
INSTITUTO NACIONAL DE PESQUISAS DA AMAZÔNIA – INPA
PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA, CONSERVAÇÃO E
BIOLOGIA EVOLUTIVA
TESTANDO LIMITES ESPECÍFICOS DOS UAKARIS PRETOS
SENSU HERSHKOVITZ (1987) (Pitheciidae: Primates)
FABRÍCIO BERTUOL
Manaus, Amazonas
Maio, 2015

FABRÍCIO BERTUOL
TESTANDO LIMITES ESPECÍFICOS DOS UAKARIS PRETOS
SENSU HERSHKOVITZ (1987) (Pitheciidae: Primates)
ORIENTADOR: Ph.D. Tomas Hrbek
Coorientador: Ph.D. Jean Philippe Boubli
Dissertação apresentada ao Instituto
Nacional de Pesquisas da Amazônia como
parte dos requisitos para obtenção do título
de Mestre em Genética, Conservação e
Biologia Evolutiva.
Manaus, Amazonas
Maio, 2015

B552t Bertuol, Fabrício Testando limites específicos dos Uakaris pretos sensu Hershkovitz
(1987) (Pitheciidae: Primates) / Fabrício Bertuol. --- Manaus: [s.n.], 2015.
44 f. : il., color.
Dissertação (Mestrado) --- INPA, Manaus, 2015.Orientador: Tomas Hrbek.Coorientador : Jean Philippe Boubli.Área de concentração:Genética, conservação e biologia evolutiva.
1. Delimitação de Espécies 2. Primatas – Uakaris pretos. 3. NGS. I.Título
CDD 599.8
Sinopse:
Foram utilizadas novas abordagens genômicas para delimitar as
três espécies dos uakaris pretos, buscando esclarecer Cacajao
ayresi, terceira espécie descrita para o grupo e contestada.
Foram encontradas duas linhagens evolutivas e dois grupos
biológicos distintos, C. ayresi fica agrupado com C. hosomi.
Palavras-chave: Cacajao, genômica, linhagens evolutivas,
grupos biológicos.
iii

Agradecimentos
Aos meus pais Denise e Aristides e irmão Lucas que apesar da distância sempre
estiveram presentes e me apoiaram nesta jornada. Meus pais sempre foram
incentivadores do estudo, sempre se sacrificaram para dar o melhor aos filhos indiferente
das situações. Fica meu eterno agradecimento;
Tomas Hrbek pela oportunidade da orientação, pelos ensinamentos aprendidos ao
longo destes anos em Manaus e pela paciência representada em algumas situações, em
diversos momentos lembrou meu pai, pois em alguns momentos de dúvida e dificuldade
sempre me dizia “tem que aprender a se virar”;
Jean Philipe Boubli pela orientação, que apesar de fisicamente distante sempre
esteve presente para responder minhas dúvidas, sempre ajudou mesmo em situações de
dificuldade e me ensinou a aprender e gostar dos primatas;
Izeni Pires Farias pelos ensinamentos aprendidos estes anos e pelas dicas
construtivas para este trabalho;
Rupert Collins pela ajuda com o sistema operacional Ubuntu sempre ajudando a
esclarecer minhas dúvidas na instalação de programas e análises estatísticas;
Mario Nunes pela ajuda no laboratório e esclarecimento de dúvidas em análises
estatísticas;
Lelé e Iracema pela ajuda e apoio em relação a obtenção de amostras da coleção
zoológica de mamíferos do INPA;
José sempre presente para discutir assuntos relacionados a área da genômica e
NGS, sempre com boas dicas e respondendo perguntas;
Deyla pelas ajudas e dicas de escrita durante esse trabalho;
Elciomar e Rommel meus camaradas durante esse período em Manaus, demos
iv

boas risadas, bebemos muitas cervejas e jogamos muita sinuca no Carlão. Tivemos boas
conversas na hora de tomar o cafezinho do seu Zé e cultivamos boa amizade durante
este tempo;
Vinícius pela camaradagem no laboratório e dicas de escrita;
Sandra e Jéssica pela ajuda e colaboração durante a nossa geração de resultados
genômicos;
Ao Conselho Nacional de Desenvolvimento Científico e Tecnológico CNPq pela
concessão da bolsa de estudo;
Ao Instituto Leônidas e Maria Deane da Fundação Oswaldo Cruz (FIOCRUZ) por
disponibilizar os equipamentos de NGS e ao Vitor por nós acompanhar e ao seu seu
chefe Felipe Naveca;
Ao projeto CNPq SISBIOTA/Rede BioPAHM, que subsidiou esse estudo;
Universidade Federal do Amazonas (UFAM) e Laboratório de Genética e Evolução
Animal (LEGAL) pelo espaço físico e estrutura para o desenvolvimento da pesquisa;
Ao Instituto Nacional de Pesquisa da Amazônia (INPA) e ao Programa de Pós-
Graduação em Genética, Conservação e Biologia Evolutiva (PPG-GCBev) pela
oportunidade de estudo;
….a todos os demais colegas do LEGAL por esse tempo compartilhado que
estivemos juntos para nosso enriquecimento pessoal.
Obrigado a todos por participarem desta etapa.
v

Resumo
Os uakaris pretos atualmente possuem três espécies válidas: Cacajao. melanocephalus,
C. hosomi e C. ayresi; que foram propostas após revisão taxonômica do grupo porém,
não existe consenso sobre esta classificação devido a discordâncias sobre a validade de
C. ayresi proposta como sendo espécie nova para o grupo. A discordância existe devido à
baixa resolução filogenética encontrada em relação a sua espécie irmã, C. hosomi. Neste
sentido, foram testados os limites específicos dos uakaris pretos para validar ou não C.
ayresi, com a utilização de filogenia de Citocromo b e sequenciamento de nova geração
para delimitar linhagens evolutivas e grupos biológicos. Foram utilizadas dez amostras
inéditas de uakaris pretos para uma nova reconstrução filogenética do Citocromo b e vinte
e uma amostras para o sequenciamento de nova geração. Todas as amostras utilizadas
na reconstrução filogenética do Citocromo b se agruparam com suas respectivas espécies
descritas, indicando uma monofilia para este marcador. Os resultados provenientes do
NGS indicam que C. hosomi e C. ayresi formam uma única linhagem evolutiva enquanto
que C. melanocephalus forma outra linhagem. Foram encontrados dois grupos biológicos,
um composto por C. melanocephalus e outro composto C. ayresi e C. hosomi. Os
resultados indicam que C. ayresi e C. hosomi são espécies distintas pela filogenia do
Citocromo b, seguindo o conceito filogenético de espécies enquanto que os resultados
provenientes do NGS indicam que C. ayresi e C. hosomi formam uma única linhagem
evolutiva por apresentarem polifilia, não sendo classificadas como espécies distintas pelo
conceito filogenético de espécies. As espécies C. ayresi e C. hosomi também se agrupam
em um mesmo grupo biológico, indicando compartilhamento do sistema de acasalamento
e por isso classificadas como pertencentes a mesma espécie pelo conceito biológico de
espécies portanto, C. hosomi e C. ayresi não podem ser classificadas como espécies
distintas.
Palavras chave: Cacajao, ddRAD, NGS, linhagem evolutiva, grupo biológico.
vi

Abstract
The black Uakari currently have three valid species: Cacajao. melanocephalus, C. hosomi
and C. ayresi; proposed after a group taxonomic revision, however there is no consensus
on this classification due to disagreements over the validity of C. ayresi proposed as new
species for the group. The disagreement exist due to the low phylogenetic resolution found
between C. ayresi and its sister species, C. hosomi. In this regard, the specific limits of
black uakaris were tested to validate or not C. ayresi, using the phylogeny of Cytochrome
b and next generation sequencing to delimit evolutionary lineages and biological groups. It
was used ten new samples of black uakaris to building a phylogenetic reconstruction of
Cytochrome b and twenty-one samples for next-generation sequencing. All samples used
in the phylogenetic reconstruction of Cytochrome b were grouped with their respective
described species, indicating a monophyletism for this marker. The results from the NGS
indicate that C. hosomi and C. ayresi form a single evolutionary lineage while C.
melanocephalus form another branch. Two biological groups were found, one composed
by C. melanocephalusand other composed by C. ayresi and C. hosomi. The results
indicate that C. ayresi and C. hosomi are distinct species by phylogenetic Cytochrome b,
following the phylogenetic species concept, while the results from the NGS indicate that C.
ayresi and C. hosomi form a single evolutionary lineage presenting polyphyly and is not
classified as distinct species by phylogenetic species concept. The species C. ayresi and
C. hosomi are also grouped in the same biological group, sharing mating system and thus
classified as belonging to the same species by the biological species concept, therefore C.
hosomi and C. ayresi can not be classified as separate species.
Keywords: Cacajao, ddRAD, NGS, evolutionary lineage, biological group.
vii

Sumário1 INTRODUÇÃO...............................................................................................................................11
1.1. CONCEITO DE ESPÉCIE.....................................................................................................111.2. UAKARIS PRETOS...............................................................................................................121. 3 NOVAS ABORDAGENS - NEXT GENERATICON SEQUENCING (NSG)......................16
2 OBJETIVOS....................................................................................................................................182.1. GERAL...................................................................................................................................182. 2. ESPECÍFICOS.......................................................................................................................18
3 MATERIAL E MÉTODOS.............................................................................................................183.1. AMOSTRAGEM....................................................................................................................183.2 EXTRAÇÃO DO DNA...........................................................................................................203.3. SEQUENCIAMENTO DO DNA MITOCONDRIAL CITOCROMO B...............................213.4. PREPARAÇÃO DA BIBLIOTECA GENÔMICA.................................................................223.5. ANÁLISE DOS DADOS........................................................................................................24
3.5.1. ANÁLISES DOS DADOS DO DNA MITOCONDRIAL..............................................243.5.2 ANÁLISES NGS – PYRAD............................................................................................253.5.3. RECONSTRUÇÃO FILOGENÉTICA...........................................................................253.5.4. GRUPOS BIOLÓGICOS................................................................................................26
4. RESULTADOS...............................................................................................................................274.1. RECONSTRUÇÃO FILOGENÉTICA COM O GENE CITOCROMO B............................274.2. FILOGENÔMICA..................................................................................................................284.3 GRUPOS BIOLÓGICOS........................................................................................................29
5. DISCUSSÃO..................................................................................................................................31
viii

