Embed Size (px)
Citation preview

Desconocemos la totalidad de los factores res-ponsables del desarrollo y de la progresión
del cáncer de próstata. La experiencia adquiridacon terapia hormonal basada en la deprivaciónandrogénica, nos ha enseñado que pasado untiempo se produce una reactivación del creci-miento del tumor, proceso que va acompañado deresistencia al tratamiento debido a la apariciónde células que no responden a los andrógenos.
De hecho, cada vez resulta más evidente que lafunción prostática no sólo depende de andróge-nos, sino que existen una gran diversidad de hor-monas peptídicas, factores de crecimiento,bucles reguladores autocrinos/paracrinos e inte-racciones entre el epitelio y el estroma que con-trolan y mantienen el funcionamiento normal dela próstata y que están implicados en el desarro-llo de sus enfermedades.
RESUMEN
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
Revisamos los mecanismos de control de crecimiento prostático basados en andrógenos y pro-
ductos de secreción neuroendocrina, prestando especial atención al papel de la somatostina (SS)
como inhibidor del crecimiento neoplásico. Las aportaciones de nuestro grupo confirman que este
efecto antiproliferativo de la SS sobre la próstata está mediado por la fosfotirosina fosfatasa SHP-1,
presente en la próstata humana. Esta enzima juega un papel en el control de la proliferación celu-
lar prostática y en la progresión del cáncer de próstata. Además, pensamos que su presencia podría
determinar el potencial terapéutico de la SS en el control del cáncer de próstata.
PALABRAS CLAVE: Neoplasia prostática. Proteína tirosina fosfatasa. SHP-1. Somatostatina. Célula neuroendocrina. Próstata.
ABSTRACT
PHOSPHOTYROSINE PHOSPHATASE SHP-1, SOMATOSTATIN AND PROSTATE CANCER
We review the mechanisms involved in prostatic growth based on androgens and product of neu-
roendocrine secretion, with special reference to the role of somatostatin (SS) in the inhibition of neo-
plastic growth. Our contributions in the field confirm the antiproliferative effect of SS on the prostate
is mediated by phosphotyrosine phosphatase SHP-1, that is present in human prostate. This enzyme
plays a role in the control of prostatic cell proliferation and in the progression of prostate cancer.
Besides, we consider its presence may determine the therapeutic potential of SS in the control of
prostate cancer.
KEY WORDS: Prostate neoplasm. Protein tyrosin phosphatase. SHP-1. Somatostatin. Neuroendocrine cell. Prostate.
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA YCÁNCER DE PRÓSTATA
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, R.M. ROPERO, R.M. MARTÍN,F.J. RODRÍGUEZ, F.J. GONZÁLEZ, J.I. LÓPEZ, J.C. ANGULO
Departamento de Bioquímica. Departamento de Cirugía. Universidad de Alcalá. Servicio de Urología.Hospital Príncipe de Asturias. Alcalá de Henares. Madrid.
Servicio de Anatomía Patológica. Hospital de Basurto. Bilbao.
Actas Urol Esp. 28 (4): 269-285, 2004
269
ACTAS UROLÓGICAS ESPAÑOLAS ABRIL 2004RREEVVIISSIIÓÓNN DDEE CCOONNJJUUNNTTOO

Así, la fase independiente de andrógenos esel centro de atención de la mayoría de las
investigaciones actuales; investigaciones queintentan sentar las bases del desarrollo de nue-vas terapias. En este sentido, sabemos quealgunos péptidos, como la somatostatina (SS),poseen un claro potencial antimitogénico pero,lamentablemente, los datos preclínicos acercade su utilización en el carcinoma prostático soncontradictorios, probablemente debido al desco-nocimiento de los mecanismos moleculares através de los cuales ejerce su acción biológica.Así, para entender el efecto antiproliferativo dela SS en el cáncer de próstata, es necesarioconocer los subtipos expresados en la próstatanormal y neoplásica, cuál de estos receptores esresponsable del efecto antiproliferativo, y elmecanismo o mecanismos celulares utilizados.Es probable que en el momento que conozcamoslas respuestas a estas preguntas se puedan rea-lizar nuevos ensayos clínicos que valoren cierta-mente la eficacia de la SS en el tratamiento deesta enfermedad.
REGULACIÓN DEL CRECIMIENTOPROSTÁTICO
El desarrollo de la próstata, y el del resto delas estructuras sexuales accesorias internasmasculinas, depende de los andrógenos. La tes-tosterona se sintetiza en las células de Leydig deltestículo diariamente y se vierte al plasma. Laesteroidogénesis testicular está controlada por eleje hipotálamo-hipofisiario. La liberación por elhipotálamo de la GnRH (hormona estimulante degonadotropinas) estimula la secreción de la LH(hormona luteinizante) por la hipófisis, que va acontrolar la secreción de testosterona a nivel tes-ticular. Además de los andrógenos testiculares,llegan a la próstata andrógenos liberados por laglándula adrenal. Esta segunda fuente de andró-genos puede adquirir relevancia en situacionesde castración, aunque su importancia en la prós-tata normal y en el desarrollo y el tratamiento delcáncer y de la hiperplasia prostática benigna(HBP) sigue siendo tema de debate1.
Dada su naturaleza lipofílica, la testosteronase transporta en sangre unida a proteínas: 58%por la globulina transportadora de hormonassexuales (SHBG), 40% por la albúmina, dejando
sólo un 2% de hormona libre disponible para sercaptada por los tejidos2. La fracción libre se veráafectada por variaciones en los niveles de lostransportadores, modificando la cantidad dispo-nible para la próstata. Situaciones como la obesi-dad, la edad, y el aumento de estrógenos o de lamisma testosterona pueden producir estas varia-ciones3.
Después de su internalización en la célulaprostática la testosterona se transforma en dihi-drotestosterona (DHT) por la enzima 5-α-reducta-sa presente en el retículo endoplasmático rugo-so4. La DHT es 10 veces más activa que la tes-tosterona. Así, la DHT se une a un receptornuclear específico que, activará o reprimirá laexpresión de determinados genes, regulando lafuncionalidad celular. Determinados antiandró-genos pueden unirse al receptor de andrógenos,pero los cambios conformacionales inducidos nopermiten que el receptor interaccione con elADN5. La metabolización final de la DHT está acargo de óxido reductasas que transforman laDHT primero en dioles y luego en trioles total-mente inactivos.
La forma en que los andrógenos controlan laproliferación y regulan la función prostática estásiendo ampliamente estudiada. La DHT regula elcrecimiento prostático, estimula la proliferacióncelular e inhibe la apoptosis o muerte celular pro-gramada. Una disminución de los niveles de DHTy de su receptor provoca involución de la prósta-ta, mientras que su aumento inicia una prolifera-ción celular activa, ambos procesos están asocia-dos a una expresión génica específica5. Sinembargo, no existen pruebas suficientes de quela DHT estimule directamente la síntesis de ADN,más bien sus efectos estarían mediados por unaserie de factores de crecimiento, cuya síntesis severía estimulada por la DHT6. Los factores de cre-cimiento, por lo tanto, serían mediadores directosde la acción de los andrógenos y pueden ser res-ponsables de mediar las interacciones entre elepitelio y el estroma7.
En condiciones normales los andrógenos y losfactores de crecimiento crean un balance queregula el crecimiento, la diferenciación, la super-vivencia y la muerte de células prostáticas. En lapróstata se ha observado la acción de 4 familiasde factores de crecimiento, producidos de manera
270
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.

local y que actúan de manera paracrina o auto-crina en el estroma o en el epitelio: familia EGF(EGF y TGF-α), familia TGF-ß (TGF-ß1, 2 y 3),familia IGF (IGF-I, IGF-II, IGF-BP) y familia FGF(bFGF y KGF)8 (Fig. 1).
Los niveles de andrógenos regulan, tanto en elepitelio como en el estroma, la secreción de losfactores de crecimiento así como la expresión desus receptores, creando un delicado equilibrioentre ambos compartimentos celulares. Duranteel desarrollo del cáncer de próstata, este equili-brio se pierde alterándose la forma y lugar desecreción de los factores de crecimiento, así comola expresión de sus receptores y proteínas rela-cionadas, como algunos proto-oncogenes y genessupresores de tumores. Este estado promueve lainvasión y la metástasis a sitios favorables parael desarrollo de la célula neoplásica. La terapia desupresión androgénica conlleva la selección delas células malignas, sobreviviendo aquellascapaces de crecer en ausencia de andrógenos. Segenera así la fase independiente de andrógenos
de la enfermedad5. En este momento sucedendiversas alteraciones moleculares9, las principa-les se reflejan en la Tabla I.
CÉLULAS NEUROENDOCRINASLas células neuroendocrinas (NE) de la prós-
tata pertenecen al sistema denominado antigua-mente como APUD (amine precursor uptake anddecarboxylation). Estas células, componenteminoritario del epitelio prostático, carecen delreceptor de andrógenos y secretan gran varie-dad de péptidos, neuropéptidos y otras molécu-las bioactivas. La mayoría de las células NEprostáticas secretan cromogranina A, enolasaespecífica de neuronas y serotonina. Ademásproducen calcitonina, gonadotropina coriónicahumana α, péptido similar a la TSH, neuroten-sina, proteína relacionada con la PTH, colecisto-quinina, bombesina y somatostatina10. Lascélulas NE, actuando de forma paracrina y/oautocrina regulan las secreciones exocrinas yendocrinas de células epiteliales y NE vecinas,así como la proliferación y diferenciación celu-lar. Estas células presentan prolongacionesextendidas hacia el lumen de las glándulas queles permiten detectar cambios de pH, concen-tración de proteínas, neuropéptidos u hormonasdel contenido luminal, y a través de señalesdeterminadas modificar o regular la secreciónde las células exocrinas11.
271
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
Estroma
TGF-β
TGF-αIGF-I, IIbFGF, KGF
BombesinaSerotoninaNeurotensinaSomatostatina
EGFTGF-αIGF-I, IIbFGF, KGF
Epitelio TGF-β
Próstata Normal
Cáncer
Células NE
FIGURA 1. Factores de crecimiento que actúan en la prós-tata normal y en el cáncer. En la próstata normal (fle-chas negras) los factores de crecimiento TGFαα, bFGF, KGF,IGF-I, IGF-II y TGF-ß, secretados por el estroma, actúan demanera paracrina sobre sus receptores ubicados en elepitelio. El TGFαα, IGF-I, IGF-II, bFGF y KGF estimulan laproliferación de las células epiteliales, mientras que elTGF-ß la inhibe. Sin embargo, en el cáncer (flechas rojas)estos factores de crecimiento, secretados por el epitelio,actúan de manera autocrina sobre estas células. En esteúltimo caso, predominan los efectos mitogénicos porquedisminuye la sensibilidad a los efectos inhibitorios delTGF-ß, potenciándose sus efectos angiogénicos y metas-tásicos.
TABLA IALTERACIONES MOLECULARES ASOCIADAS
AL DESARROLLO DEL CARCINOMA DE PRÓSTATA
– Aparición de bucles autocrinos con efectos:• Mitogénicos: EGF, TGFα, bFGF,, KGF, IGF-I,
NGF• Inmunosupresores: TGFß1 y 2• Angiogénicos: bFGF, VEGF• Metastásicos: EGF, bFGF, BMP-6
– Alteraciones en la expresión de proteínas de unión a IGF: IGFBP
– Incremento en la expresión de proto-oncogenes: EGFR, p185erbB-2, p160erbB-3
– Disminución de productos supresores de tumores: TGFßRI, TGFßRII, IGFRII
– Alteraciones en la expresión de proteínas queregulan el balance muerte/proliferación celular:bcl-2, p21Cip1, p27Kip1, p16, CDK, CDC

