Embed Size (px)
Citation preview

1
1
Phylogenie IPhylogenie I
WS 2006/2007
Genomforschung und Sequenzanalyse
Einführung in Methoden der Bioinformatik
Bernhard Lieb &Tom Hankeln
MolekulareMolekulare
2
Themen
Grundlagen und Begriffe der molekularen Phylogenie
Evolutionsmodelle

2
3
Warum Phylogenie?
=> Verständnis von phylogenetischen Zusammenhängen:
• Organismische Evolution (Systematik)• Evolution von Proteinfamilien (Funktion)• Medizin (Epidemiologie)• Forensik (CSI Miami)
=> Stammbäume
4
„Tierische Walverwandtschaft“
Wann und wie sind Hämocyanine entstanden?
Wann hat sich der moderne Mensch entwickelt?
AIDS without sex? - „The Florida Dentist“
Typische Fragestellungen der molekularen Phylogenie
3
5
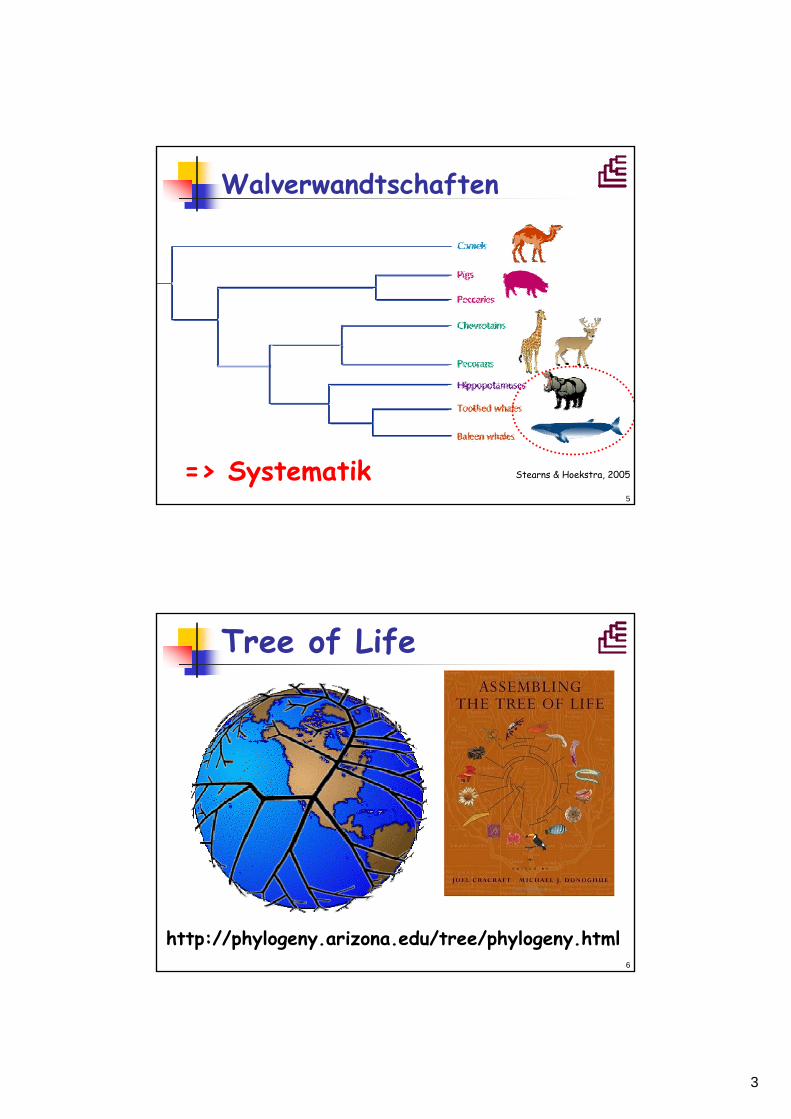
Walverwandtschaften
=> Systematik Stearns & Hoekstra, 2005
6
http://phylogeny.arizona.edu/tree/phylogeny.html
Tree of Life

4
7
Tree of Life
8
Tree of Life
“ Darwinism, an exposition of the theory of natural selection with some of its applications”
The present work treats the problem of the Originof Species on the same general lines as wereadopted by Darwin; but from the standpointreached after nearly thirty years of discussion, with an abundance of new facts and the advocacyof many new or old theories.
Alfred Russel Wallace, 1889.
The affinities of all the beings of the sameclass have somtimes been represented by a great tree….“
Charles Darwin, 1859
5
9
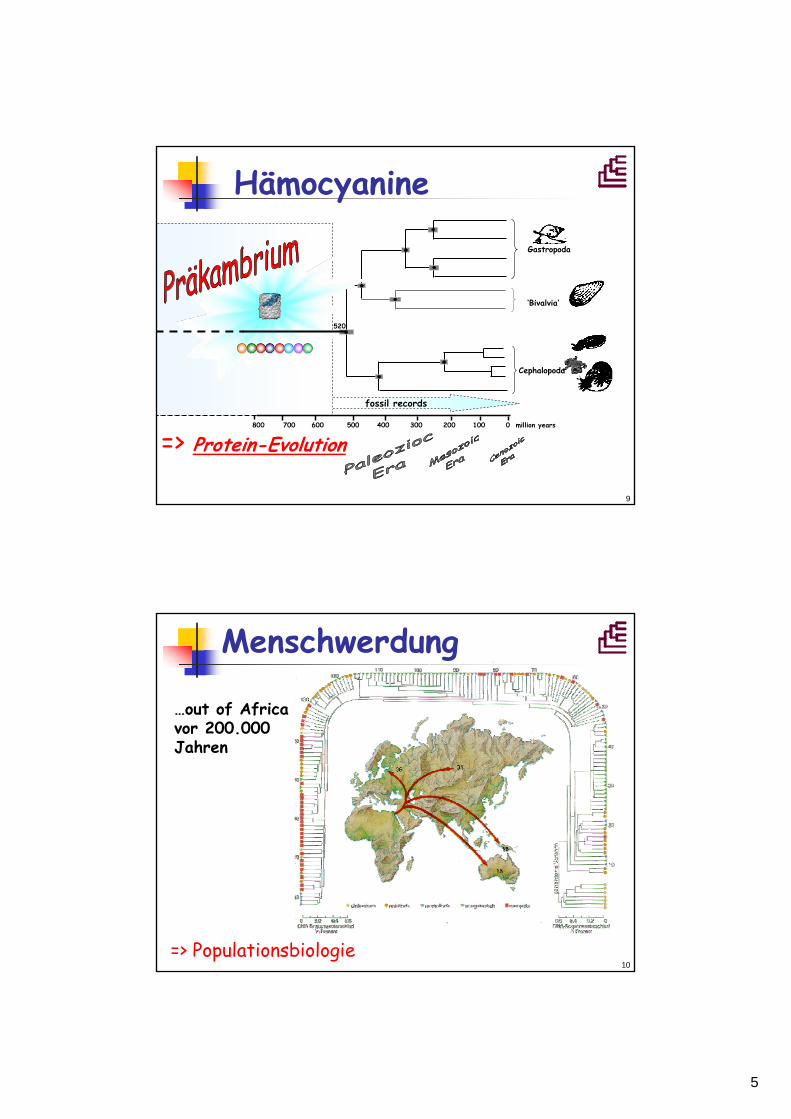
Hämocyanine
800 700 600 500 400 300 200 100 0 million years
520
fossil records
Gastropoda
‘Bivalvia‘
Cephalopoda
=> Protein-Evolution
10
Menschwerdung
…out of Africavor 200.000 Jahren
=> Populationsbiologie

6
11
HIV
=> Epidemiologie/Forensik…
Dr. Acer continued to treat patients after learning in 1986 that he was infected with H.I.V. and afterdeveloping AIDS symptoms in 1987.
12
Phylogenie, aber wie?
•Vergleich der strukturellen (morphologischen) und anatomischen Merkmale von Fossilien
•Vergleich der morphologischen, anatomischen und physiologischen Merkmale rezenter Lebewesen
•Vergleich der Ontogenese
•Analyse der DNA, RNA und/oder Proteinsequenzen

7
13
Moleküle:- DNA- RNA- Protein
Morphologie oder Moleküle?
Morphologie:- Fossilien- Merkmale- Ontogenie..
TAGAGGCATGATCCAT
UAGAGGCAUGAUCCAU
NETRQIVHHWHWMMTR
14
Die Beschreibung der Zustände der molekularen Datenist ‘stets’ eindeutig!
Gertrude Stein:“A rose is a rose is a rose is a rose.”
Vorteile der molekularen Daten

8
15
Die 3. Aminosäure im Präproinsulin des Kaninchens (Oryctolagus cuniculus) ist immer Serin, und die homologe Position im Präproinsulin des Goldhamsters (Mesocricetus auratus) ist immer Leucin ("diskrete, diskontinuierliche Charaktere").
Morphologische Beschreibungen arbeiten häufig mit Bezeichnungen wie "dünn", "reduziert", "etwas verlängert", "teilweise geschlossen"etc.
Die Beschreibung der Zustände der molekularen Daten ist stets eindeutig:
Vorteile der molekularen Daten
16
• Evolution der Sequenzen lässt sich mitModellen beschreiben (PAM, BLOSUM...).
• Molekulare Daten lassen sich relativeinfach quantifizieren.
• Sequenzen zweier Organismen lassensich ‘einfacher’ homologisieren.
• Molekulare Daten sind in fast beliebigerMenge vorhanden
Vorteile der molekularen Daten
9
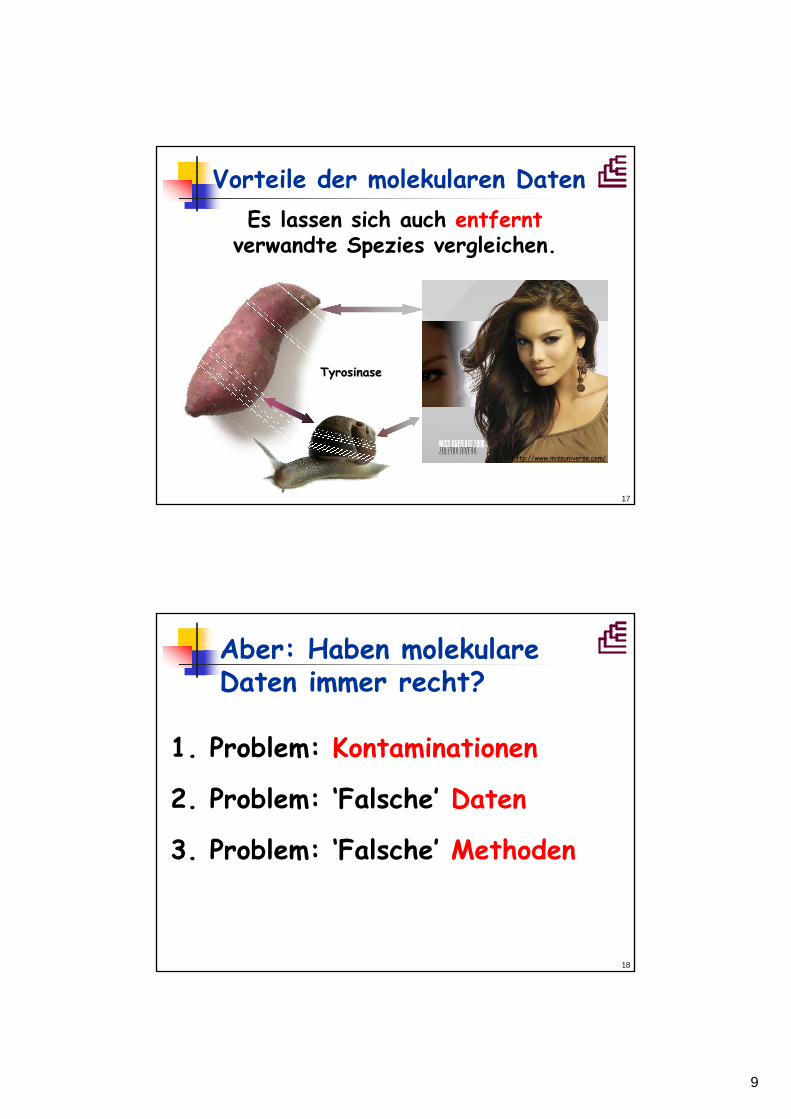
17
Es lassen sich auch entferntverwandte Spezies vergleichen.
Vorteile der molekularen Daten
http://www.missuniverse.com/
TyrosinaseTyrosinase
18
1. Problem: Kontaminationen
2. Problem: ‘Falsche’ Daten
3. Problem: ‘Falsche’ Methoden
Aber: Haben molekulareDaten immer recht?