LISTA DE FIGURAS
Figura 1 - Mapa da distribuição geográfica das três espécies de uakari preto. Polígonos
indicando os limites de distribuição. Cacajao hosomi em amarelo, C. ayresi em marrom e
C. melanocephalus em vermelho
(IUCN)................................................................................................................................14.
Figura 2 – Variação na coloração das espécies de uakaris pretos: 1 - C. melanocephalus;
2 - C. hosomi; 3 - C. ayresi................................................................................................14.
Figura 3 - Mapa de distribuição das espécies de uakaris preto e da amostragem utilizada
para a filogenia do Citocromo b no presente trabalho. Os pontos corresponde aos locais
de coleta e os polígonos às áreas de distribuição da espécie segundo a IUCN, sendo
vermelho correspondente à distribuição da espécie Cacajao melanocephalus; vermelho,
C. hosomi em amarelo , C. ayresi em
marrom...............................................................................................................................19.
Figura 4 - Mapa de distribuição das espécies de uakaris preto e da amostragem utilizada
para o sequenciamento de próxima geração no presente trabalho. Os pontos
corresponde aos locais de coleta e os polígonos às áreas de distribuição das espécies
segundo a IUCN, sendo vermelho correspondente à distribuição de Cacajao
melanocephalus, em amarelo C. hosomi e marrom C.
Ayresi.................................................................................................................................20.
Figura 5 – Árvore filogenética de verossimilhança do Citocromo b representando as
linhagens evolutivas encontradas. Cacajao melanocephalus em vermelho, Cacajao
hosomi em amarelo e Cacajao ayresi em marrom indicando a monofilia do grupo com a
exclusão da amostra MPEG 8991 de qualidade
inadequada........................................................................................................................28.
Figura 6 – Árvore filogenética de verossimilhança de RADseq representando as linhagens
evolutivas encontradas. C. melanocephalus em vermelho, suportada por 0,969. C. hosomi
ix

e C. ayresi em amarelo indicando um grupo polifilético com 0, 999 de valor de
suporte...............................................................................................................................29.
Figura 7 – Grupos biológicos encontrados. Em vermelho Cacajao melanocephalus forma
um grupo, em amarelo C. hosomi e C. ayresi, formam outro grupo biológico...................30.
Figura 8 - Resultado indicando k=2, como a melhor probabilidade para explicar os dados.
A vertical mostra a média da estimativa da distribuição das probabilidades dos dados e a
horizontal número de clusters testados (k) e o desvio padrão (SD) da probabilidade
média.................................................................................................................................31.
x

1 INTRODUÇÃO
1.1. CONCEITO DE ESPÉCIE
Dentro das áreas biológicas, espécie é a unidade principal de trabalho, porém
ainda hoje sua delimitação não possui um consenso. O que é uma espécie e quais
critérios são necessários e suficientes para reconhecer uma, tem gerado debate
desde a formulação de Mayr (1942) sobre conceito biológico de espécie.
Este debate continuou até recentemente quando uma nova proposta
prevaleceu sobre as outras, de espécies como linhagens evolutivas (de Queiroz,
1998), agrupando todos os conceitos em um sentido único. Que critério para
delimitação usar, o que é necessário e suficiente para delimitar especies prevalece
pouco claro. Ficou esclarecido as diferenças entre os conceitos de espécies
existentes (o conceito de espécie primária) e os critérios de delimitação espécies (os
conceitos de espécie secundários). O conceito de espécie linhagem generalizada de
de Queiroz (1998) é um conceito de espécie primária, enquanto o conceito biológico
de espécies (Mayr, 1942) e filogenético (Cracraft, 1983), dois entre muitos, são
conceitos de espécies secundárias ou seja, critérios para a delimitação das
espécies.
Considerando que as espécies surgem pelos processos de especiação
seguido pela divergência, os diferentes conceitos (critérios) focam em diferentes
propriedades, as quais tem tempos médios de surgimento diferentes após o evento
da especiação. Os diferentes conceitos de espécie frequentemente se referem a
diferenças no tempo durante e após a especiação (de Queiroz, 2007).
As espécies são o resultado de um processo evolutivo seguido pela
divergência, sendo assim, espécies podem ser identificadas por características
específicas que surgiram de maneira independente nas linhagens após o evento de
especiação, como por exemplo, diferenças no sistema reprodutivo, reconhecimento
sexual, diferenças na morfologia (internas e externas) e em comportamento e uso de
nicho. Portanto, para identificar linhagens divergentes e possíveis espécies é
necessário a inclusão de diferentes informações (Padial et al., 2010). Porém, parte
dos problemas observados em tentar diferenciar linhagens evolutivas ocorre pela
11

baixa quantidade de informações geradas, ou seja, possuem poucos dados para
esclarecer as relações entre as linhagens.
Fleagle (2014) indica quais são os principais conceitos utilizados dentro da
Primatologia, o Conceito Biológico de Espécies continua tendo aceitação por alguns
autores, enquanto o Conceito Filogenético de Espécies (Cracraft, 1983), possui boa
aceitação, mas o que prevalesse é o Conceito Geral de Espécies (de Queiroz, 2007)
que é indicado sendo o mais lúcido (Groves, 2014).
1.2. UAKARIS PRETOS
Os uakaris pretos são macacos do novo mundo, pertencentes à família
Pithecidae, sendo os únicos da família a apresentar cauda curta e adaptações para
a predação de sementes (Hershkovitz, 1897; Boubli et al., 2008). São primatas de
médio porte (3 - 4 kg), restritos à bacia do rio Negro e rio Orinoco/Cassiquiari
(Boubli, 1999). Vivem em grupos sociais muito grandes de até 100 indivíduos com
uma área de vida muito extensa chegando até a 1.500 ha (Boubli, 1993).
Hershkovitz (1987), revisou a taxonomia do gênero Cacajao e reconheceu
duas espécies e seis subespécies para o gênero, destas, quatro subespécies foram
encontradas dentro de uakaris brancos, sendo: Cacajao calvus calvus, C. c.
rubicundus, C. c. ucayalii, C. c. novaesi e duas dentro de uakaris pretos, C.
melanocephalus melanocephalus ocorrendo no Brasil e C. m. ouakary, restrita a
Venezuela.
Embora Hershkovitz, (1987) tenha reconhecido duas subespécies, a
subdivisão dos uakaris pretos já havia sido proposta anteriormente por Hernández-
Camacho e Cooper (1976) tendo Hershkovitz (1987) simplesmente mantido o
arranjo proposto por estes autores. Esta divisão é suportada por dados filogenéticos
inferidos por citocromo b, corroborando a separação geográfica das subespécies e
diferenças morfológicas (Boubli et al., 2008; Figueiredo-Ready et al., 2013).
A divisão dos uakaris pretos de acordo com Hershkovitz (1987) se manteve
até nova revisão taxonômica proposta por Boubli et al., (2008) baseado em
evidencias morfológicas, ecológicas, distribuição geográficas e filogenia molecular.
12

Estes autores propuseram uma reorganização da taxonomia deste grupo, elevando
os taxa anteriores a espécies e incluindo a descrição de uma terceira espécie
proveniente do rio Aracá, Cacajao ayresi, nome dado em homenagem ao
pesquisador José Márcio Ayres que realizou o primeiro estudo sobre a ecologia dos
uakaris brancos na Reserva de Desenvolvimento Sustentável, Mamirauá,
Amazonas, Brasil.
Boubli et al. (2008) reinterpretaram a origem do tipo de uakari preto descrito
por Humboldt em 1801 e sinonimizaram melanocephalus e ouakary, com isso, os
uakaris pretos da região do Pico da Neblina no Brasil, ficaram sem nome disponível
tendo estes autores atribuído novo nome para este táxon, hosomi, palavra da língua
Yanomami para uakari preto. Desta forma, Boubli et al. (2008) propuseram três
espécies válidas: C. melanocephalus, C. hosomi e C. ayresi.
A distribuição geográfica de C. melanocephalus seria delimitada ao sul pelos
rios Solimões e Japurá, no Brasil, a oeste pelo rio Apaporis e montanha de La
Macarena na Colômbia, ao norte pelo rio Guaviare também na Colômbia, rio Negro e
Canal Cassiquiari no Brasil e rio Orinoco, na Venezuela (Boubli, 1993; Boubli et al.,
2008) (Figura 1). Este táxon apresenta variação em sua coloração (Figura 2),
apresentando três faixas de cores, sendo que na cabeça e membros superiores a
espécie apresenta uma coloração preta variando para amarelada a partir da região
escapular até os quadris e se tornando marrom avermelhada nos membros
inferiores e voltando a cor preta no final dos membros inferiores e ponta da cauda,
seu peso varia entre 1.900 a 2.600 g.
Por sua vez, a espécie Cacajao hosomi é limitado ao sul e oeste pelo rio
Negro, pelo rio Marauiá no leste (Brasil), e pelo canal de Cassiquiare e rio Orinoco
ao norte (Venezuela) (Figura 1). Essa espécie possui coloração variando de preta na
cabeça e membros superiores a marrom avermelhada a partir da região escapular
(Figura 2), retornando à cor preta na ponta dos membros inferiores e ponta da
cauda. É considerada a maior espécie dos uakaris pretos com peso médio variando
de 3.100 a 4.500 g (Boubli et al., 2008).
13

Figura 1 - Mapa da da distribuição geográfica das três espécies de uakari preto. Polígonos indicando
os limites de distribuição. C. hosomi em amarelo, C. ayresi em marrom e C. melanocephalus em
vermelho (IUCN), área indicado por ?, provável área de C. melanocephalus porém, sem confirmação.
Figura 2 – Variação na coloração das espécies de uakaris pretos: 1 - C. Melanocephalus; 2 - C.
Hosomi; 3 - C. ayresi.
14
1 2 3

