
Fundamentos de Termodinámica del Equilibrio Físico y Químico
TERMODINÁMICA AVANZADA
MÓDULO I
Ing. Qco. M.Sc. CARLOS ARTURO BELLO BLANCO
UNIVERSIDAD DE SAN BUENAVENTURA
JUNIO DE 2007
CARTAGENA
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 1

Fundamentos de Termodinámica del Equilibrio Físico y Químico
INTRODUCCIÓN
Al ingeniero químico le corresponde en su perfil ocupacional, el análisis, el estudio, la investigación y la aplicación de los principios físicos, químicos y biológicos que brinda la naturaleza, con el fin de diseñar, simular, controlar y optimizar procesos fisicoquímicos de plantas químicas para el fin de obtener productos valiosos a partir de materias primas en armonía con la conservación de los recursos naturales y el medio ambiente. Para ello dispone principalmente de los siguientes principios y leyes fundamentales:
Principio de la conservación de la materia (balance de materia) Principio de la conservación de la energía (balance de energía) o primera ley de la
Termodinámica Principio del incremento de la entropía (balance de entropía) o segunda ley de la
Termodinámica Análisis exergético (balance de energía) Principio del Equilibrio de fases y equilibrio químico (balance de potenciales
químicos) Principio de la Conservación de la cantidad de movimiento (segunda ley de
Newton) Leyes de la cinética química Correlaciones Empíricas y modelos matemáticos, Principios de control, regulación y simulación de procesos.
La aplicación de estos principios está sujeta a restricciones técnicas, sociales, económicas, ambientales, éticas, jurídicas y morales.
Estos principios forman los marcos conceptuales de las áreas más importantes en la ingeniería química como son la Termodinámica, los Fenómenos de Transporte y la Cinética Química. La Termodinámica es la ciencia que estudia la energía, sus transformaciones y las propiedades involucradas en dichas transformaciones y es una de las áreas del
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 2

Fundamentos de Termodinámica del Equilibrio Físico y Químico
conocimiento más importante dentro de la formación y capacitación del profesional de la ingeniería química, ya que juega un papel preponderante en la interpretación, análisis, diseño y simulación de procesos químicos (reactores químicos) y procesos de separación (operaciones unitarias) tales como absorción, destilación, secado, evaporación, extracción líquida, lixiviación, etc.
Debido a esta importancia, el objetivo de este trabajo es brindar a los estudiantes y docentes de ingeniería química, una herramienta a la mano y de fácil comprensión de los principios y fundamentos del área de la Termodinámica del Equilibrio. Para un mejor entendimiento y comprensión de los temas de este trabajo, el lector debe conocer e interpretar los fundamentos y principios de la Fisicoquímica y de la Termodinámica Básica.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 3

Fundamentos de Termodinámica del Equilibrio Físico y Químico
1. PRINCIPIOS FUNDAMENTALES
1.1. Principio de la Conservación de la materia.
El principio de la conservación de la materia establece que la materia no se crea ni se destruye sino que se transforma. Para fluidos en procesos de flujo este principio se expresa mediante la ecuación de continuidad. La ecuación de continuidad de un elemento de volumen de control está dada por:
= - -
Considerando que entran NCE corrientes y salen NCS corrientes de flujo másico, la ecuación de continuidad queda expresada por:
0vuA
vuA
dtdmmm
dtdm NCE
1i enti
iiNCS
1j salj
jj
vc
NCE
1ii
NCS
1jj
vc ∑∑∑∑
====
=
−
+
=−+
(1-1)
donde k
kkk v
uAm = , es el flujo másico, Ak es el área de flujo, uk es la velocidad media y
vk es el volumen específico de la corriente k.
Si el proceso ocurre a estado estacionario, el término de acumulación de materia en el volumen de control es cero y la ecuación de continuidad se convierte en:
∑∑==
=
NCE
1i i
iiNCS
1j j
jj
vuA
vuA
1.2. Principio de la conservación de la cantidad de movimiento.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 4
Velocidad de acumulación de
materia en el volumen de control
Velocidad de flujo de materia que
entra al volumen de control
Velocidad de flujo de materia que sale del volumen
de control

Fundamentos de Termodinámica del Equilibrio Físico y Químico
El principio de la conservación de la cantidad de movimiento representa también la segunda ley de Newton y establece que:
=
Con la aplicación de este principio a un elemento de volumen diferencial se obtiene la siguiente ecuación de variación de la cantidad de movimiento en forma vectorial♦:
[ ] g..pDtDv ρτρ +∇+−∇=
El término de la izquierda es la derivada sustancial del vector velocidad v con respecto al tiempo y representa la acumulación de cantidad de movimiento debido a la aceleración y las fuerzas de inercia debido al movimiento. El primer término de la derecha representa la fuerza de presión. El segundo término es la matriz del esfuerzo cortante y representa las fuerzas viscosas. El tercer término representa la fuerza gravitacional debido al peso del elemento diferencial.
1.3. Primera ley de la termodinámica.
El principio de la conservación de la energía establece que la energía se conserva. La forma general de este principio es la siguiente:
= - - + + -
Los términos de transporte de energía son la energía interna, la energía cinética, la energía potencial que acompañan a la masa que fluye hacia o desde sistema. La velocidad neta de energía que entra desde los alrededores está representada por el flujo de calor, la potencia de flujo y la potencia debido al trabajo de eje a través de la frontera física del sistema.
Para el mismo volumen de control la ecuación del principio de la conservación de la energía está dada por:e Ecuación de variación de cantidad de movimiento en Fenómenos de Transporte
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 5
Acumulación de la cantidad de movimiento
Suma de fuerzas externas
Velocidad de acumulación de energía en el volumen de
control
Velocidad deenergía que entra
con la masa al volumen de
control
Velocidad deEnergía que sale con la masa del
volumen de control
Velocidad neta de energía que entra
desde los alrededores a través de las
fronteras

