Embed Size (px)
Citation preview

The Simian Virus 40 Core C Enhancer-like ElementIs a Positive Regulator in the Rat a1B AdrenergicReceptor Gene Proximal Promoter1
Ngoc Dao and Bin Gao2
Department of Pharmacology and Toxicology, Medical College of Virginia,Virginia Commonwealth University, Richmond, Virginia 23298
Received November 19, 1998
Transcription of the rat a1B adrenergic receptor(a1BAR) gene is controlled by three promoters (P1, P2,and P3), which generate 2.3-, 2.7-, and 3.3-kb tran-scripts, respectively. The expression of the 2.3-kbmRNA species is tissue-specific. To explore the under-lying mechanism, the P1 promoter was analyzed.DNase I footprinting of the P1 promoter yielded threeprotected regions: P1f1(249 to 262); P1f2 (273 to 290),and P1f3 (295 to 2115). Sequence analysis of P1f3 re-vealed the presence of an SV40 core C enhancer-likeelement. In gel mobility shift assays, P1f3 was found tobind a sequence specific protein, which was competedaway by a SV40 core C enhancer consensus oligonu-cleotide. Mutations of this enhancer-like core se-quence within P1f3 significantly reduced specific pro-tein binding to P1f3 and inhibited P1 promoteractivity. The distribution of the protein which binds toP1f3 is restricted. These findings suggest that the P1promoter is controlled by a cell-type-specific tran-scription factor, which may account for the tissue-specific expression of 2.3-kb rat a1BAR mRNAspecies. © 1998 Academic Press
The a1B adrenergic receptor (a1BAR) is a G-proteincoupled receptor that regulates a variety of physiologicprocesses, such as peripheral vasoconstriction, in-creased cardiac contractility, hepatic glycogenolysis,
and cell proliferation (1–4). Expression of the a1BARgene is regulated by hormonal and developmental fac-tors in a tissue specific manner (1–4). Northern blotanalyses of rat a1BAR mRNA have documented threemRNA species of 2.3, 2.7, and 3.3 kb in length (5–7).The 3.3-kb species is preferentially expressed in ratliver (5–7), whereas the 2.7-kb species is dominant andwidely expressed in many tissues (5–7). The 2.3-kbspecies was detected at a low abundance in the rat liver(5,8) but at high abundance in DDT1MF-2 cells (9) andin rat medullary thyroid carcinoma 623 cells (10). Inthe rat liver, the expression of these three mRNA spe-cies is likely controlled by three distinct promoterswithin the a1BAR gene: a distal promoter (P3), middlepromoter (P2) and proximal promoter (P1), respec-tively (5). The P2 promoter, which controls the expres-sion of the major 2.7-kb mRNA, has been well charac-terized in our laboratory (8, 11–13), including itsbinding of a number of transcription factors, such asAP2, NF1, SP1, CP1, and CREB. However, the P3 andP1 promoters have not been extensively studied. TheP3 promoter region contains consensus sites forsequence-specific proteins, including CCAAT/enhancerbinding protein (C/EBP), hepatocyte nuclear factor-5(HNF-5) and AP2 (5). Determining whether these bind-ing sites are functional or not requires further studies.Recently, Farber and coworkers (14) reported that theupstream (2164) region of the P1 promoter contains ahypoxia-induced factor 1 binding site, which is respon-sible for upregulation of a1BAR gene transcription insmooth muscle cells by hypoxia.
Here we analyzed the P1 promoter by DNase I foot-printing and DNA gel mobility shift assays. Our datashow that the P1 promoter contains multiple sequence-specific protein binding sites, including a simian virus40 (SV40) core C enhancer-like sequence element (15,16), which specifically binds an as yet unidentifiednuclear protein and plays a positive regulatory role inthe transcription of a1B AR gene P1 promoter. The
1 This work was supported by National Institutes of Health GrantCA72681 (to B. G.). N.D. was supported by National Cancer InstituteTraining Grant 2R25CA22032.
2 To whom correspondence should be addressed Department ofPharmacology & Toxicology, MCV/VCU, Box 980613, Richmond, VA23298. Fax: (804) 828-2117. E-mail: [email protected].
