Embed Size (px)
Citation preview

TESIS
“Polimerización In situ de Poliestireno de Alto Impacto en presencia de Nanopartículas de Sílice y Micropartículas de Mg(OH)2 y su Influencia sobre
la Cinética de Polimerización, la Morfología de las Fase Elastomérica y la Retardancia a la Flama en los Nanocompuestos Finales”
Presenta: I.Q. Andrés Ubaldo Alarcón
Para obtener el grado de:
Maestro en Tecnología de Polímeros
Asesor:
Dr. Florentino Soriano Corral
Co-Asesor:
Dr. Ramón Enrique Díaz de León Gómez
Saltillo, Coahuila Noviembre 2017


TESIS COi\ CARACTER ABIERTO
PROGRAMA: MAESTRÍA EI\ TECNOLOGÍA DE POLÍMEROS
AUTOR: ANDRÉS UBALDO ALARCÓN FrRMA /.nJl';s ü,4.
TITULO: Polimerización In Situ de Poliestireno de Alto lmpacto enpresencia de Nanopartículas de Sílice v Micropartículas de Mg (OII)z vsu Influencia sobre la Cinética de Polimerización, la Morfoloeía de laFase Elastomérica v la Retardancia a la Flama en los NanocompuestosFinales.
ASESORES: Dr. Florentino Soriano Corral FIRMA
Dr. Ramón Enrique Díaz de León Gómez FIRMA
EI Centro de lnvestigación en Química Aplicada clasilga el presentedocumento de tesis como ABIERTO.
Lln documento clasificado como Abierto se expone en los estantes delCentro de Información para su consulta. Dicho documento no puede sercopiado en ninguna modalidad sin autorización por escrito del Titular delCentro de Información o del Director General del CIQA.
Saltillo, Coahuila, a 23 de Noviembre de 2017
-^'oAcloli ur,
-\o "t
ñffiffiÉLv 5./,
.PJ
-l ^v.
¡t\Dr. Oliverio Santiago Rodríguez Fernández
Director General del CIQASello átllu rñ§iitución

CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADAPrograma de Maestría en Tecnología de Polímeros
TESIS
Polimerización In Situ de Poliestireno de Alto Impacto en presencia deNanopartículas de Sílice y Micropartículas de Mg (OH)2 y su Influencia sobre Ia
Cinética de Polimerización,la Morfología de la Fase Elastomérica yla Retardancia a la Flama en los Nanocompuestos Finales
Presentada por:
A¡IDRÉS UBALDO ALARCÓN
Para obtener el grado de:
Maestro en Tecnología de PolÍmeros
Asesorado por:
Dr. Florentino Soriano CorralDr. Ramón Enrique Díaz de León Gómez
SINODALES
t
Presidente
Dr. Luis Francisco Ramos de ValleVocal
Saltillo, Coahuila Noviembre,20lT

GENTRO DE INVESTIGAGIÓNEN QUíMICA APLIGADA
cENTRo DE rNvESTrcACróN nN euÍlrrcA ArLTcADAPrograma de Maestría en Tecnología de Polímeros
TESIS
Polimerización In Situ de Poliestireno de Alto Impacto en presencia deNanopartículas de Sílice y Micropartículas de Mg (OH)2 y su Influencia
sobre la Cinética de Polimerización, la Morfología de la FaseElastomérica y la Retardancia a la Flama en los Nanocompuestos Finales
Presentada por:
ANDRÉS UBALDO ALARCÓN
Para obtener el grado de:
Maestro en Tecnología de Polímeros
Asesorado por:
Dr. Florentino Soriano CorralDr. Ramón Enrique Díaz de León Gómez

DECLARACIÓN
Declaro que la información contenida en la Parte Experimental así como en la
Parte de Resultados y Discusiones de este documento y que forman parte de las
actividades de investigación y desarrollo realizadas durante el período que se
me asignó para llevar a cabo mi trabajo de tesis, será propiedad del Centro de
Investigación en Química Aplicada.
Saltillo, Coahuila a23 de noviembre de20L7
Nombre y Firma

Dedicatoria
A Dios
A mis padres, Andrés Ubaldo y Silvia Alarcón, por todo su amor y apoyo.
A mis hermanas Flora y Silvia
A mis abuelos
A mis amigos

Agradecimientos
A mis asesores, el Dr. Florentino Soriano Corral y el Dr. Ramón E. Díaz de León Gómez por su confianza, apoyo y dedicación en el desarrollo de este trabajo de investigación, muchas gracias. Al Dr. Oliverio Rodríguez Fernández y al Centro de Investigación en Química Aplicada (CIQA) por el apoyo brindado para la realización y desarrollo de este trabajo en las instalaciones del centro. Al Consejo Nacional de Ciencia y Tecnología (CONACYT), por la beca otorgada durante la realización de esta maestría. A mis evaluadores la M.C. Isaura Yañez Flores, el Dr. Luis Francisco Ramos de Valle, y el Dr. Saúl Sánchez Valdés por sus valiosas aportaciones para mejorar este trabajo de investigación. Al Laboratorio Nacional en Innovación y Desarrollo de Materiales Ligeros para la Industria Automotriz (LANI-Auto), se agradece el apoyo parcial a través del proyecto CONACYT 280425. Al M.C. Ricardo Mendoza Carrizales y al M.C. José Alejandro Díaz Elizondo por su paciencia y apoyo técnico en planta 2. A la Ing. María Concepción González Cantú por su apoyo y paciencia en plata 1. A la Dra. María Guadalupe y al Dr. Gustavo Soria Argüello Neira Velázquez por la facilitación del equipo de modificación de plasma frío, y por el apoyo brindado para su uso. Al LCQ Alejandro Espinoza Muñoz, a la M.C. Rosario Rangel Ramírez, a la LCQ Irma Oralia Solís de la Peña y al I.Q. Efraín Alvídrez Ramos, gracias por la facilitación de equipos y caracterizaciones de los materiales obtenidos. Al Ing. Josué de Jesús Campos Oyervides por realizar las pruebas de resistencia al impacto de los materiales. Al Dr. Enrique Díaz Barriga Castro por el apoyo técnico brindado para la caracterización morfológica mediante TEM de los materiales obtenidos en el presente trabajo. A la Lic. Miriam Lozano Estrada por el apoyo técnico brindado para la caracterización morfológica mediante SEM.
i

Resumen. En el presente trabajo de investigación se sintetizó poliestireno de alto impacto (HIPS, por
sus siglas en inglés, High Impact Polystyrene), y nanocompuestos polímericos a base de
HIPS y diferentes concentraciones de nanopartículas de SiO2 modificadas superficialmente
y sin modificar, y de micropartículas comerciales de Mg(OH)2 , mediante polimerización In
situ por radicales libres.
Previamente, las nanopartículas de SiO2 comerciales fueron modificadas superficialmente
mediante la polimerización de estireno por plasma frio, generado por radiofrecuencia.
Se caracterizaron tanto las nanopartículas de SiO2 como Mg(OH)2 modificadas
superficialmente y sin modificar en cuanto a su composición, estructura cristalina y
morfología mediante microscopia electrónica de transmisión (TEM). Adicionalmente, se
utilizó microscopía electrónica de barrido para establecer la morfología del Mg(OH)2. Por
otro lado, se realizaron análisis de espectroscopia infrarroja (IR), análisis
termogravimétrico (TGA) a fin de determinar cualitativa y cuantitativamente la
modificación realizada sobre las nanopartículas.
Una vez caracterizadas ambos tipos de partículas, estas se incorporaron a un sistema de
reacción por lotes, donde se polimerizó estireno en presencia de Polibutadieno y con
diferentes concentraciones de nanopartículas, utilizando 0.1% en peso de peróxido de
benzoílo (BPO) como iniciador. Cabe destacar que durante esta reacción se dio
seguimiento a la cinética de polimerización a través de la determinación de la conversión
en función del tiempo.
Las características de los nanocompuestos de HIPS/SiO2, HIPS/Mg(OH)2 y
HIPS/SiO2/Mg(OH)2 se determinaron mediante diversas técnicas de caracterización como:
Cromatografía de exclusión por tamaño (SEC), para evaluar pesos moleculares y
distribución de pesos moleculares (Mn, Mw y DPM), Microscopia electrónica de barrido y
transmisión (STEM) para caracterizar la morfología de la fase elastomérica, Contenido de
Gel (CG) y Grado de Injerto aparente (GI) , Resistencia al impacto IZOD y Prueba de
flamabilidad para evaluar la velocidad de quemado de los materiales finales.
iii
ii

En relación a los HIPS sintetizados, se encontró que la presencia de micro y nanopartículas
durante la polimerización In situ retarda la cinética de reacción. Las propiedades físico-
químicas de los compuestos de HIPS / SiO2 y/o Mg(OH)2 , como era de esperar, fueron
diferentes a las propiedades del blanco de HIPS ( HIPS B ; sin partículas de SiO2 y/o
partículas de Mg(OH)2); el grado de injerto del PB y el grado de entrecruzamiento
disminuyeron en comparación al HIPS B ; los pesos moleculares (Mn y Mw) se
incrementaron en la mayoría de los compuestos ; la resistencia al impacto mejoró cuando
el HIPS se sintetizó en la presencia de SiO2 modificada superficialmente ; y la morfología
también presentó un ligero cambio, todo esto asociado a los cambios presentados en la
cinética de la polimerización. En cuanto a la retardancia a la flama, la presencia de SiO2
con el Mg(OH)2 no presentó el efecto sinérgico que ha sido reportado por algunos autores
para sistemas similares, esto como consecuencia de la aglomeración y la localización de
las nanopartículas de SiO2 en la fase dispersa.
iv

ÍNDICE
Dedicatoria……………………………………………………………………………………………………………. i
Agradecimientos…………………………………………………………………………………………….... ii
Resumen………………………………………………………………………………………………………………… iii
CAPITULO I. Introducción…………………………………………………………………………………………. 1
CAPITULO II. Antecedentes………………………………………………………………………………………. 4
2.1 Poliestireno de alto impacto (HIPS)……………………………………………………………………… 4
2.1.1 Síntesis de HIPS…………………………………………………………….……….……….………………… 7
2.1.2 Proceso de Polimerización In Situ en masa de HIPS ……………………………………….. 7
2.1.2.1 Homopolimerización de Estireno………………………………………………………………….. 8
2.1.2.2 Copolimerización de injerto (CI)……………………………………………………………………. 10
2.1.2.3 Entrecruzamiento…………………………………………………………….……….………………….. 12
2.1.2.4 Cinética de polimerización en masa…………………………………………………………….. 12
2.1.3 Proceso industrial de obtención de HIPS ………………………………………………………… 15
2.1.4 Desarrollo morfológico durante el proceso de Polimerización In situ…………… 18
2.1.5 Cambio en la viscosidad durante la inversión de fases…………………………………… 20
2.1.6 Variables de síntesis…………………………………………………………….……….…………………. 21
2.1.6.1 Concentración y tipo de iniciador……………………………………………………………….... 22
2.1.6.2 Concentración y tipo de elastómero……………………………………………………………. 22
2.1.6.3 Velocidad de agitación…………………………………………………………….……….………….. 23
2.1.7 Relación de la estructura y composición con las propiedades mecánicas del
HIPS…………………………………………………………….…………………………………………………………….
23
2.1.7.1. Peso Molecular de la Matriz …………………………………………………….……….………… 25
2.1.7.2 Tipo de elastómero…………………………………………………….……….……….……………… 25
2.1.7.3 Relación de volumen entre las fases …………………………………………………………… 26
2.1.7.4 Tamaño de partícula y distribución de tamaño de partícula……………………….. 26
2.1.7.5 Grado de Injerto (GI) y contenido de gel (CG)………………………………………………. 27

2.1.7.6 Influencia del uso de aditivos ……………………………………………………..………………. 27
2.1.8 Morfologías de la fase elastomérica de los HIPS……………………………………………. 28
2.1.8.1 Morfologías más importantes en un HIPS…………………………………………………….. 29
2.1.8.2 Mecanismo de mejora al impacto …………………………………………………….…………. 30
2.2 Nanopartículas…………………………………………………….……………………………………………… 32
2.2.1 Métodos para síntesis de nanopartículas ……………………………………………………….. 35
2.2.2 Nanopartículas de sílice…………………………………………………….…………………………….. 37
2.2.3 Nanopartículas comerciales de sílice (AEROSIL)………………………………………………. 37
2.2.4 Síntesis de nanopartículas sílice…………………………………………………….…………………. 38
2.3 Retardantes a la flama………………………………………………….…………………………………….. 41
2.3.1 Flamabilidad de polímeros………………………………………………….……………………………. 41
2.3.2 Aditivos Retardantes a la flama……………………………………………………………………….. 42
2.3.2.1 Mecanismo de Acción de Retardantes a la flama…………………………………………. 43
2.3.2.2 Hidróxido de Magnesio…………………………………………………….……………………........ 45
2.3.2.3 Mecanismo de retardancia a la flama del Hidróxido de Magnesio……………….. 45
2.4 Efecto de la incorporación de nanopartículas de sílice sobre la cinética de
polimerización …………………………………………………….…………………………………………………….
47
2.5 Nanosílice como retardante a la flama ………………………………………………………………. 51
2.5.1 Nanocompuestos de polímeros estirénicos / Sílice…………………………………………… 51
2.6 Modificación superficial de nanopartículas mediante plasma generado por
radiofrecuencia………………………………………………………………………………………………………….
52
CAPITULO III. Justificación…………………………………………………….…………………………………. 54
CAPITULO IV. Hipótesis.…………………………………………………….…………………………………….. 55
CAPITULO V. Objetivos.…………………………………………………….……………………………………… 56
5.1 Objetivo general…………………………………………………….…………………………………………… 56
5.2 Objetivos Particulares …………………………………………………….…………………………………. 56
CAPITULO VI. Metodología …………………………………………………….………………………………. 57
6.1 Caracterización Morfológica por TEM de nanopartículas de SiO2 y
Micropartículas de Mg(OH)2 …………………………………………………….………………………….
57

6.2 Caracterización de micropartículas de Mg(OH)2 por Microscopía electrónica de
barrido (SEM) …………………………………………………….………………………………………………........
58
6.3 Modificación por plasma frio de nanopartículas comerciales de SiO2 ……………….. 58
6.4 Análisis por Infrarrojo (IR) de nanopartículas de SiO2 y Micropartículas de
Mg(OH)2 ……….……………………………………….………………………………………………………………….
59
6.5 Análisis termogravimétrico TGA …………………………………………………….……………....... 59
6.6 Síntesis de HIPS y nanocompuestos …………………………………………………….…………….. 59
6.7 Preparación de probetas para impacto y flamabilidad ………………………………………. 63
6.8 Resistencia al Impacto IZOD …………………………………………………….…………………………. 63
6.9 Prueba de Flamabilidad …………………………………………………….………………………………… 63
6.10 Contenido de Gel (CG) y Grado de Injerto final aparente (GI) ………………………….. 64
6.11 Cromatografía de exclusión por tamaño (SEC).………………………………………………… 65
6.12 Caracterización Morfológica por TEM de Nanocompuestos ………………………….. 65
CAPITULO VII. Resultados y discusión …………………………………………………………………….. 67
7.1 Caracterización de partículas de Mg(OH)2 y de SiO2 sin modificación y
modificadas …………………………………………………….…………………………………………………………
67
7.1.1 Microscopia electrónica de transmisión (TEM) y de barrido (SEM) ………………….. 67
7.1.2. Análisis por espectroscopia infrarroja (FTIR). ………………………………………………….. 68
7.1.3. Análisis termogravimétrico (TGA) ……………………………………………………………………. 70
7.2 Síntesis del HIPS y nanocompuestos de HIPS ………………………………………………………. 72
7.2.1 Conversión en función de tiempo de polimerización ……………………………………….. 72
7.2.2 Análisis de la caracterización físico-química.………………………………………………....... 77
7.2.2.1 Efecto de la velocidad de polimerización sobre la distribución de pesos
moleculares (DPM). …………………………………………………….…………………………………………….
77
7.2.2.2 Efecto de la velocidad de polimerización sobre el contenido de gel (CG) y
grado de injerto (GI).…………………………………………………….……………………………………………
82
7.2.3 Evolución de la morfología del HIPS ……………………………………………………………….. 84
7.2.4 Morfología final de los HIPS sintetizados …………………………………………………………. 88
7.2.5. Resistencia al impacto ……………………………………………………………………………………. 96

7.2.6 Fracción volumen de fase dispersa ………..………..………..………..………..…………….... 100
7.2.7 Flamabilidad.……………………………………………………………………………………………………. 103
CAPITULO VIII. Conclusiones…………………………………………………….………………….............. 113
CAPITULO IX. Trabajo futuro.……………………………………………….………………….................. 115
Bibliografía…………………………………………………….…………………………………………………………. 116

1
CAPITULO I. Introducción
El homopolímero de Poliestireno (PS) a temperatura ambiente es un polímero vítreo y
presenta un comportamiento frágil, ya que su temperatura de transición vítrea (Tg) se
encuentra entre 90 y 100 °C. El uso de elastómeros durante el proceso de polimerización
del estireno conduce a la obtención de una mezcla de polímeros constituida por una fase
elastomérica dispersa en una matriz continua de PS 1. El resultado de la adición de la fase
elastomérica, generalmente de Polibutadieno (PB), a la matriz vítrea de Poliestireno (PS)
da como resultado un polímero con características únicas, conocido como poliestireno de
alto impacto (HIPS). El HIPS además de presentar valores aceptables de rigidez y
estabilidad dimensional, presenta una alta resistencia al impacto proporcionada por la
fase elastomérica.
Por otro lado, el uso de micro y nanopartículas de sílice (nombre común del dióxido de
silicio, SiO2) como cargas reforzantes en la preparación de compuestos poliméricos ha
tomado gran interés en los últimos años, esto como consecuencia del aumento en la
demanda de nuevos materiales con mejores propiedades térmicas, mecánicas y de
estabilidad dimensional 3,4,5,6.
Retomando el tema del material que compete a la presente investigación, el HIPS en los
últimos años, ha manifestado una disminución significativa en cuanto a su uso, debido
principalmente al posicionamiento, en el mercado, que han logrado otros polímeros y/o
compuestos termoplásticos como el polipropileno (PP) y el poli (acrilonitrilo-butadieno-
estireno) (ABS). No obstante, la naturaleza heterogénea y versátil del HIPS presenta
potencial para superar lo ofrecido tanto por el ABS como por el PP. El uso de diferentes
tipos de nanopartículas o micropartículas reforzantes de la matriz de poliestireno puede
mejorar sus características mecánicas y propiedades como, retardancia a la flama,
actividad bactericida, resistividad eléctrica, entre otras.
Existen estudios, como los reportados por Katančić y colaboradores 3,4 y Feng Yang y
colaboradores 5,6, entre otros, sobre la influencia que presenta la incorporación de SiO2,
en las propiedades térmicas y de retardancia a la flama en nanocompuestos de

2
poliestireno homopolímero (PS) y HIPS obtenidos por extrusión. En estos nanocompuestos
de polímeros estirenicos/SiO2, ellos observaron que la presencia del SiO2 no presenta un
efecto retárdante a la flama. Sin embargo, al ser incorporado en conjunto con un agente
bromado y/o hidróxido de aluminio, como retardantes a la flama se presenta un efecto
sinérgico mejorando la retardancia a la flama del material.
Por otro lado, es bien conocido que existen diferentes procesos para la elaboración de
nanocompuestos, como: Mezclado en fundido, mezclado en solución y la incorporación de
nanopartículas durante la polimerización. En cuanto al uso de mezclado en solución y
durante la polimerización para la elaboración de nanocompuestos hay pocos estudios
reportados.
Sin embargo, se sabe que al incorporar nanopartículas durante la polimerización presenta
una importantes ventajas sobre el mezclado en solución, una de ellas asociada a las
condiciones cambiantes del proceso de polimerización como lo es la mayor viscosidad del
sistema con respecto al mezclado en solución, lo que resulta en una mejor distribución y
dispersión de las nanopartículas y, por ende, en un producto final superior. Otra ventaja,
es que del proceso de polimerización se obtiene un compuesto listo para procesarse,
mientras que en mezclado en solución existe una serie de pasos para extraer el o los
solventes utilizados en el proceso.
Durante el proceso de polimerización las nanopartículas (que en caso requerido son
tratadas con una apropiada modificación superficial) son incorporadas en el monómero y
la polimerización se lleva a cabo en presencia de ellas. Al final el nanocompuesto es
formado durante la reacción, proceso al cual algunos autores le nombran polimerización
“In situ”.
Tomando en cuenta que existe muy poca investigación respecto a la elaboración de
nanocompuestos de HIPS/SiO2/Agente retardante a la flama mediante el proceso de
polimerización In situ, el estudio del efecto de la incorporación de este tipo de partículas
sobre las características cinéticas durante la polimerización de estireno y el posible efecto
sinérgico en cuanto a retardancia a la flama que pudiera presentarse en el

3
nanocompuesto final, en presencia del hule y de las nanopartículas de SiO2 y Mg(OH)2, se
considera relevante y necesario. Cabe mencionar que este tipo de estudios sistemáticos
que analizan las diferentes variables que pueden presentarse durante y al final de la
obtención de este tipo de nanocompuesto funcionales sentará las bases para estudios
posteriores.

4
CAPITULO II. Antecedentes
2.1 Poliestireno de alto impacto (HIPS)
El Poliestireno es uno de los más antiguos polímeros termoplásticos producido
comercialmente 7. El homopolímero de Poliestireno (PS) es un polímero amorfo,
transparente, con brillo, fácilmente procesable y de bajo costo, pero con poca resistencia
mecánica debido a su fragilidad 7.
A temperatura ambiente, el PS, es un polímero vítreo y presenta una baja absorción de
energía de impacto (resistencia al impacto de 0.15 lbf-ft/in) debido a la ausencia de la
movilidad local de segmentos de cadena, ya que su Tg se encuentra entre 90 y 100 °C,
temperatura a partir de la cual ocurren movimientos de segmentos de cadena
responsables, en este caso, de la disipación de la energía. Generalmente para mejorar la
resistencia al impacto de un polímero termoplástico rígido se le incorpora cierta cantidad
de una segunda fase elastomérica, obteniendo diversos polímeros heterogéneos con un
amplio espectro de propiedades bien equilibradas 1.
El uso de elastómeros durante el proceso de polimerización de estireno conduce a la
obtención de una mezcla de polímeros In situ con dominios discretos de la fase
elastomérica dispersos en una matriz continua de PS (Ver Fig. 1) 1. La fase elastomérica se
constituye de un copolímero de injerto (CI) de polibutadieno-c-poliestireno (PB-g-St)
generado In Situ, y de polibutadieno (PB) (Ver Fig. 2) 2. El CI actúa como emulsificante y
compatibilizante dando lugar a un sistema heterogéneo 8.
Figura 1. Fase de PB dispersa en la matriz de PS

5
Las partículas de hule dispersas presentan una morfología compleja y en ocasiones con
mono o múltiples oclusiones de PS, siendo la más común (y deseada para la mejora de
resistencia al impacto) la morfología tipo salami la cual se muestra en la Fig. 2 1.
Figura 2. Interfase PS – PB en morfología tipo salami del HIPS
Las propiedades del HIPS como producto final están determinadas, en gran medida, por su
morfología 7. En la Tabla 1 se muestran las propiedades mecánicas de un HIPS utilizado
en la Industria del embalaje.
Tabla 1. Propiedades Mecánicas del HIPS Correspondientes a la Formulación
que se utiliza en la Industria del Embalaje 2, 7.
Propiedades Unidades Valores
Índice de fluidez g/ 10 min 2.5 Temperatura Vicat °C 102 Impacto IZOD J/cm (ft-lb/in ) 1.068 (2) Resistencia a la tensión kg/cm2 (psi) 274 (3900) Módulo de tensión kg/cm2 (psi) 24 607 (350000) Elongación % 60 Cantidad de hule % 7.3 Tamaño de partícula m 1.2
Distribución del tamaño de partícula - 2.4 Contenido de gel % 30 Grado de hinchamiento - 12

6
En cuanto a su comportamiento reológico, el HIPS se comporta como un sólido
viscoelástico que tiende a deformarse cuando se le expone a un esfuerzo constante. La
deformación (elongación) disminuye con el incremento del peso molecular y aumenta con
el contenido de la fase elastomérica. Cabe resaltar que la medida de la tenacidad en un
HIPS será determinada por el área bajo la curva en el ensayo de esfuerzo-deformación 2.
Por otro lado, en fundido, el HIPS se comporta como un fluido no newtoniano, donde la
viscosidad depende de la velocidad del esfuerzo de corte.
El HIPS es un polímero no polar, y se desempeña como excelente aislante eléctrico. Es
resistente al agua, a los álcalis y a los ácidos inorgánicos diluidos. Se hincha con
determinados solventes orgánicos y se disuelve con otros, y es particularmente sensible a
los hidrocarburos aromáticos. En este sentido, es importante destacar que un parámetro
de suma importancia en un HIPS lo representa el contenido de gel (CG), determinado por
la masa de la fase elastomérica entrecruzada, y definido como la fracción no soluble en
tolueno. Del CG se puede también determinar el Grado de hinchamiento (GH), que
expresa la relación volumétrica del gel hinchado respecto a su estado normal en ausencia
de solvente, es un índice de la cantidad de dominios entrecruzados que presenta el
material 2.
El HIPS es un material “commodity” importante ya que en él se combina la facilidad de
procesamiento de poliestireno con una mayor resistencia al impacto, por lo que se utiliza
en una gran variedad de aplicaciones como: embalajes, envases, electrodomésticos,
frigoríficos, juguetes, entre otros.

7
2.1.1 Síntesis de HIPS
El primer estudio documentado sobre la mejora de la resistencia al impacto del PS data de
1927, siendo una patente de producción de PS con resistencia al impacto mejorada a
partir de la polimerización de estireno en una solución hule/monómero de estireno (St). El
proceso no tuvo éxito comercial, esto se debió a que el producto formado se constituía de
una fase continúa de hule reticulado, el cual no era posible procesar por las técnicas
tradicionales de transformación de plásticos.
Los desarrollos posteriores realizados por Dow Chemical Company, en los años 40 y 50,
dieron por resultado un producto comercial de PS y copolímeros de estireno-butadieno
(SBR), obtenido por procesos de emulsión. Este producto fue llamado poliestireno de alto
impacto (HIPS). Desde 1960, los SBR comenzaron a ser reemplazados por el PB para ser
incorporados al PS, debido a que con esta formulación se obtuvieron HIPS con
propiedades mecánicas y de impacto superiores, principalmente debido a la aparición de
un mayor grado de injerto y de entrecruzamiento en el hule 1.
2.1.2 Proceso de Polimerización In Situ en masa de HIPS
La reacción de polimerización de estireno en presencia de hule se lleva a cabo mediante el
uso de iniciadores, por lo general se utiliza como iniciador peróxido de benzoílo (BPO).
El proceso discontinuo de polimerización In Situ se divide en dos etapas principales:
La primera de etapa es conocida como pre-polimerización, durante esta etapa se
fija la morfología de la fase hulosa y se genera, simultáneamente, PS y CI ;
La segunda etapa de la polimerización, ya sea en suspensión o masa estática, se
lleva a cabo a elevadas temperaturas donde continúa la homopolimerización, se
entrecruza la fase hulosa y se establecen los pesos moleculares 10,11.

8
Radical benzoílo
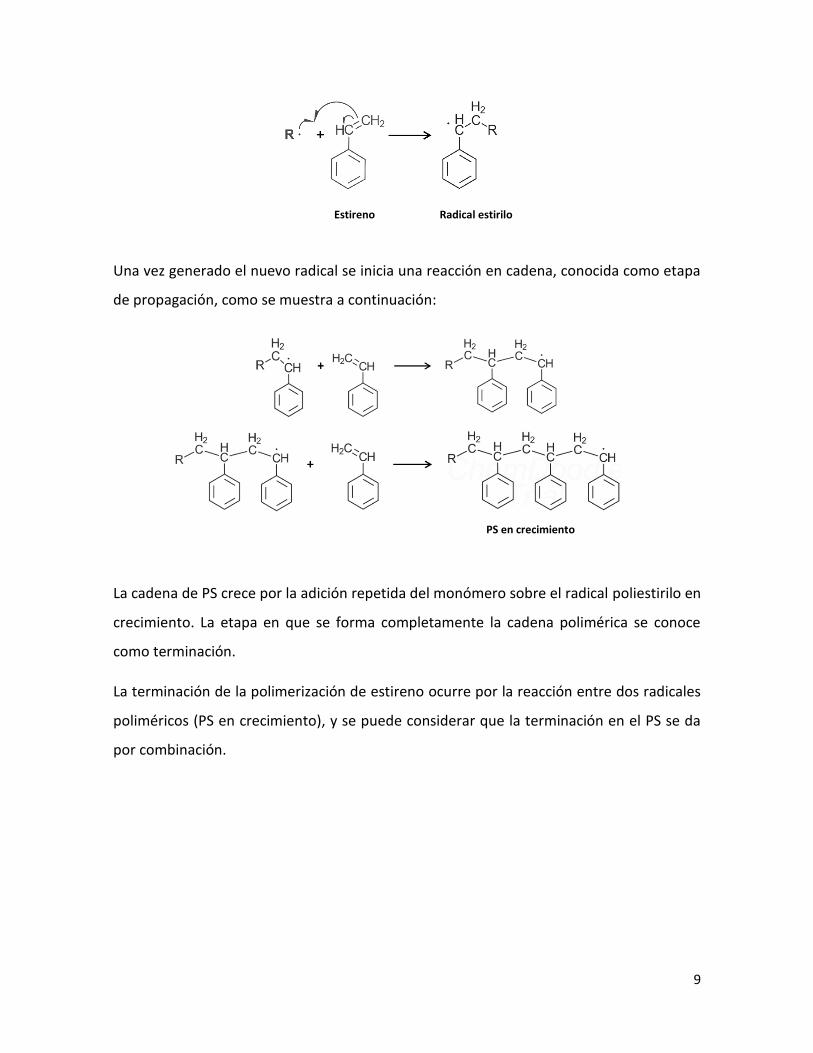
2.1.2.1 Homopolimerización de Estireno
Para iniciar la etapa de pre polimerización, se utilizan iniciadores del tipo peróxido (RO-
OR). La temperatura de reacción varía dependiendo del tiempo de vida del iniciador
usado. En el inicio, el sistema es una solución homogénea de PB en St; los radicales
formados a partir de la descomposición térmica del iniciador atacan al monómero de St
(generando PS) y a las cadenas de PB (lo cual da lugar a la reacción de formación del CI).
El homopolímero se forma por la polimerización del St por radicales libres, como se
muestra en la siguiente ecuación:
La reacción de polimerización del estireno se inicia generando los radicales libres. En el
caso del iniciador peróxido de benzoílo (BPO), este se descompone a altas temperaturas
mediante una ruptura homolítica del enlace oxígeno – oxígeno 12 como se muestra a
continuación:
El radical benzoílo generado ataca al doble enlace del grupo vinil del estireno, generando
un nuevo radical, e iniciando una reacción en cadena.
Representando el radical benzoílo ( ) como (radical primario) la
reacción con estireno se puede representar como sigue:
Peróxido de benzoílo
Estireno Poliestireno
9
Una vez generado el nuevo radical se inicia una reacción en cadena, conocida como etapa
de propagación, como se muestra a continuación:
La cadena de PS crece por la adición repetida del monómero sobre el radical poliestirilo en
crecimiento. La etapa en que se forma completamente la cadena polimérica se conoce
como terminación.
La terminación de la polimerización de estireno ocurre por la reacción entre dos radicales
poliméricos (PS en crecimiento), y se puede considerar que la terminación en el PS se da
por combinación.
PS en crecimiento
Estireno Radical estirilo

10
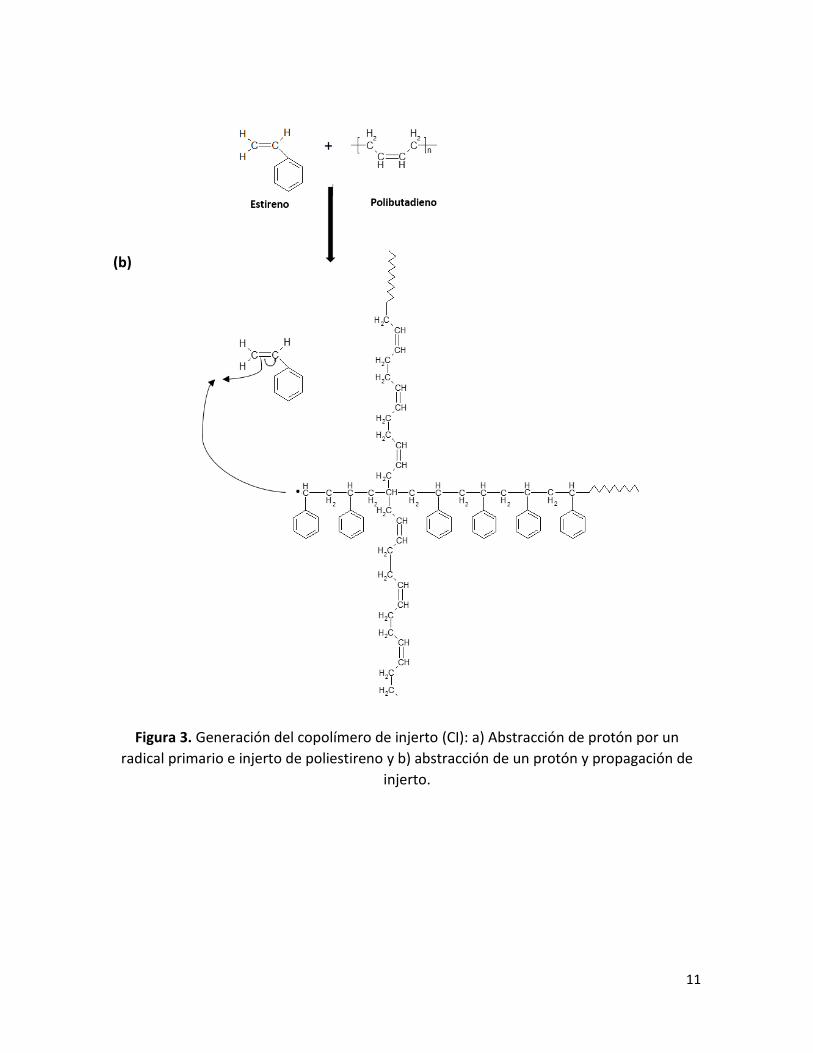
2.1.2.2 Copolimerización de injerto (CI)
El mecanismo de generación del copolímero ha sido investigado teórica y/o
experimentalmente ampliamente 2, 10, 11, 13 -20.
La reacción de formación del CI se muestra en la Fig. 3. Los sitios de injerto se generan
por la abstracción de hidrógenos alílicos del PB por: (a) radicales primarios provenientes
de la descomposición del iniciador obtenidos por iniciación térmica, que abstraen un
protón lábil desde el polibutadieno, aquí se genera un radical polibutadienilo (Ver Figura
3a) donde inicia la polimerización de monómero de St, dando lugar a injertos de PS. Y (b)
por la acción de radicales poliestirilo, quienes de la misma manera abstraen un protón
lábil, generando un radical polibutadienilo donde nuevamente se polimeriza el St (Ver
Figura 3b), dando lugar al injerto de PS 1,2.
(a)
11
(b)
Figura 3. Generación del copolímero de injerto (CI): a) Abstracción de protón por un
radical primario e injerto de poliestireno y b) abstracción de un protón y propagación de
injerto.