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( )[ ] ( )[ ] ( ) 0WQeehmeehmdt
)u.m(ds
NCE
1ientipci
NCS
1jsaljpcj
vc =−−++−+++ ∑∑==
(1-2)
Donde u = energía interna, en kJ/kmol o en BTU/lbmolh = entalpía, en kJ/kmol o en BTU/lbmol
ec = energía cinética, en kJ/kmol o en BTU/lbmolep = energía potencial, en kJ/kmol o en BTU/lbmolm = flujo molar, en kg/s o en lbmol/sQ = flujo de calor, en kJ/s o en BTU/s
sW = potencia de eje, en kJ/s o en BTU/s
Para un proceso estacionario, el término de acumulación de energía es cero:
[ ] [ ] ( ) 0WQemem s
NCE
1ientTii
NCS
1jsalTjj =−−− ∑∑
==
(1-3)
Muchos equipos y dispositivos en ingeniería operan a estado estacionario y tienen una entrada y una salida. La ecuación de la primera ley para estos equipos y dispositivos es:
[ ] [ ] ( )
[ ] 0wq
0WQemWQeem
s
sTsTentTsal
=+−
=−−=+−−
θ∆
∆
1.4. Ecuación de la energía mecánica.
De la combinación del principio de la conservación de la cantidad de movimiento y de la energía se deduce la ecuación de la energía mecánica, la cual está dada por:
0wlw)e()e(vdP sp
P
Pc
2
1
=++++∫ ∆∆ (1-4)
Donde lw es el trabajo perdido por mol debido a las irreversibilidades.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 6

Fundamentos de Termodinámica del Equilibrio Físico y Químico
La forma más simple de la ecuación de energía mecánica es la ecuación de Bernoulli aplicada a fluidos incompresibles:
0g
Zgg2)u(P
cc
2
=++∆∆
ρ∆
La ecuación de energía mecánica se usa frecuentemente en la solución de sistemas de flujo en tuberías, conductos y accesorios como válvulas, codos, etc.
1.5. Segunda ley de la termodinámica.
Con la primera ley se estima la cantidad de la energía que se transforma en un proceso determinado, pero no limita la transformación de calor en trabajo útil; la segunda ley restringe el uso de la primera ley porque establece criterios para determinar la calidad de la energía y la energía disponible, es decir, la energía que puede transformarse en trabajo útil, también determina la dirección de los procesos, establece cuando un proceso es posible, espontáneo o está en equilibrio termodinámico, y también proporciona los criterios para determinar la idealidad de un proceso y permite evaluar la eficiencia de un proceso.
Como introducción a la segunda ley se definen los conceptos de depósito térmico y máquinas cíclicas.
Por definición, un depósito térmico es aquel espacio de dimensiones relativamente grande con respecto al sistema que intercambia calor con éste. La característica más importante de un depósito térmico es que sufre procesos isotérmicos e internamente reversibles. Un depósito térmico es una fuente térmica cuando suministra calor al sistema y es un sumidero térmico cuando recibe calor del sistema.
Por otra parte, una máquina térmica es aquel proceso cíclico cuya finalidad es producir trabajo a partir de una transferencia de calor desde al menos un depósito térmico a alta temperatura hacia al menos otro depósito térmico a baja temperatura. La eficiencia de una máquina térmica se define como el cociente entre el trabajo producido y el calor suministrado desde los depósitos térmicos a alta temperatura o fuentes, es decir:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 7
Fundamentos de Termodinámica del Equilibrio Físico y Químico
∑∑
∑∑∑
∑−=
−
==
ff
ss
ff
ss
ff
ff
netotérmica Q
Q1
Q
QWη
El calor que se transfiere hacia el o los depósitos térmicos a baja temperatura o sumideros limita la conversión del calor desde las fuentes en trabajo. Esto quiere decir que no es posible operar una máquina térmica con una eficiencia de 100%.
Un refrigerador es aquel proceso cíclico cuya finalidad es mantener un depósito a temperatura por debajo de la del entorno (fuente) mediante la transferencia de calor desde ese espacio (fuente) hacia otro depósito a temperatura alta (sumidero) y el suministro de trabajo desde el entorno. El coeficiente de rendimiento de un refrigerador se define como el cociente entre el calor suministrado desde el depósito a temperatura baja o fuente y el trabajo consumido, es decir:
1Q/Q1
QQQ
WQ
COPfuentesumiderofuentesumidero
fuente
consumido
fuente
−=
−==
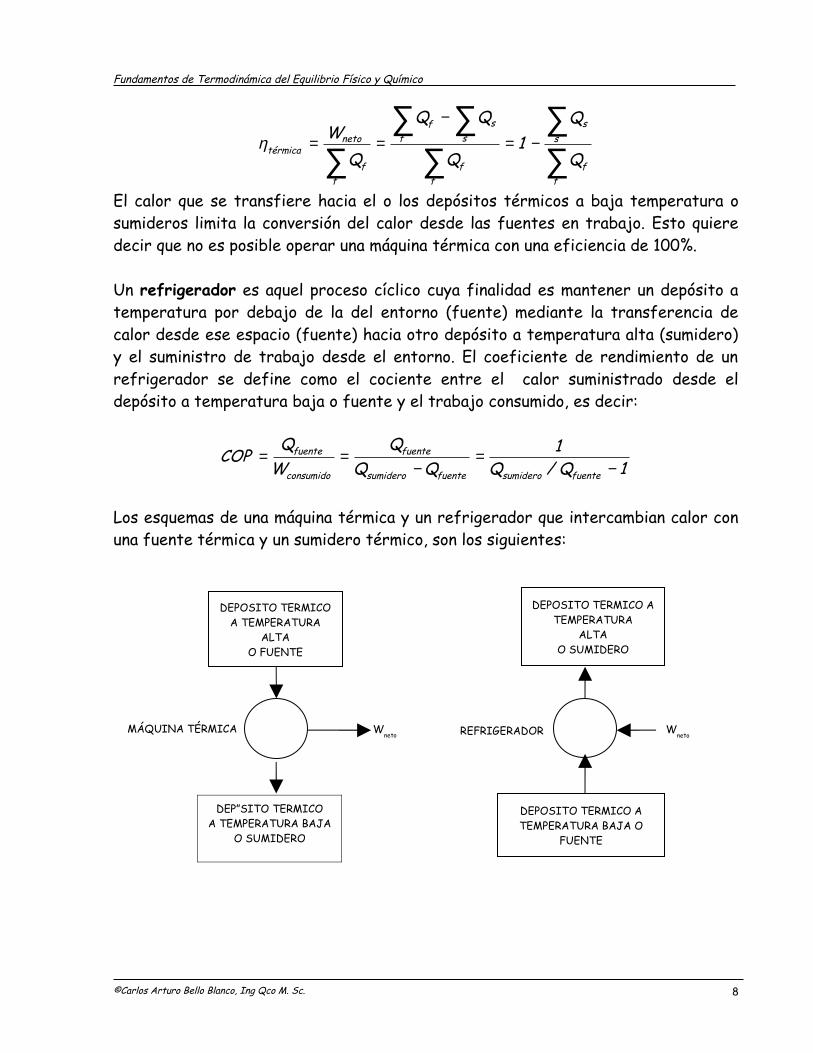
Los esquemas de una máquina térmica y un refrigerador que intercambian calor con una fuente térmica y un sumidero térmico, son los siguientes:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc.
MÁQUINA TÉRMICA Wneto
Wneto
DEPOSITO TERMICOA TEMPERATURA
ALTAO FUENTE
DEPOSITO TERMICO A TEMPERATURA BAJA O
FUENTE
REFRIGERADOR
DEPOSITO TERMICO A TEMPERATURA
ALTA O SUMIDERO
8
DEPÓSITO TERMICOA TEMPERATURA BAJA
O SUMIDERO