En los últimos años las células NE han adqui-rido cierto protagonismo al observarse que másdel 50% de los tumores de próstata, contienencélulas neoplásicas dispersas o formando focos,carentes de receptor de andrógenos y que pre-sentan un fenotipo NE12. El número de estascélulas aumenta conforme el adenocarcinomaprogresa hacia fases más avanzadas o resistentesa andrógenos. Estudios recientes parecen indicarque la diferenciación NE se correlaciona con unmal pronóstico, con la progresión del tumor y conla independencia androgénica10. Es posible queligeros cambios en el microambiente celular faci-liten la adquisición de un fenotipo NE por partede ciertas células tumorales13. Los productossecretados por estas células, actuando de formaparacrina/autocrina, activan la proliferación delas células vecinas y facilitan el crecimiento inde-pendiente de andrógenos del tumor. De hecho sehan observado células tumorales proliferando enla proximidad de focos de células NE14. Sinembargo, los resultados obtenidos al intentarestablecer una estrecha relación entre diferencia-ción NE y avance del tumor, no son concluyen-tes10.
Existen distintas poblaciones de células NEque varían en sus productos de secreción y ensus receptores. La producción de factores auto-crinos/paracrinos por las células neuroendocri-nas o por las células epiteliales tumorales dife-renciadas a neuroendocrinas, está probablemen-te influenciado por hormonas y podría cumplirun papel importante en la progresión del cáncer.Sin embargo, son necesarios más estudios paraevaluar correctamente la significación diagnósti-ca y terapéutica de la diferenciación neuroendo-crina en el cáncer de próstata.
LA SOMATOSTATINAEntre los factores sintetizados y liberados por
las células NE prostáticas se encuentra la soma-tostatina. La somatostatina es un neuropéptido,de amplia distribución en el organismo, que inhi-be una gran variedad de acciones fisiológicas,regulando la liberación de hormonas, péptidos yneurotransmisores15 y actuando como un inhibi-dor endógeno del crecimiento16. Existen princi-palmente dos formas activas circulantes del pép-tido, una de 14 aminoácidos (la somatostatina
14) y otra de 28 (la somatostatina 28). Dada sucorta vida media, que limita sus posibles aplica-ciones in vivo, se han desarrollado análogos quese utilizan en clínica: el octreotide (SMS 201-995), el lanreotide (BIM 23014) y el vapreotide(RC 160)17.
Todas las acciones de la somatostatina pasanpor la unión del péptido a receptores presentesen las membranas de las células. Estos recepto-res pertenecen a la clase de receptores acopladosa proteínas G y presentan siete dominios trans-membrana. En la actualidad se conocen cincosubtipos de receptores denominados sstr1, sstr2,sstr3, sstr4 y sstr518,19. Cada uno de estos subti-pos presenta un perfil farmacológico específico yse encuentran asociados a diferentes sistemas detransducción de señales: adenilato ciclasa, cana-les iónicos, guanilato ciclasa, fosfolipasa C, fosfo-lipasa A2, MAP quinasa, serin-treonin fosfatasasy tirosinas fosfatasas19.
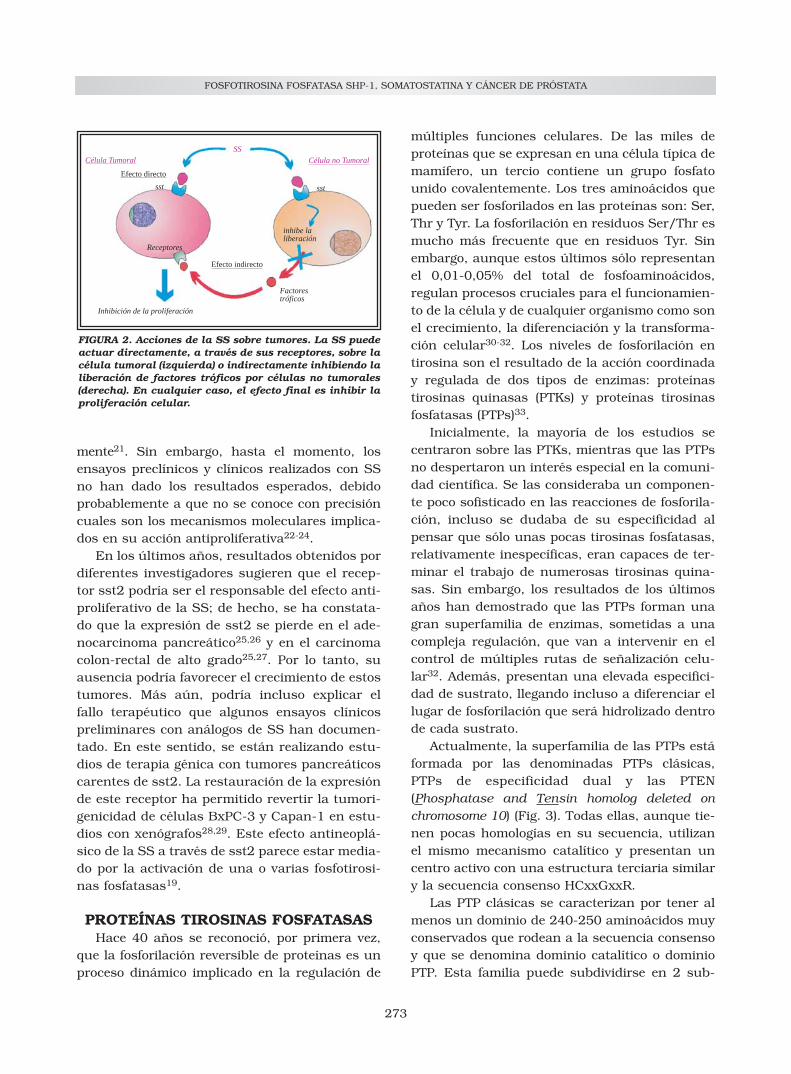
La somatostatina es uno de los pocos neuro-péptidos que inhibe la proliferación celular, invitro e in vivo, de múltiples líneas celulares ytumores experimentales. In vitro, este efecto anti-proliferativo depende de la presencia de recepto-res para la SS en las células tratadas. Sin embar-go, in vivo, la situación es diferente. La SS o susanálogos pueden inhibir la proliferación de tumo-res que carecen de receptores para este péptido.Esta observación ha permitido distinguir entreefectos antiproliferativos directos de la SS -mediados por receptores ubicados en las célulastumorales- y efectos indirectos -mediados porreceptores ubicados en células no tumorales-. Enestos últimos, la SS inhibe la secreción de facto-res de crecimiento, como el IGF-I, y de otras hor-monas tróficas que actúan sobre las célulastumorales20 (Fig. 2).
Por lo tanto, hay muchas esperanzas deposi-tadas en este péptido como un agente antitumo-ral de amplia utilización. Por una parte presentaactividad antineoplásica en diversos modelos invivo e in vitro en tumores para los cuales los tra-tamientos actuales no resultan eficaces (mama,próstata, páncreas, colon y pulmón)16. Por otrolado, su uso clínico en el tratamiento de la acro-megalia y en el síndrome carcinoide ha permitidocomprobar su buena tolerancia, si se comparacon terapias antineoplásicas utilizadas habitual-
272
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.
mente21. Sin embargo, hasta el momento, losensayos preclínicos y clínicos realizados con SSno han dado los resultados esperados, debidoprobablemente a que no se conoce con precisióncuales son los mecanismos moleculares implica-dos en su acción antiproliferativa22-24.
En los últimos años, resultados obtenidos pordiferentes investigadores sugieren que el recep-tor sst2 podría ser el responsable del efecto anti-proliferativo de la SS; de hecho, se ha constata-do que la expresión de sst2 se pierde en el ade-nocarcinoma pancreático25,26 y en el carcinomacolon-rectal de alto grado25,27. Por lo tanto, suausencia podría favorecer el crecimiento de estostumores. Más aún, podría incluso explicar elfallo terapéutico que algunos ensayos clínicospreliminares con análogos de SS han documen-tado. En este sentido, se están realizando estu-dios de terapia génica con tumores pancreáticoscarentes de sst2. La restauración de la expresiónde este receptor ha permitido revertir la tumori-genicidad de células BxPC-3 y Capan-1 en estu-dios con xenógrafos28,29. Este efecto antineoplá-sico de la SS a través de sst2 parece estar media-do por la activación de una o varias fosfotirosi-nas fosfatasas19.
PROTEÍNAS TIROSINAS FOSFATASASHace 40 años se reconoció, por primera vez,
que la fosforilación reversible de proteínas es unproceso dinámico implicado en la regulación de
múltiples funciones celulares. De las miles deproteínas que se expresan en una célula típica demamífero, un tercio contiene un grupo fosfatounido covalentemente. Los tres aminoácidos quepueden ser fosforilados en las proteínas son: Ser,Thr y Tyr. La fosforilación en residuos Ser/Thr esmucho más frecuente que en residuos Tyr. Sinembargo, aunque estos últimos sólo representanel 0,01-0,05% del total de fosfoaminoácidos,regulan procesos cruciales para el funcionamien-to de la célula y de cualquier organismo como sonel crecimiento, la diferenciación y la transforma-ción celular30-32. Los niveles de fosforilación entirosina son el resultado de la acción coordinaday regulada de dos tipos de enzimas: proteínastirosinas quinasas (PTKs) y proteínas tirosinasfosfatasas (PTPs)33.
Inicialmente, la mayoría de los estudios secentraron sobre las PTKs, mientras que las PTPsno despertaron un interés especial en la comuni-dad científica. Se las consideraba un componen-te poco sofisticado en las reacciones de fosforila-ción, incluso se dudaba de su especificidad alpensar que sólo unas pocas tirosinas fosfatasas,relativamente inespecíficas, eran capaces de ter-minar el trabajo de numerosas tirosinas quina-sas. Sin embargo, los resultados de los últimosaños han demostrado que las PTPs forman unagran superfamilia de enzimas, sometidas a unacompleja regulación, que van a intervenir en elcontrol de múltiples rutas de señalización celu-lar32. Además, presentan una elevada especifici-dad de sustrato, llegando incluso a diferenciar ellugar de fosforilación que será hidrolizado dentrode cada sustrato.
Actualmente, la superfamilia de las PTPs estáformada por las denominadas PTPs clásicas,PTPs de especificidad dual y las PTEN(Phosphatase and Tensin homolog deleted onchromosome 10) (Fig. 3). Todas ellas, aunque tie-nen pocas homologías en su secuencia, utilizanel mismo mecanismo catalítico y presentan uncentro activo con una estructura terciaria similary la secuencia consenso HCxxGxxR.
Las PTP clásicas se caracterizan por tener almenos un dominio de 240-250 aminoácidos muyconservados que rodean a la secuencia consensoy que se denomina dominio catalítico o dominioPTP. Esta familia puede subdividirse en 2 sub-
273
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
Célula Tumoral
Efecto directo
sst
Célula no Tumoral
sst
SS
Efecto indirecto
Inhibición de la proliferación
Factorestróficos
inhibe laliberación
Receptores
FIGURA 2. Acciones de la SS sobre tumores. La SS puedeactuar directamente, a través de sus receptores, sobre lacélula tumoral (izquierda) o indirectamente inhibiendo laliberación de factores tróficos por células no tumorales(derecha). En cualquier caso, el efecto final es inhibir laproliferación celular.