Abbreviations used: AR, adrenergic receptor; SV 40, simian virus40; AP2, activator protein 2; NF1, nuclear factor 1; CP1, CCAATbinding protein 1; CREB, cAMP-response element binding protein;EBP1, enhancer binding protein 1; URF, upstream repressor factor;C/EBP, CCAAT enhancer binding protein; HNF 5, hepatocyte nu-clear factor 5.
BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 253, 804–808 (1998)ARTICLE NO. RC989860
8040006-291X/98 $25.00Copyright © 1998 by Academic PressAll rights of reproduction in any form reserved.

expression of this protein is tissue restricted with adistribution pattern similar to the tissue-specific ex-pression of the 2.3-kb a1BAR mRNA species.
MATERIALS AND METHODS
Cell culture. The DDT1MF-2 hamster smooth muscle cell line andthe Hep3B human hepatocellular carcinoma cell line were obtainedfrom the American Type Culture Collection (Rockville, MD) andcultured under conditions specified by the supplier.
DNase I footprinting. Preparation of liver nuclear extracts andDNase I footprinting were described previously (5, 25). The sensestrand from p(2127, 249)/CAT was phosphorylated at the uniqueHind III site of the pCAT vector with T4 polynucleotide kinase and[g32] ATP. Subsequent digestion with XbaI yielded the end-labeledsense strand probe A. The antisense strand from p(2127, 249)/CATwas phosphorylated at the unique XbaI site of the pCAT vector withT4 polynucleotide kinase and [g-32]ATP. Subsequent digestion withHindIII yielded the end-labeled sense strand probe B.
Oligonucleotide synthesis. The synthetic oligodeoxyribonucleo-tides (oligos) were prepared on a Cyclone Plus DNA synthesizer(Milligen). After ammonium hydroxide deprotection, oligos wereevaporated to dryness by vacuum centrifugation (Savant Speed-Vac)and purified by electrophoresis on a 10% polyacrylamide, 8 M ureagel (26).
Construction of plasmids. The P1/CAT and P1m/CAT were pre-pared by subcloning P1 and P1m promoter regions into pCAT enhancerreporter vectors, respectively. The P1 promoter region was amplified byPCR using the rat a1BAR gene 59 flanking region (27) as template,primer 1 (59-CTTCGCCAGGAGGACGCTCTGGAAAGAAG-39) (2128to 2108, with the SV40 core enhancer-like sequence element under-lined) containing a PstI linker, and primer 2 (59-CTGCG-ACCAAGGCACCTCAGCTA-39) (249 to 270) containing a HindIIIlinker as 59 and 39 primers, respectively. The P1m region was amplifiedby PCR using the rat a1BAR gene 59 flanking region as a template,primer 1m (59-CTTCGCCAGGAGGACGCTCTATGCAGAAG-39) (2128to 2108, with the mutations underlined) containing a PstI linker, andprimer 2 as 59 and 39 primers, respectively. PCR was carried as de-scribed before (5). The final PCR product was purified and subclonedinto the pCAT basic vector (Promega). The mutations within the SV40core C enhancer-like element in the P1m/CAT construct were verifiedby sequencing.
Transient transfections and CAT assays. Transient transfectionsand CAT assays were performed as described previously (5).
DNA mobility shift assays (DMSA). Whole cell extracts used inDMSA were prepared as reported (28). Preparation of nuclear ex-tracts for DMSA and the assay itself have been described earlier (11).The following double-stranded oligos were used as probes in DMSA:OL1, 59-ACG CCT CTG GAA AGA AGA CCA CGG AGG GAG CAAAGT TTC AGG GTA GCT GAG GTG CCT TGG TCG CAG (2135 to249 of the rat a1BAR gene 59 flanking region [27]); AP1, 59-CGC TTGATG AGT CAG CCG GAA-39; AP2, 59-GAT CGA ACT GAC CGCCCG CGG CCC G-39; AP3, 59-CTA CTG GGA CTT TCC ACA CAT-39;TFIID, 59-GCA GAG CAT ATA AGG TGA GGT AGG A-39; NFkb,59-AGT TGA GGG GAC TTT CCC AGG C-39; SP1, 59-ATT CGA TCGGGG CGG GGC GAG C-39; NF1, 59-TAT TTT GGA TTG AAG CCAATA TGA TAA TGA-39; C/EBP, 59-AAA GAT GGT ATG ATT TTGTAA TGG GGT AGG A-39; SV40 oligo , 59-GTT AGG GTG TGG AAAGTC CCC AGG CTC-39.
