Embed Size (px)
Citation preview

87
TEORIA CINETICA DEI GAS
1. CENNI DI TEORIA CINETICA DEI GAS_____________________________ 882. LA PRESSIONE _________________________________________________ 883. LA TEMPERATURA _____________________________________________ 894. L’EQUAZIONE DI JOULE-CLAUSIUS ______________________________ 905. IL MOTO BROWNIANO__________________________________________ 956. PRINCIPIO DI EQUIPARTIZIONE DELL’ENERGIA___________________ 967. LAPRESSIONE IN FUNZIONE DELLE VARIABILI MICROSCOPICHE ___ 968. LA LIQUEFAZIONE DEI GAS, LE ESPERIENZE DI ANDREWS _________ 989. L’EQUAZIONE DI STATO (DI VAN DER WAALS) PER I GAS REALI____ 10010. LA LEGGE DI DISTRIBUZIONE DELLE VELOCITA´ DI MAXWELL-BOLTZMANN____________________________________________________ 10011. LA DISTRIBUZIONE DI MAXWELL NELLO SPAZIO DELLE FASI ____ 10612. VERIFICA SPERIMENTALE DELLA DISTRIBUZIONE DI MAXWELL _ 10813. LA TEORIA CINETICA NEI CORPI SOLIDI _______________________ 109

88
1. CENNI DI TEORIA CINETICA DEI GAS
Se consideriamo un gas come l’insieme di numerosissimi punti materiali(composto da piu’ di 1023 atomi o molecole che e’ il numero contenuto inpochi grammi di sostanza) in linea di principio sembrerebbe possibilestudiarne il comportamento macroscopico applicando la legge di Newton perdescrivere il moto di tutti i suoi componenti microscopici (atomi o molecole).Cio’ non e’ possibile fondamentalmente per due motivi: si dovrebbe risolvereun sistema di un numero enorme di equazioni differenziali (una per ogniatomo) ed inoltre si dovrebbero poter definire le condizioni iniziali, cioe’misurare in uno stesso istante tutte le posizioni e le velocita’ di ogni singoloatomo.Come sfruttare dunque la meccanica del punto materiale per descrivere icomportamenti macroscopici di un gas? La teoria cinetica dei gas risponde aquesta esigenza.La teoria cinetica dei gas e’ lo studio del comportamento microscopico dellemolecole o degli atomi, che costituiscono la materia, e delle loro interazioniche conducono alle equazioni tra quantità macroscopiche (quali la pressione,il volume, la temperatura ecc.) come ad esempio la legge dei gas perfetti.
2. LA PRESSIONE
Se una forza F agisce su una superficie S si può definire una grandezza dettapressione nel modo seguente:
†
P =
r F ⋅ r n
S=
F cosaS
=Fn
S
Figure 1dove n e’ il versore normale alla superficie S.E’ interessante notare che la pressione ha le dimensioni di una densitàd’energia. Infatti, moltiplicando e dividendo l’ultimo membro dell’equazioneprecedente per uno spostamento d si ottiene P = L / DV, dove L rappresenta illavoro compiuto dal materiale per cambiare il suo volume di una quantità DV:

89
L = P DVE’ interessante notare che la causa microscopica della pressione su di unasuperficie e’ dovuta agli urti che le innumerevoli molecole (ad esempio lemolecole di un gas), che costituiscono un corpo realizzano, nel loromovimento, sulla superficie stessa. Infatti ad ogni urto le molecole cambianodirezione di volo e dunque cambiano la loro quantita’ di moto. Per il teoremadell’impulso ad ogni cambiamento della quantita’ di moto corrispondel’azione di una forza. E’ proprio questa forza che genera la pressione. E’altrettanto evidente che minore e’ il numero delle molecole che costituisconoil corpo e minore e’ il significato di pressione. In altri termini la pressione e’una grandezza macroscopica che si riferisce ad un corpo formato da unnumero rilevante di molecole.
3. LA TEMPERATURA
Se si pongono due corpi, di differente stato termico, a contatto tra loro, essiraggiungeranno dopo un certo intervallo di tempo, l'equilibrio termico;avranno, cioe', la stessa temperatura. La temperatura, dei due corpirimarrebbe immutata se questi fossero inseriti in un sistema isolato (cioe' sefosse possibile evitare scambi di calore con l'ambiente esterno).Per lo stesso principio, due corpi in equilibrio termico con un terzo, lo sonotutti tra loro (Principio zero della termodinamica).Fatte queste premesse, possiamo definire temperatura quella grandezzafisica che serve a misurare lo stato termico di un corpo.Bisogna dunque definire un sistema di misura che si basi su criteri oggettividi valutazione, le misure di temperatura dovranno essere basate su fenomenifisici rilevati con uno strumento atto allo scopo.Lo strumento atto a misurare la temperatura di un corpo e' detto termometro.Esistono due differenti tipi di termometro: a liquido e a gas.Il termometro a liquido (figura) e' costituito da un corpo di vetro di piccolamassa (bulbo) contenente un liquido (sostanza termometrica): in genere siutilizza il mercurio, essendo questo metallo un buon conduttore di calore emantenendosi allo stato liquido sia alle basse che alle alte temperature.Il bulbo e' attaccato ad un tubo capillare chiuso alla sua estremita'. All'internodel termometro viene riprodotta la condizione di vuoto. Quando il bulboviene riscaldato, il liquido, aumentando divolume, sale all'interno del tubo capillare.Dalla figura si deduce che l'altezza L dellasostanza termometrica varia con il variaredella temperatura. Interpretandomatematicamente il suddetto fenomeno,possiamo affermare che c'e' unaproporzionalita' diretta tra la lunghezza Ldella colonna del liquido e la temperaturaT. Per misurare la proporzionalita' e'necessario tarare il termometro, bisognacioe' costruire una scala termometrica.La scala termometrica e' la graduazionedel termometro per consentire la misurazione della temperatura.La graduazione di un termometro richiede di:

90
1. segnare sulla scala termometrica dei punti di riferimento, detti punti fissi, atemperature ben determinate e riproducibili.2. fissare un'unita' di misura per la lettura delle temperature dei corpi.
Per convenzione si scelgono come punti fissi la temperatura a cui fonde ilghiaccio e la temperatura a cui bolle l'acqua alla pressione di un'atmosfera.Esistono diverse scale termometriche macroscopiche: la scala Celsius, laKelvin e la Fahrenheit.
TK = TC + 273.15TC = 5/9 (TF –32)TF = 9/5 TC + 32
Mediante la legge di Joule Clausius troveremo un’interessante interpretazionemicroscopica della temperatura.
4. L’EQUAZIONE DI JOULE-CLAUSIUS
Consideriamo un gas composto di N molecole, tutte con massa m, confinatein un contenitore con volume finito. Il teorema del Viriale applicato ad untale sistema diventa
12
i =1
N
 mvi2 = -
12
F xi xi + F y
i yi + Fzi zi( )
i=1
N
ÂFacciamo alcune ipotesi restrittive sulle forze in gioco:
a) siano trascurabili le forze a distanza tra le molecole,b) le forze in gioco durante le collisioni siano di tipo elastico , cioè´
durante l'urto tra la molecola iesima e quella jesima sia
r F i = -
r F j
c) le molecole siano monoatomiche, cioè composte di singoli atomicosi´ da poter essere assimilate a punti materiali, perciò nell'istante dell'urtotra la molecola iesima e la molecola jesima dovra’ essere
xi = xj , yi = yj , zi = zj ,d) le uniche altre forze in gioco siano quelle che agiscono durante gli
urti tra le molecole e le pareti del contenitore in cui il gas e´ confinato,e) il gas sia uniforme in modo che lo sia anche la pressione sulle pareti.
Un gas di molecole che verifica queste condizioni si chiama gas perfetto. Inqueste ipotesi di gas perfetto calcoliamo il contributo al Viriale dovuto allesole forze interne che agiscono durante l'urto tra la molecola iesima e lamolecola jesima :
F xi xi +F y
i yi +F zi zi( ) + F x
j xj +Fyj y j + Fz
j zj( ) =
F xi xi - xj( )+F y
i y i - y j( )+ Fzi zi - zj( ) = 0

91
Estendendo il calcolo agli urti tra tutte le molecole si ottiene l'importanterisultato che il contributo al Viriale dovuto alle sole forze che agisconodurante gli urti tra le molecole e´ nullo.Quindi nell'equazione (4.1) contribuiscono soltanto le forze che agisconodurante gli urti delle molecole con le pareti del contenitore. Consideriamo ilgas contenuto in un contenitore cubico di lato a come mostrato in figura, dove d
r F = - P dS r n e´ la forza esercitata dalle pareti sulle molecole che urtano
l'elemento di superficie infinitesima dS.
Figure 2Assumendo che gli urti sulle pareti siano elastici, tale forza risulta sempreperpendicolare alla parete e dunque P e´ la pressione che agisce sulle paretidel contenitore. Il contributo al Viriale sulle facce del cubo e´ dato da:
1.
-12
r r ⋅dr F
SÚ =
12
r r ⋅ r n P dSSÚ =
=12
P x cosa dS + y cosb dS + zcos g dS +
SÚ
SÚ
SÚ
Ê
Ë
Á Á
ˆ
¯
˜ ˜
avendo introdotto gli angoli di Eulero del versore normale alla superficie r n = cosa ,cosb ,cos g( ) , ed essendo l'integrale esteso a tutta la superficieesterna S del contenitore. Si noti che in figura sono stati disegnati comeesempio gli angoli d’Eulero del vettore r, infatti a e’ l’angolo tra il vettore el’asse x, b e’ l’angolo tra il vettore e l’asse y e g e’ l’angolo tra il vettore e l’assez. Si noti ancora che i tre angoli di Eulero non sono tra loro indipendenti, mavale la seguente relazione:
cos2 a + cos2 b + cos2 g = 1La superficie esterna del contenitore e´ data dalla somma delle sei facce delcubo
S ≡ Sxy + S'xy +Sxz + S'xz +Syz + S' yz
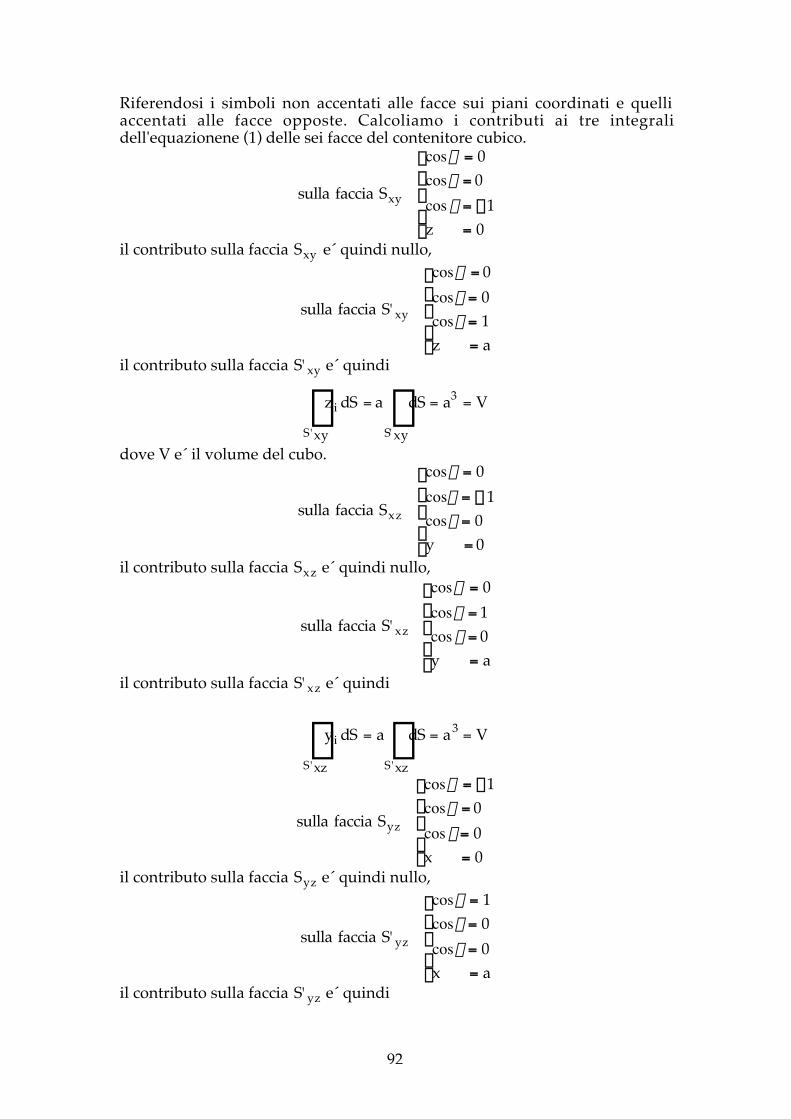
92
Riferendosi i simboli non accentati alle facce sui piani coordinati e quelliaccentati alle facce opposte. Calcoliamo i contributi ai tre integralidell'equazionene (1) delle sei facce del contenitore cubico.
sulla faccia Sxy
cosa = 0cosb = 0cos g = -1z = 0
Ï
Ì Ô
Ó Ô
il contributo sulla faccia Sxy e´ quindi nullo,
sulla faccia S' xy
cosa = 0cosb = 0cosg = 1z = a
Ï
Ì Ô
Ó Ô
il contributo sulla faccia S' xy e´ quindi
zi dS =
S'xyÚ a dS =
S' xyÚ a3 = V
dove V e´ il volume del cubo.
sulla faccia Sxz
cosa = 0cosb = - 1cosg = 0y = 0
Ï
Ì Ô
Ó Ô
il contributo sulla faccia Sxz e´ quindi nullo,
sulla faccia S' xz
cosa = 0cosb = 1cos g = 0y = a
Ï
Ì Ô
Ó Ô
il contributo sulla faccia S' xz e´ quindi
yi dS =
S'xzÚ a dS =
S'xzÚ a3 = V
sulla faccia Syz
cosa = -1cosb = 0cos g = 0x = 0
Ï
Ì Ô
Ó Ô
il contributo sulla faccia Syz e´ quindi nullo,
sulla faccia S' yz
cosa = 1cosb = 0cosg = 0x = a
Ï
Ì Ô
Ó Ô
il contributo sulla faccia S' yz e´ quindi

