Embed Size (px)
Citation preview

1
Muhammad Ghadafi
NAMA KELOMPOK :
1. INTAN AYU A (113234008)
2. MUHAMAD GHADAFI (113234019)
3. FATIHATUR RIFKA A. (113234212)
KIMIA A & B 2011

2
1.1 Pengenalan sintesis organik
Sintesis organik adalah pembangun dari senyawa organik kompleks dengan bahan
awal senyawa sederhana oleh serangkaian reaksi kimia. Senyawa yang disintesis sintesis
(hasil sintesis) di alam disebut produk alami. Alam menyediakan sejumlah besar senyawa
organik dan kebanyakan memiliki sifat kimia dan farmasi yang menarik. Contoh produk
alami termasuk kolesterol (1.1), sebuah steroid yang ditemukan di sebagian besar jaringan
tubuh, limonene (1.2), sebuah terpene yang ditemukan dalam minyak lemon dan jeruk, kafein
(1.3), sebuah purin ditemukan dalam daun teh dan biji kopi, dan morfin (1.4), alkaloid
ditemukan dalam opium.
Sintesis dari molekul organik adalah aspek yang paling penting dari kimia organik.
Ada dua bidang utama penelitian di bidang sintesis organik, yaitu sintesis total (total sythesis)
dan metodologi (metodhology). Sebuah sintesis total adalah sintesis kimia lengkap senyawa
kimia organik yang komplek dari molekul yang simpel (sederhana), yang tersedia secara
komersial atau perkusor alami. Penelitian metodologi biasanya melibatkan tiga tahapan
utama, yaitu penemuan (Discovery), optimasi dan study lingkungan (optimization and study
of scope) dan keterbatasan (Limitations). Beberapa kelompok peneliti dapat melakukan
sintesis total untuk menampilkan metodologi baru dan dengan demikian menunjukkan
aplikasinya untuk sintesis kompleks senyawa lainnya.

3
Senyawa yang disintesis mungkin memiliki kerangka karbon kecil seperti vanili (1.5)
(aroma vanila) atau mungkin memiliki kerangka karbon yang lebih kompleks sebagai
penicillin G (1.6) (antibiotik) dan taksol (1.7) (digunakan untuk pengobatan beberapa jenis
kanker). Namun, tiga tantangan yang harus dipenuhi dalam merancang sebuah sintesis untuk
senyawa tertentu: (1) kerangka atom karbon atau kerangka yang ditemukan di kompleks yang
diinginkan harus dirakit (disusun); (2) gugus fungsional yang menjadi ciri senyawa yang
diinginkan harus diperkenalkan atau dirubah (difranformasikan) dari gugus lain pada posisi
yang tepat, dan (3) jika pusat stereogenik muncul, mereka harus diperbaiki dengan cara yang
tepat.
Dengan demikian, dalam rangka memahami sintesis sebuah molekul yang kompleks, kita
perlu mengetahui ikatan karbon-karbon yang membentuk reaksi, yaitu perubahan gugus
fungsi (functional groups interconversions) dan aspek stereokimia.
Ikatan karbon-karbon membentuk reaksi adalah alat yang paling penting untuk
membangun molekul organik. Reaksi dengan gugus fungsi dikonversikan menjadi yang lain
disedut sebagai perubahan gugus fungsi (functional groups interconversions) disingkat FGI.
Penyusunan ruang dari suatu subtitusi dapat mempunyai sebuah dampak yang signifikan pada
kereaktifan dan interaksi terhadap molekul yang lain. Banyak obat yang bersifat kiral harus
dibuat dengan kemurnian enantiomer yang tinggi karena enantiomer yang lain mungkin akan
tidak aktif atau memiliki efek samping. Dengan demikian, ada kebutuhan untuk
mengembangkan metode untuk mensintesis senyawa organik sebagai salah satu enantiomer

4
murni dan menggunakan teknik-tekni ini disebut sebagai sintesis asimetris (asymmetric
synthesis) (bagian 1.5).
Oleh karena itu, reaksi pembentukan ikatan karbon-karbon, sintesis asimetris, desain
ligan kiral baru, reaksi ramah lingkungan dan sintesis ekonomis atom adalah tujuan utama
penelitian saat ini.
1.2 Analisis Retrosintesis (Disconnection Approach)
EJ Corey1,2
membawa pendekatan yang lebih formal untuk mendesain sintesis, yang dikenal
sebagai analisis retrosintesis. Analisis sintesis dengan cara terbalik disebut analisis
retrosintesis atau secara alternatif pendekatan pendekatan diskoneksi. Analisis retrosintesis
atau retrosintesis adalah sebuah teknikvuntuk memecahkan masalah dalam perencanaan
sintesis, terutama yang disajikan oleh struktur yang kompleks. Dalam pendekatan ini, sintesis
direncanakan mundur mulai dari produk yang relatif kompleks menuju bahan yang tersedia
sebagai molekul yang sederhana (bahan awal) (Skema 1.1). Pendekatan ini menghendaki
pembangunan kerangka karbon dari molekul target, menempatkan gugus fungsional dan
kontrol yang tepat dari aspek stereokimia.
Tabel 1.1
Pembeda Sintesis Retrosintesis
Langkah Reaksi Retroreaksi
Tanda yang digunakan
Struktur awal Reaktan Target

5
Struktur akhir Produk Prekursor
Substrat yang dibutuhkan Reaksi kefungsionalan Retron (Sinton)
Terminologi yang digunakan dalam analisis sintetis dan retrosintesis ditunjukkan pada Tabel
1.1. Sebuah transformasi dalam kasus teknik retrosintesis dari reaksi Wittig ditampilkan
berikut:
Dengan cara yang sama, analisis retrosynthetic dari reaksi Diels-Alder diwakili
berikut:
Langkah retrosynthetic melibatkan pemecahan ikatan (s) untuk membentuk dua (atau lebih)
sinthon disebut sebagai pemutusan (diskoneksi). Sinton adalah sebuah fragmen ideal,
biasanya kation, anion atau radikal, hasil dari diskoneksi. Satu harus memilih pemutusan
yang sesuai dengan reaksi yang menghasilkan produk paling banyak.
FGI adalah proses transformasi dari gugus fungsi satu ke gugus fungsi yang lain
untuk membantu perencanaan sintetis dan untuk memungkinkan pemutusan sesuai reaksi
yang sesuai pula. Dalam merencanakan strategi sintetis, selain merancang cara membangun
rangka karbon dengan fungsi yang diperlukan, ada faktor lain yang harus diatasi termasuk
kontrol regiokimia dan stereokimia.
Poin di atas dijelaskan dengan pembahasan analisis retrosintesis dari sikloheksanol :

6
Hidroksikarbokation dan ion hidrida yang terbentuk setelah pemutusan sikloheksanol
merupakan sinton. Kesetaraan sintesis dari hidroksikarbokation dan ion hidrida adalah
sikloheksanon dan natrium borohidrida, berturut-turut. Dengan demikian, molekul target
sikloheksanol dapat dibuat dengan mereaksikan sikloheksanon dengan natrium borohidrida.
Ikatan C-C dari sikloheksanol dapat juga didiskoneksi seperti yang ditunjukkan di bawah ini:
Sintesis yang setara untuk karbokation sikloheksil adalah bromida sikloheksil. Dengan
demikian, sikloheksanol dapat dibuat dengan reaksi bromida sikloheksil dengan ion
hidroksida.
Namun, dalam kasus ini sikloheksena juga terbentuk, dengan demikian, metode ini tidak
dapat dianggap seefektif sebelumnya.
Sebuah pohon retrosintesis adalah grafik asiklik diarahkan untuk beberapa (atau
semua) retrosintesis yang memungkinkan dari sebuah target tunggal. Analisis retrosintesis,
kemudian, terdiri dari penerapan transformasi yang akan diterapkan pada molekul target,
sehingga menghasilkan semua prekursor dari mana target itu dapat dibuat dalam satu
langkah. Analisis dapat diulang untuk masing-masing perkusor. Prekursor molekul sehingga
dihasilkan dalam beberapa cara sederhana dari target dari mana itu berasal dan kemudian
dianggap sebagai target dan dianalisis sama. Analisis berakhir ketika prekursor dijabarkan,
yang dianggap relatif sederhana atau mudah tersedia, menghasilkan pohon intermediet
sintetis.

7
Hasil akhir adalah pohon retrosintesis lengkap yang akan berisi semua kemungkinan
sintesis target yang diberikan - wajar dan tidak masuk akal, efisien dan praktis. Tentu saja,
seperti pohon akan unmanageably (tidak termanageri) besar baik bagi manusia dan komputer,
bahkan ketika jumlah tingkat prekursor terbatas. Untuk menjaga ukuran pohon retrosintesis di
bawah kontrol, memeriksa semua kemungkinan pemutusan yang secara kimiawi sudah ada
(relatif ada) (sesuai dengan reaksi terkenal,reagen, efek mengarahkan). Prinsip-prinsip
panduan untuk seleksi ini yang disebut strategi.
Beberapa pedoman untuk retrosintesis diberikan di bawah ini :
1. Hal ini lebih baik menggunakan pendekatan konvergen daripada divergen untuk
molekul yang molekul.
2. Gunakan hanya pemutusan sesuai dengan memutus ikatan C-C dan ikatan C-X selama
memunkinkan.
3. Diskoneksi dapat diketahui dengan mudah menggunakan reaksi yang telah diketahui
4. Sintesis harus pendek.
5. Hal ini lebih baik menggunakan reaksi-reaksi yang tidak membentuk campuran.
6. Fokusnya adalah pada penghilangan pusat stereo (stereocentre) dibawah kontrol
stereo (stereocontrol). Stereocontrol dapat dicapai melalui kontrol baik mekanistik
atau pengendalian substrat.
Analisis sintetik dengan bantuan komputer yang ditunjuk OCSS (organic chemical
simulation of synthesis) dan LHASA (logic and heuristics applied to synthetic analysis)
dirancang untuk membantu ahli kimia sintetis dalam analisis oleh Corey et al.3,4
. LHASA
menghasilkan pohon sintetis intermediet dari molekul target dengan analisis dalam arah
retrosintesis.
Klik kimia (Clik chemistry) adalah pendekatan sintetis modular terhadap perakitan
suatu molekular baru yang benar-benar ada. Alam memiliki preferensi keseluruhan untuk
ikatan karbon-heteroatom lebih karbon ataupun ikatan karbon melalui heteroatom yang
reversibel. Jadi mengikuti jejak alam, istilah 'klik kimia'5 diciptakan oleh Kolb, Finn dan
Sharpless pada tahun 2001 untuk sintesis terbatas pada molekul yang mudah dibuat. Klik
kimia seperti yang didefinisikan oleh Sharpless adalah reaksi yang modular, mempunyai
lingkup yang luas, menghasilkan produk yang tinggi, yang terbentuk hanya produk ofensif,
yang stereospesifik, sederhana untuk melakukan dan membutuhkan penggunaan pelarut yang
tidak berbahaya. Dari semua reaksi yang termasuk dalam payung klik kimia, Huisgen

8
sikloadisi 1,3-dipole dari alkuna dan azide untuk menghasilkan 1,2,3-triazoles tidak
diragukan lagi contoh utama dari reaksi klik. Reaksi ini dipercepat dengan katalis tembaga(I),
tidak memerlukan gugus pelindung, dan hampir selesai konversi berlangsung. Reaksi
selektif, karena hanya 1,4-disubstitusi 1,2,3-triazole adalah satu-satunya produk yang
terbentuk dan tidak ada pembentukan triazole 1,5-disubstitusi, yang juga dibentuk dalam
diinduksi termal Huisgen sikloadisi (Skema 1.2).
Skema 1.2
Karena keandalan, spesifisitas dan biokompatibilitas klik kimia, aplikasi adalah
ditemukan di hampir semua bidang kimia modern dari penemuan obat untuk ilmu material.
1.3 Strategi Umpolung
Umpolung adalah kelas umum dari reaksi di mana reaktivitas karakteristik gugus atau atom
untuk sementara terbalik. Konsep umpolung sangat membantu terutama dengan gugus
karbonil. Tetapi untuk memahami konsep ini, adalah penting untuk memahami reaktivitas
normal gugus karbonil. Sebagai contoh, di bawah kondisi normal karbonil karbon elektrofilik
dan nukleofilik-karbon karena resonansi, seperti yang ditunjukkan di bawah ini:

9
Tapi jika polaritas dari senyawa karbonil dibalik, karbon asil menjadi nukelofilik. Ini adalah
suatu konversi dari karbon karbonil (1.8), dan karbon menjadi nukleofilik. Dasar yang kuat
bisa melepaskan hidrogen berdekatan dengan sulfur dalam dithiane untuk memberikan 2-
lithio-1 ,3-dithiane (1.9). Asil anion setara (1.9) dihasilkan dalam cara bereaksi dengan alkil
halida untuk memberikan produk teralkilasi (1.10). Akhirnya, karbonil kelompok
diregenerasi dengan tanpa dithiane (Skema 1.3). Dengan demikian, jenis inversi dari
polarisasi normal atom gugus fungsional dikenal sebagai umpolung.
Skema 1.3 Konversi heksanal menjadi keton dipentyl (reaksi corey-Seebach)
Dalam Skema 1.3, heksanal pada reaksi dengan 1,3-propanedithiol memberikan turunan 1,3-
dithiane (1.8). Sebuah basa kuat seperti n-Butyllithium yang mengandung proton untuk
memberikan kepada 2-lithio-1 ,3-dithiane 1.9, yang bereaksi dengan 1-Bromopentane untuk
memberikan produk teralkilasi 1.10. Perlakukan (1.10) dengan HgO dan BF3 (Boron
trifluorida) dalam THF berair (tetrahidrofuran) menghasilkan keton dipentyl (reaksi corey-
Seebach6 ). Dengan demikian, dithianyllithium (2-lithio- 1,3-dithiane) (1.9) adalah sebuah
'asil anion' setara sintetis.
The dithiane anion (1.9) juga bereaksi dengan asil halida, keton dan aldehida untuk
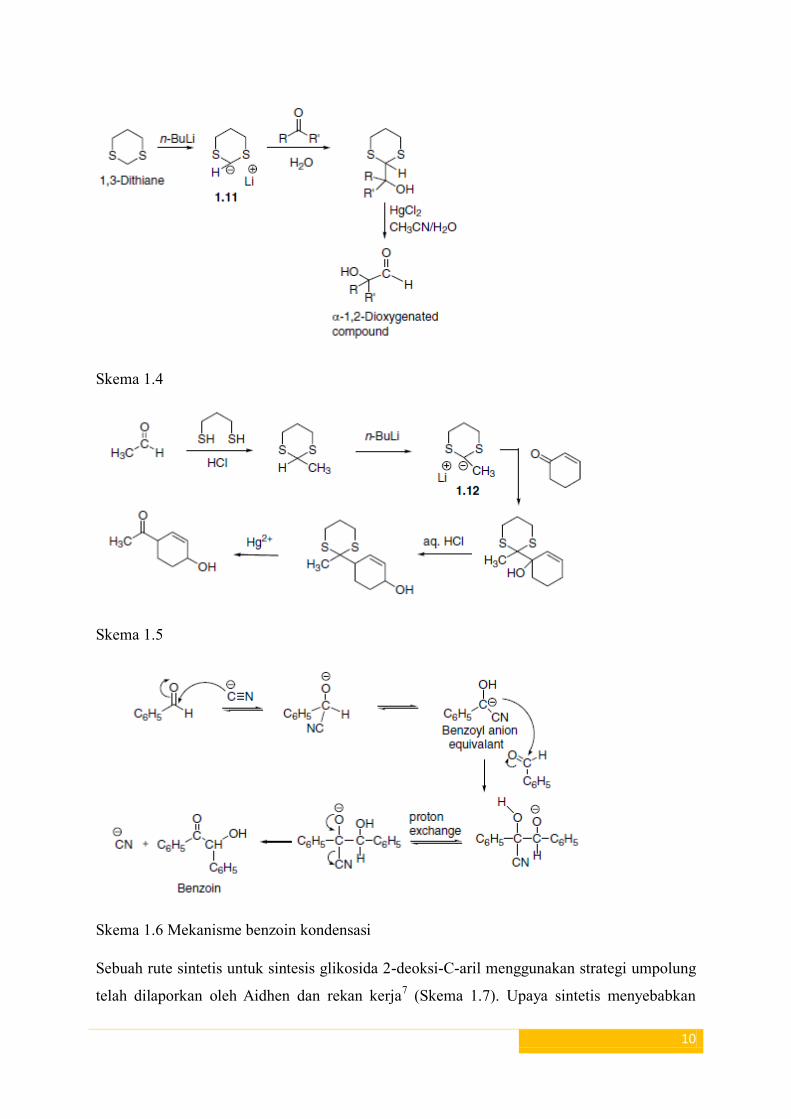
memberikan senyawa dioxygen yang sesuai. Skema (1.4) dan (1.5) menunjukkan reaksi
dithiane anion (1.11) dan (1.12) dengan keton. Contoh yang paling umum dari reaktivitas
umpolung dari gugus karbonil adalah kondensasi benzoin (Skema 1.6).
10
Skema 1.4
Skema 1.5
Skema 1.6 Mekanisme benzoin kondensasi
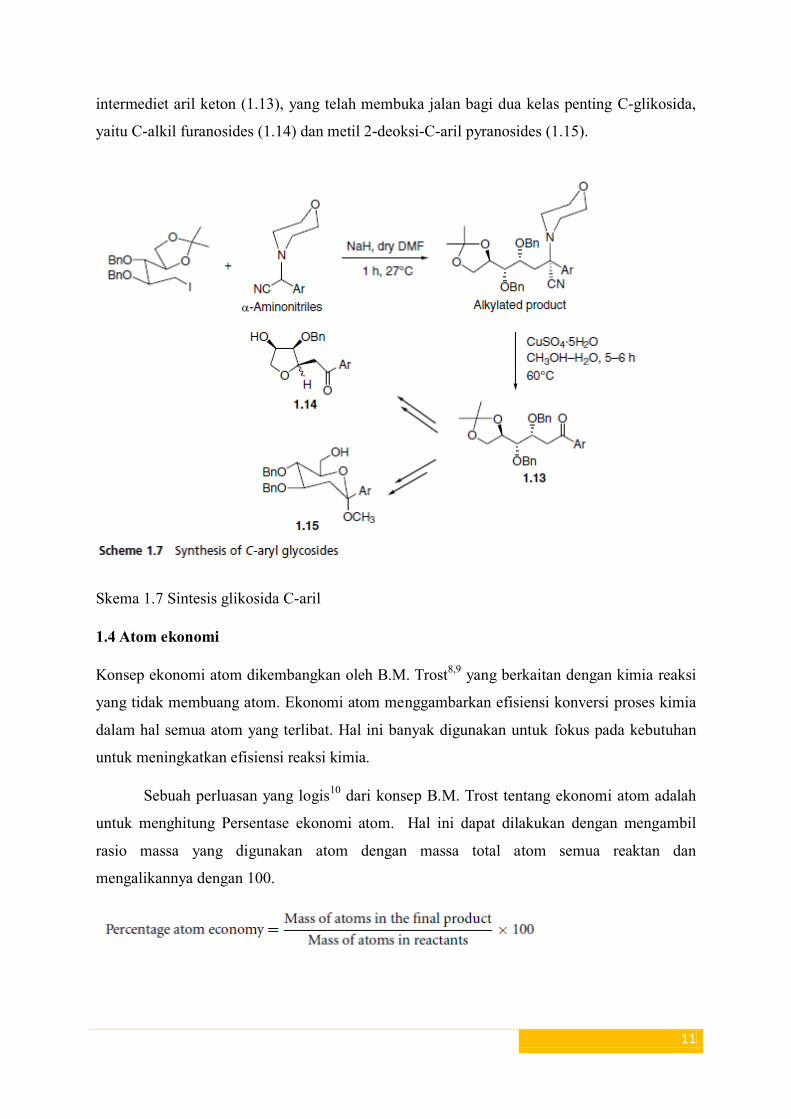
Sebuah rute sintetis untuk sintesis glikosida 2-deoksi-C-aril menggunakan strategi umpolung
telah dilaporkan oleh Aidhen dan rekan kerja7 (Skema 1.7). Upaya sintetis menyebabkan
11
intermediet aril keton (1.13), yang telah membuka jalan bagi dua kelas penting C-glikosida,
yaitu C-alkil furanosides (1.14) dan metil 2-deoksi-C-aril pyranosides (1.15).
Skema 1.7 Sintesis glikosida C-aril
1.4 Atom ekonomi
Konsep ekonomi atom dikembangkan oleh B.M. Trost8,9
yang berkaitan dengan kimia reaksi
yang tidak membuang atom. Ekonomi atom menggambarkan efisiensi konversi proses kimia
dalam hal semua atom yang terlibat. Hal ini banyak digunakan untuk fokus pada kebutuhan
untuk meningkatkan efisiensi reaksi kimia.
Sebuah perluasan yang logis10
dari konsep B.M. Trost tentang ekonomi atom adalah
untuk menghitung Persentase ekonomi atom. Hal ini dapat dilakukan dengan mengambil
rasio massa yang digunakan atom dengan massa total atom semua reaktan dan
mengalikannya dengan 100.

12
R.A. Sheldon11
telah mengembangkan konsep serupa yang disebut persentase pemanfaatan
atom. Untuk misalnya, persentase atom ekonomi dan persentase perhitungan atom yang
dimanfaatkan untuk Reaksi oksidasi benzena dengan anhidrida maleat diberikan di bawah ini:
Strategi sintetis
Bahkan jika reaksi itu untuk dilanjutkan dengan 100% hasil, hanya 44,14% (berat) dari atom
dari reaktan dimasukkan menjadi produk yang diinginkan, dengan 55,86% dari reaktan atom
berakhir sebagai produk samping.
Hal ini sering sulit untuk mengetahui struktur semua produk samping. Karena itu,
persentase atom ekonomi dapat ditentukan dengan membagi berat rumus molekul (MFW)
dari produk yang diinginkan oleh jumlah dari MFWs semua reaktan dan mengalikannya
dengan 100.
Persentase atom ekonomi reaksi ini adalah 44,14. Ini berarti bahwa 44,14% dari massa dari
reaktan berakhir pada produk yang diinginkan.
Perkembangan terbaru termasuk munculnya kimia hijau dan bahan baku yang tinggi
(minyak) harga semakin menuntut atom ekonomi yang tinggi. Dalam proses kimia yang
melibatkan tambahan yang sedrhana, dengan apa pun yang diperlukan hanya katalis, jumlah
bahan awal atau reaktan sama dengan jumlah semua produk yang dihasilkan dan tidak ada
atom yang terbuang. Itu Reaksi Diels-Alder adalah contoh dari reaksi atom berpotensi efisien.
Karena begitu sedikit dari reaksi yang digunakan ada penambahan, sintesis molekul
kompleks membutuhkan pengembangan metodologi atom ekonomi baru. Ekonomi atom
dapat ditingkatkan dengan hati-hati memilih bahan awal dan sistem katalis.

13
Sebuah contoh klasik dari meningkatkan rute ke produk komersial adalah ibuprofen
(1.16), yang merupakan analgesik (penghilang rasa sakit) dan juga efektif sebagai non-steroid
anti- inflamasi. Ibuprofen diproduksi menggunakan enam langkah (Skema 1.8) oleh Boots
Perusahaan, dengan ekonomi atom keseluruhan hanya 40%.
Skema 1.8 Sintesis ibuprofen oleh Perusahaan Boots Halaman 10
Total MFW semua reaktan yang digunakan adalah 514,5 (C20H42NO10ClN9 ) Dan total MFW
atom digunakan adalah 206 (ibuprofen; C13H18O2 ).
Pada 1990-an Hoechst Celanese Corporation (bersama-sama dengan perusahaan mereka
Boots membentuk proses BHC untuk mempersiapkan dan memesarkan ibuprofen , 1,16)
mengembangkan baru tiga tahap proses (Skema 1.9), dengan ekonomi atom dari 77,4%.

14
Skema 1.9 Sintesis ibuprofen oleh proses BHC
Total MFW semua reaktan yang digunakan adalah 266 (semua reagen; C15H22O4 ) Dan total
MFW atom digunakan adalah 206 (ibuprofen; C13H18O2 ).
Selain ekonomi atom yang lebih tinggi, dalam proses BHC, HF digunakan dalam jumlah
katalitik dan dapat terbentuk kembali dan digunakan kembali. Namun, dalam langkah
pertama dalam proses Boots, yang AlCl3 (Aluminium klorida) hidrat diproduksi dalam
jumlah besar sebagai produk samping karena AlCl3 digunakan dalam jumlah stoikiometri.
Dengan demikian, ada peningkatan yang signifikan dalam BHC proses atas proses Boots.
1.5 Selektivitas
B.M. Trost telah mencetuskan seperangkat kriteria yang proses kimianya dapat dievaluasi.
Masalah-masalah selektivitas dapat dikategorikan di bawah berikut judul: chemoselectivity,
regioselektivitas, Diastereoselektivitasnya dan enantioselectivity.
1.5.1 chemoselectivity
Chemoselectivity adalah diferensiasi antara berbagai gugus fungsional dalam polifungsional
molekul dengan reaktivitas preferensial satu kelompok fungsional atas yang lain. Sebagai
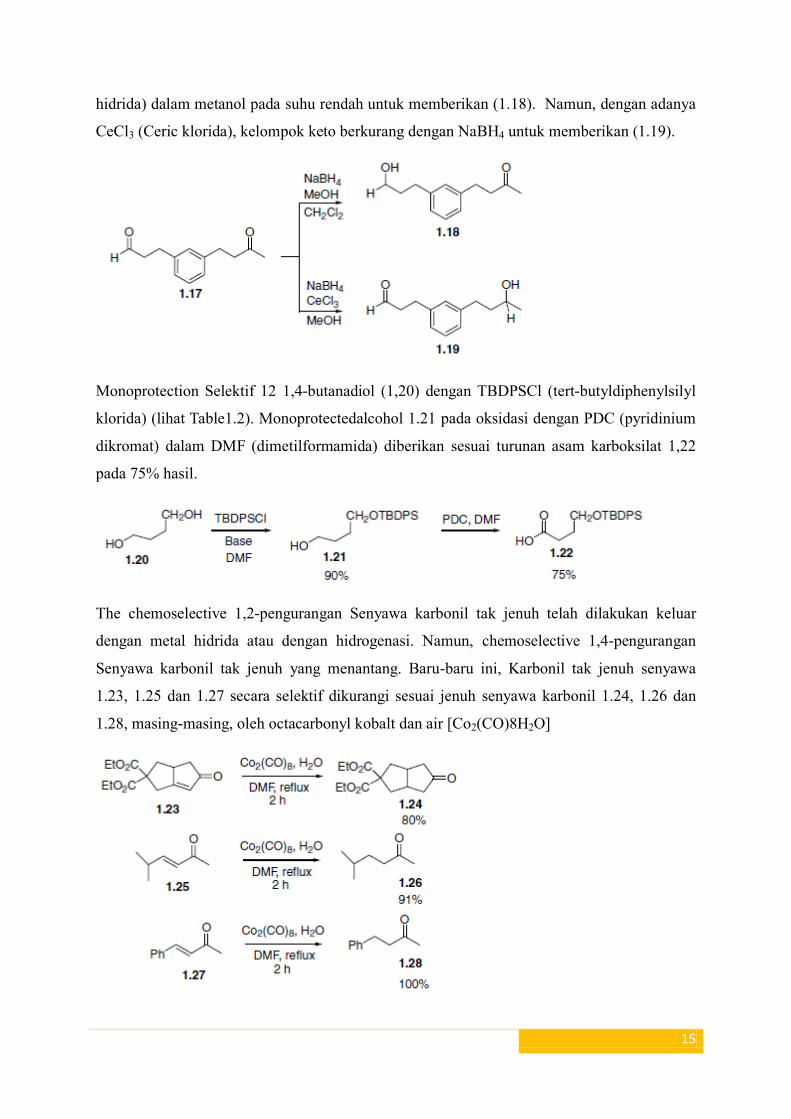
contoh, pengurangan chemoselective dari kelompok aldehida dengan NaBH4 (Natrium boro-
15
hidrida) dalam metanol pada suhu rendah untuk memberikan (1.18). Namun, dengan adanya
CeCl3 (Ceric klorida), kelompok keto berkurang dengan NaBH4 untuk memberikan (1.19).
Monoprotection Selektif 12 1,4-butanadiol (1,20) dengan TBDPSCl (tert-butyldiphenylsilyl
klorida) (lihat Table1.2). Monoprotectedalcohol 1.21 pada oksidasi dengan PDC (pyridinium
dikromat) dalam DMF (dimetilformamida) diberikan sesuai turunan asam karboksilat 1,22
pada 75% hasil.
The chemoselective 1,2-pengurangan Senyawa karbonil tak jenuh telah dilakukan keluar
dengan metal hidrida atau dengan hidrogenasi. Namun, chemoselective 1,4-pengurangan
Senyawa karbonil tak jenuh yang menantang. Baru-baru ini, Karbonil tak jenuh senyawa
1.23, 1.25 dan 1.27 secara selektif dikurangi sesuai jenuh senyawa karbonil 1.24, 1.26 dan
1.28, masing-masing, oleh octacarbonyl kobalt dan air [Co2(CO)8H2O]

16
.
1.5.2 regioselektivitas
Regioselektivitas (kontrol orientational) adalah pembentukan satu isomer konstitusional
sebagai produk utama di mana dua atau lebih isomer konstitusional dapat diperoleh. Sebagai
contoh, penambahan HBr (hydrogen bromide) untuk 1-methylcyclohexene (1.29)
memberikan 1-bromo-1- Methylcyclohexane (1.30) sebagai produk utama dan 1-bromo-2-
Methylcyclohexane (1.31) terbentuk sebagai produk minor.

17
LiAlH 4 (Litium aluminium hidrida) menyerang epoksida pada sterik kurang terhalang
C-O obligasi untuk memberikan alkohol yang sesuai.
Strategi sintetis
1.5.3 Stereoselektivitas
Stereoisomerisme adalah susunan atom dalam molekul yang konektivitas tetap sama tetapi
pengaturan mereka dalam ruang berbeda di setiap isomer (atom dihubungkan dalam urutan
yang sama). Dua jenis utama stereoisomerisme adalah cis-trans atau Z-E isomerisme dan
isomerisme optik.
Cis-trans atau Z-E isomerisme: The cis dan trans -1,2-dibromoethenes isomer tidak
dapat mudah interconverted karena pembatasan ikatan rangkap karbon-karbon. Keduanya
memiliki sama molekul rumus C2H 2Br2 , Tapi susunan atom mereka dalam ruang berbeda.
Cis-dan trans -1,2-dibromoethenes tidak bayangan cermin satu sama lain, dengan demikian,
mereka karena itu tidak enantiomer dan diastereomer.

18
Jika alkena tersebut trisubstituted atau tetrasubstituted, istilah cis dan trans biasanya tidak
diterapkan tetapi E-Z sistem tata nama diterapkan untuk diastereomer alkena. Umumnya,
Cahn Ingold-Prelog-(CIP) aturan yang digunakan untuk menetapkan prioritas untuk masing-
masing ujung ganda obligasi. Jika R 1 > R 2 dan R 3 > R 4 kemudian alkena dengan R 1 dan
R 3 di sisi yang sama ditunjuk Z dari zusammen kata Jerman, yang berarti bersama-sama.
Jika R 1 dan R 3 berada di seberang sisi kemudian alkena ditunjuk E dari Entgegen kata
Jerman, yang berarti berlawanan. Untuk contoh, (Z) - dan (E)-3-kloro-2-pentenes adalah
sebagai berikut:
Opticalisomers: Opticalisomersarestereoisomersthatcanbeformedaroundasymmetrical karbon
(s) alsoknownas kiral carbons.A stereocentre orchiralcentreinorganicchemistry umumnya
mengacu pada atom karbon dalam suatu senyawa kimia yang merupakan karbon asimetrik
atom atau karbon kiral. Senyawa adalah kiral jika itu adalah non-superimposable pada
cermin gambar. enantiomer dua isomer optik yang merupakan refleksi dari satu sama lain.
Mereka memiliki sifat fisik yang sama, kecuali untuk kemampuan mereka untuk memutar
pesawat-terpolarisasi cahaya, yang mereka lakukan dalam besaran yang sama tetapi dalam
arah yang berlawanan. Campuran dari jumlah yang sama dari kedua enantiomer dikatakan
campuran rasemat. Sebuah campuran rasemat tidak memutar pesawat- cahaya terpolarisasi.
Tugas masing-masing stereocentre baik sebagai R atau S berikut dari CIP aturan urutan.
Rincian aturan urutan CIP dapat ditemukan dalam buku teks sarjana pada kimia organik.
diastereoisomer dua isomer optik yang tidak refleksi satu sama lain atau superimposable.

