucrando a la nube π. Cl2
Cl
+ ClH
Nota: Más adelante estudiaremos cuáles son las condiciones requeridas en cada caso.
7.4. Estructura del benceno según la Teoría del Enlace de Valencia (TEV): La hibridación de los átomos de carbono es sp2 :
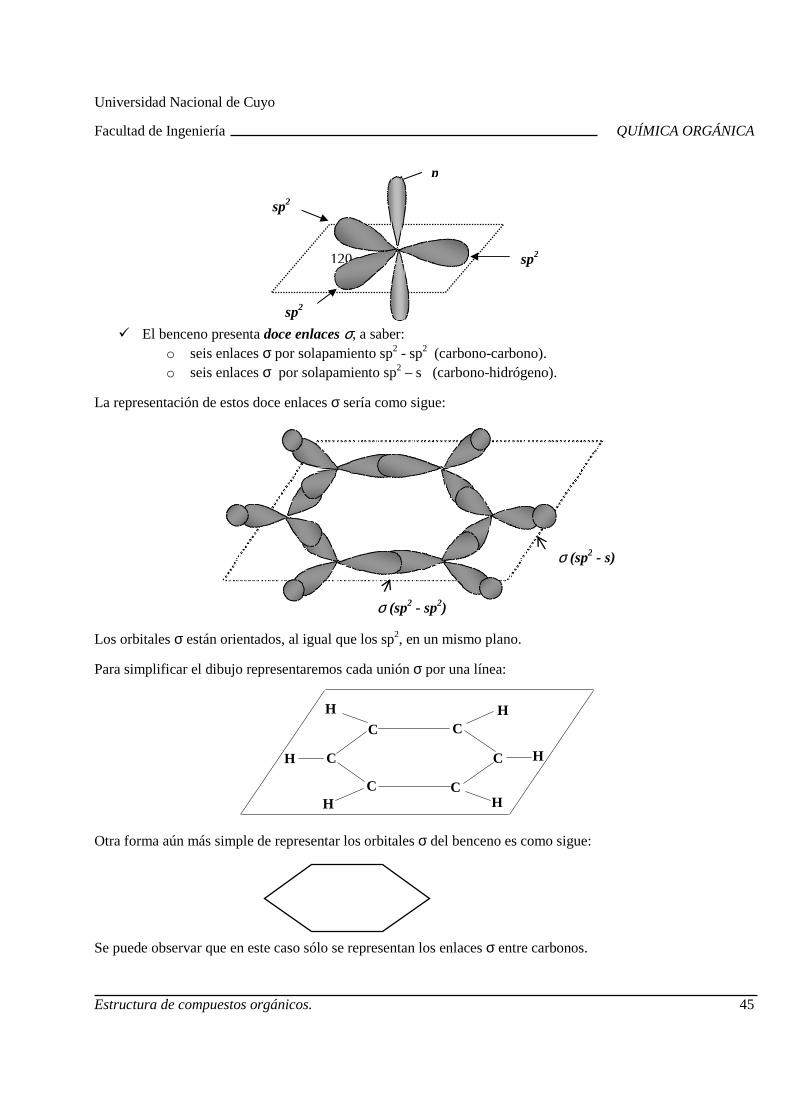
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 45
El benceno presenta doce enlaces σ, a saber: o seis enlaces σ por solapamiento sp2 - sp2 (carbono-carbono). o seis enlaces σ por solapamiento sp2 – s (carbono-hidrógeno).
La representación de estos doce enlaces σ sería como sigue:
Los orbitales σ están orientados, al igual que los sp2, en un mismo plano.
Para simplificar el dibujo representaremos cada unión σ por una línea:
C
C
CC
C
CH
H
HH
H
H
Otra forma aún más simple de representar los orbitales σ del benceno es como sigue:
Se puede observar que en este caso sólo se representan los enlaces σ entre carbonos.
σ (sp2 - sp2)
σ (sp2 - s)
p
sp2 120
sp2
sp2 sp2
sp2
sp2

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 46
nube π (hexacéntrica extendida)
C
C
C
C
C
C H
H
H H
H
H
Además de los enlaces σ, el benceno presenta una nube electrónica π deslocalizada arriba y abajo del plano del anillo:
Los orbitales p de los átomos de carbono con hibridación sp2 quedan paralelos entre sí, lo que permite que se superpongan (cada orbital p se puede superponer con los orbitales p vecinos), generándose una nube de electrones π hexacéntrica extendida, que es una nube cerrada de electrones y es la que justifica en parte la estabilidad.
En la siguiente figura se representa la molécula de benceno completa, es decir con todos los enlaces σ y la nube π extendida:
Otras formas de representación de esta nube π son las siguientes:
Hay hechos experimentales que determinan la aceptación de esta descripción del benceno:
Por difracción de rayos X y electrónica, se ha determinado que la molécula de benceno es plana y simétrica, sus enlaces son iguales y los ángulos de enlaces son todos de 120°.
Las reacciones características del benceno son las sustituciones electrofílicas (SE). Esto se justifica por lo siguiente:
o Si ocurre sustitución se mantiene intacto el sexteto de electrones π.
o Los electrones π deslocalizados están menos retenidos y por lo tanto más expuestos al ataque de buscadores de electrones.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 47
7.5. Criterios de aromaticidad Analizadas las características del benceno, veremos qué propiedades debe tener un compuesto para ser considerado Aromático, es decir, estableceremos los Criterios de Aromaticidad:
1) Desde un punto de vista experimental, son aromáticas las sustancias que:
* Tienen fórmulas moleculares que hacen suponer alto grado de insaturación, pero son resistentes a las reacciones de adición: Los compuestos aromáticos no se comportan como alquenos.
* Las moléculas de los compuestos aromáticos son planas o casi planas.
Esto permite que los orbitales p, al ser paralelos, se superpongan y se genere una nube π extendida.
* Los compuestos aromáticos dan preferentemente sustituciones electrofílicas.
* Los compuestos aromáticos tienen bajos calores de hidrogenación y de combustión (prueba de gran estabilidad).
* Los compuestos aromáticos tienen alta Energía de resonancia.
2) Desde un punto de vista teórico, la nube de electrones π debe tener (4n + 2) electrones π (Regla de Hückel), donde n es un número entero y positivo (n = 1; 2; 3....).
Hay muchos compuestos que también tienen carácter aromático, a pesar que parecen muy distintos al benceno o sus derivados. Entre ellos se encuentran:
hidrocarburos aromáticos de núcleos condensados compuestos heterocíclicos.
7.6. Hidrocarburos aromáticos de núcleos condensados a) Naftaleno
El naftaleno se considera como compuesto aromático porque, además de dar sustituciones electrofílicas, es un compuesto muy estable, lo cual lo demuestra su bajo calor de combustión que es inferior al que se podría esperar por el alto número de dobles enlaces.
Contrariamente a lo que ocurre con el benceno, en el naftaleno se observa que no todos los enlaces entre carbonos son de igual longitud. El enlace C1 - C2 (1,365A°) tiene más carácter doble, mientras que C2 - C3 es más largo (1,404A°), por tener más carácter de simple enlace (ver estructuras contribuyentes).
Además, su energía de resonancia es de 61 kcal/mol, es decir, inferior a la que correspondería para dos anillos bencénicos. Esto es correcto ya que, si se observa cualquiera de los dos núcleos en el naftaleno, se comprueba que sólo en dos formas contribuyentes equivale a un núcleo bencénico, mientras que en la restante presenta únicamente dos enlaces dobles. Es decir, cada anillo no tiene 100% de carácter bencénico.
No obstante, por razones prácticas, el naftaleno se representa de la siguiente forma:
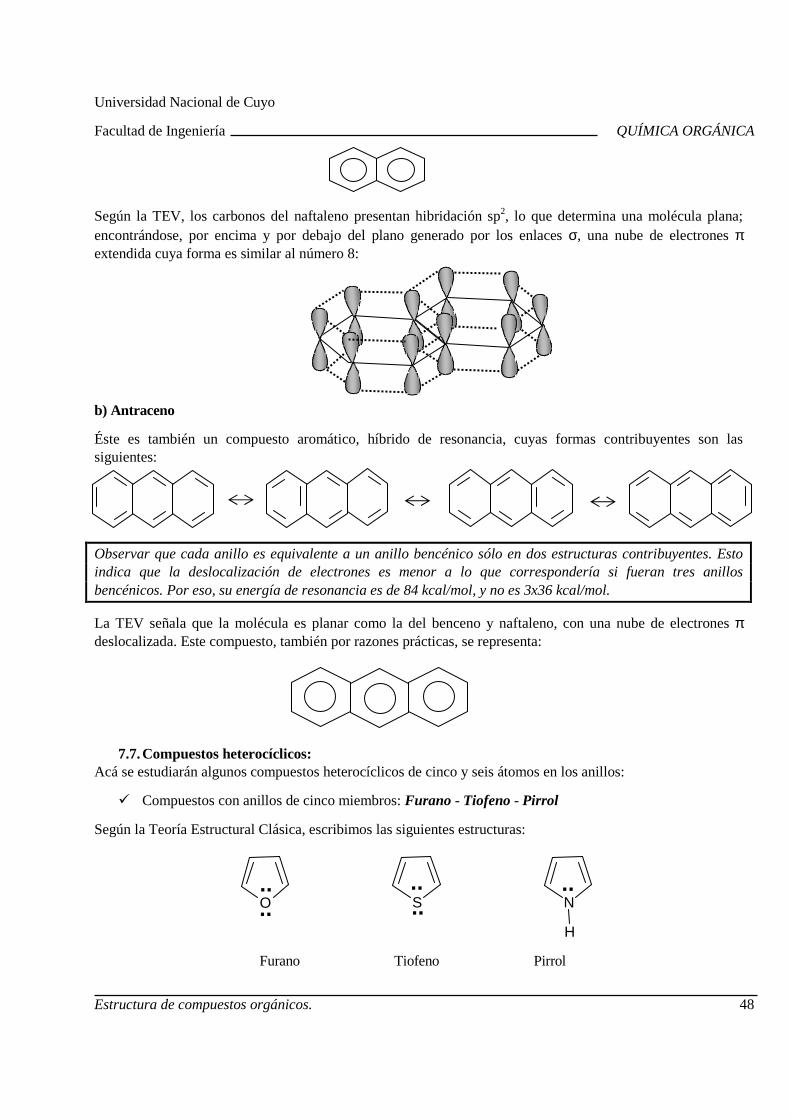
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 48
Según la TEV, los carbonos del naftaleno presentan hibridación sp2, lo que determina una molécula plana; encontrándose, por encima y por debajo del plano generado por los enlaces σ, una nube de electrones π extendida cuya forma es similar al número 8:
b) Antraceno
Éste es también un compuesto aromático, híbrido de resonancia, cuyas formas contribuyentes son las siguientes:
Observar que cada anillo es equivalente a un anillo bencénico sólo en dos estructuras contribuyentes. Esto indica que la deslocalización de electrones es menor a lo que correspondería si fueran tres anillos bencénicos. Por eso, su energía de resonancia es de 84 kcal/mol, y no es 3x36 kcal/mol.
La TEV señala que la molécula es planar como la del benceno y naftaleno, con una nube de electrones π deslocalizada. Este compuesto, también por razones prácticas, se representa:
7.7. Compuestos heterocíclicos: Acá se estudiarán algunos compuestos heterocíclicos de cinco y seis átomos en los anillos:
Compuestos con anillos de cinco miembros: Furano - Tiofeno - Pirrol
Según la Teoría Estructural Clásica, escribimos las siguientes estructuras:
O S N
H.. .... .. ..
Furano Tiofeno Pirrol
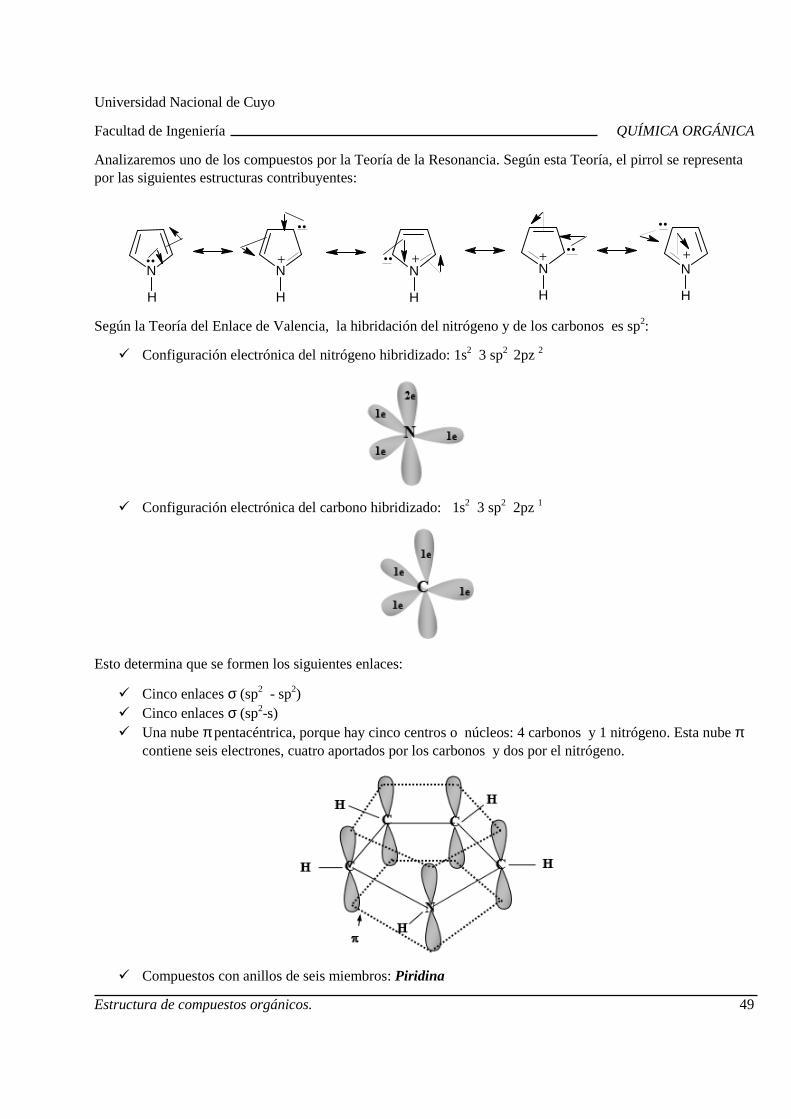
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 49
Analizaremos uno de los compuestos por la Teoría de la Resonancia. Según esta Teoría, el pirrol se representa por las siguientes estructuras contribuyentes:
N
H
.. ++ +.. ..N
H
N
H
N
H
N
H
+
.. ..
Según la Teoría del Enlace de Valencia, la hibridación del nitrógeno y de los carbonos es sp2:
Configuración electrónica del nitrógeno hibridizado: 1s2 3 sp2 2pz 2
Configuración electrónica del carbono hibridizado: 1s2 3 sp2 2pz 1
Esto determina que se formen los siguientes enlaces:
Cinco enlaces σ (sp2 - sp2) Cinco enlaces σ (sp2-s) Una nube π pentacéntrica, porque hay cinco centros o núcleos: 4 carbonos y 1 nitrógeno. Esta nube π
contiene seis electrones, cuatro aportados por los carbonos y dos por el nitrógeno.
Compuestos con anillos de seis miembros: Piridina
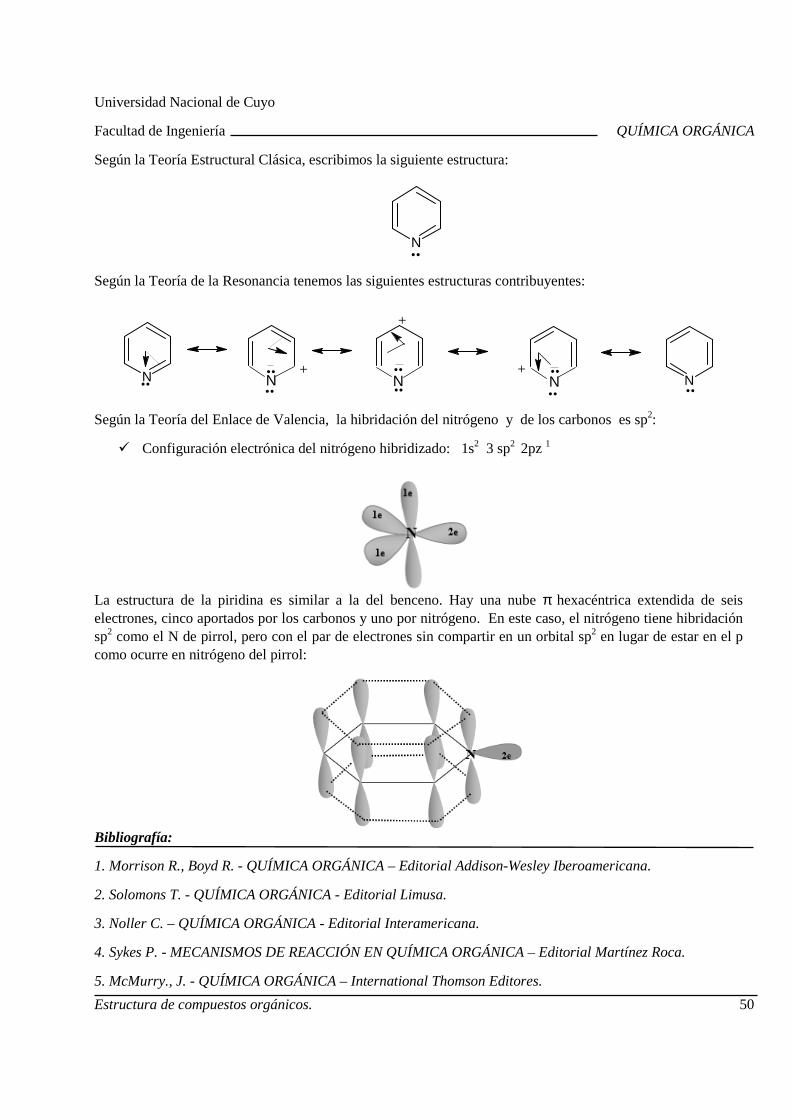
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura de compuestos orgánicos. 50
Según la Teoría Estructural Clásica, escribimos la siguiente estructura:
..N
Según la Teoría de la Resonancia tenemos las siguientes estructuras contribuyentes:
..
+
+ +...... .. .. ..
..N N N NN
Según la Teoría del Enlace de Valencia, la hibridación del nitrógeno y de los carbonos es sp2:
Configuración electrónica del nitrógeno hibridizado: 1s2 3 sp2 2pz 1
La estructura de la piridina es similar a la del benceno. Hay una nube π hexacéntrica extendida de seis electrones, cinco aportados por los carbonos y uno por nitrógeno. En este caso, el nitrógeno tiene hibridación sp2 como el N de pirrol, pero con el par de electrones sin compartir en un orbital sp2 en lugar de estar en el p como ocurre en nitrógeno del pirrol:
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Noller C. – QUÍMICA ORGÁNICA - Editorial Interamericana.
4. Sykes P. - MECANISMOS DE REACCIÓN EN QUÍMICA ORGÁNICA – Editorial Martínez Roca.
5. McMurry., J. - QUÍMICA ORGÁNICA – International Thomson Editores.

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 51
Isomería
Isomería plana o estructural: Isómeros de cadena. Isómeros de posición. Isómeros de función. Propiedades de los isómeros planos. Isomería espacial o estereoisomería: Isomería Geométrica Configuración. Isomería Óptica. Isómeros ópticos. Centro quiral. Actividad óptica. Enantiómeros. Diasterómeros. Par racémico. Forma Meso. Propiedades de los estereoisómeros.
Isómeros: Compuestos diferentes de igual fórmula molecular.
Básicamente, hay dos tipos de isomería:
1. Isomería plana o estructural
Isómeros planos son aquellos compuestos que tienen igual fórmula molecular pero distinta fórmula estructural. Es decir, escribiendo sus estructuras en un plano (ej.: papel, pizarra) determinamos que se trata de compuestos distintos.
Hay diferentes tipos de isómeros planos:
1.1. Isómeros de cadena
Estos isómeros presentan distintas cadenas carbonadas, lo que determina diferencias en sus propiedades físicas.
Ejemplo: Compuestos de fórmula molecular C4 H10
Fórmulas estructurales: CH3 CH2 CH2 CH3 CH3 CH CH3
CH3 n-butano (butano) Isobutano (metilpropano)
Los dos compuestos pertenecen a la familia de los alcanos, tienen iguales pesos moleculares e iguales fórmulas moleculares pero, al tener distintas estructuras, sus propiedades son distintas:
Propiedades n-butano Isobutano punto de ebullición, ºC 0 -12
punto de fusión, ºC -138 -159
densidad del líquido relativa a 20ºC 0,622 0,604

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 52
1.2. Isómeros de posición
Son compuestos cuyas estructuras se diferencian en la posición del Grupo Funcional (ej.: alquenos, alcoholes).
Ejemplo: Compuestos de fórmula molecular C4 H8 de cadena abierta lineal.
Fórmulas estructurales: CH2 CH CH2 CH3 CH3 CH CH CH3
1-buteno 2-buteno
En este caso, los isómeros presentan distinta posición del doble enlace, que es el grupo funcional de la familia a la que pertenecen.
Los isómeros de posición presentan distintas propiedades, principalmente físicas.
1.3. Isómeros de función
Son compuestos que tienen igual fórmula molecular, pero pertenecen a series homólogas diferentes por poseer grupos funcionales distintos (ej.: aldehídos y cetonas).
Ejemplo: Compuestos de fórmula molecular C3H6O
Fórmulas estructurales: CH3 CH2 C
H
O CH3 C CH3
O
propanal propanona
Otro ejemplo se presenta entre alcoholes y éteres.
Ejemplo: Compuestos de fórmula molecular C2 H6 O
Fórmulas estructurales: CH3 CH2
OH
CH3 O CH3
Alcohol etílico (etanol) Éter metílico (metoximetano)
En estos casos, los isómeros presentan distintos grupos funcionales, por lo que no sólo difieren en propiedades físicas, sino también en propiedades químicas.
2. Isomería espacial o estereoisomería
Son isómeros espaciales aquellos compuestos que tienen igual fórmula molecular, igual fórmula estructural, pero diferente orientación de los átomos en el espacio.
2.1. Isómeros conformacionales Construyendo un modelo molecular del etano comprobaremos que los dos grupos metilo pueden rotar fácilmente uno respecto al otro. La energía necesaria para mover los átomos de hidrógeno de un metilo respecto del otro, o barrera de rotación, es de 2.9 kcal mol-1. Este valor es tan pequeño que los químicos hablan de libre rotación de los grupos metilo. En general, hay rotación libre alrededor de todos los enlaces simples. La conformación producida por la rotación respecto al enlace carbono-carbono del etano representa un continuo entre los dos extremos que se muestran abajo: una conformación alternada o escalonada y una conformación eclipsada. Entre estos dos extremos hay una cantidad infinita de conformaciones posibles.
C C
H6
H5H4
H1H2
H3
60ºC C
H6
H5H4H1
H2
H3
“líneas y cuñas”
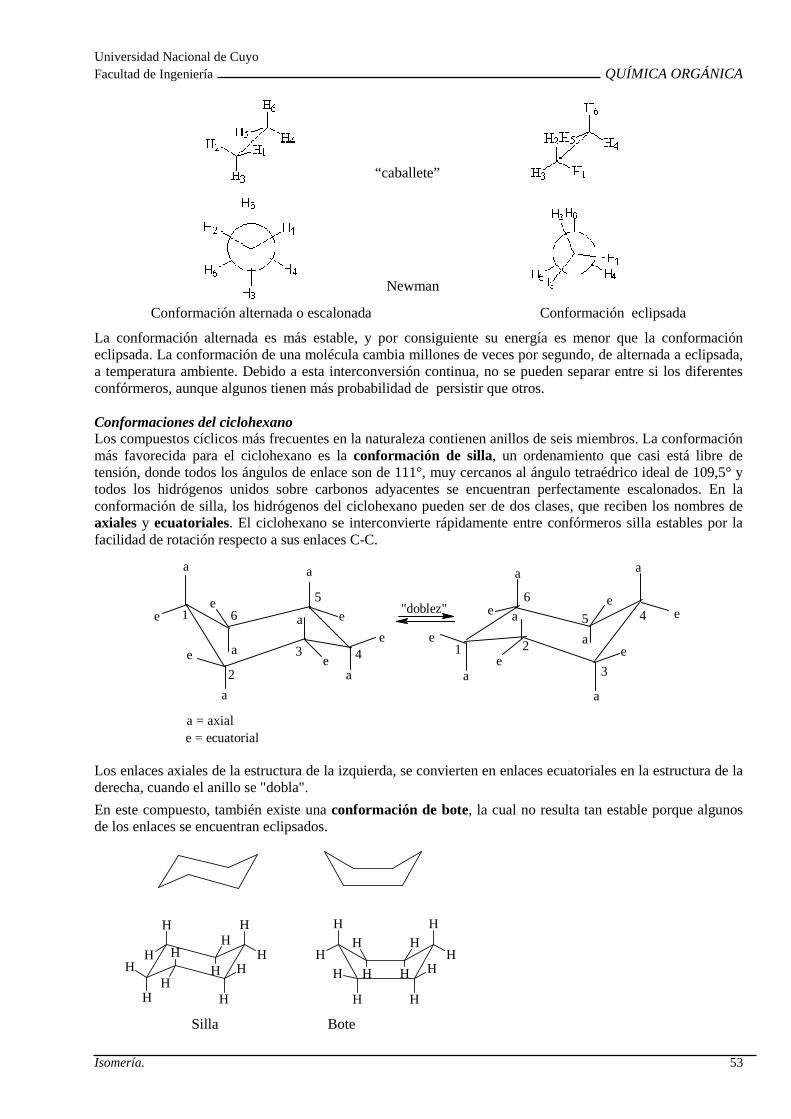
Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 53
Conformación alternada o escalonada Conformación eclipsada
La conformación alternada es más estable, y por consiguiente su energía es menor que la conformación eclipsada. La conformación de una molécula cambia millones de veces por segundo, de alternada a eclipsada, a temperatura ambiente. Debido a esta interconversión continua, no se pueden separar entre si los diferentes confórmeros, aunque algunos tienen más probabilidad de persistir que otros. Conformaciones del ciclohexano Los compuestos cíclicos más frecuentes en la naturaleza contienen anillos de seis miembros. La conformación más favorecida para el ciclohexano es la conformación de silla, un ordenamiento que casi está libre de tensión, donde todos los ángulos de enlace son de 111°, muy cercanos al ángulo tetraédrico ideal de 109,5° y todos los hidrógenos unidos sobre carbonos adyacentes se encuentran perfectamente escalonados. En la conformación de silla, los hidrógenos del ciclohexano pueden ser de dos clases, que reciben los nombres de axiales y ecuatoriales. El ciclohexano se interconvierte rápidamente entre confórmeros silla estables por la facilidad de rotación respecto a sus enlaces C-C.
1
2
3 4
56
a a
a
a
a
aee
e
e
ee
e = ecuatoriala = axial
"doblez"
1 2
3
45
6
a
a
a
a
a
a
ee
e
ee
e
Los enlaces axiales de la estructura de la izquierda, se convierten en enlaces ecuatoriales en la estructura de la derecha, cuando el anillo se "dobla".
En este compuesto, también existe una conformación de bote, la cual no resulta tan estable porque algunos de los enlaces se encuentran eclipsados.
H
HH
H
H
H
H
H
H
H
H
H
HH
H H
H H HH
H HH H
Silla Bote
“caballete”
Newman

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 54
2.2. Isómeros geométricos
Este tipo de isomería se debe al impedimento a la rotación en torno de un doble enlace (C=C o C=N). Recordemos que el enlace π se forma sólo si los orbitales p son paralelos. Para que haya rotación se debería romper el enlace π, y éste podría volver a formarse cuando los orbitales p quedaran nuevamente paralelos.
Como consecuencia del impedimento a la libre rotación en un doble enlace, las configuraciones (I) y (II) son distintas y cada una responde a un compuesto determinado:
(I) (II)
Aunque se gire (II) en torno de distintos ejes, nunca coincidirá con (I). Son dos configuraciones diferentes. Se dice que no son superponibles, y se las llama diasterómeros.
Ejemplo: Compuestos de fórmula molecular C4H8 y estructura CH3-CH=CH-CH3 (2-buteno), se presenta en dos formas diferentes:
Una representación más simple es la siguiente:
C C
H H
CH3 CH3
C C
H CH3
CH3 H Cis-2-buteno Trans-2-buteno
Cis significa “del mismo lado”, mientras que trans “de lados opuestos”. Es decir, en el cis-2-buteno los grupos metilo están del mismo lado del plano perpendicular al plano donde se orientan los enlaces σ y en el trans 2-buteno los grupos metilo están de lados opuestos respecto al mismo plano.
En la tabla podemos ver algunas diferencias en las propiedades de los isómeros:
Propiedades Cis-2-buteno Trans-2-buteno Punto de ebullición, ºC 4 1
Punto de fusión, ºC -139 -106
El mayor punto de ebullición del isómero cis se explica por la polaridad de la molécula; en el isómero trans el momento dipolar es igual a cero. Observar que los enlaces formados por solapamiento de sp2 - s y sp3 - sp2 son polares, debido a la diferencia de electronegatividad entre carbono e hidrógeno y entre carbonos con distinta hibridación. En cuanto al mayor punto de fusión del isómero trans se debe a la mayor simetría de su molécula. Con respecto a la estabilidad, el isómero trans es el más estable por presentar los grupos más voluminosos (CH3) más alejados, lo que minimiza el factor estérico o espacial.
C C
H
H 3 C
H
CH 3
C C
H
H 3 C
CH 3
H
C C
A
B
D
E
C C
A
B
E
D

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 55
Al principio hablamos del impedimento a la rotación en torno al doble enlace, pero hay otra condición para que haya isomería geométrica y es que cada carbono del doble enlace debe ir unido a dos átomos o grupos de átomos distintos. Es decir, para que haya isomería geométrica se debe cumplir:
1º Existencia de doble enlace (C=C ó C=N).
En este curso consideraremos sólo compuestos con enlaces C=C, pero debemos tener en cuenta que también puede haber isomería geométrica con unión C=N.
2º Cada carbono del doble enlace debe ir unido a dos átomos o grupos de átomos distintos.
Los ejemplos siguientes explican por qué para que haya isomería geométrica se debe cumplir también esta segunda condición:
C
C
a b
a b
C
C
a b
b a
C
C
a b
c d
C
C
a b
d c
C
C
a a
c b
C
C
a a
b c
I II III
(I) Son dos configuraciones distintas, aunque movamos según distintos ejes una de las moléculas nunca coincidirá con la otra. Se trata de dos isómeros.
(II) Son dos configuraciones distintas, aunque movamos según distintos ejes una de las moléculas nunca coincidirá con la otra. Se trata de dos isómeros.
En (III) no hay isomería, hemos representado al mismo compuesto dos veces. Si imaginamos un eje paralelo al papel y giramos la segunda molécula, observamos que coincide con la primera.
2.3. Sistema E-Z
Cuando encontramos compuestos como el 1-bromo-2-cloropropeno, en donde los cuatro sustituyentes son diferentes no se puede usar el sistema cis-trans de nomenclatura. Para este tipo de compuestos se inventó el sistema Z-E
C C
Cl
CH3H
Br
C C
CH3
ClH
Br
Para dar nombre a un isómero con esta nomenclatura se trabaja de la siguiente manera.
Se determinan las prioridades relativas de los dos grupos unidos a cada uno de los carbonos sp2. Dicha prioridad se determina según el número atómico de los átomos directamente unidos a cada uno de los C que participan del doble enlace. El átomo de mayor número atómico tiene la mayor prioridad.
Si el doble enlace presenta los dos grupos de mayor prioridad del mismo lado del plano de referencia se le asigna la configuración Z (del alemán zusammen) Cis: (Si los grupos son iguales)
Si el doble enlace presenta los dos grupos de mayor prioridad de lados opuestos del plano de referencia se le asigna la configuración E (del alemán entgegen) Trans: (Si los grupos son iguales)
Entonces en el 1-bromo-2-cloropropeno uno de los carbonos sp2 está unido a un Br y un H. El número atómico del bromo es mayor que el del hidrógeno, de modo que el Br es el de mayor prioridad. El otro

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 56
carbono sp2 está unido a un Cl y a un C. El cloro tiene el número atómico mayor, por lo que es el prioritario.
C C
Cl
CH3H
Br
C C
CH3
ClH
Br
(Z)- 1-bromo-2-cloropropeno (E)- 1-bromo-2-cloropropeno
Si los dos grupos unidos a un carbono sp2 comienzan en el mismo átomo (hay un empate) habrá que alejarse del punto de unión y tener en cuenta los números atómicos de los átomos unidos a los átomos empatados
ClH2CH2C
CH2
CH
CH2OH
CH3
CH3
Cl
ClH2C
ClH2CH2C
CH
CH2OH
CH3
CH3
Isómero Z Isómero E
El C del grupo CH2Cl está unido a Cl, H, H y el C del grupo CH2CH2Cl está enlazado con C, H, H. El Cl tiene mayor número atómico que el C, de manera que el grupo CH2Cl tiene mayor prioridad. Lo mismo ocurre con los sustituyentes del otro C con hibridación sp2 siendo el grupo prioritario CH2OH por poseer el O el mayor número atómico.
2.4. Isómeros ópticos
Los isómeros ópticos son moléculas que coinciden en todas sus propiedades excepto en la capacidad de desviar el plano de luz polarizada. Es por esto, que el tipo de isomería más interesante es el que da lugar a la actividad óptica.
A principios de siglo XIX el físico Jean-Baptiste Biot señaló que algunas sustancias orgánicas de origen natural poseían la propiedad de girar el plano de la luz polarizada. Este fenómeno consiguió explicarse cuando los químicos comenzaron a considerar la disposición tridimensional de las moléculas en el espacio y la configuración tetraédrica del átomo de carbono
Las propiedades geométricas de un carbono con hibridación sp3 hacen que, en el caso de que esté unido a cuatro átomos o grupos de átomos diferentes, la molécula no tenga plano de simetría y que existan dos maneras diferentes de ordenar a los cuatro átomos o grupos de átomos. Éstas dos configuraciones originan dos formas isoméricas especulares no superponibles llamadas enantiómeros.
A B C D E
La figura muestra ejemplos de planos de simetría: (A) el metano tiene seis (sólo se indica uno). (B) el clorometano tiene tres. (C) el diclorometano tiene sólo dos. (D) el bromoclorometano sólo uno y (E) el bromoclorofluormetano ninguno. Las moléculas quirales no pueden tener ningún plano de simetría.
El carbono con hibridación sp3 que está unido a cuatro átomos o grupo de átomos diferentes se denomina centro quiral o estereocentro. Cuando se presenta este caso se dice que la molécula es quiral y ópticamente

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 57
activa, ya que es capaz de desviar el plano de la luz polarizada.
De esta forma, se dice que dos enantiómeros se diferencian por:
1. Sus propiedades químicas son idénticas, salvo frente a reactivos ópticamente activos.
2. Sus propiedades físicas son iguales, excepto la dirección de rotación del plano de la luz polarizada.
La dirección de rotación de dos enantiómeros es distinta, pero de igual magnitud. Por lo tanto la rotación específica de uno de ellos es (+) y la del otro (-), pero el valor absoluto es igual.
Actividad óptica
La actividad óptica es la capacidad de una sustancia quiral para rotar el plano de la luz polarizada y se mide utilizando un polarímetro.
La luz normal consiste en ondas electromagnéticas que vibran en todas las direcciones. Cuando la luz pasa a través de un polarizador las ondas electromagnéticas vibran en un plano y dicho plano de oscilación coincide con el plano de propagación de la onda.
Cuando la luz polarizada pasa a través de una cubeta que contiene una sustancia quiral, se produce una rotación en el plano de polarización y por ello reciben el nombre de sustancias ópticamente activas.
Cuando un compuesto ópticamente activo, rota la luz polarizada en el sentido de las agujas del reloj, se dice que es dextrógiro y se representa por (+). Y, si por el contrario, rotan la luz en sentido contrario a las agujas del reloj, son levógiras y se representa por (-).
2.4.1. Rotación específica
Cuando un haz de luz incide sobre una molécula, los electrones de los distintos enlaces y los más cercanos a los núcleos interaccionan con el vector campo eléctrico de la luz. Cuando la luz polarizada plana atraviesa una sustancia quiral, el campo eléctrico interacciona de modo diferente con lo que podríamos llamar mitades “izquierda” y “derecha” de la molécula. Esta diferente interacción da como resultado una rotación del plano de polarización denominada rotación óptica y la muestra que la provoca se dice que es ópticamente activa.
La rotación medida (en grados) en el polarímetro, es la rotación óptica observada, α, de la muestra. Su valor es función de la concentración y de la estructura de la molécula activa, de la longitud de la cubeta que contiene la muestra, de la longitud de la onda de luz incidente, del disolvente y de la temperatura. Con el fin de evitar ambigüedades se ha establecido un valor estándar de α, el cual se conoce como rotación específica,, característica de cada compuesto.
Donde:
Rotación Haz de luz
Fuente lumínica
Onda de luz normal, oscilando en todos los planos. Analizador
El plano gira
Cubeta con: (-)-2-bromobutano
Polarizador (filtro)
Luz polarizada plana, oscilando en un único plano.

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 58
[α] = Rotación específica
λ = Longitud de onda de la luz incidente; la fuente de luz más común es la correspondiente a la línea D de una lámpara de vapor de sodio (λ = 589,3 nm).
T = temperatura en ºC.
α = Rotación observada.
L = Longitud de la cubeta que contiene la muestra, en dm.
c = concentración en gramos por mL de disolución.
Recordar: La rotación específica es una constante física característica de las moléculas ópticamente activas.
2.4.2. Sistema de nomenclatura R, S
Cuando estamos en presencia de distintos estereoisómeros de un compuesto, nos preguntamos cuáles son los nombres que identifican a cada uno de ellos. Para evitar confusiones, es necesario utilizar un sistema de nomenclatura que indique el ordenamiento de los átomos o grupos en torno al centro asimétrico. Por este motivo, en química se emplean las letras R y S. Este sistema fue inventado por R. S. Cahn, C. Ingolg y V. Prelog.
Recordar: En cada par de enantiómeros con un centro asimétrico, un miembro tendrá la configuración R y el otro la configuración S.
Antes de aplicar esta nomenclatura a un estereocentro, se deben asignar prioridades a los sustituyentes mediante el uso de reglas de secuencia.
Regla 1: Se consideran los átomos directamente unidos al estereocentro.
Un sustituyente con mayor número atómico tiene preferencia sobre los de número atómico inferior.
Cuando dos sustituyentes tienen idéntica prioridad, se exploran las cadenas de dichos sustituyentes hasta encontrar un punto de diferencia.
Los dobles o triples enlaces se tratan como si fueran sencillos, duplicando o triplicando los átomos de la cadena.
Regla 2: Orientar la molécula de modo que el átomo o grupo que tenga la prioridad menor se aleje del espectador. A continuación trazar una flecha imaginaria del grupo o átomo con la mayor prioridad hasta el grupo o átomo con la siguiente prioridad mayor.
1. Si la flecha apunta en el sentido de las manecillas del reloj, el centro asimétrico tiene la configuración R ( Rectus en latín: Derecho).
2. Si la flecha apunta en contra del sentido de las manecillas del reloj, el centro tiene una configuración S (Sinister en latín: Izquierdo).
2.4.3. Fórmulas de Fischer
Una proyección de Fischer es una forma simplificada de representar, en dos dimensiones, átomos de carbono tetraédricos y sus sustituyentes. En este método, la molécula se dibuja en forma de cruz, con el átomo central en el punto de intersección. Por convenio, las líneas horizontales representan enlaces dirigidos hacia el observador, y las verticales enlaces que se alejan de éste. Las moléculas en perspectiva deben disponerse de esta forma para facilitar su transformación en proyecciones de Fischer.
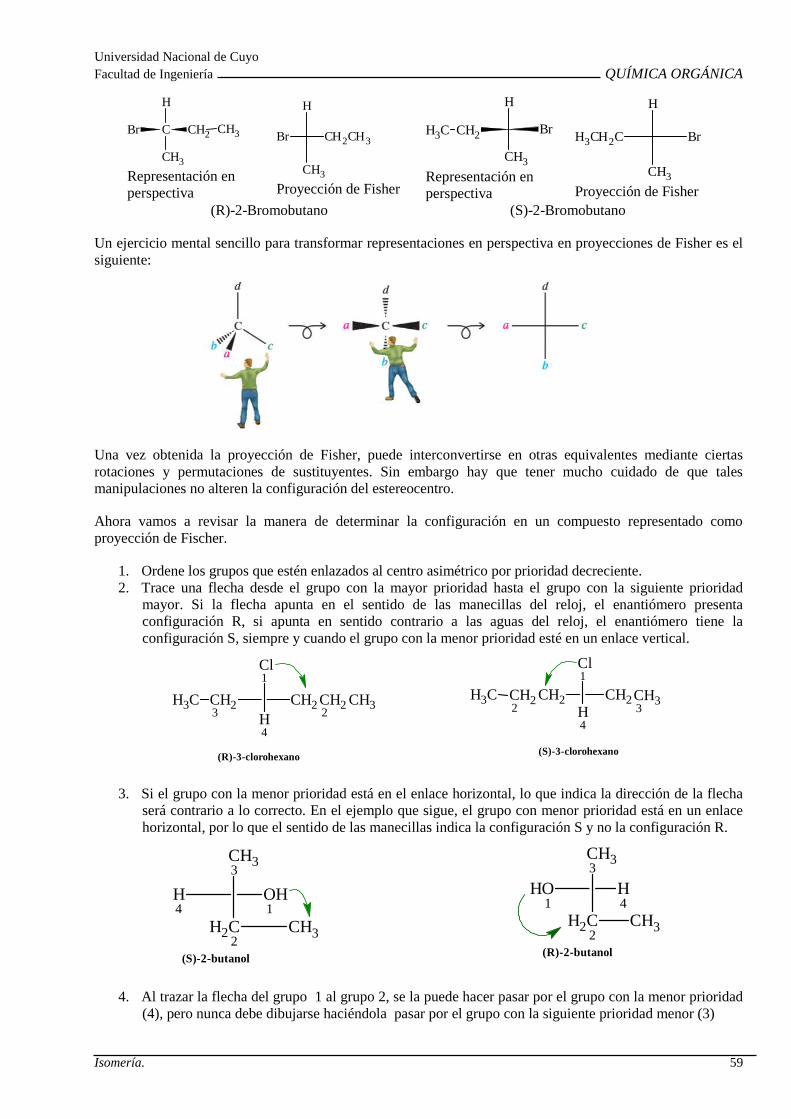
Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 59
C CH2
CH3
CH3Br
H
Representación en perspectiva
CH3
H
Br CH2CH3
Proyección de Fisher
CH2
CH3
CH3 Br
H
Representación en perspectiva
CH3
H
BrH3CH2C
Proyección de Fisher
(R)-2-Bromobutano (S)-2-Bromobutano
Un ejercicio mental sencillo para transformar representaciones en perspectiva en proyecciones de Fisher es el siguiente:
Una vez obtenida la proyección de Fisher, puede interconvertirse en otras equivalentes mediante ciertas rotaciones y permutaciones de sustituyentes. Sin embargo hay que tener mucho cuidado de que tales manipulaciones no alteren la configuración del estereocentro.
Ahora vamos a revisar la manera de determinar la configuración en un compuesto representado como proyección de Fischer.
1. Ordene los grupos que estén enlazados al centro asimétrico por prioridad decreciente. 2. Trace una flecha desde el grupo con la mayor prioridad hasta el grupo con la siguiente prioridad
mayor. Si la flecha apunta en el sentido de las manecillas del reloj, el enantiómero presenta configuración R, si apunta en sentido contrario a las aguas del reloj, el enantiómero tiene la configuración S, siempre y cuando el grupo con la menor prioridad esté en un enlace vertical.
CH3 CH23
CH2CH22
CH3
Cl1
H4
(R)-3-clorohexano
CH33
CH2CH2CH22
CH3
Cl1
H4
(S)-3-clorohexano
3. Si el grupo con la menor prioridad está en el enlace horizontal, lo que indica la dirección de la flecha será contrario a lo correcto. En el ejemplo que sigue, el grupo con menor prioridad está en un enlace horizontal, por lo que el sentido de las manecillas indica la configuración S y no la configuración R.
H4
OH1
CH33
CH22
CH3
(S)-2-butanol
H4
OH1
CH33
CH22
CH3
(R)-2-butanol
4. Al trazar la flecha del grupo 1 al grupo 2, se la puede hacer pasar por el grupo con la menor prioridad (4), pero nunca debe dibujarse haciéndola pasar por el grupo con la siguiente prioridad menor (3)

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 60
H4
CH33
COH2
OH1
O
ácido (S)-láctico
H4
CH33
COH2
OH1
O
ácido (R)-láctico
Ejemplos
1. Compuestos con un centro quiral
a. Ejemplo 1: 2-bromobutano
Fórmula estructural: CH3 CHBr CH2 CH3 C1 C2 C3 C4
Centro quiral: C2 va unido a –H, -Br, -CH3, -CH2-CH3
Configuraciones posibles:
Los enantiómeros giran el plano de la luz polarizada el mismo ángulo pero en sentidos opuestos. Así, para el 2-bromobutano el enantiómero (-) gira el plano 23,1º en sentido antihorario, y su imagen especular (+)-2-bromobutano lo hace 23,1º en sentido horario. De aquí se desprende que una mezcla 1:1 de enantiómeros (+) y (-) no presenta rotación óptica y es por lo tanto ópticamente inactiva. Este tipo de mezcla se denomina mezcla racémica.
2. Compuesto con más de un centro quiral
a. Ejemplo 1: 3-cloro-2-butanol
Muchos compuestos orgánicos tienen más de un centro asimétrico. Cuantos más centros asimétricos tenga un compuesto, más estereoisómeros puede tener.
Conociendo el número de centros asimétricos que tiene un compuesto es posible calcular el número máximo de estereoisómeros del mismo:
Un compuesto puede tener un máximo de 2n estereoisómeros, donde n es igual al número de centros asimétricos.
Fórmula estructural:
HC
CH3
H3CH2CBr
CH2CH3C
CH3
HBr
Imágenes especulares – No superponibles Enantiómeros

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 61
CH3 CH CH CH3
Cl OH
* *
C1C2C3C4
2 centros quirales: C2 y C3 Configuraciones posibles:
CH3
C C
CH3Cl
OH
H
H CH3
CC
CH3Cl
OH
H
H
CH3
CC
CH3H
OH
Cl
HCH3
C C
CH3H
OH
Cl
H
1 2 3 4
Imágenes especulares- No superponibles Imágenes especulares- No superponibles 1 y 2 son enantiómeros. 3 y 4 son enantiómeros.
1 y 3 son esteroisómeros que no son idénticos y tampoco son imágenes especulares; se llaman diasterómeros. Los diasterómeros son estereisómeros que NO son enantiómeros. Las configuraciones 1 y 4, 2 y 3, 2 y 4, también son diasterómeros.
Los diasterómeros tienen propiedades físicas distintas (distintos puntos de fusión, de ebullición, solubilidad, rotaciones específicas, entre otras) y distintas propiedades químicas.
2.4.4. Formas meso
Algunos compuestos con dos centros asimétricos tienen solamente 3 estereoisómeros en lugar de 4, es por ello que se dice que el máximo número de estereoisómeros que puede tener un compuesto con n centros asimétricos es 2n.
Un ejemplo de lo anterior es el compuesto 2,3-dibromobutano.
Fórmula estructural:
CH3 CH CH CH3Br Br
2,3-dibromobutano
Configuraciones posibles:
CH3
C
H Br
C
CH3
H Br
1
CH3
C
H Br
C
CH3
Br H
2
CH3
CBr H
C
CH3
H Br
3
El isómero faltante es la imagen especular de 1, pues 1 y su imagen especular son la misma molécula. Si esta imagen especular se gira 180º, se puede observar que 1 y su imagen especular son idénticas.
CH3
CH Br
C
CH3
H Br
CH3
CHBr
C
CH3
HBr
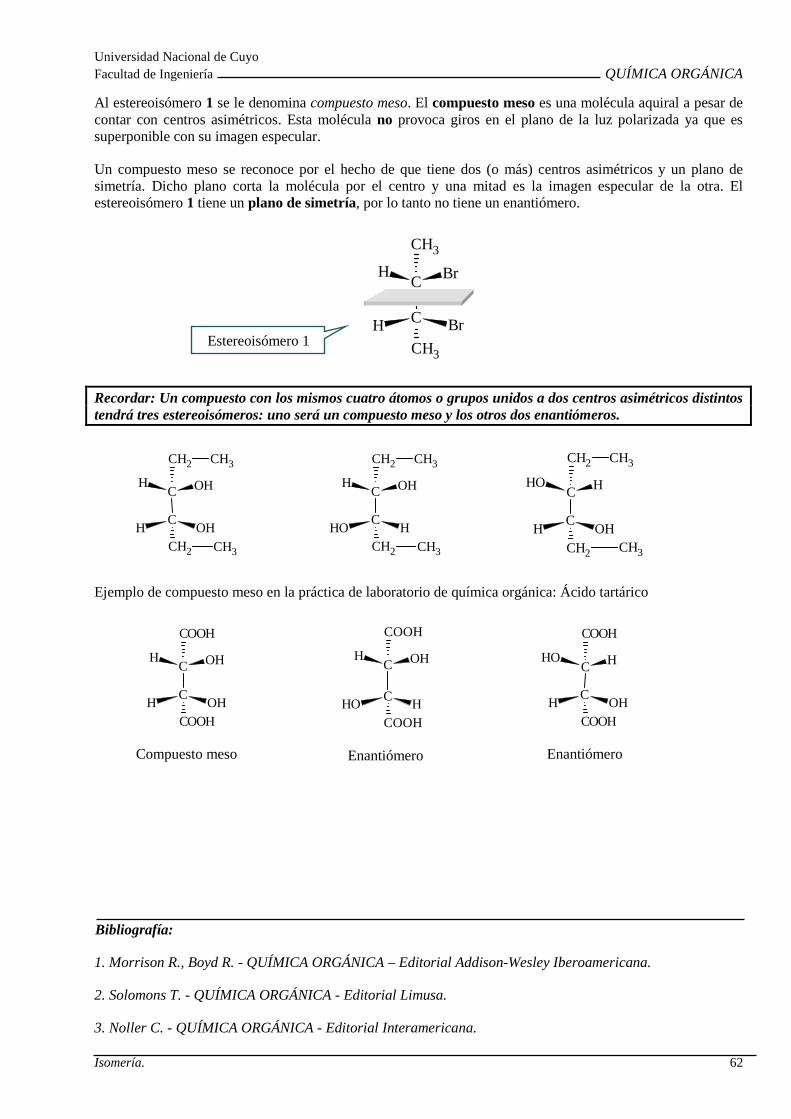
Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Isomería. 62
Al estereoisómero 1 se le denomina compuesto meso. El compuesto meso es una molécula aquiral a pesar de contar con centros asimétricos. Esta molécula no provoca giros en el plano de la luz polarizada ya que es superponible con su imagen especular.
Un compuesto meso se reconoce por el hecho de que tiene dos (o más) centros asimétricos y un plano de simetría. Dicho plano corta la molécula por el centro y una mitad es la imagen especular de la otra. El estereoisómero 1 tiene un plano de simetría, por lo tanto no tiene un enantiómero.
CH3
CH Br
C
CH3
H Br
Recordar: Un compuesto con los mismos cuatro átomos o grupos unidos a dos centros asimétricos distintos tendrá tres estereoisómeros: uno será un compuesto meso y los otros dos enantiómeros.
CH2
CH OH
C
CH2
H OH
CH3
CH3
CH2
CH OH
C
CH2
OH H
CH3
CH3
CH2
COH H
C
CH2
H OH
CH3
CH3
Ejemplo de compuesto meso en la práctica de laboratorio de química orgánica: Ácido tartárico
COOH
CH OH
C
COOHH OH
Compuesto meso
COOH
CH OH
C
COOHOH H
Enantiómero
COOH
COH H
C
COOHH OH
Enantiómero
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.
Estereoisómero 1

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 63
1. Electronegatividad La electronegatividad es una medida de la capacidad de un átomo que está unido a otro para atraer hacia sí los electrones compartidos en el enlace covalente. Cuando dos átomos de distinta electronegatividad forman un enlace covalente, los electrones no se comparten equitativamente, sino que el de mayor electronegatividad atrae con más fuerza y esto da lugar a la formación de un enlace polar.
Ejemplo:
Cl H Cl H
enlace polar
ó
El hidrógeno, que es el menos electronegativo, queda con una deficiencia de densidad electrónica, presentando una carga positiva parcial (δ+), mientras que el cloro más electronegativo se enriquece en densidad apareciendo en él una carga negativa parcial (δ-). Como la molécula de HCl tiene un extremo parcialmente positivo y otro parcialmente negativo, se dice que es una molécula polar o un dipolo.
Además, debido a que las cargas están separadas, la molécula tiene un Momento Dipolar (µ), que se expresa en unidades Debye (D). El momento dipolar es el producto de la magnitud de la carga, expresada en unidades electrostáticas, por la distancia que las separa, en Amstrong (A°):
Es importante destacar que no todas las moléculas que tienen enlaces polares, son polares. Que la molécula sea polar o no polar depende de la combinación de las polaridades de los enlaces. Esta combinación se hace en forma similar a la suma de vectores. Por ejemplo, la molécula de tetracloruro de carbono no es polar, a pesar de tener enlaces polares, debido a que la configuración de la molécula (tetraédrica) determina que no haya un dipolo resultante de la suma de los enlaces polares. En cambio la molécula de agua, por su forma no lineal es una molécula polar.
El oxígeno del agua, y los carbonos de la molécula de tetracloruro de carbono tienen hibridación sp3, con ángulos de enlaces de aproximadamente 109º. En el caso de la molécula de agua, los dos pares de electrones
sin compartir del oxígeno ocupan sendos orbitales híbridos.
Resumiendo, existen los siguientes tipos de compuestos:
Estructura y Propiedades Físicas
Electronegatividad. Polaridad de enlaces. Momento dipolar. Moléculas polares y no polares. Fuerzas intermoleculares: fuerzas de Van der Waals, atracción dipolo-dipolo, puente hidrógeno. Punto de ebullición. Punto de fusión. Solubilidad. Solvatación de iones y de moléculas covalentes. Solvatación y transformaciones orgánicas.
Cl
Cl C
Cl
Cl
H H O
2e
2e
δ− δ+ δ− δ+

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 64
2. Estructura y Propiedades Físicas Las propiedades físicas de un compuesto dan indicaciones sobre su estructura y a la vez, conociendo ésta, se pueden predecir las propiedades que presenta. Las grandes diferencias entre las propiedades físicas de los compuestos orgánicos proporcionan distintos medios de separación, caracterización e identificación de dichas sustancias
Las propiedades físicas de un compuesto dependen generalmente del tipo de enlaces que mantienen unidos a sus átomos.
La correlación entre las propiedades físicas y la estructura depende de las interacciones o fuerzas intermoleculares.
2.1. Fuerzas intermoleculares Estas son producidas por las mismas causas que las fuerzas de enlace dentro de las moléculas. Sin embargo, las interacciones intermoleculares implican distancias más grandes que las longitudes de enlaces ordinarias y las fuerzas entre las moléculas son más débiles que las que se encuentran en los enlaces químicos.
Las fuerzas intermoleculares más comunes son: atracción dipolo-dipolo, enlace por puente hidrógeno y fuerzas de Van der Waals.
2.1.1. Atracción dipolo-dipolo Dos moléculas polares se pueden asociar de manera que los polos con cargas opuestas ( δ+ y δ− ) queden próximos entre sí. El resultado de esto es una interacción atractiva neta o atracción dipolo-dipolo. Por lo tanto, la atracción dipolo-dipolo es la atracción electrostática entre estas regiones de carga positiva y carga negativa de diferentes moléculas.
Tanto en los líquidos como en los sólidos, hay una fuerte tendencia de los dipolos a alinearse y unir las moléculas.
Un tipo de atracción dipolo-dipolo fuerte es el enlace por puente de hidrógeno, que suele analizarse como otro tipo de fuerza intermolecular
- +- +
+ -
- +

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 65
2.1.2. Enlace por puente de hidrógeno (asociación puente hidrógeno) Los compuestos que contienen enlaces oxígeno- hidrógeno, nitrógeno-hidrógeno, flúor-hidrógeno, tienen una tendencia a la asociación mayor de la que podría esperarse teniendo en cuenta sus pesos moleculares y momentos dipolares.
Por ejemplo, la comparación de las propiedades físicas del agua (p.e. 100°C; p.f. 0°C) y de la acetona (p.e. 57°C; p.f. -95°C) muestran que la asociación notablemente elevada en el agua puede deberse a algunas interacciones que no están presentes en la acetona.
Evidentemente, la presencia del grupo hidroxilo tiene un pronunciado efecto sobre las propiedades físicas. El enlace oxígeno-hidrógeno es elevadamente polar, la nube electrónica se distorsiona hacia el átomo electronegativo por lo que el átomo de hidrógeno adquiere algo de carácter positivo (δ+).
Como consecuencia, el intenso campo local asociado con el hidrógeno que se une al oxígeno produce fuertes interacciones con el átomo de oxígeno (con electrones sin compartir) de una segunda molécula. Esta interacción entre el hidrógeno con δ+ de una molécula y el oxígeno con δ- de otra se conoce con el nombre de enlace por puente hidrógeno o asociación puente hidrógeno.
Por lo tanto en este enlace, el hidrógeno sirve como puente entre dos átomos electronegativos, sujetando a uno con un enlace covalente y al otro con fuerzas electrostáticas. Esta atracción tiene una fuerza de unas 5 Kcal/mol, por lo que es mucho más débil que el enlace covalente (entre 50 y 100 Kcal/mol)) que lo mantiene unido al primer átomo electronegativo, pero es más fuerte que otras atracciones dipolo- dipolo.
Para que el enlace por puente de hidrógeno sea importante, los átomos electronegativos deben ser flúor, oxígeno o nitrógeno. Sólo es suficientemente positivo un hidrógeno unido a uno de estos elementos y sólo ellos son suficientemente negativos (ya que tienen la carga negativa concentrada sobre sus átomos pequeños) para que exista la atracción necesaria.
La magnitud de esta interacción atractiva varía con la electronegatividad de los átomos unidos al hidrógeno: cuanto más electronegativo es el átomo que está unido covalentemente al átomo de hidrógeno, mayor es la atracción que experimenta ese hidrógeno hacia una densidad electrónica adicional.
La unión por puente hidrógeno se representa con líneas de puntos:
FH H F OH
H
H O
H
N
H
H
H
H N
H
H
N
H
H
H
H O
H
En las fórmulas, la unión por puente hidrógeno se representa con líneas de puntos:
En estos casos los enlaces por puente hidrógeno se forman entre varias moléculas (enlace puente hidrógeno intermolecular), pero también se pueden presentar en una misma molécula (enlace puente hidrógeno intramolecular):
HO
NR2CH2
CH2CH2
2.1.3. Fuerzas de Van der Waals Las fuerzas que existen entre las moléculas de un compuesto no polar se conocen como fuerzas de Van der Waals.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 66
Se originan porque, a pesar que no hay un momento dipolar neto debido a la distribución simétrica promedio de cargas alrededor de una molécula, los electrones se desplazan de modo que, en un instante cualquiera, esa distribución probablemente se distorsionará y habrá un pequeño dipolo. Este dipolo momentáneo afectará la distribución de electrones en otra molécula cercana; el extremo negativo del dipolo tiende a repeler electrones, y el positivo a atraerlos; es decir, el dipolo induce un dipolo de orientación opuesta en la molécula vecina:
A pesar que los dipolos momentáneos y los inducidos cambian constantemente, resulta una atracción neta entre ambas moléculas.
Estas fuerzas de Van der Waals son débiles y de corto alcance: sólo actúan entre las partes de moléculas diferentes que están en contacto, es decir de sus superficies. Por lo que la relación entre la magnitud de las fuerzas de Van der Waals y el área de las superficies moleculares es directa.
Las fuerzas de Van der Waals surgen de la interacción entre núcleos y electrones por lo que afectan a toda clase de átomos y moléculas, son débiles para moléculas pequeñas pero son especialmente importantes para moléculas no polares que nunca se licuarían si no existiesen estas fuerzas. Son el único tipo de fuerzas intermoleculares presentes en SO3, CO2, O2, N2, Br2, gases nobles, hidrocarburos, compuestos orgánicos que presentan cadenas hidrocarbonadas grandes, etc.
3. Propiedades físicas Dentro de las propiedades físicas más importantes se encuentran el punto de fusión, el punto de ebullición y la solubilidad.
3.1. Punto de Fusión El punto de fusión de un sólido es aquella temperatura a la cual el sólido se convierte en líquido.
Fusión es el cambio desde una disposición muy ordenada de partículas en el retículo cristalino al más desordenado que caracteriza a los líquidos. La fusión se produce cuando se alcanza una temperatura a la que la energía térmica de las partículas es suficientemente grande como para vencer las fuerzas intracristalinas que las mantienen en posición.
La mayoría de los compuestos orgánicos cristalinos tienen puntos de fusión característicos, lo que hace que sea la constante física generalmente más usada
En un compuesto iónico las unidades estructurales del cristal son iones. Para un sólido que cristalice en el sistema cúbico, por ejemplo cloruro de sodio, cada ión positivo está rodeado equidistantemente por seis iones negativos: uno a cada lado, uno arriba, uno abajo, uno al frente y otro detrás. A su vez cada ión negativo está rodeado de forma semejante por seis positivos. Un ión negativo no está ligado a ningún ión positivo, sino que es atraído de la misma forma por los seis iones.
El cristal, aunque presente distintas disposiciones geométricas, es una estructura muy fuerte y rígida, ya que las fuerzas electrostáticas que mantienen a cada ión en su posición son poderosas y sólo se pueden romper a altas temperaturas.
Por lo tanto, para fundir un sólido iónico hay que suministrar energía suficiente para vencer las fuerzas electrostáticas y por lo tanto tienen puntos de fusión elevados.
+ - + - + -
- + - + - +

