
Available online at www.sciencedirect.com
www.elsevier.com/locate/brainres
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4
0006-8993/$ - see frohttp://dx.doi.org/10.
Abbreviations: EA
occlusion; 2-AG, 2-a
PKCε, protein kinasnCorresponding aE-mail addresses:1Contributed equ
Research Report
Activation of STAT3 is involved in neuroprotectionby electroacupuncture pretreatment via cannabinoidCB1 receptors in rats
Heng Zhoua,1, Zhi Zhanga,1, Haidong Weia, Feng Wanga, Fan Guoa,Zijun Gaoa, Giovanni Marsicanob, Qiang Wanga,n, Lize Xionga,n
aDepartment of Anesthesiology, Xijing Hospital, Forth Military Medical University, Xi'an 710032, Shaanxi Province, ChinabINSERM U862, NeuroCentre Magendie, 33076 Bordeaux, France
a r t i c l e i n f o
Article history:
Accepted 3 July 2013
Pretreatment with electroacupuncture (EA) attenuates cerebral ischemic injury through the
endocannabinoid system, although the molecular mechanisms mediate this neuroprotection
Available online 20 July 2013
Keywords:
Neuroprotection
Middle cerebral artery occlusion
Ischemic tolerance
Stroke
Behavioral
nt matter & 2013 Elsevie1016/j.brainres.2013.07.00
, electroacupuncture; S
rachidonylglycerol; AEA,
e C epsilon; TTC, 2,3,5-truthors. Fax: +86 29 8477 [email protected] to this work.
a b s t r a c t
are unknown. It is well-known that signal transducer and activator of transcription 3 (STAT3)
plays an essential role in cell survival and proliferation. Therefore, we investigated whether
STAT3 is involved in EA pretreatment-induced neuroprotection via cannabinoid CB1 receptors
(CB1R) after transient focal cerebral ischemia in rats. Two hours after EA pretreatment, focal
cerebral ischemia was induced by middle cerebral artery occlusion (MACO) for 120min. The
expression of pSTAT3(Ser727), which is necessary for STAT3 activation, was examined in the
ipsilateral ischemic penumbra. Infarct volumes and neurological scores were evaluated at 72 h
after MACO in the presence or absence of the STAT3 inhibitor peptide (PpYLKTK). Neuronal
apoptosis and the Bax/Bcl-2 ratio were also evaluated 24 h after reperfusion. Our results
showed that EA pretreatment significantly enhanced neuronal expression of pSTAT3(Ser727)
in the ischemic penumbra 6 h after reperfusion. Moreover, EA pretreatment reduced infarct
volume, improved neurological outcome, inhibited neuronal apoptosis and decreased the
Bax/Bcl-2 ratio following reperfusion. The beneficial effects of EA were attenuated by PpYLKTK
administered 30min before MACO, and PpYLKTK effectively reversed the increase in pSTAT3
(Ser727) expression. Furthermore, CB1R antagonist or CB1R knockdown with siRNA blocked
the elevation of pSTAT3(Ser727) expression by EA pretreatment, whereas the two CB1R
agonists increased STAT3 activation. In conclusion, EA pretreatment enhances STAT3
activation via CB1R to protect against cerebral ischemia, suggesting that STAT3 activation
may be a novel target for stroke intervention.
& 2013 Elsevier B.V. All rights reserved.
r B.V. All rights reserved.6
TAT3, Signal transducers and activators of transcription 3; MCAO, middle cerebral artery
N-arach-idonoylethanolamine-anandamide; ERK1/2, extracellular signal-regulated kinases1/2;
iphenyltetrazolium chloride; TUNEL, deoxyuridine triphosphate nick-end labeling.262.n (Q. Wang), [email protected] (L. Xiong).
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4 155
1. Introduction
Stroke is one of the leading causes of long-term disability andthe first leading cause of death in China (Wu et al., 2012).Although recombinant tissue plasminogen activator (rtPA)has been shown to be an effective drug to treat stroke, it mustbe administered within 3.5 h of stroke onset, and only a fewpatients (less than 3%) can benefit from this therapy (Li et al.,2012). The physical, emotional, and financial toll strokeinflicts on the patients and their families cannot be over-stated. Therefore, the development of new methods forstroke intervention is a major imperative.
Electroacupuncture (EA), originating from Chinese tradi-tional acupuncture and modern electrotherapy, has beenproven to be effective not only in analgesia, but also for thereduction of ischemia/reperfusion injury of the heart andbrain (Xiong et al., 2003, Tsou et al., 2004, Wang et al., 2005,Zhou et al., 2012). Our previous study showed that 30 min ofEA pretreatment at the Baihui acupoint (GV 20) 2 h beforefocal cerebral ischemia was an effective method of inducingischemic tolerance (Xiong et al., 2003). Further studies foundthat EA pretreatment could modulate the endocannabinoidsystem by upregulating the production of the endocannabi-noids, including 2-arachidonylglycerol (2-AG) and N-arach-idonoylethanolamine-anandamide (AEA), which cause protec-tion through cannabinoid CB1 receptors (Wang et al., 2009).Notably, activation of extracellular signal-regulated kinases1/2(ERK1/2) and protein kinase C epsilon (PKCε) was involved in EA
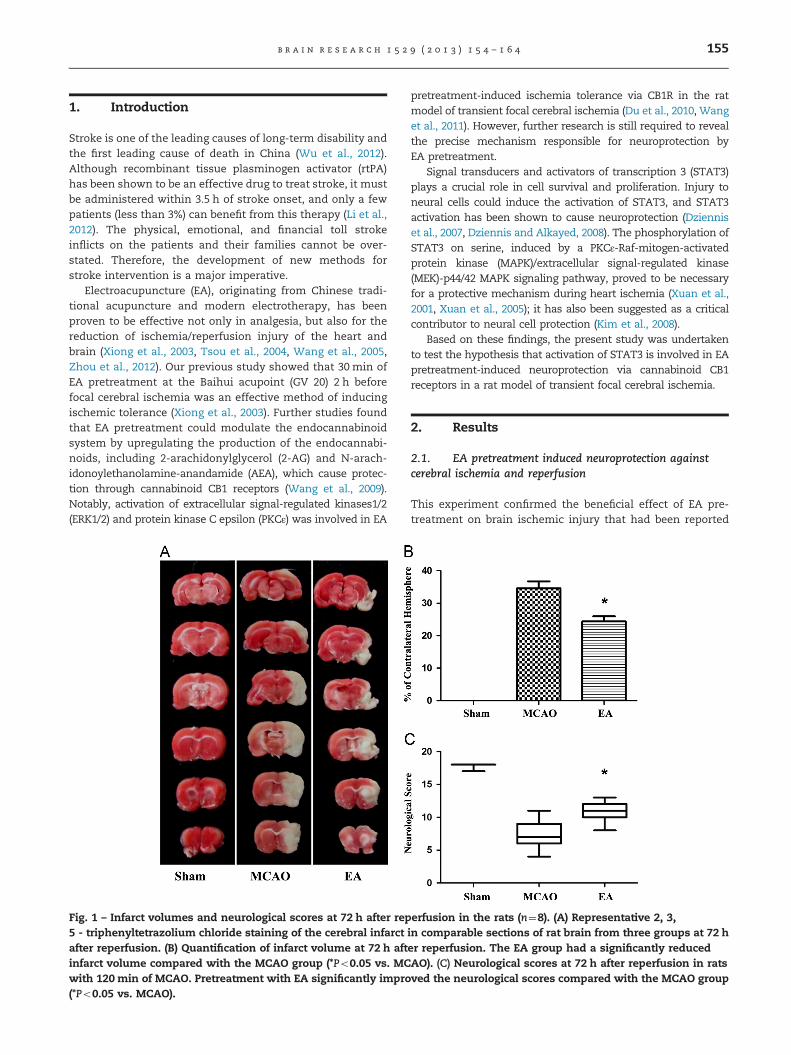
Fig. 1 – Infarct volumes and neurological scores at 72 h after rep5 - triphenyltetrazolium chloride staining of the cerebral infarctafter reperfusion. (B) Quantification of infarct volume at 72 h aftinfarct volume compared with the MCAO group (*Po0.05 vs. MCwith 120 min of MCAO. Pretreatment with EA significantly impro(*Po0.05 vs. MCAO).
pretreatment-induced ischemia tolerance via CB1R in the ratmodel of transient focal cerebral ischemia (Du et al., 2010, Wanget al., 2011). However, further research is still required to revealthe precise mechanism responsible for neuroprotection byEA pretreatment.
