
ANALES DE LA UNIVERSIDAD DE VALENCIA
ROSARIO DOMINGO SEBASTIAN
ESTUDIO TEOHICO DE LA REACCION DIELS-ALDER MEDIANTE EL METODO FEMO
VOL. XXXII - CURSO 1958-39CUADERNO II -. CIENCIAS

Esta Tesis tue presentada en Ia Facultad de Ciencias dela Universidad de Vajencia para aspirar a) grado de Doctoren Ciencias (Sección de Quimicas) y leida el 14 de diciembrede 1957 ante el tribunal formado por los catedráticos D. Ma-nuel Lora Tamayo (presidente), D. Tomás Batuecas Marugán,D. Julián Rodriguez de Velasco, D. José lgnacio FernándezAlonso (ponente) y D. José M. Viguera Lobo, obteniendo lacalificación de Sobresaliente cum laude.
El trabajo de la presente Tesis doctoral se realizó en elLaboratorio de Qulmica FIsica de la Facultad de Ciencias dela Universidad de Vaiencia, durante los años 1955 a 1957,
Expreso mi sincero agradecimiento a! catedrático de QuI-mica Fisica, Prof. José Ignacio Fernández A lonso, por haber-me sugerido el tema de Ia presente tesis, asI CO/flU POT laatención y solicitud con que me ha asistido en todo momento.
Al mismo tiempo, doy las gracias a! Consejo Superior deInvestigaciones Cientificas, por la ayuda que, como becario yAyudante de Sección, nw ha prestado a través del Instituto deQuImica FIsica "A. de G. Rocasolano".
Expreso también mi agradecimiento al instituto de Cálculo,por su muy valiosa colaboración en la resolución de numero-sas matrices.
Finalmente, agradezco a esta Facultad la acogida que meha dispensado poniendo a mi disposición todos los mediosde trabajo.

INTRODUCCION
El conocirniento de la naturaleza intipna del enlace qulmico ha sido unade las m/iximas aspiraciones de los qulmicós de todos los tiempos. Y si bienes cierto que el carácter electrostático del enlace heteropolar está ya hoyfuera de toda duda, merced a las aportaciones de diferentes investigadores,desde Berzelius hasta Kossel, no ocurrió lo mismo en cuanto al enlacehomopolar se ref iere.
Aun cuando G. N. Lewis definió este áltimo como for,nado por un parde electrones, su estructura Intina no pudo conocerse hasta el advenimientode la Mecánica cuántica. En efecto, en 1927, Heitler y London descubrieronque el enlace covalente de Ia ,nolécula de hidrógeno estaba formado por unpar de electrones y que, en virtud del "principio de identidad" de las partIcu-las elemenz'ales, la urnón tenIa su. origen en el llamado "efecto de intercam-bio", conducente a urra nueva forma de energia, desconocida para la Mecd-nica clásica: la "energIa de intercambio".
La aportación fundamental de Heitler y London constituyó la base de unnuevo e importantIsi!no campo: la QuImica cuántica, nexo entre la FIsicade las partIculas elementales (electrones) y la Qulmica.
Varios han sido los caminos seguidos para establecer esta union. Uno deellos consiste en emplear la teorIa mecanocuántica del electrOn y obtener lassoluciones de la ecuaciOn de Schradinger para el sistema correspondiente.Este fue el que iniciaron Heitler y London al explicar la naturaleza del enlacecovalente. Tam bién de esta forma se han explicado otras cuestiones de ,náxi-mo interés; entre ellas, las fuerzas de van der Waals (London), las fuerzasrepulsivas en los cristales iónicos (Born-Mayer) y las reglas de Hurne-Rotherypara las' aleaciones (Jones). Estd claro que el mayor inconveniente del métodoreside en la extraordinaria dii icultad que presenra la resoluciOn de la ecua-dOn de ondas para los sistemas de varios electrones, incluso en for,naaproximada.
—7—

ROSARIO DOMINGO SEBASTIAN
Otro de los caminos —y muy fructIfero por cierto— tue el desarrolladoper los quImicos, quienes incorporaron e interpretaron, segán la nueva Me-cánica cuántica, la vieja terminologla quImica. Resulta dif Icil saber si estemétodo tiene base real, pues, como señala muy bien Löwdin, (1) estamos enIa misma situaciOn en que nos encontrarIamos si hubiéramos construido untünel desde un lado de Ia montana sin haber alcanzado todavIa la salida ala otra ladera.
De esta forma han nacido las liamadas teorias semieinpIricas ', valiosI-sinus herramientas de trabajo para el quImico, puesto que nos han permitidocorrelacionar unos datos quImicos experi,nentales con otros también experi-ment ales, lográndose establecer inserpolaciones y extrapolaciones de losresultados quImicos. Este inétodo de estudio ha alcanzado extraordinarioéxito en la Quimica Qrgánica, particularmente en el tratamiento de lossistemas con jugados. Solo a tjtulo puramente informativo, mencionaremosla explicaciOn del enlace aromático, poder director de los sustituyentes en lossistemas aromáticos, adiciOn en los sisteinas conjugados, cálculo de memen-tos dipol ares, color de las moléculas orgánicas, etc. En resumen, el granempleo de las reorIas semienipIricas descansa en su gran simplicidad, y sisus aplicaciones no se lievan hasta el extremo, sus conclusiones cuantitativasson, de ordinario, excelentes.
Como antes hemos indicado, no es posible resolver exactamente la ecua-cidn de Shrodinger para un sistema de varios electrones; sin embargo, ellono quiere decir que no podamos obtener sus soluciones con la precisionexigida, y la existencia de las funciones propias ha sido discutida con tododetalle por varies autores.
A Löwdin (1) debemos el siguiente esquema, que recopila los diferentesinétodos desarrollados para hallar las soluciones aproximadas de ía ecuaciOnde ondas.
* En QuImica cuántica se acostumbra a distinguir entre teorfa.s "puras" y "semi-empIricas". En el sentido estricto de Ia palabra, ni en FIsica ni en Qulmica hay teorlaspuras, pues todas ellas relacionan unos datos experimentales con otros. Sin embargo,denominamos teorlas puras aquellas que permiten deducir resultados de interés quf-mico a partir de los valores fIsicos de Ia masa y carga electrOnica, constante de Planek,nimeros cuánticos y ccuación de Schrodinger.
—8—

REACCION DIELS-ALDER POR EL METODO FEMO
Resumiendo esta lntroducción, subrayarenos que todavla está lejana lameta de la QuImica cuántica: predicción de las propiedades de una mo-lécula hipotética antes de ser sintetizada y obtención de aquellas otras conpropiedades deseadas. Sin embargo, un paso iinportante en esta direcciónlo constituye el que el quImico se familiarice con un vocabulario en términosde electrones y su corn portamiento mecanocuántico.
Las investigaciones desarrolladas en la presente Tesis doctoral tienen porobjeto estudiar, desde el punto de vista mecanocuántico, un caso particularde las reacciones de adición en los hidrocarburos aromiiticos: la liamadasIntesis diénica o reacción Diels-Alder. Brown Ia estudió con todo detaile porel método LCAO MO, ilegando a conclusiones que están de completo acuer-do con los resultados experiment ales. Nosotros, queriendo confirmar estosresultados reóricos, hemos realizado un estudio análogo a! desarrollado pordicho autor, aplicando el método del electron libre (FEMO).
Por ello iniciaremos nuestro estudio exponiendo los fundamentos de dichométodo.
—9—

ECUACIÔN DE SCHRöDINGER
1Aproximacion por funciones Aproximacion por funciones Aproximaciónde ondas monoelectronjcas de ondas btelectrónjcas atómica
1Producto sencillo de Producto antisimétricofunciones de onda de funciones de onda
1Mtodo semiempfrico Método de los orbitales Método del enlace
moleculares de valencia
I
14,MTODO DEL Orbitales moleculares formados por Método
combinación lineal de orbitales semiempIricoELECTRON LIBREatómicos
Método celular Aproximación conven- Orbitales diferentes para______ cional con orbitales spines diferentes
II simple y doblernenteI
I
4, ocupadosj I
semiempio4,
Método
Inclusion del recubri- InclusiOn del recubri-I
miento y de las integra- miento y de las integra-les de varios centros les de varios centros
1.InclusiOn de estados
ionizados
InelusiOn de la interac- InclusiOn de Ia interac- InclusiOn de Ia interac- InclusiOn de estados su-ciOn -configuracional ciOn conflguracional ciOn configuracional re- periores y reducciOn demanente totalidad
SoluciOn exacta de Ia + _________ecuaciOn de Schrddin-
ger
1Movimjento nuclear y
efectos térmicos
4,Efectos relativjstas
'if"Verdad real"

PARTE I
MTODO DEL ELECTRON LIBRE (FEMO)
CONSIDERACIONES GENERALES.—Como es bien sabido, en todos los carbo-nos pertenecientes a un sistema orgánico conjugado es necesario distinguir dostipos de orbitales: o- y lr. Los primeros se forman a partir de los orbitalesatómicos 2s, 2p y 2p, originándose los hibridos trigonales, sp2, que estánsituados en un piano y forman entre si anguios de 1200. Cuando se recubrencon los orbitales is del hidrógeno, forman el esqueleto hidrocarbonado delsistema. Las funciones de onda u orbitales a- tienen simetrIa ciiIndrica segünla dirección del enlace. El cuarto electron del carbono, 2p0, al recubrirselateralmente con otro orbital analogo, forma el orbital lr, el cual tiene unnodo en el piano de los orbitales a-.
La presencia de un electrOn en cada átomo de carbono del sistemaconjugado, tiende a extender la nube r-electrónica sobre el conjunto molecu-lar, por lo que resulta difIcil localizar de manera definida los dobles enlaces.A estos electrones r se les da también el nombre de electrones móviles, pues,aun con ciertas restricciones, se mueven libremente en tomb al esqueletoformado por los electrones a-.
Las propiedades fundamentales de las moldculas dependen exciusivamen-te de estos electrones it, cuyos movimientos será interesante estudiar. Elproblema que se plantea es enorme, pues el movimiento es tan grande queno puede situarse a un determinado electron en un enlace dado. Bajo estaconsideraciOn hay que suponer a todos los electrones it como pertenecientes,en cualquier instante, a todos los átornos, lo que hace intervenir en el cálculoun gran ntImero de integrales de difIcil solución, que representarán Ia influen-cia de cada uno de los electrones sobre los distintos átomos. Recordemosa este respecto que, sOlo el benceno, ha sido estudiado en su total corn-plejidad. Una sirnplificaciOn que se aplica corrienternente es la de considerar
— 11 —

ROSAR1O DOMINGO SEBASTIAN
solamente Ia interacción entre las partIculas más próximas, aunque sin des-preciar totalmente las influencais lejanas que, bajo la forma de unas cons-tantes (integrales de resonancia y de recubrimiento), se hacen intervenir enlas ecuaciones. En esto se basa el tan conocido método de cálculo de los MOy, dentro del mismo, de la aproximación LCAO.
METODO FEMO.—En el método general MO, la aproximación más sim-ple la constituye la liamada aproximación del electron libre (FEMO), cuyosentido fIsico se tiene al considerar que los electrones r se comportan demanera análoga a los electrones libres en los metales o en los semiconduc-tores. (2) La comparación surge a! suponer que los electrones r están some-tidos a un campo de potencial periódico creado por la cadena de grupos CH,de manera analoga a los electrones en un metal; por tanto, podrán tratarsemediante la aproximación de Sommerfeld, que considera a los electronesmetálicos comportándose como un gas unidimensional Fermi, sobre los quese ejerce un cierto potencial uniforme. Esta aproximación da resultados que,Si bien no son de absoluta validez cuantitativa, son mejores de lo que cabriaesperarse dada la simplicidad del modelo.
Han sido estudiados, en principio, por Bayliss, Simpson y otros autores,diversos compuestos orgánicos conjugados. Las predicciones para las longi-tudes de onda de algunos polienos resultan un poco altas al compararlas conlas experimentales. La discrepancia (3) se debe a que en el estado fundamen-tal de una molécula, el gas ir-electrónico ocupa su primera zona de Brillouin,zona que es producida por una periodicidad de energIa potencial a lo largode la cadena, debido a la alternancia de los simples y dobles enlaces. El saltodel electron que corresponde a Ia primera banda de absorciOn, es un saltodesde lo más alto de la primera hasta lo más bajo de Ia segunda zona deBrillouin, y este proceso requiere mucha energIa, por lo que la banda corres-ponde a una longitud de onda más corta que la predicha teOricamente.
Realmente, Bayliss, en su modelo aplicado a polienos, admite que losdobles y simples enlaces no pueden permutarse por resonancia y, claro está,que Ia teorla de Sommerfeld no resulta satisfactoria en estas condiciones.Además, establece que el camino de los electrones a lo largo de una tra-yectoria puede terminar en el mismo ndcleo de carbono o más lejos, preci-samente en el átomo de hidrogeno. Por tanto, al aplicarse el modelo a sus-tancias como las polimetinas, (') no se obtienen buenos resultados, debidoa que el camino del electrOn será distinto segdn tengamos la forma cis- o
— 12 —

REACCION DIELS-ALDER POR EL !sIETODO FEMO
trans- de las mismas. Nikitine indica que, su modelo, no puede aplicarsecuantitativamente a los polienos cIclicos cis o trans. de cadenas conjugadas,
en las cuales intervienen heteroátomos, pues Ia introducción de éstos podrIadar lugar a niveles energéticos "impuros" o falseados. Kuhn (3) considera la
cadena como un conjunto de osciladores eléctricos conjugados, en la que elelectron es uria partIcula con vibraciones armOnicas airededor de los ndcleos,
que precisamente serán las posiciones de equilibrio de los átomos. Tambiénileva a cabo un estudio de moléculas con heteroátomos, pero comete el graveerror de incluir los electrones de éstos en el conjunto del gas electrOnico, de
manera que se encuentra con más electrones que dobles enlaces tiene lacadena.
El problema de las cianinas simétricas fue estudiado por Nikitine yKomoss (5) de una manera muy particular. Estos mismos autores (6) han
desarrollado el modelo metálico para tres dimensiones, pero su enfoque estotalmente distinto del seguido por los demás autores.
En su sentido más exacto (7) el método FEMO se fundamenta en lasuposiciOn de que sOlo los electrones ir son los realmente insaturados, distri-buidos en ciertos niveles de energia y con unas funciones de onda asociadasEn el estado fundamental siguen Ia estadIstica de Fermi-Dirac y cumplen elprincipio de exclusiOn de Pauli. Los niveles de energIa más altos estarándisponibles para ser ocupados en cualquier excitación molecular. El modelose puede aplicar tanto a sistemas conjugados lineales como aromáticos. Enlos priineros, los electrones r son confinados, no por las paredes de un recintoimpenetrable, sino por la atracciOn de un nUcleo cargado positivamente;por tanto, se originará un campo de energIa potencial a lo largo de losejes moleculares. La conducta de un electron en estas condiciones se deduce,segdn la mecánica cuántica, resolviendo Ia ecuación de Schrödinger:
v2 W + (8ir2m/h2) (E—V) W = 0
Si la partIcula se mueve libremente en un recinto de paredes impenetrables,entonces V 0 dentro del mismo y V c fuera, de tal suerte que no tienetendencia a salir del recinto. En consecuencia, la ecuación anterior sesimplificará:
2 W + (87r2mE/h2) 'V =0En realidad, el campo de energIa potencial debe oscilar a lo largo de los
ejes moleculares, pero puede prescindirse de esta consideración reemplazando
— 13 —

ROSARIO DOMINGO SEBASTIAN
la cadena por un recinto ideal de potencial, que tendrIa la forma de unalinea quebrada.
En nuestro estudio hemos aplicado el desarroilo matemático de Rueden-berg y Scherr (8) y Scherr, (9) que tiene una gran analogla con la aproximaciónMO LCAO. De Ia comparación del método FEMO con este iiitimo, sonevidentes las siguientes ventajas: en primer lugar, no hay que recurrir aparámetros experimentales, excepto una distancia interaómica, D, fáciimentemedible, mientras que en el MO LCAO hay que emplear dos parámetros,S y /3, integrales de resonancia y recubrimiento respectivamente, cuyos valo-res no se conocen con mucha exactitud. Por otra parte, las funciones de ondaque se obtienen son mucho más visuales, °) pero tienen el inconvenientede ser solamente válidas para los electrones r, mientras que las funcionescalculadas por la aproxiinación LCAO pueden aplicarse a todos loselectrones.
MOvIMIENIO LONGITuDINAL.—Supongamos un electron cualquiera que semueve en un recinto de longitud 1 y anchura €, de manera que 1. Enestas condiciones, tomando el eje z como perpendicular al piano molecular,el eje x será el que sigue en su movimiento el electron. La función de ondaserá comparable a las funciones propias de una cuerda tensa en vibración;por consiguiente, han de ser del tipo seno o coseno, pero como la soluciOncoseno no satisface la condiciOn lImite, en la cual = 0 para x = 0, seelimina y queda: (8)
m (x y z) = (2/l) (21€) sen (ir n xii) sen (ir m y/€) sen (n m'z/€) (1)
La función debe ser antisimétrica con respecto al plano z = €/2, piano nodalde los electrones r, y n, m y m' son los ndmeros cuánticos que toman losvalores:
n=l,2,3 m=l,2,3 ym'=2,4,6
Como los valores propios vienen dados por:
E n m m = (h2/8m) [(n/i)2 + (m/e)2 + (m'/e)2] (2)
y recordando que 1, los movimientos en las direcciones y y z quedanen su estado fundamental (m = 1, m' = 2), mientras que el movitniento enla dirección x comprende sus niveles más bajos, asI que:
— 14 —

• REACCION DIELS-ALDER POR EL METODO FEMO
E = E 1 2 = (h2/8m) [(n/i)2 + (5/e)2] = E + cte
E = (h2/8m12)n2 (3)
Por consiguiente, los movimientos segün y y z pueden despreciarse para bajasexcitaciones y emplearse para las energIas la ecuación (3) y para las funcio-nes de onda normalizadas:
(x) = (2/l) sen (r n x/l) (4)
que pueden definirse asI:
2 (x) =fdzfdY 2 (x, y, z) =fdQ 212 (5)
teniendo , (x) el mismo signo que 1fl12 (xyz) y donde Q indica la seccióndel tubo.
En el caso lImite de 0, las expresiones de ,, (x) y E dan una des-cripción correcta del sistema para todas las excitaciones finitas a lo largo deleje x, ya que las energIas de excitación segdn z e y se hacen infinitas. Porotra parte ,aunque el efecto de las excitaciones altas es cambiar la formaestructural del sistema, no las varnos a tener en cuenta y seguiremos consi-derando —claro está que por conveniencia matemática— a los electronesformando una nube que es muy larga comparada con su espesor, sin queesto responda a una realidad fIsica.
TRAYECTORIA NO LINEAL.—Este tipo de trayectoria de enlace correspondea los hidrocarburos bencenoides, para los cuales tomaremos como modeloel benceno, compuesto conjugado por excelencia. Los electrones ir se move-ran en un tubo de sección circular —toro de radio R— en vez de una tra-yectoria exagonal, la ünica hasta ahora considerada. El potencial anailticoserá de la forma:
V (r, ', z) U (2 a)_2 { (z/R)2 + [(r/R) — (R/r)]2
} (6)
U = 1i2J2mR2 (7)
— 15 —

ROSARIO DOMINGO SEMSTIAN
donde z, r y son las coordenadas cilindricas y a es una constante sin dimen-siones, tal que a << 1. Estas coordenadas corresponden a las tres clases demovimiento que el electron tendrá: z es Ia coordenada perpendicular alpiano molecular, z 0, c.' el desplazamiento angular en el piano moleculary r distancia al eje z.
Si hacemos e = aR (diámetro del tubo) y 1 = 2irR (longitud del cIrculo),encontremos que 6/1 = a/2 << 1.
Ruedenberg y Parr (11) han demostrado que el potencial (6) puede descom-ponerse en los siguientes niveies de energIa:
ur,= + Enr + E5 (8)
dond E' corresponde a las excitaciones angulares, Enr a las radiales ya las que tienen lugar fuera del piano molecular. Los "del IflOVi
miento longitudinal" son de la forma
E0 = U2 f (a n2) ; (9)
n 0, ± 1, ± 2
f(x) =[(1 + 4x) —1] /2x = 1 —x + (10)
Como despreciamos los movimientos transversales, se obtienen los siguientes
niveles lImites:
urn = (11)
que, como vemos, son prácticamente iguales a los dados por la ecuación (3).obtenida a! suponer que el electron se mueve a lo largo de una ilnea, queserá el lImite del tubo cuando 0.
Nuestra consideración general de que el potenciai es infinito en todoslos puntos, excepto en el esqueleto, equivale a la descripción del movimientoelectrónico en una dimensiOn segdn el método MQ, con lo cual la función
(x) depende ünicamente de la "coordenada de enlace" x, y la ecuaciOn deSchrödinger será de la forma:
(d/dx)2 + (2m/112) [E — V(x)] }
' (x) 0 (12)
— 16 —

REACCION DIELS-ALDER POR EL METODO FEMO
donde V (x) es el potencial apropiado en cada punto del esqueleto. Entonces.la descripción completa del movimiento del electron en las tres dimensionespodrá escribirse asI:
(xyz) (x) (21€) sen (iy/e) sen (2z/e),con (13)
0< y <e, —112€ �z <1/2€, e<<l
donde x es Ia coordenada tangencial a las trayectorias de enlace, y es per-pendicular a x en el piano molecular y z es perpenducular a éste. Se eligeel piano z = 0 como piano molecular, y la función (1) (x, y, z) es antisimé-trica respecto a este piano.
Entre el orbital molecular unidimensional p (x) y la función de ondarigurosa 't (x, y, z) existe la relaciOn:
p2 (x) = urn dO 2 (xyz) (14)€—o .1 Q(x)
siendo e el diárnetro del tubo y Q (x) su secciOn transversal en el punto x.
JUNTAS 0 PUNTOS RAMIFIcAD0S.—Aquellos puntos de la estructura con-jugada donde se reOnen tres o más ramas, presentan un problerna particularen cuanto a la consideración generalizada del movimiento en una dirección.Para simplificar la cuestiOn, Griffith (12) ha propuesto dividir la molécula ensegmentos lineales o "ramas" que pueden ser de longitud distinta. Por ejem-plo, èl naftaleno se partirIa en tres ramas (fig. 1).
—17—
z:T
Fio. 1