93
xi dS =
S'yzÚ a dS =
S' yzÚ a3 = V
In conclusione il teorema del Viriale applicato ad un gas perfetto confinato inun contenitore chiuso diventa
2.
12
i =1
N
 mvi2 =
32
PV
Si noti che questa formula (Kroenig-Clausius) stabilisce una relazioneanalitica tra tipiche grandezze microscopiche, quali l'energia cinetica, e grandezzemacroscopiche quali la pressione ed il volume ( misurate globalmente su ungrande insieme di molecole).
Per un gas perfetto vale l' equazione di stato dei gas perfetti3. PV = m RT (4.4)
essendo m il numero di moli di cui e´ composto il sistema, R la costanteuniversale dei gas e T la temperatura assoluta. Ricordiamo ancora che ilnumero di Avogadro N vale:
†
N = 6.022 ⋅10 23
e rappresenta il numero di molecole contenute in una mole di materiale.La costante universale dei gas R vale:
R = 8.315 Joule / (mole oK)
Ricordiamo che la massa atomica è un numero relativo che indica il rapportotra la massa di un atomo di tal elemento e la dodicesima parte dell’atomo dicarbonio 12C, mentre la massa molecolare è anch’essa un numero relativo,perché espresso dalla somma delle masse atomiche degli elementi contenutinella molecola di una sostanza. Una mole (o grammo-molecola) e’ unaquantità di materia pari al suo peso molecolare espresso in grammi. Adesempio una mole d’ossigeno 16O2 e’ 32 grammi. Dunque il numero di moli sipuò esprimere come:
†
m =N
Nallora l'equazione (2) diventa
†
12
m vi2
i-1
NÂ =
32
m R T =32
NRN T
che divisa per il numero totale di molecole del sistema da´
†
1N
12
m vi2
i-1
NÂ =
32
RN T =
32
k T
Il primo membro e´ l'energia cinetica media < Ec > delle molecole checompongono il sistema gassoso in esame ed abbiamo introdotto la costante kdi Boltzmann:
†
k =R
Nsi ottiene in conclusione l’equazione 4:

94
4. < Ec > =
32
kT
che rappresenta l'equazione di Joule-Clausius e che stabilisce che l'energiacinetica media delle molecole di un gas e´ proporzionale alla temperaturaassoluta.
Alcune osservazioni importanti:a) l'equazione di Joule-Clausius e´ valida, per come e´ stata dedotta, in unriferimento solidale col contenitore in cui e´ confinato il gas, altrimenti siarriverebbe all'assurdo che la temperatura assoluta di un gas dipenderebbedal moto del contenitore in cui il gas e´ confinato;b) la temperatura assoluta ammette lo zero come valore minimo checorrisponde allo stato fisico in cui tutte le molecole sono ferme. Si puo’ parlareanche di temperature assolute negative, ma questi stati particolarissimi nonsono descrivibili mediante la dinamica classica, bensi’ attraverso la meccanicaquantistica che descrive gli stati possibili per gli atomi reali. Il concetto ditemperatura negativa e’ collegato ad una proprieta’ quantistica degli atomiche si chiama spin e che rappresenta un nuovo grado di liberta’ dell’atomo(momento angolare intrinseco). Si chiama anche “temperatura degli spin” percontraddistinguerla dalla temperatura cinetica. Per raggiungere talitemperature negative si usano adeguate apparecchiature a radio frequenza.c) se il sistema e´ isolato esiste un valore massimo per l'energia cinetica dellemolecole, in tal caso esiste un valore massimo per la temperatura;d) l'equazione (4) consente un’interpretazione microscopica della temperaturaassoluta, che e’ un indice dell’energia cinetica media degli atomi checompongono il sistema.e) l'equazione (4) fornisce l'energia cinetica media per un sistema di molecolepuntuali per le quali e’ possibile parlare soltanto d’energia traslazionale. Lemolecole reali sono più complesse. In tal caso non sono più trascurabili ne’ lerotazioni ne’ le vibrazioni.f) Per le molecole reali poi non sono più trascurabili le forze d’interazione adistanza (ad esempio le forze elettriche e magnetiche).g) Si chiama energia interna U di un corpo l’energia totale in esso contenutache in generale sarà data dalla somma delle energie cinetiche delle molecolepiù l’energia potenziale. Per le molecole puntuali (gas perfetto) l’energiapotenziale e’ nulla e l’energia interna assume la forma:
†
U =32
k N T =32
m R T .Per le molecole reali l’energia potenziale non e’ più trascurabile ed e’ associataal campo di forze a distanza che agiscono tra le molecole.h) L’equazione di Joule-Clausius e’ valida anche per liquidi e solidi.

95
Figure 3
5. IL MOTO BROWNIANO
Delle particelle molto piccole sospese in un fluido si muovono casualmente intutte le direzioni anche se il fluido e’ in perfetto equilibrio macroscopico.Questo moto e’ continuo e non si interrompe ne’ si attenua mai. La scoperta e’stata fatta da Brown nel 1828.
Figure 4E’ questa la prima evidenza sperimentale che le molecole di un liquido simuovono in continuazione. A causa di tale moto le molecole, urtando leparticelle in sospensione, ne causano il moto Browniano.

96
6. PRINCIPIO DI EQUIPARTIZIONE DELL’ENERGIA
L'osservazione (e) del paragrafo precedente ci consente di estendere laformula di Joule-Clausius anche ai gas poliatomici mediantea) l'osservazione che la molecola monoatomica ha tre gradi di liberta´ eb) l'assunzione che l'energia cinetica di una molecola si ripartisca in mediaequamente tra i suoi gradi di liberta´ ndof
5.
†
< Ec >ndof
=12
k T
e´ questo il Principio d’Equipartizione dell'Energia di Boltzmann. Per i gasmonoatomici si ritrova l'equazione (4), mentre per i gas biatomici, per i qualila molecola ha 5 gradi di liberta´, si ottiene la formula
< Ec > =
52
kT
7. LAPRESSIONE IN FUNZIONE DELLE VARIABILIMICROSCOPICHE
Consideriamo un gas ideale contenuto in un recipiente cubico di lato l . Lapressione esercitata dal gas sulle pareti del contenitore e´ dovuta agli urtidelle molecole sulle pareti stesse. Infatti, ad ogni urto una molecola cambia lasua quantità´ di moto e ciò´ significa, in base al teorema
Figure 5dell'impulso, che una forza non nulla agisce durante l'urto; di fatto e´ questaforza che induce la pressione sulle pareti della scatola. Consideriamo lamolecola i-esima con quantità´ di moto
r q i = mr v i e trascuriamo per semplicità´gli urti tra le molecole ( ad esempio considerando un gas molto rarefatto)

97
avendo gia´ visto in precedenza che il contributo degli urti delle molecole traloro e´ trascurabile. Siano A1 e A2 le due pareti della scatola normali all'assex, allora la molecola varia la sua quantità di moto ad ogni urto e poiché l'urtosi suppone elastico, la molecola inverte esclusivamente la componente dellavelocità lungo l'asse x (inverte in pratica la direzione del moto) senzamodificare la componente della velocità lungo y e z, come e´ richiesto dalteorema di conservazione della quantità di moto. Dopo l'urto su A1 lamolecola percorre la distanza l con velocità´ ( lungo x) vxi prima di urtare laparete A2 , impiegando quindi un tempo
l
vxi. Dopo l'urto su A2 la molecola
ripercorrera´ la distanza l ancora nello stesso tempo prima di urtare di nuovola parete A1. Riassumendo la molecola i-esima varia la sua quantità´ di motodi una quantità´
Dqxi = - 2mi vxiin ogni intervallo di tempo
Dti =
2l
vxiScrivendo l'equazione fondamentale della dinamica nella forma
r F = D
r q Dt
si ha che la forza esercitata dalla parete A1 sulla molecola i-esima durantel'urto e´
F i = -
mi vxi2
le quindi la forza f i esercitata dalla molecola sulla parete e´
f i =
mi vxi2
lIl contributo alla pressione dovuto alla molecola i-esima e´ dunque
Pi =
fil2 =
m i vxi2
Vavendo introdotto il volume V = l3 della scatola. Il contributo alla pressionedel gas di tutte le molecole e´ dunque
P =
mV
vxi2
i =1
N
Âavendo assunto che tutte le molecole abbiano la stessa massa m e che N sia ilnumero totale delle molecole contenute nella scatola. Introducendo lavelocità´ quadratica media
< vx
2 > =1N
vxi2
i=1
N
Âsi ottiene
P =
mNV
< vx2 >
Per l'isotropia delle direzioni lungo cui si muovono disordinatamente lemolecole ( se per assurdo esistesse una direzione di moto privilegiata alloratutto il sistema tenderebbe a traslare in tale direzione, confinando tutte le

