Embed Size (px)
DESCRIPTION
notas
Citation preview

INTRODUCCIÓN Y PRINCIPIOS BÁSICOSLa Espectroscopia de Ultravioleta-Visible es la más antigua de las técnicas espectroscópicas y en alguna ocasión fue ampliamente utilizada para la determinación de estructuras.Sin embargo, las nuevas técnicas instrumentales son más reveladoras de la estructura de un compuesto y más fáciles de interpretar.Aunque ya no es muy usada para el análisis estructural, si es muy utilizada en el análisis cuantitativo ya que detecta concentraciones extraordinariamente pequeñas de los compuestos que absorben energía ultravioleta y/o visible y, por lo tanto, es posible tener alta precisión y exactitud al medir concentraciones.La Espectroscopia de Ultravioleta-Visible (UV-VIS) es un ejemplo de espectroscopia de absorción. Un espectro de absorción se obtiene colocando la sustancia entre el detector del espectrofotómetro y una fuente de energía que proporciona radiación electromagnética en el intervalo de frecuencia a estudiarse. El espectrofotómetro analiza, para una determinada frecuencia, la energía transmitida con referencia a la energía incidente. Una molécula puede absorber un cuanto de radiación en varias de las regiones del espectro electromagnético. Sin embargo, el efecto que se produce con la radiación de las distintas regiones del espectro es diferente. Por ejemplo, la absorción de un cuanto de radiación de microondas (alrededor de una caloría/mol) provoca un cambio en la molécula de un estado rotacional a otro. Cuando luz ultravioleta y/o visible es absorbida por una molécula produce cambios en los estados electrónicos, es decir, la absorción de energía luminosa por compuestos orgánicos en la región UV-VIS implica la transferencia de los electrones desde el estado fundamental a estados de mayor energía. Ahora bien, ¿cuáles son esos estados de energía? Tanto los estados fundamentales como los estados de mayor energía son descritos como orbitales moleculares. Estos son formados por la combinación lineal de dos orbitales atómicos y dicha combinación forma no uno, sino dos orbitales moleculares: uno enlazante, que es más estable que los orbitales atómicos que lo formaron, y uno antienlazante, que es menos estable que los que lo originaron.
Así, además de los orbitales moleculares sigma () y pi () que conocemos, existen orbitales antienlazantes asociados con ellos: * (sigma de antienlace) y * (pi de antienlace). De esta manera, la absorción de energía ultravioleta y/o visible implica la transferencia de los electrones de los orbitales , y n (orbitales con electrones no

enlazantes, como los que existen en el oxígeno) a orbitales * y *. Como los orbitales n no forman enlaces, no hay orbitales antienlace asociados con ellos (Figura 2).Las transiciones electrónicas comprometidas en las regiones ultravioleta y visible son: → *, → *, n →* y n → *.
Aunque el ultravioleta en el espectro electromagnético abarca desde los 10 nm hasta los 380 nm, no todo este rango es útil. El oxígeno del aire absorbe fuertemente la luz ultravioleta por debajo de los 200 nm y las mediciones espectroscópicas en esta región requieren instrumentos especiales en los que el aire se excluye por completo. Esta región del espectro se conoce como ultravioleta al vacío y tiene poca importancia en los estudios rutinarios de los laboratorios de química. Las longitudes de onda que sobrepasan el nivel de los 200 nm no son absorbidas por el oxígeno del aire y esta es la región que adquiere la máxima importancia. Los espectrofotómetros UV-VIS más comunes trabajan en el rango de los 200- 900 nm.Como consecuencia de lo anterior, no se pueden estudiar todos los tipos de transiciones electrónicas. La energía necesaria para la transición σ → σ* es muy alta; por lo tanto, los compuestos en los cuales todos los electrones de la capa de valencia están comprometidos en la formación de enlaces simples, como es el caso de los alcanos, no presentan absorción en la región del ultravioleta común. Los compuestos que contienen electrones no enlazados en átomos de oxígeno, nitrógeno, azufre o halógenos puede mostrar absorciones causadas por transiciones n → σ*. Estas implican menor energía que las transiciones σ → σ* y, aunque pueden mostrar absorción en la región del ultravioleta común, la mayoría de estos compuestos absorben por debajo o muy cerca de los 200 nm, por lo que tampoco podemos estudiarlos. Ejemplos:
CH3OH, λmax = 183 nm N (CH3)3, λmax = 227 nm CH3Cl, λmax = 173 nm λmax significa: longitud de onda a la cual el compuesto presenta su máximo de absorción.
Así pues, las transiciones electrónicas que más interesan al químico orgánico son (a) n → π*, en la que un electrón de un par no compartido pasa a un orbital π*(antienlazante) y (b) π → π*, en la que un electrón pasa de un orbital π (enlazante) a un π* (antienlazante).

Sin embargo, aunque una transición puede ocurrir incluso para un alqueno simple, como el etileno, la absorción todavía se realiza cerca de los 200 nm y no resulta práctico el análisis de alquenos simples. Entonces, ¿qué tipo de compuestos se estudian normalmente en el UV-VIS?Pues resulta que la conjugación de los dobles enlaces reduce la energía requerida para la transición, por lo que la absorción se desplaza hacia longitudes de ondas mayores, donde puede medírsele más convenientemente. Si la conjugación comprende un número suficiente de dobles enlace, la absorción se desplaza hacia la región del visible, pasando el compuesto a ser coloreado (figuras 3 y 4). Dado lo anterior se concluye que en espectroscopia de Ultravioleta-Visible aplicada a la Química Orgánica se estudian únicamente compuestos que presente conjugación: la conjugación entre dos o más dobles enlaces (o triples); entre dobles enlaces carbono-carbono y carbono-oxígeno; entre dobles enlaces y anillos aromáticos; incluso, la presencia de un anillo aromático en sí. Además, puede revelar el número y la ubicación de sustituyentes unidos a los carbonos del sistema conjugado. Antes de iniciar el estudio de la elucidación de estructuras mediante esta técnica, revisaremos los conceptos y definiciones que son más frecuentemente utilizados. Asimismo, nos detendremos brevemente para ver los componentes principales de un Espectrofotómetro de Ultravioleta-Visible.
Figura 4. Excitación electrónica de butadieno, CH2=CH-CH=CH2,y hexatrieno, CH2=CH-CH=CH-CH=CH2.