ROSARIO DOMINGO SEBASTIAN
Se elige en cada rama un sistema independiente de coordenadas y se define(x ) como Ia parte de Ia función de onda a lo largo de Ia rama B; de
manera más precisa:
5(x)= (x) Si X = x5 (en Ia rama B)
y (15)(x) = 0 Si X = XB (en Las demás ramas),
de suerte que -
(15')
CONDICIONES DE CONTINUIDAD Y CONSERVACIóN.—Vamos ahora a estable-cer dos condiciones fundamentales a cumplir por los puntos ramificados,Ilamadas de continuidad y conservación. SegUn la primera, las funciones deJas tres ramas (x1), 2 (x2), (x3) deben tomar el mismo valor en lajunta.
1(x11 ) = 2(x23 ) = 3(X3J ) (16)
Como Ia ecuación (12) es una diferencial de segundo orden, debemos tambiénobtener otra condición en la que intervenga la primera derivada de lasfunciones B(xB). Para ello, consideraremos de nuevo el sistema de tubosque corresponden al enlace del esqueleto molecular, y sea (x, y, z) la solu-ción de Ia ecuación de Schrodinger en tres dimensiones en el interior de losmismos, con Ia condición Ilmite de que (x, y, z) = 0 en las paredes.Partiendo de la relación:
div (grad ) = grad2 + (17)
y aplicando el teorema de Gauss, se obtiene:
fdV(grad2÷) =fdS(/&) = 1/2fdS(/n) , (18)
siendo S Ia superficie del volumen V elegido y (n) La derivada en la direc-cidn de la normal a S (fig.. 2). V es el volumen que comprende el area
— 18 —

REACCION DIELS-ALDER POR EL METODO FEMO
(t/hxB)JdQ42 =2J
J = fdV (grad2 ) f 4))Jvdonde XB es la coordenada tangencialde la junta.
a! tubo de Ia rama B y hacia fuera
FIG. 2
Estudiemos, en primer lugar, la condición lImite 0; en virtud de(14) y (15), el primer miembro de la ecuación (19) se convierte en:
1(i XB) 2 (x) QB =
= [(I XBJ+ = finito
urn J = finito-30
0 lo que es lo mismo
(21)
(22)
Por tanto el integrando de I se convierte en un infinito del orden —2
— 19 —
rayada, y como se anula en las paredes y Q1, Q2 y Q3 son las seccionestransversales, Ja ecuación (18) se simplifica:
(19)
(20)

ROSARIO DOMINGO SEBASTIAN
En segundo lugar, también puede demostrarse que:
limJ = 0 (23)o, 6 - o
donde 6 es la distancia de cada una de las tres superficies 0B a partir delpunto medio de la junta. Por consiguiente, la expresión (21) se convierte en:
(/ X) 2(xB) = B (x)(oj x11) B (xB) = 0
en Ia junta XB =XBJ (24)
que, en virtud de la condición de continuidad, equivale a
1 (/ xB) (xB)]. = 0 (25)B Junta
y que también puede deducirse para el caso de funciones de onda complejas.Está claro que la ecuación (25) expresa la conservación de la densidad me-canocáutica:
(ilI/2m) [ (d * / dx) — * (d /dx)] (26)
En definitiva, a la ecuación (25) se le denomina condición de conservación,que han de cumplir todas las juntas.
Resumjendo, pues, estas consideraciones, las ecuaciones fundamentalesdel mdtodo FEMO son:
1 . Ecuación de Schrodinger para cada rama:
H (x)={
(_i2/2m) (d/dx)2 + V (x) } q (x) = E (x) (27)
2. Condición de continuidad:
1(x1) = 2(x2) = 3(x3) (28)
3& Condición de conservación:
( i )j = 0 (29)
— 20 —

REACCION IDIELS-ALDER POR EL METODO FEMO
4•a Condicic$n frontera para los extremos libres:
(30)
Indicaremos que an postuladó fundamental de esta teorla es que: "el caminOdel electron libre termma, no en el nücleo del átomo final, sino una longitudde enlace más allá".
En el caso de una rama cerrada, el del benceno, las condiciones que hande cumplir las juntas pueden resumirse asI: La funciOn de ondà tiene an'periodo que es una fracciOn entera de Ia longitud de Ia rama. Es, pues, dste,otra justificación del planteamiento del problema bajo un aspecto lineal, yaque estas condiciones se pueden aplicar a ambos tipos de ramas, pues situviéramos tin punto no ramificado se podrIa considerar como una junta consolo dos ramas.
Reciente, Kuhn (14) ha modificado Ia ecuación (29) correspondiente a Iacondición de conservación, expresándola de la forma siguiente:
( g x/ x1 + x,/ t x2 + x,3) = 'Kc81
siendo x1, x2, x3 las funciones de onda unidimensionales a lo largo delas tres ramas x1, x. y x3 y tomando la constante K = 1/(O'9 x lO_8) cm—1.
Se elige este valor porque conduce a un buen acuerdo entre el modelo delgas electrónico libre calculado y el tratamiento exacto de un electron enan campo de potencial. Con esta nueva condición se ha estudiado una seriede dienos y se ha Ilegado a un mejor acuerdo entre las longitudes de ondade las máximas absorciones calculadas y Las equivalentes experimentales,que el existente entre las halladas con la ecuaciOn (29), para Ia cual K = 0.La aplicación de esta nueva condición equivale a la antigua, pero admitiendola existencia de tin potencial virtual en la junta.
HERMICIDAD, ORT000NALIDAD Y NORMALIZAcION.—Como nuestro objetono es desarrollar en toda su extension la teorIa de Ruedenberg y Scherr, (8)indicaremos sus aspectos más importantes. Sea
f(x) = ('B) (31)
una fuiición en nuestro espadio de configuracion multiconexo. Entonces, Laintegral en este espacio se define asI:
— 21 —

ROSARIO DOMINGO SEBASTIAN
Jf (x) dx =J' (xe) dxB (32)
siendo 1 la longitud total de las trayectorias del electron libre y 'B la de larama B.
Puede demostrarse que H, la hamiltoniana de la eduación de Schrödin-ger (27), es un operador hermItico. For consiguiente, los valores propios dedicha ecuación son reales y las funciones propias pertenecientes a los dife-rentes valores propios son mutuamente ortogonales. Pero no deberá perdersede vista que la validez de estas propiedades depende esencialmente de lascondiciones de continuidad y conservación antes citadas.
For tiltimo es necesario que las funciones propias estén normalizadas,para lo cual han de cumplir:
f2 (x) dx = fB2 (xB) dxB = 1 (33)
POTENCIAL CONSTANTE.—Si el potencial V es constante, la ecuación (27)admite solución. La función propia del nivel energético E, tiene la forma:
(x) = Bn (x) (34)B
Bn (IB) = a cos (k XB+ 6) (35)
E = (112/2m) k2 (36)
Para determinar los parámetros k, aB ' 8B (B 1, 2, 3 ) aplicamos lascondiciones de continuidad:
a1 cos (kx1 + 8) =.a2 cos (kx1 + 6) =(37)
= a3 cos (kx33 + 83) ,
y de conservación:
a1 sen (kx1 + 8) + a8 sen (kx2 + 8) +(38)
+ a3 sen (kx3 + 83) = 0
— 22 —

REACCION DIELS-ALDER POR EL METODO FEMO
que también pueden expresarse asI:
tg (kx1 + 8) + tg (kx21 + ) + tg (kx3 + 6) = 0 (39)
siendo XBJ Ia coordenada de la junta en la rama B. La condición de nor-malización (k 0):
1/2 a 1B =jdx2(x) = 1 (40)
Esta ecuación (40) puede considerarse como una generalización de losresultados que se obtienen para una sola rama, especialmente suponiendoque el valor medio de cos2 t = 1/2, entre 0 y ir. Observaremos que:
a cos (kx + 6B) a COS (kxB + 8B)
con
a'B = aB=
6B ±
y para evitar ambiguedades, se elige siempre &, de tal forma que:
—r/2 < < r/2 (4!)
y de este modo quedan fijados simultáneamente los signos relativos de lasamplitudes aB para las diferentes ramas.
Por otra parte, se observa que los signos de k ' 8B pueden cambiarsimultáneamente, sin que esto afecte a la función de onda o a! valor propio.Adoptaremos siempre:
k�0 (42)
que nos dará en cada caso un signo definido para 6B•La longitud de onda de De Brogue del orbital molecular (34) viene dada
por:
Ap 2r/k (43)
La consideración de un potencial constante V (x), conduce a tratar todoslos átomos como iguales; por tanto, es natural suponer que Las distancias D
— 23 —

ROSARIO DOMINGO SEBASTIAN
entre átomos próximos tambidn serán constantes en la estructura conjugada,de tal fonna que puede definirse Ia constante sin dimensiones, x:
x=kD (44)
y la ecuación (36) puede escribirse asI:
(E/E) = (a/D)2 2 (45)
siendo a = !12/e2m el radio de Bohr y EH = e2/2a el potencial de ioniza-ción del átomo de hidrogeno. Está claro que x varla asi:
ox7r (46)
VECTORES PROPIOS DE LA FUNCION PROPIA DEL ELECTRoN LIBRE.—SInentrar en Ia definiciOn matemática del vector propio del electron libre cones-pondiente a Ia función propia (x) (34), solo indicaremos cómo se operacon ellos. En la práctica, se elige el origen de coordenadas en una rama, detal modo que siempre coincida con un átomo o con el punto medio de unenlace. Si consideramos lo primero, o sea que XB = 0 coincide con unátomo P. entonces:
B (P) = a8 COS 6B ; (47)
y designando por 0 el átomo próximo a P. de tal forma que XB = D, pameste átomo se obtiene:
(Q)=U) [cos x —sen - tg 681 (48)
Mediante las ecuaciones (47) y (48) se ilega a las relaciones:
tg 8B = [cos x — PB (°)!B ()1 /sen x (49)
a2B= B (P)/cos2 S (I + tg2 6 ) 2 (P) , (50)
que utiizaremos pam calcular a28 y tg 6. El signo de aB debe ser el mismoque el de B (P).
Por otra parte, si X8 = 0 coincide con el punto medio de un enlaceentre dos átomos, P y Q, se obtiene:
— 24 —

REACCION DIELS-ALDER POR EL METODO FEMO
B (P) =aB [cos _ COS B + sen sen (51)
(0) aB [cos cos — sen —_ sen ] (52)
o bien:
B (P) + (0) 2 aB cos x/2 cos (53)
B (P)— B = 2 a8 cos xf2. cos (54)
de donde resulta para:
1+cosxtg 8,, (55)
sen x (P) + 4B
a8 = •[ (P) + 2 (Q) — 2 cos B (P) (0)1 /sen2 x (56)
Considerando tres puntos consecutivos P_1, P0 y P, sobre una rama,de coordenadas (x8 — D), (x3) y (x8 + D), respectivamente, fácilmente sedemuestra que puede escribirse la siguiente relación:
B (P_1) — 2 cos x (P0) + B (1) = 0 (57)
Si P0 es el átomo de un extremo libre, o sea (P_1) = 0, Ia ecuación anteriorse reduce a:
—2 cos B (P0) + (+) 0 (58)
Por otra parte, para una junta con los tres átomos próximos P1. P2 y P3.tomará la forma:
B=iB (P8) = 3 (1) + k—1 sen x(d BdxB)J (59)
Recordando la condición de conservación, se anula el segundo miembrode Ia ecuación anterior, resultando:
(Pr) + (P2) + q (P3) — 3 cos (J) = 0 (60)
—25 —

ROSARIO DOMINGO SEBASTIAN
APLICACION DE LA TEORIA.—Los valores que se obtienen para 2 cosconducen a un ndmero infinito de valores de x. Por definición, hemossupuesto que � 0, y a cada uno de dstos corresponde uno propio de Iaenergia E y una función de onda p, es decir, cada vector propio da lugara una serie infinita de funciones propias.
Para los niveles energéticos más bajos, o sea aquellos comprendidos enel rango (0 ir), la longitud de onda de De Brogue cumple la con-.dición A � 2D, de modo que no puede haber más queunnododecada
- fdnción de onda entre dos átornos próxirnos. El valor propio x = 0 im-plica que A = oc, o sea una función de onda constante; ello no puede ocurrirsi el camino del electrOn tiene un extremo libre, pues entonces una funciOnde onda constante tendrIa que anularse.
Cuando la molécula contiene N n electrones, en el estado fundamentalIa mitad de los N niveles más bajos están doblemente ocupados; si N esimpar, el nivel más alto estará sOlo ocupado por un electron.
Puede demostrarse que, para todos los hidrocarburos alternantes, todoslos niveles para el estado fundamental cumplen la condiciOn:
Ruedenberg y Scherr (8) han comprobado, por otra parte, que para los hidro-carburos no alternantes por ellos estudiados se cumple, asimismo, dicha con-dición.
Como es bien conocido, los espectros r electrónicos se originan al saitarlos electrones r desde los niveles más altos ocupados a los más bajos vacan-tes. La energIa de transiciOn entre dos de estos niveles, m y n, viene dadapor E = EmEn,
E =ED (x2), (61)
siendo
ED = EH (a/D)2 , (62)
y el correspondiente ndmero de ondas por
1/A = E/hc
1/A = (1/AD) (2) , (63)
— 26 —

REACCION DIELS-ALDER POR EL METODO FEMO
o tainbidn
1
1/A A6 (1fA)2. (63')2
Por x indicamos x61 — x,, A es la longitud de onda de De Brogue delorbital de un electron libre, a el radio de Bohr, EN el potencial de ioniza-ciOn del hidrógeno y Ac la longitud de Compton.
De las formulas anteriores se saca la relación:
(ED/Ec) = ( I A) = (D/D)2. (64)
Los valores de las constantes usados por nosotros son: (15)
a = O'529171 A
EH 13'59782 e. v.
D = 1'4 para sistemas conjugadosD 1'353 para el etileno
Al comparar este método con el LCAO se observa que los N nivelesdel primero corresponden a los N niveles de la aproximaciOn LCAO. Lasemejanza de las matrices para ambos métodos es notable; la Onica dife-rencia reside en el factor (2/3) , introducido para las juntas. Con el fin desimplificar el cálculo numérico, también se aplica al método FEMO los prin-cipios de Ia simetrIa molecular, lo mismo que en el método LCAO. Señala-remos que la funciOn propia del electron libre pertenece a una representacióndefinida de la simetrIa de grupo tridimensional de su molécula ; para hallaresta representación, se toma en consideración el hecho de que la funciónde onda total tridimensional del electron libre es antisimétrica con respectoa! plano molecular. Esto permite que haya una correspondencia unIvocaentre los estados del FE y los del LCAO, de acuerdo con la clasificación
teOnca de sus grupos. Hay que seflalar, no obstante, que el orden de lasenergIas no es siempre el mismo y también que las degeneraciones de tipoaccidental no tienen por qué coincidir en ambos métodos.
lJnos conceptos nuevos que se introducen en el método FEMO son lasilamadas populación atómica y populación de enlace (16)• Tienen un signifi-cado fIsico similar a q (ndmero de electrones en un átomo, o sea, la cargaelectrónica del mismo) y a p (Indice de enlace) del modelo LCAO. La coin-
— 27 —

ROSARIO DOMINGO SEBASTIAN
cidencia no es totalmente exacta en cuanto a las juntas se refiere, pues, comoya hemos dicho varias veces, el término de junta se introduce. y se usa exclu-sivamente en el método FEMO y no tiene equivalente apropiado en el LCAO.La populación total r electrónica en la molécula se expresa asI:
N =fn f dx (x) = a (P) = b (M),
donde el sumatorio sobre P contiene todos los átomos y el sumatorio de Mcontiene todos los puntos medios de enlace; a representa, pues, Ia popula-ción atómica, y b la populación de enlace, y N el nümero total de elec-trones.
SISTEMAS coNJuGADos.—El desarrollo matemático anterior nos permiteplantear toda una serie de ecuaciones capaces de resolver Ia estructura delas moléculas conjugadas. Las funciones de onda de un orbital q puedenrepresentarse per.
siendo Ia parte del orbital , localizado en La rama B, de manera que
Bi = aBI cos (. x + (65)
Para conocer estas funciones de onda, Scherr () ha propuesto dos métodos;ci primero consis.te en obtener aBI2 y tg directamente de las condicionesde junta; el segundo se basa en calcular Los vectores propios a partir de lasecuaciones seculares. Nosotros seguiremos ci primero, ya que resulta sermucho más sencillo y Mcii de aplicar.
De manera analoga al método LCAO, en el que los conceptos de Indicede enlace y densidad de carga son dos magnitudes del mayor interés quImico,puesto que sirven para caracterizar el comportamiento de las moléculas, enel método FEMO hay otras magnitudes similares, a saber: densidad de en-lace y valencia unida, que definimbs a conti.nuación.
DENSIDAD DE ENLACE.—Para un enlace cualquiera, r—s, Scherr (9) defineeste concepto mediante la expresión siguiente:
— 28 —

REACCION DIELS-ALDER P0k EL METODO FEMO
n/2
bri = 2 ' qi2 (r—s). (66)j=1
donde (r—s) es el valor de en el punto medio del enlace r—s, queviene dado por
(r—s) (r) + (s) I 2 cos ½ x (67)
También dicho autor ha dado Ia siguiente formula aproximada para de-finir bri:
nJ2
bri f[ /2 + (r) (s) (68)j =1
siendo f una cierta constante, que equivale al valor medio de 1/ cos2 ½y puede tomarse como aproximadamente igual a 1,25.
El Indice de enlace de Coulson y Longuet-Higgins (17) viene dado en elmétodo FEMO por la expresiOn:
n/2Pr1 = 2 (r) (s). 69)j1
La longitud de enlace r—s se calcula por la siguiente formula empIrica,también debida a Scherr
Rr (ii) = 1,665—0,1398 (1 + b1,). (70)
VALENCIA uNIDA.—El concepto de valencia libre, que tanta importanciatiene dentro de Ia. aproximación LCAO para los hidrocarburos alternan-tes —ya que éstos presentan una distribución de carga unitaria uniforme—,puesto que permiten hacer predicciones acerca de Ia reactividad de las mo-léculas segün la aproximación esl&ica, tiene su análogo dentro del métodoFEMO en el concepto de valencia unida, que se define asI: es igual a Iasuma de las densidades de enlace de todos aquellos que concurren en elmismo átomo. Por consiguiente, cuanto menor sea la valencia unida, Vr,tanto mayor será la reactividad del átomo r. Justamente, todo lo contrarioque para Ia valencia libre, Fr.
—29—

ROSARIO DOMINGO SEBASTIAN
RECIENTES DESARROLLOS EN EL METODO FEMO.—Aun cuando en nues-tros estudios hemos aplicado el método FEMO, tal como lo desarrollaronlos americanos Ruedenberg y Scherr en 1953, no queremos silenciar las pos-teriores modificaciones que ha experimentado, de las cuales las más impor-tantes son las que indicamos a continuación:
En 1954, Basu (18) aplicó este método al estudio de las cianinas y de losdifenilpolienos. Sin embargo, los resultados que obtuvo no fueron demasiado
-buenos, puesto que adrnitió que
-los electrones se movIan en un potencial
constante, despreciando asI la mayor electronegatividad del Nitrógèn frentéa Ia del Carbono. En una publicación posterior, (19) corrigiendo la electro-negatividad del heterondcleo, mejoró los resultados precedentes.
Lichten (20) ha introducido una modificación en ci método, al suponerque todos los electrones se mueven libremente en el recinto, pero conV 0 en el interior del mismo, aunque el potencial sigue siendo constante.Esto supone dnicamente la variación del cero para la energIa total, pero civalor de Ia energIa cinética es inalterabie
El propio Ruedenberg (21) ha modificado, en parte, su teoria. Define demanera diferente la trayectoria del electron libre, si bien emplea las mismasecuaciones de movimiento. En esencia, la modificación se reduce a consi-derar que cada átomo que no sea junta tiene como una suerte de "cola", quese prolonga justamente una longitud de enlace más. A continuación mdi-camos los dos tipos de esquema para la molécula de estireno. (Fig. 3.)
F!G.3
0 sea que, en Ia teorla modificada, cada átomo es una junta, en Ia quelas tres ramas forman ángulos de 1200 entre si. A estas supuestas juntas lasdenomina "falsas juntas", para diferenciarlas de las primitivas. En ci átomolibre del estireno solo habrá una "falsa cola".
— 30 —
I
I

REACCION DIELS-ALDER POR EL METODO FEMO
Un método afIn a éste es el recientemente desarrollado por Frost,conocido con el nombre de "Modelo de la función delta". En él se suponeque el electron se mueve en una dimension segdn una lInea que pasa porlos ndcleos, y V = 0, excepto en los mismos, donde tiende a — con lasfunciones delta negativas. La funciOn de onda se expresa como combinaciónlineal de orbitales atómicos. En este método se puede operar con hetero-ndcleos, no limita arbitrariamente la coordenada y pueden predecirse ener-glas de ionizaciOn.
Muy recientemente, Ham y Ruedenberg () han mejorado el métodoFEMO incluyendo la interacción electrónica. Las funciones de onda delelectrOn libre se consideran como orbitales moleculares que se obtienen apartir de un potencial mesoelectrOnico, y luego se introduce Ia interacciónconfiguracional. En la evaluaciOn de las integrales han precisado ciertos pará-metros semiempIricos, que los determinan a partir del espectro del benceno.En un futuro prOximo, nos proponemos aplicar estos cálculos a las moléculaspor nosotros estudiadas.
Por tiltimo, Wangh y Fessenden, (24') haciendo uso del modelo clásico delelectrOn libre de Pople, predijeron los espectros de resonancia molecular delos protones unidos a los anillos aromáticos y discutieron las modificacionesnecesarias para describir los espectros de los bencenos simplemente susti-tuidos.
— 31 —

PARTE. II
DL&GRAMAS MOLECULARES
Deseando hacer un estudio lo más amplio posible de la reacción Diels-Alder por el método FEMO, y no habidndose calculado con anterioridad—por otros autores y nosotros mismos— un niimero suficiente de hidrocar-buros aromáticos, hemos creldo conveniente realizar previamente el cálculode aquellos que más nos han interesado a este efecto.
Las moléculas estudiadas son: 3 :4 Benzofenantreno (fig. 4), Piceno (Ii-gura 5), Hexaceno (fig. 6), 1:2, 6 :7 Dibenzopireno (fig. 7), 3 :4, 9: 10 Diben-zopireno (fig. 8), Zetreno (fig. 9) y Pleiadeno (fig. 10). (La numeración utili-zada por nosotros ha sido, en general, arbitraria.)
Por no alargar Ia presente exposición, no describiremos la tdcnica delcálculo; solo indicaremos que en La resolución de las matrices molecularespara encontrar los vectores propios hemos realizado las correspondientes fac-torizaciones, de acuerdo con la simetrIa de cada molécula.
En cada una de las figuras indicamos los origenes elegidos para las dife-rentes ramas, x, en donde las fiechas seflalan las direcciones positivas. Asi-mismo, en dichas figuras se muestran los valores calculados para las valenciasunidas, Yr, y las densidades de enlace, bra.
En el Apdndice se muestran las ecuaciones seculares con indicación desus simetrIas para cada una de las moléculas estudiadas. Las Tablas I delmismo recogen las funciones de onda ir-electrónicas: cos
x1. y los corres-pondientes vectores propios normalizados; las Tablas II, las funciones deonda ir-electrónicas: a1
2. las Tablas III, las funciones de onda r-electrO-nicas: tg 3,.
La Tabla A agrupa los valores de los Indices de enlace, P.r,' calculados
segiin Ia ecuación (69), y las longitudes de enlace en Angstroms, R (A), ob-tenidas segdn la ecuación (70). En lo que se refiere a estas longitudes de
— 32 —