12
2.1.2.3 Entrecruzamiento
Una vez que la morfología es establecida en la etapa de prepolimerización, y la partícula
es estabilizada por la alta viscosidad de la matriz y el copolímero de injerto, la reacción
procede a una segunda etapa, la cual es llevada a cabo a un intervalo de temperaturas
mayores, entre 140-210°C. Bajo estas nuevas condiciones de reacción tiene lugar el
entrecruzamiento de la fase hulosa, generalmente a conversiones mayores de 90 % de
estireno, de modo que la partícula alcanza una integridad adecuada y se fija a la matriz
para soportar procesos futuros 8,26.
La morfología final de la fase elastomérica queda definida básicamente por la formulación
de reacción y por las condiciones de prepolimerización. En general, se puede disminuir el
diámetro de partículas incrementando la velocidad de agitación, incrementando la
eficiencia de injerto y reduciendo la viscosidad del sistema (por aumento de la
temperatura de reacción) 2.
2.1.2.4 Cinética de polimerización en masa
El HIPS se sintetiza vía radicales libres ya sea por iniciación química y/o térmica. La
formación del CI comienza tan pronto como el monómero empieza a polimerizar 2,37. Al
final del proceso, el PB inicial se transforma casi íntegramente en CI. El producto final es
una mezcla de PS, PB-g-PS y una pequeña fracción de PB residual.
Las reacciones involucradas en el proceso son: iniciación química y/o térmica,
propagación, transferencias al monómero, al hule, al modificador, y terminación por
combinación 20. A continuación, se detalla el mecanismo cinético global, el cual es una
extensión del mecanismo propuesto por Brydon et al. (1974) 13.

13
Mecanismo Cinético Global 2
Iniciación Química:
Iniciación Térmica:
Propagación:
Transferencia al Monómero:
Transferncia al PB o al Copolímero:
.2
2II d
k
..1
1 SStI ik
..0
2 PPI ik
.23
1
0 SSt ik
..1
n
pk
nSStS
..1
0
0PStP
pk
..1
n
pk
nPStP
..1
SSStSn
fmk
n
..1
SPStPfm
k
n
..10
SP
'
StPfm
k
..0
PSPSn
fhk
n
..0
PPPPfh
k
n

14
Transferencia al Modificador (X):
Terminacion por Combinación:
La nomenclatura utilizada en las ecuaciones del Mecanismo Cinético Global es la
siguiente2:
I2 : molécula de iniciador químico.
I⋅ : radical primario de iniciador.
X : molécula de modificador.
St : molécula de estireno
Sn : molécula de PS de longitud de cadena n.
S1⋅ : radical de estireno.
n
fxk
nSSSX
..1
PSPXfx
k
n
..1
PS
'
PXfx
k
..10
mn
tck
mnSSS
..
PSPtc
k
nn
..
PPPtc
k
nm
..
P
''
PPtc
k
n
..0
P
''
SPtc
k
n
..0
P
'
PPtc
k
..
00

15
Sn⋅ : homoradical de PS de longitud de cadena n.
P : molécula de PB o de copolímero PB-co-PS que contiene la mayoría de sus unidades Bd
no injertadas.
P0⋅ : radical primario de copolímero, generado por un ataque a una unidad de Bd sin
reaccionar de P .
P1⋅ : radical de copolímero, con una nueva cadena en crecimiento de unidades repetitivas
de St. Pn⋅ : radical no primario de copolímero con una nueva cadena en crecimiento de n
unidades repetitivas de St.
kd : constante de descomposición del iniciador.
Ki0 : constante de iniciación térmica.
ki1 , ki2 , ki3 : constantes de iniciación.
kp : constante de propagación.
k fm k´fm : constantes de transferencia de cadena al monómero.
k fh : constante de transferencia al hule.
k fX k´fX : constantes de transferencia al modificador.
ktc k´tc k”tc : constantes de terminación por combinación.
2.1.3 Proceso industrial de obtención de HIPS
Industrialmente el HIPS se produce por proceso en masa en continuo, donde básicamente
se polimeriza St en presencia de un mínimo de 6% y un máximo de 10 % en peso de PB
con la adición de pequeñas cantidades de iniciador (para producir el injerto al hule), de
solvente (para disminuir la viscosidad de la mezcla), en algunos casos de modificador (para
controlar los pesos moleculares) y otros aditivos 2.

16
La distribución del tiempo de residencia en los reactores, así como las condiciones de
reacción (velocidad de agitación, temperatura, viscosidad) juegan un papel importante en
la determinación del tamaño y la morfología de las partículas de hule y de las propiedades
finales del producto.
La producción continua del HIPS involucra las siguientes etapas: disolución,
prepolimerización, polimerización principal o finalización, volatilización o separación y
“pelletizado”. En la Fig. 4 se presenta un diagrama de flujo donde se representan las
diferentes etapas de la producción que se describen brevemente a continuación 2.
Figura 4. Diagrama de flujo para el proceso de obtención de HIPS en masa 2.
1) Disolución (Disolvedor y tanque de alimentación): En esta etapa se carga el St, el
hule (PB o copolímero de Bd) y el solvente en un tanque agitado (disolvedor). El
hule se disuelve en la mezcla St-solvente y se adicionan otros reactivos y aditivos
(como aceite, antioxidantes, agentes de transferencia, etc.). Luego, la carga del
disolvedor se transfiere al tanque de alimentación agitado, el cual alimenta al
reactor de prepolimerización 2.
2) Prepolimerización (Reactor de prepolimerización): La mezcla es polimerizada
hasta conversiones de aproximadamente 20-30%, bajo agitación, en presencia de
un iniciador químico, donde el reactor puede presentar diferentes configuraciones

17
como; reactores de tanque agitados, reactores torre, reactores agitados en
cascada (BASF) o reactores “loop” con mezcladores estáticos 2.
3) Finalización (Reactor de polimerización): Esta etapa ocurre en ausencia de
agitación y se alcanzan conversiones de aproximadamente 75-85%, a
temperaturas comprendidas entre 135 y 160 °C. Los reactores para esta etapa
presentan diseños muy variados, como por ejemplo del tipo intercambiadores
tubo-carcaza en serie, pero en todos los casos lo que se busca es maximizar la
conversión del monómero sin que disminuyan los pesos moleculares medios a
niveles indeseables 2.
4) Volatilización (Devolatilizador): En esta etapa, se separa el monómero residual y
el solvente mediante un proceso de volatilización, que consiste en calentar la
mezcla hasta aproximadamente 225°C en un intercambiador de tubo y carcaza o
precalentador, que se mantiene bajo vacío. Los vapores de solvente y St sin
reaccionar son separados, se condensan y reciclan al tanque de transferencia
inicial 2.
5) Pelletizado (Extrusora y Pelletizado): El polímero fundido se extruye en forma de
filamentos uniformes, lisos y elásticos. Dichos filamentos se enfrían mediante un
baño de agua, se secan, se pelletizan, y luego son clasificados según su tamaño, y
envasados para su despacho 2.

18
2.1.4 Desarrollo morfológico durante el proceso de Polimerización In situ.
El proceso de polimerización en masa In Situ se divide, según la evolución de la morfología
estable, en cuatro etapas: La etapa 1, se constituye de una solución homogénea de St/PB
(Polibutadieno en estireno) , mientras que las etapas 2, 3 y 4 se constituyen de soluciones
St/PB y St/PS (Poliestireno en estireno), es decir, soluciones heterogéneas. En la Figura 5
se muestra gráficamente la evolución de las diferentes etapas.
Etapa 1. Solución homogénea (St/PB) Al iniciar la polimerización, el sistema es
homogéneo. Conforme el St polimeriza y forma PS, a bajas conversiones (del 2% al 6%)
existe una separación de fases, por la incompatibilidad termodinámica entre el PS y el
hule.
Etapa 2. Separación de Fases (St/PS) y (St/PB). Esta inicia con la separación de fases,
donde la fase dispersa es rica en PS. La estabilidad de la dispersión se mejora en gran
medida mediante la formación del copolímero de injerto (CI).
Etapa 3. Inversión de Fases. A medida que avanza la polimerización, la cantidad de PS en
el sistema aumenta y la del estireno disminuye. Por lo tanto, la fracción en volúmen de la
fase rica en PS (St/PS) aumenta y la de la fase rica en PB (St/PB) disminuye. La etapa 3 se
alcanza cuando el volumen de ambas fases es similar y no se distingue cuál de las fases es
la dispersa y/o la continua, fenómeno conocido como co-continuidad de fases, durante
esta misma etapa y con la consecuente obtención de mayor volumen de PS, la fase St/PS
se transforma en la fase continua y la fase St/PB pasa a ser la fase dispersa. La etapa 3 se
conoce como inversión de fases, y es en esta donde se establece la morfología de este tipo
de materiales.
Las Etapas 1, 2 y 3 conforman la pre-polimerización y se llevan a cabo en presencia de
iniciador y con una adecuada agitación.
Etapa 4. Finalización. Esta última etapa implica la consolidación de la morfología
alcanzada en la inversión de fases. En esta etapa la polimerización continúa hasta la
conversión final, tratando de preservar la morfología, sin cambios en el número de

19
partículas y oclusiones, y procede a mayor temperatura (para favorecer la iniciación
térmica) y sin agitación para retener la morfología 22,23.
Figura 5. Etapas del proceso de polimerización del Estireno
y su respectiva evolución morfológica del HIPS
La termodinámica de la formación del HIPS ha sido estudiada por Johnston (1978) 2, 24,
donde, el proceso se describió en términos de equilibrio de fases y descomposición
espinodal bajo condiciones isotérmicas y fue representado en un sistema ternario que no
considera al CI.
El camino de reacción se representó con un vector que cruza el diagrama de fases PS-PB-
St. Este vector en un determinado momento intersecta a la curva binodal (que define el
límite de miscibilidad) y la curva espinodal (que define el límite de estabilidad difusional
en el sistema). Una vez dentro de la curva, el sistema se compone de dos fases: una rica
en PS y otra rica en hule.
En la Figura 6, se representa el desarrollo morfológico de la fase elastomérica en el
diagrama ternario PS/St/PB.
Etapa 1
Etapa 2 Etapa 3
(Inversión de Fases)
Etapa 4
C o n v e r s i ó n

20
Figura 6. Diagrama de fases para el sistema PS/St/PB y vector de reacción.
Punto A: comienzo de la polimerización, B: Separación de fases y C: período de inversión
de fases
2.1.5 Cambio en la viscosidad durante la inversión de fases
El período de inversión de fases se puede caracterizar experimentalmente por una
reducción de la viscosidad durante la reacción de polimerización (como se muestra en la
Fig. 7) 2 y ocurre alrededor de 20 - 25 % de conversión (cuando se utiliza PB como hule
precursor de la morfología) 2. La prepolimerización se lleva a cabo con una agitación
adecuada para que la inversión de fases y desarrollo de la morfología se faciliten.
En este periodo se generan emulsiones múltiples al quedar atrapadas gotas de solución de
PS/St dentro de las gotas de la fase elastomérica. El St continua polimerizando, generando
diferentes tipos de partículas con mono o múltiples oclusiones 2.
Jordhamo y colaboradores 25, desarrollaron una expresión semi-empírica para predecir la
inversión de fases en mezclas de PS-PB, donde se toma en cuenta no sólo los volúmenes
de fases, sino la viscosidad en ambas fases. El modelo reológico se basa en la siguiente
ecuación:
Ecuación 1.

21
Donde son las fracciones volumétricas y ηi las viscosidades de cada fase.
Figura 7. Cambio en la viscosidad durante la polimerización del St en presencia de PB.
2.1.6 Variables de síntesis
Las variables de síntesis como: Concentración y tipo de iniciador, velocidad de agitación,
concentración y tipo de hule precursor, principalmente; afectan la estructura final del
HIPS, es decir, inciden sobre el grado de injerto, la morfología de la fase elastomérica, el
peso molecular de la matriz de PS y el grado de entrecruzamiento; y por consiguiente
sobre las propiedades mecánicas.
Estas diversas variables o parámetros que influyen sobre la estructura final del HIPS, se
describen a continuación.

22
2.1.6.1 Concentración y tipo de iniciador
Cuando se emplean altas concentraciones de iniciador disminuye el peso molecular de la
fase de PS como consecuencia de la iniciación de mayor número de radicales
monoméricos, y además se genera un aumento en el número de injertos (los cuales
compatibilizan las fases), estabilizando las fases por la disminución en la tensión
interfacial, por lo que el tamaño de partícula disminuye 27.
Por otro lado, la eficiencia de los iniciadores para iniciar la reacción de copolimerización
de injerto depende de la estructura molecular y de la selectividad que éstos presentan.
Debido a la selectividad de los iniciadores, algunos tendrán mayor tendencia a iniciar la
reacción de copolimerización, y otros, mayor tendencia a iniciar la reacción de homo
polimerización
El tipo de iniciador también afecta sobre la morfología, así como propiedades mecánicas y
tiempos de prepolimerización 28. Algunos iniciadores producen partículas más pequeñas
de hule que otros iniciadores, debido a que generan mayor cantidad de injerto.
El tiempo de polimerización necesario también es dependiente del tipo de iniciador, por
ejemplo, con el uso de iniciadores monofuncionales de tipo peróxidos, como el BPO, se
emplean largos tiempos de prepolimerización y, por su parte, con iniciadores
multifuncionales se puede alcanzar la inversión de fases en menor tiempo, es decir,
menores tiempo de prepolimerización, manteniendo buenas propiedades mecánicas en el
producto final 28.
2.1.6.2 Concentración y tipo de elastómero
Los diferentes grados de HIPS se clasifican por su contenido de PB en: semi-resistentes al
impacto (3-6 % PB) y resistentes al impacto (6-10 % PB). La cantidad de elastómero que se
puede adicionar a un HIPS comercial obtenido por polimerización en masa se limita al 14%
en peso, debido a la alta viscosidad del medio. Sin embargo el criterio más útil
corresponde al contenido de gel (masa del PS ocluido + PS injertado en el PB + PB

23
empleado que no se injerto), el cual se obtiene respecto a la cantidad de hule utilizado, y
que por lo general es igual a tres o cuatro veces la cantidad de hule añadido, es decir la
fracción volumen de la fase elastomérica aumenta de 3 a 4 veces con respecto a la
fracción volumen del hule inicial 1,2,29.
La fracción de PS injertado en las cadenas de PB aumenta con la concentración de PB,
aunque la cantidad de PS injertado por unidad de masa de PB disminuye con la
concentración de PB, en otras palabras la cantidad de injerto total será mayor al haber
mayor cantidad de PB , pero el CI formado tendrá menor cantidad de injerto de PS sobre
el PB por unidad de masa al aumentar la cantidad de PB, por lo cual el CI formado a
mayores concentraciones de PB podría tener menor interaccion con la matriz de PS que
un CI formado a menor concentracion de PB Esto puede dar como resultado un
compatibilizador menos eficiente 8, 26.
2.1.6.3 Velocidad de agitación
La velocidad de agitación que está directamente relacionada con el esfuerzo de corte que
se aplica al sistema de reacción, juega un papel importante en la síntesis de HIPS ya que la
formación y determinación de tamaño de las partículas de hule esta en función del
esfuerzo de corte. Así mismo se ha encontrado que el tamaño de partícula puede
controlarse durante el periodo de inversión de fases mediante la velocidad de agitación, y
de forma más general, se ha visto que a mayor velocidad de agitación se obtiene un
tamaño de partícula menor y viceversa 31.
2.1.7 Relación de la estructura y composición con las propiedades mecánicas del HIPS
Como antes se mencionó, el HIPS es un sistema polimérico de dos fases, cuyas
propiedades finales y de procesamiento dependen de una relación compleja entre 7.
El peso molecular de la fase continua y la distribución de los pesos moleculares

24
El tipo de elastómero empleado
La relación entre volúmenes de las fases
El tamaño de partícula de elastómero y distribución de tamaño de partículas.
La estructura de la partícula de elastómero.
La densidad de reticulación polímero injertado.
Concentración de aditivos.
La relación entre la estructura y las propiedades de los HIPS se ilustra a continuación (Ver
Fig. 8):
Figura 8. Relación entre la estructura y las propiedades del HIPS 7
Por ejemplo, de la Figura 8 en la columna del tamaño de partícula se puede observar que
el aumento en este parámetro, disminuye por un lado la rigidez, mientras que la
tenacidad (asociada a la resistencia al impacto) se incrementa hasta un punto óptimo, que
generalmente se encuentra entre 2-5 m.

25
2.1.7.1. Peso Molecular de la Matriz
El peso molecular del PS afecta las propiedades mecánicas y reológicas. La relación entre
el peso molecular de la fase de PS y propiedades del polímero son similares a las del PS
homopolímero 7.
La viscosidad del fundido y la relación entre la arquitectura y procesabilidad son
controladas predominantemente por el peso molecular y, también, la distribución de
peso molecular. Incrementando el peso molecular de la fase continua de PS incrementa la
viscosidad del fundido.
2.1.7.2 Tipo de elastómero
En la mayoría de los casos el tipo de elastómero utilizado en la síntesis de HIPS es el PB,
aunque también pueden ser utilizados copolímeros de estireno/butadieno. La mayoría de
PBs comerciales usados presentan un peso molecular de 180 000 a 260 000 g/mol y son
de cadena larga ramificada para suprimir el flujo frio 7.
Los procesos de polimerización comercial producen un sistema polimérico que tiene un
elastómero incorporado y especies injertadas donde cadenas cortas de PS han sido unidas
a los dominios de hule.
Este anclaje de PS injertado en las partículas discretas de elastómero, produce cierta
compatibilidad entre las fases dispersa y continúa, dando lugar a una mayor resistencia al
impacto.
Cuando fuerzas externas actúan en el HIPS, las partículas de hule tienen una acción de
relajación de estrés. Para realizar este trabajo deben estar suficientemente ligados a la
matriz y tener cierta elasticidad. Esto significa que el hule usado debe ser capaz de ser
injertado y entrecruzarse. Ambas propiedades pueden ser controladas dentro de ciertos
límites por medio de la microestructura. Estos requisitos originan que se usen
predominantemente Polibutadienos medio-cis y algunas veces también alto-cis, ya que
estos proporcionan mejores propiedades a los HIPS 7.

26
2.1.7.3 Relación de volumen entre las fases
La relación entre los volúmenes de partícula (hule + oclusiones) y de la matriz de PS se
denomina relación de volúmenes de fases. Esta relación dependerá principalmente del
contenido de hule, del número y tamaño de las oclusiones y de la eficiencia de injerto 7. El
injerto en etapas tempranas de la polimerización convierte a la solución de hule en
estireno en una emulsión de una solución de PS/St, dispersado en la solución de PB/St.
Esta solución es lo suficientemente estable para que las gotas de solución de PS coalescan
solo parcialmente. De este modo, la fase de hule dispersa seguirá conteniendo inclusiones
de la solución de PS. Mientras más solución de PS es atrapada dentro de la fase de PB
después de la inversión de fases, mayor será la relación en volumen de las fases PB/PS,
donde el número y tamaño de las oclusiones dependerá de las condiciones de reacción.
2.1.7.4 Tamaño de partícula y distribución de tamaño de partícula
La distribución de tamaño de partícula de hule del HIPS está esencialmente determinado
por tres factores, a saber, el esfuerzo de corte del material durante la reacción e
inmediatamente después de la inversión de fase, la relación de viscosidad entre las fases
dispersa y la fase continua, y del grado de injerto (GI) y la tensión interfacial 7.
En cuanto el efecto de las relacion de viscosidades entre fases, Karam y Bellinger
demostraron que la deformación crítica de las gotas (deformación donde se descomponen
las gotas), alcanza un mínimo cuando la relación de viscosidad se aproxima a 1 7. Una
disminución de la viscosidad de la fase continua da como resultado un aumento en el
tamaño promedio de las partículas de la fase elastomerica y una reducción en la
estabilidad de la emulsión aceite en aceite. Respecto al GI, un aumento en el injerto da
lugar a partículas más pequeñas. Por su parte el efecto de la tensión interfacial, esta
relacionada con el GI, a mayor cantidad de GI hay menor tensión interfacial, por lo cual se
forman partículas mas pequeñas de la fase elastomérica.

27
La influencia del esfuerzo de corte se conoce desde hace algún tiempo. Con el aumento
del corte, el tamaño de partícula disminuye, es decir, el pico de distribución del tamaño de
partícula se desplaza a valores más bajos, y la proporción relativa de partículas más
pequeñas aumenta 7.
2.1.7.5 Grado de Injerto (GI) y contenido de gel (CG).
Un parámetro también importante a tomar en cuenta, es el grado de injerto (GI). Este se
define como la cantidad de PS unido química o físicamente (ocluido) a la fase del hule. El
grado de injerto influye sobre la estabilización entre las fases incrementando o
disminuyendo la tensión entre las mismas, lo cual influye en el tamaño de partícula de la
fase elastómerica 7.
El contenido de gel, CG, (fracción no soluble en tolueno) de poliestireno en el hule, indica
el volumen de fase del hule reticulado, aumenta la tenacidad hasta un punto (alrededor
del 30%) y luego es perjudicial. Por su parte, el Grado de hinchamiento, GH, (relación
volumétrica de un gel hinchado a su estado no hinchado), se asocia a la densidad de
entrecruzamiento del dominio reticulado, alcanzando una tenacidad óptima lograda
típicamente en valores alrededor del 12% de hinchamiento.
El objetivo de cualquier injerto es aumentar la eficiencia del hule es decir, la relación entre
el contenido de gel y el contenido de hule, y permitir que las partículas de hule se unan a
la fase de PS para asegurar la transmisión de fuerzas externas. El polímero de injerto actúa
como emulsificante y estabiliza las partículas de hule dispersas en el sistema bifásico 7.
2.1.7.6 Influencia del uso de aditivos
Los aditivos comúnmente utilizados en la producción de HIPS son los lubricantes y los
antioxidantes. El lubricante de elección es el aceite mineral, otras alternativas como son el
estearato de butilo y ésteres ftálicos no son capaces de mantener capacidades de
lubricación efectivas. Como un material inerte, el aceite mineral es agregado en la mayoría

28
de los casos a la alimentación de la polimerización, usualmente en concentraciones del 2-
3% en peso 32.
2.1.8 Morfologías de la fase elastomérica de los HIPS
Para lograr las propiedades mecánicas deseadas en un HIPS, se debe, principalmente,
controlar la morfología de la fase elastomérica (forma, tamaño y distribución de la fase
elastomérica en la matriz termoplástica). La morfología, como antes se ha mencionado,
se controla mediante la manipulación de las variables de síntesis, como: la temperatura de
reacción, velocidad de agitación, tipo y concentración de iniciador, tipo y contenido de
hule, entre otras 32.
Al manipular las variables de síntesis, es posible obtener diferencias estructurales en la
morfología de la fase dispersa, resultando en la obtención de diferentes tipos de
partículas (Ver Figura 9), las cuales dependiendo de su configuración (salami, núcleo-
coraza, puntos, varillas, lámelas, cerebro) y tamaño darán un buen reforzamiento o en su
caso proporcionarán mejores propiedades ópticas al material 32.
Figura 9. Estructuras morfológicas de las partículas elastoméricas presentes en los HIPS.

29
2.1.8.1 Morfologías más importantes en un HIPS
Las principales morfologías que se presentan en un HIPS y que comercialmente son
utilizadas son estructuras tipo “salame” (con múltiples oclusiones vítreas) o núcleo-coraza
(“core-shell”) monocluidas. Estas estructuras pueden observarse en la Fig. 10 que muestra
dos imágenes obtenida mediante microscopia electrónica de transmisión (TEM) 2. Las
diferencias estructurales y morfológicas entre las resinas de HIPS son consecuencia de
diferencias de proceso de producción y el contenido y el tipo de hule, por lo que puede
existir una amplia variedad de tipos de HIPS disponibles comercialmente. El tamaño de
partícula óptimo (más común) presenta un diámetro promedio (DP) de hasta 5 m y una
morfología del tipo salami. Esta morfología consiste en partículas de diferentes tamaños
con oclusiones de PS rodeadas por una membrana de hule, dispersas en una matriz de PS
y es típico de los HIPS obtenidos mediante el proceso de polimerización en masa In Situ
(Ver Fig. 10 a).
Figura 10. Imágenes obtenidas mediante Microscopía Electrónica de Transmisión (TEM) de
HIPS: a) Tipo Salame y b) Tipo núcleo coraza 1.
La morfología de los HIPS obtenida por polimerización en emulsión generalmente
presenta partículas esféricas de hule (puntos), no deseable, ya que deteriora la
transparencia y presenta una pobre resistencia al impacto. También se generan partículas
con una estructura de tipo núcleo coraza, es decir, partículas compuestas de un núcleo de

30
PS (núcleo o core) recubierto por una membrana de PB (coraza) (Ver Figura 10 b). Esta
morfología afecta significativamente a la transparencia de los HIPS, siendo una resina
semitransparente utilizada en la fabricación de envases. Las partículas de PS recubiertas
con hule son del orden 0.2 m de diámetro. Los HIPS con el tamaño de partícula de este
orden no presentan la misma resistencia a la fractura que los HIPS con morfología tipo
salami y tiene una resistencia al impacto apenas ligeramente superior a la del
homopolímero de PS 1.
2.1.8.2 Mecanismo de mejora al impacto
Los mecanismos implicados en el aumento a la resistencia al impacto del HIPS incluyen
principalmente la microfisuración (agrietamiento). Las partículas elastoméricas
promueven el crecimiento de microfisuras de deformación plástica, mejor conocidos
como “crazes” con mayor absorción y disipación de energía, por ende mayor resistencia al
impacto1. Dichos “crazes” se evidencian físicamente como un color blancuzco en la zona
de deformación. Si la concentración de esfuerzos no excede un límite que conduzca a la
formación de una fractura o grieta, el material conserva su integridad estructural. Así, la
concentración, el tamaño, la morfología y las propiedades de la fase elastomérica son muy
relevantes para el rendimiento final del material. En la Figura 11 se muestra
esquemáticamente lo que ocurre cuando un HIPS se somete a determinada energía de
impacto. Las partículas elastoméricas absorben la energía de impacto ya que primero,
actúan como iniciadores o concentradores de microfisuras (crazes) y segundo actúan
como terminadores de microfisuras, evitando la transformación rápida de microfisuras a
fractura 1.

31
Figura 11. Mecanismo de absorción de impacto de HIPS a través de microfisuramiento.
Cuando el HIPS se somete a una energía de impacto, parte de esta energía se disipa por el
hule en forma de calor, por la vibración térmica de los átomos y los movimientos de
relajación de los segmentos de la macromolécula. La energía restante es disipada con la
formación de varias microfisuras en la interfaz de la matriz de PS con el hule. Estas
microfisuras se propagarán hasta encontrar una nueva partícula elastomérica en la que de
nuevo se disipa la energía en forma de calor y nuevas microfisuras pueden ser nucleadas,
en un proceso de redistribución de la energía de impacto. 33-36 Los factores que aumentan
la formación de microfisuras, y por lo tanto la absorción de energía de impacto, son; una
buena adhesión de la fase elastomérica a la matriz, el tamaño de partícula correcto o el
alto contenido y la baja Tg del hule, la morfología esférica de las partículas elastoméricas y
el alto coeficiente de expansión térmica del hule 1.

32
2.2 Nanopartículas
Definiendo la nanoescala o el tamaño a la cual las partículas son consideradas
nanopartículas se tiene que deberán presentar un tamaño de 1-100 nm (en al menos una
de sus tres dimensiones), intervalo más aceptado para dimensionalizar los nanomateriales
38. En la Figura 12 se muestran nanopartículas que se encuentran en ese intervalo de
tamaño 39.
Figura 12. Escala de tamaños y nanopartículas en el intervalo comprendido entre 1 y 100
nm 39.
Dado al extremadamente pequeño tamaño y el consecuente gran número de electrones
libres en la superficie en las nanopartículas, las propiedades ópticas, eléctricas y
magnéticas, así como la buena reactividad química, de las nanopartículas son distintas
respecto a micropartículas (MPs) y materiales a gran escala 38.
Por otro lado, el sistema internacional de unidades, considera nanomateriales a partículas
de una dimensión menor a 10 nm 38 y muchos autores definen nanomateriales a
materiales de tamaños de varios cientos o incluso varios miles de nanómetros. En lo que
definitivamente están de acuerdo los científicos es que las extraordinarias propiedades de
los nanomateriales son resultado del efecto de tamaño cuántico a escala nanométrica, y
que el efecto es mayor en partículas con tamaños de 10 a 20 nm.