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 67
En el caso de algunos compuestos que presentan uniones covalentes y iónicas, por ejemplo en sales como acetato de sodio, benzoato de sodio, etc., las propiedades físicas están determinadas en gran medida por los enlaces iónicos, es decir, tienen puntos de fusión altos.
En un compuesto no iónico (compuesto covalente) los átomos se mantienen unidos por enlaces covalentes; forma cristales en los que las unidades estructurales son moléculas. Las fuerzas que mantienen unidas a estas moléculas son débiles comparadas con las de los compuestos iónicos y se vencen con facilidad.
Por ejemplo, los hidrocarburos de más bajo peso molecular son gases a temperatura ambiente. Cuanto mayor es el número de átomos en una cierta molécula, más alto es su peso molecular y más grande es la suma de atracciones de Van der Waals hacia otra molécula de su clase. Por consiguiente, cuando el peso molecular aumenta, por lo general las atracciones de Van der Waals se intensifican y producen interacciones moleculares más fuertes y puntos de fusión más elevados:
Etano - 172°C
n-pentano - 130°C
n-pentadecano 10°C
No sólo influye el tamaño de las moléculas, sino también su ajuste en el retículo cristalino. Entre compuestos isómeros, el que tenga la estructura molecular más compacta, más aproximadamente simétrica, es el que tiene más probabilidad de tener el mayor punto de fusión:
o- xileno - 25°C m-xileno - 48°C p-xileno 13°C
Resumiendo:
Punto de fusión
Sólidos iónicos Sólidos no iónicos
> Peso molecular < Peso molecular > Simetría <Simetría (Pesos moleculares similares)
3.2. Punto de ebullición El punto de ebullición de un líquido es aquella temperatura a la cual la presión de vapor del líquido se iguala a la presión atmosférica del sistema. Depende de la presión, ya que los cambios de volumen al pasar del estado líquido al gaseoso son muy grandes.
En un líquido iónico las partículas tienen un arreglo menos regular que en un sólido, gozan de mayor libertad de movimiento, y cada una de ellas es atraída por muchas otras. La ebullición significa la separación de moléculas individuales, o pares de iones con carga opuesta, del seno del líquido. Esto sucede cuando se alcanza una temperatura suficiente para que la energía térmica de las partículas supere las fuerzas de cohesión que las mantiene en el líquido
La unidad de un compuesto iónico es el ión, cada ión es retenido por otros de carga opuesta, Se necesita mucha energía para que un par de iones de carga opuesta pueda dejar el líquido; la ebullición se realiza entonces a altas temperaturas.
En un compuesto no iónico la unidad es la molécula. Las débiles fuerzas intermoleculares, puentes H, dipolo- dipolo y de Van der Waals, son más fáciles de vencer que las fuerzas interiónicas de los compuestos iónicos, por lo que la ebullición se produce a temperaturas mucho menores: el metano, no polar, hierve a –161,5°C mientras que el cloruro de hidrógeno, polar, lo hace a –85°C.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 68
La intensidad de las atracciones intermoleculares aumenta de manera notable a medida que crece la magnitud de los dipolos de los enlaces individuales de las dos moléculas que interactúan.
Los líquidos cuyas moléculas se mantienen unidas por puentes de hidrógeno se denominan líquidos asociados. La ruptura de estos puentes necesita mayor energía, por lo que un líquido asociado tiene un punto de ebullición anormalmente alto para un compuesto de su peso molecular y momento dipolar: el agua tiene mayor punto de ebullición que el dimetil éter que es más pesado, pero no forma puentes de hidrógeno y por lo tanto líquidos asociados.
H
OH
CH3
CH3 O
p.e. 100°C p.e. 24°C
Por ejemplo, el metanol, que como el agua es un líquido asociado, presenta un punto de ebullición muy alto para un compuesto de su tamaño y polaridad:
CH3 O
H
H O
CH3
p.e. 64,5ºC
Cuanto más grandes son las moléculas, más fuertes son las fuerzas de Van der Waals y las fuerzas dipolo-dipolo. Algunas propiedades como la polaridad, la formación de puentes hidrógeno, se conservan similares, pero el punto de ebullición aumenta con el tamaño molecular.
Los puntos de ebullición de sustancias orgánicas son altos, pero no superan los 350°C, a temperaturas mayores comienzan a romperse los enlaces covalentes dentro de las moléculas.
Por lo tanto, el punto de ebullición aumenta regularmente con el aumento del peso molecular, pero es algo menor en los compuestos isómeros con estructuras ramificadas como resultado de su forma molecular; y mientras más numerosas son éstas, menor es la temperatura correspondiente.
n- pentano 36°C
Isopentano 28°C (con una ramificación)
Neopentano 9,5°C (con dos ramificaciones)
Esta disminución de los puntos de ebullición con las ramificaciones se observa en todas las familias de los compuestos orgánicos. Con la ramificación, se debilitan las fuerzas intermoleculares que pueden ser superadas a temperaturas más bajas. Por ejemplo, en dos moléculas de n-pentano hay más fuerzas de Van der Waals debido a su mayor superficie que en dos moléculas de neopentano:
CH3
CH2CH2
CH2CH3
CH3
CH2CH2
CH2CH3
C CH3
CH3
CH3
CH3
C CH3
CH3
CH3
CH3
Las esferas tienen, para el mismo volumen, menor superficie exterior y por lo tanto menos fuerzas de atracción y menor punto de ebullición.
Resumiendo:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 69
Punto de ebullición Líquidos iónicos Líquidos no iónicos > peso molecular < peso molecular
Puentes H > Dipolo-Dipolo > Van der Waals (Pesos moleculares similares)
3.3. Solubilidad Cuando se disuelve un sólido o un líquido, las unidades estructurales, iones o moléculas, se separan unas de otras y el espacio entre ellas pasa a ser ocupado por moléculas de solvente.
Cuando una sustancia se disuelve, sus partículas componentes (iones o moléculas) se dispersan entre las del solvente, dentro de las cuales deben crearse espacios para las partículas del soluto.
La separación, tanto de las partículas del soluto como de las del solvente, requiere de vencer las fuerzas que las mantienen unidas en cada compuesto. La energía que se requiere para superar la atracción intermolecular existente proviene de la formación de nuevas formas de atracción entre el soluto y el disolvente.
Estas fuerzas son fundamentalmente las fuerzas de Van de Waals, las interacciones dipolo - dipolo o puentes de hidrógeno, dependiendo del tipo de partículas que intervengan. En síntesis, para que la disolución se realice, la energía liberada cuando se establecen las interacciones soluto-solvente debe ser similar a la energía requerida para vencer las fuerzas de atracción entre partículas soluto-soluto y solvente-solvente.
Es por ello que se dice que “lo semejante disuelve a lo semejante”. Los compuestos polares se disuelven en disolventes polares y los compuestos no polares se disuelven en disolventes no polares. La razón por la que esto ocurre es que un disolvente polar, como el agua, tiene cargas parciales que pueden interactuar con las cargas parciales de un compuesto polar. Los polos negativos de las moléculas de disolvente rodean al polo positivo del soluto polar, y los polos positivos de las moléculas de disolvente rodean al polo negativo del soluto polar. El agrupamiento de las moléculas de disolvente en torno a las del soluto separa a las moléculas del soluto y es lo que las hace disolverse. La interacción entre las moléculas del disolvente y las del soluto se llama solvatación.
Ya que los compuestos no polares carecen de carga neta, no atraen a los disolventes polares. Para que una molécula no polar se disuelva en un disolvente polar como el agua debería empujar y separar las moléculas de agua y romper sus puentes hidrógeno. Los puentes hidrógeno encierran la resistencia suficiente para impedir la entrada del compuesto no polar. En cambio, los solutos no polares se disuelven en los disolventes no polares porque las interacciones de Van der Waals entre las moléculas de disolvente y de soluto son más o menos iguales que entre las moléculas de disolvente-disolvente y entre las moléculas de soluto-soluto.
Para solutos iónicos: las fuerzas que mantienen unido al retículo iónico son poderosas y para vencerlas se necesita una gran cantidad de energía, que es suministrada por la formación de muchos enlaces ión-dipolo entre los iones y el disolvente: una molécula polar tiene un extremo positivo y otro negativo, por lo tanto hay atracción electrostática entre un ión positivo y el extremo negativo de una molécula de solvente y entre un ión negativo y la parte positiva de la molécula de solvente.
Estas atracciones son enlaces ión- dipolo. Cada uno de estos enlaces es relativamente débil, pero en conjunto aportan suficiente energía para vencer las fuerzas interiónicas del cristal.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 70
Alrededor de cada ión se acumulan muchas moléculas de solvente, con su extremo positivo hacia un ión negativo, y su extremo negativo, hacia un ión positivo. En la solución, cada ión está rodeado por muchas moléculas de solvente, por lo que se dice que está solvatado; si el solvente es agua, está hidratado.
Para que un solvente pueda disolver compuestos iónicos, debe tener una constante dieléctrica alta, es decir, tiene que tener propiedades altamente aislantes para disminuir la atracción entre iones de carga opuesta cuando están solvatados.
El agua debe sus propiedades como disolvente de sustancias iónicas a su alta polaridad, a su elevada constante dieléctrica, y a que contiene el grupo -OH , por lo que puede formar puentes de hidrógeno.
El poder de solvatación del agua, es decir la capacidad para formar enlaces fuertes con los iones disueltos, es lo que la caracteriza. Este poder no sólo depende de un momento dipolar alto sino tiene que ver con la naturaleza de los enlaces ión-dipolo formados.
Para cationes: estos son atraídos al polo negativo del agua, que no sólo es muy electronegativo sino que tiene pares de electrones sin compartir.
Con dos pequeños hidrógenos unidos, el oxígeno está bien expuesto, varios átomos de oxígeno de otras moléculas de agua pueden agruparse alrededor del catión como en la figura:
O
H
H
δ−
δ+
δ+
+
Para aniones: éstos son atraídos al polo positivo de una molécula polar, es decir hacia los hidrógenos, y los enlaces ión –dipolo que se manifiestan son puentes de hidrógeno.
Estos enlaces de hidrógeno permiten una solvatación particularmente intensa de los aniones. No sólo hay una carga positiva grande concentrada sobre un átomo pequeño, hidrógeno, sino que este sobresale de la molécula, quedando muy expuesto. El anión puede estar sujeto por puentes de hidrógeno a varias moléculas de agua como en la figura:
OH
H
δ−
δ+
δ+
-
Así el agua, debe gran parte de su poder disolvente a su grupo –OH, solvata a los cationes por el par de electrones sin compartir del oxígeno, y a los aniones, por la formación de puentes de hidrógeno.
El metanol tiene un comportamiento similar al agua, pero disminuido debido a la presencia del grupo metilo que es más grande y ocasiona mayor aglomeración que el segundo hidrógeno de esta.
A estos solventes se los denomina próticos, porque tienen hidrógeno unido a oxígeno o nitrógeno con densidad positiva alta como para formar puentes de hidrógeno.
Existen otros solventes polares llamados apróticos que presentan constante dieléctrica elevada pero no contienen hidrógenos con elevada densidad positiva, por lo que no pueden formar puentes de hidrógeno y su acción disolvente se basa sólo en la solvatación de cationes.
Para la disolución de solutos no iónicos: la solubilidad de estos compuestos está determinada por su polaridad y su capacidad para formar puentes hidrógeno. Las sustancias no polares o débilmente polares se disuelven en solventes no polares o débilmente polares. Las débiles fuerzas de Van der Waals, que mantienen unidos a las

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 71
moléculas de soluto y a las del solvente pueden ser reemplazadas fácilmente por otras similares que son las que van a unir a las moléculas de soluto con las del solvente.
Los alcanos son no polares, por lo que son solubles en disolventes no polares e insolubles en disolventes polares como el agua. Las densidades de los alcanos aumentan al incrementarse el peso molecular, sin embargo, hasta un alcano con 30 carbonos es menos denso que el agua. Eso implica que una mezcla de alcano y agua se separará con formación de dos fases, y que la de alcano, al ser menos densa, flotará sobre el agua.
Los haluros de alquilo tiene cierto carácter polar, pero sólo los fluoruros de alquilo cuentan con un átomo que puede formar puente hidrógeno con el agua. Ello quiere decir que los fluoruros de alquilo son los más solubles en agua. Los demás haluros son menos solubles en agua que los éteres o los alcoholes con la misma cantidad de carbonos. Son solubles en los disolventes orgánicos típicos, de baja polaridad, como benceno, éter, cloroformo o ligroína.
Los yoduros, bromuros y los policlorocompuestos son más densos que el agua.
Se observa entonces que los alcanos y halogenuros de alquilo tienen las propiedades físicas que podemos esperar para compuestos de baja polaridad, cuyas moléculas se mantienen unidas por fuerzas de Van der Waals o por atracciones dipolares débiles. Tienen puntos de fusión y de ebullición relativamente bajos, y son solubles en disolventes no polares e insolubles en agua.
Hay otra consecuencia que resulta de su baja polaridad: mientras los alcanos y halogenuros de alquilo son buenos disolventes para otras sustancias de baja polaridad, son incapaces de solvatar apreciablemente iones simples, de modo que no pueden disolver sales inorgánicas.
Un alcohol tiene al mismo tiempo un grupo alquilo no polar y un grupo OH muy polar y, lo que es más importante, es capaz de establecer puentes de hidrógeno: con sus moléculas compañeras o con otras moléculas.
El comportamiento de los alcoholes como solutos refleja la tendencia a formar puentes de hidrógeno. Puesto que las moléculas de los alcoholes se mantienen unidas por el mismo tipo de fuerzas intermoleculares que las de agua, puede haber mezclas de las dos clases de moléculas. La energía necesaria para romper un puente de hidrógeno entre dos moléculas de agua o dos de alcohol, es proporcionada por la formación de un puente de hidrógeno entre una molécula de agua y otra de alcohol.
Entonces, un alcohol ¿Es una molécula polar o no polar? ¿Es soluble en un solvente no polar o es soluble en agua? La respuesta depende del tamaño del grupo alquilo. Al aumentar ese tamaño y ser una fracción más importante de la molécula del alcohol, el compuesto se vuelve cada vez menos soluble en agua. En otras palabras, la molécula se parece cada vez más a la de un alcano. Los grupos formados por cuatro carbonos tienden a estar en la línea divisoria, a temperatura ambiente: los alcoholes con menos de cuatro carbonos son solubles en agua, pero los que tienen más de cuatro carbonos son insolubles en ella. Es decir un grupo OH puede hacer que unos tres o cuatro carbonos se disuelvan en agua.
La estimación de los cuatro carbonos sólo es una guía aproximada porque la solubilidad de un alcohol también depende de la estructura del grupo alquilo. Los alcoholes con grupos alquilo ramificados son más solubles en agua que los que tienen alquilos no ramificados, con la misma cantidad de carbonos, ello se debe a que la ramificación minimiza la superficie de contacto de la parte no polar de la molécula. Entonces, el alcohol ter-butílico es más soluble en agua que el alcohol n-butílico.
Los éteres presentan una solubilidad en agua comparable a la de los alcoholes: tanto el dietiléter como el alcohol n-butílico, por ejemplo, tienen una solubilidad de unos 8 g por 100 g de agua. La solubilidad de los

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura y Propiedades Físicas 72
alcoholes inferiores se debe a los puentes de hidrógeno entre moléculas de agua y de alcohol; es probable que la solubilidad de los éteres en agua se debe a la misma causa.
Los fenoles más sencillos son líquidos o sólidos. El fenol tiene cierta solubilidad en agua (9 g por 100 g de agua) probablemente debido a la formación de puentes hidrógeno con ella; la mayoría de los otros fenoles son esencialmente insolubles.
El grupo carbonílico polarizado conviertea aldehídos y cetonas en sustancias polares. Los aldehídos y las cetonas inferiores son solubles en agua, probablemente por los puentes de hidrógeno que pueden establecerse entre las moléculas de disolvente y las de soluto. La solubilidad límite se alcanza alrededor de unos cinco carbonos. Los aldehídos y cetonas son solubles en los disolventes orgánicos usuales.
Los ácidos carboxílicos son moléculas polares, y al igual que los alcoholes pueden formar puentes de hidrógeno entre sí y con otros tipos de moléculas. Por consiguiente, los primeros son miscibles con agua, el ácido de cinco carbonos es parcialmente soluble y los superiores son virtualmente insolubles. No cabe duda de que la solubilidad en agua se debe a los puentes de hidrógeno entre el ácido carboxílico y el agua. El ácido aromático más simple, el benzoico, contiene demasiados átomos de carbono como para tener una solubilidad apreciable en agua.
Los ácidos carboxílicos son solubles en disolventes orgánicos menos polares, como éter, alcohol, benceno, etc.
Los derivados de ácidos carboxilico son solubles en disolventes como éteres, cloroalcanos e hidrocarburos aromáticos. Al igual que los alcoholes y los éteres, los compuestos carbonilicos con menos de cuatro carbonos son solubles en agua.
Los esteres, las amidas N,N- disustituidas y los nitrilos se usan con frecuencia como disolventes porque son polares.
Las aminas de bajo peso molecular son solubles en agua porque pueden formar puentes hidrógeno con el agua. Al comparar las aminas con las mismas cantidades de átomos de carbono se podrá advertir que las aminas primarias son más solubles que las secundarias porque las primarias tienen dos hidrógenos que pueden formar puentes. Las aminas terciarias, tienen un par de electrones no enlazado que puede aceptar puentes de hidrógeno pero, a diferencia de las otras carecen de hidrógenos que ceder como puentes de hidrogeno. La solubilidad límite se da al tomar unos seis átomos de carbono. Son solubles en disolventes menos polares, como éter, alcohol, benceno, etc.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Noller C. – QUÍMICA ORGÁNICA - Editorial Interamericana.
4. Sykes P. - MECANISMOS DE REACCIÓN EN QUÍMICA ORGÁNICA – Editorial Martínez Roca.
5. Bruice, P - FUNDAMENTOS DE QUIMICA ORGANICA- Editorial Pearson Education.
6. Shriner, R; Reynold, F; Curtin, D - IDENTIFICACION SISTEMATICA DE COMPUESTOS ORGANICOS - Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura, reactividad y transformaciones orgánicas 73
El estudio sistemático de las reacciones orgánicas lleva a la investigación de la influencia de la estructura sobre la reactividad.
En la estructura se debe considerar al grupo funcional y, además, al resto de la molécula porque, si bien la reactividad varía ampliamente entre compuestos que contienen diferentes grupos funcionales, se ha comprobado que también difiere entre miembros distintos de una misma familia (con igual grupo funcional).
Los efectos que pueden presentar las estructuras son los siguientes:
Efecto Inductivo
Efecto de Resonancia o Mesómero
Efecto Estérico o Espacial
1. Efecto Inductivo El efecto inductivo se genera por la tendencia de un sustituyente (átomo o grupo de átomos) a liberar o atraer electrones, es decir, por su electronegatividad. Este efecto, que puede deberse a la polaridad permanente de la molécula o a la polarización inducida por interacciones con otras especies, actúa a lo largo de la cadena carbonada y se debilita gradualmente con el aumento de la distancia al sustituyente que lo generó.
1.1. Efecto de Grupos Aceptores: La mayoría de los sustituyentes del hidrógeno en una molécula orgánica contienen elementos más electronegativos que él, por lo que estos sustituyentes suelen ejercer efecto inductivo de atracción de electrones (ej.: -F, -Cl, -OH, -NH2, -NO2) y se los llama grupos aceptores.
Por ejemplo, si en el etano se sustituye un hidrógeno por cloro, el enlace C-C que no era polar en el etano se polariza por la presencia del enlace polar carbono-cloro. Es decir, el cloro genera en la molécula de cloroetano un efecto de corrimiento de las densidades electrónicas de los enlaces, denominado Efecto Inductivo.
C C
H
H
H
H
H
H
C C
H
Cl
H
H
H
H
I
Este efecto se simboliza con la letra “I”, colocándose una flecha con la punta hacia el grupo aceptor.
1.2. Efecto de Grupos Dadores: Entre los grupos dadores de electrones se destacan los grupos alquilo. Si bien hay datos experimentales que prueban el efecto dador de los grupos alquilo, aún no se ha determinado fehacientamente por qué liberan electrones.
Un ejemplo es el del cloruro de etilo visto anteriormente, en el cual el grupo etilo está actuando como dador de electrones.
1.3. Efectos sobre la reactividad La acción del efecto inductivo sobre la reactividad será vista a lo largo del desarrollo de la materia. No obstante, se dan a continuación algunos ejemplos.
Estructura y reactividad
Efectos sobre la reactividad (Efectos inductivos, estéricos y de resonancia) Análisis de efectos sobre las estructuras de compuestos orgánicos.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura, reactividad y transformaciones orgánicas 74
Acción sobre la basicidad La basicidad de una amina se debe al par de electrones libres del átomo de nitrógeno (base de Lewis). Por lo tanto, una característica estructural que contribuya a aumentar la densidad electrónica de dicho átomo reforzará esa propiedad, mientras que una que atraiga electrones la disminuirá.
Ejemplos:
N H
H
CH3
N H
H
CH2F
Compuesto más básico Compuesto menos básico
El reemplazo del hidrógeno de la metilamina por flúor, que es más electronegativo, ejerce un efecto inductivo aceptor de electrones y, por lo tanto, disminuye la basicidad del compuesto.
Acción sobre la estabilidad de carbocationes: En un carbocatión, un carbono tiene deficiencia electrónica, lo que induce a los grupos alquilo unidos a él a ceder densidad electrónica.
De acuerdo con las leyes de la electrostática, la estabilidad de un sistema cargado aumenta con la dispersión de la carga. Por lo tanto, todo factor que permita esparcir la carga positiva del carbono deficiente en electrones y distribuirla sobre el resto del ión, debe estabilizar al carbocatión. En este caso el factor estabilizante es el efecto inductivo dador de electrones de los grupos alquilo.
C+
H
H
H
< C+
H
R
H
< C+
R
R
H
< C+
R
R
R
Catión metilo Carbocatión
1º Carbocatión
2º Carbocatión
3º Cada grupo alquilo ejerce efecto inductivo dador. A mayor cantidad de grupos alquilo, mayor es el efecto inductivo ejercido sobre el C+, mayor es la dispersión de la carga y mayor es la estabilidad del ión.
Como consecuencia del efecto inductivo ejercido, los carbocationes terciarios, que tienen mayor dispersión de la carga, son más estables y se forman con mayor rapidez que los secundarios, los primarios y el catión metilo, tal como se verá en las reacciones que ocurren a través de iones.
2. Efecto mesómero o de resonancia Este efecto se presenta en moléculas con dobles enlaces conjugados y, por lo tanto, con deslocalización de electrones π.
Analizaremos primero la molécula del benceno, que posee dobles enlaces conjugados:
Según la Teoría de la Resonancia, los electrones π están deslocalizados, por lo que al compuesto se lo representa mediante formas contribuyentes :
La representación de benceno por la Teoría de Enlace de Valencia muestra mejor la deslocalización de los electrones π en una nube extendida y uniformemente distribuida. En el benceno no hay efecto mesómero.
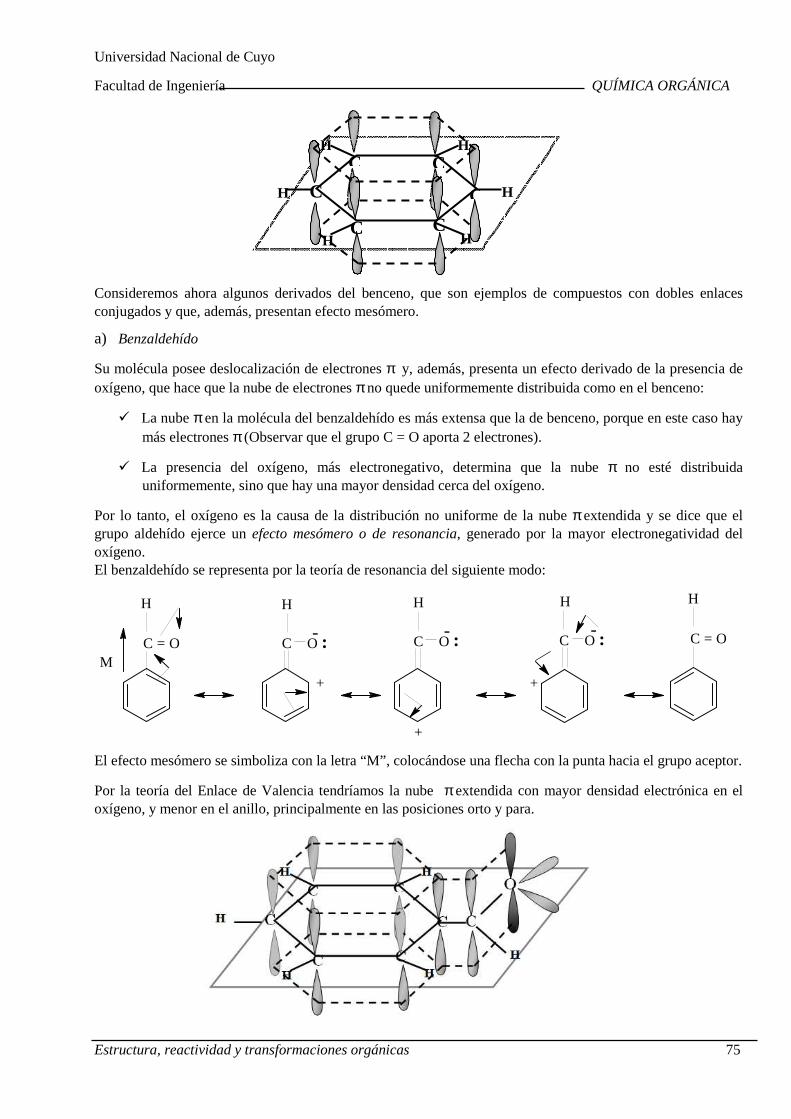
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura, reactividad y transformaciones orgánicas 75
H
C
C
C
C
H C
H
H
H
H
C
Consideremos ahora algunos derivados del benceno, que son ejemplos de compuestos con dobles enlaces conjugados y que, además, presentan efecto mesómero.
a) Benzaldehído
Su molécula posee deslocalización de electrones π y, además, presenta un efecto derivado de la presencia de oxígeno, que hace que la nube de electrones π no quede uniformemente distribuida como en el benceno:
La nube π en la molécula del benzaldehído es más extensa que la de benceno, porque en este caso hay más electrones π (Observar que el grupo C = O aporta 2 electrones).
La presencia del oxígeno, más electronegativo, determina que la nube π no esté distribuida uniformemente, sino que hay una mayor densidad cerca del oxígeno.
Por lo tanto, el oxígeno es la causa de la distribución no uniforme de la nube π extendida y se dice que el grupo aldehído ejerce un efecto mesómero o de resonancia, generado por la mayor electronegatividad del oxígeno. El benzaldehído se representa por la teoría de resonancia del siguiente modo:
C = O
H
M
+
C O :-
H
C O :-
H
C O :-
H
+
+
C = O
H
El efecto mesómero se simboliza con la letra “M”, colocándose una flecha con la punta hacia el grupo aceptor.
Por la teoría del Enlace de Valencia tendríamos la nube π extendida con mayor densidad electrónica en el oxígeno, y menor en el anillo, principalmente en las posiciones orto y para.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura, reactividad y transformaciones orgánicas 76
b) Fenol
La molécula de fenol presenta dos efectos: inductivo y mesómero.
El oxígeno, que es más electronegativo que el carbono, ejerce un efecto inductivo aceptor de electrones:
El orbital p del oxígeno, ocupado por dos electrones sin compartir, se superpone con el orbital p del C1
que es paralelo. Esto produce una extensión de la nube π deslocalizada. Es decir, el oxígeno a pesar que es más electronegativo, comparte un par de electrones que tenía sin compartir, ejerciendo así un efecto mesómero dador de electrones.
Observar que el oxígeno, más electronegativo, no puede ejercer un efecto mesómero aceptor porque tiene su octeto completo.
:O HM
+O H
..
+O H
+O H :O H
..
..
I
2.1. Efectos sobre la reactividad Cuando en una molécula hay un grupo que ejerce efecto mesómero dador de electrones, la reactividad
del compuesto frente a reactivos electrofílicos (buscadores de electrones), está favorecida (ej.: – OH, – NH2, etc.). Por ejemplo, debido al efecto mesómero que ejerce el grupo –OH, el fenol es un compuesto más reactivo que el benceno frente a reactivos buscadores de electrones.
Si el grupo ejerce el efecto mesómero aceptor de electrones es más difícil el ataque de reactivos electrofílicos (ej.: – NO2 , – C=O, – COOH, etc.).
Por ejemplo, debido al efecto mesómero que ejerce el grupo aldehido, el benzaldehido es un compuesto menos reactivo que el benceno frente a reactivos electrofílicos.
3. Efecto estérico Este efecto también llamado efecto espacial o impedimento estérico, está asociado con la presencia de grupos voluminosos en espacio insuficiente para desenvolverse normalmente.
3.1. Efectos sobre la reactividad Acción en reacciones de sustitución
Un ejemplo de la existencia de factores estéricos lo constituyen las diferencias de velocidades en reacciones de sustitución del halógeno en halogenuros de alquilo. Consideremos los siguientes casos:
bromuro de metilo
bromuro de etilo
bromuro de isopropilo
bromuro de terbutilo
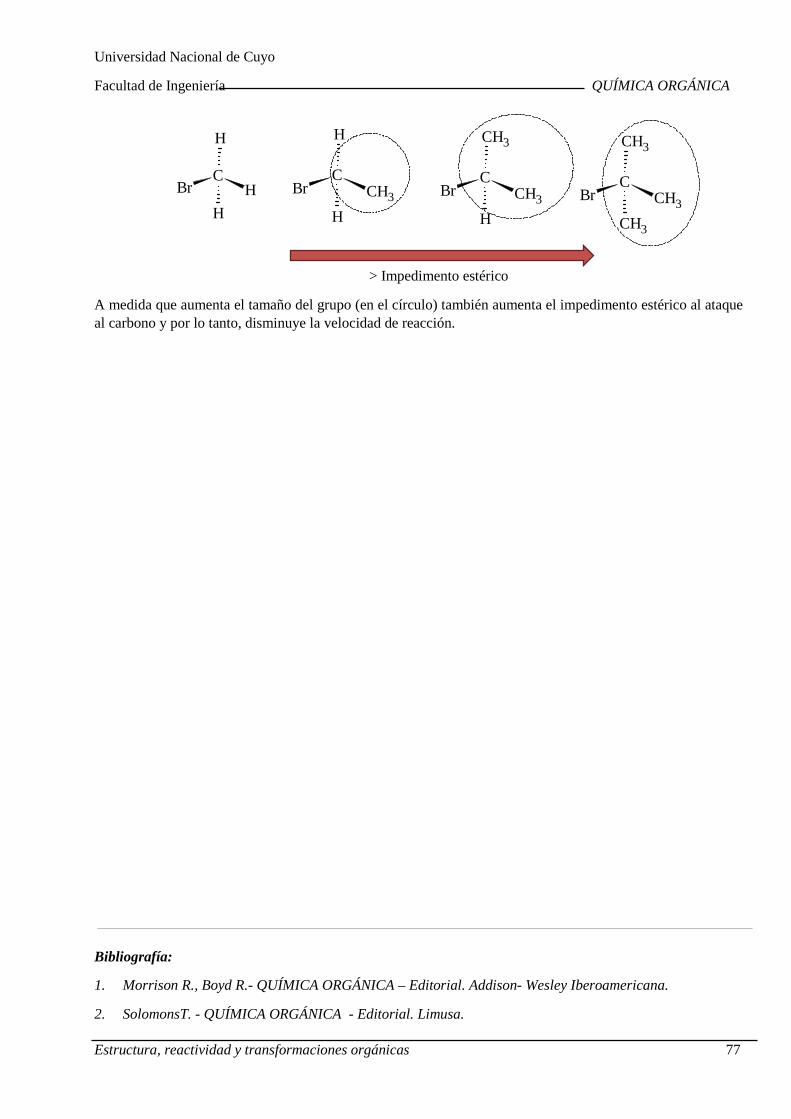
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Estructura, reactividad y transformaciones orgánicas 77
H
C
H
Br H
H
C
H
Br CH3
CH3
C
H
Br CH3
CH3
C
CH3
Br CH3
> Impedimento estérico
A medida que aumenta el tamaño del grupo (en el círculo) también aumenta el impedimento estérico al ataque al carbono y por lo tanto, disminuye la velocidad de reacción.
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial. Addison- Wesley Iberoamericana.
2. SolomonsT. - QUÍMICA ORGÁNICA - Editorial. Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 78
Los primeros químicos llamaban ácido a todo compuesto con sabor agrio. Entre los ácidos más conocidos están el ácido cítrico (presente en los limones y otras frutas cítricas), el ácido acético (que se encuentra en el vinagre), y el ácido clorhídrico (que se encuentra en los jugos gástricos del estómago, el sabor agrio asociado con el vómito). Los compuestos que neutralizan a los ácidos se denominan bases o compuestos alcalinos. Los líquidos para limpiar vidrios y para destapar cañerías son soluciones alcalinas comunes.
1. Reacción ácido-base Las reacciones de los ácidos y bases orgánicos, al igual que las reacciones semejantes de los compuestos inorgánicos, se llevan a cabo la mayoría de las veces en medio acuoso, y son normalmente rápidas y reversibles. Los términos “ácido” y “base” tienen dos definiciones, respectivamente: la de Brönsted-Lowry y la de Lewis:
1.1. Teoría de Brönsted - Lowry En la definición de Brönsted-Lowry, un ácido es una especie que dona un protón (ión hidrógeno H+), y una base es una especie que acepta un protón. En la siguiente reacción, el cloruro de hidrógeno es un ácido porque dona un protón al agua y el agua actúa como una base porque acepta un protón del cloruro de hidrógeno. A estas reacciones de un ácido con una base se las denomina reacción ácido-base.
:Cl H2O:.. -
H
ácido
H
basebase ácido
Cl: H2O:..
El agua puede aceptar un protón porque tiene dos pares de electrones no compartidos; cada par puede formar un enlace covalente con un protón. En la reacción inversa, el catión hidronio H3O
+ es un ácido porque dona un protón al anión cloruro Cl-, que actúa como base. Se observa entonces que:
Las especies que tienen un hidrógeno actúan como ácido al donar ese hidrógeno como protón H+ Las especies con pares de electrones no compartidos actúan como base al aceptar un protón H+.
Es importante destacar que el agua puede comportarse como un ácido porque tiene un protón para donar, pero también se puede comportar como una base porque tiene pares de electrones no compartidos que pueden aceptarlo.
1.2. Teoría de Lewis Lewis ofreció nuevas definiciones más amplias de ácido y base:
Ácido: especie que acepta un par de electrones para formar un enlace covalente Base: especie que dona un par de electrones para formar un enlace covalente
Todos los ácidos que donan protones (definición de Brönsted-Lowry) se apegan también a la definición de Lewis. En el ejemplo anterior, el: Cl– acepta los electrones del enlace H–Cl que quedan libres por la interacción del hidrógeno con la base (agua). Simultáneamente, el agua actúa como donador de electrones cuando un par no compartido participa en un enlace σ O–H en el ión hidronio. Pero la definición de Lewis es más amplia, porque no se limita solamente a las especies que liberan protones, sino que además abarca algunos compuestos que tienen orbitales de valencia sin llenar y, así, pueden aceptar un par de electrones. Un ejemplo es la reacción entre el tricloruro de aluminio y el amoniaco:
Acidez y basicidad de compuestos orgánicos
Ácidos y bases de Lewis. Ácidos y bases de Brönsted Lowry. Acidez de alquinos, alcoholes, fenoles y ácidos carboxílicos. Influencia de los sustituyentes sobre la acidez. Basicidad de alcoholes y aminas. Influencia de los sustituyentes sobre la basicidad.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 79
Actúa como ácido,
aún sin tener H:NH3
-
ácido base
:NH3
El aluminio sólo tiene 6 electrones en su capa externa, por ello tiende a aceptar otro par para completar su octeto y en consecuencia actúa como un ácido, aún sin tener protones para donar. Este tipo de compuesto se denomina ácido de Lewis.
El amoniaco tiene un par de electrones para compartir por lo que es una base. Todas las bases son bases de Lewis porque todas tienen un par de electrones para compartir, ya sea con un protón, o con otro tipo de átomo, como el aluminio.
1.3. Equilibrio Ácido-Base Cuando un compuesto actúa como ácido de Brönsted-Lowry y pierde un protón la especie resultante se llama base conjugada. Del mismo modo, cuando un compuesto actúa como base y acepta un protón, la especie resultante se llama ácido conjugado. Veamos por ejemplo la siguiente reacción:
ácido y base fuertes ácido y base débiles
:NH3:Cl :NH3-
H
ácido
H
baseconjugada
base ácidoconjugado
Cl:
El ácido clorhídrico es un ácido fuerte ya que tiende a entregar un protón con mucha facilidad. Esto significa que el ión cloruro es una base débil debido a que tiene poca tendencia a aceptar un protón. En cambio, el amoniaco es una base fuerte porque tiene mucha tendencia a aceptar un protón, mientras que el ión amonio es un ácido débil. En reglas generales, se establece que:
Cuanto más fuerte es un ácido, más débil es su base conjugada Cuanto más débil es un ácido, más fuerte es su base conjugada El equilibrio favorece la formación del ácido y base más débiles
2. Acidez de compuestos orgánicos La acidez de los compuestos orgánicos se observa experimentalmente en el análisis del equilibrio ácido-base, y se explica mediante el análisis de sus estructuras.
2.1. Análisis del equilibrio ácido-base
2.1.1. Constante de acidez y pKa Cuando se disuelve en agua un ácido fuerte (cloruro de hidrógeno), casi todas sus moléculas se ionizan y en el equilibrio de la reacción se ven favorecidos los productos. Por el contrario, cuando se disuelve en agua un ácido débil (ácido acético), sus moléculas se ionizan parcialmente y en el equilibrio se ven favorecidos los reactivos. El grado en que se ioniza un ácido (HA) se señala mediante la constante de acidez o constante de
disociación ácida aK (en cuyo cálculo la concentración de agua se asume constante):
AH H2O-A H3O
Ka=H3O
+A-
HA

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 80
Cuanto mayor es la constante de disociación del ácido, más fuerte es el ácido y mayor es su tendencia a donar un protón. Además, cuando más estable es el anión A- formado, más ácido es HA. Por lo tanto, la acidez tiene
una relación directa con la estabilidad del anión que se genera. La escala de aK sirve también para
correlacionar la reactividad relativa de los aniones como bases. Por conveniencia, la fuerza de un ácido suele
indicarse por medio de su valor apK donde: aa KpK log−= . Se observa que mientras más pequeño es el
apK más fuerte es el ácido.
Ácidos muy fuertes apK < 1
Ácidos moderadamente fuertes apK = 1-3
Ácidos débiles apK = 3-5
Ácidos muy débiles apK = 5-15
Ácidos extremadamente débiles apK >15
Para determinar la posición de equilibrio de una reacción ácido-base es necesario comparar el valorapK del
ácido que se encuentra a la izquierda con el valorapK del ácido que se encuentra a la derecha. En el
equilibrio se favorece la reacción del ácido fuerte y la formación del ácido débil. Es decir, el fuerte reacciona para formar uno débil.
2.1.2. Reactividad frente a metales activos El carácter ácido de un compuesto orgánico, definido como la tendencia a liberar protones hidrógeno, se evidencia al ponerlo en presencia de un metal activo. En estas reacciones se desprende gas hidrógeno y se forma la sal correspondiente.
Alquinos terminales HC ≡ C– H + Na → HC ≡ C:– Na+ + ½ H2
acetiluro de sodio
Alcoholes R – O – H + Na → R – O:– Na+ + ½ H2
alcóxido de sodio
Fenoles Ar – O – H + Na → Ar – O:– Na+ + ½ H2
fenóxido de sodio
Ácidos Carboxílicos R – COO – H + Na → R – COO:– Na+ + ½ H2
carboxilato de sodio
Si esta reacción ocurre para un compuesto dado, significa que esa sustancia presenta carácter ácido. Sin embargo, y en líneas generales, una sustancia con carácter ácido podrá actuar como tal siempre que no esté en presencia de un ácido más fuerte que ella misma. Analizamos entonces la acidez relativa de los distintos compuestos.
2.1.3. Acidez relativa. Análisis del equilibrio ácido-base Para determinar la acidez relativa de un compuesto (ej. HA) con respecto a otro (ej.: HB) se tiene que demostrar que el ácido más fuerte tiene capacidad para desplazar de sus sales al segundo. Experimentalmente, se lleva a cabo para cada par de ácidos la reacción de uno de ellos (HA) con la sal del otro (MB) y se determina hacia donde está desplazado el equilibrio, lo que permite deducir cómo es la acidez de uno con respecto al otro.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 81
HA M AM B -- HB
HA actúa como ácido de izquierda a derecha, mientras que HB lo hace de derecha a izquierda. Si el primero es el más ácido, el equilibrio estará desplazado a la derecha. Si HB es más fuerte, el equilibrio se desplazará a la izquierda.
A continuación se muestran reacciones en las que están involucrados compuestos orgánicos que se comportan como ácidos de Brönsted- Lowry:
Alquinos: Acidez con respecto al amoniaco: Se analiza el equilibrio de la reacción entre el acetileno (un alquino) con amiduro de litio (una sal del amoniaco cuando actúa como ácido):
H– C ≡ CH + H2N: – Li + → H – NH2 + HC ≡ C: – Li +
La reacción de acetileno con amiduro de litio forma amoniaco y acetiluro de litio, pero la reacción inversa es muy difícil, por lo tanto el acetileno es más ácido que el amoniaco.
Acidez con respecto al agua: HO – H + HC ≡ C:– Li + H – C ≡ C – H + HO: – Li+
El ácido más débil H – C ≡ CH es desplazado de su sal por el ácido más fuerte H-OH. Por lo tanto, la acidez relativa es:
Acidez relativa: H2O > HC ≡ CH > NH3
Alcoholes: Acidez con respecto al agua:
R– O: – Na+ + H – OH R – OH + NaOH
En la reacción de un alcóxido (la sal del alcohol, ej. Alcóxido de sodio) con agua se obtiene el hidróxido y el alcohol correspondientes, lo que conduce a afirmar que los alcoholes son menos ácidos que el agua (con excepción del metanol).
Acidez con respecto al acetileno: R – OH + HC ≡ C: – Na+ HC ≡ C–H + R – O: – Na+
El acetileno es desplazado de su sal por el alcohol, entonces:
Acidez relativa: H2O > R – OH > HC ≡ CH
Fenoles: Acidez con respecto al agua:
Ar O – H + NaOH Ar O: – Na + + H2O
El equilibrio se desplaza hacia la derecha, es decir que el agua no puede desplazar al fenol de su sal: el fenol es más ácido.
Acidez con respecto a alcoholes: Ar O – H + R – O: – Na+ Ar O: – Na + + R – OH

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 82
Acidez con respecto al ácido carbónico: H2CO3 + Ar O: – Na + Ar O – H + CO3H – Na+
El equilibrio está desplazado a la derecha, lo que indica que el ácido carbónico es más fuerte que fenol, por lo tanto la acidez relativa es:
Acidez relativa: H2CO3 > Ar – OH > H2O > R – OH
Ácidos carboxílicos: Son los más ácidos dentro de los compuestos orgánicos. Reaccionan con hidróxidos y con carbonatos ácidos. En este último caso se observa el desprendimiento de dióxido de carbono en forma gaseosa.
RCOO – H + NaOH RCOO: – Na + + H2O
RCOO – H + NaHCO3 RCOO: – Na + + H2O + CO2
En base al análisis anterior, ordenamos los compuestos estudiados de acuerdo a su acidez relativa
Acidez relativa: RCOOH > H2CO3 > ArOH > H2O > ROH > HC ≡ CH > NH3
Recordamos que un ácido fuerte da una base conjugada débil (no acepta el H+ con facilidad) y que si el ácido es débil su base conjugada es fuerte (acepta H+ con facilidad). Entonces, analizando las bases conjugadas de estos ácidos, la basicidad relativa será:
Basicidad relativa: RCOO– < HCO3– < ArO– < HO– < RO– < HC ≡ C– < NH2
–
La misma conclusión se obtiene al observar las constantes de disociación ácida de los compuestos (a mayor
aK mayor acidez)
Compuesto Ka
ácidos carboxílicos RCOOH 10-4 a 10-5 ácido carbónico H2CO3 10-7 fenoles Ar OH 10-10 alcoholes R OH 10-16 a 10-18 agua H2O 2.10-16 acetileno HC≡CH 10-25 amoniaco NH3 10-38
2.2. Análisis de estructuras Alquinos:
La acidez del acetileno y de los alquinos con el triple enlace en el extremo de la cadena (R-C≡CH) es una propiedad única que diferencia estos hidrocarburos de los alcanos y alquenos. Un carbono con triple enlace es más electronegativo que el carbono con enlace simple o doble. Por lo tanto, un hidrógeno unido a un carbono con triple enlace, como en el acetileno o en otro alquino con triple enlace en el extremo de la cadena, tiene una acidez considerable.
HC ≡ CH H+ + HC ≡ C: –
acetileno ión acetiluro

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 83
La teoría del enlace de valencia explica este fenómeno basándose en la influencia que tiene la hibridación del átomo de carbono unido a un hidrógeno sobre la acidez: con más carácter s en el orbital híbrido (sp > sp2 > sp3), más cerca del núcleo están los electrones compartidos con el átomo de hidrógeno. Esto hace que, por un lado, aumente la electronegatividad del C, y por ende la polaridad del enlace C-H dejando más expuesto el H. Por otro lado, el anión que se forma al romper el enlace C-H es más estable cuanto más carácter s tiene el orbital híbrido del carbono, porque puede acomodar mejor la carga. Además, el anión formado es una base más débil porque los electrones están menos disponibles al aumentar el carácter s, es decir que su ácido conjugado es más fuerte.
Alcanos Alquenos Alquinos sp3 sp2 sp
25% carácter s 33% carácter s 50% carácter s
Aumento de: electronegatividad del C polaridad del enlace C-H
fuerza con que los electrones son retenidos estabilidad del anión
acidez Alcoholes
Los alcoholes son sustancias que pueden actuar como ácidos o como bases.
Como ácidos, los alcoholes presentan acidez similar al agua. El hidrógeno está unido al oxígeno (muy electronegativo), y la polaridad del enlace O-H facilita la separación de un protón, mientras que el oxígeno acomoda muy bien la carga negativa de los electrones dejados.
R – O – H → R– O: – + H+
La diferencia entre el alcohol y el agua es el grupo alquilo:
Estado Base Anión Formado
H – O – H R → O – H I
H – O:– R → O:– I
En el agua, al no haber grupo alquilo y por consiguiente efecto inductivo, el enlace O-H es más polar que en el alcohol. Además, el anión del agua es más estable que el del alcohol, en el cual el efecto inductivo aumenta la densidad negativa sobre el oxígeno, haciéndolo menos estable. Por otro lado, la solvatación del anión contribuye a la estabilización del mismo. A mayor tamaño del grupo alquilo, la solvatación se dificulta por el impedimento estérico, lo cual reduce la estabilidad del anión.
Alcohol Primario Alcohol Secundario Alcohol Terciario
Estado Base
H
R C O H
H
I I
H
R C O H
R
I I
I
R
R C O H
R
I I
I
I
El O atrae los electrones del enlace O-H dejando al
H más expuesto
Al aumentar el número de radicales, aumenta el efecto inductivo y la densidad electrónica sobre el O, que retiene con más fuerza el protón H y disminuye la
acidez