FIG. 4.—El dibujo superior muestra las ramas elegidas y las direcciones positivas, asIcomo Ia numeración seguida. El central indica las densidades do enlace. Finalmente,Ia figura inferior muestra las valencias unidas. (Esta notación se hace extensiva a las
restantes figuras.)5
3:4 - BENZOFENANTRENO
lv
I
8
t
•1
Iv'
F. 92Q
I f97$
79p

x,
• HEXACENO
ly
—•x.
s4 4
2.D27
ly,
FIG. 6
— 35 —

3:4, 9:10 - DIBENZOPIRENO
x
Fio. 8
— 37 —
2.°'7
1.929
l.898
9
I 14
'13
12

U
to
PLEJADENO
f. 992
FIG. 10
— 39 —

ROSARIO DOMINGO SEBASTIAN
enlace calculadas, puede observarse que, en general, ,están en buen acuerdocon las halladas mediante Ia curva de Coulson, Daudel y Robertson, () Sibien son un poco más altas —aproximadamente 0,02 A—, lo que coincidecon lo obtenido por Scherr. ()
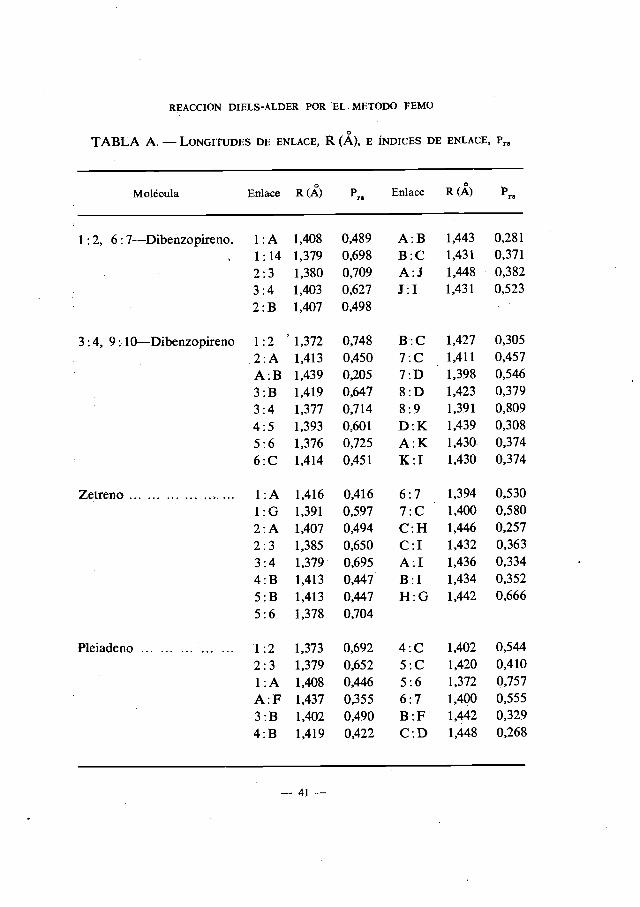
TABLA A. —LONGITUDES DE ENLACE, R (A), E INDICES DE ENLACE, P,
Molécula Enlace R (A) Enlace R (A) 'rg
3 : 4—Benzofenantreno ... 1:22:33:44:AA:BB:C
1,3771,4061,3771,4101,4361,428
0,7200,6050,7160,4780,3210,388
11: C11:1212:D1:DA:D
1,4181,381
1,418
1,4121,434
0,4170,7790,4120,4660,353
Piceno 1: 14l:AA:BA:DB:2B:C2:3
1,3721,4191,4391,4291,4091,4331,377
0,7480,4500,3030,3770,4840,3560,713
3 :44:55:C6:C6:77:DD:E
1.392
1,3771,4121,4191,3681,4171,436
0,6080,7180,4670,4100,7750,4290,324
Hexaceno 1:22: 31: AA:D
16: A
1,3701,3971,4231,4431,401
0,7610,5620,4250,2910,524
15 :BB: E
16: B15:CC: F
1,4031,4461,4071,4331,447
0,5070,2620,4820,4920,263
— 40 —
REACCION DIELS-ALDER POR EL METODO FEMO
TABLA A. — LONGITUDES DE ENLACE, R (A), E INDICES DE ENLACE, rs
0 0
Molécula Enlace R (A) 1r, Enlace R (A) P,
2:3 1,380 0,7093:4 1,403 0,6272:B 1,407 0,498
3:4, 9: 10—Dibenzopireno 1:2 1,372 0,7482:A 1,413 0,450A:B 1,439 0,2053:B 1,419 0,6473:4 1,377 0,7144:5 1,393 0,6015 :6 1,376 0,7256:C 1,414 0,451
Zetreno 1:A 1,416 0,4161:G 1,391 0,5972: A 1,407 0,4942: 3 1,385 0,6503:4 1,379 0,6954:B 1,413 0,4475:B 1,413 0,4475:6 1,378 0,704
Pleiadeno 1:2 1,373 0,6922: 3 1,379 0,6521:A 1,408 0,446A:F 1,437 0,3553:B 1,402 0,4904:B 1,419 0,422
A:J 1,448 0,382J:I 1,431 0,523
B: C 1,427 0,3057:C 1,411 0,4577:D 1,398 0,5468: D 1,423 0,3798:9 1,391 0,809D:K 1,439 0,308A:K 1,430 0,374K:I 1,430 0,374
1:2, 6: 7—Dibenzopireno. 1: A 1,408 0,489 A: B 1,443 0,28 1
1:14 1,379 0,698 B:C 1,431 0,371
6:77:CC:HC:IA:IB:IH:G
4:C5:C5:66:7B:FC:D
1,394 0,5301,400 0,5801,446 0,2571,432 0,3631,436 0;3341,434 0,3521,442 0,666
1,402 0,5441,420 0,4101,372 0,7571,400 0,5551,442 0,3291,448 0,268
— 41 —

ROSARLO DOMINGO SEBASTIAN
REACTIVIDAD QuIMIcA.—El análisis de los diagrarnas moleculares obteni-dos nos permitirá establecer conclusiones acerca del comportamiento qulmicode estas moléculas, prediciendo las posiciones .y enlaces más reactivos, dadospor los Indices Vr Y br,, que compararemos con los Indices de valencialibre, Fr, y de enlace, p,, calculados por el método LCAO. Los valores co-rrespondientes a estos tiltimos parámetros los hernos tornado, para el3:4 Benzofenantreno y Piceno, de Coulson y Daudel (25); para el 3 :4, 9: 10Dibenzopireno, de Chalvet- y Peltier (26); para el Zetreno, de Coulson yMoser (27); finalmente, para el Pleiadeno, de Estellés, Fernández Alonso yMira (28), Por lo que respecta al Hexaceno y al 1:2, 6:7 Dibenzopireno, nadahemos encontrado en la bibliografIa. La Tabla B agrupa los resultados deestas comparaciones.
TABLA B.—COMPARACION DE LOS INDICES ESTRUCTURALES ESTATICOS PARA
LAS POSICIONES Y ENLACES MAS REACTIVOS, SEGUN EL METODO FEMO y LCAO
Molécula v,, (irio) Fmax (LcAo)
POSICIONES
bmax M0) 1rnax
ENLACE
(LcAo)
3: 4-—-BenzofenantrenoPiáeno
2 2
13 5
1' :2'5:6
1:25:6
Hexaceno 6 — 1:2 —
1:2, 6:7—Dibenzopireno ...
3:4, 9:10.—Dibenzopireno..Zetreno
3 —
7 8
5 5
3:41:27: 8
—
6:77: 8
Pleiadeno 4 5 5 :6 5 :6
Se observa que, en general, ambos rnétodos asignan la maxima reactivi-dad a las mismas posiciones y enlaces. Las mayores discrepancias se pre-sentan en el 3 : 4, 9: lO—dibenzopireno, Si bien hernos de seflalar que lasdiferencias nurnéricas para las diversas posiciones y enlaces son tan pequenasque podernos aceptar como satisfactorios los resultados precedentes.
EsPECm0s.—Mediante la ecuación (61) hemos calculado las energIas co-rrespondientes a las transiciones N -3 V1.
— 42 —

REACCION DIELS-ALDER POR EL METODO FEMO
TABLA C.FRECUENCIAS CALCULADAS (pog LOS METODOS FEMO 'i LCAO)Y EXPERIMENTALES PARA LAS TRANSICIONES N — V,
Molécula-
FEMO
v (cm—')
LCAO Experimentales4
3: 4—Benzofenantreno 25.529 26.600' 26.880
Piceno '. 22.538 23.400' 26.600
Hexaceno 8.118 —
1:2, 6:7—Dibenzopireno ... 23.514 — 25.300
3:4, 9-10—Dibenzopireno ... 15.430 — 23.090Zetreno 9.247 9.75 12 16.077*
Pleiadeno 8.534 1O.087 —
1 A. PULLMAN: Comp. Rend. 229, 887 (1949).2 C. A. COULSON y C. M. MOSER: I. Chem. Soc., 1341 (1953).
B. PULLMAN y A. PULLMAN, G. BERTh1ER y J. Powris: J. Chim. phys, 49, 21(1952).
E. CLu: "Aromatische Kohienwasserstoffe". Springer, Berlin, 2.& ed. 1952.* Este valor, como indica Coulson y Moser (bc. cit.), no parece que hay forma
de correlacionarlo con las transiciones predichas.
Puede observarse que el acuerdo entre los valores calculados por elmétodo FEMO y los experimentales es francamente bueno, tanto más, puestoque, como indican Ruedenberg y Scherr (8), en este método hay un soloparámetro ajustable, D, mientras que en el LCAO, hay dos, S. integral derecubrimiento, y /3, integral de resonancia.
La mayor diferencia corresponde al 3 :4, 9: 10—Dibenzopireno, que tam-bién presentaba las máximas discrepancias en relación con la reactividadquImica.
— 43 —

PARTE III
CONSIDERAIONES GENERALES SOBRELA REACION DIELSALDER
Antes de realizar el estudio teórico de esta reacción nos ha parecido con-veniente dar una idea general de sus caracterIsticas experimentales más im-portantes. ()
En el siglo pasado se conocieron ejemplos aislados de esta reacción. AsI,Zincke (29f) descubrió y desarrolló correctamente la dimerización de la tetra-clorociclopentadienona. A principios del actual, Euler y Josephson (29) reco-nocieron la adición del jsopreno a la p-benzoquinona. Pero fue unos añosmás tarde cuando Diels y Alder (30) demostraron la generalidad de la adicióndidnica..
Se define como "reacción Diels-Alder" o "sIntesis diénica" la adicion decompuestos etilénicos o acetildnicos a las posiciones 1:4 de un dieno con-jugado, tal como se especifica a continuación:
HC HC"cH co.I
+ IJ I __ 0
HC HC_,Dieno. Aducto.
1. :111.
FIG. 11
Tanto el dieno como el filodieno pueden ser otras moléculas de las aquIindicadas, si bien respondiendo a la misma constitución. Hemos de seflalar,
—44—
Filodieno.II.

REACCION DIELS-ALDER POR EL METODO FEMO
sin embargo, que nuestras consideraciones cuantitativas posteriores se refe-rirán siempre a un solo filodieno, el anhIdrico maleico (II). En cuanto alos dienos (I) considerados, se indicarán posteriormente.
Se ha observado que, frecuentemente, los aductos de la adición diénicason termodinámicamente inestables. Refiriéndonos exciusivamente a! anhI-drido maleico, Bachman y Kloetzel, (31) por una parte, y Wassermann, (32) potra, se vieron en Ia necesidad de establecer en sus reacciones con los ben-zólogos del antraceno el siguiente equilibrio.
Dieno + anhIdrido maleico==> Aducto
Lógicamente, la inestabilidad de ciertos aductos, a que nos hemos referidoantes, se explica admitiendo que el anterior equilibrio está muy desplazadoa Ia izquierda.
Por otra parte, la sIntesis en si viene regida por una pronunciada estereo-especificidad segdn la cual la formación del aducto no tiene lugar de unamanera cualquiera, sino siguiendo unas ciertas leyes, conocidas con el nom-bre de reglas de Alder: ()
1.0 La adición de un filodieno es necesariamente cis con respectoa! dieno y la parte no saturada de aquél; es decir, cis con respectoa ambos reaccionantes.
2.° En el caso de la adición de un dieno cIclico (ciclópentadieno, fu-rano), pueden formarse teóricamente dos tipos de aductos: endo-aducto, Si los grupos originales del filodieno se orientan en el aductohacia el doble enlace superviviente del dieno, y exo-aducto, Si laorientación es al contrario. Si bien en su casi totalidad la configu-raciOn resultante es la endo-cis, se conocen algunos casos (coffin enla adición del furano y anhIdrido maleico en éter, (34)) en que seforma la exo-cis.
En cuanto a! mecanismo de la reacción Diels-Alder, (35) es objeto deamplia discusión, Si bien el estudio del efecto de los Sustituyentes sobre elcurso de la velocidad de la adición nos ha proporcionado una valiosa thfor-macion. En general, puede establecerse que el átomo de carbono más nucleO-fib del dieno reacciona con el carbono más electrOfilo del filodieno. (36)
A su vez, el principio de Ia adiciOn cis- y el que a veces los dienos noreactivos onginen la isomerización del ácido maleico en. ácido fumárico, (37)proporcionan fuerte apoyo a la suposiciOn de que los dos nuevos enlacesC—C se forman simultáneamente.
— 45 —

ROSARIO DOMINGO SEBASTIAN
Esta reacción casi siempre transcurre de acuerdo con un inecanismo polaro heterolItico, pero bajo determinadas circunstancias (por ejemplo, en fasegaseosa) puede realizarse por un mecanismo apolar u homolitico.
ESTUDIO TEORICO DE BROWN SOBRE LA REACCIONDIELS-ALDER
Brown () ha efectuado un detallado estudio de esta reacción aplicandoel método de los orbitales moleculares, en la aproximación habitual LCAO.Como ya se especificó en Ia Introducción, nuestra investigación desarrollaun estudio paralelo al realizado por el autor precitado, pero. utilizando elmétodo del electron libre.
Basu (39) utilizO con anterioridad el método FEMO para el estudio de dichareacción, pero se limitó a aprovechar los resultados de Scherr, (9) a los cualesaplicó la teorla de Brown; como ya podrá comprenderse, este estudio noera sistemático. Además, convendrá aclarar que no solo para La reacciónDiels-Alder, sino para todos aquellos cálculos de energIas de para-localiza-ción en los cuales el etileno es una de las moléculas residuales, nuestrosresultados. difieren totalmente de los obtenidos por Basu. En efecto, al calcu-lar Ia energIa ir-electrónica —concepto que será aclarado más tarde— deletileno, hemos supuesto que la trayectoria del electrOn libre se prolonga unenlace más por cada extremo, de acuerdo con el postulado de Ruedenbergy Scherr, (8) obteniendo para su energfa el valor 0,222 h2 / 8ml2, donde 1 esla longitud del enlace C—C, mientras Basu admite que solo se prolonga 1/21.
Como nuestras bases teóricas de la sIntesis diOnica son las mismas quelas expuestas por Brown. (38) vamos a indicar, con cierto detalle, los funda-mentos desarrollados por este Oltimo. Sin embargo, convendrá anticipar algu-nos conceptos de la teorIa de las velocidades absolutas de reacción, debidaprincipalmente a Eyring. (40)•
Segtmn esta teorla, la constante de velocidad k' viene dada por la expresión
k' = kT/h . eG*/RT
(71)
o lo que es lo mismo
k' = kT/h exp (S*/R) exp (— H*/RT) (72)
— 46 —

REACCION DIELS-ALDER POR EL METODO FEMO
donde es el coeficiente de transmisión, kT/h (k es la constante de Boltz-mann y h la constante de Planck) el factor de frecuencia, constante universalque aparece en la ecuación de la velocidad absoluta de las reacciones quImi-cas ordinarias y S y H* son las diferencias de entropla y entalpIa entreci complejo activado y las sustancias reaccionantes, respectivBmente, referidastodas eflas a sus estados standard.
Ciertos resultados estereoquImicos y cinéticos (41) permiten sentar la hipO-tesis de que, en la sIntesis diénica, el complejo activado tiene, en general,una configuración analoga a la del producto final de la reacción. Es decir,podemos escribir:
—
FIG. 12
Ahora bien, Ia formación de un aducto supone la localización de doselectrones ir en el hidrocarburo conjugado para formar los dos nuevos enla-ces o-, C—C, que, segdn Bergmann y Bergmann, (37) lo hacen simultánea-mente, es decir, en un proceso de una sola etapa. Y ahora es cuandoBrown (38) postula la hipótesis fundamental de su teorfa, a saber: "La faci-lidad de Ia formación de un aducto en un par de átomos m y n del hidro-carburo depende solamente de la energIa requerida para localizar dos elec-trones r en los átomos m y n, siempre que éstos estén orientados favorable-mente". Esto equivale a admitir que los átomos m y n estén en posición
— 47 —
I.RE ACCIONANT ES COMPLEJO ACTIVADO
ifi.PRODUCTOS

ROSARIO DOMINGO SEBASTIAN
para, por lo que proponen para Ia energIa de localización de dichos dec-trones el nombre de energIa de para-localización, P.
Examinando con detenimiento la ecuación (72) observamos que el coefi-ciente de transmisión es casi siempre la unidad para todas las reaccionesqulmicas; por tanto, no lo tendremos en cuenta, asI como tampoco consi-deraremos el factor kT/h, constante universal. En consecuencia, nuestra aten-ción se centrará exciusivamente en los otros dos factores, los exponenciales,en ios que figuran S—y-- H*. La- hipótesis de Brown (38a) supone que la -entropIa de activación, S", es constante para un filodieno dado, y que laparte variable importante de la energIa potencial de activación es la energIade para-localización, P. Si tales consideraciones son ciertas, es verosImil ad-mitir que la energIa de para-localización es la parte variable importante dela energIa libre de reacción, G, o, lo que es lo mismo, sus diferenciasse deben exciusivamente a las habidas en el calor de reacción, H*. Dicho.de otra manera, ello conduce a admitir que los productos formados másrápidamente serán los más estables. Esta deducción es realmente impor-tante, pues experimentalmente es imposible discernir si los productos obte-nidos son los más rápidamente formados o los más estables desde el puntode vista del equilibno. La teorla de Brown interpreta cualitativamente ambosaspectos del problema, ya que, en resumen, se describe el sistema reaccio-nante en función de las variaciones habidas en la energIa de resonancia.
Otras aproximaciones implica esta teorla: la energIa de activación es Iasuma de la energia de formación de los enlaces o- en el filodieno y de lasvariaciones energéticas que acompañan a las posibles alteraciones en la con-figuracion espacial del hidrocarburo; además, supone que todos estos fac-tores son constantes al pasar de una molécula a otra. Por otra parte, seadmite, desde un punto de vista cualitativo, que la energIa de conjugaciónaumenta con el ndmero de sistemas separados, es decir, con el ndmero demoléculas residuales.
Finalmente, Brown (38d) demuestra que la suma de los Indices de valen-cia libre, F, correspondientes a las posiciones para elegidas, pueden tomarsecomo criterio de reactividad en la reacción Diels-Alder, si bien hay que seña-lar que es más exacto el de Ia energIa de para-localización. Dado que en elmétodo FEMO Ia valencia unida, V. equivale a! indice de valencia libre, F,en el LCAO, en nuestras consideraciones emplearemos conjuntamente elIndice V y la energIa de p-localización.
Aunque los resultados obtenidos por nosotros serán comparados con los
—48—

REACCION DIELS-ALDER POR EL METODO FEMO
hallados por otros autores mediante el método LCAO, hay que seflalar quese han utilizado distintas aproximaciones para obtener estas energIas. AsI,Dewar () calculó las energIas de para-localización de doce hidrocarburosaromáticos por el mdtodo NBMO y sus valores coincidieron totalmente conLos obtenidos por Brown. ()
Indicaremos de pasada que, en estos ditimos aos, ha comenzado a estu-diarse la importancia del factor estérico en el filodieno, habiendo desarrolladoVaughan y Andersen (43) una teorla general acerca de esta cuestión.
Cálculo de la energia de p-localización.—La energia de p-localización, P.viene definida por la expresión:
P=ErE+2i (73)
donde E es la energIa r electrónica del sistema conjugado original, 2a la.energIa de dos electrones aislados y Er La energIa ir electrOnica total deuno o más sistemas conjugados que se separan cuando dos electrones r hansido localizados en los átomos m y n, que, conjuntamente, constituyen Lamolécula residual.
Aun cuando La energfa viene dada por la expresión:
E = h2/8m12n2 (74)
nosotros, siguiendo a Basu, (39) La expresaremos en La forma siguiente:
P=2+nK (75)
donde 2a es La energIa de los dos electrones lr, n es el factor numérico calcu-Lado por nosotros y K = h2/8ml2, es constante en todas las reacciones ytiene un valor aproximado de 440 Kcal/mol. La ecuación (75) es muy apro-piada y cómoda de manéjar a estos fines.
Las energias ir electrónicas totales se calculan segdn La Parte I, distri-buyendo Los N electrones ir del sistema en N/2 niveles, que serán los demás baja energIa, y teniendo en cuenta que para un ndmero N impar deelectrones el iuitimo nivel estarIa ocupado solamente por un electron. Hayque senalar que esto ocurre muy raramente.
4 —49-—
ROSARIO DOMINGO SEBASTIAN
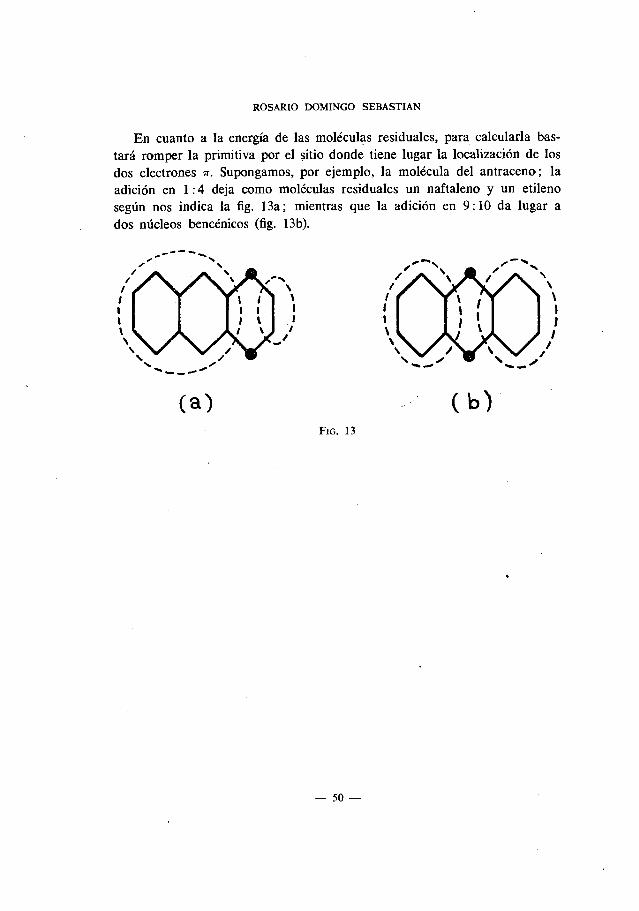
En cuanto a Ia energIa de las moléculas residuales, para calcularla bas-tará romper Ia primitiva por el sitio donde tiene lugar Ia Iocalización de losdos electrones r. Supongamos. por ejemplo, la molécula del antraceno; laadición en 1:4 deja como moléculas residuales un naftaleno y un etilenosegün nos indica la fig. 13a; mientras que la adición en 9: 10 da lugar ados mIcleos bencénicos (fig. 13b).
(a)FIG. 13
— 50 —
(b)
/I
//
SI
— — — ——
I

