Embed Size (px)
DESCRIPTION
studio di attività infrarossa e raman di strutture a nanotubo
Citation preview

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 1
STUDIO DELL’ATTIVITA’ INFRAROSSA E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Raffaella Esposito, Francesca Santoro, Marzia Antonella Scelsi
L’analisi svolta in questa relazione si prefigge di: studiare la geometria di alcune strutture molecolari a nanotubo presenti in letteratura, lavorando sia nel gruppo di simmetria globale della molecola sia attraverso la simmetria locale del campo di forza molecolare; ricavarne teoricamente la rappresentazione dei modi vibrazionali, distinguendoli tra stretching e bending; identificare i modi vibrazionali attivi in spettroscopia infrarossa e Raman, per quanto riguarda sia le transizioni fondamentali sia gli overtones, per giungere ad una predizione qualitativa della struttura a bande dei due tipi di spettri e confrontarla, ove possibile, con i risultati riportati in letteratura. Le strutture analizzate sono di due tipi: nanotubi di MgO (Chen et al., Physica E 41: 2009, 852-855) e strutture a tubo di isomeri del C60F60 (Jia et al., J. Am. Chem. Soc. 2008, 130, 3985-3988).
PARTE I: NANOTUBI DI MgO In Chen et al., Physica E 41: 2009, 852-855 si trovano due possibili strutture di nanotubi di MgO, riportate in seguito: una struttura a sezione quadrata composta da due molecole di MgO (indicata con il simbolo (MgO)2n) e una struttura a sezione esagonale perché composta da tre molecole di MgO ((MgO)3n). Si analizzano di seguito entrambe le strutture, considerando per ciascuna anche i sottocasi di struttura formata da un numero pari o dispari di strati; si utilizza la simbologia adottata nell’articolo citato (MgO)mn, dove m indica il numero di molecole che costituiscono la sezione del tubo e n il numero di strati che formano il tubo stesso.
Strutture (MgO)2n in alto e (MgO)3n in basso. Gli atomi in rosso sono di Ossigeno, quelli in bianco di Magnesio.
A. (MgO)3n
A tutte le strutture del tipo (MgO)3n, con n dispari, l’articolo citato assegna simmetria C2v. Tutte queste strutture presentano quindi come elementi di simmetria un asse C2 (in blu) e due piani verticali σxz (in verde) e σyz (in giallo), come si osserva nel disegno sottostante.

R. Esposito, F. Santoro, M. A. Scelsi Pag. 2
La struttura più semplice è formata da tre strati per un totale di 18 atomi: in via preliminare ci aspettiamo di trovare dalla nostra analisi 3N-6 = 48 modi vibrazionali. Partiamo dal calcolo della rappresentazione riducibile di spostamento Γd
rid della molecola, individuando il numero di atomi che restano fissi in ciascuna delle operazioni di simmetria e moltiplicandolo per il corrispondente carattere χ = 2 cos α ± 1 (χ è la traccia della matrice di rotazione di un angolo α) o, equivalentemente, per il corrispondente carattere nella rappresentazione traslazionale Γtrasl.
1 Il passo successivo è di scomporre la Γdrid come somma diretta delle
rappresentazioni irriducibili del gruppo C2v moltiplicate per degli opportuni fattori interi ai che si ricavano dalla seguente formula:
ai = (1/h) ΣR (χR ∙ χiR ∙ CR)
dove h è l’ordine del gruppo (numero di operazioni di simmetria presenti nel gruppo), χR è il carattere dell’operazione R nella rappresentazione riducibile Γrid, χi
R è il carattere dell’operazione R nella rappresentazione irriducibile Γi, CR è il numero di operazioni nella classe cui R appartiene. Questo procedimento è teoricamente giustificato dalle proprietà di ortonormalità delle rappresentazioni irriducibili. Il tutto è condensato nella tavola che segue.
C2v E C2 σxz σyz
# atomi fissi 18 2 6 6 Χ 3 -1 1 1
Γdrid 54 -2 6 6 Σ/4
A1 54 -2 6 6 16 A2 54 -2 -6 -6 10 B1 54 2 6 -6 14 B2 54 2 -6 6 14
Γd = 16*A1 ⊕ 10*A2 ⊕ 14*B1 ⊕ 14*B2
1 Il carattere di un’operazione nella rappresentazione di spostamento è dato dalla traccia della matrice dell’operatore
stesso in uno spazio a 3N dimensioni (N numero di atomi, 3 perché immagino di centrare su ogni atomo una terna
cartesiana e vedo come si trasformano i vettori di base sotto l’operazione considerata). A questa traccia
contribuiscono però solo i vettori di base che non cambiano posizione nella molecola, perciò devo contare il numero
degli atomi fissi. Poi, dal momento che gli unici moti possibili di un atomo sono le traslazioni, per sapere come
cambiano i vettori di base sotto le operazioni del gruppo di simmetria, devo moltiplicare il numero di atomi fissi in
ogni operazione per il carattere dell’operazione stessa nella rappresentazione traslazionale.

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 3
Per confermare la forma della Γd, studiamo la simmetria locale del campo di forza molecolare, individuando gli elementi di simmetria che passano per ciascun atomo della molecola e quindi collocando ciascun atomo in un gruppo locale. Studieremo poi le correlazioni tra i gruppi locali e quello globale della molecola, il C2v. Il numero di siti equivalenti è calcolato come rapporto tra l’ordine del gruppo globale della molecola hGM e l’ordine del gruppo locale hGL via via considerato. Si è calcolato dapprima il numero di siti e di atomi equivalenti di Ossigeno, e si è successivamente notato che all’atto pratico gli atomi di Magnesio hanno lo stesso comportamento in simmetria locale; i risultati sono pertanto così riassunti:
2 atomi ∈ C2v; 2 siti equivalenti → 2 atomi equivalenti (1 atomo Mg e 1 atomo O);
4 atomi ∈ Cs (σxz); 2 siti equivalenti → 2 atomi equivalenti (1 atomo Mg e 1 atomo O);
4 atomi ∈ Cs (σyz); 2 siti equivalenti → 2 atomi equivalenti (1 atomo Mg e 1 atomo O);
8 atomi ∈ C1; 4 siti equivalenti → 2 atomi equivalenti (1 atomo Mg e 1 atomo O), perciò:
ΓGM = 2 * ΓGL = C2v ⊕ 2 * ΓGL = Cs (σxz) ⊕ 2 * ΓGL = Cs(σyz) ⊕ 2 * ΓGL = C1 (*) Compiliamo quindi la tavola delle correlazioni discendenti tra il C2v e i gruppi locali Cs (σxz), Cs (σyz), C1:
C2v E σxz Cs (σxz) E σyz Cs (σyz) E C1
A1 1 1 A’ 1 1 A’ 1 A A2 1 -1 A’’ 1 -1 A’’ 1 A B1 1 1 A’ 1 -1 A’’ 1 A B2 1 -1 A’’ 1 1 A’ 1 A
Adesso possiamo costruire i diagrammi a frecce discendenti, ricordando anche che gli unici movimenti possibili di un atomo sono le traslazioni (indicate con Tx, Ty e Tz): C2v (k) Cs (σxz) (j) C2v (k) Cs (σyz) (j) C2v (k) C1 A1 A’ (Tx, Ty) A1 A’ (Tx, Ty) A1 A A2 A2 A2
B1 A’’ (Tz) B1 A’’ (Tz) B1 B2 B2 B2
Sfruttando ora il teorema delle correlazioni (v. pag. 10) è possibile costruire i diagrammi a frecce ascendenti come segue: Cs (σxz) (j) C2v (k) Cs (σyz) (j) C2v (k) C1 C2v (k) A’ A1 A’ A1 A A1 A2 A2 A2 A’’ B1 A’’ B1 B1 B2 B2 B2 Infine si riassume l’analisi di correlazione nella tabella seguente: Cs (σxz)
C2v
A’ x,y
A” z
n=Σj akj
Cs (σyz) C2v
A’ x,y
A” z
n=Σj akj
C1
C2v
A x,y,z
n=Σj akj
A1 2 - 2 2 - 2 3 3 A2 - 1 1 - 1 1 3 3 B1 2 - 2 - 1 1 3 3 B2 - 1 1 2 - 2 3 3
La correlazione tra il C2v e se stesso è banale: basta considerare la simmetria delle coordinate x, y e z. In definitiva avremo:

R. Esposito, F. Santoro, M. A. Scelsi Pag. 4
ΓGL = C2v = A1 ⊕ B1 ⊕ B2 ΓGL = Cs (σxz) = 2*A1 ⊕ A2 ⊕ 2*B1 ⊕ B2 ΓGL = Cs (σyz) = 2*A1 ⊕ A2 ⊕ B1 ⊕ 2*B2
ΓGL = C1 = 3*(A1 ⊕ A2 ⊕ B1 ⊕ B2) Sommando queste rappresentazioni secondo la (*), ritroviamo la rappresentazione di spostamento ricavata lavorando nel gruppo globale della molecola. Se dalla rappresentazione di spostamento, che descrive tutti i possibili moti molecolari, sottraiamo le rappresentazioni di traslazione e di rotazione (leggibili direttamente dalla tavola dei caratteri del C2v), otteniamo la rappresentazione vibrazionale, che descrive solo i moti di vibrazione della molecola
Γtrasl = A1 ⊕ B1 ⊕ B2
Γrot = A2 ⊕ B1 ⊕ B2
Γvibr = 15*A1 ⊕ 9*A2 ⊕ 12*B1 ⊕ 12*B2
che contiene effettivamente 48 modi di vibrazione, come ci aspettiamo. Inoltre si può notare che il numero e la simmetria dei modi di vibrazione coincidono con quelli trovati con metodo computazionale dagli autori dell’articolo sopra citato. A questo punto introduciamo l’analisi delle coordinate interne (lunghezze dei legami e angoli tra gli stessi) della molecola per discernere, tra i modi vibrazionali, quali siano vibrazioni di stiramento (stretching) e quali di piegamento (bending) della molecola. Questa analisi è utile dal momento che le vibrazioni di stiramento e quelle di piegamento hanno solitamente energie abbastanza diverse tra loro, e quindi le relative righe saranno collocate in regioni degli spettri abbastanza lontane tra loro. In particolare, è sufficiente lavorare con le lunghezze dei legami per ottenere la rappresentazione dei modi di stretching, e ricavare poi la rappresentazione del bending per sottrazione dalla Γvibr, in modo da evitare il problema delle ridondanze nel calcolo diretto della rappresentazione di bending. Nel seguito si ricava la rappresentazione riducibile relativa ai modi di stretching Γstretch considerando il numero di legami che si conservano per ciascuna della operazioni di simmetria.
C2v E C2 σxz σyz
Γstretch 30 0 6 4 Σ/4
A1 30 0 6 4 10 A2 30 0 -6 -4 5 B1 30 0 6 -4 8 B2 30 0 -6 4 7
Γstretch = 10*A1 ⊕ 5*A2 ⊕ 8*B1 ⊕ 7*B2
che corrisponde a 30 modi di stretching e
Γbend = 5*A1 ⊕ 4*A2 ⊕ 4*B1 ⊕ 5*B2
che corrisponde a 18 modi di vibrazione di deformazione angolare (bending). Gli autori dell’articolo a questo punto focalizzano la propria attenzione su tre modi vibrazionali precisi, indicati con ASM (Axial Stretching Mode, modo di stiramento assiale, di simmetria B2), RBM (Radial Breathing Mode, modo di respiro radiale, di simmetria A1) e RTM (Radial Twisting Mode, modo di avvitamento radiale, di simmetria A2). È possibile visualizzare direttamente i movimenti atomici che caratterizzano ciascuna di queste vibrazioni molecolari attraverso l’uso delle coordinate di simmetria Si, definite come:
Si = ΣR *χiR ∙ R̂(Δs)+
dove χiR è il carattere dell’operazione R nella rappresentazione irriducibile Γi e R̂ (Δs) è il risultato
dell’applicazione dell’operazione R ad un vettore Δs che rappresenta la variazione di lunghezza di un

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 5
legame tra due atomi. Utilizzando graficamente delle frecce per indicare il Δs, è possibile avere una visualizzazione immediata ed intuitiva di come si spostano gli atomi all’interno della molecola.
Rappresentazione grafica con le coordinate di simmetria del modo RBM (Ag)
Rappresentazione grafica con le coordinate di simmetria del modo RTM (Au)
Rappresentazione grafica con le coordinate di simmetria del modo ASM (Bu)
Come si può notare, nei disegni sono rappresentati nanotubi del tipo (MgO)36, quindi con n pari; tuttavia la rappresentazione con le coordinate di simmetria è la stessa anche se, in questo caso, la molecola appartiene al gruppo C2h e le vibrazioni RBM, RTM e ASM hanno simmetria Ag, Au e Bu. Una transizione vibrazionale è attiva nell’infrarosso se il momento di dipolo della molecola cambia durante la vibrazione; analogamente, una transizione vibrazionale è attiva in Raman se la polarizzabilità della molecola varia durante la transizione. Infatti la probabilità di transizione, per esempio nell’IR, è data dal modulo quadro dell’integrale del momento di transizione
Mνν' = ∫ ψ*(ν') μ ψ(ν) dx e se il momento di dipolo μ fosse costante potrebbe essere portato fuori dell’integrale, il cui risultato sarebbe zero per l’ortonormalità delle autofunzioni dello stato iniziale e finale. Il momento di dipolo è una grandezza vettoriale definita come μ = ex, dove e indica la carica elementare e x la separazione tra le cariche; pertanto μ è un vettore a tre componenti che si trasformano come x, y e z e che quindi formano una base per le rappresentazioni irriducibili del gruppo di simmetria della molecola; la polarizzabilità α

R. Esposito, F. Santoro, M. A. Scelsi Pag. 6
invece è un tensore simmetrico di rango tre, quindi ha sei componenti indipendenti la cui simmetria è riportata nelle colonne dei prodotti di ordine due x2, xy, etc. nelle tavole dei caratteri. Supponiamo che ψ(ν') formi una base per la rappresentazione irriducibile Γν' (stato finale), che ψ(ν) formi una base per la rappresentazione irriducibile Γν (stato iniziale) e che le componenti di μ formino una base per la rappresentazione Γμ. Una proprietà interessante è che la probabilità di transizione, legata all’integrale di cui sopra, è diversa da zero (e quindi la transizione è permessa) se e solo se nel prodotto diretto Γν' x Γμ x Γν compare almeno una volta la rappresentazione totalsimmetrica. Lo stesso dicasi per le transizioni Raman. Detto questo, non resta che stabilire la simmetria delle funzioni d’onda dello stato iniziale e finale delle transizioni che vogliamo considerare. Nell’approssimazione dell’oscillatore armonico, lo stato fondamentale è descritto sempre da una funzione d’onda pari, che quindi forma una base della rappresentazione totalsimmetrica di qualunque gruppo; lo stesso dicasi per le funzioni d’onda che descrivono stati eccitati con numero quantico n pari, perché queste funzioni hanno una dipendenza dalle potenze pari della coordinata di spostamento q; infine le funzioni d’onda che descrivono stati eccitati con numero quantico dispari hanno la stessa simmetria della vibrazione. A questo punto, enunciamo una regola molto utile per determinare le transizioni fondamentali permesse negli spettri IR e Raman ed per rendere immediata questa determinazione. Si noti che nel prodotto diretto Γν' x Γμ x Γν (essendo Γν la rappresentazione totalsimmetrica) l’unico modo per far comparire nel risultato la rappresentazione totalsimmetrica è che una delle componenti di Γμ abbia la stessa simmetria di Γν'. Quindi le uniche transizioni fondamentali attive in IR (o equivalentemente in Raman) sono quelle che hanno la stessa simmetria di una delle componenti di μ (o equivalentemente di α). In tal caso le componenti del momento di dipolo μ e della polarizzabilità α sono le seguenti:
Γμ = (A1, B1, B2) Γα = (A1, A2, B1, B2) Esplicitiamo dunque il primo prodotto diretto Γν' x Γμ x Γν relativo, ad esempio, alla transizione fondamentale 01 in simmetria A1 (i calcoli sono identici per gli overtones):
IR:
2
1
1
1
2
1
1
1B
B
A
A
B
B
A
A attiva in spettroscopia IR
Raman:
2
1
2
1
1
2
1
2
1
1
B
B
A
A
A
B
B
A
A
A attiva in spettroscopia Raman
Per overtones si intendono transizioni dallo stato fondamentale del modo vibrazionale a stati eccitati superiori al primo. Così il primo overtone sarà la transizione 0 2, il secondo sarà la transizione 0 3, e così via. La funzione d’onda del secondo stato eccitato di ogni modo vibrazionale non degenere avrà simmetria A1, come è stato spiegato in precedenza; invece le funzioni d’onda del terzo stato eccitato avranno la stessa simmetria del modo, rispettivamente A1, B1 e B2. Avendo a disposizione tutto il background teorico necessario, risulta veramente semplice compilare la seguente tabella per la molecola (MgO)3n con i modi vibrazionali attivi nei due tipi di spettri:
Spettro infrarosso Spettro Raman
Fondamentale A1, B1, B2
39 bande attese, 39 modi attivi (per n=3)
A1, A2, B1, B2 48 bande attese, 48 modi attivi
(per n=3)
Primo overtone (tutti) (tutti)
Secondo overtone A1, B1, B2 (tutti)