10
19
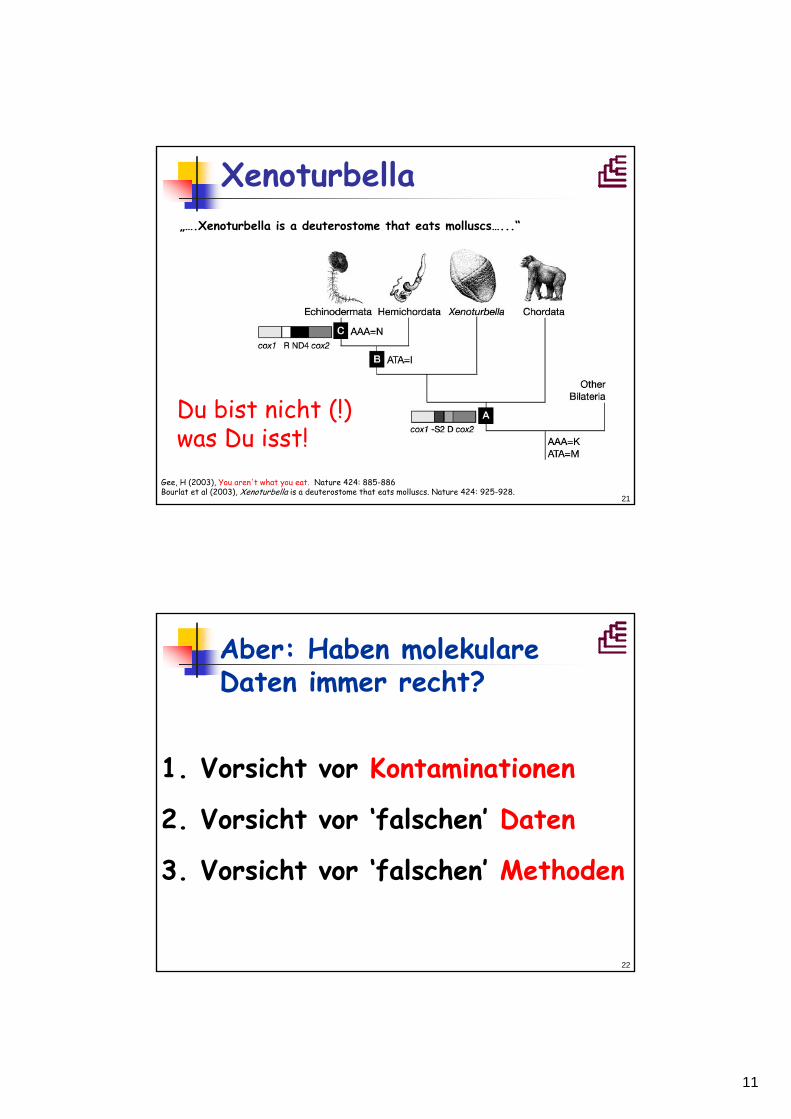
Xenoturbella
18S rRNA(SSU rDNA))
M. Norén, U. Jondelius Nature 390, 31 – 32 (1997)
20
Xenoturbella
"Cladistic analyses of oogenesisindicates that Xenoturbella bocki is a sister group or a subgroup of protobranch bivalves."
OocytenNucula sulcataXenoturbella bocki
Israelsson (1997)...and molluscan embryogenesis Vol 390 No 6655 p 32
11
21
Xenoturbella
Du bist nicht (!) was Du isst!
„….Xenoturbella is a deuterostome that eats molluscs…...“
Gee, H (2003), You aren't what you eat. Nature 424: 885-886Bourlat et al (2003), Xenoturbella is a deuterostome that eats molluscs. Nature 424: 925-928.
22
1. Vorsicht vor Kontaminationen
2. Vorsicht vor ‘falschen’ Daten
3. Vorsicht vor ‘falschen’ Methoden
Aber: Haben molekulareDaten immer recht?