PARTE IV
ENERGIAS DE PARA-LOCALIZACION Y SU APLICACIONAL ESTUDIO DE LA REACCION DJELS-ALDER
CONSIDERACIONES CENERALES. — Nuestro estudio teórico de la reacciónDiels-Aider abarca 28 hidrocarburos aromáticos, cuyos nombres se indicanen las Tablas correspondientes, agrupados de la forma siguiente: la Tabia Irecopila los resultados correspondientes a los poliacenos; Ia Tabla II agrupalos referentes a los hidrocarburos aromáticos angulares; la Tabia III, loscalculados para los benzólogos del antraceno; finalmente, en la Tabla IV,se indican los resultados para una serie de hidrocarburos aromáticos que noencajan en ninguno de los grupos precedentes.
Antes de proseguir con nuestras consideraciones, convendrá dar una ex-plicación de los resultados consignados en dichas Tablas. Los datos flume-ricos referentes al método LCAO son los hallados por Brown (38a)• Este autorcalcula dos tipos de energIa de para-localización: P(—/3) y P'(—y). Las pri-meras corresponden a la aproximación habitual más simple, es decir, aquellaen que no se tiene en cuenta ningUn refinamiento del método; las segundasson las energIas calculadas tomando en consideración la integral de recubri-miento. Son los valores de P'(—y) los ünicos que Brown convierte enKcals/mol, adoptando como factor de conversion el dado por Dewar, ()y —34 kcai. Las constantes de velocidad, k0, que se consignan en las Ta-bIas I y III, tambiCn se deben a Brown, (38a) las cuales expresan valores rela-tivos de La velocidad con respecto a la adición 9: 10 del antraceno con cianhIdrido maleico, a Ia cual se le asigna el valor unidad.
Por iuitimo, queremos hacer una observación referente a los valores deP(—/3) y P'(—y), caiculados por Brown. Ciertamente, este autor ha obtenidomuchas de estas energIas por métodos aproximados, sin resolver directamentelas ecuaciones que resultan al efectuar las polarizaciones correspondientes.
— 51 —

ROSARLO DOMINGO SEBASTIAN
Sin embargo, los valores numéricos calculados por nosotros para las energIasde polarización, P, han sido obtenidos resolviendo Integramente las ecuacio-nes planteadas, sin recurrir a ningün artificio de aproximación. Para dar unaidea de lo laborioso de los cálculos realizados, indicaremos que hemos tenidonecesidad de resolver, en muchos casos, ecuaciones de grado 22, aproximandohasta la sexta decimal. Por ello, el Instituto de Cálculo de Madrid se vio enla necesidad de solicitar el concurso del Instituto Nazionale per le Applica-zioni del Calcolo (INAC), de Roma. Desgraciadamente, dado lo elevado delimporte de estos cálculos, redujimos al minimo dicha colaboración. (*)
POLIACENOS.—En la Lámina I se indican las moléculas estudiadas, asIcomO las diferentes polarizaciones consideradas. La Tabla I recoge los resul-tados numéricos calculados por nosotros, asI como los obtenidos porBrown. () En ci análisis de esta ditima resaltan dos hechos fundamentales.En primer término, se observa que la energIa de para-locaiizacion corres-pondiente al par de posiciones más reactivas en una molécula dada dismi-nuye al aumentar el tamaflo de Ia molécula. Por otra parte, para una moléculadeterminada, la reactividad de los sucesivos pares de posiciones "meso"(tales como, por ejemplo, los 1:4, 5: 16 y 6: 15, en el hexaceno) aumenta amedida que nos aproximamos al centro de la molécula.
No hemos tenido en cuenta otros pares de posiciones que los consignados,porque solamente éstos dan lugar a moléculas residuales suficienternenteestables.
Comenzaremos por indicar los resultados experimentales referentes a Iareacción de adición entre los poliacenos y el anhIdrido maleico. El bencenono lo adiciona. (45) A este efecto, convendrá recordar el hecho importantesiguiente: () con un filodieno determinado, la facilidad de Ia adición pareceestar gobernada por el "carácter" de los dobles enlaces que forman ci dieno.Un ejemplo de ello lo constituye el que, si bien el 1: 2-dihidrobenceno fun-ciona como dieno en esta reacción, ci benceno no. Esto se debe a que enel primer caso los enlaces son etilénicos y originan un dieno reactivo; porel contrario, en ci benceno todos los enlaces son aromáticos y, por consi-guiente, relativamente inactivos.
(*) Solo a tItulo puramente informativo, diremos que en la resniuciOn de las tresünicas ecuaciones de grado 22 que se enviaron, en cuya resoiución la máquina elec-trOnica trabajó durante 30 horas, el INAC ha pasado una factura de 1.500.000 L.
— 52 —

o 0Benceno.
Haftaleno.
ococoAntraceno.
o:rx OOOIlaftaceno.
OXXX O3OO coc DOoPentaceno.
(xX'X 000000 00O0 000000Hexaceno.
Lámina 1.—P0LIAcEN0s
Los puntos seflatados indican las pa1 arizaciones consideradas, cuyas energIay depara-localización se indican en la Tabta I

TA
BL
A 1
.—P0
LIA
cEN
0s
Mol
écul
aPo
sici
ones
FL
CA
O
P(—
/3)
P'(—
y) P
'(Kca
ls)
FEM
OV
P
.
Vel
ocid
ad'
rila
tiva
Ben
ceno
1:4
0,79
74'
4,00
22,
672
912
4,00
02a
c. a
. 10"
Naf
tale
no1:
40,
9056
'3,
682
2,40
282
23,
746
2a—
0,17
3Kc.
a. 1
0A
ntra
ceno
1:4
9:10
0,91
86'
1,04
02
3,63
2
3,31
2,34
2
2,07
802
703,
742
3,46
82a
—0,
181K
2a—
O,3
54K
c. a
. 10
1
Naf
tace
no1:
45:
12
0,92
14'
1,05
90
3,62
2
3,25
2,33
2
2,01
792
68
3,74
23,
458
2c,—
0,18
2K2a
—0,
363K
— 30
Pent
acen
o1:
45:
146:
13
0,92
22'
1,06
36
1,08
02
3,61
2
3,23
3,18
2,33
2
1,99
1,94
792
68 66
3,74
23,
458
3,44
8
2a—
0,18
3K2a
—0,
365K
2—0,
373K
— 90
1600
Hex
acen
o1:
45:
16
6:15
— — —
3,61
2
3,23
3,16
2,33
'1,
99
1.92
792
68 65
3,68
83,
474
3,17
4
2a—
0,18
3K2a
—O
,366
K2a
—O
,376
K
— 90
5000
C, 0 z r-'
11) 0 t,1 -l 0 'ii m 0
1C
.A
. CO
UL
SON
y R
. DA
UD
EL
: Dic
tionn
aire
des
Gra
ndeu
rs th
éori
ques
des
crip
tive2
des
mol
ecul
es. V
ol. I
I. M
athe
-m
atic
al I
nstit
ute,
Oxf
ord
y C
entr
e de
Chi
mie
Thé
oriq
ue d
e Fr
ance
. Par
is.
2R
.D
. BR
OW
N: 1
. Che
m. S
oc..
691
(195
0).
'C
.W
. Scn
iu: J
. Che
mP
hys.
21,
158
2(1
953)
.

ROSARIO DOMINGO SEBASTIAN
Tampoco ci naftaleno adiciona anhIdrido maleico. (45) Sin embargo, men-cionaremos el hecho siguiente: (47) cuando el naftaleno se funde a 1000 Cdurante 24 horas con 30 partes moleculares de anhIdrido maleico, la reac-ción tiene lugar en proporciOn inferior al 1 %; ahora bien, en presenciade grupos aiquflicos convenientemente distribuidos, se ilegan a obtener aduc-tos con un rendimiento de hasta el 90 %.
El antraceno es ci primero de los poliacenos que forma un aducto conel anhIdrido maleico, reacción que tiene lugar en las posiciones 9: 10 (fig. 12).
La pronunciada reactividad de las posiciones mesoantracénicas viène mdi-cada por ci hecho de que ci 2-isopropenilantraceno experimenta la adiciónexciusivamente en las posiciones 9: 10. ()
Como la reaccidn anterior es un verdadero equiiibrio, se ha sugerido quepuede utilizarse como medio de purificacidn del antraceno al realizarse lainversion piroiItica. Afladiremos que Diels y Alder ('fl) sugieren que la ante-rior reacción constituye una valiosa prueba en favor de la estructura Arm-strong-Hinsberg, propuesta para Ia molécula del antraceno.
Finalmente, seflalaremos que la presencia de grupos meso-metilo aumen-tan Ia velocidad de reacciOn; por el contrario, los grupos meso-fenilo laretardan extraordinariamente. (50)
Como la energIa de para-localizaciOn calcuiada por nosotros en las posi-ciones 9: 10 vale: P 2 a — 0,354 K, éste será ci valor umbra! que, a partirde ahora, nos indicará a priori cuándo la formaciOn de un aducto procedecon velocidad apreciable. Recordaremos, a este efecto, que los valores cr1-ticos hallados por Brown () son P'(—--)/) = 2,07, o bien 70 Kcai/mol.
Por lo que se refiere a los restantes poliacenos, naftaceno, pentaceno yhexaceno, todos ellos adicionan con facilidad anhfdrido maleico en las posi-ciones meso-centrales, facilidad que aumenta rápidamente a! ir avanzandoen la serie homóloga. AsI, por ejemplo, ci antraceno lo hace muy fácilmente;ci pentaceno y hexaceno, casi instantáneamente. (51) Como se observará con-sultando Ia Tabia I, los valores numéricos caiculados por nosotros para estasmolécuias se encuentran por debajo del valor umbra! antes definido.
Resumiendo nuestras consideraciones sobre los resultados consignados,diremos que entre los valores teóricos calcuiados por nosotros y los halladospor Brown existe un total y perfecto acuerdo, asI como con los resultadosexperimentales.
— 54 —

dl3Ib dbFenantreno.
R5b SbCriseno.
3 — Benz ofenantreno.
Pi ce no.
Lámina II.—HrnRocRBuRos ANGULARES
Los puntos senalados indican las polarizaciones consideradas, cuyas energIas depara-localización se recogen en Ia Tabla ii

TA
BL
A 1
1.—
HID
RO
CA
RB
UR
OS
AN
GU
LA
RE
S
LC
AO
FEM
O—
—--
-.--
-.z
Mol
écul
aPo
sici
ones
FP(
—/3
)P'
(—i)
P'(K
cals
)V
P
C,,
Fen
antr
eno
1: 4
0,89
08'
3,77
22,
472
842
3,76
12—
0,15
3 K
9: 1
20,
5909
4,46
2,88
983,
986
2L—
0,19
4 K
2:12
0,54
184,
512,
9199
4,11
12a
—O
,127
K
4: 5
0,88
12
4,37
——
3,77
22a
—0,
062
K
Cri
seno
3 : 6
0,89
413,
742
2,45
283
23,
781k
2a—
0,15
7 K
8: 1
60,
602
4,33
——
3,95
72a
—0,
162
K
7: 1
30,
578
4,52
2,94
100
4,02
82a
—0,
179
K
6:7
0,88
24,
57—
3,82
!

TABLA II.—HrnRocARBuItos ANGULARES (c0NTINUAcION)
0
I
C. A. Coulson y R. Daudel, "Dictionnaire des Grandeurs théoriquesmatical Institute, Oxford, y Centre de Chimie Théorique de France, Paris.
2 R. D. Brown, J. Cliem. Soc., 691 (1950).3 C. W. Scherr, J. Chem. Phys., 21, 1582 (1953).4 J. I. Fernández Alonso, J. Mira y J. L. Oliete. J. Chim. prys., 54, 822 (1957).
descriptives des molecules. Vol. II. Mathe-
C'
Molécula Posiciones F P(—f)
LCAO---------P'(—i) P'(Kcals)
FEMO-V p .
3 :4 Benzofenantreno 1' :4'2: 121:4
4':132' :4
0,89510,63 1
0,5800,5770,536
3,742
4,254,525—
2,452
2,702,93——
832
92
100——
3,758k 2—0,1583,764 2a—0,2853,909 2a—0,1793,993 2a—0,1124,030 2a—0,1 15
KKKKK
Piceno 1:46: 166:71: 14
15 :22
0,8931
0,5800,8840,8870,288
c.ac.a
3,752
4,51
3,803,97—
2,462
2,92———
842
99———
3.760k 2a—0,1554,002 2a—0,1813,806 —
3,743 —
4,120 2a—0,251
KK
K

REACCION DIELS-ALDER POR EL METODO FEMO
HIDROCARBUROS ANGULARES.—Las moléculas consideradas dentro de estegrupo son las representadas en la Lámina II. Los resultados numdricos obte-nidos para las diferentes posiciones estudiadas se muestran en la Tabla II.
Tomando como base el valor umbral antes definido, el análisis de flues-tros resultados numéricos —lo mismo que los de Brown— sugiere que nm-guna de estas moléculas formará aducto con el anhIdrido maleico. Esto estáen total acuerdo con los resultados experimentales referentes al fenantrenoy criseno. (52) Nada podemos decir con respecto al 3 : 4-benzofenantreno ypiceno, para los cuales no hemos encontrado datos en la bibliografia.
En resumen, nuestros resultados numéricos coinciden totalmente con losde Brown y los experimentales.
BENZOLOGOS DEL ANTRACENO.—La Lámina III muestra los cuerpos estu-diados en esta serie, cuyos resultados numéricos se recogen en la Tabla III.
En términos generales, puede indicarse que todos estos cuerpos adicionananhIdrido maleico, preferentemente en las posiciones meso-antracdnicas.
En total acuerdo con los resultados teóricos de Brown, de los nuestrosse deduce que la facilidad de reacción con el anhIdrido maleico sigue el ordensiguiente:
1: 2-benzonaftaceno > antraceno > 4: 5-benzocriseno > 1: 2-benzan-traceno > pentafeno > 1:2, 3 : 4-dibenzantraceno > 1 :2, 7: 8-diben-zantraceno = 1:2, 5 : 6-dibenzantraceno.
El 1: 2—benzantraceno reacciona con el anhIdrido maleico en las posicio-nes meso-antracénicas, pero lo hace con menos facilidad que el propio antra-ceno (53), en franco contraste con su isómero, el naftaceno.
Como hace observar Brown, el 1: 2-benzantraceno y el naftaceno nosproporcionan un magnIfico ejemplo del diferente efecto originado por lasanillaciones angular y lineal sobre la energIa de resonancia de un hidrocar-buro policIclico —la molécula residual es Ia misma para la adición mesoen los dos hidrocarburos—, hecho que está relacionado con la diferenciaen las densidades de enlace de los enlaces 1—2 y 2—3 del antraceno, 1,094 y0,921 respectivamente, (9) resultado coincidente con lo hallado por Brown.()(Los valores de los Indices de enlace, LCAO, son a su vez 0,7376 y0,5858 (25))
Los I : 2, 5 : 6- y 1: 2, 7: 8-Dibenzantracenos reaccionan en las posicionesmeso-antracénicas, mucho más difIdilmente que el antraceno y el 1:2-Ben-zantraceno. (54) Convendrá ilamar la atención de que, Si empleáramos un
—57—

ROSARIO DOMINGO SEBASTIAN
mayor nümero de cifras significativas en el valor de P. nos encontrarlamoscon que el derivado 1: 2, 5 :6- reacciona ligerIsimamente con más facilidadque el 1: 2, 7: 8-Dibenzantraceno. Este hecho, también observado por Brown,pone de manifiesto que Ia velocidad de reacción dcpenderá de algdn otrofacfr,r no tenido en cuenta por nosotros, como, por ejemplo, Ia energIa deactivación.
El Pentafeno (2 : 3, 6 : 7-Dibenzofenantreno) adiciona dos moléculas deanhIdrido maleico en las posiciories 1: 4 y 5: 14, pero mucho más lentamenteque el antraceno. (55) Respecto a este cuerpo, hace observar Brown que laadición de la primera molécula de anhIdrico maleico en las posiciones 5: 14
da origen a un aducto que contiene una molécula de antraceno; por con-siguiente, la segunda adición debe ocurrir en las posiciones ,neso-antracé-nicas, que corresponden a las 8: 13 del Pentafeno, y debemos esperar, enconsecuencia, que esta adición se realice más rápidamente que la de laprimera molécula del filodieno.
Por tiltimo, se sabe experimentalmente (56) que la reactividad de los cuer-pos considerados en este grupo y hasta ahora no estudiados con el anhIdridomaleico, sigue el orden siguiente:
1: 2-Benzonaftaceno> 4 : 5-Benzocriseno> 1: 2, 3 : 4-Dibenzoantraceno
En este hecho experimental se basa precisamente un método para la separa-ción de los mismos.
Queremos liamar la atención acerca de los valores teóricos dados parael 1: 2-Benzonaftaceno y 4: 5-Benzocriseno que, de acuerdo con lo indicadoen la Tabla III, se han obtenido por interpolación de los correspondientesa las posiciones meso-antracénicas de los cuerpos de este grupo (fig. 16). Porello, no les damos el mismo peso que a los otros calculados directamente;sin embargo, el hecho de que sigan el orden correcto esperado nos inclinaa aceptarlos como buenos.
En resumen, de la comparación de nuestros resultados teóricos con losexperimentales podemos establecer que para que haya reacción con el anhI-drido maleico la energIa de para-localización debe tener un valor P<2a—0,300 K. Asimismo, que el orden de la facilidad de reacción predicha teóri-camente coincide totalmente con el experimental.
— 58 —

001:2. —BenzanEraceno.
I 41
6cx91:2, 5:6—Dibenzantraceno.
1:2,7:8 —Dibenz8ntraceno.
PentaFeno.
1:2.— BenzonaF€aceno.
4:5— Benzocriseno.
Lámina III.—AN-rRAcENO Y SUS NOMÔLOGOS
Los puntos señalados indican 1a polarizaciones consideradas, cuyas energIas depara-localización se recogen en hi Tabla III
1:2., 3:4— Dibenzantraceno.

TA
BLA
JIL
—A
Nm
AcE
N0
Y S
US
BE
NZ
óLO
GO
S
C z 11) ri C t71
tTi - C C C
Mol
ecul
a
L C
A 0
—--
--—
---
Pos
icio
nes
FP(
—/3
)P'
(—y)
F E
M 0
—--
--P
'(Kca
ls)
VP
Vel
ocid
adre
lativ
aka
2
Ant
race
no1:
40,
9186
13,
63
9:10
1,04
023,
31
2,34
2,07
802
703,
742
2a—
0,18
1 K
3,46
82a
—0,
354
K
— I
1: 2
-Ben
zant
race
no1'
: 4'
0,89
113,
782
9:10
1,01
73,
41
5:8
0,91
43,
64
2,49
2
2,17
2,36
852
74 80
3,94
042a
—0,
149
K3,
646
2a—
0,33
0 K
3,97
12a
—0,
176
K
—
0,00
3—
1:2,
3 :4
-Dib
enza
ntra
ceno
1':4
'0,
8791
3,83
2
9:10
0,99
83,
495:
80,
914
3,70
2,55
2
2,24
—
872
76 —
3,77
552c
i—0,
137
K3,
452
2a—
0,31
4 K
3,59
62a
—0,
174
K
—
0,00
006
—
1:2,
5:6-
Dib
enza
ntra
ceno
1':4
'0,
8911
3,79
2
9:10
0,99
63,
51
2,49
2
2,26
85 77
3,76
52a
—0,
151
K3,
138
2a—
0,30
8 K
—
0,00
002
1: 2
, 7: 8
-Dib
enza
ntra
ceno
1' :4
'0,
8911
3,79
2
9:10
0,99
63,
51
2,49
2
2,26
852
77
3,8
142—
0, 1
51 K
3,50
72a
—0,
308
K
—
0,00
002
Pent
afen
o1:
40,
9141
3,67
5: 1
41,
015
3,45
2,38
2
2,20
812
75
3,75
Q4
2—0,
175
K3,
490
2a—
0,32
4 K
—
0,00
006

TABLA III (CONTINUACION)
* Estas energIas de para-localización Se han obtenido por interpolación, de acuerdo con los datos de la Tabla V(figura 16).
1 C. A. Coulson y R. Daudel, "Dictionnaire des Grandeurs théoriques descriptives des molecules". VoL II. Mathe-matical Institute, Oxford, y Centre de Chimie Thëorique de France, Paris.
2 R. D. Brown, J. Chern. Soc., 691 (1950).3 C. W. Scherr, I. Chem. Phys., 21, 1582 (1953).4 J. I. Fernández Alonso, J. Mira y I. L. Oliete, I. Chi,n. phys.. 54, 822 (1957).5 J. I. Fernández Alonso y J. L. Oliete, trabajo no publicado.
0
0
z
tn
U)-I-4
z
0
Molécula .
Posiciones FL
_________P(—/1)
A 0P'(—y) P'(Kca!s)
F E M 0——-'---VVelocidad
relativaka2
1: 2-Benzonaftaceno 1' :4'5:126: 117:10
————
3,802
3,36
3,283,74
2,492
2,112,032,34
852
72
6979
—— —— 2a—0,355 Kt— —
—
0,1
10—
4 : 5-Benzocriseno 9: 12 — 3,742 2,452 832 —
3:61':4'
——
3,383,64
2,142,36
7380
— 2a—,0,337 Kt— —
0,02—

Bifenilo.
Pireno.
0-0
c8
c D
82Perileno.
Lámjna IV.—Orgos HIDROCARBUROS
Los punt os señalados indican las polarizaciones consideradas, cuyas energias depara-localizacion se recogen en la Tabla III
5
TriPe nile no.
1:2. ;6:7- Dibentopireno.
t: z—Benzopireno.