33
Respecto a la nanotecnología, definida como la manipulación y control de la materia a
escala nanométrica, se está extendiendo rápidamente a través de todos los campos vitales
de la ciencia y la tecnología como la electrónica, la industria aeroespacial, de defensa,
médica y dental. Esta también implica el diseño, la síntesis, caracterización y aplicación de
materiales y dispositivos en la escala nanométrica. Como antes se mencionó, en la
nanoescala, las propiedades físicas, químicas y biológicas son diferentes a la de la escala
atómica y al de las moléculas de la materia en escala micrométrica, por lo tanto, ofrece la
oportunidad de desarrollar nuevas clases de materiales avanzados que satisfagan las
demandas de aplicaciones de alta tecnología 40.
La síntesis y el ensamblado modular de nanopartículas permiten explotar sus propiedades
únicas, lo que puede llevar a nuevas aplicaciones en catálisis, electrónica, fotónica,
magnetismo, así como aplicaciones biológicas 41.
Tabla 2. Principales tipos de nanopartículas orgánicas 39
Nanopartículas Orgánicas
Nanopartículas poliméricas
Utilizadas en aplicaciones médicas. No obstante, presentan una biodistribución (distribución hacia órganos y tejidos) no específica y una baja capacidad de carga.
Dendrímeros
Macromoléculas definidas, que presentan una estructura altamente ramificada formada por un núcleo y múltiples ramas. Su estructura molecular les permite cargar diferentes agentes activos.
Liposomas
Nanosistemas esféricos formados por una bicapa lipídica que permiten la encapsulación de fármacos hidrófilos e hidrófobos. A pesar de estar ampliamente estudiados, una de sus principales desventajas es su rápida eliminación.
Micelas
Agregados coloidales de moléculas amfifílicas. Las micelas están formadas por una parte interna hidrófoba y una externa hidrófila que permite la internalización de fármacos de carácter apolar para su vehiculización.
Generalmente, las nanopartículas se pueden clasificar atendiendo a su naturaleza
orgánica u inorgánica 35. En las Tabla 2 y 3 se mencionan algunos tipos de nanopartículas
34
con apliaciones biológicas dentro de la clasificación orgánica e inorgánica,
respectivamente. Las nanopartículas orgánicas están formadas por materiales tales como
polímeros, estructuras repetitivas, bicapas lipídicas, mientras que los materiales
inorgánicos estarían formados por metales y materiales inertes como el dióxido de titanio,
la hidroxiapatita o la sílice 39.
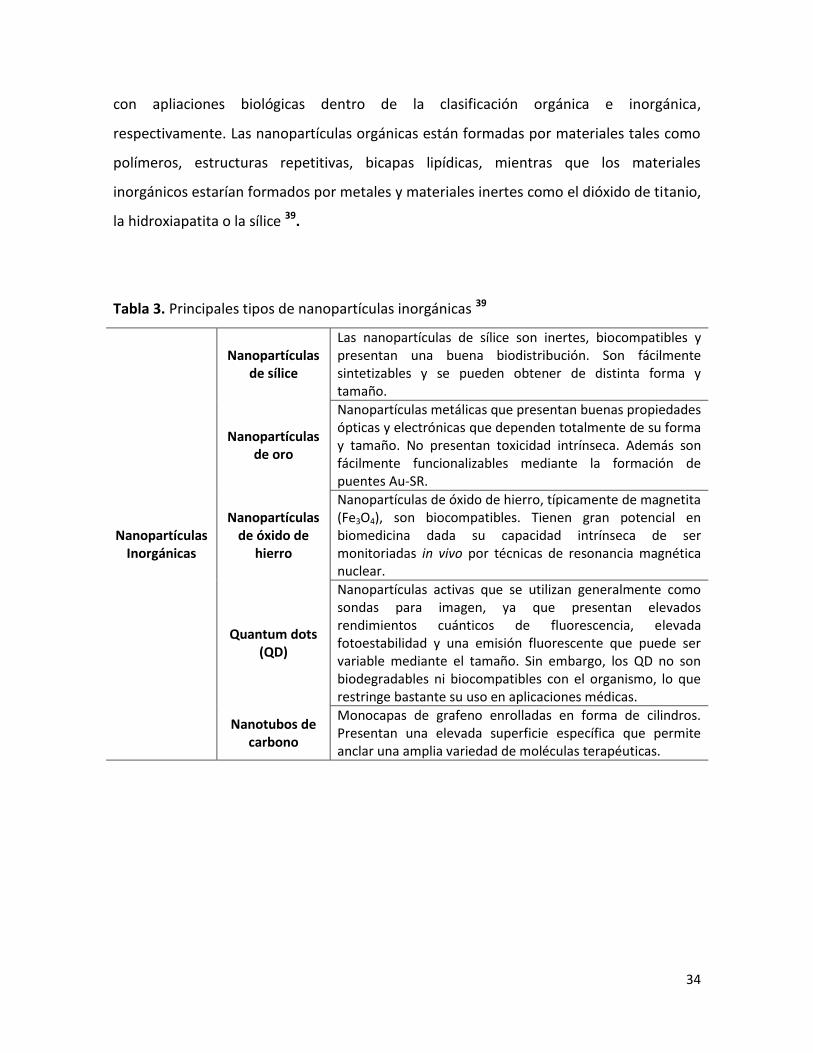
Tabla 3. Principales tipos de nanopartículas inorgánicas 39
Nanopartículas Inorgánicas
Nanopartículas de sílice
Las nanopartículas de sílice son inertes, biocompatibles y presentan una buena biodistribución. Son fácilmente sintetizables y se pueden obtener de distinta forma y tamaño.
Nanopartículas de oro
Nanopartículas metálicas que presentan buenas propiedades ópticas y electrónicas que dependen totalmente de su forma y tamaño. No presentan toxicidad intrínseca. Además son fácilmente funcionalizables mediante la formación de puentes Au-SR.
Nanopartículas de óxido de
hierro
Nanopartículas de óxido de hierro, típicamente de magnetita (Fe3O4), son biocompatibles. Tienen gran potencial en biomedicina dada su capacidad intrínseca de ser monitoriadas in vivo por técnicas de resonancia magnética nuclear.
Quantum dots (QD)
Nanopartículas activas que se utilizan generalmente como sondas para imagen, ya que presentan elevados rendimientos cuánticos de fluorescencia, elevada fotoestabilidad y una emisión fluorescente que puede ser variable mediante el tamaño. Sin embargo, los QD no son biodegradables ni biocompatibles con el organismo, lo que restringe bastante su uso en aplicaciones médicas.
Nanotubos de carbono
Monocapas de grafeno enrolladas en forma de cilindros. Presentan una elevada superficie específica que permite anclar una amplia variedad de moléculas terapéuticas.

35
2.2.1 Métodos para síntesis de nanopartículas Los métodos para obtener nanopartículas que se han utilizado se suelen agrupar en dos
categorias: “top-down” (de arriba hacia abajo) y “bottom-up” (de abajo hacia arriba) 41. La
primera consiste en la reducción de las dimensiones del tamaño original de sólidos
másicos en tamaños más pequeños. Este enfoque puede involucrar la molienda o el
desgaste, métodos químicos, y la volatilización de un sólido seguido por la condensación
de los componentes volatilizados.
La segunda categoría,” de abajo hacia arriba”, consiste en la fabricación de nanopartículas
a través de la condensación de átomos o entidades moleculares en una fase gaseosa o en
solución. Este último enfoque es mucho más popular en la síntesis de nanopartículas,
donde, las nanopartículas pueden ser soportadas o no. Dicho soporte da estabilidad a las
nanopartículas, además de que les puede conferir propiedades específicas.
La manipulación de las condiciones de síntesis permite el control racional de la morfología
de las partículas y provee los medios para adaptar las propiedades de los materiales
durante el proceso de síntesis. Otro aspecto fundamental de la síntesis de nanopartículas
es su estabilización, de tal manera que pueda mantenerse su tamaño y su forma en
función del tiempo.
Existen varios métodos que utilizan el enfoque “top-down” o “de arriba hacia abajo” 41, los
más representativos son:
a) Evaporación térmica
b) Deposición química en fase vapor (CVD)
c) Preparación de clusters gaseosos
d) Implantación de iones
e) Molienda
A excepción de la molienda, varios de los métodos que se utilizan en el enfoque “de arriba
hacia abajo” requieren de instrumentación compleja y complicada, lo cual los hace

36
costosos, y por tanto, muchas veces se prefieren los métodos que utilizan el enfoque “de
abajo hacia arriba”.
Existen diversos métodos que utilizan la metodología de “abajo hacia arriba” para la
síntesis de nanopartículas, los más empleados son aquellos que utilizan procedimientos
químicos. Por lo general, inician con la reducción de los iones metálicos a átomos
metálicos, seguido por la agregación controlada de estos átomos. El método químico es el
más conveniente para la obtención de nanopartículas uniformes y pequeñas.
Los métodos más representativos de esta metodología son 41 :
a) El método coloidal
b) Reducción fotoquímica y radioquímica
c) Irradiación con microondas
d) Utilización de dendrímeros
e) Síntesis solvotermal
f) Método sol-gel
En la Figura 13 se muestran imágenes obtenidas mediante SEM de nanpartículas
sintetizadas por el método de “abajo hacia arriba”, especificamente, por el método sol-
gel, el cual se caracteriza por la obtención de nanopartículas monodispersas en tamaño 37.
Figura 13. Imágenes de microscopía electrónica de barrido de pequeñas esferas de óxido
de silicio sintetizadas por el método sol-gel (Escala de la barra 1 m) 37.

37
2.2.2 Nanopartículas de sílice
Por su parte, la aplicación de nanopartículas de sílice (SiO2) como refuerzo en la
preparación de nanocompuestos de polímeros, ha llamado mucho la atención, debido a la
mayor demanda de nuevos materiales con prop1iedades térmicas, mecánicas, físicas, y
químicas mejoradas. Los recientes desarrollos en la síntesis de nanopartículas
monodispersas, con distribución estrecha de tamaños (Ver Figura 13), mediante el
método de sol-gel proporcionan un impulso significativo al desarrollo de nanocompuestos
poliméricos con sílice 40.
Comunmente, las partículas de sílice extraídas de recursos naturales contienen impurezas
metálicas y no favorables para aplicaciones científicas e industriales avanzadas. La sílice
sintética (sílice coloidal, geles de sílice, sílice pirogénica y sílice precipitada), es pura y se
produce en su mayoría en forma de polvo amorfo en comparación con sílice mineral
natural que está en formas cristalinas (cuarzo, tridimita, cristobalita) 40.
2.2.3 Nanopartículas comerciales de Sílice (AEROSIL)
En 1941 Degussa patentó un proceso de hidrólisis a alta temperatura de óxidos metálicos
para producir partículas de óxido extremadamente finas. Este proceso fue escalado a
producción en los 1950s y se ha convertido en el proceso para la preparación de
nanopartículas de dióxido de silicio, oxido de alumnio y dióxido de titanio. Bajo análisis
TEM, las partículas primarias de los tres óxidos muestran formas cubicas con esquinas
redondeadas. El dióxido de sílicio, que domina el mercado mundial, se presenta bajo la
marca comercial de AEROSIL™ 42.
El AEROSIL es sílice muy pura, altamente dispersa, amorfa, y con una distribución de
tamaño de partícula monodispersa producida por hidrolisis a alta temperatura de
tetracloruro de silicio en una llama de gas oxi-hidrógeno. Es un polvo esponjoso blanco
que consiste de partículas primarias de forma esférica.
* En este trabajo se utilizaran los términos SiO2 (dióxido de silicio) o sílice de manera indisitinta

38
Las partículas primarias interactúan a la flama para desarrollar agregados, que se unen de
forma reversible para formar aglomerados. La Figura 14 muestra una micrografía TEM de
AEROSIL 300 donde se observan partículas primarias, agregados, y aglomerados.
Figura 14. Micrografía TEM de AEROSIL 300 muestra las partículas en agregados 42.
Respecto a algunas propiedades del AEROSIL, este comienza a sinterizar y se convierte en
vidrio por encima de 1200 °C. La cristalización solo ocurre después de un tratamiento
térmico. El AEROSIL es casi insoluble en agua e insoluble en ácidos. Sin embargo se
disuelve en medios alcalinos fuertes para formar silicatos.
2.2.4 Síntesis de nanopartículas de sílice
Para producir nanopartículas de sílice generalmente se utiliza los métodos de “abajo hacia
arriba” o enfoque químico que implica una ruta común que se utiliza para producir
nanopartículas desde de la escala atómica o molecular 38.
Algunos de los métodos más utilizados para sintetizar nanopartículas de sílice son:
Microemulsión inversa, síntesis por condensación de vapor químico y el proceso sol-gel 40.

39
Síntesis por microemulsión inversa
En microemulsión inversa, las moléculas de los agentes tensoactivos disueltos en
disolventes orgánicos forman micelas esféricas. En presencia de agua, los grupos de
cabeza polares se organizan para formar microcavidades que contienen agua. Los
principales inconvenientes del enfoque microemulsión inversa son alto costo y las
dificultades en la eliminación de los tensoactivos en los productos finales 41.
Sin embargo, el método se aplicó con éxito para el recubrimiento de las nanopartículas
con diferentes grupos funcionales para diversas aplicaciones.
Síntesis por condensación de vapor químico (CVC)
Las nanopartículas de sílice también se pueden producir a través de la descomposición de
los precursores metal-orgánicos a alta temperatura por llama. Este proceso también se
conoce como la condensación de vapor químico (CVC) 39. En un proceso típico CVC,
nanopartículas de sílice se producen por reacción de tetracloruro de silicio, SiCl4 con
hidrógeno y oxígeno. La dificultad en el control del tamaño de partícula, la morfología y la
composición de la fase es la principal desventaja de la síntesis por llama. Sin embargo,
este es el método más destacado que se ha utilizado para producir comercialmente
nanopartículas de sílice en forma de polvo.
Síntesis por el proceso sol-gel
El proceso sol-gel se aplica ampliamente para producir sílice, vidrio, y materiales
cerámicos debido a su capacidad para formar productos puros y homogéneos en
condiciones suaves.
El proceso implica la hidrólisis y condensación de alcóxidos de metales (Si (OR) 4), tales
como ortosilicato de tetraetilo (TEOS, Si (OC2H5) 4) o sales inorgánicas tales como silicato
de sodio (Na2SiO3) en presencia de ácido mineral (por ejemplo, HCl) o base (por ejemplo,

40
NH3) como catalizador. Un diagrama general de flujo para el proceso de sol-gel que
conduce a la producción de sílice utilizando alcóxidos de silicio (Si (OR)4) se muestra en la
Figura 15 40,41.
Figura 15. Diagrama de flujo de proceso típico de sol-gel.
Las reacciones generales de TEOS que conduce a la formación de partículas de sílice en el
proceso de sol-gel se pueden escribir como 40 :

41
La hidrólisis de las moléculas TEOS forma grupos silanol. La condensación / polimerización
entre los grupos silanol o entre los grupos silanol y los grupos etoxi crea puentes de
siloxano (Si-O-Si) que forman toda la estructura del sílice. La formación de partículas de
sílice se puede dividir en dos etapas: la nucleación y el crecimiento. Dos modelos, la
adición de monómero y la agregación controlada, se han propuesto para describir el
mecanismo de crecimiento de sílice.
El modelo de adición del monómero describe que, después de una explosión inicial de
nucleación, el crecimiento de partículas se produce a través de la adición de monómeros
hidrolizadas, en la superficie de las partículas (primarias)40.
Por el contrario, el modelo de agregación elabora que la nucleación se produce
continuamente durante toda la reacción y los núcleos resultantes (partículas primarias) se
agregan entre sí para formar dímeros, trímeros, y partículas más grandes (partículas
secundarias). Ambos modelos conducen a la formación ya sea de red esférica o gel,
dependiendo de las condiciones de reacción.
2.3 Retardantes a la flama
2.3.1 Flamabilidad de polímeros
Los polímeros se utilizan en una amplia variedad de productos presentes en nuestra vida
diaria, como aparatos electrónicos, computadoras, muebles, ropas y vehículos. Sin
embargo, los polímeros son productos a base de hidrocarburos, lo que hace que sean
combustibles y/o flamables 43, 44,45.
Los materiales poliméricos, por lo tanto, plantean un riesgo de incendio en general. De
hecho, según las estadísticas proporcionadas por la Asociación Nacional de Protección
contra Incendios (NFPA, por sus siglas en inglés), los primeros artículos que típicamente se
incendian en una casa involucran productos hechos con algún polímero 43.

42
A pesar de que los polímeros representan un riesgo de incendio, no es ni práctico, ni
posible, eliminar el uso de polímeros; por lo que se buscan alternativas para proteger
estos materiales contra el fuego. La seguridad contra incendios de los materiales
poliméricos puede mejorarse aumentando la resistencia a la ignición, reduciendo la
liberación de calor, disminuyendo la cantidad de productos tóxicos y de humo, etc..
El uso de polímeros inherentemente ignífugos o polímeros térmicamente estables es una
manera potencial de reducir la posibilidad de una propagación del fuego y el consiguiente
daño asociado con el fuego. Sin embargo, debido al alto costo, esta alternativa no suele
ser una opción viable. Teniendo en cuenta los gastos involucrados y la facilidad de
procesamiento, la aplicación de aditivos retardantes de llama a los polímeros es una
manera eficaz de mitigar el riesgo de incendio 43.
2.3.2 Aditivos Retardantes a la flama
Los retardantes a la flama, son sustancias que tienen el objetivo de inhibir la combustión o
reducir su velocidad, de tal forma que la tasa de liberación de calor sea lo más baja
posible. Los retardantes a la flama mejoran el comportamiento al fuego de varias maneras
posibles, todas ellas relacionadas con los factores que afectan al mantenimiento o
autosostenimiento de la combustión 43, 44.
El ciclo de combustión autosostenido de un polímero se observa en la Figura 16 Cuando el
polímero se somete al fuego, e inicia la combustión, la acumulación de calor sobre el
material provoca la descomposición del polímero y la emisión de volátiles combustibles,
que al mezclarse con el oxígeno del aire darán el mayor aporte a la combustión. Cuando el
calor desprendido por la flama mantiene la velocidad de degradación del polímero por
encima de la requerida para mantener la concentración de combustible volátil, entonces,
se establece un ciclo de autosostenido de la combustión, ya que el suministro de energía
es constante y es igual a la energía requerida para llevar a cabo la combustión 45, 46, 47.

43
Figura 16. Esquema del ciclo de combustion autosostenido de un polímero
El polímero seguirá degradándose por el proceso de combustión hasta que uno o varios de
los factores limiten la continuidad del mismo, es decir el calor generado sea menor al
necesario para la ignición del combustible volátil, o se reduzca la cantidad de combustible
volátil o reducir la cantidad de oxigeno disponible para la combustión 45, 46, 47.
2.3.2.1 Mecanismo de Acción de Retardantes a la flama
Uno de los métodos, para retardancia o inhibición de flama, consiste en el uso de
inhibidores de llama (afectan a las reacciones radicalarias) como átomos de cloro, bromo
o fósforo. Otra opción supone la disposición de barreras al paso del oxígeno a la zona de la
llama o introducir elementos que previenen que el calor llegue al polímero, cortando así el
ciclo de combustión. Existe también la opción de modificar la estructura molecular de los
polímeros y, a consecuencia modificar la química de descomposición de estos. En la Figura
17 se muestra un diagrama esquemático con los diferentes mecanismos de acción de los
retardantes de flama y su efecto sobre el ciclo de combustión 44 ,45,46, 47.

44
En la Figura 17 se muestran las diferentes clasificaciones de los agentes retardantes a la
flama en función del mecanismo de retardo o inhibición de flama y el lugar en donde
actúa. Esta clasificación se detalla a continuación:
a. Barreras a la transferencia al calor
Reduce el flujo de calor al polímero para evitar la pirólisis. Esto puede conseguirse
mediante la introducción de un disipador de calor, que se descompone
endotérmicamente o disponiendo de una barrera, capa de “char” o
recubrimientos intumescente, que se forma cuando el polímero se expone a
condiciones de fuego.
Reduce el calor generado por debajo del requerido para mantener la combustión.
Figura 17. Esquema de los diferentes principios de accion de los retardantes a la flama.

45
Modificación química de la superficie
• Formación de recubrimientos protectores que constituyen barreras físicas al paso
de calor y/o sustancias volátiles inflamable lo que dificulta o inhibe el proceso de
combustión 46.
Inhibidores de Llama
Introduciendo en las formulaciones compuestos que liberen átomos de cloro o
bromo. El cloro y particularmente los átomos de bromo son inhibidores de llama
muy eficientes.
Aislando la llama del suministro de oxígeno / aire 46.
2.3.2.2 Hidróxido de Magnesio
Existen diversas clases de materiales que son útiles como retardantes de flama.
Comúnmente, los minerales inorgánicos, organofosfatos y compuestos halogenados se
utilizan en todos los polímeros por su capacidad para inhibir la combustión y el humo
generado durante la combustion de plásticos y otros materiales. Actualmente una
pequeña parte de este gran mercado, lo constituye el Hidróxido de Magnesio que está
atrayendo la atención debido a su rendimiento, precio, baja corrosividad, y baja
toxicidad48.
Dentro de los Hidróxidos metálicos usados como retardantes a la llama, el trihidróxido de
aluminio ( Al(OH)3 ) constituye la solución más empleada a la hora de mejorar las
propiedades de resistencia al fuego de las poliolefinas. Los niveles de adición típicos de
esta sustancia oscilan entre el 30 y el 60 % del peso total del material 46,47, 48.
El uso del hidróxido de magnesio ( Mg(OH)2 ) está reservado para las aplicaciones donde
se requiere una mayor estabilidad térmica que la del Al(OH)3 ya que su precio es más
elevado. Además, existen diversos grados en el mercado con diferentes propiedades lo
que se traduce en diferentes comportamientos en el material final 48.

46
Al igual que el Al(OH)3 , el Mg(OH)2 necesita de 30 a 60 % en peso, aproximadamente,
para proporcionar un efecto retardante a la llama notable, y conseguir una clasificación
UL94 V0 en poliolefinas; los tratamientos que se aplican a estas partículas junto con el
empleo de agentes sinergísticos, como el borato de zinc, pueden disminuir
considerablemente estas cantidades 46,47, 48.
2.3.2.3 Mecanismo de retardancia a la flama del Hidróxido de Magnesio
El Hidróxido de magnesio (Mg(OH)2), es un retardante de llama anti ácido y libre de
halógenos. Se descompone endotérmicamente cuando se calienta de acuerdo con el
siguiente mecanismo de reacción 46, 47 ,48.
Teóricamente, la fase gaseosa de agua envuelve la flama, reduciendo el oxígeno y
diluyendo los gases inflamables. Similar a la función del “char” formado por los
retardantes de llama que contienen fósforo. Un material aislante del calor se puede
formar en la superficie del plástico en contacto con la flama, reduciendo el flujo de
productos de descomposición, potencialmente inflamables, a la fase de gas, donde
ocurren las reacciones de combustión, los productos de descomposición no son tóxicos y
el MgO es alcalino, reduciendo la probabilidad de que salgan gases ácidos del plástico 48.
En la Tabla 4 se muestran las propiedades físicas y químicas del hidróxido de magnesio y
Al(OH)3. El hidróxido de magnesio presenta una temperatura de descomposición 100 °C
mayor que Al(OH)3, (230 °C) lo que permite su procesamiento a una temperatura mayor
mediante extrusión del compuesto polimérico. Además, el hidróxido de magnesio adsorbe
mayor energía durante el proceso de descomposición.
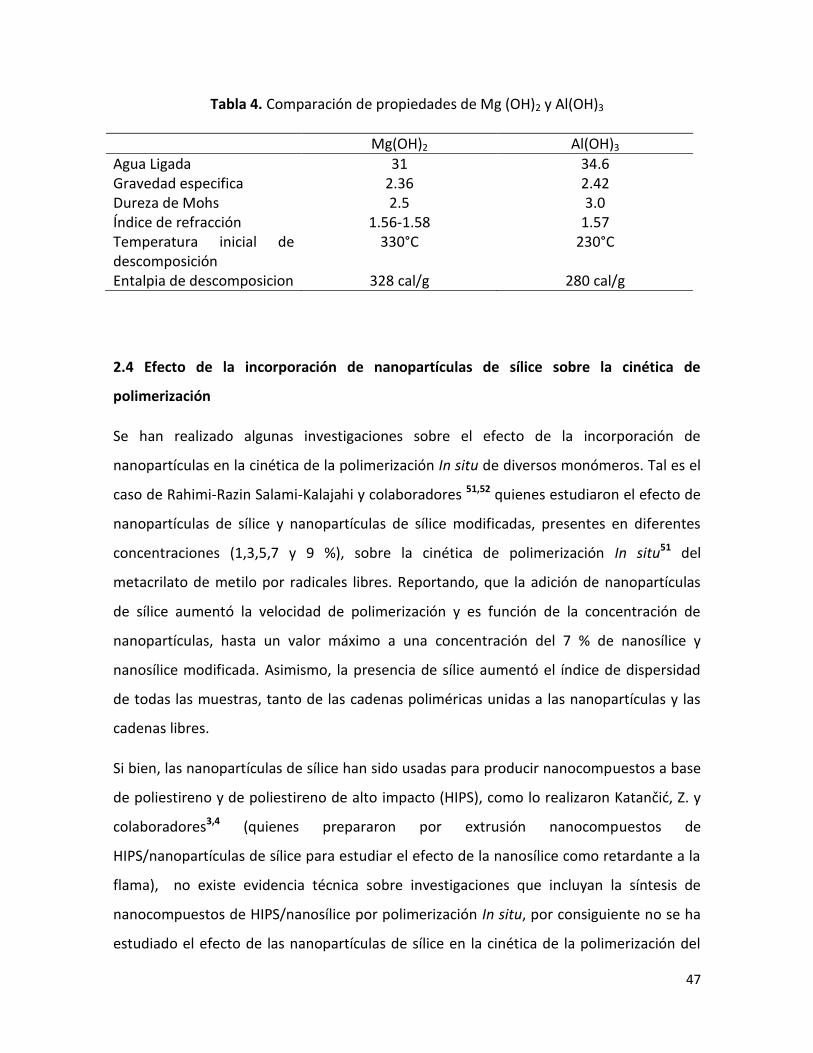
47
Tabla 4. Comparación de propiedades de Mg (OH)2 y Al(OH)3
Mg(OH)2 Al(OH)3
Agua Ligada 31 34.6 Gravedad especifica 2.36 2.42 Dureza de Mohs 2.5 3.0 Índice de refracción 1.56-1.58 1.57 Temperatura inicial de descomposición
330°C 230°C
Entalpia de descomposicion 328 cal/g 280 cal/g
2.4 Efecto de la incorporación de nanopartículas de sílice sobre la cinética de
polimerización
Se han realizado algunas investigaciones sobre el efecto de la incorporación de
nanopartículas en la cinética de la polimerización In situ de diversos monómeros. Tal es el
caso de Rahimi-Razin Salami-Kalajahi y colaboradores 51,52 quienes estudiaron el efecto de
nanopartículas de sílice y nanopartículas de sílice modificadas, presentes en diferentes
concentraciones (1,3,5,7 y 9 %), sobre la cinética de polimerización In situ51 del
metacrilato de metilo por radicales libres. Reportando, que la adición de nanopartículas
de sílice aumentó la velocidad de polimerización y es función de la concentración de
nanopartículas, hasta un valor máximo a una concentración del 7 % de nanosílice y
nanosílice modificada. Asimismo, la presencia de sílice aumentó el índice de dispersidad
de todas las muestras, tanto de las cadenas poliméricas unidas a las nanopartículas y las
cadenas libres.
Si bien, las nanopartículas de sílice han sido usadas para producir nanocompuestos a base
de poliestireno y de poliestireno de alto impacto (HIPS), como lo realizaron Katančić, Z. y
colaboradores3,4 (quienes prepararon por extrusión nanocompuestos de
HIPS/nanopartículas de sílice para estudiar el efecto de la nanosílice como retardante a la
flama), no existe evidencia técnica sobre investigaciones que incluyan la síntesis de
nanocompuestos de HIPS/nanosílice por polimerización In situ, por consiguiente no se ha
estudiado el efecto de las nanopartículas de sílice en la cinética de la polimerización del

48
HIPS. Aunque para nanocompuestos de poliestireno/nanosílice, existen algunos estudios
sobre el efecto de la presencia de nanopartículas de sílice en la cinética de polimerización
In situ del estireno 51.
En este sentido, Lopez y colaboradores49 sintetizaron materiales híbridos por
polimerización In situ de estireno con diferentes concentraciones de nanopartículas de
sílice mesoporosa (0, 5, 10 y 15%), utilizando AIBN como iniciador. En la experimentación
que realizaron, la presencia de nanopartículas de sílice mesoporosa en la polimerización
de estireno afecto el peso molecular; obteniendose pesos moleculares menores en los
nanocompuestos que en el poliestireno puro. Observaron además, que la presencia de
nanopartículas durante la síntesis del poliestireno incrementa la viscosidad del sistema
limitando los procesos difusionales. Por tanto en los materiales reforzados la terminación
de la polimerización se da probablemente por transferencia y no por combinación, por lo
que se obtuvieron menores pesos moleculares 51.
Por otro lado, Bera y colaboradores 53 realizaron una investigación sobre la polimerización
In situ del estireno en presencia de nanopartículas de sílice. Para llevar a cabo este
estudio, prepararon una serie de nanocompuestos por polimerización In situ de estireno
con diferentes contenidos de sílice (1, 3 y 5 % en peso) con un tamaño promedio de
partícula de 7 nm. Mientras la cinética de reacción fue seguida mediante calorimetría
diferencial de barrido isotérmica (DSC) a 70, 80 y 90°C. Estos autores, llegaron a la
conclusión de que la adición de nanopartículas de sílice no afecta las constantes de
velocidad de reacción, y que estas solo dependían de la temperatura a la cual se realizaba
la reacción 53.
Por su parte, Sobani y colaboradores (2012) 54 estudiaron la cinética de la polimerización
de estireno mediante polimerización por radicales libres y por adición fragmentación
reversible (RAFT) con diferentes concentraciones de partículas de sílice nanoporosas. La
conversión de monómero, el peso molecular y el índice de dispersidad de cada sistema de
polimerización se monitorearon durante la polimerización para investigar la cinética de la
reacción. De acuerdo a los resultados que obtuvieron en ambos sistemas de

49
polimerización, por radicales libres y RAFT, resulto en la disminución de la velocidad de
reacción, conversión y peso molecular, dado que la máxima velocidad de polimerización
fue observada en muestras sin partículas de sílice. Según esta investigación, la
disminución en la velocidad de polimerización con una mayor incorporación de partículas
se ha atribuyó a una menor estabilidad del sistema debido a la formación de agregados.
Además, los radicales requieren de movimiento, para la búsqueda de otros radicales libres
y monómeros, teniendo que vencer las resistencias de los reactivos y partículas de sílice
nanoporosa. Por lo tanto, en un medio con gran cantidad de nanopartículas de sílice, el
movimiento de los macroradicales se suprime, lo que conduce a la reducción de la
velocidad de polimerización y de la conversión. Otro factor que provocó la reducción en la
cinética de polimerización en presencia de las nanopartículas de sílice, según Sobani 54, se
atribuyó a la ligera disminución en la concentración de monómero en masa por absorción
física de monómero en las partículas de sílice. Lo anterior también reduce la movilidad de
las cadenas de polímero (aumento en la viscosidad del sistema) y disminuye la velocidad
de propagación y por consiguiente hay una disminución en los valores de peso molecular.
Por otro lado, Sobani y colaboradores, encontraron en las muestras sintetizadas por
radicales libres, que el aumento de valores de índice de dispersidad con la conversión fue
debido a fenómenos físicos. El aumento de conversión conduce a un aumento de la
viscosidad del sistema, lo que ejerce un mecanismo de polimerización controlado por la
difusión en el medio y en consecuencia, conduce a un aumento de valores de índice de
dispersidad. Mientras que en los sistemas donde se utilizó RAFT, la adición de partículas
de sílice nanoporosa no tuvo ningún efecto significativo en los valores de índice de
dispersidad.
Otro trabajo donde se investigó el efecto de nanopartículas de sílice sobre la cinética de
polimerización In situ de estireno fue el hecho por Khezri y Mahdavi 55. Ellos estudiaron el
efecto de la incorporación de nanopartículas hidrofóbicas de sílice sobre la polimerización
radicálica por transferencia de átomo (ATRP) In situ de estireno. Se obtuvó como
resultado que al adicionar el 3% en peso de nanopartículas de sílice disminuye la velocidad

50
de reacción, el peso molecular de las cadenas de poliestireno disminuyó de 11,095 hasta
8,755 g.mol-1 y el valor del índice de dispersidad se incrementó de 1.15 a 1.46.
La variación que estos autores reportan sobre la conversión, los pesos moleculares, y los
valores de índice de dispersidad mediante la adición de sílice aerogel modificada la
atribuyeron a varias razones:
i) Las nanopartículas de sílice actúan como una impureza, por lo que aumenta la
transferencia de cadena y la terminación de las reacciones de los radicales que
se propagaban, y por lo tanto amplió la distribución del peso molecular de los
polímeros resultantes.
ii) La movilidad de los radicales que crecieron en la solución fue restringida por la
sílice nanoporosa aerogel especialmente a mayores cargas. Por lo tanto, los
valores de la velocidad de polimerización y de conversión disminuyen por la
adición de contenido de aerogel de sílice nanoporoso.
iii) Se observó en los datos cinéticos la reacción irreversible entre los radicales
crecientes y los restos funcionales en la superficie de sílice nanoporosa aerogel
lo que redujó la eficiencia de la polimerización.
De acuerdo a las diversas investigaciones antes descritas existe evidencia importante del
efecto que puede presentar la incorporación de sílice en sistemas reactivos,
independientemente de la naturaleza de los mismos. Sin embargo, también sigue
existiendo polémica sobre la forma en la que pueden actuar dichas nanopartículas sobre
la cinética de polimerización, lo que dió la pauta para establecer una buena estrategía en
el presente trabajo de investigación.

51
2.5 Nanosílice como retardante a la flama
En búsqueda de nuevos materiales que sirvan como retardantes a la flama, varios grupos
de investigación han encontrado que la sílice como carga en nanocompuestos ayuda a
mejorar la estabilidad térmica 3-6, 43, 49, en algunos polímeros presenta un efecto
retardante a la flama 50, en presencia de un agente retardante a la flama tradicional, la
sílice presenta un efecto sinérgico 3-6. Dado a que es mejor estudiar el efecto retardante a
la flama en un sistema polimérico específico, en este caso HIPS, ya que lo que funciona en
un sistema polimérico no es necesariamente tan eficiente en otro, a continuación se
presentan investigaciones donde se estudio el efecto como retardante a la flama de
nanopartículas de sílice en compuestos con base de HIPS y PS homopolímero.
2.5.1 Nanocompuestos de polímeros estirénicos / Sílice (SiO2)
Respecto a nanocompuestos a base de polímeros estirénicos, se han estudiado
propiedades mecánicas, térmicas y de flamabilidad por grupos de investigadores, como el
de Katančić y colaboradores 3,4, quienes estudiaron el efecto de la sílice como co-agente
retardante a la llama en el HIPS. Ellos realizaron dos estudios, preparando en ambos casos
nanocompuestos de HIPS/nanopartículas de Sílice/agente retardante a la flama, por
extrusión; en un estudio utilizaron polifosfato organico 3 como agente retardante a la
flama, y en el otro estudio utilizaron hidróxido de aluminio Al (OH)3 y fosfato de difenil 2-
etilhexilo (DPO) como agente retardante a la flama4. En ambos estudios encontraron que
la sílice presenta un efecto sinérgico con los agentes retardantes a la llama tradicionales.
Por su parte, el grupo de Feng Yang y colaboradores 5,6, prepararon nanocompuestos de
PS/nanopartículas de Sílice, encontrando que la sílice proporciona una mejora en la
estabilidad térmica, pero por si sola no presentó una mejora en la resistencia a la flama,
sin embargo, en combinación con poliestireno bromado, se obtuvo un efecto retardante a
la flama utilizando menos poliestireno bromado del utilizado convencionalmente,
logrando mejores propiedades mecánicas.