grupos, dependiendo de la localización celular:las PTPs no receptor o intracelulares y las PTPstransmembrana o similares a un receptor. LasPTP no receptor son enzimas intracelulares quetienen un solo dominio catalítico. En su extremoN-terminal o C-terminal aparecen diferentesdominios estructurales de longitud variable yhomólogos a los que aparecen en otras proteínas.Estos segmentos no catalíticos controlan la fun-ción de estas PTPs, dirigiéndolas a compartimen-tos subcelulares específicos o modulando direc-tamente la actividad enzimática. Además, la acti-vidad de la enzima se regula por cambios confor-macionales causados por fosforilación en resi-duos de Ser/Thr o Tyr, o a través de la interac-ción del dominio catalítico con los propios domi-nios no catalíticos adquiriendo una conformacióninactiva, como ocurre en las SHP-1 y SHP-232.Las PTP transmembrana presentan una estruc-tura similar a la de un receptor típico de mem-brana: un dominio extracelular variable, undominio transmembrana y uno o dos dominioscatalíticos intracelulares. Los dominios extrace-lulares pueden interaccionar con ligandos queregulan la actividad de los dominios catalíticos.Estos dominios presentan una gran variabilidad
estructural, posiblemente reflejo de sus diferen-tes funciones y ligandos fisiológicos. En la mayo-ría de los casos, en estos dominios aparecensecuencias similares a las presentes en proteínasimplicadas en procesos de adhesión celular, loque sugiere que estas PTPs pueden participaractivamente en los procesos de interacción célu-la-célula y célula-matriz extracelular31. Las fosfa-tasas de especificidad dual (DSP) poseen capaci-dad de desfosforilar residuos de pSer/pThr ypTyr. Al igual que otras fosfatasas, muestran unaalta selectividad de sustratos regulando vías deseñalización celular importantes como la deMAPK34.
La PTEN se identificó inicialmente como ungen supresor de tumores localizado en el cromo-soma 10q23, región que está alterada en el 70%de los glioblastomas y en el 60% de los cánceresde próstata avanzados. La proteína que codificaeste gen es similar a las PTPs de especificidaddual, pero además de desfosforilar residuos depTyr, desfosforila fosfatidilinositol-3,4,5-trifosfato(PIP3). Este lípido es un componente clave en laactivación de la Ser/Thr quinasa Akt, una de lasprincipales rutas de control del crecimiento celu-lar. La pérdida de la PTEN, durante la tumorogé-
274
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.
SUPERFAMILIA DE LAS PTP (HCX5R)
PTP CLÁSICAS PTP DUALES PTEN
PTP no receptor PTP similares a receptor
PTP1B
SHP-1SHP-2
MEG2
PEST
PTPH1
CD45PTPλ
LAR PTPα PTPγ
PTPβ VHR MKP KAP
FYVE-DSP
cdc25
PTEN
FIGURA 3. Superfamilia de las fosfotirosinas fosfatasas.

nesis, mantendría la Akt permanentemente acti-va, permitiendo a las células proliferar sin con-trol35. Existen clasificaciones anteriores en lasque se incluía a esta PTP junto con las de especi-ficidad dual. Actualmente se separa este grupo deenzimas porque poseen especificidad por sustra-tos de diferente naturaleza: lípidos y proteínasfosforiladas en Tyr. Otro miembro de este grupoes la miotubularina (MTM)36.
SHP-1En 1986, Sadowski y cols.37 identificaron un
dominio N-terminal que estaba altamente conser-vado entre las proteínas tirosinas quinasas cito-plasmáticas de la familia Src. A este dominio N-terminal, de aproximadamente 100 aminoácidos,lo denominaron SH2 (dominio 2 de homologíacon Src). Años después, se descubrió la presen-cia de dominios SH2 en proteínas que intervienenen la transducción de señales como la fosfolipasaCγ1, la proteína estimuladora de la actividadGTPasa (GAP), la fosfatidil inositol 3 quinasa(PI3K) y la proteína de unión a actina (tensina).
Los dominios SH2 unen específicamente resi-duos tirosina fosforilados, y la secuencia adya-cente a este residuo es la que determina la espe-cificidad de unión. Por lo tanto, estos dominiosjuegan un papel importante en las interaccionesentre los receptores tirosina quinasa para los fac-tores de crecimiento y las proteínas implicadasen la transducción de señales. Los receptorespara factores de crecimiento son activados tras launión de sus ligandos, dando lugar a una auto-fosforilación en múltiples residuos tirosina situa-dos en su domino intracelular, creando sitios deunión específicos para proteínas citoplasmáticascon dominios SH2. Estas proteínas, como resul-tado de esa unión, pueden ver modificada suactividad y su especificidad de sustrato.
Hasta el momento se conocen tres PTPs condominios SH2: SHP-1 (anteriormente llamadaPTP1C)38, SH-PTP139, Hcp o SHP40 presente enmamíferos, SHP-2 (anteriormente llamadaPTP1D41, PTP2C42, SH-PTP243, Syp44, SH-PTP345,o (SAP-246 también presente en mamíferos, yCorkscrew o csw en Drosophila. La SHP-1 seexpresa principalmente en células hematopoyéti-cas y epiteliales, mientras que la SHP-2 y la cswson más ubicuas. La estructura de todas ellas es
muy similar (Fig. 4). Poseen dos dominios SH2ubicados en el extremo N-terminal, un dominiocatalítico y en el extremo C-terminal dos sitios defosforilación en Tyr que le permiten interaccionarcon otras proteínas con dominios SH247.
La SHP-1 es una enzima citosólica de aproxi-madamente 66 kDa que se expresa preferente-mente en células hematopoyéticas, aunque exis-te una variante en algunos tipos de células epite-liales48. La existencia de dos cepas de ratones(motheaten (me) y motheaten viable (mev))49 quecarecen de SHP-1 o expresan una forma deficien-te de la enzima con sólo un 20% de actividad, hanpermitido demostrar que esta PTP es un regula-dor negativo de múltiples señales en célulashematopoyéticas, entre las que se incluyen lasactivadas por interleuquinas, factores de creci-miento, adhesión e inmunoreceptores50.
En células no-hematopoyéticas, SHP-1 actúaregulando procesos activados por factores decrecimiento, neuropéptidos y citoquinas. Laactivación de receptores tirosina quinasas (EGF,PDGF, IGF-1) o de receptores que activan tirosi-na quinasas tipo no-receptor (Src, Jak, Syk)producen la fosforilación de un gran número desustratos, activando sistemas de señalizaciónintracelular que regulan el crecimiento celular,la diferenciación, la migración y la apoptosis51.Las tirosinas fosfatasas cumplen un papel deci-sivo desactivando muchas de estas señales.Diversos estudios demuestran que SHP-1 escapaz de unirse al receptor del EGF fosforilado einactivarlo38,52,53. Este efecto es específico de
275
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
Y536 Y564
Y543 Y580
Y541
Y548
SHP-1
SHP-2
CSW
Inserto PTP
Dominio catalíticoDominios SH2
FIGURA 4. Estructura de las PTPs con dominios SH2. Lastres PTPs de este grupo poseen dos dominios SH2 ubica-dos en el extremo N-terminal, un dominio catalítico en elextremo C-terminal y dos sitios de fosforilación en Tyr.