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 84
Anión Formado
H
R C O:–
H
I I
H
R C O:–
R
I I
I
R
R C O:–
R
I I
I
I
Al aumentar el número de radicales, el anión se desestabiliza por el efecto inductivo (dificultad para acomodar la carga) y por menor solvatación (impedimento
estérico): disminuye la acidez
Fenoles Los fenoles son compuestos ácidos, siendo más ácidos que los alcoholes y el agua.
ArOH ArO:– + H+
Observando las estructuras del alcohol, del fenol y de los respectivos aniones, se deduce:
Estado base Anión formado
R O–H R O:-
(I) (II)
: :-
(IV)
:
(III )
:
o En (I) el efecto inductivo de R aumenta la densidad electrónica sobre el oxígeno, lo que hace que se fortalezca el enlace O - H y, por lo tanto se dificulte su ruptura.
o En el ión alcóxido (II), el efecto inductivo de R inestabiliza al anión. Si es menos estable es más difícil de formar
o En (III) el efecto mesómero provoca un déficit de densidad electrónica en el oxígeno, lo que significa una mayor atracción por parte del oxígeno de los electrones que comparte con el hidrógeno. Esto facilita la ruptura de dicho enlace.
o En el ión fenóxido (IV), hay una gran estabilización por resonancia, es decir, la carga negativa está dispersa en el anión, lo que favorece su estabilización.
A continuación se muestran estructuras contribuyentes del fenol y el anión correspondiente: estructuras V a VII para fenol y VIII al X para el anión.
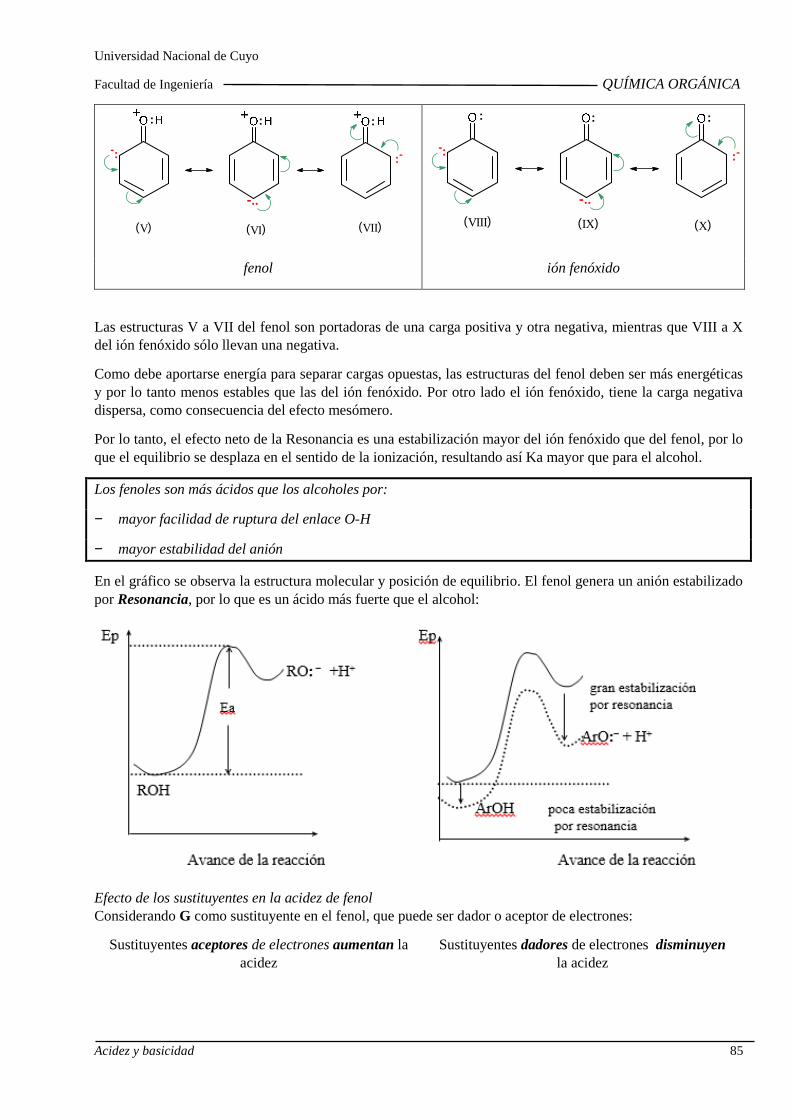
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 85
(VII )
:
(VI)(V)
:--:
::
-..
(X)
:
:-
(VIII )
-:
(IX)
::
-..
fenol ión fenóxido
Las estructuras V a VII del fenol son portadoras de una carga positiva y otra negativa, mientras que VIII a X del ión fenóxido sólo llevan una negativa.
Como debe aportarse energía para separar cargas opuestas, las estructuras del fenol deben ser más energéticas y por lo tanto menos estables que las del ión fenóxido. Por otro lado el ión fenóxido, tiene la carga negativa dispersa, como consecuencia del efecto mesómero.
Por lo tanto, el efecto neto de la Resonancia es una estabilización mayor del ión fenóxido que del fenol, por lo que el equilibrio se desplaza en el sentido de la ionización, resultando así Ka mayor que para el alcohol.
Los fenoles son más ácidos que los alcoholes por:
− mayor facilidad de ruptura del enlace O-H
− mayor estabilidad del anión
En el gráfico se observa la estructura molecular y posición de equilibrio. El fenol genera un anión estabilizado por Resonancia, por lo que es un ácido más fuerte que el alcohol:
Efecto de los sustituyentes en la acidez de fenol Considerando G como sustituyente en el fenol, que puede ser dador o aceptor de electrones:
Sustituyentes aceptores de electrones aumentan la acidez
Sustituyentes dadores de electrones disminuyen la acidez
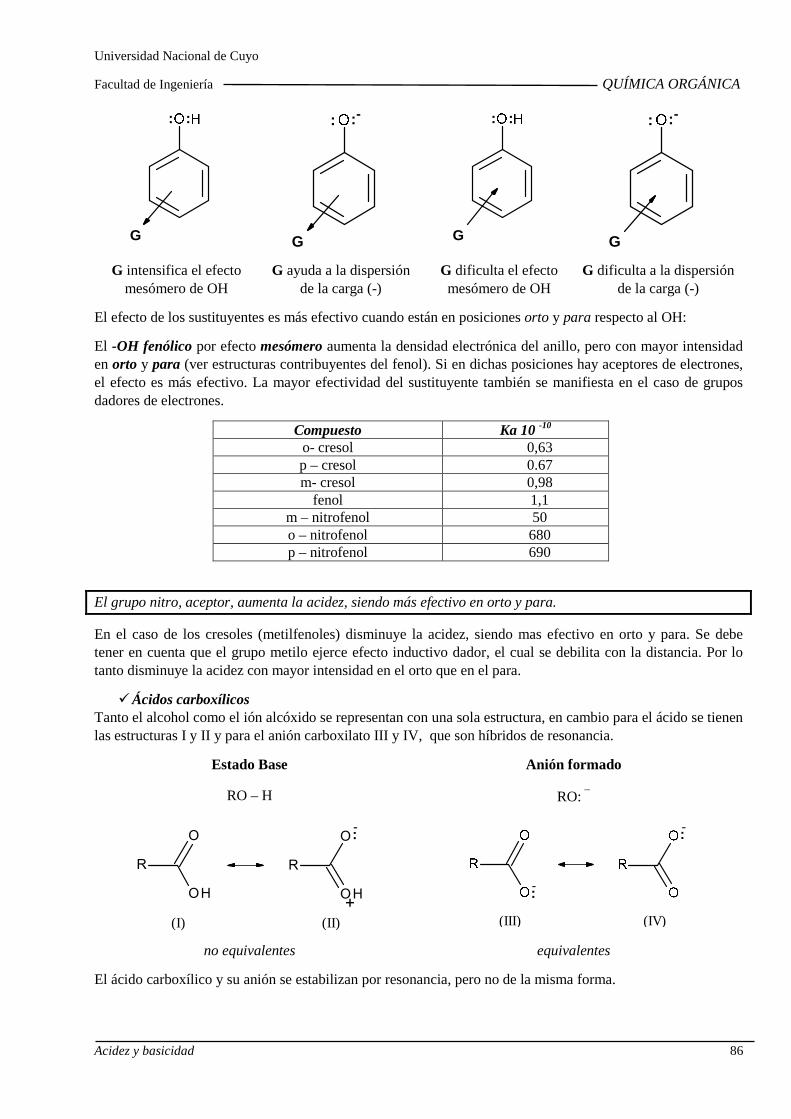
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 86
:
G
:
G
:-:
:
G
:
G
:-:
G intensifica el efecto mesómero de OH
G ayuda a la dispersión de la carga (-)
G dificulta el efecto mesómero de OH
G dificulta a la dispersión de la carga (-)
El efecto de los sustituyentes es más efectivo cuando están en posiciones orto y para respecto al OH:
El -OH fenólico por efecto mesómero aumenta la densidad electrónica del anillo, pero con mayor intensidad en orto y para (ver estructuras contribuyentes del fenol). Si en dichas posiciones hay aceptores de electrones, el efecto es más efectivo. La mayor efectividad del sustituyente también se manifiesta en el caso de grupos dadores de electrones.
Compuesto Ka 10 -10 o- cresol 0,63 p – cresol 0.67 m- cresol 0,98
fenol 1,1 m – nitrofenol 50 o – nitrofenol 680 p – nitrofenol 690
El grupo nitro, aceptor, aumenta la acidez, siendo más efectivo en orto y para.
En el caso de los cresoles (metilfenoles) disminuye la acidez, siendo mas efectivo en orto y para. Se debe tener en cuenta que el grupo metilo ejerce efecto inductivo dador, el cual se debilita con la distancia. Por lo tanto disminuye la acidez con mayor intensidad en el orto que en el para.
Ácidos carboxílicos Tanto el alcohol como el ión alcóxido se representan con una sola estructura, en cambio para el ácido se tienen las estructuras I y II y para el anión carboxilato III y IV, que son híbridos de resonancia.
Estado Base Anión formado
RO – H RO: –
-
(I)
:
(II)
O
O
R
HHO
O
R
+
(III) (IV)
-
-:
:
no equivalentes equivalentes
El ácido carboxílico y su anión se estabilizan por resonancia, pero no de la misma forma.
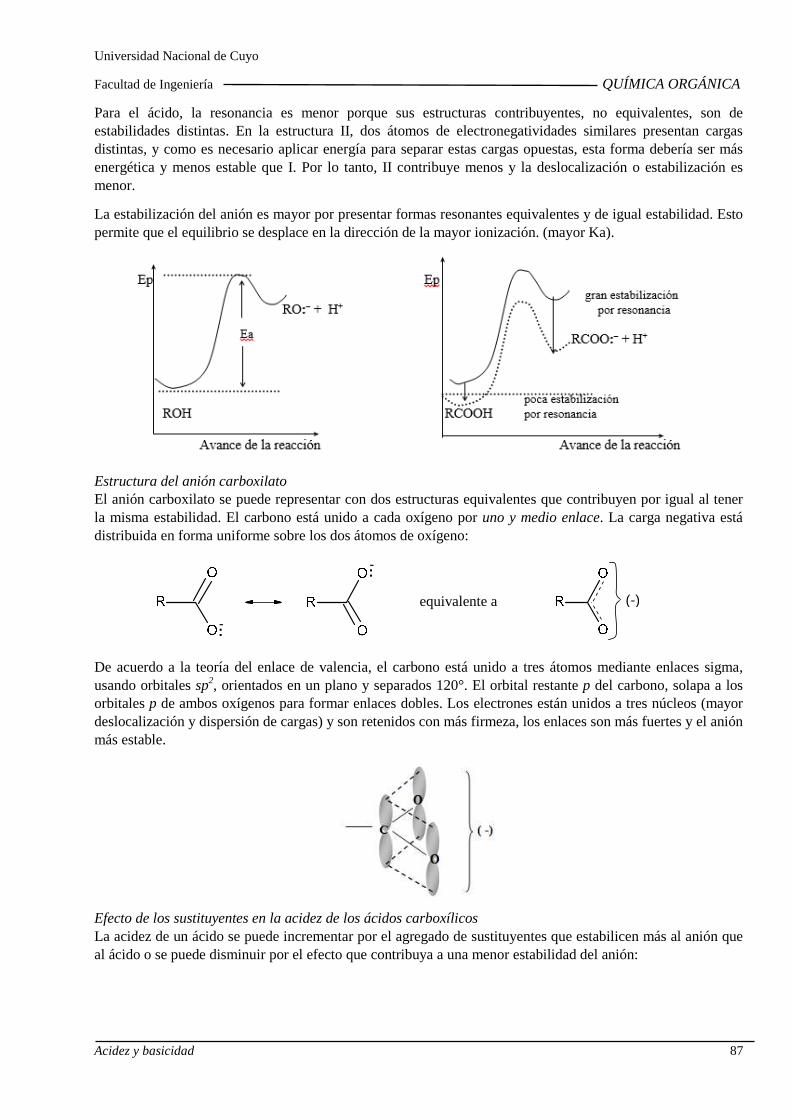
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 87
Para el ácido, la resonancia es menor porque sus estructuras contribuyentes, no equivalentes, son de estabilidades distintas. En la estructura II, dos átomos de electronegatividades similares presentan cargas distintas, y como es necesario aplicar energía para separar estas cargas opuestas, esta forma debería ser más energética y menos estable que I. Por lo tanto, II contribuye menos y la deslocalización o estabilización es menor.
La estabilización del anión es mayor por presentar formas resonantes equivalentes y de igual estabilidad. Esto permite que el equilibrio se desplace en la dirección de la mayor ionización. (mayor Ka).
Estructura del anión carboxilato El anión carboxilato se puede representar con dos estructuras equivalentes que contribuyen por igual al tener la misma estabilidad. El carbono está unido a cada oxígeno por uno y medio enlace. La carga negativa está distribuida en forma uniforme sobre los dos átomos de oxígeno:
-
-:
:
equivalente a (-)
De acuerdo a la teoría del enlace de valencia, el carbono está unido a tres átomos mediante enlaces sigma, usando orbitales sp2, orientados en un plano y separados 120°. El orbital restante p del carbono, solapa a los orbitales p de ambos oxígenos para formar enlaces dobles. Los electrones están unidos a tres núcleos (mayor deslocalización y dispersión de cargas) y son retenidos con más firmeza, los enlaces son más fuertes y el anión más estable.
Efecto de los sustituyentes en la acidez de los ácidos carboxílicos La acidez de un ácido se puede incrementar por el agregado de sustituyentes que estabilicen más al anión que al ácido o se puede disminuir por el efecto que contribuya a una menor estabilidad del anión:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 88
G (-)
G atrae electrones:
estabiliza al anión
aumenta la acidez
G (-)
G libera electrones:
desestabiliza al anión
disminuye la acidez
En los ácidos alifáticos, los sustituyentes que atraen electrones aumentan su acidez. Sin embargo, si el efecto ejercido es el inductivo, a medida que el sustituyente se aleja en la cadena del carboxilo, su efecto se disminuye y deja de ser importante a través de más de cuatro carbonos.
Ácido Ka x 10-5 Fórmico 17,7 Acético 1,75
Cloroacético 136 Tricloroacético 23200
Butanoico 1,52 2-clorobutanoico 139 3-clorobutanoico 8,9
En los ácidos aromáticos los efectos de los sustituyentes actúan de la misma forma. Los grupos que aumentan la acidez son los aceptores y los grupos que disminuyen la acidez son los dadores.
Los grupos que ejercen mayores efectos sobre la reactividad en sustituciones electrofílicas o nucleofílicas, activando o desactivando, también los tienen sobre la acidez.
Ácido Ka x 10-5 benzoico 6,3
p-nitrobenzoico 36 m-nitrobenzoico 32 o-nitrobenzoico 670 p-metilbenzoico 4,2 m-metilbenzoico 5,4 o-metilbenzoico 12,4
p-metoxibenzoico 3,3 m-metoxibenzoico 8,2 o-metoxibenzoico 8,2 p-hidroxibenzoico 2,6 m-hidroxibenzoico 8,3 o-hidroxibenzoico 105
Se ha comprobado que los grupos –OH y –OCH3 ejercen distinta influencia de acuerdo a sus dos tipos de efectos: desde la posición meta un efecto inductivo atractivo de electrones que aumenta la acidez y desde la posición para un efecto de resonancia liberador de electrones que disminuye la acidez.
En los ácidos aromáticos orto- sustituídos no se cumplen las reglas dadas: casi todos los sustituyentes localizados en la posición orto ejercen un efecto del mismo tipo - aumentan la acidez - atraigan o liberen electrones.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 89
3. BASICIDAD DE COMPUESTOS ORGÁNICOS La basicidad de un compuesto se demuestra por su reacción frente a ácidos, de los que toman el protón para formar un catión:
-B: B: HH A -
ácido débilbase fuerte
A:
Se puede determinar la fuerza de una base mediante la siguiente reacción de equilibrio:
B: B: HH OH -
base más débil
base más fuerte
HO:
La constante de equilibrio para esta reacción es:
Por ejemplo, la aplicación de esta ecuación a una amina primaria, metilamina (H3CNH2) da:
H3CNH2 H3CNH3H2O-
base más débil
base más fuerte
HO:
Alcoholes Los alcoholes son bases de Lewis de fuerza similar a la del agua. Como el agua, contienen oxígeno que presenta pares de electrones sin compartir, lo que hace que puedan actuar como bases:
..
..R O H R O H
Por su basicidad, los alcoholes se pueden comportar como reactivos nucleófilos (buscadores de centros positivos) y como sustratos en reacciones orgánicas (ej.: SN y E).
Los alcoholes como reactivos nucleófilos: Aceptan protones de carbocationes, con los que pueden provocar una eliminación:
..R OH R OH2
Los alcoholes como sustratos: Aceptan protones de ácidos para generar alcoholes protonados, y pueden actuar como sustratos en la Sustitución Nucleofílica y en la Eliminación.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 90
a) Sustitución Nucleofílica:
:
alcohol halogenuro de alquilo
alcohol protonado
H2O..
R OH2 R OH R X
b) Eliminación:
alcohol alcohol protonado alqueno
-H2O..
A mayor número de radicales alquilos, mayor efecto inductivo, mayor densidad electrónica sobre el oxígeno y mayor basicidad.
Por lo tanto, el orden de basicidad de los alcoholes es inverso al de acidez:
Basicidad: R3CO-H > R2CHO-H > RCH2O-H
Aminas Las aminas poseen un par de electrones libres sobre nitrógeno, por lo tanto son bases de Lewis con un comportamiento químico semejante al del amoniaco. La tendencia del nitrógeno a compartir este par de electrones determina el comportamiento químico de las aminas: la basicidad, la acción nucleofílica y en el caso de las aminas aromáticas también la alta reactividad del anillo.
Las aminas reaccionan con ácidos minerales convirtiéndose en sales, de acuerdo a: ..
RNH2
..[RNH3]H : A -A:
La basicidad depende de la posibilidad de acomodar una carga positiva. Comparando la fuerza como base de compuestos tales como un hidróxido, una amina primaria y el agua, experimentalmente resulta:
HO:– + H+ H2O Kb = 1,0.10-2
H3CNH2 + H+ H3CNH3+ Kb = 4,5. 10-4
H2O + H+ H3O+ Kb = 1,0. 10-16
Por lo tanto:
Basicidad: HO:– > RNH2 > H2O
Las aminas también son más básicas que los alcoholes, éteres, ésteres, etc, porque el nitrógeno es menos electronegativo que el oxígeno y, además de ser mejor dador de electrones, puede acomodar mejor la carga positiva del catión.
Análisis de las estructuras de aminas alifáticas, aminas aromáticas y amoniaco: Para analizar la basicidad de las aminas, se debe comparar las estabilidades de las mismas, con las de sus iones; mientras más estables sean estos en relación con las aminas de las que derivan, más básicas son aquellas.
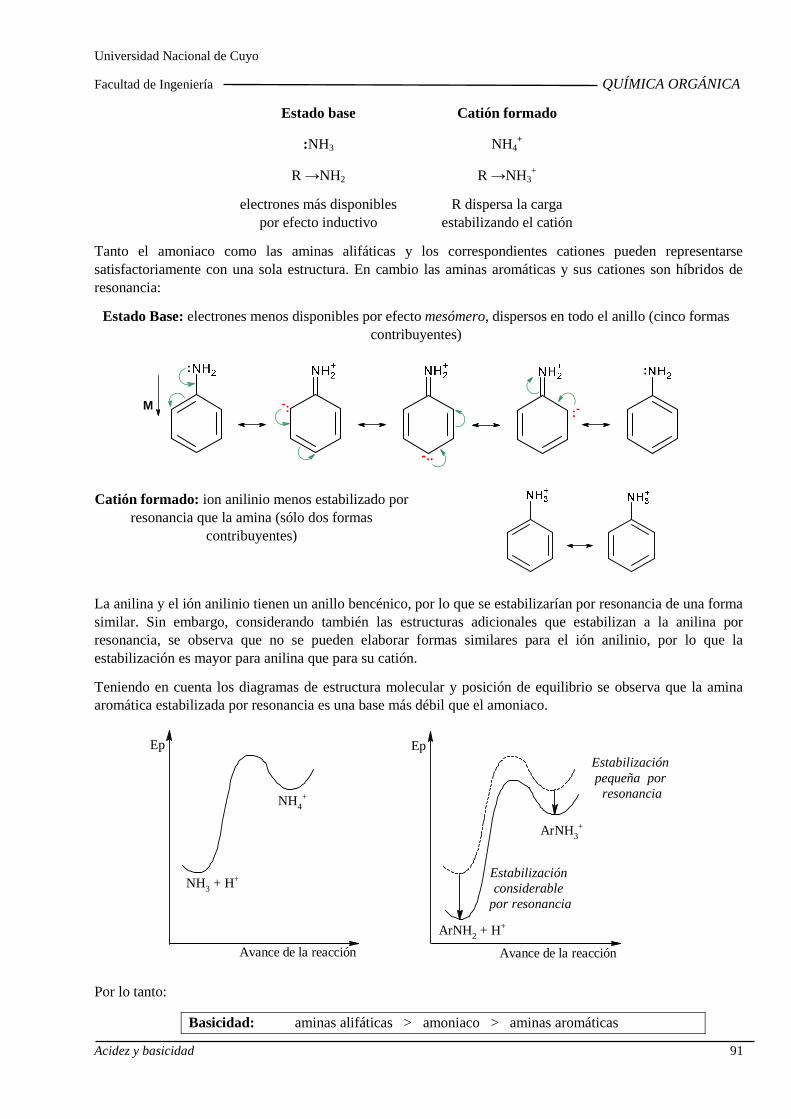
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 91
Estado base Catión formado
:NH3 NH4+
R →NH2
electrones más disponibles por efecto inductivo
R →NH3+
R dispersa la carga estabilizando el catión
Tanto el amoniaco como las aminas alifáticas y los correspondientes cationes pueden representarse satisfactoriamente con una sola estructura. En cambio las aminas aromáticas y sus cationes son híbridos de resonancia:
Estado Base: electrones menos disponibles por efecto mesómero, dispersos en todo el anillo (cinco formas contribuyentes)
M
:
:-
:
-:
-..
Catión formado: ion anilinio menos estabilizado por resonancia que la amina (sólo dos formas
contribuyentes)
La anilina y el ión anilinio tienen un anillo bencénico, por lo que se estabilizarían por resonancia de una forma similar. Sin embargo, considerando también las estructuras adicionales que estabilizan a la anilina por resonancia, se observa que no se pueden elaborar formas similares para el ión anilinio, por lo que la estabilización es mayor para anilina que para su catión.
Teniendo en cuenta los diagramas de estructura molecular y posición de equilibrio se observa que la amina aromática estabilizada por resonancia es una base más débil que el amoniaco.
Avance de la reacción
Ep
NH3 + H+
NH4+
Avance de la reacción
Ep
ArNH2 + H+
ArNH3+
Estabilización considerable
por resonancia
Estabilización pequeña por resonancia
Por lo tanto:
Basicidad: aminas alifáticas > amoniaco > aminas aromáticas
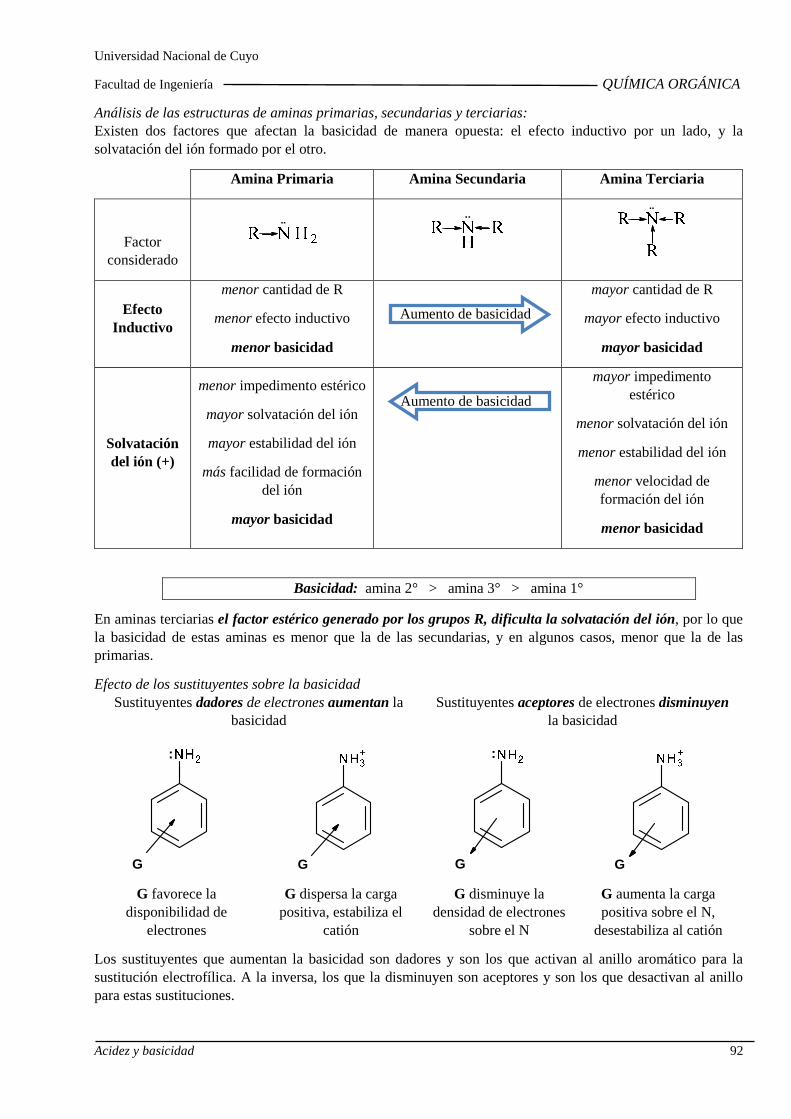
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 92
Análisis de las estructuras de aminas primarias, secundarias y terciarias: Existen dos factores que afectan la basicidad de manera opuesta: el efecto inductivo por un lado, y la solvatación del ión formado por el otro.
Amina Primaria Amina Secundaria Amina Terciaria
Factor considerado
..
..
..
Efecto Inductivo
menor cantidad de R
menor efecto inductivo
menor basicidad
mayor cantidad de R
mayor efecto inductivo
mayor basicidad
Solvatación del ión (+)
menor impedimento estérico
mayor solvatación del ión
mayor estabilidad del ión
más facilidad de formación del ión
mayor basicidad
mayor impedimento estérico
menor solvatación del ión
menor estabilidad del ión
menor velocidad de formación del ión
menor basicidad
Basicidad: amina 2° > amina 3° > amina 1°
En aminas terciarias el factor estérico generado por los grupos R, dificulta la solvatación del ión, por lo que la basicidad de estas aminas es menor que la de las secundarias, y en algunos casos, menor que la de las primarias.
Efecto de los sustituyentes sobre la basicidad Sustituyentes dadores de electrones aumentan la
basicidad Sustituyentes aceptores de electrones disminuyen
la basicidad
G
:
G G
:
G
G favorece la disponibilidad de
electrones
G dispersa la carga positiva, estabiliza el
catión
G disminuye la densidad de electrones
sobre el N
G aumenta la carga positiva sobre el N,
desestabiliza al catión
Los sustituyentes que aumentan la basicidad son dadores y son los que activan al anillo aromático para la sustitución electrofílica. A la inversa, los que la disminuyen son aceptores y son los que desactivan al anillo para estas sustituciones.
Aumento de basicidad
Aumento de basicidad

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 93
G es más efectivo cuando está en orto y para respecto al grupo amino porque son esas posiciones las que ven aumentadas las densidades electrónicas por el efecto mesómero del grupo amino. Si en esas posiciones hay un aceptor, este refuerza el efecto mesómero del grupo amino, disminuyendo la basicidad.
Los grupos dadores de electrones generan un efecto opuesto al anterior, es decir aumentan la basicidad.
Sin embargo, se ha observado lo siguiente:
los sustituyentes dadores de electrones disminuyen la basicidad cuando están en posición orto.
los sustituyentes aceptores de electrones, que disminuyen la basicidad, lo hacen con más fuerza cuando están en posición orto.
A este efecto de los sustituyentes en orto se lo denomina efecto orto. Probablemente, se deba al efecto estérico en orto, que perjudica la solvatación del ión (+).
En la Tabla siguiente se muestran las Kb de anilina, metilanilinas y nitroanilinas:
Compuesto Kb
p-toluidina 12 x 10-10
m-toluidina 5 x 10-10
anilina 4,2 x 10-10
o-toluidina 2,6 x 10-10
m-nitroanilina 0,029 x 10-10
p-nitroanilina 1 x10-12
o-nitroanilina 6 x 10-15
Se observa que o-toluidina tiene menos basicidad que anilina, a pesar de tener un grupo dador; del mismo modo o-nitroanilina es menos básica que p- nitroanilina.
Bases Heterocíclicas
o Pirrol El pirrol es una base muy débil, Kb = 2,5 x 10-14, comparada con la mayoría de las aminas:
..N
H
El pirrol es un híbrido de resonancia de las siguientes estructuras contribuyentes, donde la donación de electrones del nitrógeno al anillo queda indicada por las estructuras iónicas, en las que el nitrógeno tiene una carga positiva y los carbonos del anillo una negativa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 94
.. ..:-
-:
-:
-..
El par de electrones del nitrógeno, responsable de la basicidad de los compuestos nitrogenados, está comprometido en la nube π, por lo que no puede ser compartido con H+. Por este motivo, el pirrol presenta menor carácter básico que otros compuestos nitrogenados.
o Piridina La piridina es una base, Kb = 2,3 x 10-9, más fuerte que pirrol:
La piridina tiene un par de electrones en un orbital sp2 que puede compartir con H+ mientras que el pirrol sólo puede aceptar un ácido a expensas del carácter aromático del anillo.
..
+
+ +...... .. .. ..
..N N N NN
Es menos básica que las aminas alifáticas, ya que en éstas el par de electrones responsable de la basicidad ocupa un orbital sp3, con mayor carácter p que s. Al haber alto carácter p, el par de electrones es retenido menos firmemente, por lo que está más disponible para la reacción con un ácido.
En la piridina el par de electrones responsable de la basicidad se encuentra en un orbital sp2 y es retenido con más fuerza debido a su menor carácter p y mayor s que está más cerca del núcleo.
Resumiendo:
Basicidad:
aminas alifáticas > amoniaco > piridina > aminas aromáticas > pirrol > alcoholes > agua
Compuesto Constante de basicidad RNH2,; R2NH; R3NH, 10-3 a 10-4
NH3 10-5 ArNH2 4,2 x 10-10
C5H5N (piridina) 2,3 x 10-9 C4H5N(pirrol) 2,5 x 10-14
H2O 1,0 x 10-16 ROH ≈1,0 x 10-16

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Acidez y basicidad 95
RESUMEN
Definición de Brönsted-Lowry Ácido: compuesto dador del ión hidrógeno (H+)
Base: compuesto aceptor del ión hidrógeno (H+)
Definición de Lewis Ácido: especie que acepta un par de electrones para formar un enlace covalente
Base: especie que dona un par de electrones para formar un enlace covalente
Ácido Fuerte → Base conjugada Débil Ácido Débil → Base conjugada Fuerte El equilibrio favorece la formación de base y ácido débiles ACIDEZ Un compuesto es más ácido que otro si desplaza de sus sales al segundo. Ácidos fuertes tienen bases conjugadas (aniones) estables. La estabilización por resonancia de la base conjugada da la posibilidad de dispersar la carga
negativa del anión y aumenta la acidez Los sustituyentes aceptores de electrones aumentan la acidez BASICIDAD La capacidad de acomodar una carga positiva aumenta la basicidad La mayor estabilidad en el catión formado aumenta la basicidad Los sustituyentes aceptores de electrones disminuyen la basicidad Tanto la acidez como la basicidad aumentan con la solvatación del ión formado.
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial Addison - Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA – Editorial Limusa.
3. Yurkanis Bruice P.- FUNDAMENTOS DE QUÍMICA ORGÁNICA – Editorial Pearson

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción a la reactividad de los compuestos orgánicos 96
1. Ruptura y formación de enlaces
1.1. Homólisis y Heterólisis Cuando en una molécula se rompe un enlace, dando dos átomos o un grupo de átomos, se pueden presentar dos tipos de ruptura:
Ruptura homolítica u homólisis Ruptura heterolítica o heterólisis
1.1.1. Homólisis Es propia de dos átomos que no tienen una gran diferencia en electronegatividad. Cada átomo "se lleva" un electrón de cada par de electrones de enlace, dando lugar a radicales libres.
Por ejemplo, si consideramos una molécula A:B:
A B A B+
Un electrón queda en A y el otro en B. Generalmente, la ruptura homolítica ocurre en fase gaseosa.
Ejemplos:
H H H + H
Hidrógeno Radicales hidrógeno(átomos de hidrógeno)
CH3 H CH3 + H
Metano Radical metilo Radical hidrógeno(átomo de hidrógeno)
1.1.2. Heterólisis Es propia de átomos cuya electronegatividad es diferente. El átomo más electronegativo "se lleva" los dos electrones de enlace, formándose iones.
A BA
-B
++A
+B
-+
Los electrones se separan con A o con B. Ocurre en solventes ionizantes y requiere más energía que la ruptura homolítica.
Ejemplos:
CH3 Br CH3+ + Br
-
Bromometano Ión metilonio Ión bromuro
Introducción a la reactividad de los compuestos orgánicos
Ruptura y formación de enlaces. Homólisis y heterólisis. Electrófilos y nucleófilos. Mecanismos de reacción. Avance de la reacción: cambios de energía.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción a la reactividad de los compuestos orgánicos 97
Cl Cl Cl+ + Cl
-
Cloro Ión cloronio Ión cloruro
1.2. Nucleófilos y electrófilos La mayoría de las reacciones que estudiaremos son tratadas como procesos iónicos, porque sus reactivos iniciales son ricos o pobres en electrones. Los reactivos pueden ser iones o moléculas (neutras) y, según su carácter electrónico, pueden dividirse en dos categorías:
Los que son ricos en electrones se denominan nucleófilos o reactivos nucleofílicos (buscadores de núcleos).
Los que son deficientes en electrones se denominan electrófilos o reactivos electrofílicos (buscadores de electrones).
Reactivos Nucleofílicos Buscadores de núcleos :NH3 :OH
- :Cl
- :Br-
Electrofílicos Buscadores de electrones Cl+ H+ R+
1.3. Mecanismos de reacción No sólo es importante saber qué sucede en una reacción química, sino también cómo sucede, es decir, conocer no solo los hechos, sino también la teoría. Por ejemplo, sabemos que, bajo la influencia del calor o la luz, el metano y el cloro forman cloruro de metilo y cloruro de hidrogeno. ¿Cómo, exactamente, se convierte una molécula de metano en otra de cloruro de metilo? ¿Implica esta reacción más de un paso? Y en ese caso, ¿cuáles son los pasos? ¿Cuál es la función del calor o de la luz?
La respuesta a tales preguntas, es decir, la descripción detallada, paso a paso, de una reacción química se denomina mecanismo. Es solamente una hipótesis propuesta para explicar los hechos. A medida que se descubren nuevos hechos, el mecanismo también debe explicarlos o debe ser modificado para incluirlos; incluso puede ser necesario descartar un mecanismo para reemplazarlo por otro.
Sería difícil sostener que alguna vez haya sido demostrado un mecanismo. Sin embargo, sí explica satisfactoriamente una amplia gama de hechos, si nos permite hacer predicciones que luego se cumplen, si es consistente con mecanismos para otras reacciones relacionadas, entonces se dice que tal mecanismo está bien fundado y pasa a formar parte de la teoría de la química orgánica.
¿Por qué nos interesan los mecanismos de las reacciones? Como parte importante de la teoría de la química orgánica, contribuyen a erigir el marco dentro del cual colocamos los hechos que observamos. La comprensión de los mecanismos nos facilitará el descubrimiento de un patrón en la compleja y desconcertante maraña de las reacciones orgánicas. Descubriremos que muchas reacciones, aparentemente sin relación, tienen lugar por el mismo mecanismo o por mecanismos análogos, de modo que la mayor parte de lo aprendido sobre una reacción puede aplicarse directamente a muchas otras.
Sabiendo cómo tiene lugar una reacción, podemos modificar las condiciones experimentales—no por tanteo, sino lógicamente— para que mejore el rendimiento del producto que nos interesa o para cambiar por completo el curso de la reacción y obtener un resultado diferente. A medida que aumenta nuestro conocimiento de las reacciones, también aumenta nuestra capacidad para controlarlas.
La descripción debe incluir:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción a la reactividad de los compuestos orgánicos 98
a) Los movimientos de electrones (que producen la ruptura y formación de enlaces)
b) Las relaciones espaciales de los átomos durante dichas transformaciones.
Es una hipótesis teórica, que si luego se reafirma con hechos experimentales, se concluye como cierta.
1.4. Avance de la reacción: cambios de energía Las relaciones de energía pueden apreciarse más claramente en diagramas de energía potencial, como los de las figuras 1 y 2, que informan de qué manera se lleva a cabo una reacción en términos de energía de los reactivos, de los productos, de los estados intermedios y estados de transición. El avance de la reacción se representa por un movimiento horizontal desde los reactivos, a la izquierda, hacia los productos, a la derecha. La energía potencial (es decir, toda energía, excepto la cinética) en cualquier etapa de la reacción está indicada por la altura de la curva.
La figura 1 es el diagrama de energía para una reacción en un paso. Partimos de un valle de energía potencial con las moléculas de los reactivos. Estas partículas se mueven, por lo que poseen energía cinética, además de la energía potencial indicada. La cantidad exacta de energía cinética varía con cada par de partículas, puesto que algunas se mueven más velozmente que otras. AI chocar, la energía cinética se convierte en energía potencial. Con el aumento de la energía potencial comienza la reacción y empezamos a remontar la curva energética. Si se convierte suficiente energía cinética, alcanzamos la cima y empezamos a descender hacia la derecha.
Figura 1
Durante el descenso se reconvierte energía potencial en cinética hasta que alcanzamos el nivel de los productos, que contienen algo más de energía potencial que los reactivos, y así nos encontramos en un valle ligeramente más elevado que el que habíamos dejado. Con este incremento neto de energía potencial debe haber una disminución correspondiente de energía cinética. Las partículas nuevas se distancian y, puesto que se mueven más lentamente que las partículas de las cuales se formaron, observamos un descenso de la temperatura: se absorbe calor del entorno. El calor de reacción ∆H es la diferencia de energía entre los reactivos y los productos (diferencia de nivel entre ambos valles).
Una reacción exotérmica sigue un curso muy similar. En este caso, los productos con menos energía potencial que los reactivos terminan en un valle más bajo que el que habíamos dejado. Puesto que esta vez las partículas nuevas contienen más energía cinética que las de origen, por lo que se mueven con mayor velocidad, observamos un aumento de la temperatura: se entrega calor al entorno. Cuando se libera energía, el contenido calórico (entalpia), H de las propias moléculas debe disminuir, en consecuencia, al cambio del contenido calórico ∆H, se le da un signo negativo. En el caso de una reacción

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción a la reactividad de los compuestos orgánicos 99
endotérmica, durante la cual se absorbe calor, el aumento del contenido calórico de las moléculas se indica por un ∆H positivo.
La energía mínima que debe proporcionar una colisión para que se produzca reacción se llama energía de activación, Ea, cuya fuente es la energía cinética de las partículas en movimiento. La mayoría de los impactos liberan menos energía que este mínimo, estas colisiones son estériles, por lo que nuevamente terminamos en el valle primitivo. Sólo las colisiones violentas entre partículas, de las cuales una o ambas se mueven con velocidad desusada, son suficientemente energéticas como para permitir la reacción. La diferencia de nivel entre el valle de los reactivos y la cima es la energía de activación Ea y es la que determina la velocidad de la reacción.
Muchas colisiones proporcionan energía suficiente, pero suceden con las moléculas mal orientadas; esto nos permite subir una colina energética, pero nos encontramos fuera de ruta: podemos subir mucho sin encontrar el paso que permite el acceso al valle siguiente.
En general, una reacción química requiere colisiones de energía suficiente (Ea) y de orientación apropiada.
El valor de Ea siempre es positivo, y su magnitud depende de la energía relativa del estado de transición. El término estado de transición o complejo activado implica que esta configuración es la transición entre los reactivos y los productos, y las moléculas pueden transformarse en productos o volver a ser reactivos. Su contenido de energía corresponde al máximo de la curva de energía.
El concepto de estado de transición es útil porque podemos analizar su estructura como si se tratara de una molécula e intentar estimar su estabilidad. Todo factor que estabiliza el estado de transición en relación con los reactivos tiende a disminuir la energía de activación; es decir, todo factor que rebaja la cima de la colina energética más que el valle de los reactivos reduce la altura neta que debe vencer durante la reacción. Pero el estado de transición es sólo una disposición pasajera de átomos que, por su naturaleza intrínseca encontrándose en la cima de la colina energética, no puede ser aislado y examinado.
En la figura 2 se representa un diagrama de energía para una reacción en dos etapas. Cuando las reacciones ocurren en más de una etapa se denomina intermedio al compuesto que es producto de una etapa y luego es reactivo de la etapa siguiente. En este caso, la reacción tiene dos estados de transición, de los cuales el más energético es el primero, por lo tanto, la primera etapa es la determinante de velocidad de la reacción. Entre los dos estados de transición hay un valle en el que se ubica un intermediario. A diferencia de los estrados de transición que no son aislables, los intermediarios sí pueden aislarse, tienen tiempos de vida media finitos y son más estables que los estados de transición, pero menos que los reactivos o productos.
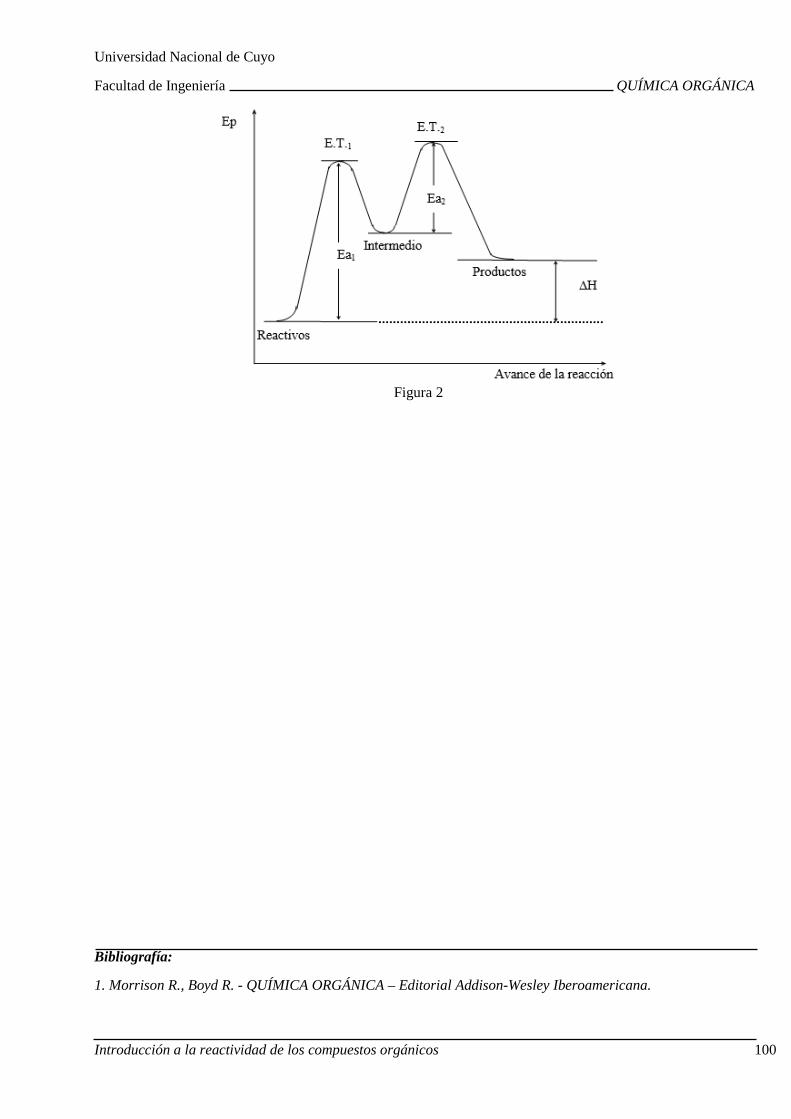
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Introducción a la reactividad de los compuestos orgánicos 100
Figura 2
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 101
Como se vio anteriormente, una reacción de sustitución implica el reemplazo de un átomo o grupo de átomos por otro, y la podemos representar como sigue:
AB + S A S + B
Sustrato Grupo
atacante Producto
Grupo saliente
donde:
Sustrato es la especie química en la que se produce la sustitución Grupo atacante es el que sustituye en el sustrato al grupo saliente Grupo saliente es el que es desplazado por el grupo atacante Producto es el compuesto que se forma al sustituirse en el sustrato el grupo saliente por el grupo
atacante
Las Sustituciones por Radicales Libres, son un tipo particular de reacciones de sustitución y en ellas tanto el grupo saliente como el entrante son radicales libres, debido a que las rupturas y formaciones de enlaces son homolíticas. Estas reacciones, en las que intervienen radicales libres, ocurren generalmente en fase gaseosa.
1. 1. Halogenación de alcanos Un ejemplo de Sustitución por Radicales libres es la Halogenación de Alcanos, en la cual un hidrógeno de un alcano se reemplaza por un halógeno. Esta reacción se puede representar de la siguiente manera:
R:H + X:X R:X + X:H
Alcano Halógeno Haluro de
alquilo
Haluro de hidrógeno
Tanto la ruptura de los enlaces C-H y X-X, como la formación de los enlaces C-X y X-H, son homolíticas.
1.1. Cloración del metano El caso de Sustitución por Radicales libres que vamos a estudiar es la cloración del metano, ya que el mecanismo de esta reacción también es válido para la cloración y la bromación de otros alcanos, como así también para compuestos que, no siendo alcanos, poseen cadenas carbonadas que se pueden halogenar (ej.: halogenación de ácidos carboxílicos, para obtener ácidos carboxílicos halogenados).
Reacción: El metano, el alcano más simple, posee cuatro enlaces C-H que podrían reaccionar en una halogenación. Cuando se mezcla cloro gaseoso con metano, en presencia de luz ultravioleta (hν) o calor (250-400ºC), se forman clorometano (cloruro de metilo) y cloruro de hidrógeno:
CH4 + Cl2 CH3Cl + ClHhv o calor
cloruro de metilo
Pero, conforme se forma el cloruro de metilo, éste puede competir por el cloro con el metano que no ha reaccionado y, por consiguiente, también se forma diclorometano (cloruro de metileno):
+ Cl2 + ClHhv o calor
cloruro de metileno
CH3Cl CH2Cl2
Reacciones de Sustitución: Sustituciones por Radicales Libres
Halogenación de alcanos. Intermedios. Estados de transición. Cinética. Diagramas de energía. Energía de activación. Calor de reacción. Importancia de las sustituciones por radicales libres.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 102
El cloruro de metileno tiene dos enlaces C-H y puede reaccionar con cloro, al igual que el metano y el cloruro de metilo, formando triclorometano (cloroformo), que a su vez puede transformarse en tetraclorometano (tetracloruro de carbono):
+ Cl2 + ClHhv o calor
cloroformo
Cl2CH2 Cl3CH
+ Cl2 + CH4hv o calor
tetraclorurode carbono
Cl3 CH Cl4C
Resumiendo:
CH4Cl2/hv o calor
ClHCH3Cl Cl2/hv o calor
ClHCl2CH2
Cl2/hv o calor
ClHCl3CH Cl2/hv o calor
ClHCl4CH
Se ha comprobado que aun mezclando metano con cloro en una relación molar 1:1, se forman los cuatro productos orgánicos.
La polihalogenación es una desventaja de la halogenación de alcanos para la síntesis de compuestos específicos. Como ocurre con muchas reacciones potencialmente útiles, el desafío de la halogenación está en su control:
Si se desea obtener cloruro de metilo, es necesario favorecer la primer reacción (metano + cloro) trabajando con exceso de metano, de tal manera que haya más probabilidad que el cloro reaccione con el metano que con el clorometano.
CH4 Cl2 CH3Cl ClHhv o calor+ +
La obtención de tetracloruro de carbono a partir de metano implica el reemplazo de los cuatro
hidrógenos, por lo cual se requieren cuatro moles de cloro por cada mol de metano. Así, si el metano y el cloro se combinan en una relación 1:4, se puede obtener tetracloruro de carbono en forma casi exclusiva:
4Cl2hv o calor
+ +CH4 4HClCl4C
Mecanismo de reacción: La reacción de monocloración del metano es la siguiente:
CH3H + Cl Clhv o calor
CH3Cl + ClH
Es decir, para que ocurra esta reacción se debe romper un enlace C-H y uno Cl-Cl y se deben formar un enlace C-Cl y uno Cl-H. Veamos cómo transcurre esta reacción, es decir, cual es el mecanismo de reacción:
a) Cuando se mezclan Cl2 y metano en un reactor, en presencia de luz o calor, podría ocurrir la ruptura del enlace Cl-Cl, cuya Energía de Disociación (D) es 58 kcal/mol, o bien, la ruptura del enlace C-H, cuya energía de disociación (D) es 104 kcal/mol.
Como el enlace Cl-Cl es más débil por tener menor D, es la molécula de cloro la que se rompe (homolíticamente) por acción de la luz o el calor, para formar dos átomos de cloro (radicales libres):

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 103
+hv o calor
Cl Cl
átomos de cloro (radicales libres)
Cl Cl
(1)
Nota: Para simplificar, no representaremos los pares de electrones sin compartir del cloro.
b) Cada átomo de cloro es muy reactivo y busca de inmediato una forma de completar su octeto. En el medio se encontrará con moléculas de cloro y metano en alta concentración y con muy pocos átomos de cloro (radicales de vida breve). Por lo tanto, es más probable la colisión con moléculas:
Si colisiona con moléculas de cloro, la reacción es muy probable pero improductiva (los productos y reactivos son iguales):
Cl + Cl Cl Cl Cl + Cl
(probable e improductiva)
La colisión con metano es también muy probable pero, además, es productiva. Para completar su octeto, el átomo de cloro extrae un átomo de hidrógeno del metano, rompiéndose el enlace C-H y formándose radical metilo y cloruro de hidrógeno, que es uno de los productos de reacción:
Cl + CH3H CH3 + Cl H
radical metilo
c) El grupo metilo, muy reactivo, ha quedado con un electrón no apareado y busca completar el octeto. Las reacciones más probables son con moléculas de cloro y de metano, y muy poco probables con átomos de cloro o radicales metilo:
Si colisiona con metano, la reacción es muy probable pero improductiva (los productos y reactivos son iguales):
CH3 + CH4 CH4+ CH3
(probable e improductiva)
La colisión productiva es con la molécula de cloro. Un carbono del radical metilo es ahora el que no tiene su octeto completo, por lo que extrae un átomo de una molécula de cloro para formar otro átomo de cloro y el producto final, cloruro de metilo:
CH3 + Cl Cl CH3Cl + Clcloruro de metilo
d) El átomo de cloro puede atacar otra molécula de metano, se repite la etapa (2) y se genera radical metilo.
A la vez, éste puede atacar a otra molécula de cloro, se repite la etapa (3), formándose más radical cloro.
Así, sucesivamente se repiten las etapas (2) y (3).
e) La reacción no continúa indefinidamente, sino que ocurren pasos de terminación de la cadena, que estadísticamente son menos probables que los de propagación. Esto da por resultado el consumo de radicales libres, sin producción de otros nuevos para que continúen la cadena. Los pasos de terminación serían los siguientes, considerándose como más probable el tercero en el se forma Cl2 :
CH3 CH3 CH3 CH3+
(2)
(3)

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 104
CH3 + Cl CH3Cl
Cl ClCl Cl+
Por lo tanto, la cloración del metano, y la halogenación de alcanos en general, es una Reacción en Cadena de radicales libres, siendo el mecanismo completo como sigue:
Iniciación:
+hv o calor
Cl Cl Cl Cl
Propagación:
C
H
H
HH Cl H+ +Cl C
H
H
H
(2)
+ Cl Cl C
H
H
ClH + ClC
H
H
H
(3)
Terminación:
CH3 CH3 CH3 CH3+CH3 + Cl CH3Cl
Cl ClCl Cl+
Resumiendo:
− La reacción es iniciada por la ruptura de una molécula de cloro, inducida por luz o calor.
− Una vez iniciada la reacción procede en ausencia de luz o calor, lo cual se debe a la formación alterna de radicales libre metilo y cloro en los dos pasos de propagación. Cada paso genera un producto y un radical libre (intermedio) que participa en el otro paso.
− La reacción no continúa indefinidamente, sino que ocurren pasos de terminación de la cadena, donde hay consumo de los radicales libres remanentes, sin producción de otros nuevos como para continuar.
Calores de reacción: Empleando los valores de las Energías de Disociación de Enlaces (D) se pueden calcular los cambios energéticos (∆H), ya sea en la reacción, o bien en cada una de las etapas del mecanismo de reacción:
a) Reacción:
CH3H + Cl Cl CH3 Cl + ClH
+104 +58 -84 -103
(1)

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 105
Consideramos sólo aquellos enlaces que se rompen y los que se forman en la reacción. Además, debemos tener en cuenta que para romper un enlace hay que entregar energía (+D) y que cuando se forma un enlace se libera energía (-D). Por lo tanto, para la reacción de cloración del metano, el ∆ H se calcula como sigue:
Como el Calor de Reacción es menor que cero, la reacción es exotérmica.
b) Mecanismo de Reacción:
Cl Cl Cl2
(reacción endotérmica) +58
Cl + CH3H CH3 + ClH
(reacción endotérmica)
+104 -103
CH3 + Cl Cl CH3Cl + Cl
(reacción
exotérmica) +58 -84
Los valores de los ∆H, tanto de la reacción, como de las etapas del mecanismo que ocurre, explican porqué la cloración del metano, a pesar de ser exotérmica (∆ H = - 25 kcal/mol), sólo tiene lugar si se provee energía (luz ultravioleta o calor).
El paso que inicia la cadena es muy endotérmico (∆ H = + 58 kcal/mol) y la reacción no ocurriría si no tiene lugar esa etapa. Una vez formados los átomos de cloro, los pasos de propagación se suceden sin dificultad.
Diagramas de Energía A continuación se muestran los Diagramas de Energía para las etapas de iniciación y de propagación:
Iniciación:
Cl Cl Cl2
Ep
Cl2
Avance reacción
Cl2

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 106
Propagación: Para dibujar el Diagrama de Energía consideraremos el Estado de Transición o Complejo Activado de cada etapa, que es el estado de máxima energía, en el cual se están formando y rompiendo los enlaces para pasar de los reactivos a los productos:
Cl CH3H CH3+ +Cl H CH3ClH
(1)
Cl ClCH3 + + ClCH3ClCl Cl CH3
(2)
(1) (2)
1.2. Reacción del metano con otros halógenos a) Consideremos los ∆H (kcal/mol) de las 2 etapas de propagación de la halogenación del metano con los
halógenos:
Propagación ∆H (kcal/mol)
X: Flúor X: Cloro X: Bromo X: Iodo
Primera Etapa:
X . + CH4
. CH3 + XH
- 32
+ 1
+16
+33
Segunda Etapa:
. CH3 + X2 XCH3 + X .
- 70
-26
-24
-20
CH4 + X2 XCH3 + XH -102 -25 - 8 +13
El balance energético para la cloración y la bromación es razonablemente exotérmico y la reacción es practicable.
Ea
Avance de la reacción Avance de la reacción
Ep
∆H=-26 kcal/mol ∆H= +1 kcal/mol Cl : H +
. CH3
. Cl + CH4
Cl .....
H.....
CH3
Ea = +4kcal/mol
Ep
Cl . + Cl:CH3
. CH3 + Cl2
Cl .....
Cl.....
CH3
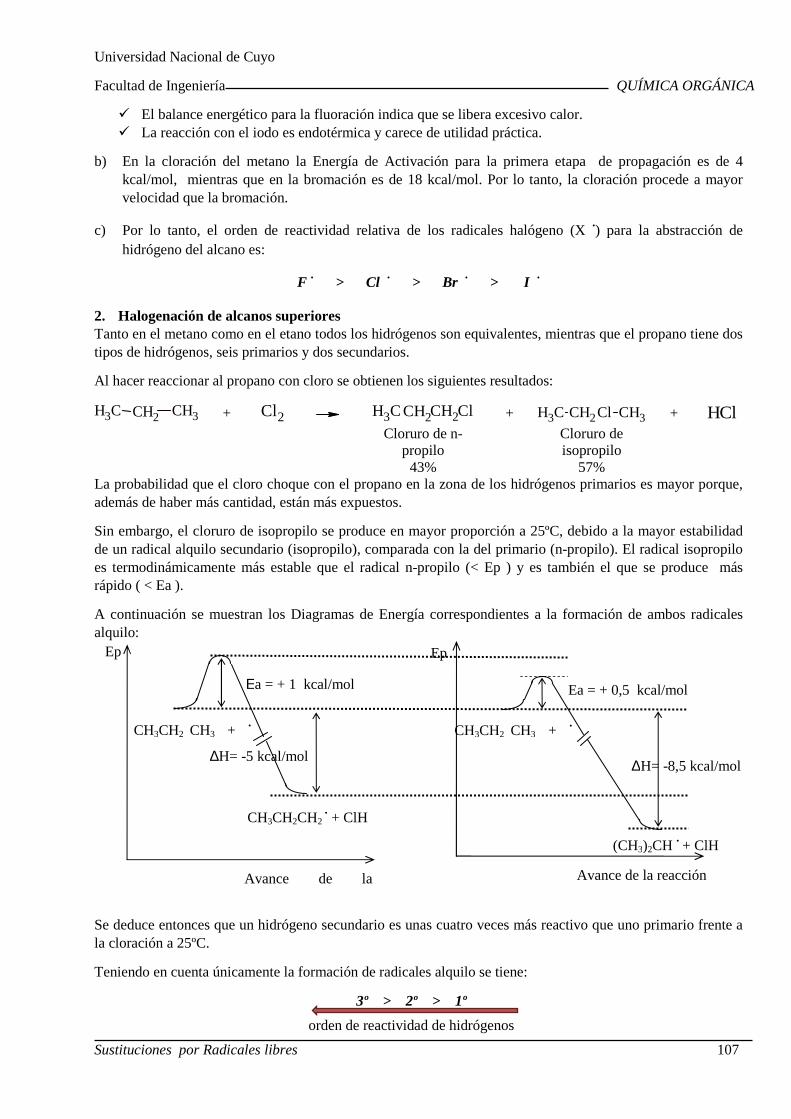
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 107
El balance energético para la fluoración indica que se libera excesivo calor. La reacción con el iodo es endotérmica y carece de utilidad práctica.
b) En la cloración del metano la Energía de Activación para la primera etapa de propagación es de 4 kcal/mol, mientras que en la bromación es de 18 kcal/mol. Por lo tanto, la cloración procede a mayor velocidad que la bromación.
c) Por lo tanto, el orden de reactividad relativa de los radicales halógeno (X .) para la abstracción de
hidrógeno del alcano es:
F . > Cl
. > Br
. > I
.
2. Halogenación de alcanos superiores Tanto en el metano como en el etano todos los hidrógenos son equivalentes, mientras que el propano tiene dos tipos de hidrógenos, seis primarios y dos secundarios.
Al hacer reaccionar al propano con cloro se obtienen los siguientes resultados:
CH3 CH2 CH3 + Cl2 CH3 CH2CH2Cl + CH3 CH2Cl CH3 + ClH
Cloruro de n-
propilo
Cloruro de isopropilo
43% 57% La probabilidad que el cloro choque con el propano en la zona de los hidrógenos primarios es mayor porque, además de haber más cantidad, están más expuestos.
Sin embargo, el cloruro de isopropilo se produce en mayor proporción a 25ºC, debido a la mayor estabilidad de un radical alquilo secundario (isopropilo), comparada con la del primario (n-propilo). El radical isopropilo es termodinámicamente más estable que el radical n-propilo (< Ep ) y es también el que se produce más rápido ( < Ea ).
A continuación se muestran los Diagramas de Energía correspondientes a la formación de ambos radicales alquilo:
Se deduce entonces que un hidrógeno secundario es unas cuatro veces más reactivo que uno primario frente a la cloración a 25ºC.
Teniendo en cuenta únicamente la formación de radicales alquilo se tiene:
3º > 2º > 1º
orden de reactividad de hidrógenos
CH3CH2CH2 . + ClH
∆H= -5 kcal/mol
Ep
CH3CH2 CH3 + .
Εa = + 1 kcal/mol
Ep
(CH3)2CH . + ClH
CH3CH2 CH3 + .
Ea = + 0,5 kcal/mol
∆H= -8,5 kcal/mol
Avance de la reacción
Avance de la reacción

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones por Radicales libres 108
Cuanto más tipos de hidrógenos, mayor es el números de productos. En general, la cloración de alcanos da lugar a mezclas complejas, por lo que en estos casos la reacción carece de utilidad en síntesis orgánica.
3. Importancia de las sustituciones por radicales libres La halogenación de alcanos es especialmente útil para aquellos compuestos que tienen un solo tipo de hidrógeno (ej.: metano, etano, ciclohexano, etc.). No obstante, un inconveniente lo constituye el hecho que es posible la sustitución de más de un hidrógeno, obteniéndose mezcla de productos. No obstante, muchas veces es usada directamente la mezcla, lo cual no implica un costo adicional derivado de la separación de los compuestos puros (ej.: uso de mezcla de productos como solventes).
Puede convenir la separación de compuestos, si cada uno tiene mercado propio. Por ejemplo, los derivados clorados del metano tienen diversas aplicaciones (Ej.: cloruro de metileno, cloroformo y tetracloruro de carbono son usados como solventes).
Los derivados monohalogenados de alcanos generalmente se obtienen, como veremos mas adelante, por otras vías.
Los más usados son los derivados clorados y bromados. El cloro es barato, pero presenta baja selectividad y problemas de polihalogenación. El bromo es líquido, más fácil de manipular y altamente selectivo, pero es relativamente caro.
El flúor es caro y corrosivo. Mientras que la iodación es muy lenta, el flúor reacciona violentamente.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA - Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 109
1. Sustituciones nucleofílicas alifáticas Las sustituciones nucleofílicas constituyen métodos útiles para preparar diversos tipos de compuestos, ya que mediante las mismas es posible, utilizando distintos reactivos, convertir un compuesto en otros, los que a la vez también pueden ser transformados.
En una reacción de sustitución un átomo o un grupo de átomos de una molécula se reemplazan por otro. Cuando el reactivo que ataca al núcleo del carbono (o atacante) es rico en electrones, reactivo nucleofílico o nucleófilo, la reacción es una Sustitución Nucleofílica. Por ejemplo, la sustitución del cloro en el cloruro de metilo por un grupo oxhidrilo:
CH3 Cl + CH3 OH+cloruro de metilo metanol
OH Cl
Este es un fenómeno heterolítico, el ión cloruro saliente se lleva el par de electrones que comparte con el carbono y el ión hidróxido entrante aporta el par de electrones necesarios para la unión con el carbono.
El ión halogenuro es una base muy débil, y de la misma forma que en el ácido libera el protón, en los halogenuros de alquilo libera el carbonio hacia otras bases. Estas bases poseen un par de electrones sin compartir y buscan un centro relativamente positivo, es decir buscan un núcleo con el cual compartir sus electrones.
Representación General:
+ X+SustratoReactivo
nucleofílicoGrupo saliente
R X R ZZ
El compuesto carbonado que sufre la sustitución se denomina sustrato y es el que posee un grupo saliente, que es desplazado del carbono y se aleja de la molécula llevándose el par de electrones.
Si bien vamos a estudiar con mayor profundidad las sustituciones nucleofílicas en haluros de alquilo, otros compuestos pueden sufrir estas reacciones (ej.: preparación de haluros de alquilo a partir de alcoholes, obtención de derivados de ácidos, etc.).
El nucleófilo: Z, puede ser neutro o tener carga negativa:
:OH- :OH2
R'COO:- :NH3
:OR- :NH2R’
:I- :NHR’2
Reacciones de Sustitución: Sustituciones Nucleofílicas Alifáticas
Sustituciones nucleofílicas en haluros de alquilo y alcoholes. Cinética. Orden de reacción y molecularidad. Diagramas de energía. Influencia de la estructura y la solvatación sobre la reactividad. Reordenamientos o transposiciones de iones carbonio.
Aplicaciones de las sustituciones nucleofílicas alifáticas en síntesis orgánica.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 110
A continuación se dan algunos ejemplos de Sustituciones Nucleofílicas de haluro de alquilo (RX) con distintos nucleófilos:
RX + :OH- R:OH + :X- Alcohol
RX + H2O R:OH + HX Alcohol
RX + :OR- R:OR' + :X- Éter
RX + :I- R:I + :X- Ioduro de alquilo
RX + :CN- R:CN + :X- Nitrilo
RX + R'COO- R'COOR + :X- Ester
RX + :NH3 R:NH2 + HX Amina Primaria
RX + :NH2R' R:NHR' + HX Amina Secundaria
RX + :NHR'2 R:NR'2 + HX Amina Terciaria
RX + :SH- R:SH + :X- Tiol
1.1. Carácter nucleofílico: Un nucleófilo eficaz es el que tiene una densidad electrónica elevada, y si bien todos los nucleófilos son bases, su reactividad no siempre es similar a su fuerza básica.
El poder nucleófilo es cuestión de velocidad: de dos nucleófilos, el más poderoso es el que ataca más rápidamente al carbono
Un anión es más reactivo como nucleófilo que la especie neutra correspondiente. Así el HO:- es más nucleófilo que el agua.
Entre especies con la misma carga, un átomo menos electronegativo con un par de electrones sin compartir es más nucleófilo que uno más electronegativo, porque dona con más facilidad su par de electrones al aproximarse al estado de transición.
Dentro de un solo grupo o un solo periodo del sistema periódico, la reactividad de los nucleófilos aumenta a medida que disminuye la electronegatividad. Así el HO:- es mejor nucleófilo que el F:- porque el oxígeno es menos electronegativo que el flúor
En el caso de especies de diferentes periodos y grupos del sistema periódico, el carácter nucleofílico se determina experimentalmente.
La reactividad relativa de algunos nucleófilos comunes es:
1.2. Grupo Saliente: La naturaleza del grupo saliente también afecta la velocidad de las reacciones de sustitución. Los mejores grupos salientes son los que tienen enlaces débiles con el carbono y los que pueden soportar con facilidad una carga negativa. El cloro, el bromo y el yodo son buenos grupos salientes.
1.3. Cinética de la Sustitución Nucleofílica La velocidad de una reacción química se puede analizar considerando:
Los distintos factores energéticos:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 111
Se analizan los valores de Energía de Activación, y las estabilidades relativas de los Estados de Transición
El cambio de concentración de las sustancias reaccionantes:
Un aumento de concentración de los reactivos produce un aumento en el número total de colisiones, y la reacción es más rápida.
Cuando se estudia la siguiente reacción:
CH3 Br CH3 OH ++ NaOH NaBr
Se observa experimentalmente que al duplicar la concentración de NaOH, se duplica la velocidad de la reacción; y que al duplicar la concentración de CH3Br , también se duplica la velocidad de la reacción.
Se deduce entonces, que la reacción entre el Bromuro de metilo e hidróxido de sodio sigue una cinética de segundo orden, ya que la velocidad depende de la concentración elevada a la primera potencia de los dos reactivos .La velocidad de la reacción depende tanto de la concentración de CH3Br como de la concentración de OH- y viene expresada por:
v = k x CH3Br NaOH
k es la constante específica de velocidad
Del mismo modo se estudia la siguiente reacción:
NaOH NaBrC CH3
CH3
BrCH3 ++ C CH3
CH3
OHCH3
Experimentalmente se observa que al duplicar la concentración de R – Br, se duplica la velocidad de la reacción; y que al duplicar la concentración de NaOH, no hay cambio en la velocidad de reacción.
Por lo tanto:
v = k RBr
Se deduce por lo tanto que la reacción del Bromuro de t-butilo con OH- sigue una cinética de primer orden puesto que la velocidad de la reacción depende de la concentración elevada a la primera potencia de un sólo reactivo.
Para explicar estas diferencias de orden cinético, se ha propuesto que la Sustitución Nucleofílica se puede realizar por medio de dos mecanismos distintos:
Sustitución Nucleofílica Bimolecular: SN 2
Sustitución Nucleofílica Monomolecular: SN 1
1.4. Sustitución Nucleofílica Bimolecular: SN2
Cinética: Para la siguiente reacción:
NaOHCH3 Br CH3 OH ++ NaBr
v = k x CH3Br NaOH