Signal transducers and activators of transcription 3 (STAT3)plays a crucial role in cell survival and proliferation. Injury toneural cells could induce the activation of STAT3, and STAT3activation has been shown to cause neuroprotection (Dzienniset al., 2007, Dziennis and Alkayed, 2008). The phosphorylation ofSTAT3 on serine, induced by a PKCε-Raf-mitogen-activatedprotein kinase (MAPK)/extracellular signal-regulated kinase(MEK)-p44/42 MAPK signaling pathway, proved to be necessaryfor a protective mechanism during heart ischemia (Xuan et al.,2001, Xuan et al., 2005); it has also been suggested as a criticalcontributor to neural cell protection (Kim et al., 2008).
Based on these findings, the present study was undertakento test the hypothesis that activation of STAT3 is involved in EApretreatment-induced neuroprotection via cannabinoid CB1receptors in a rat model of transient focal cerebral ischemia.
2. Results
2.1. EA pretreatment induced neuroprotection againstcerebral ischemia and reperfusion
This experiment confirmed the beneficial effect of EA pre-treatment on brain ischemic injury that had been reported
erfusion in the rats (n¼8). (A) Representative 2, 3,in comparable sections of rat brain from three groups at 72 her reperfusion. The EA group had a significantly reducedAO). (C) Neurological scores at 72 h after reperfusion in ratsved the neurological scores compared with the MCAO group
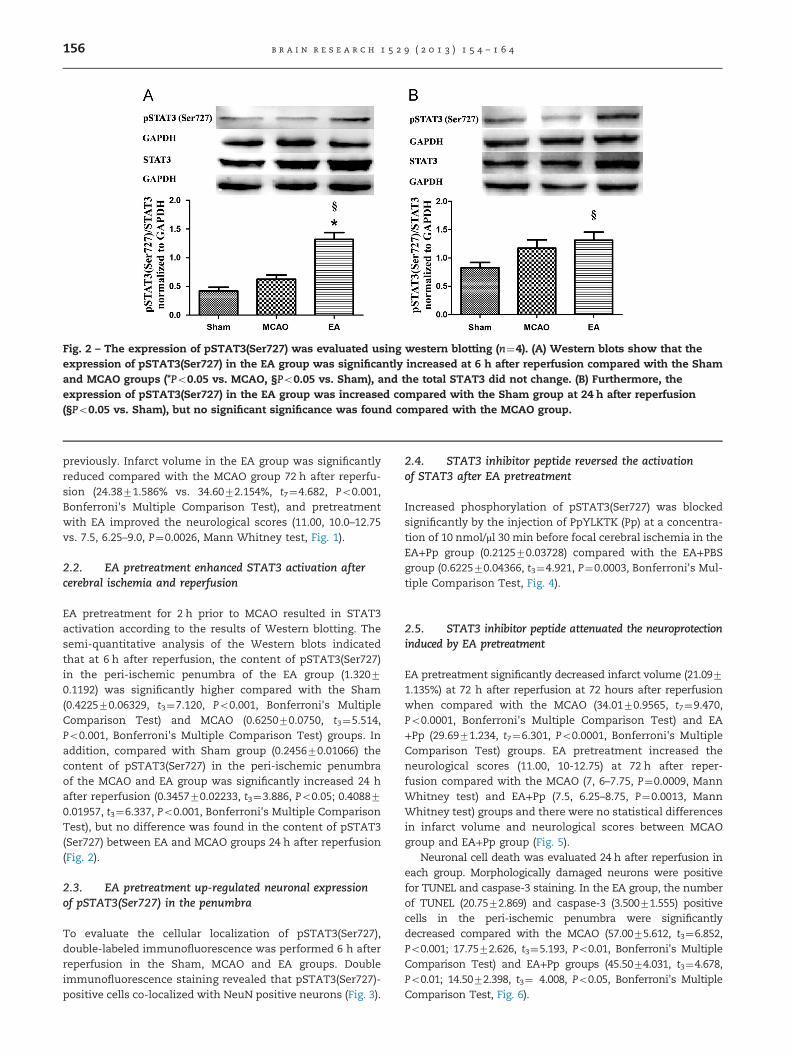
Fig. 2 – The expression of pSTAT3(Ser727) was evaluated using western blotting (n¼4). (A) Western blots show that theexpression of pSTAT3(Ser727) in the EA group was significantly increased at 6 h after reperfusion compared with the Shamand MCAO groups (*Po0.05 vs. MCAO, §Po0.05 vs. Sham), and the total STAT3 did not change. (B) Furthermore, theexpression of pSTAT3(Ser727) in the EA group was increased compared with the Sham group at 24 h after reperfusion(§Po0.05 vs. Sham), but no significant significance was found compared with the MCAO group.
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4156
previously. Infarct volume in the EA group was significantlyreduced compared with the MCAO group 72 h after reperfu-sion (24.3871.586% vs. 34.6072.154%, t7¼4.682, Po0.001,Bonferroni's Multiple Comparison Test), and pretreatmentwith EA improved the neurological scores (11.00, 10.0–12.75vs. 7.5, 6.25–9.0, P¼0.0026, Mann Whitney test, Fig. 1).
2.2. EA pretreatment enhanced STAT3 activation aftercerebral ischemia and reperfusion
EA pretreatment for 2 h prior to MCAO resulted in STAT3activation according to the results of Western blotting. Thesemi-quantitative analysis of the Western blots indicatedthat at 6 h after reperfusion, the content of pSTAT3(Ser727)in the peri-ischemic penumbra of the EA group (1.32070.1192) was significantly higher compared with the Sham(0.422570.06329, t3¼7.120, Po0.001, Bonferroni's MultipleComparison Test) and MCAO (0.625070.0750, t3¼5.514,Po0.001, Bonferroni's Multiple Comparison Test) groups. Inaddition, compared with Sham group (0.245670.01066) thecontent of pSTAT3(Ser727) in the peri-ischemic penumbraof the MCAO and EA group was significantly increased 24 hafter reperfusion (0.345770.02233, t3¼3.886, Po0.05; 0.408870.01957, t3¼6.337, Po0.001, Bonferroni's Multiple ComparisonTest), but no difference was found in the content of pSTAT3(Ser727) between EA and MCAO groups 24 h after reperfusion(Fig. 2).
2.3. EA pretreatment up-regulated neuronal expressionof pSTAT3(Ser727) in the penumbra
To evaluate the cellular localization of pSTAT3(Ser727),double-labeled immunofluorescence was performed 6 h afterreperfusion in the Sham, MCAO and EA groups. Doubleimmunofluorescence staining revealed that pSTAT3(Ser727)-positive cells co-localized with NeuN positive neurons (Fig. 3).