SHP-1 porque al utilizar quimeras SHP-1/SHP-2, con el dominio catalítico de SHP-2, no se pro-duce esta inactivación53. El mismo resultado seha observado con HER, perteneciente a lamisma familia de receptores tirosina quina-sas41. También se ha visto que la SHP-1 estáimplicada en la regulación de las señales activa-das por NGF54, por insulina55 y por el factor decrecimiento de hepatocitos56.
En cuanto a las citoquinas, SHP-1 regula lasseñales activadas por IL-357, por eritropoyetina58
y por INF-α59. En muchos de estos casos el meca-nismo de acción podría consistir en la regulaciónde quinasas de la familia Janus (Jak1 y Jak3), yasea desfosforilando al receptor y/o a las propiasJak quinasas60. Particularmente, interesantesson los hallazgos que involucran a la SHP-1 en elpapel antiproliferativo de algunos péptidos comola angiotensina II61 y la somatostatina62,63. Sinembargo, no en todos los casos SHP-1 cumple unpapel inhibidor. Su y colaboradores64, describie-ron que la sobre-expresión de SHP-1 catalítica-mente inactiva, en células 293 (células prove-nientes de riñón de embriones), inhibe las seña-les mitogénicas inducidas por el EGF y el suero,disminuyendo marcadamente la fosforilación deproteínas que integran la vía de las MAPK. Alañadir la SHP-1 normal, se revierte este efectoindicando que esta enzima podría cumplir unpapel estimulador en estas células.
SHP-1, SOMATOSTATINAY PROLIFERACIÓN CELULAR
Como ya hemos mencionado la activación dePTPs está relacionada con las propiedades anti-proliferativas de la SS. Muchos estudios se hancentrado en este sistema de señalización, a tra-vés del cual la SS podría atenuar las señalesdesencadenadas por factores de crecimiento ypor otros mitógenos. Sin embargo, los resulta-dos obtenidos hasta el momento no son conclu-yentes. Aunque se haya podido demostrar laparticipación de una actividad fosfotirosina fos-fatasa en este efecto antiproliferativo, no entodos los casos se ha identificado la enzima res-ponsable de esa actividad ni el subtipo o subti-pos de receptores de SS asociados25,66,67. Sinembargo, resultados obtenidos por diferentesinvestigadores en los últimos años sugieren que
sería la SHP-1 la enzima responsable del efectoantiproliferativo de la SS, en la mayoría de loscasos a través del receptor sst2.
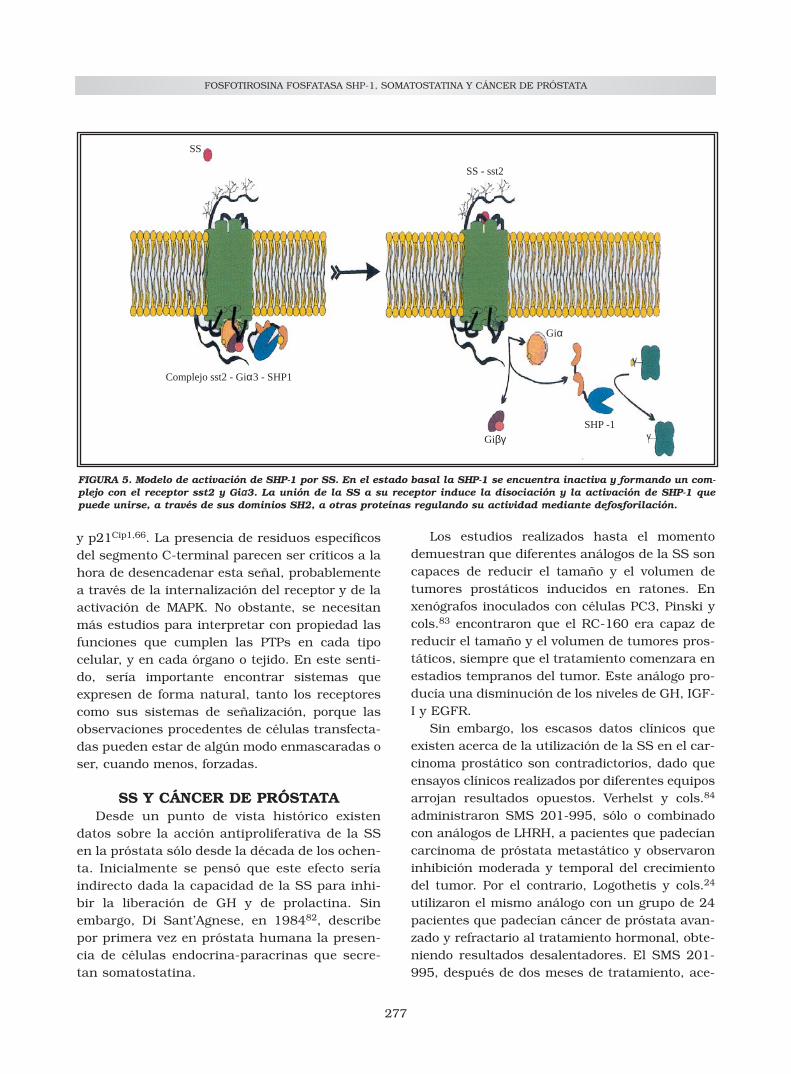
La SHP-1 inmunoprecipita con sst2 y Giα3, loque demuestra que están constitutivamente aso-ciadas68. De hecho, la SS y sus análogos inducenla disociación de SHP-1, su fosforilación y suactivación62 (Fig. 4). Una vez activada SHP-1puede desfosforilar e inactivar sustratos que esti-mulan la proliferación celular. En este sentido,Bousquet y cols.69 demuestran que la SS aceleray amplifica los efectos de SHP-1 sobre la ruta detransducción de señales de la insulina, inhibien-do sus efectos mitogénicos. Concretamente SS, através de SHP-1, desfosforila e inactiva el recep-tor de insulina y sus sustratos: IRS-1 y Shc.
En 1996, Rauly y cols.70 crearon un modelocon células NH3T3 transfectadas con el receptorsst2 y observaron que la expresión de este recep-tor inducía la aparición de un bucle autocrinoque involucraba también a la SHP-1, y que pro-vocaba la inhibición de la proliferación celular.Hallazgos más recientes en ratones que expresanSHP-1 catalíticamente inactiva demuestran quela SS, a través de sst2 y SHP-1, induce paradadel ciclo celular en G1 por aumento de los nivelesde p27Kip1 (inhibidor de la ciclina quinasa)71,72.En este sentido, estudios en células FTRL-5, deri-vadas de células foliculares tiroideas, han revela-do que la SS inhibe los efectos proliferativos de laTSH restableciendo los niveles de p27Kip1(73,74).
Además de los mecanismos citostáticos obser-vados, parece evidente que la activación de SHP-1 induce también apoptosis. La translocación deSHP-1 a la membrana es un fenómeno tempranoy esencial en la acidificación y en la apoptosisinducida por la SS75 (Fig. 5). Estos efectos pare-cen estar inducidos vía sst3 y proteínas Gi, albloquearse por la toxina pertussis (PTX), y seacompañan tanto de un aumento de p53 como dela proteína proapoptótica Bax76. Estudios poste-riores confirman la activación de endonucleasasácidas insensibles a cationes77 y de la caspasa-887. SHP-1 induce un descenso del pH intracelu-lar mediante la inhibición del intercambiadorNa+/H+ (NHE) y la bomba H+-ATPasa79-81. Al con-trario de lo que ocurre con sst3, los otros subti-pos de receptores no inducen apoptosis, sinoparada del ciclo en G1 mediante el aumento de Rb
276
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.
y p21Cip1,66. La presencia de residuos específicosdel segmento C-terminal parecen ser críticos a lahora de desencadenar esta señal, probablementea través de la internalización del receptor y de laactivación de MAPK. No obstante, se necesitanmás estudios para interpretar con propiedad lasfunciones que cumplen las PTPs en cada tipocelular, y en cada órgano o tejido. En este senti-do, sería importante encontrar sistemas queexpresen de forma natural, tanto los receptorescomo sus sistemas de señalización, porque lasobservaciones procedentes de células transfecta-das pueden estar de algún modo enmascaradas oser, cuando menos, forzadas.
SS Y CÁNCER DE PRÓSTATADesde un punto de vista histórico existen
datos sobre la acción antiproliferativa de la SSen la próstata sólo desde la década de los ochen-ta. Inicialmente se pensó que este efecto seríaindirecto dada la capacidad de la SS para inhi-bir la liberación de GH y de prolactina. Sinembargo, Di Sant’Agnese, en 198482, describepor primera vez en próstata humana la presen-cia de células endocrina-paracrinas que secre-tan somatostatina.
Los estudios realizados hasta el momentodemuestran que diferentes análogos de la SS soncapaces de reducir el tamaño y el volumen detumores prostáticos inducidos en ratones. Enxenógrafos inoculados con células PC3, Pinski ycols.83 encontraron que el RC-160 era capaz dereducir el tamaño y el volumen de tumores pros-táticos, siempre que el tratamiento comenzara enestadios tempranos del tumor. Este análogo pro-ducía una disminución de los niveles de GH, IGF-I y EGFR.
Sin embargo, los escasos datos clínicos queexisten acerca de la utilización de la SS en el car-cinoma prostático son contradictorios, dado queensayos clínicos realizados por diferentes equiposarrojan resultados opuestos. Verhelst y cols.84
administraron SMS 201-995, sólo o combinadocon análogos de LHRH, a pacientes que padecíancarcinoma de próstata metastático y observaroninhibición moderada y temporal del crecimientodel tumor. Por el contrario, Logothetis y cols.24
utilizaron el mismo análogo con un grupo de 24pacientes que padecían cáncer de próstata avan-zado y refractario al tratamiento hormonal, obte-niendo resultados desalentadores. El SMS 201-995, después de dos meses de tratamiento, ace-
277
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
SS
Complejo sst2 - Giα3 - SHP1
SS - sst2
Giβγ
Giα
SHP -1
FIGURA 5. Modelo de activación de SHP-1 por SS. En el estado basal la SHP-1 se encuentra inactiva y formando un com-plejo con el receptor sst2 y Giαα3. La unión de la SS a su receptor induce la disociación y la activación de SHP-1 quepuede unirse, a través de sus dominios SH2, a otras proteínas regulando su actividad mediante defosforilación.