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 112
Experimentalmente se demuestra que la velocidad depende de la concentración de los dos reactivos por lo que corresponde a una cinética de segundo orden (n = 2) Mecanismo de reacción: Esta reacción ocurre en una sola etapa. Cuando el ión hidróxido choca con una molécula de bromuro de metilo, por el lado más alejado del Br y con energía suficiente, se forma un enlace C - OH y se rompe el enlace C – Br, liberándose el ión Bromuro:
+ C BrH
H
H
C Br
H
HH
OH
δ− δ−
Br-+COH H
H
H
Estado de transición
HO
El Estado de Transición puede presentarse como una estructura en la que el carbono está parcialmente unido
al OH- y al Br
-, aún no se ha formado por completo el enlace C-OH, ni se ha roto en su totalidad el enlace C-
Br .
Ha disminuido la carga negativa del hidróxido porque ha comenzado a compartir sus electrones con el carbono. El bromo ha empezado a desarrollar una carga negativa parcial, ya que está separándose del carbono con el par de electrones compartido.
El OH- y el Br
- se ubican lo más lejos posible, los enlaces de los tres hidrógenos con el carbono se orientan en
un mismo plano y todos sus ángulos de enlace son de 120°, por lo que los enlaces C - H se distribuyen como los rayos de una rueda, con los enlaces C - OH y C - Br a lo largo de su eje.
Este es el mecanismo llamado SN2: Sustitución Nucleofílica Bimolecular (SN2)porque en la única etapa del mismo (etapa que determina la velocidad de reacción) se produce el choque de dos partículas que sufren ruptura o formación de enlaces.
SN2
Reacción de sustitución
Reactivo atacante nucleófilo
Molecularidad 2
Estereoquímica: Para entender mejor el mecanismo de una reacción, conviene analizarla espacialmente o estereoquímicamente:
HO + C BrH
H
H
C Br
H
HH
OHδ−
δ−
Br-+C HOH
H
H
sp2sp3
sp3
Se observa que debido a la forma del ataque del nucleófilo, los átomos adoptan una posición invertida en el espacio para formar el producto, o sea que esta reacción procede con Inversión de la configuración o Inversión de Walden.
El diagrama de Energía para esta Reacción es el siguiente:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 113
Reactividad: A diferencia de las reacciones de Radicales Libres y de iones Carbonio, la estructura del Estado de Transición no es intermedia entre la de los Reactivos y la de los Productos, de manera que no se puede suponer que factores que estabilicen al Producto lo hagan también con el Estado de Transición.
Se debe por lo tanto analizar la influencia de:
a) Efectos Electrónicos
b) Efectos Estéricos
c) Naturaleza del Reactivo Atacante
d) Características del Solvente
a) Efectos Electrónicos:
Si se compara el Estado de Transición con los Reactivos, se observa que en el Estado de Transición hay un
enlace formado parcialmente entre el C y el OH- y otro parcialmente roto entre el C y el Br
-.
El OH- aporta electrones al carbono, mientras que el bromo se los quita, de manera que la carga neta sobre el carbono del Estado de Transición es similar a la que posee el carbono del reactivo.
La atracción o entrega de electrones por los sustituyentes afecta al Estado de Transición de la misma forma que al reactivo, por lo que tiene poca influencia sobre la velocidad de reacción ya que no afecta la barrera de Energía de Activación ( Ea ).
b) Efectos Estéricos:
Tanto en reactivos como en productos el carbono tiene hibridación sp3, mientras que en el Estado de Transición el carbono tiene hibridación sp2 y está unido a 5 átomos.
Analizando los distintos H y/o grupos alquilo unidos al carbono, se pueden dar algunos casos tales como:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 114
C X
H
HH
Z
metilo
C X
H
CH3H
Z
etilo
C X
H
CH3CH3
Z
isopropilo
C X
CH3
CH3CH3
Z
t-butilo
Se observa que se va produciendo una aglomeración cada vez mayor alrededor del C, lo que obliga a los -CH3 a juntarse con Z y X. Esta aglomeración es mayor en el Estado de Transición que en los reactivos, por lo que se eleva la energía del Estado de Transición en mayor grado que en los reactivos y la reacción es más lenta (aumenta Ea)
Por lo tanto, las diferencias en la velocidad de reacción se deben solamente a factores estéricos. Se puede establecer entonces, el siguiente Orden de Reactividad:
c) Naturaleza del Reactivo Atacante:
El reactivo atacante debe ser fuerte para ir por este mecanismo, ya que debe desplazar a X-. Además, debe
estar en alta concentración para aumentar la probabilidad de colisiones con el sustrato y así posibilitar que ocurra la reacción.
d) Característica del Solvente:
OH- + R X R XOH
δ− δ−
R OH + X
Estado de transición
Debido a que las cargas están concentradas en los reactivos, mientras que en el estado de transición hay una carga negativa dispersa, se solvatan más los reactivos que el estado de transición, de manera que los reactivos se estabilizan más que el Estado de Transición por solvatación
Por lo tanto, el aumento de polaridad del Solvente estabiliza más a Reactivos que a ET, aumenta la barrera de Energía de Activación y disminuye la velocidad de la reacción.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 115
1.5. Sustitucion nucleofilica unimolecular: SN1 Cinética: Al estudiar la siguiente reacción:
C CH3
CH3
Br
CH3 ++Na OH C CH3
CH3
OH
CH3 NaBr
Experimentalmente se demuestra que la velocidad de la reacción depende solamente de la concentración de un reactivo, R -X y es independiente de la concentración de HONa por lo que corresponde a una cinética de primer orden (n = 1)
C CH3
CH3
Br
CH3v=k
Mecanismo: El mecanismo de esta reacción ocurre en dos etapas:
Primera etapa:
C CH3
CH3
Br
CH3
lentaC
+CH3
CH3
CH3Br
-+
Segunda etapa:
HOC+
CH3
CH3
CH3 + rápidaC CH3
CH3
OH
CH3
Debido a que la etapa determinante de la velocidad sólo depende de una molécula, se tiene un mecanismo monomolecular, en dicha etapa hay ruptura de enlace de una sola especie, R-X. Por lo tanto, el simbolismo es:
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 116
SN1
Reacción de sustitución
Molecularidad 1
Reactivo atacante nucleófilo
Estereoquímica: Primera etapa: lenta
C Br
CH3
CH3
CH3
sp3
C Br
CH3
CH3
CH3
δ−δ+
C+
CH3
CH3CH3
sp2
+ Br-
Segunda etapa: rápida
HOC+
CH3
CH3CH3
+
C OH
CH3
CH3
CH3
δ−δ+
C OH
CH3
CH3
CH3
COH
CH3
CH3
CH3
δ− δ+
COH
CH3
CH3
CH3
Producto sin inversión de la configuración
Producto con inversión de la configuración
Se obtiene una mezcla de productos, con y sin inversión de configuración.
El diagrama de Energía para esta reacción es el siguiente:
Los Estados de Transición, denominados también Complejos activados son estados de máxima energía, no aislables, donde se supone que se están formando y/o rompiendo enlaces.
Los Intermedios, especies generalmente aislables, son productos en una reacción y reactivos en la siguiente.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 117
Reactividad: Para analizar la Reactividad de una reacción se tienen que tener en cuenta los siguientes efectos:
a) Efectos Electrónicos
b) Naturaleza del Reactivo Atacante
c) Características del Solvente
a) Efectos electrónicos:
En la etapa determinante de la velocidad de reacción, hay formación de ion carbonio (+) o carbocatión, de manera que factores que estabilicen al catión, también favorecen la reacción.
Los iones carbonio poseen un átomo de carbono con 6 electrones, cargado positivamente.
En un carbonio, el carbono deficiente de electrones está unido a tres átomos, para cuyos enlaces emplea orbitales sp2; las uniones son trigonales, dirigidas hacia los vértices de un triángulo equilátero, ubicándose todos los átomos en un plano.
C+
R
R
R
La característica de un ión carbonio es su carbono deficiente de electrones por lo que está con carga positiva. Su estabilidad relativa depende de la mejor posibilidad de acomodar esa carga.
Se considera a los cationes como 1°, 2° y 3°, de acuerdo a la cantidad de grupos alquilo unidos al carbono deficiente de electrones.
Algunos de los carbocationes más utilizados son:
Estructura Nombre Clase
H3C+ catión metilo
metilonio Primario
H3C – H2C+
catión etilo etilonio
Primario
H3C – H2C – H2C+
catión n-propilo n- propilonio
Primario
H3C – HC+ – H3C catión i- propilo
i-propilonio Secundario
H3C – C+ – C H3 |
CH3
catión terbutilo terbutilonio
Terciario
De acuerdo a las leyes de la electrostática “la estabilidad de un sistema cargado aumenta con la dispersión de la carga”. Por lo tanto, mientras más dispersa esté la carga positiva, más estable es el catión.
Un sustituyente que libera electrones tiende a reducir la carga (+) en el carbono deficiente de electrones, el propio sustituyente se vuelve algo (+), estabilizando al ión carbonio por la dispersión de la carga.
Un sustituyente que atrae electrones tiende a aumentar la carga (+) en el carbono deficiente de electrones, por lo que le disminuye la estabilidad del ión carbonio.
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 118
La estabilidad de un sistema cargado aumenta con la dispersión de carga, por lo tanto, todo factor que tiende a esparcir la carga (+) del carbono deficiente de electrones y distribuirla sobre el resto del ión, debe estabilizar al carbocatión.
Si se considera a un sustituyente G unido al carbono deficiente de electrones, donde G puede liberar o aceptar electrones:
Por lo que, mientras mayor es el número de grupos alquilo (dadores por efecto inductivo), más estable es el carbocatión.
Cuanto más estable es un carbocatión, más rápido se forma.
Este orden, que es coincidente con el mecanismo propuesto, se debe a la facilidad de formación y a la estabilidad de iones carbonio.
El producto de la etapa determinante de la velocidad de reacción, la etapa lenta, es un ión carbonio y el Estado de Transición (ET) es un carbocatión en formación. De manera que factores que estabilicen al carbocatión estabilizan también al ET disminuyendo su energía potencial (Ep) y bajando la Ea, por lo que resulta más rápida la reacción.
Carbocatión primario Carbocatión secundario Carbocatión terciario Al formarse un catión más estable, es más estable también el catión incipiente del ET y baja la barrera de Energía de Activación (Ea1) y la velocidad de la primera etapa es mayor.
Además, los carbocationes presentan la posibilidad de la Transposición, tema que se verá más adelante.
Por lo tanto, el orden de reactividad es:
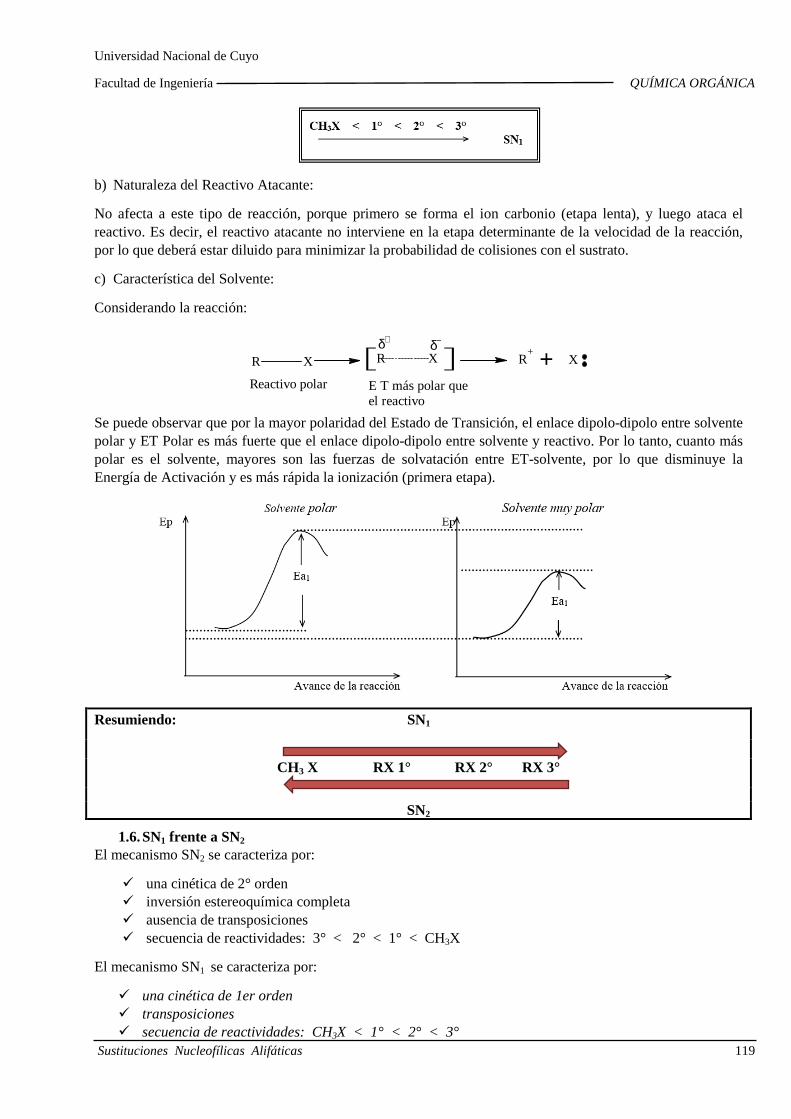
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 119
b) Naturaleza del Reactivo Atacante:
No afecta a este tipo de reacción, porque primero se forma el ion carbonio (etapa lenta), y luego ataca el reactivo. Es decir, el reactivo atacante no interviene en la etapa determinante de la velocidad de la reacción, por lo que deberá estar diluido para minimizar la probabilidad de colisiones con el sustrato.
c) Característica del Solvente:
Considerando la reacción:
R X R X + X
E T más polar que el reactivo
δ−δ+
R+
Reactivo polar
Se puede observar que por la mayor polaridad del Estado de Transición, el enlace dipolo-dipolo entre solvente polar y ET Polar es más fuerte que el enlace dipolo-dipolo entre solvente y reactivo. Por lo tanto, cuanto más polar es el solvente, mayores son las fuerzas de solvatación entre ET-solvente, por lo que disminuye la Energía de Activación y es más rápida la ionización (primera etapa).
Resumiendo: SN1
CH3 X RX 1° RX 2° RX 3°
SN2
1.6. SN1 frente a SN2 El mecanismo SN2 se caracteriza por:
una cinética de 2° orden inversión estereoquímica completa ausencia de transposiciones secuencia de reactividades: 3° < 2° < 1° < CH3X
El mecanismo SN1 se caracteriza por:
una cinética de 1er orden transposiciones secuencia de reactividades: CH3X < 1° < 2° < 3°

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 120
Por lo tanto, considerando:
Efectos electrónicos:
si XR es 3°, se favorece SN1
si XR es 1°, se desfavorece SN1
si XR es 2° puede ser SN1 o SN2
Efectos estéricos:
si XR es 3° se desfavorece SN2
si XR es 1° se favorece SN2
si XR es 2° puede ser SN1 o SN2
Observaciones
− Los halogenuros de alquilo además de Sustituciones Nucleofílicas pueden presentar reacciones de Eliminación. Estos dos tipos de reacciones compiten entre sí (el desarrollo del tema se verá en Reacciones de Eliminación Iónicas).
1.7. Otras sustituciones nucleofílicas alifáticas No sólo los haluros de alquilo dan este tipo de reacciones, sino que otros compuestos orgánicos también pueden sufrir sustituciones nucleofílicas.
Ejemplos:
R OH+ X H R X + OH2
C OHR
O
X H+ C XR
O
OH2+
C OHR
O
H OR'+ C OR'R
O
OH2+
C OHR
O
+ C NH2R
O
OH2+H NH2
1.8. Preparación de haluros de alquilo a partir de alcoholes Este es uno de los métodos utilizados para preparar haluros de alquilo, los que también se obtienen a partir de alquenos por adición electrofílica.
R OH+ R Cl+ OH2ClH
Mecanismos de reacción
SN1
El carácter de base de Lewis del alcohol permite la adición del H+ en una primer etapa, generándose un ión oxonio o alcohol protonado (R-OH2
+), donde el enlace C-O es más polar que el del alcohol. Esto favorece la ruptura heterolítica de dicho enlace, con formación del ión carbonio.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 121
R OH+ R ClClH
Cl-
R OH2+
OH2
R+ Cl
-
SN1
SN2
Los alcoholes primarios reaccionan muy lentamente con ácidos por su débil carácter básico. No obstante, si forman el ión oxonio, éste sigue generalmente un mecanismo bimolecular por la dificultad de transformarse en ión carbonio primario poco estable.
R OH+ R ClClH
Cl-
R OH2+
Cl-
SN2
R OH2+
Clδ− δ+
OH2+
1.9. Preparación de éteres a partir de alcoholes (deshidratación intermolecular): Los alcoholes, al ser calentados en medio ácido, se deshidratan inter e intramolecularmente, formando éteres y alquenos, respectivamente. En el primer caso la reacción es una sustitución nucleofílica, mientras que en el segundo ocurre eliminación iónica.
R OH+ + OH2R' OHH+/φ
R O R'
Éter
Mecanismo SN
+ OH2H+/φ
Alqueno
Mecanismo EC C C C
OHH
Mecanismos de deshidratación intermolecular: SN1
Esta reacción se realiza en medio ácido, para formar ión oxonio con enlace C-O más polar y, por lo tanto más fácil de romper heterolíticamente. El protón consumido en la primera etapa es liberado en la última, por lo que el ácido se requiere en muy baja concentración, actuando como catalizador.
O
H
R'
SN1
R OH + H B R OH2 R+ R O R ́ R O R ́ + HB
B-
+
H2O
H
+
B-
SN2
O R'
H
SN2
δ+ δ+R OH + H B R OH2 R ́ O R OH2 R ́ O R + H2O R ́ O R + HB
B-
+
H H
+
B-

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 122
2. Reacciones de transposición Las reacciones de transposición ocurren como etapas intermedias en el desarrollo de mecanismos a través de iones carbonio, como por ejemplo las SN1. Es importante tenerlas en cuenta ya que explican porqué en muchos casos no se obtienen los resultados esperados, formándose otros productos.
Cuando un ión carbonio pueda convertirse en un ión más estable, mediante la migración de un grupo alquilo o de un hidrógeno de un átomo de carbono adyacente, ocurrirá una reagrupación. Esto sucede cuando un átomo de hidrógeno o un grupo alquilo migra con un par de electrones desde un carbono adyacente al que es portador de la carga positiva (+). El carbono que pierde el grupo migratorio adquiere la carga (+).
Una migración de hidrógeno se llama desplazamiento de hidruro (H:-), y una migración de grupo alquilo, desplazamiento de anión metilo o metiluro (H3C:-). Estos son los casos más comunes de transposiciones, también llamados desplazamientos 1,2; en donde el grupo que migra se desplaza de un átomo al vecino inmediato.
En cualquiera de los dos casos un catión menos estable se transforma en otro más estable: un carbocatión primario se transforma en uno secundario o en uno terciario, uno secundario en uno terciario.
- Desplazamiento de hidruro:
C C+
C+
H
C C+
C
H
C C+
C
H+
- Desplazamiento de metiluro:
C C+
C+
C C+
C C C+
C
CH3
+CH3 CH3
Resumiendo:
C C+
R
H
C C+
R
R
C C+
H
H
Si hay H y alquilo, migra hidruro.
3. Aplicaciones de las sustituciones nucleofílicas alifáticas Estas reacciones han sido muy estudiadas en química orgánica y se prestan muy bien para estudios de mecanismos, además de tener una enorme utilidad en síntesis. Hemos visto la preparación de alcoholes a partir de haluros de alquilo y de éteres y haluros de alquilo a partir de alcoholes.
El método de obtención de haluros de alquilo mediante sustituciones nucleofílicas es más útil que el de la halogenación de alcanos (SRL), ya que en este último caso es muy difícil obtener derivados monohalogenados.
Ejemplos útiles del uso de estas reacciones lo constituye la preparación de ésteres y amidas, reacciones que ocurren en muchas polimerizaciones por condensación.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Alifáticas 123
R C
O
O H
NH2H
OR'H
R C
O
NH2
R C
O
OR'
+ OH2
+ OH2
medio ácidoAmida
Éstermedio ácido
Cuando en una misma molécula hay más de un grupo amida o éster, ocurre la polimerización (ej.: poliésteres, poliamidas).
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial. Addison- Wesley Iberoamericana.
2. SolomonsT. - QUÍMICA ORGÁNICA - Editorial Limusa. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.
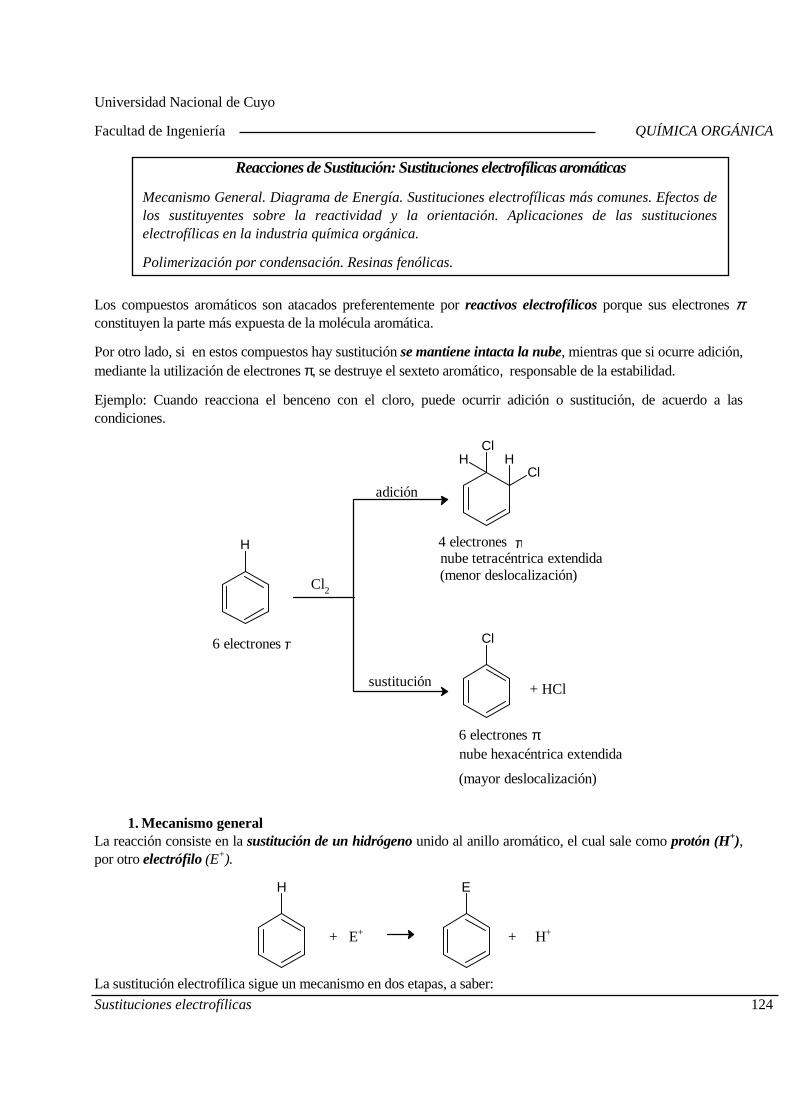
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 124
Los compuestos aromáticos son atacados preferentemente por reactivos electrofílicos porque sus electrones π constituyen la parte más expuesta de la molécula aromática.
Por otro lado, si en estos compuestos hay sustitución se mantiene intacta la nube, mientras que si ocurre adición, mediante la utilización de electrones π, se destruye el sexteto aromático, responsable de la estabilidad.
Ejemplo: Cuando reacciona el benceno con el cloro, puede ocurrir adición o sustitución, de acuerdo a las condiciones.
H
π 6 electrones
adición
Cl2
sustitución
Cl
ClHH
4 electrones π nube tetracéntrica extendida(menor deslocalización)
Cl
+ HCl
6 electrones π nube hexacéntrica extendida
(mayor deslocalización)
1. Mecanismo general La reacción consiste en la sustitución de un hidrógeno unido al anillo aromático, el cual sale como protón (H+), por otro electrófilo (E+).
H
+ E+ + H+
E
La sustitución electrofílica sigue un mecanismo en dos etapas, a saber:
Reacciones de Sustitución: Sustituciones electrofílicas aromáticas
Mecanismo General. Diagrama de Energía. Sustituciones electrofílicas más comunes. Efectos de los sustituyentes sobre la reactividad y la orientación. Aplicaciones de las sustituciones electrofílicas en la industria química orgánica.
Polimerización por condensación. Resinas fenólicas.
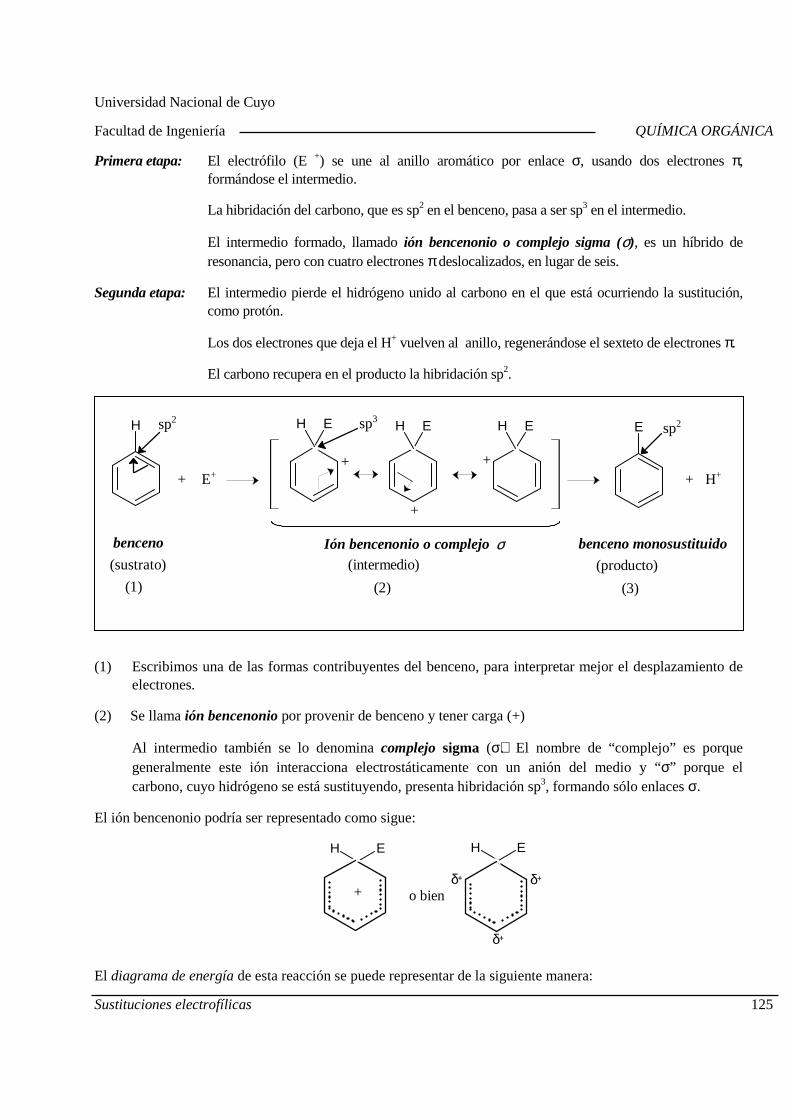
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 125
Primera etapa: El electrófilo (E +) se une al anillo aromático por enlace σ, usando dos electrones π, formándose el intermedio.
La hibridación del carbono, que es sp2 en el benceno, pasa a ser sp3 en el intermedio.
El intermedio formado, llamado ión bencenonio o complejo sigma (σ), es un híbrido de resonancia, pero con cuatro electrones π deslocalizados, en lugar de seis.
Segunda etapa: El intermedio pierde el hidrógeno unido al carbono en el que está ocurriendo la sustitución, como protón.
Los dos electrones que deja el H+ vuelven al anillo, regenerándose el sexteto de electrones π.
El carbono recupera en el producto la hibridación sp2.
(1) Escribimos una de las formas contribuyentes del benceno, para interpretar mejor el desplazamiento de electrones.
(2) Se llama ión bencenonio por provenir de benceno y tener carga (+)
Al intermedio también se lo denomina complejo sigma (σ). El nombre de “complejo” es porque generalmente este ión interacciona electrostáticamente con un anión del medio y “σ” porque el carbono, cuyo hidrógeno se está sustituyendo, presenta hibridación sp3, formando sólo enlaces σ.
El ión bencenonio podría ser representado como sigue:
o bien+
EH EH
δ+
δ+
δ+
El diagrama de energía de esta reacción se puede representar de la siguiente manera:
H sp2
+ E+ + H++
+
+
EH EH EH E sp2
benceno
(sustrato)
(1)
Ión bencenonio o complejo σ (intermedio)
(2)
benceno monosustituido
(producto)
(3)
sp3

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 126
Ep
Avance de la reacción
Ea1
Ea1 Ea2
H(1)
(2)
(4)
(5)
(3)
∆
1 Sustrato (benceno)
2 Intermedio (complejo )
3 Producto (benceno monosustituido)
4 Estado de transición o complejo activado
5 Estado de transición o complejo activado
σ
En el diagrama se observa un valle, donde se encuentra el intermedio (2), o sea, el producto de la primera etapa y reactivo de la segunda.
Las cimas corresponden a los estados de transición o complejos activados de las dos etapas, (4) y (5) respectivamente. Es decir, donde se está rompiendo el sexteto de electrones π (se pierde deslocalización o estabilización por resonancia) y, el segundo, donde se está regenerando el mismo.
Por esa razón la Energía de activación de la primera etapa es mucho mayor que la de la segunda. Por consiguiente, la primera etapa es la etapa lenta.
En los complejos activados la nube π está deformada por la acción del electrófilo entrante (E+) o saliente (H+), el cual está parcialmente unido al anillo.
δ +δ
δ δ E+ +
H E H
+
(4) (5)
Si consideramos que la reacción avanzó hasta el intermedio, éste podrá retroceder, formándose nuevamente benceno, o bien seguir hacia la derecha, formándose benceno sustituido.
Que la reacción avance hacia la izquierda o hacia la derecha desde el intermedio, depende de cómo es Ea1 (Energía de activación de la primera etapa, considerada de derecha a izquierda) con respecto a Ea2 (Energía de activación de la segunda etapa, considerada de izquierda a derecha):

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 127
Ea1 = Ea2 La reacción es reversible (ej: sulfonación)
Ea1 < Ea2 La reaccón se revierte hacia la izquierda.
Ea1 > Ea2 La reacción sigue avanzando hacia la derecha
La reacción no es productiva
(ej: nitración)
2. Aspectos particulares de las sustituciones electrofílicas más comunes
Las sustituciones electrofílicas más comunes son: nitración, sulfonación, halogenación y alquilación. Es de destacar que estas reacciones generalmente requieren la presencia de catalizadores ácidos (ej.: SO4H2, Cl3Al, Cl3Fe, etc.), lo que permite la generación de electrófilos suficientemente reactivos que ataquen al benceno a velocidad conveniente.
2.1 Nitración del benceno
H NO2
+ HNO3 + H2OH2SO4
nitrobenceno
La reacción se realiza con ácido nítrico (NO3H), a partir del cual se forma el electrófilo atacante (+NO2: ión
nitronio), en presencia de ácido sulfúrico concentrado (catalizador), que es el que aumenta la velocidad de la reacción al favorecer la formación del electrófilo. En una primera etapa el NO3H actúa como base, aceptando un protón del SO4H2 (ácido más fuerte). En la segunda, el ácido nítrico protonado se disocia, generándose el ión nitronio:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 128
O2N-OH + H:O-SO3H O2N-OH2 + :O-SO3H
O2N-OH2 O2N + :OH2
:OH2 + H:O-SO3H H:OH2 + :O-SO3H
O2N-OH + 2 H:O-SO3H O2N + 2 :O-SO3H + OH3
O2N-OH : ácido nítrico
H:O-SO3H : ácido sulfúrico
:O-SO3H : sulfato ácido
O2N-OH2 : ácido nítrico protonado
OH3 : ión hidronio
O2N : ión nitronio
. . + -
+ +
+ -
++ -
-
+
+
+
Veremos la estructura del ión nitronio, que es el electrófilo atacante (E+), pero antes recordemos las siguientes fórmulas estructurales:
H O N O
O
ácido nítrico
H O N O
O
ácido nítrico protonado
H
+
Al separar H2O del ácido nítrico protonado, se forma el ión nitronio, que es un híbrido de resonancia:
+ N O
O
+ N O
O
(La covalencia dativa entre nitrógeno y oxígeno, se puede representar como N O , o como N O)
Ya hemos visto la formación del electrófilo, veamos ahora el mecanismo de la sustitución electrofílica:
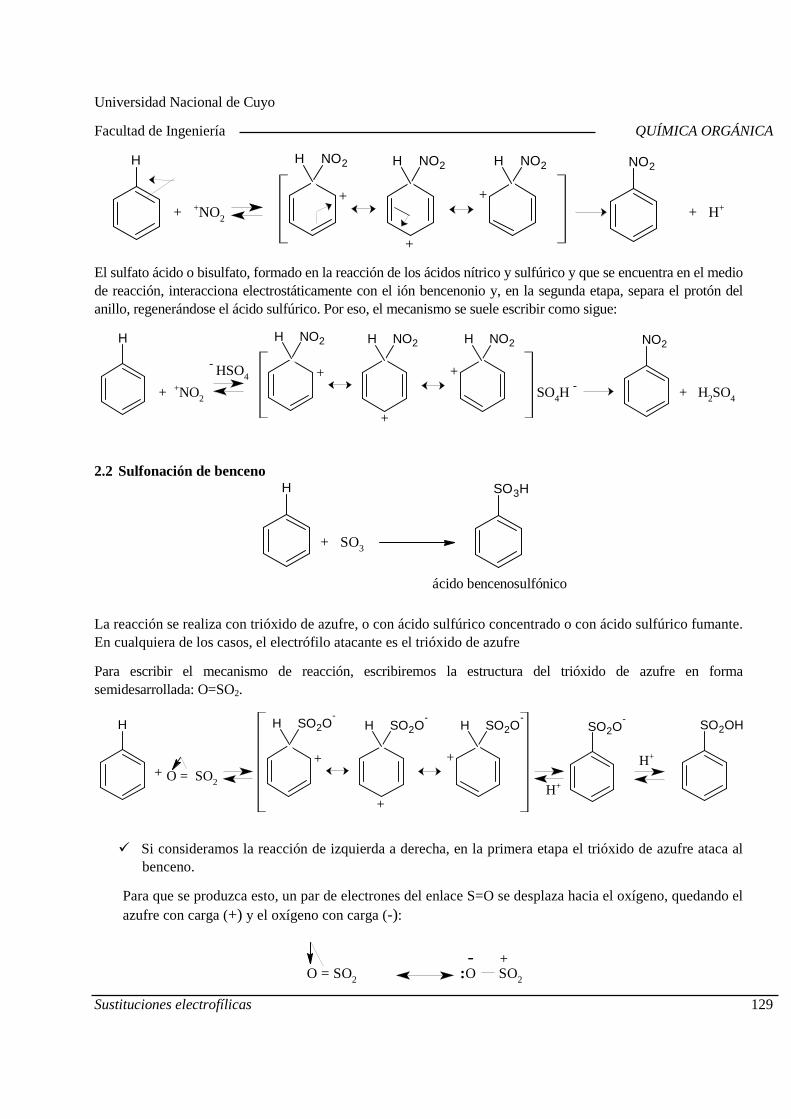
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 129
H
+ +NO2 + H++ +
NO2H NO2H NO2H NO2
+
El sulfato ácido o bisulfato, formado en la reacción de los ácidos nítrico y sulfúrico y que se encuentra en el medio de reacción, interacciona electrostáticamente con el ión bencenonio y, en la segunda etapa, separa el protón del anillo, regenerándose el ácido sulfúrico. Por eso, el mecanismo se suele escribir como sigue:
H
+ +NO2 SO4H - + H2SO4
+ +
NO2H NO2H NO2H NO2
- HSO4
+
2.2 Sulfonación de benceno H
+ SO3
SO3H
ácido bencenosulfónico
La reacción se realiza con trióxido de azufre, o con ácido sulfúrico concentrado o con ácido sulfúrico fumante. En cualquiera de los casos, el electrófilo atacante es el trióxido de azufre
Para escribir el mecanismo de reacción, escribiremos la estructura del trióxido de azufre en forma semidesarrollada: O=SO2.
H
+ + +
SO2O-
H SO2O-
H SO2O-
H SO2O-
+
O = SO2H+
H+
SO2OH
Si consideramos la reacción de izquierda a derecha, en la primera etapa el trióxido de azufre ataca al
benceno.
Para que se produzca esto, un par de electrones del enlace S=O se desplaza hacia el oxígeno, quedando el azufre con carga (+) y el oxígeno con carga (-):
O = SO2 :O SO2
+-

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 130
Por eso el trióxido de azufre se une al anillo a través del azufre, neutralizando su carga + y quedando como:
SO2O-
SO3- -:O SO2
ó ó
Se ha comprobado que la sulfonación es reversible en todas las etapas, debido a lo siguiente:
Energía de activación de la primer etapa:
Si se considera la primera etapa de derecha a izquierda, la energía de activación de la primera etapa es Ea1
Si se considera la segunda etapa de izquierda a derecha, la energía de activación de la segunda etapa es Ea2
Se ha comprobado que Ea1 = Ea2 , por lo que la primera etapa es reversible.
Segunda etapa (considerada de izquierda a derecha): Se separa el H+ del ión bencenonio, formándose el ión bencenosulfonato.
Tercera etapa (considerada de izquierda a derecha): el H+ formado en la segunda etapa se une al ión bencenosulfonato, formándose el ácido bencenosulfónico.
El ácido bencenosulfónico, que tiene carácter ácido, se ioniza y se regenera el bencenosulfonato (tercera etapa de derecha a izquierda). Es decir, en la tercera etapa hay un equilibrio ácido-base, que explica la reversibilidad de la tercera etapa:
ión bencenosulfonato + H+ ácido bencenosulfónico
Segunda etapa (considerada de derecha a izquierda): el H+ separado del ácido bencenosulfónico (ver etapa 3) es un electrófilo que puede atacar al ión bencenosulfonato y formar el ión bencenonio.
2.3 Halogenación de benceno H
+ X2 + HX
X
clorobenceno si X = cloro
AlX 3 o FeX3
La halogenación se realiza con halógeno (Cl2 o Br2), en presencia de un ácido de Lewis (Al Cl3 - Fe Cl3 - Fe Br3), que es un aceptor de electrones. Esta reacción no es útil para realizar fluoración o yodación porque el flúor, que es muy reactivo, da mezcla de productos, mientras que el iodo da una reacción reversible, por lo que el rendimiento es muy bajo.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 131
La función del catalizador es polarizar la molécula de halógeno que es no polar, porque a través del aluminio o del hierro (ácidos de Lewis: aceptores de electrones) interacciona con los electrones no compartidos del halógeno, permitiendo la ruptura heterolítica de la molécula de halógeno.
:. .X X Al X
. .
. . . .: : :
. .
. .X
X
Habrían dos opciones: que el benceno ataque al complejo (1), o que el ión halonio formado (X+) ataque al benceno. Nosotros consideraremos la segunda opción.
Ya hemos visto la formación del electrófilo, veamos ahora el mecanismo de la sustitución electrofílica (ej.: cloración):
X X + AlX3 X ............ X .............AlX 3 X+ + X AlX3
:-
: δ + -
(1)
cloronio o
bromonio
tetracloruro de Al
(si X = cloro)
:δ
H
+ Cl+ + H++ +
ClH ClH ClH Cl
+
El tetracloruro de aluminio (formado por la reacción de Cl2 con Cl3Al), que se encuentra en el medio de reacción, interacciona electrostáticamente con el ión bencenonio y, en la segunda etapa separa el protón del anillo, que forma ácido clorhídrico con un cloruro, regenerándose el cloruro de aluminio. Por eso, el mecanismo se suele escribir como sigue:
H
+ Cl+ + HCl + AlCl3 + +
ClH ClH ClH Cl
+
AlCl4-
AlCl4-
2.4 Alquilación del benceno Las alquilaciones se pueden realizar con haluros de alquilo, en presencia de ácidos de Lewis (ej: AlCl3).
H
+ R X + HX
R
alquilbenceno
AlX 3
haluro de alquilo

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 132
La función del catalizador es similar a la de la halogenación. El catalizador forma con el haluro de alquilo un complejo que transfiere el grupo alquilo al anillo. También se ha propuesto que se forma el ión carbonio y éste es el que ataca al anillo. Consideraremos el segundo caso, o sea, la formación del ión carbonio y posterior ataque al anillo aromático.
R X + AlX3 R ............ X .............AlX 3 R+ + X AlX3
:-
: δ + -
(1)
carbonio tetracloruro de Al
(si X = cloro)
:δ
Veamos el mecanismo de reacción:
H
+ R + + H++ +
RH RH RH R
+
El tetracloruro de aluminio, que se encuentra en el medio de reacción, interacciona electrostáticamente con el ión bencenonio y, en la segunda etapa separa el protón del anillo, que forma ácido clorhídrico con un cloruro, regenerándose el cloruro de aluminio. Por eso, el mecanismo se suele escribir como sigue:
H
+ R + + HCl + AlCl3 + +
RH RH RH R
+
AlCl4-
AlCl4-
Nota: Se ha comprobado que ocurren transposiciones, por lo que se estima que se llega a formar ión carbonio,
siendo éste el que ataca al benceno.
3. Efectos de los grupos sustituyentes Hemos visto la sustitución de un hidrógeno en benceno, con la formación de un benceno monosustituido, mediante un mecanismo de sustitución electrofílica:
H
+ E+ + H+
E
benceno benceno monosustituido
Observar que se forma un solo producto monosustituido del benceno, porque sus seis hidrógenos son equivalentes.
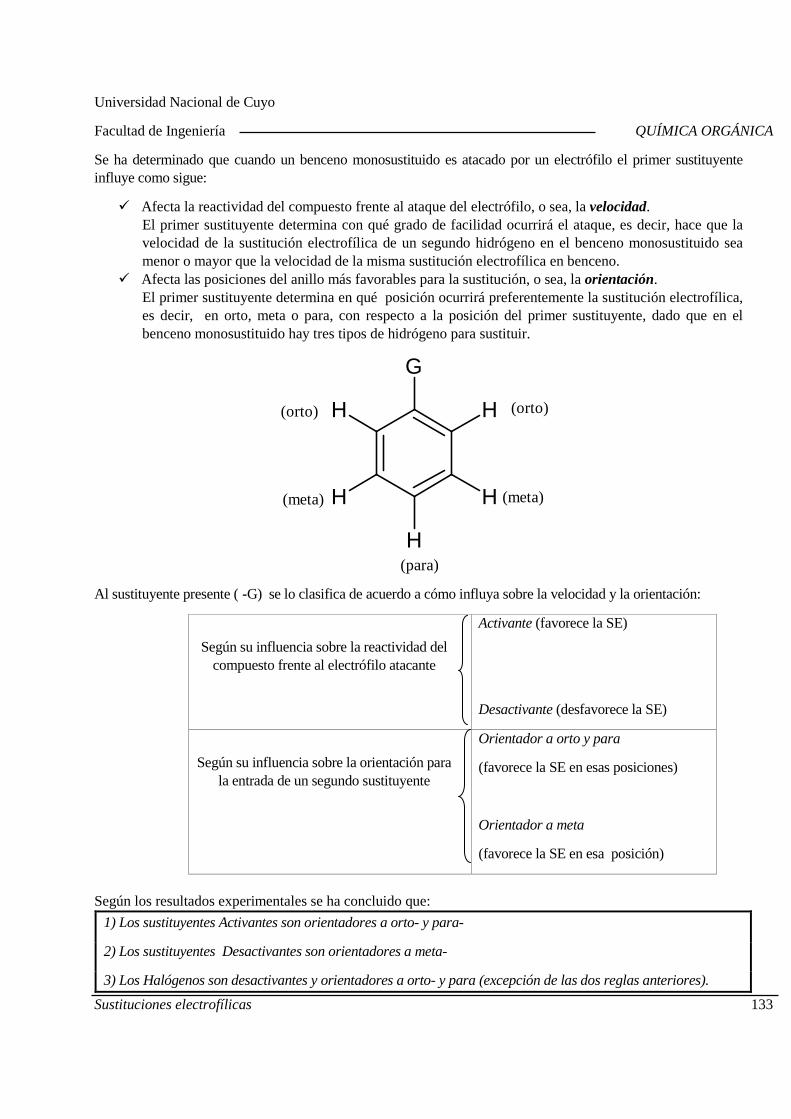
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 133
Se ha determinado que cuando un benceno monosustituido es atacado por un electrófilo el primer sustituyente influye como sigue:
Afecta la reactividad del compuesto frente al ataque del electrófilo, o sea, la velocidad. El primer sustituyente determina con qué grado de facilidad ocurrirá el ataque, es decir, hace que la velocidad de la sustitución electrofílica de un segundo hidrógeno en el benceno monosustituido sea menor o mayor que la velocidad de la misma sustitución electrofílica en benceno.
Afecta las posiciones del anillo más favorables para la sustitución, o sea, la orientación. El primer sustituyente determina en qué posición ocurrirá preferentemente la sustitución electrofílica, es decir, en orto, meta o para, con respecto a la posición del primer sustituyente, dado que en el benceno monosustituido hay tres tipos de hidrógeno para sustituir.
G
H
H
H
H
H (orto)(orto)
(meta) (meta)
(para)
Al sustituyente presente ( -G) se lo clasifica de acuerdo a cómo influya sobre la velocidad y la orientación:
Según su influencia sobre la reactividad del compuesto frente al electrófilo atacante
Activante (favorece la SE)
Desactivante (desfavorece la SE)
Según su influencia sobre la orientación para la entrada de un segundo sustituyente
Orientador a orto y para
(favorece la SE en esas posiciones)
Orientador a meta
(favorece la SE en esa posición)
Según los resultados experimentales se ha concluido que:
1) Los sustituyentes Activantes son orientadores a orto- y para-
2) Los sustituyentes Desactivantes son orientadores a meta-
3) Los Halógenos son desactivantes y orientadores a orto- y para (excepción de las dos reglas anteriores).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 134
3.1 Influencia del sustituyente sobre la velocidad: Determinación de la reactividad relativa
Vamos a ver ahora cómo influye sobre la velocidad el sustituyente presente (-G) o primer sustituyente cuando el
benceno monosustituido es atacado por un electrófilo (E+).
Se ha comprobado que la reactividad del anillo bencénico monosustituido depende de la tendencia del sustituyente presente (-G) a atraer o ceder electrones, lo que hará por efecto inductivo y/o mesómero.
Los estudios se realizan a través de determinaciones experimentales y teóricas.
a) Determinaciones experimentales
Se realizan reacciones competitivas usando cantidades equimoleculares en iguales condiciones. Es decir, por un lado se hace reaccionar al benceno y, por otro lado, en iguales condiciones se hace la misma reacción partiendo de un benceno monosustituido. Luego, se determinan los rendimientos, considerando que se ha partido de cantidades equimoleculares, concluyéndose si el sustituyente en el benceno monosustituido favoreció o perjudicó a la reacción.
Por ejemplo, cuando se hace la nitración de cantidades equimoleculares de benceno y de tolueno, se obtiene una cantidad de productos mononitrados de tolueno 25 veces mayor que el mononitrado de benceno, con lo cual se concluye que el tolueno es 25 veces más reactivo que el benceno. Por lo tanto, el grupo metilo favoreció la sustitución electrofílica, es un activante.
Para clasificar como activantes o desactivantes a otros sustituyentes se hacen experiencias con otros bencenos monosustituidos y, en cada caso, se comparan la cantidad de productos resultantes de la reacción del benceno monosustituido y del benceno. Así, se obtiene un listado de los sustituyentes Activantes y otro de los Desactivantes.
b) Determinaciones teóricas
Se realiza el análisis del estado base o reactivo (benceno monosustituido) y del intermedio de la sustitución electrofílica.
− Estado base o reactivo (benceno monosustituido)
G G
G cede densidad electrónica al anillo. Los electrones π del anillo están más disponibles que en benceno sin sustituyente.
G quita densidad electrónica al anillo. Los electrones π del anillo están menos disponibles que en benceno sin sustituyente.
Mayor reactividad que benceno frente a un reactivo electrofílico (E
+)
Menor reactividad que benceno frente a un reactivo electrofílico (E
+)
− Intermedio formado cuando el benceno monosustituido es atacado por un electrófilo
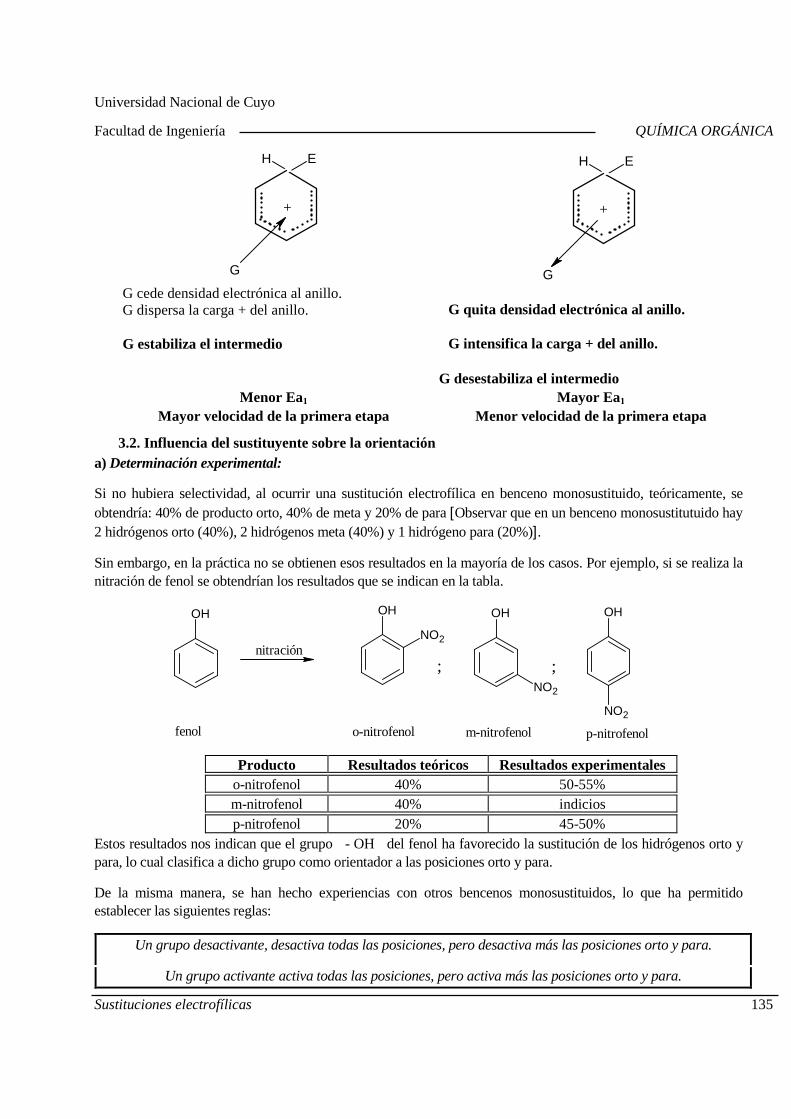
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 135
+
EH
+
EH
G G G cede densidad electrónica al anillo. G dispersa la carga + del anillo. G estabiliza el intermedio
G quita densidad electrónica al anillo.
G intensifica la carga + del anillo. G desestabiliza el intermedio
Menor Ea1 Mayor Ea1
Mayor velocidad de la primera etapa Menor velocidad de la primera etapa
3.2. Influencia del sustituyente sobre la orientación a) Determinación experimental:
Si no hubiera selectividad, al ocurrir una sustitución electrofílica en benceno monosustituido, teóricamente, se obtendría: 40% de producto orto, 40% de meta y 20% de para [Observar que en un benceno monosustitutuido hay 2 hidrógenos orto (40%), 2 hidrógenos meta (40%) y 1 hidrógeno para (20%)].
Sin embargo, en la práctica no se obtienen esos resultados en la mayoría de los casos. Por ejemplo, si se realiza la nitración de fenol se obtendrían los resultados que se indican en la tabla.
OH
nitración
OH
NO2
OH
NO2
OH
NO2
; ;
fenol o-nitrofenol m-nitrofenol p-nitrofenol
Producto Resultados teóricos Resultados experimentales o-nitrofenol 40% 50-55% m-nitrofenol 40% indicios p-nitrofenol 20% 45-50%
Estos resultados nos indican que el grupo - OH del fenol ha favorecido la sustitución de los hidrógenos orto y para, lo cual clasifica a dicho grupo como orientador a las posiciones orto y para.
De la misma manera, se han hecho experiencias con otros bencenos monosustituidos, lo que ha permitido establecer las siguientes reglas:
Un grupo desactivante, desactiva todas las posiciones, pero desactiva más las posiciones orto y para.
Un grupo activante activa todas las posiciones, pero activa más las posiciones orto y para.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 136
(Recordar que habíamos dicho que los activantes son orientadores orto-para y los desactivantes son orientadores meta)
b) Determinación teórica:
El estudio teórico se hace mediante el análisis del estado base, con lo cual se determina qué posiciones tienen mayor disponibilidad de densidad electrónica. Además, se lleva a cabo el análisis de los intermedios que se formarían cuando el benceno monosustituido es atacado en o-, m- y p-, para deducir cuál es el más estable y, por lo tanto, el que se formará más rápido. En el análisis se debe tener en cuenta el efecto (inductivo o mesómero) que ejerce el primer sustituyente, para determinar si es dador o aceptor de electrones.
Recordemos cómo los grupos sustituyentes se podrían reunir según cedan o atraigan electrones, lo que permitiría definir su influencia en las reacciones:
Grupos que
atraen electrones
Por efecto inductivo
Por efecto mesómero
Grupos que ceden electrones
Por efecto inductivo
Por efecto mesómero
H4N+
CF F
F
SO OH
O
Cl F
O N = O HO C = O H C = O C = O
R (Grupos alquilo)
Grupos del tipo -X = Y, donde Y es más electronegativo que carbono
: NH3 : OH : F : Cl
Grupos que poseen dos electrones sin compartir en el átomo que va unido al anillo
Análisis de estados bases e intermedios para las sustituciones electrofílicas sobre bencenos monosustituidos
A continuación se consideran los cuatro tipos de sustituyentes indicados en la tabla anterior:
a’) Grupo aceptor por efecto inductivo (ej: - CF3 )
− Análisis del estado base:
El grupo -CF3, aceptor de electrones, disminuye la disponibilidad de electrones π del anillo del estado base (trifluorfenilmetano).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 137
C F
F
F
I
− Análisis de los intermedios:
Si analizamos los tres intermedios que se formarían cuando el compuesto (trifluorfenilmetano) es atacado por un electrófilo (E+), se puede observar que tanto el intermedio o- como el p- presentan una forma contribuyente con la carga + en el carbono al que va unido el grupo CF3, aceptor de electrones.
Es decir, en los intermedios o- y p- el grupo CF3 le debe quitar densidad electrónica a un C+, lo cual
desestabiliza más a dichos intermedios.
En cambio, en el m- dicho sustituyente no ejerce su efecto directamente sobre C+, por lo cual, si bien desestabiliza a este intermedio, lo hace en menor medida que en los otros casos.
+
++
I
CF3
orto
meta
para
H
E
H
E E
HCF3
CF3 CF3
+
++H
E
H
E
H
CF3 CF3 CF3
+ +
+
H E EH EH
CF3 CF3 CF3
E
E+
Por lo tanto, cuando el primer sustituyente es un grupo aceptor por efecto inductivo:
− Los intermedios o- y p- son menos estables que el m-.
− Las Ea para la formación de o- y p- son mayores.
− Los intermedios o- y p- se forman a menor velocidad.
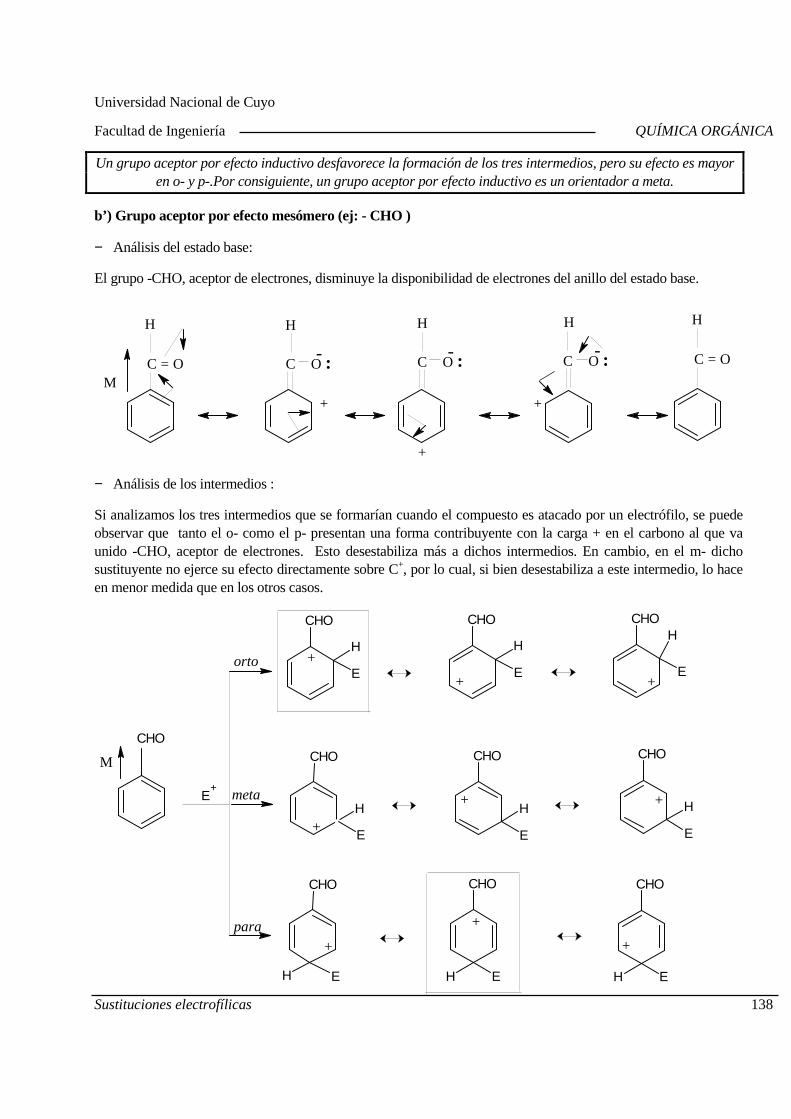
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 138
Un grupo aceptor por efecto inductivo desfavorece la formación de los tres intermedios, pero su efecto es mayor en o- y p-.Por consiguiente, un grupo aceptor por efecto inductivo es un orientador a meta.
b’) Grupo aceptor por efecto mesómero (ej: - CHO )
− Análisis del estado base:
El grupo -CHO, aceptor de electrones, disminuye la disponibilidad de electrones del anillo del estado base.
C = O
H
M
+
C O :-
H
C O :-
H
C O :-
H
+
+
C = O
H
− Análisis de los intermedios :
Si analizamos los tres intermedios que se formarían cuando el compuesto es atacado por un electrófilo, se puede observar que tanto el o- como el p- presentan una forma contribuyente con la carga + en el carbono al que va unido -CHO, aceptor de electrones. Esto desestabiliza más a dichos intermedios. En cambio, en el m- dicho sustituyente no ejerce su efecto directamente sobre C+, por lo cual, si bien desestabiliza a este intermedio, lo hace en menor medida que en los otros casos.
+
++
M
CHO
orto
meta
para
H
E
CHO
H
E
CHO
E
HCHO
+
++H
CHO
E
H
CHO
E
H
CHO
+ +
+
H E
CHO
EH
CHO
EH
CHO
E
E+

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 139
Por lo tanto, cuando el primer sustituyente es un grupo aceptor por efecto mesómero:
− Los intermedios o- y p- son menos estables que el m-.
− Las Ea para la formación de o- y p- son mayores.
− Los intermedios o- y p- se forman a menor velocidad.
Un grupo aceptor por efecto mesómero desfavorece la formación de los tres intermedios, pero su efecto es mayor en o- y p-. Por consiguiente, un grupo aceptor por efecto mesómero es un orientador a meta.
c’) Grupo dador por efecto inductivo (ej: - R )
− Análisis del estado base:
El grupo -R, dador de electrones, aumenta la disponibilidad de electrones del anillo del estado base.
R
I
− Análisis de los intermedios:
Si analizamos los tres intermedios que se formarían cuando el compuesto es atacado por un electrófilo, se puede observar que tanto el o- como el p- presentan una forma contribuyente con la carga + en el carbono al que va unido -R, dador de electrones. Esto estabiliza más a dichos intermedios. En cambio, en el m- dicho sustituyente no ejerce su efecto directamente sobre C+, por lo cual, si bien estabiliza a este intermedio, lo hace en menor medida que en los otros casos.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 140
+
++
R
orto
meta
para
H
E
H
E E
H
+
++H
E
H
E
H
+ +
+
H E EH EH
E
R R R
R R R
R R R
E+
Por lo tanto, cuando el primer sustituyente es un grupo dador por efecto inductivo:
− Los intermedios o- y p- son más estables que el m-.
− Las Ea para la formación de o- y p- son menores.
− Los intermedios o- y p- se forman a mayor velocidad.
Un grupo dador por efecto inductivo favorece la formación de los tres intermedios, pero su efecto es mayor en o- y p-. Por consiguiente, un grupo dador por efecto inductivo es un orientador a orto y para
d’) Grupo dador por efecto mesómero (ej: - OH )
− Análisis del estado base:
El grupo -OH, dador de electrones, aumenta la disponibilidad de electrones del anillo del estado base.
:O HM
+O H
..
+O H
+O H :O H
..
..