12
23
Die Zeitskala
rRNA SNP, RFLP..Phylogenetische Studien über neun Zehnerpotenzen der Zeitskala hinweg
Genrearrangement – Proteine – DNA – Spacer/Introns
Daten und Methoden
24
Voraussetzungen der molekularen Phylogenie
1. Evolution vollzieht sich durch Veränderungen.2. Verwandte Spezies stammen von einem gemeinsamen Vorfahren
ab.3. Die Speziesbildung vollzog sich durch hierarchische Auftrennung.4. Deren Verlauf lässt sich durch Stammbäume darstellen.5. Es gibt nur einen historisch korrekten Stammbaum.6. Organismen sind historisch. Sowohl die Morphologie als auch die
DNA- und Aminosäuresequenzen speichern die Informationen über die Vergangenheit.
7. Die Methoden der molekularen Evolution erlauben die Extraktion der in der DNA bzw. den Proteinen gespeicherten Informationen.
13
25
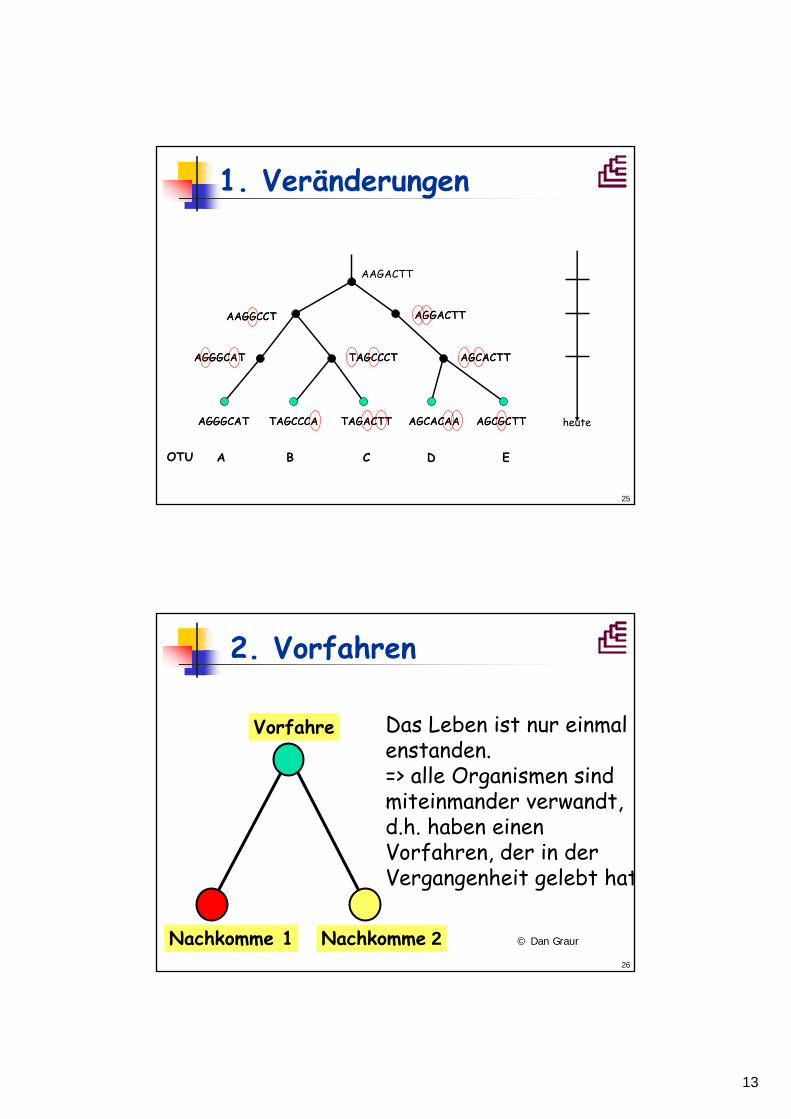
1. Veränderungen
AAGACTT
TAGCCCT AGCACTT
AAGGCCT AGGACTT
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT heute
AGGGCAT
A B C D EOTU
TAGCCCT AGCACTT
AAGGCCT AGGACTT
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT
AGGGCAT
26
Das Leben ist nur einmalenstanden.=> alle Organismen sindmiteinmander verwandt, d.h. haben einenVorfahren, der in der Vergangenheit gelebt hat
Vorfahre
Nachkomme 1 Nachkomme 2
2. Vorfahren
© Dan Graur
14
27
Vorfahre
Einige Organismenhaben einen
Vorfahren, der erstvor kurzem gelebt
hat.
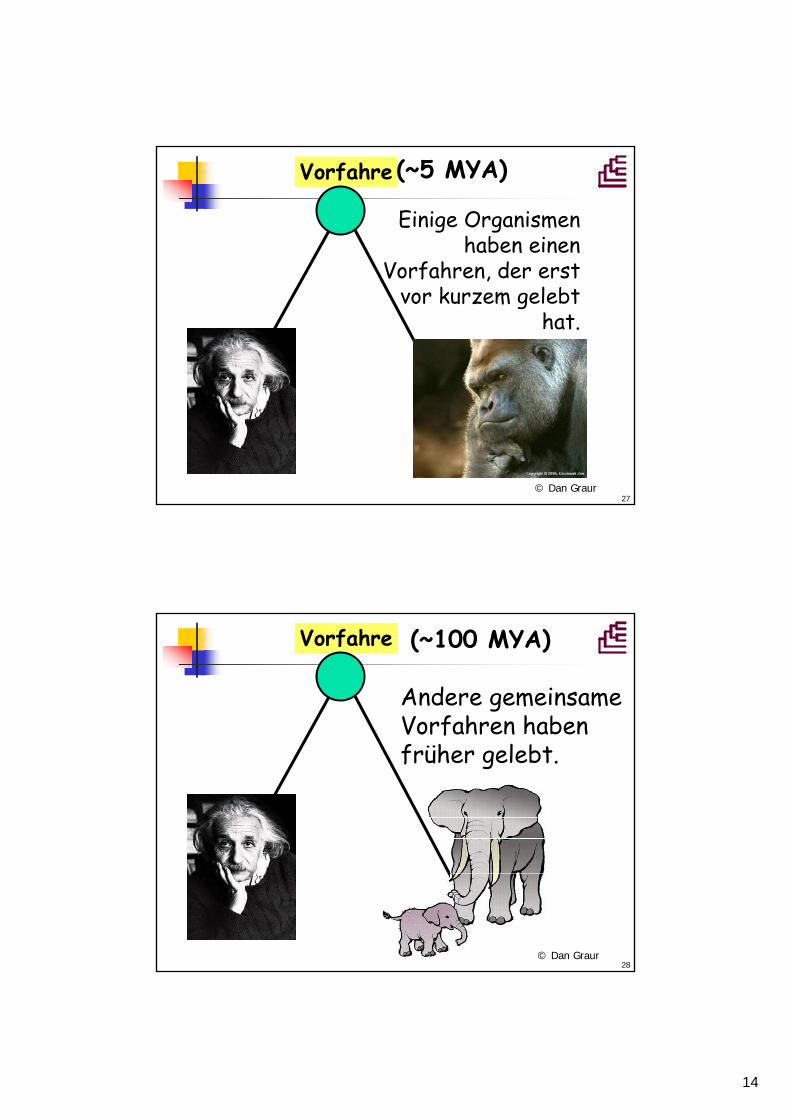
(~5 MYA)
© Dan Graur
28
Vorfahre
© Dan Graur
(~100 MYA)
Andere gemeinsameVorfahren habenfrüher gelebt.
15
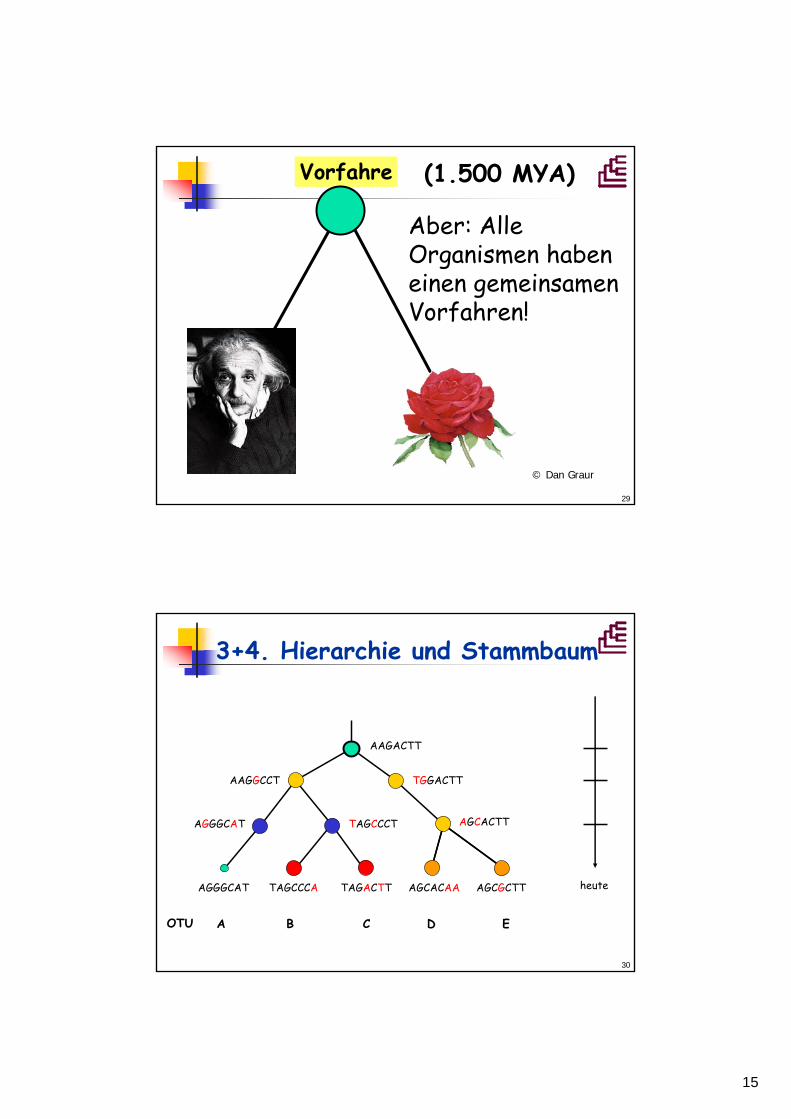
29
Vorfahre
© Dan Graur
(1.500 MYA)
Aber: AlleOrganismen habeneinen gemeinsamenVorfahren!
30
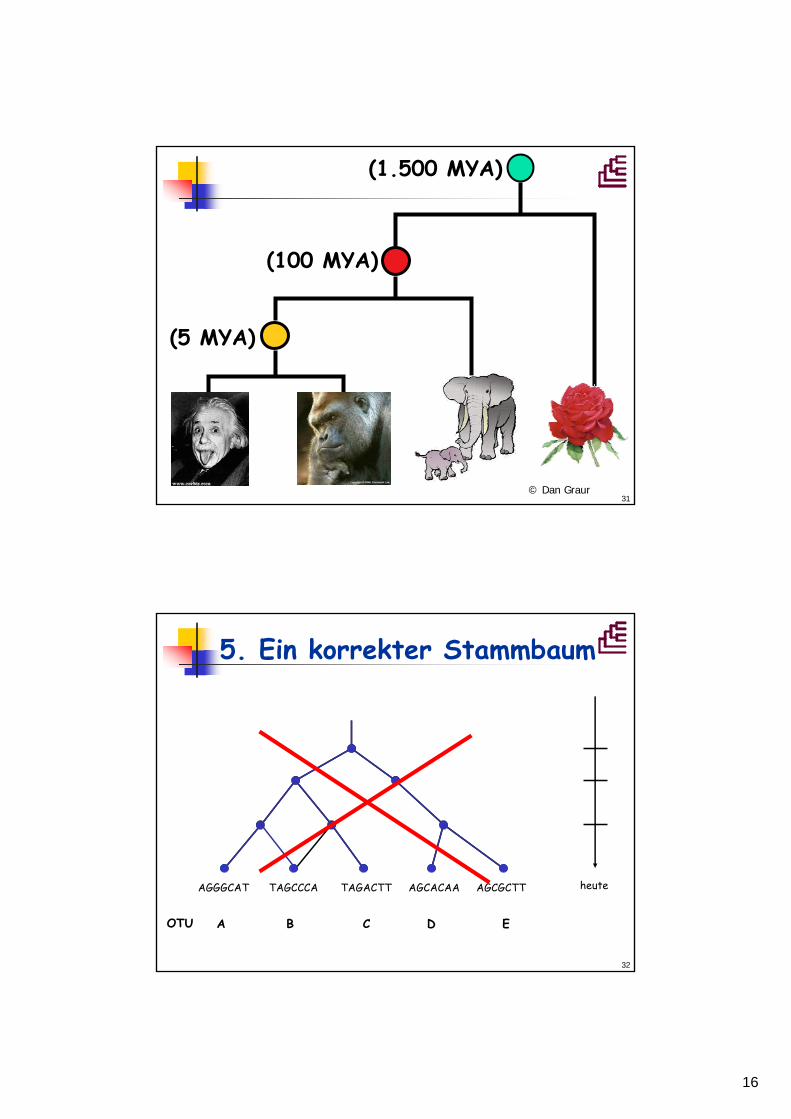
AAGACTT
TAGCCCT
AAGGCCT TGGACTT
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT heute
AGGGCAT
3+4. Hierarchie und Stammbaum
A B C D E
AGCACTT
OTU
16
31
(1.500 MYA)
(100 MYA)
(5 MYA)
© Dan Graur
32
5. Ein korrekter Stammbaum
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT heute
A B C D EOTU

17
33
Horizontaler Gentransfer
34
Horizontaler GentransferHinweise auf LGT des Alkohol adh Gens
Andersson 2005, CMLS
LGT erklärt die 6 Klassen besser als adh-Duplikation und
differntiellen Verlust von Paralogen
18
35
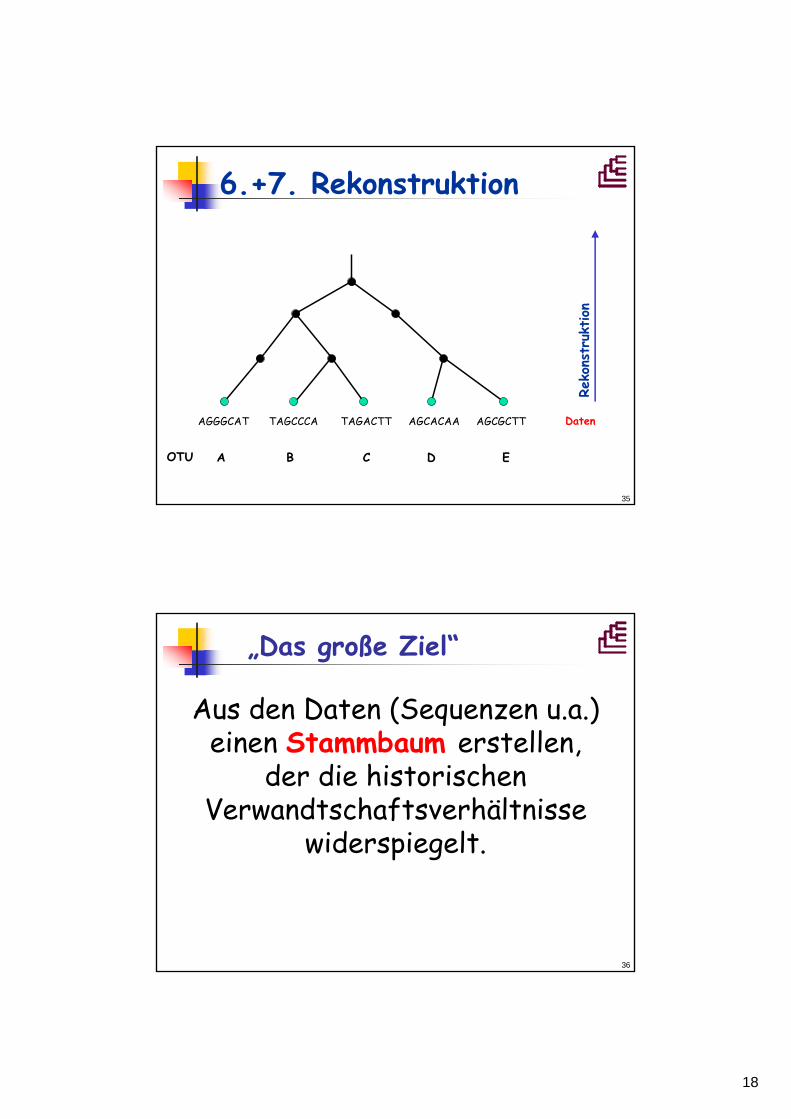
6.+7. Rekonstruktion
AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT Daten
A B C D EOTU
Reko
nstr
uktion
36
„Das große Ziel“
Aus den Daten (Sequenzen u.a.) einen Stammbaum erstellen,
der die historischen Verwandtschaftsverhältnisse
widerspiegelt.
19
37
Schwestergruppen
Was ist ein Stammbaum?
Darstellung der Verwandtschaftsverhältnisse
ABC
A – F auch "operational taxonomic units" (OTUs)
DEF
AB
CD
EF
t t
Taxon/Taxa
38
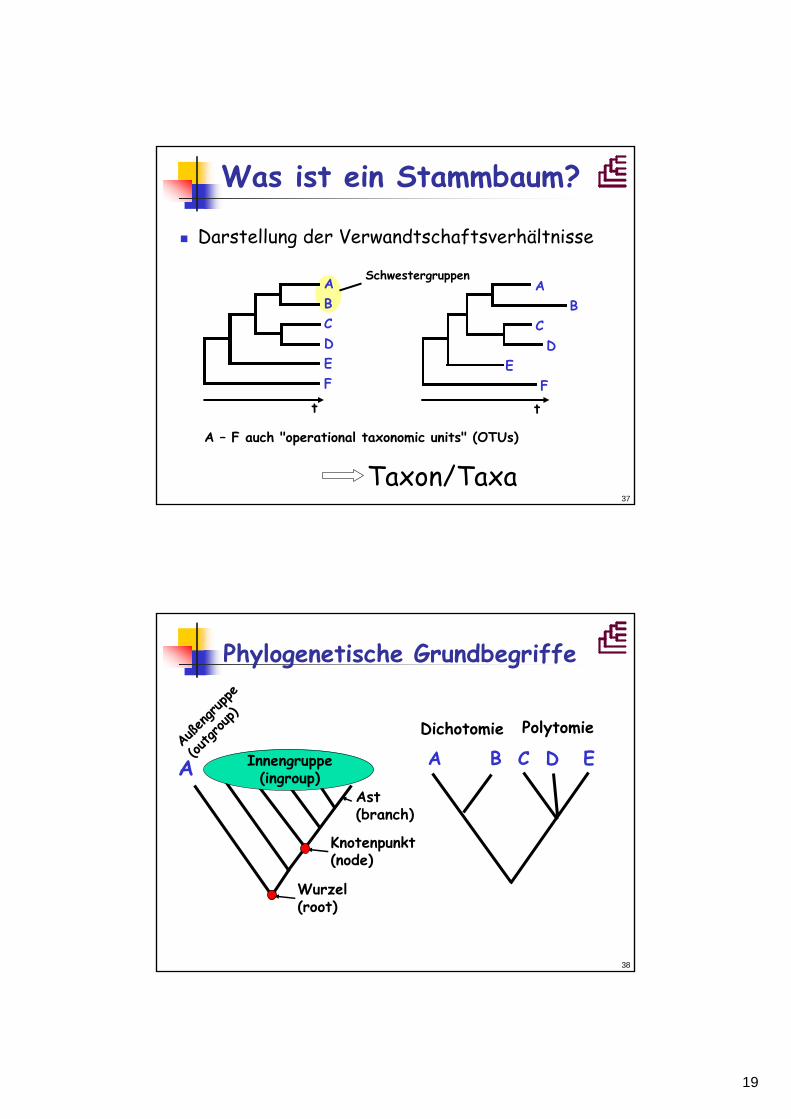
Phylogenetische Grundbegriffe
A B C D E A B C D EDichotomie Polytomie
Ast(branch)
Knotenpunkt(node)
Außengruppe
(outgroup)
Wurzel(root)
Innengruppe(ingroup)