CONCEPTOS UTILIZADOS EN ESPECTROFOTOMETRÍA ULTRAVIOLETA-VISIBLECROMÓFORO. Los diferentes libros de texto dan definiciones de grupo cromóforo que difieren un poco entre sí; así tenemos que el Dyer lo define como un grupo funcional aislado, no conjugado con ningún otro grupo, que muestra una absorción característica en las regiones ultravioleta o visible, ejemplos: C=C (absorción máxima a 171 nm para el etileno) y C=O (máximos de absorción a 166, 189 y 279 nm para la cetona).Para Silverstein/Bassler/Morril Cromóforo es un grupo insaturado que es responsable de la absorción electrónica, definición muy similar a la anterior.Lambert/Shurvell/Lightner/Cooks hacen la observación de que aunque las transiciones electrónicas se dan entre los estados fundamental y excitado de la molécula completa, la mayor parte de la acción puede ser asignada a la parte de la molécula donde los electrones son excitados y a dicha parte se le denomina cromóforo. De esta manera hablamos de la transición electrónica en un cromóforo, el cual en una molécula orgánica generalmente es un grupo funcional. Esta definición se ajusta a las dos anteriores pero hay un detalle: no menciona que el cromóforo deba ser un grupo insaturado (aunque los ejemplos que da el texto incluyen solo grupos funcionales insaturados).Por otro lado, Creswell/Runquist/Campbell lo definen solo como el sistema responsable de la absorción de luz, de tal manera que el grupo C-C (enlace carbono-carbono simple) es un cromóforo ya que es responsable de la transición σ → σ*, lo que no encaja con las primeras dos definiciones. Para propósitos de nuestro curso tomaremos como Cromóforo a un grupo insaturado responsable de la absorción en el uv-vis (esto debido a que son solo compuestos insaturados los que se estudian en esta región del espectro electromagnético). Sin embargo, hay que tener presente que podemos encontrar textos que se refieran a un Cromóforo como cualquier grupo (insaturado o no) responsable de una transición electrónica en una molécula.AUXOCROMO. La discusión anterior está muy relacionada con la definición de grupo auxócromo.Dyer: Los auxócromos son grupos que no muestran absorción selectiva por sí mismos sobre los 200 nm, pero asociados a un determinado sistema cromóforo, causan un desplazamiento en la absorción para longitudes de onda mayores y un aumento en la intensidad del pico de absorción. Los grupos auxócromos comunes son: OH, NH2, SH y los Halógenos. Todos estos grupos contienen electrones no enlazados y las transiciones que implican estos electrones n son responsables de estos efectos.Silverstein/Bassler/Morril: Grupo saturado que, cuando se encuentra unido a un cromóforo, altera tanto la longitud de onda, como la intensidad del máximo de absorción. Ejemplos: OH, NH2 y Cl.Lambert/Shurvell/Lightner/Cooks: grupos no cromóforos, que contienen electrones no enlazados. Podemos tomar cualquiera de estas definiciones, tomando en cuenta siempre estas dos consideraciones: son grupos saturados y con electrones n. Por supuesto que en el texto de Creswell/Runquist/Campbell no encontraremos la definición de auxócromo, ya que para ellos los grupos OH, NH2, Cl, etc. también serían cromóforos.

DESPLAZMIENTO BATOCRÓMICO. Es el desplazamiento de la absorción a una mayor longitud de onda debido a un efecto de solvente o de una substitución (desplazamiento rojo).DESPLAZAMIENTO HIPSOCRÓMICO. Es el desplazamiento de la absorción a una menor longitud de onda (desplazamiento azul).EFECTO HIPERCRÓMICO. Es un aumento en la intensidad de la absorción.EFECTO HIPOCRÓMICO. Es una disminución en la intensidad de la absorción.
LEY DE BEER Y COEFICIENTE DE EXTINCION MOLAREl tratamiento cuantitativo de la absorción de la energía radiante por la materia depende del principio general conocido como Ley de Beer. Supongamos que tenemos un recipiente de vidrio con caras planas que se llena con una sustancia absorbente disuelta en un solvente no absorbente (no se consideran las pérdidas causadas por la reflexión en las superficies ni por la absorción en el vidrio) y que es atravesado por una radiación monocromática. La radiación disminuirá en intensidad entre más penetre el líquido y entre mayor sea la concentración de soluto. Expresado esto de manera más general, resulta que la disminución en la intensidad es proporcional al número de moléculas absorbentes en la trayectoria del haz. La enunciación cuantitativa de esta relación es la ley de Beer (que también se conoce como ley de Lambert-Beer o Bouguer-Beer, debido a las importantes contribuciones de estos investigadores): incrementos sucesivos en el número de moléculas idénticas absorbentes que se encuentran en la trayectoria de un haz de radiación monocromática, absorben fracciones iguales de la energía radiante que las atraviesa.En términos de cálculo, esto puede enunciarse como
dI/dn = -kI, donde I es la intensidad del haz de radiación, n es el número de moléculas absorbentes y k es una constante de proporcionalidad. El reacomodo que sigue a la integración entre los límites da
ln [I/Io] = -kNEn esta ecuación Io representa la intensidad de la radiación al entrar a la celda, I es la intensidad de la radiación al salir de la celda y N representa el número de partículas efectivas que absorben la radiación.Una medida más conveniente de expresar esta relación es colocar N como el producto de la concentración c y la longitud de la trayectoria b, de modo que quede
ln [I/Io] = -k'bcLa constante k' se expresa ahora en unidades de masa por unidad de área transversal. En otras palabras, para un área transversal dada la absorción es proporcional a la cantidad de material absorbente en la trayectoria. Por conveniencia reemplazaremos a k' por la constante a, que incluye el factor de conversión de los logaritmos naturales a los comunes. Además, invertiremos la relación I/Io para eliminar el signo negativo.
log [Io/I] = abcLa cantidad log [Io/I] es tan importante que tiene un símbolo especial A llamado absorbancia. Por otra parte la relación I/Io es denominada transmitancia (T), de tal manera que A = log [1/T]. Es más común emplear el porcentaje de transmitancia (%T = 100