leró el crecimiento del tumor y produjo un rápidoaumento del PSA apareciendo nuevas metástasisóseas y viscerales. Maulard y cols.23 analizaron elefecto del tratamiento con lanreotide (BIM 32014)sobre pacientes con cáncer prostático resistentea hormonas, observando cierta mejoría en elestado general y disminución o estabilización delos niveles de PSA. Por otro lado, Figg y cols.22 noencontraron ninguna mejora clínica al tratarpacientes con cáncer de próstata metastásico condosis crecientes de somatulina (SMS 201-995).Vainas y cols.85 realizaron ensayos clínicos defase II, administrando SMS 201-995 a pacientescon bloqueo androgénico total, encontrando queel tratamiento mejora el estado general y prolon-ga la fase libre de síntomas. Recientemente,Berruti y cols.86 han encontrado efectos positivosdel lanreotide administrado a pacientes en faseindependiente de andrógenos. El análogo produ-jo una disminución de los niveles plasmáticos decromogranina A e IGF-I, sin observarse cambiosen los niveles de PSA. La razón de estas discre-pancias y, en muchos casos, del escaso efectoterapéutico puede deberse a modalidades de apli-cación inadecuadas (dosis, régimen, tipo de aná-logos utilizados), a la diferente eficacia de la SSdependiendo de los sitios de actuación (sistemaendocrino, inmune, neurovascular y el propiotumor) o al desconocimiento de los subtipos dereceptores para SS y de los diferentes sistemas detransducción de señales utilizados que, en últimocaso, determinarán las funciones celulares quevan a ser modificadas.
Diversos grupos de investigación han demos-trado la presencia de receptores de SS en célulasprostáticas normales y tumorales. Reubi ycols.87 mediante hibridación in situ combinadacon técnicas autoradiográficas, demostraron lapresencia de sst2 en próstata normal y de sst1en cáncer de próstata. En ambos casos losreceptores estaban localizados exclusivamenteen células estromales. Posteriormente, Sinisi ycols. en 199788, mediante RT-PCR realizada conRNA procedente de células epiteliales y estroma-les cultivadas, demuestran que sólo aparecenreceptores para somatostatina en las células epi-teliales. Así, mientras que las células proceden-tes de próstata normal expresan sst2, sst4 ysst5, las procedentes de cáncer de próstata
expresan sst1, sst4 y sst5. Halmos y cols.89
encontraron sst1, sst2 y sst5 en 80 muestras deprostatectomía radical analizadas por RT-PCR.Desde un punto de vista cuantitativo la expre-sión del receptor sst2 fue minoritaria (14%),mientras que sst5 y sst1 aparecieron de formadominante (64% y 86%, respectivamente) lo queparecía demostrar que en el cáncer de próstata,al igual que en otros tipos de tumores, se perdíasst2, aunque los propios autores compruebanque la expresión de los receptores para SS no seve afectada por la progresión del tumor.Recientemente, nuestro grupo de investigaciónha demostrado que las líneas tumorales prostá-ticas humanas PC3 y LNCaP expresan sst2 ysst5; pero además de tener los citados receptoresestas células producen y secretan SS. La expre-sión conjunta por la misma célula del péptido yde sus correspondientes receptores generaba unbucle autocrino que demostró ser funcional,regulando de forma negativa la proliferacióncelular63.
Por lo tanto, aunque se ha demostrado clara-mente la presencia de receptores para somatos-tatina en células prostáticas normales o tumora-les, los resultados obtenidos sobre su localiza-ción, los subtipos que se expresan y su relacióncon diferentes situaciones fisiopatológicas no sonconcluyentes.
Son muchas las preguntas sin respuestas res-pecto a la implicación de la SS en la fisiopatolo-gía prostática. Creemos que para poder dilucidartodos estos interrogantes resulta imprescindibleconocer los receptores que expresa la próstatanormal y el cáncer de próstata, cual de estos esresponsable del efecto antiproliferativo, y elmecanismo o mecanismos empleados; así comola contribución relativa de sus efectos directos eindirectos sobre el control de la proliferacióncelular de la próstata y en la diferenciación neu-roendocrina prostática.
SHP-1 Y CÁNCER DE PRÓSTATAA pesar de la gran cantidad de estudios que
existen sobre los efectos de la SS y de sus análo-gos en el cáncer de próstata, la mayoría de éstosse centran en investigar la presencia o ausenciade los diversos receptores, y en la realización deensayos clínicos para evaluar el potencial valor
278
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.

terapéutico de este péptido. Nuestro grupo es unode los pioneros en dilucidar los mecanismosmoleculares utilizados por la SS para regular elcrecimiento de la próstata. Creemos que estudiosde este tipo son de suma importancia, dado quela presencia de los receptores no garantiza que elpéptido pueda realizar sus efectos. De hecho,debemos ser conscientes que la presencia deun/os receptor/es no garantiza necesariamentesu funcionamiento ni su empleo terapéutico.Sabemos que en ocasiones se produce un dese-quilibrio entre los receptores y los mecanismosintracelulares que haría ineficaz la utilización deanálogos de SS, o incluso que podría producirresultados adversos.
En 1993, Brevini y cols.90 sugirieron que lasomatostatina era capaz de inhibir directamentela secreción y la proliferación de células LNCaP.El mecanismo de este efecto parecía estar media-do por la activación de una fosfotirosina fosfata-sa no identificada. Recientemente nuestro grupode investigación ha demostrado de forma originalla presencia de SHP-1 en próstata ventral derata91 y en próstata humana63,92.
SHP-1 también se ha encontrado en las líneastumorales prostáticas humanas PC3 y LNCaP93.Las observaciones indican que la SS, producidaendógenamente por estas líneas celulares,aumenta la cantidad y la actividad de SHP-1; dehecho, el bloqueo de este péptido aumenta la pro-liferación celular coincidiendo con una disminu-ción de la actividad y la cantidad de SHP-1. Porlo tanto, la SHP-1 es un componente más delbucle autocrino para SS detectado en estas líne-as celulares, capaz de inhibir la proliferacióncelular. Sin embargo, estas líneas celularesexpresan otras PTPs como la SHP-2 y la PTP1 B94
(Fig. 6), que también podrían participar en el con-trol de la proliferación celular como sugierenalgunos autores95.
La SHP-1 se localiza en las células epitelialesductales y acinares de la próstata humana y de lapróstata ventral de rata63,94. Tanto en la próstatade la rata como en la humana, la ubicación de laenzima se restringe a la zona apical de las célulasepiteliales ductales, mientras que en las célulasacinares la distribución es menos restrictiva. Esprobable que la distribución de esta enzima coin-cida con la localización de receptores o moléculas
con las que SHP-1 va a interaccionar, por lo quepara dilucidar su significado será necesario cono-cer con detalle los procesos celulares en los queestá involucrada. En cualquier caso, las célulasepiteliales prostáticas pasan a integrar la limita-da lista de células no hematopoyéticas que expre-san SHP-1.
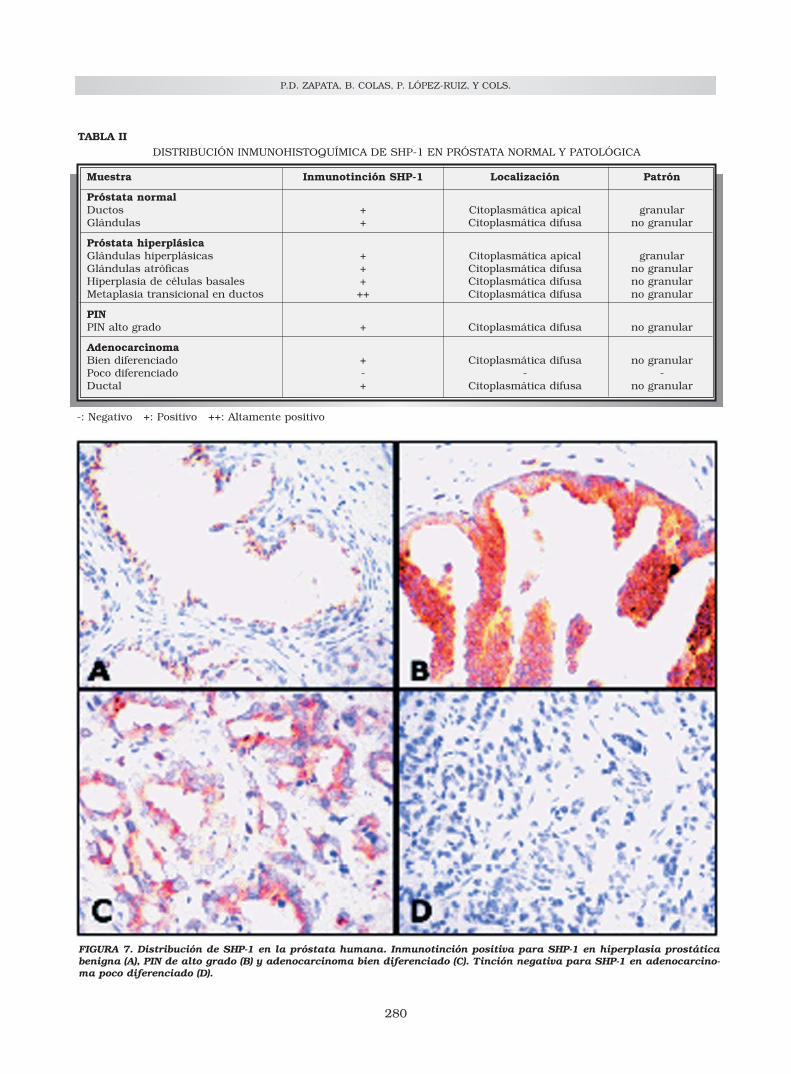
Asímismo, se ha realizado un estudio entumores prostáticos humanos de la expresión ylocalización de SHP-1, dependiendo del grado dediferenciación del tumor (Tabla II). Nuestroshallazgos indican que la enzima está presente enalteraciones no malignas, así como en adenocar-cinomas en estadios tempranos; pero que desa-parece a medida que el tumor es más indiferen-ciado. Es decir, tumores con patrón de Gleasonmás alto muestran menos expresión de SHP-1que tumores con bajo Gleason63 (Fig. 7). Estosresultados están en el mismo sentido que losobtenidos en las líneas celulares: la cantidad deSHP-1 expresada en LNCaP es mayor que en PC3.Las células LNCaP que representan una fase tem-prana del cáncer de próstata y son más diferen-ciadas, tienen más SHP-1 que las PC3. Además,cuando las células PC3 se inyectan en ratones atí-micos demuestran una agresividad e invasividadsuperior a las LNCaP al formar tumores más indi-ferenciados. Por lo tanto, la pérdida de expresiónde SHP-1 puede relacionarse estrechamente conel desarrollo y la progresión del cáncer de prósta-ta. En otros tipos de tumores también se hadetectado baja expresión de SHP-1; entre otros enadenocarcinomas pancreáticos96, linfomas97-99 y
279
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA
SHP-1sst
SS
SS
FIGURA 6. Bucle autocrino antiproliferativo. La SS secre-tada por la célula epitelial prostática actúa de formaautocrina sobre receptores sst2 y sst5, presentes en lasmismas células, e inhibe la proliferación celular median-te la activación de la fosfotirosina fosfatasa SHP-1.
280
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.
TABLA IIDISTRIBUCIÓN INMUNOHISTOQUÍMICA DE SHP-1 EN PRÓSTATA NORMAL Y PATOLÓGICA
Muestra Inmunotinción SHP-1 Localización Patrón
Próstata normalDuctos + Citoplasmática apical granularGlándulas + Citoplasmática difusa no granular
Próstata hiperplásicaGlándulas hiperplásicas + Citoplasmática apical granularGlándulas atróficas + Citoplasmática difusa no granularHiperplasia de células basales + Citoplasmática difusa no granularMetaplasia transicional en ductos ++ Citoplasmática difusa no granular
PINPIN alto grado + Citoplasmática difusa no granular
AdenocarcinomaBien diferenciado + Citoplasmática difusa no granularPoco diferenciado - - -Ductal + Citoplasmática difusa no granular
-: Negativo +: Positivo ++: Altamente positivo
FIGURA 7. Distribución de SHP-1 en la próstata humana. Inmunotinción positiva para SHP-1 en hiperplasia prostáticabenigna (A), PIN de alto grado (B) y adenocarcinoma bien diferenciado (C). Tinción negativa para SHP-1 en adenocarcino-ma poco diferenciado (D).