3:4, 9:lO—Dibenzopireno.
BiPenileno.
A S—Indaceno.
Pleiadeno.
Zeeno.
Lámjna IV.—Oiiws HIDROCARBUROS
Los puntos señalados indicon las polarizaciones consideradas, cuyas energias de- para-localización se recogen en la Tabla HI

TA
RL
A I
V.—
OT
Ros
HID
RO
CA
RB
UR
OS
—L
'CA
O—
-FE
MO
----
Mo1
cu1a
Pos
icio
nes
FP
(—/3
)P'
(—i)
P'(K
cals
)W
P
Bif
enilo
.
1:4
2:5
2:2'
0,53
6'0,
831
0,87
2
4,38
2
3,96
4,33
2,78
2
2.64
3,01
952
90 102
4,11
13,
898
4,43
0
2a—
O,0
92 K
2a—
0,07
0 K
2a—
0,07
1 K
Z
Tri
feni
leno
Pire
no
1:4
2:15
13:1
61:
12
3: 1
46:
16
0,87
82'
0,54
480,
2794
0,87
82
0,57
911
0,60
08
3,79
2
4,42
5,19
4 4,38
2
4,50
— — — 2,79
2
2,93
— — — — 952
100
3,77
24,
113
4,20
43,
772
4,09
73,
868
•
2a—
0,14
0 K
2a—
0,13
5 K
2a—
0,22
3 K
2a—
0,11
8 K
2a—
0,15
1 K
—
G
Peri
leno
11: 1
2
1: 1
2
0,22
16
0,90
61
5,43
3,92
2
— 2,60
— —
4,47
0
3,74
43
2—0,
076
K
2a—
0,12
4 K
1:2,
6 :
7-D
ibcn
zopi
reno
1: 2
-Ben
zopi
reno
1:41
1' :4
'4'
: 3 2:2'
5: 1
23:
14
0,87
80,
894
0.54
40,
606
0,56
5
— — -— — — —
— — — — —
3,76
8
3,77
44
3,81
94,
115
3,90
73,
971
2a—
0,14
1 K
2o—
0,13
9 K
2a—
0,08
5 K
2a—
0,13
0 K
2a—
0,22
3 K
2—0,
136
K
0
TA
BL
A I
V (
c0N
TIN
uAcI
ON
)
0 (I, 0 z C) 0 C',
rn C', z
.
LC
AO
FEM
O—
—-
—--
----
Mol
écul
aPo
sici
ones
FP(
—#)
P'(—
y)P'
(Kca
ls)
VP
3 :4
. 9: 1
0-D
iben
zopi
reno
1: 4
'0,
885
——
—3,
718
2a—
O,0
59K
1' :4
'0,
899
——
3,68
92a
—O
,159
K
Bif
enile
no1:
41:
8
2:11
0,85
61
0,85
60,
641
— —— —
— —
3,79
26
3,79
24,
104
2a—
0,09
32a
—O
,073
2L—
O,1
73
K K K
as-I
ndac
eno
3:4
1,11
6—
——
3,66
462a
—0,
241
K
Plei
aden
o5:
84:
91,
3668
1,15
41
—— —
— —3,
694
3,29
62a
—0,
184
2a—
0,38
4
K K

TA
BL
A I
V (
CO
NT
INU
AC
ION
)
LC
AO
FEM
O—
Mo1
cula
Posi
cion
esF
P(—
/3)
P'(—
,)P'
(Kca
)s)
VP
Zet
reno
5 :6
0,99
43,
5910
2,33
13,
575
2a—
0,26
2 K
8:21
0,6
174,
212,
853,
990
2a—
0,23
1 K
9:23
0,59
64,
293,
004,
119
2a—
0,16
2 K
11:2
40,
588
4,53
3,43
4,10
82a
—0,
126
K6:
240,
569
4,56
3,48
4,09
07:
220,
549
4,61
3,58
3,95
42a
—0,
178
K3:
16
0,54
84,
643,
684,
017
2—0,
17O
K16
:20
0,30
55,
014,
104,
086
2a—
0,31
8 K
20:2
30,
258
5,36
4,29
4,39
12n
—0,
202
K12
:21
0,66
64,
403,
473,
876
2—O
,1l8
K
1C
. A. C
ouls
on y
R. D
aude
l,"D
ictio
nnai
rede
s G
rand
eurs
théo
riqu
es d
escr
iptiv
es d
es m
o1cu
1es"
. Vol
. II.
Mat
he-
mat
ical
Ins
titut
e, O
xfor
d, y
Cen
tre
de C
him
ie T
hori
que
de F
ranc
e, P
arIs
.2
R.
D. B
row
n, 1
. Che
ni.
Soc
.,69
1 (1
950)
.3
C. W
. Sch
err,
J. C
heni
. Phy
s., 2
1, 1
582
(195
3).
4J.
I. F
erná
ndez
Alo
nso,
J. M
ira
y 1.
Olie
te, I
. Chi
m. p
hys.
, 54,
822
(19
57).
50.
Cha
lvet
y J
. Pel
tier,
Com
p.R
end.
,24
0, 1
709
(195
5).
6J
J. F
erná
ndez
Alo
nso
y R
. Dom
ingo
, Ana
l. Fi
s. y
Qui
m. B
51,
447
(19
55).
7J.
I. F
erná
ndez
Alo
nso
y J.
L. S
anto
s L
ucas
, ibi
d., B
50,
143
(19
54).
8I.
Est
e11s
. J.
I. F
erná
ndez
Alo
nso
y J.
Mir
a, ib
id.,
B 5
0, 1
1 (1
954)
.9
C.
A. C
ouls
on y
C. M
. Mos
er, 1
. Che
m. S
oc.,
1341
(19
53).
10C
.A
. Cou
lson
, C. M
. Mos
er y
M. P
. Bar
nett,
ibid
., 31
08 (
1954
).
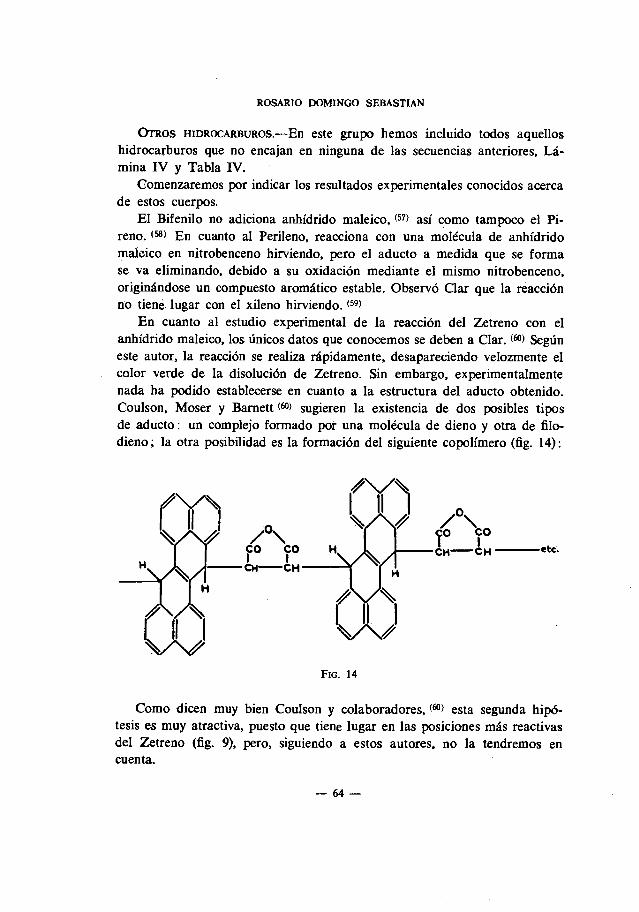
ROSARIO DOMINGO SEBASTIAN
Omos HrnRocARBuRos.—En este grupo hemos incluido todos aquelloshidrocarburos que no encajan en ninguna de las secuencias anteriores, La-mina IV y Tabla IV.
Comenzaremos por indicar los resultados experimentales conocidos acercade estos cuerpos.
El Bifenilo no adiciona anhIdrido maleico, (> asI como tampoco el Pi-reno. () En cuanto a! Perileno, reacciona con una molécula de anhfdridomaleico en nitrobenceno hirviendo, pero el aducto a medida que se formase va eliminando, debido a su oxidación mediante el mismo nitrobenceno,originándose un compuesto aromático estable. Observó Oar que la réacciónno tiené lugar con el xileno hirviendo. (59)
En cuanto al estudio experimental de Ia reacción del Zetreno con cianhIdrido maleico, los ünicos datos que conocemos se deben a Clar. 0) Següneste autor, la reacción se realiza rápidamente, desapareciendo veiozmente elcolor verde de la disolución de Zetreno. Sin embargo, experimentalmentenada ha podido establecerse en cuanto a la estructura del aducto obtenido.Coulson, Moser y Barnett (60) sugieren la existencia de dos posibles tiposde aLlucto: un complejo formado por una molécula de dieno y otra de fib-dieno; Ia otra posibilidad es la formacidn del siguiente copolImero (fig. 14):
Como dicen muy bien Coulson y colaboradores, (60) esta segunda hipó-tesis es muy atractiva, puesto que tiene lugar en las posiciones más reactivasdel Zetreno (fig. 9), pero, siguiendo a estos autores, no la tendremos encuenta.
— 64 —
FIG. 14

REACCION DIELS-ALDER POR EL METODO FEMO
Del análisis de los valores de P (Tabla IV), se observa una franca dis-crepancia entre los nuestros y los hallados por Coulson y colaboradores. 0)Segün estos autores, la adicion tiene lugar en la posición 5 :6 (o su equi-valente 12: 13), aunque con velocidad muy pequena con respecto a Ia delantraceno, y en contraposición a Oar, (60) que halló un valor bastante apre-ciable para la misma. SegUn nuestros cálculos, la posiciOn más favorecidaes la 16 :20, que ocupa el pendltimo lugar entre las diez posiciones consi-deradas por Coulson como posibles lugares de adición del anhIdrido' ma-leico. El valor que hemos calculado para la posición 5:6, P=2a—O,262 K,muestra que la velocidad en reiación con la correspondiente a la posiciónmeso del antraceno es nula. For otra parte, nuestros resultados concuerdancon los de Coulson y colaboradores en seflalar que Ia posición meso-antra-cdnica de esta molécula no es la más reactiva, condición que Brown hablaadmitido que se cumplia siempre.
Para los restantes cuerpos de esta serie no hemos encontrado en la biblio-grafIa ningdn resultado experimental.
El análisis de nuestros valores teóricos pone de manifiesto que, salvo laposición 4:9 del Pleiadeno y la 16:20 del Zetreno (indicadas anteriormente),ninguno de estos cuerpos deberán formar aductos con el anhIdrido maleico.Si bien es cierto que no hay acuerdo entre los resultados teOricos y experi-mentales para el Perileno, no hemos de perder de vista que el aducto for-mado por este dltimo es tan inestable que evoluciona inmediatamente al for-marse, como ya hemos dicho anteriormente. En relación con el Pleiadeno,baremos observar que, hasta ahora, no ha podido ser sintetizado. ()
Queremos hacer una breve referencia a! empleo de los indices estáticos,valencias libres y valencias unidas, en la predicción de la reacción Die1sAider. Podrá observarse que, en general, las variaciones de F y V sonparalelas a las de P'(—7) y P y, por consiguiente, coincidentes con los resul-tados experimentales. Sm embargo, existen discrepancias con las variacionesde los Indices dinámicos, discrepancias que hacemos mInimas supeditandosiempre nuestras conclusiones, en estos casos, a las que se deducen del aná-lisis de las energIas de para-localización, que, como hemos puesto de mani-fiesto en páginas anteriores, coinciden plenamente con los resultados expe-rimentales.
—65—

ROSARIO DOMINGO SEBASTIAN
TABLA V.—ENERGIAS DE PARA-LOCALIZACION Y VELOCIDADES RELATIVAS
PARA LAS POSICIONES meso DE LOS BENZOLOGOS DEL ANTRACENO
VelocidadiBenzologo P'(—.y)l(LCAO) P(FEMO) relativa
1:2, 5: 6-Dibenzantraceno (I) 2,26 2a—0,308 K 0,00002
1:2, 7: 8-Dibenzantraceno (II) 2,26 2a—0,308 K 0,00002
1:2, 3: 4-Dibenzantraceno (III) 2,24 2a—0,314 K 0,00006
Pentafeno (IV) 2,20 2a—0,324 K 0,00006
1: 2-Benzantraceno (V) 2,17 2a—0,330 K 0,003
4: 5-Benzocriseno (VI) 2,14 2cL—0,337 K* 0,02
Antraceno (VII) 2,07 • 2a—0,353 K 1
1: 2-Benzonaftaceno (VIII) 2,03 2—0,355 K* 10
Naftaceno (IX) 2,01 2u—0,363 K 30
Pentaceno (X) 1,94 2a—0,373 K 1600
Hexaceno (XI) 1,92 2a—0,376 K 5000
1 R. D. Brown, J. Chem. Soc., 691 (1950).* Estos valores se han obtenido por interpolación de Ia fig. 15.
— 66 —
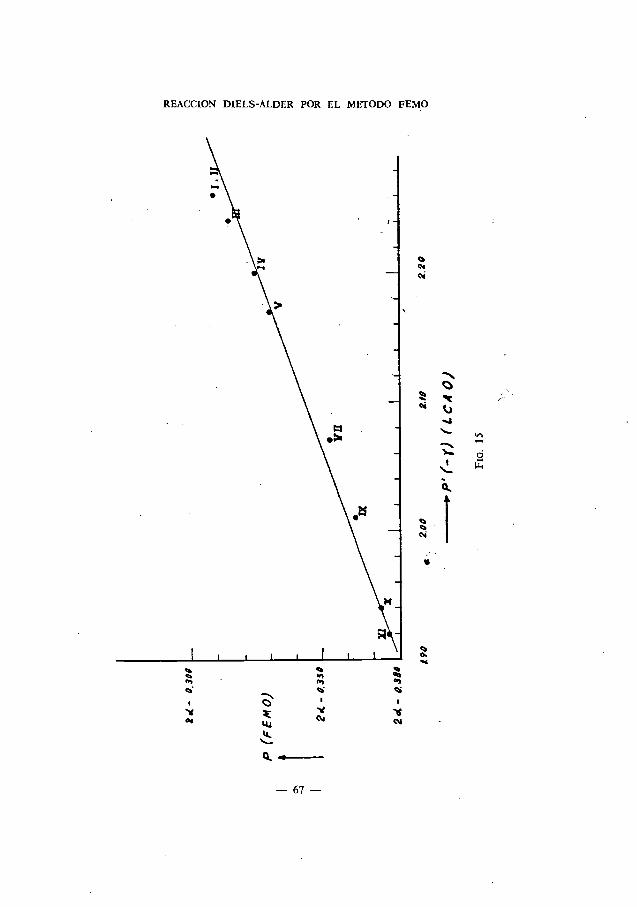
REACCION DIELS-ALDER POR EL METODO FEMO
U-
— 67 —
I
'4
-1
M
04
-I.

ROSARIO DOMINGO SEBASTIAN
TABLA VI.—ENERGIAS DE PARA-LOCALIZACION, ffiDICES DE VALENCIA LIBRE
Y DE VALENCIA UNIDA, Y VELOCIDADES RELATIVAS PARA LAS POSICIONES meso
DE LOS POLIACENOS
LCAO FEMO .
Velocidad2Aceno R P'(—y) W P relativa
Benceno (1) 0,7974 2,672 4,00G3 2 c.a. l0—'Naftaleno (II) 0,9056 2,402 3,74.6 2a—0,173 K c.a. 10Antraceno (III) 1,0402 2,072 3,468 2a—0,353 K 1
Naftaceno (IV) 1,0590 2,012 3,458 2a—0,363 K 30
Pentaceno (V) 1,0802 1,942 3,448 2a—0,373 K 1600
Hexaceno (VI) — 1,92 3,174 2a—0,376 K 5000
1 C. A. Coulson y R. Daudel, "Dictionnaire des Grandeurs théoriques descriptivesdes molecules". Vol. 11. Mathematical Institute, Oxford, y Centre de Chimie Théo-rique de France, ParIs.
2 R. D. Brown, J. Che,n. Soc., 691 (1950).3 C. W. Scherr, J. Chem. Phys., 21, 1582 (1953).
—68--

P (FEMO)
t.
REACCION DIELS-ALDER POR EL METODO FEMO
2.(.. -0200
2.L
2.-0 400
P'(-y) (L.CAO)
FIG. 16
— 69 —
21..
2.( —0.180
1.50 211 2.50

ROSARIO DOMINGO SEBASTIAN
ENERGfAS DE PARA-LOCALIZACION Y VELOCIDADES RELATIVAS PARA LAS P0-SICIONES MESO DE LOS BENZLOGOS DEL ANTRACENO.—La Tabla V recoge losvalores de P'(—y) y P en las posiciones ineso de los benzologos del antra-ceno, valores que se comparan con las velocidades relativas de adición delanhIdrido maleico, calculadas por Brown. )
Por otra parte, se ha representado (figura 15) la variación de ambosIndices dinámicos (métodos FEMO y LCAO), y se puede observar que eslineal.
En cuanto a las variaciones de dichos Indices con respecto a Ia velocidadde adición, también siguen una marcha paralela (Tabla V).
ENERGIAS DE PARA-LOCALIZACIóN, INDIcES DE VALENCIA LIBRE Y DE VALEN-
CIA UNIDA, Y VELOCIDADES RELATIVAS PARA LAS POSICIONES MESO DE LOS
POLIACENOS.—En la Tabla VI se muestran los valores de F y P'(—y)(LCAO) y V y P (FEMO), asI como Las velocidades relativas para las posi-ciones meso de los poliacenos.
Estos resultados numéricos, que nos dan una indicación de la "reactivi-dad" de los acenos, siguen una marcha paralela a los resultados experimen-tales, indicados por las velocidades relativas de Ia adición con anhIdridomaleico.
A medida que aumenta el ndmero de anillos bencénicos lineales, aumen-tan los valores de F y disminuyen los de V, mientras que los valores deP'(—y) y P disminuyen (fig. 16).
Como podrá observarse, también hay un buen acuerdo para la variaciónentre los valores de los indices estáticos y de los dinámicos calculados porambos métodos (LCAO y FEMO).
ENERGfAS DE PARA-LOCALIZACION Y POTENCIALES POLAROGRAFICOS DE AL-
GUNOS HIDROCARBUROS POLINUCLEARES. 6t)—Los hidrocarburos aromáticospolinucleares en disolución de dioxano al 75%, a la que se adicionó 175 Mde yoduro de tetrabutilamonio, son reducidos en el electrodo de gotas dernercurio. (62) Como muestran los correspondientes polarogramas, se trata deun proceso de adicion de dos electrones en una sola etapa y similar a lareducción de dichos hidrocarburos por los metales alcalinos o de sus amal-gamas con los alcoholes (2'), hecho que, por otra parte, tiene fácil expli-cación.
— 70 —

REACCION DIELS-ALDER POR EL METODO FEMO
Basu y Bhattacharva (63) han admitido que, análogamente a la reducciónquImica, en la polarográfica tiene lugar la adición de dos átomos de hidró-geno en posiciones para—. De esta forma han establecido una relación entreIa energIa de para-localización P(—f3), y los potenciales polarográficos —E,1.Representando los valores de P(—13), calculados por Brown (), versuslos de —E,1, determinados por Wawzonek y Laitinen (62), para un cie rtonilmero de hidrocarburos polinucleares obtienen una buena Ilnea recta (63)•
Puede formularse, sin embargo, ciertos reparos a esta gráfica. En efecto,el valor que Basu y Bhattacharva (63) han utilizado para P(—/3) en la posición1:4 del Naftaleno, no es 4,00, como por error han consignado, sino 3,68.Con este valor, la gráfica P(—13)— (—E11) no resulta tan buena. En cambio,representando P'(—y) frente a —E,1,, Tabla VII, la gráfica es bastantemejor (fig. 17).
Representando las energIas de para-localización, calculadas por el métodoFEMO para los mismos hidrocarburos (Tabla VII), se obtiene una gráficaexcelente (fig. 18).
TABLA VII.—VARIAcION DE LOS POTENCIALES POLAROGRAFICOS DE SEMIONDA
CON LAS ENERGfAS DE PARA-LOCALTZAC1N
Compuesto Posiciones P(FEMO) P'(—7)'—E,12
Naftaleno (I) 1:4 2a—0,173 K 2,40 2,50
Antraceno (II) 9:10 2a—0,354K 2,07 1,94
Fenantreno (III) 1:4 2a—0,153 K 2,47 2,46
1: 2-benzantraceno (IV)1: 2, 5 : 6-dibentraceno (V)
5: 10
5: 10
2a—0,330 K
2a—0,308 K
2,17
2,26
2,03
2,07
Bifenilo (6) 1:4 2a—0,092 K 2,78 2,70
1 R. D. BROWN: I. Chern Soc., 691 (1950).2 H. A. LArnNEN y S. WAWZONEK: J. Chen. Soc. 64, 1765 (1948).
—71—