Cacajao ayresi habita uma pequena área que abrange a bacia do rio
Curunduri e áreas adjacentes (Figura 1). A espécie também possui coloração
variando entre preta e marrom avermelhada e quebra da coloração na região
escapular, porém a coloração na região marrom avermelhada não é uniforme como
em C. hosomi, possuindo pelos com as pontas enegrecidas, assim, destacando uma
clara diferença morfológica entre as duas espécies (mais escura que C. hosomi)
(Figura 2) (Boubli et al., 2008).
A separação entre as espécies que ocupam a mesma margem do rio Negro,
C. hosomi e C. ayresi, foi proposta por uma barreira, imposta por exclusão
competitiva, pela espécie Chiropotes israelita (cuxiú), impedindo assim o contato
entre as duas espécies de uakaris (Boubli et al., 2008). Uakaris e cuxiús ocupam
nichos ecológicos similares, portanto, se excluiriam ecologicamente (Ayres, 1989).
De acordo com Boubli et al. (2008), C. ayresi é encontrado principalmente em
áreas de igapó e buritizais, enquanto a espécie C. hosomi é encontrado
principalmente em áreas de terra firme podendo inclusive chegar a altitudes
superiores a 1.500 m no maciço do Pico da Neblina e C. melanocephalus
essencialmente ocupa áreas de igapó na margem direita do rio Negro.
Apesar da nova proposta para delimitar os táxons (Boubli et al., 2008), a
diferenciação genética entre C. ayresi e C. hosomi foi questionada por Ferrari et al.
(2014), que consideraram baixa a diferenciação genética encontrada entre os dois
táxons (0,5 %). Ferrari et al. (2014) se baseiam também em dados morfológicos e
zoogeográficos para delimitar somente uma única espécie existente em cada lado
do rio Negro, porém elevando as subespécies de uakaris pretos ao status de
espécie, C. ouakari na margem direita do rio Negro e C. melanocephalus na margem
esquerda com uma subespécie, C. m. ayresi.
A proposta de Boubli et al. (2008) se suporta no argumento de que a
separação dos táxons ocorreu em uma escala evolutiva muito recente (0,5 a 0,3
milhões de anos) e que por isso os níveis de divergência genética seriam baixos.
Conforme os dados de Citocromo b utilizados por Boubli et al. (2008), C. ayresi
apresenta um caractere diagnóstico em relação a C. hosomi na posição 909 do
Citocromo b, uma transição de uma citosina, onde C. hosomi apresenta uma timina.
Adicionalmente, C. ayresi apresenta um caractere exclusivo (timina) no sítio 720 em
15

relação a todas as outras espécies do gênero que apresenta uma citosina na mesma
posição.
Figueiredo et al. (2013) baseados em sequências de Citocromo b, também
não suportaram a espécie C. ayresi, por estar aninhada dentro do grupo de C.
hosomi. Nesse trabalho, uma das amostras classificada como C. hosomi coletada no
rio Curunduri, limite da distribuição de C. ayresi, apresentou-se como grupo irmão de
C. ayresi, tornando assim C. hosomi parafilético ou anulando C. ayresi como uma
espécie válida. A amostra utilizada pelos autores provem de material antigo de
museu, sendo assim a sequência obtida pelos autores apresentaram ‘missing data’ o
que é um problema quando usadas em análises filogenéticas de espécies com
divergência recente.
Apesar das criticas, as novas argumentações levantadas (Figueiredo et al.,
2013; Ferrari et al., 2014) não trazem nenhuma informação nova sobre as relações
filogenéticas dos uakaris pretos. Situação similar foi observada em uma recém
descrita espécie de anta, Tapirus kabomani (Cozzuol et al., 2013) que recebeu
criticas de sua real existência (Voss et al., 2014) e levantando a contestação de qual
informação seria necessária para delimitar uma nova espécie (Cozzuol et al., 2014).
1. 3 NOVAS ABORDAGENS - NEXT GENERATICON SEQUENCING (NSG)
O desenvolvimento do sequenciamento de próxima geração (Next Generation
Sequencing – NGS) permite a obtenção de enorme quantidade de sequências e loci,
com velocidade, precisão e custo reduzido. Estas novas abordagens estão
revolucionando a geração de sequências e a descoberta de polimorfismos de
nucleotídeos único (single nucleotide polymorphisms - SNPs) ao longo de todo o
genoma (Helyar et al., 2011), que são variações em apenas uma base na sequência
do DNA. A utilização de NGS para a obtenção dos marcadores moleculares SNPs
chega a ser da ordem de milhares e estas variações de DNA entre indivíduos podem
ser usadas para inúmeros fins, como exemplo em nível populacional (Hohenlohe et
al., 2010, 2011, 2012).
Esta grande quantidade de informações proporcionaram a expansão dos
16

estudos no âmbito da genômica populacional que agora permitem o
desenvolvimento de estudos em organismos não modelo (Helyar et al., 2011). Estes
estudos permitiram explicar o encontro de adaptações paralelas (Hohenlohe 2010;
2012) e hibridização entre espécies de peixes (Hohenlohe, 2011), e agora estão
possibilitando estudos em escala filogenômica, definido como o estudo das relações
evolutivas baseados em análises comparativas de dados em escala genômica (Chan
e Ragan, 2013).
Como parte dos problemas observados em tentar delimitar linhagens
evolutivas são relacionados à baixa quantidade de informações geradas, a
filogenômica surge como uma nova ferramenta com capacidade para resolver as
relações entre os táxons (Prasad et al., 2008; Philippe et al., 2009, Cariou et al.,
2013) em virtude da geração de uma enorme quantidade de dados representativos
de todo o genoma do organismo e não apenas de regiões específicas.
Os poucos estudos com filogenômica, estão apresentando expectativas
animadoras em resolver problemas filogenéticos em escala fina e possibilitando o
esclarecimento das relações entre as linhagens evolutivas divergentes. Esta nova
abordagem permitiu, por exemplo, resolver problemas filogeográficos prévios em
mosquitos da espécie Wyeomyia smithii da América do Norte, problema este não
resolvido com dados de citocromo oxidase I (COI) (Emerson et al., 2010).
Recentemente, os dados NGS conseguiram suportar a monofilia das espécies
dos ciclídios do Lago Vitória (Wagner et al., 2013) que por muito tempo ficou mal
esclarecida, mostrando assim que a técnica permite resolver filogenias difíceis em
grupos com divergências recentes (< 15 mil anos) assim como identificar evolução
reticulada em peixes do gênero Xiphophorus da família Poeciliidae (Cui et al., 2013).
Análises genômicas praticamente resolveram a filogenia de macacos do novo
mundo (Perelman et al., 2011). Entretanto, as relações das espécies dentro dos
gêneros, não foram resolvidas para a maioria dos táxons.
Sendo assim, no presente trabalho, reexaminamos as hipóteses taxonômicas
de Hershkovitz (1987) e Boubli et al. (2008) utilizando dados do Citocromo b e
sequenciamento de nova geração para as espécies Cacajao melanocephalus, C.
hosomi e C. ayresi, o que permitirá melhorar a delimitação entre as linhagens
evolutivas com divergência recente. Além disso, a abordagem filogenômica auxiliará
17

delimitar de maneira mais refinada as relações filogenéticas entre as três espécies e
confirmar ou não o status de espécie para C. ayresi.
Também usamos a abordagem da genômica populacional com utilização de
marcadores SNPs, uma abordagem nova para os uakaris pretos, já que nunca foram
utilizadas informações a níveis populacionais para delimitar grupos biológicos, desta
forma procurando a existência de compartilhamento de sistema de acasalamento
entre os indivíduos de C. ayresi e C. hosomi.
2 OBJETIVOS
2.1. GERAL
Testar os limites específicos das espécies de uakaris pretos validando ou não
a espécie Cacajao ayresi por meio de dados do citocromo b e de sequenciamento
de nova geração.
2. 2. ESPECÍFICOS
1) Realizar uma nova reconstrução filogenética com o citocromo b, com maior
número de amostras, com o intuito de aumentar a compreensão das relações entre
as espécies.
2) Verificar a formação de grupos biológicos distintos dos indivíduos de C.
ayresi em relação às outras espécies irmãs, C. hosomi e C. melanocephalus com
dados de sequenciamento de nova geração.
3) Inferir se os indivíduos de C. ayresi formam uma linhagem evolutiva independente
de C. Hosomi.
18

3 MATERIAL E MÉTODOS
3.1. AMOSTRAGEM
Foram utilizadas vinte e uma amostras de tecido muscular fresco, estas
compostas por cinco espécimens de C. ayresi, quatro de C. hosomi, onze de C.
melanocephalus e uma de C. calvus rubicundus para o sequenciamento de próxima
geração (Tabela 1, Anexo 1, Figura 3) e dez novas amostras para filogenia do
Citocromo B com a utilização de amostras depositadas no GenBank (Tabela 1,
Anexo 1, Figura 4). As amostras de tecido foram provenientes de coletas
anteriormente realizadas por Boubli e em expedições do projeto SISBIOTA –
BIOPHAM realizadas no município de Santa Isabel do Rio Negro e Japurá,
Amazonas. Estas amostras encontram-se depositadas na Coleção de Tecido do
Laboratório de Evolução e Genética Animal (LEGAL) na Universidade Federal do
Amazonas (UFAM) e na coleção de mamíferos do Instituto Nacional de Pesquisas
da Amazônia (INPA).
Figura 3 - Mapa de distribuição das espécies de uakaris preto e da amostragem utilizada para a
filogenia do Citocromo B no presente trabalho. Os pontos corresponde aos locais de coleta e os
polígonos às áreas de distribuição da espécie segundo a IUCN, sendo vermelho correspondente à
distribuição da espécie C. melanocephalus; vermelho, C. hosomi em amarelo , C. ayresi em marrom.
19