Fundamentos de Termodinámica del Equilibrio Físico y Químico
La primera ley aplicada a una máquina térmica o un refrigerador reversible o irreversible que intercambia calor con una fuente térmica y un sumidero térmico, está expresada por:
∫ ∫ ∫ ==− 0dEWQ sistemaδδ
Pero netosumiderofuentesumiderofuente QQQQQQ =+=+=∫ ∫∫ δδδ
∫ = netoWWδ
Reemplazando en la ecuación de la primera ley queda:
netoinadolimeistradominsuneto WQQQ =+=
El resultado anterior indica que en una máquina térmica no es posible convertir el calor suministrado en trabajo neto.
Los enunciados de Kelvin-Planck y de Clausius expresan las limitaciones de las máquinas térmicas y los refrigeradores.
El enunciado de Kelvin y Planck enmarca la limitación de la primera ley:
Es imposible construir una máquina térmica cuyo único efecto sea el intercambio de calor con una sola fuente inicialmente en equilibrio y la producción de trabajo neto.
De acuerdo con este enunciado es imposible alcanzar una eficiencia térmica del 100% en la transformación de calor en trabajo neto.
El enunciado de Clausius establece que:
Es imposible operar una máquina cíclica de tal manera que el único efecto externo a la máquina sea la transferencia de calor desde un depósito a otro depósito a una temperatura mayor.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 9

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Así, es imposible transferir calor desde una temperatura menor a una mayor a menos que se suministre trabajo al sistema.
La desigualdad de Clausius establece que en un proceso cíclico:
La integral cíclica de la cantidad δQ/T es menor o igual a cero.
∫ ≤ 0TQδ
La igualdad se cumple para procesos cíclicos reversibles y la desigualdad para procesos cíclicos irreversibles. Un proceso cíclico que no cumple con la desigualdad de Clausius es un proceso imposible.
1.5.1.Procesos reversibles. Un proceso reversible es aquel que al retornar a su estado inicial no deja consecuencias en los alrededores.
La máquina de Carnot es un ejemplo de máquina térmica reversible, por lo tanto cumple la igualdad de Clausius.
Reemplazando las dos etapas no adiabáticas de la máquina de Carnot, en la ecuación
de Clausius, ésta se convierte en:
∫∫ ∫ =
+
=
1
2 sumideroL
L2
1 fuenteH
H 0TQ
TQ
TQ δδδ
La primera integral del término derecho representa el proceso de intercambio de calor QH con el depósito térmico a temperatura alta TH o fuente, y la segunda, el proceso de intercambio de calor QL con el depósito térmico a temperatura baja TL o sumidero.
Debido a que los procesos en los depósitos térmicos son reversibles e isotérmicos,
las integrales de la ecuación anterior se resuelve y se obtiene:
0TQ
TQ
sumideroL
L
fuenteH
H =
+
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 10

Fundamentos de Termodinámica del Equilibrio Físico y Químico
En general: 0TQ
TQ NS
1s s
sNF
1f f
f =
+
∑∑==
Donde NF y NS son el número de fuentes y el número de sumideros.
Ya que por convención el calor suministrado a un sistema es positivo y el calor eliminado es negativo, la ecuación anterior se convierte en:
L
L
H
H
TQ
TQ
=
El resultado anterior es una característica de la máquina de Carnot. Esta característica de los procesos reversibles es necesaria para la definición de la propiedad termodinámica denominada entropía.
Considere un ciclo que consta de dos etapas internamente reversibles A y B. En este caso el proceso cumple la igualdad de la integral cíclica de δQ/T:
∫∫
∫∫ ∫
=
=
+
=
2
1 B
2
1 A
1
2 B
2
1 A
TQ
TQ
0TQ
TQ
TQ
δδ
δδδ
Este resultado permite ver que el valor de la integral es independiente de la trayectoria A o B o cualquiera otra y solo depende de los estados inicial y final; por lo tanto, esta integral representa el cambio de una propiedad termodinámica o variable de estado. Esta propiedad es la entropía designada por S y se define como:
∫
=−=
=
2
1 rev1
rev TQS
TQdS δ∆δ
2S S Integrando ; (A)
1.5.2.Procesos irreversibles. Un proceso irreversible, contrario a uno reversible, deja consecuencias en los alrededores para que pueda retornar a su punto de partida. Considérese ahora un ciclo irreversible donde la trayectoria A es
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 11
1
2A
B