2.4. STAT3 inhibitor peptide reversed the activationof STAT3 after EA pretreatment
Increased phosphorylation of pSTAT3(Ser727) was blockedsignificantly by the injection of PpYLKTK (Pp) at a concentra-tion of 10 nmol/μl 30 min before focal cerebral ischemia in theEA+Pp group (0.212570.03728) compared with the EA+PBSgroup (0.622570.04366, t3¼4.921, P¼0.0003, Bonferroni's Mul-tiple Comparison Test, Fig. 4).
2.5. STAT3 inhibitor peptide attenuated the neuroprotectioninduced by EA pretreatment
EA pretreatment significantly decreased infarct volume (21.0971.135%) at 72 h after reperfusion at 72 hours after reperfusionwhen compared with the MCAO (34.0170.9565, t7¼9.470,Po0.0001, Bonferroni's Multiple Comparison Test) and EA+Pp (29.6971.234, t7¼6.301, Po0.0001, Bonferroni's MultipleComparison Test) groups. EA pretreatment increased theneurological scores (11.00, 10-12.75) at 72 h after reper-fusion compared with the MCAO (7, 6–7.75, P¼0.0009, MannWhitney test) and EA+Pp (7.5, 6.25–8.75, P¼0.0013, MannWhitney test) groups and there were no statistical differencesin infarct volume and neurological scores between MCAOgroup and EA+Pp group (Fig. 5).
Neuronal cell death was evaluated 24 h after reperfusion ineach group. Morphologically damaged neurons were positivefor TUNEL and caspase-3 staining. In the EA group, the numberof TUNEL (20.7572.869) and caspase-3 (3.50071.555) positivecells in the peri-ischemic penumbra were significantlydecreased compared with the MCAO (57.0075.612, t3¼6.852,Po0.001; 17.7572.626, t3¼5.193, Po0.01, Bonferroni's MultipleComparison Test) and EA+Pp groups (45.5074.031, t3¼4.678,Po0.01; 14.5072.398, t3¼ 4.008, Po0.05, Bonferroni's MultipleComparison Test, Fig. 6).
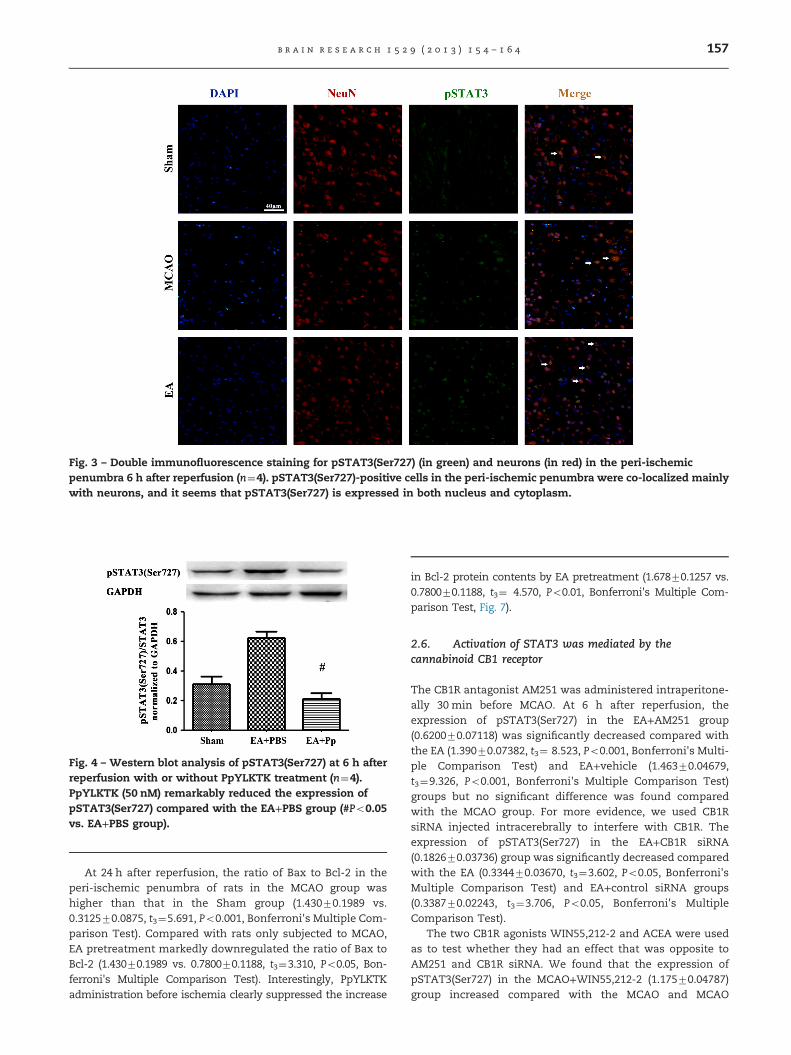
Fig. 4 – Western blot analysis of pSTAT3(Ser727) at 6 h afterreperfusion with or without PpYLKTK treatment (n¼4).PpYLKTK (50 nM) remarkably reduced the expression ofpSTAT3(Ser727) compared with the EA+PBS group (#Po0.05vs. EA+PBS group).
Fig. 3 – Double immunofluorescence staining for pSTAT3(Ser727) (in green) and neurons (in red) in the peri-ischemicpenumbra 6 h after reperfusion (n¼4). pSTAT3(Ser727)-positive cells in the peri-ischemic penumbra were co-localized mainlywith neurons, and it seems that pSTAT3(Ser727) is expressed in both nucleus and cytoplasm.
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4 157
At 24 h after reperfusion, the ratio of Bax to Bcl-2 in theperi-ischemic penumbra of rats in the MCAO group washigher than that in the Sham group (1.43070.1989 vs.0.312570.0875, t3¼5.691, Po0.001, Bonferroni's Multiple Com-parison Test). Compared with rats only subjected to MCAO,EA pretreatment markedly downregulated the ratio of Bax toBcl-2 (1.43070.1989 vs. 0.780070.1188, t3¼3.310, Po0.05, Bon-ferroni's Multiple Comparison Test). Interestingly, PpYLKTKadministration before ischemia clearly suppressed the increase
in Bcl-2 protein contents by EA pretreatment (1.67870.1257 vs.0.780070.1188, t3¼ 4.570, Po0.01, Bonferroni's Multiple Com-parison Test, Fig. 7).
2.6. Activation of STAT3 was mediated by thecannabinoid CB1 receptor
The CB1R antagonist AM251 was administered intraperitone-ally 30 min before MCAO. At 6 h after reperfusion, theexpression of pSTAT3(Ser727) in the EA+AM251 group(0.620070.07118) was significantly decreased compared withthe EA (1.39070.07382, t3¼ 8.523, Po0.001, Bonferroni's Multi-ple Comparison Test) and EA+vehicle (1.46370.04679,t3¼9.326, Po0.001, Bonferroni's Multiple Comparison Test)groups but no significant difference was found comparedwith the MCAO group. For more evidence, we used CB1RsiRNA injected intracerebrally to interfere with CB1R. Theexpression of pSTAT3(Ser727) in the EA+CB1R siRNA(0.182670.03736) group was significantly decreased comparedwith the EA (0.334470.03670, t3¼3.602, Po0.05, Bonferroni'sMultiple Comparison Test) and EA+control siRNA groups(0.338770.02243, t3¼3.706, Po0.05, Bonferroni's MultipleComparison Test).