20
39
Mono-, Para- und Polyphylie
Monophyletische Taxa:
Alle Nachkommen einer gemeinsamen Stammform
Polyphyletische Taxa:
Keine gemeinsame Stammform (unterschiedliche Vorfahren)
Paraphyletische Taxa:
Nicht alle Nachkommen einer gemeinsamen Stammform
A B C D E F
40
Paraphylumaufgrund von homologen
(ursprünglichen) MerkmalenVögel
"Reptilien"
Schildkröten KrokodileEidechsen
+Schlangen
Phylogenetische Grundbegriffe
aber nicht alle Nachkommen werden erfasst
21
41
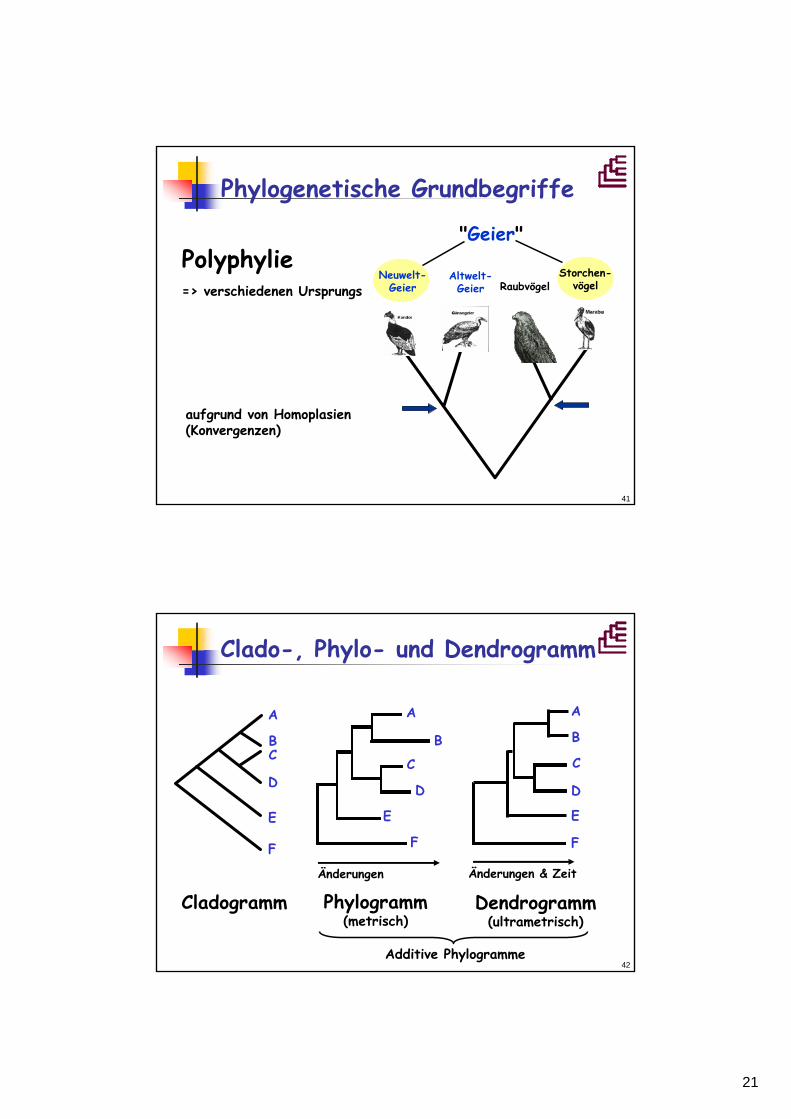
"Geier"
Neuwelt-Geier Raubvögel
Polyphylie=> verschiedenen Ursprungs
aufgrund von Homoplasien(Konvergenzen)
Altwelt-Geier
Storchen-vögel
Phylogenetische Grundbegriffe
42
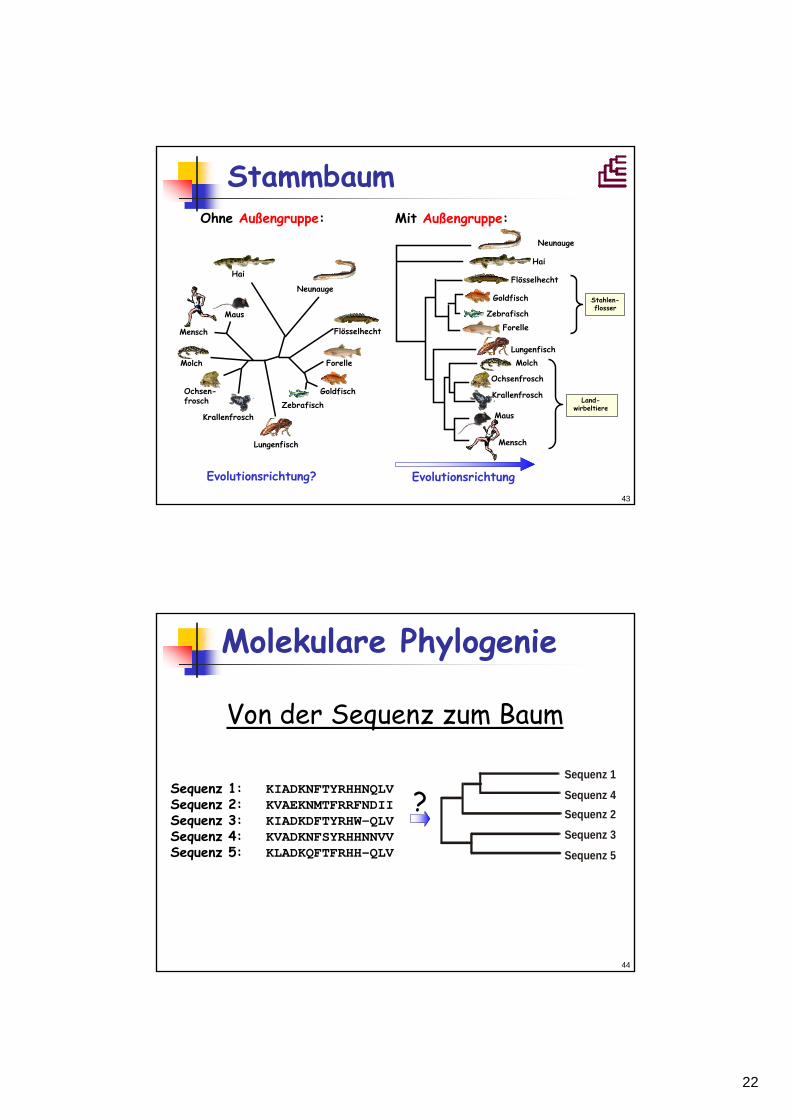
Clado-, Phylo- und Dendrogramm
B
A
C
D
E
F
Änderungen
A
BC
D
E
F
Cladogramm
Additive Phylogramme
A
B
C
D
E
F
Änderungen & Zeit
Phylogramm(metrisch)
Dendrogramm(ultrametrisch)
22
43
StammbaumOhne Außengruppe: Mit Außengruppe:
Evolutionsrichtung
Neunauge
Hai
Goldfisch
Flösselhecht
ZebrafischForelle
LungenfischMolch
Ochsenfrosch
Krallenfrosch
Mensch
Maus
Stahlen-flosser
Land-wirbeltiere
Flösselhecht
Goldfisch
Neunauge
Hai
Zebrafisch
Lungenfisch
Maus
Mensch
ForelleMolch
Ochsen-frosch
Krallenfrosch
Evolutionsrichtung?
44
Molekulare Phylogenie
Sequenz 1: KIADKNFTYRHHNQLVSequenz 2: KVAEKNMTFRRFNDIISequenz 3: KIADKDFTYRHW-QLVSequenz 4: KVADKNFSYRHHNNVVSequenz 5: KLADKQFTFRHH-QLV Sequenz 5
Sequenz 3Sequenz 2Sequenz 4
Sequenz 1
Von der Sequenz zum Baum
?