52
En otro estudio realizado por López y colaboradores 49, se sintetizaron materiales híbridos
por polimerización In situ de estireno con nanopartículas de sílice mesoporosa, variando
su contenido entre 0-15%. Se observó que los nanocompuestos presentaron buenas
propiedades térmicas y mecánicas debido a la alta área superficial y la estructura porosa
de la nanosílice que puede atrapar cadenas de polímero dentro de sus poros aumentando
su interacción.
Ahmed y colaboradores 43 sintetizaron nanocompuestos basados en PS y sílice,
preparados mediante polimerización In situ, con cargas de 1 y 3 % de nano sílice,
obteniendo como resultado un aumento del 80 % en la formación de “char”, existiendo
también una mejora significativa en la estabilidad térmica. Sin embargo, con el aumento
del contenido de nanosílice, aumentó la presencia de aglomerados de nanosícile y se
observó una reducción significativa en la dureza.
2.6 Modificación superficial de nanopartículas mediante plasma generado por
radiofrecuencia
Como en la mayoría de los nanocompuestos poliméricos, un problema recurrente es la
dispersión y distribución de las nanoestructuras en la matriz polimérica y el caso de las
nanopartículas de sílice no es la excepción. De aquí que existan diversos métodos para la
modificación superficial. Entre estos métodos se pueden mencionar los siguientes: i)
método físico, consistente en la adición de surfactantes o bien agentes compatibilizantes
de las nanopartículas con la matriz polimérica y ii) métodos químicos donde materia
orgánica es injertada iónica y/o covalentemente sobre las nanopartículas. Un método
novedoso que ha ganado interés entre la comunidad científica es la modificación de
nanopartículas mediante plasma de radiofrecuencia. La tecnología de plasma ofrece una
nueva alternativa para funcionalizar diferentes tipos de nanopartículas y promete ser una
excelente técnica debido a sus atributos versatilidad, rapidez y favorable al medio
ambiente. Este método es conocido como polimerización por plasma, se pueden emplear

53
gases polimerizables donde es posible elegir un gas con un grupo funcional en particular,
con el fin de mejorar la compatibilidad y la dispersión en matrices poliméricas.
La técnica se basa en disociar un gas en moléculas ionizadas y radicales, esto mediante un
campo electromagnético generado por radiofrecuencia bajo condiciones de vacío56. Estas
moléculas pueden ocasionar en la superficie de las nanopartículas erosión y/o remoción
y/o depósito, este último se ve favorecido al utilizar gases polimerizables. Empleando esta
metodología es posible formar capas y/o depósitos poliméricos más delgados (de hasta 1
nm) que con otras técnicas. Lo anterior, permite que las nanopartículas tratadas se anclen
fuertemente a la matriz polimérica debido a la delgada capa de polímero formada en la
superficie de las partículas, la que puede encontrarse anclada física y/o covalentemente.

54
CAPITULO III. Justificación
Considerando los avances tanto científicos como tecnológicos, hoy en día, se requiere
realizar estudios enfocados a la obtención de nuevos y/o mejores materiales poliméricos
funcionales. Hasta ahora el uso de poliestireno de alto impacto (HIPS) ha presentado una
disminución en diversas aplicaciones debido a la presencia del polipropileno y del ABS en
el mercado. Para desarrollar nuevas características funcionales del HIPS sobre sus
competidores, se requiere la generación de conocimiento básico que siente las bases para
poder alcanzar dicho objetivo. En este sentido, existe muy poca investigación respecto a la
elaboración de nanocompuestos de HIPS con agentes retardantes a la flama mediante el
proceso de polimerización In situ; por lo cual es de suma importancia realizar estudios
sistemáticos sobre la síntesis de HIPS (mediante el proceso de polimerización In situ en
masa) en presencia de agentes retardantes a la flama, que permita, en este caso, elucidar
el efecto de la incorporación de nanopartículas de sílice y micropartículas de hidróxido de
magnesio sobre la cinética de polimerización. Cabe mencionar que la cinética de
polimerización de estireno en presencia de hule se encuentra directamente relacionada
con el desarrollo morfológico de este tipo de materiales, a través de diversos fenómenos
termodinámicos que se presentan durante la polimerización como lo es, la separación de
fases, la co-continuidad de fases, y la inversión de fases donde se establece la morfología,
por lo que la influencia de parámetros externos como la inclusión de nanopartículas de
sílice y micropartículas de hidróxido de magnesio puede modificar la morfología final del
HIPS, y a su vez, la resistencia al impacto de los nanocompuestos finales.

55
CAPITULO IV. Hipótesis
Se considera que la incorporación de materia inorgánica, nanopartículas de SiO2 y
micropartículas de Mg(OH)2, durante la polimerización In situ modificará la cinética de
polimerización, disminuyendo la velocidad de reacción y por lo tanto modificará la
morfología de la fase elastomérica típica de este tipo de materiales, lo que repercutirá
sobre la resistencia al impacto final de los HIPS nanocompuestos.
Adicionalmente se espera que la presencia del SiO2 presente un efecto sinérgico en cuanto
a las propiedades de retardancia a la flama en conjunto con el Mg(OH)2, cuando se
incorporan ambos materiales durante la polimerización In situ.

56
CAPITULO V. Objetivos
De acuerdo al protocolo de tesis y a lo brevemente expuesto en la introducción en este
documento, se trazó el objetivo general y los objetivos específicos de esta investigación.
5.1 Objetivo general
Estudiar y analizar el efecto de la incorporación de nanopartículas de sílice y
micropartículas de Mg(OH)2 sobre la cinética de polimerización, por radicales libres, de
estireno en presencia de hule. Adicionalmente se observa el comportamiento de
retardancia a la flama de los nanocompuestos de HIPS/SiO2/Mg(OH)2 obtenidos “In situ”.
5.2 Objetivos Particulares
Modificar nanopartículas de sílice mediante el proceso de polimerización de
estireno por plasma de radiofrecuencia.
Caracterizar morfología, composición química, y estabilidad térmica de
nanopartículas de SiO2 modificadas y sin modificación superficial.
Analizar el efecto de la incorporación de nanopartículas de SiO2 y Mg(OH)2 durante
la polimerización por radicales libres de estireno en presencia de hule, sobre la
cinética de polimerización.
Caracterizar el desarrollo morfológico, estabilidad térmica, retardancia a la flama y
resistencia al impacto de los nanocompuestos HIPS/SiO2/Mg(OH)2.

57
CAPITULO VI. Metodología
La parte experimental se dividió en dos secciones; la primera es la modificación superficial
de nanopartículas de SiO2 y, la caracterización de nanopartículas de SiO2 y micropartículas
de Mg(OH)2 ; la segunda, la síntesis y caracterización de nanocompuestos de HIPS.
Respecto a los reactivos utilizados en este desarrollo, se utilizó monómero de estireno (St)
de Poliformas plásticas S.A. de C.V., Polibutadieno medio-cis de Dynasol, Peróxido de
benzoílo de Sigma-Aldrich, nanopartículas de SiO2 con un área superficial específica de
120 m2/g de Nanostructured & Amorphous Materials Inc. y micropartículas de Mg(OH)2 de
Marinswerk GmbH (MAGNIFIN H-5 HV) modificadas superficialmente con un tratamiento
polimérico, un área superficial específica de 2-5 m2/g y una estructura cristalina regular.
6.1 Caracterización Morfológica por TEM de nanopartículas de SiO2 y Micropartículas de
Mg(OH)2
Para el análisis de las nanopartículas de SiO2 (con y sin modificación superficial con
estireno) y micropartículas de Mg(OH)2 por TEM se utilizó un microscopio TITAN 80-300
kV, para lo cual las partículas se dispersaron en acetona mediante ultrasonido, durante 30
minutos.
Las nanopartículas fueron depositadas en una rejilla Lacey Carbon 300 Mesh Copper
mediante la inserción en la solución de acetona que contenia las partículas.
Posteriormente, la rejilla se dejó secar durante 15 min. A las imágenes obtenidas
mediante TEM de las partículas, se realizó la medición del eje menor y mayor con el
programa ImageJ, para obtener el diámetro promedio aparente.

58
6.2 Caracterización de micropartículas de Mg(OH)2 por Microscopía electrónica de
barrido (SEM)
La caracterización morfológica de las partículas de Mg(OH)2 , se realizó mediante
Microscopía Electrónica de Barrido (SEM), en un microscopio electrónico de barrido de
emisión de campo (FESEM), JEOL JSM-7401F. Las partículas se colocaron sobre una cinta
de carbón y se recubrieron con Au/Pd. Estas fueron observadas a magnificaciones de
50000X, 100000X y 150000X.
6.3 Modificación por plasma frio de nanopartículas comerciales de SiO2
Para realizar la modificación superficial se utilizó un sistema de polimerización de plasma
frio con vacío, para polimerizar el monómero de estireno en la superficie de las
nanopartículas de sílice.
El sistema de polimerización de plasma frio consta de las siguientes partes (ver Figura 18):
1) Fuente de energía de 13.56 MHz a 600 W, 2) Controlador de radio frecuencia, 3)
Contenedor de monómero, 4) Evaporador rotatorio, 5) Reactor de plasma de 500 ml en el
cual se adicionan 2 g de las partículas a modificar, 6) Bobina de cobre, 7) Medidor de
presión, 8) Válvula de control general, 9) Trampa de vacío y 10) Bomba de vacío.
Figura 18. Sistema de polimerización por plasma.
El tratamiento superficial Se realizó durante 1 hora, con una presión de vacío estabilizada
de 1.4X10-1 mbar a 45 rpm.

59
6.4 Análisis por Infrarrojo (IR) de nanopartículas de SiO2 y Micropartículas de Mg (OH)2
Se utilizó espectroscopia infrarroja (IR) para observar los grupos funcionales, presentes en
las nanopartículas de SiO2 con y sin modificación superficial y micropartículas de Mg(OH)2,
para verificar la presencia del recubrimiento superficial base estireno. La preparación de la
muestra consistió en triturar finamente cierta cantidad de partículas con KBr grado
espectroscópico para formar una pastilla que fue utilizada para el análisis. Los espectros
de IR se obtuvieron en un espectrofotómetro Thermo Nicolet NEXUS 670 FT-IR usando un
detector DTGS en rango extendido en el intervalo de 4000 a 400 cm-1 con una resolución
de 0.09cm-1.
6.5 Análisis termogravimétrico TGA
Con la finalidad de establecer el porcentaje en peso de modificación en las nanopartículas,
se utilizó un analizador termogravimétrico (TGA) de TA Instruments en un intervalo de
temperaturas de 30 a 600 °C en atmósfera de nitrógeno, posteriormente, de 600 a 800°C
se introdujo oxígeno para eliminar el residuo carbonoso, generado por la materia orgánica
de la modificación, en ambos casos se utilizó un flujo de 50 mL/min.
6.6 Síntesis de HIPS y nanocompuestos
El proceso de polimerización In situ se llevó a cabo en dos etapas:
En la primera, conocida como prepolimerización, se genera PS homopolímero y
copolímero de injerto (CI) y se dispersan las partículas elastoméricas en la matriz
de PS, ensamblándose y estableciéndose la morfología final (ya que en esta fase se
termina de generar el CI y, principalmente, porque ocurre la inversión de fases).
Una vez que se estableció la morfología en la primera etapa, y que las partículas
son estabilizadas por la alta viscosidad de la matriz y el copolímero de injerto,

60
procede la segunda etapa de polimerización. Esta etapa se realizó en masa estática
a una temperatura de 155°C. Bajo estas nuevas condiciones de reacción se
continúa con la homopolimerización y tiene lugar el entrecruzamiento de la fase
hulosa y se establecen los pesos moleculares, de modo que la partícula se fija a la
matriz para soportar los procesos futuros.
Formulación
Para elaborar los nanocompuestos, el HIPS se polimerizó en presencia de nanopartículas
de Mg(OH)2 y de SiO2. Para sintetizar el blanco de HIPS ( HIPS B ) se utilizó la siguiente
formulación: [St] = 90.9% en peso, [PB] = 8% en peso y [BPO] = 0.1% en peso. Para los
nanocompuestos se conservó esta misma proporción de estos reactivos y se añadieron las
nanopartículas en las concentraciones planteadas en las formulaciones diseñadas.
Las nanopartículas SiO2 y micropartículas Mg(OH)2 se agregaron al cargar el reactor a las
concentraciones indicadas en la tabla 5:
Tabla 5. Concentraciones de nanopartículas en los HIPS nanocompuestos
Formulación
[SiO2 NP´s]
% en peso 1
[Mg(OH)2 MP´s] %
en peso 2
[SiO2 NP´s mod. sup.]
% en peso 3
HIPS B 0 0 0
HIPS S1 2.5 0 0
HIPS S2 5 0 0
HIPS M1 0 10 0
HIPS M2 0 20 0
HIPS S1 Mod 0 0 2.5
HIPS S2 Mod 0 0 5
1 SiO2 NP´s : Nanopartículas de SiO2,
2Mg(OH)2 MP´s : Micropartículas de Mg(OH)2,
3SiO2 NP´s mod. sup. :
Nanopartículas de SiO2 modificadas superficialmente con plasma de estireno.

61
La concentración de la adición combinada de las nanopartículas de SiO2 y micropartículas
de Mg(OH)2 se muestra en la tabla 6.
Tabla 6. Concentraciones de nanopartículas SiO2 y micropartículas Mg(OH)2 combinadas en nanocompuestos
Formulación
[SiO2 NP´s] %
en peso
[Mg(OH)2 MP´s] %
en peso
HIPS M1-S1 2.5 10
HIPS M1-S2 5 10
HIPS M2-S1 2.5 20
HIPS M2-S2 5 20
Las nanopartículas de SiO2 modificadas superficialmente y las micropartículas de Mg(OH)2
se adicionaron en la concentración indicada en la tabla 7
Tabla 7. Concentraciones de nanopartículas SiO2 modificadas y micropartículas Mg(OH)2 combinadas en nanocompuestos
Formulación [SiO2 NP´s mod. sup.]
% en peso
[Mg(OH)2 MP´s] %
en peso
HIPS M1-S1 Mod 2.5 10
HIPS M1-S2 Mod 5 10
HIPS M2-S1 Mod 2.5 20
HIPS M2-S2 Mod 5 20
Proceso de Polimerización
La reacción de Pre-polimerización se llevó a cabo en un reactor Parr de acero inoxidable
con capacidad de 1L dispuesto de un sistema de agitación ancla-turbina.; se cargó con

62
estireno (St), PB , y nanopartículas comerciales de SiO2 y micropartículas comerciales de
Mg(OH)2.
Para sintetizar el HIPS B se adicionaron 276 gramos de St y 24 gramos de PB en el reactor,
18 horas antes de llevar a cabo la reacción, para disolver el hule en el estireno. Se montó
el reactor en el sistema de transmisión de agitación, y a través del controlador se inició el
calentamiento para llegar a 90°C con una agitación de 100 rpm (Fig. 19). Cuando se llegó a
la temperatura fijada de 90 2 °C, se inició la polimerización adicionando el BPO, se
controló esta temperatura durante la reacción y durante un lapso de aproximadamente 3
horas se tomaron muestras hasta llegar a un 25% de conversión (20- 25% de conversión es
el rango en que se encuentra reportada la inversión de fases para HIPS utilizando como
elastómero PB medio cis).
Figura 19. Sistema de Pre-polimerización
Una vez alcanzada la conversión donde ocurre la inversión de fases, la mezcla en reacción
(de St, PS, PB y CI de Bd-g-St) fue colocada en frascos de vidrio y colocados en un reactor
Parr de 1 galón donde se continuó la polimerización en masa estática durante 18 horas a
155°C hasta terminar la reacción de polimerización ( Conversión ≈ 1)

63
En la polimerización de HIPS con nanopartículas In situ, las nanopartículas se agregaron al
cargar el reactor, junto con el St y el PB.
Para sintetizar el HIPS con nanopartículas se agregaron los reactivos necesarios para
preparar 300 gramos de HIPS (276 gr de St y 24 gramos de PB) y la concentración de micro
y/o nanopartículas establecida para cada formulación.
6.7 Preparación de probetas para impacto y flamabilidad
Los distintos HIPS tanto el HIPS B como aquellos preparados con diferentes
concentraciones de nanopartículas se trituraron en un molino de cuchillas. El material fue
secado en una estufa durante 30 minutos a 100 °C.
Se prepararon placas de 15 cm × 15 cm y espesor de 3 mm (3 × 150 × 150 mm3) mediante
moldeo por compresión a una temperatura de 185 °C y una carga máxima de 15
toneladas.
Posteriormente, las placas fueron cortadas en una sierra de cinta, de acuerdo a las
especificaciones requeridas en la norma ASTM D-256 y la norma UL 94HB, para la prueba
de resistencia al impacto y de flamabilidad, respectivamente.
6.8 Resistencia al Impacto IZOD
La resistencia al impacto de las probetas de los HIPS se determinó utilizando un medidor
de impacto CSI modelo 137, siguiendo la metodología descrita en la norma ASTM D-256.
6.9 Prueba de Flamabilidad
La prueba de flamabilidad se basó en la norma UL 94HB para la prueba de combustión
horizontal. Antes de iniciar la prueba las probetas se acondicionaron durante 48 horas a
una temperatura de 23°C y a una humedad relativa de 50 %. En esta prueba se determinó

64
el tiempo de quemado de una muestra de plástico fijada horizontalmente, en contacto
con una flama de 20 mm de altura con mechero Bunsen durante 30 segundos.
La probeta es marcada de la manera que se muestra en la figura 19:
Figura 20. Esquema de probeta para prueba de flamabilidad
Dentro del estándar UL 94, la prueba de combustión horizontal UL 94HB, en general, es la
más fácil de aprobar, siendo así, la clasificación HB la más baja de todas las de el
estándar UL 94.
La clasificación HB indica que el material puesto a prueba se quema a una velocidad
menor que el máximo especificado (75 mm/min o 3 in/min cuando la probeta tiene un
espesor de 3mm), o cuando la flama se extingue antes de la línea final.
Los materiales con clasificación HB se consideran “autoextinguibles”. Dado a que se
utilizaron bajas concentraciones de agentes retardantes a la flama, se decidió utilizar la
prueba UL 94HB, ya que con estas se podrían apreciar los cambios en la velocidad de
quemado por los efectos de retardancia a la flama.
6.10 Contenido de Gel (CG) y Grado de Injerto final aparente (GI)
Para determinar el contenido de gel se disolvió 1 gramo de producto final de HIPS en 20
ml de tolueno dejándolo disolverse durante 24 horas. Después, se centrifugó utilizando
una ultracentrífuga Allegra 64 R de Beckman Coulter a 20,000 rpm y una temperatura de –
18 °C durante 90 minutos. El líquido sobrenadante (fracción de PS libre) se separó por
1 in 3 in
Parte donde se sujeta
la probeta
Línea inicial Línea final

65
decantación y posteriormente se precipitó en metanol y se utilizó para determinar la
distribución de pesos moleculares.
El gel hinchado se secó en una estufa a 60°C hasta peso constante y se determinó el CG
gravimétricamente.
Teniendo determinado el contenido de gel se empleó la ecuación de Gasperowicks 57,58
para determinar el Grado de injerto de los materiales:
Ecuación 2.
Donde
6.11 Cromatografía de exclusión por tamaño (SEC).
Para determinar la distribución de pesos moleculares se empleó cromatografía de
permeación en gel utilizando un equipo Alliance GPC 2000 Series, dispuesto de tres
columnas Ultrahydrogel. Las muestras fueron disueltas, previamente, en tetrahidrofurano
(THF) en una relación de 1 mg/ml (7mg de muestra en 7 ml de THF), después de ser
disueltas se filtraron utilizando filtros de 0.2 m. Se empleó como solvente de transporte
THF grado cromatográfico a una temperatura de 40°C. Para la curva de calibración se
emplearon estándares de PS.
6.12 Caracterización Morfológica por STEM de Nanocompuestos
La morfología de los nanocompuestos de HIPS se analizó mediante microscopía
electrónica de transmisión y barrido (STEM), en un microscopio electrónico FEI modelo
TITAN 80-300. Las muestras analizadas fueron primero cortadas utilizando un
ultramicrotomo LEICA Ultracut, dispuesto con cuchilla de diamante , a una temperatura de
-120°C ,con una velocidad de corte de 2 mm/min, con el propósito de evitar la

66
deformación de la morfología de la fase elastómerica, y con un espesor de corte de 90 nm
aproximadamente. Los cortes fueron depositados en una rejilla Lacey Carbon 300 Mesh
Copper.
Para contrastar la fase elastómerica de la fase de poliestireno, los cortes fueron teñidos
con tetróxido de osmio de osmio (OsO4) durante 1 h. El diámetro de partícula aparente
(Dp) y fracción volumen (Φ), parámetros característicos de la morfología, fueron
determinados a partir de las imágenes obtenidas por TEM utilizando el software
analizador de imágenes Image J.

67
CAPITULO VII. Resultados y discusión
7.1 Caracterización de partículas de Mg(OH)2 y de SiO2 sin modificación y modificadas
7.1.1 Microscopia electrónica de transmisión (TEM) y de barrido (SEM)
A fin de corroborar la morfología y composición de los materiales de partida se realizó un
análisis mediante TEM. En la Figura 21 se muestran las imágenes obtenidas mediante TEM
tanto del SiO2 como del Mg(OH)2, en estas se observa una morfología quasi-esférica de las
nanopartículas de SiO2 (Figura 21a) con un diámetro promedio de 15 nm, mientras que en
la Figura 21b se denota una morfología hexagonal para las partículas de Mg(OH)2, con una
distribución de tamaño de partícula amplia en un intervalo de 150 nm hasta 1500 nm, con
un promedio de tamaño de partícula de 864 nm.
(a) (b)
Figura 21. Imágenes obtenidas mediante TEM de a) nanopartículas esféricas de SiO2 y b)
nanopartículas hexagonales de Mg(OH)2.
Por otro lado, considerando la transmisión de electrones a través de la muestra de
Mg(OH)2 donde se observó la presencia de partículas traslapadas, se dedujo un morfología
de tipo disco hexagonal, lo cual se corroboró mediante su observación por microscopia
electrónica de barrido (SEM).

68
En la Figura 22, se muestra la imagen correspondiente al Mg(OH)2, donde se observa que
evidentemente la morfología de este material es de tipo disco y presenta una distribución
de espesores en el intervalo de 50-200 nm, donde dicha distribución es función del
tamaño de partícula, es decir, nanopartículas con mayor tamaño presentan espesores de
aproximadamente 200 nm.
Figura 22. Imagen obtenidas mediante SEM de las nano/micropartículas de Mg(OH).
Del mismo análisis por TEM utilizando difracción de electrones se estableció una
microestructura completamente amorfa de las nanopartículas de SiO2. Mientras que para
el caso de las nano/micropartículas de Mg(OH)2, se presentó una estructura cristalina de
tipo trigonal de acuerdo a la medición de los valores dimensionales de los planos
cristalográficos (d), donde el valor principal de dicha estructura fue de 0.243
correspondiente al plano cristalino (h, k, l) 1, 0, 1.
7.1.2. Análisis por espectroscopia infrarroja (FTIR).
Como se mencionó en la sección 6.3, las nanopartículas de sílice se modificaron mediante
el sistema de polimerización por plasma frío, utilizando St como monómero, para
depositar una capa de PS en la superficie. Para verificar la modificación superficial de las
nanopartículas de SiO2, se caracterizaron antes y después del tratamiento por plasma frio
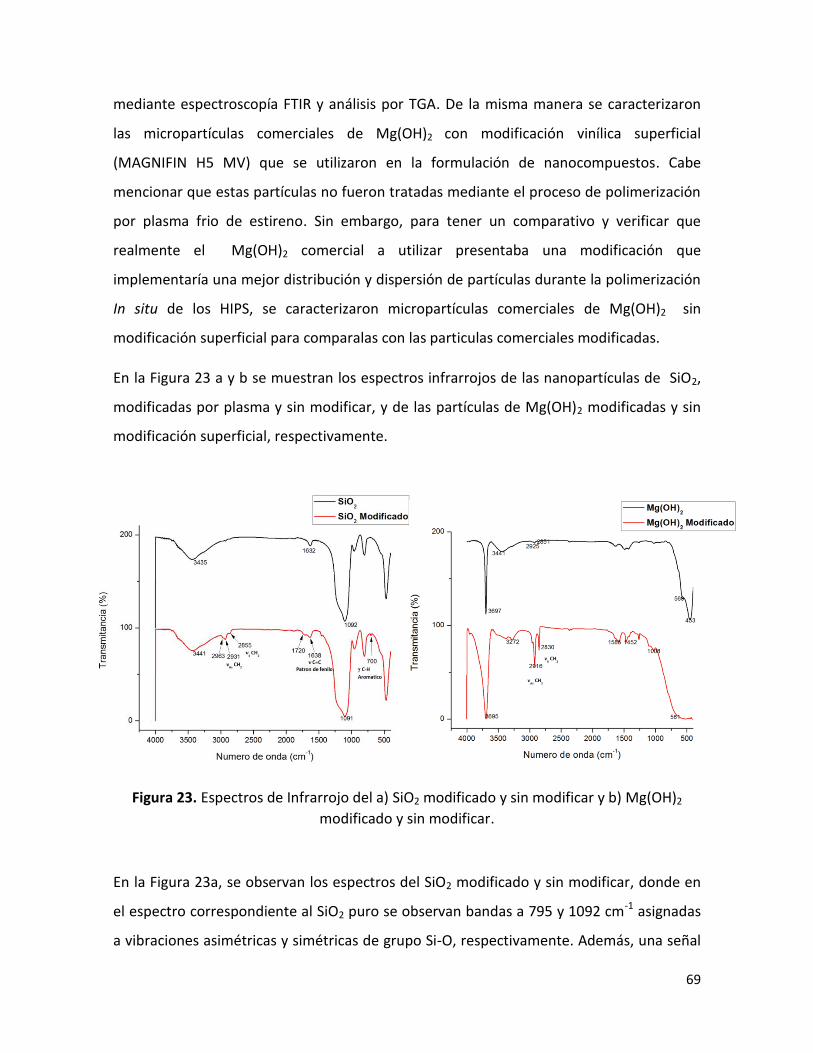
69
mediante espectroscopía FTIR y análisis por TGA. De la misma manera se caracterizaron
las micropartículas comerciales de Mg(OH)2 con modificación vinílica superficial
(MAGNIFIN H5 MV) que se utilizaron en la formulación de nanocompuestos. Cabe
mencionar que estas partículas no fueron tratadas mediante el proceso de polimerización
por plasma frio de estireno. Sin embargo, para tener un comparativo y verificar que
realmente el Mg(OH)2 comercial a utilizar presentaba una modificación que
implementaría una mejor distribución y dispersión de partículas durante la polimerización
In situ de los HIPS, se caracterizaron micropartículas comerciales de Mg(OH)2 sin
modificación superficial para comparalas con las particulas comerciales modificadas.
En la Figura 23 a y b se muestran los espectros infrarrojos de las nanopartículas de SiO2,
modificadas por plasma y sin modificar, y de las partículas de Mg(OH)2 modificadas y sin
modificación superficial, respectivamente.
Figura 23. Espectros de Infrarrojo del a) SiO2 modificado y sin modificar y b) Mg(OH)2
modificado y sin modificar.
En la Figura 23a, se observan los espectros del SiO2 modificado y sin modificar, donde en
el espectro correspondiente al SiO2 puro se observan bandas a 795 y 1092 cm-1 asignadas
a vibraciones asimétricas y simétricas de grupo Si-O, respectivamente. Además, una señal

70
a 950 cm-1 correspondiente a la vibración asimétrica del grupo Si-OH. Asimismo, se
observa una señal entre 3300 y 3500 cm-1 correspondiente al grupo O-H asociado a agua
fisicamente absorbida, este mismo grupo se puede corroborar con una vibración a 1635
asociada a agua molecular en el SiO2 59,60. Respecto a las nanopartículas de SiO2
modificadas, en la misma Figura 6b se observan las mismas bandas correspondientes al
SiO2 sin modificar y adicionalmente, se observan bandas a 2931 y 2855 cm-1 características
del estiramiento asimétrico del grupo CH2, así como una señal a 1638 cm-1
correspondiente al grupo fenilo del estireno depositado sobre la superficie de las
nanopartículas de SiO2.
Lo anteriormente descrito demuestra cualitativamente la presencia de la modificación
realizada mediante polimerización por plasma de estireno sobre las partículas estudiadas.
Por su parte, en la Figura 23b se observan los espectros correspondientes a las partículas
de Mg(OH)2, sin y con modificación superficial, en las primeras se observan señales de
1420, 1490 y 3700 cm-1 que se asocian a los grupos hidroxilos (OH), típicamente presentes
en las partículas de Mg(OH)2 61.
En el espectro correspondiente a las partículas de Mg(OH)2 comerciales modificadas
utilizadas, se muestran nuevas señales en 2916 y 2830 cm-1 correspondientes a
estiramientos simétricos y asimétricos de grupos CH2 presentes ahora en la muestra
debido a la modificación superficial con tratamiento polimérico. En este mismo espectro,
se presenta una banda de poca intensidad a 1005 cm-1, correspondiente alquenos
monosustituidos, que son parte del polímero utilizado para la modificación superficial de
este Mg(OH)2.
7.1.3. Análisis termogravimétrico (TGA)
Con el fin de evaluar la cantidad de poliestireno depositado sobre las nanopartículas de
SiO2, estas se evaluaron mediante TGA. En la Fig. 24a se muestra el termograma obtenido
mediante TGA del SiO2 puro y modificado superficialmente. En el SiO2 sin modificación
(línea negra) se observa una pérdida de masa total del 7.98 % en peso, esta pérdida se
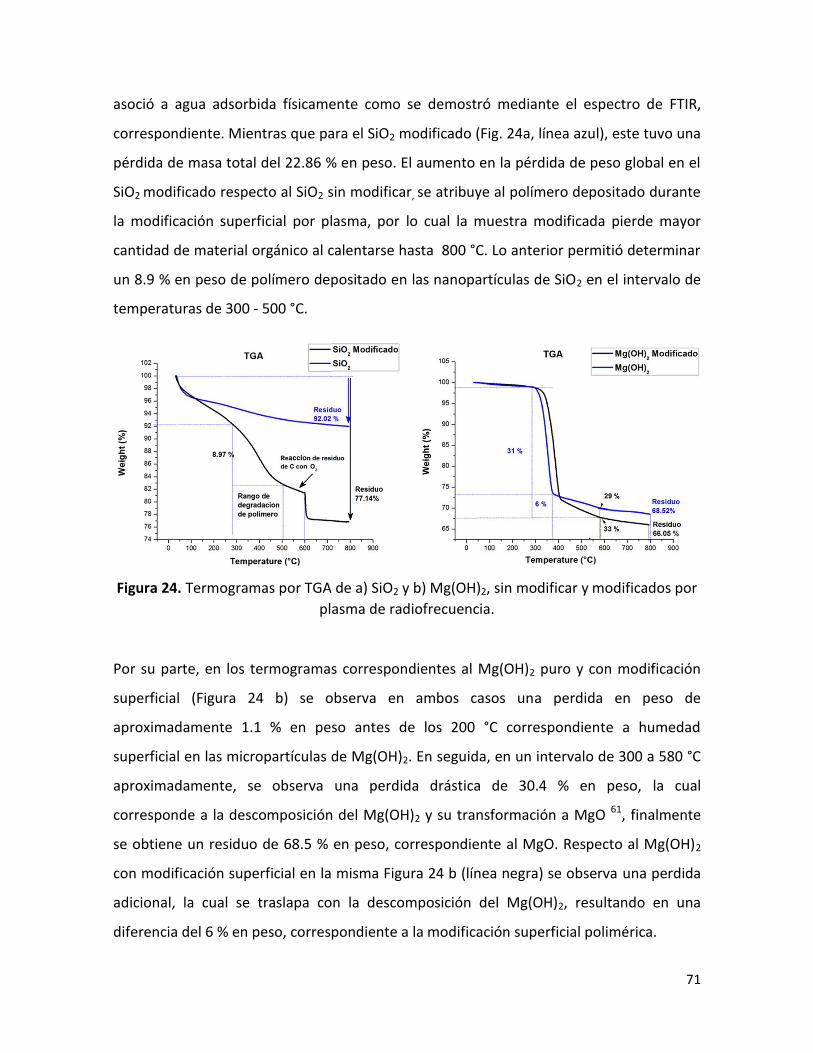
71
asoció a agua adsorbida físicamente como se demostró mediante el espectro de FTIR,
correspondiente. Mientras que para el SiO2 modificado (Fig. 24a, línea azul), este tuvo una
pérdida de masa total del 22.86 % en peso. El aumento en la pérdida de peso global en el
SiO2 modificado respecto al SiO2 sin modificar, se atribuye al polímero depositado durante
la modificación superficial por plasma, por lo cual la muestra modificada pierde mayor
cantidad de material orgánico al calentarse hasta 800 °C. Lo anterior permitió determinar
un 8.9 % en peso de polímero depositado en las nanopartículas de SiO2 en el intervalo de
temperaturas de 300 - 500 °C.
Figura 24. Termogramas por TGA de a) SiO2 y b) Mg(OH)2, sin modificar y modificados por
plasma de radiofrecuencia.
Por su parte, en los termogramas correspondientes al Mg(OH)2 puro y con modificación
superficial (Figura 24 b) se observa en ambos casos una perdida en peso de
aproximadamente 1.1 % en peso antes de los 200 °C correspondiente a humedad
superficial en las micropartículas de Mg(OH)2. En seguida, en un intervalo de 300 a 580 °C
aproximadamente, se observa una perdida drástica de 30.4 % en peso, la cual
corresponde a la descomposición del Mg(OH)2 y su transformación a MgO 61, finalmente
se obtiene un residuo de 68.5 % en peso, correspondiente al MgO. Respecto al Mg(OH)2
con modificación superficial en la misma Figura 24 b (línea negra) se observa una perdida
adicional, la cual se traslapa con la descomposición del Mg(OH)2, resultando en una
diferencia del 6 % en peso, correspondiente a la modificación superficial polimérica.