Figura 4 - Mapa de distribuição das espécies de uakaris preto e da amostragem utilizada para o
sequenciamento de próxima geração no presente trabalho. Os pontos corresponde aos locais de
coleta e os polígonos às áreas de distribuição da espécie segundo a IUCN, sendo vermelho
correspondente à distribuição da espécie C. melanocephalus em vermelho, C. hosomi em amarelo e
C. ayresi em marrom.
3.2 EXTRAÇÃO DO DNA
Os procedimentos laboratoriais foram realizados no Laboratório de Evolução
e Genética Animal (LEGAL) na Universidade Federal do Amazonas (UFAM). A
extração do DNA genômico total procedeu conforme o método CTAB 2% (Doyle e
Doyle, 1987). Foram utilizados aproximadamente 3 a 5 mg de tecido fresco, que
foram incubados para a digestão com auxílio de 500 µL de tampão de lise CTAB 2%
e 15 µL de proteinase K (10 mg/mL). Após a digestão, foram realizadas lavagens
com 500 µL de clorofórmio - álcool isoamílico (24:1), posteriormente o DNA foi
precipitado com 500 µL de etanol absoluto, seguido com 500 µL de etanol 70 % e
ressuspendido em 50 µL de água ultrapura.
A integridade e pureza do DNA assim como sua concentração foram
checadas em Nanodrop 2000 (Thermo Scientific, USA) para posterior diluição.
20

3.3. SEQUENCIAMENTO DO DNA MITOCONDRIAL CITOCROMO B
O gene mitocondrial citocromo b foi amplificado pela reação em cadeia da
polimerase (PCR) para as dez amostras cujas sequências não constavam depositas
no GenBank.
Foram realizados duas PCRs para cada amostra com auxílio de primers
internos com sobreposição, com a finalidade de otimizar o tamanho do fragmentos
gerados. Os primers utilizados na primeira reação foram os primer forward L14725
(5'– CGAAGCTTGATATGAAAAACCATCGTTG - 3') (Kocher et al., 1990) em
conjunto com o primer Gina 538 reverse (5'– AGGCAAGATAAAGTGGAAG – 3')
(Figueiredo, 2013). Para a segunda reação de PCR, foram utilizados os primers
forward Jeff – 353 (5'– TTTTATTACTTACAACTATAGC - 3') (Figueiredo, 2013) junto
com o reverse H15915 (5' – AACTGCAGTCATCTCCGGTTTACAAGAC - 3') (Irwin et
al., 1991).
O volume final do PCR foi de 15 µL, contendo 6,2 µL de água ultrapura, 1,2
µL de dNTP (2,5 mM), 1,8 µL de MgCl2 (25 mM), 1,5 µL de tampão 10 X, 1,5 µL de
primer forward e reverse (2 pmol), 0,3 µL de taq DNA polimerase (1 U/µL) e 1 µL de
DNA (30 ng/µL).
As condições de ciclagem seguiram as seguintes condições: um ciclo de
desnaturação inicial a 95 ºC durante 2 minutos, seguido de 40 ciclos de
desnaturação a 95 ºC por 15 segundos, anelamento a 48 ºC por 30 segundos,
extensão por 72 ºC por 1 minuto e para finalizar um ciclo de extensão final a 72 ºC
por 5 minutos.
As amplificações foram checadas via eletroforese em gel de agarose a 1%
corado com GelRed™ (Biotium, Inc.) e purificados com uma solução de
Exonuclease I e Fosfatase alcalina de camarão (ExoSap-IT®) (USB Corporation)
(Fermentas) (Werle et al., 1994).
Após a purificação, os fragmentos de PCR foram sequenciados com uma
reação de 4,4 μL de água ultrapura; 1,3 μL de tampão de sequenciamento 5X; 0,3 μL
de BigDye Terminator v 3.1 (Applied Biosystems®); 2,0 μL dos primers forward e
reverse e 2,0 μL do produto de PCR purificado (20-50 ng/μL). As reações de
sequenciamento seguiram as recomendações indicadas pelo fabricante com a
21

temperatura de anelamento de 50 ºC (Platt et al., 2007). Posteriormente os produtos
sequenciados foram purificados com a adição de 2,5 µL de EDTA (125 mM) e 30 µL
de álcool absoluto e então resuspendidos em Hi-DiTM formamida e injetado em
sequenciador automático ABI 3500 (Applied Biosystems®).
3.4. PREPARAÇÃO DA BIBLIOTECA GENÔMICA
Para a construção da biblioteca genômica foi utilizada a técnica de ddRADseq
(double digest restriction-site-associated DNA) (Peterson et al., 2012), que faz uso
de duas enzimas de restrição, sendo uma de corte raro e outra de corte frequente,
com o objetivo de padronizar e otimizar o tamanho dos fragmentos gerados. Esta
etapa sofreu algumas modificações para ser utilizado na plataforma Ion TorrentTM.
Para essa etapa, foram utilizadas os DNAs das 21 amostras das espécies C.
ayresi, C. hosomi, C. melanocephalus e C. calvus rubicundus. O processo de
digestão do DNA, ocorreu de forma individual em um volume final de reação de 50
µL. Foram utilizados 4,0 µL de DNA (totalizando 200 ng), que foram digeridas com
0,1 µL (1 U) das enzimas de restrição (Thermo Scientific) SdaI (corte frequente) e
Csp6I (corte raro), respectivamente. Essas enzimas criam pontas coesivas em
ambas as extremidades das moléculas de DNA digerida, que permitem a ligação aos
adaptadores.
O adaptador P1 (comum a todas as amostras), utilizado 2,0 µl (0,1 µM), se
liga a ponta gerada pela enzima SdaI e o adaptador AY 2,0 µl (5 uM) (barcode único
de cada amostra), 2,0 µL, se liga a ponta gerada pela enzima Csp6I. Estes
adaptadores se ligam inicialmente por pontes de hidrogênio à molécula de DNA
digerida. A enzima T4 ligase 0,5 µl (5 U), permite a ligação fosfodiester com o auxílio
de 0,5 µl ATP (5 mmol) e 5 µl Tampão Tango 10X.
Este processo de digestão e ligação ocorre de forma simultânea. Após a
digestão, os adaptadores que possuem um nucleotídeo alterado em suas
sequencias, se ligam a molécula de DNA e desta forma alteram o sítio de
reconhecimento de corte das enzimas, sem ocorrer ligação e digestão simultânea.
As condições do tempo de digestão foram de 37 oC durante 90 minutos, para a
22

realização do processo de digestão do DNA pelas enzimas e posterior desnaturação
destas a 68o durante 15 minutos.
Após a digestão foi realizado um PCR teste para a verificação do
funcionamento da digestão. A PCR foi realizada em um volume final de 15 µL
contendo 5.25 µL de H2O, 1.5 µL de NH4SO4 10X, 1.5 µL do primer P1 (2 mM), 1.5
µL do primer A-amp (2 mM), 1.5 µL BSA (Bovinum Serum Albumin), 1,5 µL dNTPs
(10 mM), 1.5 µl MgCl2 (25 mM), 0,35 µl da taq DNA (1 U) polimerase e 1 µL de DNA
digerido. As condições do PCR foi de um ciclo inicial a 94 ºC por 2 minuto seguido
por 35 ciclos de 94 ºC por 15 segundos, 55 ºC por 35 segundos e 68 ºC por 1 minuto
e 30 segundos com ciclo único e final de 68 ºC por 1 minutos e 30 segundos. As
amplificações foram checadas via eletroforese em gel de agarose a 1% corado com
GelRed™ (Biotium, Inc.). Posteriormente, foi realizado a etapa de enriquecimento.
Cada uma das amostras digeridas contribui para a geração de cinco PCRs
distintas com finalidade de aumentar numero dos fragmentos de DNA que realizaram
somente a ligação com os adaptadores P1 e AY.
Na etapa de enriquecimento as condições de PCR ocorreram em volume final
de 25 µL, sendo 12.4 µL de H2O, 2,0 µL MgCl2 (25 mM), 2.0 µL dNTPs (10 mM), 2.5
µL de tampão NH4SO4 10X (Fermentas), 2.5 µL de primer P1 (2 mM), 2.5 µL de
primer A-amp (2 mM), 0,1 µL de Taq (0,5 U Klentaq DNA Polymerase Technology) e
1 µL de DNA digerido. As reações de PCR ocorreram em um ciclo inicial de 68 ºC
por 1 minuto seguido por 18 ciclos iniciando a 93 ºC por 10 segundos, 52 ºC por 35
segundos, 68 ºC por 1 min e 30 segundos e um ciclo final e único de 68 ºC por 7
minutos.
Após o término, cada uma das cinco amostras de PCR foram agrupadas em
um único tubo para totalizar um volume final de 100 µL. Estas amostras foram
purificadas em coluna do kit Illustra GFX PCR DNA e Gel Band Purification kit (Ge
Healthcare Life Science, USA) de acordo com as instruções do fabricante.
Posteriormente todas as amostras contribuíram de forma equimolar para a
formação de um pool genômico em um único tubo, estas foram designadas para
corte e seleção dos fragmentos alvos a serem sequenciados. Estes são definidos
por modelos computacionais que levam em consideração um genoma completo
como referência, sendo que no nosso caso, nós usamos o genoma das espécies
23

Callitrix jaccus e Saimiri boliviensis, espécies filogeneticamente próximas,
depositadas no GenBank.
Esta relação do tamanho do genoma e corte das enzimas indicou a seleção
dos fragmentos a serem utilizados indicando o modelo de corte range com tamanho
dos fragmentos variando entre 350 – 450 pb, esta etapa foi realizada no seletor de
fragmentos em gel de agarose 2 % Pippin Prep (Sage Science).
Nesta etapa é importante selecionar o tamanho correto dos fragmentos para
produzir fragmentos em tamanho ótimo compatíveis com a capacidade da geração
de dados do chip utilizado no sequenciador, já que se perde informações com
fragmentos maiores ou menores.
Após a construção da biblioteca foram seguidas as recomendações dos
fabricantes para o sequenciamento no sequenciador Ion Torrent (PGM, Life
Technologies) com a utilização do kit de sequenciamento 318 Ion PGM chip de 400
pb.
3.5. ANÁLISE DOS DADOS
3.5.1. ANÁLISES DOS DADOS DO DNA MITOCONDRIAL
Para a construção da filogenia com o gene citocromo b, utilizamos
sequências de trabalhos prévios depositadas no GenBank (Boubli et al. 2008; 2015;
Figueiredo-Ready et al., 2013) e as sequências geradas no presente estudo,
totalizando 41 sequências (Tabela 1, Anexo 1).
Os eletroferogramas foram importados para o programa Geneious 6.1.7
(Kearse et al., 2012) e as sequências consensos foram geradas. Cada sequência
consenso foi traduzida in silico, quando apropriado e posteriormente editada e
alinhada. Para essa etapa, nós incluímos as 10 amostras sequenciadas no presente
estudo com total de 1095 pb, juntamente com amostras depositadas no GenBank.
Para encontrar o melhor modelo molecular de substituição a ser usado na
reconstrução filogenética, foi utilizado o programa JModeltest 2.1.5 (Darriba et al.,
2012), que indicou HKY 85 (Hasegawa et al., 1985), como melhor modelo.
A reconstrução filogenética pelo método de máxima verossimilhança foi
24