x T) en lugar de la absorbancia, si la radiación transmitida es de mayor interés que la naturaleza química del material absorbente. Dado lo anterior, el enunciado más corto de la ley de Beer es entonces:
A = abcLa constante a de la ecuación se denomina absortividad, y es característica de una combinación particular del soluto y el disolvente para una determinada longitud de onda. Sus unidades dependen de las de b y c, y el símbolo varía de acuerdo con ellas. Nosotros vamos a utilizar la concentración en moles/litro y la longitud de la celda en centímetros por lo que el símbolo utilizado es E denominado Coeficiente de Extinción Molar (el nombre correcto actualmente es Absortividad Molar, pero aún no es adoptado completamente por los textos de espectroscopia), por lo que la expresión de la ley de Beer que vamos a manejar es A = εbcEl hecho de que el Coeficiente de extinción sea una constante dentro de la ecuación, significa que es independiente tanto de la concentración, como de la trayectoria y de la intensidad de la radiación incidente (como se acaba de mencionar, varía dependiendo de la naturaleza de la muestra, del solvente utilizado y de la longitud de onda de la radiación). La magnitud del coeficiente de extinción molar de una absorción particular es directamente proporcional a la probabilidad de que se de la transición electrónica en particular; mientras más probable sea una transición dada, mayor su coeficiente de extinción. Aunque las transiciones n → π* ocurren a menudo con poca energía (longitud de onda más larga) que las transiciones π → π* son menos probables. Esta característica distintiva importante en las transiciones n → π* se origina en la singularidad crítica de que los electrones del par no enlazante tienden a concentrarse en una región diferente del espacio en relación a los electrones π (ver Figura 5).
Un cuanto dado de luz que origina una transición n → π* debe encontrar muchas más moléculas antes de que sea absorbido en relación al caso de los cuantos de luz necesarios para una transición π → π*. Esta diferencia se manifiesta experimentalmente en un espectro de absorción como una diferencia de intensidad; las absorciones π → π* son por lo común mucho más intensas (absorción fuerte) que las del tipo n → π* (absorción débil). La diferencia de intensidad es de 2 a 3 órdenes de magnitud. Como ejemplos veamos el espectro del óxido de mesitilo, (CH3)2C=CHCOCH3, en una misma celda de 1 cm. (Figura 6).

Figura 6. Espectros de absorción ultravioleta del óxido de mesitilo, (CH3)2C=CHCOCH3.
Se emplea una solución sumamente diluida para la absorción a 235 nm. El coeficiente de extinción se calcula como sigue:
ε = A/cb = 1.18/(9.37x10-5)(1) = 12,600 M-1cm-1
En esta solución diluida, la absorción debida a la transición n → π* es tan débil que apenas es discernible; se tiene que utilizar una solución más concentrada.
ε = 0.47/(9.37x10-3)(1) = 50 M-1cm-1
La relación de los 2 coeficientes de extinción, 12600/50 = 252 es típica para compuestos carbonilos. En general, las transiciones π → π* tienen un ε de aproximadamente 104, en tanto que las n → π* es más o menos 10-100 M-1cm-1.
ESPECTROFOTOMETO DE ULTRAVIOLETA-VISIBLEEl espectrofotómetro de UV-VIS es un instrumento que emite radiación electromagnética en forma continua, empezando desde el extremo mayor en longitud de onda del visible hasta el extremo de menor longitud de onda del ultravioleta común (200 nm). El haz de luz pasa a través de la muestra y el instrumento mide la cantidad de radiación absorbida. Los espectrofotómetros actuales tienen dos cámaras a través de las cuales son pasados los rayos de radiación electromagnética: una para la muestra y otro para el solvente en el cual se disolvió la muestra. Cuando se utilizan dos cámaras, se dice que el espectrofotómetro es de doble haz. Estos modelos substraen automáticamente la absorción debida al solvente. La Figura 7 muestra un diagrama esquemático simplificado de un espectrofotómetro Ultravioleta-Visible. La fuente de radiación para la región visible es una lámpara incandescente de tungsteno y la fuente para el ultravioleta son tubos de descarga de hidrógeno o deuterio. La fuente de deuterio es más cara pero más durable. Las fuentes producen un haz multicromático (haz con todas las longitudes de onda) y éste pasa al monocromador.El monocromador divide el haz multicromático de fotones en grupos de fotones dentro de un angosto intervalo de longitudes de onda. La longitud de onda del haz cuasimonocromático que se permite que salga del monocromador está determinada por los ángulos al que se colocan los prismas. Cambiando continuamente dichos ángulos, se