REACCION DIELS-ALDER POR EL METODO FEMO
2.- OCO
P (FE Mo)
t2.t 1O
2d.i2O
FIG. 18
— 73 —6
9O 2.00 2.50
—-
Dl.
U

CONCLUSIONES
De tcxlo lo que acabamos de exponer en la presente Tesis Doctoral, cabededucir las siguientes conclusiones:
PRIMERA. Se han calculado, mediante el método del electron libre(FEMO), los diagramas moleäulares de los siguientes hidrocarburos poliben-cénicos: 3-4-benzofenantreno, piceno, hexaceno, 1: 2, 6: 7-dibenzopireno,3:4, 9: 10-dibenzopireno, zetreno y pleaideno.
SEGUNDA. Los indices de valencia unida obtenidos para los anterioreshidrocarburos, parámetros que nos permiten hacer consideraciones sobre lareactividad de los diferentes carbonos de una molécula, coinciden muy biencon los indices de valencia libre que, para estos mismos cuerpos, han calcu-lado diferentes autores por el método LCAO.
TERCERA. Las densidades de enlace hakladas por nosotros, indices quenos consienten, hacer consideraciones acerca de la mayor o menor propor-ción del carácter del doble enlace de un enlace y de su reactividad, coincidenmuy bien con los Indices de enlace, calculados por la aproximación LCAO.
CUARTA. Las longitudes de enlace que hemos determinado para losdiferentes enlaces de los hidrocarburos precitados, son ligeramente superio-
res —del orden de 0,02 A— a las obtenidas por el método LCAO, b que estáen consonancia con los resultados hallados con anterioridad por el métodoFEMO.
QUINTA. La predicciOn de las posiciones y enlaces más reactivos delos hidrocarburos estudiados en la presente Tesis, realizada a partir del aná-ljsjs de los indices de valencia unida y de las densidades de enlace, coincidenbastante bien con las que se deducen segdn Ia aproximaciOn LCAO, esdecir, empleando los Indices de valencia libre y de enlace.
SEXTA. Realizando un desarrollo anábogo a! de Brown en el cálculo delas energIas de para-localización, segdn Ia aproximación LCAO, calculamosestas energIas mediante el método FEMO para un conjunto de 28 hidro-
— 74 —

REACCION DIELS-ALDER POR EL METODO FEMO
carburos, y las hemos empleado para predecir teóricamente la posibilidadde Ia adición de dichos cuerpos con el anhIdrido maiéico en las posicicrnesmás favorables estéricamente. -
S1PTIMA. De La comparación de las energIas de para-localización calcu-.ladas para los seis primeros términos de la sene poliacénica con los resulta-dos experimentales, y teniendo presente que ci antraceno es el prmero dela :serie que adiciona anhIdrido maléico y adoptando la velocidad de estareacción como unidad, establecemos como valor umbral teórico en La síi-tesis diénica de estos hidrocarburos, la siguiente energia de para-localización,.obtenida para la posicion 9: 10, P = 2 — 0, 354 K. Como los términos supe-riores al antraceno tienen valores de P inferiores a ,éste, adicionarán anhL-drido maléico con velocidades crecientes, en total acuerdo con los hechosexperimentales. Nuestros resultados concuerdan plenamente con los deBrown, segdn el método LCAO.
OCTAVA. Como para los cuatro hidrocarburos angulares estudiados,nuestras energIas de para-localización son superiores al valor umbral pre-citado, do indica que ninguno es capaz de adicionar anhIdrido maldico, loque coincide con lo hallado por Brown y con los resuitados experimentales.
NOVENA. Comparando las energIas de para-localización caiculadas paralos cinco benzologos del antraceno con los resultados experimentales, y ob-servando que los 1:2, 5 :6- y 1:2, 7: 8-dibenzantracenos adicionan anhIdridomaléico, aunque lo hacen con mucha menor facilidad que el antraceno, defi-nimos como valor umbral de la adición en estos hidrocarburos, ci siguientevalor crItico: P = 2a-0,308 K. Por tanto, los restantes tdrminos verificaránesta reacción con más facilidad. Para los otros hidrocarburos incluidos eneste grupo (1: 2-benzonaftaceno y 4: 5-benzocriseno), no hemos calculadolos valores de P. y los consignados en la Tabla III se han obtenido porinterpolación, indicando que tambidn hay adición. Todos nuestros resultadoscoinciden plenamente con los de Brown.
DECIMA. Para los restantes once hidrocarburos aromáticos, agrupadosbajo el nombre de "Otros hidrocarburos", nuestras energIas de para-localiza-cion, teniendo en cuenta que para P > 2-0,300 K, aproximadamente, nohay reacción, indican que, salvo en una posición en ci Zetreno y otra en elPleiadeno, estos cuerpos no reaccionarán con el anhidrido maléico, aunqueel valor óptimo para la reacción no se da en la misma posición segdn Coulsony colaboradores que por nuestros cálculos. En cuanto al Pleiadeno nadapuede confirmarse por no haber sido sintetizado todavIa.
— 7.5 -

ROSARIO DOMINGO SEBASTIAN
UND1CIMA. La comparación de las energias de para-localización calcu-ladas por nosotros con las de Brown y con las velocidades relativas de laadiciôn del anhIdrido maleico, en los benzdlogos del Antraceno, pone demanifiesto que existe una variación unIvoca entre ambos conjuntos de resul-tados teóricos, P y P'(—y).
Du0DEcIMA.—Comparando las energias de para-localizacidn y las vaiencias unidas calculadas por nosotros, con las energias de para-iocalización ylos Indices de valencia libre, hallados mediante la aproximación LCAO, asIcOmo con las velocidades relativas para la reacción de adición en las posicio-nes mesô' de los poliacenos con el anhIdrido maleico, se pone de manifiestoqué existe una variación paralela de todos estos resultados numéricos.
DECIMOTERCERA. Representando gráficamente las energias de para-ioca-lización calculadas por nosotros para un conjunto de hidrocarburos polinu-cleares frente a los potenciales de dioxano, se muestra que esta vanación esmejor que Ia obtenida por Basu al comparar estos potenciales polarograficoscon las energias de para-localizacidn obtenidas segdn la aproxirnación LCAO.
DECIMOCUARTA. Finaimente puede estabiecerse que ci empleo del méto-do FEMO en el cálculo de las energIas de para-localización y su aplicaciOna la prediccidn de la posibilidad de la reacción de adición de los hidrocar-buros aromáticos policfclicos con el anhIdrido maleico, goza de la mismavaiidez y generalidad que el método LCAO.
— 76 —

BIBLIOGRAF1A
(1) P. P.. LiJWDIN: 1. Phys. Chem., 61, 55 (1957).(2) N. S. BAYLIsS: I. Chem. Phys., 16, 286 (1948).(3) H. KuHr'4: ibid., 16, 840 (1948).(4) S. NIKrrIr.a: J. Chi,n. phys., 47, 614 (1950).(5) S. NIKrrIIta y S. G. KoMoss: ibId., 47, 798 (1950).(6) 5. NIKmr'a y S. 6. KoMoss: ibid., 51, 129 (1954).(7) N. S. BAYuss: Quart. Rev., 6, 319 (1952).(8) K. RUEDENBEItO y C. W. ScHERK: 1. Chetn. Phys., 21, 1565 (1953).(9) C. W. SCHERR: Ibid., 21, 1582 (1953).(10) J. R. PLArF: Ibid., 21, 1597 (1953).(11) K. RUENDENBERO y R. G. PAI: ibid., 19, 1268 (1951).(12) S. GRIFFITH: Trans. Faraday Soc., 49, 345 (1953).(13) H. KUHN: HeIv. Chim. Ada, 31, 1441 (1948).(14) H. KUHN: 1. Chem. Phys., 21. 293 (1956).(15) "Fundamental formulas of Physics". Prentice. Hall. Inc., New York, 1955.
Cap. 4, pág. 145.(16) K. RUEDENBERG: 1. C/,em. Phys., 22, 1878 (1954).(17) C. A. COULSON y H. C. LONGIJET-HIGrnNS: Proc. Roy. Soc., A 191, 39 (1947).(18) S. BASU: I. Chem. PIzys., 22, 1270 (1954).(19) S. BASU: Ibid., 22, 1625 (1954).(20) W. LIcInEN: ibid., 22, 1278 (1954).(21) K. RUEDENBERG: ibid., 22, 1878 (1954);(22) A. A. FROST: ibid., 25, 1150 (1956).(23) N. S. HAM y K. RJEDENBERG: ibid., 25, 1 (1956).(24) C. A. CoYisON, R. DAUDEL y J. M. ROBERTSON: Proc. Roy. Soc., A 207,
307 (1951).J. S. WAUG: y R. W. FESSENDEN: 1. Am. Chem. Soc., 79, 846 (1957).J. A. POPLE: 1. Chem. Phys., 24, 1111 (1956).
(25) C. A. COULSON y R. DAUDEL: Dictionaire des grandeurs théoriques descrip-fives des molecules. Mathematical Institute, Oxford, Centre de ChimieThéorique de France. Paris, 1956, Vol. II.
(26) 0. CHALVET y J. PELTIER: Co,np. Rend., 240, 1709 (1955).(E7) C. A. COULSON y C. M. MosER: I. Chem. Soc., 1341(1953).(28) I. ESTELLS, J. I. FERNÁNDEZ ALONSO y J. Miit: Anal. Fis. y Quim., B 50,
11(1954).(29) a) K. ALDER: Newer Methods of Preparative Organic Shemistry. Interscien-
cie, New York, 1948, pág. 381-511.b) M. L. KLOSTZEL: Organic Reactions. J. Wiley, New York, 1948. Vol. 4,
Cap. I, pág. 1-59.
—77—

ROSARIO DOMINGO SEBASTIAN
c) H. L. HOLMES: Organic Reactions, J. Wiley, New York, 1948, Vol. 4,Cap. II, Pág. 60-173.
d) L. W. BUTZ y A. W. RYTINA: Organic Reactions, J. Wiley, New York,1949, Vol. 3, Cap. , pág. 136-92.
e) E. CLAR: Aromatische Kohienwasserstoffe, 2te. Auflage, Springer Verlag,Berlin, 1952.
e') Esrte Auflage. 1941.j) C. K. ING0LD: Structure and Mechanims in Organic Chemistry. G. Bell
and Sans, London, 1953, pág. 711-721.g) 0. M. BADGER: The Structures Reactions of the Aromatic Co,npounds.
Cambridge University Press, 1954, pág. 334-363.(30) 0. DIELS y K. ALDER: Ann., 460, 98 (1928) y trabajos posteriores.(31) W. E. BACKMAN y M. C. KLOETZEL: J. Am. Chem. Soc., 60, 481 (1938).(32) A. WASSERMANN: Trans. Faraday Soc., 34, 128 (1938).(33) a) K. ALDER y 0. SmIN: Angew. Chem., 47, 837 (1934); 50, 510 (1937);
b) F. BERGMAN y.H. E. E5cHINAZI: 1. Am. Chem. Soc., 65, 1405 (1943).(34) R. B. WOODWARD y H. BR: I. Am. Chem. Soc., 70, 1161 (1948).(35) R. ALVAREZ P.-OsoRio y M. SANZ: Alinidad, 31, 523 (1954).(36) Ref. 29 g., pág. 361.(37) F. BERGMANN y E. BERGMANN: I. A,n. Chem. Soc., 59, 1443 (1937).(38) R. D. BROWN, J. Chein. Soc. (a) 691; (b) 2730 (1950); (c) 1612; (d) 3129.
(1951).(39) S. BASU: 1. .Chem. Phys., 23, 1548 (1955).(40) Véase, a este efecto, S. GEASSTONE, K. J. LAIDLERY. H. EYING: The Theory
of Rate Processes. Mc. Graw-Gill, New-Yoork, 1941.(41) A. WASSERMAN: 1. Chem. Soc., 6121 (1942).(42) M. J. S. DEWAR: 1. Am. Chem. Soc., 74, 3358 (1952).(43) W. R. WAUGHAN y H. S. ANDERSEN: 1. Org. Chem., 21, 672 (1956).(44) M. J. S. DEWAR: Trans. Faraday Soc., 42, 767 (1946).(45) MAMELI, PANCOTrO y CREsTI: Gazzetta, 67, 669 (1937) (tornado de Brown,
Ref. 38 a).Ref. 29 a, pág. 465.Ref. 29 e', pág. 12.
(46) Ref. 29 g., pág. 334.(47) Ref. 29 g., pág. .347.(48) F BERGMANN y E. BERGMANN: 1. Am. Chem. Soc., 62, 1699 (1940).(49) E. CLAR: Ber., 64, 1682 (1931).
Ref.: 29 e', pág. 126.0. DIELS y K. ALDER: Ann., 486, 191 (1931).
(50) Ref. 29 g., pág. 351.• Ref 29 b, pág. 28.
(51) E. CLAi: Ber., 64, 2194 (1931); 65, 503 (1932); 72, 1817 (1939); Ref.. 29 e.(52) R. N. JONES, C. J. GOGEK y R.V. SHARPE: Can. 1. Research, B. 26, 719 (1948).
Ref. 29 a, pág. 485.E. CLAR: Ber;, 65. 853 (1932).Ref. 29 g, pág. 349.
•
(53) E. CLAR Ber.., 65, 519 (1932); Ref. 29 e', pág. 133.Ref. 29 g, pág. 350.
• (54) CooK: J. Chem. Soc., 3273 (1931).Ref. 29 g, pág. 350.
78 —

REACCION DIELS-ALDER POR EL METODO FEMO
(55) E. CL: Ber., 64, 2194 (1931).(56) E .CL y L. Lo?.IBARDI: Ber., 65. 1411 (1931).(57) Aauzov, SALMINA y SHARPSRIMSKAYA: Trans. Butlerc'v. Inst. Chem. Tech.
Kazan, 9, nüm. 2 (1934) (tornado de Brown, ref. 38 a).(58) E. CL: Ber., 69, 1683 (1936).
Ref. 29 g, pág. 349.(59) E. CLAR: Ber., 65, 846 (1932).
Ref. 29 g, pág. 347.(60) C. A. COULSON, C. M. MOSER y M. P. BERNETT: I. Chem. Soc., 3108 (1954).(61) 1. 1. FERNANDEZ-ALONSO y R. DOMINGO: Na!ure, 179, 829 (1957).(62) S. WAWZONEK y LArrINEN: J. Am. Chern. Soc., 64, 2365 (1942).(63) S. BASU y R. BATrACHARVA: 1. Chem. Phys., 25, 596 (1956).
R0SARI0 DOMINGO
Valencia, 1 de junio de 1957.
— 79 —

APENDICE NUMERICO

3:4 - BENZOFENANTRENO

S,Y
'° —
20,5
Y8
+ 1
45,3
75 Y
' —43
2,42
1875
Y4
+ 5
20,1
7187
5 Y
2 —
182,
25=
0 (Y
=3y
)
A,
576
y8 —
928
y6+
456
y4 —
79
y2 +
4 =
0
TA
BL
A I
.—Fu
NcI
oNs
DE
ON
DA
7r-
EL
EC
TR
ON
ICA
S: C
OS
Xy
LOS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LIZ
AD
OS
Cos
. xSi
m.
4) (
1)'I>
(2)
(I)
(3)
4) (
4)41
) (A
)
1S
70,
2182
1802
1821
80,
2182
180,
2182
180,
2182
18
0,93
5605
A,
0,29
0072
0,31
9004
0,30
6851
0,25
5179
0,17
0642
0,80
9583
S,
—0,
1602
15—
0,27
8990
—0,
2915
15—
0,19
3021
—0,
0210
19
0,66
5499
A,
0,04
3981
—0,
1789
41—
0,28
2151
—0,
1966
020,
0204
75
0,58
9438
S,
0,29
7530
0,22
2925
—0,
0347
29—
0,26
3866
—0,
2763
37
0,45
8847
S,
—0,
1500
240,
1711
440,
3070
810,
1106
63—
0,20
5527
0,45
3701
A,
0,35
9903
0,31
2817
—0,
0760
52—
0,38
1827
—0,
2704
18
0,29
4991
A,
0,18
5458
—0,
1355
38—
0,26
5423
—0,
0210
570,
2530
00
0,25
3723
S,
0,25
6272
0,21
3448
—0,
1479
69—
0,28
8534
0,00
1553
cI t1

C U, C z C 11)
tI1 U,
TA
BL
A 1
.—Fu
NC
I0N
E5
DE
ON
DA
-E
LE
CT
RO
NIC
AS:
CO
SY
- L
OS
CO
RR
ESP
ON
DL
EN
TE
S V
EC
TO
RE
S PR
OPI
OS
NO
RM
AL
JZA
DO
S
00
Cos
. xSi
m.
1 (B
)(C
)t'
(11)
t(12
)'t'
(D
)
1S
0,21
8218
0,21
8218
0,21
8218
0,21
8218
0,21
8218
0,93
5605
A1
00
0,08
9461
0,16
7401
0,22
3782
0,80
9583
S1
0,12
2397
0,33
9310
0,35
0852
0,22
8776
0,01
9574
0,66
5499
A1
00
0,30
7793
0,40
9672
0,23
7480
0,58
9438
S1
—0,
3526
11—
0,07
0854
0,11
3660
0,20
4845
0,12
7827
0,45
8847
S.
—0,
0847
590,
2943
800,
2449
92—
0,06
9552
—0,
3088
200,
4537
01A
10
0—
0,07
7903
—0,
0706
900,
0137
590,
2949
91A
10
0—
0,37
5743
—0,
2216
820,
2449
550,
2537
23S
10,
3731
15.
0,28
0896
.—
0,07
9652
.
—0,
3213
16—
0,08
3398

C)
C) C z C C tIl
-o
TA
BL
A I
L—
FuN
cIO
NE
S D
E O
ND
A-E
LE
CT
RóN
ICA
S: a
B.9
00 -4
Orb
.Si
m.
B1
B2
B3
B4
B5
IS,
0,04
7619
0,04
7619
0,04
7619
0,04
7619
0,04
7619
2.
A0,
0642
10—
0,10
2327
0,06
2112
0
3S,
0,13
1961
0,18
2449
0,09
0342
0,00
4326
0,05
6848
4A
0,17
0049
-.—
0,07
9749
0,09
0367
0
5S.
0,04
2038
0,15
3083
0,09
1989
0,20
5874
0,13
1523
6S.
0,10
1954
0,14
7876
0,09
5456
0,10
0530
0,04
2356
7A
0,00
7642
—0,
1576
840,
0879
230
8-A
0,15
4640
—0,
0739
720,
0957
840
9S
0,10
3248
0,17
6282
0,08
9226
0,00
7506
0,14
8482

TA
BL
A 1
11.—
FuN
cI0N
Es
DE
ON
DA
21-
EL
EC
TR
ON
ICA
S: tg
Orb
.Si
m.
B1
B2
B3
B4
B5
1S,
00
00
0I
2A
7—
1,13
6371
—0,
7559
530,
4902
07—
3S
1,23
3421
0,76
4662
1,19
3668
3,20
8504
1,67
1728
4A
70,
1149
74—
1,03
1146
0,77
6104
—5
S—
0,04
2607
—5,
4308
89—
0,56
6745
3,40
5788
—0,
2404
646
S—
4,48
0839
0,84
0473
2,60
6692
—0,
2326
10—
2,21
2678
7A
70,
7275
28—
—0,
2856
07—
1,54
5296
—8
A7
1,46
5174
—12
,883
291
—0,
7722
15—
x9
S7
—0,
0060
25—
1,11
0931
0,26
7870
0,28
1557
0,25
8003
0 I

7
PICENO
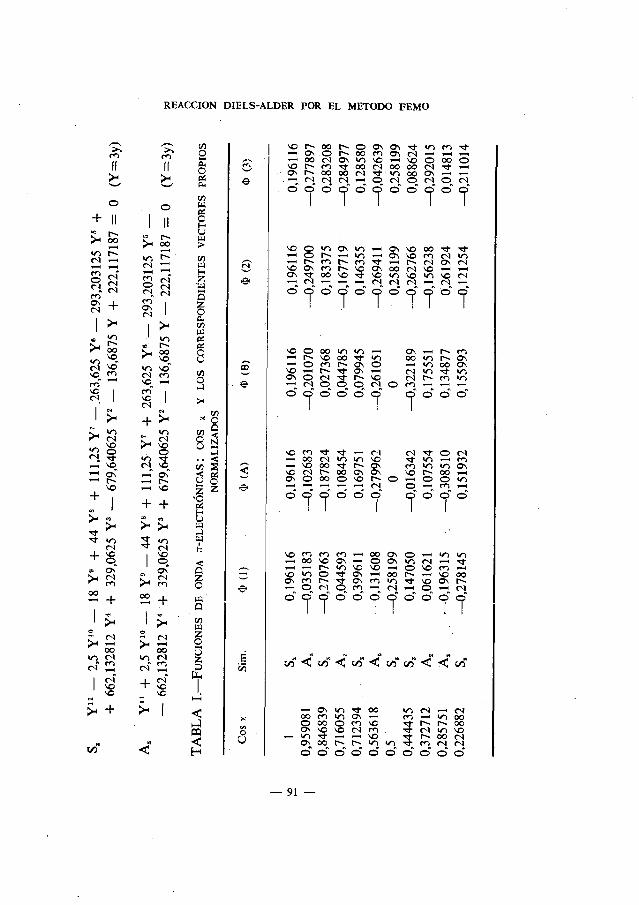
S,Y
' —2,
5yi
o —
18Y
9 +
44
Y8
+ 1
11,2
5 Y
7 —
.263
,625
Y6
—29
3,20
3125
Y +
+ 6
62,1
3281
2 Y
4 +
329
,062
5 Y
3 —
679,
6406
25Y
2 —
136,
6875
Y +
222
,117
187
=0
(Y=
3y)
71 C z 51 11 C T1 C
A,
Y"
+ 2
,5 Y
'° —
18Y
9 —
44Y
8 +
111
,25
Y7
+ 2
63,6
25 Y
629
3,20
3125
Y5
——
662
,132
812
Y H
- 32
9,06
25 Y
3 +
679
,640
625
Y2
—13
6,68
75Y
—22
2,1
1718
7 =
0(Y
=3y
)
TA
BL
A 1
.—Fu
NcI
0NE
S D
E O
ND
A T
-EL
EC
TR
ON
ICA
S:C
OS
Y L
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LJZ
AD
OS
'.0
Cos
xSi
m.
1' (
1)'1
(A)
1 (B
)1
(2)
'1(3
)
IS,
0,19
6116
0,19
6116
0,19
6116
0,19
6116
0,19
6116
0,95
9081
A,
—0,
0351
83—
0,10
2683
—0,
2010
70—
0,24
9700
—0,
2778
970,
8468
39S,
,—
0,27
0763
—0,
1878
240,
0273
680,
1833
750,
2832
080,
7160
55A
0,04
4593
0,10
8454
0,04
4785
—0,
1677
19—
0,28
4977
0,71
2394
.
S,0,
3996
110,
1697
510,
0799
450,
1463
550,
1285
800,
5636
18A
,—
0,13
1608
—0,
2799
62—
0,26
1051
—0,
2694
11—
0,04
2639
0,5
S,—
0,25
8199
00
0,25
8199
0,25
8199
0,44
4435
S,0,
1470
50—
0,01
6342
—0,
3221
89—
0,26
2766
0,08
8624
0,37
2712
A,
0,06
1621
0,10
7554
0,17
5551
—0,
1562
38—
0,29
2015
0,28
5751
A,
—0,
1963
15—
0,30
8510
0,13
4877
0,26
1924
0,01
4813
0,22
6882
S,—
0,27
8145
0,15
1932
0,15
5993
—0,
1212
54—
0,21
1014