98
molecole in un canto della scatola, cosa mai verificata sperimentalmente) siha:
< vx2 > = < vy
2 > = < vz2 >
e poiché´
< v2 > = < vx2 > + < vy
2 > + < vz2 >
si conclude
P =
13
NmV
< v2 >
in altre parole´
P =
13
r < v2 >
avendo introdotto la densità´ r =
NmV
del gas. Questa formula connette lapressione esercitata da un gas perfetto sulle pareti del recipiente con lavelocità´ quadratica media delle molecole che compongono il gas stesso.Siricordi che tale formula e´ stata derivata nell'ipotesi di gas perfetto. Sipotrebbe dimostrare che l'ipotesi della forma cubica del recipiente non e´limitativa della validità´ della formula.
Esercizio: Calcoliamo la radice quadrata vrms della velocità´ quadraticamedia, che e´ una buona approssimazione della velocità´ media, nell'ipotesiche nella scatola sia contenuta dell'aria rarefatta a 0
0 C .
vrms = < v2 > =
3Pr
con
P = 1ATM @ 104 Kgpeso / m2( )
r = 1.3 Kgmassa / m3( ) =
1.39.8
Kgpeso s2 / m3( ) @ 0.13 Kgpesos2 / m4( )
vrms =
3 ¥ 104 Kgpeso / m2( )0.13 Kgpesos2 / m4( )
@ 23 ¥ 104 m2 / s2( ) @ 480 m / s( )
8. LA LIQUEFAZIONE DEI GAS, LE ESPERIENZE DIANDREWS
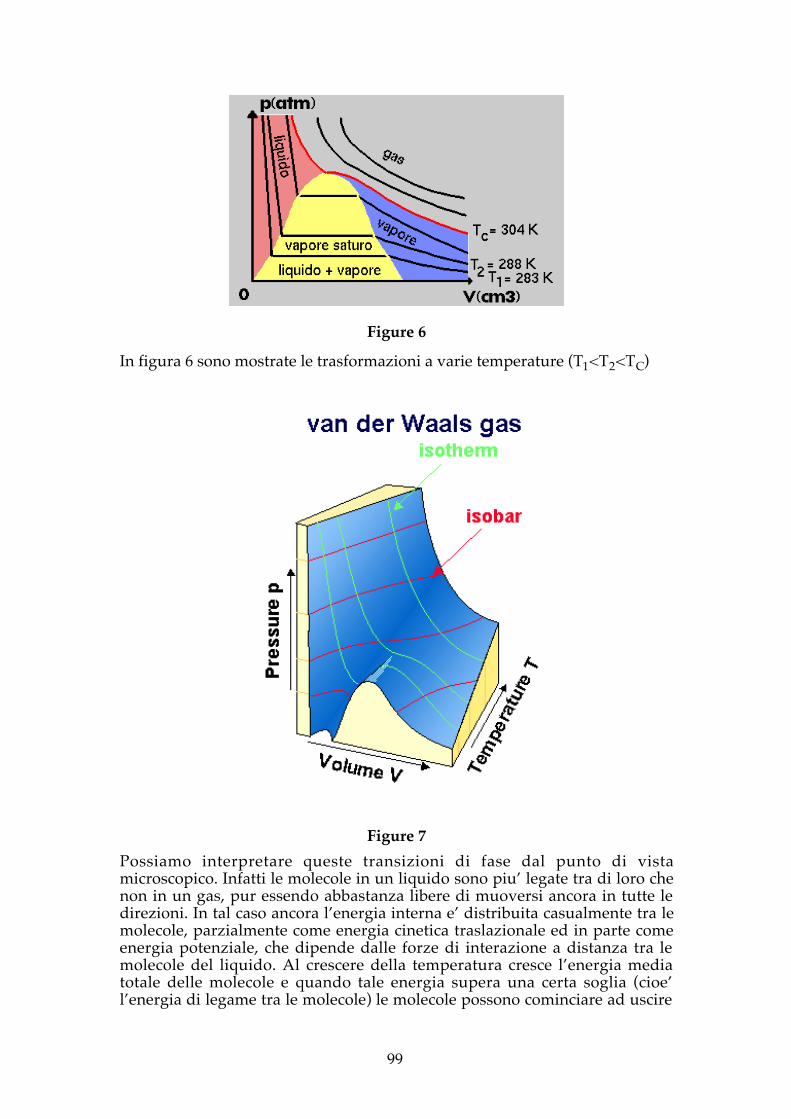
I gas reali si comportano solo approssimativamente come i gas perfettisoltanto a temperature elevate. A temperature inferiori ad un certo valoretipico di quel gas (detto temperatura critica TC) si verifica un fenomeno ditransizione di fase (dallo stato aeriforme a quello liquido) per volumidecrescenti (compressione a temperatura costante) o per pressioni crescenti.
99
Figure 6
In figura 6 sono mostrate le trasformazioni a varie temperature (T1<T2<TC)
Figure 7Possiamo interpretare queste transizioni di fase dal punto di vistamicroscopico. Infatti le molecole in un liquido sono piu’ legate tra di loro chenon in un gas, pur essendo abbastanza libere di muoversi ancora in tutte ledirezioni. In tal caso ancora l’energia interna e’ distribuita casualmente tra lemolecole, parzialmente come energia cinetica traslazionale ed in parte comeenergia potenziale, che dipende dalle forze di interazione a distanza tra lemolecole del liquido. Al crescere della temperatura cresce l’energia mediatotale delle molecole e quando tale energia supera una certa soglia (cioe’l’energia di legame tra le molecole) le molecole possono cominciare ad uscire