Fundamentos de Termodinámica del Equilibrio Físico y Químico
irrevesible mientras que la B es reversible. La integral cíclica de δQ/T es menor que cero de acuerdo a la ecuación de Clausius, de tal manera que:
∫ ∫∫ ∫ <−+
=
+
=
1
221
2
1 irrev,Arev,B
2
1 irrev,A0)SS(
TQ
TQ
TQ
TQ δδδδ
De esta manera: ∫
>−=
2
1 irrev1 T
QS δ∆ 2S S (B)
Las ecuaciones (A) y (B) representan la segunda ley de la Termodinámica. La combinación de estas ecuaciones da el cambio de entropía de cualquier proceso es:
∫≥−=TQSSS 12
δ∆
(C)
Una máquina térmica puede operar si satisface la siguiente desigualdad:
0TQ
TQ
sumideroL
L
fuenteH
H ≤
+
En general: ∑=
≤
ND
1d d
d 0TQ
(D)
La ecuación (D) es una expresión algebraica, de manera que debe tenerse en cuenta el signo del calor de cada depósito térmico.
Ejemplo 1.1. Una máquina térmica intercambia calor con un depósito (fuente) a 1000 K y un depósito (sumidero) a 300 K. Desde la fuente se suministra 2000 kJ de calor (QH). Calcule la eficiencia térmica, el trabajo producido y el calor eliminado al sumidero, en kJ, si:
a) la máquina opera como una máquina de Carnot,b) la máquina es irreversible y su eficiencia térmica es el 75% de la eficiencia de la máquina de
Carnot.Demuestre si cada máquina satisface la desigualdad de Clausius.
Solución:Balance de energía Wneto = QH – QL; La eficiencia térmica está definida por ηt = 1 – QL/QH = Wneto/QH
a) máquina de Carnot: De la ecuación (D), (ηtCarnot) = 1 – QL/QH = 1 – TL/TH
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 12

Fundamentos de Termodinámica del Equilibrio Físico y Químico
ηt = 1 – QL/QH = 1 – TL/TH = Wneto/QH = 1 – 300/1000 = 0.7; Wneto = ηt *QH = 0.7 *2000 = 1400 kJ; QL = 600 kJ.
Desigualdad de Clausius: (QH/TH) + (QL/TL) = (2000/1000) + (-600/300) = 0 (Ok) b) máquina irreversible: ηt = 0.75*(ηtCarnot) = 0.75*0.7 = 0.525 = 1 – (QL/QH); (QL/QH) = 0.475; QL
= 950 kJWneto = 1050 kJ;
Desigualdad de Clausius: (QH/TH) + (QL/TL) = (2000/1000) + (-950/300) = -1.167 < 0 (Ok)
Los resultados demuestran que ambas máquinas cumplen con la desigualdad de Clausius, por lo tanto, el proceso es posible.1.5.3. Principio del Incremento de la entropía del universo. Por definición, un sistema es una porción del universo que se somete a un análisis termodinámico. Los alrededores junto con el sistema forman un sistema compuesto adiabático que es el universo. El cambio de entropía del universo, según la ecuación anterior, es mayor o igual cero. Esto significa que la entropía no se conserva sino que se incrementa debido a las irreversibilidades de los procesos reales, es decir:
(∆S)universo = Sgen = (∆S)sistema + (∆S)alrededores ≥ 0 (1-5)
Los procesos ideales son aquellos que no tienen incremento de la entropía y suelen compararse con los procesos reales bajo las mismas condiciones para medir la eficiencia de éstos. Así, la eficiencia de un proceso, ηproceso se define como:
)reversible proceso el en esperada (Energíareal) proceso el en esperada (Energía
=procesoη
Considerando el mismo volumen de control de la primera ley, la segunda ley establece:
= + - + ≥ 0
La expresión de la segunda ley, conforme a la ecuación anterior, es la siguiente:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 13
Velocidad de
generación de entropía
en el universo
Velocidad de
acumulación de entropía
en el volumen de
control
Velocidad de entropía que sale con la masa del volumen de
control
Velocidad de entropía que entra con la masa al volumen de control
Velocidad del cambio de entropía debido a la transferencia de calor con los alrededores

Fundamentos de Termodinámica del Equilibrio Físico y Químico
[ ] [ ] 0TQsmsm
dt)s.m(dS
ND
1d d
dNCE
1ientii
NCS
1jsaljj
vcgen ≥+−+= ∑∑∑
===
(1-6)
Los tres primeros términos de la derecha de la ecuación (1-6) representan el cambio de entropía en el sistema, siendo positivo cuando el proceso es de calentamiento o evaporación y negativo para enfriamiento o condensación; el último término es la suma del cambio de entropía de los depósitos térmicos, incluyendo los alrededores, con los que el sistema intercambia calor. Obsérvese que la producción o el consumo de trabajo no están asociados a la generación de entropía. Esto es debido a que el trabajo es una forma de energía con un alto nivel de calidad.
La suma de los cambios de entropía de todos los depósitos térmicos incluyendo los alrededores está dada por:
∑∑−
==
+=1ND
1k k
k
o
oND
1d d
d
TQ
TQ
TQ
En forma general, para procesos reales la ecuación (1-6) indica que la entropía siempre se incrementa y no es conservativa. Este es el principio del incremento de la entropía del universo.
1.6. Análisis exergético (de disponibilidad) de procesos.
La combinación de la primera y segunda leyes de la Termodinámica permite encontrar la potencia o el trabajo útil en un proceso en función del cambio de la energía, el cambio de la entropía y la entropía generada. Para un proceso reversible, esta potencia o trabajo útil sería máximo si se produce o mínimo si se suministra.
Cuando este proceso se lleva a cabo reversiblemente desde el estado inicial del sistema hasta un estado a Po y To, la potencia útil se denomina exergía. La exergía se define como la máxima potencia o el trabajo máximo (o el mínimo) que se produciría o se consumiría si se llevara a cabo dicho proceso reversiblemente; así, la exergía es la disponibilidad o energía disponible que conforma la totalidad de la energía de un sistema:
Energía total = energía disponible + energía no disponible
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 14