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 7
Alle strutture del tipo (MgO)3n, con n pari, l’articolo assegna simmetria C2h: queste strutture presentano quindi come elementi di simmetria un asse C2, un piano orizzontale σh e un centro di simmetria i. Per semplicità si è analizzata una struttura (MgO)34, quindi formata da quattro anelli esagonali sovrapposti, per un totale di 24 atomi nella molecola. Come sarà confermato dall’analisi nel seguito, ci aspettiamo dunque 3N-6 = 66 modi vibrazionali possibili della molecola. Compiliamo la tabella per ricavare la rappresentazione riducibile di spostamento Γd
rid e scomporla nelle rappresentazioni irriducibili del gruppo, infine sottraiamo le rappresentazioni traslazionale e rotazionale per ottenere quella vibrazionale:
C2h E C2 I σh
# atomi fissi 24 0 0 8 χ 3 -1 -3 1
Γdrid 72 0 0 8 Σ/4
Ag 72 0 0 8 20 Bg 72 0 0 -8 16 Au 72 0 0 -8 16 Bu 72 0 0 8 20
Γd = 20*Ag ⊕ 16*Bg ⊕ 16*Au ⊕ 20*Bu
Si esegue dunque l’analisi della molecola in simmetria locale per verificare la correttezza dell’espressione di Γd ottenuta. Osservando una struttura del tipo (MgO)3n a 4 strati, si vede che:
8 atomi ∈ Cs (σh); 2 siti equivalenti → 4 atomi equivalenti (2 di ossigeno, 2 di magnesio);
16 atomi ∈ C1; 4 siti equivalenti → 4 atomi equivalenti (2 di ossigeno, 2 di magnesio); perciò:
ΓGM = 4 * ΓGL = Cs ⊕ 4 * ΓGL = C1 (**) Compiliamo quindi la tavola delle correlazioni discendenti tra il C2h e i gruppi locali Cs (σh), C1:
C2h E σh Cs (σh) C1
Ag 1 1 A’ A Bg 1 -1 A” A Au 1 -1 A” A Bu 1 1 A’ A
In base al teorema delle correlazioni (v. pag. 8), possiamo quindi compilare la seguente tabella:
Cs (σh)
C2h
A’ x,y
A” z
n=Σj akj
C1
C2h
A x,y,z
n=Σj akj
Ag 2 - 2 Ag 3 3 Bg - 1 1 Bg 3 3 Au - 1 1 Au 3 3 Bu 2 - 2 Bu 3 3
In definitiva avremo:
ΓGL = Cs = 2*Ag ⊕ Bg ⊕ Au ⊕ 2*Bu ΓGL = C1 = 3*(Ag ⊕ Au ⊕ Bg ⊕ Bu)

R. Esposito, F. Santoro, M. A. Scelsi Pag. 8
Sommando queste rappresentazioni secondo la (**), ritroviamo la rappresentazione di spostamento ricavata lavorando nel gruppo globale della molecola.
Si ricava quindi la rappresentazione dei modi vibrazionali:
Γtrasl = Au ⊕ 2*Bu
Γrot = Ag ⊕ 2*Bg
Γvibr = 19*Ag ⊕ 14*Bg ⊕ 15*Au ⊕ 18*Bu
Il numero di modi vibrazionali e la loro ripartizione tra le varie simmetrie coincidono con i risultati esposti nell’articolo citato. Con la stessa analisi di coordinate interne del (MgO)33 ricaviamo le rappresentazioni di stretching e bending.
C2h E C2 I σh
Γstretch 42 0 0 6 Σ/4
Ag 42 0 0 6 12 Bg 42 0 0 -6 9 Au 42 0 0 -6 9 Bu 42 0 0 6 12
Γstretch = 12*Ag ⊕ 9*Bg ⊕ 9*Au ⊕ 12*Bu
Γbend = 7*Ag ⊕ 5*Bg ⊕ 6*Au ⊕ 6*Bu
Da ultimo, per studiare l’attività IR e Raman, leggiamo dalla tavola dei caratteri la simmetria delle componenti del momento di dipolo μ e della polarizzabilità α:
Γμ = (Au, Bu) Γα = (Ag, Bg) perciò avremo come modi attivi nei due tipi di spettri:
Spettro infrarosso Spettro Raman
Fondamentale Au, Bu
33 bande attese, 33 modi attivi (per n=4)
Ag, Bg 33 bande attese, 33 modi attivi
(per n=4)
Primo overtone (nessuno) (tutti)
Secondo overtone Au, Bu Ag, Bg
Il risultato è coerente con la regola generale secondo cui, se un gruppo di simmetria contiene al suo interno l’operazione di inversione, si verifica un fenomeno chiamato esclusione IR-Raman: i modi vibrazionali attivi in IR non possono esserlo in Raman e viceversa; possiamo tuttavia avere l’esclusione IR – Raman in alcuni gruppi che non contengono l’inversione, ad esempio il D5h.
B. (MgO)2n In strutture di tipo (MgO)2n con un numero pari di strati è possibile individuare i seguenti elementi di simmetria: un asse C2 (in verde nella figura seguente), due assi C’2 perpendicolari tra loro e perpendicolari all’asse precedente (in blu), due piani di riflessione σd ortogonali tra loro (in giallo) e due roto-riflessioni S4 relativi ai due assi C’2; il gruppo di simmetria a cui appartiene a molecola è pertanto il D2d, di ordine h=8.

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 9
Elementi di simmetria del nanotubo (MgO)2,4.
La struttura presa in esame è costituita da N=16 atomi (8 di magnesio e 8 di ossigeno), quindi ci si aspettano 3N-6=42 modi di vibrazione. Come già mostrato in precedenza, ricaviamo la rappresentazione riducibile di spostamento Γd
rid della molecola:
D2d E 2S4 C2 2C’2 2σd
# atomi fissi 16 0 0 0 8 Χ 3 -1 -1 -1 1
Γdrid 48 0 0 0 8 Σ/8
A1 48 0 0 0 16 8 A2 48 - - - -16 4 B1 48 - - - -16 4 B2 48 - - - 16 8 E 96 - - - 0 12
Γd = 8*A1 ⊕ 4*A2 ⊕ 4*B1 ⊕ 8*B2 ⊕ 12*E
Si noti che, tenendo conto che la rappresentazione irriducibile E è doppiamente degenere, il numero totale di modi di spostamento della molecola 8+4+4+8+2*12=48 è pari al numero di gradi di libertà g.d.l.=3*N=48 della molecola stessa, e ciò rappresenta una prima conferma della corretta riduzione della Γd
rid nelle rappresentazioni irriducibili del gruppo D2d. Al fine di ottenere un’ulteriore conferma si è proceduto all’analisi della struttura della molecola in simmetria locale, identificando quali atomi appartengono agli elementi di simmetria del gruppo globale D2d; osservando la figura a pag. 7 si deduce che ciascuno degli atomi del nanotubo è contenuto in uno dei due piani di riflessione σd, mentre nessuno di essi appartiene agli assi di rotazione C2. Pertanto il gruppo locale a cui siamo interessati è il CS, in quanto contiene soltanto la trasformazione identica E ed una riflessione rispetto al piano σ; si riporta di seguito la tavola dei caratteri relativa a tale gruppo:
CS E σ
A’ 1 1
A” 1 -1
Si è quindi studiata la correlazione discendente tra gruppo globale GM= D2d e gruppo locale GL= CS, attraverso la seguente tabella:

R. Esposito, F. Santoro, M. A. Scelsi Pag. 10
D2d E Σ CS
A1 1 1 A’
A2 1 -1 A”
B1 1 -1 A”
B2 1 1 A’
E 2 0 A’ ⊕ A”
Ora, applicando il teorema della correlazione è possibile passare dal gruppo locale a quello globale: secondo tale teorema, infatti, il numero di volte aj
k che la k-esima rappresentazione irriducibile in GM è generato dalla j-esima rappresentazione in GL, è pari al numero di volte ak
j che la rappresentazione j-esima è contenuta nella rappresentazione globale k-esima. Completando la tabella delle correlazioni ascendenti dal gruppo locale CS al gruppo globale D2d è quindi possibile ricavare la rappresentazione di spostamento della molecola Γd
rid a partire dalla simmetria locale della molecola: CS
D2d A’ x,y
A” z
n=Σj akj
A1 2 - 2
A2 - 1 1
B1 - 1 1
B2 2 - 2
E 2 1 3
Calcoliamo quindi il numero di atomi equivalenti:
(2 atomi equivalenti di ossigeno e 2 atomi equivalenti di magnesio)
Quindi
ΓdGM = 4*Γd
GL = 4*(2*A1 ⊕ A2 ⊕ B1 ⊕ 2*B2 ⊕ 3*E) Si è quindi ulteriormente verificata la riduzione della Γd
nelle rappresentazioni irriducibili del gruppo globale D2d. Se dalla rappresentazione di spostamento così ottenuta si sottraggono quelle relative ai modi di traslazione e rotazione otteniamo la seguente rappresentazione vibrazionale:
Γtrasl = B2⊕ E
Γrot = A2 ⊕ E
Γvibr = 8*A1 ⊕ 3*A2 ⊕ 4*B1 ⊕ 7*B2 ⊕ 10*E
In cui è possibile riconoscere effettivamente 8+3+4+7+2*10=42 modi di vibrazione, come ci si aspetta. Si procede dunque con l’analisi in coordinate interne, al fine di distinguere modi di vibrazione di stretching e di bending. In particolare si ricava la rappresentazione riducibile relativa ai modi di stretching Γstretch