100
dal liquido producendo vapore. Inizia cosi’ la fase in cui il liquido vaporizza.Se la temperatura cresce ancora il liquido si trasforma completamente in gas.
9. L’EQUAZIONE DI STATO (DI VAN DER WAALS) PER I GASREALI
Se si considerano i gas reali, le forze attrattive a distanza tra le molecole nonsono affatto trascurabili a temperature sufficientemente basse. Si manifestanosostanzialmente due tipi di attrazioni intermolecolari: le forze di dispersionedi van der Waals e le interazioni dipolo-dipolo. Sono queste delle forze di tipoelettrico e dunque trascendono l’argomento di questo corso.Per i gas reali si puo’ riscrivere il teorema del Viriale nel modo seguente,ricordando che per i gas reali le forze interne non sono piu’ trascurabili:
†
12
 mivi2 +
12
Fipressioneri +
12
Fiinterneri = 0Â
32
N kT -32
pV +12
FiVan der Vaalsri = 0Â
N kT = pV -13
FiVan der VaalsriÂ
introducendo la forza di Van der Vaals si ottiene
N kT = pV - bp -aV
+abV 2
Ê
Ë Á
ˆ
¯ ˜
da cui
m RT = p +a
V 2Ê
Ë Á
ˆ
¯ ˜ V - b( )
Quest’ultima e’ l’equazione di van der Waals per i gas reali.
10. LA LEGGE DI DISTRIBUZIONE DELLE VELOCITA´ DIMAXWELL-BOLTZMANN
Consideriamo un gas di molecole e supponiamo che la probabilità P ditrovare una molecola con una velocità compresa tra
r v e r v + dr v , cioè
compresa nel volumetto dV = dvx dvy dvz dello spazio delle velocità, siaesprimibile in forma fattorizzata come il prodotto di tre probabilitàindipendenti. Si assume cioe’ che la probabilità di avere una certacomponente della velocità lungo un asse sia indipendente dal valore che lavelocità assume sugli altri assi.

101
6.
d3Pdvxdvydvz
= y vx, vy ,vz( ) = f vx( )g vy( )h vz( )
Dove y ,f, g, h sono densità´ di probabilità. Nell'assunzione che le molecole simuovano disordinatamente, che cioè non esistano direzioni privilegiate nelmoto delle stesse, vale la proprietà seguente:
f = g = hDifferenziando allora logaritmicamente l'equazione (6), cioe’ facendo laderivata del logaritmo dell’equazione, si ottiene
y ' r v ( )y
r v ( )dv =
f' vx( )f vx( )
dvx +f' vy( )f vy( )
dvy +f' vz( )f vz( )
dvz
poiche’ il moto delle molecole e’ caotico, cioe’ non ci sono direzioni preferite edunque possiamo trascurare gli angoli, consideriamo soltanto quelle molecoleche hanno modulo della velocità costante, allora dv = 0 e dunque
7.
f' vx( )f vx( )
dvx +f' vy( )f vy( )
dvy +f' vz( )f vz( )
dvz = 0
ma essendo anche:8.
0 = dv2 = 2r v ⋅ dr v = 2 vxdvx + vydvy + vzdvz( )Le equazioni (7) e (8) sono entrambe verificate se
f' vx( )f vx( )
= avx
f' vy( )f vy( )
= a vy
f' vz( )f vz( )
= a vz
Ï
Ì
Ô Ô Ô
Ó
Ô Ô Ô
essendo a una costante arbitraria. Integrando si ottiene
ln f vx( ) =a2
vx2 +Bx
ln f vy( ) =a2
vy2 +By
ln f vz( ) =a2
vz2 +Bz
Ï
Ì
Ô Ô
Ó
Ô Ô
essendo Bx ,By ,Bz costanti d'integrazione, cioè
9.
f vx( ) = Ax ea2
vx2
f vy( ) = Ay ea2
vy2
f vz( ) = Az ea2
vz2
Ï
Ì
Ô Ô
Ó
Ô Ô
essendo Ax,y,z = eBx ,y,z . Si noti che la costante a deve essere negativa
altrimenti la probabilità sarebbe crescente con la velocità v, in altre parole nel

102
sistema gassoso le molecole tenderebbero spontaneamente ad aumentare lapropria velocità, con conseguente aumento della temperatura del gas, ciò chee´ in contrasto con quanto avviene in natura. Poniamo quindi
a2
= - a 2 . Sinoti inoltre che la somma di tutte le probabilità deve essere uguale ad 1.Questa ovvia proprietà´ si chiama anche unitarietà:
1 = f u( )
-•
+•
Ú du = A e-a 2u 2
-•
+•
Ú du = A pa
da cui
Ax = Ay = Az =
ap
L'equazione (6) diventa allora
d3Pdvxdvydvz
= y vx, vy ,vz( ) =a 3
p32
e-a 2v2
da cui possiamo calcolare la probabilità che una molecola abbia velocitàcompresa nel volumetto dV :
d3P r v ( ) =
a 3
p32
e-a 2v2dvxdvydvz
Passiamo in coordinate sferiche, come mostrato nella fig. 8
X Y
Z
O
q
f
fig.19a
Figure 8Calcolo dell’elemento di Volume in coordinate sferiche.Le legge di trasformazione da coordinate sferiche a coordinate cartesiane è
vx = v senq cosf
vy = vsenqsenf
vz = vcosq
Ï
Ì Ô
Ó Ô
Vale il teorema dello Jacobiano

103
dvx dvy dvz = J
vx vy dvz
v q f
Ê
Ë Á
ˆ
¯ ˜ dvdqdf
dove lo Jacobiano della trasformazione è:
Jvx vy dvz
v q f
Ê
Ë Á
ˆ
¯ ˜ =
∂vx∂v
∂vx∂q
∂vx∂f
∂vy
∂v∂vy
∂q
∂vy
∂f
∂vz∂v
∂vz∂q
∂vz∂f
=
calcoliamo il determinante:
∂vx∂v
∂vx∂q
∂vx∂f
∂vy∂v
∂vy∂q
∂vy∂f
∂vz∂v
∂vz∂q
∂vz∂f
=
senq cosf v cosqcosf - v senq senf
senq senf vcosq senf vsenq cosf
cosq - v senq 0=
= vsenq senqcosf vsenq cosf + vsenqsenfsenq senf( ) +
+ cosq vcosq cosf vsenqcosf + v senq senfv cosq senf( ) =
= v2 sen3q cos2 f + v2 sen3q sen2f + v2 cos2 q senq cos2 f + v2 cos2 qsenqsen2f =
= v2 sen3q + v2 cos2 q senq =
= v2 senqsi ottiene quindi l'elemento di volume dv
dvx dvy dvz = v2 senq dvdqdf
con
0£q < p
0£f < 2pÈ
Î Í ˘
˚ ˙
Si ottiene dunque in coordinate sferiche
d3P r v ( ) =
a 3
p32
e-a 2v2dvsenq dq df
Integrando sugli angoli q,f si ha
dP v( ) =a 3
p32
v2e-a 2v2dv senq dq
0
p
Ú df
0
2p
Ú10.
dP v( ) = 4 a 3
pv2e-a 2v2
dv