19
Diastereomer dapat memiliki sifat fisik yang berbeda dan reaktivitas yang berbeda. Senyawa
dapat memiliki 2n stereoisomer, dimana n adalah jumlah stereocentres. Asam tartarat berisi
dua pusat asimetris, tapi dua dari konfigurasi adalah sama dan bersama-sama disebut
senyawa meso. Suatu senyawa meso secara optik tidak aktif (atau akiral) karena
mengandung sebuah pesawat simetri internal. Sisa dua konfigurasi adalah (+) - dan (-) -
gambar cermin, sehingga enantiomer. Bentuk meso adalah diastereomer dari bentuk-bentuk
lain.
Dalam reaksi stereoselektif, satu stereoisomer terbentuk dalam jumlah besar daripada yang
lain. Ketika stereoisomer adalah enantiomer selektivitas ini dikenal sebagai
enantioselectivity. The tingkat kemurnian enansiomer dari suatu larutan diukur dengan
kelebihan enansiomer nya, atau ee. Persentase enansiomerik kelebihan ditemukan dengan
membagi rotasi optik yang diamati oleh rotasi optik enansiomer murni secara berlebihan dan
mengalikannya dengan 100.
Misalnya, rotasi spesifik diamati dari campuran rasemat adalah 8,52 derajat rotasi. Rotasi
spesifik murni S-enansiomer -15.00 derajat rotasi. Karena murni S-enantiomer memiliki -
15.00 ◦ dan rotasi spesifik campuran adalah 8,52 ◦ maka isomer R-dikonfigurasi dengan
rotasi spesifik 15,00 ◦ adalah berlebihan.
The 0% ee berarti campuran rasemat 50:50, sedangkan 50% ee berarti campuran 75:25.
Dengan demikian, Enon- kelebihan tiomeric atau ee adalah ukuran untuk berapa banyak satu
enantiomer hadir dibandingkan yang lain. Sebagai contoh, dalam sebuah sampel dengan 40%
ee di R, sisanya 60% adalah rasemat dengan 30% dari R dan 30% dari S, sehingga jumlah
total R adalah 70%. Dengan demikian, persentase kelebihan enansiomer juga ditulis sebagai:

20
Baru-baru ini, theenantiomericexcessof-aminoacidesterhydrochlorideshasbeendetermined
langsung dengan menggunakan FAB (pemboman atom cepat) spektrometri massa tanpa
kromatografi- pemisahan grafis dari enansiomer 14.
Ketika masing-masing reaktan stereoisomeric membentuk produk stereoisomeric
berbeda reaksition dikenal sebagai reaksi stereospesifik. Misalnya, penambahan: cbr 2
(Dibromo- karbena, dibuat dari bromoform dan basis) ke cis-2-butena memberikan cis -2,3-
dimetil-1, 1 - dibromocyclopropane (1.32), sedangkan penambahan: cbr 2 ke trans-isomer
eksklusif menghasilkan trans-siklopropana 1.33.
Brominasi alkena juga merupakan reaksi stereospesifik.
Oleh karena itu, semua reaksi stereospesifik juga reaksi stereoselektif. Namun, semua reaksi
stereoselektif tidak selalu stereospesifik.
Ketika molekul yang sudah berisi setidaknya satu stereocentre mengalami reaksi
mana stereocentre baru dibuat, ada kemungkinan pembentukan dua (atau lebih) produk
stereoisomeric. Misalnya, pengurangan 1.34 mengarah ke diastereoisomer produk 1.35 dan
1.36 dengan Diastereoselektivitasnya (de) 83% (selain wajah si) (1.35). Untuk lebih rincian
tentang ulang atau penambahan wajah si, lihat Bab 6, Bagian 6.4.2.
1.5.4 sintesis asimetrik atau sintesis kiral

21
Achiralsubstanceis enansiomer murni atau whenonlyoneoftwopossibleenantiomers
homochiral hadir. Sebuah zat kiral enantioenriched atau heterochiral ketika lebih dari satu
enansiomer hadir tapi tidak dengan mengesampingkan yang lain. Jika produk yang
diinginkan adalah enansiomer, reaksi harus cukup stereoselektif bahkan ketika ekonomi atom
adalah 100%. Untuk penggunaan biologis kita hampir perlu satu enansiomer dan dalam
kemurnian tinggi. Ini karena ketika senyawa kiral biologis aktif berinteraksi dengan situs
reseptor yang adalah kiral, dua enansiomer dari molekul kiral berinteraksi secara berbeda dan
dapat menyebabkan kimia yang berbeda. Sebagai contoh, salah satu enansiomer asparagines
(1.37) pahit sementara lain adalah manis. Sejauh aplikasi obat yang bersangkutan, suatu
enansiomer tertentu dari obat mungkin efektif sementara yang lain tidak aktif atau berpotensi
membahayakan. Misalnya, salah satu enansiomer ethanbutol (1.38) digunakan sebagai
antibiotik dan penyebab lain kebutaan.
Meskipun penting, kemampuan untuk memperoleh molekul kiral dalam bentuk enansiomer
murni adalah tantangan yang sulit. Salah satu strategi untuk membuat enantiomer murni
untuk menghasilkan rasemat tersebut campuran dan kemudian memisahkan kedua
enantiomer dan efektif membuang yang tidak diinginkan enansiomer. Pemisahan enantiomer
adalah usaha yang sangat sulit, dan menghancurkan setengah produk reaksi pada setiap
langkah stereogenik tidak layak sebagai hasil dalam multi-langkah sintesis menurun secara
eksponensial.
Sintesis kiral, juga disebut sintesis asimetris, adalah sintesis yang mempertahankan
atau memperkenalkan kiralitas diinginkan. Pada prinsipnya, ada tiga metode yang berbeda
untuk menginduksi asimetri dalam reaksi. Ada dapat berupa satu atau beberapa pusat
stereogenik tertanam dalam merangsang kiralitas substrat dalam reaksi (kontrol substrat) atau
eksternal sumber menyediakan induksi kiral (kontrol reagen). Dalam kedua kasus yang

22
diperoleh Stereoselektivitas mencerminkan perbedaan energi antara transisi diastereomerik
dasar.
Pendekatan yang jelas untuk sintesis kiral akan menemukan bahan awal kiral, seperti
asam amino alami, karbohidrat, asam karboksilat atau terpene. Sumber utama bahan-bahan
tersebut mulai kiral kadang-kadang disebut chirons adalah alam itu sendiri. Sintesis senyawa
kimia enansiomer murni kompleks dari zat enansiomer murni tersedia seperti asam amino
alami yang dikenal sebagai sintesis kiral kolam renang. Sebagai contoh, lithium kiral amida
15a 1.39 yang digunakan untuk beberapa jenis sintesis asimetris enantioselektif dapat
disiapkan dalam kedua bentuk enansiomer mulai dari yang sesuai optik aktif asam amino, dan
ini sering tersedia secara komersial.
Namun, chiralpoolsynthesis dibatasi oleh jumlah kemungkinan enansiomerik mulai senyawa
murni dan membutuhkan sejumlah stoikiometri dari bahan awal, yang mungkin menjadi
langka dan mahal.
Pembantu kiral senyawa optik aktif yang digunakan untuk mengarahkan asimetris
sintesis. The kiral bantu sementara dimasukkan ke dalam sintesis organik yang
memperkenalkan kiralitas dalam senyawa rasemat sebaliknya. Ini stereocentre sementara
kemudian memaksa pembentukan asimetris dari stereocentre kedua. Sintesis demikian
diastere- oselective, daripada enantioselektif. Setelah penciptaan stereocentre kedua original
tambahan dapat dihapus pada langkah ketiga dan didaur ulang. EJ Corey pada tahun 1975,
BM Trost pada tahun 1980 dan JK Whitesell pada tahun 1985 memperkenalkan kiral
pembantu 8-phenylmenthol 15b (1.40), asam mandelic kiral 15c (1.41) dan trans-2-fenil-1-
sikloheksanol 15c (1.42), masing-masing.

23
Dalam rangka untuk memaksimalkan Diastereoselektivitasnya diamati untuk pembantu,
maka akan muncul wajar bahwa kelompok fungsional stereocontrolling berada dalam posisi
dalam ruang sedekat mungkin dengan pusat stereogenik baru terbentuk. Pembantu imida kiral
seperti Evans ' N-acyloxazolidinones (1.43) digunakan untuk alkilasi asimetris dan asimetris
aldol kondensasi (Skema 1.10).
Banyak varian struktural N-acyloxazolidinones telah dilaporkan dan mantan- hibit
pembelahan reaktivitas yang berbeda atau Diastereoselektivitasnya gratis dibandingkan
dengan N-acyloxazolidinone (1.43).
Beberapa contoh pembantu kiral 16 yang mengandalkan relatif jauh stereogenik cen-
tres kontrol Diastereoselektivitasnya dikenal. Misalnya, alkilasi dari enolates dari 1.44 dan
1.46-1.45 dan 1.47 dikendalikan melalui 1,4 - 1,3 dan induksi-asimetris, masing-masing.

24
Faktor-faktor sterik dan elektronik menggabungkan untuk mentransfer atau informasi
stereokimia estafet dari pusat stereogenik ke situs reaksi. Perubahan kecil di sudut obligasi
atau het- eroatom hibridisasi dapat mengakibatkan perubahan besar dalam
diastereoselectivities. Sebagai contoh, mengubah gugus pelindung nitrogen dalam
imidazolidinone yang diturunkan pembantu 1.48 hasil dalam perbaikan yang signifikan
dalam diastereoselectivities diamati selama 1.49 enolat alkilasi.
Kelompok conformationally fleksibel berfungsi untuk kedua relay dan memperkuat
stereokimia Informasi dari pusat stereogenik yang ada, sehingga memungkinkan kontrol yang
efisien diastereos- Elektivitas (Skema 1.11).
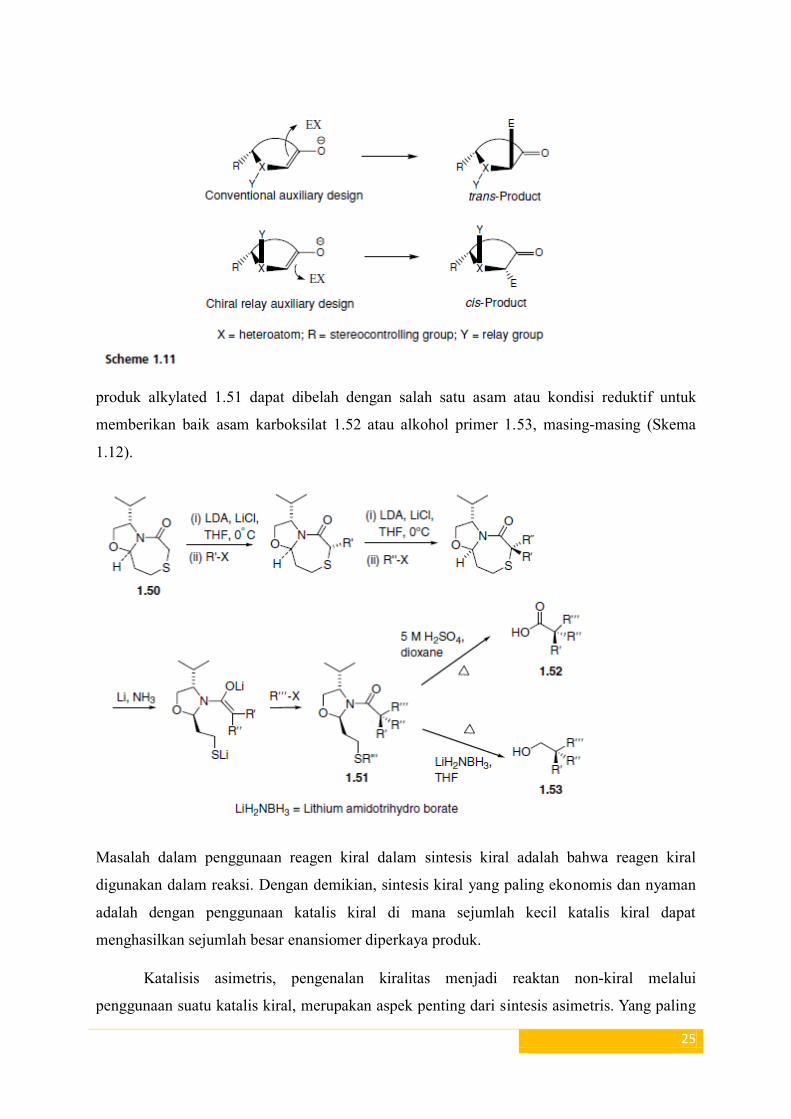
Metode tambahan kiral dapat digunakan untuk sintesis semua karbon kuartener
stereocentres 17 . Dengan demikian, kiral bisiklik thioglycolate laktam 1.50 yang dialkilasi
tiga kali
25
produk alkylated 1.51 dapat dibelah dengan salah satu asam atau kondisi reduktif untuk
memberikan baik asam karboksilat 1.52 atau alkohol primer 1.53, masing-masing (Skema
1.12).
Masalah dalam penggunaan reagen kiral dalam sintesis kiral adalah bahwa reagen kiral
digunakan dalam reaksi. Dengan demikian, sintesis kiral yang paling ekonomis dan nyaman
adalah dengan penggunaan katalis kiral di mana sejumlah kecil katalis kiral dapat
menghasilkan sejumlah besar enansiomer diperkaya produk.
Katalisis asimetris, pengenalan kiralitas menjadi reaktan non-kiral melalui
penggunaan suatu katalis kiral, merupakan aspek penting dari sintesis asimetris. Yang paling