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 141
− Análisis de los intermedios :
Si analizamos los tres intermedios que se formarían cuando el compuesto es atacado por un electrófilo, se puede observar que tanto el o- como el p- presentan una forma contribuyente con la carga + en el carbono al que va unido -OH, dador de electrones. Esto permite que se forme otra forma contribuyente por el pasaje del par de electrones del oxígeno hacia el anillo, con la formación de doble enlace C=O. Al haber otra forma contribuyente, hay más deslocalización de electrones, mayor dispersión de la carga + y, por lo tanto, mayor estabilización de dichos intermedios. En cambio, en el m- dicho sustituyente no ejerce su efecto directamente sobre C+, por lo cual, si bien estabiliza a este intermedio, lo hace en menor medida que en los otros casos.
OH
+
++
M
orto
meta
para
H
E
OH
H
E
OH
E
HOH
+
++H
OH
E
H
OH
E
H
OH
+ +
+
H E
OH
EH
OH
EH
OH
E
:
: OH
H
+
E
:
: : :
: +
EH
OH: :
:
E+
Por lo tanto, cuando el primer sustituyente es un grupo dador por efecto mesómero:
− Los intermedios o- y p- son más estables que el m-.
− Las Ea para la formación de o- y p- son menores.
− Los intermedios o- y p- se forman a mayor velocidad.
Un grupo dador por efecto mesómero favorece la formación de los tres intermedios, pero su efecto es mayor en o- y p-. Por consiguiente, un grupo dador por efecto mesómero es un orientador a orto y para.
e’) Halógenos
Los halógenos deben analizarse aparte, porque se ha determinado que los halógenos son desactivantes y orientadores a orto y para.
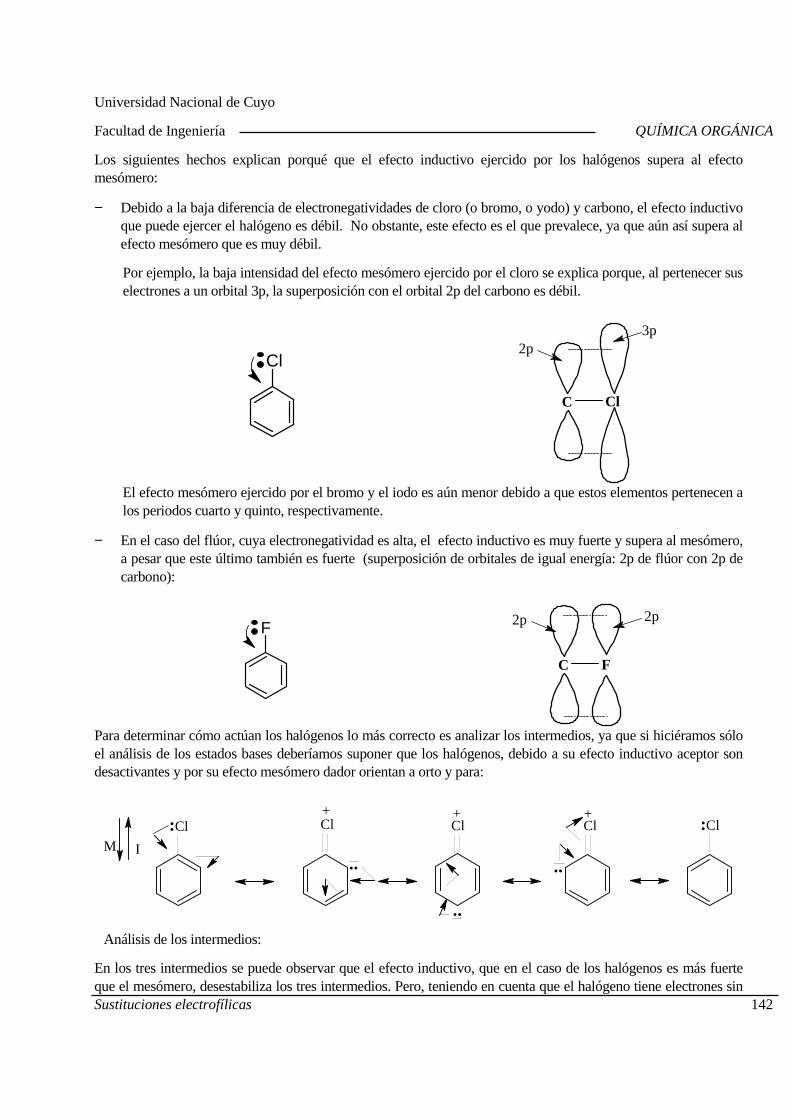
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 142
Los siguientes hechos explican porqué que el efecto inductivo ejercido por los halógenos supera al efecto mesómero:
− Debido a la baja diferencia de electronegatividades de cloro (o bromo, o yodo) y carbono, el efecto inductivo que puede ejercer el halógeno es débil. No obstante, este efecto es el que prevalece, ya que aún así supera al efecto mesómero que es muy débil.
Por ejemplo, la baja intensidad del efecto mesómero ejercido por el cloro se explica porque, al pertenecer sus electrones a un orbital 3p, la superposición con el orbital 2p del carbono es débil.
Cl
C Cl
2p3p
El efecto mesómero ejercido por el bromo y el iodo es aún menor debido a que estos elementos pertenecen a los periodos cuarto y quinto, respectivamente.
− En el caso del flúor, cuya electronegatividad es alta, el efecto inductivo es muy fuerte y supera al mesómero, a pesar que este último también es fuerte (superposición de orbitales de igual energía: 2p de flúor con 2p de carbono):
F
C F
2p 2p
Para determinar cómo actúan los halógenos lo más correcto es analizar los intermedios, ya que si hiciéramos sólo el análisis de los estados bases deberíamos suponer que los halógenos, debido a su efecto inductivo aceptor son desactivantes y por su efecto mesómero dador orientan a orto y para:
:Cl
M
+Cl
..
+Cl
+Cl :Cl
..
..I
Análisis de los intermedios:
En los tres intermedios se puede observar que el efecto inductivo, que en el caso de los halógenos es más fuerte que el mesómero, desestabiliza los tres intermedios. Pero, teniendo en cuenta que el halógeno tiene electrones sin
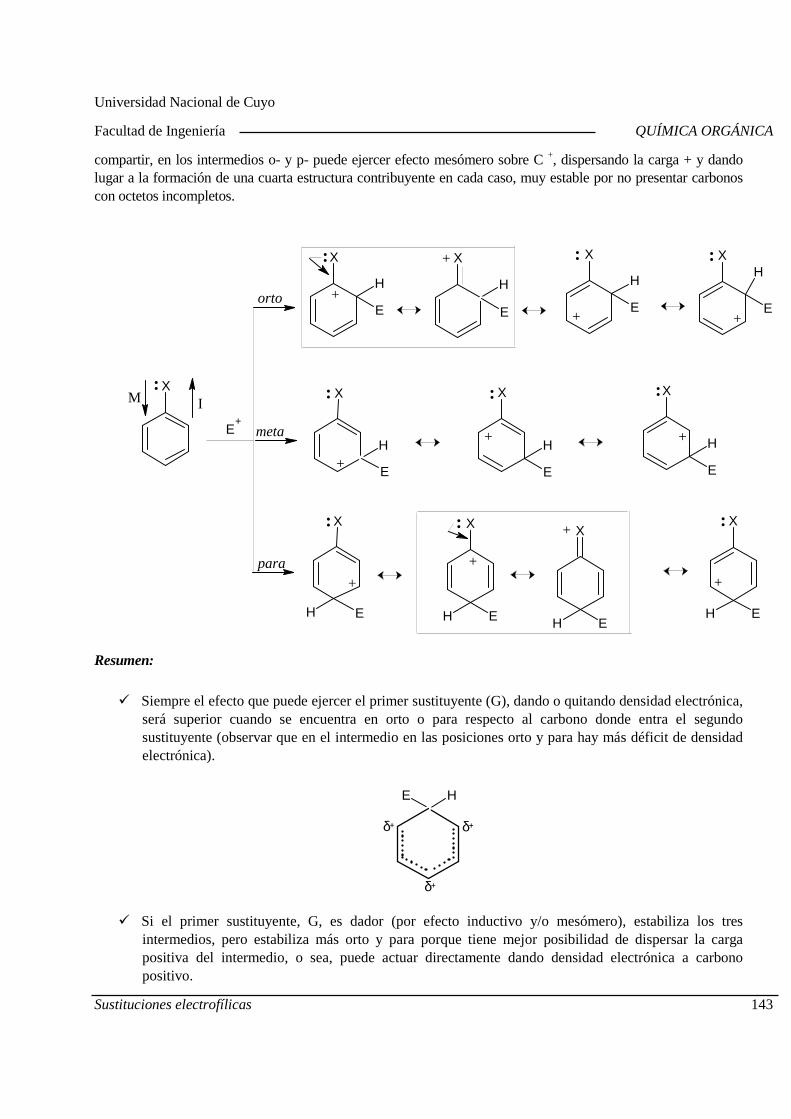
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 143
compartir, en los intermedios o- y p- puede ejercer efecto mesómero sobre C +, dispersando la carga + y dando lugar a la formación de una cuarta estructura contribuyente en cada caso, muy estable por no presentar carbonos con octetos incompletos.
X
+
++
M
orto
meta
para
H
E
X
H
E
X
E
HX
+
++H
X
E
H
X
E
H
X
+ +
+
H E
X
EH
X
EH
X
E
:
: X
H
+
E
:
: : :
:+
EH
X: :
:
I
E+
Resumen:
Siempre el efecto que puede ejercer el primer sustituyente (G), dando o quitando densidad electrónica, será superior cuando se encuentra en orto o para respecto al carbono donde entra el segundo sustituyente (observar que en el intermedio en las posiciones orto y para hay más déficit de densidad electrónica).
HE
δ+
δ+
δ+
Si el primer sustituyente, G, es dador (por efecto inductivo y/o mesómero), estabiliza los tres intermedios, pero estabiliza más orto y para porque tiene mejor posibilidad de dispersar la carga positiva del intermedio, o sea, puede actuar directamente dando densidad electrónica a carbono positivo.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 144
HE
δ+
δ+
δ+ G G
G
Si el primer sustituyente, G, es aceptor (por efecto inductivo y/o mesómero), desestabiliza los tres intermedios, pero su acción desestabilizadora es mayor cuando está en orto o para, respecto al carbono donde se produce la sustitución. En esas posiciones aparece mayor carga positiva que en meta.
HE
δ+
δ+
δ+ G G
G
Un grupo sustituyente afecta a las Sustituciones Aromáticas Nucleofílica y Electrofílica de forma opuesta.
o Los grupos aceptores son activantes en las sustituciones nucleofílicas, y desactivantes en las sustituciones electrofílicas.
o Los grupos dadores son desactivantes en las sustituciones nucleofílicas y activantes en las sustituciones electrofílicas.
4. Sustituciones electrofílicas en hidrocarburos polinucleares de núcleos condensados
Las sustituciones electrofílicas en el naftaleno, al igual que en el benceno, abren el camino para la síntesis de sus derivados. En cambio, para introducir diferentes grupos en el antraceno no es frecuente recurrir a las sustituciones electrofílicas, dado que estas reacciones dan, en la mayoría de los casos, mezclas de compuestos de sustitución y también de adición.
4.1. Naftaleno
Se ha determinado que, salvo en sulfonación donde el electrófilo puede atacar las posiciones α y ß, las sustituciones electrofílicas en naftaleno ocurren preferentemente en posición α. Esto se puede deducir del análisis de los intermedios que resultan de ambos ataques electrofílicos, ya que el que se forma por ataque en α es más estable porque presenta dos formas contribuyentes con sexteto de electrones π en un anillo (anillo bencénico inalterado), lo cual no ocurre con el otro intermedio.
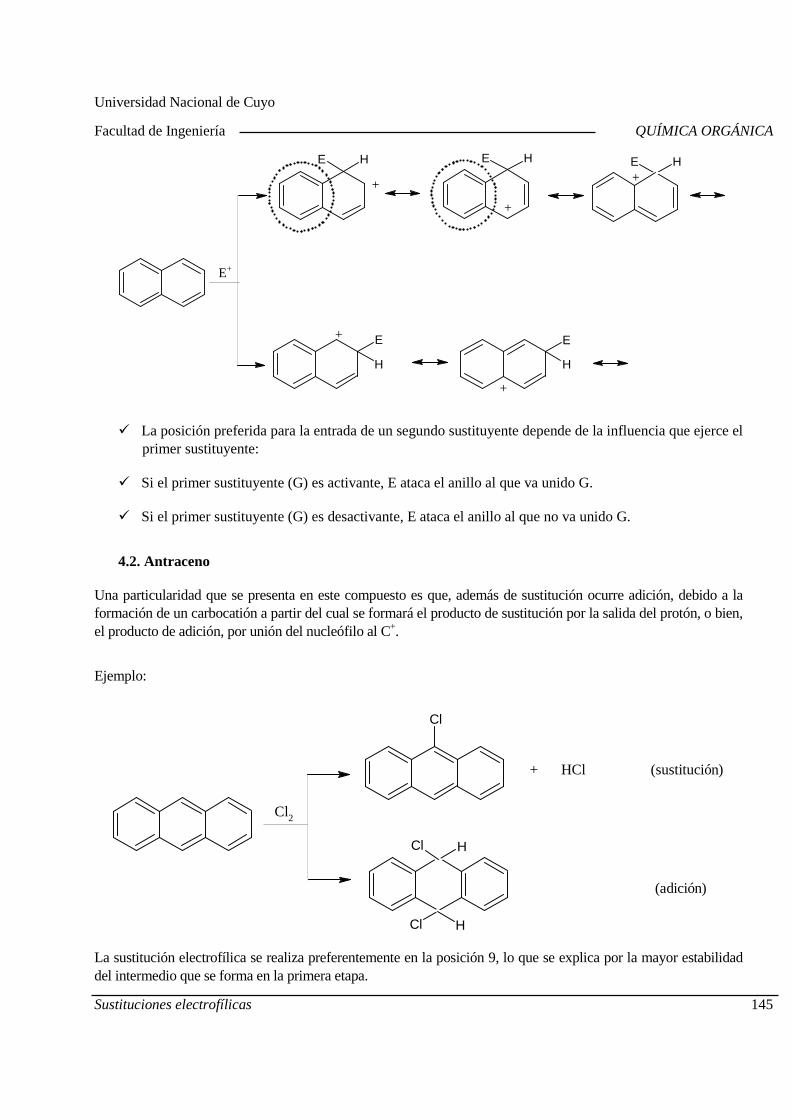
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 145
HE
+
HE HE
+
+
E
H
+ E
H
+
E+
La posición preferida para la entrada de un segundo sustituyente depende de la influencia que ejerce el primer sustituyente:
Si el primer sustituyente (G) es activante, E ataca el anillo al que va unido G.
Si el primer sustituyente (G) es desactivante, E ataca el anillo al que no va unido G.
4.2. Antraceno
Una particularidad que se presenta en este compuesto es que, además de sustitución ocurre adición, debido a la formación de un carbocatión a partir del cual se formará el producto de sustitución por la salida del protón, o bien, el producto de adición, por unión del nucleófilo al C+.
Ejemplo:
Cl2
Cl
+ HCl (sustitución)
HCl
Cl H
(adición)
La sustitución electrofílica se realiza preferentemente en la posición 9, lo que se explica por la mayor estabilidad del intermedio que se forma en la primera etapa.
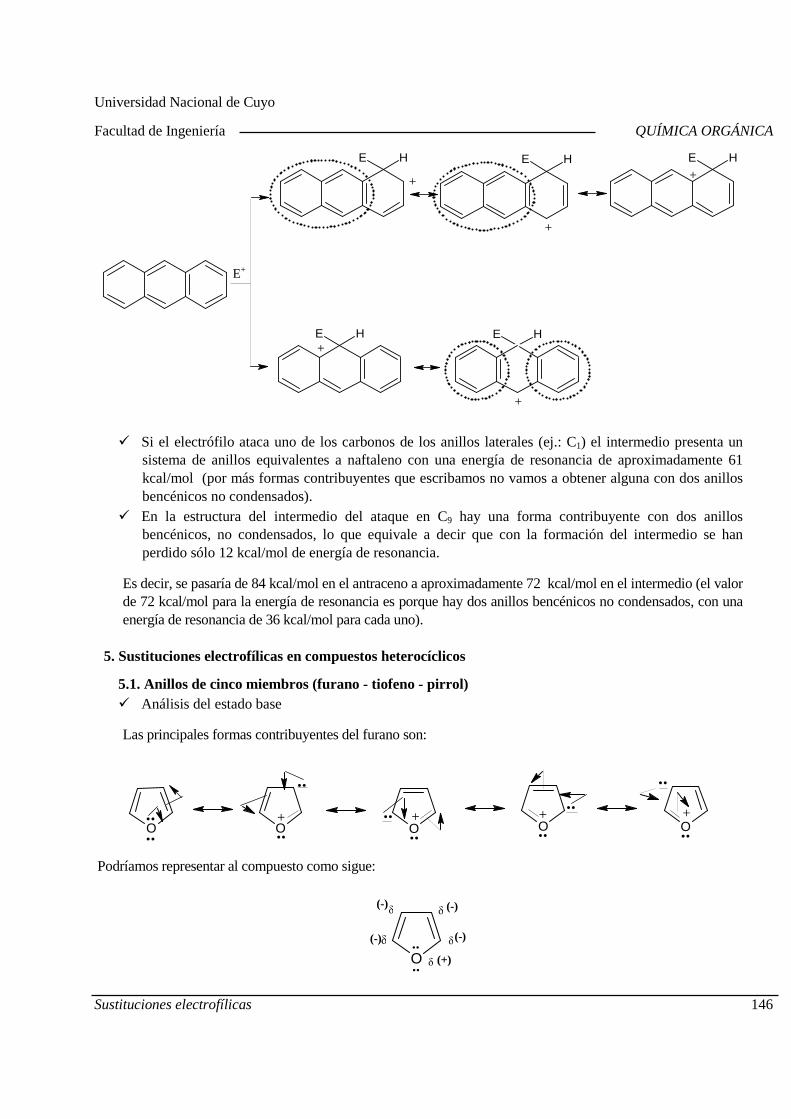
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 146
+
HE HE
+
+
+
+
E+
HE HE HE
Si el electrófilo ataca uno de los carbonos de los anillos laterales (ej.: C1) el intermedio presenta un sistema de anillos equivalentes a naftaleno con una energía de resonancia de aproximadamente 61 kcal/mol (por más formas contribuyentes que escribamos no vamos a obtener alguna con dos anillos bencénicos no condensados).
En la estructura del intermedio del ataque en C9 hay una forma contribuyente con dos anillos bencénicos, no condensados, lo que equivale a decir que con la formación del intermedio se han perdido sólo 12 kcal/mol de energía de resonancia.
Es decir, se pasaría de 84 kcal/mol en el antraceno a aproximadamente 72 kcal/mol en el intermedio (el valor de 72 kcal/mol para la energía de resonancia es porque hay dos anillos bencénicos no condensados, con una energía de resonancia de 36 kcal/mol para cada uno).
5. Sustituciones electrofílicas en compuestos heterocíclicos
5.1. Anillos de cinco miembros (furano - tiofeno - pirrol) Análisis del estado base
Las principales formas contribuyentes del furano son:
O.... ++ +.. ..
O O O O.. .. .. ..+
.. ..
Podríamos representar al compuesto como sigue:
(-)δ
Oδ
δ
δ (-)
(-)
(-)
..
..δ (+)
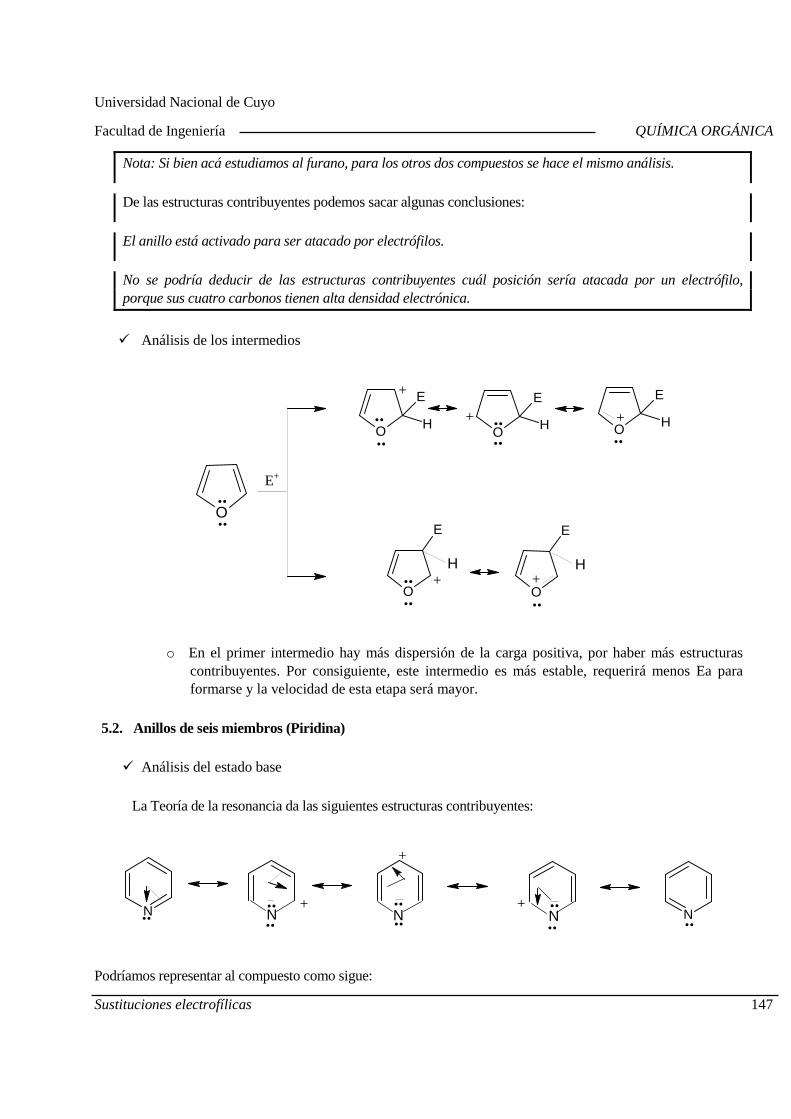
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 147
Nota: Si bien acá estudiamos al furano, para los otros dos compuestos se hace el mismo análisis.
De las estructuras contribuyentes podemos sacar algunas conclusiones:
El anillo está activado para ser atacado por electrófilos.
No se podría deducir de las estructuras contribuyentes cuál posición sería atacada por un electrófilo, porque sus cuatro carbonos tienen alta densidad electrónica.
Análisis de los intermedios
O....
+
+ +
+
E+
O H
E
O H
E
O H
E
O
E
O
E
....
..
....
H H
..
....
+
o En el primer intermedio hay más dispersión de la carga positiva, por haber más estructuras contribuyentes. Por consiguiente, este intermedio es más estable, requerirá menos Ea para formarse y la velocidad de esta etapa será mayor.
5.2. Anillos de seis miembros (Piridina)
Análisis del estado base
La Teoría de la resonancia da las siguientes estructuras contribuyentes:
..
+
+ +...... .. .. ..
..N N N NN
Podríamos representar al compuesto como sigue:
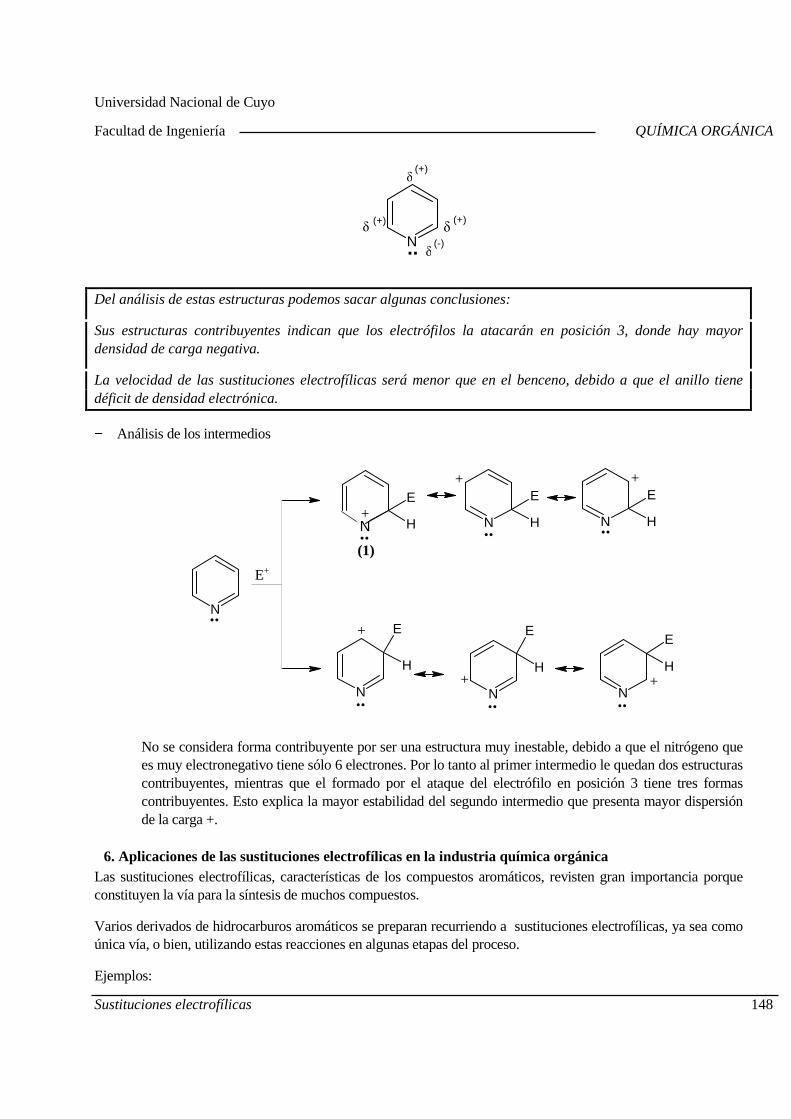
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 148
δ (+)
N..
δ
δ
δ
(+)
(+)
(-)
Del análisis de estas estructuras podemos sacar algunas conclusiones:
Sus estructuras contribuyentes indican que los electrófilos la atacarán en posición 3, donde hay mayor densidad de carga negativa.
La velocidad de las sustituciones electrofílicas será menor que en el benceno, debido a que el anillo tiene déficit de densidad electrónica.
− Análisis de los intermedios
..
..+
+
+
+
E+
....
..
..
+
N
H
E
N H
E
N H
E
N
H
E
N
H
E
N
E
H
+
..N(1)
No se considera forma contribuyente por ser una estructura muy inestable, debido a que el nitrógeno que es muy electronegativo tiene sólo 6 electrones. Por lo tanto al primer intermedio le quedan dos estructuras contribuyentes, mientras que el formado por el ataque del electrófilo en posición 3 tiene tres formas contribuyentes. Esto explica la mayor estabilidad del segundo intermedio que presenta mayor dispersión de la carga +.
6. Aplicaciones de las sustituciones electrofílicas en la industria química orgánica Las sustituciones electrofílicas, características de los compuestos aromáticos, revisten gran importancia porque constituyen la vía para la síntesis de muchos compuestos.
Varios derivados de hidrocarburos aromáticos se preparan recurriendo a sustituciones electrofílicas, ya sea como única vía, o bien, utilizando estas reacciones en algunas etapas del proceso.
Ejemplos:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 149
A partir del benceno, se prepara etilbenceno por alquilación, el cual es usado para la preparación de estireno. El estireno se polimeriza para dar poliestireno, que veremos en polimerización por adición.
La sulfonación, conjuntamente con la alquilación del benceno, son la vía para la preparación de detergentes (alquilbencenosulfonatos sódicos), que estudiaremos mas adelante.
La anilina es un compuesto orgánico de gran importancia industrial, por lo tanto de alta demanda, que no puede prepararse directamente a partir de benceno. Pero es posible mediante una sustitución electrofílica (nitración del benceno) obtener nitrobenceno, el cual luego es reducido a anilina. Es decir, una sustitución electrofílica permite preparar anilina a partir de benceno.
Benceno Nitrobenceno Anilinanitracion reducción
Ya vimos la preparación de fenoles a partir de haluros de arilo, que se obtienen por cloración del benceno. Por otra parte, la preparación de resinas fenólicas son polimerizaciones por condensación, que ocurren por sustituciones electrofílicas en fenol.
Las resinas fenol-formaldehido fueron los primeros polímeros sintéticos que se produjeron comercialmente. Son polímeros económicos, que poseen alta resistencia térmica y dieléctrica. No obstante, son de color oscuro y frágiles, dependiendo de la carga o relleno la resistencia del producto acabado.
Aproximadamente un tercio de las resinas fenólicas se usan para fines de moldeo y gran parte del resto se destina como aglutinante en la manufactura de madera terciada y otros materiales laminados, y como adhesivos impermeables para otros fines.
Estas polimerizaciones son catalizadas por ácidos o por bases, siendo mucho más rápidas cuando se usan catalizadores alcalinos. La reacción de polimerización para producir una unidad de resina fenólica comienza en algunos casos como sigue:
Catálisis ácida
En medio ácido se protona el aldehido, transformándose en un electrófilo mas fuerte para atacar al fenol en las posiciones orto y para.
H2C = O H2C = O:HH+
+OH
CH2OHHOH
+H+
CH2OH
OH
CH2OH
OHAlcohol
o-hidroxibencílico
Alcohol p-hidroxibencílico
y
SE
Catálisis básica
En medio básico el fenol está como fenóxido, con un anillo muy activado para el ataque del electrófilo.
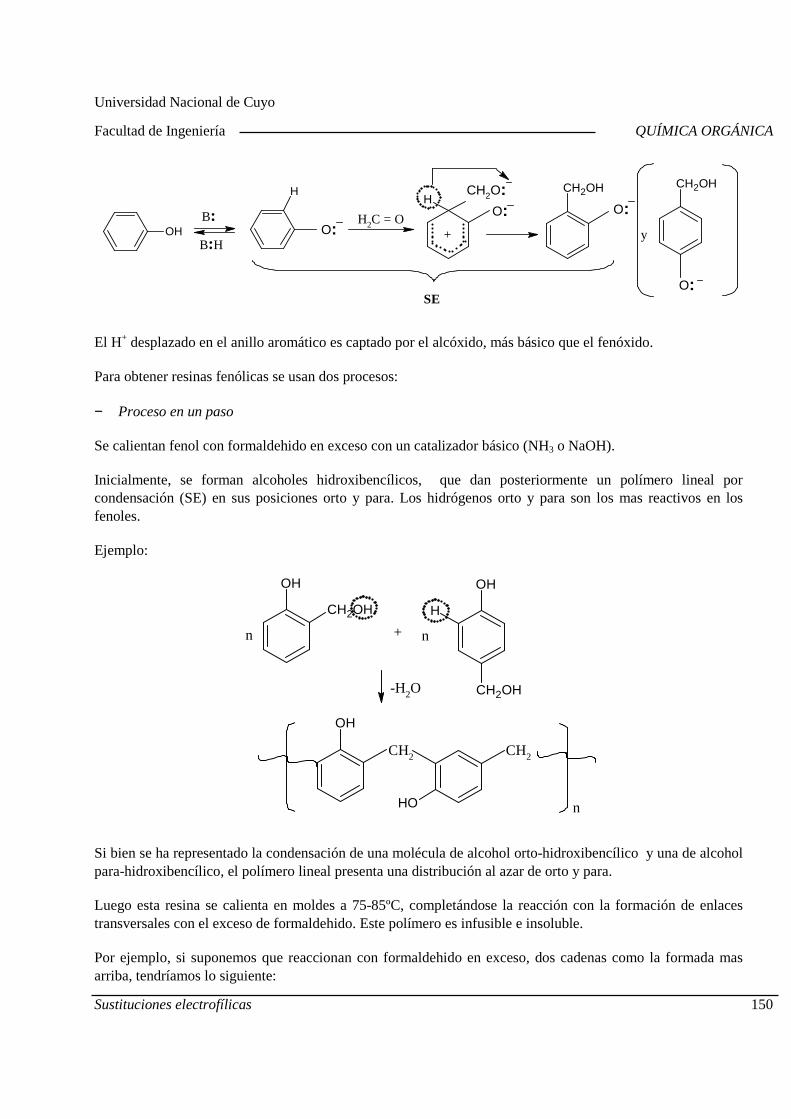
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 150
B:OH +
SE
B:H
H
O:H2C = O
HCH2O:
O:CH2OH
O:
y
CH2OH
O:
El H+ desplazado en el anillo aromático es captado por el alcóxido, más básico que el fenóxido.
Para obtener resinas fenólicas se usan dos procesos:
− Proceso en un paso
Se calientan fenol con formaldehido en exceso con un catalizador básico (NH3 o NaOH).
Inicialmente, se forman alcoholes hidroxibencílicos, que dan posteriormente un polímero lineal por condensación (SE) en sus posiciones orto y para. Los hidrógenos orto y para son los mas reactivos en los fenoles.
Ejemplo:
OH
CH2OH
n + n
H
OH
CH2OH-H2O
OH
OH
CH2 CH2
n
Si bien se ha representado la condensación de una molécula de alcohol orto-hidroxibencílico y una de alcohol para-hidroxibencílico, el polímero lineal presenta una distribución al azar de orto y para.
Luego esta resina se calienta en moldes a 75-85ºC, completándose la reacción con la formación de enlaces transversales con el exceso de formaldehido. Este polímero es infusible e insoluble.
Por ejemplo, si suponemos que reaccionan con formaldehido en exceso, dos cadenas como la formada mas arriba, tendríamos lo siguiente:
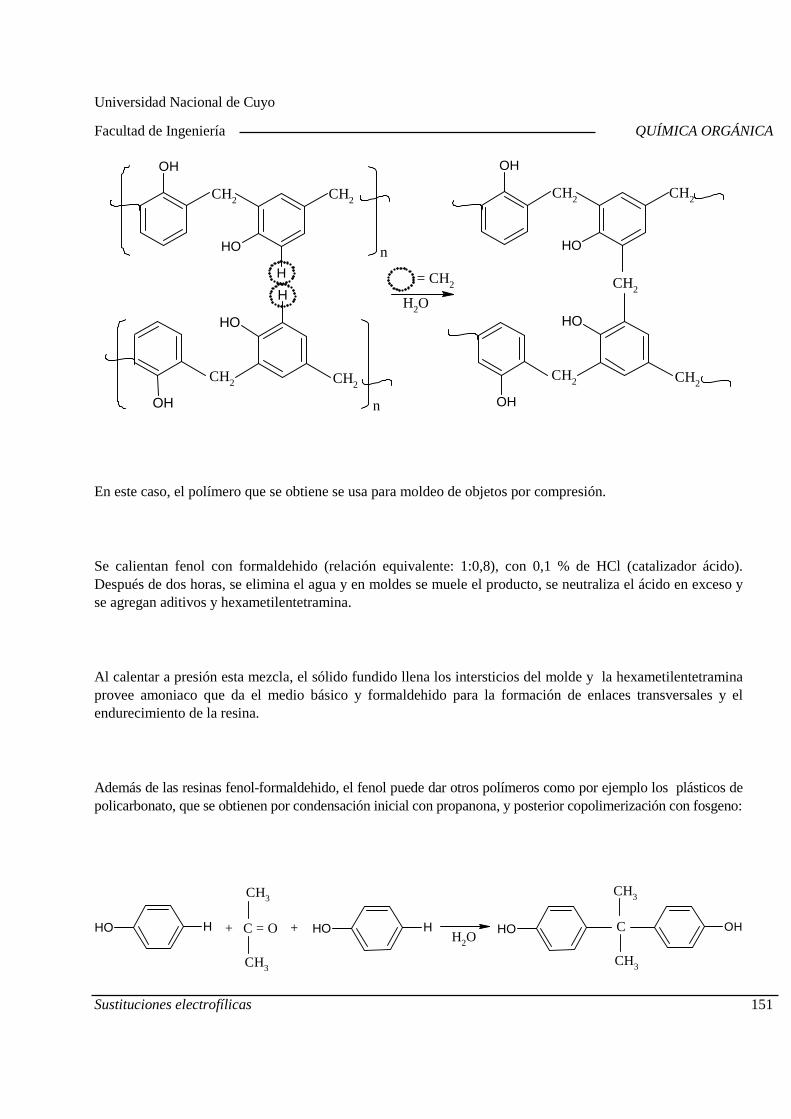
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 151
OH
OH
H
CH2 CH2
n
OH
OH
H
CH2
n
CH2
= CH2
H2O
OH
OH
CH2 CH2
CH2
OH
OH
CH2 CH2
En este caso, el polímero que se obtiene se usa para moldeo de objetos por compresión.
Se calientan fenol con formaldehido (relación equivalente: 1:0,8), con 0,1 % de HCl (catalizador ácido). Después de dos horas, se elimina el agua y en moldes se muele el producto, se neutraliza el ácido en exceso y se agregan aditivos y hexametilentetramina.
Al calentar a presión esta mezcla, el sólido fundido llena los intersticios del molde y la hexametilentetramina provee amoniaco que da el medio básico y formaldehido para la formación de enlaces transversales y el endurecimiento de la resina.
Además de las resinas fenol-formaldehido, el fenol puede dar otros polímeros como por ejemplo los plásticos de policarbonato, que se obtienen por condensación inicial con propanona, y posterior copolimerización con fosgeno:
OH H + C = O
CH3
CH3
+ OH HH2O
OH C
CH3
CH3
OH
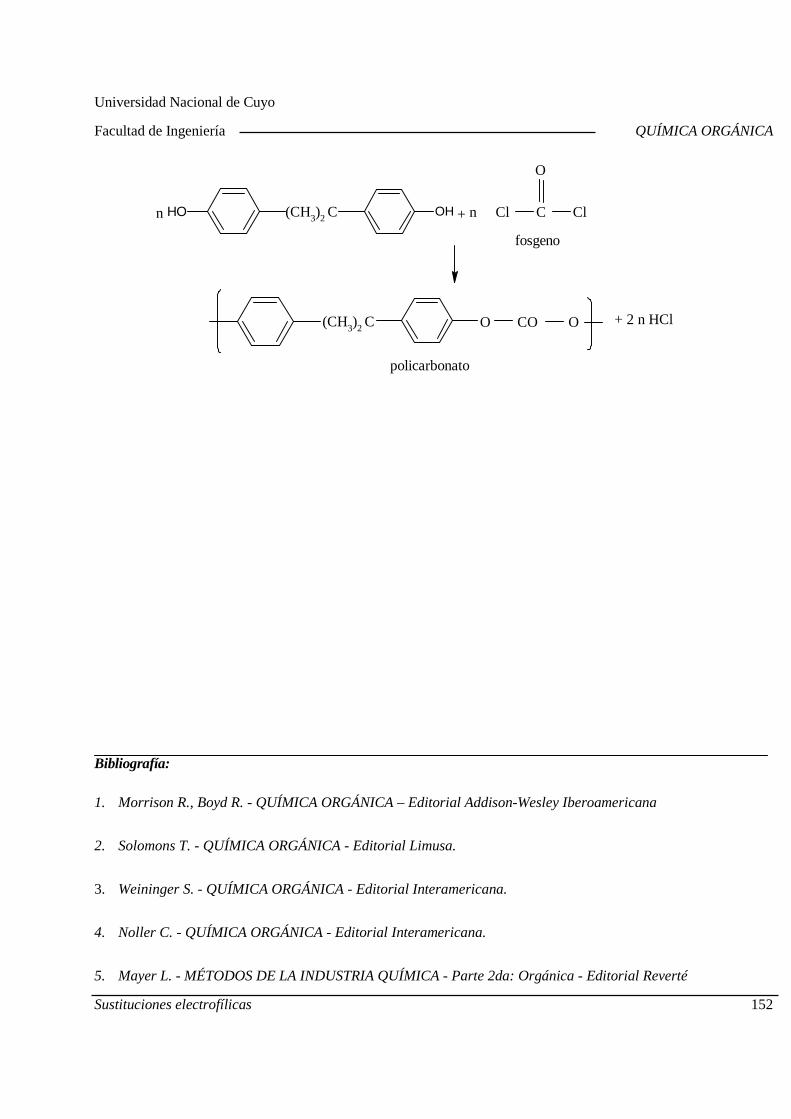
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones electrofílicas 152
n OH (CH3)2 C OH + n Cl C Cl
O
(CH3)2 C O CO O + 2 n HCl
fosgeno
policarbonato
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Weininger S. - QUÍMICA ORGÁNICA - Editorial Interamericana.
4. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.
5. Mayer L. - MÉTODOS DE LA INDUSTRIA QUÍMICA - Parte 2da: Orgánica - Editorial Reverté

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 153
La sustitución nucleofílica aromática ocurre cuando el sustrato es un derivado del benceno cuyo sustituyente es aceptor de electrones (ej.: halogenuros de arilo, nitroderivados). En este curso estudiaremos las sustituciones nucleofílicas en halogenuros de arilo.
Se ha comprobado que los halogenuros de arilo presentan muy baja reactividad frente a reactivos nucleofílicos tales como -OH, -OR, -NH2 y CN−. Sin embargo, la Sustitución Nucleofílica ocurre más fácilmente si el anillo aromático contiene además del halógeno, ciertos grupos que atraen electrones ubicados en posiciones orto o para con respecto al halógeno. Además, es conveniente utilizar reactivos nucleofílicos fuertes o temperaturas elevadas.
1. Reacción General:
Ar-
X + Z Ar-
Z + X
Como ejemplo de reacciones de sustitución aromática está la obtención del fenol a partir de clorobenceno:
Cl
NaOH 6-8%
350 ºC - 300atm
OH
clorobenceno fenol
Se observan efectos similares usando otros reactivos nucleofílicos y condiciones enérgicas de trabajo. Sin embargo, la reacción procede con facilidad si el anillo tiene grupos aceptores de electrones en posición orto o para.
La dificultad en el desarrollo de la reacción, comparándola con la Sustitución Nucleofílica Alifática, puede deberse a las siguientes causas:
a) Efecto mesómero en el haluro de arilo
b) Diferentes energías de enlace debido a las distintas hibridaciones del carbono
a) Efecto mesómero en el haluro de arilo
Si bien se ha comprobado que los halógenos unidos a un anillo aromático (ej.: clorobenceno) ejercen un efecto inductivo aceptor que supera al efecto mesómero dador, este último explicaría la dificultad de la ruptura heterolítica del enlace carbono-halógeno. Las formas contribuyentes siguientes muestran que dicho enlace tiene carácter doble.
-
Cl Cl+
Cl+
-
Cl+
Cl
-
Reacciones de Sustitución: Sustituciones Nucleofílicas Aromáticas
Sustituciones nucleofílicas en compuestos aromáticos. Cinética. Diagramas de energía. Efecto de los sustituyentes sobre la reactividad. Aplicaciones de las sustituciones nucleofílicas aromáticas.
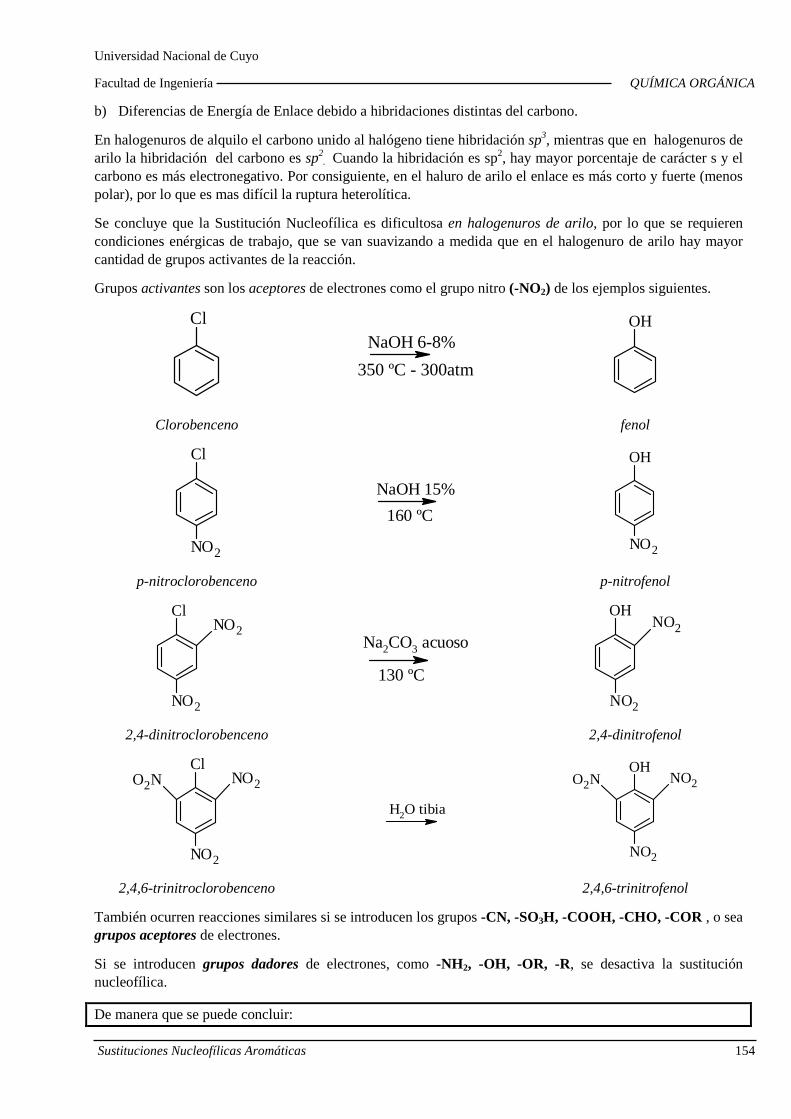
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 154
b) Diferencias de Energía de Enlace debido a hibridaciones distintas del carbono.
En halogenuros de alquilo el carbono unido al halógeno tiene hibridación sp3, mientras que en halogenuros de arilo la hibridación del carbono es sp2
. Cuando la hibridación es sp2, hay mayor porcentaje de carácter s y el
carbono es más electronegativo. Por consiguiente, en el haluro de arilo el enlace es más corto y fuerte (menos polar), por lo que es mas difícil la ruptura heterolítica.
Se concluye que la Sustitución Nucleofílica es dificultosa en halogenuros de arilo, por lo que se requieren condiciones enérgicas de trabajo, que se van suavizando a medida que en el halogenuro de arilo hay mayor cantidad de grupos activantes de la reacción.
Grupos activantes son los aceptores de electrones como el grupo nitro (-NO2) de los ejemplos siguientes.
Cl
NaOH 6-8%
350 ºC - 300atm
OH
Clorobenceno fenol
Cl
NO2
NaOH 15%
160 ºC
OH
NO2
p-nitroclorobenceno p-nitrofenol
Cl
NO2
NO2
Na2CO3 acuoso
130 ºC
OH
NO2
NO2
2,4-dinitroclorobenceno 2,4-dinitrofenol
Cl
NO2
NO2O2N
H2O tibia
OH
NO2
NO2O2N
2,4,6-trinitroclorobenceno 2,4,6-trinitrofenol
También ocurren reacciones similares si se introducen los grupos -CN, -SO3H, -COOH, -CHO, -COR , o sea grupos aceptores de electrones.
Si se introducen grupos dadores de electrones, como -NH2, -OH, -OR, -R, se desactiva la sustitución nucleofílica.
De manera que se puede concluir:
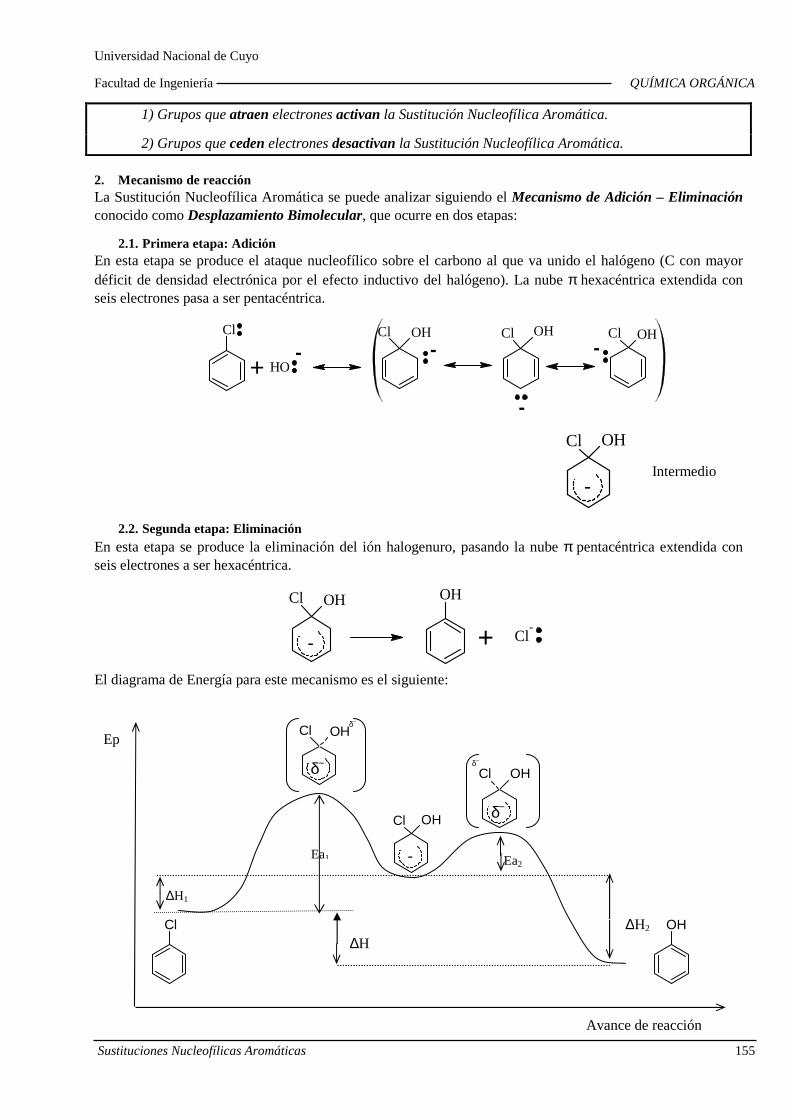
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 155
1) Grupos que atraen electrones activan la Sustitución Nucleofílica Aromática.
2) Grupos que ceden electrones desactivan la Sustitución Nucleofílica Aromática.
2. Mecanismo de reacción La Sustitución Nucleofílica Aromática se puede analizar siguiendo el Mecanismo de Adición – Eliminación conocido como Desplazamiento Bimolecular, que ocurre en dos etapas:
2.1. Primera etapa: Adición En esta etapa se produce el ataque nucleofílico sobre el carbono al que va unido el halógeno (C con mayor déficit de densidad electrónica por el efecto inductivo del halógeno). La nube π hexacéntrica extendida con seis electrones pasa a ser pentacéntrica.
Cl OHCl OHCl Cl OH
+ OH- -
-
-
Cl OH
-
Intermedio
2.2. Segunda etapa: Eliminación En esta etapa se produce la eliminación del ión halogenuro, pasando la nube π pentacéntrica extendida con seis electrones a ser hexacéntrica.
Cl OH
-
OH
+ Cl-
El diagrama de Energía para este mecanismo es el siguiente:
Ea1
∆H1
Ea2
∆H2
Avance de reacción
Ep
Cl OH
-
OHCl
δ−
δ−
Cl OH
δ−
δ−
OHCl∆H

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 156
3. Reactividad: Al estudiar la etapa determinante de la velocidad se observa que en el Estado de Transición hay una carga negativa en desarrollo, mientras que en el producto de la etapa (Carbanión) hay una carga negativa completa.
Los factores que estabilicen al carbanión, dispersando la carga negativa (-), deben estabilizar también al carbanión del Estado de Transición. Por lo tanto, se analiza el carbanión (intermedio) y se sacan conclusiones acerca del carbanión en formación (ET).
Suponiendo las estructuras:
Z Cl
(-)
Z Cl
(-)
G
Z Cl
(-)
G
I II III
En la estructura II, el grupo G aceptor de electrones dispersa la carga negativa (-) del carbanión. El mismo efecto ejerce G en el carbanión incipiente (ET) y lo estabiliza, con lo que disminuye la barrera de Energía de Activación y la reacción aumenta su velocidad.
En la estructura III, el grupo G dador de electrones, desestabiliza al intermedio. El mismo efecto ejerce G en el carbanión incipiente (ET), aumentando la barrera de Energía de Activación y por lo tanto diminuyendo la velocidad de la reacción
Los grupos sustituyentes se podrían reunir según cedan o atraigan electrones (por efecto inductivo y/o mesómero) y esto permitiría definir su influencia en las reacciones.
Grupos que atraen electrones
(SN: activantes)
(SE: desactivantes)
Por efecto inductivo
Por efecto mesómero
Grupos que ceden electrones
(SN: desactivantes)
(SE: activantes)
Por efecto inductivo
Por efecto mesómero
Nota: En el caso del grupo ácido sulfónico, que ejerce efectos inductivo y mesómero, consideraremos sólo el primero.
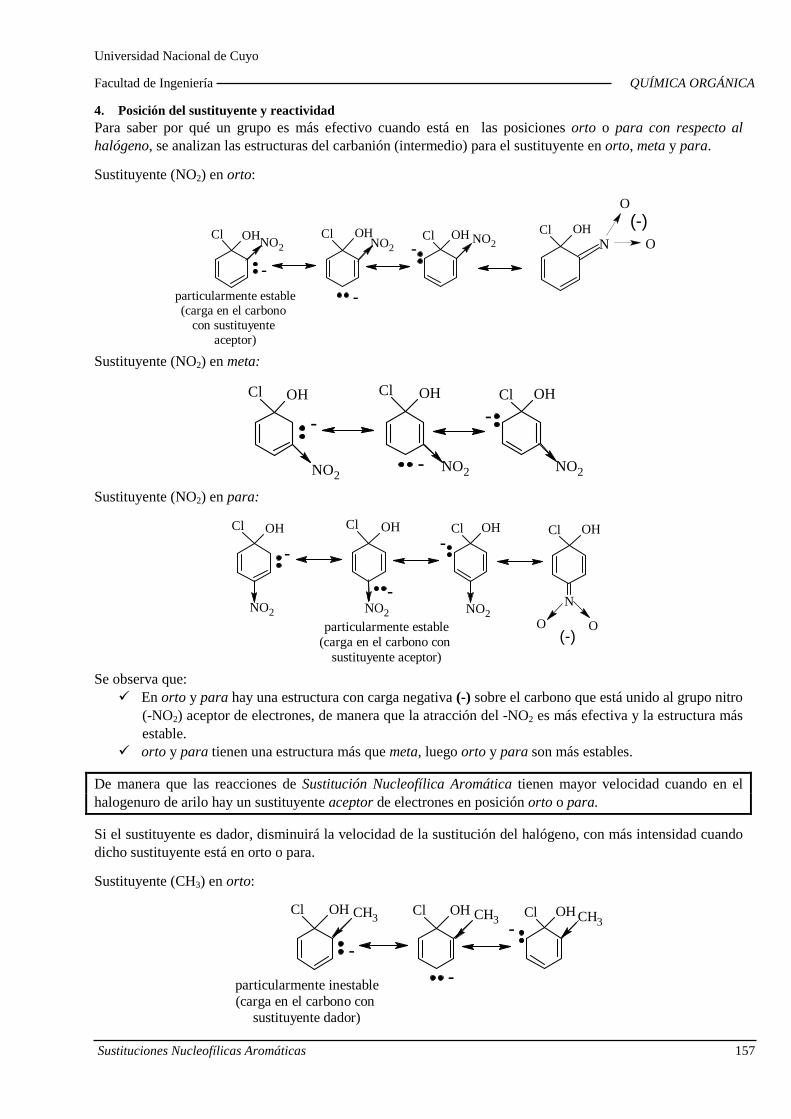
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 157
4. Posición del sustituyente y reactividad Para saber por qué un grupo es más efectivo cuando está en las posiciones orto o para con respecto al halógeno, se analizan las estructuras del carbanión (intermedio) para el sustituyente en orto, meta y para.
Sustituyente (NO2) en orto:
Sustituyente (NO2) en meta:
-
OHCl
NO2
OHCl
NO2
Cl OH
NO2
-
-
Sustituyente (NO2) en para:
-
OHCl
NO2
OHCl
NO2
Cl OH
NO2
-
-
OHCl
N
O-
Oparticularmente estable(carga en el carbono con
sustituyente aceptor)
(-)
Se observa que:
En orto y para hay una estructura con carga negativa (-) sobre el carbono que está unido al grupo nitro (-NO2) aceptor de electrones, de manera que la atracción del -NO2 es más efectiva y la estructura más estable.
orto y para tienen una estructura más que meta, luego orto y para son más estables.
De manera que las reacciones de Sustitución Nucleofílica Aromática tienen mayor velocidad cuando en el halogenuro de arilo hay un sustituyente aceptor de electrones en posición orto o para.
Si el sustituyente es dador, disminuirá la velocidad de la sustitución del halógeno, con más intensidad cuando dicho sustituyente está en orto o para.
Sustituyente (CH3) en orto:
-
OHCl CH3OHCl CH3
Cl OHCH3-
-particularmente inestable(carga en el carbono con
sustituyente dador)
-
OHClNO2
OHClNO2
Cl OH NO2-
-particularmente estable(carga en el carbono
con sustituyente aceptor)
Cl OHN
O(-)
O

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Sustituciones Nucleofílicas Aromáticas 158
Sustituyente (CH3) en meta:
-OHCl
CH3
OHCl
CH3
Cl OH
CH3
-
-
Sustituyente (CH3) en para:
-OHCl
CH3
OHCl
CH3
Cl OH
CH3
-
-
particularmente inestable(carga en el carbono con
sustituyente dador)
Se observa que en orto y para hay una estructura con carga negativa (-) sobre el carbono que está unido al grupo metilo dador de electrones, de manera que la estructura es menos estable.
Por lo tanto, las reacciones de Sustitución Nucleofílica Aromática tienen menor velocidad cuando en el halogenuro de arilo hay un sustituyente dador de electrones. El efecto es mayor cuando el sustituyente está en posición orto o para.
5. Aplicaciones de las sustituciones nucleofílicas aromáticas Las sustituciones nucleofílicas aromáticas en haluros de arilo constituyen una vía importante para la preparación de fenoles.
La mayoría de los fenoles se preparan industrialmente por los mismos métodos del laboratorio. Sin embargo, hay procesos especiales para obtener algunos de estos compuestos a escala comercial. Por la cantidad que se produce, el fenol ocupa uno de los lugares más altos en la lista de compuestos aromáticos sintéticos, ya que se utiliza en la manufactura de resinas. Del alquitrán de hulla se obtiene cierta cantidad de fenol y cresoles, pero gran parte es sintética, encontrándose entre los procesos empleados la preparación con clorobenceno e hidróxido de sodio, a 360ºC.
Entre los usos de los fenoles se destacan su aprovechamiento como reveladores fotográficos, la preparación de polímeros (resinas fenólicas) y las aplicaciones medicinales. Además, los fenoles son antioxidantes en alimentos y cosméticos, oxidándose ellos en lugar de la sustancia protegida.
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial. Addison- Wesley Iberoamericana.
2. SolomonsT. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Noller C - QUÍMICA ORGÁNICA - Editorial Interamericana.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones por radicales libres 159
Una reacción de adición es aquella en la cual dos moléculas se unen entre sí (se adicionan) para formar una mayor. Nada se pierde en este proceso, ya que todos los átomos de las moléculas originales están formando parte de la nueva molécula obtenida. Además, para que estas reacciones tengan lugar es necesario que al menos uno de los reactivos posea un doble o un triple enlace.
C C
H H
H H+ YZ C C
Y2+
Z
H
HH
H
Las reacciones de adición pueden ser homolíticas (por radicales libres) y heterolíticas (iónicas). Acá estudiaremos la adición por radicales libres en alquenos. En el caso de los alquinos el mecanismo es igual pero, en lugar de la adición de una molécula, se pueden adicionar dos. Veremos la hidrogenación de alquenos, como ejemplo de reacción de adición por radicales libres.
1. Hidrogenación de alquenos La reacción general es la siguiente:
C C
H H
H H+ H2 C C
H H
H
HH
HPt, Pd o Ni
Recordemos que los alquenos poseen un doble enlace que consiste en una unión σ fuerte y una π débil, por lo que es de esperar que los radicales libres puedan utilizar electrones de la nube π.
En el caso de los alquinos el mecanismo es igual pero, al poseer dos enlaces π se pueden adicionar dos moléculas de H2 en lugar de una.
La hidrogenación puede realizarse por dos métodos distintos (reacción heterogénea y reacción homogénea). En ambos casos, el catalizador provoca la adición de hidrógeno molecular al doble enlace.
1.1. Hidrogenación heterogénea (dos fases) Es el método clásico, todavía muy utilizado. El catalizador es un metal finamente dividido, por lo común platino, paladio o níquel.
Se agita una solución del alqueno bajo una ligera presión de hidrógeno en presencia de una pequeña cantidad del catalizador. La reacción es rápida y suave y, una vez completada, se separa mediante filtración el producto saturado del catalizador.
Reacciones de adición: Adiciones por radicales libres
Hidrogenación de alquenos. Calores de hidrogenación. Diagramas de energía. Aplicaciones de las adiciones por radicales libres.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones por radicales libres 160
La molécula de hidrógeno se escinde en la superficie del metal, formándose dos átomos
(radicales libres) muy reactivos. La nube π del doble enlace es así atacada fácilmente por éstos,
obteniéndose el alcano correspondiente.
1.2. Hidrogenación homogénea (una fase) Este método es mucho más moderno y ofrece una flexibilidad imposible de alcanzar con los catalizadores antiguos. Mediante modificaciones en los catalizadores, puede llevarse a cabo la hidrogenación con una gran selectividad.
Los catalizadores son complejos orgánicos de metales de transición, como rodio o iridio (ej.: catalizador de Wilkinson). Los mismos son solubles en disolventes orgánicos y la hidrogenación se efectúa así en una sola fase. El inconveniente del método es la dificultad de separación del catalizador y el producto una vez terminada la reacción. Sin embargo, se están realizando estudios para resolver esta dificultad: el catalizador va incorporado químicamente a un polímero sólido insoluble, lo que permite una filtración fácil al final de la reacción. De esta manera, la hidrogenación homogénea se convierte en heterogénea, pero el modo de acción parece permanecer igual.
2. Calor de hidrogenación La hidrogenación es exotérmica, porque los dos enlaces σ (C-H) que se forman liberan más energía que la que se absorbe al romperse los enlaces σ (H-H) y π. La cantidad de calor desprendida al hidrogenar un mol de un compuesto se llama calor de hidrogenación; es simplemente el ∆H de la reacción.
El Calor de Hidrogenación de casi todo alqueno se aproxima bastante a un valor de 30 kcal por cada doble enlace del compuesto, lo que puede observarse en la tabla siguiente:
Alqueno Calor de hidrogenación, kcal/mol eteno 32,8
propeno 30,1 metilpropeno 28,4
1-buteno 30,3 1-penteno 30,1
3-metil-1-buteno 30,3 cis-2-buteno 28,6
trans-2-buteno 27,6 cis-2-penteno 28,6
trans-2-penteno 27,6 La hidrogenación procede a una velocidad muy baja en ausencia de un catalizador, aún a temperaturas elevadas, a pesar de ser una reacción exotérmica, por lo que el proceso no catalizado debe tener una energía de activación muy alta. La función del catalizador es reducir la energía de activación (Ea), de modo que la