72
7.2 Síntesis del HIPS y nanocompuestos de HIPS
En esta sección se abordaran los resultados obtenidos durante el proceso de
polimerización In situ del HIPS y de los materiales compuestos obtenidos, discutiendo
fundamentalmente, el efecto de la presencia de nanopartículas de SiO2 y micropartículas
de Mg(OH)2 sobre la cinética de polimerización de St.
Como se mencionó en la metodología para sintetizar los compuestos de HIPS, por
polimerización In situ, se utilizaron nanopartículas de SiO2, sin y con modificación
superficial (mediante el proceso de polimerización por plasma frio de estireno) y
micropartículas comerciales de Mg(OH)2 modificadas superficialmente.
Cabe señalar que si bien ya se han hecho investigaciones del efecto de la incorporación de
nanopartículas de SiO2 durante la polimerización In situ de estireno sobre la cinética de
polimerización y sobre las propiedades de los nanocompuestos de PS finales, no hay
estudios que evidencien el efecto de la presencia de SiO2 y Mg(OH)2 durante la
polimerización In situ del HIPS sobre la cinética de polimerización y su repercusión en las
propiedades morfológicas y mecánicas de los HIPS finales. Importante mencionar que la
síntesis de HIPS es más compleja que la síntesis del PS ya que implica reacciones de homo
y copolimerización, así como la formación de una fase dispersa debido a la presencia de
hule.
7.2.1 Conversión en función de tiempo de polimerización
En la Figura 25 se muestra la conversión en función del tiempo de reacción de la síntesis
del HIPS B. Cabe mencionar que para establecer el tiempo de reacción al cual se alcanza el
20 % de conversión se trazó una línea base punteada, perpendicular al eje “Y”
correspondiente a un 20 % de conversión. Proyectando esta línea pero ahora
perpendicular al eje “X” correspondiente al tiempo de reacción, se puedo establecer el
tiempo requerido para alcanzar el 20 % de conversión durante la etapa de
prepolimerización. En el caso del HIPS B se requirieron 80 minutos de reacción para
alcanzar al 20 % de conversión. Cabe mencionar que este tiempo de reacción fue
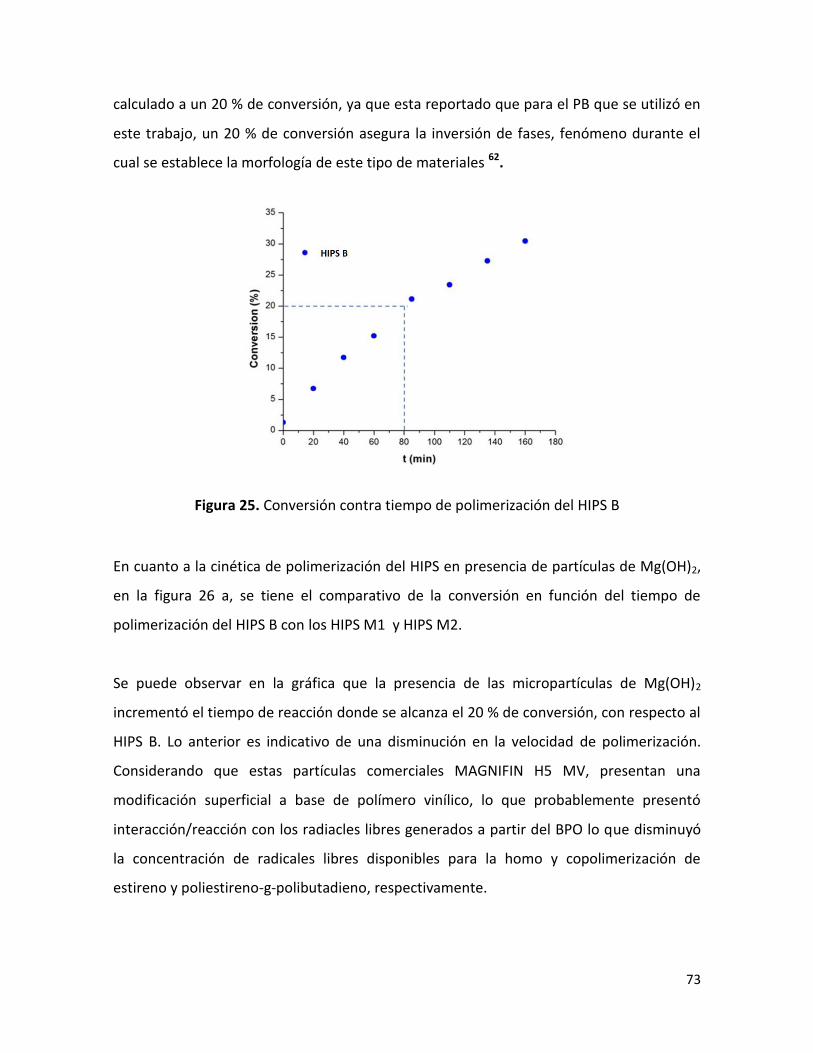
73
calculado a un 20 % de conversión, ya que esta reportado que para el PB que se utilizó en
este trabajo, un 20 % de conversión asegura la inversión de fases, fenómeno durante el
cual se establece la morfología de este tipo de materiales 62.
Figura 25. Conversión contra tiempo de polimerización del HIPS B
En cuanto a la cinética de polimerización del HIPS en presencia de partículas de Mg(OH)2,
en la figura 26 a, se tiene el comparativo de la conversión en función del tiempo de
polimerización del HIPS B con los HIPS M1 y HIPS M2.
Se puede observar en la gráfica que la presencia de las micropartículas de Mg(OH)2
incrementó el tiempo de reacción donde se alcanza el 20 % de conversión, con respecto al
HIPS B. Lo anterior es indicativo de una disminución en la velocidad de polimerización.
Considerando que estas partículas comerciales MAGNIFIN H5 MV, presentan una
modificación superficial a base de polímero vinílico, lo que probablemente presentó
interacción/reacción con los radiacles libres generados a partir del BPO lo que disminuyó
la concentración de radicales libres disponibles para la homo y copolimerización de
estireno y poliestireno-g-polibutadieno, respectivamente.
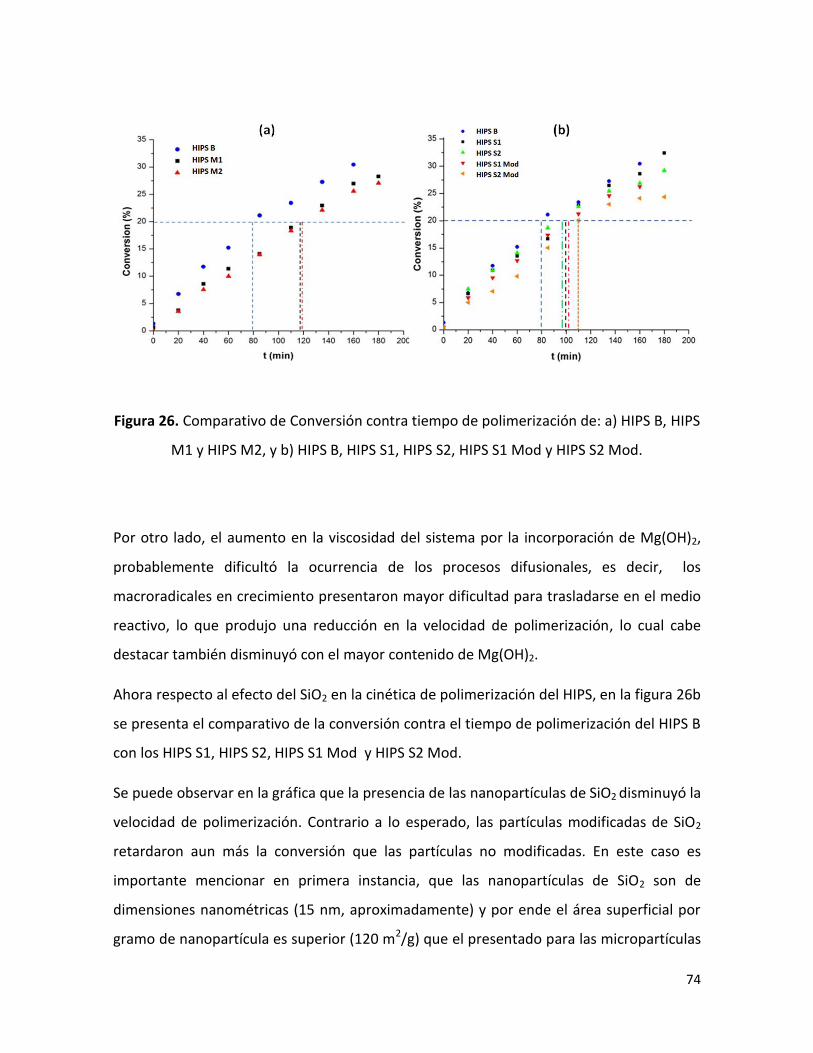
74
Figura 26. Comparativo de Conversión contra tiempo de polimerización de: a) HIPS B, HIPS
M1 y HIPS M2, y b) HIPS B, HIPS S1, HIPS S2, HIPS S1 Mod y HIPS S2 Mod.
Por otro lado, el aumento en la viscosidad del sistema por la incorporación de Mg(OH)2,
probablemente dificultó la ocurrencia de los procesos difusionales, es decir, los
macroradicales en crecimiento presentaron mayor dificultad para trasladarse en el medio
reactivo, lo que produjo una reducción en la velocidad de polimerización, lo cual cabe
destacar también disminuyó con el mayor contenido de Mg(OH)2.
Ahora respecto al efecto del SiO2 en la cinética de polimerización del HIPS, en la figura 26b
se presenta el comparativo de la conversión contra el tiempo de polimerización del HIPS B
con los HIPS S1, HIPS S2, HIPS S1 Mod y HIPS S2 Mod.
Se puede observar en la gráfica que la presencia de las nanopartículas de SiO2 disminuyó la
velocidad de polimerización. Contrario a lo esperado, las partículas modificadas de SiO2
retardaron aun más la conversión que las partículas no modificadas. En este caso es
importante mencionar en primera instancia, que las nanopartículas de SiO2 son de
dimensiones nanométricas (15 nm, aproximadamente) y por ende el área superficial por
gramo de nanopartícula es superior (120 m2/g) que el presentado para las micropartículas

75
de Mg(OH)2 (2-5m2/g), lo que produce una mayor interacción con el medio reactivo,
provocando, un incremento en la viscosidad, lo que disminuyó los procesos difusionales, y
por lo tanto, la velocidad de polimerización. En este mismo sentido, al disminuir la tensión
interfacial entre el medio reactivo y la nanosílice mediante la modificación de las
nanopartículas, el efecto antes descrito (incremento en la viscosidad) se presentó en
mayor magnitud, dada la mejora en compatibilidad de las nanopartículas con el sistema
estireno/hule así como la dispersión y distribución de las mismas en el sistema, debido a la
deposición de polímero tipo estirénico sobre la superficie de las nanopartículas.
En cuanto a la parte experiemental, cabe mencionar que debido al aumento de la
viscosidad al incorporar las nanopartículas de SiO2 al St, se utilizaron las nanopartículas en
una concentración máxima de 5 % en peso. En un inicio se planteó y realizó la
incorporación de nanopartículas de SiO2 en una concentración de 7.5 % en peso, lo que
resultó en una solución con alta viscosidad (Ver figura 27), en la cual el PB ya no pudo ser
incorporado, de aquí que se optó por reducir dicho contenido.
Fig. 27. Efecto de la incorporación de nanopartículas de SiO2
en la viscosidad del Estireno.
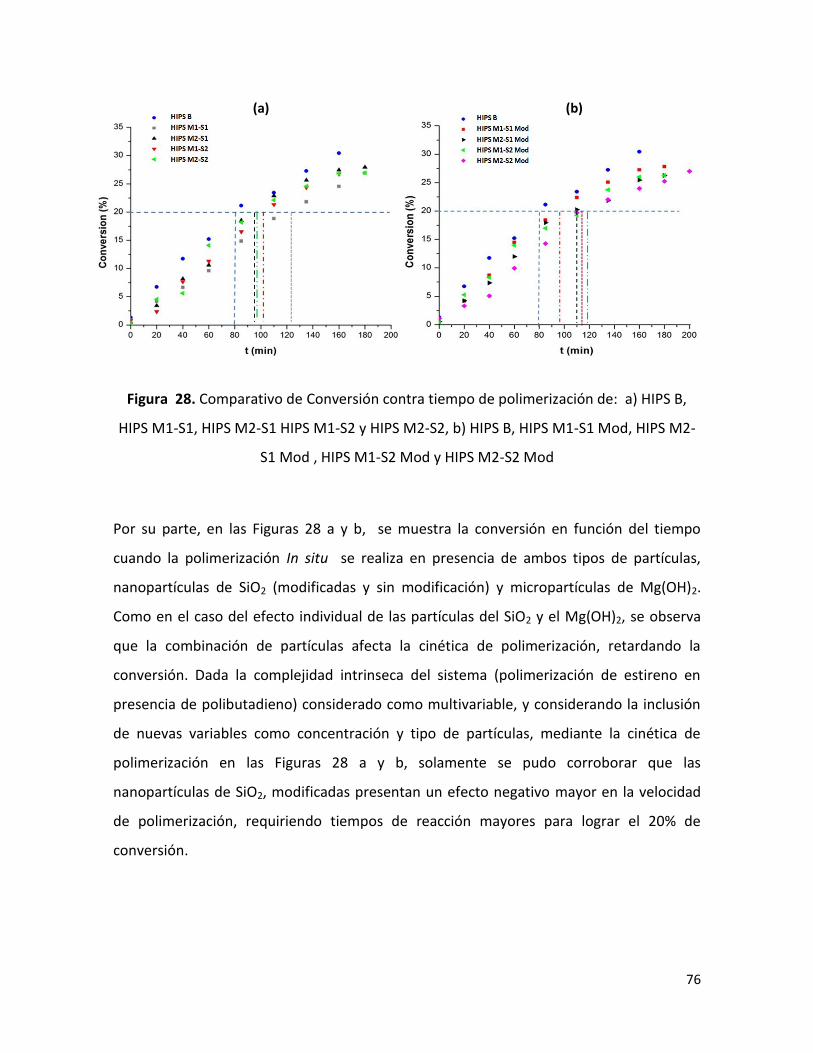
76
Figura 28. Comparativo de Conversión contra tiempo de polimerización de: a) HIPS B,
HIPS M1-S1, HIPS M2-S1 HIPS M1-S2 y HIPS M2-S2, b) HIPS B, HIPS M1-S1 Mod, HIPS M2-
S1 Mod , HIPS M1-S2 Mod y HIPS M2-S2 Mod
Por su parte, en las Figuras 28 a y b, se muestra la conversión en función del tiempo
cuando la polimerización In situ se realiza en presencia de ambos tipos de partículas,
nanopartículas de SiO2 (modificadas y sin modificación) y micropartículas de Mg(OH)2.
Como en el caso del efecto individual de las partículas del SiO2 y el Mg(OH)2, se observa
que la combinación de partículas afecta la cinética de polimerización, retardando la
conversión. Dada la complejidad intrinseca del sistema (polimerización de estireno en
presencia de polibutadieno) considerado como multivariable, y considerando la inclusión
de nuevas variables como concentración y tipo de partículas, mediante la cinética de
polimerización en las Figuras 28 a y b, solamente se pudo corroborar que las
nanopartículas de SiO2, modificadas presentan un efecto negativo mayor en la velocidad
de polimerización, requiriendo tiempos de reacción mayores para lograr el 20% de
conversión.
(a) (b)

77
Tabla 8. Tiempo de polimerización aproximado para obtener 20 % de conversión
7.2.2 Análisis de la caracterización físico-química.
7.2.2.1 Efecto de la velocidad de polimerización sobre la distribución de pesos
moleculares (DPM).
En la Tabla 9 se muestran los valores de peso molecular promedio en número y en peso
(Mn y Mw) y la dispersidad (D) de los HIPS obtenidos. En primera instancia se observa que
el HIPS B alcanza valores de Mn=37,000 g/mol, Mw=218,500 g/mol y una D=5.9. Respecto a
los compuestos de HIPS, la presencia de las partículas de SiO2 sin modificar y modificadas
afectó de manera significativa el peso molecular de los HIPS finales con respecto al blanco
de referencia, en ambas concentraciones incorporadas. Para la serie de HIPS con SiO2, se
alcanzó un incremento en el peso molecular promedio en número (Mn) de
Muestra HIPS t (min) al 20% de Conversión
HIPS B 80 HIPS S1 100 HIPS S2 96
HIPS S1 Mod 103 HIPS S2 Mod 110
HIPS M1 117 HIPS M2 119
HIPS M1-S1 122 HIPS M2-S1 94
HIPS M1-S2 103
HIPS M2-S2 95 HIPS M1-S1 Mod 96 HIPS M2-S1 Mod 110 HIPS M1-S2 Mod 118
HIPS M2-S2 Mod 114
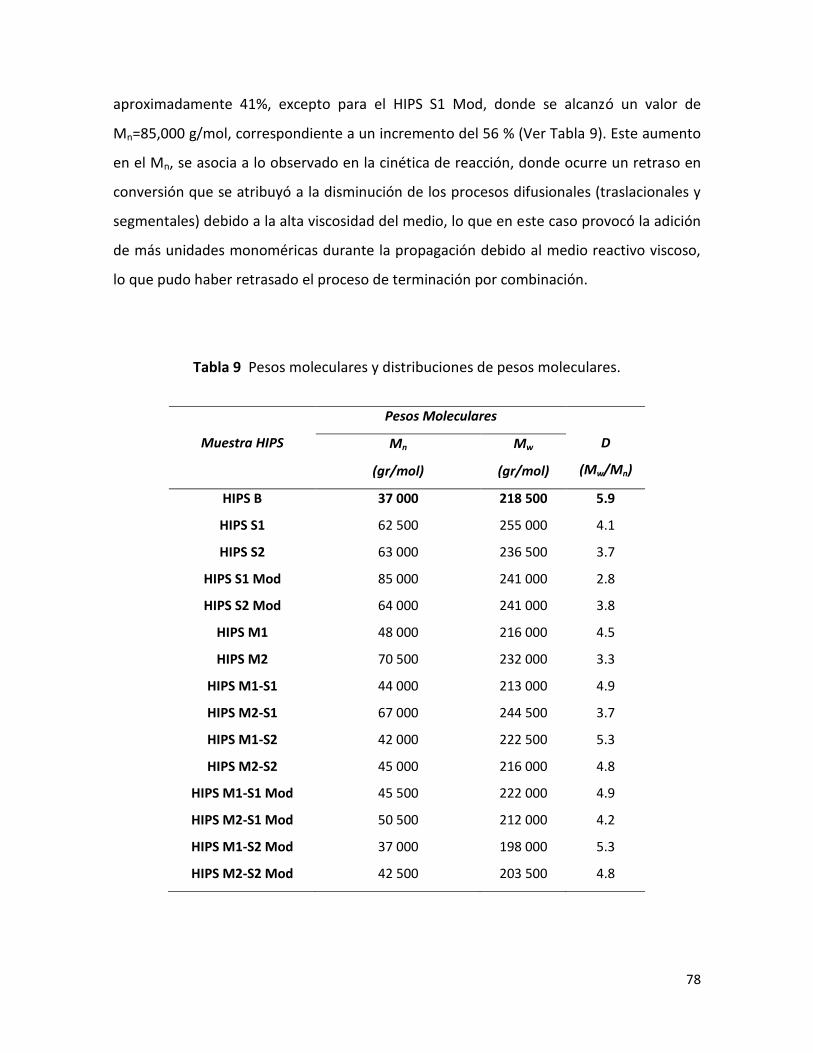
78
aproximadamente 41%, excepto para el HIPS S1 Mod, donde se alcanzó un valor de
Mn=85,000 g/mol, correspondiente a un incremento del 56 % (Ver Tabla 9). Este aumento
en el Mn, se asocia a lo observado en la cinética de reacción, donde ocurre un retraso en
conversión que se atribuyó a la disminución de los procesos difusionales (traslacionales y
segmentales) debido a la alta viscosidad del medio, lo que en este caso provocó la adición
de más unidades monoméricas durante la propagación debido al medio reactivo viscoso,
lo que pudo haber retrasado el proceso de terminación por combinación.
Tabla 9 Pesos moleculares y distribuciones de pesos moleculares.
Muestra HIPS
Pesos Moleculares
D
(Mw/Mn)
Mn Mw
(gr/mol) (gr/mol)
HIPS B 37 000 218 500 5.9
HIPS S1 62 500 255 000 4.1
HIPS S2 63 000 236 500 3.7
HIPS S1 Mod 85 000 241 000 2.8
HIPS S2 Mod 64 000 241 000 3.8
HIPS M1 48 000 216 000 4.5
HIPS M2 70 500 232 000 3.3
HIPS M1-S1 44 000 213 000 4.9
HIPS M2-S1 67 000 244 500 3.7
HIPS M1-S2 42 000 222 500 5.3
HIPS M2-S2 45 000 216 000 4.8
HIPS M1-S1 Mod 45 500 222 000 4.9
HIPS M2-S1 Mod 50 500 212 000 4.2
HIPS M1-S2 Mod 37 000 198 000 5.3
HIPS M2-S2 Mod 42 500 203 500 4.8

79
Tambien de la Tabla 9 se puede observar un incremento en Mn para los HIPS que
contienen Mg(OH)2 (HIPS M1 y HIPS M2), incremento esperado dada la disminución en la
conversión. En este sentido, se corrobora que además de la presencia de grupos vinílicos
en el modificador superficial, aunado al incremento en la viscosidad, provoca una
disminución en la conversión y un incremento en el Mn, el cual es más evidente al
incrementar el contenido de Mg(OH)2 desde 10 a 20 % en peso.
Peso molecular promedio en número (Mn)
En el caso de la presencia combinada de partículas de SiO2 e Mg(OH)2 el mayor impacto en
el peso molecular promedio en número ( Mn ) se observó en la concentración de 2.5 % en
peso de SiO2 y 20 % en peso de Mg(OH)2 , en el HIPS M2-S1 , aumentando de los 37,000
gr/mol en el HIPS B , a los 67,000 gr/mol. Mientras en los otros compuestos, HIPS M1-S1,
HIPS M1-S2 y HIPS M2-S2 se alcanzaron valores de Mn de 44,000, 42,000 y 45,000 gr/mol
respectivamente, es decir presentaron un ligero incremento.
Por último, el efecto en el Mn con presencia combinada de partículas de SiO2 modificadas
superficialmente y de Mg(OH)2 provocó también un incremento en el Mn, sin embargo, no
se observó una tendencia que pudiera evidenciar un efecto sinérgico sobre de ambas
partículas sobre la diferencia existente en el Mn de los diversos compuestos.
Como se puede observar en los resultados de Mn, en el caso de las combinaciones de
nanopartículas de SiO2 tanto modificadas como sin modificar; con micropartículas de
Mg(OH)2, con excepción de HIPS M2-S1, se obtuvo un aumento menor en Mn que en la
incorporación individual de partículas en las correspondientes concentraciones. Lo que
pudiera estar relacionado con la dispersión y distribución de ambas partículas en el medio
de reacción, donde probablemente se formaron aglomerados de partículas lo que
disminuyó el área superficial global y por ende la interacción de las partículas con el
sistema reactivo.

80
Peso molecular promedio en peso ( Mw )
Como se puede apreciar en la tabla 10, respecto al Mw= 218,500 gr/mol del HIPS B, el Mw
aumento en presencia de 2.5 y 5 % en peso SiO2 modificado, HIPS S1 Mod y HIPS S2 Mod
respectivamente, quedando prácticamente igual en ambos casos (241,000 gr./mol).
También se puede apreciar que en concentración de 20 % en peso de Mg(OH)2 (HIPS M2)
también hubo un aumento en el Mw. En un contenido de 20 % en peso de Mg(OH)2 y 2.5
% de SiO2 (HIPS M2-S1) se observó el mismo efecto. El mayor aumento en el Mw se
presentó en presencia de SiO2 (sin modificar), en el HIPS S1 y HIPS S2. En el caso de los
HIPS M1, HIPS M1-S1, HIPS M1-S2, HIPS M2-S2, HIPS M1-S1 Mod y HIPS M2-S1 Mod el Mw
se mantiene prácticamente igual al del HIPS B. Finalmente en los HIPS M1-S2 Mod y HIPS
M2-S2 Mod el Mw fue de 198,000 y 203,500 gr/mol, respectivamente, menor a los
218,500 gr/mol del HIPS B.
Dispersidad (D)
La dispersidad disminuyó prácticamente en todos los pesos moleculares de los
compuestos de HIPS con partículas de SiO2 y/o Mg(OH)2 , y en el caso de los HIPS M1-S2 y
HIPS M1-S2 Mod, con una dispersidad de 5.3 y 5.3 respectivamente, se mantuvo
prácticamente igual a la dispersidad del HIPS B (5.905).
Lopez y colaboradores49 atribuyeron la disminución del Mn en la síntesis de
nanocompuestos PS/Sílice al favorecimiento de la reacción de terminación por
transferencia ( La transferencia se considera un tipo de terminacion aunque la molecula ,
inicialmente estable, con la que reacciona el macroradical , queda activada como radical ),
en su caso, por el aumento de viscosidad y disminución de procesos difusionales 49,54,55 de
traslación, lo que aumenta las reacciones de terminación por transferencia y se generan
cadenas poliméricas de menor peso molecular. En este sentido en la síntesis de
nanocompuestos de HIPS realizado en este trabajo, la presencia de nanopartículas de SiO2

81
y micropartículas de Mg(OH)2 disminuyó las reacciones de terminación por transferencia,
51,54,debido a una disminución de los procesos difusionales 49,54,55 tanto de traslación como
segmentales, provocando un aumento en el Mw y Mn, evitando la transferencia al
monómero, al PB o al CI. Lo anterior disminuye la presencia de moléculas de menor
tamaño por lo que el Mn aumentó en mayor proporción que el Mw, lo que ocasionó una
disminución en la dispersidad, D. El efecto en la dispersidad no sigue una tendencia
general, pero la mayoría de los compuestos de HIPS tuvieron un aumento, respecto al
HIPS B, mayor en el Mn que en el Mw, provocado por la disminución de terminación por
transferencia 51,54.
En resumen en la Figura 27 y en la Tabla 8 se puede ver el comparativo de la conversión
en función del tiempo del HIPS S1, HIPS S2, HIPS S1 Mod y HIPS S2 Mod con el HIPS B. Se
puede observar que en la síntesis de los HIPS S1 Mod y HIPS S2 Mod (con SiO2 modificado)
hubo una mayor reducción en la velocidad de polimerización que en los HIPS S1 y HIPS S2;
a pesar de que estos últimos contienen SiO2 sin modificar por lo que se esperaba mayor
impacto a la velocidad de polimerización, ya que al no tener modificación, el iniciador
podría interactuar químicamente con las partículas de SiO2, lo que reduciría la cantidad de
radicales de iniciador, y aunque no se descarta una interacción entre el BPO y las
nanopartículas, la velocidad de polimerización fue, aparentemente, menos afectada.
De igual manera, comparando el tiempo de polimerización para llegar al 20% de
conversión de los HIPS M1 y HIPS M2 con el tiempo de los HIPS S1 y HIPS S2 de la Tabla 8,
se puede ver que se requirió mayor tiempo en los HIPS M1 y HIPS M2, con 10 y 20 %
Mg(OH)2 (con modificación superficial) respectivamente; es decir, la velocidad de
polimerización fue menor ; aunque no son comparables del todo ya que son distintos tipos
de partículas respecto a naturaleza química y área superficial.
Regresando a los HIPS con SiO2 modificado y sin modificar, el Mw (ver tabla 9) aumentó en
mayor proporción en los HIPS S1 y HIPS S2 que en los HIPS S1 Mod y HIPS S2 Mod;
entonces, tanto el Mw como la velocidad de polimerización fueron mayores en presencia
de SiO2 sin modificar. Esto se puede explicar debido al aumento en la viscosidad que

82
causaron las partículas de SiO2 (sin modificar) en el sistema St/PB/PS, este aumento en la
viscosidad causo problemas de difusión49,54,55 tanto traslasional como segmental,
restringiendo el movimiento de los macro radicales, lo que hace más lentas las reacciones
de terminación, esto a su vez provoca un aumento en la velocidad global de la reacción
(de manera parecida como ocurre en el fenómeno conocido como autoaceleración o
efecto Trommsdorff). Estos problemas difusionales, explicarían la menor velocidad de
polimerización y mayor Mw.
7.2.2.2 Efecto de la velocidad de polimerización sobre el contenido de gel (CG) y grado
de injerto (GI).
El contenido en gel (CG) definido como el % de Insolubles en un HIPS y el Grado de Injerto,
considerado como el PS injertado químicamente u ocluido en la fase elastomérica, son
parámetros que evidentemente se relacionan con el proceso de polimerización, es decir,
pueden ser directamente influenciados por la cinética de polimerización. Al respecto en la
Tabla 10 se muestran los valores del CG y GI para el HIPS B y todos los compuestos
sintetizados. En primera instancia se puede observar que el HIPS B, presentó un CG de 50
% y GI de 529 %, valores considerados normales en un material sintetizado con la receta
de síntesis y condiciones de proceso utilizadas en este trabajo. Sin embargo, al incluir las
diferentes partículas y sus combinaciones se presenta una disminución drástica en ambos
parámetros físico-químicos.
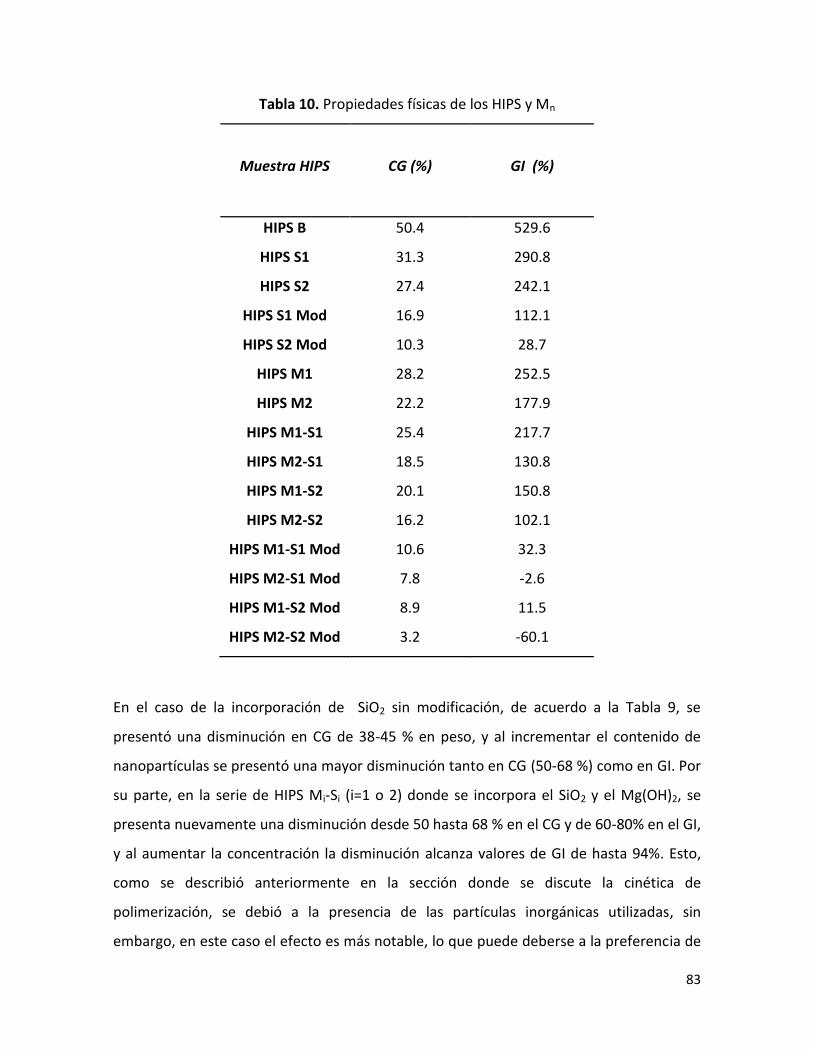
83
Tabla 10. Propiedades físicas de los HIPS y Mn
Muestra HIPS CG (%) GI (%)
HIPS B 50.4 529.6
HIPS S1 31.3 290.8
HIPS S2 27.4 242.1
HIPS S1 Mod 16.9 112.1
HIPS S2 Mod 10.3 28.7
HIPS M1 28.2 252.5
HIPS M2 22.2 177.9
HIPS M1-S1 25.4 217.7
HIPS M2-S1 18.5 130.8
HIPS M1-S2 20.1 150.8
HIPS M2-S2 16.2 102.1
HIPS M1-S1 Mod 10.6 32.3
HIPS M2-S1 Mod 7.8 -2.6
HIPS M1-S2 Mod 8.9 11.5
HIPS M2-S2 Mod 3.2 -60.1
En el caso de la incorporación de SiO2 sin modificación, de acuerdo a la Tabla 9, se
presentó una disminución en CG de 38-45 % en peso, y al incrementar el contenido de
nanopartículas se presentó una mayor disminución tanto en CG (50-68 %) como en GI. Por
su parte, en la serie de HIPS Mi-Si (i=1 o 2) donde se incorpora el SiO2 y el Mg(OH)2, se
presenta nuevamente una disminución desde 50 hasta 68 % en el CG y de 60-80% en el GI,
y al aumentar la concentración la disminución alcanza valores de GI de hasta 94%. Esto,
como se describió anteriormente en la sección donde se discute la cinética de
polimerización, se debió a la presencia de las partículas inorgánicas utilizadas, sin
embargo, en este caso el efecto es más notable, lo que puede deberse a la preferencia de