26
ekstensif dipelajari reaksi katalisis asimetris adalah bahwa hidrogenasi alkena. Selain reaksi
hidrogenasi, kompleks kelompok logam platinum efektif dapat digunakan untuk
hydrosilations asimetris, alkylations allylic, isomerizations, hydroformyla- tions dan
carbonylations. Semua anggota kelompok logam platinum telah sukses sepenuhnya
digunakan. Reaksi karbonat 1.54 dengan natrium malonat dimetil dengan katalis [Mo (CO) 3
C 7 H 8 ] Dan kiral ligan 1.55 memberikan bercabang produk 1.56 pada 95% hasil dan 95%
ee 18 .
Ligan kiral sekali melekat pada bahan awal fisik mendikte lintasan untuk serangan, hanya
menyisakan trayektori yang diinginkan terbuka. Salah satu ligan kiral yang banyak digunakan
untuk memperkenalkan kiralitas dalam kombinasi dengan ruthenium atau rodium BINAP (2,2
- bis (diphenylphosphino) -1,1-binaphthyl) (1,57). Kedua (S)-BINAP dan (R)-BINAP adalah
tersedia secara komersial. Ini terdiri dari dua kelompok naftil dihubungkan oleh ikatan
tunggal dengan kelompok diphenylphosphino pada akhir setiap kelompok naftil. Rotasi
tentang single ikatan mengikat dua kelompok naftil dibatasi karena kekakuan sistem p
mereka. Oleh karena itu, sudut yang dibuat oleh dua pesawat p adalah tetap untuk sekitar 90 ◦
dan dua terpisah enantiomer ada. BINAP dibuat dari Binol (1,1-bis-2-naftol) (lihat Bab 6,
Bagian 6.1.1, Skema 6.3).
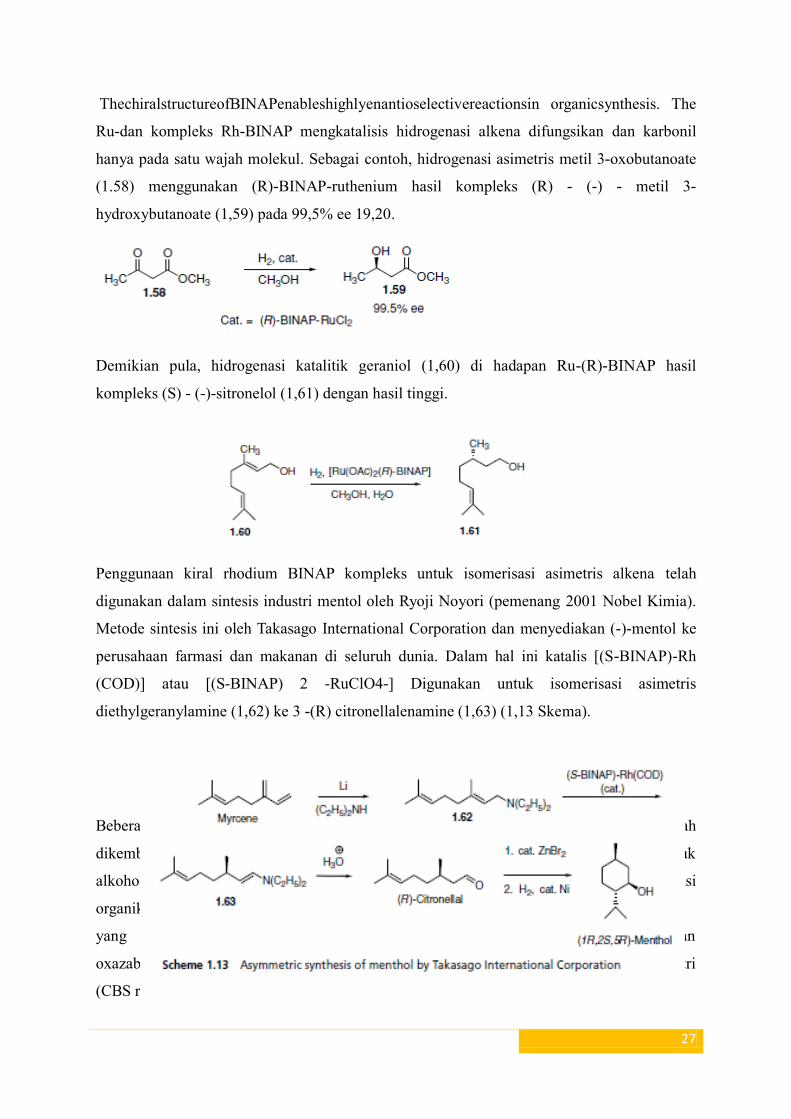
27
ThechiralstructureofBINAPenableshighlyenantioselectivereactionsin organicsynthesis. The
Ru-dan kompleks Rh-BINAP mengkatalisis hidrogenasi alkena difungsikan dan karbonil
hanya pada satu wajah molekul. Sebagai contoh, hidrogenasi asimetris metil 3-oxobutanoate
(1.58) menggunakan (R)-BINAP-ruthenium hasil kompleks (R) - (-) - metil 3-
hydroxybutanoate (1,59) pada 99,5% ee 19,20.
Demikian pula, hidrogenasi katalitik geraniol (1,60) di hadapan Ru-(R)-BINAP hasil
kompleks (S) - (-)-sitronelol (1,61) dengan hasil tinggi.
Penggunaan kiral rhodium BINAP kompleks untuk isomerisasi asimetris alkena telah
digunakan dalam sintesis industri mentol oleh Ryoji Noyori (pemenang 2001 Nobel Kimia).
Metode sintesis ini oleh Takasago International Corporation dan menyediakan (-)-mentol ke
perusahaan farmasi dan makanan di seluruh dunia. Dalam hal ini katalis [(S-BINAP)-Rh
(COD)] atau [(S-BINAP) 2 -RuClO4-] Digunakan untuk isomerisasi asimetris
diethylgeranylamine (1,62) ke 3 -(R) citronellalenamine (1,63) (1,13 Skema).
Beberapa katalis baru di mana borohidrida dikomplekskan dengan ligan kiral difungsi telah
dikembangkan dan digunakan untuk pengurangan enantioselektif keton prokiral untuk
alkohol kiral. Corey-Bakshi-Shibata reduction23, 24 (pengurangan CBS) adalah reaksi
organik
yang mengurangi keton enantioselectively menjadi alkohol dengan menggunakan
oxazaborolidines kiral dan BH3 · THF atau catecholborane sebagai pereduksi stoikiometri
(CBS reagen, 1,64) (lihat juga Bab 6, Bagian 6.4.2).

28
Sebagai contoh, (S)-2-metil-CBS-oxazaborolidine mengikat reversibel dengan diborane
untuk membentuk reaktif mengurangi spesies 1,64. Koordinasi oksigen keton dengan Lewis
mengarahkan boron asam dan mengaktifkan gugus karbonil untuk transfer hidrida.
Mekanisme transfer hidrida intramolekul berfungsi sebagai model untuk mencapai enantio
selektif reduksi (lihat Skema 6.20).
Naproxen, obat anti-inflamasi, disintesis dengan memanfaatkan enantio selektif
asimetris hidrosianin dari vinil naftalen 1,65 memanfaatkan ligan kiral 1.66. Karena S-
enansiomer diinginkan secara medis sedangkan R-enansiomer menghasilkan efek kesehatan
yang berbahaya, enantio selektivitas reaksi ini penting. Sintesis naproxen nitril (1.67)
ditunjukkan di bawah ini menghasilkan S-(-)-enansiomer dengan 75% ee.
Pada tahun 2001, KB Sharpless memenangkan Hadiah Nobel dalam Kimia untuk
karyanya pada asimetris amino hidroksil 25-27 dan asimetris epoxida 28-30. Ini oksidasi
stereoselektif reaksi kuat metode asimetris katalitik yang telah merevolusi sintetis kimia
organik.
Sharpless asimetris oksidasi 28-30 adalah epoksidasi enantio selektif dari allil alkohol
dengan terrsier butil hidroperoksida (t-BuOOH), titanium tetra iso propoksida [Ti (O-IPR) 4]
dan (+) - atau (-)-dietil tartrat [(+) - atau (-)-DET] untuk menghasilkan epoksida optik aktif
dari akiral aklik alkohol. Reaksi diastereo selektif untuk mensubstitusi alkohol alilik.
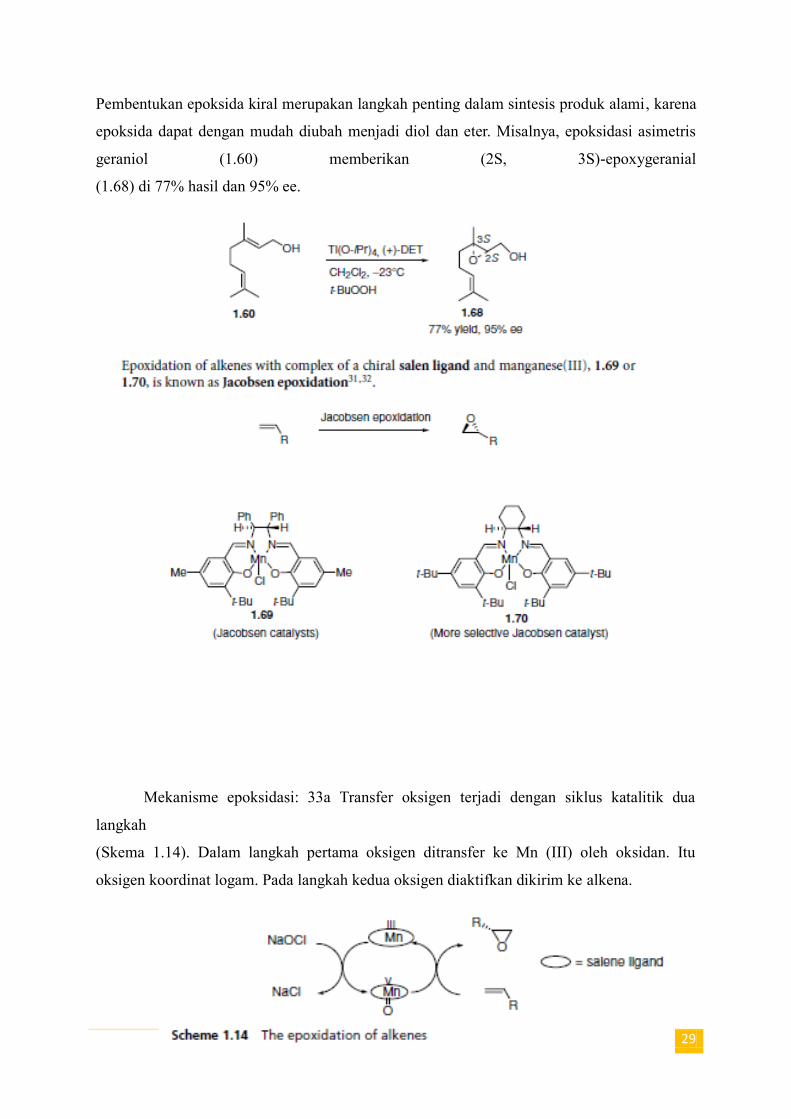
29
Pembentukan epoksida kiral merupakan langkah penting dalam sintesis produk alami, karena
epoksida dapat dengan mudah diubah menjadi diol dan eter. Misalnya, epoksidasi asimetris
geraniol (1.60) memberikan (2S, 3S)-epoxygeranial
(1.68) di 77% hasil dan 95% ee.
Mekanisme epoksidasi: 33a Transfer oksigen terjadi dengan siklus katalitik dua
langkah
(Skema 1.14). Dalam langkah pertama oksigen ditransfer ke Mn (III) oleh oksidan. Itu
oksigen koordinat logam. Pada langkah kedua oksigen diaktifkan dikirim ke alkena.

30
Pemindahan oksigen ke alkena dapat terjadi melalui beberapa mekanisme yang
berbeda (Skema 1,15). Radikal oksigen dapat dibentuk sebagai perantara ketika kelompok-
kelompok radikal menstabilkan yang terpasang. Mekanisme ini didukung oleh fakta bahwa
cis-alkena memberikan kedua cis dan trans-epoksida. Pembentukan metallaoxetane sebagai
perantara juga diusulkan oleh Norrby et al. Namun, pengiriman oksigen terpadu juga telah
diusulkan.
Selektivitas transfer oksigen dari kompleks okso-mangan untuk alkena tergantung
pada orientasi relatif dari katalis diaktifkan dan alkena. Pendekatan alkena ligan salen
sedemikian rupa sehingga menghindari mengangkat kelompok butil dan turunannya,
substituen jauh dari ligan salen.

31
Dihidroksi alkena dengan sejumlah katalis osmium ferri dari stoikiometri pereaksi
oksidasi, seperti barium klorat, ters-butil hidroperoksida (T-BuOOH), N-metil-N-okso-
morfolin (NMO), natrium peroxo disulfat (Na2S2O8), yodium (I2) atau kalium ferricyanide
[K3Fe(CN)6], merupakan metode penting untuk produksi diol. Reaksi stereospesifik sebagai
syn-diol diperoleh. Namun, Stereoselektivitas bervariasi tergantung pada struktur alkena.
Sharpless memberikan
metode asimetris dihydroxylation 34 dari alkene. Ketika osmium ferri dan oksidasi agen
NMO atau K3Fe(CN)6 digunakan dalam kiral kina alkaloid, kelebihan enansiomer (ee%) dari
produk diol terbentuk. Ini adalah kiral kina alkaloid yang menyediakan komponen optik aktif
untuk katalis. Reaksi dilakukan dalam solusi buffer untuk memastikan pH yang stabil. NMO
atau K3Fe (CN)6 digunakan untuk regenerasi osmium (VIII).
Reagen ini tersedia secara komersial dicampur: campuran asimetris dihydroxyl (AD-
mix). AD-mix tersedia dalam dua variasi: AD-mix α adalah (DHQ) 2PHAL + K2OsO2(OH)4
+ K3Fe (CN)6 dan AD-campuran adalah (DHQD) 2PHAL + K2OsO2(OH)4 + K3Fe(CN)6.
Ligan (DHQ) 2PHAL adalah 1,4-bis (9-O-dihydroquinine) phthalazine (1,71) dan ligan
(DHQD) 2PHAL adalah 1,4-bis (9-O-dihydroquinidine) phthalazine (1,72).
Reaksi AD Sharpless sangat berguna dan efisien untuk dihydroxylation asimetris
alkena.

32
Mekanisme reaksi dari dihydyroxyl Sharpless diberikan dalam Bab 7, Bagian 7.5.
Rutenium katalisis memungkinkan dihydroxyl menyediakan akses mudah ke syn-diol, tapi
over-oksidasi adalah reaksi samping yang umum. Peningkatan protokol untuk Ru-katalis syn-
dihydroxylation hanya menggunakan 0,5 mol katalis% di bawah kondisi asam yang
memberikan produk di hasil yang tinggi dengan formasi hanya kecil sisi products.
Sharpless dan rekan kerja pertama melaporkan aminohydroxyl 25-27 dari alkena pada
tahun 1975 dan juga telah diperpanjang reaksi menjadi efisien satu langkah katalitik asimetris
aminohydroxyl. Reaksi ini menggunakan katalis osmium [K2OsO2(OH)4], garam chloramine
(seperti chloramine T, lihat Bab 7, Bagian 7.6) sebagai oksidan dan kina alkaloid 1,71 atau
1,72 sebagai ligan kiral. Misalnya, aminohydroxyl asimetris dari stirena (1,73) bisa
menghasilkan dua alkohol amino regioisomeric 1,74 dan 1.75. Menggunakan aminohydroxyl
asimetris Sharpless, (1R)-N-etoksikarbonil-1-fenil-2-hydroxyethylamine (1,74) diperoleh
dengan O'Brien et al.36 sebagai produk utama dan dengan kelebihan enansiomer yang tinggi
daripada rekan regioisomeric (R)-N-etoksikarbonil-2-fenil-2-hydroxyethylamine (1,75). Yang
sesuai alkohol amino bebas diperoleh oleh deproteksi etil karbamat (urethane) derivatif.
Sintesis sisi taxol chain 37 melibatkan reaksi aminohydroxyl asimetris (Skema 1.16).
Kemampuan untuk memilih kondisi reaksi untuk memberikan senyawa enansiomer
murni penting dalam industri. Metode untuk meningkatkan hasil enantiomer tertentu dengan
biaya rendah dalam permintaan tinggi, dan ini telah menghasilkan dalam penelitian untuk
mengembangkan blok bangunan kiral baru, ligan kiral dan katalis. Masalah tambahan yang
dihadapi dalam industri adalah bahwa katalis sering mahal dan sulit untuk pulih, dan
penelitian tentang pemulihan katalis penting.

33
1.6 Kelompok Melindungi
Ketika reaksi kimia yang akan dilakukan secara selektif pada satu situs reaksi dalam
multifungsi senyawa organik (molekul organik mengandung dua atau lebih dari dua reaktif
kelompok) dan kami ingin reaksi pada satu tempat yang reaktif, maka tempat reaktif lain
harus sementara diblokir atau diprotek. Langkah ini disebut deproteksi. Perlindungan dan
deproteksi gugus fungsional telah mendapat perhatian dalam beberapa tahun terakhir bukan
hanya karena kepentingan fundamentalnya, tetapi juga karena peran mereka dalam multi-
langkah sintesis. Penyusunan molekul organik kompleks menuntut tersedianya berbagai
gugus untuk memungkinkan kelangsungan hidup kelompok fungsional reaktif selama
berbagai operasi sintesis, akhirnya mengakibatkan produksi selektif dari molekul target.
Misalnya, dalam konversi etil 5-okso-hexanoat (1.76) menjadi 6-hidroksi-2-hexanon (1.77),
diperlukan untuk memblokir gugus keton pertama dan kemudian gugus ester berkurang
dengan LiAlH4. Gugus keton dilindungi sebagai asetal karena gugus asetal tidak bereaksi
dengan reduktor LiAlH4. Pada tahap akhir gugus asetal dihilangkan dengan pnambahan asam.
Keseluruhan skema transformasi ini diberikan dalam Skema 1.17.