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones por radicales libres 161
reacción pueda proceder rápidamente a temperatura ambiente. Por supuesto, el catalizador no afecta al cambio neto de energía del proceso total, ya que sólo rebaja la barrera energética entre los reactivos y el estado de transición.
Un catalizador rebaja la Ea, permitiendo que la reacción proceda de un modo distinto, es decir, por medio de un mecanismo diferente. En ese caso, los reactivos se adsorben en la enorme superficie del metal sólido dividido finamente, o se unen temporalmente a un ión metálico soluble. En estas condiciones, la reacción es muy distinta a la que tendría lugar en otro caso. Se cree, por ejemplo, que la superficie catalítica rompe el enlace π del alqueno antes de la reacción con el hidrógeno. En la hidrogenación homogénea, el complejo del ión metálico rompe el enlace hidrógeno-hidrógeno y transfiere estos átomos, de uno en uno, al doble enlace.
3. Calor de hidrogenación y estabilidad de los alquenos A menudo, los calores de hidrogenación pueden brindarnos información valiosa acerca de las estabilidades relativas de compuestos no saturados.
En los 2-butenos isómeros, por ejemplo, el cis tiene un calor de hidrogenación de 28.6 kcal, y el trans, de 27.6 kcal. Ambas reacciones consumen un mol de hidrógeno y dan el mismo producto, n-butano. Por lo tanto, si el isómero trans libera 1 kcal menos de energía que el cis, sólo puede significar que contiene 1 kcal menos de energía. En otras palabras, el isómero trans es más estable que el cis en 1 kcal.
De los alquenos simples disustituidos, el isómero trans generalmente es el más estable; los dos sustituyentes mayores están más separados que en el isómero cis, por lo que hay menos impedimento y menor tensión de Van der Waals.
Sin catalizador
Con catalizador
C = C + H2
CC
Ep
Avance de la reacción
n-
trans-2-buteno cis-2-buteno
∆H = 28,6 kcal/mol
∆H = 27,6 kcal/mol
Ep
Avance de la reacción

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones por radicales libres 162
Los calores de hidrogenación indican que la estabilidad de un alqueno también depende de la ubicación del doble enlace. Son característicos los ejemplos siguientes:
CH3 CH2CH CH2 CH3 CH CHCH3
CH2CH2CH CH2CH3 CH2CH CHCH3CH3
CH3 CH CH CH2
CH3
CH2 C
CH3
CH2 CH3 CH3 C CH CH3
CH3
Cada grupo de alquenos isómeros da el mismo alcano. En consecuencia, las diferencias en calores de hidrogenación deben proceder de diferencias en estabilidades. En cada caso, se observa que cuanto mayor es el número de grupos alquilo unidos a los carbonos del doble enlace, menor es el calor de hidrogenación y más estable es el alqueno.
R2C=CR2 > R2C=CHR > R2C=CH2, RCH=CHR > RCH=CH2 > H2C=CH2
4. Aplicaciones de las adiciones por radicales libres La hidrogenación es un modo útil de preparar alcanos. Pero no se limita a la síntesis de alcanos, sino que es un método general para convertir un doble enlace carbono-carbono en uno simple, prácticamente en todo tipo de compuestos.
Empleando el mismo equipo, igual catalizador y condiciones muy parecidas, podemos convertir un alqueno en un alcano, un alcohol no saturado en uno saturado, un éster no saturado en uno saturado, etc.
Modificando el catalizador y las condiciones, podemos hidrogenar selectivamente un enlace múltiple sin tocar en la misma molécula otro enlace. Por ejemplo: un doble enlace carbono-carbono, pero no uno carbono-oxígeno; uno triple, pero no uno doble; incluso un doble enlace carbono-carbono, pero no otro. Como veremos, podemos convertir incluso un compuesto no saturado ópticamente inactivo en un producto activo.
Una de las aplicaciones mas importantes de las adiciones por radicales libres es la polimerización por radicales libres, que veremos mas adelante.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-WesleyIberoamericana
2. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 163
Entre las reacciones más importantes del doble y triple enlace carbono-carbono se encuentran las reacciones de adición electrofílica.
Por ejemplo, los ácidos fuertes de Brönsted, tales como HCl, HBr, HI y H2SO4, se adicionan rápidamente al doble enlace carbono-carbono de los alquenos, formándose nuevos enlaces covalentes con el H y las bases conjugadas de los ácidos. Por ejemplo, si el eteno reacciona con ácido sulfúrico, se forma sulfato ácido de etilo:
CH2 CH2 + CH2 CH2
H OSO3H
H-O-S-O3H
En general, estas reacciones son exotérmicas ya que el enlace π es relativamente débil (~63 kcal/mol) en relación a los enlaces σ formados entre los átomos o grupos de átomos de los reactivos atacantes y los carbonos del alqueno o alquino. Como ejemplo analicemos la siguiente reacción:
C C
F
F
F
F
+ H Br C CH
H
H
H
H
Br
Enlaces rotos: Enlaces formados:
π C=C
C-H
Br-H
Br-C
Total 150,5 kcal/mol Total 167 kcal/mol
1. Mecanismo de reacción Los reactivos analizados hasta ahora (HBr, H2SO4) han sido ácidos fuertes de Brönsted, por lo tanto como primer paso parece sensato determinar la base con la cual el ácido puede reaccionar. Ya que estos ácidos no reaccionan con los alcanos, podemos deducir que son los electrones π quienes dan el carácter básico al compuesto con doble enlace.
Reacciones de Adición: Adiciones Electrofílicas
Adiciones electrofílicas en alquenos y alquinos. Mecanismos de reacción. Aplicaciones de las adiciones electrofílicas.
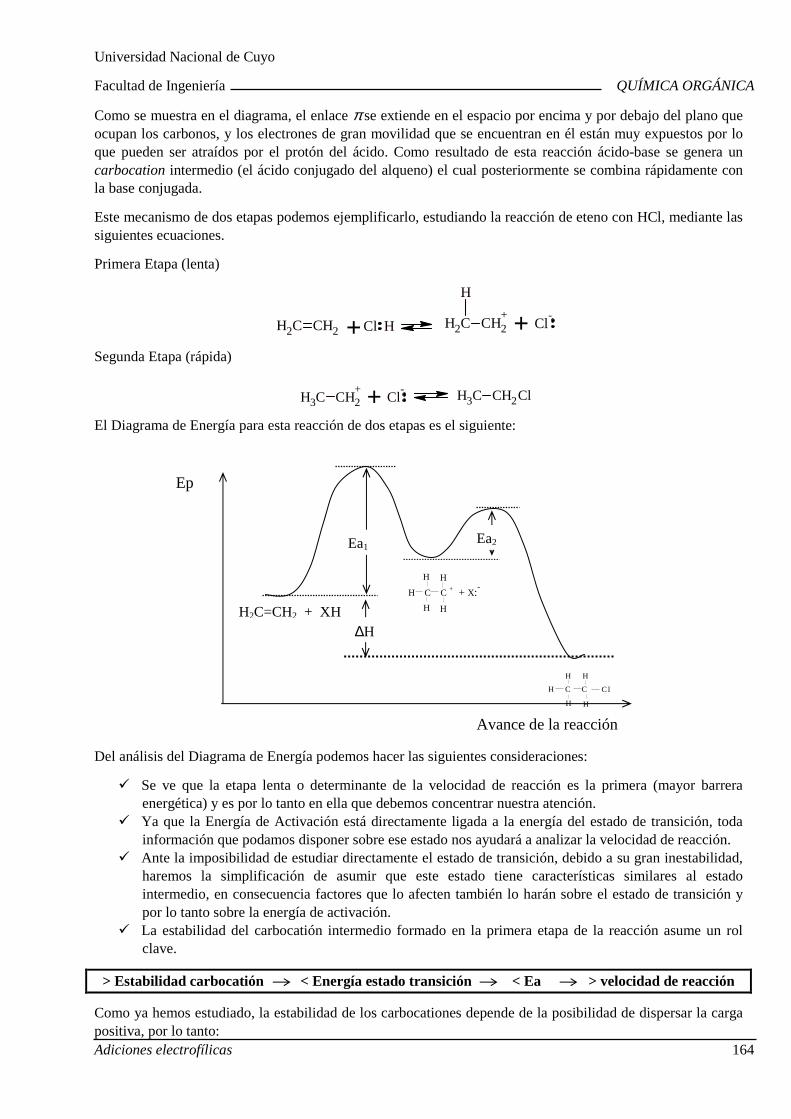
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 164
Como se muestra en el diagrama, el enlace π se extiende en el espacio por encima y por debajo del plano que ocupan los carbonos, y los electrones de gran movilidad que se encuentran en él están muy expuestos por lo que pueden ser atraídos por el protón del ácido. Como resultado de esta reacción ácido-base se genera un carbocation intermedio (el ácido conjugado del alqueno) el cual posteriormente se combina rápidamente con la base conjugada.
Este mecanismo de dos etapas podemos ejemplificarlo, estudiando la reacción de eteno con HCl, mediante las siguientes ecuaciones.
Primera Etapa (lenta)
CH2 CH2 +Cl H CH2 CH2+
H
Cl-+
Segunda Etapa (rápida)
CH3 CH2+
Cl-+ CH3 CH2Cl
El Diagrama de Energía para esta reacción de dos etapas es el siguiente:
Del análisis del Diagrama de Energía podemos hacer las siguientes consideraciones:
Se ve que la etapa lenta o determinante de la velocidad de reacción es la primera (mayor barrera energética) y es por lo tanto en ella que debemos concentrar nuestra atención.
Ya que la Energía de Activación está directamente ligada a la energía del estado de transición, toda información que podamos disponer sobre ese estado nos ayudará a analizar la velocidad de reacción.
Ante la imposibilidad de estudiar directamente el estado de transición, debido a su gran inestabilidad, haremos la simplificación de asumir que este estado tiene características similares al estado intermedio, en consecuencia factores que lo afecten también lo harán sobre el estado de transición y por lo tanto sobre la energía de activación.
La estabilidad del carbocatión intermedio formado en la primera etapa de la reacción asume un rol clave.
> Estabilidad carbocatión < Energía estado transición < Ea > velocidad de reacción
Como ya hemos estudiado, la estabilidad de los carbocationes depende de la posibilidad de dispersar la carga positiva, por lo tanto:
H C C
H
H
H
H
C l
H2C=CH2 + XH
H C C + + X:-
H
H
H
H
∆H
Ea2 Ea1
Ep
Avance de la reacción

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 165
CH3(+) < CH3CH2
(+) < (CH3)2CH(+) ≅ CH2=CH-CH2(+) < C6H5CH2
(+) ≅ (CH3)3C(+)
Estabilidad Carbocationes
A efectos de ejemplificar lo dicho, analicemos la adición de HCl a propeno.
C CCH3
H H
H+ H Cl
C C+
H
H
H
CH3
H+ Cl
-C C
H
H
CH3
H
HCl
C+
CH H
CH3 H
H Cl-+ C C
Cl
CH3
HH
HH
Intermedio más estable Producto principal
El carbocatión secundario más estable se forma preferencialmente y luego la base conjugada del ácido (-:Cl) se une rápidamente al carbonio intermedio para formar el producto final, en este caso el 2-cloropropano.
Se puede formar algo de 1-cloropropano, pero a menor velocidad debido a la menor estabilidad del ión carbonio intermedio (que requiere mayor Ea).
El siguiente diagrama de energía resume lo planteado:
1.1. Transposiciones
La formación de carbocationes es muchas veces acompañada por transposiciones. Este reordenamiento estructural, que tiene lugar por desplazamiento de grupos metiluro o hidruro adyacentes al carbono con carga positiva, ocurre cuando el carbocatión transpuesto es más estable que el inicial.
Por ejemplo la adición de HCl a 3,3-dimetil-1-buteno da como producto principal 2-cloro-2,3-dimetilbutano en lugar del esperado 3-cloro-2,2 dimetil butano.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 166
CH3 C CH CH2
CH3
CH3H
+
CH3 C CH+
CH3
CH3
CH3
CH3 C+
CH CH3
CH3
CH3
Cl-
Cl-
CH3 C CH CH3
CH3
CH3 Cl
CH3 C CH CH3
CH3
CH3Cl
Transposición
3- cloro- 2,2 - dimetilbutano 2 – cloro – 2,3 - dimetilbutano
1.2. Regioselectividad y Regla de Markovnicov Sólo se obtiene un producto en las reacciones de adición cuando los alquenos son simétricos (eteno, ciclohexeno, etc.). Sin embargo, si el doble enlace no une carbonos estructuralmente equivalentes, como en el propeno, 1-buteno, 2-metil-2-buteno y 1-metil-ciclohexeno, el reactivo electrofílico se puede adicionar en dos formas diferentes:
(CH3)2C = CH CH3 + HCl
(CH3)2CH CHCl CH3 y (CH3)2CCl CH2 CH3
2-metil-2-buteno 3-metil-2-clorobutano 2-metil-2-cloro-butano
Si bien ambos productos son posibles, uno de los isómeros se obtiene preferencialmente. En este caso, el 2-metil-2-clorobutano es prácticamente el producto principal.
En forma similar el 1-buteno reacciona con HBr dando como producto principal 2-bromobutano.
CH3 CH2 CH CH2+ BrH
CH3 CH2 CHBr C H3 y CH3 CH2 CH2 CH2 Br
2-bromobutano
Producto principal 1-bromobutano
Cuando una reacción genera un isómero estructural en mayor proporción que otro, se dice que es regioselectiva. Este comportamiento regioselectivo fue advertido por el químico ruso Vladimir Markovnicov, quien después de estudiar numerosas reacciones de adición como las precedentes, advirtió una tendencia a obtener productos de adición favorecidos por una determinada estructura. En virtud de esto enunció una regla empírica, conocida actualmente como Regla de Markovnicov:
“Cuando un ácido de Brönsted, XH, se adiciona a un doble enlace asimétrico, el hidrogeno del ácido se une al carbono del doble enlace que mas hidrógenos tiene unidos a él”
La regla de Markovnicov es una guía útil para el alumno, ya que permite determinar rápidamente el producto principal de una adición electrofílica. Pero, no es la razón por la cual las reacciones siguen ese camino. Se requieren muchos más fundamentos para poder explicar lo que está sucediendo, como es el análisis de los iones carbonio intermedios realizado anteriormente.
Otra evaluación más simple que se suele llevar a cabo es la del estado base (alqueno), que analiza el efecto inductivo ejercido por cada uno de los grupos alquilo sobre los carbonos del doble enlace. Esto permite inferir hacia cual carbono se desplazarán los electrones π para permitir el enlace con el electrófilo en la primera etapa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 167
C C
H
CH3
CH3
CH3 Es más probable que los electrones π se desplacen al carbono con menor grupos alquilo (con menor densidad electrónica).
2. Adiciones electrofílicas más comunes
2.1. Adición de ácidos fuertes de Brönsted Como se planteó anteriormente, los ácidos fuertes de Brönsted, tales como HCl, HBr, HI y H2SO4, se adicionan rápidamente al doble enlace. Por ejemplo, si el ciclohexeno reacciona con dichos ácidos, deberían esperarse los productos siguientes:
2.2. Adición de agua
Los ácidos débiles como el agua normalmente no se adicionan a los alquenos. Sin embargo, una pequeña cantidad de ácido fuerte (ej.: SO4H2) sirve para catalizar la adición de agua, obteniéndose de esta manera alcoholes a partir de alquenos.
Si ácido sulfúrico se disuelve en agua, se ioniza completamente de la siguiente forma:
H2SO4 + OH2 OH3++HSO4
-
El ion hidronio es un ácido fuerte y es el que reacciona con el alqueno:
CH2 CH2 OH3++
OH2
CH2 CH2+
HOH H
OH+
H
CH2 CH2
H
H+
OH
CH2 CH2
H
Por lo tanto, si se quiere adicionar un ácido fuerte a un alqueno (ej.: H2SO4, HCl, HBr, etc.), no debe usarse diluido en agua o alcohol; ya que estos ácidos fuertes se ionizarán en el solvente dando como producto final un alcohol. Usando el ácido concentrado o solventes como, hexano, benceno, etc., se evita la competencia del solvente en la adición y es el ácido el que se adiciona.
2.3. Adición de halógenos A diferencia de los alcanos, los alquenos reaccionan rápidamente a temperatura ambiente y en ausencia de luz con bromo líquido puro o con una solución de bromo en algún solvente orgánico como el tetracloruro de carbono. El doble enlace se rompe y los átomos de bromo se adicionan a cada uno de los carbonos. El color rojo amarronado característico del bromo se pierde, obteniéndose un líquido incoloro. Por ello, esta es una reacción útil para distinguir alcanos de alquenos. Los productos de estas reacciones son dihaluros vecinales.
R2C CR2 + X2R2C CR2
X X

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 168
Los otros halógenos, excepto el flúor, se comportan en forma similar. (El flúor reacciona con todos los hidrocarburos en forma explosiva dando carbón y fluoruro de hidrógeno)
El mecanismo de reacción podemos interpretarlo de la siguiente manera:
La molécula de bromo, que no es polar, es muy polarizable. Al acercarse a los electrones π del alqueno, se repelen los electrones del enlace bromo-bromo hacia el extremo más alejado. Esto origina un dipolo inducido en la molécula de bromo.
Dipolo inducido significa que ha sido creado por alguna influencia externa (en este caso la aproximación del enlace π) y no que exista realmente.
Los electrones π se desplazan hacia el átomo de bromo ligeramente positivo, mientras que los electrones del enlace Br-Br se desplazan hacia el otro átomo de bromo, hasta producir la rotura heterolítica del enlace, generando el ion bromuro.
CH3 CH CH2
Br
Br
δ+
δ−
CH3 CH+
CH2
Br
Br-
Nótese la forma en que los electrones π se han desplazado. El primer átomo de bromo se ha unido al carbono de la derecha y de esta forma se ha formado un carbocatión secundario. Este es más estable que el primario que se hubiera obtenido si los electrones π se hubieran desplazado en sentido opuesto.
En la segunda etapa, el ion bromuro es fuertemente atraído por el carbocatión:
CH3 CH+
CH2
Br
Br-
CH3 CH CH2
BrBr
Una versión, probablemente más precisa de este mecanismo, es la siguiente:
Al igual que lo visto anteriormente, los electrones π se desplazan hacia el extremo positivo del dipolo inducido Br-Br. Pero ahora el átomo de bromo se une a ambos átomos de carbono, formándose un ión bromonio (la carga positiva se encuentra sobre el bromo).
CH3 CH CH2
Br
Br
δ+
δ−
CH3 CH CH2
Br+
Br-

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones electrofílicas 169
El ion bromonio es luego atacado, por atrás por el ion bromuro.
CH3 CH CH2
Br+
Br-
CH3 CH CH2
Br
Br
2.4. Formación de halohidrinas
En el caso del ácido hipobromoso o hipocloroso (XOH), que son ácidos débiles de Brönsted, no reaccionan como dadores de protones. Ya que el oxígeno es más electronegativo que el cloro o el bromo, el electrófilo
será el catión del halógeno (X+). La especie nucleofílica que se une al carbocatión intermedio es por lo tanto
el ion hidróxido. El producto es denominado halohidrina.
CH3 CH CH2+ Cl OH CH3 CH CH2
ClOH 1-cloro-2-propanol (halohidrina)
3. Aplicaciones de las adiciones electrofílicas Las adiciones electrofílicas revisten gran importancia en la preparación de sustancias orgánicas. A continuación se dan algunos ejemplos:
La adición de ácido sulfúrico a alquenos de cadena larga da sulfatos ácidos de alquilo, que en forma de sales sódicas son usados como detergentes aniónicos: R O SO3 :
- Na +
La adición de ácido clorhídrico a alquenos es un método muy usado en la preparación de haluros de alquilo, compuestos de gran importancia en síntesis orgánica.
La adición de agua a alquenos se usa como método de preparación de alcoholes. Las aplicaciones más importantes de estas reacciones corresponden a la fabricación de polímeros. El cloruro de vinilo, usado en la preparación de PVC, se obtiene por adición de ácido clorhídrico al
acetileno.
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial Addison- Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones nucleofílicas 170
Los aldehídos y cetonas sufren con facilidad reacciones de adición nucleofílica al doble enlace carbono-oxígeno del grupo carbonilo, debido a las características estructurales del mismo.
1. Estructura del grupo carbonilo El carbono del carbonilo tiene hibridación sp2, por lo que los tres átomos unidos a él son coplanares, con ángulos de enlace de aproximadamente 120º. Un modelo de los enlaces σ y π, como el indicado en el siguiente gráfico, muestra una buena descripción de la distribución electrónica en el grupo funcional
Puesto que el oxígeno es más electronegativo que el carbono, es de esperar una mayor densidad electrónica π en el extremo oxigenado del grupo carbonilo y una carga parcial positiva bastante pronunciada sobre el carbono. Esto lo confirma la existencia de un gran dipolo de enlace.
C O
F
Fδ+ δ−
Resumiendo, esta distribución de carga, que justifica la existencia de reacciones de adición nucleofílica, se puede explicar considerando la presencia de dos efectos:
La gran capacidad del oxígeno, por su electronegatividad, para adquirir una carga negativa. La existencia de una forma contribuyente iónica en el híbrido de resonancia.
C O
F
F-+
C O
2. Mecanismo de adición nucleofílica De acuerdo al análisis hecho al grupo carbonilo podemos decir:
La distribución coplanar de los grupos alrededor del carbono del carbonilo implica que este carbono es accesible tanto por arriba como por abajo.
La carga positiva del carbono del carbonilo lo hace fácilmente atacable por nucleófilos. La carga negativa del oxígeno del carbonilo implica que las reacciones de adición nucleofílica son
susceptibles a los catalizadores ácidos.
Por lo que podemos interpretar que las reacciones de adición nucleofílica siguen los siguientes mecanismos:
Reacciones de adición: Adiciones nucleofílicas
Adiciones nucleofílicas en aldehídos y cetonas. Cinética. Reactividades de aldehídos y cetonas. Importancia de las adiciones nucleofílicas. Autopolimerización del formaldehído.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones nucleofílicas 171
a) En una primera etapa, el nucleófilo (N:) utiliza su par de electrones para unirse al carbono del carbonilo, simultáneamente el par de electrones π del doble enlace carbono oxígeno, se desplaza hacia el oxígeno del carbonilo, cambiando el estado de hibridación del carbono de sp2 a sp3. Como ya dijimos, aquí juega un papel importante la capacidad del oxígeno de acomodar un par de electrones.
En una segunda etapa el oxígeno, ahora mucho más básico, se une a un electrófilo, que es generalmente un protón.
C O
R'
R
δ+ δ−
H+
CN O
R
R´
+-
CN OH
R
R´
Trigonal coplanar Tetraédrica Tetraédrica
b) Un segundo mecanismo se produce cuando la adición nucleofílica es catalizada por ácidos.
En una primera etapa el ácido ataca a los electrones sin compartir del oxígeno del carbonilo. La carga positiva del ión oxonio aumenta aún más la deficiencia de electrones alrededor del átomo de carbono, como se evidencia en la segunda forma contribuyente del híbrido de resonancia representado.
C O
R
R´
+ H+C O
R
R´
+H C O H
R
R´
+
Posteriormente se produce el rápido ataque del nucleófilo.
+ N - O HCN
R
R´
C
R
R´
+O H
Generalmente las adiciones en los compuestos carbonilos son catalizadas por ácidos para facilitar la ruptura heterolítica del enlace π y, en consecuencia, aumentar la velocidad de adición del nucleófilo.
2.1. Nucleófilos Los nucleófilos atacantes pueden ser iónicos o neutros. Los neutros poseen generalmente un protón que puede ser eliminado en la segunda etapa de la reacción.
Nucleófilos iónicos:
HO:- (oxhidrilo) H:- (hidruro) R3C:- (carbanión) RO:- (alcóxido) N≡C:- (cianuro)
Nucleófilos neutros:
H-O-H (agua) R-O-H (alcohol) H3N: (amoníaco) R-NH2 (amina)

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones nucleofílicas 172
2.2. Reversibilidad de las adiciones nucleofílicas Los dobles enlaces del carbonilo son unas 30 kcal/mol más fuertes que los dobles enlaces carbono-carbono, posiblemente debido a la mayor estabilidad por resonancia que involucra un contribuyente iónico
C O
F
F-+
C O
Esta gran energía de enlace es aún más sorprendente si se la compara con la energía correspondiente al enlace sencillo. La energía de enlace del doble enlace carbonilo en aldehídos y cetonas es de 176 a 179 kcal/mol, más del doble de la del enlace simple carbono-oxígeno (aprox. 86 kcal/mol).
Las consecuencias de estas relaciones de energía son importantes: debido a su gran dipolo, el doble enlace carbonilo adiciona rápidamente reactivos polares tales como el agua. Sin embargo, la reacción inversa de eliminación es también muy rápida, y en muchos casos la constante de equilibrio favorece al aldehído y a la cetona sobre su forma hidratada; es decir, el doble enlace carbonilo fuerte hace al aldehído o cetona el reaccionante favorecido termodinámicamente
C O
R'
R
+ OH Hrápido
muy rápido
C
R
R'
OH
OH
2.3. Reactividad relativa de aldehídos y cetonas Los aldehídos en general son más reactivos que las cetonas, por dos razones:
2.3.1. Efectos estéricos Con un hidrógeno unido al carbonilo en el aldehído, la aproximación del nucleófilo está menos restringida estéricamente. El complejo activado del aldehído se forma más rápidamente.
CN O
R
R´
δ−δ−
(R y R’ radicales alquilo y/o H)
2.3.2. Efectos electrónicos Los aldehídos están más polarizados que las cetonas. Para poder explicar ésto podemos analizar los estados bases o los complejos activados:
a) Si se consideran los estados bases, podemos ver que el menor efecto inductivo en los aldehídos implica mayor densidad positiva sobre el carbono y por lo tanto mayor reactividad
C
O
HRI
C
O
R'R
I I
Aldehido
Mayor δ+ en C Más reactivo
Cetona Menor δ+ en C Menos reactivo

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones nucleofílicas 173
Por otro lado, los aldehídos aromáticos son menos reactivos que los alifáticos, e incluso, que las cetonas. Esto se debe al efecto mesómero, por el cual la carga positiva se dispersa en el anillo:
C = O
H
M
+
C O :-
H
C O :-
H
C O :-
H
+
+
C = O
H
b) Si se consideran los complejos activados, en los aldehídos el efecto inductivo es menor, por lo tanto la
carga negativa sobre el oxígeno es menor y el complejo es más estable.
3. Adiciones nucleofílicas comunes
3.1. Adición de Agua Los aldehídos y cetonas reaccionan con el agua para dar 1,1 dioles.1
C
H
O
H
C
H H
OH
OH
OH H
Hidrato de formaldehído(1,1-metanodiol)
C
CH3
O
CH3
C
CH3 CH3
OH
OH
Hidrato de acetona(2,2- propanodiol)
OH2
Estas reacciones son muy lentas si la solución es neutra, pero se acelera notablemente si pequeñas cantidades de ácido o base es agregada. A continuación veremos uno de los mecanismos (adición de agua catalizada por ácido), con el objeto de comprender las mayores velocidades por acción de los catalizadores:
C
O
CH3 H
H++
CH3
CH
O H+
+ C
O HOH2
CH3 H
C
CH3HOH
OH2+
H+
+
+
C
CH3HOH
OH
Aldehído Aldehído protonado Hidrato de
acetaldehído En general los productos de las reacciones de hidratación no son aislables, ya que el equilibrio se encuentra fuertemente desplazado en dirección del compuesto carbonílico (observar que son dos grupos oxhidrilo, con alta densidad electrónica, unidos a un mismo carbono). No obstante, cuando el grupo carbonilo está unido a grupos aceptores fuertes (ej.: -CCl3), el carbonilo se hace menos estable (posee mayor δ+), y la forma hidratada puede predominar en el equilibrio.
C
CCl 3 H
O
OH H
C
CCl 3 H
OH
OH Cloral Hidrato de cloral
1 Significa que los dos grupos oxhidrilo están unidos al mismo carbono, que no siempres es C1

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Adiciones nucleofílicas 174
3.2. Adición de alcoholes Los alcoholes son nucleófilos debido a los pares libres de electrones en el átomo de oxígeno; pero son nucleófilos relativamente débiles y requieren de una catálisis ácida para reaccionar.
C
O
R H
H++
RC
H
O H+
+ C
O H
R H
R'OH
+HOR´
HC
OHR H +
C
RHOH
OR'
Aldehído Aldehído protonado Hemiacetal
Los hemiacetales pueden reaccionar posteriormente con un exceso de alcohol para formar acetales. Esta reacción es equivalente a la sustitución del grupo oxhidrilo del hemiacetal por el alcohóxido del alcohol. Es una reacción SN1 y el mecanismo es el siguiente:
H+ R'OH
C
RH OH
OR'
C
RH OH2+
OR'
OH2C
+
RH
OR'
H+ C
RH
OR'
OR'
+
++ +
OR'C
OR´
HR
H
+
Hemiacetal Acetal
Todos los pasos en esta reacción son reversibles, y es posible controlar el sentido de la misma añadiendo un exceso de uno de los reaccionantes o removiendo uno de los productos a medida que se va formando. Así, para que una cetona (o aldehído) se pueda transformar en un cetal (o acetal) es necesaria la remoción del agua formada durante la reacción. La mayoría de los hemiacetales de cadena abierta no son lo suficientemente estables como para ser aislados. Pero aldehídos que tienen grupos hidroxilos y cuatro a cinco átomos de carbono aparte del grupo carbonilo, pueden formar hemiacetales cíclicos estables por adición nucleofílica intramolecular, lo que veremos detalladamente al estudiar hidratos de carbono.
4. Importancia de las adiciones nucleofílicas La formación de hemiacetales y acetales explica las propiedades de productos naturales como los carbohidratos, que existen generalmente en las formas hemiacetálicas o acetálicas. El formaldehido, posee un grupo carbonilo altamente polarizado sin impedimento estérico, lo que lo hace muy reactivo. Si este compuesto se almacena sin diluir, se autopolimeriza (autoadición) dando productos muy resistentes, que son usados en cojinetes, partes para bombas, etc.:
CH2 O CH2F O Fnn
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA - Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones por Radicales Libres 175
Reacciones de Eliminación: Eliminaciones por Radicales Libres
Craqueo térmico de alcanos. Cinética. Diagramas de energía. Importancia industrial.
Los métodos más utilizados para la síntesis de alquenos o para introducir enlaces dobles o triples carbono – carbono en todo tipo de moléculas se basan en las reacciones de eliminación de átomos o grupos atómicos de carbonos adyacentes unidos por simple o doble enlace. Con menos frecuencia intervienen centros no adyacentes, formándose anillos.
Las reacciones de eliminación pueden ocurrir por radicales libres o por iones. Como ejemplo de las primeras, estudiaremos la pirólisis de alcanos (cracking térmico). Es importante aclarar que en las refinerías de petróleo también se llevan a cabo el hidrocraking y el cracking catalítico, con hidrógeno y catalizadores, respectivamente.
1. Craking térmico de alcanos A temperatura suficientemente elevada en ausencia de oxígeno, empieza en los alcanos la ruptura homolítica de los enlaces, seguida de la recombinación de los radicales libres para dar alcanos de menor tamaño y alquenos.
Este proceso se conoce como cracking térmico o pirólisis y se emplea para fabricar combustibles a partir de las fracciones del petróleo más pesadas. También el proceso es útil para la preparación de alquenos que son importantes desde el punto de vista industrial.
A pesar de la gran importancia de este método en la industria del petróleo, tiene escasa utilidad en la escala del laboratorio ya que los alquenos sencillos que se pueden preparar son productos que se consiguen en el mercado a bajo precio. Otra razón es que en los procesos de craqueo se obtienen mezclas complejas de las que tiene que aislarse el producto deseado, casi siempre con rendimiento bajo.
Ejemplo:
CH3 CH2 CH3
cracking
CH2 CH2 CH4 CH2 CH CH3 H2+ + +
Las reacciones de pirólisis se explican por mecanismo de radicales libres:
Los radicales libres se forman por ruptura de enlace carbono-carbono y/o carbono- hidrógeno. Los radicales mayores que el etilo se descomponen en una o más moléculas de alqueno y un radical
libre metilo o etilo. Cada radical libre (o cada átomo de hidrógeno) formado comienza una reacción en cadena provocando
la eliminación de un átomo de hidrógeno perteneciente a una molécula de un alcano, formando de este modo nuevos radicales libres.
La facilidad de eliminación de un hidrógeno de un alcano por un radical libre aumenta en el siguiente orden:
primario < secundario < terciario (ver Sustituciones por Radicales Libres)
Las reacciones en cadena terminan mediante la combinación de radicales libres o átomos. La naturaleza y las cantidades relativas de los productos están determinadas principalmente por los
radicales libres formados en las reacciones en cadena y no por los radicales formados por pirólisis directa ni por los productos de combinación de los radicales o de los átomos.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones por Radicales Libres 176
2. Pirólisis de metano El metano da carbono e hidrógeno como productos finales. Este proceso es endotérmico y consume 190 kcal/mol.
Para que esta reacción sea rápida se necesitan temperaturas de más de 1.200º C. Sin embargo, otros hidrocarburos se descomponen a temperaturas más bajas (500-600º).
3. Pirólisis de etano
3.1. Iniciación En la descomposición térmica del etano son posibles dos reacciones de iniciación: la disociación del enlace C-C, que da dos radicales metilo, o la disociación de C-H que da un radical etilo y un átomo de H. La primera reacción es más rápida y es la reacción de iniciación:
(1) CH3 CH3 CH32
(2) CH3 CH3 CH3 CH2 + H
3.2. Propagación Una vez formados los radicales metilo, pueden ocurrir fácilmente otras reacciones, ya que los radicales libres son muy reactivos y, por lo tanto, los cambios de energía necesarios son relativamente pequeños.
(3) CH3 CH3 CH3+ CH4+ CH3 CH2
(4) CH3 CH2 +CH2 CH2 H
(5) CH3 CH3+ H CH3 CH2+ H2
Luego se repiten alternadamente (4) y (5), dando los productos principales: etileno e hidrógeno. Cuando H·,
que se genera en la reacción (4), no encuentra más etano, se produce el cese. La reacción total es endotérmica.
Los perfiles de energía de los diferentes pasos son los siguientes:
El gráfico I corresponde a la disociación de etano con formación de radicales metilo, de acuerdo a la reacción (1)
El gráfico II corresponde a la transferencia de átomos de hidrógeno de acuerdo a la reacción (3) El gráfico III es la descomposición de radicales etilo con formación de eteno y átomos de hidrógeno
como lo indica la reacción (4) El gráfico IV corresponde a la reacción de átomos de hidrógeno con etano conforme a la reacción (5)
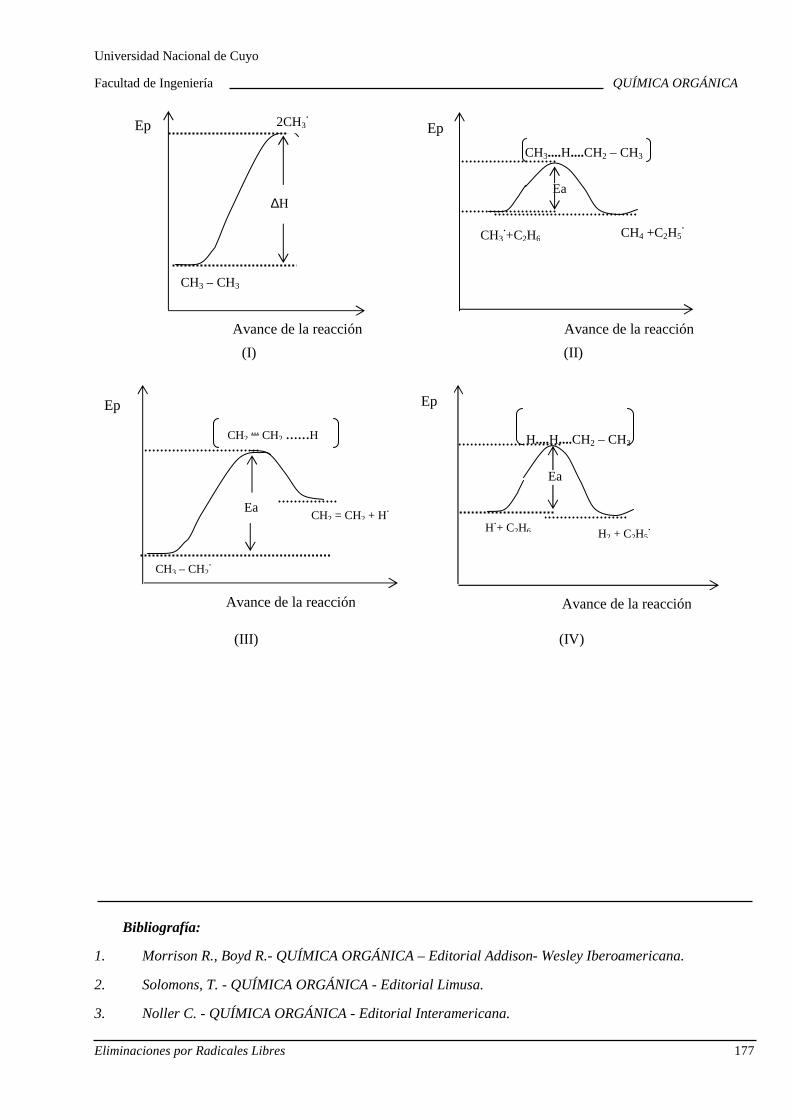
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones por Radicales Libres 177
(I) (II)
(III) (IV)
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial Addison- Wesley Iberoamericana.
2. Solomons, T. - QUÍMICA ORGÁNICA - Editorial Limusa.
3. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.
∆H
Ep
Avance de la reacción
CH3 – CH3
2CH3·
CH3....H....CH2 – CH3
Ea
Ep
CH3·+C2H6 CH4 +C2H5
·
Avance de la reacción
Ea
Avance de la reacción
Ep
CH3 – CH2·
CH2 = CH2 + H·
CH2 … CH2 ……H
H....H....CH2 – CH3
Ea
Avance de la reacción
Ep
H·+ C2H6 H2 + C2H5·

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 178
Casi todos los compuestos que sufren sustitución nucleofílica (es decir los que tienen un grupo saliente en un átomo con hibridación sp3) también pueden sufrir eliminación, con formación de dobles o triples enlaces.
En una eliminación típica, un sustrato que tiene dos grupos (A y B) en carbonos adyacentes experimenta la ruptura de los dos enlaces que unen el esqueleto de carbono a dichos grupos. Dos de los cuatro electrones de los enlaces σ aparecen en el producto como un enlace π. Los dos electrones restantes quedan ya sea como un par de electrones localizados en A o B, con producción de un anión y un catión, o como un enlace covalente entre los dos fragmentos. Al mismo tiempo, los dos átomos de carbono a los cuales A y B estaban unidos cambian su hibridación de sp3 a sp2
Formación de un doble enlace:
C C
F
F
F
F+ +BH
-A
+C C
H
A
H
B
Formación de un triple enlace:
C CF F+ +BH-
A+C C
A B
Los métodos de eliminación iónica, también conocidos como eliminación 1,2 más importantes por su aplicabilidad general son: deshidrohalogenación de halogenuros de alquilo y deshidratación de alcoholes.
C C + H B + Zeliminación
C C
H Z
BH-
+
Sustrato base AlquenoBase protonada
Grupo saliente
-
El sustrato, generalmente halogenuro de alquilo o alcohol, contiene un grupo saliente, átomo o grupo que abandona la molécula llevándose un par de electrones. En una posición β con respecto al grupo saliente, el sustrato contiene un átomo o grupo de átomos, casi siempre hidrógeno, que puede ser sacado por la base, dejando su par de electrones, con el que se forma el segundo enlace entre los carbonos.
1. Reactividad nucleofílica: Se cumplen las mismas consideraciones que para la Sustitución Nucleofílica.
1.1. Cinética de la Eliminación: La velocidad de una reacción química se puede analizar siguiendo los mismos considerandos que en la Sustitución Nucleofílica. Por ejemplo, cuando se estudia la reacción:
C C + ZeliminaciónC C
H Z
+ B-
+H B-
Se pueden dar dos casos:
Reacciones de Eliminación: Eliminaciones Iónicas
Reacciones de eliminación en haluros de alquilo. Cinética. Diagramas de energía. Reactividad de haluros de alquilo frente a reactivos nucleofílicos. Competencia con las sustituciones nucleofílicas. Importancia industrial de las eliminaciones iónicas.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 179
Caso 1: Se observa experimentalmente que al duplicar la concentración de base, se duplica la velocidad de la reacción; y que al duplicar la concentración de R – X también se duplica la velocidad de la reacción, por lo tanto la velocidad de la reacción depende tanto de la concentración de R-X como de la concentración de :B y viene expresada por:
Se deduce entonces, que la reacción sigue una cinética de segundo orden ya que la velocidad depende de la concentración elevada a la primera potencia de los dos reactivos.
Caso 2: Experimentalmente se observa que al duplicar la concentración de R – X, se duplica la velocidad de la reacción. Y que al duplicar la concentración de la base no hay cambio en la velocidad de reacción. Por lo tanto la velocidad de la reacción sólo depende de la concentración de R-X y viene expresada por:
Se deduce por lo tanto que la reacción sigue una cinética de primer orden puesto que la velocidad de la reacción depende de la concentración elevada a la primera potencia de un solo reactivo.
Para explicar estas diferencias de orden cinético, se ha propuesto para las reacciones de Eliminación dos mecanismos distintos:
Eliminación Bimolecular: E2
Eliminación Monomolecular : E1
2. Eliminación en halogenuros de alquilo: Deshidrohalogenación de halogenuros de alquilo
CH3 CH2 Cl + CH2 CH2 + + OH2NaOH NaCl
Esta reacción significa la eliminación del átomo de halógeno como halogenuro (X:-) y de un hidrógeno como
protón (H+) de un carbono adyacente al que pierde el halógeno.
El reactivo necesario es una base, neutra o iónica que debe extraer un H+ unido al carbono y, simultáneamente, se separa el ión halogenuro, formándose el doble enlace C = C. La ruptura de enlaces es heterolítica. El solvente utilizado es un solvente polar (ej.: alcohol).
Reacción general:
Las rupturas de enlaces se pueden interpretar por un mecanismo general de Eliminación:
C CC C
X
H OH-
O representado como:
C C + XalcoholC C
H X
+ B-
+H B-

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 180
C CC C
X
H OH -
+ H2O + X -
La energía necesaria para romper los enlaces C- H y C-X es provista por:
La formación del enlace entre H+ y la base fuerte HO: -. La formación de un enlace π que, aunque débil, proporciona alrededor de 70 kcal/mol. La energía provista por la solvatación de los iones halogenuros por las moléculas de alcohol, las que al ser polares se orientan formando enlaces ión-dipolo, que son débiles separadamente pero en conjunto liberan una energía considerable.
Al igual que el HO: – desprende al H+ de la molécula original, las moléculas de solvente le sustraen el ión halogenuro.
Mecanismo: Hay dos mecanismos propuestos para explicar la deshidrohalogenación de halogenuros de alquilo:
Eliminación bimolecular: E2
Eliminación monomolecular: E1
2.1. Eliminación bimolecular: E2
2.1.1. Molecularidad: Se considera un mecanismo de Eliminación Bimolecular, E2, porque en la etapa que determina la velocidad de reacción, dos especies químicas sufren ruptura o formación de enlaces (RX y -:OH).
2.1.2. Cinética: El estudio cinético de la reacción de halogenuros de alquilo con una base, en determinadas condiciones muestra:
Al duplicar la concentración de halogenuro, se duplica la velocidad de reacción. Al duplicar la concentración de la base, se duplica la velocidad de reacción.
La ecuación cinética es entonces:
La reacción responde a una cinética de 2° orden (1 respecto a halogenuro y 1 respecto a la base); la velocidad depende de las concentraciones de las dos especies.
2.1.3. Mecanismo: Se puede proponer un mecanismo en una sola etapa en la que intervienen las dos especies:
X-
H O
R
H
O R
O HR
H
OR
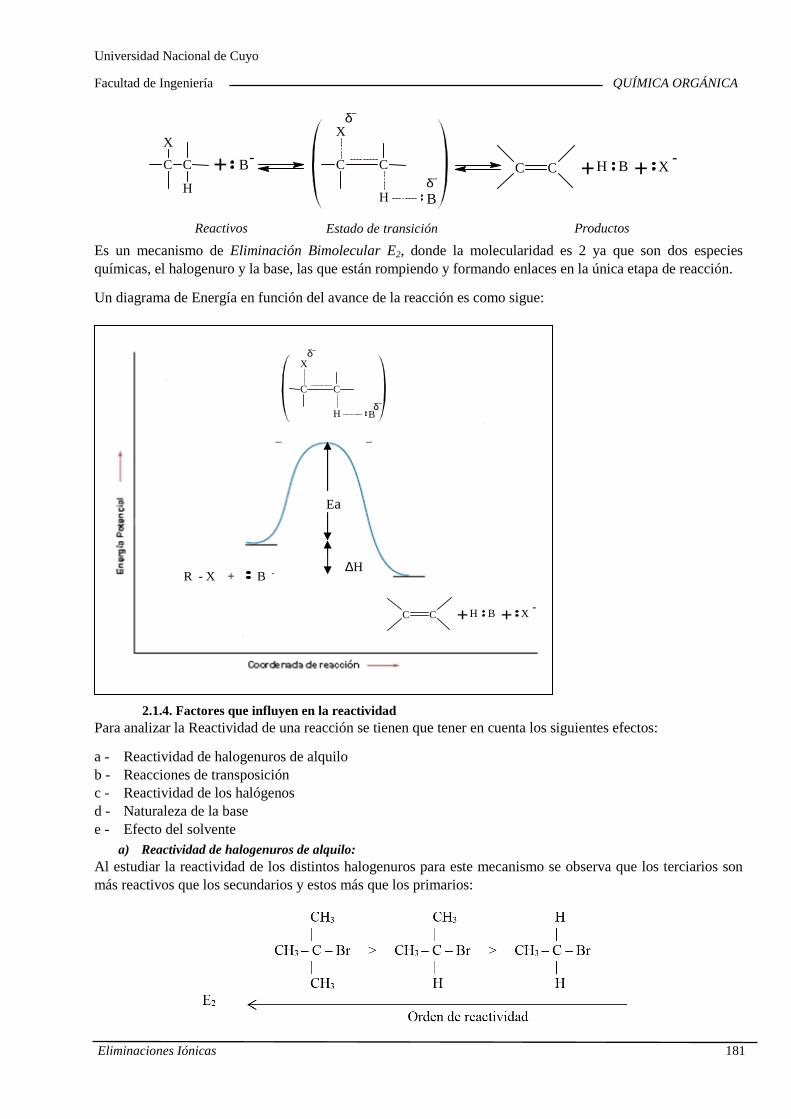
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 181
C C + + XC C
X
H
+
Reactivos Productos
C C
X
H
δ−
δ−
Estado de transición
H B-B -
B
Es un mecanismo de Eliminación Bimolecular E2, donde la molecularidad es 2 ya que son dos especies químicas, el halogenuro y la base, las que están rompiendo y formando enlaces en la única etapa de reacción.
Un diagrama de Energía en función del avance de la reacción es como sigue:
2.1.4. Factores que influyen en la reactividad Para analizar la Reactividad de una reacción se tienen que tener en cuenta los siguientes efectos:
a - Reactividad de halogenuros de alquilo b - Reacciones de transposición c - Reactividad de los halógenos d - Naturaleza de la base e - Efecto del solvente
a) Reactividad de halogenuros de alquilo: Al estudiar la reactividad de los distintos halogenuros para este mecanismo se observa que los terciarios son más reactivos que los secundarios y estos más que los primarios:
C C
X
H
δ−
B-δ−
R - X + B -
C C + + XH B -
Ea
∆H
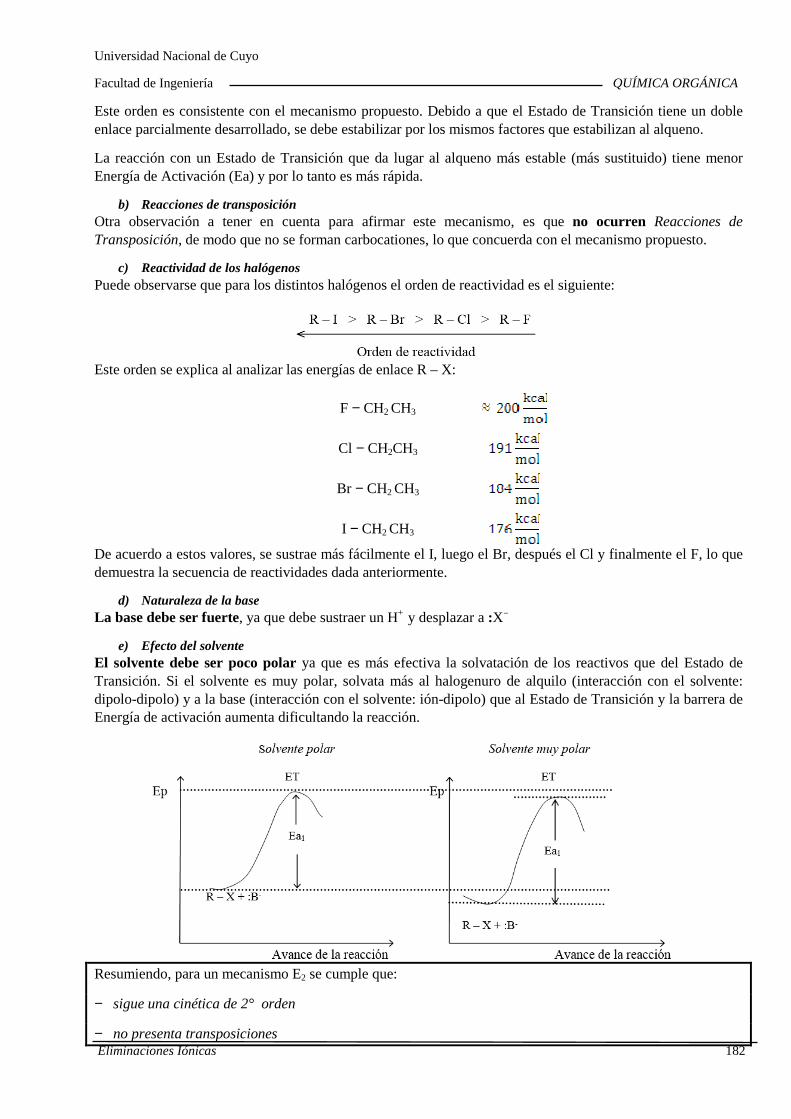
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 182
Este orden es consistente con el mecanismo propuesto. Debido a que el Estado de Transición tiene un doble enlace parcialmente desarrollado, se debe estabilizar por los mismos factores que estabilizan al alqueno.
La reacción con un Estado de Transición que da lugar al alqueno más estable (más sustituido) tiene menor Energía de Activación (Ea) y por lo tanto es más rápida.
b) Reacciones de transposición Otra observación a tener en cuenta para afirmar este mecanismo, es que no ocurren Reacciones de Transposición, de modo que no se forman carbocationes, lo que concuerda con el mecanismo propuesto.
c) Reactividad de los halógenos Puede observarse que para los distintos halógenos el orden de reactividad es el siguiente:
Este orden se explica al analizar las energías de enlace R – X:
F − CH2 CH3
Cl − CH2CH3
Br − CH2 CH3
I − CH2 CH3
De acuerdo a estos valores, se sustrae más fácilmente el I, luego el Br, después el Cl y finalmente el F, lo que demuestra la secuencia de reactividades dada anteriormente.
d) Naturaleza de la base La base debe ser fuerte, ya que debe sustraer un H+ y desplazar a :X−
e) Efecto del solvente El solvente debe ser poco polar ya que es más efectiva la solvatación de los reactivos que del Estado de Transición. Si el solvente es muy polar, solvata más al halogenuro de alquilo (interacción con el solvente: dipolo-dipolo) y a la base (interacción con el solvente: ión-dipolo) que al Estado de Transición y la barrera de Energía de activación aumenta dificultando la reacción.
Resumiendo, para un mecanismo E2 se cumple que:
− sigue una cinética de 2° orden
− no presenta transposiciones

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 183
− el orden de reactividad de halogenuros de alquilo: 3° > 2° > 1° , debido a que se forma el alqueno más estable.
2.2. Eliminación monomolecular: E1
2.2.1. Molecularidad: Este mecanismo es una Eliminación Monomolecular, puesto que la molecularidad es 1. Es decir, en la etapa lenta, determinante de la velocidad de reacción, una sola especie sufre ruptura de enlace. La especie que rompe una unión covalente es el halogenuro de alquilo y forma un carbocatión.
2.2.2. Cinética: El estudio cinético de la reacción de halogenuros de alquilo con una base, en determinadas condiciones muestra:
Al duplicar la concentración de halogenuro, se duplica la velocidad de reacción. Al variar la concentración de la base, no varía la velocidad de reacción.
La velocidad es directamente proporcional a la concentración del halogenuro y la ecuación cinética es entonces:
La reacción responde a una cinética de 1° orden (1° orden respecto a halogenuro). La velocidad depende de la concentración de una de las dos especies.
2.2.3. Mecanismo: Para esta reacción debe existir una etapa en la que intervenga sólo el halogenuro de alquilo. Esta es la etapa determinante de la velocidad de reacción, es decir, la etapa lenta.
Se puede proponer un mecanismo en dos etapas:
Primera etapa
C+
C + X
Intermedio
C C
Xδ−
δ+
Estado de transición
lenta C C
X
-
Segunda etapa
C C +B-+
rápida δ−
Estado de transición
C+
C
H
δ+
C C
H B
HB
Los Estados de Transición son estados de máxima energía, no aislables, donde se supone que se están formando y/o rompiendo enlaces. Los Intermedios, especies generalmente aislables, son productos de una etapa y reactivos de la siguiente.
Un diagrama de Energía potencial en función del avance de la reacción muestra lo siguiente:
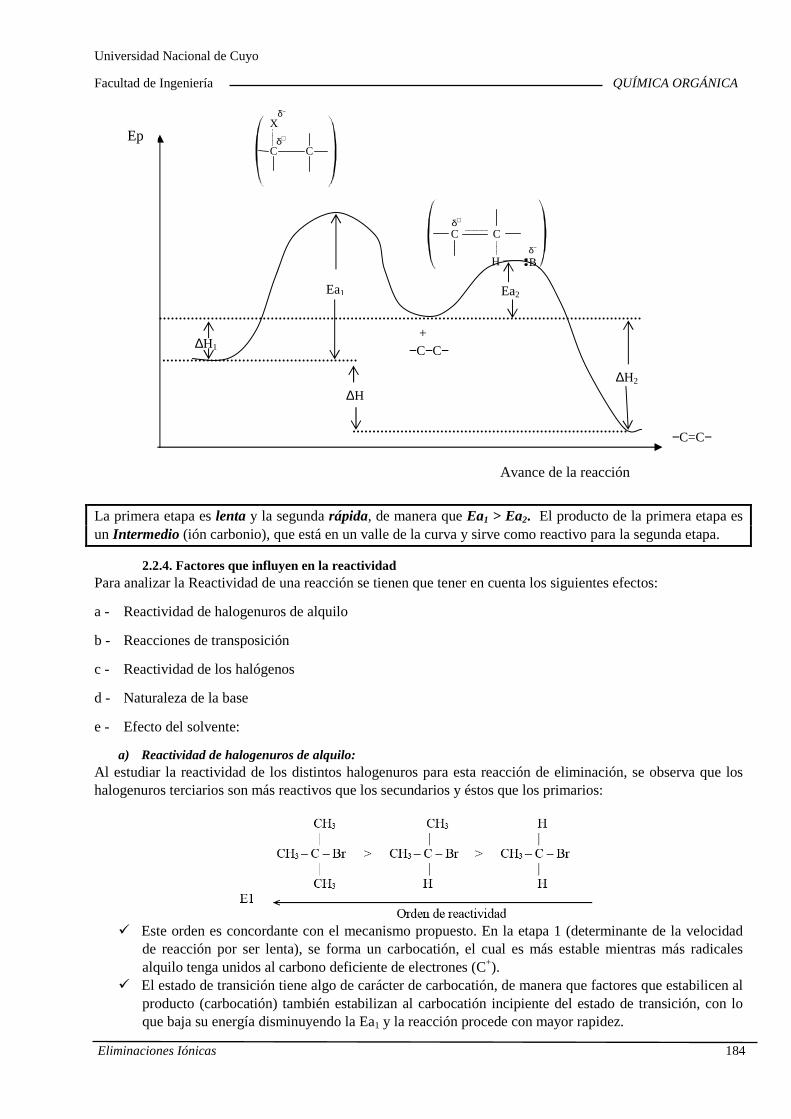
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 184
Ea1 Ea2
∆H1
∆H ∆H2
C C
Xδ−
δ+
δ−
δ+
C C
H B
La primera etapa es lenta y la segunda rápida, de manera que Ea1 > Ea2. El producto de la primera etapa es un Intermedio (ión carbonio), que está en un valle de la curva y sirve como reactivo para la segunda etapa.
2.2.4. Factores que influyen en la reactividad Para analizar la Reactividad de una reacción se tienen que tener en cuenta los siguientes efectos:
a - Reactividad de halogenuros de alquilo
b - Reacciones de transposición
c - Reactividad de los halógenos
d - Naturaleza de la base
e - Efecto del solvente:
a) Reactividad de halogenuros de alquilo: Al estudiar la reactividad de los distintos halogenuros para esta reacción de eliminación, se observa que los halogenuros terciarios son más reactivos que los secundarios y éstos que los primarios:
Este orden es concordante con el mecanismo propuesto. En la etapa 1 (determinante de la velocidad
de reacción por ser lenta), se forma un carbocatión, el cual es más estable mientras más radicales alquilo tenga unidos al carbono deficiente de electrones (C+).
El estado de transición tiene algo de carácter de carbocatión, de manera que factores que estabilicen al producto (carbocatión) también estabilizan al carbocatión incipiente del estado de transición, con lo que baja su energía disminuyendo la Ea1 y la reacción procede con mayor rapidez.
Avance de la reacción
+ −C−C−
−C=C−
Ep