Fundamentos de Termodinámica del Equilibrio Físico y Químico
La energía no disponible es aquella parte de la energía total que no puede convertirse en trabajo útil y representa el trabajo perdido o potencial destruido durante el proceso. Este trabajo perdido se conoce también como la irreversibilidad del proceso, I, de modo que:
IWWW útilrevperdida =−=
El estado a Po y To se define como el estado muerto en donde el sistema no tiene exergía o capacidad de realizar un trabajo. El estado de los alrededores con energía cinética y potencial despreciables, se puede considerar como el estado muerto.
1.7. Equilibrio de fases.
El criterio del equilibrio de fase establece que si una mezcla gaseosa de composición molar yi (i=1, 2, 3, ...NC) se encuentra en equilibrio con una mezcla líquida de composición molar xi (i=1, 2, 3, ....NC) a las mismas condiciones de temperatura y presión, la fugacidad de cada componente es igual para cada fase:
Gi
Li ff =
donde Lif = fugacidad del componente i en la fase líquida,
Gif = fugacidad del componente i en la fase gaseosa.
NC = número de componentes
La fugacidad y los coeficientes de fugacidad y la aplicación de este principio se tratarán más adelante en la sección de equilibrio de fase.
1.8. Equilibrio químico.
Este principio establece que en un sistema reaccionante de NR reacciones con NEQ especies químicas en una fase homogénea, el cambio de la energía de Gibbs total a temperatura y presión constantes tiende a un mínimo a medida que se aproxima el equilibrio químico.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 15

Fundamentos de Termodinámica del Equilibrio Físico y Químico
∑ ∑= =
=NEQ
1i
NR
1r
)r(ii 0)(νµ
donde )r(iν = coeficiente estequiométrico de la especie i en la reacción r,
iµ = potencial químico de la especie i.
Los coeficientes estequiométricos de los productos son positivos mientras que los de los reactivos son negativos.
Por ejemplo, considere las reacciones simultáneas independientes:
H2 + ½ O2 ←→ H2O (r = 1)C + ½ O2 ←→ CO (r = 2) C + O2 ←→ CO2 (r = 3)
Los coeficientes estequimétricos de cada especie aparecen en la siguiente tabla:
Coeficientes estequiométricos de las especies químicas.especie qca. ( →)
tipo de reacción ( ↓ )H2 O2 C CO H2O CO2
1 -1 -½ 0 0 1 02 0 -½ -1 1 0 03 0 -1 -1 0 0 1
El principio del equilibrio químico expresado en función de los potenciales químicos de cada especie está dado, para el sistema en cuestión, por:
Para la reacción (1):
0)()1()1()(NEQ
1iO2
1HOH
)1(ii 222
=−+−+=∑=
µµµνµ
Para la reacción (2):
0)()1()1()(NEQ
1iO2
1CCO
)2(ii 2
=−+−+=∑=
µµµνµ
Para la reacción (3):
0)1()1()1()(NEQ
1iOCCO
)3(ii 22
=−+−+=∑=
µµµνµ
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 16

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Más adelante, en la sección de equilibrio químico se estudiará y aplicará este principio en sistemas reaccionantes en fase homogénea donde los potenciales químicos están relacionados con la actividad y la fugacidad.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 17

Fundamentos de Termodinámica del Equilibrio Físico y Químico
2. ANALISIS TERMODINÁMICO DE PROCESOS
El análisis termodinámico de procesos se basa en la combinación de la primera con la segunda ley de la termodinámica. La primera ley está restringida por la segunda ley ya que no establece los criterios para determinar la dirección de los procesos, la irreversibilidad, el trabajo máximo reversible y en los procesos cíclicos, por la eficiencia térmica se compara con el 100% y no la eficiencia térmica del proceso óptimo.
Para un proceso dado, el potencial de trabajo máximo se encuentra si el proceso ocurriera reversiblemente. La diferencia entre el trabajo reversible y el trabajo real representa la irreversibilidad del proceso.
Es necesario encontrar la potencia (o el trabajo) real útil combinando las dos leyes.
- Sistemas cerrados.
Primera ley:
+−=+==− ∑
kkooutil QQQ;VPWWUWQ ; ∆∆
Segunda ley: ∑++=k k
k
o
ogen T
QTQSS ∆
La combinación de las dos ecuaciones y eliminación del calor de los alrededores, Qo, da el siguiente resultado:
( ) genok k
okoutil ST
TT1QSTUW −
−−−−= ∑∆∆ (2-1)
El trabajo útil reversible se obtiene si no existe irreversibilidad (Sgen = 0); en tal caso:
( ) ∑
−−−−=
k k
okorev T
T1QSTUW ∆∆ (2-2)
- Sistemas abiertos.Para un volumen de control la potencia útil está dada por:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 18

Fundamentos de Termodinámica del Equilibrio Físico y Químico
[ ] [ ] ∑∑∑ −
−−−+
−=
== kgeno
k
ok
NCS
1jsaljj
NCE
1ientii
vcútil ST
TT1Qmm
dtdW ϕϕΦ
(2-3)
Donde Φ = U – Uo + Po(V – Vo) – To(S – So) y ϕ = (h – ho) + ec + ep – To(s – so).
La suma neta de los flujos de calor entre el sistema y los depósitos térmicos incluyendo los alrededores está dado por:
∑ ∑=
−
=
+=ND
1d
1ND
1kkod QQQ
El signo de Qd se asigna con respecto al depósito térmico d.
Las propiedades con el subíndice º corresponden al estado muerto. A menudo Po y To
se toman iguales a 100 kPa y 300 K, respectivamente, en caso que no se especifique otra condición.
La potencia máxima (o mínima) se encuentra en el caso de que el proceso sea reversible; en este caso, la entropía generada es cero. De la ecuación (2-3):
[ ] [ ] ∑∑∑
−−−+
−=
== k k
ok
NCS
1jsaljj
NCE
1ientii
vcrev T
T1QmmdtdW ϕϕΦ
(2-4)
La diferencia entre la potencia reversible y la potencia útil es la irreversibilidad I o exergía destruída Θdest:
destruidagenoSTI Θ == (2-5)
El término (dΦ/dt)vc se aplica a sistemas no estacionarios y es la velocidad de acumulación de exergía en el volumen de control. Para procesos en estado estable la potencia reversible queda expresada como:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 19