RESULTS
As shown in Fig. 1, DNase I footprinting of the sense(A) and antisense strands (B) of the P1 promoter
yielded three protected regions: P1f1 (249 to 262);P1f2 (273 to 290) and P1f3 (295 to 2115). Proteinbinding to these footprints is specific, as it can beprevented by excess unlabeled oligo OL1 (Fig. 1A),whereas double-stranded oligos containing consensusbinding sites for SP1, AP1, AP2, AP3, TFIID, NFkBand CTF/NF1 did not prevent these footprints (Fig.1B). A summary of the footprinting data is presented inFig. 1C. A computer-based search of the transcriptionfactor pattern data base (TFD) (GCG, Genetics Com-puter Group, Madison, WI) of several thousandsequence-specific response elements yielded no homol-ogies with footprints P1f1 or P1f2, but a perfect matchto the SV40 core C enhancer sequence (59 TGGAAAG39) (15, 16) was established for P1f3.
Proteins binding to P1f3 were analyzed by DNA mo-bility shift assays using nuclear proteins from DDT1MF-2 cells which, in preliminary experiments, yieldedthe strongest shifted bands. As shown in Fig. 2, the32P-labeled P1f3 oligo binds a complex specifically, as itis competed away by unlabeled P1f3 oligo. This com-plex is also abolished by unlabeled SV40 core C en-hancer oligo, but not by oligos containing consensusbinding sites for SP1, AP1 and TFIID (Fig. 2B), andAP2, AP3, NFkB, CTF/NF1 and C/EBP (data notshown). This suggests that the factor that binds to P1f3also binds to the SV40 core C enhancer sequence. Tofurther verify this, we mutated this core sequencewithin P1f3 to generate oligo P1f3m, which was thenused as a probe in DNA gel mobility shift assays. Asshown in Fig. 3, the excess P1f3 but not the P1f3m oligoabolished specific protein binding to the 32P-labeledP1f3 oligo. This confirmed the role of the SV40enhancer-like core sequence in protein binding to P1f3.
The SV40 core C enhancer-like element has beenreported to function either as a positive or a negativetranscriptional regulator (19, 24). To determine itsfunctional role in the a1BAR P1 promoter, we intro-duced the above mutations in P1f3m into the P1 pro-moter, to generate the P1m/CAT construct, and thentransfected it into DDT1 MF-2 cells. As shown in Fig. 3,the activity of the P1m/CAT construct was approxi-mately 50% of that of the wild type P1. These resultsindicate that the SV40 core enhancer-like sequenceelement acts as a positive regulator of P1 promoteractivity.