34
Sebuah kelompok pelindung harus memenuhi sejumlah persyaratan. Sebuah gugus
pelindung yang baik harus mudah untuk mengenakan, tanpa generasi pusat stereogenik baru,
dan mudah untuk dihilangkan. Untuk melindungi kelompok harus memiliki minimal fungsi
tambahan untuk menghindari tempat lanjut reaksi. Gugus pelindung harus membentuk
turunan kristal dengan reaksi yang tinggi, hasil yang dapat dengan mudah dipisahkan dari
produk samping. Gugus pelindung seharusnya tidak mengganggu reaksi yang dilakukan
sebelum dihilangkan. Gugus pelindung dapat dibelah dalam berbagai kondisi termasuk
solvolisis dasar, asam, logam berat, ion fluorida, eliminasi reduktif, eliminasi, hidrogenolisis,
oksidasi, reduksi melarutkan logam, substitusi nukleofilik, transisi katalis logam, cahaya dan
enzim. Metode elektrolit dan dibantu fotolisis penting dalam metode untuk menghilangkan
gugus pelindung. Gugus Photolabile disebut senyawa dikurung atau phototriggers, dilindungi
dari radiasi pada panjang gelombang 254-350nm dengan tinggi hasil kuantum.
Gugus pelindung harus tetap melekat sepanjang sintesis dan mungkin dihapus setelah
selesai sintesis. Namun, kelompok-kelompok pelindung tidak dimasukkan ke dalam produk
akhir, dengan demikian, penggunaannya membuat reaksi kurang atom ekonomis. Dengan
kata lain, penggunaan gugus pelindung kelompok harus dihindari sebisa mungkin. Berbagai
gugus pelindung kelompok saat ini tersedia untuk fungsional yang berbeda kelompok.
Sebuah gambaran yang sangat singkat yang paling umum digunakan melindungi kelompok
diberikan dalam bab ini. Mereka diklasifikasikan menurut kelompok fungsional mereka..
1.6.1 hidroksi umum melindungi kelompok
Kelompok hidroksil harus dilindungi selama oksidasi, asilasi, halogenasi, dehidrasi
dan reaksi lain yang rentan. Gugus hidroksil dilindungi dengan membentuk eter alkil mereka,
eter alkoksialkil, eter silil dan ester. Namun, eter lebih disukai atas ester karena stabilitas
mereka dalam asam asetat dan kondisi dasar.
Alkil eter dan alkoksialkil
Alkil eter umumnya disiapkan dengan penambahan asam-katalis dari alkohol ke
alkena atau Sintesis eter Williamson (Skema 1,18).
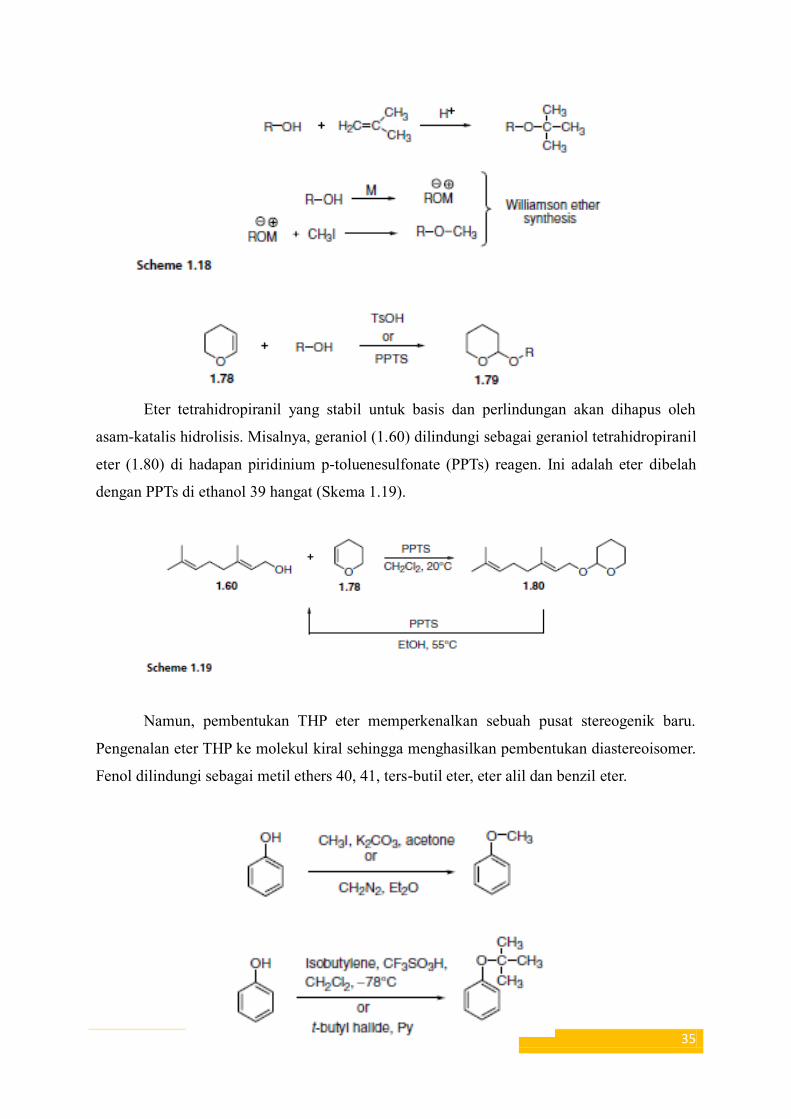
35
Eter tetrahidropiranil yang stabil untuk basis dan perlindungan akan dihapus oleh
asam-katalis hidrolisis. Misalnya, geraniol (1.60) dilindungi sebagai geraniol tetrahidropiranil
eter (1.80) di hadapan piridinium p-toluenesulfonate (PPTs) reagen. Ini adalah eter dibelah
dengan PPTs di ethanol 39 hangat (Skema 1.19).
Namun, pembentukan THP eter memperkenalkan sebuah pusat stereogenik baru.
Pengenalan eter THP ke molekul kiral sehingga menghasilkan pembentukan diastereoisomer.
Fenol dilindungi sebagai metil ethers 40, 41, ters-butil eter, eter alil dan benzil eter.

36
Miura dan co-workers 42 melaporkan perlindungan fenol oleh alkohol alil di hadapan
jumlah katalitik paladium (II) asetat dan titanium (IV) isopropoksida. Reaksi ini sangat
umum, namun gagal dalam kasus 3,5-dimethoxyphenol karena pembentukan eksklusif
produk C-allylated.
Eter biasanya dapat dihapus oleh asam, dengan derivatif THP 1,79 bereaksi lebih
cepat dibandingkan dengan eter tert-butil. Benzil eter dihapus dalam berbagai kondisi seperti
hidrogenolisis, melarutkan reduksi logam (Na di NH3) dan HBr (ringan). Metil eter yang
cleaved43 dengan refluks dengan EtSNa dan DMF. eter tert-Butil dapat dibelah dengan
Asam trifluroacetic (CF3COOH) pada 25 ◦ C.
Pembelahan nukleofilik eter alkil aril memberikan sesuai fenol dengan hanya 1 equiv.
dari thiophenol di hadapan N-metil-2-pyrrolidinone (NMP) dalam katalitik jumlah potasium
carbonate. Nitro aromatik dan substituen kloro yang mengungsi dengan thiolates
stoikiometrik yang diawetkan dengan metode ini. Selain itu senyawa karbonil tidak jenuh
penambahan undergo Michael dari tiolat bawah kondisi ini.
Tetrahydropyranylation alkohol dalam kondisi bebas pelarut secara efisien dikatalisis
oleh bismuth triflat (0,1 mol%). Prosedur percobaan sederhana dan bekerja dengan baik
dengan berbagai alkohol dan fenol. Katalis tidak sensitif terhadap jumlah udara dan

37
kelembaban, mudah untuk menangani dan relatif tidak beracun. Deproteksi THP eter juga
dikatalisasi oleh bismuth triflate45 (1,0 mol%). Benzil (Bn) atau p-metoksibenzil kelompok
(PMB) dapat dihapus di bawah mengurangi kondisi pembelahan (Skema 1,20).
Logam alkali (seperti Li) dalam amonia cair biasanya diterapkan untuk deproteksi benzil (Bn)
ethers46. Lithium naphthalenide dibuat dari lithium dan naftalena distoikiometri amount47
atau katalitik amount48 sering digunakan untuk Deprotect eter benzil.
Hwu melaporkan pembelahan selektif dari benzil (Bn) eter dengan diisopropilamida
lithium (LDA) di hadapan kelompok metoksi, namun, dengan menggunakan natrium bis
(trimetilsilil) amida [NaN (SiMe3) 2], dimetoksibenzena ini mengalami selektif
mono-O-demethylation (Skema 1.21).
\

38
Hirota melaporkan penghapusan selektif dari benzil (Bn) dengan
Hidrogenolisis Pd-C-katalis fenol dilindungi PMB. Penghapusan kelompok PMB dihambat
oleh kehadiran piridin.
Klasik Prosedur untuk menghilangkan kelompok alil melibatkan urutan dua langkah,
di mana kelompok alil pertama kali diisomerisasikan ke fungsi propenil sesuai dengan basa
kuat seperti kalium tert-butoksida (t-BuOK) atau katalis logam seperti Pd-C, di ikuti oleh
konversi dari kelompok propenil menjadi alkohol gratis. Namun, baru-baru ini beberapa
metode dimana lainnya dilaporkan dalam literatur untuk menghilangkan kelompok alil
menggunakan berbagai
reagen seperti DDQ, CeCl3 · 7H2O/NaI, Ti (O-IPR)4 atau p-TsOH. Metode lain yang efisien
untuk deproteksi alil adalah dengan menggunakan DMSO (dimetilsulfoksida)/NaI reagen.
Benzil, etil dan kelompok yang melindungi tert-butil yang cukup stabil reaksi dibawah ini.
Aril eter Propargylic (juga ester) yang dibelah oleh benzil trietil ammonium tetra tiomolibdat
dalam asetonitril di kamar dengan temperatur 52. Ester alil tidak dibelah di bawah kondisi ini.
Electroreduction di hadapan Ni-bipiridin kompleks seperti katalis lain metode untuk
mempengaruhi deproteksi dari propargil ethers53 (Skema 1.22).
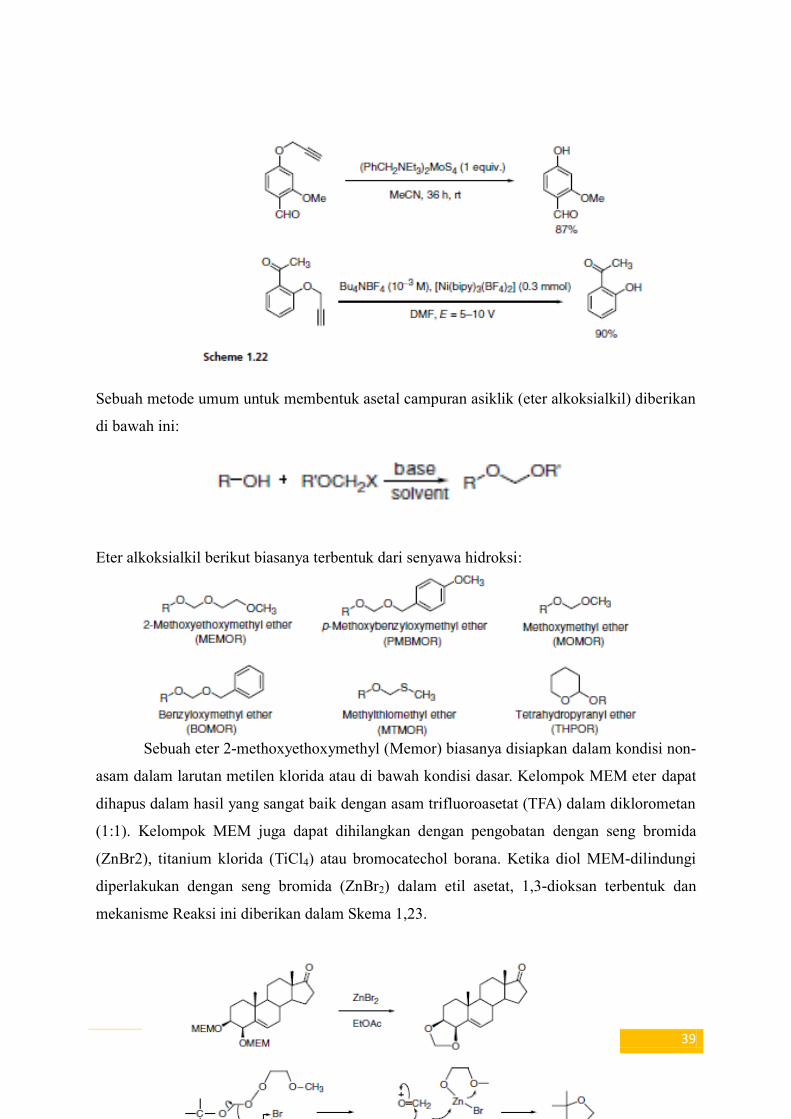
39
Sebuah metode umum untuk membentuk asetal campuran asiklik (eter alkoksialkil) diberikan
di bawah ini:
Eter alkoksialkil berikut biasanya terbentuk dari senyawa hidroksi:
Sebuah eter 2-methoxyethoxymethyl (Memor) biasanya disiapkan dalam kondisi non-
asam dalam larutan metilen klorida atau di bawah kondisi dasar. Kelompok MEM eter dapat
dihapus dalam hasil yang sangat baik dengan asam trifluoroasetat (TFA) dalam diklorometan
(1:1). Kelompok MEM juga dapat dihilangkan dengan pengobatan dengan seng bromida
(ZnBr2), titanium klorida (TiCl4) atau bromocatechol borana. Ketika diol MEM-dilindungi
diperlakukan dengan seng bromida (ZnBr2) dalam etil asetat, 1,3-dioksan terbentuk dan
mekanisme Reaksi ini diberikan dalam Skema 1,23.
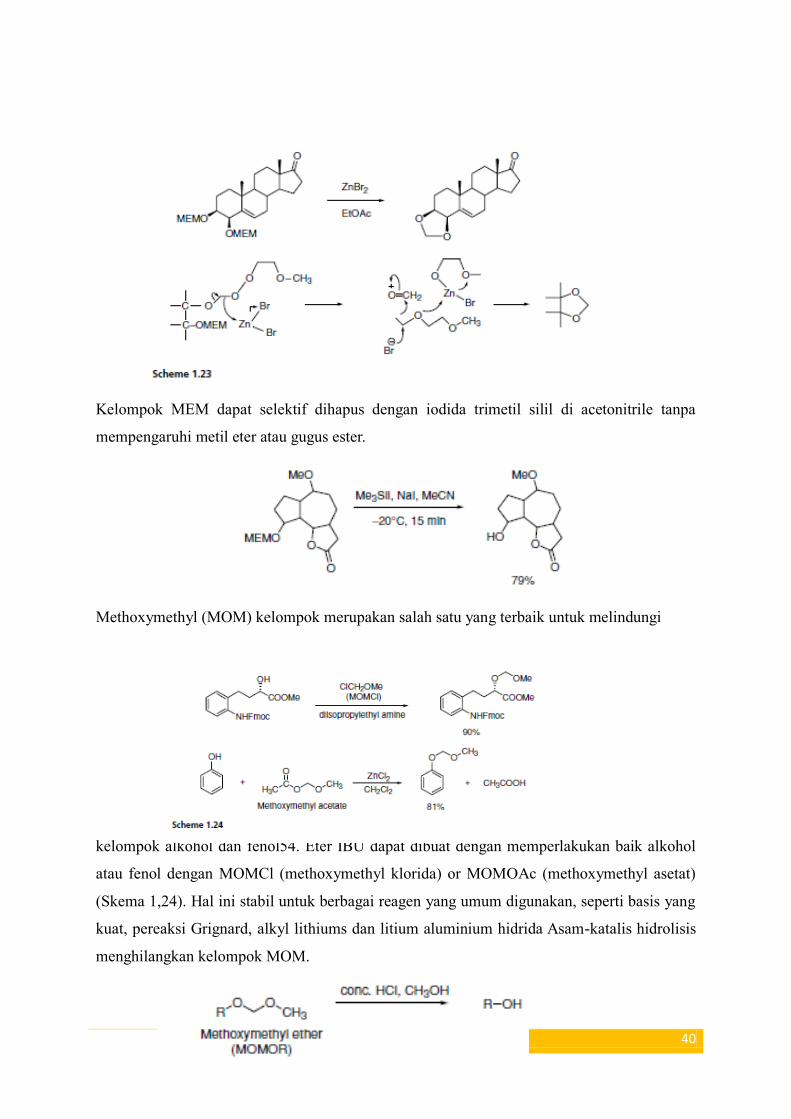
40
Kelompok MEM dapat selektif dihapus dengan iodida trimetil silil di acetonitrile tanpa
mempengaruhi metil eter atau gugus ester.
Methoxymethyl (MOM) kelompok merupakan salah satu yang terbaik untuk melindungi
kelompok alkohol dan fenol54. Eter IBU dapat dibuat dengan memperlakukan baik alkohol
atau fenol dengan MOMCl (methoxymethyl klorida) or MOMOAc (methoxymethyl asetat)
(Skema 1,24). Hal ini stabil untuk berbagai reagen yang umum digunakan, seperti basis yang
kuat, pereaksi Grignard, alkyl lithiums dan litium aluminium hidrida Asam-katalis hidrolisis
menghilangkan kelompok MOM.