permite que salga una secuencia de longitudes de onda, lo que produce un barrido de longitudes de onda de un extremo al otro de la región estudiada. El interruptor o chopper divide la luz monocromática en pequeños impulsos los cuales son dirigidos alternativamente hacia las celdas de la muestra y el blanco.
El detector mide y compara la radiación que llega hasta él. La diferencia en intensidad de los haces del blanco y la muestra es convertida en impulsos eléctricos y es enviada al graficador, donde se traza el espectro de las sustancia que se está analizando. La mayoría de los espectrofotómetros entregan gráficas que relacionan la longitud de onda (en nanómetros) contra Absorbancia o Transmitancia. Aunque teóricamente la absorbancia puede variar de cero a infinito, las absorbancias mayores de 2 ó 3 se usan rara vez, y el rango en que la precisión analítica es adecuada es aún más limitado, por tanto, los valores exactos permisibles se determinan en parte por el tipo de instrumento empleado. El valor de absorbancia comúnmente registrado es de 0 - 2.0.
TÉCNICAS DE PREPARACIÓN DE LA MUESTRALas muestras para Ultravioleta-Visible generalmente consisten de soluciones. La concentración de la solución debe ser tal que se produzca una absorbancia de

aproximadamente 1.0 en el máximo de la banda de absorción. Al respecto cabe aclarar lo siguiente:Si se grafica la absorbancia contra la concentración, debe resultar una línea recta que pase por el origen (de acuerdo con la ecuación de la ley de Beer). Las desviaciones a esta ley se designan como positivas o negativas, según sea el caso. La forma de la curva de absorción puede variar a veces con los cambios en la concentración de la solución, y a menos que se tomen precauciones en la observación parecerá que la ley de Beer falla. Este fenómeno puede deberse a la interacción de las moléculas del soluto entre ellas mismas o con las del solvente, o puede deberse también a factores instrumentales.Aun para un sistema que no presente desviación de la ley de Beer, el rango de concentración en el cual son útiles los análisis fotométricos es limitado, tanto para valores bajos como en los altos. En las concentraciones altas del material absorbente penetra tan poca energía radiante, que la sensibilidad del fotómetro resulta inadecuada; por otro lado, en las concentraciones bajas el error inherente en la lectura del registrador es muy grande en comparación con la cantidad que se está midiendo. Se ha encontrado que el valor óptimo (donde es menor el error o desviación de la ley de Beer) para la absorbancia es de 0.4343 (que corresponde a una transmitancia de 36.8 %), aunque no hay mucha diferencia en un rango de absorbancia de 0.2 a 1.0. Esto es de mayor importancia cuanto se realizan trabajos cuantitativos, aunque las alturas relativas de las bandas de absorción para trabajos cualitativos también son mejor medidas con estos valores de absorbancia. Si conocemos el valor del coeficiente de extinción del compuesto a analizar, la concentración que necesitamos para tener una absorbancia de 1.0 se puede calcular a partir de la ley de Beer. Si la sustancia es desconocida o no se conoce su coeficiente de extinción, se tienen que realizar diluciones a prueba y error hasta obtener una solución con una concentración tal que me dé 1.0 de absorbancia. Para muestras que tienen más de una longitud de onda máxima (λmax), es decir, que presenten dos o más bandas de absorción, para las cuales los coeficiente de extinción difieren considerablemente, la información correcta solo se puede obtener corriendo el espectro a más de una concentración (como en el ejemplo del óxido de mesitilo). A veces aparece solo un máximo, y cuando corremos otra vez el espectro a una concentración más alta, puede observarse otro máximo. La preparación de la solución es un aspecto muy importante, ya que al requerirse soluciones muy diluidas, generalmente se tiene que preparar una solución madre y de ella hacer diluciones hasta obtener la solución requerida. Nos ocuparemos de este tema en una sección que veremos más adelante.SolventesIdealmente, un solvente para espectroscopia de ultravioleta-visible debe ser transparente (no absorber) en la región que va a ser examinada. Existen varios solventes que solo poseen enlaces sigma y por lo tanto no absorben en los rangos del visible y del ultravioleta cercano. Buenos solventes son el ciclohexano, etanol, metanol y el agua. Se deben usar solventes grado espectral o muy puros, y manejarlos de tal manera que no se contaminen. Ya que algunas impurezas absorben intensamente, pequeñas cantidades de ellas introducidas en el solvente en que se disolvió la muestra pueden producir señales que alterarían la apariencia del espectro del compuesto de interés.

CeldasSe pueden encontrar de varios tamaños, ya sea de forma cilíndrica o rectangular. Las celdas rectangulares de 1 cm. Son las más comunes y requieren de aproximadamente 3 ml de solución. Para la región visible, las celdas de vidrio son apropiadas, pero para trabajos en el ultravioleta, se requieren las celdas de cuarzo (muy caras) ya que el vidrio ordinario presenta una absorción considerable en la región ultravioleta. Se debe tener mucho cuidado en la limpieza de las celdas. Deben lavarse varias veces con el solvente antes de usarse y entre sucesivas determinaciones. Después de terminado los análisis deberán limpiarse inmediatamente enjuagando varias veces con el solvente que se utilizó. Con celdas que se encuentren muy sucias puede usarse detergente o ácido nítrico caliente. No deben tallarse con abrasivos ni con ningún implemento que pueda rayarlas. Las celdas rectangulares generalmente presentan dos lados esmerilados y dos lados transparentes (que llamamos ventanas), siendo el manejo de las mismas a través de los lados esmerilados. Cuando se llenan las celdas no debe permitirse que la solución escurra por las paredes exteriores y después del llenado hay que taparlas inmediatamente para prevenir la evaporación del solvente que cambiaría la concentración de la solución. Cuando se inserten las celdas en el contenedor del instrumento los lados esmerilados deben quedar paralelos al haz de radiación. Las ventanas nunca deben entrar en contacto con los sujetadores del contenedor ya que pueden rayarse.
DETERMINACIÓN DE ESTRUCTURAS DE COMPUESTOS ORGÁNICOS MEDIANTEESPECTROSCOPIA ULTRAVIOLETA-VISIBLEAnteriormente revisamos que la característica estructural de los compuestos que normalmente se estudian en con esta técnica es la conjugación. Pues bien, se han obtenidos reglas empíricas (Reglas de Woodward) para calcular la longitud de onda al cual presentan el máximo de absorción tres tipos de compuestos conjugados: Dienos (y polienos) conjugados, Cetonas α, β-insaturadas y derivados de Bencenos substituidos con grupo Carbonilo. El valor de estas reglas en los estudios estructurales de los compuestos orgánicos será evidente al revisarlas y considerar algunos ejemplos de aplicación.