Cos
xS
im.
'1(4
)4
(5)
b (6
)(7
)c1
(C)
1 (D
)
-
1S
,0,
1961
160,
1961
160,
1961
160,
1961
160,
1961
160,
1961
16
0,95
9081
A,
—0,
2833
52—
0,26
5617
—0,
2261
43—
0,18
2984
—0,
1267
68—
0,05
9180
0,84
6839
S.0,
2962
900,
2186
140,
0739
71—
0,05
8060
—0,
1723
01—
0,23
3762
0,71
6055
A,
—0,
2404
00—
0,05
9302
0,15
5472
0,34
8497
0,34
3614
0,14
3597
0,71
2394
S,0,
0368
45—
0,07
6085
—0,
1452
49—
0,31
4284
—0,
3025
39—
0,11
6769
0,56
3618
A0,
2213
470,
2921
490,
1079
7401
5147
00,
0627
68—
0,08
0716
0,5
S.
0—
0,25
8199
—0,
2581
99—
0,12
9099
0,12
9099
0,25
8199
0,44
4435
S.0,
3415
410,
2149
61—
0,15
0468
—0,
0933
930,
0674
540,
1533
5 1
0,37
2712
A,
—0,
0614
370,
2462
190,
2449
740,
1478
55—
0,35
5189
—0,
1169
12
0,28
5751
A,
—0,
2534
58—
0,15
9665
0,16
2209
0,16
3843
—--
0,06
8572
—0,
2030
32
0,22
6882
S0,
0255
040,
2225
870,
0754
98—
0,32
7192
—0,
2239
670,
2255
64
TA
BL
A I
.—Fu
NcI
oNE
s D
E O
ND
Ar-
EL
EC
TR
6NIC
AS:
CO
Sx
yN
OR
MA
LIZ
AD
OS
LO
S C
OR
RE
SPO
ND
IEN
TE
S V
EC
TO
RE
S PR
OPI
OS
0 (I) I

TA
BL
A 1
1.—
FuN
cloN
Es
DE
ON
DA
-E
LE
CT
RO
NIC
AS:
a13
Orb
.Sl
imB
1B
2B
3B
4B
5B
6B
,
1S6
0,03
8461
0,03
8461
0,03
8461
0,03
8461
0,03
8461
0,03
8461
0,03
8461
2A
,0,
0605
300,
1418
280,
0807
590,
0542
620,
0646
410,
0298
080,
1711
853
S0,
0793
930,
158.
146
0,09
1476
0,00
9870
0,05
6973
0,05
5008
0,05
9176
4:A
60,
0140
020,
0139
80.
0,08
3928
0,03
3263
0,13
9611
0,02
0685
:0,
1452
405
S,0,
1865
090,
0322
260,
0226
210,
0894
060,
1113
340,
1435
390,
0159
256
A,
0,05
9669
0,09
4005
0,09
0060
0,16
3526
0,02
3693
0,08
7084
0,01
4930
7S,
0,08
8889
0,08
8889
00,
0666
630,
0888
890,
0888
898
5,.
0,02
9941
0,12
3830
0,12
1622
0,19
3872
0,02
3517
0,03
2413
0,03
2562
9A
0,01
2106
0,03
2879
0,08
7883
0,06
8255
0,12
6437
0,04
0192
0,04
3579
10A
,0,
1079
050,
1493
350,
0725
200,
0355
600,
0413
440,
1095
360,
1154
2711
S,0,
1261
160,
0386
520,
0502
040,
0260
300,
1306
910,
0615
820,
0829
400

TA
BL
A 1
11.—
FuN
cloN
Es
DE
ON
DA
ir-E
LE
CT
RO
NIC
AS:
tg
0 0 z 0 (I,
tTl z
Orb
.Si
m.
B1
283
B4
B5
8,87
1S
00
00
00
02
A,
—6,920509
—3,528601
0,380359
—0,
5849
341,738530
—2,740766
3S
0,287970
1,866243
0,956062
—3,489724
—0,958680
0,081518
0
4A,
—2,458339
0,434234
4,781574
—3,947402
0,427126
—0,056173
5S,
0,40
9820
0,34
4040
—1,
7051
573,
6040
670,
4651
483,
0866
270
6A
,—
1,56
3623
—0,
4465
080,
2348
961,
1830
342,
2390
74—
3,51
6623
7S,
0,57
7347
—0,
5773
47—1,732042
0,57
7347
0
8S
0,620186
—21,512403
1,277522
—0,025210
—2,041713
0,61
5089
0
9A,
—1,479291
—1,
3573
03—
0,67
0548
—1,
1021
630,
0469
401,393046
10
A,
—1,341577
0,75
4326
1,358199
—0,977092
—2,791252
—1,287329
11
S,
0,793816
—0,821264
—0,
1153
09—0,263985
1,26
7054
—0,458643
0

HEXACENO

432
y —
648
y6 —
180
y5 +
524
y —
22 y
-12
2y2
+ 9
y +
7 =
0
l44y
—16
8y—
68y4
+ 9
6y3
+ lO
y2—
l2y—
l0
432
yT +
648
y6
—18
0y5
—52
4y4
—22
y3
+ 1
22 y
2 +
9y
—7
=0
l44y
6 +
168
y5
—68
y4 —
96y3
+ b
y2 +
12
y —
10
t•i1 r) r) tn U)
tn 0 'TI
tnt 0
sxsy
S1A
AS7
AA
7
TA
BL
A 1
.—Fu
NcI
ON
ES
DE
ON
DA
-E
LE
CT
RO
N!C
AS:
CO
S x
YL
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
S
NO
RM
AL
IZA
DO
S
'.0
Cos
xSi
m.
1 (1
)1
(2)
1 (A
)1
(16)
11' (
B)
't'(1
5)D
(C
)
1SS
0,17
9605
0,17
9605
0,17
9605
0,17
9605
0,17
9605
0,17
9605
0,17
9605
0,97
4496
SA0,
2412
370,
2542
030,
2159
650,
1741
700,
1234
910,
0633
63—
0,90
0045
0,20
3905
0,25
4852
0,11
2195
—0,
0131
59—
0,13
5881
0,21
7858
0,25
6283
0,78
2971
S1A
10,
1448
920,
2560
20—
0,02
9127
—0,
1841
82—
0,25
9291
—0,
1655
79—
0,65
0244
AS
0,14
8280
0,06
4456
0,12
8381
0,23
0537
0,17
1430
0,27
5306
0,18
6602
0,63
4786
S1S
0,06
9363
0,25
7309
—0,
1692
47—
0,22
2424
—0,
1131
350,
1201
090,
2656
220,
5991
07A
1A7
0,25
4342
0,11
5704
0,18
9052
0,27
4498
0,13
9855
0,11
6721
—
0,51
0651
AS7
0,30
0845
0,14
8837
0,15
8416
0,10
0258
—0,
0560
23—
0,24
2105
0,19
1239
0,47
7215
SA0,
0115
070,
2525
07—
0,26
3489
—0,
1022
270,
1659
210,
1738
46—
0,38
6555
A2A
70,
3016
570,
1701
290,
0630
85—
0,16
5414
—0,
1909
69—
0,24
7014
—
0,36
2006
SS—
0,05
1058
.0,
1850
02—
0.22
1969
0,03
1965
0,24
5112
—0,
0108
810,
2529
900,
2359
43A
1S0,
2386
530,
1621
41—
0,04
9525
—0,
3232
32—
0,10
3004
0,14
7318
0,17
2531
0,08
2354
A2A
70,
1463
410,
1256
46—
0,10
1542
—0,
2729
710,
0565
820,
3435
32—

TA
BL
A 1
1.—
FuN
cIO
NE
S D
E O
ND
A 7
r-E
LE
CT
RO
NIC
AS:
Orb
ital
Sim
etrI
aB
1B
2B
5B
4B
5B
6
1S1
S,0,
0322
580,
0322
580,
0322
580,
0322
580,
0322
580,
0322
582 3 4
S1A
S1S,
SA
0,06
5454
0,06
8366
0,07
3524
0,04
7243
0,01
3250
0,00
0952
0,07
2780
0,08
0461
0,06
8149
0,0
1544
7
0,01
9435
0,07
5416
0,07
9707
0,06
6548
0,07
0853
—
0,07
9136
—
5 6A
S0,
0237
570,
0809
990,
0942
470,
0350
440,
0539
500,
0507
900,
1680
500,
0156
590,
0758
930,
0744
950,
1991
12
0,08
6317
0,
7A
A0,
0667
880,
1783
050,
0762
930,
0975
790,
0212
52—
8 9 10
AS
SA AA
0,09
0538
0,08
6324
0,09
4365
0,10
2567
0,09
3996
0,01
2975
0,02
5604
0,07
0150
0,04
6333
0,01
2828
0,03
7273
0,11
8899
0,06
4798
0,03
9135
0,07
1735
0,14
9473
— —Z
11S1
S,0,
0502
580,
0723
500,
0637
410,
0882
230,
0715
090,
0939
8512
A1S
0,06
8816
0,00
6420
0,10
5236
0,02
7772
0,04
1801
0,07
7918
13A
1A0,
0344
070,
0224
720,
0808
060,
0069
780,
1188
20—
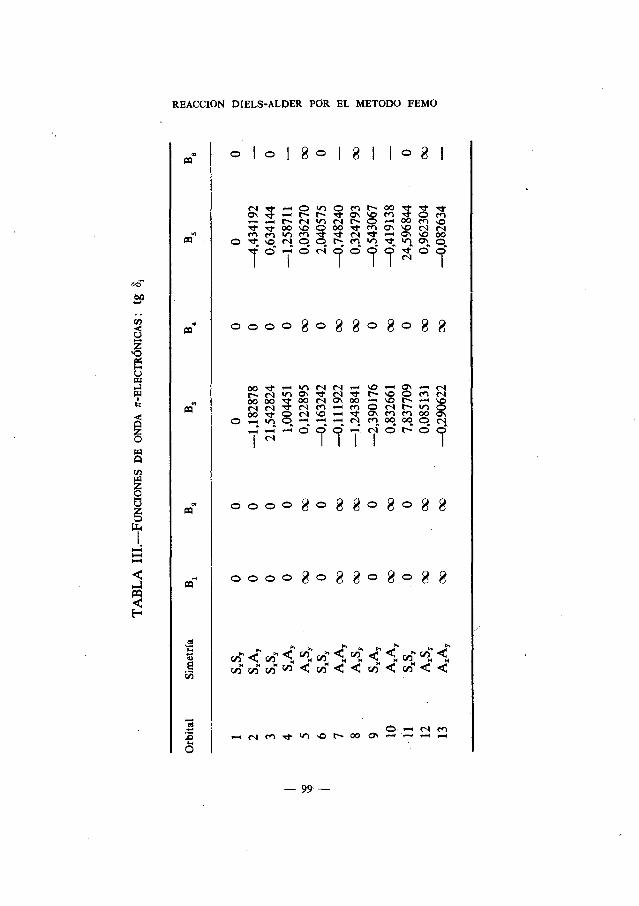
TA
BL
A I
II.—
Fu1c
IoN
Es
DE
ON
DA
ir-E
LE
cTR
ON
ICA
S: tg
6
23 C)
C)
U)
23 T1 - 0 0
Orb
ital
Sim
etri
aB
B2
B3
B4
B5
B4
IS4
S70
U0
00
02
S1A
00
—1,
1828
780
—4,
4341
92—
30
021
,542
824
00,
6341
440
4SA
00
1,00
4451
0—
1,25
8711
—
5A
1So
0,12
2895
X0,
0362
70oe
6SS
00
—0,
1632
420
2,04
0575
0
7A
1A—
0,11
1922
—0,
7482
40—
8A
S—
1,24
3841
cv0,
3247
93cv
9S1
A0
0—
2,39
0176
0—
0,54
3067
—
10A
1Acv
cv0,
8326
61cv
—0,
4191
38—
11S1
S0
07,
8377
090
24,5
9684
40
12A
3Scv
cv0,
0851
31cv
0,96
2304
cv
13A
3Acv
cv—
0,29
0622
cv—
0,08
2634
—

1:2, 6:7 - DIBENZOPIRENO

432
yT—
504
y6 —
420
y5 +
564
y4
+ 6
8 y3
—15
2y2
+ 2
y +
10
=0
432
y7+
504
y6
—42
0 y'
—56
4y4
+68
y3+
152
y2+
2y
—10
= 0
72 y
5+
60
y4—
38 y
3—
26 y
2+
5y
+ 2
= 0
TA
BL
A 1
.—Fu
Ncl
oNE
s D
EO
ND
A 7
r-E
LEC
TR
ON
ICA
S:
CO
S x
Y L
OS
CO
RR
ESP
ON
DL
EN
TE
S V
EC
TO
RE
S PR
OPI
OS
NO
RM
AL
IZA
DO
S
rn (I)
rn 0 0 '71
711 0
sxsy AS
A1A
S1A
72 y
—60
y4 —
38y3
+ 2
6 y2
+ S
y —
2=
0
0
Cos
xSi
m.
4) (
1)(1
' (A
)4)
(B
)(I
) (2
)(1
) (3
)'I)
(14
)'1
) (J
)
1S,
S.0,
1856
950,
1856
950,
1856
950,
1856
950,
1856
950,
1856
950,
1856
95
0,93
6861
SA0,
0471
120,
0882
730,
2009
920,
2756
370,
3154
760
0
0,86
8833
AS
0,32
6481
0,19
1545
0,06
6560
0,04
8502
0,01
7721
0,37
5769
0,10
6222
0,85
4806
S1S.
0,20
9155
0,11
2893
—0,
0639
76—
0,21
2979
0,30
0134
0,24
4681
0,14
4325
0,59
7469
SA0,
1869
310,
2233
710,
2134
41—
0,05
4239
—0,
2782
530
0
0,58
6988
0,23
5984
—0,
1249
86—
0,12
7586
0,02
7897
0,16
0337
0,40
2025
—0,
3284
93
0,56
8489
AA
0,19
3694
0,22
0226
0,18
1894
0,27
1882
0,12
7230
00
0,50
5476
AS
0,10
6917
—0,
1034
29—
0,18
1557
—0,
3534
46—
0,17
5760
0,21
1517
—0,
0822
03
0,37
7472
0,01
0947
—0,
0207
360,
2787
720,
0576
500,
2352
490,
0290
01—
0,31
3202
0,36
9045
A1A
0,26
5488
0,19
5954
—0,
0485
400,
2982
35—
0,17
1584
00
0,27
8290
AS
0,07
975
1—
0,24
2187
—0,
0179
630,
2092
280,
1344
150,
2865
75—
0,26
3983
0,23
6538
S1A
0,30
2458
0,14
3086
—0,
2009
22—
0,08
4741
0,16
0834
00

TA
BL
A 1
1.—
FuN
cIO
NE
s D
E O
ND
A 2
r-E
LE
CT
RO
NIC
AS:
Orb
ital
Sim
etrI
aB
1B
3B
3B
4B
5B
6
IS1
S0,
0344
830,
0344
830,
0344
830,
0344
830,
0344
830,
0344
832
S1A
0,01
8144
0,12
2215
0,06
3716
00,
0417
150,
1027
703
AS
0,14
1202
0,07
7365
0,05
1472
0,17
2042
0,06
7551
0,00
4788
S1S1
0,05
9869
0,10
8375
0,02
1238
0,02
2460
0,00
4413
0,09
7132
5S1
A0,
0543
400,
0598
420,
0775
920
0,05
7036
0,09
6934
6S1
S0,
1616
230,
0201
07—
0,11
4925
0,13
5991
0,02
0514
0,03
2399
°7
A1A
0,05
5431
0,05
3248
0,07
1657
00,
1533
470,
0750
278
A1S
10,
0447
380,
0331
440,
0119
010,
0273
290,
1333
110,
1249
349
S1S,
0,00
0840
0,09
6174
0,10
8928
0,14
2428
0,1
1283
50,
0803
5310
A1S
0,08
1597
0,05
5304
0,04
4452
00,
0074
680,
0933
2211
AS
0,08
2122
0,06
1366
0,10
0543
0,19
3116
0,00
0894
0,05
0068
12S1
A0,
0969
030,
0788
560,
0216
870
0,06
5295
0,04
1839

TA
BL
A 1
11.—
FuN
cloN
Es
DE
ON
DA
r-E
LE
CT
RO
NIC
AS:
tg8
(I)
Orb
ital
Sim
etri
aB
1B
2B
3B
4B
5B
6
1S1
S0
00
00
02
S1A
—2,
6788
951,
4231
252,
6790
040
00
3A
1S0,
5698
47—
4,05
7575
0,63
4766
4S
1S7
0,60
7093
5,04
7589
—0,
8163
040
00
5S1
A—
0,74
5055
—0,
5599
780,
7450
550
00
61,
3792
40—
0,48
4976
—2,
5213
220
00
7A
1A—
0,69
1013
—0,
7806
690,
6910
120
00
8A
1S1,
7069
83—
0,07
4408
—0,
3352
899
SS
2,45
0292
0,48
7380
—15
,884
621
00
010
A1A
,—
0,39
7074
4,74
0636
0,39
7074
00
011
A1S
73,
4514
10—
13,7
4727
4—
0,84
5088
12S1
A—
0,24
3447
0,97
6390
0,24
3446
00
0
'TI 0

3:4, 9:10 - DIBENZOPIRENO

S1Y
'2 +
4 Y
" —
14,5
Y1°
—70
.25
Y9
+ 5
3,62
5 Y
8 +
425
,062
5 Y
7 +
29,
5781
25 Y
6 —
1108
,546
87—
445,
5Y
+ 1
237,
1484
37 Y
3 +
639
,773
475
Y2
—47
8,40
625
Y —
239,
2031
25=
0(Y
=3y
).
A1
Y'2
—4
Y"
—14
,5Y
'° +
70,
25 Y
° +
53,
625
Y8
—42
5,06
25Y
7 +
29,
5781
25 Y
6 +
110
8,54
687
Y5
—44
5,5
Y4
—12
37,1
4843
7Y
3 +
639
,773
4375
Y2
+ 4
78,4
0625
Y —
239,
2031
25=
0(Y
=3y
).
TA
BL
A 1
.—Fu
NcI
0NE
S D
E O
ND
A 2
--E
LE
CT
RO
NIC
AS:
CO
S x
y LO
SC
OR
RE
SPO
ND
IEN
TE
S V
EC
TO
RE
S PR
OPI
OS
NO
RM
AL
IZA
DO
S
Los
xSi
m.
'I> (
2)(A
)1
(B)
1(3
)1'
(4)
(5)
1S1
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,95
2717
A1
0,03
4132
0,09
9159
0,20
2358
0,25
8281
0,28
9779
0,29
3873
0.86
7871
S—
0,12
2240
—0,
0899
480,
0757
560,
2062
640,
2822
600.
2836
420,
7831
33S1
0,44
1987
0,25
0280
0,08
8949
0,03
3159
—0,
0370
12—
0,09
1130
0,68
8573
A.
—0,
0519
71—
0,12
3542
—0,
0714
440,
1494
140,
2772
090,
2323
430,
5632
54A
10,
1300
750,
2766
050,
2576
090,
2717
380,
0485
06—
0,21
7096
0,54
4998
S1—
0,00
9277
—0,
0008
530,
0472
61—
0,23
7739
—0,
3064
21—
0,09
6263
0.45
8087
S10,
3335
51—
0,02
7961
—0,
2626
10—
0,28
8004
—0,
0012
520,
2868
570.
4078
02S.
—0,
2068
130,
0381
36—
0,15
7400
—0,
1327
990,
0490
890,
1728
360,
3932
57A
1—
0,05
1567
—0,
0921
240,
1601
710,
1464
970,
2753
930,
0701
040,
2868
25A
1—
0,19
8641
—0,
3125
920,
1217
330,
2681
870.
0321
13—
0,24
9766
0,15
6069
S10,
1954
32—
0,13
4430
0,19
0611
0,09
2233
0,21
9400
—0,
0237
49
0 0 11 C,)

TA
BL
A 1
.—Fu
Ncl
oNE
s D
E O
ND
A n
--E
LE
CT
RO
NIC
AS:
CO
S X
YL
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LIZ
AD
OS
0
S10,
1856
950,
1856
950,
1856
950,
1856
950,
1856
950,
1856
95A
10,
2701
770,
2209
320,
1589
200,
0818
800,
0281
820,
0469
23°
S10,
2100
770,
0810
02—
0,07
4928
—0,
2110
97—
0,28
6926
—0,
1877
71S1
-—-0
,105
723
—0,
0744
60—
0,15
8161
—0,
1732
64—
0,30
5975
0,05
7070
A1
0,04
2761
—0,
1734
55—
0,32
9625
—0,
2804
88—
0,11
7993
—0,
1317
90S
A1.
—0,
2930
66—
0,11
3045
—0,
1555
63—
0,06
2197
—0,
0292
480,
0797
13
S,,
0,20
1511
0,31
5901
0,26
7709
—0,
0241
01—
0,26
7822
—0,
0393
03
510,
2640
64—
0,04
4929
—0,
0631
98—
0,01
2971
0,15
4739
—0,
1093
66
S10,
0918
77—
0,09
7901
—0,
0542
490,
0536
55—
0,29
0977
0,41
0868
Z
A1
—0,
2202
56—
0,24
3338
0,09
3343
0,31
6754
0,17
7303
0,10
3052
A1
—0,
1753
910,
1491
530,
1820
00—
0,04
4748
—0,
0284
36—
0,19
2069
S—
0,22
6813
—0,
0470
480,
3953
960,
1704
66—
0,24
7820
—0,
0677
63
Cos
xSi
m.
1 (6
)t'
(C)
4' (
7)D
(D
)1
(8)
1 (K
)
I
0,95
2717
0,86
787
1
0,78
3133
0,68
8573
0,56
3254
0,54
4998
0,45
8087
0,40
7802
0,39
3257
0,28
6825
0,15
6069