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 185
Mientras más radicales estén unidos al C+ mas estable es el intermedio y mas estable es también el estado de transición.
b) Reacciones de transposición Otro de los hechos que avalan este mecanismo es que pueden ocurrir Transposiciones, las que son características de mecanismos vía formación de iones carbonio:
Como ejemplo de este análisis se observa en el siguiente caso:
CH2 CH2 ClCH2CH3KOH/alcohol CH CH2CH2CH3
CH CH3CHCH3
(20%)
(80%)
Esto se debe a que en la primera etapa se forma un carbocatión que sufre transposiciones.
Si el carbocatión 1° formado en la primera etapa no sufre transposición, el alqueno que se obtiene es el 1-buteno:
CH2 CH2 ClCH2CH3 CH2 CH2+
CH2CH3 CH CH2CH2CH3
Si el carbocatión sufre transposición de H: - (hidruro) se pueden formar 1-buteno y 2-buteno:
CH CH2CH2CH3
CH CH3CHCH3Producto principal
C C+
CH2CH3
H
H
H
H
CH+
CH3CH2CH3más estable
Recordar que los alquenos más estables son los más ramificados en el doble enlace:
R2C = CR2 > R2C = CHR > R2C = CH2, RHC = CHR > RHC = CH2 > H2C=CH2
Estabilidad de alquenos
c) Reactividad de los halógenos: Se siguen las mismas consideraciones que para el mecanismo de Eliminación Bimolecular.
d) Naturaleza de la base: En este mecanismo la característica de la base no afecta ya que no interviene en la etapa determinante de la velocidad de reacción, por lo que generalmente se trabaja con baja concentración de la misma.
e) Efecto del solvente: El solvente debe ser muy polar para solvatar los iones X: - y carbonio producidos en la primera etapa.
Además si se analizan las estructuras de los reactivos y del Estado de Transición (ET), el halogenuro de alquilo (reactivo) es polar, pero más polar es el ET. El agregado de un solvente muy polar estabiliza más al ET que a los reactivos bajando la Ea y acelerando la reacción.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 186
Por lo tanto para un mecanismo E1 se considera que:
- sigue una cinética de primer orden
- presenta transposiciones
- el orden de reactividad de halogenuros de alquilo es 3° > 2° > 1° , debido a que se forma el carbocatión más estable.
3. Competencia entre eliminación y sustitución: a) Para las siguientes condiciones:
Solvente poco polar
Reactivo (:Z) fuertemente nucleofílico
La Sustitución es por un mecanismo SN2 de manera que los halogenuros primarios reaccionan más rápidamente.
La Eliminación es por un mecanismo E2 y como la velocidad de reacción depende de la estabilidad del alqueno formado, entonces los halogenuros terciarios (que forman alquenos ramificados más estables), reaccionan más rápidamente.
C C
X
H
Z
SN2
E2
Por lo tanto:
Resumiendo:
Partiendo de un halogenuro de alquilo primario, en las condiciones de solvente poco polar y reactivo fuertemente nucleofílico, por mecanismo SN2 se puede obtener alcohol primario.
Partiendo de un halogenuro de alquilo terciario, en las condiciones de solvente poco polar y reactivo fuertemente nucleofílico, por mecanismo E2 se puede obtener alqueno.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 187
b) Para las siguientes condiciones:
Solvente muy polar
Reactivo (:Z) débilmente nucleofÍlico
La Sustitución es por un mecanismo SN1 de manera que los halogenuros terciarios reaccionan más rápidamente (debido a la mayor estabilidad de los carbocationes terciarios).
La Eliminación es por un mecanismo E1, de manera que los halogenuros terciarios reaccionan más rápidamente (debido a la mayor estabilidad de los carbocationes terciarios).
C+
C
H
Z
SN1
E1
Por lo tanto:
Resumiendo:
Partiendo de un halogenuro de alquilo terciario, en las condiciones de solvente muy polar y reactivo débilmente nucleofílico, por mecanismos SN1 y E1 se formarán rápidamente, tanto alcohol como alqueno.
Partiendo de un halogenuro de alquilo primario, en las condiciones de solvente muy polar y reactivo débilmente nucleofílico, por mecanismos SN1 y E1 se formarán muy lentamente, tanto alcohol como alqueno.
4. Reacciones de Eliminación en alcoholes: Deshidratación intramolecular Un alcohol se convierte en alqueno por deshidratación en presencia de un ácido y calor:
C C
H OH
H+/calorC C + OH2
Es una reacción de Eliminación y procede generalmente por un mecanismo Monomolecular: E1. En algunos casos los alcoholes primarios, por la inestabilidad de los iones carbonio que formarían en un mecanismo E1, siguen un mecanismo E2.
El agregado de ácido es para facilitar la formación del ión carbonio en la etapa determinante de la velocidad de reacción. En este paso, el H+ es transferido desde el ión bisulfato a la base más fuerte, el alcohol, para generar ión oxonio:
CH3 CH2 O
H
+ H2SO4 CH3 CH2 O+
H
H
HSO4-+
(1) Este alcohol protonado permite que el ión carbonio se forme con mayor facilidad, debido a la mayor polaridad del enlace C – O.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 188
CH3 CH2 O+
H
H
CH3 C+
H
H+ OH2 etapa fácil
(2) De no formarse el alcohol protonado el enlace C – O es menos polar:
CH3 CH2 O H CH3 C+
H
H + etapa muy difícilHO-
La reacción (1) implica la separación de una partícula cargada R+ de otra neutra, H2O. Este proceso requiere menor energía que la formación del ión carbonio del alcohol (2), pues en este caso se separaría una especie cargada positivamente de otra especie cargada negativamente.
Una vez formado el ión oxonio la reacción sigue un mecanismo de Eliminación Monomolecular E1
Primera etapa:
C CH
H
H
H
H
O+
H
H
lenta
δ+
δ+
C C+
H
H
H H
H
+ OH2
Reactivos Estado de transición Intermedio
H C C H
H H
H O
H
H
Segunda etapa:
C C+
H
H
H H
H
OH C2H5
rápida
δ+
δ+
Estado de transición
C C
H
H
H
H
+ OH+
H5C2HH C C H
H H
H
OH C2H5
En la segunda etapa el H+ es transferido a la base más fuerte que es el alcohol.
Cuando se estudian las reactividades de los alcoholes primarios, secundarios y terciarios, se observa que se deshidratan más fácilmente los alcoholes terciarios, luego los secundarios y finalmente los primarios:
Este orden, que es coincidente con el mecanismo de deshidratación propuesto, se debe a la facilidad de formación y estabilidad de iones carbonio.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Eliminaciones Iónicas 189
El producto de la etapa determinante de la velocidad de reacción es un ión carbonio y el estado de transición tiene carácter de carbocatión; de manera que factores que estabilicen al carbocatión estabilizan también al ET disminuyendo su energía, bajando la Ea y haciendo más rápida la reacción.
El agregado de grupos alquilos sobre el C+ , deficiente de electrones, estabilizan el ión carbonio, y también el ET con lo que baja la Ea y se acelera la reacción.
Otro hecho concordante con este mecanismo es la factibilidad de que ocurran transposiciones, que es explicable por la formación de iones carbonios que son los que producen este tipo de reacción.
Por ejemplo, la deshidratación de n-butanol puede producir dos productos:
CH2 CH2 OHCH2CH3 CH CH2CH2CH3
CH CH3CHCH3
H+/calor
Producto principal
CH2 CH2OHCH2CH3 + H+
CH2 CH2OH2+
CH2CH3
CH2 CH2 OH2+
CH2CH3 CH CH2+
CH2CH3
H
CH+
CH3CCH3
H
H
CH CH3CHCH3Producto principal por ser el alqueno
más estable
CH CH2CH2CH3
Hasta ahora hemos visto la deshidratación intramolecular que sigue un mecanismo E1. Paralelamente, ocurre deshidratación intermolecular, mediante un mecanismo SN1, con formación de éter.
Ejemplo:
CH3 CH2 OHH
+
CH3 CH2 OH2+
OH2CH3 CH2
+CH2 CH2
OH CH2 CH3CH3 CH2 O
+CH2 CH3
H
H+
CH3 CH2 O CH2 CH3
+ CH3 CH2 OH2+
5. Importancia industrial de las eliminaciones iónicas. Las reacciones de eliminación son de gran utilidad en la preparación de alquenos puros.
La formación de dobles enlaces por eliminaciones es una vía para obtener compuestos no saturados diferentes a alquenos, como por ejemplo, ácidos carboxílicos no saturados.
Bibliografía:
1. Morrison R., Boyd R.- QUÍMICA ORGÁNICA – Editorial Addison- Wesley Iberoamericana
2. SolomonsT. - QUÍMICA ORGÁNICA – Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 190
Oxidación y reducción son términos familiares por haberlos utilizado en el estudio de la química inorgánica, en donde la oxidación y la reducción se refieren a cambios en los números de oxidación. En los compuestos orgánicos, donde también estas reacciones tienen que ver con variaciones de números de oxidación, simplificaremos el estudio empleando los términos como sigue:
Oxidación: Es el aumento de átomos de oxígeno o pérdida de átomos de hidrógeno en una molécula orgánica.
Reducción: Es el aumento de átomos de hidrógeno o pérdida de átomos de oxígeno en una molécula orgánica.
1. Oxidación de compuestos orgánicos La secuencia general de oxidación de moléculas orgánicas es:
C
CH3
C O C O
OH
C
OH
CO2 + OH2
Hidrocarburo Alcohol Aldehído o cetona
Ácido
Los agentes oxidantes comúnmente utilizados son:
Agentes Oxidantes
O2/ φ
O2/V2O5/ φ
KMnO4 / H+ / φ
KMnO4 / OH-
K2Cr2O7/ H+ / φ
CrO3
Reactivo de Fehling y Reactivo de Tollens
De estos agentes oxidantes tenemos que el oxígeno y el oxígeno con pentóxido de vanadio, ambos en caliente, son los oxidantes más enérgicos que se conocen, ya que permiten oxidar directamente hasta llegar a ácidos carboxílicos o a dióxido de carbono y agua.
El permanganato de potasio y el dicromato de potasio, si se utilizan en medio ácido y en caliente, se consideran como oxidantes fuertes, o dicho de otro modo estaríamos trabajando en condiciones enérgicas.
El óxido crómico y el dicromato de potasio y el permanganato de potasio en medio alcalino y en frío, son oxidantes más suaves.
Oxidación y reducción de compuestos orgánicos
Oxidación de hidrocarburos. Combustión. Calor de combustión. Oxidación de compuestos orgánicos oxigenados.
Reducción de hidrocarburos alifáticos. Reducción de hidrocarburos aromáticos. Reducción de compuestos orgánicos oxigenados. Reducción de compuestos orgánicos nitrogenados: Reducción de nitrocompuestos aromáticos.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 191
Tanto el reactivo de Fehling como el reactivo de Tollens son oxidantes muy débiles capaces de oxidar aldehídos y que se utilizan principalmente para diferenciar aldehídos de cetonas.
1.1. Reacciones de combustión La combustión es la oxidación con oxígeno y calor de un compuesto orgánico. Esta reacción puede ser completa o incompleta, en función de la cantidad de oxígeno disponible.
Para el caso de los compuestos constituidos por carbono, hidrógeno y/u oxígeno, si la combustión es completa, los productos son dióxido de carbono y agua, mientras que la incompleta produce monóxido de carbono también y, en algunos casos carbono (partículas sólidas). A continuación, veremos casos de combustión completa de hidrocarburos:
Combustión de alcanos Ejemplos:
El mecanismo de esta reacción es muy complicado y aún no se conoce bien. Sin embargo, parece indudable que se trata de una reacción en cadena de radicales libres.
Es un proceso exotérmico y, sin embargo, para su iniciación se requiere una temperatura muy elevada, como es la de una llama. Como en la cloración, se necesita mucha energía para romper los enlaces que generan las partículas reactivas iniciales; una vez vencida esta barrera energética, los pasos siguientes, propagadores de la cadena, proceden sin dificultad y desprendimiento de calor.
La cantidad de calor que se genera al quemar un mol de compuesto a dióxido de carbono y agua es el calor de combustión.
Para los alcanos de cadena abierta cada grupo metileno –CH2- contribuye con un valor muy cercano a 157,4 kcal/mol al calor de combustión. El hecho que la combustión de los hidrocarburos sea exotérmica hace que se utilicen como combustibles.
Por otro lado, la inflamabilidad depende de la volatilidad del hidrocarburo. Las mezclas de aire y vapores de hidrocarburos en proporciones adecuadas hacen explosión al inflamarse y ésto hace que se utilicen en los motores de combustión interna.
Combustión de cicloalcanos Como los calores de hidrogenación, los de combustión a menudo pueden proporcionar información valiosa acerca de las estabilidades relativas de los compuestos orgánicos. Veamos si los calores de combustión de los cicloalcanos apoyan la proposición de que “los anillos menores o más grandes que el ciclopentano y el ciclohexano serían inestables”
El examen de muchos compuestos ha demostrado que el calor de combustión de un hidrocarburo alifático concuerda muy bien con el que se puede calcular, si se considera una cierta contribución característica por cada unidad estructural. Para los alcanos de cadena abierta cada grupo metileno, -CH2-, contribuye con un valor muy cercano a 157,4 kcal/mol al calor de combustión.
La tabla siguiente contiene los calores de combustión determinados experimentalmente para algunos de los cicloalcanos.
Tamaño del anillo Calor de combustión por CH2 (kcal/mol)
3 166,6

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 192
4 164,0 5 158,7 6 157,4 7 158,3 8 158,6
Cadena abierta 157,4
Observamos lo siguiente:
El calor de combustión por grupo metileno (–CH2 –) para el ciclopropano es superior en 9 kcal al valor para la cadena abierta de 154,7 kcal.
El calor de combustión por grupo metileno (–CH2 –) para el ciclobutano es superior en 7 kcal al valor para la cadena abierta de 154,7 kcal.
Cualquiera que sea el compuesto en el cual aparezca el grupo –CH2– siempre da los mismos productos por combustión: dióxido de carbono y agua.
Si el ciclopropano y el ciclobutano liberan más energía por grupo –CH2– que un compuesto de cadena abierta, esto sólo puede significar que contiene más energía por grupo –CH2–. Esto último se explica porque ambos compuestos presentan una elevada tensión angular y tensión torsional, por lo que el ciclopropano y el ciclobutano son menos estables que las sustancias de cadena abierta.
Tensión angular: Se produce por la desviación de los ángulos normales de 109º (carbonos con hibridación sp3) cuando se forman ciclos donde los ángulos de enlaces son distintos a dicho valor. Esto se presenta en los casos de ciclopropropano y ciclobutano donde los ángulos son de 60º y 90º, respectivamente:
Tensión torsional: Se produce cuando los enlaces de carbonos distintos quedan eclipsados, es decir, se ubican tan cerca entre si, que se producen repulsiones de nubes electrónicas y, por lo tanto, mayor energía en la molécula. Esto se presenta en los casos de ciclopropropano y ciclobutano donde todos los enlaces C-H están eclipsados. Es mucho más estable la molécula cuando los enlaces están alternados
En el ciclopentano el calor de combustión por grupo –CH2– es próximo al de cadena abierta, esto se explica por la falta de tensión angular (estas moléculas no son planas y los ángulos de enlaces son próximos a 109°).
En el ciclohexano el calor de combustión por grupo –CH2– es igual al de cadena abierta, esto se explica por la falta de tensión angular (estas moléculas no son planas y los ángulos de enlaces son de 109°).
Alifáticos 157,4 kcal/mol
157,4 kcal/mol
158,7 kcal/mol
164,0 kcal/mol
166,6 kcal/mol
CO2 + H2O
Avance de la reacción
Ep

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 193
En los cicloalcanos de más de siete átomos de carbono el calor de combustión por grupo –CH2– es similar al de cadena abierta, esto se explica por la falta de tensión angular (estas moléculas no son planas y los ángulos de enlaces son próximos a 109°).
Ejemplos de combustión de otros hidrocarburos
Oxidación química de alquenos Estudiaremos la oxidación de alquenos, los que con agentes oxidantes suaves da derivados dihidroxilados y con agentes oxidantes enérgicos forma ácidos carboxílicos.
Hidroxilación de alquenos La hidroxilación de alquenos es el método más importante para la síntesis de glicoles, algunos de los cuales son usados como anticongelantes. Esta oxidación se realiza agitando el alqueno junto con un oxidante suave a temperatura ambiente (ej.: MnO4K en medio alcalino). Se evitan el calor y el medio ácido para impedir la ruptura del enlace π:
CH2 CH2condiciones suaves
CH2 CH2
OH OH
etileno etilenglicol(1,2-etanodiol)
Ruptura oxidativa de alquenos Al oxidarse con un oxidante fuerte (ej.: MnO4K en medio ácido / caliente), los alquenos sufren la ruptura tanto del enlace π como del σ, formando sales de ácidos carboxílicos. Esta reacción puede ilustrarse con la descomposición oxidativa del cis- o del trans-2-buteno que produce dos moles de ácido acético:
CHCH3CHCH3
condiciones enérgicas
2-buteno
CCH3 OH
O
2
Ácido acético
En condiciones enérgicas el grupo CH2 terminal de un 1-alqueno se oxida completamente a dióxido de
carbono y agua.
CH CH2CH2CH3
condiciones enérgicas
CCH2 OH
O
CH3
Ácido propanoico
+ CO2 + OH2
Con frecuencia la descomposición oxidativa de los alquenos se utiliza para localizar las posiciones del doble enlace C=C.
La oxidación de hidrocarburos ramificados da como resultado un ácido carboxílico y una cetona, o bien, una o dos cetonas.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 194
Nota: Veremos más adelante cómo las cetonas se pueden oxidar en condiciones enérgicas formando ácidos carboxílicos y, en algunos casos, bióxido de carbono.
Oxidación química de Alquinos Cuando los alquinos se tratan con un oxidante fuerte (ej.: permanganato de potasio en medio ácido y calor) sufren una ruptura oxidativa en forma similar a la que se produce en alquenos.
C CR R'
condiciones enérgicas
RCOOH +R'COOH
Los productos de la reacción son ácidos carboxílicos y, si el triple enlace es terminal, bióxido de carbono.. Los ácidos carboxílicos son fáciles de identificar y sus identidades permiten localizar la posición del triple enlace en un alquino.
4. Oxidación química de hidrocarburos aromáticos mononucleares En general el benceno es bastante inerte frente a los agentes oxidantes usuales como permanganato de potasio o dicromato de potasio. Pero en los alquilbencenos, el anillo bencénico sensibiliza bastante las cadenas laterales alifáticas para la oxidación. La cadena lateral se oxida completamente quedando sólo un grupo carboxilo en lugar de la cadena original.
Generalmente se emplea un oxidante fuerte (ej.: permanganato de potasio) para este fin, sin embargo la oxidación de una cadena lateral requiere de un tratamiento prolongado.
CH2CH3CH2CH2
condiciones enérgicas
COOH + CO2
Butilbenceno Ácido benzoico
Esta reacción se utiliza con dos fines:
a) Síntesis de ácidos carboxílicos.
C CH3CHCH3
CH3condiciones
enérgicasCOOHCH3
Ácido etanoico
+ CH3 C
CH3
O
Propanona
C CH3CCH3
CH3 CH3condiciones
enérgicas2 CH3 C
CH3
O
Propanona
C CH3CCH2
CH3 CH3
CH3
condiciones enérgicas
CH3 C
CH3
O
Propanona
CH2 C
CH3
OCH3
Butanona
+
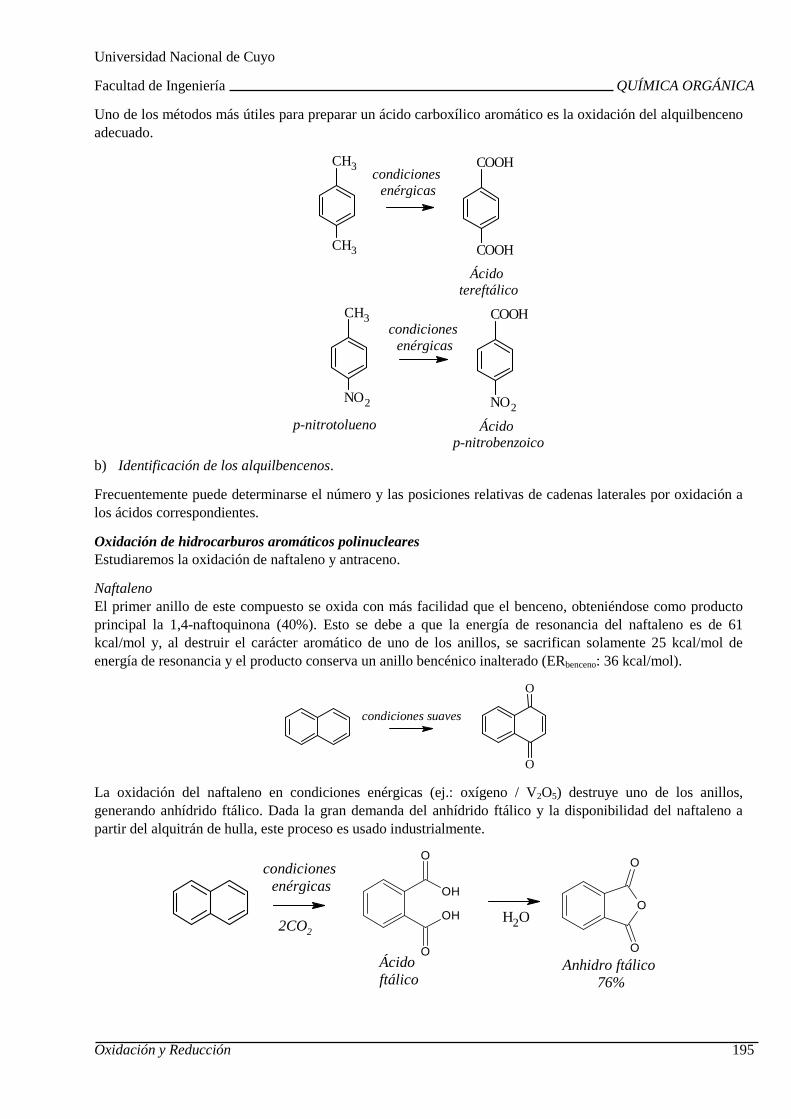
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 195
Uno de los métodos más útiles para preparar un ácido carboxílico aromático es la oxidación del alquilbenceno adecuado.
CH3
CH3
COOH
COOH
condiciones enérgicas
Ácido tereftálico
CH3
NO2
COOH
NO2
condiciones enérgicas
p-nitrotolueno Ácido p-nitrobenzoico
b) Identificación de los alquilbencenos.
Frecuentemente puede determinarse el número y las posiciones relativas de cadenas laterales por oxidación a los ácidos correspondientes.
Oxidación de hidrocarburos aromáticos polinucleares Estudiaremos la oxidación de naftaleno y antraceno.
Naftaleno El primer anillo de este compuesto se oxida con más facilidad que el benceno, obteniéndose como producto principal la 1,4-naftoquinona (40%). Esto se debe a que la energía de resonancia del naftaleno es de 61 kcal/mol y, al destruir el carácter aromático de uno de los anillos, se sacrifican solamente 25 kcal/mol de energía de resonancia y el producto conserva un anillo bencénico inalterado (ERbenceno: 36 kcal/mol).
condiciones suaves
O
O
La oxidación del naftaleno en condiciones enérgicas (ej.: oxígeno / V2O5) destruye uno de los anillos, generando anhídrido ftálico. Dada la gran demanda del anhídrido ftálico y la disponibilidad del naftaleno a partir del alquitrán de hulla, este proceso es usado industrialmente.
condiciones enérgicas
2CO2OH2
Ácido ftálico
Anhidro ftálico 76%
O
O
O
OH
O
O
OH

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 196
Antraceno El antraceno es menos resistente a la oxidación que el naftaleno y que el benceno. Este compuesto se oxida principalmente a 9,10–antraquinona, producto que tiene importancia industrial (ej.: Preparación de colorantes de la antraquinona).
Tanto la orientación como la facilidad relativa con que se realiza esta reacción es comprensible en base a las estructuras implicadas. Un ataque a las posiciones 9 y 10 del antraceno, cuya Energía de Resonancia es de 84 kcal/mol, deja intactos dos anillos bencénicos, lo que implica un sacrificio de sólo 12 kcal de energía de resonancia.
Oxidación de alcoholes El compuesto que resulte de la oxidación de un alcohol depende del número de hidrógenos unidos al carbono enlazado con el grupo –OH, es decir, depende si se trata de un alcohol primario, secundario o terciario.
Alcoholes primarios Un alcohol primario tiene dos hidrógenos unidos a carbono (R−CH2OH), de modo que puede perder hidrógeno para dar un aldehído o, incluso, un ácido. En ambos casos, conserva la cadena carbonada intacta.
C OR
OH
Ácido
OHCH2R
Alcohol primario
C OR
H
Aldehído
Oxidación a aldehídos Utilizando condiciones suaves (ej.: CrO3), se pueden obtener aldehídos pero, debido a la facilidad de éstos para oxidarse a ácidos, deben ser eliminados de la mezcla reaccionante.
OHCH2R
Alcohol primario
C OR
H
Aldehído
condiciones suaves
Ejemplo:
CH3 CH2CH2OH
condiciones suaves
CH3 CH2 C OH
1-propanol Propanal
Oxidación a ácidos carboxílicos La oxidación de alcoholes primarios a ácidos se suele hacer en condiciones enérgicas (ej.: permanganato de potasio y calor).
Antraceno 9,10-antraquinona
Condiciones suaves
O
O

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 197
OHCH2R
Alcohol primario
C OR
OH
Ácido
condiciones enérgicas
Alcoholes secundarios Un alcohol secundario puede perder su único hidrógeno (R2CHOH) para transformarse en una cetona, manteniendo inalterada la cadena carbonada.
OHCRH
R
Alcohol secundario
C OR
R
Cetona
condiciones suaves
La oxidación de un alcohol secundario a cetona se realiza en condiciones suaves (ej.: CrO3), ya que en condiciones enérgicas puede continuar la oxidación con ruptura de la cadena carbonada (ver Oxidación de cetonas).
Ejemplo:
CH3CCH3
H
OH
2-Propanol
C OCH3
CH3
Propanona
condiciones suaves
Veremos más adelante que las cetonas se pueden oxidar a ácidos carboxílicos y, en algunos casos, formar bióxido de carbono.
Alcoholes terciarios Un alcohol terciario (R3COH) no tiene hidrógenos unidos al carbono al que va unido el –OH, de manera que no es oxidado con métodos comunes, ya que debería romperse la cadena carbonada.
RCR
OH
R
Un alcohol 3°
No hay oxidación
Para oxidar un alcohol terciario se realiza primero su deshidratación en medio ácido/calor, formándose alqueno, que luego se oxida como vimos anteriormente.
Ejemplo:
CH3CCH3
OH
CH3C CH2CH3
CH3
Compuesto que puede ser oxidado
H+/φ

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 198
Oxidación de aldehídos y cetonas Los aldehídos y cetonas se distinguen fácilmente mediante ensayos químicos, puesto que los aldehídos se oxidan con facilidad a ácidos carboxílicos en condiciones suaves, mientras que las cetonas son relativamente resistentes a la mayoría de los reactivos oxidantes.
Oxidación de aldehídos
Los aldehídos son más fáciles de oxidar que las cetonas, debido principalmente a que los aldehídos pueden pasar a ácidos carboxílicos manteniendo intacta la cadena carbonada.
Para estos compuestos pueden usarse agentes de oxidación muy suaves dándonos así un ensayo altamente selectivo de este grupo funcional. Dos de estos reactivos son el reactivo de Tollens y el reactivo de Fehling.
Reactivo de Tollens
El reactivo de Tollens está constituido por:
Nitrato de plata: provee los cationes plata que oxidan al aldehído.
Hidróxido de amonio: da el medio básico necesario para que ocurra la reacción.
Aunque el ión plata (Ag+) es un agente oxidante muy débil oxidará a los aldehídos a iones carboxilato. Al hacerlo, el catión plata se reduce a plata metálica. Si la velocidad de la reacción es lenta y las paredes del recipiente están limpias, la plata metálica se deposita sobre las paredes del tubo de ensayo en forma de un espejo; en caso contrario, se deposita como un precipitado gris o negro.
C OR
H
Aldehído
+ Ag+
C OR
O-
ión carboxilato
+ Ag
C OR
R
Cetona
+ Ag+
No reacciona
Reactivo de Fehling El reactivo de Fehling contiene:
Sulfato cúprico: provee los iones cúpricos (Cu++) que oxidan al aldehído. Hidróxido de sodio: da el medio básico. Tartrato de sodio y potasio: acompleja a los iones cúpricos y los mantiene en solución.
La oxidación con Reactivo de Fehling presenta un cambio característico de color, desde el azul del complejo cúprico hasta un pardo rojizo del precipitado (óxido cuproso).
C OR
H
Aldehído
+ Cu2+
C OR
O-
ión carboxilato
+ Cu2O
Azul Rojo ladrillo
Oxidación de cetonas En el caso de las cetonas, para que ocurra la oxidación se debe romper la cadena carbonada, por lo que la oxidación sólo sucede en condiciones enérgicas.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 199
Además, para comprender la oxidación de las cetonas debemos estudiar previamente el equilibrio tautomérico o cetoenólico:
Una característica de los compuestos de carbonilo (aldehídos y cetonas) es la marcada acidez de los hidrógenos en los carbonos adyacentes al grupo carbonilo, debido a la polaridad del enlace C-H, por el efecto inductivo generado por dicho grupo. Estos hidrógenos se denominan por lo general hidrógenos α y el carbono al que están unidos es el carbono α.
C C
HO
α
En un compuesto carbonílico, al romperse ese enlace C-H, se separa un protón y el anión que se produce está estabilizado por resonancia.
C C
HO
C C
O
C C
O-
-
Aquí se ve que es posible escribir dos estructuras de resonancia para el anión. En la primera estructura, la carga negativa está sobre el carbono y en la segunda está sobre el oxígeno. El híbrido puede representarse en la siguiente forma:
δ−
δ−
C C
O
Cuando este anión, estabilizado por resonancia, acepta el protón puede hacerlo de la siguiente forma:
o Puede adicionar el protón en el carbono, formando el compuesto carbonilo original. o Puede aceptar el protón en el oxígeno para formar un enol.
C C
HO
C C
O-
H+
H+
C C
OH
Compuesto carbonílico
Alcohol no saturado(enol)
C C
O -
Este equilibrio está muy desplazado hacia la forma carbonílica (izquierda), debido a que en el anión es más probable que sea el carbono el que capte el protón, ya que este elemento es mejor dador de electrones (menos electronegativo) que el oxígeno.
Este equilibrio tautomérico explica la oxidación de las cetonas, ya que el alcohol no saturado es el que sufre la oxidación en el doble enlace carbono-carbono, obteniéndose generalmente mezclas de ácidos carboxílicos (ver oxidación de alquenos) y, en algunos casos, bióxido de carbono.
Ejemplo:
C
O
CH3 CH
HH
Propanona
Equilibrio tautomérico
COH
CH3 C HH
Oxidación enérgica C
OH
CH3 O + CO2
2-propenol
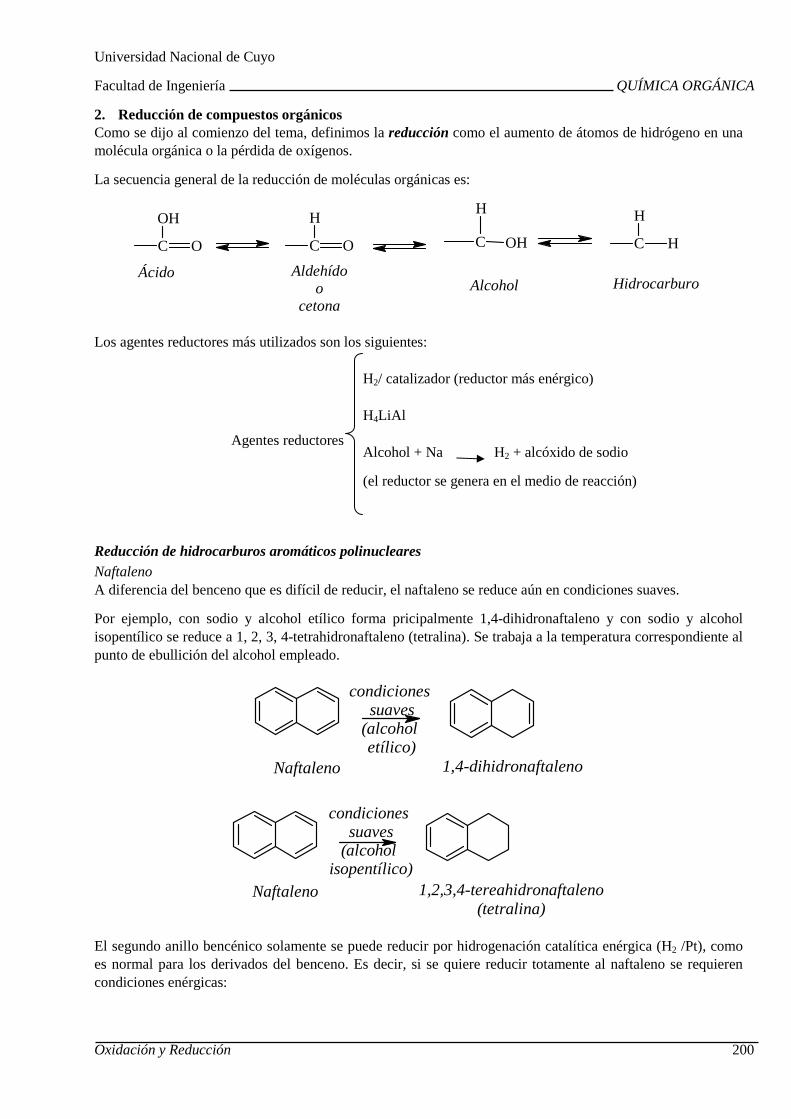
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 200
2. Reducción de compuestos orgánicos Como se dijo al comienzo del tema, definimos la reducción como el aumento de átomos de hidrógeno en una molécula orgánica o la pérdida de oxígenos.
La secuencia general de la reducción de moléculas orgánicas es:
C O
OH
Ácido
C O
H
Aldehídoo
cetona
OHC
H
Alcohol
C H
H
Hidrocarburo
Los agentes reductores más utilizados son los siguientes:
Agentes reductores
H2/ catalizador (reductor más enérgico)
H4LiAl
Alcohol + Na H2 + alcóxido de sodio
(el reductor se genera en el medio de reacción)
Reducción de hidrocarburos aromáticos polinucleares Naftaleno A diferencia del benceno que es difícil de reducir, el naftaleno se reduce aún en condiciones suaves.
Por ejemplo, con sodio y alcohol etílico forma pricipalmente 1,4-dihidronaftaleno y con sodio y alcohol isopentílico se reduce a 1, 2, 3, 4-tetrahidronaftaleno (tetralina). Se trabaja a la temperatura correspondiente al punto de ebullición del alcohol empleado.
condiciones suaves
(alcohol etílico)
Naftaleno 1,4-dihidronaftaleno
condiciones suaves
(alcohol isopentílico)
Naftaleno 1,2,3,4-tereahidronaftaleno(tetralina)
El segundo anillo bencénico solamente se puede reducir por hidrogenación catalítica enérgica (H2 /Pt), como es normal para los derivados del benceno. Es decir, si se quiere reducir totamente al naftaleno se requieren condiciones enérgicas:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Oxidación y Reducción 201
condiciones energéticas
Naftaleno decahidronaftaleno(decalina)
Decalina y tetralina son compuestos que se usan como solventes.
Antraceno En este compuesto se observa una mayor reactividad en el anillo central, debido a que ello permite la formación de productos con dos anillos bencénicos aislados, no condensados, lo que equivale a decir que en el producto la energía de resonancia es de 72 kcal/mol. Si se reduce otro de los anillos, el producto presenta dos anillos condensados con una energía de resonancia de 61 kcal/mol.
condiciones suaves
Antraceno 9,10-dihidroantraceno
Reducción de alquenos y alquinos La reducción de alquenos se realiza con H2 en presencia de catalizador (ver Adición por Radicales Libres).
Los alquinos dan como productos alquenos o alcanos, según se adicionen una o dos moléculas de hidrógeno por molécula de alquino, procediendo la reacción en la misma forma que la reducción de alquenos.
Reducción de nitrocompuestos aromáticos La reducción de nitrocompuestos aromáticos es el método más útil para preparar aminas, puesto que emplea materias primas de fácil acceso y genera el tipo más importante de aminas, las aminas primarias aromáticas (ej.: anilina), las que son muy importantes desde el punto industrial.
Para la reducción del grupo nitro se utilizan metales como el cinc, hierro y estaño, usualmente en presencia de ácido. La producción comercial de anilina a partir de nitrobenceno utiliza chatarra de hierro como reductor.
NO2 NH2
Fe/H+
Debe observarse que la reducción de los grupos nitro por metales puede detenerse en estados intermedios (ej.: nitrosobenceno, fenilhidroxilamina), dependiendo del pH del medio.
Bibliografía:
1. Morrison R., Boyd R. – QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. – QUÍMICA ORGÁNICA – Editorial Limusa.
3. Noller C. – QUÍMICA ORGÁNICA – Editorial Interamericana

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 202
Los polímeros son sustancias de peso molecular muy elevado, es decir, son macromoléculas formadas a partir de pequeñas unidades químicas simples llamadas monómeros. La reacción por la cual los monómeros se unen entre sí se denomina polimerización.
Ejemplo:
polimerización etileno polietileno
(monómero) (polímero) Esta reacción se podría representar por la siguiente ecuación química:
CH2 CH2 CH2 CH2+ ++... ...
CH2 CH2 CH2 CH2... ...
Etileno Polietileno La reacción general para la polimerización en cadena de monómeros de etileno a polietileno puede escribirse como:
C C
H
H
H
H
npolimerización
C C
H
H
H
Hn
unidad repetitiva(unidad estructural)
monómero polímero
grado de polimerización(número de moléculas de monómeros que se combinan para formar la macromolécula)
1. Clasificación de polímeros Los polímeros pueden ser naturales o sintéticos.
1.1. Polímeros naturales También son llamados biológicos e incluyen materiales como caucho, lana, celulosa, almidón, proteínas, alquitrán y resinas.
Todas estas sustancias son muy importantes en la vida diaria, pero tienen dos desventajas fundamentales:
Sus propiedades físicas están subordinadas a la naturaleza del material en particular y normalmente no pueden variarse.
La obtención de estas materias primas está sujeta a la explotación de recursos naturales, los cuales son de lenta reposición.
1.2. Polímeros sintéticos El primer polímero sintético, de la familia de las resinas fenol - formaldehido, fue sintetizado a partir de 1907 y se conoció con el nombre de Baquelita. Este producto se usó para receptores telefónicos, aislantes eléctricos y asas de utensilios de cocina.
Polímeros
Generalidades. Clasificación: Polímeros naturales y sintéticos. Características estructurales. Peso molecular. Polimerización por condensación. Polamidas, poliésteres y poliuretanos. Copolimerización. Polimerización por adición. Polimerización por radicales libres, aniónica y catiónica. Caucho natural. Cauchos sintéticos. Configuración de los polímeros. Propiedades.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 203
Las propiedades físicas de los polímeros sintéticos pueden estructurarse de acuerdo con las necesidades de casi cualquier aplicación. Además, las materias primas para muchos de ellos se extraen de productos derivados del petróleo y/o carbón.
Inicialmente, la química de los polímeros sintéticos se limitaba a sustituir polímeros naturales de importancia comercial, como los casos clásicos ejemplo del descubrimiento del nylon en sustitución de la seda y del caucho sintético para reemplazar al caucho natural. Luego se empezó a diseñar polímeros con características diferentes y más ventajosas que las de los productos naturales a los que debían reemplazar. El avance en el conocimiento de las reacciones de polimerización y en la mayor complejidad de las técnicas empleadas, han hecho posible la fabricación de polímeros con propiedades físicas de gran interés (ej.: polietileno, poliestireno, baquelita, cauchos sintéticos, nylon, PVC, teflón, polipropileno, etc.).
2. Características estructurales de los polímeros
2.1. Esqueleto covalente Como ya dijimos, los polímeros están constituidos por secuencias de unidades repetitivas, unidas entre sí por enlaces covalentes.
2.1.1. Homoplímeros Cuando todas las unidades de monómeros son idénticas, se forma un homopolímero (ej.: polietileno).
nA A A A A A ...
2.1.2. Copolímeros Los copolímeros (ej.: Nylon 6 6) se forman a partir de más de una clase de monómero, siendo posible una gran variedad de ordenamientos.
nA A B B A A A B A A A ...n BH+
copolímero desordenado
nA A B A B A B A B A B ...n BH+
copolímero alternado
nA A A A A A B B B B ...n BH+
copolímero de bloques
2.2. Polímeros lineales y ramificados
2.2.1. Polímeros lineales Los homopolímeros y copolímeros, representados anteriormente, son lineales. Esto implica que tienen un ordenamiento lineal de unidades de monómeros. En un polímero lineal, cada monómero (excepto las unidades terminales) debe formar un enlace en cada uno de sus extremos.
2.2.2. Polímeros ramificados Los polímeros ramificados son copolímeros que se forman con algún monómero con tres o más sitios reactivos, por los que puede unirse a otras unidades mediante enlaces covalentes.
Los enlaces entre cadenas poliméricas, que se conocen como entrecruzamientos, pueden formarse durante el proceso inicial de polimerización o mediante una reacción posterior de las cadenas poliméricas.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 204
En algunos casos, los grupos funcionales de los monómeros forman los entrecruzamientos, mientras que en otros se adicionan moléculas pequeñas para que promuevan y participen en el proceso de entrecruzamiento.
A B B A B B A
A B B A B B A
X
X
polímero ramificado
3. Peso molecular de los polímeros Para determinar el tamaño de un polímero se utiliza el peso molecular del mismo. Para calcularlo realizaremos el siguiente razonamiento:
Si un polímero de etileno contiene 1000 moléculas, el peso molecular del polímero será:
28 x 1000 = 28000
PM del etileno Unidades repetitivas
PM del polímero
Varias propiedades del polímero, tales como resistencia, dureza, elasticidad y solubilidad, son muy afectadas por el peso molecular promedio del polímero.
Para un peso molecular promedio menor que 1000 los polímeros son generalmente líquidos oleoviscosos.
Para pesos moleculares de 1000 a 10000, los polímeros amorfos son sólidos suaves y cerosos. Por encima de 10000, los polímeros amorfos pueden ser sólidos, ya sea duros, quebradizos, blandos
y flexibles, dependiendo de la naturaleza del polímero y del grado de enlaces transversales que contenga.
Ejemplos:
El polietileno de peso molecular aproximado a 1000000 presenta las propiedades físicas que observamos en las botellas y vajillas de este material.
El nylon de peso molecular aproximado de 50000 posee las propiedades físicas de un hilo o hebra.
4. Mecanismos de Polimerización Los mecanismos de polimerización se dividen en dos clases generales:
Polimerización por condensación Polimerización por adición
5. Polimerización por condensación La polimerización por condensación es también llamada Polimerización en Etapas e involucra la reacción de condensación entre dos moléculas polifuncionales, generalmente con la eliminación de una pequeña molécula (ej.: H2O).
Los monómeros reaccionan unos con otros para dar dímeros, trímeros y oligómeros mayores, cada uno de los cuales contiene aún grupos funcionales reactivos. Las cadenas seguirán creciendo y reaccionando con el monómero y entre sí, para formar un polímero de peso molecular creciente, hasta que eventualmente se termine la polimerización cuando uno o todos los grupos reactivos se terminen.
Mientras que en la polimerización por adición cada reacción depende de la anterior, que origina el centro activo, en la polimerización por condensación cada reacción es esencialmente independiente de la precedente.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 205
Si bien la polimerización por condensación involucra generalmente la eliminación de una molécula sencilla, hay casos (ej.: poliuretano) en que el polímero se forma sólo por la adición directa de dos moléculas polifuncionales.
Los grupos reactivos más comunes en los polímeros de condensación son:
C
OH
C O
OH
NH2
Alcohol Ácido carboxílico Amina primaria
N C O C O
Cl
C O
H
Isocianato Cloruro de acilo Aldehído
A continuación se muestran ejemplos de dos tipos de polímeros por condensación, las poliamidas y los poliésteres, que se forman mediante sustituciones nucleofílicas:
Poliamidas Una amina reacciona con un ácido carboxílico o con un haluro de acilo para formar amida:
C OHR
O
+ NH
H
R' CR
O
N
H
R' + OH2
Grupo amidaÁcido Amina
Los polímeros llamados poliamidas se forman haciendo reaccionar una diamina con un diácido. Un ejemplo de este tipo de polímeros es el nylon tipo 6/6, que es el producto de polimerización de hexametilendiamina con ácido adípico (ácido hexanodioico). La reacción de estos compuestos es como sigue:
N
H
H
(CH2)6 NH
H
+
C
O
OH
(CH2)4 CO
OH OH2
N
H
H
(CH2)6 N
H
C
O(CH2)4 C
O
OH
hexametilendiamina(1,6-diaminohexano)
ácido adípico(ácido hexanodioico)
amida
El producto de esta reacción posee en sus extremos dos grupos activos (amino y carboxilo), lo que permite la reacción en un extremo (grupo amino) con una segunda molécula de ácido adípico y por el otro extremo (grupo carboxilo) con una segunda molécula de hexametilendiamina, formándose un nuevo producto que también tiene dos grupos activos. Las sucesivas reacciones llevan a la formación del polímero nylon 66:
N
H
H
(CH2)6 N
H
H+ C
O
OH(CH2)4C
O
OH OH2
N
H
(CH2)6 N
H
CO
(CH2)4 C
O
Nylon 66
n n
n
n
El nombre nylon 6-6 se debe al hecho que ambos reactivos tienen seis carbonos. Es una de las fibras sintéticas más importantes y se usa, entre otras cosas, en ropa, velas de embarcaciones, paracaídas, cepillos, alfombras, cojinetes, etc.
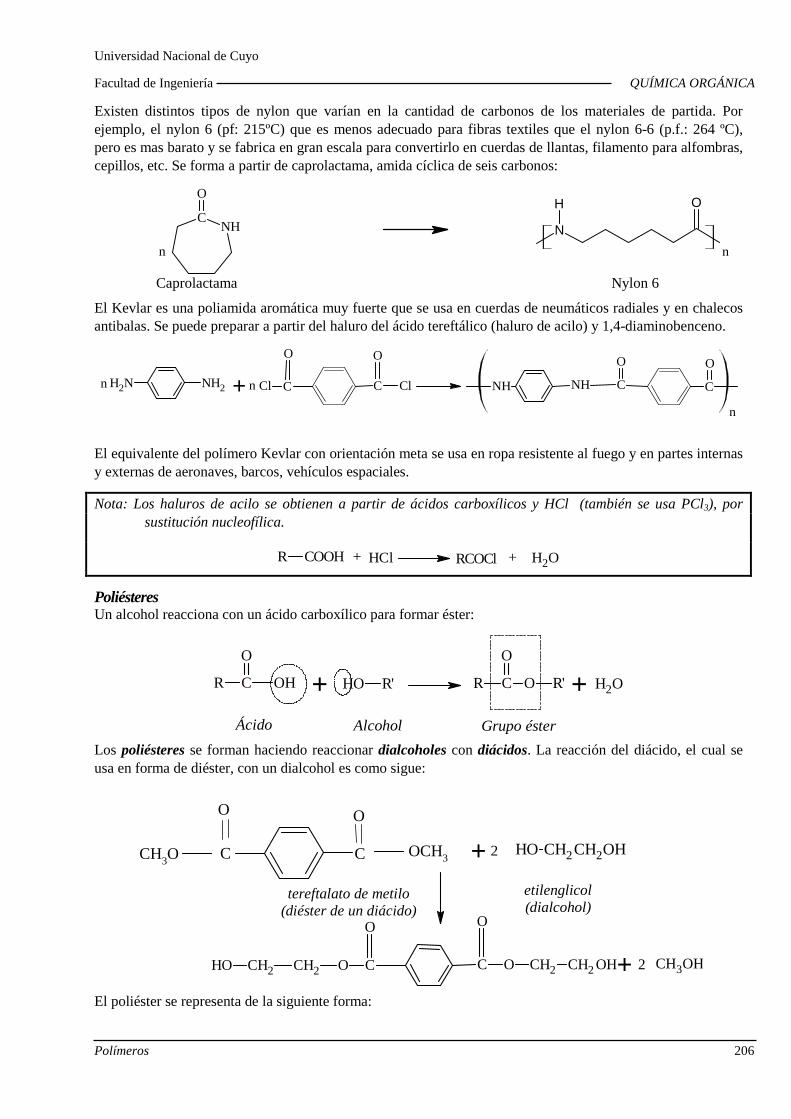
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 206
Existen distintos tipos de nylon que varían en la cantidad de carbonos de los materiales de partida. Por ejemplo, el nylon 6 (pf: 215ºC) que es menos adecuado para fibras textiles que el nylon 6-6 (p.f.: 264 ºC), pero es mas barato y se fabrica en gran escala para convertirlo en cuerdas de llantas, filamento para alfombras, cepillos, etc. Se forma a partir de caprolactama, amida cíclica de seis carbonos:
CNH
O
n
n
N
H O
Caprolactama Nylon 6
El Kevlar es una poliamida aromática muy fuerte que se usa en cuerdas de neumáticos radiales y en chalecos antibalas. Se puede preparar a partir del haluro del ácido tereftálico (haluro de acilo) y 1,4-diaminobenceno.
NH2 NH2n + Cl C
O
C
O
Cln NH NH C
O
C
O
n
El equivalente del polímero Kevlar con orientación meta se usa en ropa resistente al fuego y en partes internas y externas de aeronaves, barcos, vehículos espaciales.
Nota: Los haluros de acilo se obtienen a partir de ácidos carboxílicos y HCl (también se usa PCl3), por sustitución nucleofílica.
R COOH + ClH RCOCl + OH2
Poliésteres Un alcohol reacciona con un ácido carboxílico para formar éster:
+C OHR
O
Ácido
OH R'
Alcohol
CR
O
O R' + OH2
Grupo éster
Los poliésteres se forman haciendo reaccionar dialcoholes con diácidos. La reacción del diácido, el cual se usa en forma de diéster, con un dialcohol es como sigue:
+ OH CH2CH2OH2
C
O
O C
O
O CH2 CH2OHOH CH2 CH2 + 2 CH3OH
tereftalato de metilo(diéster de un diácido)
etilenglicol(dialcohol)
C
O
OCH3C
O
CH3O
El poliéster se representa de la siguiente forma:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 207
C
O
O C
O
O CH2 CH2
n
Las fibras textiles que se conocen como Dacrón y las películas transparentes registradas con el nombre Mylar son poliésteres que se fabrican a partir del éster dimetílico del ácido tereftálico y etilenglicol, como en el ejemplo anterior.
Los envases PET (polietilentereftalato), usados en botellas grandes para gasesosas, se fabrican por polimerización por condensación. Estos envases reciclables generalmente poseen códigos de reciclado de plásticos.
Poliuretanos Un uretano se forma por condensación del grupo isocianato con el oxhidrilo de alcohol, mediante una reacción de adición.
R N C O + O R'H R N C
H
O R'
O
uretano
Si los reactivos son bifuncionales, se forman poliuretanos:
O R'H O HNCO R N C O+ nn R N C
H
O R'
O
NC
H
O
O
n
poliuretano
Un diisocianato usado es el diisocianato de difenilmetano (O=C=N-C6H4-CH2-C6H4-N=C=O .
Los poliuretanos se emplean sobre todo en la fabricación de pinturas, material esponjoso y elastómeros. Sus propiedades mecánicas pueden ser variadas en gran medida por el empleo de diferentes isocianatos o dioles (ej.: polietilenglicol).
6. Polimerización por adición En la polimerización por adición, también llamada de reacción en cadena, el monómero se polimeriza directamente mediante una reacción en cadena, en la cual los activadores son radicales libres, aniones, o cationes.
Las reacciones de este tipo involucran estados sucesivos de iniciación, propagación y terminación que son comunes a las reacciones de cadena en general. En este mecanismo cada etapa depende de la anterior, que es la que origina el centro activo.
La polimerización de alquenos y alquenos sustituidos (ej.: cloruro de vinilo) ocurre por adición. La especie iniciadora se adiciona al alqueno para dar un intermedio muy reactivo (iniciación), el que dispara las rapidísimas adiciones sucesivas de muchas moléculas del alqueno (propagación). La polimerización se interrumpirá cuando una especie reactiva se una al extremo donde la cadena está creciendo o bien este extremo expulse una especie química de pequeño tamaño (terminación).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 208
La combinación de los monómeros se genera por la reacción entre dobles enlaces C=C. La mayor parte de las reacciones de adición son de tipo etilénico. Un enlace C=C se rompe al exponerlo a la acción de un iniciador apropiado (radical libre, catión o anión), produciéndose así la serie de reacciones en cadena que generan finalmente la cadena polimérica. De acuerdo a cuál sea el iniciador, la polimerización será por radicales libres, catiónica o aniónica.
7. Polimerización por Radicales Libres En esta polimerización, el iniciador es un radical libre. Entre los radicales libres que pueden usarse como catalizadores iniciadores de la polimerización, podemos mencionar al peróxido de hidrógeno o agua oxigenada (H2O2) y al peróxido de benzoilo [(C6H5 CO)2 O2], cuyas moléculas pueden romperse homolíticamente dando radicales libres mediante los siguientes procesos:
H O O H calor
+H O O H
Peróxido de hidrógeno Radicales libres
CH5C6 O O C C6H5
O O
calor
COH5C6 O O OC C6H5+
Peróxido de benzoilo Radicales libres
(benzoilo) En general, los radicales libres más comunes están formados por oxígeno y un átomo o un grupo de átomos unido al mismo, que puede ser hidrógeno, alquilo, acilo, etc. (ej: °OH ; °O-R ; Ar-COO°).
El radical libre, que representaremos como R’• se adiciona al alqueno dando un nuevo radical. Las sucesivas adiciones a otras moléculas del alqueno (propagación en cadena) producen un radical polímero.
R' + C C
F
F
F
F
C CR'
H
H
H
H
C C
F
F
F
F
n
C CR'
H
H
H
H
C C C C
H
H
H
H H
H
H
H
n-1
Cadena en desarrollo (radical libre) La propagación termina a través de cualquier reacción en la que se consuman los radicales libres. A continuación se dan algunas alternativas:
Pueden unirse dos cadenas en desarrollo (Aº y Bº):
A B2+
A B3++
Puede separarse un radical libre Hº de una cadena en desarrollo remanente:
C CR'
H
H
H
H
C C C
H
H
H
H H
C
H
Hn-1
Puede unirse un radical libre Hº a una cadena en desarrollo remanente:
C CR'
H
H
H
H
C C C C
H
H
H
H H
H
H
H
H
n-1