Fundamentos de Termodinámica del Equilibrio Físico y Químico
[ ] [ ] 0WTT1Qmm rev
k k
ok
NCE
1isalii
NCS
1jentjj =
−−− ∑∑∑
==
- ϕϕ
Para cualquier proceso a estado estable:
[ ] [ ] ∑∑∑ =−
−−−
== kgenoútil
k
ok
NCS
1jsaljj
NCE
1ientii STW
TT1Qmm ϕϕ (2-6)
La ecuación (2-6) representa la ecuación general del balance de exergía de un sistema sometido a un proceso en estado estable. Una forma condensada del balance de exergía es:
destruidaprodsum ΘΘΘ =− (2-7)
La exergía suministrada, Θsum, es la suma de las exergías que entran al sistema. La exergía producida, Θprod, es la exergía que se recupera y es la suma de las exergías que salen, por ejemplo, la potencia útil producida en el sistema.
La eficiencia termodinámica de un proceso se define basada en la segunda ley, como la relación entre la exergía producida y la exergía suministrada, es decir:
sum
destruida
sum
prodII 1
ΘΘ
ΘΘ
η −== (2-8)
Las ecuaciones de balance de materia, energía, entropía y exergía tienen términos correspondientes en cada una de ellas: los términos de acumulación en el volumen de control, los términos que acompañan al flujo de materia, los términos que acompañan a la transferencia de calor y la potencia a través de la frontera y los términos de conservación para la materia y la energía, y no conservación para la entropía y la exergía.
- Balance de materia:
0mmdtdm NCE
1ii
NCS
1jj
vc =−+
∑∑
==
- Balance de energía:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 20

Fundamentos de Termodinámica del Equilibrio Físico y Químico
[ ] [ ] 0WQmmdt
)u.m(ds
NCE
1ientii
NCS
1jsaljj
vc =+−−+
∑∑
==
θθ
- Balance de entropía:
[ ] [ ] gen
ND
1d d
dNCE
1ientii
NCS
1jsaljj
vc STQsmsm
dt)s.m(d
=+−+
∑∑∑
===
- Balance de exergía:( ) [ ] [ ] ∑∑∑ −=+
−+−+
== kdestruidaútil
k
ok
NCS
1jentjj
NCE
1isalii
vc WTT1Qmm
dtd ΘϕϕΦ
Ejemplo 2.1. Se comprime aire de manera estable mediante un compresor de 5 kW desde 100 kPa y 37º C hasta 600 kPa y 167º C a una relación másica de 1.6 kg/min. Durante este proceso hay una transferencia de calor entre el compresor y los alrededores que están a 37º C. Haga un análisis termodinámico del proceso y establezca la eficiencia termodinámica.Datos: Cp del aire = 1.0 kJ/kg-K. Talred = 37º C = 310 K = To
Solución:Análisis de la primera ley:
Δh = Cp(T2 – T1) = 130 kJ/kg
Q = WC + mΔh = -5 + (1.6/60)*130 = -1.533 kW
Análisis de la segunda ley:
Δs = Cpln(T2/T1) – Rln(P2/P1) = 1.0*ln(440/310) – 0.287*ln(600/100) = -0.164 kJ/kg-K
Sgen = m Δs + (-Q/Talred) = (1.6/60)(-0.164) + 1.533/310 = 5.71*10-4 kW/K
Análisis exergético:Θsum = Wc = 5 kW; Θprod = m[Δh - To Δs] = (1.6/60)[130 – 310*(-0.164)] = 4.822 kW
Θdestruida = Θsum - Θprod = ToSgen = 0.177 kW
La eficiencia termodinámica del proceso es: ηII = 1.0 – 0.177/5 = 0.965
El análisis de equipos como intercambiadores de calor y mezcladores basado en la segunda ley permite encontrar su respectiva eficiencia termodinámica.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 21

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Para un intercambiador de calor la exergía suministrada es aportada por el fluido caliente (disminuye la energía térmica, la entropía y la exergía); mientras que el fluido frío recupera (produce) exergía (aumenta la energía térmica, la entropía y la exergía). La eficiencia termodinámica estaría dada por:
( ) ( )( ) fcfc
ffffICII m
mϕ∆ϕ∆
η
−=
Para un mezclador donde entran NCE corrientes y sale una corriente, la exergía suministrada es aportada igualmente por las corrientes calientes (comparada con la corriente de salida) y la exergía es recuperada por las corrientes frías. La eficiencia termodinámica del mezclador con K corrientes frías y M corrientes calientes, estaría dada por:
( )( )[ ]
( )[ ]∑∑
−
−−= M
mmccsalcc
K
kkcfsalcf
mezcII
m
m
ϕϕ
ϕϕη
Ejemplo 2.2. Para la planta termoeléctrica con turbina de gas que opera con fuego indirecto (combustión externa) que se muestra en la figura, a) haga un análisis termodinámico de acuerdo con la primera ley; b) haga un análisis termodinámico de acuerdo con la segunda ley.Tanto el compresor como la turbina operan adiabáticamente. Desprecie las caídas de presión en el calentador de aire. Además de las especificaciones mostradas en la figura, tome las siguientes informaciones:Eficiencias adiabáticas: ηC = ηT = 0.85; relación de presión en el compresor y en la turbina: rPC = rPT
= P2/P1 = P3/P4 = 7Suponga que el aire es un gas ideal con Cp = 1.045 kJ/kg y k = Cp/Cv = 1.36; Ra = 0.287 kJ/kgK; Po = 100 kPa, To = 288 K
Solución:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 22
compresor turbina
4100 kPa
3 800º C
qent
AireT
1 = 15° C,
P1 = 100 kPa 1
2
Fuente de calor a 1200 K
calentador