Unlike the 2.7-kb mRNA species of the a1B AR genewhich is ubiquitously expressed, expression of the2.3-kb mRNA is tissue specific, with low amountsfound in rat liver (5, 8), and high levels in DDT1 MF-2cells (9) and rat medullary thyroid carcinoma 623 cells(10). It was of interest, therefore, to test whether theexpression of the protein that bound to P1f3 displayssimilar tissue specificity. To estimate the relative tis-sue levels of this protein, we performed DNA gel mo-bility shift assays using P1f3 as a probe and nuclearextracts from different tissues and cells. As shown in
Vol. 253, No. 3, 1998 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
805
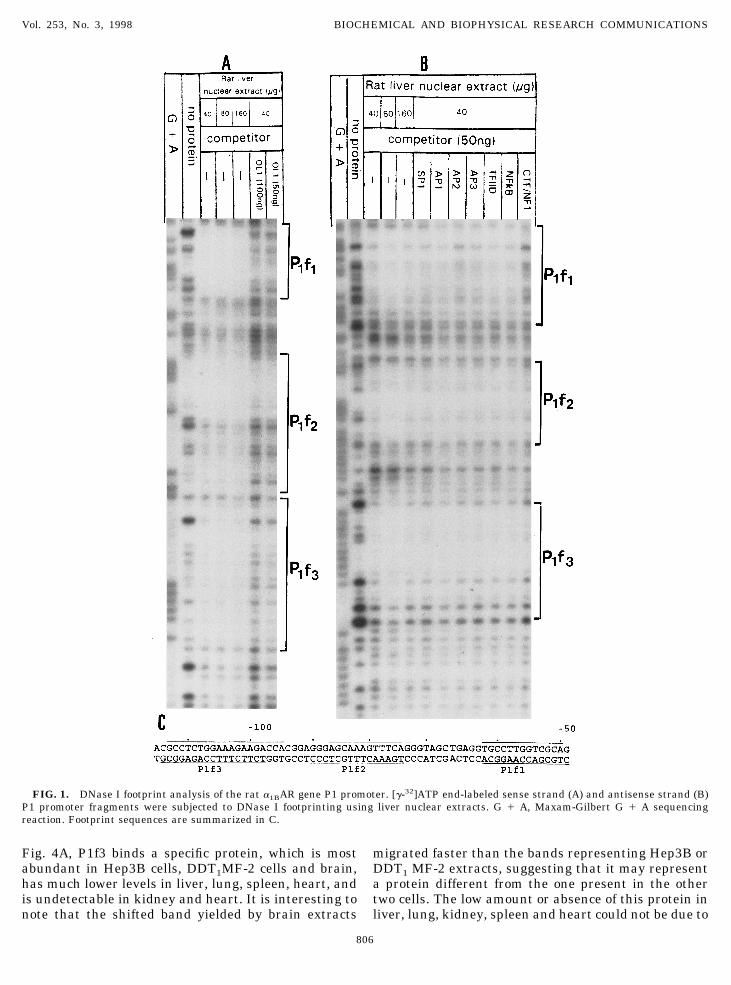
Fig. 4A, P1f3 binds a specific protein, which is mostabundant in Hep3B cells, DDT1MF-2 cells and brain,has much lower levels in liver, lung, spleen, heart, andis undetectable in kidney and heart. It is interesting tonote that the shifted band yielded by brain extracts
migrated faster than the bands representing Hep3B orDDT1 MF-2 extracts, suggesting that it may representa protein different from the one present in the othertwo cells. The low amount or absence of this protein inliver, lung, kidney, spleen and heart could not be due to
FIG. 1. DNase I footprint analysis of the rat a1BAR gene P1 promoter. [g-32]ATP end-labeled sense strand (A) and antisense strand (B)P1 promoter fragments were subjected to DNase I footprinting using liver nuclear extracts. G 1 A, Maxam-Gilbert G 1 A sequencingreaction. Footprint sequences are summarized in C.
Vol. 253, No. 3, 1998 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
806

possible technical problems with the nuclear extracts,because NF1 complexes could be detected at muchhigher levels in these extracts than in those fromHep3B or DDT1MF-2 cells (Fig. 4B).
DISCUSSION
We have previously identified three promoters of therat a1B AR gene and extensively studied the P2 pro-moter (5, 8, 11–13). Here, we further characterized theproximal P1 promoter and identified its threesequence-specific binding sites: P1f1 (249 to 262);P1f2 (273 to 290) and P1f3 (295 to 2115). The P1f1sequence reveals no apparent homologies with the con-sensus sequences of known transcription factors, andin competition assays the only consensus oligo thatproduced weak protection was that for NF1/CTF1 (Fig.1B). However, binding of NF1/CTF1 is unlikely to beresponsible for this footprint, because in gel shift as-says the NF1/CTF1 oligo did not abolish the shiftedband (Dao and Gao, unpublished data), and cotrans-fection of an NF1 expression vector was earlier foundnot to affect P1 promoter activity (29). The P1f2 se-quence also did not contain apparent homologies withknown consensus binding sites, and the P1f2 footprintwas not inhibited by a number of consensus oligostested (Fig. 1B). This suggests that the factors bindingto P1f2 and, possibly, to P1f1, are novel proteins thatremain to be identified.