23
45
Vergehensweise
Stammbaumberechnung
Multiple Sequence Alignment
Auswahl der Methode
Auswahl des Algorithmus
Ergebnisüberprüfung (output)
Sequenzen (Input)
46
SequenzalignmentsGutes Alignment Voraussetzung
für korrekten Stammbaum!
* 20 * 40YPQTKIYFPHF-DLSHGSAQIRAHGKKVFAALHEAVNHID : 39YPQTKIYFPHF-DMSHNSAQIRAHGKKVFSALHEAVNHID : 39FPQTKTYFSHF-DVHHGSTQIRSHGKKVMLALGDAVNHID : 39FPSTKTYFSHF-DLGHNSTQVKGHGKKVADALTKAVGHLD : 39FPTTKTYFPHF-DLSHGSAQVKGHGKKVADALTNAVAHVD : 39MPTTRIYFPAK-DLSERSSYLHSHGKKVVGALTNAVAHID : 39YPQTKTYFSHWADLSPGSGPVKKHGKTIMGAVGEAISKID : 40YPQTKTYFSHWADLSPGSAPVKKHGGVIMGAIGNAVGLMD : 40
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

24
47
Stammbaumerstellung________________________________________________
________________________________________________2 13 34 155 1056 9547 10.3958 135.1359 2.027.02510 34.459.42515 213.458.046. 676.87520 8.200.794.532.637 891.559.375.
________________________________________________
Zahl der möglichen gewurzelten BäumeZahl der Taxa
48
Stammbaumerstellung
Bei der Untersuchung von 20 20 TaxaTaxa gibt es:
⇒ 8.200.794.532.637.891.559.374 falscheStammbäume, aber nur
⇒ EINEN richtigen!
Aber: Es gibt nur einen historisch korrekten Stammbaum:
25
49
Stammbaumerstellung
Methoden der molekularen Phylogenie müssen erlauben, trotz der vielen möglichen Stammbäumen einen diskreten Stammbaum zu berechnen, der die Evolution der Sequenzen widerspiegelt.
Ab einer gewissen Anzahl von Sequenzen ist es nicht möglich, alle theoretischen Stammbäume zu berechnen!
=> d.h., wir brauchen "intelligente" Algorithmen.
50
Programmpaket: PHYLIP
Joe Felsenstein

26
51
Programm: PAUP* 4.0
David Swofford
PAUP: Phylogenetic Analysis UsingParsimony (and other methods)
52
上機嫌で酔ってます。
http://www.megasoftware.net/index.html
Sudhir Kumar
Koichiro Tamura ?
Masatoshi Nei

27
53
http://www.staff.uni-mainz.de/lieb/
http://molgen.biologie.uni-mainz.de/
http://evolution.genetics.washington.edu/phylip/software.html
….or google it ☺
Software links
54
Stammbaumerstellung1. Distanz-orientierte Methoden• UPGMA (Unweighted Pair-Group Method with
Arithmetric Means)• Neighbor-joining• Minimal Evolution
=> Sequenzen werden in Distanzmatrix konvertiert
2. Charakter-orientierte Methoden• Parsimony• Maximum Likelihood
=> jede Position wird als informative Einheit betrachtet
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

28
55
Matrix-orientierte Methoden
Zwei Schritte:
1.Berechnen der paarweisenAbstände zwischen den Sequenzen
2. Erstellen eines Stammbaums anhand dieser Abstandsdaten
Aus ‘jedem‘ Datensatz kann eine Distanzmatrix erstellt werden
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen
56
Sequenzevolution
Ursprungssequenz
Sequenz A Sequenz B
ZeitMutationen
Unterschied = Divergenz = Distanz
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

29
57
Berechnung einer Distanzmatrix
Sequenz 1 TATAAGCATG ACTAGTAAGCSequenz 2 TATTAGCATG ACTGGTAACCSequenz 3 TATTGGCATG ACTAGCAGGC Sequenz 4 TGTTGCCACG ATTAGCTACC Sequenz 5 CGTAGCTATG ACCAACGGGC
Distanz = Durchschnittliche Änderung pro Position
hier: 20 Positionen; => Wieviele beobachtete Änderungen?
3 von 20 Positionen verändert => Distanz zwischen 1 und 2 : 0,15
58
1 2 3 4 5Sequenz 1 0.00 0.15Sequenz 2 Sequenz 3 Sequenz 4 Sequenz 5
1 2 3 4 5Sequenz 1 0.00 0.15Sequenz 2 Sequenz 3 Sequenz 4 Sequenz 5
Distanzmatrix
1 2 3 4 5 Sequenz 1 0.00 0.15 0.20 0.45 0.50Sequenz 2 0.00 0.25 0.40 0.65Sequenz 3 0.00 0.35 0.40Sequenz 4 0.00 0.50Sequenz 5 0.00
1 2 3 4 5 Sequenz 1 0.00 0.15 0.20 0.45 0.50Sequenz 2 0.00 0.25 0.40 0.65Sequenz 3 0.00 0.35 0.40Sequenz 4 0.00 0.50Sequenz 5 0.00
Abstand zwischen Sequenz 1 und Sequenz 2, ausgedrückt in durchschnittlichen Änderungen pro Nukleotidposition (unkorrigierte Hamming-Distanz).

30
59
Abstand gegen Zeit!
t
%
beobachteter Abstand
tatsächlicher Abstand zweier Sequenzen= Anzahl der Mutationen
=> Abstand wird unterschätzt!
Sättigung
60
Warum?
13 Mutationen=>
3 Unterschiede
EinzelsubstitutionSequentielle SubstitutionZufallssubstitutionParallele SubstitutionKonvergente Substitution
Rücksubstitution

31
61
Korrektur der Distanzen
t
%
beobachteter Abstand
tatsächlicher Abstand= Anzahl der Mutationen
Korrektur
62
Korrektur der Distanzen
Frage: Wie korrigieren wir? Wir wollen die tatsächliche Anzahl der evolutiven Ereignisse rekonstruieren.
Wir brauchen also ein Evolutionsmodell, welches die Wahrscheinlichkeit von multiplen Austauschen, Rückmutationen etc. berücksichtigt.

32
63
DNA-Evolutionsmodelle
1969: Jukes & Cantor (JC)1980: Kimura 2-Parameter (K2P)1981: Felsenstein 81 (F81)1985: Hasegawa, Koshino & Yano (HKY85)1990: General Reversible Model (REV)etc…
64
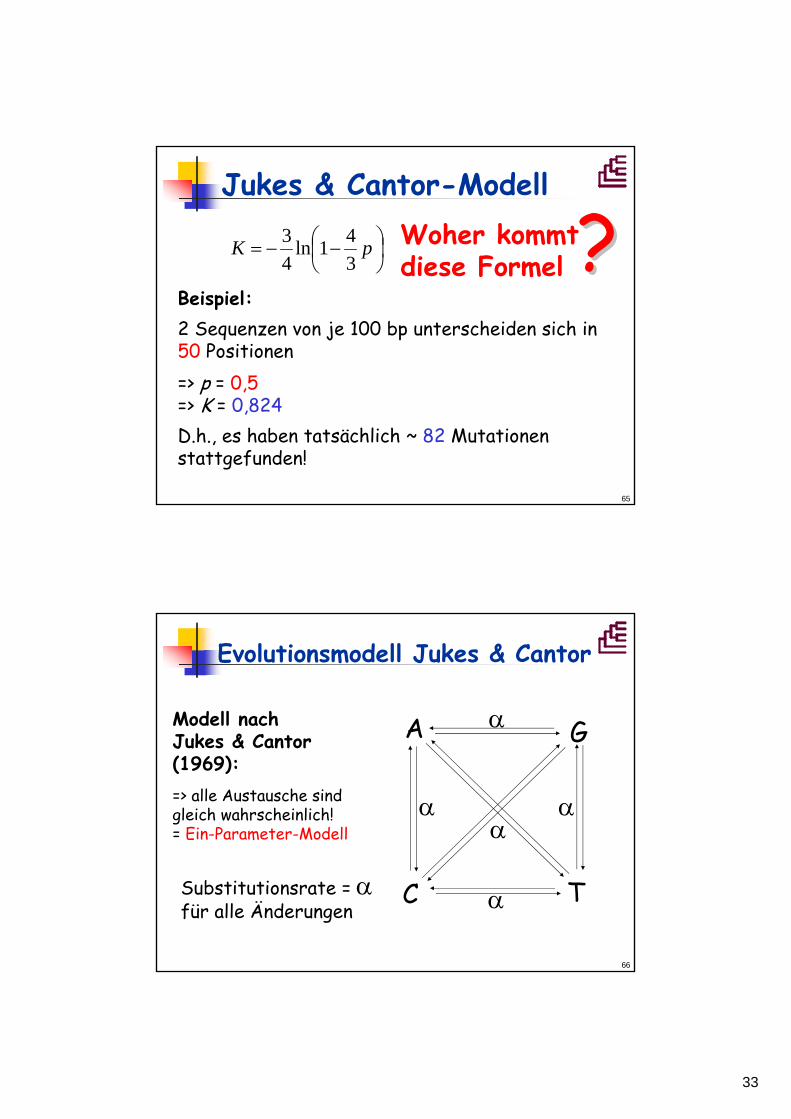
Evolutionsmodell Jukes & Cantor
⎟⎠⎞
⎜⎝⎛ −−= pK
341ln
43
K ist der berechnete Abstand (Anzahl der tatsächlichen Substitutionen), p der beobachteteAbstand zwischen zwei Sequenzen.
Korrigierte Distanz nach Jukes & Cantor:
33
65
Jukes & Cantor-Modell
Beispiel:2 Sequenzen von je 100 bp unterscheiden sich in 50 Positionen=> p = 0,5=> K = 0,824D.h., es haben tatsächlich ~ 82 Mutationenstattgefunden!
⎟⎠⎞
⎜⎝⎛ −−= pK
341ln
43 Woher kommt
diese Formel ??
66
Evolutionsmodell Jukes & Cantor
Modell nach Jukes & Cantor(1969):=> alle Austausche sind gleich wahrscheinlich!= Ein-Parameter-Modell
A G
C T
α
α
α
α
α
Substitutionsrate = αfür alle Änderungen