Por homoanular y heteroanular entenderemos que la doble ligadura conjugada se encuentra en un mismo anillo, mientras que en un sistema heteroanular la doble ligadura conjugada se encuentra en diferentes anillos, es decir:
A continuación se dan algunos ejemplos de la forma en que se aplican los datos de esta tabla.

Notas para el uso de las tablas 2 y 3:1) Los valores calculados generalmente quedan dentro de ± 5 nm de los valores observados experimentalmente.

2) La rectificación por solvente es necesaria porque la relación de puentes de hidrógeno del estado excitado de la transición relativa al estado fundamental varía según la polaridad del solvente empleado. Las absorciones de compuestos no polares (como los dienos) no sufren desplazamiento con cambio de solvente.3) Para aldehídos insaturados existe una correlación empírica similar. Las posiciones de absorción son generalmente 5 nm más bajas que las correspondientes cetonas.A continuación se dan ejemplos de cómo utilizar los datos de las tablas 2 y 3.
Sistemas Conjugados Cruzados. Son aquellos que contienen un sistema conjugado "ramificado", por ejemplo:
Puede estimarse que hay dos grupos cromóforos insaturados sobrepuestos; cada uno origina transiciones que se espera alcancen un elevado coeficiente de extinción, y aunque esto realmente sucede, debido a la naturaleza de la banda del espectro, solo se observan aquellas que absorben a longitud de onda mayores y poseen mayor intensidad. Para calcular la longitud de onda del máximo de absorción de acuerdo a las reglas de Woodward, lo que se hace es tomar cada uno de los sistemas por separado y calcular su λmax (como si no existiera el otro sistema). La longitud de onda encontrada será la que tenga mayor valor. Ejemplo:

La conjugación cruzada tiene poco efecto en la λmax de la colesta-1,4-dieno-3-ona. Los cálculos utilizados fueron para una enona (β, β-disustituída) sin la corrección requerida por el doble enlace 1,2 y el grupo β’. Los cálculos correspondientes utilizando el sistema Δ1, 2 resultan en 227 nm como λmax de predicción; ésta es una indicación de que, cuando se presenta tal selección, la predicción de mayor confianza se deriva del sistema que es más altamente sustituido (transición π → π* de mayor longitud de onda).
DERIVADOS DEL BENCENOEl benceno manifiesta tres bandas de absorción, las cuales se originan de transiciones π → π*:
184 nm (ε = 60,000)204 nm (ε = 7,900)256 nm (ε = 200)
La banda a 184 nm se debe a una transición permitida. Las bandas a 204 y 256 nm tienen coeficientes de extinción relativamente bajos para una transición π → π*. Estos valores bajos se deben a la gran simetría del benceno que origina que las absorciones sean de una baja probabilidad. Se dice que este género de absorciones están prohibidas por simetría.Se han utilizado diferentes notaciones para designar las bandas de absorción del benceno. Nosotros utilizaremos la siguiente:
184 nm Banda E1204 nm Banda E2256 nm Banda B
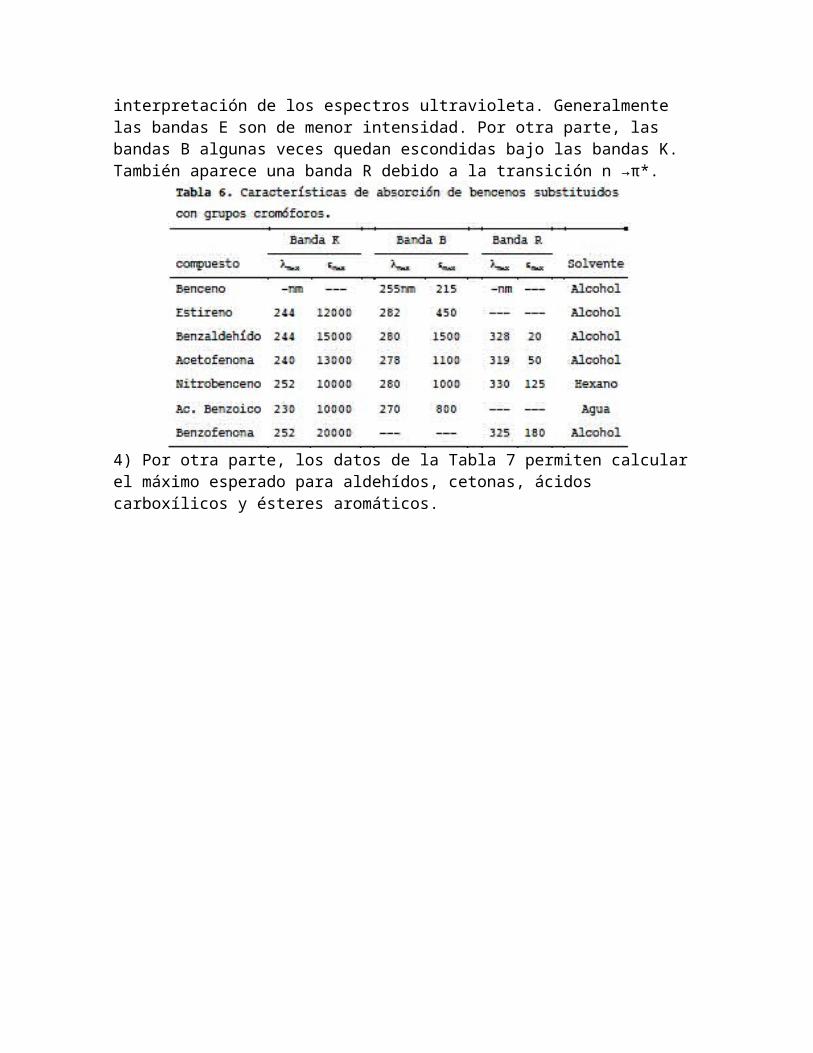
Enseguida revisaremos los efectos de las substituciones en el anillo de benceno:1) La substitución de grupos alquilo en el anillo de benceno produce el desplazamiento batocrómico de la banda B, pero el efecto sobre las bandas E no se ha definido con precisión (Tabla 4).
2) La substitución en el anillo de benceno de grupos auxocrómicos desplaza las bandas E y B hacia longitudes de onda mayores, frecuentemente con la intensificación de la banda B (Tabla 5).