In contrast, P1f3 contains a perfect match (59 TG-GAAAG 39) to the core C enhancer sequence element ofSV40, which has been located in many viral and mam-malian genes (15–24). In gel mobility shift assays, P1f3
bound a sequence specific protein, which is competedaway by an oligo corresponding to the SV40 enhancerelement core sequence but not by other consensus oli-
FIG. 3. Mutations of the SV40 core enhancer-like sequence ele-ment within P1f3 abolishes specific protein binding to P1f3 andinhibits P1 promoter activity. (A) DNA sequences of P1f3 or mutatedP1f3m from the P1 promoter of the rat a1B AR. Mutated sequences inP1f3m are underlined. (B) DNA mobility shift assay performed with32P-labeled oligo P1f3 plus DDT1 MF-2 cell extracts. One ng oflabeled P1f3 was incubated with 10 mg of DDT1 MF-2 cell extract inthe absence (lane 1) or presence of 100 ng of competitor oligos P1f3and P1f3m (lanes 2 and 3), as indicated above the lanes. (C): Muta-tions of core sequences within P1f3 inhibit P1 promoter activity. Themutations in oligo P1f3 (A) were introduced into the P1 promoterconstruct to produce the P1m construct, as described under Materialand Methods. This construct was then transiently transfected intoDDT1 MF-2 cells and CAT activities were measured and expressedas percent of the P1 promoter activities. Means 6 S.E. from fourexperiments are shown. *, significant difference (P , 0.01) comparedwith wild type P1 promoter activity.
FIG. 4. The protein binding to P1f3 is tissue specific. Ten micro-grams of nuclear extracts was prepared from different tissues andcells were subjected to DNA mobility shift assay using 32P-labeledoligo P1f3 (top) or NF1 (bottom) as a probe.
FIG. 2. DNA mobility shift analyses of the specific proteins in-teracting with the P1f3 footprint. DNA mobility shift assays wereperformed with 32P-labeled oligo P1f3 plus DDT1 MF-2 cell extracts.One ng of labeled P1f3 was incubated with 10 mg of DDT1 MF-2 cellextract in the absence (lanes 1 and 9) or presence of different com-petitor oligos (lanes 2–8 and lanes 10–13), as indicated above thelanes.
Vol. 253, No. 3, 1998 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
807

gos (Fig. 2). Several nuclear factors, including EBP1(15, 16), C/EBP (23), URF (20) and NFkB (21) havebeen shown to bind the SV40 core C enhancer-likeelement. Of these, we could exclude C/EBP and NFkBbased on the inability of either consensus oligo to com-pete in either DNase I footprinting or DMSA. Whetherthis protein is EBP1or URF requires further studies todetermine.
The findings that mutations of the SV40 enhancer-like sequence within P1f3 reduced protein binding toP1f3 as well as inhibited P1 promoter activity indicatesthat this core sequence plays a positive role in control-ling transcription of the rat a1BAR gene. This is con-sistent with our previous finding that deletion of region2127 to 2105 of the a1BAR 59-flanking region, whichcontains half of the SV40 core enhancer-like element,abolished P1 promoter activity in Hep3B and DDT1MF-2 cells (5). Of considerable interest is the presentfinding that the expression of the protein binding toP1f3 is restricted, and the pattern of its tissue-specificity fits remarkably well the tissue-specific ex-pression of the 2.3 kb a1BAR mRNA species, as re-viewed above. The notable exceptions are the brain andHep3B cells, which express very low levels of 2.3 kba1BAR mRNA (6), but high amounts of protein bindingto P1f3. However, the complex formed in brain nuclearextracts migrated faster than those from Hep3B andDDT1 MF-2 extracts, suggesting that it may representa different protein which may not serve as a positiveregulator for the P1 promoter. The Hep3B cells expressvery high levels of P1 promoter activity (5) and a 4.5 kba1BAR mRNA (30). However, these cells do not expressthe 2.3 kb a1BAR mRNA and are devoid of functionala1BAR as measured by radioligand binding (30, 31).The reasons for this are not yet clear.
In summary, the data here demonstrate that the P1promoter of the rat a1BAR gene contains multiplesequence-specific protein binding sites including anSV40 core enhancer-like sequence element, whichplays a positive regulatory role in P1 promoter activity.The protein binding to this core sequence is cell-typespecific, which may explain the tissue-restricted ex-pression of 2.3 kb a1BAR mRNA.
ACKNOWLEDGMENT
We thank Dr. George Kunos for critical reading of the manuscript.
REFERENCES
1. Graham, R. M., Perez, D. M., Hwa, J., and Piascik, M. T. (1996)Circ. Res. 78, 737–749.
2. Mineman, K. P., and Esbenshade, T. A. (1994) Annu. Rev. Phar-macol. Toxicol. 34, 117–133.
3. Minemen, K. P. (1988) Pharmacol. Rev. 40, 87–119.4. Kunos, G., Ishac, J., Gao, B., and Jiang, L. (1995) Ann. N.Y.
Acad. Sci. 757, 261–270.5. Gao, B., and Kunos, G. (1994) J. Biol. Chem. 269, 15762–15767.6. MeGehee, R. E., Jr., Rossby, S. P., and Cornett, L. E. (1990) Mol.