84
las partículas de ubicarse en la fase elastomérica, lo que se discutirá más adelante en la
sección de análisis morfológico. Respecto a los HIPS Si Mod y los correspondientes a la
serie de HIPS Si-Mi Mod, se observa una disminución de 66-80 % de CG y un 79-94 % en el
GI, efecto similar a lo visto para la cinética de polimerización y distribución de pesos
moleculares. Además, se confirma que a mayor concentración de nanopartículas de SiO2
con o sin modificación, afecta negativamente en mayor magnitud ambos parámetros
analizados.
Por otro lado, es importante mencionar que los valores de GI reportados para los HIPS
M2-S1 Mod y HIPS M2-S2 Mod, que presentan valores negativos se debe específicamente
a que el calculo de GI se realiza considerando el CG y el contenido de hule inicial. En este
caso es muy probable que la fase elastomérica en forma de partículas al encontrarse
parcialmente entrecruzadas al exponerse a extracción con solventes, tanto el hule PB no
entrecruzado como el PS ocluido son extraidos y por lo tanto el GI disminuye
drasticamente, lo que da un valor de GI aparentemente negativo.
7.2.3 Evolución de la morfología del HIPS
Para dar seguimiento a la evolución de la morfología durante la etapa de pre
polimerización y verificar los fenómenos termodinámicos involucrados,
fundamentalmente, el fenómeno de inversión de fases, se observaron y analizaron
imágenes adquridas por TEM a diferentes conversiones y tiempos de polimerización de los
HIPS que presentarón una mayor disminución en cuanto a velocidad de polimerización
(HIPS M1-S1 Mod, HIPS M1-S2 Mod, HIPS M2-S1 Mod y HIPS M2-S2 Mod) y el HIPS B.
En la Figura 29 se muestran imágenes obtenidas mediante TEM del HIPS B a diferentes
conversiones y tiempos de polimerización. Se puede apreciar en las micrografías el
seguimiento del desarrollo morfológico en función de la conversión, a un 12 % de
conversión se observa que las fases se encuentran separadas y que la fase St/PS (área

85
X = 0.12 ( 40 min.)
Co-continuidad
X = 0.15 ( 60 min.)
Intérvalo de Inversión de fases
X = 0.21 ( 85 min.)
Establecimiento morfológico
X = 0.225 ( 110 min. )
Inversión de fases
X = 0.28 ( 180 min.)
Morfología final
X = 0.145 ( 60 min.)
Co-continuidad
clara en la micrografia) continua dispersa en la fase continua de St/PB (área obscura en la
micrografia). Con el avance de la reacción hasta una conversión de 15% se observa una
morfología en transición a la inversión de fases; mientras que al 21% la inversión de fases
ha ocurrido completamente, y la fase St/PB pasó a ser la fase dispersa en el sistema y asi
se establece la morfología que en este caso, fue del tipo core-shell.
Figura 29. Evolución morfológica del HIPS B (Escala de la barra 0.5 m)
Figura 30. Evolución de la morfología del HIPS M1-S1 Mod (Escala de la barra 0.5 m)

86
En la Figura 30 se muestran las imágenes correspondientes a la evolución morfológica del
HIPS M1-S1 Mod. En este caso se observa que la inversión de fases ocurre a un 22.5 % de
conversión (conversión esperada).
Como se indicó en la sección de antecedentes, la inversión de fases del sistema estudiado
ocurre, generalmente, cuando el volumen de la fase St/PS es mayor al volumen de la fase
St/PB, y que el volumen de la fase St/PS se incrementa con la conversión, en este caso
aproximadamente a un 20 % de conversión. Sin embargo, el tiempo requerido para que
esto ocurra en el HIPS M1-S1 Mod se incremento notablemente alcanzando la inversión
de fases hasta los 110 min, 25 min más que para el HIPS B. Lo anterior se debió a la
disminución de la velocidad de polimerización evidenciada en la sección 7.2.1, y que es
provocada por la presencia del SiO2 y Mg(OH)2.
Por su parte, en las Figuras 31, 32 y 33, se muestran las imágenes obtenidas mediante
TEM para los compuestos, HIPS M1-S2 Mod, HIPS M2-S1 Mod y HIPS M2-S2 Mod. Para los
tres casos ocurre un comportamiento similar al descrito para el HIPS M1-S1, la inversión
de fase ocurre a conversiones aproximadas al 20 %, sin embargo, el tiempo para alcanzar
dicha conversión se incrementa como consecuencia de la disminución en la velocidad de
polimerización.
Figura 31. Evolución de la morfología del HIPS M1-S2 Mod (Escala de la barra 0.5 m)

87
Figura 32. Evolución de la morfología del HIPS M2-S1 Mod (Escala de la barra 0.5 m)
Figura 33. Evolución de la morfología del HIPS M2-S2 Mod (Escala de la barra 0.5 m)
X = 0.12 ( 60 min.)
Co-continuidad
X = 0.18 ( 85 min.)
Inversión de fases
X = 0.27 ( 180 min.)
Morfología final
X = 0.10 ( 60 min.)
Co-continuidad
X = 0.20 ( 110 min.)
Co-continuidad
X = 0.27 ( 200 min.)
Morfología final

88
7.2.4 Morfología final de los HIPS sintetizados
La morfología de la fase elastomérica de los HIPS se determinó mediante imágenes
obtenidas mediante TEM. El tipo de morfología observada en los HIPS fue
predominantemente núcleo coraza, aunque se observaron de manera escasa estructuras
tipo salami en algunos nanocompuestos de HIPS.
En la Figura 34 se muestra la imagen obtenida mediante TEM del HIPS B, se observa
principalmente morfología del tipo núcleo coraza y algunas partículas de forma irregular
con múltiples oclusiones, parecidas a estructuras salami pero sin forma de elipse.
Figura 34. Morfología final del HIPS B (Escala de la barra 0.5 m).
Este tipo de morfología se debió a que se utilizó una velocidad de agitación de 100 rpm, y
es bien sabido que el tamaño de partícula disminuye cuando se incrementa la velocidad
de agitación en la etapa de prepolimerización, lo que favorece que se desarrolle
morfología tipo núcleo coraza en lugar de morfología tipo salami32. La velocidad de
agitación más adecuada para una morfología tipo salami y mayores tamaños se encuentra
aproximadamente en 50 rpm.

89
Tabla 12. Diámetro de partícula aparente y fracción volumen de hule del HIPS B
Muestras HIPS 1Dp (nm) 2Φ
HIPS B 227 0.22 1Dp: diámetro promedio de partícula,
2Φ: fracción volumen
Respecto a los HIPS con SiO2 sin modificación superficial, en la Figura 35 a y b se observan
las imágenes obtenidas por TEM de los HIPS S1 y HIPS S2 respectivamente. En las
imágenes se puede observar una morfología de la fase elastomérica tipo núcleo coraza
principalmente y aglomerados de SiO2 encapsulados en una membrana de PB, es decir,
dentro de la fase elastomérica. Ademas en el HIPS S2 (Ver fig. 35 b) se observan
aglomerados de nanopartículas en la matriz de PS, cercanos a la fase elastomérica;
también se distinguen estructuras elastoméricas de forma alargada.
El diámetro de partícula promedio (Dp) presentó un aumento (ver tabla 13) en los HIPS S1
y HIPS S2 en comparación con el HIPS B, esto se puede explicar por el cambio de relación
de viscosidades entre fase dispersa y la fase continua y la disminución en el grado de
injerto.
No obstante, a que un mayor Mw de los HIPS S1 y HIPS S2 pudo ocasionar una mayor
viscosidad de la fase St/PS, del mismo modo que la presencia de las partículas de SiO2, lo
que puede disminuir el diámetro de partícula, este cambio en la relación de viscosidades
de la solución heterogénea St/PB/St/PS no presentó un efecto sobre este factor como se
puede observar en la Tabla 13. Lo anterior podría indicar que la contribución respecto al
diámetro de partícula, que en este caso se incrementó, se debe fundamentalmente a una
disminución en el grado de injerto (GI) , ya que al disminuir el grado de injerto se
incrementa la incompatibilidad entre las fases lo que provoca un aumento en el tamaño
de partícula.
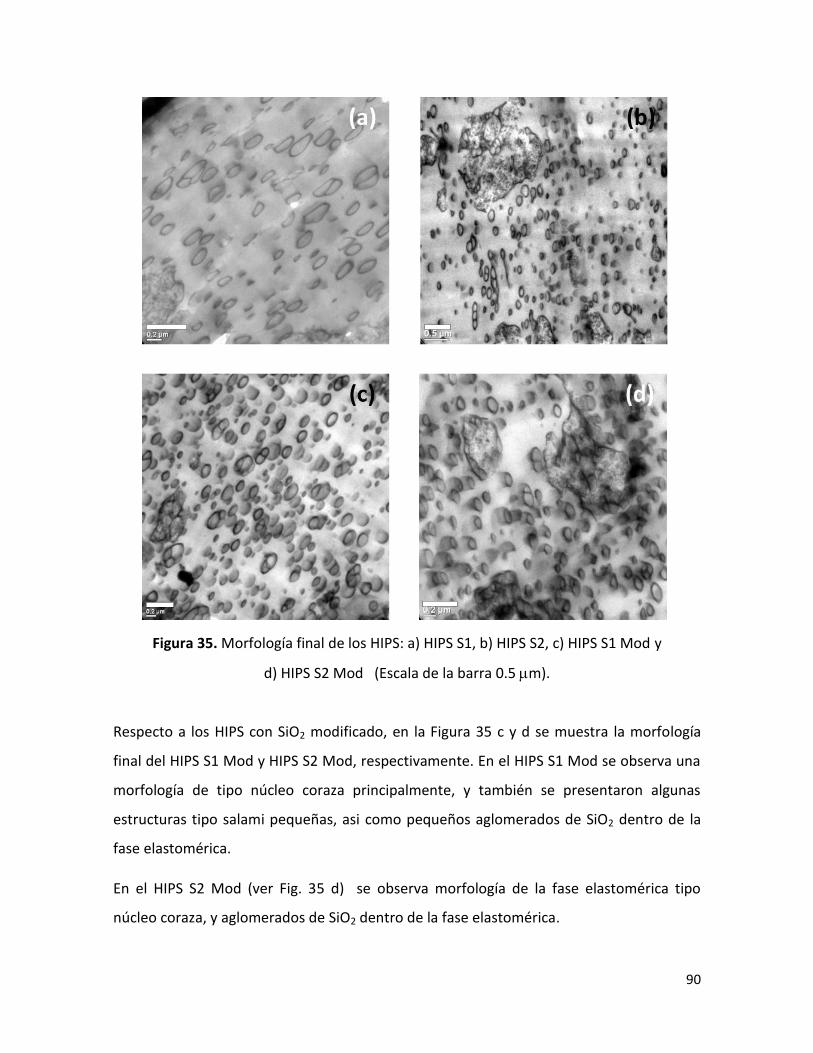
90
Figura 35. Morfología final de los HIPS: a) HIPS S1, b) HIPS S2, c) HIPS S1 Mod y
d) HIPS S2 Mod (Escala de la barra 0.5 m).
Respecto a los HIPS con SiO2 modificado, en la Figura 35 c y d se muestra la morfología
final del HIPS S1 Mod y HIPS S2 Mod, respectivamente. En el HIPS S1 Mod se observa una
morfología de tipo núcleo coraza principalmente, y también se presentaron algunas
estructuras tipo salami pequeñas, asi como pequeños aglomerados de SiO2 dentro de la
fase elastomérica.
En el HIPS S2 Mod (ver Fig. 35 d) se observa morfología de la fase elastomérica tipo
núcleo coraza, y aglomerados de SiO2 dentro de la fase elastomérica.

91
Con relación al Diámetro de partícula promedio en el HIPS S1 Mod el diámetro de
partícula disminuyó (ver tabla 13) en comparacion al HIPS B, contrario a los HIPS S1 y HIPS
S2. El aumento de la viscosidad, asociado al incremento en Mw, durante la
prepolimerización en el HIPS S1 Mod ocasionó la disminución en el tamaño de partícula.
Respecto al GI, aunque no se puede asegurar que el GI fue mayor en el HIPS S1 Mod que
en el HIPS B, se puede concluir que fue mayor que el GI obtenido para los HIPS S1 y el HIPS
S2. Comportamiento similar se presentó en el HIPS S2 Mod, ya que el diámetro de
partícula también es menor que el del HIPS B.
Tabla 13. Diámetro de partícula aparente y fracción volumen de hule de los HIPS HIPS B, HIPS S1, HIPS S2, HIPS S1 Mod y HIPS S2 Mod
Muestras HIPS Dp (nm) Φ
HIPS B 226 0.22
HIPS S1 246 0.36
HIPS S2 229 0.26
HIPS S1 Mod 209 0.17
HIPS S2 Mod 188 0.21
En la Figura 36 a y b, se observan las imágenes obtenidas por TEM de los HIPS M1 y HIPS
M2, respectivamente. En las imágenes se puede observar una morfología de la fase
elastomérica tipo núcleo-coraza principalmente. En el HIPS M1 (Ver Fig. 36 a) además de
núcleo coraza se ven algunas pequeñas estructuras salamis, mientras en la Figura 36b
correspondiente al HIPS M2 predomina la morfología de tipo núcleo-coraza.
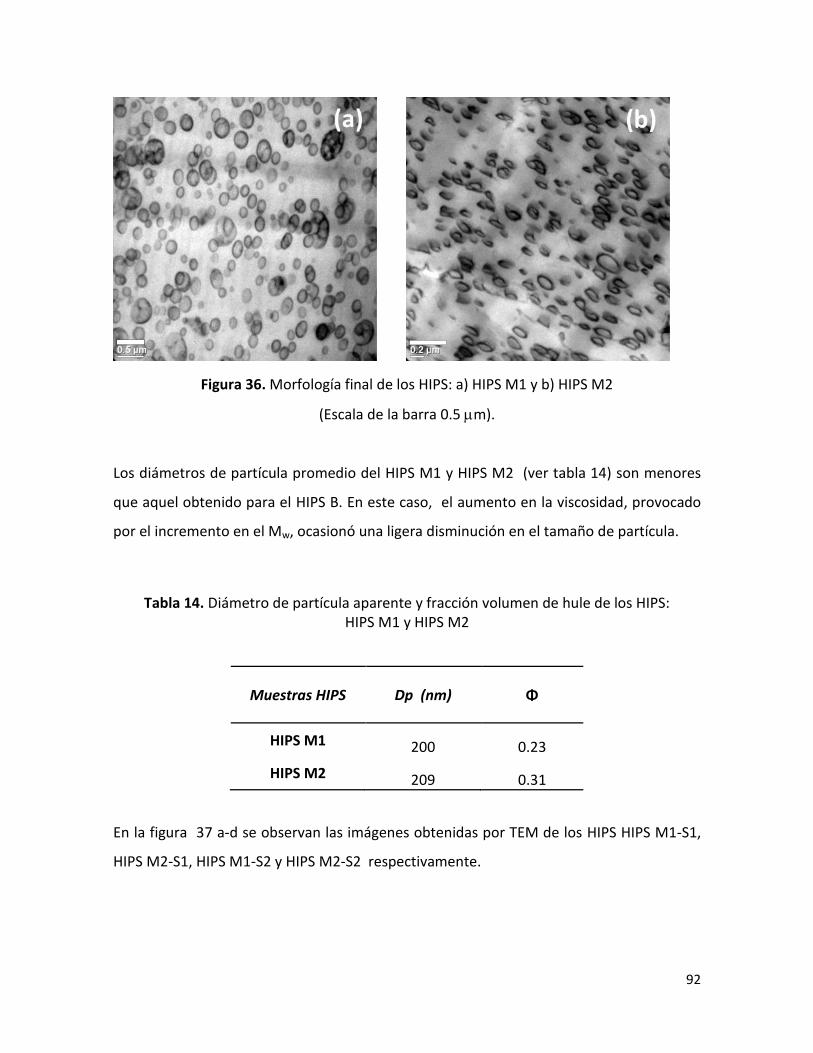
92
Figura 36. Morfología final de los HIPS: a) HIPS M1 y b) HIPS M2
(Escala de la barra 0.5 m).
Los diámetros de partícula promedio del HIPS M1 y HIPS M2 (ver tabla 14) son menores
que aquel obtenido para el HIPS B. En este caso, el aumento en la viscosidad, provocado
por el incremento en el Mw, ocasionó una ligera disminución en el tamaño de partícula.
Tabla 14. Diámetro de partícula aparente y fracción volumen de hule de los HIPS: HIPS M1 y HIPS M2
Muestras HIPS Dp (nm) Φ
HIPS M1 200 0.23
HIPS M2 209 0.31
En la figura 37 a-d se observan las imágenes obtenidas por TEM de los HIPS HIPS M1-S1,
HIPS M2-S1, HIPS M1-S2 y HIPS M2-S2 respectivamente.
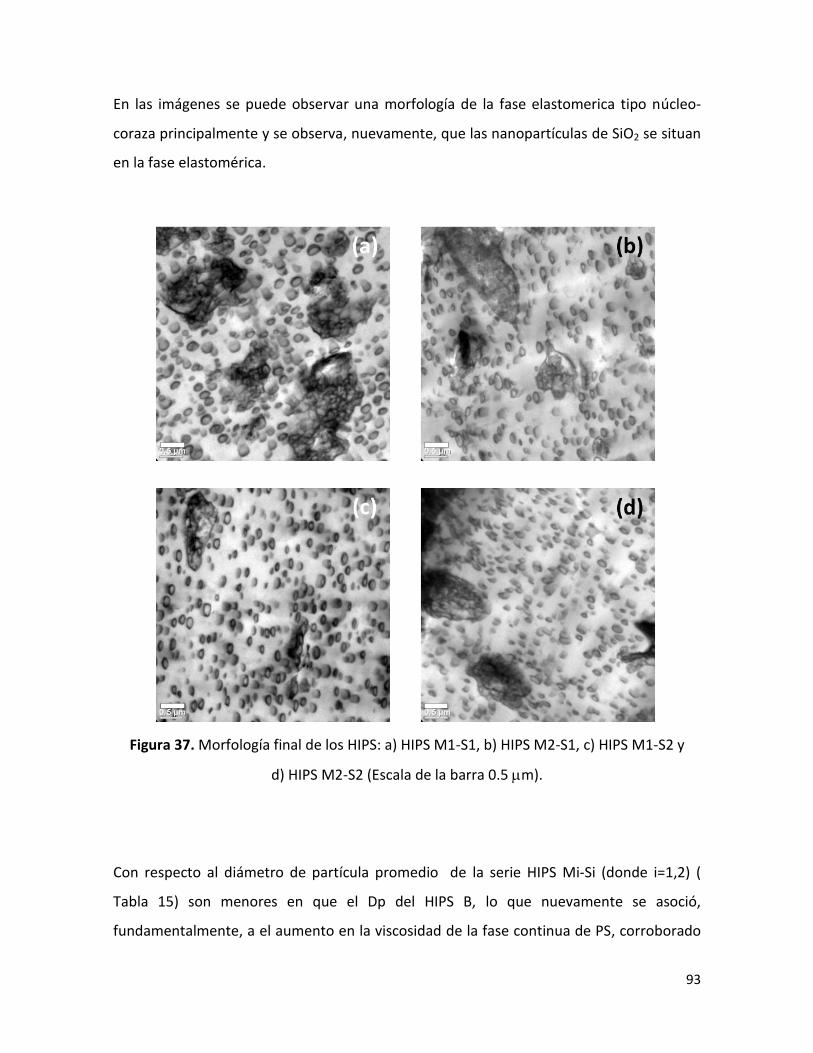
93
En las imágenes se puede observar una morfología de la fase elastomerica tipo núcleo-
coraza principalmente y se observa, nuevamente, que las nanopartículas de SiO2 se situan
en la fase elastomérica.
Figura 37. Morfología final de los HIPS: a) HIPS M1-S1, b) HIPS M2-S1, c) HIPS M1-S2 y
d) HIPS M2-S2 (Escala de la barra 0.5 m).
Con respecto al diámetro de partícula promedio de la serie HIPS Mi-Si (donde i=1,2) (
Tabla 15) son menores en que el Dp del HIPS B, lo que nuevamente se asoció,
fundamentalmente, a el aumento en la viscosidad de la fase continua de PS, corroborado

94
por el incremento en el Mw de la fase de PS. Aunque en este caso el efecto de la
combinación de nano y micropartículas se potencializó, dando lugar a menores diámetros
de partícula respecto a los HIPS Mi y Si.
Tabla 15. Diámetro de partícula aparente y fracción volumen de hule de los HIPS: HIPS M1-S1, HIPS M2-S1, HIPS M1-S2 y HIPS M2-S2
Finalmente, respecto a la morfología de los compuestos finales, en la Figura 38 a-d se
muestran las imágenes obtenidas por TEM de los HIPS M1-S1 Mod, HIPS M2-S1 Mod, HIPS
M1-S2 Mod y HIPS M2-S2 Mod, respectivamente.
En las imágenes se puede observar una morfología de la fase elastomérica tipo núcleo-
coraza principalmente y se observan aglomerados de partículas en la fase elastomérica,
especialmente a mayor contenido de SiO2 en el HIPS M1-S2 Mod. Por su parte, en el HIPS
M2-S2 Mod se observan algunas estructuras de forma varilla y partículas agrupadas de
SiO2 en la fase continua.
Muestras HIPS Dp (nm) Φ
HIPS M1-S1 194 0.25
HIPS M2-S1 191 0.32
HIPS M1-S2 186 0.28
HIPS M2-S2 165 0.27
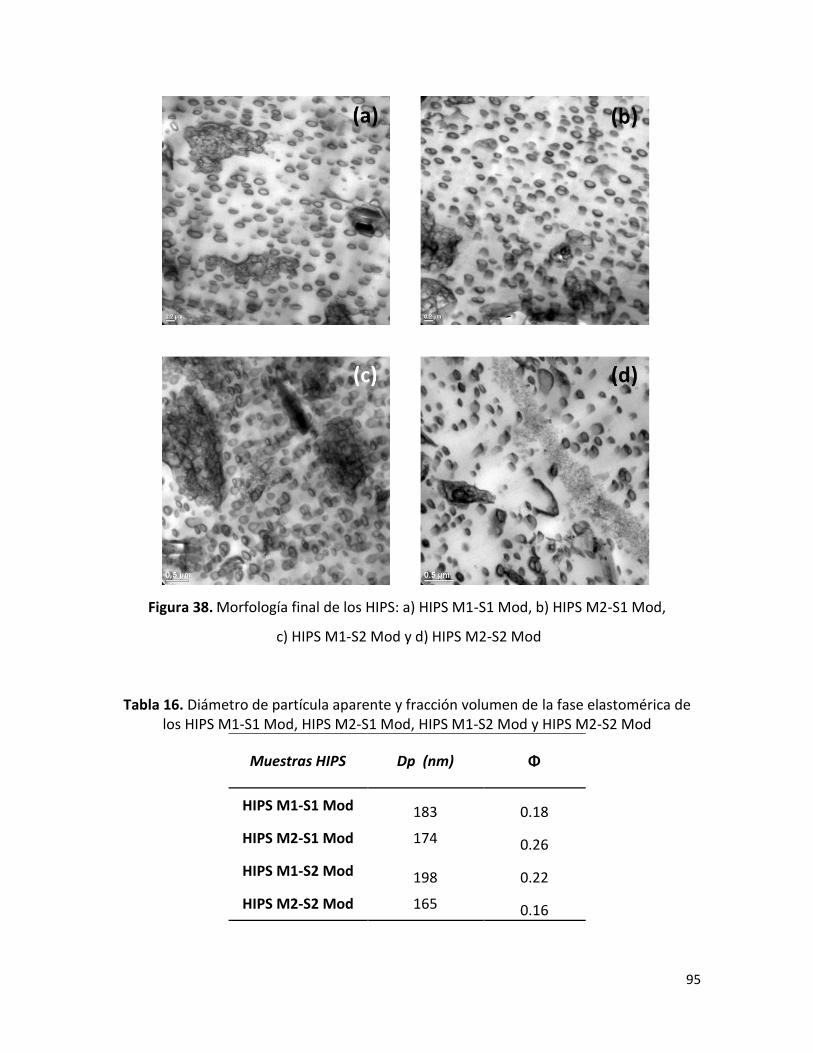
95
Figura 38. Morfología final de los HIPS: a) HIPS M1-S1 Mod, b) HIPS M2-S1 Mod,
c) HIPS M1-S2 Mod y d) HIPS M2-S2 Mod
Tabla 16. Diámetro de partícula aparente y fracción volumen de la fase elastomérica de los HIPS M1-S1 Mod, HIPS M2-S1 Mod, HIPS M1-S2 Mod y HIPS M2-S2 Mod
Muestras HIPS Dp (nm) Φ
HIPS M1-S1 Mod 183 0.18
HIPS M2-S1 Mod 174 0.26
HIPS M1-S2 Mod 198 0.22
HIPS M2-S2 Mod 165 0.16

96
Como se observó en las imágenes obtenidas por TEM, ocurrieron algunos cambios en la
morfología de la fase elastomérica con la adición de partículas de SiO2 e Mg(OH)2. Dichos
cambios en el tamaño y morfología de partículas elastoméricas se debio ,
fundamentalmente a la combinación de dos factores como el incremento en la viscosidad
de la solución St/PS, por el aumento en Mw, lo que cambio la relación de viscosidades de
las soluciones St/PB y St/PS , asi como cambios en la tensión superficial que provocaron
una cambio el grado de injerto de la fse hulosa, ambos factores provocados por la
modificación de la cinética de polimerización debido a la presencia de las nano y
micropartículas.
7.2.5. Resistencia al impacto.
Si bien la morfología de un HIPS, asi como sus propiedades fisicoquímicas como, el GI, el
CG y la DPM, definen la resistencia al impacto y la tenacidad de este tipo de materiales, en
el caso de compuestos con nanopartículas, como es este caso, no esta completamente
dilucidado. En el caso de la morfología de los materiales antes descrita y parámetros como
Dp y , no existe la correlación típica con la resistencia al impacto debido a la presencia
de las nanopartículas. Por ejemplo, de la Tabla 17 se puede observar que el HIPS S1, a
pesar de que presentó valores de Dp y mayores que el HIPS B, la resistencia al impacto
decae aproximadamente un 36 %, cuando lo normal es que debe presentar un
incremento. Por otro lado, el HIPS S1 Mod presenta un decremento tanto en Dp como en
, sin embargo su resistencia al impacto se incrementó hasta un 153 %,
aproximadamente, con respecto al HIPS B. Evidentemente la presencia de las
nanopartículas en este material provocaron un incremento en la resistencia al impacto.
En la Tabla 17 se puede apreciar que los nanocompuestos con un contenido menor de
nano partículas de SiO2 modificadas superficialmente (2.5 % en peso de nanopartículas de
SiO2 modificadas), presentaron una más alta resistencia al impacto en comparación al
HIPS B; este mismo efecto no se observó con la presencia de las nanopartículas de SiO2 sin
modificar superficialmente. Tratando de explicar este efecto, por la acción que las
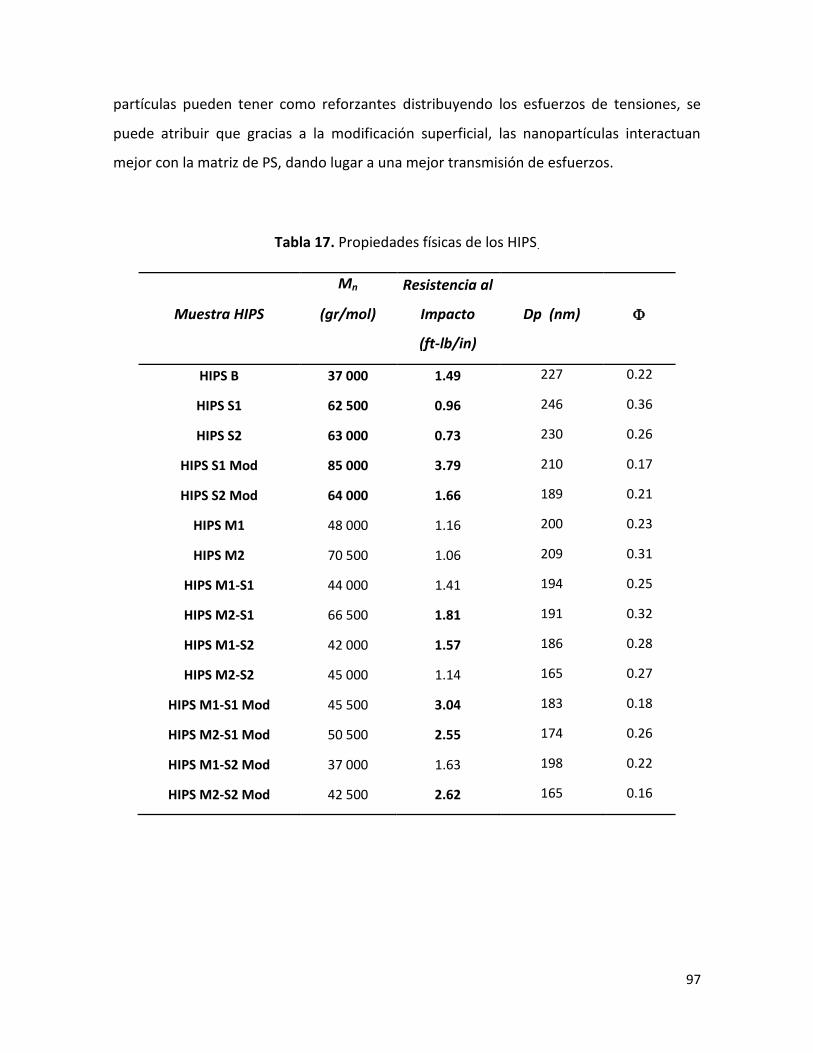
97
partículas pueden tener como reforzantes distribuyendo los esfuerzos de tensiones, se
puede atribuir que gracias a la modificación superficial, las nanopartículas interactuan
mejor con la matriz de PS, dando lugar a una mejor transmisión de esfuerzos.
Tabla 17. Propiedades físicas de los HIPS.
Muestra HIPS
Mn Resistencia al
Impacto
(ft-lb/in)
(gr/mol) Dp (nm)
HIPS B 37 000 1.49 227 0.22
HIPS S1 62 500 0.96 246 0.36
HIPS S2 63 000 0.73 230 0.26
HIPS S1 Mod 85 000 3.79 210 0.17
HIPS S2 Mod 64 000 1.66 189 0.21
HIPS M1 48 000 1.16 200 0.23
HIPS M2 70 500 1.06 209 0.31
HIPS M1-S1 44 000 1.41 194 0.25
HIPS M2-S1 66 500 1.81 191 0.32
HIPS M1-S2 42 000 1.57 186 0.28
HIPS M2-S2 45 000 1.14 165 0.27
HIPS M1-S1 Mod 45 500 3.04 183 0.18
HIPS M2-S1 Mod 50 500 2.55 174 0.26
HIPS M1-S2 Mod 37 000 1.63 198 0.22
HIPS M2-S2 Mod 42 500 2.62 165 0.16