3) El acoplamiento directo de un grupo insaturado (cromóforo) en el anillo de benceno produce un desplazamiento batocrómico intenso de la banda B, y la aparición de una banda K (ε > 10,000) en la región de 200 a 250 nm (Tabla 6).El traslape de las posiciones de absorción de la banda K y el desplazamiento de las bandas E de los bencenos substituidos auxocrómicamente puede dar lugar a confusiones en la interpretación de los espectros ultravioleta. Generalmente las bandas E son de menor intensidad. Por otra parte, las bandas B algunas veces quedan escondidas bajo las bandas K. También aparece una banda R debido a la transición n →π*.
4) Por otra parte, los datos de la Tabla 7 permiten calcular el máximo esperado para aldehídos, cetonas, ácidos carboxílicos y ésteres aromáticos.
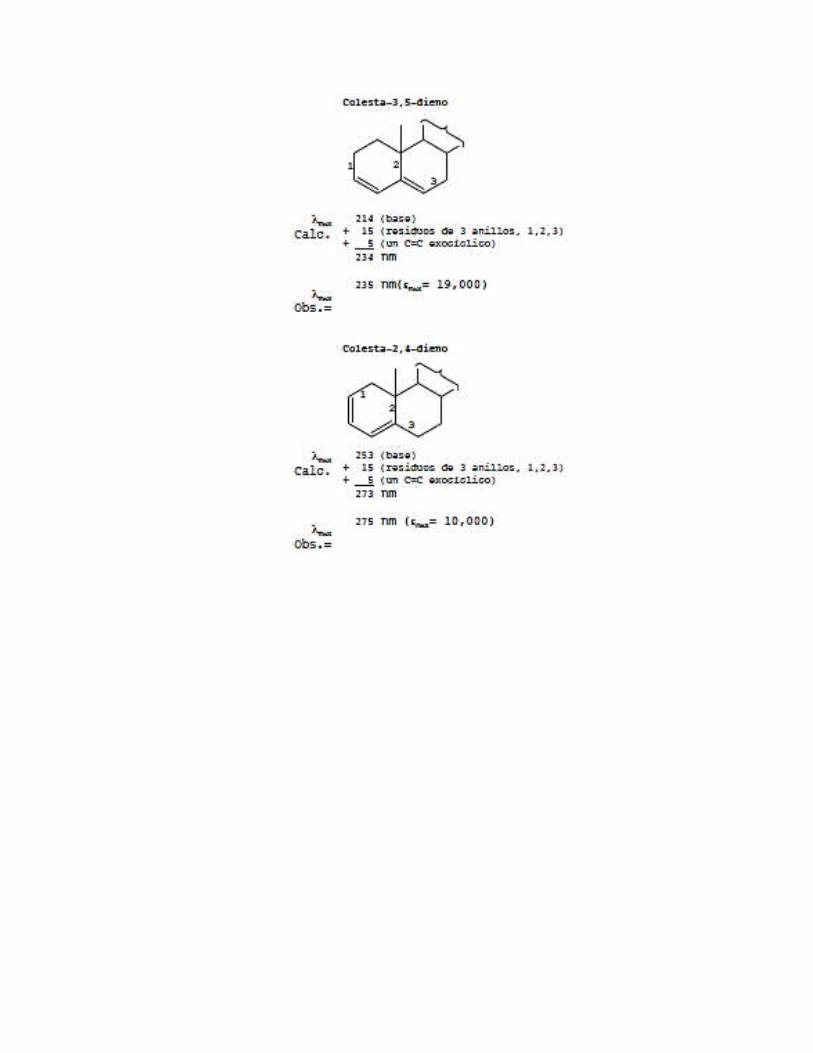
A continuación se dan ejemplos de cómo utilizar los datos de la Tabla 7.