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 209
8. Polimerización por adición: Polimerización catiónica La polimerización catiónica se inicia por la adición de un ácido (especie fuertemente electrofílica) al alqueno. Los ácidos empleados frecuentemente para iniciar la polimerización catiónica son: ácido sulfúrico, ácido fluorhídrico, cloruro de aluminio, trifluoruro de boro.
A+ + C C
F
F
F
F
C C+
A
H
H
H
H
C C
F
F
F
FC C
F
F
F
F C CA
H
H
H
H
C C C C+
H
H
H
H H
H
H
H
n-1
C CA
H
H
H
H
C C+
H
H
H
H
n
H+-
C CA
H
H
H
H
C C C
H
H
H
H H
C
H
Hn-1
En esta polimerización, el extremo por el que crece la cadena es un catión.
Para que la polimerización catiónica tenga éxito, el alqueno debe llevar sustituyentes dadores de electrones, de manera que el catión que está aumentando de tamaño sea suficientemente estable como para sobrevivir lo necesario para adicionarse a otra molécula del alqueno, y así sucesivamente hasta generar un catión polímero.
La terminación podrá ocurrir por pérdida de un protón en el extremo donde crecía la cadena.
9. Polimerización por adición: Polimerización aniónica
En esta polimerización el iniciador es un reactivo fuertemente nucleofílico (ej.::NH2-). Su adición al alqueno
origina una carbanión y las sucecivas adiciones a otras moléculas del alqueno dan un anión de peso molecular elevado.
B- + C C
F
F
F
F
C C-
BH2
H
H
H
H
C C
F
F
F
FC C
F
F
F
F C CBH2
H
H
H
H
C C C C
H
H
H
H H
H
H
H
n-1
C CBH2
H
H
H
H
C C-
H
H
H
H
n
H+
C CBH2
H
H
H
H
C C C
H
H
H
H H
C
H H
Hn-1
La polimerización aniónica es útil cuando el alqueno lleva como sustituyentes grupos que estabilizan los carbaniones (grupos aceptores).
Cualquier reacción en la que se destruya el carbanión en el extremo de la cadena, terminará la polimerización.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 210
10. Ejemplos de polímeros por adición
C CA
H
H
H
H
H
n
Para obtener polietileno sólido se realiza la polimerización a 100ºC, en presencia de radicales libres iniciadores. El polímero posee grupos metilo cada 8 a 10 carbonos de la cadena, probablemente por desplazamientos 1,2-durante la polimerización. No obstante, se han desarrollado catalizadores que reducen las ramificaciones, además de permitir que la reacción se realice a presión atmosférica. En este caso se forma un polímero denso y rígido llamado polietileno de alta densidad.
Los polietilenos son termoplásticos, porque se ablandan y fluyen por acción del calor. Además, por ser saturados son inertes y, por sus altos pesos moleculares, son insolubles en la mayoría de los solventes.
Polietileno
Pf: 110-137 ºC
C CA
H
H
H
CH3
H
n
El propileno puede polimerizarse con un ordenamiento al azar (polímero atáctico), con atracciones débiles entre las cadenas. Esto hace que el producto sea aceitoso o semisólido.
En cambio, si se emplean los catalizadores usados en la preparación de polietileno de alta densidad, se puede obtener polipropileno con ordenamiento muy regular (polímero isotáctico). Estos polímeros son productos útiles, que se destinan a diversos fines, como por ejemplo la fabricación de fibras sintéticas, hojas y artículos moldeados.
Polipropileno
Pf: 150-243 ºC
C CA
H
H
H
C6H5
H
n
El poliestireno es un polímero lineal termoplástico, que se prepara por calentamiento de estireno a 85-100ºC, en presencia de peróxido de benzoilo. Es liviano, aislante eléctrico, muy transparente, de gran resistencia.
Cuando el estireno se copolimeriza con divinilbenceno, este último enlaza transversalmente las cadenas, formándose un producto (red tridimensinal) que se usa como lecho de las resinas de intercambio iónico.
CH2 CH CH2 CHH
C6H5
H
CHCH2 CH CH2HH5C6
H
Unidad divenilbenceno
Poliestireno
Pf: 150-243 ºC
Si se sulfona este copolímero y se lo trata con solución de NaOH, muchos grupos sulfonatos sódicos (−SO3Na) quedan unidos a los anillos aromáticos, obteniéndose una resina que se usa para ablandar el agua.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 211
HH SO3 SO3H H+- SO3H Na
+-Na OH
resina
Cuando un agua dura pasa a través de la resina, los cationes del agua (ej.: Ca++, Mg++) son retenidos y los cationes Na+ pasan al agua.
SO3H Na+- + Me
+ SO3H Me+- + Na
+
aguaagua
Ejemplos de otros polímeros por adición:
C CA
F
F
F
F
H
n
C CA
H
H
H
Cl
H
n
C CA
H
H
CH3
COOCH3
H
n
politetrafluoretileno (teflón) policloruro de vinilo (PVC) polimetacrilato de metilo
(plexiglás)
Se ablanda a T > 250°C. Resistente al ataque de todos los
reactivos. Se despolimeriza a 600-800ºC sin carbonizarse.
pf: aprox. 204°C Resina dura y frágil.
pf: 160°C Polímero termoplástico muy
transparente, de alto índice de refracción.
Se usa como sustituto del vidrio. Tanto el caucho natural, como los cauchos artificiales se pueden considerar polímeros por adición. El caucho natural fue probablemente el primer polímero termoestable usado por el hombre. El látex de caucho que se extrae del árbol del hule es principalmente poliisopreno, polímero lineal con enlaces dobles (C=C) en su esqueleto carbonado.
CH C
CH3
CH2 CH2H Hn
isopropeno o metil-1,3 - butadieno (hibrido de resonancia)
Poliisopropeno
n H2C CH C CH2 H2C CH C CH2
CH3CH3
El caucho natural tiende a ser pegajoso cuando está tibio y quebradizo cuando está frío. En 1839 Good Year descubrió que ese látex podía endurecerse mezclándolo con azufre y calentándolo, llamando al proceso vulcanización. De hecho, el caucho vulcanizado no funde.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 212
CH C
CH3
CH2 CH2H H
CH CCH2 CH2H H
CH3
S2-+
CH C
CH3
CH2 CH2H H
SH
S
S
CH CCH2 CH2H H
CH3S
El contenido de azufre puede variar desde 0,5 a 50 partes en peso de caucho. El aumento de azufre produce más entrecruzamientos, resultando un caucho más duro. Con un contenido de azufre de 30 a 50 partes, el producto es un plástico rígido conocido como caucho duro o ebonita.
En años recientes se ha usado el entrecruzamiento con radicales libres para reemplazar el azufre en la vulcanización, lo cual mejora algunas propiedades, en especial la resistencia a la oxidación.
En la Tabla siguiente se dan algunos ejemplos de cauchos sintéticos, todos preparados por adición 1,4 (poliisopreno) o 1,2.
Monómeros Estructura del polímero Nombre
isobutileno / butadieno CH2 C
CH3
CH3
CH2 CH CH CH2 HH
yx
caucho butílico
butadieno / estireno H CH2 CH CH CH2 CH2 CH
C6H5
H
yx
buna S
butadieno / acrilonitrilo H CH2 CH CH CH2 CH2 CH
C
H
Nyx
buna N
cloropreno CH C
Cl
CH2 CH2H Hn
neopreno
La mayoría de los cauchos se usan principalmente en la fabricación de neumáticos. Sin embargo, el neopreno que es más resistente a los aceites, productos químicos, calor y aire que la mayoría, tiene usos especiales, como en mangueras para bombas de combustibles y tuberías en motores de automóviles.
11. Configuración de los polímeros En el polipropileno y en muchos otros polímeros hay posibilidad de distintas configuraciones. Esta situación no se da en el polietileno donde la unidad repetitiva es simétrica, en contraste con la mayoría de los compuestos de vinilo que no son simétricos. Los compuestos polivinílicos tienen la siguiente composición:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 213
C CA
H
H
H
R
H
n
donde R puede ser:
H (ej.: etileno) OH (ej.: alcohol vinílico) Cl (ej.: cloruro de vinilo) CH3 (ej.: propileno) OCOCH3 (ej.: acetato vinílico) C≡N (ej.: acrilonitrilo) (ej.: estireno)
Consideremos el caso del polipropileno:
En la molécula del monómero propileno, los carbonos del doble enlace van unidos a dos hidrógenos en el caso de C1 y a un hidrógeno y un metilo en el caso de C2. Cuando estas moléculas se polimerizan, pueden ocurrir distintos ordenamientos, dando lugar a diferentes estereoisómeros, a saber:
CC
CC
CC
CC
CC
HHH
HCH3
H
H
H
H
CH3H H H H H
CH3 H CH3 CH3 H
Isómero atáctico
Los grupos metilo están distribuidos aleatoriamente a uno u otro lado de la
cadena principal de los átomos de carbono.
CC
CC
CC
CC
CC
HHH
HHH
H
H
H
HH H H H H
CH3 CH3 CH3 CH3 CH3
Isómero isotáctico
Los grupos metilo se encuentran del mismo lado de la cadena principal de los átomos de
carbono.
CC
CC
CC
CC
CC
HHH
HCH3
H
H
CH3
H
HH H H H H
CH3 H CH3 H CH3
Isómero sindiotáctico
Los grupos metilo están ubicados de modo regular y alternado en uno y otro lado de la cadena principal de los átomos de carbono.
Las diferencias en la configuración de las cadenas de polímeros tienen efecto directo sobre sus propiedades y por lo tanto, sobre los usos que se le da al polímero en particular.
12. Propiedades de los polímeros En general, podemos decir que las propiedades físicas de un polímero están determinadas por:
Naturaleza y ordenamiento de las unidades del monómero. Fuerzas intermoleculares más débiles entre cadenas poliméricas (puentes H, Van der Walls,
interacciones electrostáticas). Peso molecular. Configuración.
A continuación se dan las propiedades más importantes de los polímeros:
12.1. Comportamiento frente al calor Los polímeros se suelen clasificar por su comportamiento frente al calor:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 214
12.1.1. Termoplásticos: Estos polímeros al ser calentados se ablandan o funden y, si luego se los enfría, solidifican y recuperan sus propiedades originales.
Por esto, los polímeros de este tipo pueden ser recalentados y reformados varias veces sin sufrir cambios significativos en sus propiedades, incluso pueden reciclarse.
Son básicamente polímeros lineales, que sólo tienen atracciones secundarias (Van der Waals) entre las cadenas y que disminuyen al calentar. Así, cuando se eleva la temperatura, las moléculas pueden responder a la presión, fluyendo unas sobre otras, razón por la que es posible inyectar estos materiales en moldes cuando están calientes, o bien, darles formas de láminas o fibras (ej.: polietileno, vinilos, acrílicos, nylon, etc.).
12.1.2. Termoestables: Estos polímeros se ablandan o funden al ser calentados y, al enfriarse, se convierten en sólidos infusibles, es decir, no vuelven a fundirse al aplicarles calor.
Los termoestables se alteran tanto química como estructuralmente durante el procesamiento térmico (curado). Poseen estructura reticular, por lo que poseen más centros activos, lo que genera polímeros tridimensionales o entrecruzados, con gran número de entrecruzamientos entre cadenas.
Se vuelven rígidos al calentarlos y aplicarles presión (curado), como consecuencia de una polimerización adicional o de entrecruzamiento, con lo que el plástico gana rigidez antes que le quiten el calor y la presión.
La producción de estos polímeros es baja comparada con la de los termoplásticos, ya que requiere un tiempo importante para el curado y, además, el material sobrante no puede ser reciclado porque la reacción de formación de cadenas es irreversible. Sin embargo, estos polímeros tienen ventajas cuando se utilizan en productos en los que las temperaturas de servicios son elevadas (ej.: epóxidos, fenólicos, poliésteres insaturados, etc.)
12.2. Otras propiedades de los polímeros A continuación veremos, a modo de información, otras propiedades de los polímeros.
12.2.1. Cristalinidad Un polímero cristalino es el que tiene una estructura ordenada, tanto en las cadenas individuales como en la configuración tridimensional de las mismas. En general, las regiones cristalinas son escasas y ningún polímero es completamente cristalino, ya que entre los cristalitos (largos segmentos poliméricos lineales orientadas de manera regular unos respecto a otros) se extienden regiones no cristalinas amorfas, que constituyen defectos en la estructura cristalina.
Cuando un polímero no contiene grupos laterales grandes, las cadenas quedan cercanas entre sí y las fuerzas de atracción son más intensas, con lo que las cadenas orientadas quedan fijas en sus posiciones, o sea, cristalizadas. Esto explica que los polímeros lineales puedan cristalizar con más facilidad que los ramificados.
La cristalinidad tiende a aumentar la dureza, resistencia y fragilidad del polímero.
Las fibras sintéticas como el nylon son polímeros característicos, con una cristalinidad del 90% o más, mientras que el polietileno lineal tiene una cristalinidad de aproximadamente el 90%.
12.2.2. Punto de fusión El punto de fusión de un polímero cristalino se define como la temperatura a la que desaparece toda la cristalinidad. Está determinado principalmente por la flexibilidad de la cadena y sus fuerzas internas, por ser éstas causas las que determinan la facilidad de la separación de cadenas en las regiones cristalinas. El peso molecular, salvo si es muy bajo, influye poco en el punto de fusión.
Los polímeros amorfos y muchos plásticos terminados no muestran puntos de fusión definidos. Cuando estos materiales se calientan, se alcanza una temperatura a la cual la energía térmica de las moléculas es suficiente

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 215
para originar una descomposición de la cadena del polímero. Esta temperatura se denomina temperatura de degradación.
Existen dos tipos de degradaciones que pueden darse individual o simultáneamente:
Degradación fortuita La ruptura de la cadena es al azar, obteniéndose una mezcla de productos de degradación.
Depolimerización de la cadena. La ruptura se sucede mediante el desprendimiento de unidades sucesivas del monómero desde uno de los extremos de la cadena.
En general, la temperatura de fusión aumenta con el entrelazamiento.
12.2.3. Transición vítrea La temperatura a la cual el material se transforma en suave y flexible se conoce como temperatura de transición vítrea. En general, la cristalinidad, la unión transversal y la adición de grupos rígidos, aumentarán dicha temperatura.
A bajas temperaturas los polímeros amorfos son duros, vidriosos y frágiles. Esto se debe a que la energía térmica es muy baja y los átomos de las cadenas sólo pueden vibrar con respecto a sus posiciones de equilibrio. Las cadenas individuales se congelan en su posición y el polímero se vuelve duro y quebradizo.
A medida que aumenta la temperatura, el aumento de temperatura térmica permite mayores movimientos de algunos segmentos del polímero y el material se vuelve flexible y menos frágil.
A temperaturas aún más altas, hay suficiente energía como para permitir un movimiento libre de las cadenas, siendo el material elástico.
12.2.4. Transparencia Esta propiedad es función de la cristalinidad. Los polímeros no cristalinos (ej.: poliestireno, polimetacrilato de metilo, etc.) tienen excelente transparencia. Otros, van desde aspecto lechoso hasta opacos (ej.: el teflón, que tiene una cristalinidad del 60%, es muy opaco).
12.2.5. Solubilidad Esta propiedad es importante debido a que la mayoría de las polimerizaciones ocurren en solución. La solubilización de un polímero generalmente es un proceso lento que ocurre en dos etapas:
a) El solvente se mezcla gradualmente con el polímero, para producir un gel esponjoso.
b) El gel se desintegra gradualmente para formar una solución verdadera.
o En general, la solubilidad se favorece por similitudes físicas y químicas entre polímero y solvente. Así, los polímeros polares requieren solventes polares. o La solubilidad disminuye con el aumento del peso molecular del polímero. o Los polímeros cristalinos son casi insolubles y se ha determinado que la solubilidad es menor cuanto más alto es el punto de fusión cristalino. o Generalmente, los polímeros que poseen muchas uniones transversales son insolubles o poco solubles. o En el caso de polímeros cristalinos y con uniones transversales, sólo ocurre el primer paso de la solubilización, sin llegarse a la solución verdadera.
12.2.6. Inflamabilidad Las estructuras que producen residuos carbonosos (ej.: aromáticos de enlaces múltiples) reducen la inflamabilidad. La presencia de oxígeno e hidrógeno, la aumentan, mientras que cloro, flúor, bromo y fósforo la disminuyen.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Polímeros 216
Los plásticos entrecruzados son más resistentes a la llama que los termoplásticos similares, porque se requiere más energía para desintegrarlos.
12.2.7. Absorción de humedad Carbono, hidrógeno y flúor determinan bajas absorciones de humedad, mientras que el oxígeno y el cloro aumentan la absorción ligeramente y con nitrógeno el aumento es significativo.
12.2.8. Resistencia a la intemperie Varios factores influyen en esta propiedad, tales como: radiación UV, agua, ozono, variaciones de temperatura.
La luz UV produce efectos muy pronunciados en los polímeros. La degradación del material se manifiesta de varias formas (ej.: fragilidad, cuarteadoras, manchas blancas superficiales, etc.).
Los polímeros saturados son más resistentes al ataque de radiación UV y de ozono. Cuando los polímeros absorben esa radiación en la superficie, aparecen manchas blancas. Si penetra la radiación, el polímero se degrada por debajo de la superficie y se observan fragilidad, cuarteaduras, oscurecimientos, etc.
Para aumentar la resistencia a la intemperie, se pueden usar cargas y absorbentes UV.
12.2.9. Elasticidad Los elastómeros son aquellos polímeros que pueden estirarse rápidamente bajo tensión hasta alcanzar un tamaño de varias veces el tamaño original, con una pequeña pérdida de energía como calor (ej.: poliuretanos).
Todos los elastómeros deben tener alta resistencia a la tensión, se deben retraer con rapidez a sus dimensiones originales y su deformación, una vez retirada la tensión, debe ser pequeña.
Los elastómeros son polímeros amorfos, con uniones transversales para evitar una movilidad excesiva de las cadenas lineales. Sin embargo, los segmentos individuales de las cadenas deben tener suficiente movilidad para permitir la extensión y contracción de las cadenas, sin un cambio permanente en las dimensiones del material.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana
2. Noller C. - QUÍMICA ORGÁNICA - Editorial Interamericana.
3. Mayer L. - MÉTODOS DE LA INDUSTRIA QUÍMICA - Parte 2da: Orgánica - Editorial Reverté
4. SolomonsT. - QUÍMICA ORGÁNICA - Editorial Limusa.

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 217
Grasas y Aceites Definición. Estructura. Ácidos grasos omega. Grasas trans.
Jabones Hidrólisis de las grasas. Acción limpiadora de los jabones. Desventajas de los jabones.
Detergentes Estructura. Clasificación. Ventajas de los detergentes.
1. Grasas y aceites
1.1. Definición. Estructura Una grasa es una mezcla de triglicéridos, estos son compuestos en los que cada uno de los tres grupos OH de la glicerina forma un éster con un ácido graso. Por lo tanto, desde el punto de vista químico, las grasas son ésteres de la glicerina y ácidos grasos superiores.
CH2
CH
CH2
O
O
O
C
C
C
R
R'
R''
O
O
O
Los tres ácidos grasos que componen el triglicérido pueden ser iguales o diferentes. Los ácidos grasos pueden ser saturados o insaturados, los que tienen más de un doble enlace se llaman poliinsaturados. Generalmente, son cadenas lineales de entre 5 y 18 átomos de carbono.
Los triglicéridos que son sólidos o semisólidos a temperatura ambiente se llaman grasas. La mayor parte de las grasas se obtiene de animales y está formada sobre todo por triglicéridos largos compuestos por ácidos grasos que son saturados o que solo cuentan con un doble enlace.
Ejemplo:
CH3 (CH2)14 COOH
CH3 (CH2)16 COOH
Ácido palmítico Ácido esteárico
Las colas de los ácidos grasos saturados se ajustan bien entre sí y por ello los triglicéridos tienen puntos de fusión más altos, lo que los hace ser sólidos a temperatura ambiente.
Los triglicéridos líquidos a temperatura ambiente se llaman aceites. Provienen de productos vegetales como maíz, soja, aceitunas. Se componen principalmente de ácidos grasos insaturados, por ello presentan puntos de fusión bajos y son líquidos a temperatura ambiente.
Ejemplo:
CH3 (CH2)7 CH CH (CH2)7 COOH
Ácido oleico
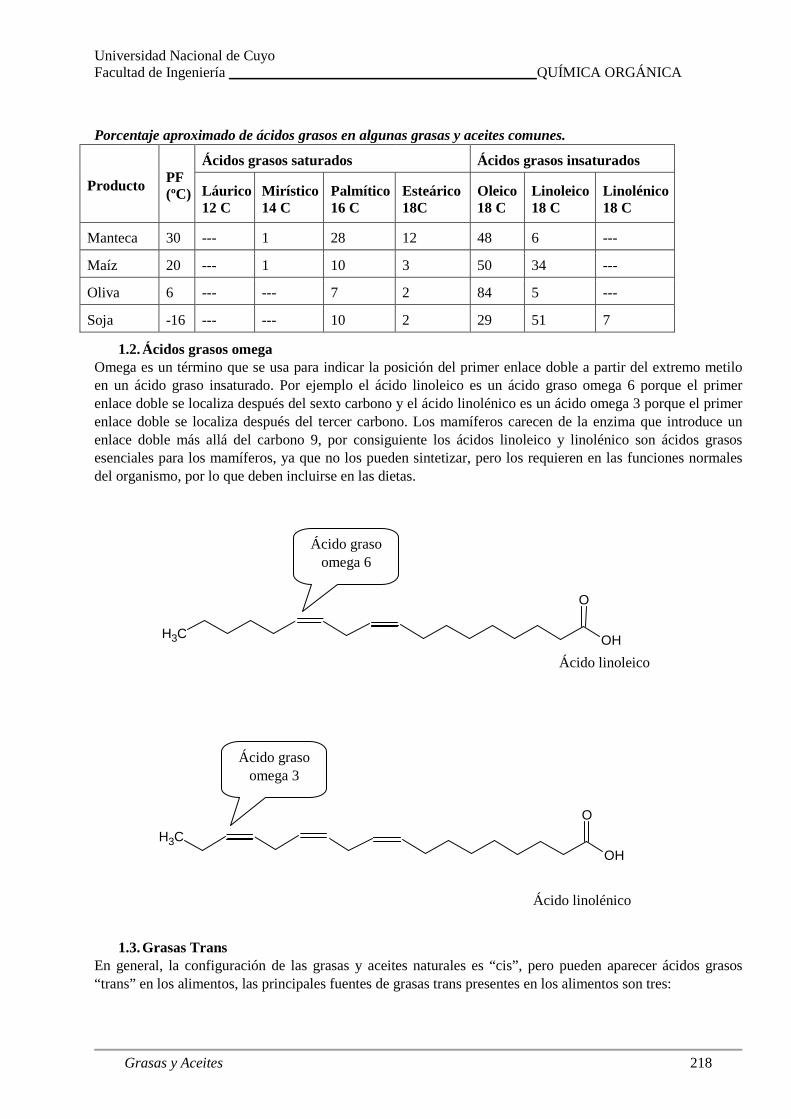
Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 218
Porcentaje aproximado de ácidos grasos en algunas grasas y aceites comunes.
Producto PF (ºC)
Ácidos grasos saturados Ácidos grasos insaturados
Láurico 12 C
Mirístico 14 C
Palmítico 16 C
Esteárico 18C
Oleico 18 C
Linoleico 18 C
Linolénico 18 C
Manteca 30 --- 1 28 12 48 6 ---
Maíz 20 --- 1 10 3 50 34 ---
Oliva 6 --- --- 7 2 84 5 ---
Soja -16 --- --- 10 2 29 51 7
1.2. Ácidos grasos omega Omega es un término que se usa para indicar la posición del primer enlace doble a partir del extremo metilo en un ácido graso insaturado. Por ejemplo el ácido linoleico es un ácido graso omega 6 porque el primer enlace doble se localiza después del sexto carbono y el ácido linolénico es un ácido omega 3 porque el primer enlace doble se localiza después del tercer carbono. Los mamíferos carecen de la enzima que introduce un enlace doble más allá del carbono 9, por consiguiente los ácidos linoleico y linolénico son ácidos grasos esenciales para los mamíferos, ya que no los pueden sintetizar, pero los requieren en las funciones normales del organismo, por lo que deben incluirse en las dietas.
CH3
O
OH
O
OHCH3
1.3. Grasas Trans En general, la configuración de las grasas y aceites naturales es “cis”, pero pueden aparecer ácidos grasos “trans” en los alimentos, las principales fuentes de grasas trans presentes en los alimentos son tres:
Ácido linoleico
Ácido graso omega 6
Ácido linolénico
Ácido graso omega 3

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 219
Se encuentran en pequeñas cantidades en la carne y productos lácteos enteros de animales rumiantes, como vacas y ovejas, generándose a causa de la acción de ciertas bacterias contenidas en su estómago. Sin embargo, esta situación no es la que preocupa a los especialistas.
Se obtienen mediante calentamiento y cocción de aceites a altas temperaturas. Son producidas por proceso de hidrogenación industrial o solidificación de aceites para su uso en
margarinas y grasas para pastelería.
Este último procedimiento tiene la finalidad de lograr la obtención de grasas vegetales sólidas que dan frescura y mejor textura a los alimentos industrializados; adicionalmente, permite que las grasas no se tornen rancias y con ello se incrementa la estabilidad del sabor y fecha de caducidad de productos como manteca vegetal, margarina, pan, pasteles, galletas y aderezos para ensalada. También se utilizan en la preparación de comida rápida, como hamburguesas, panchos y papas y pollo fritos.
Antes del año 2000 se consideraba más saludable el consumo de margarina que de manteca de origen animal, porque el consumo de grasas saturadas provenientes de alimentos de origen animal eleva los niveles de colesterol LDL (lipoproteína de baja densidad), que comúnmente se llama colesterol “malo” pues transporta mayor cantidad de colesterol en sangre y, en exceso, se acumula en las paredes de las arterias y las endurece. Además, disminuye el colesterol HDL (lipoproteína de alta densidad) comúnmente conocido como colesterol “bueno”, ya que ayuda a que el LDL no se acumule en exceso, recogiéndolo de las paredes de las arterias y llevándolo de vuelta a hígado para su metabolismo. Los niveles elevados de colesterol LDL y de HDL bajos, promueven cambios en las arterias y en el corazón, con consecuente desarrollo de enfermedades del corazón, infartos y derrames cerebrales.
A partir del 2001, una gran cantidad de estudios reveló que tanto las grasas saturadas como las trans incrementan las concentraciones de colesterol ‘malo’ y reducen los niveles del ‘bueno’; en consecuencia, aumenta el riesgo de padecer enfermedades cardiacas.
2. Jabones
2.1. Hidrólisis de las grasas Las grasas, por ser ésteres, pueden ser hidrolizadas, la hidrólisis alcalina produce glicerina y sales de ácidos carboxílicos:
CH2
CH
CH2
O
O
O
C
C
C
R
R'
R''
O
O
O
+ Na OH3
CH2
CH
CH2
O
O
O
H
H
H
+
O-
C R
O
O-
C R''
O
O-
C R'
O
Na+
Na+
Na+
Cuando las sales de ácidos carboxílicos que se forman poseen de 10 a 18 carbonos se usan como jabones y la reacción es denominada reacción de saponificación.
Un jabón es una mezcla de sales sódicas o potásicas, de ácidos de cadenas largas, formada por hidrólisis alcalina de una grasa sólida o líquida.
Cada grasa se caracteriza por poseer un índice de saponificación, que son los miligramos de hidróxido de potasio necesarios para saponificar un gramo de grasa.
Supongamos que el peso de la grasa sea de A gramos y se requieran B mg de KOH para saponificarla. Calculamos el índice de saponificación como sigue:

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 220
2.2. Acción limpiadora de los jabones Una molécula de jabón está constituida por dos partes:
Grupo hidrófilo: Grupo carboxilato que es polar y soluble en agua. Grupo hidrófobo: Grupo R – que no es polar y es insoluble en agua.
Ejemplos:
CH3 (CH2)14 COO:- Na+
Palmitato de sodio
CH3 (CH2)7 CH CH (CH2)7 COO:- K+
Oleato de potasio
Al poseer estas dos fracciones, el jabón ejerce su acción limpiadora de la siguiente manera: 1º. En medio acuoso, las moléculas de jabón se congregan en micelas (similares a esferas), pudiendo
contener cada una centenares de moléculas de jabón.
Por la afinidad con el agua, la cabeza (grupo iónico) se ubica en la superficie de la micela, mientras que la cola (cadenas hidrocarbonadas), se reúnen en el centro. Los cationes Na+ están disueltos en el agua (ión-dipolo) pero no participan en la acción del jabón.
2º. Las micelas se rechazan entre sí, debido a las cargas negativas (por ej: -COO-) de sus superficies. 3º. Las sustancias no polares que componen la suciedad, como ser grasas y aceites, son afines a la
fracción hidrófoba del jabón (interior de la micela), lo que hace que se ubiquen en el interior de las micelas. Se forma así una emulsión de grasas y aceite en agua, separándose de esta manera la suciedad de la superficie que se está limpiando.

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 221
2.3. Desventajas de los jabones Los jabones forman sales insolubles con los iones calcio y magnesio, que están presentes en las aguas
duras. Por esta razón el jabón en este tipo de aguas es menos efectivo como agente limpiador.
Ejemplo:
Na+ + Ca
2+Ca
2+
2+ Na
+
jabónión en agua
dura sal de calcio
Soluble en agua Insoluble en agua
22 H3C-(CH2)14-COO:-
2 H3C-(CH2)14-COO:-
3. – Detergentes
3.1. - Estructura Igual que los jabones, los detergentes tienen una cadena saturada no polar muy larga (grupo alquilo) que es la fracción hidrófoba y, además, un extremo polar soluble en agua.
Ejemplos:
Sales de sulfatos ácidos alquílicos provenientes de alcoholes de 12 a 18 carbonos:
H23C11CH2 O HHOSO3H
OH2OH2
Na OHH23C11CH2 O SO3H H23C11CH2 O SO3
-Na
+
Hidrófoba Hidrófila
Nota: Es conveniente que –R no sea muy ramificado porque impide la biodegradabilidad del detergente.
La biodegradabilidad o degradación por acción de microorganismos es esencial para evitar la presencia de detergentes en masas de aguas naturales (ríos, lagos, etc.) o en plantas de tratamiento de agua (ej.: plantas potabilizadoras, plantas de tratamiento de líquidos cloacales o industriales).
3.2. – Clasificación de los detergentes Se los suele clasificar de acuerdo a las características químicas:
Detergentes Características Ejemplo
Neutros Sus moléculas son polares no
iónicas
H17C8 O CH2 CH2O H
n
Octil fenol polietoxilado
Aniónicos El extremo polar tiene
carga negativa Lauril sulfato de sodio
OS
O-Na
+
OO

Universidad Nacional de Cuyo Facultad de Ingeniería QUÍMICA ORGÁNICA
Grasas y Aceites 222
Na+
CH3 (CH2)11 S
O
O
O-
Dodecil benceno sulfonato de sodio
Catiónicos El extremo polar tiene
carga positiva
N+
CH3
CH3R
R
Cl-
Cloruro de dimetil dialquil amonio
3.3. – Ventajas de los detergentes La mayoría de los alquilbencenosulfonatos y alquilsulfatos metálicos son solubles en agua, por lo que
se pueden usar sin inconvenientes en aguas duras. Los alquilbencenosulfonatos y alquilsulfatos metálicos, que son sales de ácidos fuertes, dan
soluciones neutras. Los jabones, por ser sales de ácidos débiles, dan soluciones ligeramente alcalinas.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA – Editorial Limusa.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Aminoácidos y proteinas 223
1. Aminoácidos
1.1. Definición. Estructura. Los aminoácidos son los productos finales de la hidrólisis de las proteínas. Son estructuras que contienen uno o más grupos amino (-NH2) y uno o más grupos carboxilo (-COOH).
En general, podemos representar un aminoácido como sigue:
CR COOH
NH2
H
El grupo -R puede ser de distinta naturaleza química (ej.: un grupo fenólico, un grupo bencénico, un grupo heterocíclico, etc.).
El grupo amino de los aminoácidos se ubica en el Cα , que es el carbono adyacente al grupo carboxilo. Sólo en dos aminoácidos, prolina e hidroxiprolina, esto no se cumple.
Ejemplos:
CH2 COOH
NH2
CH3 CH COOH
NH2
CH2 CHCOOH
NH2
HOOC Glicina Alanina Ácido aspártico
1.2. Aminoácidos naturales y esenciales Existen 26 aminoácidos naturales, los cuales han sido encontrados en las proteinas, de los cuales diez se denominan aminoácidos esenciales, no porque sean los únicos necesarios para el funcionamiento de la especie, sino porque se hace indispensable ingerirlos a través de la dieta porque nuestras células no pueden sintetizarlos. El resto de los aminoácidos pueden ser sintetizados en nuestras células a partir de compuestos sencillos que contengan carbono, oxígeno, hidrógeno y nitrógeno.
1.3. Configuración de los aminoácidos En todos los α-aminoácidos, excepto la glicina, el Cα es centro quiral, lo cual implica que todo aminoácido es ópticamente activo.
CR COOH
NH2
Hcentro quiral
Los estudios estereoquímicos de estos aminoácidos naturales han demostrado que todos tienen la misma configuración en el carbono Cα que el L-(-) gliceraldehido.
COOH
H
R
H2N
CHO
H
CH2OH
HO
L-Aminoácido L-Gliceraldehido
Aminoácidos y proteínas
Estructura de aminoácidos. Propiedades ácido-base. Punto isoeléctrico. Unión peptídica. Péptidos y proteínas. Importancia.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Aminoácidos y proteinas 224
Por analogía con la nomenclatura ordinaria de los carbohidratos se suele decir que pertenecen a la familia L, pero en el caso de los α-aminoácidos, el centro quiral de menor orden es el que determina la familia, mientras que en el caso de los carbohidratos es el centro quiral de mayor orden el que determina la familia.
1.4. Propiedades físicas y químicas Son sólidos cristalinos no volátiles. Son insolubles en solventes no polares (ej.: benceno, éter, etc.), mientras que son apreciablemente
solubles en agua. Tienen elevado momento dipolar. Como los aminoácidos contienen potencialmente grupos amino y carboxilo libres, son electrolitos
anfóteros.
El aminoácido sólido es una sal interna o ion dipolar o switerión.
CR COO
NH2
H
CR COO
NH3+
H H
Observar que el H+ del grupo carboxilo (carácter ácido) es captado por el grupo amino (carácter básico). En solución acuosa se establece un equilibrio:
CHRNH2 COO H
+
OH-
CHRNH3
+COO
H+
OH-
CHRNH3
+COOH
(II) (I) (III) El aminoácido sólido es una sal interna I (ión dipolar o switerión). Cuando la solución es alcalina, hay más aniones II que cationes III (equilibrio desplazado hacia la
izquierda). En soluciones ácidas, en cambio, se encuentran más cationes III (equilibrio desplazado hacia la
derecha).
Por lo tanto, cuando una solución de aminoácido se coloca en un campo eléctrico, la migración del mismo hacia uno u otro electrodo dependerá del pH de la solución.
En solución alcalina la migración se producirá hacia el ánodo (el aminoácido está como anión: II). En solución ácida habrá migración neta del aminoácido hacia el cátodo (el aminoácido está como
catión: III). Si II y III se equilibran exactamente, no hay migración neta, ya que cualquier movimiento de la
molécula como ión positivo hacia el cátodo, será anulado por un movimiento igual y contrario de iones negativos hacia el ánodo.
El pH de la solución para la cual un aminoácido no migra en un campo eléctrico se conoce como punto isoeléctrico de dicho aminoácido.
1.5. Clasificación de los aminoácidos De acuerdo a que el número de grupos básicos sea igual, mayor o menor que el de grupos acídicos, los aminoácidos pueden ser:
* neutros
* básicos
* ácidos
Ejemplos:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Aminoácidos y proteinas 225
CH2 COO
NH3+
CH COONH3
+CH2CH2HOOC
CH COO
NH3+
CH2CH2CH2CH2NH3+
Glicina (neutro)
Ácido glutámico (ácido)
Lisina (básico)
2. Proteinas Un péptido es la unión de varios aminoácidos por medio de enlaces peptídicos. Según sea el número de las unidades de aminoácidos por molécula, se los conoce como dipétidos,
tripéptidos, etc, hasta llegar a los polipéptidos. Convencionalmente, se consideran como polipéptidos los que tienen pesos moleculares de hasta
10000. Proteinas son los polipéptidos cuyos pesos moleculares son superiores a 10000.
2.1. Unión peptídica La unión peptídica es una unión de tipo amida entre el grupo carboxilo de un aminoácido y el grupo α-amino de otro aminoácido, con pérdida de una molécula de agua.
CHR
NH2
CO
OH + CHNH
R
CO
OHHOH2
CHR
NH2
C
O
NH CH
R
CO
OH
unión peptídica (grupo amida)
aminoácido dipéptidoaminoácido
2.2. Estructura de las proteínas La estructura de las proteínas es una determinada organización de sus elementos constitutivos, los aminoácidos. Podemos considerar a la estructura de las proteínas en varios niveles: primaria, secundaria, terciaria y cuaternaria.
La estructura primaria es la secuencia particular de aminoácidos unidos entre sí por enlaces peptídicos. Esta estructura, característica de cada proteina, tiene relación directa con su actividad fisiológica.
La estructura secundaria es la disposición de la secuencia de aminoácidos en el espacio. Los aminoácidos, a medida que van siendo enlazados durante la síntesis de proteínas adquieren una disposición espacial estable (ej.: helicoidal).
La estructura terciaria informa sobre la disposición de la estructura secundaria de un polipéptido al plegarse sobre sí misma originando una conformación globular, que es la que facilita la solubilidad en agua y así realizar funciones de transporte, enzimáticas, hormonales, etc. Esta conformación globular se mantiene estable gracias a la existencia de enlaces entre los grupos unidos a las cadenas de polipéptidos (ej.: puente disulfuro –S-S- , puentes hidrógeno).
La estructura cuaternaria es más compleja. Se describe como muchas subunidades, no siempre idénticas, que se reunen para formar complejos más grandes. No todas las proteínas poseen esta estructura. Informa de la unión, mediante enlaces débiles no covalentes de varias cadenas polipeptídicas con estructura terciaria, para formar un complejo proteico.
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA – Editorial Limusa.
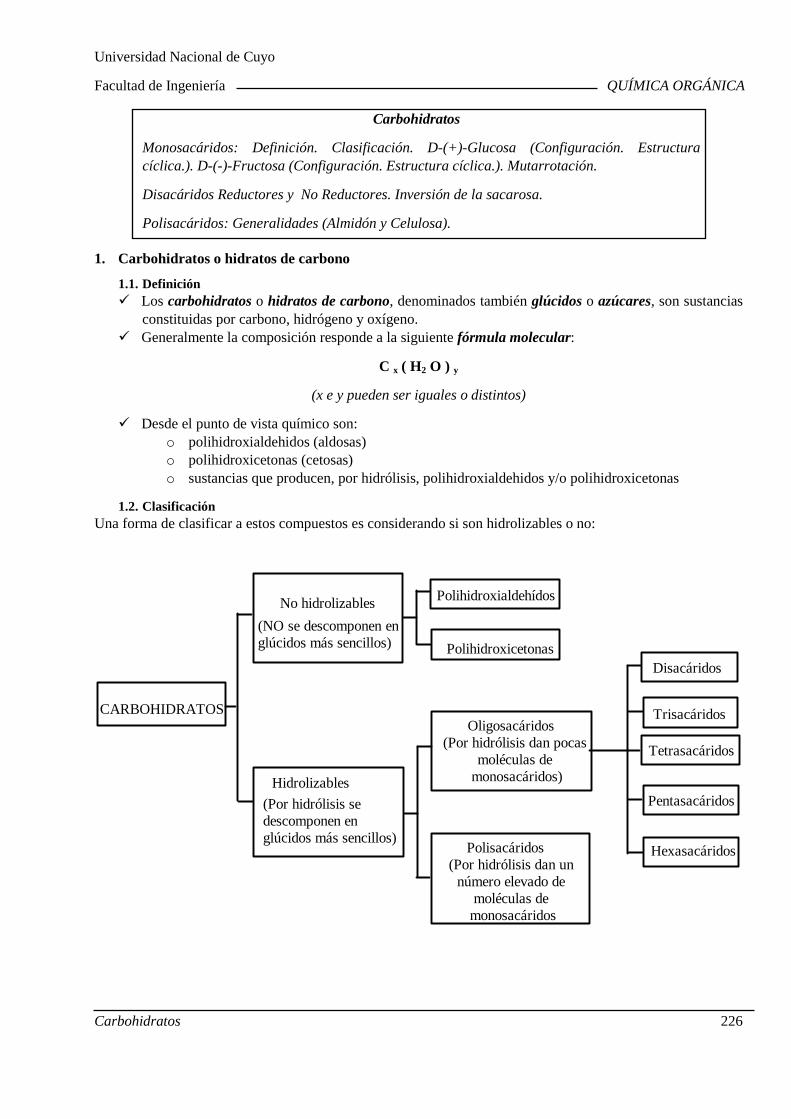
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
226
1. Carbohidratos o hidratos de carbono
1.1. Definición Los carbohidratos o hidratos de carbono, denominados también glúcidos o azúcares, son sustancias
constituidas por carbono, hidrógeno y oxígeno. Generalmente la composición responde a la siguiente fórmula molecular:
C x ( H2 O ) y
(x e y pueden ser iguales o distintos)
Desde el punto de vista químico son: o polihidroxialdehidos (aldosas) o polihidroxicetonas (cetosas) o sustancias que producen, por hidrólisis, polihidroxialdehidos y/o polihidroxicetonas
1.2. Clasificación Una forma de clasificar a estos compuestos es considerando si son hidrolizables o no:
CARBOHIDRATOS
No hidrolizables
(NO se descomponen en glúcidos más sencillos)
Hidrolizables
(Por hidrólisis se descomponen en glúcidos más sencillos)
Polihidroxialdehídos
Polihidroxicetonas
Oligosacáridos (Por hidrólisis dan pocas
moléculas de monosacáridos)
Polisacáridos (Por hidrólisis dan un
número elevado de moléculas de monosacáridos
Disacáridos
Trisacáridos
Tetrasacáridos
Pentasacáridos
Hexasacáridos
Carbohidratos
Monosacáridos: Definición. Clasificación. D-(+)-Glucosa (Configuración. Estructura cíclica.). D-(-)-Fructosa (Configuración. Estructura cíclica.). Mutarrotación.
Disacáridos Reductores y No Reductores. Inversión de la sacarosa.
Polisacáridos: Generalidades (Almidón y Celulosa).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
227
1.2.1. Monosacáridos
1.2.1.1. Definición. Estructura. Los monosacáridos son carbohidratos no hidrolizables. Los monosacáridos son polihidroxialdehidos o polihidroxicetonas. Según contengan grupo aldehido o cetona se los clasifica en:
o Aldosas o Cetosas
Los monosacáridos pueden contener de tres a siete átomos de carbono, y se los denomina triosas, tetrosas, pentosas, hexosas y heptosas, respectivamente.
Ejemplos:
Cantidad de carbonos
Carbohidrato con grupo aldehido
Carbohidrato con grupo cetona
3
CHO
CHOH
CH2OH
CH2OH
C
CH2OH
O
aldotriosa cetotriosa
4
CHO
CHOH
CH2OH
CHOH
CH2OH
C
CHOH
O
CH2OH
Aldotetrosa cetotretosa
1.2.1.2. Propiedades químicas de los monosacáridos a) Oxidación con Reactivo de Fehling o de Tollens
Esta reacción se produce por la oxidación de un grupo aldehído libre en las moléculas de monosacáridos.
CH O
CHOH
CH2OH
Cu++ Fehling
o Ag+ Tollens
COOH
CHOH
CH2OH
Aldosa Ácido aldónico (ej.: ácido glucónico)
A pesar que las cetonas no reducen a dichos reactivos, las cetosas sí lo hacen, por lo que este ensayo no sirve para diferenciar aldosas de cetosas.
Se ha comprobado que las cetosas pueden reducir Fehling o Tollens debido a que, mediante un equilibrio tauomérico, pueden transformarse en aldosas.
b) Equilibrio tautomérico
Recordemos que los aldehídos y las cetonas están en equilibrio con alcoholes no saturados:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
228
C
C
H
F
F
OF
C
C
F
F
F OH
En aldosas y cetosas se da una situación similar. Si consideramos la aldosa o la cetosa, estará en equilibrio tautomérico con un alcohol no saturado (enodiol). Este alcohol, al poseer oxhidrilos unidos a los dos carbonos, estará en equilibrio con ambos carbohidratos.
A continuación se representa el equilibrio, indicándose con flechas los desplazamientos de electrones y H+ , para la transformación desde aldosa y cetosa a enodiol.
C
C
H O
H OH
F
C
C
F
OH
O
H
H
C OH H
C O
H
F
Aldosa Enodiol Cetosa
Este equilibrio explica por qué las cetosas, a pesar de no tener grupo aldehido, reducen los reactivos de Fehling y Tollens.
1.2.1.3. Glucosa (azúcar de uva) La glucosa es un monosacárido, cuya fórmula molecular posee seis átomos de carbono. Además, es
un polihidroxialdehido, por lo que podemos decir que este carbohidrato es una aldohexosa. Fórmula molecular: C6 H12 O6 Fórmula estructural:
CHO
CHOH
CHOH
CHOH
CHOH
CH2OH
Configuración:
Podemos observar que C2 , C3 , C4 y C5 son centros quirales. Por consiguiente habrían 16 estereoisómeros que responden a esa estructura (recordemos que el número máximo de isómeros es 2 n, donde n es el número de centros quirales).
A la glucosa dextrógira, por una serie de estudios realizados, se le ha asignado la siguiente configuración:

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
229
C
C
C
CC
CH2OH
OH
H
H
H
H
OH
OH
OH
OH
(+) glucosa
Esta representación se denomina representación en cruz (Representación de Fisher), donde se considera que los enlaces horizontales de cada centro quiral estarían hacia adelante del plano, mientras que los verticales estarían hacia atrás (recordemos que cada centro quiral está en el centro de un tetraedro).
El enantiómero de la (+) glucosa es la (-) glucosa, imagen especular de la primera:
CCC
CC
CH2OH
OH
OHOH
OHOH
HH
HH
(-) glucosa
1.2.1.4. - Familia D y familia L Para asignar la configuración D o L a un carbohidrato, se considera la configuración del centro quiral de mayor orden (en la glucosa es C5) y se lo compara con la configuración del 2,3-dihidroxipropanal (gliceraldehido). Si observamos la estructura, comprobamos que hay un centro quiral (C2).
HOH2C CHOH CHO Por consiguiente, hay dos configuraciones distintas (enantiómeros). Las actividades ópticas son de igual valor absoluto, pero de signos opuestos. Arbitrariamente, se le ha asignado al gliceraldehido dextrógiro la configuración (I) y se lo considera el progenitor de la familia D, mientras que, a su enantiómero, gliceraldehido levógiro, se le da la configuración (II) y sería el progenitor de la familia L:
CO
C
CH2OH
H OH
H
CO
C
CH2OH
OH H
H
(I) (II)
D (+) gliceraldehído L (-) gliceraldehído En el caso de la glucosa, para determinar a qué familia pertenece, se analiza la posición del oxhidrilo del centro quiral de mayor orden (C5 ), es decir, el más alejado del grupo carbonilo (es importante aclarar que la asignación D o L es independiente de la actividad óptica).

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
230
C
C
C
CC
CH2OH
OH
H
H
H
H
OH
OH
OH
OH
CCC
CC
CH2OH
OH
OHOH
OHOH
HH
HH
D (+) glucosa L (-) glucosa
1.2.1.5. - Tautomería de la glucosa Por el equilibrio tautomérico, la glucosa se transforma en un enodiol, que a la vez puede pasar a glucosa, o bien, a fructosa:
C
C
C
CC
CH2OH
OH
H
H
H
H
OH
OH
OH
OH
C
C
C
C
C
CH2OH
OH
H
H
H
OH
OH
OH
OH
H
CC
CC
CH2OH
H
HH
OHO
OHOH
CH2OH
D (+) glucosa enodiol D (-) fructosa o levulosa Así, la D (-) fructosa y la D (+) glucosa se pueden transformar mutuamente, a través de un enodiol. Este equilibrio explica porqué la D (-) fructosa (cetohexosa), a pesar de no poseer grupo aldehido, reduce los reactivos de Fehling o Tollens.
1.2.1.6. - Formación de hemiacetales (estructuras cíclicas de la glucosa) El hecho que la glucosa no dé algunas reacciones características de los aldehidos se explica porque, al igual que otros carbohidratos, su estructura acíclica está en equilibrio con dos estructuras cíclicas hemiacetálicas.
Recordemos que los aldehidos pueden reaccionar con los alcoholes para dar hemiacetales (ver Adiciones nucleofílicas):
C O
H
R + H O R' C
H
R
OH
O R'
aldehído alcohol hemiacetal
En solución acuosa, la glucosa reacciona intramolecularmente (la misma molécula tiene grupo aldehido y grupo alcohol), para dar hemiacetales cíclicos. Al reaccionar el grupo aldehido con el grupo alcohol de C5 se forma una estructura cíclica de seis miembros (un oxígeno y cinco carbonos).
Existen dos formas hemiacetálicas de la glucosa debido a que, al pasar de la forma abierta (aldehídica) a la cíclica (hemiacetálica), el C1 pasa de no quiral a quiral y, por consiguiente, debemos tener en cuenta su configuración. Así, la ciclación da lugar a la formación de un par de isómeros, llamados anómeros, que sólo se diferencian en la configuración de C1, denominado carbono anomérico y al –OH unido al mismo se lo denomina oxhidrilo hemiacetálico.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
231
Por lo expuesto, podemos considerar que la D (+) glucosa se presenta como un equilibrio entre la forma abierta (aldehídica) y dos formas cíclicas anómeras (hemiacetálicas).
Se ha convenido en llamar α al isómero en el cual el -OH del carbono anomérico está a la derecha en la representación de Fisher (1), y β al que tiene -OH a la izquierda.
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
OH
OH
O
C
C
C
CC
CH2OH
OH
H
H
H
H
OH
OH
OH
OH
C
C
C
C
C
CH2OH
HO H
H
H
H
H
OH
OH
OH
O
α - D (+) glucosa D (+) glucosa β - D (+) glucosa
(forma hemiacetálica) (forma abierta) (forma hemiacetálica) La forma β es la más estable porque el -OH del carbono anomérico está más alejado del –OH de C2 , por lo que la interacción entre ambos es menor.
¿Por qué la glucosa forma un ciclo de seis miembros? ¿Por qué no se forma hemiacetal por reacción de los grupos alcohólicos de los otros carbonos?
La razón es que un anillo de seis miembros es más estable que uno de tres, cuatro, cinco o siete miembros, porque se mantienen los ángulos de aproximadamente 109°, característicos de la hibridación sp3 (observemos que tanto los carbonos como el oxígeno del ciclo presentan hibridación tetraédrica).
Las representaciones de Fisher, que no tienen en cuenta los ángulos de enlaces no son las correctas, a pesar de lo cual se siguen usando. Haworth propuso otro tipo de representación que se acercaría más a la realidad. Para ello consideró lo siguiente:
El ciclo se representa por un hexágono en cuyos vértices se ubican los cinco carbonos y el oxígeno. En cada centro quiral, los grupos que en la fórmula de Fisher están a la derecha en la de Haworth se
ubican hacia abajo. En cada centro quiral, los grupos que en la fórmula de Fisher están a la izquierda en la de Haworth se
ubican hacia arriba.
Por lo tanto, tenemos las siguientes representaciones para las formas hemiacetálicas de la D (+) glucosa:
CC
C1C
OH
OH
CH2OH
H
OH
H
H
OH
OH
H
CC
C1C
OCH
OH
CH2OH
H
OH
H
H
OH
H
OH
α - D (+) glucosa β - D (+) glucosa
Como cada ciclo es similar al del pirano, las estucturas hemiacetálicas se suelen denominar como sigue:
− α - D (+) glucopiranosa
− β - D (+) glucopiranosa

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
232
La representación de Haworth es plana, por lo que los ángulos serían de 120°, muy superiores a 109° y, por lo tanto, habría mucha tensión angular.
Pero no hay tensión angular debido a que estos ciclos no son planos, sino que son plegados. Es decir, la forma del ciclo (conformación) es como sigue:
Con esta forma del ciclo, todos los ángulos son de aproximadamente 109°, no existiendo tensión angular por desviación del ángulo normal de enlace, que es lo que ocurriría si fuera plano. A esta forma se la denomina conformación silla.
1.2.1.7. - Mutarrotación de la glucosa Por ser hemiacetales, α-D(+) glucopiranosa y β-D(+) glucopiranosa se hidrolizan fácilmente. En solución acuosa, cada uno de estos anómeros se convierte, como ya vimos, en una mezcla en equilibrio de ambos isómeros cíclicos.
Cuando se disuelven cristales de α - D (+) glucosa (pura) en agua, con el tiempo disminuye gradualmente su rotación específica de +112° inicial hasta +52,7°.
Cuando se disuelven cristales de β - D (+) glucosa (pura) en agua, con el tiempo aumenta gradualmente su rotación específica de +19° inicial hasta +52,7°.
Este fenómeno se explica porque, tanto la α - D (+) glucosa pura como la β - D (+) glucosa pura, al cabo de un tiempo no están solas, sino que hay una mezcla de α, β y la forma abierta, debido a que se presenta el equilibrio visto anteriormente:
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
CC
CH2OH
OH
H
H
H
H
OH
OH
OH
OH
C
C
C
C
C
CH2OH
HO H
H
H
H
H
OH
HO
OH
O
α - D (+) glucopiranosa D (+) glucosa β - D (+) glucopiranosa
A este fenómeno de variación de la actividad óptica con el tiempo se lo denomina mutarrotación.
La proporción de los isómeros de la glucosa en una mezcla en equilibrio es como sigue:
− α - D (+) glucopiranosa ≅ 37 %
− β - D (+) glucopiranosa ≅ 62 %
− D (+) glucosa (forma abierta) ≅ 1 %

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
233
1.2.1.8. Formación de acetales En general, cuando un aldehído reacciona con un alcohol se forma un hemiacetal, que al reaccionar con una segunda molécula de alcohol forma un acetal.
C O
H
R + H O R' C
H
R
OH
O R'OH2
H O R'C
H
R
O
O R'R'
aldehído alcoholhemiacetal acetal
Los carbohidratos no requieren dos moléculas de alcohol por molécula, para formar el acetal. Como se vio, el hemiacetal es el resultado de una reacción intramolecular, sin consumo de alcohol.
Ejemplos:
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
CH3OH
OH2
C
C
C
C
C
CH2OH
H OCH3
H
H
H
H
OH
HO
OH
O
α - D (+) glucopiranosa
(hemiacetal)
metil- α - D (+) glucósido
(acetal)
C
C
C
C
C
CH2OH
HO H
H
H
H
H
OH
HO
OH
O
CH3OH
OH2
C
C
C
C
C
CH2OH
H3CO H
H
H
H
H
OH
HO
OH
O
β - D (+) glucopiranosa
(hemiacetal) metil- β - D (+) glucósido
(acetal) Un acetal, al no poseer oxhidrilo hemiacetálico no está en equilibrio con la forma abierta. Esto le confiere las siguientes características:
Un acetal no sufre mutarrotación, por lo que es estable en solución acuosa. Un acetal es no reductor.
1.3. Fructosa o levulosa La fructosa es una cetohexosa, que presenta tres centros quirales (C3, C4 y C5):

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
234
CC
CC
CH2OH
H
HH
OHO
OHOH
CH2OH
D (-) fructosa
La fructosa se puede ciclar, por reacción de carbonilo con grupo alcohólico. Se ha comprobado que forma un ciclo de cinco miembros, similar al del furano, denominándose fructofuranosas a las formas hemiacetálicas.
1.3.1. Representación según Fisher:
CH2OH
C
C
C
C
CH2OH
H
H
H
OH
HO
OH
O
CC
CC
CH2OH
H
HH
OHO
OHOH
CH2OH
CH2OH
C
C
C
C
CH2OH
H
H
H
HO
HO
OH
O
α - D (-) fructofuranosa (hemiacetal)
β - D (-) fructofuranosa (hemiacetal)
La fructosa sufre mutarrotación, lo que se explica por el equilibrio anterior. La fructosa puede formar acetales que, como en el caso de la glucosa, no presentan mutarrotación. La fructosa reduce los Reactivos de Fehling y Tollens, ya que por el equilibrio tautomérico forma
glucosa que posee grupo aldehido reductor.
1.3.2. Representación según Haworth:
OH CH2OH
HOH2C
OH H
OH
H OH
OH OH
HOH2C
OH H
CH2OH
H OH
α - D (-) fructofuranosa β - D (-) fructofuranosa
Al igual que en glucosa, estos ciclos no son planos, por lo que los ángulos de enlace son muy próximos a 109º (sp3). 2. Disacáridos Los disacáridos son oligosacáridos que por hidrólisis dan dos moléculas de monosacáridos. Podríamos considerar que se forman por condensación de dos moléculas de monosacáridos, con eliminación de una molécula de agua. Según se use para formar agua uno de los –OH hemiacetálicos o los dos –OH hemiacetálicos, los disacáridos se clasifican como sigue:
− Disacáridos reductores
− Disacáridos no reductores

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
235
2.1. Disacárido reductor
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
O
OH
O
+ H2O
Monosacárido (reductor)
Monosacárido (reductor)
Disacárido (reductor)
Observar que la segunda unidad ha mantenido su oxhidrilo hemiacetálico, lo cual permite el equilibrio con la forma abierta correspondiente, que es la responsable de la reducción de los reactivos de Fehling o Tollens.
Ejemplo:
Como ejemplo de disacárido reductor tenemos la maltosa o azúcar de malta, que se obtiene por unión 1–4 de dos unidades de glucosa:
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
O
O
+ H2OHO
α - D (+) glucopiranosa α - D (+) glucopiranosa 4-O-(α-D-glucopiranosil)-α-D-glucopiranosa
(α-maltosa)
Este es un disacárido que posee un oxhidrilo hemiacetálico. Por lo tanto, es reductor. La unidad reductora puede tener configuración α o β, por lo que tendremos α - maltosa o β - maltosa. La rotación específica de la α - maltosa es +168° y la de la β-maltosa es +112°. En solución acuosa,
sufren mutarrotación, llegando al equilibrio con una rotación de +136°.

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
236
2.2. Disacárido no reductor
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H
H
H
H
H
OH
O
OH
O
+ H2OHO
Observar que ninguna de las dos unidades ha mantenido su oxhidrilo hemiacetálico, lo cual no permite el equilibrio con la forma abierta correspondiente, por lo que no reduce los reactivos de Fehling o Tollens.
Ejemplo:
Como ejemplo de disacárido no reductor tenemos la sacarosa o azúcar de caña, que se obtiene por unión 1–2 de α - D (+) glucopiranosa y β - D (-) fructofuranosa:
C
C
C
C
C
CH2OH
H OH
H
H
H
H
OH
HO
OH
O
C
C
C
C
C
CH2OH
H
H
H
H
H
OH
HO
OH
O
+ H2O
CH2OH
C
C
C
C
CH2OH
H
H
H
HO
HO
OH
O +
CH2OH
C
C
C
C
CH2OH
H
H
H
O
HO
OH
O
α - D (+) glucopiranosa β - D (-) fructofuranosa sacarosa
Este es un disacárido que no posee un oxhidrilo hemiacetálico. Por lo tanto, no tiene grupo aldehido libre y n es reductor.
Las unidades están unidas a través de sus carbonos anoméricos, por lo que en este caso no hay disacáridos anómeros, ni mutarrotación.
2.3. Azúcar invertido Al realizar la hidrólisis ácida o enzimática de la sacarosa se obtiene una mezcla de D (+) glucosa y D (-) fructosa, denominado azúcar invertido, porque se pasa de un valor positivo rotación específica a un valor neto negativo. Esto se explica porque la sacarosa posee una rotación específica de +66,5º, mientras que la mezcla resultante de su hidrólisis tiene una rotación específica de –39° (glucosa: 52,7º y fructosa: -92,4º)
hidrólisis
Sacarosa α - D (+) glucosa + β - D (-) fructosa
(+66,5º) (+52,7º) (-92,4º)
(-39º)
azúcar invertido

Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
237
El azúcar invertido es más dulce que la sacarosa, por la presencia de fructosa libre, que es el más dulce de los azúcares.
3. Polisacáridos Los polisacáridos son compuestos cuyas moléculas contienen más de diez unidades de monosacáridos, unidas por enlaces glicosídicos, y que por hidrólisis total dan monosacáridos. Se clasifican en homopolisacáridos y heteropolisacáridos:
Polisacáridos
Homopolisacáridos: Están formados por un solo tipo de monosacáridos (ej.: almidón, celulosa).
Heteropolisacáridos: Están formados por más de un tipo de monosacáridos (ej.: gomas).
3.1. Almidón El almidón se halla en forma de gránulos, cuyo tamaño y forma son característicos de la planta que lo
contiene. Está formado por dos fracciones o carbohidratos distintos, constituidos por unidades de α - D (+)
glucosa, que difieren en tamaño y forma molecular: o Amilosa: Es soluble en agua y constituye el 20% del gránulo de almidón. o Amilopectina: Es insoluble en agua y constituye el 80% del gránulo de almidón.
Por hidrólisis ácida o enzimática, ambas fracciones lentamente dan el disacárido (+) maltosa, que está constituido por dos unidades de glucosa, con unión α-glicosídica, y por posterior hidrólisis se forma el monosacárido D (+) glucosa.
hidrólisis hidrólisis
almidón …………. maltosa glucosa
Al almidón lo podríamos representar como un polímero, que contiene n-unidades de maltosa:
3.2. Celulosa Es el componente principal de las fibras de madera y de las plantas. Está constituida por cadenas de unidades de D (+) glucosa, unidas por enlaces β-glicosídicos. La hidrólisis ácida da celobiosa, constituida por dos unidades de glucosa con unión β-glicosídica, y
finalmente D (+) glucosa como único monosacárido.
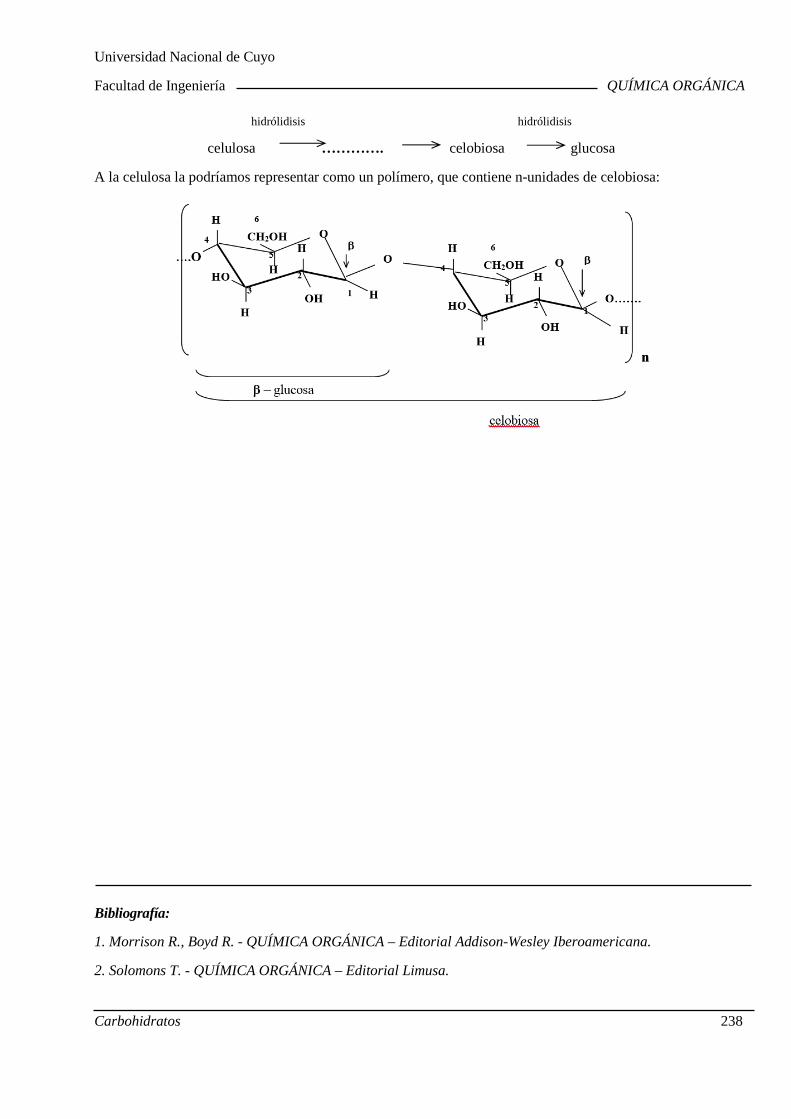
Universidad Nacional de Cuyo
Facultad de Ingeniería QUÍMICA ORGÁNICA
Carbohidratos
238
hidrólidisis hidrólidisis
celulosa …………. celobiosa glucosa
A la celulosa la podríamos representar como un polímero, que contiene n-unidades de celobiosa:
Bibliografía:
1. Morrison R., Boyd R. - QUÍMICA ORGÁNICA – Editorial Addison-Wesley Iberoamericana.
2. Solomons T. - QUÍMICA ORGÁNICA – Editorial Limusa.





