TA
BL
A 1
1.—
FuN
cloN
Es
DE
ON
DA
lr-E
LE
CFR
6NIC
AS:
a12
c-i
c-i
(I)
:8- 0 ri '-1 0 0 I1 t1 C
Orb
.Si
m.
B1
B3
B3
B4
LB
6B
7B
8B
8
1S1
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
2A
10,
0492
780,
0931
320,
0335
950,
0343
220,
0171
720,
0776
110,
1358
920,
0495
270,087402
3S1
0,01
6000
0,03
7752
0,08
8150
0,05
6858
0,03
7575
0,09
2015
0,10
3963
0,00
6682
0,08
5723
4S1
0,219111
0,003653
0,105007
0,112556
0,126105
0,031326
0,092276
0,061624
0,011357
SA1
0,017346
0,111542
0,089410
0,019414
0,085831
0,114099
0,015616
0,03
4465
0,080092
6A1
0,077480
0,029098
0,003917
0,084987
0,023153
0,025147
0,091692
0,163965
0,089852
7S1
0,00
0111
0,00
2000
-0,
0928
520,
0021
470,
0015
550,112779
0,00
4454
0,12
2009
0,10
1019
85
0,15
2606
0,01
6406
0,03
2843
0,01
2581
0,01
3706
0,00
4317
0,07
9774
0,04
7155
0,10
4560
9S
0,06
0764
0,23
9824
0,12
0283
0,18
8902
0,18
4372
0,00
9831
0,03
7324
0,02
6138
0,03
0420
10A1
0,008765
0,035006
0,103623
0,031436
0,100883
0,101487
0,054117
0,136659
0,077568
11A
10,
1106
550,
1034
540,
0022
670,
1091
400,
0370
060,
0433
660,
1464
060,
0290
390,
0741
1312
S0,
0660
750,
0079
440,
1062
470,
0203
150,
0381
880,
1684
610,
0639
590,
0423
770,
0515
83

TA
BL
A 1
11.—
FUN
cIO
NE
s D
E O
ND
A ir
-EL
EC
TR
ON
ICA
S: tg
Orb
. Sun
.B
1B
2B
3B
4B
B9
B7
B9
B9
01
S10
00
00
00
00
2A
1cv
cv—
3,81
9242
—2,
6073
57 —
1,43
9786
—3,
5806
94—
0,45
7683
0,44
4239
3S1
00
00,
7827
05—
0,25
6355
3,92
3048
3,44
2274
—0,
4053
550,
9708
09
4S1
00
0—
5,79
2941
6,14
1522
0,50
2284
0,68
7839
2,60
5506
0,12
6782
5A
.cv
cvcv
—0,
3431
50—
1,98
5358
0,22
3884
0,15
2069
—2,
3984
356,
5432
33
6A
.cv
cvcv
3,51
7862
1,62
5970
—0,
1977
88—
0,44
5452
1,21
2751
0,21
4843
7S1
00
00,
6241
33—
0,08
1356
—0,
7573
8566
,732
215
—7,
3221
91—
1,21
9728
8S1
00
00,
2277
220,
3819
14 —
0,28
4437
—10
,050
504
0,32
2871
0,70
6749
9S
00
00,
3449
720,
3036
05—
1,52
9848
4,96
6909
—0,
2345
781,
6136
42
10A
.cv
cvcv
1,4.
0001
5—
2,91
5370
3,26
3094
2,31
8727
2,08
0093
—0,
7738
93
11A
1cv
cvcv
—1,
3994
750,
0562
08—
0,55
6061
0,70
5916
—-0
,979
581
1,18
7107
12S1
00
0—
1,85
0436
2,70
4840
0,27
8471
1,59
3519
0,40
7895
—0,
0519
99

ZETRENO

ri C z rn C,) r rn rn - C C '21 rn C
S Y
'2 +
Y"
—22
Y'°
—19
,5Y
9 +
178
,25
Y8
+ 1
37,2
5 Y
7 —
664,
6875
Y6
—43
7,06
25Y
5 +
+ 1
162,
2656
25 Y
4 +
659
,390
625
Y3
—85
0,5
Y2
—38
1,58
5937
Y +
153
,773
437
=0
(Y =
3y).
A2
Y'2
—Y
"—
22Y
'° +
19,
5 Y
9 +
178
,25
Y8—
137
,25
Y7
—66
4,68
76
+43
7,06
25 Y
5 +
+ 1
162,
2656
25 Y
465
9,39
0625
Y3
—85
0,5
Y2
+ 3
81,5
8593
7 Y
+ 1
53,7
7343
7 =
0.(Y
=3y
).
TA
BLA
1.—
FUN
CIO
NE
S D
E O
ND
A r
-EL
EC
TR
ON
ICA
S: C
OS
Y L
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LIZ
AD
OS
U'
Cos
. xSi
m.
4) (
1)4)
(A
)4)
(2)
4) (
3)4)
(4)
4) (
B)
0.S
0;18
5695
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,95
7297
A2
0,07
8552
0,16
4371
0,20
9741
0,23
7197
0,24
4397
0,23
0722
0,80
7836
A2
—0,
1716
06—
0,18
1095
—0,
2686
13—
0,25
2898
—0,
1399
870,
0267
280,
7883
14S8
—0,
0064
250,
1185
500,
2878
330,
3358
750,
2421
100,
0453
220,
7715
54S.
0,28
6858
0,15
3765
0,04
3231
—0,
0870
61—
0,17
7572
—0,
1869
490,
5866
90A
,0,
2978
570,
2894
810,
0582
40—
0,22
1143
—0,
3177
25—
0,15
1679
0,49
5250
A2
—0,
2671
47—
0,03
4983
0,10
8473
0,14
2426
0,03
2600
—0,
1101
350,
4299
13S
——
0,03
3933
0,06
2214
0,29
8245
0,19
4225
—0,
1312
45—
0,30
7073
0,42
3037
A2
0,00
7537
—0,
0248
580,
2500
230,
2363
96—
0,05
0013
—0,
2787
120,
3849
85S,
0,06
8985
0,31
4404
0,09
4779
—0,
2414
27—
0,28
0670
0,02
5319
0,32
2453
S.—
0,28
4951
—0,
0225
47—
0,00
1007
0,02
1898
0,01
5129
—0,
0121
410,
0937
75A
,0,
3167
18—
0,11
2317
—0,
2982
630,
0563
780,
3088
370,
0015
44
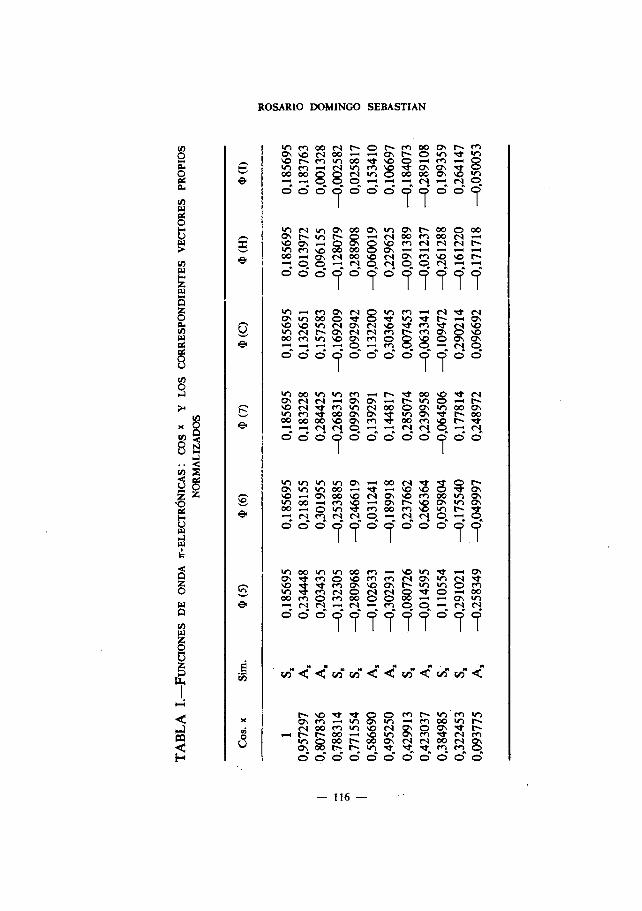
TA
BL
A I
.—Fu
NcI
os D
E O
ND
A 7
r-E
LE
CT
RóN
ICA
S: C
OS
Y L
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LIZ
AD
OS
1S,
0,18
5695
0,18
5695
0,95
7297
A,
0,23
4448
0,21
8155
0,80
7836
A1
0,20
3435
0,30
1955
0,78
8314
S,
—0,
1323
05—
0,25
3885
0,77
1554
S,
—0,
2809
68—
0,24
6619
0,58
6690
A,
—0,
1026
330,
03 1
241
0,49
5250
A,
—0,
3029
31—
0,18
9918
0,42
9913
S,
—0,
0807
260,
2376
62
0,42
3037
A,
—0,
0145
950,
2663
64
0,38
4985
S,
0,11
0554
0,05
9804
0,32
2453
S,
—0,
2910
21—
0,17
5540
0,09
3775
A,
—0,
2583
49—
0,04
9997
0,18
5695
0,18
5695
0,18
5695
0,18
5695
0,18
3228
0,13
2651
0,01
3972
0,18
3763
°.0,
2844
250,
1575
830,
0961
550,
0013
28—
0,26
8315
—0,
1692
09—
0,12
8079
—0,
0025
820,
0995
930,
0929
420,
2 88
908
0,02
5817
0,13
9291
0,13
2200
—0,
0600
190,
1534
10
0,14
4817
0,30
3645
0,22
9625
0,10
6697
0,28
5074
0,00
7453
—0,
0913
89—
0,18
4073
0,23
9958
—0,
0633
41—
0,03
1237
—0,
2891
08Z
—0,
0645
06—
0,10
9472
—0,
2612
880,
1993
59
0,17
7814
0,29
0214
—0,
1612
200,
2641
47
0,24
8972
0,09
6692
—0,
1717
18—
0,05
0053
Cos
. xS
im.
(5)
11'
(6)
11' (
7)1
(C)
1 (H
)1'
(I)

TA
BL
A 1
1.—
FUN
cIO
NE
s D
E O
ND
A ir
-EL
EC
TR
óNIC
AS:
a
(l -4 C 0
—3
Orb
. .Si
m.
B1
B2
B3
.B4
B5
B6
B7
B,
1S,
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
0,03
4483
2A
,0,
1013
000,
0598
550,
0697
060,
0353
630,
0561
620,
0554
390,
1702
140,
0091
333
A,
0,03
4640
0,07
5862
0,00
1895
0,09
5524
0,07
0511
0,09
5895
0,02
7625
0,09
6230
4S
,0,
0401
310,
1138
580,
0059
310,
0384
170,
0738
310,
0767
210,
0287
050,
0183
465
S,
0,09
3566
0,37
695
0,10
6410
0,04
4933
0,01
3842
0,01
0559
0,12
5206
0,09
4231
6A
,0,
1087
910,
1027
890,
1 12
594
0,08
4210
0,02
6250
0,02
3288
0,04
6336
0,01
3828
7A
,0,
0839
160,
0221
950,
0465
780,
0216
070,
0947
280,
0922
310,
1005
210,
2089
258
S,
0,00
8384
0,09
4304
0,09
7618
0,05
8398
0,04
2655
0,09
6559
0,01
1032
0,01
1682
9A
,,0,
0010
890,
0894
120,
1160
150,
1180
510,
1345
330,
0970
780,
0042
630,
0035
6110
S,
0,10
2033
0,09
9660
0,04
2846
0,10
6051
0,08
0456
0,01
2571
0,06
8364
0,09
8587
11S
.0,
0865
620,
0005
520,
0801
220,
0826
640,
1 16
693
0,09
2142
0,15
6681
0,03
9309
12A
,0,
1206
570,
0961
320,
0021
350,
0141
910,
0128
750,
0674
080,
0423
220,
0650
74

TA
BL
A I
IL—
FuN
cLO
NE
s D
E O
ND
A lr
-EL
EcT
RóN
ICA
S: tg
6
Orb
. Sim
.B
1B
2B
3B
4B
B6
B7
,B
,
1S
,0
00
00
00
02
A, —
3,92
6599
—1,
1024
390,
5562
88 —
0,55
5776
—1,
4804
62 —
1,46
6513
—29
,527
832
6,77
0111
3A
, —0,
4198
44 —
1,14
5957
1,28
6293
1,38
3032
1,35
6294
—1,
6916
76 —
1,40
9903
3,06
7212
4S
131
,269
789
—2,
6648
551,
3738
201,
3166
271,
2564
28 —
1,29
5976
—0,
8659
73—
0,34
4048
5S
.0,
3702
240,
7708
801,
4299
050,
9489
030,
7761
85 —
0,47
1590
0,70
7137
—0,
3591
016
A, —
0,47
5653
0,47
6040
1,97
3511
0,07
0068
—0,
7084
98 —
0,57
6614
3,44
4420
1,95
9336
,,7
A,
0,41
9336
4,13
9239
1,68
5209
4,08
0802
0,16
5597
0,01
8321
—0,
9520
541,
7211
418
S,
2,50
6827
—4,
8333
92 —
0,18
7768
3,75
3148
27,8
3085
2—41
,888
072
0,56
6485
—0,
6314
149
A.
4,25
5689
11,9
8663
2 —
0,70
2498
13,7
8481
95,
7037
504,
8163
30 —
1,83
5226
1,62
7448
10S
,—
4,52
1069
0,09
0505
—8,
1143
59 —
0,26
9904
2,39
0327
—0,
2213
21 —
0,03
6824
—0,
6663
7911
S,
0,25
7057
0,29
3466
23,3
2489
312
,717
092
—0,
6208
90 —
0,30
6624
2,24
2332
—0,
7157
7812
A,
0,45
0374
—2,
5730
3032
,654
416
—0,
3534
110,
6141
16 —
2,49
2028
0,65
9749
1,09
8582

PLEIADENO
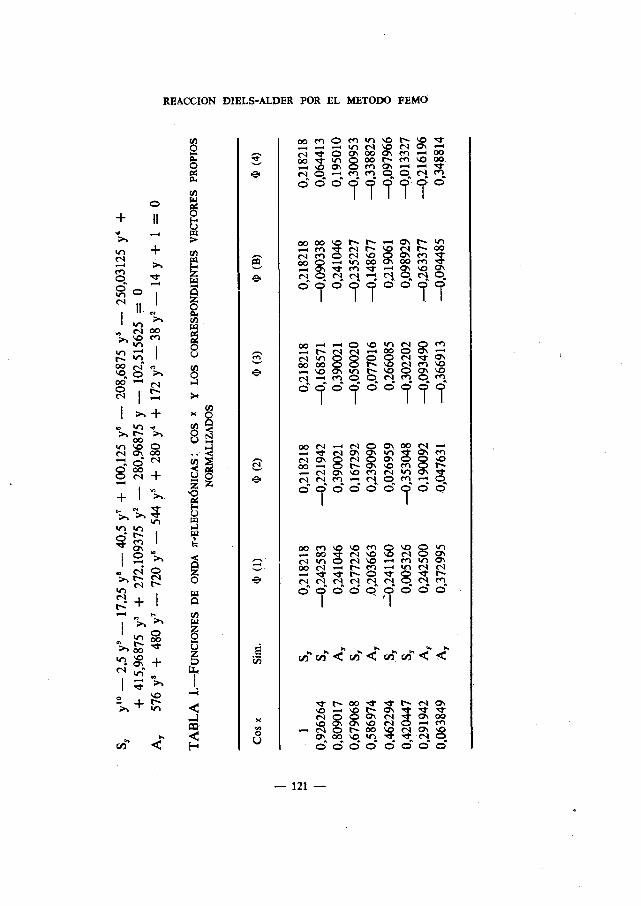
S7
y'°
— 2
,5 y
9 —
17,
25y8
— 4
0,5
yT +
100,
125
y'—
208
,687
5y5
— 2
50,0
312
5 y'
++
415
,968
75 y
3+
272,
1093
75 y
2—
280
,968
75y
— 1
02,5
1562
5=
0
A7
576
y8 +
480
y7
— 7
20ya
— 5
44 y
5 +
280
y4
+ 1
72 y
3 —
38
y2—
14
y+
10
C
TA
BL
A I
.—Fu
NcI
oNE
s D
E O
ND
A 7
r'EL
EC
TR
ON
ICA
S: C
OS
Xy
LOS
CO
RR
ESP
ON
DIE
NT
J3S
VE
CT
OR
ES
PRO
PIO
S
NO
RM
AL
IZA
DO
S
Cos
,cSi
m.
(1)
'I (2
)I
(3)
I (B
)(4
)
1S7
0,218218
0,218218
0,218218
0,218218
0,218218
0,92
6264
S7
—0,242583
—0,221942
—0,168571
—0,090338
0,064413
0,809017
A7
0,241046
0,39
0021
0,390021
0,241046
0,195010
0,679068
5,
0,277226
0,167292
—0,050020
—0,235227
—0,
3009
53
0,586974
A,
0,203663
0,239090
0,077016
—0,148677
—0,338825
0,462294
S,
—0,241 160
0,026959
0,266085
0,219061
—0,097966
0,420447
S,
0,005326
—0,353048
—0,302202
0,098929
—0,013327
0,291942
A,
0,242500
0,190092
—0,093490
—0,263377
—0,216196
0,063849
A,
0,372995
0,047631
—0,366913
—0,094485
0,348814

TA
BL
A I
.—Fu
NcI
ON
ES
DE
ON
DA
r-E
LE
CT
RO
NIC
AS:
CO
S X
YL
OS
CO
RR
ESP
ON
DIE
NT
ES
VE
CT
OR
ES
PRO
PIO
SN
OR
MA
LIZ
AD
OS
I')
Cos
xSi
m.
b (C
)'I
(5)
t (6)
tI (
A)
(F)
1S
0,21
8218
0,21
8218
0,21
8218
0,21
8218
0,21
8218
0,92
6264
S0,
2096
630,
3085
350,
3619
07—
0,22
7452
—0,
1468
740,
8090
17A
70,
0744
870,
0602
610,
0230
180
00,
6790
68S
7—
0,17
3509
0,12
0989
0,33
7828
0,20
9218
—0,
1282
320,
5869
74A
7—
0,24
9085
—0,
3488
81—
0,16
0482
00
0,46
2294
S7
—0,
3096
39—
0,02
1828
0,28
9457
—0,
2499
320,
1356
920,
0420
447
S7
—0,
1101
36—
0,01
5456
0,09
7140
0,35
7527
0,44
0311
0,39
1942
A7
0,09
3905
0,42
0517
0,23
5731
00
0,06
3849
A7
0,13
9065
—0,
1831
56—
0,16
2417
00

TA
BL
A 1
1.—
FuN
cIO
NE
s D
E O
ND
A 'r
-EL
EC
TR
ON
ICA
S:
1S,
0,04
7619
20,
1625
653
A0,
0581
034
S0,
0923
405
A0,
0632
770,
1186
476
S,0,
0825
340,
0984
697
S0,
1533
630,
0134
818
A0,
0694
760,
0844
469
0,13
9695
0,13
5359
0,04
7619
0,04
7619
0,04
7619
0,03
628
10,
0803
990,
0456
410,
1681
72—
0,05
8103
0,05
7174
0,17
9356
0,03
5859
0,03
372
4—
0,30
0433
0,04
9495
0,14
2739
0,13
1132
0,20
2900
0,22
9978
0,01
7079
0,08
1957
—0,
0290
04
0,00
8963
—0,
0413
16
Orb
ital
Sim
ctrI
aB
1B
2B
3B
4B
5B
6
0,04
76 1
9
0,05
8902
0,16
8175
0,07
7670
T1 (I,
0,04
7619
0,13
5990
0,00
5548
0,13
5942
0,12
4717
0,1
1459
4
0,01
3286
0,18
2776
0,05
6357
'TI ri
____
____
C

TA
BL
A I
II.—
FuN
cIoN
Es
DE
ON
DA
lr-E
LE
CFR
óNJC
AS:
tg
0 z 0 (I)
(I) - z
Orb
ital
Sim
etrI
aB
1B
2B
5B
4B
5B
8
1S,
00
00
00
2S
,1,
0357
876.
1791
40—
1,85
6235
—1,
6513
720
03
A7
0,32
4916
—0,
7265
351,
3763
61—
L4
S1—
5,48
1157
—0.
1396
840,
1824
433,
1476
620
05
A1
3,10
9474
0,18
3018
0,72
5014
—
6S,
0,40
7094
3,04
3099
—0,
1772
052,
5985
580
07
S70,
8241
958,
6449
714,
4420
27—
0,43
1536
00
8A
7—
2,63
6141
0,89
8150
0,08
1957
—
9A
7—
0,19
4060
0,33
5409
0,06
3980
—oo
oe

INDICEPágs.
INTRODiJCCTON 7
PARTE IMETODO DEL ELECTRON LIBRE
Consideraciones generates 11
MétodoFEMO 12Movimiento longitudinal 14Trayectoria no lineal 15Juntas o puntos ramificados 17Condiciones de continuidad y conservación 18Hermicidad, Ortogonalidad y Normalización 21Potencial constante 22Vectores propios de Ia función propia del electron libre 24AplicaciOn de la teorIa 26Sistemas conjugados 28Densidad de enlace 28Valencia unida 29Recientes desarrollos en ci método FEMO 30
PARTE II
DIAGRAMAS MOLECULARES
Diagramas moleculares 32Reactividad qulmica 42Espectros 42
PARTE III
REACCION DIELS-ALDER
Conskleraciones generales ... . 44Estudio teOrico de BROWN sobre la reacciOn DIELS-ALDER 46Cáiculo de la energIa de p-locaiizaciOn 49
— 125 —

ROSARIO DOMINGO SEBASTIAN
PARTh IV
ENERGIAS DE P-LOCALIZACION Y SU APLICACION AL ESTUDIODE LA REACCION DIELS-ALDER Pág.
Consideraciones generales 51
Poliacenos 52
Hidrocarburos angulares 57
Beflzologos del Antrceno 57-
Otros hidrocarburos 64EnergIas de p-localización y velocidades relativas para las posiciones meso de
los benzólogos del antraceno 70ErergIas de p1oca1ización, Indices de valencia libre y de valencia unida, y
velocidades relativas para las posiciones meso de los poliacenos 70EnergIas de p-localizacián y potenciales polarográficos de algunos hidrocarburos
polinucleares 70
CoNcLusIo!us 74
BIBLIoGRAFIA - 77
Arnca NUMERICO 81
— 126 —
Recommended
















![(2p/page) [.pdf]](https://img.pdfslide.tips/doc/110x75/585c7c3c1a28abed218de30a/2ppage-pdf.jpg)