AROMATICOS POLINUCLEARESLos espectros de hidrocarburos aromáticos polinucleares de estructura lineal conservan las bandas típicas del núcleo bencénico. Aumentando el número de anillos condensados, la absorción se desplaza a longitudes de onda mayores. El naftaleno y el antraceno no absorben en la región visible, pero el naftaceno (λmax = 480 nm, ε = 11,000) es amarillo y el pentaceno (λmax = 580 nm, ε = 12600) es azul.
El espectro de hidrocarburos aromáticos de estructura angular, de los cuales el primer miembro es el fenantreno, son más complicados que los isómeros lineales. Sin embargo, todos muestran intensa absorción en la región ultravioleta, y si el número de núcleos aromáticos es suficiente, la absorción se extiende hasta la región visible. ESTEREOQUIMICAEl estudio de los espectros ultravioleta-visible nos puede dar información valiosa en la comparación de isómeros cis y trans.Recordemos que las absorciones de compuestos que contienen conjugaciones extendidas se deben a transiciones π → π*, causadas por los sistemas de electrones deslocalizados presentes. Las intensidades y posiciones de estos picos dependen de la extensión de los sistemas conjugados: más largo es el sistema, mayor es la longitud de onda de absorción y mayor es el coeficiente de extinción. Si el sistema electrónico está impedido para lograr coplanaridad, la sobreposición del sistema de electrones π disminuye y se produce un marcado efecto en el espectro ultravioleta. En un par de isómeros geométricos se espera que la forma cis tenga un impedimento estérico mayor y que por lo tanto la forma trans

logre la coplanaridad del sistema electrónico con mayor facilidad. El resultado de esto es que los isómeros trans absorben a longitudes de onda mayores y tienen coeficientes de extinción más altos. El 1,2-difeniletileno o estilbeno nos permite llevar a cabo una comparación de isómeros cis y trans.
El trans-estilbeno no posee ninguna interacción estérica importante. El compuesto se caracteriza por un sistema coplanar extendido. Sin embargo, en el cis-estilbeno los dos grupos fenilo se encuentran en el mismo lado del enlace doble e interfieren estéricamente el uno con el otro. Los anillos no pueden ser coplanares con el enlace doble, de modo que la conjugación no es tan eficaz como lo es en el isómero trans y éste último presenta una absorción a mayor longitud de onda y la diferencia entre los coeficientes de extinción es el doble. Podemos generalizar diciendo que los sistemas π — π que no llegan a alcanzar coplanaridad presentan cambios notables, sobre todo en las intensidades de absorción.Otro ejemplo adicional lo tenemos en el bifenilo, que la molécula padre de una serie de compuestos en los que dos anillos aromáticos se encuentran en conjugación. El impedimento rotacional en los bifenilos se estudia fácilmente por espectroscopia ultravioleta. La transición π → π*, que rápidamente logra coplanaridad, tiene en este compuesto una absorción a 246 nm (εmax= 16,300). Si grupos voluminosos ocupan posiciones orto, la coplanaridad se restringe y la longitud del sistema electrónico π, disminuye.
PREPARACIÓN DE SOLUCIONES PARA ANÁLISIS ULTRAVIOLETA-VISIBLEDado que para obtener absorbancias adecuadas se utilizan concentraciones muy pequeñas, generalmente es difícil preparar directamente la solución de concentración deseada, por lo que hay que preparar una solución madre y hacer diluciones hasta llegar al valor requerido. Además, hay que tener en cuenta otras dos consideraciones: 1) Sólo necesitamos tres mililitros de solución para llenar la celda y, 2) Hay que evitar el gasto innecesario de solvente. Muestras SólidasEjemplo 1: Ácido Benzoico