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Análisis según la primera ley:T1 = 288 K; T3 = 1073 K. Sea rT = (rPC)(k-1)/k
= 7(0.36/1.36 = 1.6738Compresor: wcs = h2s – h1 = Cp(T2s – T1); T2s = T1(rpc)(k-1)/k = 288*1.6738 = 482.05 K; wcs = CpT1(rT – 1) = 1.045(288)(1.6738 – 1) = 202.785 kJ/kg;
wc = wcs/ηC = 202.785/0.85 = 238.57 kJ/kg
Temperatura de salida del compresor: T2 = T1 + wC/Cp = 288 + 238.57/1.045 = 516.3 K
Turbina: wTs = h3 – h4s = Cp(T3 – T4s); T4s = T3(1/rpT)(k-1)/k = 1073*(1/1.6738) = 641.06 K; wcs = CpT3(1 – (1/rpT)(k-1)/k ) = 1.045(1073)(1 – 1/1.6738) = 451.38 kJ/kg;
wT = wTs*ηT = 451.38*0.85 = 383.67 kJ/kg
Temperatura de salida de la turbina: T4 = T3 - wT/Cp = 1073 – 383.67/1.045 = 705.85 K
Trabajo neto: wneto = wT - wC = 383.67 – 238.57 = 145.1 kJ/kgCalor de entrada: : qent = h3 – h2 = Cp(T3 – T2) = 1.045(1073 – 516.3) = 581.75 kJ/kg
Eficiencia térmica según la primera ley: ηII = 145.1/581.75 = 0.25 (25%)
Análisis según la segunda ley:Exergía que entra a la planta = exergía debido a la transferencia de calor desde el depósito de calor a 1200 K
φentrada = qent(1 – To/TH) = 581.75(1 – 288/1200) = 442.13 kJ/kg
Exergía que entra con el aire a 15º C y 100 kPa = 0Exergía total que entra = 442.13 kJ/kg
Exergía recuperada por el aire en el calentador = φrecup = φ3 - φ2 = (h3 - h2) - To(s3 - s2); (h3 - h2) = Cp((T3 - T2) = 1.045*(1073 – 516.3) = 581.75 kJ/kg; (s3 - s2) = Cpln(T3/T2) - Raln(P3/P2) = 1.045*ln(1073/516.3) = 0.76444 kJ/kgK
φrecup = 581.75 – 288*0.76444 = 361.6 kJ/kg
Exergía destruida en el calentador = φentrada - φrecup = 442.13 – 361.6 = 80.53 kJ/kg
Exergía destruida en el compresor = Tosgen = To(s2 – s1) = To(Cpln(T2/T1) – Rln(P2/P1)) = 288*(1.045*ln(516.3/288) – 0.287*ln(7)) = 14.84 kJ/kg
Exergía destruída en la turbina = Tosgen = To(s4 – s3) = To(Cpln(T4/T3) – Rln(P4/P3)) = 288*(1.045*ln(705.85/1073) – 0.287*ln(1/7)) = 34.8 kJ/kg
Exergía destruída total = 80.53 + 14.84 + 34.8 = 130.17 kJ/kg
Exergía que sale con el aire a 100 kPa y 705.85 K = φ4 - φ0 = (h4 - h0) - T0( s4 - s0)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 23

Fundamentos de Termodinámica del Equilibrio Físico y Químico
= Cp(T4 - T0) - To(Cpln(T4/T0) – Rln(P4/P0)) = 1.045(705.85 - 288) - 288(1.045*ln(705.85/288) – 0.287*ln(1)) = 166.86 kJ/kg
Exergía producida = Trabajo neto = 145.1 kJ/kg
Exergía total que entra = exergía producida + exergía que sale + exergía destruida = 145.1 + 130.17 + 166.86 = 442.13 kJ/kg
Eficiencia para el ciclo = ηciclo = φprod/φrecup = 145.1/361.6 = 0.4 (40%)
Eficiencia para la planta = ηplanta = φprod/φentra = 145.1/442.13 = 0.328 (32.8%)
Distribución de la exergía destruida. Compresor: 14.84/130.17 = 0.114 (11.4%); Turbina: 34.80/130.17 = 0.267 (26.7%) Calentador: 80.53/130.17 = 0.619 (61.9%)
Los resultados muestran que el calentador de aire es el equipo con más pérdidas (61.9% del total).
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 24

Fundamentos de Termodinámica del Equilibrio Físico y Químico
3. ECUACIONES DE ESTADO
Las ecuaciones de estado son modelos matemáticos de predicción del comportamiento PvT de una sustancia pura o de una mezcla en fase homogénea.
Las ecuaciones que se usan en este trabajo son la ecuación virial para la fase vapor, las ecuaciones de estado cúbicas que son modificaciones de la ecuación de van der Waals, para las fases líquida y vapor y la ecuación de Racket(*) para líquidos saturados.
3.1. ECUACION VIRIAL
La forma más usual de la ecuación virial es la siguiente:
RTBP1
RTPvZ +== (3-1)
Donde Z = factor de compresibilidad, v = volumen molar, B = segundo coeficiente virial, en volumen/mol, R = constante universal de los gases ideales.
El segundo coeficiente virial B se evalúa con la correlación propuesta por Pitzer :
[ ])1()0(
c
c wBBP
RTB += (3-2)
Donde B(0) y B(1) son funciones de la temperatura reducida, (Tr) y del factor acéntrico (w), mediante las siguientes expresiones:
6.1r
)0(
T422.0083.0B −= (3-3)
2.4r
)1(
T172.0139.0B −= (3-4)
HAYDEN, J. G. and O’Conell. Ind. Eng. Chem., Process Des. Develop. Vol. 14 (1975); p. 209-216
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 25