The two CB1R agonists WIN55,212-2 and ACEA were usedas to test whether they had an effect that was opposite toAM251 and CB1R siRNA. We found that the expression ofpSTAT3(Ser727) in the MCAO+WIN55,212-2 (1.17570.04787)group increased compared with the MCAO and MCAO

Fig. 5 – Effect of pSTAT3(Ser727) inhibition with PpYLKTK on EA pretreatment-induced neuroprotection (n¼8). (A, B) Theinfarct volume at 72 h after reperfusion in the rats with 2 h MCAO. Pretreatment with EA significantly reduced the infarctvolume, whereas PpYLKTK (50 nM) abolished the protective effect of EA pretreatment (#Po0.05 vs. EA+PBS group). (C)Pretreatment with EA significantly increased the neurological scores at 72 h after reperfusion, whereas PpYLKTK (50 nM)abolished the protective effect of EA pretreatment (#Po0.05 vs. EA+PBS group).
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4158
+vehicle groups (0.490070.06014, t3¼9.511, Po0.001; 0.742570.04328, t3¼6.005, Po0.001 Bonferroni's Multiple ComparisonTest). The expression of pSTAT3(Ser727) in the MCAO+ACEAgroup (0.498170.04738) was increased compared with theMCAO and MCAO+vehicle groups (0.220370.02722, t3¼5.035,Po0.01; 0.281670.03975, t3¼3.923, Po0.05 Bonferroni's Multi-ple Comparison Test, Fig. 8).
3. Discussion
Ischemic tolerance can be induced by sublethal attacks, andthere are several pretreatments that have been shown toinduce neuronal protection against ischemia-reperfusioninjury. Many of these treatments activate extacellular orintracellular signaling pathways by triggering endogenousprotective responses (Durukan and Tatlisumak, 2010). There-fore, further research is required to define the precise mole-cular crosstalk and to optimize a novel way of dealing withcerebral ischemia.
We reported that EA pretreatment could produce rapidischemic tolerance against lethal ischemia in a previous study(Xiong et al., 2003). Further research found that the adenosine A1receptor and the endocannabinoid system are involved inischemic tolerance induced by EA pretreatment (Wang et al.,2005, Wang et al., 2009). In addition, activation of ERK1/2 andPKCε plays an important role in EA pretreatment-inducedneuronal protection via the endocannabinoid system (Du et al.,2010, Wang et al., 2011). However the mechanism of this
protection is not fully understood, and more evidence is neededbefore EA pretreatment can be used in the clinical setting.
Several cytokines and growth factors released after injuryin the body are believed to function as key regulators for theactivation of STAT3 during cerebral ischemia (Planas et al.,2006). These molecules combine with different receptorsand activate kinases to phosphorylate different residues ofSTAT3 (Darnell, 1997, Li and Shaw, 2004, Aziz et al., 2007).Two different phosphorylation residues of STAT3 with dif-ferent mechanisms have been established, and these twophosporylation sites can both modulate STAT3 activation(Kim et al. 2008). The phosphorylation of tyrosine on STAT3by JAKs has been found to increase after cerebral ischemiaand provide neuronal protection (Dziennis et al., 2007). Theother phosphorylation residue is serine that most likelyincreases STAT3 activity through an association with thePKCε-Raf-MEK(MAPK/ERK)–ERK pathway (Xuan et al., 2001,Xuan et al., 2005). STAT3 has been shown to be activated incerebral ischemia (Suzuki et al., 2001, Dziennis et al., 2007).Interestingly, ischemic preconditioning for cardiac protectionhas also been shown to be modulated by both the JAK-STATand PKCε-Raf-MEK (MAPK/ERK)–ERK pathways (Xuan et al.,2001, Xuan et al., 2005). An experiment in vivo showed thatoxygen–glucose deprivation could induce pSTAT3(Ser727) andinduce ischemic tolerance though the mediation of COX-2expression in cultured cortical neurons (Kim et al., 2008).However, the significance of serine phosphorylation of STAT3after cerebral ischemia and its contribution to the EApretreatment-induced ischemic tolerance has not yet beenclarified.
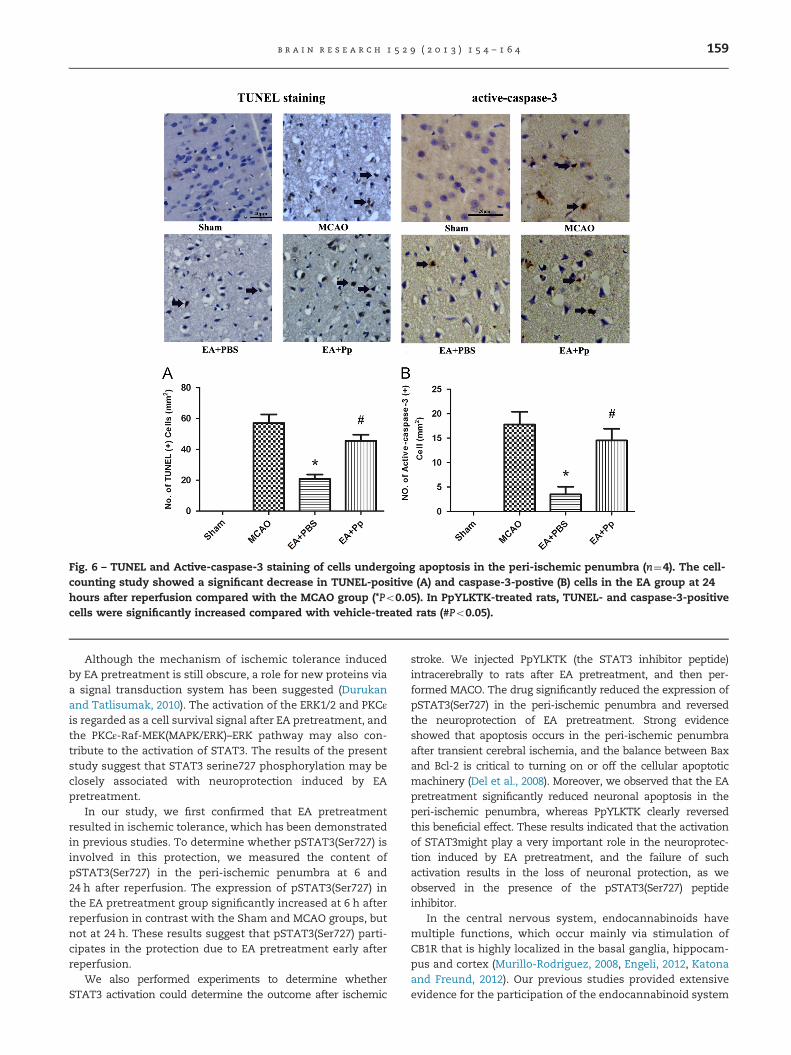
Fig. 6 – TUNEL and Active-caspase-3 staining of cells undergoing apoptosis in the peri-ischemic penumbra (n¼4). The cell-counting study showed a significant decrease in TUNEL-positive (A) and caspase-3-postive (B) cells in the EA group at 24hours after reperfusion compared with the MCAO group (*Po0.05). In PpYLKTK-treated rats, TUNEL- and caspase-3-positivecells were significantly increased compared with vehicle-treated rats (#Po0.05).
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4 159
Although the mechanism of ischemic tolerance inducedby EA pretreatment is still obscure, a role for new proteins viaa signal transduction system has been suggested (Durukanand Tatlisumak, 2010). The activation of the ERK1/2 and PKCεis regarded as a cell survival signal after EA pretreatment, andthe PKCε-Raf-MEK(MAPK/ERK)–ERK pathway may also con-tribute to the activation of STAT3. The results of the presentstudy suggest that STAT3 serine727 phosphorylation may beclosely associated with neuroprotection induced by EApretreatment.