98
Comparando la resistencia al impacto de los nanocompuestos de HIPS con baja
concentración de SiO2 Modificado con la resistencia del HIPS B se tiene lo siguiente:
En el HIPS S1 Mod la resistencia fue 3.79 ft-lb/in, alcanzando un 153.5 % de
incremento en la resistencia al impacto con respecto al HIPS B.
En el HIPS M1-S1 Mod la resistencia fue 3.04 ft-lb/in, alcanzando un 103.5 % de
incremento en la resistencia al impacto con respecto al HIPS B.
En el HIPS M2-S1 Mod la resistencia fue 2.55 ft-lb/in, alcanzando un 70.5 % de
incremento en la resistencia al impacto con respecto al HIPS B.
En contraparte, la resistencia del HIPS B en comparación con los nanocompuestos de HIPS
con alta concentración de SiO2 modificado se tiene que:
En el HIPS S2 Mod la resistencia fue de 1.66 ft-lb/in, un 11.3 % de incremento en la
resistencia al impacto con respecto al HIPS B.
En el HIPS M1-S2 Mod la resistencia fue de 1.63 ft-lb/in, un 8.9 % de incremento en
la resistencia al impacto con respecto al HIPS B.
En el HIPS M2-S2 Mod la resistencia fue de 2.63 ft-lb/in, un 75.7 % de incremento
en la resistencia al impacto con respectoal HIPS B.
El efecto de aumento a la resistencia al impacto de los HIPS nano y microcompuestos,
además de los combinados, descrita anteriormente, se puede deber a que las
nanopartículas modificadas se ubican preferentemente en la fase elastomérica y
presentan una mejor interacción con el PS ocluido, dicha oclusión de nanopartículas de
SiO2 en la fase elastomérica probablemente se debe al aumento en la viscosidad del
sistema (efecto gel, debido a la gran área superficial de las partículas) y al momento de la
inversión de fases, dichos dominios altamente viscosos fueron ocluidos con el PB de
menor viscosidad, durante la etapa de pprepolimerización. Por lo anterior, se puede decir
que a menores concentraciones, las nanopartículas de SiO2 se distribuyeron mejor en el

99
sistema interactuando mejor con las cadenas poliméricas por lo que mejoró la resistencia
al impacto.
Por otro lado, al intentar relacionar la resistencia al impacto con el Mn (Ver tabla 17) se
puede observar en el HIPS S1 Mod y el HIPS S2 Mod un peso molecular de 85,000 y 64,000
gr/mol, respectivamente, valores superioriores al Mn de 37,000 g/mol del HIPS B.
Mientras que la resistencia al impacto para los HIPS S1 Mod y HIPS S2 Mod fue de 3.8 y
1.7 ft-lb/in, respectivamente, ambos presentaron una mayor resistencia al impacto
respecto al HIPS B, sin embargo, se puede observar que el HIPS S1 Mod con mayor Mn
presentó una resistencia al impacto superior a la presentada por el HIPS B y el HIPS S2
Mod. Con lo anterior podríamos atribuir que el aumento en el Mn también contribuyó a
un aumento en la resistencia al impacto.
Por otro lado, en el caso del HIPS S1 y el HIPS S2 sus Mn fueron de 62,500 y 63,000 gr/mol
respectivamente, también mayores al del HIPS B, sin embargo presentaron valores de
resistencia al impacto menores al HIPS B; con lo cual no podemos relacionar , al menos no
de manera directa, el cambio de la resistencia al impacto con el Mn, pero si con la
presencia de la modificación superficial en las nanopartículas de SiO2.
Al relacionar el cambio de la resistencia al impacto de los compuestos de HIPS, con el
tamaño de partícula de la fase elastomerica y el GI, la presencia de particulas durante la
sintesis disminuye el GI y cambia el tamaño de particula, como se vio en la seccion 7.2.6.
Relacionando la resistencia al impacto con el tamaño de particula y el GI (ver la seccion
7.2.6.), se puede concluir que en algunos casos la presencia de nano y micro partículas de
SiO2 e Mg(OH)2 disminuye el GI lo que puede afectar la resistencia al impacto. Como se
mencionó en la sección 7.2.6 en los HIPS S1 y HIPS S2 el GI disminuyó por lo que el CI
provee de un menor efecto compatibilizante, por lo que la resistencia al impacto puede
disminuir por los valores bajos de este parámetro en todos los HIPS nano y/o
microcompuestos. El HIPS S1 y el HIPS S2 presentaron una resistencia al impacto de 0.96 y
0.73 ft-lb/in, respectivamente, menor al 1.50 ft-lb/in del HIPS B. En cambio en el HIPS S1
Mod y el HIPS S2 Mod hubo disminución en el Dp, especialmente en el HIPS S1 Mod, por

100
lo que se puede concluir que el HIPS S1 Mod presentó un mayor GI y por lo tanto una
mayor resistencia al impacto (3.79 ft-lb/in). Una relación compleja entre el cambio en el
GI, DPM y la morfología provocó que aumentara, o disminuyera, la resistencia al impacto.
Por su parte, en el caso de la incorporación de micropartículas de Mg(OH)2 modificadas, se
observó (Tabla 17) que la resistencia al impacto disminuyó, efecto esperado por la escala
dimensional de dichas partículas, lo que hace que presenten menor área superficial y por
lo tanto menor interacción con la matriz, actuando como generadores de fallas. Sin
embargo, al realizar las combinaciones de micro y nanopartículas, en general las
formulaciones que contienen SiO2 sin modificar presentaron un decremento en la
resistencia al impacto, corroborando lo discutido anteriormente respecto a los HIPS S1 y
S2, donde se utilizaron nanopartículas sin modificar y la resistencia al impacto decreció.
Por su parte, en la serie de HIPS Si-Mi Mod se observó un aumento al impacto del 75 %. En
este caso la combinación de las nanopartículas causo un efecto de aumento en resistencia
al impacto, que se atribuye específicamente a la presencia de las nanopartículas de SiO2
modificadas en las diferentes combinaciones y que es evidente que la presencia de las
micropartículas de Mg(OH)2 inhibió ligeramente dicho incremento. Y nuevamente se
corroboró que el incremento en el contenido de nanopartículas de SiO2 da lugar a un
aumento en resistencia al impacto menor con respecto al proveido cuando se incorpora
un bajo contenido de las mismas.
7.2.6 Fracción volumen de fase dispersa.
Para determinar el contenido de la fase elastómerica dispersa por otra forma, diferente al
contenido de gel y grado de injerto, se midió la fracción volumen de las partículas de hule
en imágenes obtenidas por TEM. Considerando que las partículas presentan forma de
elipse, se determinó el diámetro mayor y menor, y se obtuvo el promedio de ambos

101
valores; se calculó el área ocupada por las partículas de hule en el área total del HIPS
(matriz y fase dispersa).
Fracción volumen con Contenido de Gel
Comparando la fracción volumen de hule con el contenido de gel (Ver Tabla 18) se
observa que, en todos los casos, la fracción volumen de la fase dispersa es mayor al 8%
(cantidad de hule adicionada inicialmente), mientras que el contenido de Gel que se
obtuvo después de centrifugar la muestra es mucho menor al volumen de la fase
elastomérica que tiene el HIPS antes de la centrifugación.
Este fenómeno se podría deber al efecto observado por Yogesha Subbaiah y
colaboradores 63; ellos observaron que durante la estimación del contenido de gel puede
haber pérdida de partículas cuando los dominios de hule son menores a 1 m durante la
ultracentrifugación a 20 000 rpm, ya que algunos quedan suspendidos en el tolueno (entre
mas pequeños son los dominios son mas propensos a quedar suspendidos),
sedimentándose una menor cantidad de partículas de la fase elastomérica a la cantidad de
partículas presente en la muestra evaluada; por lo que al hacer el análisis gravimétrico
(secando y posteriormente pesando la muestra) se determina un contenido de gel (hule
con oclusiones de PS) inferior al contenido de gel que realmente presenta la muestra. Sin
embargo, todos los diámetros de partículas están en el rango de 160 a 240 nm, por lo que
si los bajos contenidos de gel se debieran al efecto de pérdida de partículas, al quedar
suspendidas o disolverse, en todos los nanocompuestos de HIPS, y en el HIPS de
referencia, sintetizados se hubiera presentado bajo contenido de gel.
Otra explicación podría ser que el hule de los dominios se disolvió, para lo cual el hule
debió estar pobremente entrecruzado.
Peng 17 estudió las reacciones de injerto y entrecruzamiento iniciadas térmicamente y
encontró que la densidad de injerto y el grado de entrecruzamiento se incrementan con el
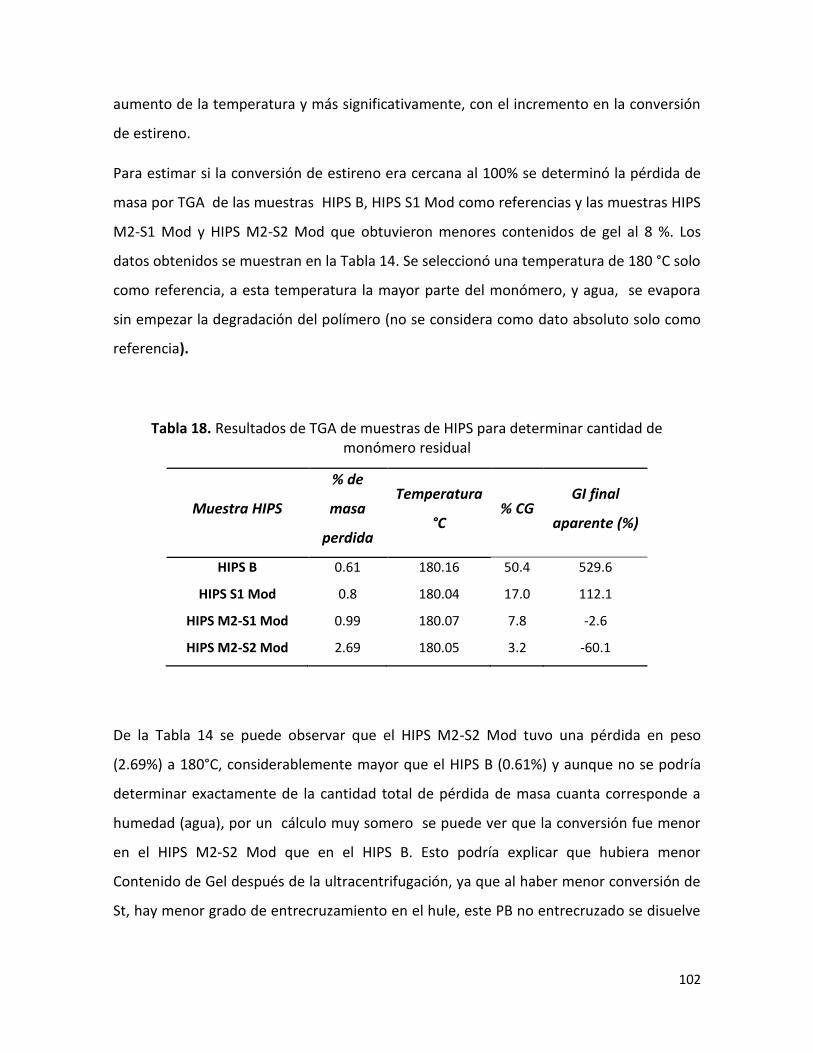
102
aumento de la temperatura y más significativamente, con el incremento en la conversión
de estireno.
Para estimar si la conversión de estireno era cercana al 100% se determinó la pérdida de
masa por TGA de las muestras HIPS B, HIPS S1 Mod como referencias y las muestras HIPS
M2-S1 Mod y HIPS M2-S2 Mod que obtuvieron menores contenidos de gel al 8 %. Los
datos obtenidos se muestran en la Tabla 14. Se seleccionó una temperatura de 180 °C solo
como referencia, a esta temperatura la mayor parte del monómero, y agua, se evapora
sin empezar la degradación del polímero (no se considera como dato absoluto solo como
referencia).
Tabla 18. Resultados de TGA de muestras de HIPS para determinar cantidad de monómero residual
Muestra HIPS
% de
masa
perdida
Temperatura
°C % CG
GI final
aparente (%)
HIPS B 0.61 180.16 50.4 529.6
HIPS S1 Mod 0.8 180.04 17.0 112.1
HIPS M2-S1 Mod 0.99 180.07 7.8 -2.6
HIPS M2-S2 Mod 2.69 180.05 3.2 -60.1
De la Tabla 14 se puede observar que el HIPS M2-S2 Mod tuvo una pérdida en peso
(2.69%) a 180°C, considerablemente mayor que el HIPS B (0.61%) y aunque no se podría
determinar exactamente de la cantidad total de pérdida de masa cuanta corresponde a
humedad (agua), por un cálculo muy somero se puede ver que la conversión fue menor
en el HIPS M2-S2 Mod que en el HIPS B. Esto podría explicar que hubiera menor
Contenido de Gel después de la ultracentrifugación, ya que al haber menor conversión de
St, hay menor grado de entrecruzamiento en el hule, este PB no entrecruzado se disuelve

103
en el tolueno en lugar de solo hincharse como el PB entrecruzado, por lo cual solo
precipito una pequeña porción de la fase dispersa de PB y CI (la que logro entrecruzarse).
Por lo anterior el contenido de gel y el grado de hinchamiento, con excepción del HIPS B,
no serían valores fiables. La presencia de partículas reduce la velocidad de la
polimerización y el grado de injerto; estas (las partículas) interaccionan con los radicales
libres, disminuyendo la concentración de especies reactiva, lo que afecta el grado de
injerto y el contenido de gel. Tambien se concluyó que la presencia de las nano y
micropartículas dificulta las reacciones de entrecruzamiento del PB, por lo cual al
ultracentrifugar, el hule queda disuelto / suspendido en el Tolueno.
Aunque al parecer el SiO2 ocasiona mayor pérdida en el grado de entrecruzamiento, para
determinar que partícula ( Mg(OH)2 , SiO2 o SiO2 Mod ) ocasiona mayor pérdida de grado
de entrecruzamiento se necesita realizar un estudio más detallado, para verificar si esto se
puede compensar con un mayor tiempo de polimerización en masa estática, mayor
concentración de iniciador o mayor temperatura.
7.2.7 Flamabilidad
Tiempo de quemado (Prueba de quemado Horizontal)
En la Tabla 19 se muestran los resultados obtenidos de la prueba de flamabilidad. La
adición de 2.5 % en peso de nanopartículas de sílice modificada superficialmente ( HIPS S1
Mod) presentó una disminución en el tiempo necesario para que se consumiera la
probeta de prueba, aumentando la velocidad de quemado, mientras con la adición de 5 %
en peso de sílice modificada superficialmente (HIPS S2 Mod) se mantuvo el tiempo de
quemado . Feng Yang y colaboradores5 observaron que en la prueba de quemado
horizontal de nanocompuestos de poliestireno con Sílice modificada superficialmente se
quemaron en menor tiempo que el polímero puro, similar a lo observado en el

104
nanocompuestos de menor concentración de sílice modificada (HIPS S1 Mod). Sin
embargo la adición de sílice sin modificación superficial aumento de 104.6 segundos (en
el HIPS B) a 127 y 130 para las concentración de 2.5 % (HIPS S1) y 5 % en peso (HIPS S2),
repectivamente.
Se esperaba que los tiempos de quemado para los nanocompuestos con SiO2 sin
modificación superficial fueran similares a los de los nanocompuestos con SiO2
modificada superficialmente, sin embargo los tiempos de quemado fueron diferentes para
los dos tipos de nanopartículas de SiO2, similar a lo ocurrido en los resultados de la
prueba de la resistencia al impacto (los nanocompuestos con SiO2 modificada
superficialmente presentaron mayores valores de resistencia al impacto en los productos
finales que los nanocompuestos con SiO2 sin modificación superficial). Como se menciono
en el párrafo anterior, los nanocompuestos con SiO2 modificado superficialmente
presentaron menores tiempos de quemado (o mayores velocidades de quemado) que los
nanocompuestos con SiO2 sin modificación superficial, lo cual podría explicarse que al
tener en su composición un porcentaje de polímero, la cantidad de SiO2 es menor en las
nanopartículas modificadas superficialmente; sin embargo el nanocompuesto con mayor
concentración de SiO2 modificado (HIPS S2 Mod) presentó menor tiempo de quemado
que el nanocompuesto con menor concentración de SiO2 sin modificación superficial (HIPS
S1 Mod). Un mecanismo al que se le atribuye el efecto de retardancia a la flama al
incorporar nanopartículas de SiO2 a polímeros, es el aumento, que ocasionan las
partículas, en la viscosidad del fundido del polímero 50. Por lo anterior, se puede atribuir
que las nanopartículas sin modificación superificial causan un mayor aumento de
viscosidad del fundido, ya que la modificación superficial cubre los nanoporos de las
partículas de SiO2, al introducirse cadenas de PS en los poros de la sílice, por lo cual, las
partículas modificadas, presentan una menor área superficial.
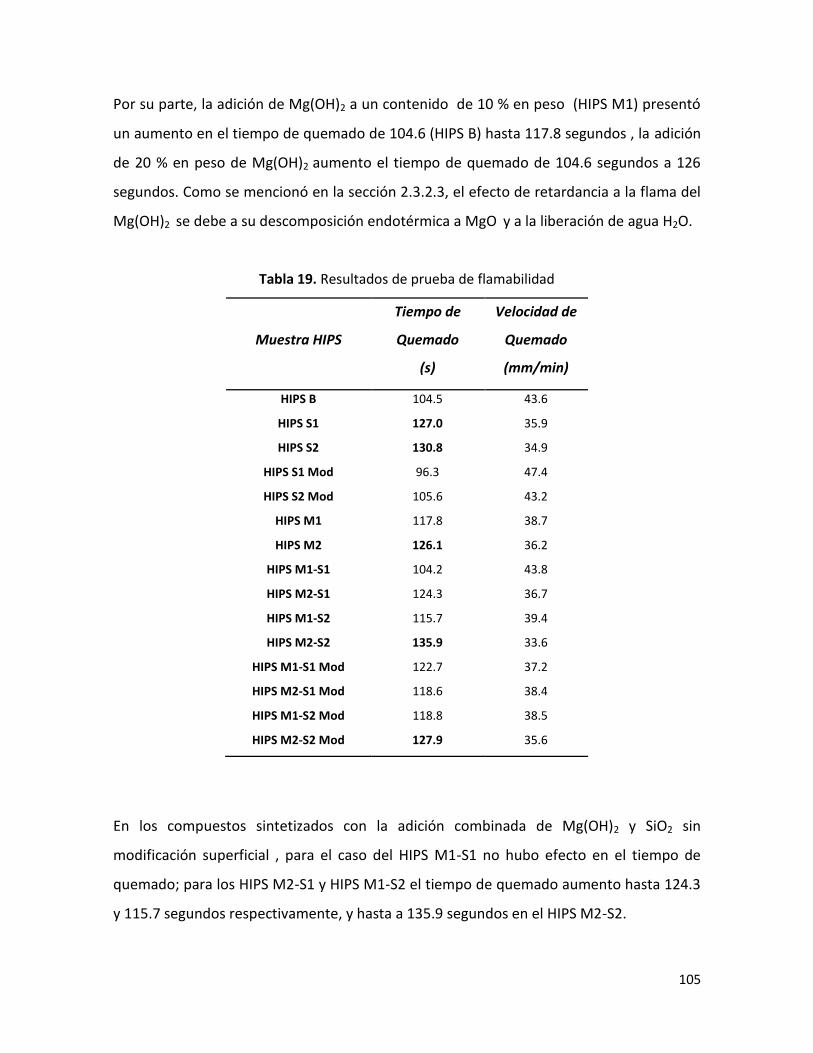
105
Por su parte, la adición de Mg(OH)2 a un contenido de 10 % en peso (HIPS M1) presentó
un aumento en el tiempo de quemado de 104.6 (HIPS B) hasta 117.8 segundos , la adición
de 20 % en peso de Mg(OH)2 aumento el tiempo de quemado de 104.6 segundos a 126
segundos. Como se mencionó en la sección 2.3.2.3, el efecto de retardancia a la flama del
Mg(OH)2 se debe a su descomposición endotérmica a MgO y a la liberación de agua H2O.
Tabla 19. Resultados de prueba de flamabilidad
Muestra HIPS
Tiempo de
Quemado
(s)
Velocidad de
Quemado
(mm/min)
HIPS B 104.5 43.6
HIPS S1 127.0 35.9
HIPS S2 130.8 34.9
HIPS S1 Mod 96.3 47.4
HIPS S2 Mod 105.6 43.2
HIPS M1 117.8 38.7
HIPS M2 126.1 36.2
HIPS M1-S1 104.2 43.8
HIPS M2-S1 124.3 36.7
HIPS M1-S2 115.7 39.4
HIPS M2-S2 135.9 33.6
HIPS M1-S1 Mod 122.7 37.2
HIPS M2-S1 Mod 118.6 38.4
HIPS M1-S2 Mod 118.8 38.5
HIPS M2-S2 Mod 127.9 35.6
En los compuestos sintetizados con la adición combinada de Mg(OH)2 y SiO2 sin
modificación superficial , para el caso del HIPS M1-S1 no hubo efecto en el tiempo de
quemado; para los HIPS M2-S1 y HIPS M1-S2 el tiempo de quemado aumento hasta 124.3
y 115.7 segundos respectivamente, y hasta a 135.9 segundos en el HIPS M2-S2.

106
En los nanocompuestos de HIPS con SiO2 modificada superficialmente y Mg(OH)2 , el HIPS
M1-S1 Mod tuvo un tiempo de quemado de 122.7 segundos , el HIPS M2-S1 Mod dio un
tiempo de quemado de 118.6 segundos , el HIPS M1-S2 Mod obtuvo un tiempo de
quemado de 118.8 segundos y el HIPS M2-S2 Mod tuvo un tiempo de quemado de 128
segundos .
Con los datos descritos anteriormente se tuvo como resultado que las nanopartículas de
SiO2 no presentaron un efecto sinérgico en combinación con las micropartículas de
Mg(OH)2, esto atribuido a la encapsulación y aglomeración de las nanopartículas de SiO2
en la fase elastomérica. El nanocompuesto con un mayor tiempo de quemado fue el HIPS
M2-S2 (135.9 segundos), mayor que el Mg(OH)2 y el SiO2 por separado (126 segundos para
el HIPS M2, y 130.8 segundos para el HIPS S2) ; pero en la mayoría de nanocompuestos
con combinación de nanopartículas de SiO2 (modificadas y sin modificar) con
micropartículas de Mg(OH)2 no se observa un aumento sustancial en el tiempo de
quemado.
Cabe mencionar que es difícil hacer una comparación rigurosa con los estudios
presentados por los grupos de investigación de Katančić 3,4 y de Feng Yang 5,6, ya que
utilizaron concentraciones y/o agentes retardantes a la flama de diferente naturaleza y
prepararon los nanocompuestos en mezclado en fundido.
Feng Yang y colaboradores5, utilizaron una concentración de 10 % de nanopartículas de
SiO2 y 35% de BrPS como agente retardante a la flama en la prueba donde apreciaron un
mayor efecto sinérgico del SiO2 de retardancia a la flama. Estas concentraciones son
mayores a las utilizadas en este trabajo.
Por su parte en los trabajos de Katančić 3,4 y colaboradores, en un estudio3 prepararon
nanocompuestos de HIPS con 4 % de sílice y 8 % de agente retardante a la flama
(polifosfato de amonio y polifosfato orgánico), los cuales tienen un mecanismo de acción
muy diferente al Mg(OH)2 . Y en otro estudio4 utilizaron 18 % de EVA (etilenvinilacetato),
18 % de Al(OH)3 y 8% de SiO2 prepararando los nanocompuestos mediante mezclado en
fundido, para evaluar las propiedades de retardancia a la flama. Como se observa las