41
Bromocatechol borana atau LiBF4 (lithium fluoroborate) dalam asetonitril (CH3CN) dan air
juga telah digunakan untuk deproteksi kelompok MOM. Eter Benzyloxymethyl (BOMOR)
biasanya dibuat dengan mereaksikan BOMCl (benzyloxymethyl klorida) dengan alkohol di
bawah kondisi dasar dan dapat selektif dihapus dengan H2, Pd-C atau Na/NH3.
Sebuah p-methoxybenzyloxymethyl (PMBM) kelompok dihapus oleh hidrolisis asam atau
reduksi.
The methylthiomethyl eter (MTMOR): Tersier gugus hidroksil, yang rentan terhadap asam-
katalis dehidrasi, dapat dengan mudah dilindungi sebagai MTM eter dan sembuh dalam
kondisi baik menghasilkan. MTM eter dari gugus hidroksil dapat dibentuk baik oleh
Williamson khas sintesis eter atau reaksi dengan dimetilsulfoksida (DMSO) dan anhidrida
asetat
(Ac2O). Dalam kasus terakhir, reaksi berlangsung dengan Pummerer rearrangement56-58
(Skema 1,25).
Mekanisme penataan ulang Pummerer diberikan dalam Skema 1,26. Sebuah

42
methylthiomethyl (MTM) kelompok dihapus oleh asam atau dapat dibelah dengan
pengobatan ringan
dengan perak berair atau garam merkuri (netral merkuri klorida) yang kebanyakan eter yang
stabil, sebagai hasilnya, deproteksi selektif molekul polifungsional menjadi mungkin
menggunakan MTM eter untuk gugus hidroksi.
Eter silil
Perlindungan gugus hidroksil melalui pembentukan eter silil telah banyak digunakan
dalam sintesis organik. Eter silil tahan terhadap oksidasi, sudah baik termal stabilitas,
viskositas rendah dan mudah diperoleh dari senyawa awal mereka. Banyak metode yang
dapat digunakan untuk sintesis eter trialkilsilil (Skema 1,27). Alkohol bereaksi cepat dengan
trialkilsilil klorida (R3SiCl) untuk memberikan trialkilsilil ethers59 (ROSiR3) dengan adanya
basis amina seperti trietilamina, piridin, imidazole atau 2,6-lutidine (Tabel 1.2).
Tidak seperti 3-alkil halida, klorida trialkilsilil (R3SiCl) menjalani substitusi nukleofilik
dengan mekanisme yang mirip dengan SN2 tersebut. Anion enolat yang diperoleh dari
alkohol bereaksi dengan klorida trialkilsilil (R3SiCl), menghasilkan eter trialkilsilil (R3SiOR)
oleh substitusi pada oksigen. Kekuatan luar biasa dari Si-O obligasi dikombinasi lagi C-Si

43
panjang ikatan (Crowding kurang sterik) berfungsi untuk menstabilkan transisi seperti
yang ditunjukkan dalam Skema 1,28.
Dengan kelompok bulkier, suchas TBS, mungkin untuk membedakan antara primer dan
menengah alkohol. Ini adalah contoh dari regio control (lihat bagian 1.5).
Mono protection Selektif 1,4-butanadiol (1,20) dengan TBDPSCl memberikan hasil 90% dari
terkait alkohol 1,2112. Penghapusan kelompok TMS umumnya dilakukan dengan adanya
katalis
termasuk besi (III) dan timah (II) klorida, tembaga (II) nitrat, cerium (III) nitrat, asam sitrat
dan natrium hidroksida atau berbagai turunan fluoro. Derivatif TMS agak mudah dihidrolisis
menjadi prekursor alkohol mereka, tetapi bulkier silil eter lebih tahan dan stabil pada kisaran
pH yang lebar. Kelompok-kelompok pelindung mudah dibelah oleh fluoride anion, sering
diperkenalkan sebagai garam tetra alkyl ammonium seperti tetra butyl ammonium fluoride
(TBAF).
Maiti dan Roy60 melaporkan metode selektif untuk deproteksi dari allylic primer, benzilik,
homoallylic dan aril eter TBS menggunakan DMSO berair pada 90 ◦ C. Semua lainnya TBS
yang dilindungi kelompok, eter benzil, THP eter serta metil eter tetap tidak terpengaruh.

44
Ester
Asilasi alkohol merupakan reaksi penting bagi ahli kimia organik sintetik, itu secara
historis digunakan untuk derivatisasi dan karakterisasi alkohol. Asilasi biasanya dilakukan
dengan menggunakan asil klorida atau anhidrida yang sesuai di hadapan dari dasar seperti
trietilamina atau piridin (Skema 1,29). Laju reaksi cepat dapat dicapai dengan menambahkan
4 -(dimethylamino) piridin (DMAP) sebagai co-katalis.
Dengan kondisi tersebut, substrat dasar-sensitif dapat mengalami dekomposisi. Untuk
menghindari Kelemahan ini, protik dan Lewis asam dapat dimanfaatkan, seperti asam p-
toluenasulfonat, seng klorida, kobalt klorida atau triflat skandium.
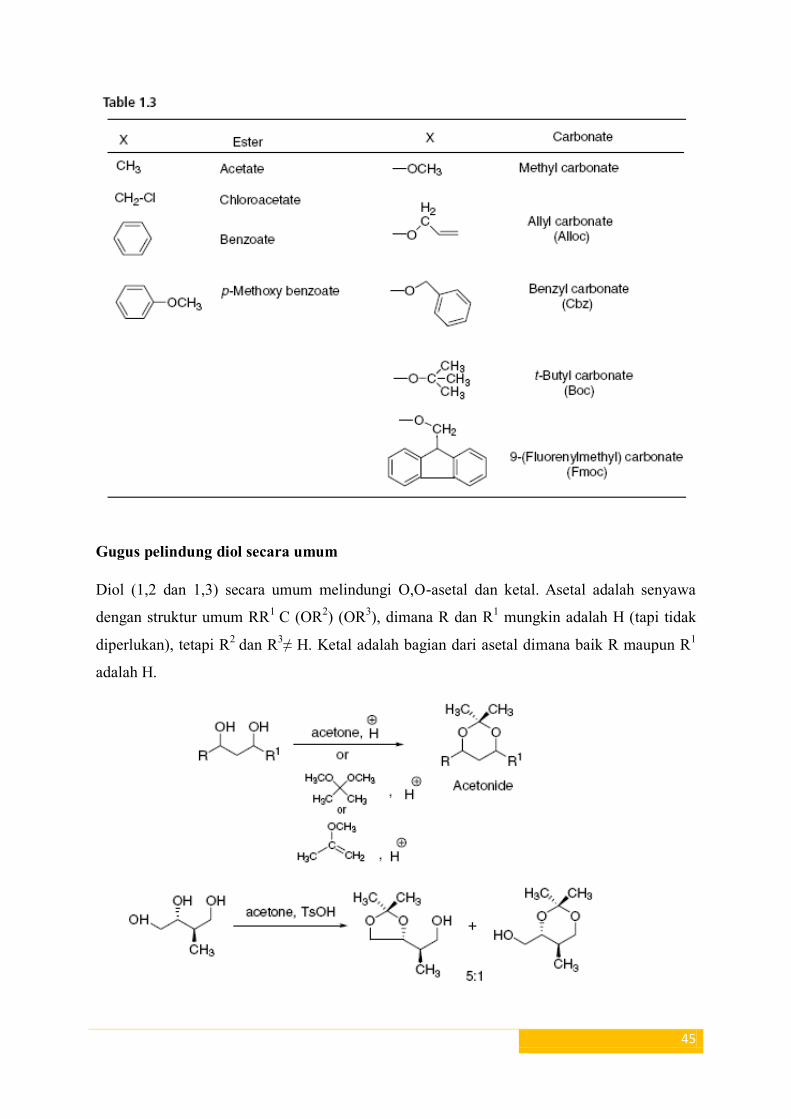
Asetat, chloroacetate, benzoat, p-metoksi benzoat, benzil karbonat (Cbz), tertbutyl
karbonat (Boc) dan 9 - (Fluorenylmethyl) karbonat (Fmoc) biasanya disiapkan untuk
melindungi gugus hidroksil (Tabel 1.3).
Karbonat metil yang dibelah di bawah kondisi dasar (K2CO3/MeOH). Fmoc dapat dibelah
dengan basis seperti Et3N, Py, morfolin atau diisopropiletilamina. Karbonat alil dapat
Berikut adalah tabel mekanisme reaksi dalam sintesis organik
45
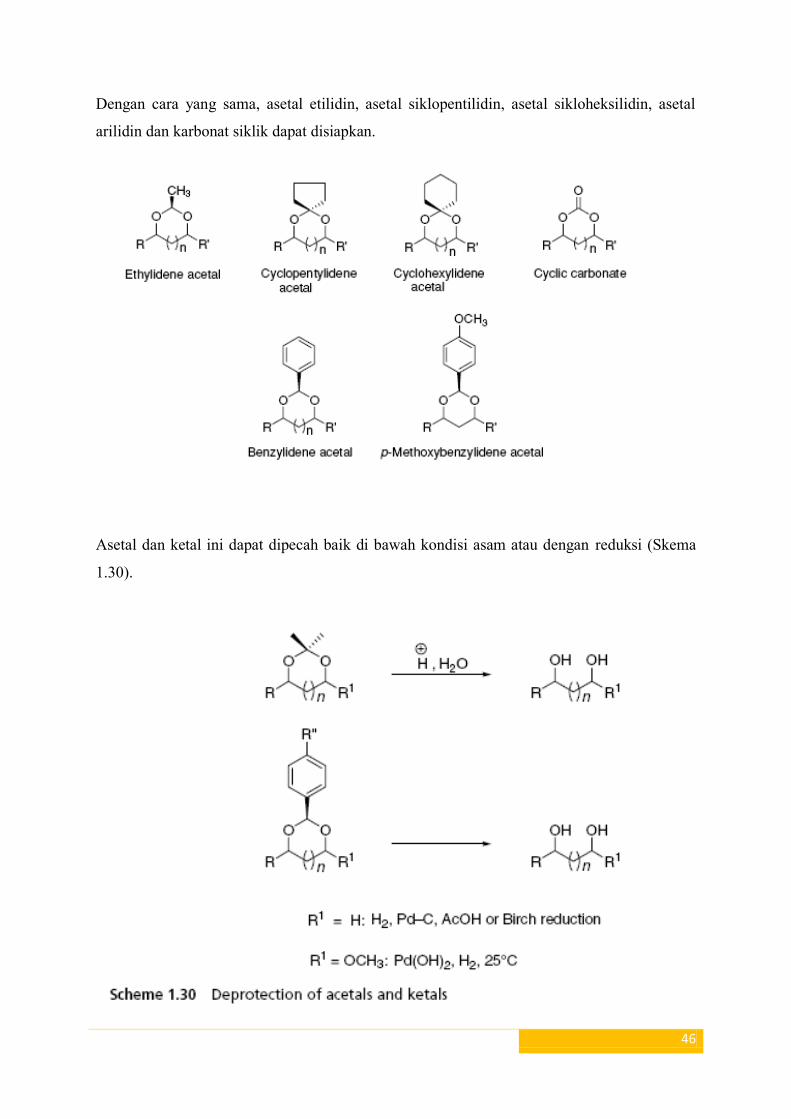
Gugus pelindung diol secara umum
Diol (1,2 dan 1,3) secara umum melindungi O,O-asetal dan ketal. Asetal adalah senyawa
dengan struktur umum RR1
C (OR2) (OR
3), dimana R dan R
1 mungkin adalah H (tapi tidak
diperlukan), tetapi R2
dan R3≠ H. Ketal adalah bagian dari asetal dimana baik R maupun R
1
adalah H.
46
Dengan cara yang sama, asetal etilidin, asetal siklopentilidin, asetal sikloheksilidin, asetal
arilidin dan karbonat siklik dapat disiapkan.
Asetal dan ketal ini dapat dipecah baik di bawah kondisi asam atau dengan reduksi (Skema
1.30).