realizada no programa PHYML (Guindon e Gascuel 2003) utilizando bootstrap de
1000 replicas, um método que consiste em um algorítimo simples que ajusta as
topologias das árvores e comprimento dos ramos simultaneamente. Este algoritmo
começa de uma árvore inicial construída por um rápido método baseado em
distância e modifica esta árvore para melhorar sua probabilidade a cada iteração.
Devido aos ajustes simultâneos na topologia e comprimento dos braços, somente
poucas interações são suficientes para atingir o ótimo (Guindon e Gascuel 2003).
Foram utilizadas as espécies Chiropotes chiropotes, Chiropotes utahicki, Cacajao
calvus rubicundus e Cacajao calvus calvus como grupos externos.
3.5.2 ANÁLISES NGS
Para esta etapa foi utilizado o programa pyRAD (Eaton, 2014), desenvolvido
para análises a nível populacional e filogenético. Este programa é um pipeline que
agrupa alinhamentos de novo para RADseq loci com a aplicabilidade de uso para
diferentes técnicas de preparo de bibliotecas genômicas.
Este programa separa inicialmente os RADseq de cada indivíduos por
barcode e, posteriormente, realiza alinhamento de novo (sem genoma de referência)
procurando sequencias ortólogas segundo padrão de similaridade de 88%, sendo
que cada alelo deve ter uma cobertura mínima de sete leituras (sequencias).
Posterior à construção de loci de novo o pipeline realizou a construção de
sequencia concatenadas únicas por indivíduo as quais são homólogas entre elas e a
realização de uma matriz de SNPs não ligados para análises populacionais.
3.5.3. RECONSTRUÇÃO FILOGENÉTICA
A construção filogenética dos RADseq para as sequencias concatenadas foi
construída no programa PhyML implementado no programa Geneious, com a
utilização de 1000 réplicas de Bootstrap e suporte pelo modelo evolutivo
Shimodaira-Hasegawa (1999) com a utilização de C. calvus novaesi como grupo
externo.
25

3.5.4. GRUPOS BIOLÓGICOS
Para determinar a existência de grupos biológicos distintos entre C. ayresi, C.
hosomi e C. melanocephalus foi utilizada uma abordagem Bayesiana de
agrupamento probabilístico em um modelo em que há K populações, cada uma das
quais é caracterizada por um conjunto de frequências alélicas em cada locus
(Pritchard et al., 2000). Esta abordagem permite atribuir indivíduos às populações
com base em seus genótipos multilocos e estima simultaneamente a frequência dos
alelos nas populações. Este método pode ser aplicado a diversos marcadores como
por exemplo SNPs (Pritchard et al., 2000; Falush et al., 2003).
No caso dos uakaris, ocorrerá a busca das frequências alélicas de casa
indivíduo para cada locus na tentativa de diminuir o desvio do equilíbrio de Hardy-
Weinberg com objetivo de determinar a existência de grupos biológicos (Falush et al.
2003) utilizado os modelos de mistura (admixture model) e de frequências alélicas
correlacionadas entre populações (correlated allele frequencies).
O admixture model, permite que cada indivíduo possa apresentar uma
característica genética não necessariamente exclusiva à apenas uma população
biológica, uma vez que esses organismos podem trazer consigo uma herança
genética de ancestrais recentes derivados de mais de uma população biológica
(Falush et al., 2003).
O correlated allele frequencies, assume que as frequências alélicas em
populações proximamente relacionadas devem ser similares, sendo ele capaz de
identificar populações que são subestruturadas uma vez que a estrutura
populacional gera desequilíbrio de ligação (Falush et al. 2003). Todos esses modelos
foram então implementados no programa STRUCTURE versão 2.3.4 (Pritchard et al.
2000; Falush et al. 2003; Falush et al., 2007).
Para cada valor de K testado, foram realizadas 1.000.000 réplicas sob o
algoritmo Monte Carlo via Cadeias de Markov (MCMC) com valores de corte (burn-
in) de 100.000 permutações. Os outputs produzidos foram importados dentro do
programa STRUCTURE HARVESTER 0.6.92 (Earl e VonHoldt, 2012), onde os
dados obtidos foram sumarizados. Atribui cada indivíduo sua composição genética
no programa CLUMP 1.1.2 (Jakobsson & Rosenberg, 2007), que avalia a
26

similaridade de tais resultados e produz uma matriz final dos dados.
Tal matriz foi analisada dentro do programa DISTRUCT 1.1 (Rosenberg,
2004), onde foi elaborada a representação gráfica dos mesmos. O valor de K
escolhido foi aquele maximizado pelo modelo log-likelihood (log(P(X/K)), por esta ser
frequentemente a melhor escolha para o número de populações de acordo com os
modelos testados (Falush et al. 2003).
4. RESULTADOS
4.1. RECONSTRUÇÃO FILOGENÉTICA COM O GENE CITOCROMO B
Os resultados da reconstrução filogenética do citocromo b mostram a
existência de uma monofilia recíproca existente entre as três espécies estudadas
(Figura 4). Para isso deve-se excluir a amostra duvidosa portadora de “missing data”
causadora da parafilia, MPEG 8991.
A espécie C. melanocephalus teve maior representação com vinte uma
sequências utilizadas destas, sete novas sequencias incluídas aumentando a
distribuição da amostragem na filogenia com indivíduos do alto rio Japurá e todos os
indivíduos utilizados se agrupam com alto valor de suporte. Na margem esquerda
são encontradas as espécies C. hosomi com oito sequências, sendo uma sequência
nova e C. ayresi com sete sequencias, duas novas, todas sequências se agrupam
em seus respectivos grupos taxonômicos.
27

Figura 5 – Árvore filogenética de verossimilhança do Citocromo b representando as linhagens
evolutivas encontradas. Cacajao melanocephalus em vermelho, Cacajao hosomi em amarelo e
Cacajao ayresi em marrom indicando a monofilia do grupo com a exclusão da amostra MPEG 8991
de qualidade inadequada.
4.2. FILOGENÔMICA
A construção filogenética com a utilização dos RADseq, foi realizada com a
utilização de 626 loci RADseq, destes 1.235 sendo sítios variáveis e 568
28

parcimoniosos. Os resultados indicam a existência de duas linhagens distintas para
as três espécies estudas, com >95% de valor de bootstrap. Foi delimitada uma
linhagem na margem direita do rio Negro composta pelos onze indivíduos de C.
melanocephalus, e na margem esquerda uma linhagem que agrupa os quatro
indivíduos da espécie C. hosomi junto com os cinco indivíduos da espécie C. ayresi
(Figura 6).
Figura 6 – Árvore filogenética de verossimilhança de RADseq representando as linhagens evolutivas
encontradas. Cacajao melanocephalus em vermelho, suportada por 0,969. Cacajao hosomi e
Cacajao ayresi em amarelo indicando um grupo polifilético com 0,999 de valor de suporte.
29

4.3 GRUPOS BIOLÓGICOS
Os resultados foram construídos a partir de uma matriz de SNPs de 477
SNPs não ligados. Foram testados K=5, o que melhor representa a formação de
grupos biológicos foi de K=2 (Figura 6), indicados pelos valores de probabilidade
posterior lnPr=-2324.23 (Figura 7). Os resultados dos grupos biológicos indicam que
as três espécies estudadas se agrupam em dois grupos biológicos distintos, cada
grupo ocupa uma margem do Rio Negro, sendo este barreira para os grupos. Os
onze indivíduos indicados pertencentes a espécie C. melanocephalus formam um
grupo biológico, ocupando a margem direita, enquanto que os quatro indivíduos
indicados serem C. hosomi e os cinco indivíduos indicados como C. ayresi, formam
um grupo biológico na margem esquerda, o grupo da margem esquerda compõe as
duas espécies, indícios da existência de compartilhamento de sistema de
acasalamento.
Dois indivíduos do grupo biológico da margem esquerda, classificados como
C. hosomi, são híbridos (indivíduos que possuem acima de 5% do seu genoma
proveniente de outra espécie). Estes possuem mistura genética com o grupo
biológico da margem direita composto por C. melanocephalus. Estes resultados
indicam a possibilidade de quebra de barreira nas cabeceiras do Rio Negro e
provável existência de uma área de contato com uma zona de hibridização entre os
dois grupos biológicos.
C. melanocephalus C. hosomi C. ayresi
Figura 7 – Grupos biológicos encontrados. Em vermelho C. melanocephalus forma um grupo, em
amarelo C. hosomi e C. ayresi, formam outro grupo biológico.
30

Figura 8 - Resultado do indicando k=2, como a melhor probabilidade para explicar os dados. A
vertical mostra a média da estimativa da distribuição das probabilidades dos dados e a horizontal
número de clusters testados (k) e o desvio padrão (SD) da probabilidade média.
5. DISCUSSÃO
No momento, existem três espécies válidas de uakaris preto, Cacajao
melanocephalus na margem direita do rio Negro, C. hosomi e C. ayresi na margem
esquerda do rio Negro. Estas espécies foram descritas em uma revisão taxonômica
do grupo, baseada em critérios morfológicos (morfometria e coloração), ecológicos
(utilização do ambiente), da distribuição geográfica (ocorrência alopátrica) e filogenia
molecular (Citocromo b) (Boubli et al., 2008). Após esta proposta que reorganizou a
antiga taxonomia que reconhecia apenas uma espécie separada em duas
subespécies (C. m. ouakari e C. m. melanocephalus) (Hershkovitz, 1987), surgiram
contestações e atualmente não existe consenso sobre quantas espécies de uakaris
pretos existem: as três espécies C. melanocephalus (margem direita do rio Negro) e
C. hosomi e C. ayresi (margem esquerda do rio Negro) sensu Boubli et al., (2008);
ou C. ouakari (margem direita do rio Negro) e C. melanocephalus (margem esquerda
do rio Negro) senso Ferrari et al. (2014).
31