34
67
Jukes & Cantor-Modell
Substitutions-Wahrscheinlichkeits Matrix!
- PAC PAG PATPCA - PCG PCTPGA PGC - PGTPTA PTC PTG -
P =
Gesamtwahrscheinlichkeit =1 => Pii= 1-∑Pijj
j≠i
1-∑Pij PAC PAG PATPCA 1-∑Pij PCG PCTPGA PGC 1-∑Pij PGTPTA PTC PTG 1-∑Pij
P =
68
Jukes & Cantor-Modell
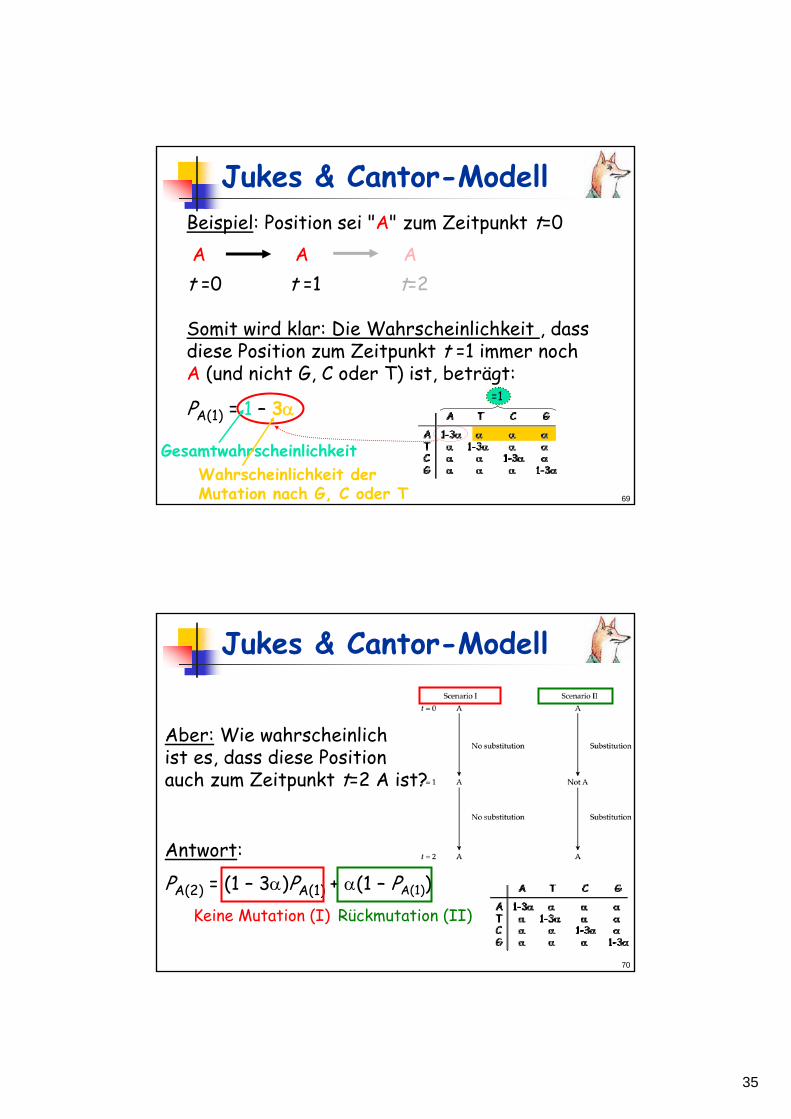
A T C G
1-3α α α αα 1-3α α αα α 1-3α αα α α 1-3α
ATCG
Ein-Parameter-Modell!
35
69
Jukes & Cantor-ModellBeispiel: Position sei "A" zum Zeitpunkt t=0A A A
t =0 t =1 t=2
Somit wird klar: Die Wahrscheinlichkeit , dass diese Position zum Zeitpunkt t =1 immer noch A (und nicht G, C oder T) ist, beträgt:
PA(1) = 1 – 3α
Wahrscheinlichkeit der Mutation nach G, C oder T
=1
Gesamtwahrscheinlichkeit
70
Jukes & Cantor-Modell
Aber: Wie wahrscheinlich ist es, dass diese Position auch zum Zeitpunkt t=2 A ist?
Antwort:
PA(2) = (1 – 3α)PA(1) + α(1 – PA(1))Keine Mutation (I) Rückmutation (II)

36
71
Jukes & Cantor-Modell
Beispiel rechnen:
Annahme: α = 0,01
Wie gross ist PA(1)? => PA(1) = 1 – 3α = 0,97
Wie gross ist PA(2)? => PA(2) = (1 – 3α)PA(1) + α(1 – PA(1)) =
0,97 · 0,97 + 0,01(1 – 0,97) =
0,9412
Wahrscheinlichkeit, dass, wenn betrachtetes Nukleotid zum Zeitpunkt t=0 "A" ist, bei t=1 immer noch "A" ist.
=3α=0,03
72
Jukes & Cantor-Modell
PA(2) = (1 – 3α)PA(1) + α(1 – PA(1))
=> PA(t+1) = (1 – 3α)PA(t) + α(1 – PA(t))
=> PA(t+1) = PA(t) – 4αPA(t) + α
Pro Zeiteinheit:
=> PA(t+1) – PA(t) = – 4αPA(t) + α
=> = – 4αPA(t) + αdPA(t) dt <= Differentialgleichung 1. Ordnung
Verallgemeinerung
{Bsp.: P zum Zeitpunkt 2 minus P zum Zeitpunkt 1}PA(2) – PA(1) = – 4αPA(1) + α
ausrechnen
ausrechnen

37
73
Jukes & Cantor-Modell
te α441
41 −⎟
⎠⎞
⎜⎝⎛ −+= )0A()A( PtP
Lösung der Differentialgleichung 1. Ordnung:
αα +−= )()( 4A
tt P
dt
dP
Annahme: PA(0) = 1 (Ursprungssequenz bei t = 0) =>
te α443
41 −+=)A(tP
74
Jukes & Cantor-Modell
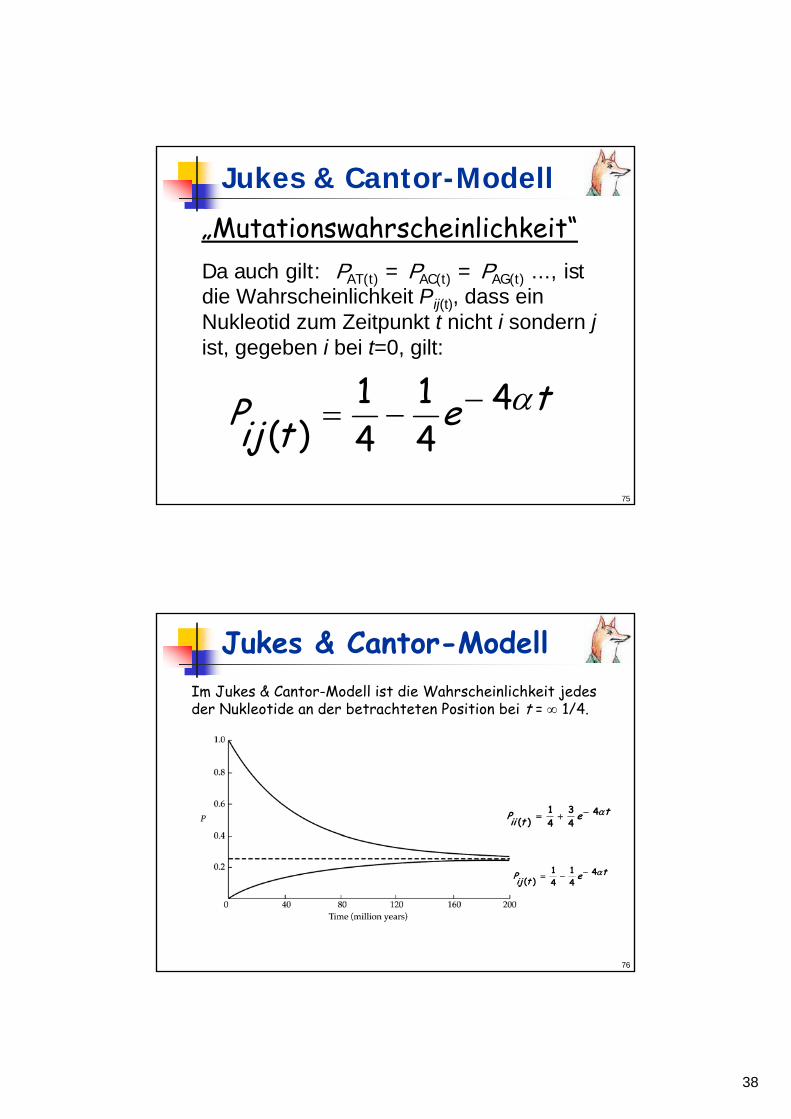
Da bei JC gilt: PAA(t) = PGG(t) = PTT(t) = PCC(t)kann die Wahrscheinlichkeit Pii(t), dass einNukleotid i nach t Zeiteinheiten i bleibtbeschrieben werden als:
tetiiP α443
41
)(−+=
38
75
Jukes & Cantor-Modell
Da auch gilt: PAT(t) = PAC(t) = PAG(t) ..., istdie Wahrscheinlichkeit Pij(t), dass einNukleotid zum Zeitpunkt t nicht i sondern jist, gegeben i bei t=0, gilt:
tetijP α441
41
)(−−=
„Mutationswahrscheinlichkeit“
76
Jukes & Cantor-Modell
tetijPα4
41
41
)(−−=
Im Jukes & Cantor-Modell ist die Wahrscheinlichkeit jedesder Nukleotide an der betrachteten Position bei t = ∞ 1/4.
tetiiPα4
43
41
)(−+=
39
77
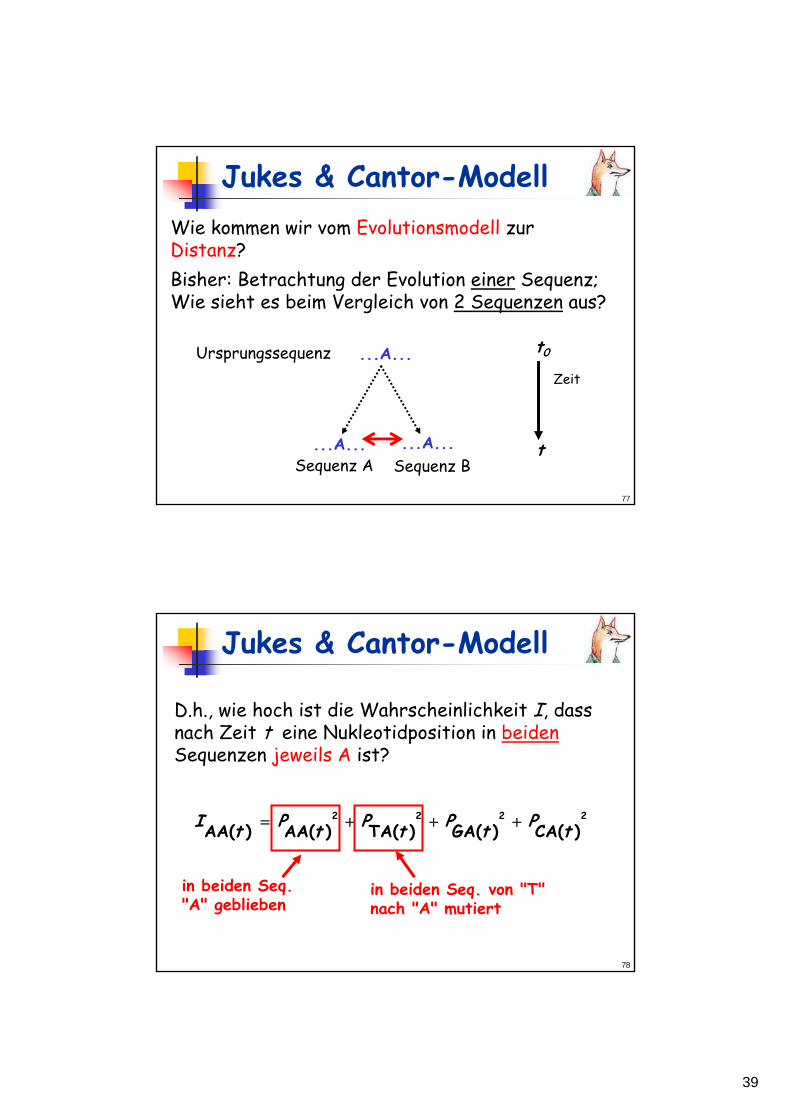
Jukes & Cantor-ModellWie kommen wir vom Evolutionsmodell zur Distanz?Bisher: Betrachtung der Evolution einer Sequenz;Wie sieht es beim Vergleich von 2 Sequenzen aus?
Ursprungssequenz
Sequenz A
Zeit
...A...Sequenz B...A...
...A... t0
t
78
Jukes & Cantor-Modell
D.h., wie hoch ist die Wahrscheinlichkeit I, dass nach Zeit t eine Nukleotidposition in beidenSequenzen jeweils A ist?
2222
)(CA)(GA)(TA)(AA)(AA tPtPtPtPtI +++=
in beiden Seq. "A" geblieben
in beiden Seq. von "T" nach "A" mutiert