In our study, we first confirmed that EA pretreatmentresulted in ischemic tolerance, which has been demonstratedin previous studies. To determine whether pSTAT3(Ser727) isinvolved in this protection, we measured the content ofpSTAT3(Ser727) in the peri-ischemic penumbra at 6 and24 h after reperfusion. The expression of pSTAT3(Ser727) inthe EA pretreatment group significantly increased at 6 h afterreperfusion in contrast with the Sham and MCAO groups, butnot at 24 h. These results suggest that pSTAT3(Ser727) parti-cipates in the protection due to EA pretreatment early afterreperfusion.
We also performed experiments to determine whetherSTAT3 activation could determine the outcome after ischemic
stroke. We injected PpYLKTK (the STAT3 inhibitor peptide)intracerebrally to rats after EA pretreatment, and then per-formed MACO. The drug significantly reduced the expression ofpSTAT3(Ser727) in the peri-ischemic penumbra and reversedthe neuroprotection of EA pretreatment. Strong evidenceshowed that apoptosis occurs in the peri-ischemic penumbraafter transient cerebral ischemia, and the balance between Baxand Bcl-2 is critical to turning on or off the cellular apoptoticmachinery (Del et al., 2008). Moreover, we observed that the EApretreatment significantly reduced neuronal apoptosis in theperi-ischemic penumbra, whereas PpYLKTK clearly reversedthis beneficial effect. These results indicated that the activationof STAT3might play a very important role in the neuroprotec-tion induced by EA pretreatment, and the failure of suchactivation results in the loss of neuronal protection, as weobserved in the presence of the pSTAT3(Ser727) peptideinhibitor.
In the central nervous system, endocannabinoids havemultiple functions, which occur mainly via stimulation ofCB1R that is highly localized in the basal ganglia, hippocam-pus and cortex (Murillo-Rodriguez, 2008, Engeli, 2012, Katonaand Freund, 2012). Our previous studies provided extensiveevidence for the participation of the endocannabinoid system
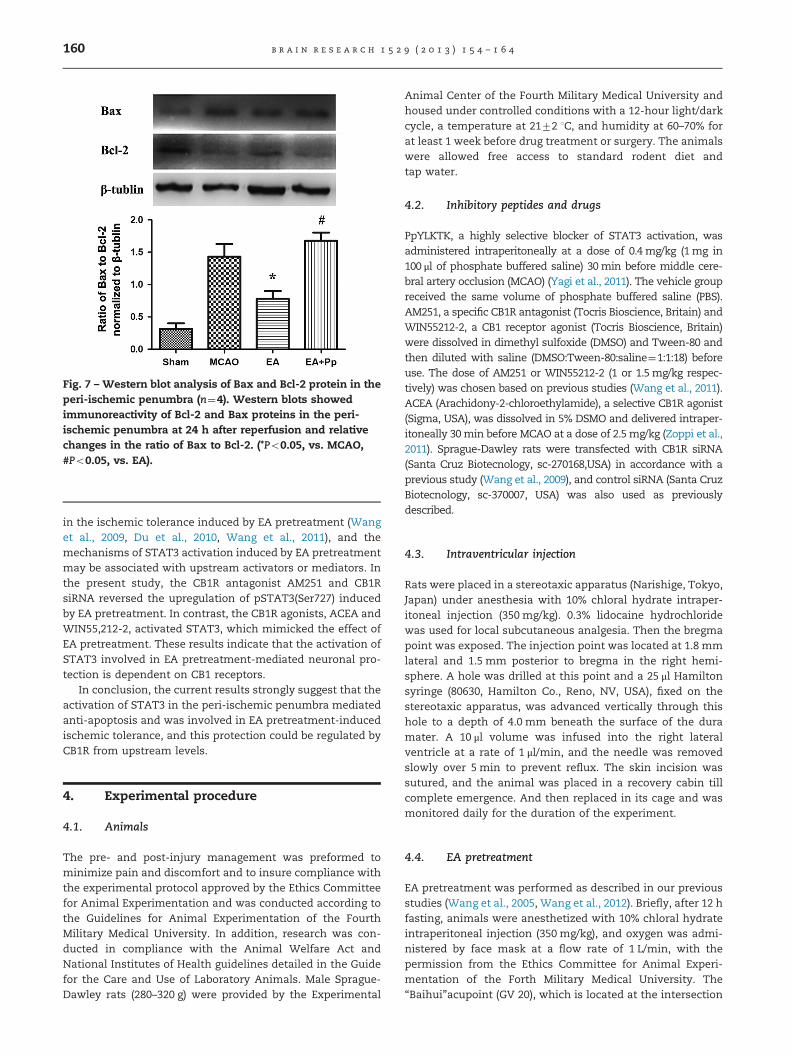
Fig. 7 – Western blot analysis of Bax and Bcl-2 protein in theperi-ischemic penumbra (n¼4). Western blots showedimmunoreactivity of Bcl-2 and Bax proteins in the peri-ischemic penumbra at 24 h after reperfusion and relativechanges in the ratio of Bax to Bcl-2. (*Po0.05, vs. MCAO,#Po0.05, vs. EA).
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4160
in the ischemic tolerance induced by EA pretreatment (Wanget al., 2009, Du et al., 2010, Wang et al., 2011), and themechanisms of STAT3 activation induced by EA pretreatmentmay be associated with upstream activators or mediators. Inthe present study, the CB1R antagonist AM251 and CB1RsiRNA reversed the upregulation of pSTAT3(Ser727) inducedby EA pretreatment. In contrast, the CB1R agonists, ACEA andWIN55,212-2, activated STAT3, which mimicked the effect ofEA pretreatment. These results indicate that the activation ofSTAT3 involved in EA pretreatment-mediated neuronal pro-tection is dependent on CB1 receptors.
In conclusion, the current results strongly suggest that theactivation of STAT3 in the peri-ischemic penumbra mediatedanti-apoptosis and was involved in EA pretreatment-inducedischemic tolerance, and this protection could be regulated byCB1R from upstream levels.
4. Experimental procedure
4.1. Animals
The pre- and post-injury management was preformed tominimize pain and discomfort and to insure compliance withthe experimental protocol approved by the Ethics Committeefor Animal Experimentation and was conducted according tothe Guidelines for Animal Experimentation of the FourthMilitary Medical University. In addition, research was con-ducted in compliance with the Animal Welfare Act andNational Institutes of Health guidelines detailed in the Guidefor the Care and Use of Laboratory Animals. Male Sprague-Dawley rats (280–320 g) were provided by the Experimental
Animal Center of the Fourth Military Medical University andhoused under controlled conditions with a 12-hour light/darkcycle, a temperature at 2172 1C, and humidity at 60–70% forat least 1 week before drug treatment or surgery. The animalswere allowed free access to standard rodent diet andtap water.