Cell. Endocrinol. 74, 1–9.7. Lomansney, J. W., Cotecchia, S., Lorenz, W., Leung, W.-Y.,
Schwinn, D. A., Yang-Feng, T. L., Brownstein, M., Lefkowitz,R. J., and Caron, M. G. (1991) J. Biol. Chem. 266, 6365–6369.
8. Gao, B., Jiang, L., and Kunos, G. (1996) Mol. Cell. Biol. 16,5997–6008.
9. Hu, Z.-W., Shi, X.-Y., Sakaue, M., and Hoffman, B. B. (1993)J. Biol. Chem. 268, 3610–3615.
10. Esbenshade, T. A., Theroux, T. L., and Minneman, K. P. (1994)Mol. Pharmacol. 45, 591–598.
11. Gao, B., Spector, M., and Kunos, G. (1995) J. Biol. Chem. 270,5614–5619.
12. Gao, B., Chen, J., Johnson, C., and Kunos, G. (1997) Mol. Phar-macol. 52, 1019–1026.
13. Chen, J., Spector, M., Kunos, G., and Gao, B. (1997) J. Biol.Chem. 272, 23144–23150.
14. Eckhart, A., Yang, N., Xin, X., and Faber, J. (1997) Proc. Natl.Acad. Sci. USA 914, 9487–9492.
15. Clark, L., Nicholson, J., and Hay, R. (1989) J. Mol. Biol. 206,615–626.
16. Clark, L., Pollock, R., and Hay, R. (1988) Genes Dev. 2, 991–1002.17. Sassone-Corsi, P., and Borrelli, E. (1986) Trends Genet. 8, 215–
219.18. Zintz, C., and Inana, G. (1990) Exp. Eye Res. 50, 759–770.19. Zhang-Keck, Z., Kibbe, W., Moye-Rowley, W., and Parker, C.
(1991) J. Biol. Chem. 266, 21362–21367.20. Pan, J., and McEver, R. P. (1993) J. Biol. Chem. 268, 22600–
22608.21. Clark, L., Matthews, J., and Hay, R. (1990) Mol. Cell. Biol. 64,
1335–1344.22. Costa, R., Grayson, D., Xanthopoulos, K., and Darnell, J. E.
(1988) Proc. Natl. Acad. Sci. USA 85, 3840–3844.23. Landschulz, W. H., Johnson, P. F., Adashi, B., Graves, B. J., and
McKnight, S. L. (1988) Genes Dev. 2, 786–800.24. Burglin, T. R. (1991) Cell 66, 11–12.25. Garabedian, M. J., Labaer, J., Liu, W., and Thomas, J. (1993) in
Gene Transcription—A Practical Approach (Hames, B. D., andHiggins, S. J., Ed.), pp. 260–272, IRL Press, Oxford, New York,Tokyo.
26. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) MolecularCloning: A Laboratory Manual, 2nd ed., Cold Spring HarborLaboratory Press, Cold Spring Harbor, NY.
27. Gao, B., and Kunos, G. (1993) Gene 131, 243–247.28. Andrews, N. C., and Faller, D. V. (1991) Nucleic Acids Res. 19,
2499.29. Gao, B., Jaffe, H., and Kunos, G. (1998) Mol. Cell. Biochem. 178,
187–196.30. Kost, D. P., DeFrances, M. C., Lee, C. R., and Michalopoulos,
G. K. (1992) Pathobiology 80, 303–308.31. Auer, K., Spector, M., Tombes, R., Seth, P., Fisher, P., Gao, B.,
Dent, P., and Kunos, G. (1998) FEBS Lett. 436, 131–138.
Vol. 253, No. 3, 1998 BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS
808
![Immunoreaction Enhancer Solutionlifescience.toyobo.co.jp/user_data/pdf/products/manual/...-1 - [1] はじめに Can Get Signal® Immunoreaction Enhancer Solutionは、ウェスタンブロッティングや](https://img.pdfslide.tips/doc/110x75/5e3e66223b7bec42fd7fa1f9/immunoreaction-enhancer-1-1-can-get-signal-immunoreaction.jpg)