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 11
considerando il numero di legami che si conservano per ciascuna della operazioni di simmetria; infine si ricava la rappresentazione relativa a i modi di bending come differenza tra Γvibr e Γstretch.
D2d E 2S4 C2 2C’2 2σd
Γstretch 28 0 0 0 6 Σ/8
A1 28 - - - 12 5
A2 28 - - - -12 2
B1 28 - - - -12 2
B2 28 - - - 12 5
E 56 - - - 0 7
Γstretch = 5*A1 ⊕ 2*A2 ⊕ 2*B1 ⊕ 5*B2 ⊕ 7*E
Γbend = Γvibr- Γstretch = 3*A1 ⊕ A2 ⊕ 2*B1 ⊕ 2*B2 ⊕ 3*E
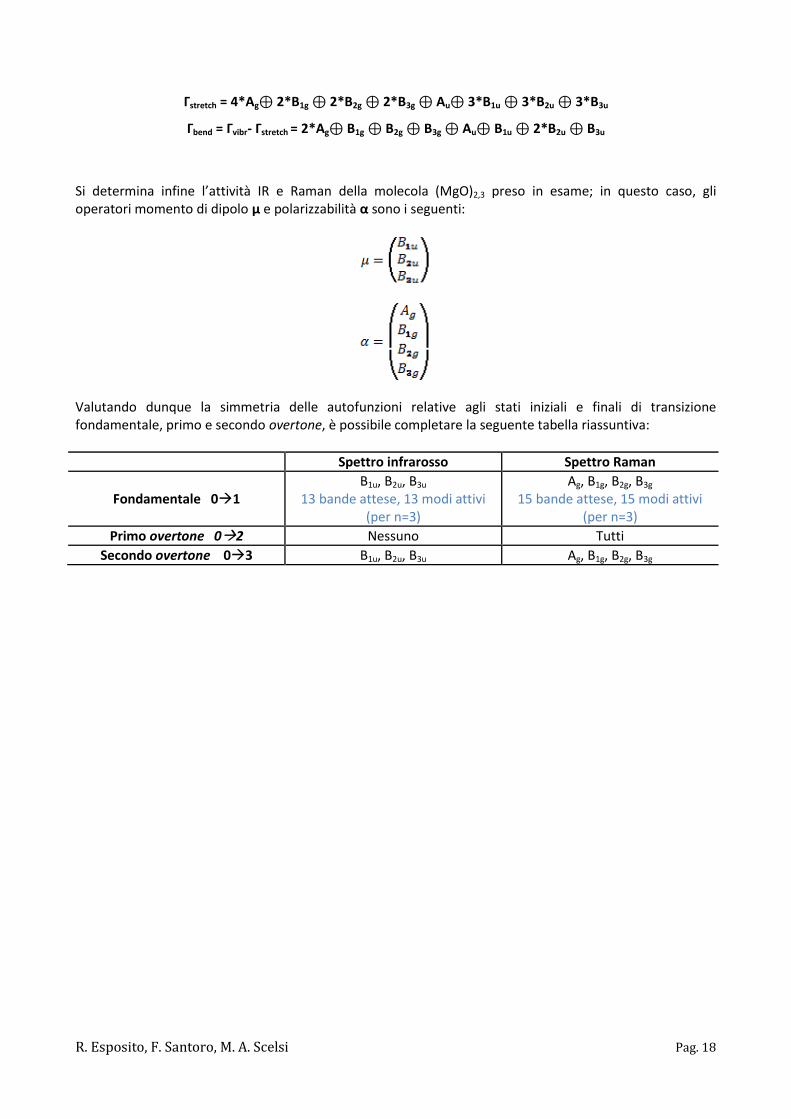
Si determina infine l’attività IR e Raman delle transizioni vibrazionali del nano tubo (MgO)2,4 preso in esame, secondo la regola già enunciata. Si noti, a tal proposito, che gli operatori momento di dipolo μ e polarizzabilità α sono i seguenti:
Valutando dunque la simmetria delle autofunzioni relative agli stati iniziali e finali di transizione fondamentale, primo e secondo overtone, è possibile completare la seguente tabella riassuntiva:
Spettro infrarosso Spettro Raman
Fondamentale 01 B2,E
17 bande attese, 27 modi attivi (per n=4)
A1, B1, B2, E 29 bande attese, 39 modi attivi
(per n=4)
Primo overtone 02 E A1, A2, B1, B2, E
Secondo overtone 03 B2,E A1, B1, B2, E
Si noti che nel calcolo degli overtones è necessario valutare come si trasformano le funzioni d’onda dei livelli eccitati relativi a modi vibrazionali di simmetria E, ovvero doppiamente degeneri. Si potrebbe pensare che la simmetria di tali funzioni sia ricavabile, per esempio per il primo overtone, semplicemente dal
prodotto diretto E E; tuttavia questo prodotto diretto è di ordine 4 e quindi dà luogo a 4 funzioni d’onda, mentre ad un’analisi più attenta si scopre che il secondo stato eccitato è descritto solo da 3 funzioni, secondo lo schema seguente: Transizione 02: E’a(0) E’b(2) E’a(0) E’b(0) E’a(1) E’b(1) E’a(2) E’b(0)

R. Esposito, F. Santoro, M. A. Scelsi Pag. 12
L’analisi corretta degli overtones delle vibrazioni degeneri prevede quindi l’uso della seguente formula ricorsiva:
χv(R)=1/2[χ(R)χv−1(R)+χ(Rv)]
( 1(R) = (R) e 0(R) = 1) per calcolare il carattere, sotto l’operazione di simmetria R, del ν-esimo livello energetico del modo vibrazionale doppiamente degenere. La combinazione di caratteri così ottenuta viene scomposta nella somma diretta delle rappresentazioni irriducibili del gruppo di simmetria, la quale fornisce appunto le simmetrie delle funzioni d’onda che rappresentano lo stato. Di seguito si esplicita il calcolo per primo e secondo overtone, quindi rispettivamente per le transizioni dallo stato fondamentale verso gli stati eccitati ν=2 e ν=3.
D2d E 2S4 C2 2C’2 2σd
χ(R) 2 0 -2 0 0
R2 E C2 E E E
χ(R2) 2 -2 2 2 2
χ2(R) 3 -2 3 1 1 A1 ⊕ B1⊕ B2
R3 E C’2 C2 C’2 σd
χ(R3) 2 0 -2 0 0
χ3(R) 4 0 -4 0 0 2 E
Da cui si ricava:
ψν=2E ~ Γ₂ = A1 ⊕ B1⊕ B2
ψν=3E ~ Γ₃ = 2E
A questo punto, valutando il prodotto diretto Γν' x Γμ/α x Γν si può concludere che le transizioni 02 e 03 in simmetria E, sono attive sia in simmetria Raman che IR. Esplicitiamo, ad esempio, la transizione 02 in IR:
EA
EA
EB
AE
B
B
B
A
1
2
2
12
2
1
1
Si analizza ora la struttura (MgO)2n con numero n dispari di strati (vedi Fig. seguente).

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 13
Gli elementi di simmetria della molecola sono tre assi di rotazione C2 lungo tre direzioni ortogonali (in blu in figura), e tre piani di riflessione σxy, σxz, σyz anch’essi perpendicolari tra loro; il gruppo di simmetria a cui appartiene la molecola presa in esame è dunque il D2h (h=8). Il nanotubo a tre strati è costituito da 12 atomi, pertanto ci si aspetta 12*3-6=30 modi di vibrazione della molecola. Ricaviamo dunque, come già mostrato, la rappresentazione riducibile di spostamento:
D2h E C2(z) C2(y) C2(x) i σ(xy) σ(xz) σ(yz)
# atomi fissi 12 2 0 2 0 6 4 6 Χ 3 -1 -1 -1 -3 1 1 1
Γdrid 36 -2 0 -2 0 6 4 6 Σ/8
Ag 36 -2 - -2 - 6 4 6 6 B1g 36 -2 - 2 - 6 -4 -6 4 B2g 36 2 - 2 - -6 4 -6 4 B3g 36 2 - -2 - -6 -4 6 4 Au 36 -2 - -2 - -6 -4 -6 2 B1u 36 -2 - 2 - -6 4 6 5 B2u 36 2 - 2 - 6 -4 6 6 B3u 36 2 - -2 - 6 4 -6 5
Γd
rid = 6*Ag⊕ 4*B1g ⊕ 4*B2g ⊕ 4*B3g ⊕ 2*Au⊕ 5*B1u ⊕ 6*B2u ⊕ 5*B3u
Γtrasl =B1u ⊕ B2u ⊕ B3u
Γrot = B1g ⊕ B2g ⊕ B3g
Γvibr = 6*Ag⊕ 3*B1g ⊕ 3*B2g ⊕ 3*B3g ⊕ 2*Au⊕ 4*B1u ⊕ 5*B2u ⊕ 4*B3u
Si noti che la Γvibr è costituita effettivamente da 30 modi di vibrazione secondo le previsioni. Ora si procede con l’analisi in simmetria locale per verificare la correttezza dell’espressione della Γd
rid:
2 atomi (asse z) ∈ C2V (Z) (atomi O)
2 atomi (asse x) ∈ C2V (X) (atomi Mg)
4 atomi ∈ CS(σyz) (atomi Mg)
4 atomi ∈ CS(σxy) (atomi O)
Calcoliamo quindi il numero di atomi equivalenti:

R. Esposito, F. Santoro, M. A. Scelsi Pag. 14
(1 atomo di O, 1 atomo di Mg)
(1 atomo di O, 1 atomo di Mg)
La rappresentazione riducibile di spostamento relativa al gruppo globale si può scrivere dunque come segue:
)()()(2)(2 xySyzSxVzV Cd
Cd
Cd
Cd
GMridd
(Eq. 1)
Studiamo le correlazioni tra gruppo globale GM= D2h e gruppi locali.
Correlazione D2h- C2V (Z)
D2h E C2 (z) σ(xz) σ’(yz) C2V (Z)
Ag 1 1 1 1 A1
B1g 1 1 -1 -1 A2
B2g 1 -1 1 -1 B1
B3g 1 -1 -1 1 B2
Au 1 1 -1 -1 A2
B1u 1 1 1 1 A1
B2u 1 -1 -1 1 B2
B3u 1 -1 1 -1 B1
C2V(Z)
D2h A1 z
A2 (*) B1 x
B2 y
n=Σj akj
Ag 1 - - - 1
B1g - - - - 0
B2g - - 1 - 1
B3g - - - 1 1
Au - - - - 0
B1u 1 - - - 1
B2u - - - 1 1
B3u - - 1 - 1
(*) : Si trascura la simmetria A2, poiché non è un modo di spostamento per la molecola.
Correlazione D2h- C2V (X)

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 15
D2h E C2 (x) σ(xy) σ’(xz) C2V (X)
Ag 1 1 1 1 A1
B1g 1 -1 1 -1 B1
B2g 1 -1 -1 1 B2
B3g 1 1 -1 -1 A2
Au 1 1 -1 -1 A2
B1u 1 -1 -1 1 B2
B2u 1 -1 1 -1 B1
B3u 1 1 1 1 A1
C2V(X)
D2h A1 z
A2 (*) B1 x
B2 y
n=Σj akj
Ag 1 - - - 1
B1g - - 1 - 1
B2g - - - 1 1
B3g - - - - -
Au - - - - -
B1u - - - 1 1
B2u - - 1 - 1
B3u 1 - - - 1
(*) : Si trascura la simmetria A2, poiché non è un modo di spostamento per la molecola.
Correlazione D2h- CS (σyz)
D2h E σ(yz) CS (σyz)
Ag 1 1 A’
B1g 1 -1 A”

R. Esposito, F. Santoro, M. A. Scelsi Pag. 16
B2g 1 -1 A”
B3g 1 1 A’
Au 1 -1 A”
B1u 1 1 A’
B2u 1 1 A’
B3u 1 -1 A”
CS (σyz)
D2h A’ x,y
A” z
n=Σj akj
Ag 2 - 2
B1g - 1 1
B2g - 1 1
B3g 2 - 2
Au - 1 1
B1u 2 - 2
B2u 2 - 2
B3u - 1 1
(*) : Si trascura la simmetria A2, poiché non è un modo di spostamento per la molecola.
Correlazione D2h- CS (σxy)
D2h E σ(xy) CS (σxy)
Ag 1 1 A’
B1g 1 1 A’
B2g 1 -1 A”
B3g 1 -1 A”
Au 1 -1 A”
B1u 1 -1 A”

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 17
B2u 1 1 A’
B3u 1 1 A’
CS (σxy)
D2h A’ x,y
A” z
n=Σj akj
Ag 2 - 2
B1g 2 - 2
B2g - 1 1
B3g - 1 1
Au - 1 1
B1u - 1 1
B2u 2 - 2
B3u 2 - 2
(*) : Si trascura la simmetria A2, poiché non è un modo di spostamento per la molecola.
A questo punto è possibile sommare i termini relativi ai quattro differenti gruppi locali, tenendo conto dell’ equazione (Eq. 1):
Γdrid-GM = 6*Ag⊕ 4*B1g ⊕ 4*B2g ⊕ 4*B3g ⊕ 2*Au⊕ 5*B1u ⊕ 6*B2u ⊕ 5*B3u
Si osserva che tale espressione coincide con la Γd ricavata in precedenza. Si procede dunque con l’analisi in coordinate interne, al fine di distinguere modi di vibrazione di stretching e di bending.
D2h E C2(z) C2(y) C2(x) i σ(xy) σ(xz) σ(yz)
Γdstretch 20 0 0 0 0 4 4 4 Σ/8
Ag 20 - - - - 4 4 4 4 B1g 20 - - - - 4 -4 -4 2 B2g 20 - - - - -4 4 -4 2 B3g 20 - - - - -4 -4 4 2 Au 20 - - - - -4 -4 -4 1 B1u 20 - - - - -4 4 4 3 B2u 20 - - - - 4 -4 4 3 B3u 20 - - - - 4 4 -4 3
R. Esposito, F. Santoro, M. A. Scelsi Pag. 18
Γstretch = 4*Ag⊕ 2*B1g ⊕ 2*B2g ⊕ 2*B3g ⊕ Au⊕ 3*B1u ⊕ 3*B2u ⊕ 3*B3u
Γbend = Γvibr- Γstretch = 2*Ag⊕ B1g ⊕ B2g ⊕ B3g ⊕ Au⊕ B1u ⊕ 2*B2u ⊕ B3u
Si determina infine l’attività IR e Raman della molecola (MgO)2,3 preso in esame; in questo caso, gli operatori momento di dipolo μ e polarizzabilità α sono i seguenti:
Valutando dunque la simmetria delle autofunzioni relative agli stati iniziali e finali di transizione fondamentale, primo e secondo overtone, è possibile completare la seguente tabella riassuntiva:
Spettro infrarosso Spettro Raman
Fondamentale 01 B1u, B2u, B3u
13 bande attese, 13 modi attivi (per n=3)
Ag, B1g, B2g, B3g
15 bande attese, 15 modi attivi (per n=3)
Primo overtone 02 Nessuno Tutti
Secondo overtone 03 B1u, B2u, B3u Ag, B1g, B2g, B3g

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 19
PARTE II: ISOMERI DEL C60F60 Data la crescente attenzione che negli ultimi anni la comunità scientifica rivolge alle forme allotropiche del grafene e ai fullereni, abbiamo incluso nel nostro studio alcuni isomeri della molecola C60F60 le cui strutture a tubo sono state ricavate computazionalmente da Jia et al. (J. Am. Chem. Soc. 2008, 130, 3985-3988) e classificate per stabilità in base al loro valore dell’entalpia di formazione. Il software utilizzato ha dato come risultato interessante il fatto che le strutture a tubo, caratterizzate da anelli di pentagoni adiacenti alle estremità del tubo stesso, dovrebbero essere più stabili della struttura a gabbia derivata immediatamente dal fullerene C60, che obbedisce invece alla regola del pentagono isolato (IPR). Gli autori hanno notato che la stabilità degli isomeri varia a seconda del numero di legami C – F che si trovano all’interno del tubo; ma anche con tutti i legami C – F all’esterno del tubo, sono possibili due strutture, ovviamente di simmetria e stabilità diverse. Nel seguito si analizzeranno quindi le due possibili strutture a tubo con tutti i legami C – F all’esterno (C60F60), una struttura con due legami C – F all’interno del tubo (F2@C60F58) e una struttura con quattro legami C – F all’interno del tubo (F4@C60F56).
A. C60F60
Una delle due possibili strutture a tubo di C60F60 è quella riportata nella figura seguente:
Tubo C60F60, gruppo di simmetria ed entalpia di formazione. A sinistra, vista laterale; a destra, vista frontale. Gli atomi
in nero sono di carbonio, quelli in verde di fluoro.
Questa struttura appartiene al gruppo D6h (h = 24), le cui operazioni di simmetria sono evidenziate nella figura seguente:

R. Esposito, F. Santoro, M. A. Scelsi Pag. 20
Gli assi in rosa e arancio nella vista frontale sono in realtà i tre piani di riflessione σv ed i tre piani di riflessione σd. In vista laterale è stato riportato solo uno dei tre assi C’2 che passano per il centro di inversione della molecola e per due legami C – C diametralmente opposti; non sono stati riportati (per difficoltà di disegno) i tre assi C’’2, che passano per il centro di inversione e per il centro di ogni coppia di esagoni di carbonio diametralmente opposti. (In altre parole, gli assi C’2 sono individuati dalle intersezioni dei 3 piani σv con il piano σh; gli assi C’’2 invece sono individuati dalle intersezioni dei tre piani σd con il piano σh.) Lavorando con la solita procedura nel gruppo della molecola D6h, la rappresentazione di spostamento della molecola risulta essere:
Γd = 20(A1g ⊕ A2u) ⊕ 10(A2g ⊕ A1u) ⊕ 14(B1g ⊕ B2u) ⊕ 16 (B2g ⊕ B1u) ⊕ 30(E1g ⊕ E2g ⊕ E1u ⊕ E2u) ed essendo le rappresentazioni di traslazione e rotazione:
Γtrasl = A2u ⊕ E1u Γrot = A2g ⊕ E1g
si ricava la rappresentazione vibrazionale: Γvibr = 20*A1g ⊕ 9*A2g ⊕ 14(B1g ⊕ B2u) ⊕ 16(B2g ⊕ B1u) ⊕ 29(E1g ⊕ E1u) ⊕ 10*A1u ⊕ 19*A2u ⊕ 30(E2g ⊕
E2u) che consta di 3N-6 = 354 modi vibrazionali, considerando la doppia degenerazione di tutte le rappresentazioni irriducibili di tipo E. Data la complessità sia della struttura molecolare che del gruppo di simmetria, si è ritenuto opportuno lavorare anche in simmetria locale per avere un riscontro sulla forma della rappresentazione di spostamento. Osservando la molecola notiamo che tutti i 120 atomi appartengono ad un gruppo di simmetria locale Cs, però:
48 atomi ∈ Cs (σd); 12 siti equivalenti → 4 atomi equivalenti (2 atomi C e 2 atomi F);
72 atomi ∈ Cs (σv); 12 siti equivalenti → 6 atomi equivalenti (3 atomi C e 3 atomi F), perciò:
ΓGM = 4 * ΓGL = Cs (σd) ⊕ 6 * ΓGL = Cs (σv) (Eq. 2) Costruiamo quindi la tavola delle correlazioni discendenti.
D6h E σd Cs (σd) E σv Cs (σv)
A1g 1 1 A’ 1 1 A’ A2g 1 -1 A’’ 1 -1 A’’ B1g 1 1 A’ 1 -1 A’’ B2g 1 -1 A’’ 1 1 A’ E1g 2 0 A’⊕A’’ 2 0 A’⊕A’’ E2g 2 0 A’⊕A’’ 2 0 A’⊕A’’ A1u 1 -1 A’’ 1 -1 A’’ A2u 1 1 A’ 1 1 A’ B1u 1 -1 A’’ 1 1 A’ B2u 1 1 A’ 1 -1 A’’ E1u 2 0 A’⊕A’’ 2 0 A’⊕A’’ E2u 2 0 A’⊕A’’ 2 0 A’⊕A’’
Cs (σd)
D6h
A’ x,y
A” z
n=Σj akj
Cs (σv) D6h
A’ x,y
A” z
n=Σj akj
A1g 2 - 2 2 - 2 A2g - 1 1 - 1 1 B1g 2 - 2 - 1 1

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 21
B2g - 1 1 2 - 2 E1g 2 1 3 2 1 3 E2g 2 1 3 2 1 3 A1u - 1 1 - 1 1 A2u 2 - 2 2 - 2 B1u - 1 1 2 - 2 B2u 2 - 2 - 1 1 E1u 2 1 3 2 1 3 E2u 2 1 3 2 1 3
ΓGL= Cs (σd) = 2*A1g ⊕ A2g ⊕ 2*B1g ⊕ B2g ⊕ 3*E1g ⊕ 3*E2g ⊕ A1u ⊕ 2*A2u ⊕ B1u ⊕ 2*B2u ⊕ 3*E1u ⊕ 3*E2u
ΓGL= Cs (σv) = 2*A1g ⊕ A2g ⊕ B1g ⊕ 2*B2g ⊕ 3*E1g ⊕ 3*E2g ⊕ A1u ⊕ 2*A2u ⊕ 2*B1u ⊕ B2u ⊕ 3*E1u ⊕ 3*E2u
Da cui, applicando l’equazione 2, si ritrova effettivamente la rappresentazione di spostamento calcolata nel gruppo globale della molecola. Avendo a disposizione la rappresentazione vibrazionale, prima di passare alla previsione degli spettri, è opportuno distinguere i modi di stretching da quelli di bending: per far ciò, utilizziamo la consueta procedura dell’analisi delle coordinate interne, in particolare delle lunghezze dei legami.
D6h E 2C6 2C3 C2 3C’2 3C’’2 i 2S3 2S6 σh 3σd 3σv
Γstretch 150 0 0 0 2 0 0 0 0 6 16 18 Σ/24
A1g 150 - - - 6 - - - - 6 48 54 11
A2g 150 - - - -6 - - - - 6 -48 -54 2
B1g 150 - - - 6 - - - - -6 48 -54 6
B2g 150 - - - -6 - - - - -6 -48 54 6
E1g 300 - - - 0 - - - - -12 0 0 12
E2g 300 - - - 0 - - - - 12 0 0 13
A1u 150 - - - 6 - - - - -6 -48 -54 2
A2u 150 - - - -6 - - - - -6 48 54 10
B1u 150 - - - 6 - - - - 6 -48 54 7
B2u 150 - - - -6 - - - - 6 48 -54 6
E1u 300 - - - 0 - - - - 12 0 0 13
E2u 300 - - - 0 - - - - -12 0 0 12
Γstretch = 11*A1g ⊕ 2*A2g ⊕ 6*(B1g ⊕ B2g) ⊕ 12*E1g ⊕ 13*E2g ⊕ 2*A1u ⊕ 10*A2u ⊕ 7*B1u ⊕ 6*B2u ⊕
13*E1u ⊕ 12*E2u
Γbend = Γvibr - Γstretch = 9*A1g ⊕ 7*A2g ⊕ 8*B1g ⊕ 10*B2g ⊕ 17*E1g ⊕ 17*E2g ⊕ 8*A1u ⊕ 9*A2u ⊕ 9*B1u ⊕
8*B2u ⊕ 16*E1u ⊕ 18*E2u

R. Esposito, F. Santoro, M. A. Scelsi Pag. 22
Il vettore momento di dipolo elettrico e il tensore polarizzabilità elettrica hanno componenti di simmetria:
Si può quindi immediatamente affermare che le transizioni fondamentali attive in infrarosso saranno quelle dei modi vibrazionali di simmetria A2u ed E1u, e daranno origine a 19 bande singole (A2u) e a 29 doppietti (bande doppiamente degeneri per i modi E1u); le transizioni fondamentali attive in Raman saranno invece quelle dei modi vibrazionali di simmetria A1g, E1g e E2g, quindi lo spettro conterrà 20 bande singole (A1g) e 59 doppietti (29 per l’E1g e 30 per l’E2g). Si noti che i modi vibrazionali di simmetrie A2g, B1g, B2g, A1u, B1u, B2u, E2u sono completamente silenti sia in spettroscopia infrarossa che in Raman: per evidenziarne l’esistenza e studiarli sarà pertanto necessario ricorrere ad altri metodi spettroscopici. Per quanto riguarda gli overtones, la situazione è leggermente più complessa, dovendo valutare come si trasformano le funzioni d’onda dei livelli eccitati (superiori al primo) di modi vibrazionali doppiamente degeneri. A questo scopo è possibile avvalersi della seguente formula ricorsiva:
χv(R)=1/2[χ(R)χv−1(R)+χ(Rv)]
per calcolare il carattere, sotto l’operazione di simmetria R, del ν-esimo livello energetico del modo vibrazionale doppiamente degenere. Una volta ottenuta la stringa dei caratteri di tutte le operazioni, la si scompone, secondo la procedura usuale, nella somma diretta di rappresentazioni irriducibili del gruppo di simmetria. Da questa rappresentazione si calcola, tramite il prodotto diretto Γν' x Γμ x Γν, se la probabilità di transizione è uguale o diversa da zero e quindi se la transizione è permessa o proibita. Consideriamo dapprima lo spettro IR. Dal momento che la molecola contiene un gran numero di atomi, tipicamente le righe corrispondenti agli overtones avranno intensità molto minore rispetto ai fondamentali, ma li analizziamo ugualmente. Per quello che riguarda i modi di vibrazione di simmetria A2u, tutti gli overtones saranno visibili. Per quanto riguarda i modi di vibrazione due volte degeneri di simmetria E1u, applichiamo la formula sopra riportata:
D6h E 2C6 2C3 C2 3C’2 3C’’2 i 2S3 2S6 σh 3σd 3σv
R E C6 C3 C2 C’2 C’’2 i S3 S6 σh σd σv
R2 E C3 C3 E E E E C3 C3 E E E
R3 E C2 E C2 C’2 C’’2 i σh i σh σd σv
χ(R) 2 1 -1 -2 0 0 -2 -1 1 2 0 0
χ(R2) 2 -1 -1 2 2 2 2 -1 -1 2 2 2
χ2(R) 3 0 0 3 1 1 3 0 0 3 1 1
χ(R3) 2 -2 2 -2 0 0 -2 2 -2 2 0 0
χ3(R) 4 -1 1 -4 0 0 -4 1 -1 4 0 0
Dalle righe evidenziate si ricava:

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 23
ψν=2E1u ~ Γ₂ = A1g ⊕ E2g
ψν=3E1u ~ Γ₃ = B1u ⊕ B2u ⊕ E1u
da cui si comprende come il primo overtone, cioè la transizione 0 -> 2 del modo vibrazionale E1u, sia proibito e invece il secondo, ovvero la transizione 0 -> 3, sia permesso. Per quel che riguarda i modi attivi in Raman, sicuramente saranno visibili tutti gli overtones del modo vibrazionale A1g, ma restano da analizzare le due vibrazioni doppiamente degeneri di simmetria E1g ed E2g. Si potrebbe applicare il procedimento appena descritto, ma esistono anche delle regole dovute a Wilson, Decius e Cross che permettono di giungere al risultato desiderato in maniera più veloce. Grazie ad esse troviamo subito:
ψν=2E1g ~ Γ2 = A1g ⊕ E2g
ψν=3E1g ~ Γ₃ = B1g ⊕ B2g ⊕ E1g
ψν=2E2g ~ Γ2 = A1g ⊕ E2g
ψν=3E2g ~ Γ₃ = A1g ⊕ A2g ⊕ E2g
Poiché tutte queste rappresentazioni contengono almeno una o due rappresentazioni irriducibili contenute anche nel tensore polarizzabilità, sia la transizione 0 -> 2 che quella 0 -> 3 sono permesse e, in maniera meno intensa rispetto alle righe dei fondamentali, visibili nello spettro.
Un’altra struttura a tubo del C60F60 è riportata nella figura seguente:
Gli elementi di simmetria della molecola sono: 5 rotazioni di 180° i cui assi sono evidenziati in verde nella figura seguente; 5 piani di simmetria individuati da un vertice della sezione pentagonale e dal punto medio del lato opposto al vertice stesso ; due rotazioni di 72°, una in senso orario e una in senso antiorario, e due rotazioni di 144° intorno ad un asse che attraversa la molecola, evidenziato in blu nella figura; considerando questo stesso asse si possono ottenere delle roto-riflessioni e in particolare una inversione.

R. Esposito, F. Santoro, M. A. Scelsi Pag. 24
La struttura appartiene quindi al gruppo D5d e l’ordine del gruppo è h= 20.
Si ricava la rappresentazione riducibile di spostamento Γdrid:
D5d E 2C5 2C5
(2)
5C2 i 2S10 (3)
2S10 5σd
# atomi fissi
120 0 0 0 0 0 0 24
Χ 3 1+2cos72° 1+2cos144° -1 -3 -1-2cos72° -1-2cos144° 1 Γd
rid 360 0 0 0 0 0 0 24 Σ/20
A1g 360 0 0 0 0 0 0 120 24 A2g 360 0 0 0 0 0 0 -120 12 E1g 720 0 0 0 0 0 0 0 36 E2g 720 0 0 0 0 0 0 0 36 A1u 360 0 0 0 0 0 0 -120 12 A2u 360 0 0 0 0 0 0 120 24 E1u 720 0 0 0 0 0 0 0 36 E2u 720 0 0 0 0 0 0 0 36
Γdrid = 24*A1g⊕ 12*A2g ⊕ 36*E1g ⊕ 36*E2g ⊕ 12*A1u⊕ 24*A2u ⊕ 36*E1u ⊕ 36*E2u
Γtrasl = A2u ⊕ E1u Γrot = A2g ⊕ E1g
Γvibr = 24*A1g⊕ 11*A2g ⊕ 35*E1g ⊕ 36*E2g ⊕ 12*A1u⊕ 23*A2u ⊕ 35*E1u ⊕ 36*E2u
La Γvibr è costituita da 354 modi di vibrazione come previsto, dove è stata considerata la doppia degenerazione delle rappresentazioni di tipo E. Per verificare la correttezza di tale espressione applichiamo il metodo delle correlazioni discendenti e procediamo con l’analisi in simmetria locale. Tutti gli atomi della molecola considerata appartengono ai piani σd, quindi il gruppo locale da considerare è GL=Cs :

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 25
102
20
GL
GM
h
h siti equivalenti 12
10
120 atomi equivalenti ( 6 atomi di C e sei di F );
quindi la rappresentazione riducibile di spostamento si può scrivere come segue: d
C
d
GMrid s 12
Si studiano ora le correlazioni tra il gruppo globale GM = D5d e il gruppo locale Cs:
D5d E σ CS
A1g 1 1 A’
A2g 1 -1 A”
E1g 2 0 A’ + A”
E2g 2 0 A’ + A”
A1u 1 -1 A”
A2u 1 1 A’
E1u 2 0 A’ + A”
E2u 2 0 A’ + A”
CS D5d
A’ x
A’ y
A” z
n=Σj akj
A1g 1 1 - 2
A2g - - 1 1
E1g 1 1 1 3
E2g 1 1 1 3
A1u - - 1 1
A2u 1 1 - 2
E1u 1 1 1 3
E2u 1 1 1 3
Si noti che bisogna considerare separatamente i modi di spostamento x e y.
Γdrid-GM = 12*(2*A1g⊕ A2g ⊕ 3*E1g ⊕ 3*E2g ⊕ A1u⊕ 2*A2u ⊕ 3*E1u ⊕ 3*E2u)
Tale espressione coincide effettivamente con quella ricavata precedentemente. Analisi delle coordinate interne:
D5d E 2C5 2C5
(2) 5C2 i 2S10
(3)
2S10 5σd
# legami
fissi 150 0
0 2 0 0 0 20

R. Esposito, F. Santoro, M. A. Scelsi Pag. 26
Γstretch 150 0 0 10 0 0 0 100 Σ/20
A1g 150 - - 10 - - - 100 13
A2g 150 - - -10 - - - -100 2
E1g 300 - - 0 - - - 0 15
E2g 300 - - 0 - - - 0 15
A1u 150 - - 10 - - - -100 3
A2u 150 - - -10 - - - 100 12
E1u 300 - - 0 - - - 0 15
E2u 300 - - 0 - - - 0 15
Γstretch = 13 A1g ⊕ 2 A2g ⊕ 15 E1g ⊕ 15 E2g ⊕ 3 A1u ⊕ 12 A2u ⊕ 15 E1u ⊕ 15 E2u
Γbend = Γvibr - Γstretch = 11 A1g ⊕ 9 A2g ⊕ 20 E1g ⊕ 21 E2g ⊕ 9 A1u ⊕ 11 A2u ⊕ 20 ⊕ E1u ⊕ 21 E2u
Si analizza ora l’attività infrarossa e Raman della molecola considerata. Gli operatori momento di dipolo e polarizzabilità sono rispettivamente:
u
u
A
E
2
1
g
g
g
E
E
A
2
1
1
;
Spettro infrarosso Spettro Raman
Fondamentale 01 E1u, A2u
58 bande attese, 93 modi attivi A1g, E1g, E2g
95 bande attese, 166 modi attivi
Primo overtone 02 nessuno A1g, E1g, E2g, E1u
Secondo overtone 03 A2u, E1u A1g, E1g, E2g
Per quanto riguarda gli overtones, è evidente che il modo vibrazionale A1g è presente in Raman, mentre per i modi doppiamente degeneri il risultato è stato ottenuto studiando come si trasformano le funzioni d’onda dei livelli eccitati di tali modi vibrazionali: Modi di vibrazione E1g :
D5d E C5 C52 C2 i S10
3 S10 σd
R E C5 C52 C2 i S10
3 S10 σd
R2 E C52 C5 E E C5
2 C5 E