4.2. Inhibitory peptides and drugs
PpYLKTK, a highly selective blocker of STAT3 activation, wasadministered intraperitoneally at a dose of 0.4 mg/kg (1mg in100 μl of phosphate buffered saline) 30min before middle cere-bral artery occlusion (MCAO) (Yagi et al., 2011). The vehicle groupreceived the same volume of phosphate buffered saline (PBS).AM251, a specific CB1R antagonist (Tocris Bioscience, Britain) andWIN55212-2, a CB1 receptor agonist (Tocris Bioscience, Britain)were dissolved in dimethyl sulfoxide (DMSO) and Tween-80 andthen diluted with saline (DMSO:Tween-80:saline¼1:1:18) beforeuse. The dose of AM251 or WIN55212-2 (1 or 1.5mg/kg respec-tively) was chosen based on previous studies (Wang et al., 2011).ACEA (Arachidony-2-chloroethylamide), a selective CB1R agonist(Sigma, USA), was dissolved in 5% DSMO and delivered intraper-itoneally 30min before MCAO at a dose of 2.5mg/kg (Zoppi et al.,2011). Sprague-Dawley rats were transfected with CB1R siRNA(Santa Cruz Biotecnology, sc-270168,USA) in accordance with aprevious study (Wang et al., 2009), and control siRNA (Santa CruzBiotecnology, sc-370007, USA) was also used as previouslydescribed.
4.3. Intraventricular injection
Rats were placed in a stereotaxic apparatus (Narishige, Tokyo,Japan) under anesthesia with 10% chloral hydrate intraper-itoneal injection (350 mg/kg). 0.3% lidocaine hydrochloridewas used for local subcutaneous analgesia. Then the bregmapoint was exposed. The injection point was located at 1.8 mmlateral and 1.5 mm posterior to bregma in the right hemi-sphere. A hole was drilled at this point and a 25 μl Hamiltonsyringe (80630, Hamilton Co., Reno, NV, USA), fixed on thestereotaxic apparatus, was advanced vertically through thishole to a depth of 4.0 mm beneath the surface of the duramater. A 10 μl volume was infused into the right lateralventricle at a rate of 1 μl/min, and the needle was removedslowly over 5 min to prevent reflux. The skin incision wassutured, and the animal was placed in a recovery cabin tillcomplete emergence. And then replaced in its cage and wasmonitored daily for the duration of the experiment.
4.4. EA pretreatment
EA pretreatment was performed as described in our previousstudies (Wang et al., 2005, Wang et al., 2012). Briefly, after 12 hfasting, animals were anesthetized with 10% chloral hydrateintraperitoneal injection (350 mg/kg), and oxygen was admi-nistered by face mask at a flow rate of 1 L/min, with thepermission from the Ethics Committee for Animal Experi-mentation of the Forth Military Medical University. The“Baihui”acupoint (GV 20), which is located at the intersection
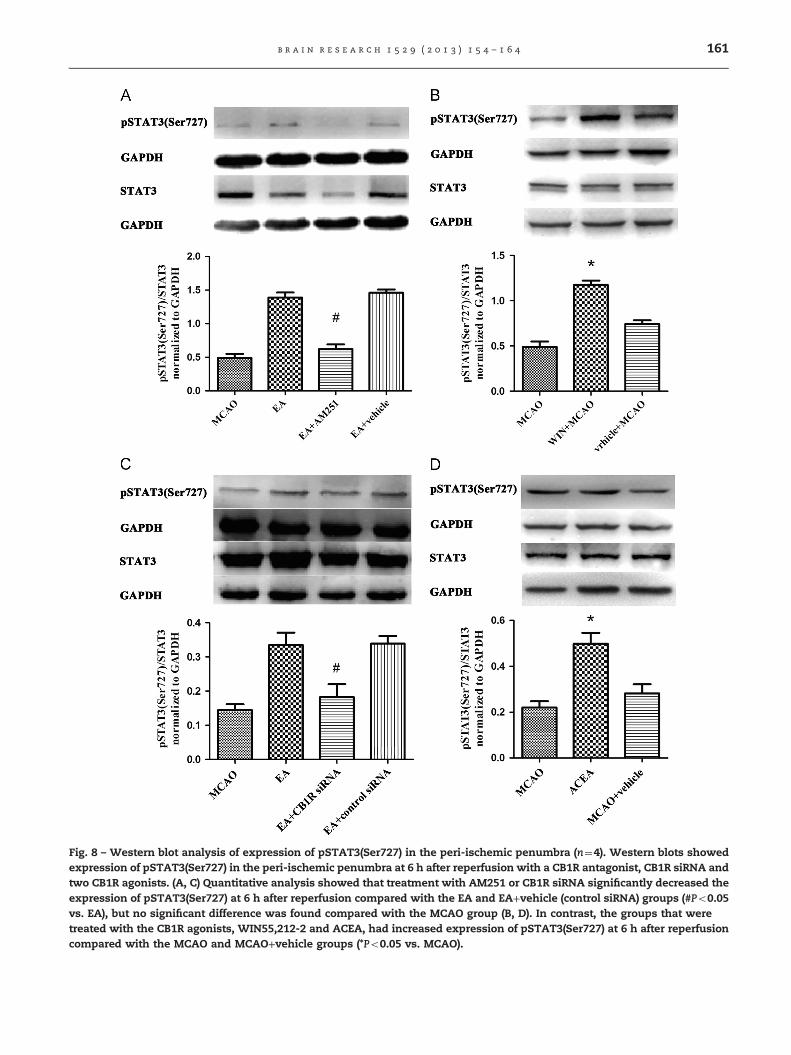
Fig. 8 – Western blot analysis of expression of pSTAT3(Ser727) in the peri-ischemic penumbra (n¼4). Western blots showedexpression of pSTAT3(Ser727) in the peri-ischemic penumbra at 6 h after reperfusion with a CB1R antagonist, CB1R siRNA andtwo CB1R agonists. (A, C) Quantitative analysis showed that treatment with AM251 or CB1R siRNA significantly decreased theexpression of pSTAT3(Ser727) at 6 h after reperfusion compared with the EA and EA+vehicle (control siRNA) groups (#Po0.05vs. EA), but no significant difference was found compared with the MCAO group (B, D). In contrast, the groups that weretreated with the CB1R agonists, WIN55,212-2 and ACEA, had increased expression of pSTAT3(Ser727) at 6 h after reperfusioncompared with the MCAO and MCAO+vehicle groups (*Po0.05 vs. MCAO).
b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4 161

b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4162
of the sagittal midline and the line linking the 2 ears, wasstimulated with the intensity of 1 mA and frequency of2/15 Hz for 30 min using an G6805-2 EA instrument (ModelNo.227033; Qingdao Xinsheng Ltd, China). The core tempera-ture of all the rats was maintained (Spacelabs Medical Inc,USA) at 37.070.5 1C during EA pretreatment by surface heat-ing or cooling. The right femoral artery was cannulated forcontinuous monitoring of blood pressure and for arterialblood sampling. Arterial blood gases and plasma glucosewere measured at the onset of EA, 15 min after EA and atthe end of EA. pO2, pCO2, and pH were quantified using ablood gas analyzer (ABL700, Radiometer, Denmark), andplasma glucose was measured with a blood glucose meter(OneTouch UltraEasy, Johnson & Johnson, USA).
4.5. Rat model of transient focal cerebral ischemia
At 2 h after EA pretreatment, focal cerebral ischemia wasinduced by MCAO in rats as described previously (Hata et al.,1998, Wang et al., 2005). Briefly, anesthesia was induced with3% isoflurane. First, the right common carotid artery and rightexternal carotid artery were exposed and ligated proximally.Then, a 3-0 nylon monofilament suture with its tip roundedby heating near a flame was inserted through an arteriotomyin the common carotid artery just below the carotid bifurca-tion and advanced into the internal carotid artery approxi-mately 17–18 mm distal to the carotid bifurcation until a mildresistance was felt, thereby occluding the origins of theanterior cerebral artery, the middle cerebral artery and theposterior communicating artery. Reperfusion was accom-plished by withdrawing the suture after 120 min of ischemiaunder anesthesia with 3% isoflurane. Rectal temperature wasmonitored (Spacelabs Medical Inc., Redmond, WA, USA) andmaintained at 37.0–37.5 1C by surface heating or coolingduring surgery, which usually lasted 10–15 min. Cerebralblood flow was measured by a transcranial laser Dopplerflowmeter (PeriFlux 5000, Perimed AB, Sweden), and only ratswith 480% flow reduction during the ischemic period and470% flow recovery within the first 10 min reperfusion wereincluded in the study. For the Sham group, the vessels werevisualized and cleared of connective tissue, and the twocarotid arteries were ligated without inserting the suture.After surgery, animals were placed in the recovery cabin tokeep warm and oxygen supply.