40
79
Jukes & Cantor-Modell
tetI α843
41
)(−+=
Lösung:
Wobei I der Anteil identischer Nukleotide zweierSequenzen ist, die sich vor t Zeiteinheiten getrennt haben.
22224
41
414
41
414
41
414
43
41
)( ⎟⎠⎞
⎜⎝⎛ −−+⎟
⎠⎞
⎜⎝⎛ −−+⎟
⎠⎞
⎜⎝⎛ −−+⎟
⎠⎞
⎜⎝⎛ −+= tetetetetiiI αααα
Verallgemeinerung:
A->A A->G A->T A->C+ + +
80
Jukes & Cantor-ModelltetI α8
43
41
)(−+=
⎟⎠⎞⎜
⎝⎛ −−= tep α81
43
Wie gross ist jetzt aber nun der Anteil unterschiedlicher Nukleotide nach t Zeiteinheiten
(Distanz!)?
„Ganz einfach“: p = 1 – I(t). Daraus folgt:
Beobachtete Substitutionen
I der Anteil identischer Nukleotidezweier Sequenzen, die sich vor t Zeiteinheiten getrennt haben.

41
81
Jukes & Cantor-Modell
AberAber: Wir kennnen in der Praxis weder α noch t ! Daherberechnen wir den korrigierten Abstand K zwischen zwei
Sequenzen. Nach Jukes & Cantor:
K = 2t · 3α
2 x t, da für jede der zwei Sequenzen die Zeit t vergangen ist
Jedes der Nukleotide kann in drei andere umgewandelt werden
mit Substitutionsrate α.
⎟⎠⎞⎜
⎝⎛ −−= tep α81
43
Seq A
Seq B t
82
Jukes & Cantor-Modell
⎟⎠⎞
⎜⎝⎛ −−= pK
341ln
43
K = 2t · 3α => K = 6αt ; einsetzen in:
=>
K ist der berechnete Abstand (Anzahl der tatsächlichen Substitutionen), p der beobachteteAbstand zwischen zwei Sequenzen.
⎟⎠⎞
⎜⎝⎛ −−= pt
341ln8α=>⎟
⎠⎞⎜
⎝⎛ −−= tep α81
43

42
83
Jukes & Cantor-Modell
Beispiel2 Sequenzen von je 100 bp unterscheiden sich in 50 Positionen=> p = 0,5=> K = 0,824D.h., es haben tatsächlich ~ 82 Mutationenstattgefunden!
⎟⎠⎞
⎜⎝⎛ −−= pK
341ln
43
84
Aber:
Modell ist zu einfach!Denn jeder Basenaustausch wird gleich bewertet.In der Natur aber nicht so beobachtet.In der Praxis sind meist bessere Modelle notwendig.

43
85
DNA-Evolutionsmodelle
1969: Jukes & Cantor (JC)1980: Kimura 2-Parameter (K2P)1981: Felsenstein 81 (F81)1985: Hasegawa, Koshino & Yano (HKY85)1990: General Reversible Model (REV)uvm…
86
Transitionen vs. Transversionen
Vergleich DNA verschiedener Huftiere
=> Transitionen (A <=> G, C <=> T) sind wesentlich häufiger als Transversionen (A,G <=> C,T)
44
87
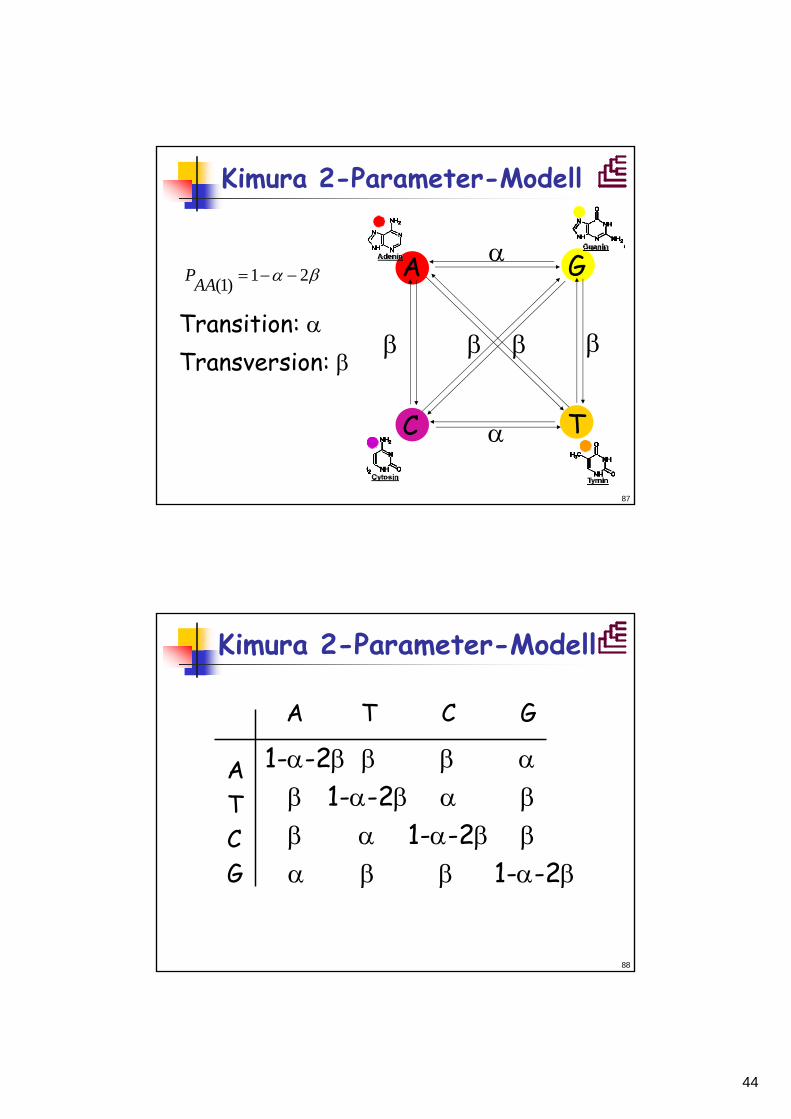
Kimura 2-Parameter-Modell
βα 21)1( −−=AAP
Transition: αTransversion: β
A G
C T
α
β
α
ββ β
88
A T C G
1-α-2β β β αβ 1-α-2β α ββ α 1-α-2β βα β β 1-α-2β
ATCG
Kimura 2-Parameter-Modell
45
89
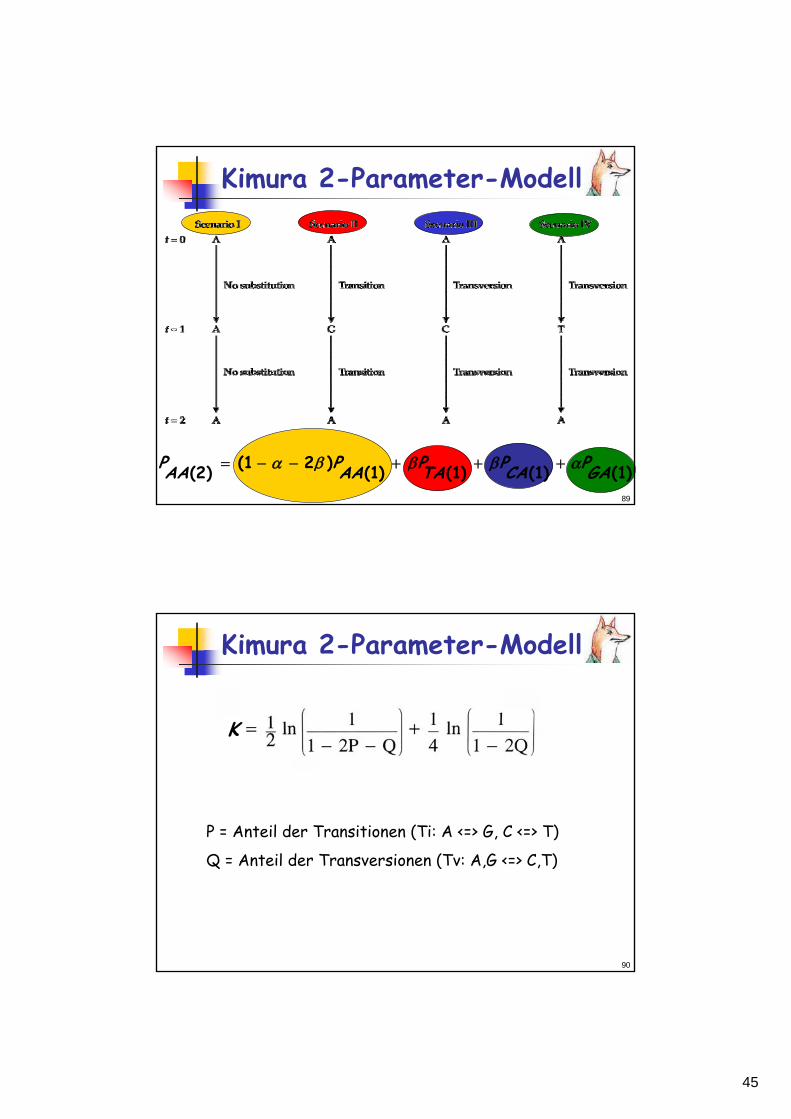
Kimura 2-Parameter-Modell
)1()1()1()1()21()2( GAPCAPTAPAAPAAP αβββα +++−−=
90
Kimura 2-Parameter-Modell
K
P = Anteil der Transitionen (Ti: A <=> G, C <=> T)
Q = Anteil der Transversionen (Tv: A,G <=> C,T)

46
91
JC und K2P im Vergleich
Beispiel 1: 2 Seqenzen mit je 100 bpDivergenz: 10 Ti, 2 Tvp (unkorrigiert) = 12 / 100 = 0,12K (JC) = 0,131K (K2P) = 0,134
Beispiel 2: 2 Seqenzen mit je 100 bpDivergenz 25 Ti, 10 Tvp (unkorrigiert) = 35 / 100 = 0,35K (JC) = 0,471K (K2P) = 0,514
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen
92
Felsenstein 81 Model
Alle Substitutionen gleich häufig; die
erwartete Nukleotidzusammen
-setzung ist identitsch.
Transitionen und Trans-versionenunterschiedlich
häufig; die erwartete
Nukleotidzusammen-setzung ist identitsch.
Erwartete Nukleotid-
zusammensetzungunterschiedlich.