Se requiere preparar para su análisis en el Ultravioleta- Visible una solución de Ácido Benzoico con una concentración tal que la banda de mayor intensidad presente una absorbancia de 1.0.1) Necesitamos conocer las longitudes de onda a las cuales el compuesto a analizar presenta máximos de absorción y sus respectivos coeficientes de extinción. En este caso son a 270 nm (ε = 800) y a 230 (ε = 10,000).2) ¿Qué volumen vamos a preparar? Si se dispone de matraces volumétricos de 10 ó 25 ml, ésta cantidad sería suficiente. Generalmente no contamos con ellos, por lo que prepararemos 50 ó 100 ml. Este ejercicio lo vamos a realizar con 100 ml.3) Aplicamos la ley de Beer, A= εbc. El valor del coeficiente de extinción que se utiliza es 10,000, ya que nos interesa la banda de mayor intensidad. La longitud de la celda es de 1 cm.
1.0 = (10,000) (1) (c)c = 1.0 / 10,000
c = 1 x10-4 moles/litro4) Necesitamos entonces 100 ml de una solución con concentración de 1 x10-4 moles/litro. ¿Qué cantidad de ácido benzoico debo pesar y aforar a 100 mL?Si se requieren de 0.0001 moles en 1000 mL entonces necesitamos 0.00001 moles en 100mL
n = g/PM g = (n) (PM) g = (0.00001) (122)g = 0.00122
Pesamos entonces 0.00122 g, los pasamos a un matraz de 100 ml y aforamos con el solvente apropiado. Sin embargo, hay un problema: la cantidad a pesar es muy pequeña y es un principio general que la muestra tomada no debe ser demasiado pequeña (siempre que sea posible), para que los errores de pesada den lugar a errores relativos bajos (menores del 1%). Así pues, debe pesarse una cantidad relativamente grande de la muestra, disolverse en un volumen conocido y tomar alícuotas de esta solución madre para efectuar diluciones hasta llegar a la concentración deseada.Entonces, la cantidad a pesar no es 0.00122 g sino una que nosotros consideremos conveniente, digamos 0.05 g. ¿A qué volumen aforamos? También los seleccionamos nosotros, recordando que hay que gastar lo menos que se pueda de solvente. En nuestro ejemplo lo vamos a aforar a 250 mL.Tenemos ahora una solución que contiene 0.05 g de ácido benzoico en 250 ml de solvente y de aquí vamos a obtener una solución que contenga 0.00122 g en 100 mL (lo cual me da una concentración de 1 x10-4 moles/litro).5) El planteamiento es el siguiente: tengo 0.05 g en 250 mililitros, ¿qué volumen debo tomar para que en éste se encuentren 0.00122 g?
0.05 g ---------- 250 mL0.00122 g ---------- X
X = 6.1 mLTomo dicha alícuota de la solución madre, la aforo a 100 mL y obtengo la solución de concentración 1 x10-4 M.En ocasiones, aquí se puede presentar otro problema: al igual que no debo pesar cantidades muy pequeñas, tampoco los volúmenes tomados deben ser demasiado
pequeños. Incluso, probablemente no cuente con pipetas de esos volúmenes. Por otra parte, los volúmenes de las alícuotas no deben ser muy grandes, ya que resultaría poco práctico e implicaría errores adicionales de lectura y drenaje si utilizamos pipetas comunes (5 y 10 mL). Vamos a considerar como volúmenes razonables las alícuotas mayores de 5 y menores de 20 mL.6) En resumen, en el laboratorio hacemos lo siguiente: pesamos 0,05 g de ácido benzoico y los aforamos con el solvente adecuado a 250 mL; de esta solución tomamos 6.1 mL y los pasamos a un matraz de 100 ml, aforamos con el solvente y ya tenemos lista nuestra solución de trabajo.Muestras LíquidasEjemplo 2: BenzaldehídoVamos a preparar 100 mL de una solución de Benzaldehído que presente un máximo de absorción de 1.0 para la banda de mayor intensidad.1) Este compuesto presenta tres bandas de absorción: a 244 (ε = 15,000 M -1cm-1), 280 (ε = 1,500 M-1cm-1) y 328 nm (ε = 20 M-1cm-1). Aplicando la Ley de Beer tenemos que
c = A / ε bc = 1.0 / (15,000) (1)
c = 6.666 x 10-5 moles/litro2) Como necesito 100 mL de solución entonces requiero de 6.666 x10-6 moles. ¿Qué volumen de benzaldehído voy a tomar? Calculo la cantidad de gramos y luego convierto, por medio de la densidad, a mililitros. La densidad del benzaldehído a 20°C es 1.0447.
g = n x PM; g = (6.666 x10-6) (106.12)Gr = 7.0746 x10-4
δ = g/ml mL = gr/ δml = 7.0746 x10-4 / 1.0447
mL = 6.77 x10-4
Esta cantidad es demasiado pequeña, por lo que vamos a preparar una solución madre y de ésta se obtiene, mediante diluciones, la solución de trabajo.3) La cantidad de benzaldehído que se toma para preparar la solución madre, así como el volumen al cual vamos a aforar lo seleccionamos nosotros, tomando en cuenta las indicaciones mencionadas en el primer ejemplo. Para este ejercicio vamos a considerar 5 mL de benzaldehído en un matraz de 250 mL.4) Entonces necesito tomar un volumen tal de esta solución madre que en él vayan 6.77 x10-4 mL de benzaldehído.
5 mL ---------- 250 ml sol.6.77 x10-4 ---------- X
X = 0.03385 mL de soluciónTomaría dicha alícuota, la aforaría a 100 mL y ya tendría una solución de concentración 6.66 x10-5 M. Sin embargo, este volumen sigue siendo muy pequeño (recordemos que vamos a considerar alícuotas mayores de 5 y menores de 20 ml) por lo que vamos a preparar una dilución intermedia.5) Tomamos 5 ml de la solución madre y aforamos a 100 ml
250 mL sol. ---------- 5 mL benzaldehído5 mL sol. ---------- X
X = 0.1 mL de benzaldehídoAhora tengo una nueva dilución que contiene 0.1 mL de benzaldehído en 100 mL de solución. Repito el mismo planteamiento: ¿cuánto volumen de esta nueva solución tengo que tomar para que en él se encuentren 6.77 x10-4 mL de benzaldehído?
100 mL de sol. ---------- 0.1 benzaldehídoX ---------- 6.77 x10-4
X = 0.677 mL de soluciónEste volumen sigue siendo muy bajo, por lo que se prepara otra dilución.6) De nuevo tomo 5 mL (podrían ser 10 mL u otro valor entre 5 y 20) y lo vuelvo aforar a 100 mL (también ésta cantidad puede cambiar, según lo consideremos conveniente).
100 mL sol. ---------- 0.1 benzaldehído5 mL sol. ---------- X
X = 0.005 mL de benzaldehídoLa dilución recién preparada tiene 0.005 mL de benzaldehído en 100 mL de solución. Y vuelvo al mismo planteamiento:
100 mL de sol. ---------- 0.005 benzaldehídoX ---------- 6.77 x10-4
X = 13.54 mL de sol.
Ahora sí, es un volumen razonable. Tomamos aproximadamente 13.54 mL de alícuota, aforamos a 100 mL y tenemos nuestra solución de trabajo.7) En resumen, el trabajo de laboratorio consiste en lo siguiente: Tomamos 5 mL del frasco reactivo de benzaldehído y aforamos a 250 mL (solución madre). De aquí tomo una alícuota de 5 mL y se pasan a un matraz de 100 mL (dilución intermedia uno). De esta dilución tomo una alícuota de 5 ml y se afora a 100 mL (dilución intermedia dos). Finalmente de esta última dilución tomo aproximadamente 13.5 mL y los llevo a un matraz de 100 mL, aforo con el solvente y obtengo la solución de trabajo, cuya concentración es 6.66 x10-5 M y que presentará en el espectro de ultravioleta-visible una absorbancia de aproximadamente 1.0.














![Espectroscopia UV-Visible e IR[1]](https://img.pdfslide.tips/doc/110x75/577c79a51a28abe054937f38/espectroscopia-uv-visible-e-ir1.jpg)