4.6. Neurobehavioral evaluation and infarct assessment
At 72 h after reperfusion, an observer, who was blinded to thetreatment group, assessed the rats neurologically based onthe method of Garcia (Garcia et al., 1995), which is an 18points neurological scoring system contains spontaneousactivity, side stroking, vibrissa touch, limb symmetry, climb-ing and forelimb walking. After the last neurobehavioralevaluation, animals were anesthetized with overdose chloralhydrate and decapitated, and 2-mm-thick coronal sectionsthroughout the brain were stained with 2% 2,3,5-triphenylte-trazolium chloride (TTC) at 37 1C for 20 min to evaluate theinfarct volume as described previously (Hata et al., 1998,Wang et al., 2005, Wang et al., 2008).
4.7. TUNEL staining
At 24 h after reperfusion, deeply anesthetized animals (n¼5)were perfused with 4% paraformaldehyde (PFA). The brainswere sectioned at 5 μm. To investigate DNA fragmentation, weperformed terminal deoxynucleotidyl transferase-mediateddeoxyuridine triphosphate nick-end labeling (TUNEL) stainingusing a commercial kit (Roche, Mannheim, Germany) accordingto a previously-described protocol (Wang et al., 2008, Wanget al., 2009). The TUNEL staining was quantitatively evaluatedwith the method described (Wang et al., 2002). Briefly, 32 pixelsof 0.10 mm2 were viewed under a light microscope at 100�magnification, and the total number of positive-stained cells inthese pixels was counted and expressed as cells per mm2.
4.8. Active-caspase-3 staining
The slides were washed in 0.01 M PBS, quenched for 10 min ina solution of methanol containing 3% hydrogen peroxide, andthen incubated for 1 h in blocking solution (2% bovine serumalbumin, 0.2% milk, 0.1% Triton X-100 in PBS), followed byincubation overnight in rabbit anti-active caspase-3 anti-serum (Cell Signaling Technology, Beverly, MA, USA) diluted1:400 in blocking solution, while the negative control withblocking solution only. Following incubation with primaryantiserum, the sections were incubated for 90 min in second-ary antiserum (goat anti-rabbit, 1:200 in blocking solution),and then reacted in the dark with ABC reagents (standardVectastain ABC Elite Kit, USA; Vector Laboratories, Burlin-game, CA, USA) for 90 min. The sections were then washedtwice with PBS followed by diaminobenzidine (DAB) staining.Images were viewed with a fluorescence light microscope.Active caspase-3-positive cells in the different regions of eachsection were counted by observers blinded to the treatmentconditions.
4.9. Western blot analysis
The ipsilateral peri-ischemic penumbra was homogenized inRIPA lysis buffer (Beyotime, Nantong, China) with 1� Rochecomplete protease inhibitor cocktail on ice. Protein concen-tration was quantified by the Bradford reagent after centrifu-ging and extraction of the suspension. Samples (50 μg) wereloaded onto polyacrylamide-SDS gels. The gels were electro-phoresed and then transferred to PVDF membranes. Afterthat, the membranes were blocked with a blocking bufferusing bovine serum albumin (Sigma, 3%) for 60 min andprobed with primary antibodies overnight at 4 1C, and thenincubated for 1 h at room temperature with appropriatesecondary horseradishperoxidase–conjugated goat-anti-rabbit antibodies (Beyotime,Nantong, China; 1:5000 dilution).The following primary antibodies were used in this study:anti-pSTAT3 (Ser727) rabbit polyclonal antibody (Cell signal-ing technology, USA; 1:1000 dilution), anti-STAT3 rabbitpolyclonal antibody (Cell signaling technology, USA; 1:1000dilution).
Rabbit anti-Bcl-2antibody (Cell Signaling Technology, USA;1:1000 dilution) and rabbit anti-Bax antibody (Epitomics, USA;1:500 dilution) were used in this study as primary antibodies.Semi-quantitative analysis of the blots was performed using

b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4 163
densitometry. Each sample was subjected to immune-blotting 3 times, and the final optical density value (relativeto that of the internal standard) represents the average ofthese 3 separate analyses.
4.10. Immunofluorescent double staining
To evaluate the co-localization of pSTAT3(Ser727) andneuron-specific nuclear protein (NeuN), we performed doubleimmunofluorescent staining. The sections fixed by 4% PFA,washed 3 times with PBS. After incubated for 1 h in blockingsolution (2% bovine serum albumin, 0.2% milk, 0.1% TritonX-100 in PBS), the slides were simultaneously incubated withtwo of following primary antibodies: anti-pSTAT3(Ser727)(Cell Signaling Technology, USA; 1:200 dilution), anti-NeuN(Chemicon International, Temecula, CA, USA; 1:2000 dilution)and a negative control absent of the primary antibodypSTAT3(Ser727). For double labeling, the primary antibodieswere detected with FITC-conjugated secondary antibody(1:200; Jackson ImmunoResearch Laboratories) and Cy3-conjugated secondary antibody (1:200; Jackson ImmunoRe-search, West Grove, PA, USA). The sections were placed onslides and then covered with DAPI (4',6-diaminodino-2-phe-nylindole) (Vector Laboratories, Burlingame, CA, USA). Thesections were examined under a fluorescence microscope(Olympus, Japan).
4.11. Statistics
SPSS 13.0 for Windows was used to perform all statisticalanalyses. All values, except for neurological scores, are pre-sented as means7SEM and compared among groups by one-way analysis of variance. If the ANOVA was significant, thena Bonferroni's Multiple Comparison Test was used as a post-hoc test to compare between-group differences. The neurolo-gical deficit scores were expressed as the median (range) andwere analyzed with a Kruskal-Wallis test followed by theMann–Whitney U-test for multiple comparisons. Type I errorwas defined aso0.5, Values of Po0.05 were consideredstatistically significant.
Acknowledgments
This work was supported by the Major Program of the NationalNatural Science Foundation of China (Grant 30930091), theNational Natural Science Foundation of China (Grant81072888, 81035375), and the Overseas, Hong Kong & MacaoScholars Collaborated Researching Fund (Grant 81228022). Eng-lish words in this work have been professionally edited byShanghai BIOON Info-tech Co. Ltd before its submission.
r e f e r e n c e s
Aziz, M.H., Manoharan, H.T., Verma, A.K., 2007. Protein kinase Cepsilon, which sensitizes skin to sun's UV radiation-inducedcutaneous damage and development of squamous cellcarcinomas, associates with Stat3. Cancer Res. 67, 1385–1394.
Darnell, J.J., 1997. STATs and gene regulation. Science 277,1630–1635.
Del, P.G., Bruno, A., Del, P.M., Venditti, A., Maurillo, L., Buccisano, F.,Stasi, R., Neri, B., Luciano, F., Siniscalchi, A., de Fabritiis, P.,Amadori, S., 2008. Deregulation of the mitochondrial apoptoticmachinery and development of molecular targeted drugs inacute myeloid leukemia. Curr. Cancer Drug Targets 8, 207–222.