47
93
HKY85 und GTR-Modelle
Transitionen und Trans-versionen und
Nukleotid-zusammensetzung
sind unterschiedlich häufig.
Alle Parameter (Austausche und
Austauschrichtungen) und
Nukleotidzusammensetzung dürfen variieren.
94
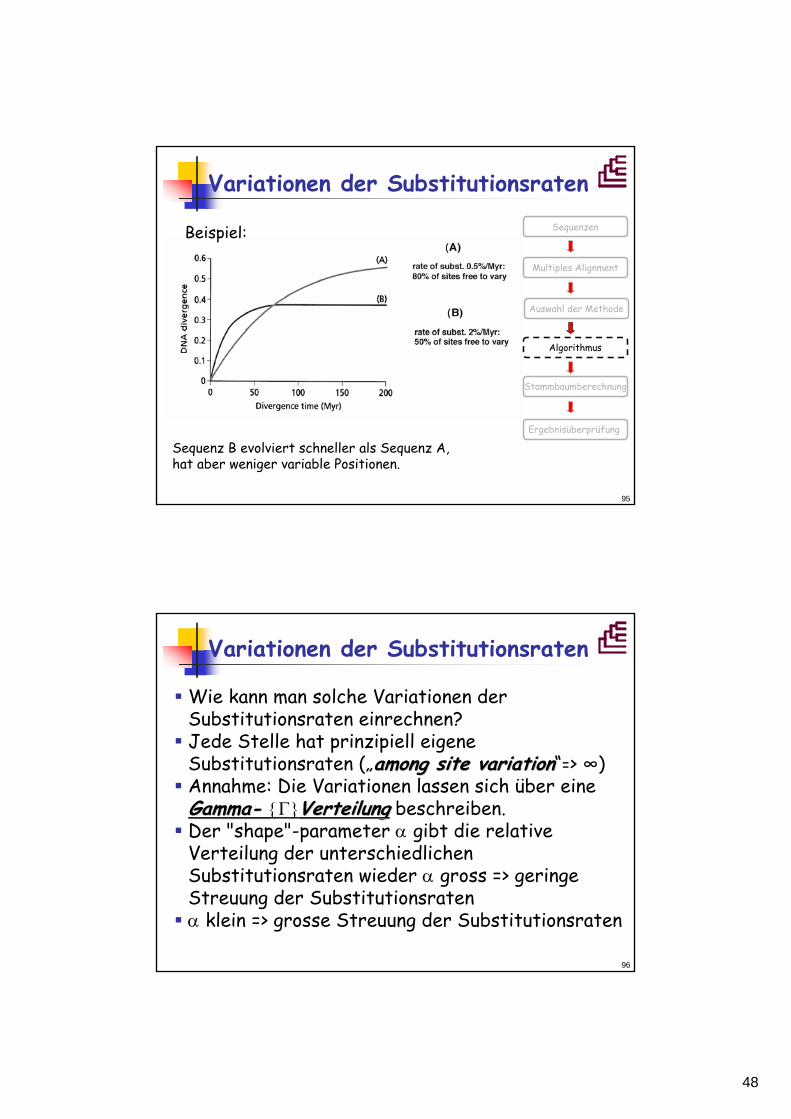
Variationen der Substitutionsraten
Bisher nahmen wir an, dass die Evolutionsraten innerhalb einer Sequenz gleich sind.Kann das stimmen? NeinNein: Evolutionsrate ist abhängig von der Funktion: 4-fach degenerierte, synonymesynonyme Codonpositionenevolvieren wesentlich schneller als nichtnicht--synonymesynonymeCodonpositionen (3. Position).Dies hat natürlich Einfluss auf die Abschätzung der Substitutionen.
48
95
Variationen der Substitutionsraten
Beispiel:
Sequenz B evolviert schneller als Sequenz A,hat aber weniger variable Positionen.
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen
96
Variationen der Substitutionsraten
Wie kann man solche Variationen der Substitutionsraten einrechnen?Jede Stelle hat prinzipiell eigene Substitutionsraten („amongamong sitesite variationvariation“=> ∞)Annahme: Die Variationen lassen sich über eine GammaGamma-- {Γ}VerteilungVerteilung beschreiben.Der "shape"-parameter α gibt die relative Verteilung der unterschiedlichen Substitutionsraten wieder α gross => geringe Streuung der Substitutionsratenα klein => grosse Streuung der Substitutionsraten

49
97
Variationen der Substitutionsraten
shape-parameter αα=0,5
α=2
α=5
α=100
α=50
shape-parameter α
Substitutionsrate
Häu
figk
eit
98
Variationen der Substitutionsraten

50
99
Welches Modell ist das beste?
Je komplexer das Modelle (mehr Annahmen), desto genauer und realistischer unsere Berechnung der Substitutionsrate.
ABERABER:Zusätzliche Parameter müssen aus den Daten abgeschätzt werden. Je mehr Annahmen man trifft, desto größer wird der statistische Fehler (Varianz) der erhaltenen Werte!
=> möglichst "gute" Daten=> möglichst einfaches Modell, das dennoch exakt ist
100
Welches Modell ist das beste?
Wer sagt mir, welches Modell das beste für meine Daten ist?
=> Wir können und müssen die Modelle testen!
•• Modeltest Modeltest : Berechnet "vernünftigen" Stammbaum, und daraus hierarchisch die Parameter.
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

51
101
Welches Modell ist das beste?
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
SequenzenModeltest:
hierarchischer Vergleich der Likelihood (->hLRT)
102
Welches Modell ist das beste?
Modeltest:
hierarchischer Likelihood ratiotest (->hLRT)

52
103
FindModel und Modeltesthttp://hcv.lanl.gov/content/hcv-db/findmodel/findmodel.htmlThe Hepatitis C Virus (HCV) database project
Analyse ausgehend von einem „alignmentalignment“
http://darwin.uvigo.es/software/modeltest.html
ModeltestAnalyse ausgehend von einer „scorescore matrixmatrix“
Modelle:• Hierarchischer likelihood ratio tests (hLRT),• Akaike Information Criterion (AIC = -2 lnL + 2K; Akaike 1974),• Korrigiertes AIC (AICc = AIC + 2K(K+1)/(N-K-1); Hurvich and
Tsai 1989, Sugiura 1978) or• Bayesian Information Criterion (BIC = -2lnL + KlogN; Schwarz
1978) [ L = model likelihood, K = number of estimatableparameters, N = sample size].
104
‚inputinput ‘#NEXUS[Johan Nylander 2004-07-01]
[! ***** MrModeltest block -- Modified from MODELTEST 3.0 *****]
[The following command will calculate a NJ tree using the JC69 model of evolution]
BEGIN PAUP;Log file= mrmodelfit.log replace;DSet distance=JC objective=ME base=equal rates=equal pinv=0subst=all negbrlen=setzero;NJ showtree=no breakties=random;
End;[!***** BEGIN TESTING 24 MODELS OF EVOLUTION ***** ]
BEGIN PAUP;Default lscores longfmt=yes; [Workaround for the bug in PAUP 4b10]Set criterion=like;
[!** Model 1 of 24 * Calculating JC **]lscores 1/ nst=1 base=equal rates=equal pinv=0scorefile=mrmodel.scores replace;[!** Model 2 of 24 * Calculating JC+I **]lscores 1/ nst=1 base=equal rates=equal pinv=estscorefile=mrmodel.scores append;

53
105
‚scoresscores ‘
Tree -lnL1 9208.05402262Tree -lnL p-inv1 8635.42079401 0.38584637Tree -lnL gamma shape1 8523.87431700 0.410427Tree -lnL p-inv gamma shape1 8511.67616714 0.31115986 1.152060Tree -lnL freqA freqC freqG freqT1 9189.14230391 0.26897917 0.27091908 0.20020177 0.25989998Tree -lnL freqA freqC freqG freqT p-inv1 8605.49496982 0.27495452 0.28357511 0.19131684 0.25015353 0.38613047Tree -lnL freqA freqC freqG freqT gamma shape1 8492.98507240 0.26735048 0.28390116 0.19003283 0.25871554 0.402591Tree -lnL freqA freqC freqG freqT p-inv gamma shape1 8480.73674522 0.26918511 0.28348312 0.18971056 0.25762120 0.31535868 1.166148Tree -lnL ti/tv ratio1 9064.14374435 1.19014600Tree -lnL ti/tv ratio p-inv
106
‚outputoutput ‘** Log Likelihood scores **
+I +G +I+GJC = 9208.0537 8635.4209 8523.8740 8511.6758F81 = 9189.1426 8605.4951 8492.9854 8480.7363K80 = 9064.1436 8476.4131 8342.2559 8327.3379HKY = 9042.0869 8435.8828 8307.2676 8289.9834SYM = 9026.4961 8452.5811 8305.9883 8293.9131GTR = 9023.1475 8427.3691 8295.7188 8281.5918Run settingsUsing the standard AIC (not the AICc)Not using branch lengths as parametersRunning all four hierarchies for the hLRTPrinted parameter values are from the hLRT1 hierarchy
---------------------------------------------------------------* HIERARCHICAL LIKELIHOOD RATIO TESTS (hLRTs) *---------------------------------------------------------------Equal base frequencies
Null model = JC -lnL0 = 9208.0537Alternative model = F81 -lnL1 = 9189.14262(lnL1-lnL0) = 37.8223 df = 3 P-value = <0.000001
Ti=TvNull model = F81 -lnL0 = 9189.1426Alternative model = HKY -lnL1 = 9042.08692(lnL1-lnL0) = 294.1113 df = 1 P-value = <0.000001
Unequal Tv and unequal TiNull model = HKY -lnL0 = 9042.0869Alternative model = GTR -lnL1 = 9023.14752(lnL1-lnL0) = 37.8789 df = 4 P-value = <0.000001

54
107
‚‚commandcommandoutputoutput ‘‘
PAUP* Commands Block:If you want to implement the previous estimates as likelihod settings in PAUP*, attach the next block of commands after the data in your PAUP file:[!Likelihood settings from best-fit model (GTR+I+G) selected by hLRT in MrModeltest 2.2]
BEGIN PAUPBEGIN PAUP;Lset Base=(0.2810 0.2726 0.2122) Nst=6 Rmat=(1.6097 3.7278 1.5969 1.0437 6.1587) Rates=gamma Shape=0.8258 Pinvar=0.2910;END;--MrBayes Commands Block:If you want to implement a "best" model in MrBayes, attach the next block of commands after the
data in your NEXUS file:(NOTE: In a Bayesian analysis, the Markov chain is integrating over the uncertainty in parameter values. Thus, you usually do NOT want to use the parameter values estimated by the commands in MrModeltestor Modeltest. You rather want to specify the general "form" of the model (such as nst=1 etc.)[!MrBayes settings for the best-fit model (GTR+I+G) selected by hLRT in MrModeltest 2.2]
BEGIN MRBAYESBEGIN MRBAYES;Prset statefreqpr=dirichlet(1,1,1,1);Lset nst=6 rates=invgamma;END;--





