47
Asetal juga dibelah oleh besi klorida baik diserap atau tidak diserap pada silika gel.
Kelompok TBS itu (lihat Tabel 1.2) tidak deproteksi di bawah kondisi ini. Tergantung pada
nomor ekivalen besi klorida yang digunakan, deproteksi selektif pada salah satu atau kedua
kelompok asetal dapat dicapai 61-63 (Skema 1,31).
Gugus pelindung amina secara umum
Perlindungan Nitrogen terus menarik banyak perhatian dalam berbagai bidang kimia, seperti
peptida, nukleosida, polimer dan sintesis ligan. Tetapi, dalam beberapa tahun terakhir,
sejumlah gugus pelindung nitrogen telah digunakan sebagai pembantu kiral. Dengan
demikian, desain baru, lebih ringan dan metodenya lebih efektif untuk perlindungan nitrogen
masih aktif dalam topik sintesis kimia.
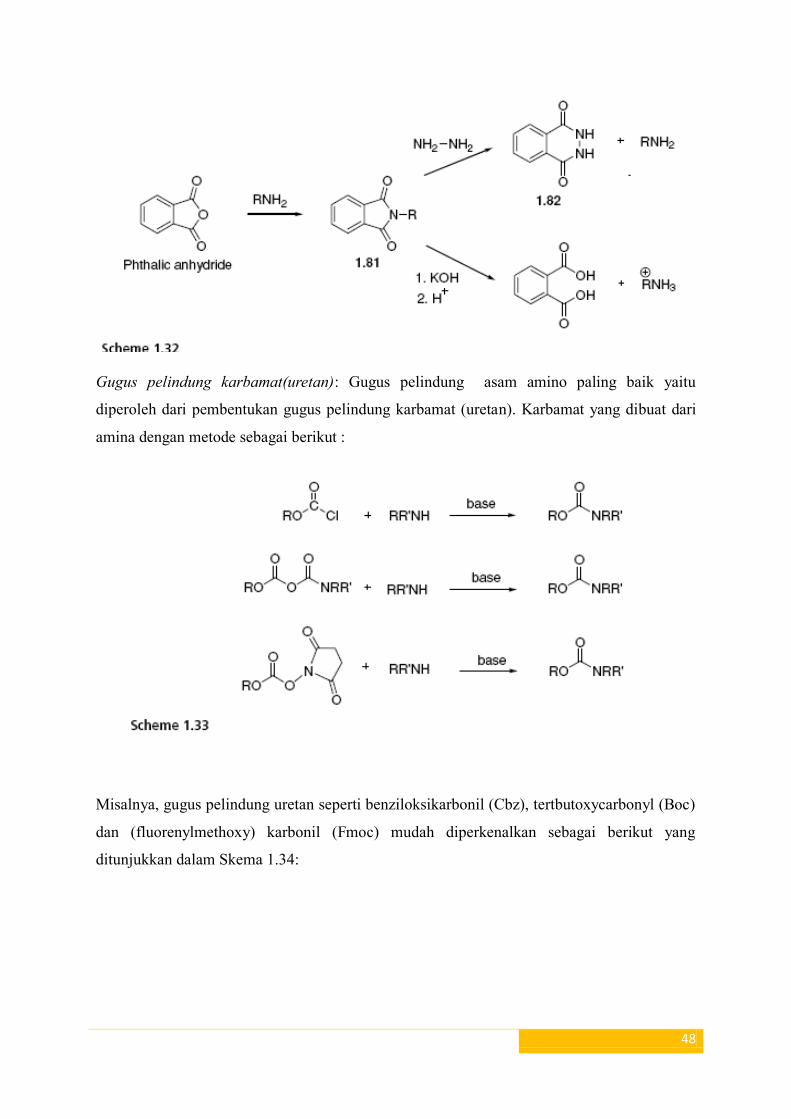
Gugus pelindung Imida dan amida: Kelompok ftalimida telah berhasil digunakan
untuk melindungi gugus amino. Pembelahan dari N-alkilftalimida (1,81) mudah dilakukan
dengan hidrazin, dalam larutan panas atau dalam dingin untuk waktu yang lama untuk
memberikan 1,82 dan amina. Basa-katalis hidrolisis N-alkilftalimida 1.81 juga memberikan
yang sesuai amina (Skema 1,32).
48
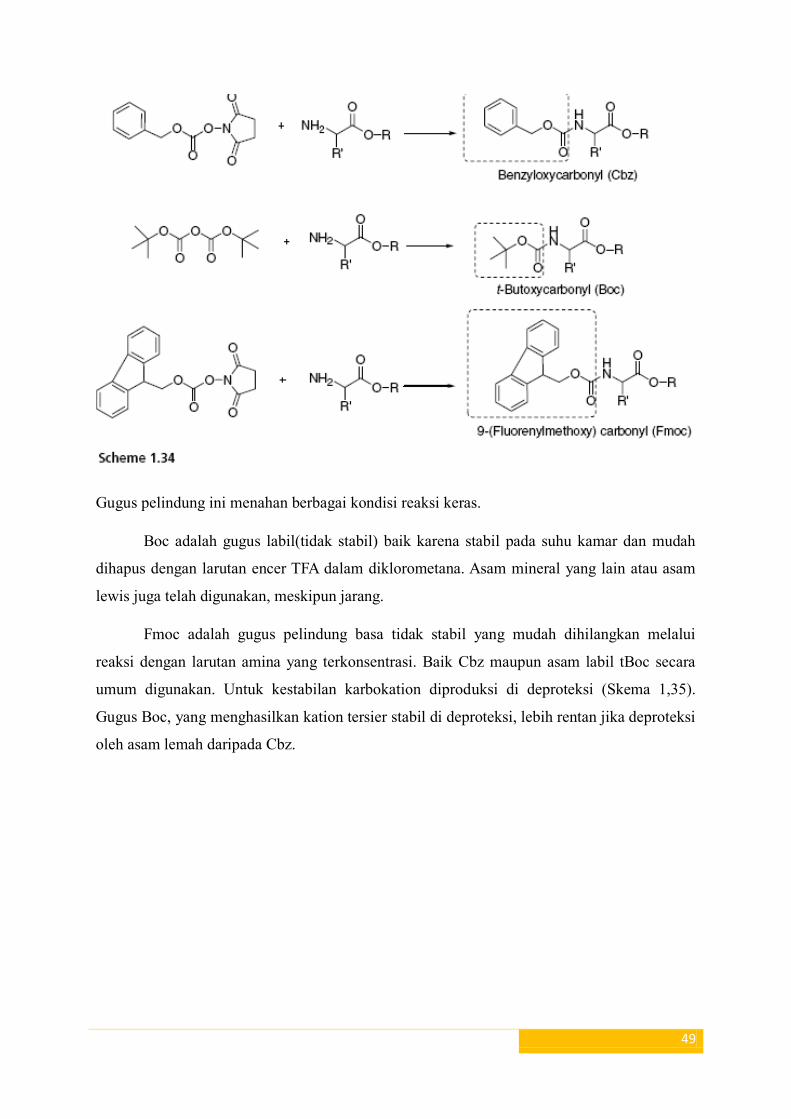
Gugus pelindung karbamat(uretan): Gugus pelindung asam amino paling baik yaitu
diperoleh dari pembentukan gugus pelindung karbamat (uretan). Karbamat yang dibuat dari
amina dengan metode sebagai berikut :
Misalnya, gugus pelindung uretan seperti benziloksikarbonil (Cbz), tertbutoxycarbonyl (Boc)
dan (fluorenylmethoxy) karbonil (Fmoc) mudah diperkenalkan sebagai berikut yang
ditunjukkan dalam Skema 1.34:
49
Gugus pelindung ini menahan berbagai kondisi reaksi keras.
Boc adalah gugus labil(tidak stabil) baik karena stabil pada suhu kamar dan mudah
dihapus dengan larutan encer TFA dalam diklorometana. Asam mineral yang lain atau asam
lewis juga telah digunakan, meskipun jarang.
Fmoc adalah gugus pelindung basa tidak stabil yang mudah dihilangkan melalui
reaksi dengan larutan amina yang terkonsentrasi. Baik Cbz maupun asam labil tBoc secara
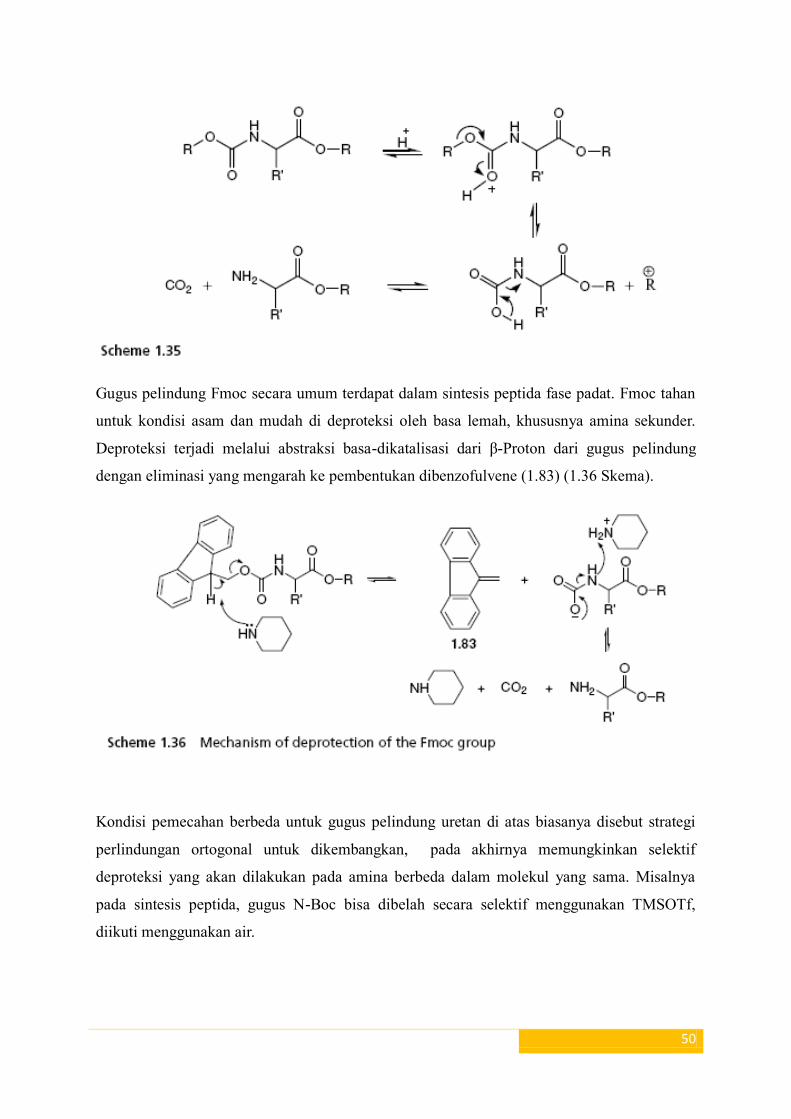
umum digunakan. Untuk kestabilan karbokation diproduksi di deproteksi (Skema 1,35).
Gugus Boc, yang menghasilkan kation tersier stabil di deproteksi, lebih rentan jika deproteksi
oleh asam lemah daripada Cbz.
50
Gugus pelindung Fmoc secara umum terdapat dalam sintesis peptida fase padat. Fmoc tahan
untuk kondisi asam dan mudah di deproteksi oleh basa lemah, khususnya amina sekunder.
Deproteksi terjadi melalui abstraksi basa-dikatalisasi dari β-Proton dari gugus pelindung
dengan eliminasi yang mengarah ke pembentukan dibenzofulvene (1.83) (1.36 Skema).
Kondisi pemecahan berbeda untuk gugus pelindung uretan di atas biasanya disebut strategi
perlindungan ortogonal untuk dikembangkan, pada akhirnya memungkinkan selektif
deproteksi yang akan dilakukan pada amina berbeda dalam molekul yang sama. Misalnya
pada sintesis peptida, gugus N-Boc bisa dibelah secara selektif menggunakan TMSOTf,
diikuti menggunakan air.
51
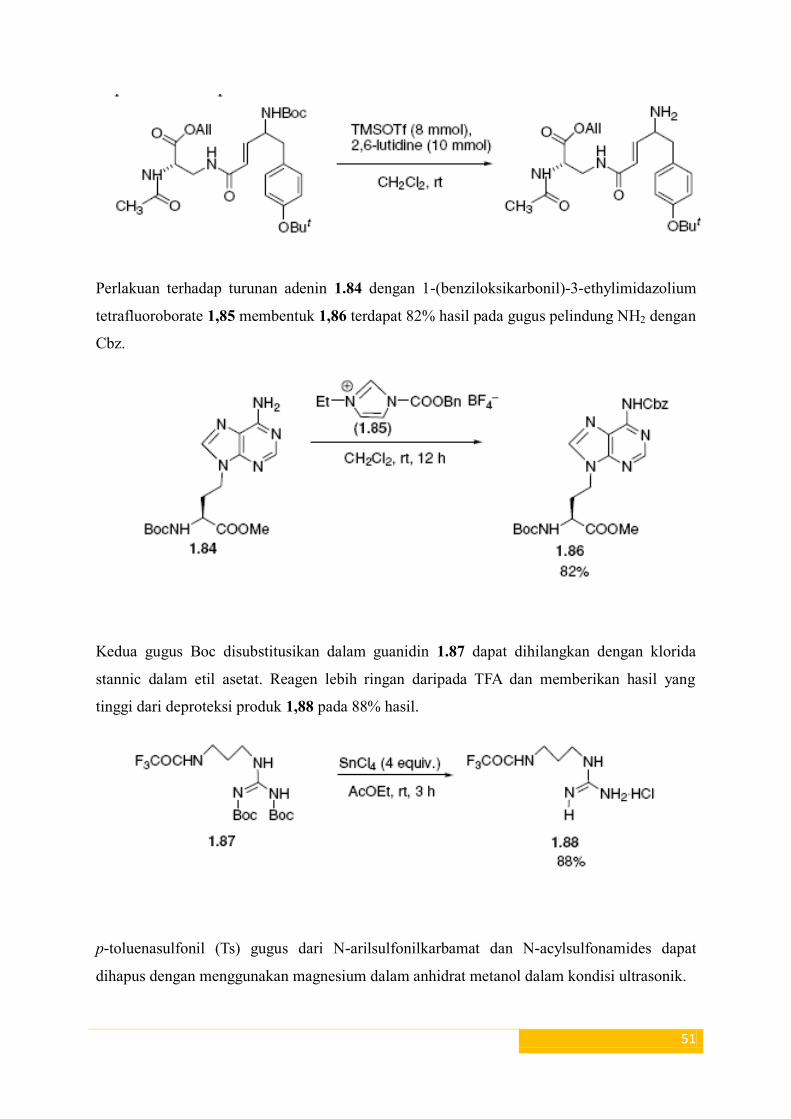
Perlakuan terhadap turunan adenin 1.84 dengan 1-(benziloksikarbonil)-3-ethylimidazolium
tetrafluoroborate 1,85 membentuk 1,86 terdapat 82% hasil pada gugus pelindung NH2 dengan
Cbz.
Kedua gugus Boc disubstitusikan dalam guanidin 1.87 dapat dihilangkan dengan klorida
stannic dalam etil asetat. Reagen lebih ringan daripada TFA dan memberikan hasil yang
tinggi dari deproteksi produk 1,88 pada 88% hasil.
p-toluenasulfonil (Ts) gugus dari N-arilsulfonilkarbamat dan N-acylsulfonamides dapat
dihapus dengan menggunakan magnesium dalam anhidrat metanol dalam kondisi ultrasonik.
52
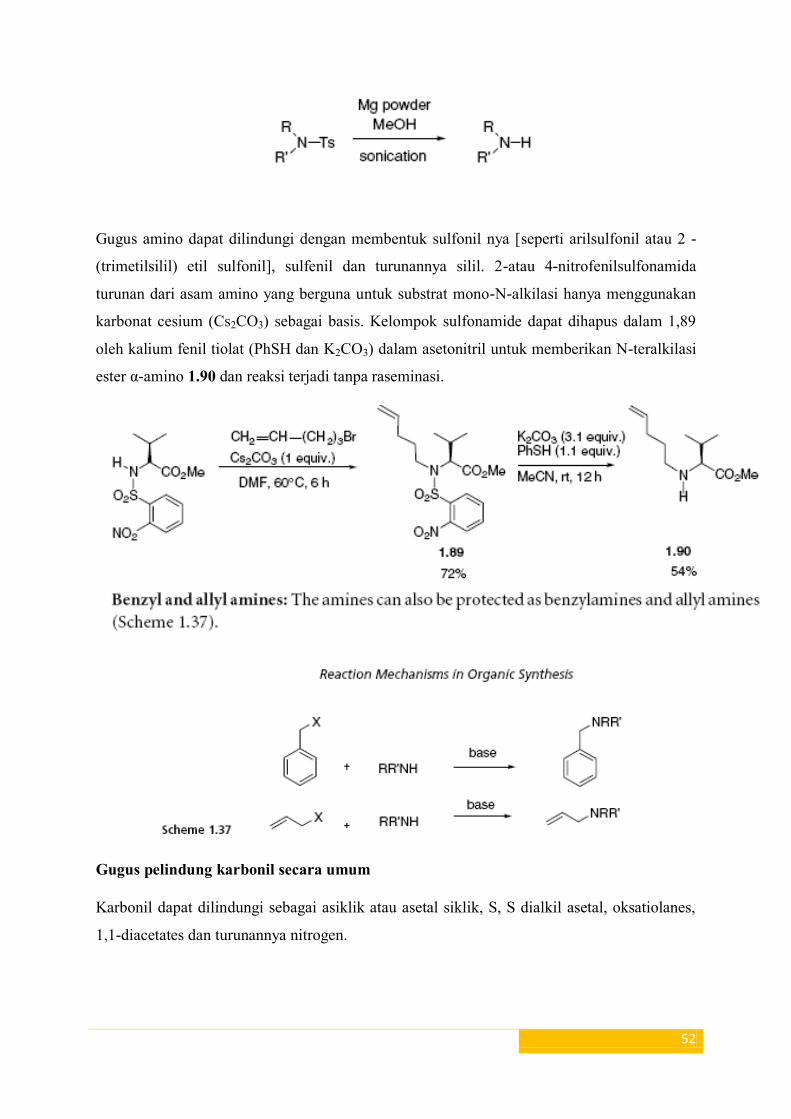
Gugus amino dapat dilindungi dengan membentuk sulfonil nya [seperti arilsulfonil atau 2 -
(trimetilsilil) etil sulfonil], sulfenil dan turunannya silil. 2-atau 4-nitrofenilsulfonamida
turunan dari asam amino yang berguna untuk substrat mono-N-alkilasi hanya menggunakan
karbonat cesium (Cs2CO3) sebagai basis. Kelompok sulfonamide dapat dihapus dalam 1,89
oleh kalium fenil tiolat (PhSH dan K2CO3) dalam asetonitril untuk memberikan N-teralkilasi
ester α-amino 1.90 dan reaksi terjadi tanpa raseminasi.
Gugus pelindung karbonil secara umum
Karbonil dapat dilindungi sebagai asiklik atau asetal siklik, S, S dialkil asetal, oksatiolanes,
1,1-diacetates dan turunannya nitrogen.