104
Se N e´ il numero totale delle molecole che compongono il sistema e n(v) e´ ilnumero delle molecole con velocità´ v, poiché´
P v( ) =
n v( )N
si ha
11. n v( ) = N 4a3
pv2 e-a 2v2
Determiniamo ora il parametro a calcolando la velocità´ quadratica media
v2 =
n v( )0
•
Ú v2 dv
N
v2 =4a 3
pv4
0
•
Ú e-a 2v2dv
Ricordando che
x2n
0
•
Ú e-ax2dx =
1 ⋅ 3 ⋅ 5 ⋅ ⋅ ⋅ 2n - 1( )2n+1an
pa
si ottiene
v2 =
32a2
ed utilizzando l'equazione di Joule-Clausius (4) si ottiene
v2 =
3km
Tin conclusione
a 2 =
m2kT
La legge di distribuzione (10) si può´ allora scrivere
12. dP v( ) =
4p
m2kT
Ê Ë
ˆ ¯
32 v2 e
- mv 22kT dv
che e´ nota come legge di distribuzione delle velocità´ di Maxwell.

105
Si ricordi che k = R / N , con N numero d’Avogadro. Dunque R/M = k / m.Essendo M = m N la massa molare cioè la massa di una mole di gas.Ricordando che l’energia cinetica di una molecola e’ E = 1/2 mv2, si puòscrivere la densita’ di probabilità che una molecola abbia energia cinetica E
†
f E( ) = A e-
EkT
E’ questa la legge di distribuzione di Boltzmann, che ci dice che al cresceredell’energia cinetica il numero delle molecole decresce con leggeesponenziale.Come applicazione calcoliamo l’energia cinetica media <E>.Prima però determiniamo il parametro A. A questo scopo basta imporre lacondizione d’unitarietà della probabilità totale:
†
Ae-
EkT dE =1
0
•
Ú
= -AkT e-
EkT d E
kTÈ
Î Í ˘
˚ ˙ 0
•
Ú
= -AkT e-
EkT
È
Î Í Í
˘
˚ ˙ ˙
0
•
= AkT
da cui si ricava
A =1
kTadesso il valor medio dell’energia e’ per definizione:

106
†
< E >= E f E( )0
•
Ú dE
e dunque ricaviamo:
†
< E >=1
kTE e
-EkT dE
0
•
Ú
ed essendo:
†
x e-axdx = -1a2 +
xa
Ê
Ë Á
ˆ
¯ ˜ Ú e-ax
x e-axdx =0
•
Ú1a2
in conclusione si ha:
†
< E >=1
kTkT( )2
= kT
Si noti che qui abbiamo usato l’approssimazione di un motomonodimensionale delle molecole, così l’espressione e’ in accordo con laformula di Joule-Clausius per tre gradi di libertà. Si noti però che manca iltermine 1/2.
11. LA DISTRIBUZIONE DI MAXWELL NELLO SPAZIO DELLEFASI
Consideriamo il moto di una singola molecola contenuta nella scatola cubicadella fig. 9.
a
a
a
z
x
y
fig.19 b
Figure 9Questa molecola ha una posizione confinata

107
0 £ x < a0 £ y < a0 £ z < a
È
Î
Í Í
˘
˚
˙ ˙
ed una velocità v0 costante in valore assoluto, ma che cambia segno ad ogniurto contro le pareti. Allora lo stato rappresentativo del moto della molecolasi muove nello spazio delle fasi come mostrato in fig. 10.
vx0
-v x0
0a x
vx
fig.19c
Figure 10E´ immediato rappresentare allora l'insieme di tutte le molecole checompongono il gas confinato nella scatola cubica come mostrato in fig. 11.

108
xa
dNdx
vx
dN dvx x
vx
-vx
0
0
fig19 d
Figure 11
12. VERIFICA SPERIMENTALE DELLA DISTRIBUZIONE DIMAXWELL
Consideriamo l'apparato di misura rappresentato nella fig. 12 (metodo diZartmann). Esso e´ costituito da un tubo a vuoto in cui e´ posta una sorgented’atomi prodotti per riscaldamento di un granulo metallico; un fascettod’atomi e´ costretto a percorrere il tratto OP mediante un sistema dicollimatori t1,...t4; un disco ruotante attorno ad un’asse parallela al fascettoraccoglie gli atomi che sono passati attraverso la finestrella f.