policitemia vera100. En algunos estudios la pér-dida de SHP-1 se asocia a mayor sensibilidad alos efectos mitogénicos de los factores de creci-miento.
Como ya hemos mencionado, los factores decrecimiento están directamente implicados en elcontrol del crecimiento de la próstata. Una de lasalteraciones más frecuentes durante el desarrollodel carcinoma de próstata, es la aparición debucles autocrinos para factores de crecimiento, loque sin duda representa una adaptación a ladeprivación androgénica. Esta deprivación tam-bién podría generar un bucle autocrino para laSS, que sería incapaz de inhibir la proliferacióncelular en ausencia de SHP-1. Recientemente,Halmos y cols.89 han demostrado que la expre-sión de los receptores para SS no se ve afectadapor la progresión del tumor. Por lo tanto, altera-ciones post-receptor, como la pérdida de la SHP-1, podrían implicarse en la progresión del cáncerde próstata al permitir que persistan las señalesgeneradas por los factores de crecimiento. Eneste sentido Douziech y cols.96 demuestran quela SS tiene efectos opuestos sobre la proliferaciónde células derivadas de cáncer de páncreas,dependiendo de la presencia o no de SHP-1. Enausencia de esta PTP, la SS no inhibe la prolife-ración celular.
La distribución difusa de SHP-1 en tumoresbien diferenciados puede indicar pérdida de lapolaridad celular y alteraciones iniciales de losmecanismos en los que se involucra esta enzima.Por otro lado, la pérdida de expresión de SHP-1 enfases avanzadas de la enfermedad, junto con elconocimiento de la relación que existe entre SHP-1 y SS, y la observación clínica de que la enferme-dad neoplásica prostática hormonorresistente serelaciona con fenómenos neuroendocrinos localespoco conocidos a día de hoy, nos llevan a conside-rar que la proteína fosfotirosina fosfatasa objeto deesta revisión (SHP1) está definitivamente implica-da en la progresión del cáncer de próstata. En estemomento estamos estudiando un mayor númerode muestras humanas, con intención de aclarar supapel en la patología humana y comprender mejorel significado de los patrones de distribución quepreceden a su pérdida total.
El conjunto de todos los resultados comenta-dos demuestran que los efectos antiproliferativos
de la SS sobre la próstata están, al menos enparte, mediados por la fosfotirosina fosfatasaSHP-1, presente en estas células y activada acorto y largo plazo por la SS. Esta enzima tam-bién está presente en muestras de próstatahumana con hiperplasia prostática benigna y concáncer bien diferenciado. Ahora bien, está ausen-te en cáncer de próstata avanzado poco diferen-ciado. Pensamos que SHP-1 juega un papel claveen el control de la proliferación celular prostáticay su presencia determinará el potencial terapéu-tico de la SS en el control del cáncer de próstata.
INVESTIGACIONES DE FUTUROEstos resultados abren a su vez la puerta a
nuevas investigaciones. Se necesita estudiarmejor si la expresión de SHP-1 puede utilizarsecomo marcador de pronóstico o para clasificar lostumores, tal y como sucede en el caso de los lin-fomas de células B pequeñas97. Un estudio para-lelo de la expresión de SS y de sus receptores pro-porcionará una visión global de todos los elemen-tos del bucle autocrino que hemos descubierto, yla participación del mismo en el cáncer de prós-tata. Es posible que el patrón de expresión dereceptores para SS en líneas celulares prostáti-cas humanas no sea representativo del que seencuentra in vivo. En cualquier caso, estas líneascelulares son útiles a la hora de analizar los efec-tos de pérdida o ganancia de un determinado tipode receptor de SS o de SHP-1 sobre el fenotipocelular. Recientemente se han publicado estudiosde terapia génica con tumores pancreáticos quecarecen de sst2. La restauración de la expresiónde este receptor induce un bucle autocrino parala SS mediado por SHP-1, a través del cual semodula la proliferación, adherencia e invasióncelular28,101.
Es posible que un mejor conocimiento de losmecanismos moleculares implicados en el efectoantiproliferativo de la SS permita explicar losresultados contradictorios de los ensayos clínicosrealizados hasta el momento o que, por lo menos,permita una mejor planificación de futuros estu-dios. El desarrollo de nuevas terapias, o de tera-pias combinadas, para el carcinoma avanzadoque no responde a la deprivación androgénica esuno de los principales retos que tiene hoy laoncología urológica.
281
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA

Agradecimientos: A la Fundación para laInvestigación en Urología por las diversas becasconcedidas, becas que han hecho posible la con-solidación de este grupo y la financiación de susaportaciones a la literatura.
REFERENCIAS11. DENIS LJ.: Maximal androgen blockade: facts and
fallacies. Endocr Rel Cancer 1998; 5: 353-356.12. GRIFFITHS K.: Molecular control of prostate
growth. In textbook of benign prostatic hyperpla-sia, 1 edn. Edited by Kirby R, Mc Connell JD,Fitzpatrick JM, Roehrborn CG, Boyle P. Oxford(UK): Isis Medical Media 1996: 23-55.
13. RUIJTER E, VAN DE KAA C, MILLER G, RUITER D,DEBRUYNE F, SCHALKEN J.: Molecular geneticsand epidemiology of prostate cancer. Endoc Rev1999; 20: 22-45.
14. RUSSELL DW, WILSON JD.: Steroid 5α reductase:two genes/two enzymes. Rew Biochem 1994; 63: 25.
15. JENSTER G.: The role of the androgen receptor inthe development and progression of prostate can-cer. Semin Oncol 1999; 26: 407-421.
16. BERTHON P, WALLER AS, VILLETTE JM.:Androgen are not a direct requirement for the pro-liferation of human prostatic epithelium in vitro. IntJ Cancer 1997; 73: 910-916.
17. BYRNE RL, LEUNG H, NEAL DE.: Peptide growthfactors in the prostate as mediators of stromalepithelial interactions. Br J Urol 1996; 77: 627-633.
18. RUSSELL P, BENNETT S, STRICKER P.: Growthfactor involvement in progression of prostate can-cer. Clin Chem 1998; 44: 705-723.
19. DJAKIEW D.: Dysregulated expression of growthfactors and their receptors in the development ofprostate cancer. Prostate 2000; 42: 150-160.
10. ABRAHAMSSON PA.: Neuroendocrine differentia-tion in prostatic carcinoma. Prostate 1999; 39:135-148.
11. APRIKIAN A, HAN K, GUY L, LANDRY F, BEGIN L,CHEVALIER S.: Neuroendocrine differentiation andthe bombesin/gastrin-releasing peptide family ofneuropeptides in the progression of humanprostate cancer. Prostate Suppl 1998; 8: 52-61.
12. JIBORN T, BJARTELL A, ABRAHAMSSON PA.:Neuroendocrine differentiation in prostatic carcino-ma during hormonal treatment. Urology 1998; 51:585-589.
13. BONKHOFF H.: Neuroendocrine cells in benign andmalignant prostate tissue: morphogenesis, prolifer-ation, and androgen receptor status. Prostate 1998;8: 18-22.
14. DI SANT’AGNESE PA.: Neuroendocrine differentia-tion in prostatic carcinoma. Hum Pathol 1992; 23:287-296.
15. PATEL YC.: General aspects of the biology andfunction of somatostatin. In Basic and cliniclaaspects of neuroscience, 1992 edn. Edited by WeilC, Muller EE, Thorner MO. Berlin: Springer-Verlag1992: 1-16.
16. POLLAK MN, SCHALLY AV.: Mechanisms of anti-neoplastic action somatostatin analogs. Proc SocExp Biol Med 1998; 217: 1431-1452.
17. LAMBERTS SWJ, KRENNING EP, REUBI JC.: Therole of somatostatin and its analogs in the diagno-sis and treatment of tumors. Endocrine Rev 1991;12: 450-482.
18. YAMADA Y, POST SR, WANG K et al.: Cloning andfunctional characterization of a family of humanand mouse somatostatin receptors expressed inbrain, gastrointestinal tract, and kidney. Proc NatlAcad Sci USA 1992; 89: 251-255.
19. PATEL YC.: Somatostatin and its receptor family.Front Neuroendocrinol 1999; 20: 157-198.
20. WECKBECKER G, RAULF F, BODMER D, BRUNSC.: Indirect antiproliferative effect of the somato-statin analog octreotide on MIA PaCa-2 humanpancreatic carcinoma in nude mice. Yale J Biol Med1998; 70: 549-554.
21. LAMBERTS SW, VAN DER LELY A, DEHARDERWW, HOFLAND LJ.: Drug therapy: octreotide. NewEng J Med 1996; 334: 246-254.
22. FIGG WD, THIBAULT A, COOPER MR et al.: Aphase I study of the somatostatin analogue somat-uline in patients with metastatic hormone refracto-ry prostate cancer. Cancer 1995; 15: 2159-2164.
23. MAULARD C, RICHAUD P, DROZ JP, JESSUELD D,DUFOUR-ESQUERRE F, HOUSSET M.: Phase I-IIstudy of the somatostatin analogue lanreotide inhormone-refactory prostate cancer. Cancer Chemo-ther Pharmacol 1995; 36: 259-262.
24. LOGOTHETIS CJ, HOSSAN EA, SMITH TL.: SMS201-995 in the treatment of refractory prostaticcarcinoma. Anticancer Res 1994; 14: 2731-2734.
25. BUSCAIL L, DELESQUE N, ESTEVE JP et al.:Stimulation of tyrosine phosphatase and inhibitionof cell proliferation by somatostatin analogues:mediation by human somatostatin receptor subty-pes SST1 and SST2. Proc Natl Acad Sci USA 1994;91: 2315-2319.
26. PATEL YC, GREENWOOD MT, KENT G, PANETTAR, SRIKANT CB.: Multiple gene transcripts of thesomatostatin receptor SSTR2: tissue selective dis-tribution and cAMP regulation. Biochem BiophysRes Commun 1993; 192: 288-294.
27. BUSCAIL L, SAINT-LAURENT N, CHASTRE E et al.:Loss of sst2 somatostatin receptor gene expressionin human pancreatic and colorectal cancer. CancerRes 1996; 56: 1823-1827.
28. ROCHAIX P, DELESQUE N, ESTEVE JP et al.:Gene therapy for pancreatic carcinoma: local anddistant antitumor effects after somatostatin recep-tor sst2 gene transfer. Hum Gene Ther 1999; 10:995-1008.
29. DELESQUE N, BUSCAIL L, ESTEVE JP et al.: sst2somatostatin receptor expression reverses tumori-genicity of human pancreatic cancer cells. CancerRes 1997; 57: 956-962.
30. TONKS NK, CHARBONNEAU H.: Protein tyrosinedephosphorylation and signal transduction. TIBS1989; 14: 497-500.
31. FAUMAN EB, SAPER MA.: Structure and functionof the protein tyrosine phosphatases. TIBS 1996;21: 413-417.
32. TONKS NK, NEEL BG.: Combinatorial control ofthe specificity of protein tyrosine phosphatases.Curr Opin Cell Biol 2001; 13: 182-195.
282
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.