107
formulaciones son diferentes a las utilizadas en este trabajo, aun asi se consideran como
buenos antescedentes del efecto sinérgico que tiene el SiO2 con agentes retardantes a la
flama tradicionales; por lo cual a una concentración mayor de agente retardante a la
flama (Mg(OH)2) pueda presentarse un efecto sinérgico , pero mediante mezclado durante
la polimerización In situ , es algo difícil aumentar las concentraciones de micropartículas
de Mg(OH)2 y ,especialmente, de las nanopartículas de SiO2 por el aumento en la
viscosidad (la incorporación de SiO2 aumentaba drásticamente la viscosidad) y la
tendencia de las nanopartículas de SiO2 de aglomerarse y situarse en la fase elastomérica.
Efecto de la aglomeración y localización de las nanopartículas de SiO2 en el tiempo de
quemado del nanocompuesto final.
En las morfologías presentadas en la sección 7.2.6 de los HIPS sintetizados se observo en
algunas secciones, partículas aglomeradas y dentro de la fase elastomérica. Las imágenes
obtenidas mediante TEM, indican que las particulas de SiO2 sin y con modificación
superficial presentaron el mismo problema. La aglomeración nanopartículas de SiO2 se
puede atribuir a la incompatibilidad de fases de PB/St y PS/St; cuando ocurre la separación
de las fases, las nanopartículas de SiO2, especialmente las modificadas mediante plasma
con estireno, se dispersan, probablemente, solo en la fase de PS/St por lo que tienden
mas facilmente a agruparse ; al ir aumentando la fase de PS/St, también aumenta la
viscosidad y, las nanopartículas no logran dispersarse bien en toda esta fase que esta
aumentando de volumen , y al ocurrir el proceso de inversión de fases las nanopartículas
se aglomeran y quedan atrapadas en las oclusiones de PS al ensamblarse o formarse las
partículas de hule, quedando las nanopartículas dentro de la fase elastomérica. Para
confirmar este mecanismo, o elucidar cual fue el mecanismo que ocasionó la
aglomeración de las nanopartículas y la localización dentro de la fase elastomérica, se
requiririá realizar otros estudios.
Para asegurar si las nanopartículas de SiO2 solo se distribuyen en la fase de PS/St al ocurrir
la inversión de fases se podría hacer la siguiente prueba: mezclar y distribuir las
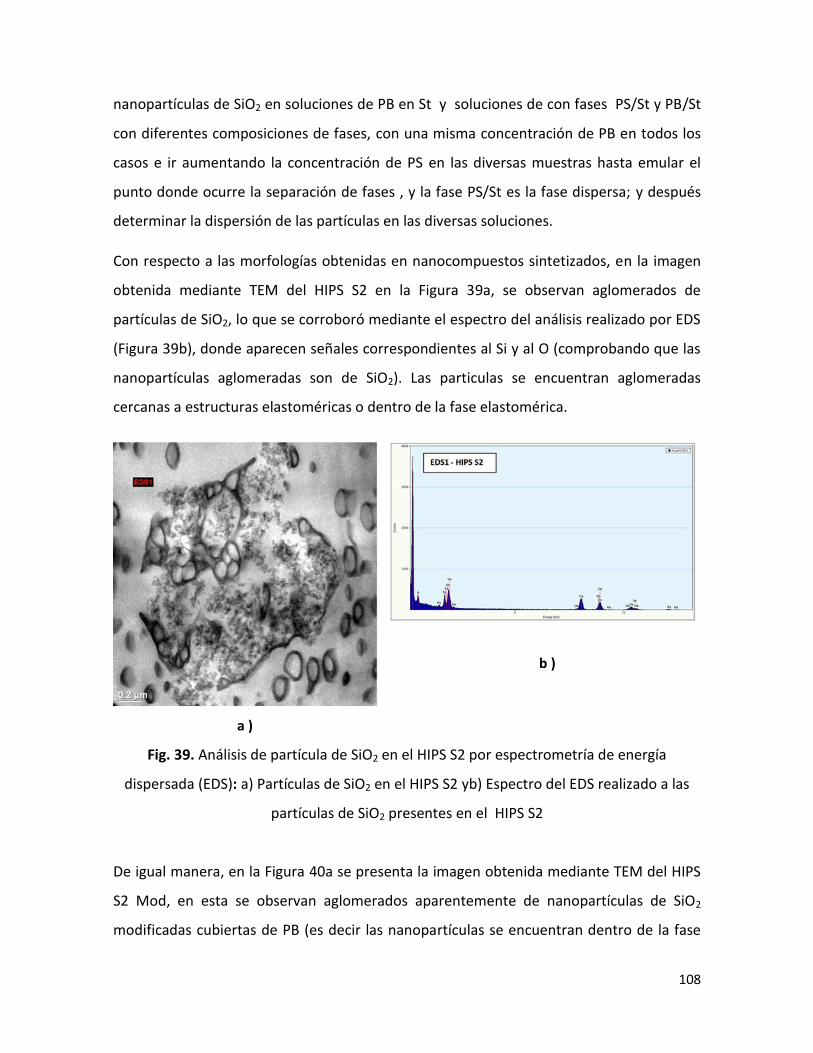
108
nanopartículas de SiO2 en soluciones de PB en St y soluciones de con fases PS/St y PB/St
con diferentes composiciones de fases, con una misma concentración de PB en todos los
casos e ir aumentando la concentración de PS en las diversas muestras hasta emular el
punto donde ocurre la separación de fases , y la fase PS/St es la fase dispersa; y después
determinar la dispersión de las partículas en las diversas soluciones.
Con respecto a las morfologías obtenidas en nanocompuestos sintetizados, en la imagen
obtenida mediante TEM del HIPS S2 en la Figura 39a, se observan aglomerados de
partículas de SiO2, lo que se corroboró mediante el espectro del análisis realizado por EDS
(Figura 39b), donde aparecen señales correspondientes al Si y al O (comprobando que las
nanopartículas aglomeradas son de SiO2). Las particulas se encuentran aglomeradas
cercanas a estructuras elastoméricas o dentro de la fase elastomérica.
Fig. 39. Análisis de partícula de SiO2 en el HIPS S2 por espectrometría de energía
dispersada (EDS): a) Partículas de SiO2 en el HIPS S2 yb) Espectro del EDS realizado a las
partículas de SiO2 presentes en el HIPS S2
De igual manera, en la Figura 40a se presenta la imagen obtenida mediante TEM del HIPS
S2 Mod, en esta se observan aglomerados aparentemente de nanopartículas de SiO2
modificadas cubiertas de PB (es decir las nanopartículas se encuentran dentro de la fase
a )
b )
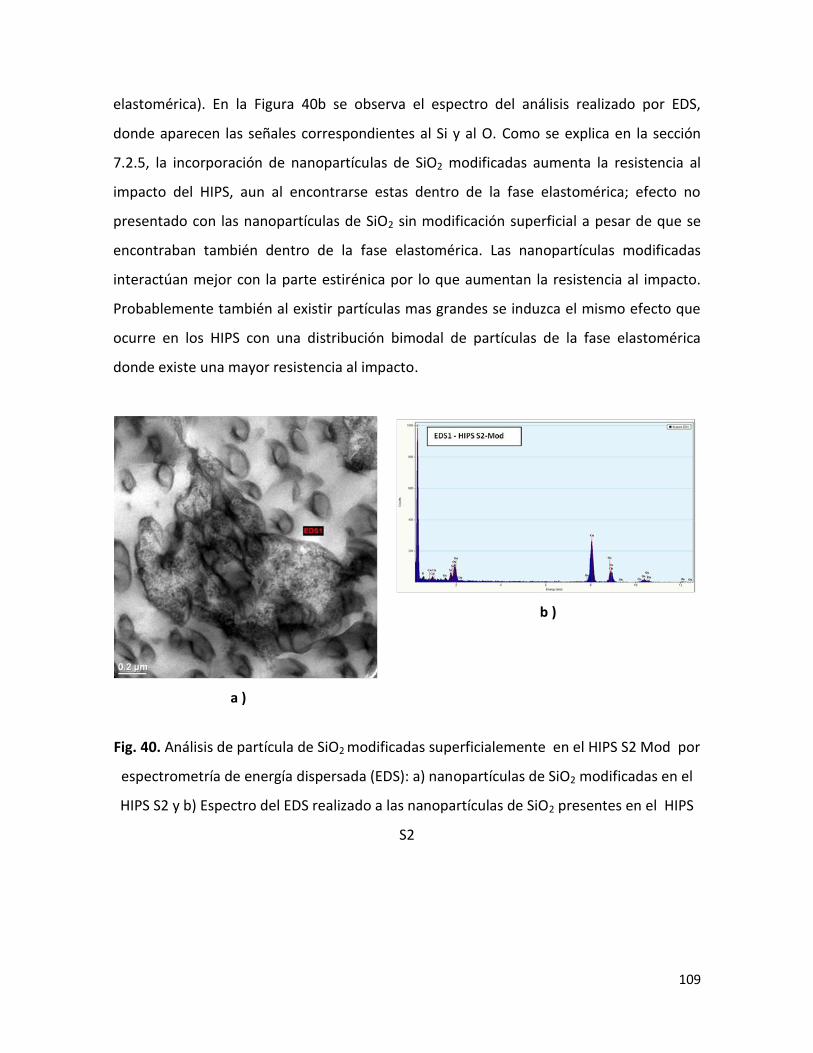
109
elastomérica). En la Figura 40b se observa el espectro del análisis realizado por EDS,
donde aparecen las señales correspondientes al Si y al O. Como se explica en la sección
7.2.5, la incorporación de nanopartículas de SiO2 modificadas aumenta la resistencia al
impacto del HIPS, aun al encontrarse estas dentro de la fase elastomérica; efecto no
presentado con las nanopartículas de SiO2 sin modificación superficial a pesar de que se
encontraban también dentro de la fase elastomérica. Las nanopartículas modificadas
interactúan mejor con la parte estirénica por lo que aumentan la resistencia al impacto.
Probablemente también al existir partículas mas grandes se induzca el mismo efecto que
ocurre en los HIPS con una distribución bimodal de partículas de la fase elastomérica
donde existe una mayor resistencia al impacto.
Fig. 40. Análisis de partícula de SiO2 modificadas superficialemente en el HIPS S2 Mod por
espectrometría de energía dispersada (EDS): a) nanopartículas de SiO2 modificadas en el
HIPS S2 y b) Espectro del EDS realizado a las nanopartículas de SiO2 presentes en el HIPS
S2
a )
b )
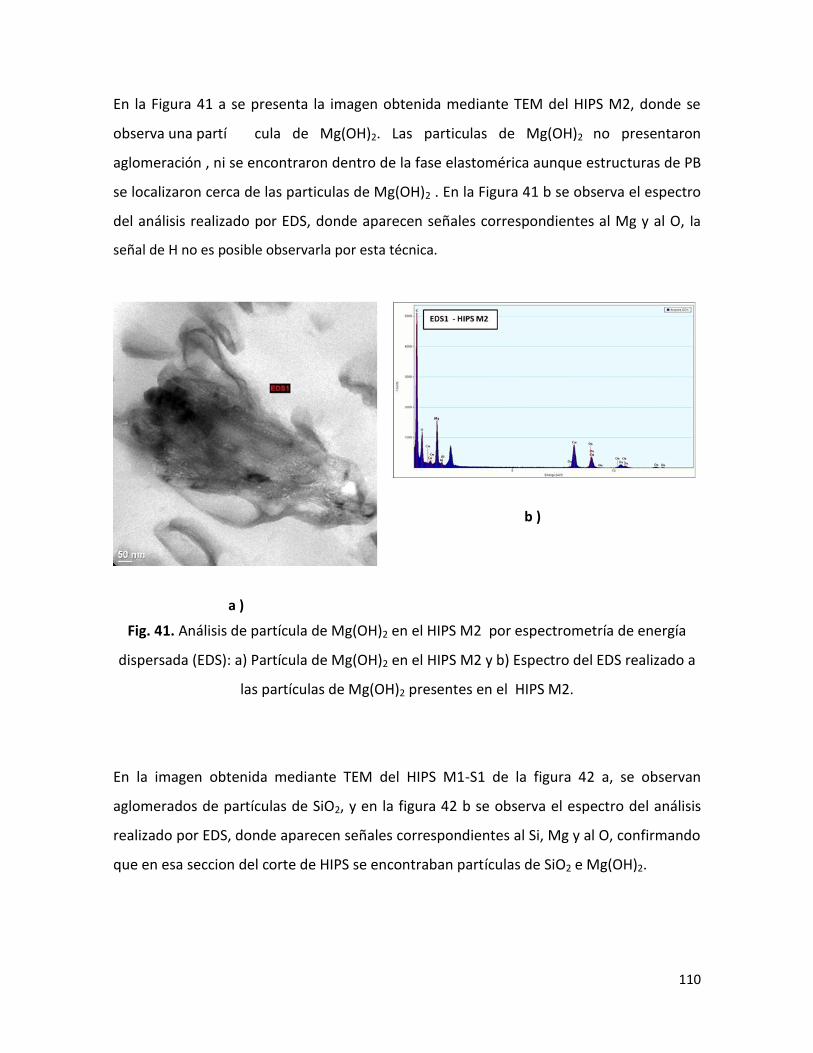
110
En la Figura 41 a se presenta la imagen obtenida mediante TEM del HIPS M2, donde se
observa una partí cula de Mg(OH)2. Las particulas de Mg(OH)2 no presentaron
aglomeración , ni se encontraron dentro de la fase elastomérica aunque estructuras de PB
se localizaron cerca de las particulas de Mg(OH)2 . En la Figura 41 b se observa el espectro
del análisis realizado por EDS, donde aparecen señales correspondientes al Mg y al O, la
señal de H no es posible observarla por esta técnica.
Fig. 41. Análisis de partícula de Mg(OH)2 en el HIPS M2 por espectrometría de energía
dispersada (EDS): a) Partícula de Mg(OH)2 en el HIPS M2 y b) Espectro del EDS realizado a
las partículas de Mg(OH)2 presentes en el HIPS M2.
En la imagen obtenida mediante TEM del HIPS M1-S1 de la figura 42 a, se observan
aglomerados de partículas de SiO2, y en la figura 42 b se observa el espectro del análisis
realizado por EDS, donde aparecen señales correspondientes al Si, Mg y al O, confirmando
que en esa seccion del corte de HIPS se encontraban partículas de SiO2 e Mg(OH)2.
a )
b )
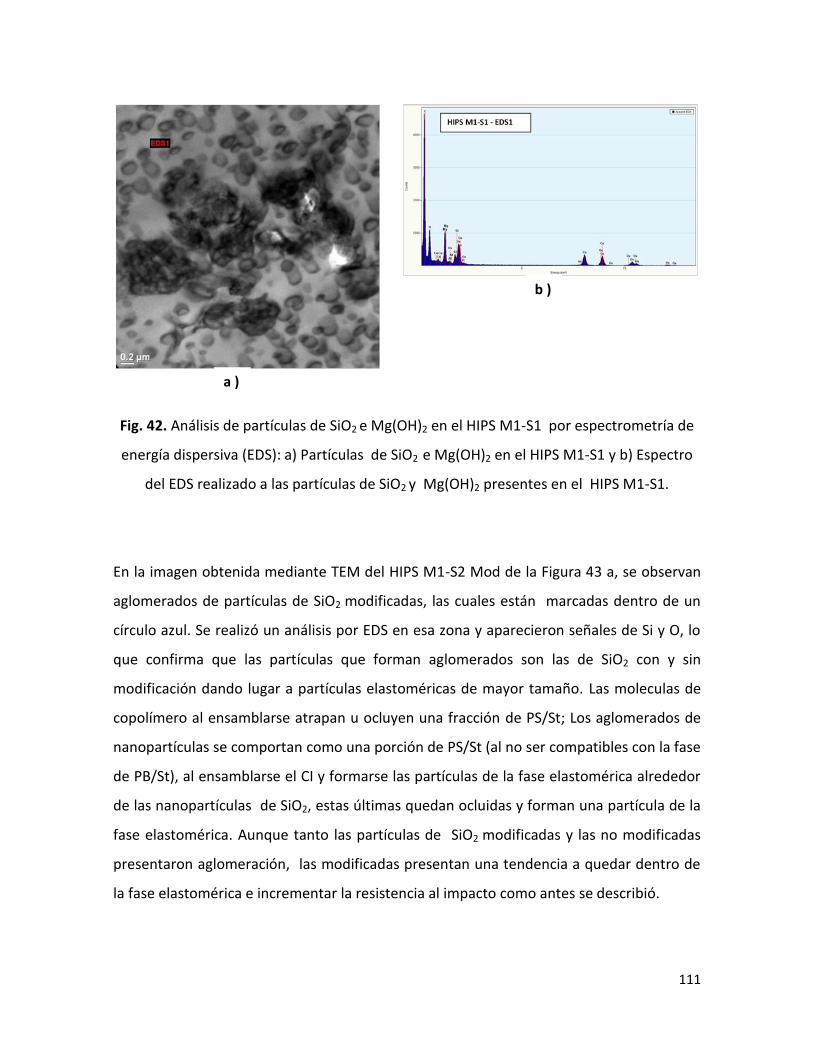
111
Fig. 42. Análisis de partículas de SiO2 e Mg(OH)2 en el HIPS M1-S1 por espectrometría de
energía dispersiva (EDS): a) Partículas de SiO2 e Mg(OH)2 en el HIPS M1-S1 y b) Espectro
del EDS realizado a las partículas de SiO2 y Mg(OH)2 presentes en el HIPS M1-S1.
En la imagen obtenida mediante TEM del HIPS M1-S2 Mod de la Figura 43 a, se observan
aglomerados de partículas de SiO2 modificadas, las cuales están marcadas dentro de un
círculo azul. Se realizó un análisis por EDS en esa zona y aparecieron señales de Si y O, lo
que confirma que las partículas que forman aglomerados son las de SiO2 con y sin
modificación dando lugar a partículas elastoméricas de mayor tamaño. Las moleculas de
copolímero al ensamblarse atrapan u ocluyen una fracción de PS/St; Los aglomerados de
nanopartículas se comportan como una porción de PS/St (al no ser compatibles con la fase
de PB/St), al ensamblarse el CI y formarse las partículas de la fase elastomérica alrededor
de las nanopartículas de SiO2, estas últimas quedan ocluidas y forman una partícula de la
fase elastomérica. Aunque tanto las partículas de SiO2 modificadas y las no modificadas
presentaron aglomeración, las modificadas presentan una tendencia a quedar dentro de
la fase elastomérica e incrementar la resistencia al impacto como antes se describió.
a )
b )
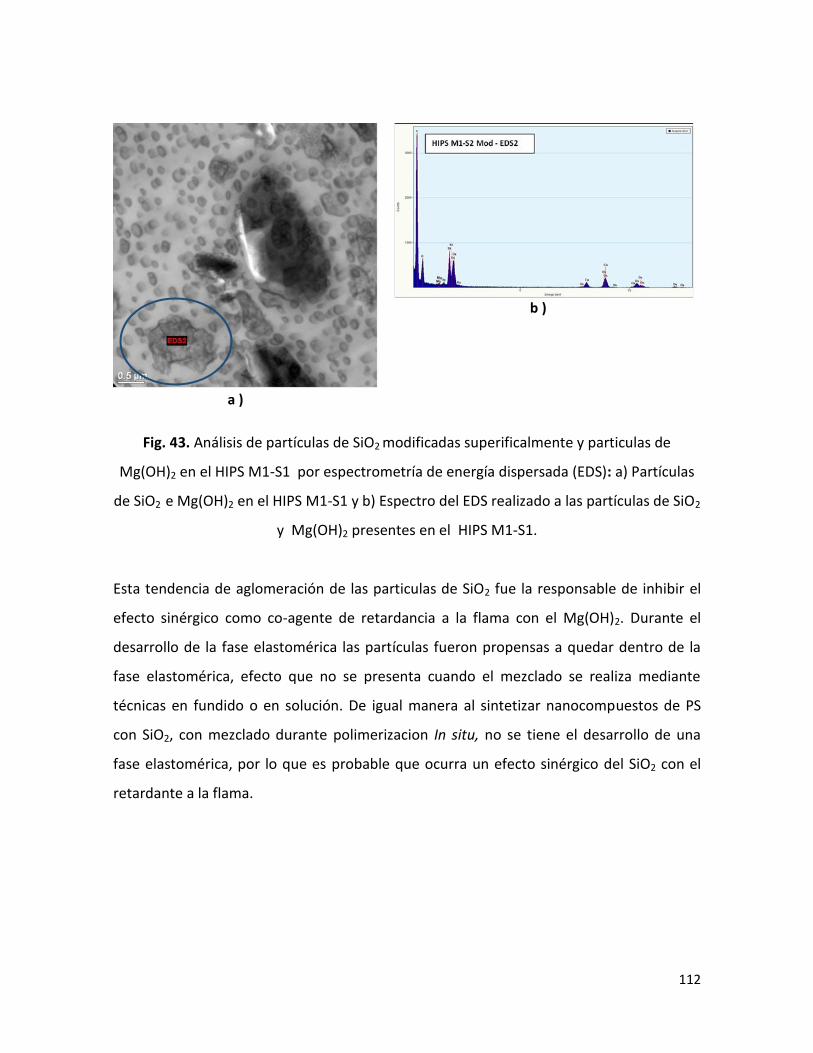
112
Fig. 43. Análisis de partículas de SiO2 modificadas superificalmente y particulas de
Mg(OH)2 en el HIPS M1-S1 por espectrometría de energía dispersada (EDS): a) Partículas
de SiO2 e Mg(OH)2 en el HIPS M1-S1 y b) Espectro del EDS realizado a las partículas de SiO2
y Mg(OH)2 presentes en el HIPS M1-S1.
Esta tendencia de aglomeración de las particulas de SiO2 fue la responsable de inhibir el
efecto sinérgico como co-agente de retardancia a la flama con el Mg(OH)2. Durante el
desarrollo de la fase elastomérica las partículas fueron propensas a quedar dentro de la
fase elastomérica, efecto que no se presenta cuando el mezclado se realiza mediante
técnicas en fundido o en solución. De igual manera al sintetizar nanocompuestos de PS
con SiO2, con mezclado durante polimerizacion In situ, no se tiene el desarrollo de una
fase elastomérica, por lo que es probable que ocurra un efecto sinérgico del SiO2 con el
retardante a la flama.
a )
b )

113
CAPITULO VIII. Conclusiones
Se logró modificar superficialmente las nanopartículas de SiO2 polimerizando
estireno en la superficie mediante el sistema de modificación por plasma frío. Los
resultados obtenidos por TGA evidenciaron un déposito de polímero en la
superficie de las nanopartículas de SiO2 aproximadamente de 8.9 % en peso.
Se determinó que la presencia de nanopartículas de SiO2 y micropartículas de
Mg(OH)2 durante la polimerización In situ tuvo efecto en la cinética de
polimerización, retardando la velocidad de reacción e incrementando Mw y Mn. Lo
anterior fue ocasionado por la disminución de procesos difusionales, ya que las
partículas en el sistema de reacción aumentaron la viscosidad, y por La
disminución de la concentración de radicales del iniciador.
En los nanocompuestos, el tiempo requrido para llegar al rango de conversión
donde ocurre la inversión de fases fue mayor que para el HIPS B, en los
nanocompósitos HIPS Mi-Si Mod el tiempo requerido para alcanzar la inversión de
fases fue en promedio de 110 min. , mientras para el HIPS B fue de 85 min.
La morfología de la fase elastomérica de los compuestos de HIPS también tuvo un
ligero cambio con respecto al del HIPS B; en el HIPS B la morfologia fue
principalemte núcleo-coraza con unas estructuras similares a salamis pero sin
forma de elipse; en los compuestos esta ultima morfología desapareció, y en
algunos casos aparecieron pequeños salamis o estructuras alargadas; una porción
de las partículas de SiO2 se aglomeraron y se localizaron dentro de la fase
elastomérica, y el diámetro promedio de partícula, con excepción del HIPS S1 y el
HIPS S2 , fue menor en todos los casos.

114
La incorporación de nanopartículas de SiO2 modificadas superficialmente tuvo un
efecto, de manera general, de mejora en la resistencia al impacto, efecto no
presentado por las nanopartículas de SiO2 sin modificar.
La presencia de partículas redujo el grado de Injerto; las partículas disminuyen la
velocidad de la polimerización y el contenido de injerto en las cadenas de PB;
también reduce la conversión total final del St y el grado de entrecruzamiento; por
lo cual el PB no entrecruzado, después de la centrifugación, quedo suspendido en
el Tolueno dando un muy bajo contenido de gel.
Las nanopartículas de SiO2 no presentaron un efecto sinérgico en combinación con
el Mg(OH)2, esto al parecer se debió a que hubo cierta aglomeración de las
nanopartículas de SiO2, y que estas tuvieron la tendencia a alojarse dentro de las
estructuras de la fase elastomérica, no distribuyéndose en la matriz por lo que no
interactuaron con el Mg(OH)2 de la manera adecuada.

115
CAPITULO IX. Trabajo futuro
Preparar nanocompuestos de HIPS en fundido, mediante extrusión, con las
concentraciones de Mg(OH)2 y SiO2 utilizadas en este trabajo, así como
concentraciones mayores, ayudaría a comprobar el efecto sinérgico (en
retardancia la flama) del SiO2 y el Mg(OH)2.
Como se mencionó anteriormente, hacer pruebas para determinar la causó de la
aglomeración de las nanopartículas de SiO2 , se podría realizarse la prueba descrita
en la sección 7.2.7 , :
Mezclar y dispersar las nanopartículas de SiO2 en soluciones de PB en St y
soluciones de con fases PS/St y PB/St con diferentes composiciones de fases, con
una misma concentración de PB en todos los casos e ir aumentando la
concentración de PS en las diversas muestras hasta emular el punto donde ocurre
la separación de fases, y la fase PS/St es la fase dispersa; y después determinar la
dispersión de las partículas en las diversas soluciones, para determinar si las
nanopartículas se aglomeran al separarse las fases de PS y PB.
Tambien se podrían preparar los nanocompuestos de PS con iguales
concentraciones, a las utilizadas en este trabajo, de micropartículas de Mg(OH)2 y
nanopartículas de SiO2 mediante polimerización In situ de estireno (en presencia
de las partículas) ; así no se tendría el efecto de la separación de fases de PB/St y
PS/St y del desarrollo morfológico de la fase elastomérica; en caso de no
presentarse aglomeración de SiO2 en estos nanocompuestos, se confirmaría que
,en el HIPS, se aglomeran las partículas por la presencia de la fase elastomérica.
Tambien sería interesante realizar mas pruebas de flamabilidad, como la prueba
mediante el cono calorimétrico, para seguir mas detalladamente el efecto de
retardancia a la flama del SiO2 y el Mg(OH)2.

116
X. Bibliografía.
1. Grassi, V.G. ; C. Forte, M.M. & Dal Pizzol M.F. Aspectos Morfológicos e Relação
Estrutura - Propiedades de Poliestireno de Alto Impacto. Polímeros: Ciência e Tecnologia
2001 ; 11 ; p. 158 - 168 .
2. Casis, N. Síntesis, Caracterización Y Modelado Matemático Del Poliestireno de Alto
Impacto; Tesis Doctoral en Ingeniería Química; Instituto de Desarrollo Tecnológico para la
Industria Química (INTEC) ; 2006.
3. Katančić, Z. ; Sejdić, J.T. & Murgić, Z.H. Study of flammability and thermal
properties of high-impact polystyrene nanocomposites; Polymer Degradation and Stability
2011; 96; p. 2104 - 2111.
4. Katančić, Z. ; Sejdić, J.T. ; Jelenčić, J. & Murgić , Z.H. Thermal decomposition of
fire-retarded high-impact polystyrene and high-impact polystyrene/ethylene–vinyl acetate
blend nanocomposites followed by thermal analysis ; Journal of Elastomers & Plastics
2014 ; 46 ; 233 -252.
5. Y., Feng ; Yngard, R. & Nelson G.L. Flammability of Polymer–Clay and Polymer–
Silica Nanocomposites ; Journal of Fire Sciences 2005 ; 23 ; p. 209-226.
6. Y., Feng; & Nelson G.L. Polymer/silica nanocomposites prepared via extrusión;
Polymers for Advanced Technologies 2006 ; 17; p. 320–326.
7. Scheirs , J. & Priddy, D. B. Modern Styrenic Polymers: Polystyrenes and Styrenic
Copolymers (Martin, M. F. ; Viola, J. P. & Wuensch, J. R. Preparation, Properties and
Applications of High-impact Polystyrene ) ; Wiley Series in Polymer Science ; 2003.

117
8. Fischer , M. & Hellmann, G.P. On the Evolution of Phase Patterns during the High
Impact-Modified Polystyrene Process ; Macromolecules 1996 ; 29 ; p. 2498-2509.
9. Leal, G.P. & Asua, J.M. Evolution of the morphology of HIPS particles ; Polymer
2009 ;50 ; p. 68-76.
10. Huang, N.J. & Sundberg, D.C. Fundamental Studies of Grafting Reactions in Free
Radical Copolymerization. II. Grafting styrene, acrylate, and methacrylate monomers onto
cis-polybutadiene using AIBN initiator in solution polymerization; Polymer Science Part A:
Polymer Chemistry 1995 ; 33; p. 2551–2570.
11. Huang, N.J. & Sundberg, D.C. Fundamental Studies of Grafting Reactions in Free
Radical Copolymerization. III. Grafting of styrene, acrylate, and methacrylate monomers
onto cis-polybutadiene using benzoyl peroxide initiator in solution polymerization ;
Polymer Science Part A: Polymer Chemistry 1995 ; 33 ; 2571–2586.
12.https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry_Textbook_Maps/Map
%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/17%3A_Radical_reacti
ons/17.2%3A_Radical_chain_reactions (14/septiembre/2017)
13. Brydon A. ; Burnett, G.M. ; & Cameron, G.G. Free-radical grafting of monomers to
Polydienes. II. Kinetics and mechanism of Styrene grafting to Polybutadiene; Polymer
Science 1974; 12; p. 1011–1021.
14. Manaresi, P. ; Passalacqua, V. & Pilati, F. Kinetics of graft polymerization of
styrene on cis-1,4-polybutadiene ; Polymer 1975; 16; p. 520 – 526.

118
15. Sundberg, D.C. ; Arndt, J. & Tang, M. Y. Grafting of Styrene Onto Polybutadiene
Latices in Batch and Semi-Continuous Reactors; Dispersion Science and Technology 1984;
5; p. 433-445.
16. Shyan Chern , C. & Poehlein ,G. W. Kinetics of Grafting in solution Polimerization ;
Chemical Engineering Communications 1987; 60 ; p. 101-117.
17. Peng, F.M. Polybutadiene grafting and crosslinking in high-impact polystyrene bulk
thermal process; Applied Polymer Science 1990 ; 40 ; p. 1289–1302.
18. Huang. N.J. & Sundberg D.C. Fundamental Studies of Grafting Reactions in Free
Radical Copolymerization. I. A detailed kinetic Model for Solution Polymerization; Polymer
Science Part A: Polymer Chemistry 1995; 33; p. 2533–2549.
19. Huang. N.J. & Sundberg D.C. Fundamental Studies of Grafting Reactions in Free
Radical Copolymerization. IV. Grafting of styrene, acrylate, and methacrylate monomers
onto vinyl-polybutadiene using benzoyl peroxide and AIBN initiators in solution
polymerization ; Polymer Science Part A: Polymer Chemistry 1995; 33; p. 2587–2603.
20. Estenoz, D. A. ; González, I. M. ; Oliva, H. M. & Meira, G. R. Polymerization of
Styrene in the Presence of Polybutadiene: Determination of the Molecular Structure;
Applied Polymer Science 1999; 74; p. 1950–1961.
21. Estenoz, D.A. ; Leal, G.P. ; López, Y.R. ; Oliva, H.M.; & Meira, G.R. Bulk
polymerization of styrene in the presence of polybutadiene. The use of bifunctional
initiators; Journal of Applied Polymer Science 1996, 62, p. 917-939.

119
22. Luciani, C. ; Estenoz, D. ; Meira, G.; Díaz de León, R. & Morales, G. Poliestireno de
Alto Impacto (HIPS). Predicción de las Propiedades Moleculares, Morfológicas y
Mecánicas; Congreso CONAMET/SAM 2004.
23. Luciani, C. V. ; Estenoz, D. A. & Meira, G. R. Mathematical Model of an
Heterogeneous Polymerization for the Production of High-Impact Polystyrene; 2nd
Mercosur Congress on Chemical Engineering; 4th Mercosur Congress on Process Systems
Engineering; ENPROMER 2005.
24. Johnston, G.J. Thermodynamics of formation of high impact polystyrene; Polymer
1978 ; 19, p. 227-228.
25. Jordhamo, G. ; Manson J. & Sperling L. Phase continuity and inversion in polymer
blends and simultaneous interpenetrating networks; Polymer Engineering & Science 1986,
26, p. 517-524.
26. Stein D. J. ; Fahrbach G. & Adler H. Crosslinking Reactions in High Impact
Polystyrene Rubber Particles; Advances in Chemistry 1975, 142, p. 148–158.
27. Braun, D. ; Cherdron , H. ; Rehahn ,M.; Ritter ,H. & Voit ,B. Polymer Synthesis:
Theory and Practice. Fundamentals, Methods, Experiments ; Springer ; 2013.
28. Acuña, P. & Morales, G. Síntesis de poliestireno de alto impacto (HIPS) a partir de
diferentes iniciadores multifuncionales: efecto de la estructura y del contenido de oxígeno
activo del iniciador; Revista Iberoamericana de Polímeros 2011 ; 12 , p. 107-115.
29. Elias, H.G. & Mülhaupt , R. Ullmann's Encyclopedia of Industrial Chemistry;
Plastics, General Survey, 2. Production of Polymers and Plastics ; Wiley & Sons ; 2002.

120
30. Moore, J.D. An electron microscope study of the microstructure of some rubber-
reinforced polystyrenes ; Polymer 1971 ; 12 ; p. 478–86.
31. Chen C. C. Continuous production of solid polystyrene in back-mixed and linear-
flow reactors; Polymer Engineering & Science 2000 ; 40; p. 441 - 464.
32. González Vega, R.E. Efecto de los parámetros de proceso sobre la morfología y
propiedades en Poliestireno reforzado obtenido mediante extrusión reactiva; Tesis
Maestría en Tecnología de Polímeros; CIQA ; 2008.
33. Okamato, Y.; Miyagi, H.; Kakugo, M.; & Takahashi, K. Impact Improvement
Mechanism of HIPS with Bimodal Distribution of Rubber Particle Size; Macromolecules
1991; 24; p. 5639 - 5644 .
34. Brown, H.R. Effect of a diblock copolymer on the adhesion between incompatible
polymers ; Macromolecules 1989 ; 22; p. 2859 - 2860.
35. Gebizlioglu, O. S. ; Beckham, H. W.; Argon, A.S.; Cohen, R.E. & Brown,H.R. A new
mechanism of toughening glassy polymers. 1. Experimental procedures; Macromolecules
1990 ; 23 ; p. 3968 - 3974.
36. Kuboki, T.; Ben Jar , P.Y. ; Takahashi, K. & Shinmura, T. Observation of “Black” and
“White” Crazes in High-Impact Polystyrene under Transmission Electron Microscopy;
Macromolecules 2000 ; 33 ; p. 5740-5742.
37. Molau, G.E. & Keskkula, H Heterogeneous polymer systems. IV. Mechanism of
rubber particle formation in rubber-modified vinyl polymers; Polymer Science 1966; 4; p.
1595–1607 .

121
38. Li, B. & Zhong, W.H. Nanoscience and Nanomaterials: Synthesis, Manufacturing
and Industry Impacts ; DEStech Publications, Inc. ; 2011.
39. Llinàs, M.C. & Sanchez G.,D. Nanopartículas de sílice: preparación y aplicaciones
en biomedicina ; Afinidad ; 71; 20 - 31 (2014).
40. Rahman, I. A. & Padavettan , V. Synthesis of Silica Nanoparticles by Sol-Gel: Size-
Dependent Properties, Surface Modification, and Applications in Silica-Polymer
Nanocomposites—A Review ; Journal of Nanomaterials 2012 ; 2012 ; p. 1 - 15.
41. Zanella, R. Metodologías para la síntesis de nanopartículas: controlando forma y
tamaño; Mundo Nano 2012 ; 5 ; p. 69 – 81.
42. Koo, H. J. Polymer Nanocomposites: Processing, Characterization, and
Applications ; McGraw-Hill Nanoscience and Technology Series ; 2006.
43. Ahmed, L. ; Zhang ,B. ; Hawkins ,S. ; Mannan , M. S. & Cheng, Z. Study of
thermal and mechanical behaviors of flame retardant polystyrene-based nanocomposites
prepared via in-situ polymerization method ; Journal of Loss Prevention in the Process
Industries 2017 ; 49 ; p. 228-239.
44. Alaee, M.; Arias, P.; Sjodin, A. & Bergman, A. An overview of commercially used
brominated flame retardants, their applications, their use patterns in different
countries/regions and possible modes of release ; Environment International 2003 ; 29 ; p.
683– 689.
45. Morgan, A.B. & Wilkie, C.A. Flame Retardant Polymer Nanocomposites. John Wiley
& Sons, Inc. ; 2007.

122
46. Horrocks, A. R. & Price; , D. Fire Reatardant Materials ; Woodhead Publishing ; 1ra
Edición ; 2001.
47. Mouritz, P. & Gibson, A. G. Fire Properties of Polymer Composite Materials;
Springer; 1era Edición ; 2006.
48. Walter, M.D. & Wajer, M.T. Overview of Flame Retardants Including Magnesium
Hydroxide ; Martin Marietta Magnesia Specialties LLC 2015 ; Version 1.2.15
(http://magnesiaspecialties.com/wp-content/uploads/MagShield-Overview-of-Flame-
Retardants-Including-MgOH2.pdf (07/septiembre/2017)).
49. Lopez, J.F.; Perez, L.D. & Lopez, B.L. Síntesis y Caracterización de Nanohíbridos
Poliestireno / Sílica Mesoporosa MCM-41 ; Scientia et Technica 2007 ; 1 ; p. 971 -976.
50. Kashiwagi, T. ; Shields, J. R. ; Harris, R.H. Jr & Davis, R. D. Flame-Retardant
Mechanism of Silica: Effects of Resin Molecular Weight ; Applied Polymer Science 2003 ; 87
; p. 1541 - 1553.
51. Rahimi-Razin, S. ; Salami-Kalajahi, M. ; Haddadi-Asl, V. ; Behboodi-Sadabad, F. ;
Roghani-Mamaqani, H & Hemmati, M. Investigating the effect of pristine and modified
silica nanoparticles on the kinetics of methyl methacrylate polymerization ; Chemical
Engineering Journal 2011 ; 174 ; p. 368 - 375.
52. Rahimi-Razin,S. ; Salami-Kalajahi, M. ;Haddadi-Asl, V. & Roghani-Mamaqani, H. ;
Effect of different modified nanoclays on the kinetics of preparation and properties of
polymer-based nanocomposites ; Journal of Polymer Research 2012 ; 19 ; p. 1-16.

123
53. Bera, O. ; Pavličević, J. ; Jovičić, M. ; Pilić, B. ; Stoiljkovic, D. & Radičević, R. The
Influence of Nanosilica on Styrene Free Radical Polymerization Kinetics ; Polymer
Composites 2012 ; 33 ; p. 262-266.
54. Sobani,M.; Haddadi-Asl, V.; Mirshafiei-Langari,S.A.; Salami-Kalajahi, M; Roghani-
Mamaqani, H. & Khezri, K.; A kinetics study on the in situ reversible addition–
fragmentation chain transfer and free radical polymerization of styrene in presence of
silica aerogel nanoporous particles; Designed Monomers and Polymers 2014 ; 17; p. 245–
254.
55. Khezri,K. & Mahdavi, H.; Polystyrene-silica aerogel nanocomposites by in situ
simultaneous reverse and normal initiation technique for ATRP ; Microporous and
Mesoporous Materials 2016 ; 228 ; p. 132-140.
56. Yavuz, Ö. & Saka, C. Surface modification with cold plasma application on kaolin
and its effects on the adsorption of methylene blue ; Applied Clay Science 2013 ; 85 ; p. 96
-102.
57. Soriano C. , F. ; Morales, G. ; Acuña, P. ; Diaz B. , E. ; Arellano, B. ; Vargas , C.
& De la Paz , O. Synthesis and Characterization of High Impact Polystyrene from a
Heterogeneous Styrene-Rubber-Polystyrene Solution: Influence of PS Concentration on
the Phase Inversion, Morphology and Impact Strength ; Macromolecular Symposia 2013 ;
325-326 ; p. 177 – 183.
58. Gasperowicz, A. & Łaskawski, W. Grafting of styrene onto low molecular weight
polymers and copolymers of butadiene ; Journal of Polymer Science 1976 ; 14 ; p. 2875-
2886.

124
59. B.H. Patel & P.N. Patel; Synthesis and Characterization of Silica Nano-Particles by
Acid Leaching Technique ; Research Journal of Chemical Sciences 4(5), 52-55 (2014).
60. Beganskiene, A.; Sirutkaitis, V. ; Kurtinaitiene, M. ; Juskenas,R.; Kareiva, A. ; FTIR,
TEM and NMR Investigations of Stöber Silica Nanoparticles ; Materials Sciencie; 10 , 287-
290 (2004)
61. Kwon , H. & Gon Park D.; Infra-Red Study of Surface Carbonation on Polycrystalline
Magnesium Hydroxide ; Bull. Korean Chem. Soc., 30, No. 11, 2567-2573(2009).
62. Vonka, M. ; Šeda, L. & Kosek, J. Modelling of the High-Impact Polystyrene
Morphogenesis; Macromolecular Symposia 2011 ; 302 ; p. 151 – 160.
63. Subbaiah, Y. ; Raghavendra, V.B. ; Puttamadappa, S. ; Bandyopadhyay, S. ;
Mukhopadhyay, P. & Kumar, A. Influence of submicron tiny gel rubber particles on the
estimation of grafting and rubber efficiency in high-impact polystyrene ; Journal of
Elastomers & Plastics 2016 ; publicado 21 de diciembre ; p. 1 - 12.





![tilngï n Il üLL5J-nJl änll-wg üLLSJ-n-ll ti ñLLnJl üLîLQ!-iJlg üLîg1n]lairqualityandmobility.org/usedvehicles/Arabic.pdf · 2020. 11. 11. · ISO 9001 Jl..i-i-nJl Jl.n.ó](https://img.pdfslide.tips/doc/110x75/60e21bfc39065323ac21da36/tilng-n-il-ll5j-njl-nll-wg-llsj-n-ll-ti-llnjl-llq-ijlg-lg1nla.jpg)


![بورصة الأوراق المالية والضرائب · äIJgiJI ä.DJg.4 u51ÞJIg äxJlnJl iljßl ¾.ådl 911áPlg ñLilÄ]l u.i4iJ ¥il.i nJl Ece Eze 08028543482](https://img.pdfslide.tips/doc/110x75/5e0ee355882c4f38e959fab5/-ijgiji-djg4-u51jig.jpg)




