109
+-
t1 t2 t3 t4
d
fP
fig.20
discorotante
Figure 12Aprendo la finestrella f in un tempuscolo molto piccolo si seleziona ungrappolo d’atomi di cui si vuole misurare la distribuzione delle velocità. Gliatomi del grappolo raggiungono il disco ruotante in tempi inversamenteproporzionali alla loro velocità. Misurando la densità del deposito d’atomisul disco a vari angoli si può risalire alla distribuzione delle velocità degliatomi prodotti per riscaldamento della sorgente. Sia v la velocità di un atomodel grappolo, allora il tempo impiegato da esso per raggiungere il disco e´
t =
dv
dove d e´ la distanza tra la finestrella ed il disco stesso. Sia w la velocitàangolare costante con cui ruota il disco, allora l'angolo di cui e´ ruotato ildisco prima che un atomo con velocità v lo raggiunga e´
q = w tda cui si ricava la relazione tra la velocità di un atomo e l'angolo d’arrivo suldisco. Il risultato sperimentale e´ in perfetto accordo con la distribuzione diMaxwell. Si noti che poiché q v = cos tan te allora si ha
Dvv
=Dqq
in pratica l'errore percentuale della velocità uguaglia l'errore percentualedell'angolo. Questa relazione permette di determinare l'accuratezza con cui sipuò eseguire la misura della velocità.
13. LA TEORIA CINETICA NEI CORPI SOLIDI
Nei corpi solidi, contrariamente a quanto accade nei gas, gli atomi sono legatiin una struttura sostanzialmente rigida (struttura cristallina), cioèmantengono una posizione media fissa gli uni rispetto agli altri secondo unastruttura di tipo cristallino o amorfo che dipende dagli atomi checompongono il corpo solido.

110
Figure 13In figura 13 e’ mostrato uno schema di corpo solido dove gli atomi sono liberidi oscillare attorno alle loro posizioni medie che sono fisse. Anche qui’ alcrescere della temperatura aumentano le energie cinetiche degli atomi, e cio’corrisponde a moti oscillanti sempre piu’ vigorosi. Supponiamo ad esempioche ogni atomo del reticolo solido si muova di moto armonico, in tal caso gliatomi interagiscono tra di loro e posseggono l’energia potenziale di unoscillatore armonico, oltre che l’energia cinetica. L’andamento delle energie diun singolo atomo sono mostrate nella figura 14, dove il moto dell’atomo e’schematizzato come dovuto all’azione di una molla elastica.
Figure 14In ogni istante l’energia potenziale elastica e l’energia cinetica variano, mal’energia totale, che e’ la loro somma, resta costante.
†
Etot = T +U =12
m w2A2
cioe'
Etot = p m n2A2
111
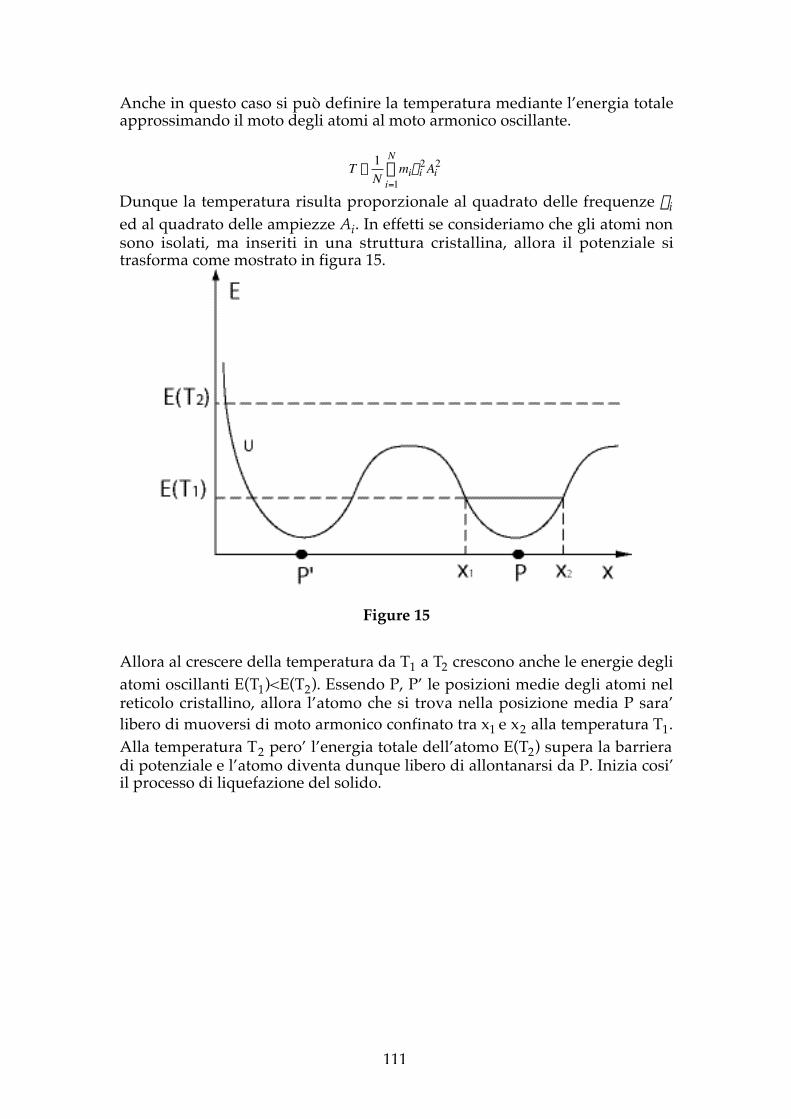
Anche in questo caso si può definire la temperatura mediante l’energia totaleapprossimando il moto degli atomi al moto armonico oscillante.
†
T ª1N
mini2Ai
2
i=1
NÂ
Dunque la temperatura risulta proporzionale al quadrato delle frequenze nied al quadrato delle ampiezze Ai. In effetti se consideriamo che gli atomi nonsono isolati, ma inseriti in una struttura cristallina, allora il potenziale sitrasforma come mostrato in figura 15.
Figure 15
Allora al crescere della temperatura da T1 a T2 crescono anche le energie degliatomi oscillanti E(T1)<E(T2). Essendo P, P’ le posizioni medie degli atomi nelreticolo cristallino, allora l’atomo che si trova nella posizione media P sara’libero di muoversi di moto armonico confinato tra x1 e x2 alla temperatura T1.Alla temperatura T2 pero’ l’energia totale dell’atomo E(T2) supera la barrieradi potenziale e l’atomo diventa dunque libero di allontanarsi da P. Inizia cosi’il processo di liquefazione del solido.





