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 27
R3 E S10 C5 C2 i S10 S103 σd
χ(R) 2 2cos72° 2cos144° 0 2 2cos72° 2cos144° 0
χ(R2) 2 2cos144° 2cos72° 2 2 2cos144° 2cos72° 2
χ2(R) 3 2cos144°+1 2cos72°+1 1 3 2cos144°+1 2cos72°+1 1
χ(R3) 2 2cos144° 2cos72° 0 2 2cos144° 2cos72° 0
χ3(R) 4 -1 -1 0 4 -1 -1 0
Dalla tabella si ottengono le funzioni seguenti:
ψν=2E1g ~ Γ₂ = A1g ⊕ E2g
ψν=3E1g ~ Γ₃ = E1g⊕ E2g
Modi di vibrazione E2g:
ψν=2E2g ~ Γ₂ = A1g ⊕ E1g
ψν=3E2g ~ Γ₃ = E1g⊕ E2g
Quindi i modi di vibrazione E1g e E2g sono attivi in Raman in entrambi gli overtones considerati. Modi di vibrazione E1u :
ψν=2E1u ~ Γ₂ = A1g ⊕ E2g
ψν=3E1u ~ Γ₃ = E1u ⊕ E2u
Pertanto il modo di vibrazione a simmetria E1u è attivo in spettroscopia IR nel secondo overtone.
B. F2@C60F58:
struttura con due legami C – F all’interno del tubo

R. Esposito, F. Santoro, M. A. Scelsi Pag. 28
Gli elementi di simmetria della molecola sono: una rotazione di 180° , una riflessione e una inversione come indicato nella figura seguente:
La struttura appartiene al gruppo di simmetria C2h, il cui ordine è h=4. Gli atomi in totale sono 120 e i modi vibrazionali attesi sono 354. Si può ricavare la rappresentazione riducibile di spostamento direttamente tramite la seguente tavola di correlazione e dalla rappresentazione riducibile ottenuta per la molecola precedente :
D5d E C2 i σ C2h
A1g 1 1 1 1 Ag
A2g 1 -1 1 -1 Bg
E1g 2 0 2 0 Ag⊕ Bg
E2g 2 0 2 0 Ag⊕ Bg
A1u 1 1 -1 -1 Au
A2u 1 -1 -1 1 Bu
E1u 2 0 -2 0 Au⊕ Bu
E2u 2 0 -2 0 Au⊕ Bu
Γdrid = 24*A1g⊕ 12*A2g ⊕ 36*E1g ⊕ 36*E2g ⊕ 12*A1u⊕ 24*A2u ⊕ 36*E1u ⊕ 36*E2u =
=24*Ag ⊕ 12*Bg ⊕ 72*Ag ⊕ 72*Bg ⊕ 12*Au⊕ 24*Bu ⊕ 72*Au ⊕ 72*Bu = = 96*Ag⊕ 84*Bg ⊕ 84*Au ⊕ 96*Bu
Tale espressione è confermata lavorando nel gruppo della molecola e in seguito anche applicando il metodo delle correlazioni:

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 29
C2h E C2 i
σh
# atomi fissi
120 0 0 24
Χ 3 -1 -3 1 Γd
rid 360 0 0 24 Σ/4
Ag 360 0 0 24 96 Bg 360 0 0 -24 84 Au 360 0 0 -24 84 Bu 360 0 0 24 96
Γdrid = 96*Ag⊕ 84*Bg ⊕ 84*Au ⊕ 96*Bu
Γtrasl = Au ⊕ 2Bu Γrot = Ag ⊕ 2Bg
Γvibr = 95*Ag⊕ 82*Bg ⊕ 83*Au ⊕ 94*Bu La Γvibr è costituita da 354 modi di vibrazione come previsto. Procediamo con l’analisi in simmetria locale per verificare la correttezza di tale espressione : Osservando la molecola notiamo che :
24 atomi ∈ Cs ; 22
4
GL
GM
h
h siti equivalenti → 12
2
24 atomi equivalenti (6 atomi di C, 5 atomi
esterni di Fluoro e 1 atomo interno di Fluoro) ;
96 atomi ∈ C1 ; 41
4
GL
GM
h
h siti equivalenti → 24
4
96 atomi equivalenti (12 atomi di C e 12
atomi di F esterni ). Γrid-GM = 12 * ΓGL = Cs ⊕ 24 * ΓGL = C1
Si studiano ora le correlazioni tra il gruppo globale GM = D2h e il gruppo locale Cs:
C2h E σ CS
Ag 1 1 A’
Bg 1 -1 A”
Au 1 -1 A”
Bu 1 1 A’
CS C2h
A’ x
A’ y
A” Z
n=Σj akj
Ag 1 1 - 2
Bg - - 1 1
Au - - 1 1
Bu 1 1 - 2

R. Esposito, F. Santoro, M. A. Scelsi Pag. 30
ΓGL = Cs = 2 *Ag⊕ Bg ⊕ Au ⊕ 2*Bu Studiando allo stesso le correlazioni tra il gruppo globale GM = D2h e il gruppo locale C1 ,si ottiene :
ΓGL = C1 =3 *(Ag⊕ Bg ⊕ Au ⊕ Bu )
E quindi l’espressione finale: Γrid-GM = 12*(2 *Ag⊕ Bg ⊕ Au ⊕ 2*Bu ) + 24 * 3 *(Ag⊕ Bg ⊕ Au ⊕ Bu )
che coincide con l’espressione ottenuta precedentemente.
Si procede quindi con l’analisi in coordinate interne:
C2h E C2 i σh
# legami
fissi 150 2 0 20
Γstretch 150 2 0 20 Σ/4
Ag 150 2 - 20 43
Bg 150 -2 - -20 32
Au 150 2 - -20 33
Bu 150 -2 - 20 42
Γstretch = 43 Ag ⊕ 32 Bg ⊕ 33 Au ⊕ 42 Bu
Γbend = Γvibr - Γstretch = 50 Ag ⊕ 52 Bg ⊕ 52 Au ⊕ 50 Bu
Si analizza infine l’attività infrarossa e Raman della molecola considerata; in tal caso gli operatori momento di dipolo e polarizzabilità sono rispettivamente:
u
u
A
B
g
g
B
A ;
Spettro infrarosso Spettro Raman
Fondamentale 01 Au , Bu
177 bande attese, 177 modi attivi Ag, Bg
177 bande attese, 177 modi attivi
Primo overtone 02 nessuno Tutti Secondo overtone 03 Au , Bu Ag, Bg

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 31
C. F4@C60F56 :
struttura con quattro legami C – F all’interno del tubo
La molecola presenta come solo elemento di simmetria, oltre all’identità, una rotazione di 180° come mostrato nella seguente figura:
La molecola appartiene quindi al gruppo C2 di ordine h=2. Si ricava la rappresentazione riducibile di spostamento dai calcoli precedenti:
D5d E C2 C2
A1g 1 1 A
A2g 1 -1 B
E1g 2 0 A⊕ B
E2g 2 0 A⊕ B

R. Esposito, F. Santoro, M. A. Scelsi Pag. 32
A1u 1 1 A
A2u 1 -1 B
E1u 2 0 Au⊕ Bu
E2u 2 0 A⊕ B
Γdrid = 24*A1g⊕ 12*A2g ⊕ 36*E1g ⊕ 36*E2g ⊕ 12*A1u⊕ 24*A2u ⊕ 36*E1u ⊕ 36*E2u =
=24*A ⊕ 12*B ⊕ 72*A ⊕ 72*B ⊕ 12*A⊕ 24*B ⊕ 72*A ⊕ 72*B = = 180*A ⊕ 180*B
Conferma:
C2 E C2
# atomi fissi
120 0
Χ 3 -1 Γd
rid 360 0 Σ/2
A 360 0 180 B 360 0 180
Γdrid = 180*A ⊕ 180*B
Γtrasl = A ⊕ 2B Γrot = A ⊕ 2B
Γvibr = 178*A ⊕ 176*B La Γvibr è costituita da 354 modi di vibrazione come previsto. Si procede con l’analisi in simmetria locale per verificare la correttezza di tale espressione : poiché nessun atomo appartiene all’asse C2 il gruppo locale da considerare è GL=C1 .
21
2
GL
GM
h
h siti equivalenti → 60
2
120 atomi equivalenti;
Γrid-GM = 60 * ΓGL
CS C2
A x
A y
A Z
n=Σj akj
C2 E CS
A 1 A
B 1 A

STUDIO DELL’ATTIVITA’ IR E RAMAN DI STRUTTURE MOLECOLARI A NANOTUBO Pag. 33
A 1 1 1 3
B 1 1 1 3
ΓGL = 3*A ⊕ 3*B
Γrid-GM = 60 * (3*A ⊕ 3*B)
C2h E C2
# legami
fissi 150 2
Γstretch 150 2 Σ/2
A 150 2 76
B 150 -2 74
Γstretch = 76 A ⊕ 74 B
Γbend = Γvibr - Γstretch = 102 A ⊕ 102 B
Concludiamo con lo studio di attività infrarossa e Raman; gli operatori momento di dipolo e polarizzabilità sono i seguenti:
A
B
B
A
;
Spettro infrarosso Spettro Raman
Fondamentale 01 A , B
354 bande attese, 354 modi attivi A , B
354 bande attese, 354 modi attivi
Primo overtone 02 A , B A , B
Secondo overtone 03 A , B A , B
Quindi tutti i modi sono attivi sia in IR che in Raman.





