Du, J., Wang, Q., Hu, B., Peng, Z., Zhao, Y., Ma, L., Xiong, L., Lu, Y.,Zhu, X., Chen, S., 2010. Involvement of ERK 1/2 activation inelectroacupuncture pretreatment via cannabinoid CB1receptor in rats. Brain Res. 1360, 1–7.
Durukan, A., Tatlisumak, T., 2010. Preconditioning-inducedischemic tolerance: a window into endogenous gearing forcerebroprotection. Exp. Transl. Stroke Med. 2, 2.
Dziennis, S., Alkayed, N.J., 2008. Role of signal transducer andactivator of transcription 3 in neuronal survival andregeneration. Rev. Neurosci. 19, 341–361.
Dziennis, S., Jia, T., Ronnekleiv, O.K., Hurn, P.D., Alkayed, N.J., 2007.Role of signal transducer and activator of transcription-3 inestradiol-mediated neuroprotection. J. Neurosci. 27, 7268–7274.
Engeli, S., 2012. Central and peripheral cannabinoid receptors astherapeutic targets in the control of food intake and bodyweight. Handb. Exp. Pharmacol., 357–381.
Garcia, J.H., Wagner, S., Liu, K.F., Hu, X.J., 1995. Neurologicaldeficit and extent of neuronal necrosis attributable to middlecerebral artery occlusion in rats. Statistical validation. Stroke26 (627–634), 635.
Hata, R., Mies, G., Wiessner, C., Fritze, K., Hesselbarth, D., Brinker,G., Hossmann, K.A., 1998. A reproducible model of middlecerebral artery occlusion in mice: hemodynamic, biochemical,and magnetic resonance imaging. J. Cereb. Blood Flow Metab.18, 367–375.
Katona, I., Freund, T.F., 2012. Multiple functions ofendocannabinoid signaling in the brain. Annu. Rev. Neurosci.35, 529–558.
Kim, E.J., Raval, A.P., Perez-Pinzon, M.A., 2008. Preconditioningmediated by sublethal oxygen-glucose deprivation-inducedcyclooxygenase-2 expression via the signal transducers andactivators of transcription 3 phosphorylation. J. Cereb. BloodFlow Metab. 28, 1329–1340.
Li, L., Shaw, P.E., 2004. A STAT3 dimer formed by inter-chaindisulphide bridging during oxidative stress. Biochem. Biophys.Res. Commun. 322, 1005–1011.
Li, X., Luo, P., Wang, Q., Xiong, L., 2012. Electroacupuncturepretreatment as a novel avenue to protect brain againstischemia and reperfusion injury. Evid. Based Complement.Alternat. Med. 2012, 195397.
Murillo-Rodriguez, E., 2008. The modulatory role ofendocannabinoids in sleep. Rev. Neurol. 46, 160–166.
Planas, A.M., Gorina, R., Chamorro, A., 2006. Signalling pathwaysmediating inflammatory responses in brain ischaemia.Biochem. Soc. Trans. 34, 1267–1270.
Suzuki, S., Tanaka, K., Nogawa, S., Dembo, T., Kosakai, A.,Fukuuchi, Y., 2001. Phosphorylation of signal transducer andactivator of transcription-3 (Stat3) after focal cerebralischemia in rats. Exp. Neurol. 170, 63–71.
Tsou, M.T., Huang, C.H., Chiu, J.H., 2004. Electroacupuncture onPC6 (Neiguan) attenuates ischemia/reperfusion injury in rathearts. Am. J. Chin. Med. 32, 951–965.
Wang, K.C., Koprivica, V., Kim, J.A., Sivasankaran, R., Guo, Y.,Neve, R.L., He, Z., 2002. Oligodendrocyte-myelin glycoproteinis a Nogo receptor ligand that inhibits neurite outgrowth.Nature 417, 941–944.
Wang, Q., Gou, X., Xiong, L., Jin, W., Chen, S., Hou, L., Xu, L., 2008.Trans-activator of transcription-mediated delivery of NEP1-40protein into brain has a neuroprotective effect against focalcerebral ischemic injury via inhibition of neuronal apoptosis.Anesthesiology 108, 1071–1080.

b r a i n r e s e a r c h 1 5 2 9 ( 2 0 1 3 ) 1 5 4 – 1 6 4164
Wang, Q., Li, X., Chen, Y., Wang, F., Yang, Q., Chen, S., Min, Y.,Li, X., Xiong, L., 2011. Activation of epsilon protein kinaseC-mediated anti-apoptosis is involved in rapid toleranceinduced by electroacupuncture pretreatment throughcannabinoid receptor type 1. Stroke 42, 389–396.
Wang, Q., Peng, Y., Chen, S., Gou, X., Hu, B., Du, J., Lu, Y., Xiong, L.,2009. Pretreatment with electroacupuncture induces rapidtolerance to focal cerebral ischemia through regulation ofendocannabinoid system. Stroke 40, 2157–2164.
Wang, Q., Wang, F., Li, X., Yang, Q., Li, X., Xu, N., Huang, Y.,Zhang, Q., Gou, X., Chen, S., Xiong, L., 2012.Electroacupuncture pretreatment attenuates cerebralischemic injury through alpha7 nicotinic acetylcholinereceptor-mediated inhibition of high-mobility group box 1release in rats. J Neuroinflammation 9, 24.
Wang, Q., Xiong, L., Chen, S., Liu, Y., Zhu, X., 2005. Rapid toleranceto focal cerebral ischemia in rats is induced bypreconditioning with electroacupuncture: window ofprotection and the role of adenosine. Neurosci. Lett. 381,158–162.
Wu, P., Zuo, X., Ji, A., 2012. Stroke-induced microRNAs: Thepotential therapeutic role for stroke. Exp. Ther. Med. 3,571–576.
Xiong, L., Lu, Z., Hou, L., Zheng, H., Zhu, Z., Wang, Q., Chen, S.,2003. Pretreatment with repeated electroacupuncture
attenuates transient focal cerebral ischemic injury in rats.Chin. Med. J. (Engl) 116, 108–111.
Xuan, Y.T., Guo, Y., Han, H., Zhu, Y., Bolli, R., 2001. An essentialrole of the JAK-STAT pathway in ischemic preconditioning.Proc. Natl. Acad. Sci. USA 98, 9050–9055.
Xuan, Y.T., Guo, Y., Zhu, Y., Wang, OL., Rokosh, G., Messing, RO.,Bolli, R., 2005. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signaltransducers and activators of transcription 1 and 3 andinduction of cyclooxygenase-2 after ischemic preconditioning.Circulation 112, 1971–1978.
Yagi, T., Yoshioka, H., Wakai, T., Kato, T., Horikoshi, T.,Kinouchi, H., 2011. Activation of signal transducers andactivators of transcription 3 in the hippocampal CA1 region ina rat model of global cerebral ischemic preconditioning. BrainRes. 1422, 39–45.
Zhou, W., Ko, Y., Benharash, P., Yamakawa, K., Patel, S., Ajijola, O.A.,Mahajan, A., 2012. Cardioprotection of electroacupunctureagainst myocardial ischemia-reperfusion injury by modulationof cardiac norepinephrine release. Am. J. Physiol. Heart. Circ.Physiol. 302, H1818–H1825.
Zoppi, S., Perez, N.B., Madrigal, J.L., Manzanares, J., Leza, J.C.,Garcia-Bueno, B., 2011. Regulatory role of cannabinoidreceptor 1 in stress-induced excitotoxicity andneuroinflammation. Neuropsychopharmacology 36, 805–818.
Recommended

