53
Asetal asiklik dan siklik stabil untuk basa, tetapi dihapus dengan asam. Aldehida alifatik yang
lebih reaktif daripada aldehida aromatik, dan lebih reaktif daripada keton. Dimetil Asetal
dapat dibuat dalam kondisi yang berbeda dari aldehida dan ketone65, 66
seperti yang
ditunjukkan di bawah ini:
Pembentukan asetal siklik dari αβ-karbonil tak jenuh biasanya lebih lambat dari pada
karbonil jenuh. Dengan demikian, keton jenuh dapat selektif dilindungi di hadapan αβ-keton
tak jenuh dengan etilena glikol dan sejumlah stoikiometri dari p-TsOH dan air67
.
Tetapi, αβ-keton tak jenuh juga selektif dilindungi seperti yang ditunjukkan68
:
54
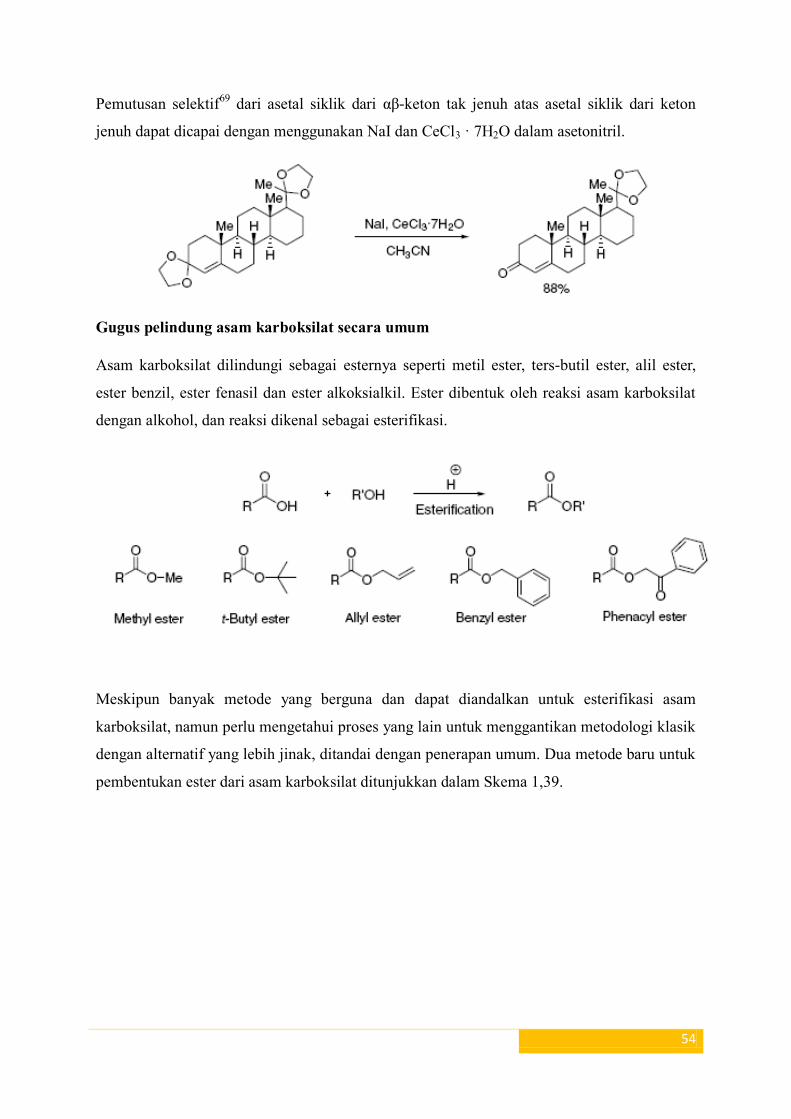
Pemutusan selektif69
dari asetal siklik dari αβ-keton tak jenuh atas asetal siklik dari keton
jenuh dapat dicapai dengan menggunakan NaI dan CeCl3 · 7H2O dalam asetonitril.
Gugus pelindung asam karboksilat secara umum
Asam karboksilat dilindungi sebagai esternya seperti metil ester, ters-butil ester, alil ester,
ester benzil, ester fenasil dan ester alkoksialkil. Ester dibentuk oleh reaksi asam karboksilat
dengan alkohol, dan reaksi dikenal sebagai esterifikasi.
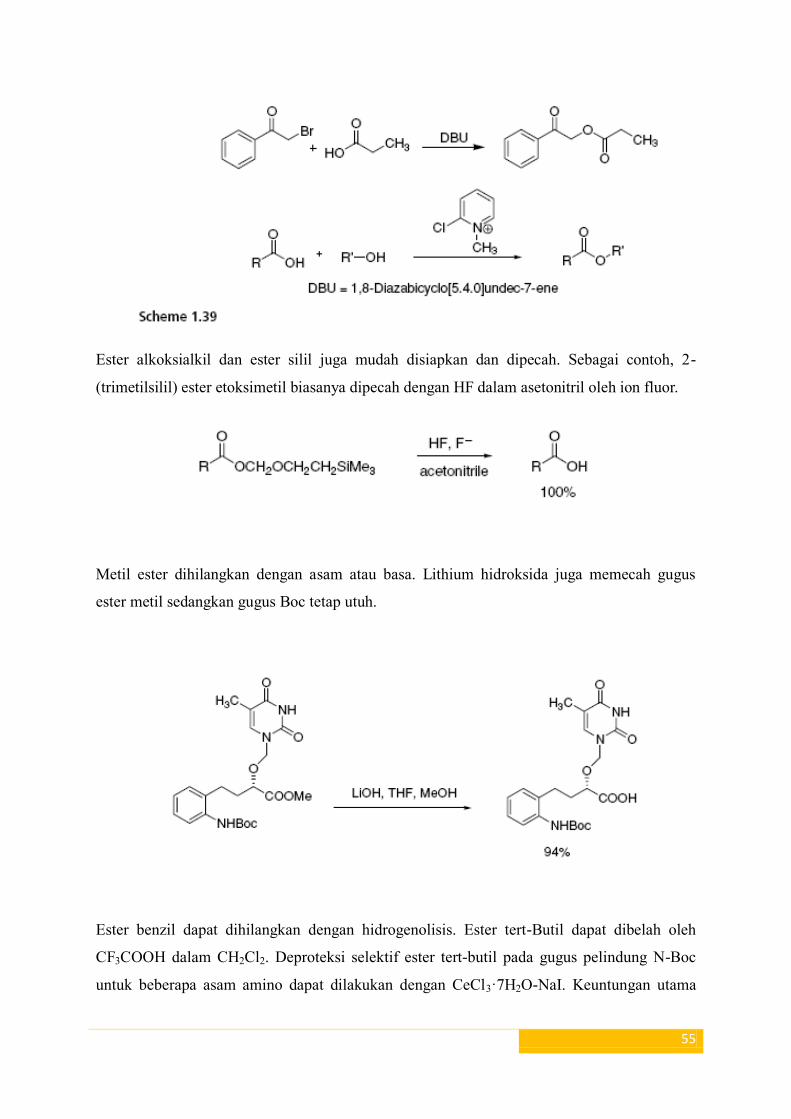
Meskipun banyak metode yang berguna dan dapat diandalkan untuk esterifikasi asam
karboksilat, namun perlu mengetahui proses yang lain untuk menggantikan metodologi klasik
dengan alternatif yang lebih jinak, ditandai dengan penerapan umum. Dua metode baru untuk
pembentukan ester dari asam karboksilat ditunjukkan dalam Skema 1,39.
55
Ester alkoksialkil dan ester silil juga mudah disiapkan dan dipecah. Sebagai contoh, 2-
(trimetilsilil) ester etoksimetil biasanya dipecah dengan HF dalam asetonitril oleh ion fluor.
Metil ester dihilangkan dengan asam atau basa. Lithium hidroksida juga memecah gugus
ester metil sedangkan gugus Boc tetap utuh.
Ester benzil dapat dihilangkan dengan hidrogenolisis. Ester tert-Butil dapat dibelah oleh
CF3COOH dalam CH2Cl2. Deproteksi selektif ester tert-butil pada gugus pelindung N-Boc
untuk beberapa asam amino dapat dilakukan dengan CeCl3·7H2O-NaI. Keuntungan utama
56
dari metode ini adalah biaya reagen murah dan sifat ringan dari interaksi klorida Ceric
dibandingkan dengan asam Lewis lainnya.
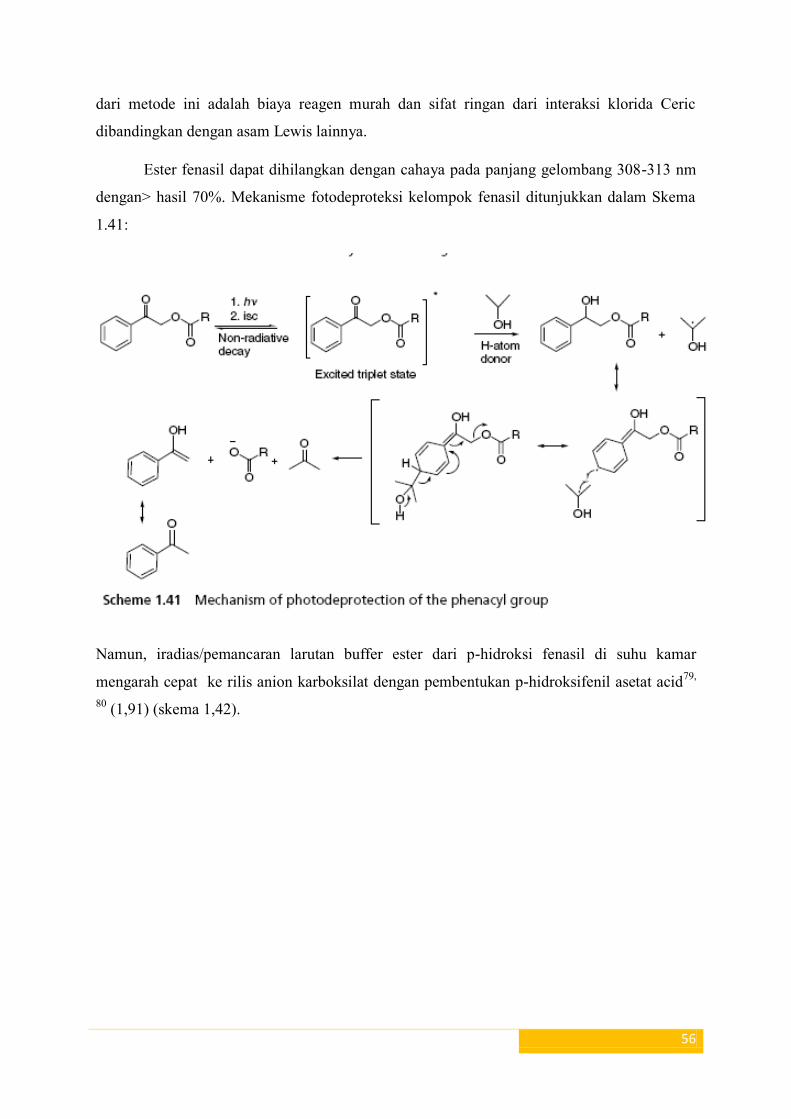
Ester fenasil dapat dihilangkan dengan cahaya pada panjang gelombang 308-313 nm
dengan> hasil 70%. Mekanisme fotodeproteksi kelompok fenasil ditunjukkan dalam Skema
1.41:
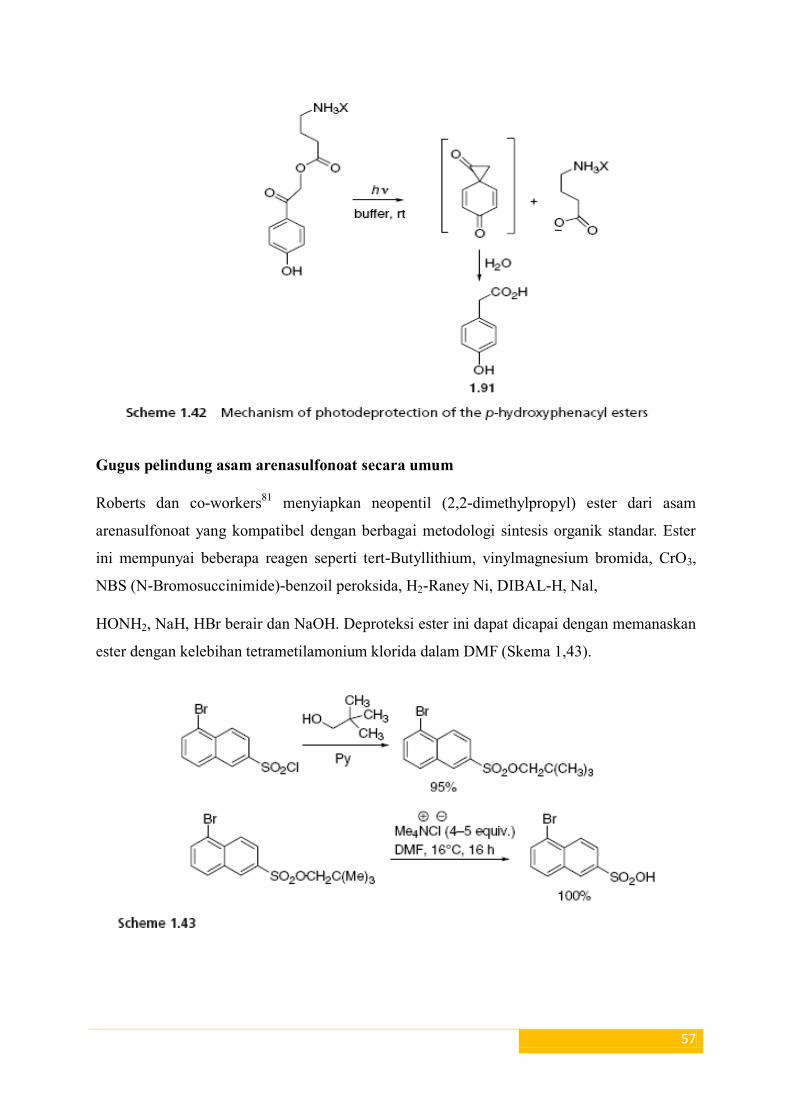
Namun, iradias/pemancaran larutan buffer ester dari p-hidroksi fenasil di suhu kamar
mengarah cepat ke rilis anion karboksilat dengan pembentukan p-hidroksifenil asetat acid79,
80 (1,91) (skema 1,42).
57
Gugus pelindung asam arenasulfonoat secara umum
Roberts dan co-workers81
menyiapkan neopentil (2,2-dimethylpropyl) ester dari asam
arenasulfonoat yang kompatibel dengan berbagai metodologi sintesis organik standar. Ester
ini mempunyai beberapa reagen seperti tert-Butyllithium, vinylmagnesium bromida, CrO3,
NBS (N-Bromosuccinimide)-benzoil peroksida, H2-Raney Ni, DIBAL-H, Nal,
HONH2, NaH, HBr berair dan NaOH. Deproteksi ester ini dapat dicapai dengan memanaskan
ester dengan kelebihan tetrametilamonium klorida dalam DMF (Skema 1,43).

58
Gugus pelindung alkuna secara umum
Alkuna dapat dilindungi sebagai derivatif silil dan yang paling umum gugus silil TMS, TES,
TIPS dan TBS diperkenalkan dengan mereaksikan alkuna dengan trialkilsilil sesuai klorida
(lihat Tabel 1.2 untuk struktur R´3SiCl).
Pemecahan trialkilsililalkuna dapat dicapai dengan menggunakan TBAF di hadapan THF.
Pemecahan trimetilsililalkuna juga dapat dilakukan dengan menggunakan KF / MeOH,
AgNO3 / 2,6 - lutidine atau K2CO3/MeOH.





