A primeira contestação surge com uma nova construção filogenética do grupo
com a utilização de novas amostras provenientes de peles de museu (Figueiredo et
al., 2009). Uma das amostras proporciona uma parafilia entre C. hosomi e C. ayresi
porém, esta sequencia possui qualidade baixa (Figueiredo et al., 2009). A segunda
contestação surge propondo uma nova organização taxonômica se baseando nos
resultados filogenéticos da primeira contestação, com a adição de novas
informações morfológicas e biogeográficas (Ferrari et al. 2014). Propõe o
reconhecimento de duas espécies válidas uma de cada lado do rio Negro,
reestabelecendo a nomenclatura anterior sensu Hershkovitz (1987) e classificando
C. ayresi como subespécie de C. melanocephalus.
A nova proposta da reconstrução filogenética do presente trabalho com o
Citocromo b com a inclusão de novas amostras propõe a existência de uma
monofilia recíproca, esta encontrada com a exclusão da amostra MPEG 8991
identificada como portadora de “missig data”, que causa a parafilia entre C. hosomi e
C. ayresi.
Cada uma dessas três espécies classificadas como válidas, C.
melanocephalus, C. hosomi e C. ayresi; possui todos seus indivíduos agrupados no
mesmo clado e por isso classificadas como espécies válidas sob o conceito
filogenético de espécies (Cracraft, 1983). Este conceito filogenético identifica
espécies sob o critério da monofilia, sendo que os grupos monofilético são definidos
como linhagens que contém todos os descendentes conhecidos de um ancestral
comum. Desta forma, as espécies são identificadas estimando-se a filogenia de
populações estreitamente relacionadas e encontrando os menores grupos
monofiléticos, para serem espécies filogeneticamente separadas. Essas populações
ficaram separadas durante um tempo suficiente o que permitiu a evolução das
características diagnósticas.
Em contrapartida, as novas abordagens para a construção filogenética
baseadas em dados genômicos, utilizadas no presente trabalho, trouxeram novas
interpretações sobre os uakaris pretos como observado na filogenia dos RADseq.
Com dados encontrados, fica clara a existência de duas linhagens evolutivas
distintas, com os indivíduos classificados como C. melanocephalus formando uma
linhagem na margem direita do rio Negro, enquanto os indivíduos classificados como
32

C. hosomi e C. ayresi formando outra linhagem na margem esquerda. Desta forma,
existe uma polifilia na linhagem evolutiva composta por C. hosomi e C. ayresi, sendo
assim e levando em consideração o conceito filogenético de espécies proposto por
Cracraft (1983), as duas espécies não poderiam ser classificadas como espécies
distintas e, portanto, não estão seguindo trajetórias evolutivas independentes.
Os resultados da formação dos grupos biológicos são inéditos para os uakaris
pretos, ainda não tendo sido testados a nível populacional. Estes dados indicam a
existência de dois grupos biológicos distintos, sendo um composto pelos indivíduos
de C. melanocephalus e outro composto pelos indivíduos de C. hosomi e C. ayresi.
De acordo com o conceito biológico de espécies, o isolamento reprodutivo é o
critério essencial na identificação da independência evolutivas das espécies, porque,
confirma a ausência de fluxo gênico. Se estas populações de organismos não se
hibridizam regularmente na natureza, ou quando ocorre, incapaz de produzir prole
fértil, estas estão isoladas reprodutivamente e são consideradas espécies distintas.
Entre C. hosomi e C. ayresi foi observado que estas possuem compartilhamento no
sistema de acasalamento, formam um mesmo grupo biológico e por isso não são
válidas sob este critério.
Separação entre as linhagens mitocondriais também foram relatadas em
Gato leopardo (Prionailurus bengalensis) na Índia. Estas separadas entre linhagens
ao norte e sul dentro da mesma espécie, com suas respectivas populações bem
estruturadas em cada região. As variações climáticas foram indicadas como
prováveis explicação para a separação entre as duas linhagens. Nesse caso ocorreu
uma separação ecológica entre as duas linhagens (Mukherjee et al., 2010).
Discordâncias entre os resultados do DNA mitocondrial e nuclear como
encontrados no presente estudo são relatadas na literatura. Como a taxa de
evolução do genoma mitocondrial é estimada em 5, 7 X 10 -8 dez vezes mais rápida
que o nuclear (Brown et al. 1982), pode-se encontrar divergência entre genes em
uma mesma espécie assim como separação mitocondrial entre a mesma espécie.
Discordâncias dos dados do DNA mitocondrial e nuclear foi encontrado em
um estudo realizado com peixes do gênero Xiphophorus. Este gênero possui uma
divisão na morfologia que separa as duas formas (platis e espadas) porém, as
espécies X. clemenciae e X. monticolus se agrupam pelo DNA mitocondrial
33

juntamente com os platis porém, sua morfologia indica pertencer aos espadas.
Diferentemente, com a utilização de genes nucleares foi encontrada incongruência,
sendo que a nova resolução filogenética agrupou as duas espécies com seus
respectivos grupos morfológicos, indicando que estes surgiram por evento de
hibridização (Kang et al., 2013). Posteriormente, este trabalho foi reexaminado com
a utilização de RADseq que corroborou com a nova classificação das espécies por
genes nucleares (Jones et al., 2013).
Dados de RADseq também foram utilizados por Cui et al., (2014) para
esclarecer as relações filogenéticas em presença de hibridização dos peixes
espadas e platis. Os resultados demonstraram que a hibridação foi
surpreendentemente generalizada na história evolutiva do gênero confirmando
também a classificação de X. clemenciae e X. monticolus.
Argumentações atuais sugerem a importância da inclusão de dados nucleares
para a melhor resolução das relações filogenéticas das espécies e indicando que o
DNA mitocondrial é uma ferramenta extremamente útil, mas que em certos casos
pode ser limitada (Barrowclough e Zink, 2009) ou apresentar resultados
discordantes. Sendo assim, a inclusão de informações geradas por NGS são de
grande importância para a resolução de difícil resolução baseados apenas em um
tipo de marcador, como o mitocondrial.
Um exemplo, é o trabalho de Wagner et al., (2013) que por meio de dados de
NGS conseguiram encontrar resoluções ao nível de espécie para dezesseis
espécies de peixes ciclídeos do Lago Vitória provenientes de uma mesma
comunidade de uma ilha rochosa com radiação adaptativa, onde trabalhos
anteriores baseados apenas em análises filogenética não encontraram nenhuma
resolução.
Outro exemplo é o trabalho de Emerson et al. (2010), onde os autores
conseguiram delimitar as linhagens evolutivas do mosquito Wyeomyia smithi, que
possuíam relações mal esclarecidas com outros dados, pois divergiram
recentemente. Neste trabalho foi encontrada separação da espécie em duas
principais linhagens na América do Norte, uma ao norte e outra ao sul com divisões
internas para três formas ao norte e duas ao sul.
Porém alguns trabalhos com a utilização de RADseq indicam indícios da
34

separação filogenética entre as linhagens de subespécies de borboletas do gênero
Heliconius e formação de grupos biológicos similares entre as linhagens indicadas
como divergentes (Nadeau et al., 2013).
As duas linhagens evolutivas dos uakaris encontradas neste estudo com
dados de RADseq se agruparam em dois grupos biológicos distintos, um em cada
margem do rio Negro e desta maneira, fica clara a importância do rio na separação
destas linhagens, sendo um agente importante no processo de diversificação destes
grupos. O rio Negro é conhecido por ser uma das grandes barreiras biogeográficas
da Amazônia e anteriormente já tinha sido proposto como agente vicariante entre as
linhagens dos uakaris pretos assim como para outros primatas (Boubli et al., 2015) e
aves não voadoras do gênero Psophia (Ribas et al., 2012). Porém, o indício da
existência de híbridos entre os dois grupos biológicos encontrados indica uma
provável quebra na barreira nas cabeceiras, que anteriormente já havia sido
proposta para aves (Naka et al., 2012). Casos de hibridização para macacos do
novo mundo já foram relatadas anteriormente em macaco-aranha, guaribas e
calitriquideos (Arnould et al., 2009).
CONCLUSÃO
Diante das novas evidências apresentadas, C. hosomi e C. ayresi não podem
ser classificadas como espécies distintas, por serem agrupadas por dois critérios de
delimitação de espécies. Estas se encontram em polifilia em uma mesma linhagem
evolutiva bem suportada e ficarem agrupadas em um mesmo grupo biológico e
desta forma compartilham sistema de acasalamento, sendo assim, C. hosomi e C.
ayresi não podem existir como espécies distintas, pois os seus respectivos genomas
se encontram em equilíbrio de Hardy-Weinberg. Porém, também pode ser um caso
de estruturação intraespecífica ou um caso de especiação adaptativa incipiente, um
processo em início de especiação, e desta forma necessita a busca de genes sobre
seleção associados com a divergência.
35

REFERÊNCIAS
Arnold, M. L,; Meyer, A. 2006. Natural hybridization in primates: One evolutionary
mechanism. Zoology, 109: 261-276.
Ayres, J. M. 1989. Comparative feeding ecology of the uakari and bearded saki,
Cacajao and Chiropotes. Journal of Human Evolution, 18: 697–716.
Boubli, J. P. 1993. Southern expansion of the geographical distribution of Cacajao
melanocephalus melanocephalus. International Journal of Primatology, 14(6): 933–
937.
Boubli, J. P. 1999. Feeding ecology of black-headed uacaris (Cacajao
melanocephalus melanocephalus) in Pico da Neblina National Park, Brazil.
International Journal of Primatology, 20(5): 719-749.
Boubli, J. P.; da Silva, M. N.; Amado, M. V.; Hrbek, T.; Pontual, F. B.; Farias, I. P.
2008. Taxonomic Reassessment of Cacajao melanocephalus Humboldt (1811), with
the Description of Two New Species. International Journal of Primatology, 29: 723–
741.
Boubli, J. P.; Ribas, C.; Lynch Alfaro, J. W.; Alfaro, M. E.; da Silva, M. N. F.; Pinho, G.
M.; Farias, I. P. 2015. Spatial and temporal patterns of diversification on the Amazon:
A test of the riverine hypothesis for all diurnal primates of Rio Negro and Rio Branco
in Brazil. Molecular Phylogenetics and Evolution, 82: 400–412.
Brown, W. M.; Prager, E. M.; Wang, W.; Wilson, C. 1982. Mitochondrial DNA
Sequences of Primates: Tempo and Mode of Evolution, Journal of Molecular
Evolution 18: 225-239
Cozzuol, M. A.; Clozato, C. L.; Holanda, E. C.; Rodrigues, F. H. G.; Nienow, S.;
Thoisy, B.; Redondo, R. A. F.; Santos, F. R. 2013. Journal of Mammalogy, 94(6):
36