33. SUN H, TONKS NK.: The coordinated action of pro-tein tyrosine phosphatases and kinases in cell sig-naling. TIBS 1994; 19: 480-485.
34. KEYSE S.M.: Protein phosphatases and the regula-tion of mitogen activated protein kinase signaling.Curr Opin Cell Biol 2000; 12: 186-192.
35. WISHART MJ, TAYLOR GS, SLAMA JT, DIXON JE.:PTEN and myotubularion phosphoinositide phos-phatases: bringing bioinformatics to the lab bench.Curr Opin Cell Biol 2001; 13: 172-181.
36. BLONDEAU F, LAPORTE J, BODIN S, SUPERTI-FURGA G, PAYRASTRE B, MANDEL J.: Myorubu-larin, a phosphatase deficient in myotubular myo-pathy, acts on phosphatidylinositol 3-kinase andphosphatidylinositol 3-phosphate pathway. HumMol Genet 2000; 9: 2223-2229.
37. SADOWSKI Y, STONE JC, PAWSON T.: A non cat-alytic domain conserved among cytoplasmic pro-tein-tyrosine kinases modifies the kinase functionand transforming activity of Fujinami sarcomavirus P130 gag-fps. Mol Cell Biol 1986; 6: 4396-4408.
38. SHEN S, BASTIEN L, POSNER BI, CHRETIEN P.: Aprotein-tyrosine phosphatase with sequence simi-larity to the SH2 domain of the protein-tyrosinekinases. Nature 1991; 352: 736-739.
39. PLUTZKY J, NEEL BG, ROSENBERG RD.: Isolationof a src homology 2-containing tyrosine phospha-tase. Proc atl Acad Sci USA 1992; 89: 1123-1127.
40. MATTHEWS RJ, BOWNE DB, FLORES E, THOMASML.: Characterization of hematopoietic intracellu-lar protein tyrosine phosphatases: description of aphosphatase containing an SH2 domain andanother enriched in proline-, glutamic acid-, ser-ine-, and threonine-rich sequences. Mol Cell Biol1992; 12: 2396-2405.
41. VOGEL W, LAMMERS R, HUANG J, ULLRICH A.:Activation of a phosphotyrosine phosphatase bytyrosine phosphorylation. Science 1993; 259:1611-1614.
42. AHMAN S, BANVILLE D, ZHAO Z, FISCHER EH,SHEN SH.: A widely expressed human protein tyro-sine phosphatase containing src homology 2domains. Proc Natl Acad USA 1994; 90: 2197-2201.
43. FREEMAN JRRM, PLUTZKY J, NEEL BG.:Identification of a human src homology 2-contai-ning protein tyrosine-phosphatase. Proc Natl AcadSci USA 1992; 89: 11239-11243.
44. FENG GS, HUI CC, PAWSON T.: SH2-containingphosphotyrosine phosphatase as a target of proteintyrosine kinase. Science 1993; 259: 1607-1611.
45. ADACHI M, SEKIYA M, MIYACHI T et al.: Molecularcloning and sequencing of a novel protein-tyrosinephosphatase SH-PTP3 with sequence similarity tothe src-homology 2 region. FEBS Let 1993; 314:335-339.
46. MATOZAKI T, UCHIDA T, FUJIOKA Y, KASUGA M.:Src kinase tyrosine phosphorylates PTP1C, a proteintyrosine phosphatase containing Src homology-2domains that down-regulates cell proliferation.Biochem Biophys Res Commun 1994; 204: 871-881.
47. MATOZAKI T, KASUGA M.: Roles of protein-tyro-sine phosphatases in growth factor signaling. CellSignal 1996; 8: 13-19.
48. BANVILLE D, STOCCO R, SHEN SH.: Human pro-tein tyrosine phosphatase 1C (PTPN6) gene struc-ture: alternate promoter usage and exon skippinggenerate multiple transcripts. Genomics 1995; 27:165-173.
49. SHULTZ LD, GREEN MC.: Motheaten, an immunod-eficient mutant of the mouse. II. Depressed immunecompetence and elevated serum immunoglobulins. JImmunol 1976; 116: 936-943.
50. SHULTZ LD, RAJAN TV, GREINER DL.: Severedefects in immunity and hematopoietic caused bySHP-1 protein-tyrosine phosphatase deficiency.Trends Biotechnol 1997; 15: 302-307.
51. SCHWARTZ MA, BARON V.: Interactions betweenmitogenic stimuli, or, a thousand and one connec-tions. Curr Opin Cell Biol 1999; 11: 197-202.
52. TOMIC S, GREISER U, LAMMERS R et al.:Association of SH2 domain protein tyrosine phos-phatases with the epidermal growth factor receptorin human tumor cells. Phosphatidic acid activatesreceptor dephosphorylation by PTP1C. J Biol Chem1995; 270: 21277-21284.
53. TENEV T, KEILHACK H, TOMIC S et al.: Both SH2domain are involved in interaction of SHP-1 withthe epidermal growth factor receptor but cannotconfer receptor-directed activity to SHP-1/SHP-1chimera. J Biol Chem 1997; 272: 5966-5973.
54. VAMBUTAS V, KAPLAN DR, SELLS MA, CHER-NOFF J.: Nerve growth factor stimulates tyrosinephosphorylation and activation of Src homology-containing protein-tyrosine phosphatase 1 in PC12cells. J Biol Chem 1995; 270: 25629-25633.
55. UCHIDA T, MATOZAKI T, NOGUCHI T et al.:Insulin stimulates the phosphorylation of Tyr538
and the catalytic activity of PTP1C, a protein tyro-sine phosphatase with Src homology-2 domains. JBiol Chem 1994; 269: 12220-12228.
56. MACHIDE M, KAMITORI K, KOHSAKA S.:Hepatocyte growth factor-induced differential acti-vation of phospholipase Cγ1 and phosphatidylinos-itol 3-kinase is regulated by tyrosine phosphataseSHP-1 in astrocytes. J Biol Chem 2000; 275:31392-31398.
57. YI T, MUI ALF, KRISTAL G, IHLE JN.: Hemato-poietic cell phosphatase associates with theinterleukin-3 (IL-3) receptor b chain and down-regulates IL-3 -induced tyrosine phosphorylationand mitogenesis. Mol Cell Biol 1993; 13: 7577-7585.
58. KLINGMÜLLER U, LORENZ U, CANTLEY LC, NEELBG, LODISH HF.: Specific recruitment of SH-PTP1to the erythropoietin receptor causes inactivation ofJAK2 and termination of proliferative signals. Cell1995; 80: 729-738.
59. DAVID M, CHEN HE, LING L, GOELZ S, LARNERAC, NEEL BG.: Differential regulation of the a/binterferon-stimulated Jak/Stat pathway by theSH2-domain containing tyrosine phosphataseSHPTP1. Mol Cell Biol 1995; 15: 7050-7058.
60. JIAO H, BERRADA K, YANG W, TABRIZI M, PLA-TANIAS LC, YI T.: Direct association with anddephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphataseSHP-1. Mol Cell Biol 1996; 16: 6985-6992.
283
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA

61. BEDECS K, ELBAZ N, SUTREN M et al.:Angiotensin II type 2 mediate inhibition of mitogen-activated protein kinase cascade and functionalactivation of SHP-1 tyrosine phosphatase. BiochemJ 1997; 325: 449-454.
62. LÓPEZ F, ESTEVE JP, BUSCAIL L y cols.: The tyro-sine phosphatase SHP-1 associates with the sst2somatostatin receptor and is an essential compo-nent of sst2-mediated inhibitory growth signaling.J Biol Chem 1997; 272: 24448-24454.
63. ZAPATA P, ROPERO RM, VALENCIA AM y cols.:Autocrine regulation of PC3 prostate cell proliferationby somatostatin through the modulation of SHP-1. JClin Endocrinol Metabolism 2002; 87: 915-926.
64. SU L, ZHAO ZJ, BOUCHARD P, BANVILLE D, FIS-CHER EH, KREBS EG, SHEN S.: Positive effect ofoverexpressed protein-tyrosine phosphatase PTP1Con mitogen-activated signaling in 293 cells. J BiolChem 1996; 271: 10385-10390.
65. FLORIO T, YAO H, CAREY KD, DILLON TJ, STORKPJS.: Somatostatin activation of mitogen-activatedprotein kinase via somatostatin receptor 1 (SSTR1).Mol Endoc 1999; 13: 24-37.
66. SHARMA K, PATEL YC, SRIKANT CB.: C-terminalregion of human somatostatin receptor 5 isrequired for induction of Rb and G1 cell cyclearrest. Mol Endoc 1999; 13: 82-90.
67. REARDON DB, DENT P, WOOD SL, KONG T,STURGILL TW.: Activation in vitro of somatostatinreceptor subtypes 2, 3, or 4 stimulates proteintyrosine phosphatase activity in membranes fromtransfected ras- transformed NIH 3T3 cells: coex-pression with catalitically inactive SHP-2 blocksresponsiveness. Mol Endoc 1997; 11: 1062-1069.
68. ZEGGARI M, ESTEVE JP, RAULY I y cols.: Co-purifi-cation of a protein tyrosine phosphatase with acti-vated somatostatin receptors from rat pancreaticacinar membranes. Biochem J 1994; 303: 441-448.
69. BOUSQUET C, DELESQUE N, LÓPEZ F y cols.:sst2 somatostatin receptor mediates negative regu-lation of insulin receptor signaling through thetyrosine phosphatase SHP-1. J Biol Chem 1998;273: 7099-7106.
70. RAULY I, SAINT-LAURENT N, DELESQUE N ycols.: Induction of a negative autocrine loop byexpression of sst2 somatostatin receptor in NIH3T3 cells. J Clin Invest 1996; 97: 1874-1883.
71. PAGÈS P, BENALI N, SAINT-LAURENT N y cols.:sst2 somatostatin receptor mediates cell cyclearrest and induction of p27Kip1. J Biol Chem 1999;274: 15186-15193.
72. LÓPEZ F, FERJOUX G, CORDELIER P y cols.:Neuronal nitric oxide synthase is a SHP-1 sub-strate involved in sst2 somatostatin receptorgrowth inhibitory signaling. FASEB J 2001.
73. MEDINA DL, VELASCO JA, SANTISTEBAN P.:Somatostatin is expressed in FRTL-5 thyroid cellsand prevents thyrotropin-mediated down-regula-tion of the cyclin-dependent kinase inhibitorp27kip1. Endocrinology 1999; 140: 87-95.
74. MEDINA DL, TORO MJ, SANTISTEBAN P.:Somatostatin interferes with thyrotropin-inducedG1-S transition mediated by cAMP-dependent pro-tein kinase and phosphatidylinositol 3-kinase. JBiol Chem 2000; 275: 15549-15556.
75. SRIKANT CB, SHEN S.: Octapeptide somatostatinanalog SMS 201-995 induces translocation ofintracellular PTP1C to membranes in MCF-7human breast adenocarcinoma cells. Endocrinology1996; 137: 3461-3468.
76. SHARMA K, PATEL YC, SRIKANT CB.: Subtypeselective induction of wild type p53 and apoptosis,but not cell cycle arrest, by human somatostatinreceptor 3. Mol Endoc 1996; 10: 1688-1696.
77. SHARMA K, SRIKANT CB.: Induction of wild-typep53, Bax, and acid endonuclease during somato-statin-signaled apoptosis in MCF-7 human breastcancer cells. Int J Cancer 1998; 76: 259-266.
78. LIU D, MARTINO G, THANGARAJU M y cols.:Caspase-8-mediated intracellular acidification pre-cedes mitochondrial dysfunction in somatostatin-induced apoptosis. J Biol Chem 2000; 275: 9244-9250.
79. SHARMA K, SRIKANT CB.: G protein coupledreceptor signaled apoptosis is associated with acti-vation of a cation insensitive acidic endonucleaseand intracellular acidification. Biochem BiophysRes Commun 1998; 242: 134-140.
80. THANGARAJU M, SHARMA K, LEBER B,ANDREWS DW, SHEN S, SRIKANT CB.: Regulationof acidification and apoptosis by SHP-1 and Bcl-2.J Biol Chem 1999; 274: 29549-29557.
81. THANGARAJU M, SHARMA K, LIU D, SHEN SH,SRIKANT CB.: Interdependent regulation of intra-cellular acification and SHP-1 in apoptosis. CancerRes 1999; 59: 1649-1654.
82. DI SANT’AGNESE PA, DE MESY JENSEN KL.:Somatostatin and/or somatostatin like immunore-active endocrine-paracrine cells in the humanprostate gland. Arch Pathol Lab Med 1984; 108:693-696.
83. PINSKI J, SCHALLY AV, HALMOS G, SZEPESHAZIK.: Effect of somatostatin analog RC-160 andbombesin/gastrin releasing peptide antagonist RC-3095 on growth of PC-3 human prostate cancerxenografts in nude mice. Int J Cancer 1993; 55:963-967.
84. VERHELST J, DE LONGUEVILLE M, ONGENA P,DENIS L, MAHLER C.: Octreotide in advanced pro-static cancer relapsing under hormonal treatment.Acta Urol Belg 1994; 62: 83-88.
85. VAINAS G, PASAITOU V, GALAKTIDOU G y cols.: Therole of somatostatin analogues incomplete antiandro-gen treatment in patients with prostatic carcinoma. JExp Clin Cancer Res 1997; 16: 119-126.
86. BERRUTI A, DOGLIOTTI L, MOSCA A y cols.:Effects of the somatostatin analog lanreotide on thecirculating levels of chromogranin-A, prostate-spe-cific antigen and insulin-like growth factor-1 inadvanced prostate cancer patients. Prostate 2001;47: 205-211.
87. REUBI JC, WASER B, SCHAER J, MARKWALDERR.: Somatostatin receptors in human prostate andprostate cancer. J Clin Endocrinol Metab 1995; 80:2806-2814.
88. SUNISI AA, BELLASTELLA A, PREZIOSO D y cols.:Different expression patterns of somatostatinreceptor subtypes in cultured epithelial cells fromhuman normal prostate and prostate cancer. J ClinEndocrinol Metab 1997; 82: 2566-2569.
284
P.D. ZAPATA, B. COLAS, P. LÓPEZ-RUIZ, Y COLS.

89. HALMOS G, SCHALLY AV, SUN B, DAVIS R, BOST-WICK DG, PLONOWSKI A.: High expression ofsomatostatin receptors and messenger ribonucleicacid for its receptor subtypes in organ-confinedand locally advanced human prostate cancers. JClin Endocrinol Metab 2000; 85: 2564-2571.
90. BREVINI TA, BIANCHI R, MOTTA M.: Directinhibitory effect of somatostatin on the growth ofthe human prostatic cancer cell line LNCaP: possi-ble mechanism of action. J Clin Endocinol Metab1993; 77: 626-631.
91. VALENCIA AM, OLIVA JL, ANGULO JC, PRIETOJC, COLÁS B.: Identification of PTP1C in prostatetumor cells. Regulation by EGF. 92nd AnnualMeeting Amer. Urolog. Assoc., New Orleans,Louisiana (USA) 1997.
192. LÓPEZ JI, ANGULO J, COLÁS B y cols.: Identifi-cation of SHP-1 in the prostate and its possibleimplication in the development and progression ofprostate cancer. 91st Annual Meeting of theUnited States and Canadian Academy ofPathology, Chicago, IL, february 23 to march 1,2002.
193. ZAPATA P, ROPERO RM, LÓPEZ-RUIZ MP, LÓPEZJI, ANGULO J, COLAS B.: Somatostatina y cáncerde próstata. Revisiones en Urología 2000; 1: 7-16.
194. VALENCIA AM, OLIVA JL, BODEGA G y cols.:Identification of a protein-tyrosine phosphatase(SHP1) different from that associated with acidphosphatase in rat prostate. FEBS Let 1997; 406:42-48.
195. FLORIO T, THELLUNG S, ARENA S, CORSARO A,BAJETTO A, SCHETTINI G, STORK PJS.:Somatostatin receptor 1 (SSTR1)-mediated inhibi-tion of cell proliferation correlates with the activa-tion of the MAP kinase cascade: role of the phos-photyrosine phosphatase SHP-2. J Physiol 2000;94: 239-250.
196. DOUZIECH N, CALVO E, COULOMBE Z et al.:Inhibitory and stimulatory effects of somatostatinon two human pancreatic cancer cell lines: a pri-mary role for tyrosine phosphatase SHP-1.Endocrinology 1999; 140: 765-777.
197. KOSSEV PM, RAGHUNATH PN, BAGG A, SCHUS-TER S, TOMASZEWSKI JE, WASIK NA.: SHP-1expression by malignant small B-cell lymphomasreflects the maturation stage of their normal B-cellcounterparts. Am J Surg Pathol 2001; 25: 949-955.
198. ZHANG J, SOMANI A-K, SIMINOVITCH KA.: Rolesof the SHP-1 tyrosine phosphatase in the negativeregulation of cell signaling. Semin Immunol 2000;12: 361-378.
199. DELIBRIAS CC, FLOETTMANN JE, ROWE M,FEARON DT.: Down regulated expression of SHP-1 in Burkitt lymphomas and germinal center Blymphocytes. J Exp Med 1997; 186: 1575-1583.
100. WICKREMA A, CHEN F, NAMIN F, YI T et al.:Defective expression of SHP-1 phosphatase in poly-cythemia vera. Exp Hematol 1999; 27: 1124-1132.
101. BENALI N, CORDELIER P, CALISE D, PAGÈS P,ROCHAIX P, NAGY A, ESTEVE JP, POUR PM,SCHALLY AV, VAYSSE N, SUSINI C, BUSCAIL L.:Inhibition of growth and metastatic progression ofpancreatic carcinoma in hamster after somatostatinreceptor subtype 2 (sst2) gene expression and admi-nistration of cytotoxic somatostatin analog AN-238.Proc Natl Acad Sci USA 2000; 97: 9180-9185.
Dr. Javier C. AnguloServicio de Urología. Hospital Príncipe de AsturiasCtra. Acalá-Meco, s/n28805 Alcalá de Henares (Madrid)
(Trabajo recibido el 7 octubre de 2002)
285
FOSFOTIROSINA FOSFATASA SHP-1, SOMATOSTATINA Y CÁNCER DE PRÓSTATA















![RECEPTORES de membrana - Guía de Bioquímica · I. Con receptores intracelulares •Andrógenos •Calcitriol (1,25[OH] 2-D 3) •Estrógenos •Glucocorticoides •Mineralocorticoides](https://img.pdfslide.tips/doc/110x75/5ba0f21e09d3f2c06a8b5ae9/receptores-de-membrana-guia-de-bioquimica-i-con-receptores-intracelulares.jpg)