1331–1345.
Cozzuol, M. A.; Thoisy, B.; Fernades-Ferreira, H.; C.; Rodrigues, F. H. G.; Santos, F.
R. How much evidence is enough evidence for a new species? Journal of
Mammalogy, 95(4) :899–905.
Cui, R.; Schumer, M.; Kruesi, K.; Walter, R.; Andolfatto, P.; Rosenthal, G. G. 2013.
Phylogenomics Reveals Extensive Reticulate Evolution in Xiphophorus Fishes.
Evolution, 67-8: 2166–2179.
Cracraft, J. 1983. Species concepts and speciation analysis. Current Ornithology, 1:
159-187.
Cariou, M.; Duret, L.; Charlat, S. 2013. Is RAD-seq suitable for phylogenetic
inference? An in silico assessment and optimization. Ecology and Evolution, 3(4):
846–852.
Chan, C. X.; Ragan, M. A. Next-generation phylogenomics. Biology Direct 2013, 8: 3.
Darriba D.; Taboada, G. L.; Doallo, R.; Posada, D. 2012.jModelTest 2: more models,
new heuristics and parallel computing. Nature Methods, 9(8): 772.
Doyle, J. J.; Doyle, J. L. 1987. A rapid DNA isolation procedure for small quantities of
fresh leaf tissue. Phytochemical Bulletin, 19: 11-15.
De Queiroz K (1998) The general lineage concept of species, species criteria, and
the process of speciation: A conceptual unification and terminological
recommendations. In: Howard D. J., Berlocher S.H., (eds) Endless Forms: Species
and Speciation. Oxford University Press, Oxford, England, 57–75.
de Queiroz, K. 2007. Species Concepts and Species Delimitation. Systematic
Biology, 56: 879 – 886.
37

Earl, D. A.; von Holdt, B. M. 2012. STRUCTURE HARVESTER: a website and
program for visualizing STRUCTURE output and implementing the Evanno method.
Conservation Genetics Resources, 4: 359–361.
Eaton, D. A. R. 2014. PyRAD: assembly of de novo RADseq loci for phylogenetic
analyses. Bioinformatics, 30(13): 1844-1849.
Emerson, K. J.; Merz, C. R.; Catchen, J. M.; Hohenlohe, P. A.; Cresko, W. A.;
Bradshaw, W. E.; Holzapfel, C. M. 2010. Resolving postglacial phylogeography using
high-throughput sequencing. Proceedings of the National Academy of Sciences of
the United States of America, 107(37): 16196-16200.
Falush D, S. M.; Pritchard, J. K. 2003. Inference of population structure using
multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164:
1567–1587.
Falush D, S. M.; Stephens, M.; Pritchard, J. K. 2007. Inference of population
structure using multilocus genotype data: dominant markers and null alleles.
Molecular Ecology Notes, 7: 574–578.
Ferrari, S.; Guedes, P. G.; Figueiredo-Ready, W. M. B.; Barnett, A. A. 2014.
Reconsidering the taxonomy of the Black-Faced Uacaris, Cacajao melanocephalus
group (Mammalia: Pitheciidae), from the northern Amazon Basin. Zootaxa 3866 (3):
353–370.
Figueiredo-Ready, W. M. B.; Schneider, H.; Ferrari, S. F.; Haralda, M. L.; da Silva, J.
M. C.; Silva Júnior, J. S.; Bates, J. M. 2013. A molecular phylogeography of the
uacaris (Cacajao). In: Veiga, E.; Ferrari, S. F.; Narconk, M. A. Evolutionary Biolgy and
Conservation of Tits, Sakis and Uacaris. Cambrige University Press, p. 23-27.
Fleagle, J. G. 2014. Identifying Primate Species: Themes and Perspectives.
38

Evolutionary Anthropology, 23: 39–40.
Groves,C. 2014. The Species in Primatology. Evolutionary Anthropology, 23: 2–4.
Guindon, S.; Gascuel, O. 2003. A Simple, Fast, and Accurate Algorithm to Estimate
Large Phylogenies by Maximum Likelihood. Systematic biology, 52(5): 696–704.
Hasegawa, M.; Kishino, H.; Yano, T. 1985. Jounal of Molecular Evolution, 22: 160-
174.
Hernandéz-Camacho, J.; Cooper, R. W. 1976. The nonhuman primates of Colombia.
In Thorington Jr, R. W. e Heltne, P. G. Neotropical Primates. Field Studies and
Conservation. Washington, DC: National Academy of Science, p. 35–69.
Hershkovitz, P. 1987. Uacaries, New World Monkeys of the Genus Cacajao
(Cebidae, Platyrrhini): A Preliminary Taxonomic Review With the Description of a
New Subspecies. American Journal of Primatology, 12: 1-53.
Helyar, S. J.; Hemmer-Hansen, J.; Bekkevold, D.; Taylor, M. I.; Ogden, R.; Limborg,
M. T.; Cariani, A.; Maes, G. E.; Diopere, E.; Carvalho, G. R.; Nielsen, E. E. 2011.
Application of SNPs for population genetics of nonmodel organisms: new
opportunities and challenges. Molecular Ecology Resources, 11: 123–136.
Hohenlohe, P. A.; Bassham, S.; Etter, P. D.; Stiffler, N.; Johnson, E. A.; Cresko, W. A.
2010. Population genomics of parallel adaptation in threespine stickleback using
sequenced RAD Tags. PLoS Genetics, 6: 1000862.
Hohenlohe, P. A.; Amish, S. J.; Catchen, J. M.; Allendorf, F. W.; Luikart, G. 2011.
Next-generation RAD sequencing identifies thousands of SNPs for assessing
hybridization between rainbow and westslope cutthroat trout. Molecular Ecology
Resources, 11: 117–122.
39

Hohenlohe, P. A.; Bassham, S.; Currey, M.; Cresko, W. A. 2012. Extensive linkage
disequilibrium and parallel adaptive divergence across threespine stickleback
genomes. Philosophical Transactions of the Royal Society B-Biological Sciences ,
367: 395–408.
Irwin, D. M.; Kocher, T. D.; Wilson, A. C. 1991. Evolution of cytochrome b gene in
mammals. Journal of Molecular Evolution 32: 128-144.
Jakobsson, M.; Rosenberg, N.A., 2007. CLUMPP: a cluster matching and
permutation program for dealing with label switching and multimodality in analysis of
population structure. Bioinformatics, 23(14): 1801–1806.
Kocher, T. D.; Thomas, W. K.; Meyer, A.; Edwards S.V.; Pääbo, S.; Villablanca, F.X.
1989. Dynamics of mtDNA evolution in animals: amplification and sequencing with
conserved primers. Proceedings of Natinal Academy of Sciences. USA 86: 6196–
6200.
Mayr, E. 1942. Systematics and the origin of species. Columbia University Press,
New York.
Nadeau, N. J.; Martin, S. H.; Krzysztof, M. K.; Salazar, C.; Dasmahapatra, K. K.;
Davey, J. W.; Baxter, S. W.; Blaxter, M. L.; Mallet, J.; Jiggins, C. D. 2013. Genome-
wide patterns of divergence and gene flow across a butterfly radiation. Molecular
Ecology, 22, 814–826.
Naka, L. N.; Bechtoldt, C. L.; Henriques, L. M. P.; Brumfiel, R. T. 2012. The Role of
Physical Barriers in the Location of Avian Suture Zones in the Guiana Shield,
Northern Amazonia. The American Naturalist, 179(4): E115-E132.
Padial, J. M.; Miralles, A.; De la Riva, I.; Vences, M. Review The integrative future of
taxonomy. Frontiers in Zoology 2010, 7:16.
40

Perelman, P.; Johnson, W. E.; Roos, C., Seuanez, H. N.; Horvath, J. E.; Moreira, M.
A. M.; Kessing, B., Pontius, J.; Roelke, M.; Rumpler, Y.; Schneider, M. P.; Silva, A.;
O’Brien, S. J.; Pecon-Slattery, J. 2011 A Molecular Phylogeny of Living Primates.
Plos Genetics, 7(3): e1001342.
Peterson, B. K.; Weber, J. N.; Kay, E. H.; Fisher, H. S.; Hoekstra, H. E. Double Digest
RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in
Model and Non-Model Species. PLoS ONE, 7: 37135.
Philippe, H.; Derelle, R.; Lopez, P.; Pick, K.; Borchiellini, C.; Boury-Esnault, N.;
Vacelet, J.; Renard, E.; Houliston, E.; Quéinnec, E.; Da Silva, C.; Wincker, P.; Le
Guyader, H.;Leys, S.; Jackson, D. J.; Schreiber, F.; Erpenbeck, D.; Morgenstern, B.;
Worheide, Manuel, M. 2009. Phylogenomics Revives Traditional Views on Deep
Animal Relationships. Current Biology, 19: 706–712.
Platt, A., Woodhall, R. & George Jr, A., 2007. Improved DNA sequencing quality and
efficiency using an optimized fast cycle sequencing protocol. BioTechniques, 43(1):
58–62.
Pritchard, J. K. Stephens, M. Donnelly, P. 2000. Inference of population structure
using multilocus genotype data. Genetics, 155: 945–959.
Prasad, A. B.; Allard, M. W.; Green, E. D. Program NCS. 2008. Confirming the
phylogeny of mammals by use of large comparative sequence data sets. Molecular
Biology and Evolution, 25, 1795–1808.
Ribas, C. C.; Aleixo, A.; Nogueira, A. C. R.; Miyaki, C. Y.; Cracraft J. 2011. A
palaeobiogeographic model for biotic diversification within Amazonia over the past
three million years. Proceedings of the Royal Society, 279 (1729), 681–689.
Rosemberg, N. A. 2004. DISTRUCT : a program for the graphical display of
population structure. Molecular Ecology Notes, 4: 137–138.
41

Shimadaira, H.; Hasegawa, M. 1999. Multiple Comparisons of Log-Likelihoods with
Applications to Phylogenetic Inference. Molecular Biology and Evolution, 16(8):1114–
1116.
Thompson, J. D.; Higgins, D. G.; Gibson, T. J .CLUSTAL W: improving the sensitivity
of progressive multiple sequence alignment through sequence weighting, position-
specific gap penalties and weight matrix choice. Nucleic Acids Research, 22(22):
4673-4680.
Voss, R. S.; Helgen, K. M.; Jansa, S. A. 2014. Extraordinary claims require
extraordinary evidence: a comment on Cozzuol et al. (2013) .Journal of Mammalogy,
95(4) :893–898.
Wagner, C. E.; Keller, I.; Wittwer, S.; Selz, O. M.; Waiko, W.; Greuter, L.; Sivasundar,
A.; Seehausen, O. 2013 .Genome-wide RAD sequence data provide unprecedented
resolution of species boundaries and relationships in the Lake Victoria cichlid
adaptive radiation. Molecular Ecology Resources, 22: 787–798.
Werle, E, Scneider, C. 1994. Convenient single-step, one tube purification of PCR
products for direct sequencing. Nucleic Acids Research, 22: 4354–4355.
42
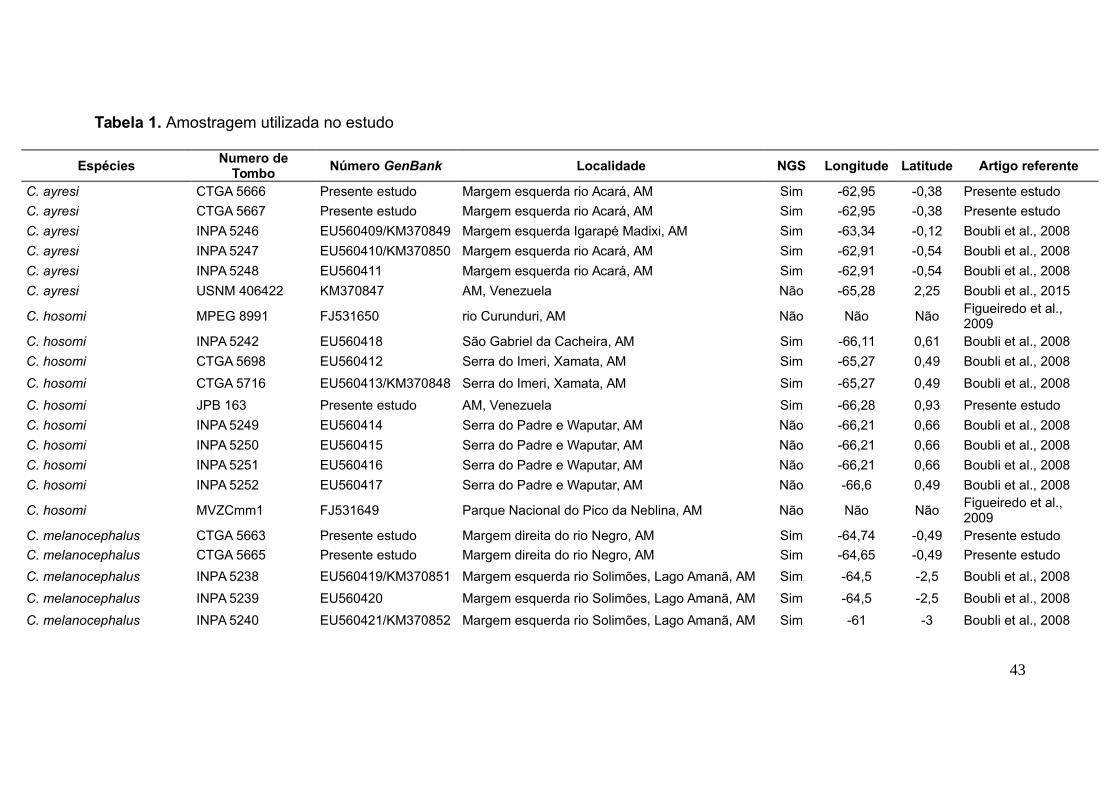
Tabela 1. Amostragem utilizada no estudo
EspéciesNumero de
TomboNúmero GenBank Localidade NGS Longitude Latitude Artigo referente
C. ayresi CTGA 5666 Presente estudo Margem esquerda rio Acará, AM Sim -62,95 -0,38 Presente estudo
C. ayresi CTGA 5667 Presente estudo Margem esquerda rio Acará, AM Sim -62,95 -0,38 Presente estudo
C. ayresi INPA 5246 EU560409/KM370849 Margem esquerda Igarapé Madixi, AM Sim -63,34 -0,12 Boubli et al., 2008
C. ayresi INPA 5247 EU560410/KM370850 Margem esquerda rio Acará, AM Sim -62,91 -0,54 Boubli et al., 2008
C. ayresi INPA 5248 EU560411 Margem esquerda rio Acará, AM Sim -62,91 -0,54 Boubli et al., 2008
C. ayresi USNM 406422 KM370847 AM, Venezuela Não -65,28 2,25 Boubli et al., 2015
C. hosomi MPEG 8991 FJ531650 rio Curunduri, AM Não Não NãoFigueiredo et al., 2009
C. hosomi INPA 5242 EU560418 São Gabriel da Cacheira, AM Sim -66,11 0,61 Boubli et al., 2008
C. hosomi CTGA 5698 EU560412 Serra do Imeri, Xamata, AM Sim -65,27 0,49 Boubli et al., 2008
C. hosomi CTGA 5716 EU560413/KM370848 Serra do Imeri, Xamata, AM Sim -65,27 0,49 Boubli et al., 2008
C. hosomi JPB 163 Presente estudo AM, Venezuela Sim -66,28 0,93 Presente estudo
C. hosomi INPA 5249 EU560414 Serra do Padre e Waputar, AM Não -66,21 0,66 Boubli et al., 2008
C. hosomi INPA 5250 EU560415 Serra do Padre e Waputar, AM Não -66,21 0,66 Boubli et al., 2008
C. hosomi INPA 5251 EU560416 Serra do Padre e Waputar, AM Não -66,21 0,66 Boubli et al., 2008
C. hosomi INPA 5252 EU560417 Serra do Padre e Waputar, AM Não -66,6 0,49 Boubli et al., 2008
C. hosomi MVZCmm1 FJ531649 Parque Nacional do Pico da Neblina, AM Não Não NãoFigueiredo et al., 2009
C. melanocephalus CTGA 5663 Presente estudo Margem direita do rio Negro, AM Sim -64,74 -0,49 Presente estudo
C. melanocephalus CTGA 5665 Presente estudo Margem direita do rio Negro, AM Sim -64,65 -0,49 Presente estudo
C. melanocephalus INPA 5238 EU560419/KM370851 Margem esquerda rio Solimões, Lago Amanã, AM Sim -64,5 -2,5 Boubli et al., 2008
C. melanocephalus INPA 5239 EU560420 Margem esquerda rio Solimões, Lago Amanã, AM Sim -64,5 -2,5 Boubli et al., 2008
C. melanocephalus INPA 5240 EU560421/KM370852 Margem esquerda rio Solimões, Lago Amanã, AM Sim -61 -3 Boubli et al., 2008
43

C. melanocephalus CTGA 5705 EU560422/KM370846 Margem esquerda rio Solimões, Lago Amanã, AM Sim -65,17 -0,47 Boubli et al., 2008
C. melanocephalus CTGA 65 Presente estudo Margem direita do rio Negro, Igarapé Parati, AM Sim -64,91 -0,58 Presente estudo
C. melanocephalus CTGA 98 Presente estudo Margem direita rio Negro, Igarapé Aiuanã, AM Sim -64,93 -0,62 Presente estudo
C. melanocephalus CTGA 756 Presente estudo Margem esquerda rio Japurá, AM Sim -69.20 -1.69 Presente estudo
C. melanocephalus CTGA 757 Presente estudo Margem esquerda rio Japurá, AM Sim -69.20 -1.69 Presente estudo
C. melanocephalus CTGA 775 Presente estudo Margem esquerda rio Japurá, AM Sim -69.34 -1.66 Presente estudo
C. melanocephalus FMNH 88250 FJ531640 Alto rio Inirida, Colômbia Não -70,4 2,3Figueiredo et al., 2009
C. melanocephalus FMNH 88251 FJ531641 Alto rio Inirida, Colômbia Não -70,4 2,3Figueiredo et al., 2009
C. melanocephalus FMNH 89470 FJ531642 Barracon, Alto Cano Itilla, Colômbia Não -72,69 1,61Figueiredo et al., 2009
C. melanocephalus FMNH 89471 FJ531643 Cano Miraflores, rio Vaupés, Colômbia Não -72 1,5Figueiredo et al., 2009
C. melanocephalus FMNH 89468 FJ531644 Lago el Dorado, rio Vaupés, Colômbia Não -70,45 1Figueiredo et al., 2009
C. melanocephalus FMNH 89469 FJ531645 Lago el Dorado, rio Vaupés, Colômbia Não -70,45 1Figueiredo et al., 2009
C. melanocephalus MVZ-016 FJ531646 rio Manacapuru, AM Não -40,76 -73,984Figueiredo et al., 2009
C. melanocephalus MVZ-017 FJ531647 rio Manacapuru, AM Não -40,76 -73,984Figueiredo et al., 2009
C. melanocephalus UFPA-Cmo1 FJ531648 Desconhecido Não Não NãoFigueiredo et al., 2009
C. calvus novaesi INPA 5241 EU560408 Sacado do Turucá, AM Sim Não Não Boubli et al., 2008
Chiropotes chiropotes UFPA-Csa3056 FJ531667 rio Trombetas, Pará Não Não NãoFigueiredo et al., 2009
44
Recommended

