Fundamentos de Termodinámica del Equilibrio Físico y Químico
El factor acéntrico es una constante de cada reportada en la literatura y puede determinarse mediante la siguiente ecuación:
1)Plog(w 7.0TSAT
r r−−= =
Donde c
SATSAT
r PPP =
)T(fP SAT = T = 0.7*Tc
La ecuación virial para una mezcla gaseosa de NC componentes, tiene la misma forma de la ecuación (3-1), reemplazando B por Bm, donde Bm es el correspondiente segundo coeficiente virial para mezclas. Bm se calcula con la siguiente ecuación:
)Byy(B ijj
NC
1i
NC
1jim ∑∑
= =
= (3-5)
donde Bij es el segundo coeficiente virial de interacción binaria entre los componentes i y j y se evalúa con la ecuación (3-2) reemplazando Pc por Pcij , Tr por Trij, w por wij, B(0) por B(0)
ij, B(1) por B(1)ij. Estas constantes y funciones se obtienen
con las siguientes expresiones:
[ ])0(ijij
)0(ij
cij
cijij BwB
PRT
B += (3-4)
6.1ijr
)0(ij T
422.0083.0B −= (3-6)
2.4ijr
)1(ij T
172.0139.0B −= (3-7)
Donde Trij está definida por: cij
rij TTT =
Las propiedades críticas de interacción binaria i,j se calculan a través de las siguientes expresiones:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 26

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( ) 5.0jcicijc T*TT =
2ww
w jiij
+=
cij
cijijcij v
RTZcP = (3-8)
33/1jc
3/1ic
ijc 2vv
v
+=
2ZZ
Z jcicijc
+= (3-9)
La ecuación virial se aplica a la fase gaseosa con la restricción de que vm/vcm ≥ 2, donde vm es el volumen molar de la mezcla y vcm es el volumen molar crítico.
El factor de compresibilidad, Z y volumen molar de la mezcla se determinan con las siguientes ecuaciones derivadas de la ecuación (3-1):
RTPB1Z m
m += ; PRTZv m
m =
)vyy(v cijj
NC
1i
NC
1jicm ∑∑
= =
=
siendo yi la fracción molar del componente i.
3.2. ECUACIONES DE ESTADO CUBICAS.
Las ecuaciones de estado cúbicas que se usan en este trabajo son las ecuaciones de Redlich-Kwong (RK), Soave-Redlich-Kwong (SRK) y Peng-Robinson (Peng-R), las cuales son modificaciones de la ecuación de van der Waals (VW),
2va
)bv(RTP −−
=
Donde c
c
c
2c
P)RT(
81b;
P)RT(
6427a ==
3.2.1. Ecuación Cúbica General. Los investigadores Redlich y Kwong modificaron la ecuación de van der Waals y propusieron que el parámetro a dependía de la
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 27

Fundamentos de Termodinámica del Equilibrio Físico y Químico
temperatura y modificaron el término a/v2 por a/(v(v + b). Posteriormente Soave, y Peng y Robinson plantearon sus modificaciones de la ecuación de Redlich-Kwong.
La siguiente ecuación cúbica general resume las modificaciones que cada uno de los investigadores propusieron en sus trabajos. Cabe destacar que las ecuaciones de estado cúbicas se han modificado para algunos sistemas o mezclas específicas, encontrándose parámetros que sólo se aplican a dichas mezclas.
La ecuación cúbica general está en función de dos parámetros u y t de cada ecuación en particular. Los valores de estos parámetros aparecen en la tabla 1.
La ecuación explícita en la presión está dada por:
)tbvubv(a
)bv(RTP 2
mmm2
m
m
mm −+−
−= (3-10)
donde am y bm son los parámetros de la sustancia pura o de la mezcla, los cuales dependen de las propiedades críticas, de la temperatura, del factor acéntrico y de la fracción molar de los componentes zFi (xi para fase líquida y yi para la fase vapor).
Las expresiones generales para estos parámetros son obtenidas a partir de las reglas de mezclado:
( ) ( )2
NC
1jjFj
NC
1i
NC
1jjiFjFim azaazza
== ∑∑∑
== =
(3-11)
∑=
=NC
1iiFim )bz(b (3-12)
ciii aa α= (3-13)
ic
2ic
ai P)RT(
Ωα = (3-14)
ic
icbi P
)RT(b Ω= (3-15)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 28

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Donde Ωa y Ωb son los coeficientes característicos de cada ecuación los cuales
aparecen en la tabla 1.
aci es una función de la temperatura reducida y del factor acéntrico de la especie i.
Para la ecuación de Redlich Kwong (RK), tiene la forma irci T/1a = , mientras que
para las ecuaciones de Soave (SRK) y Peng Robinson (Peng-R) está dado por:
[ ]2iriwci )T1(f1a −+= (3-16)
Dondefwi = 0.48 + 1.57wi - 0.176wi2, aplicable sólo para (SRK)
fwi = 0.37464 + 1.54226*wi - 0.26992*wi2 , aplicable sólo para (Peng-R)
wi = factor acéntrico del componente i
Tabla 1. Parámetros de las ecuaciones de estado cúbicas.Ecuación u t Ωa Ωb
van der Waals (vW) 0 0 27/64 1/8Redlich-Kwong (RK) 1 0 0.42748 0.08664
Soave- Redlich-Kwong (SRK) 1 0 0.42748 0.08664Peng-Robinson (Peng-R) 2 1 0.45724 0.0778
La ecuación cúbica general (ecuación (3-10)) en términos del factor de compresibilidad Z se transforma en la siguiente ecuación como una función de dos parámetros, AM, BM:
( ) ( ) ( ) 0tBtBBAZtBuBuBAZuBB1Z 3M
2MMM
2M
2MMM
2MM
3 =−−−−−−+−+− (3-17)
Donde los parámetros son:
2NC
1i ri
riciFia2
mM T
Paz
)RT(PaA
== ∑
=
Ω (3-18)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 29

Fundamentos de Termodinámica del Equilibrio Físico y Químico
∑=
==
NC
1i ri
riFib
mM T
Pz)RT(
PbB Ω (3-19)
;ci
rici
ri TTT
PPP ==
Las expresiones de cada ecuación específica (vW), (RK), (SRK) y (Peng-R) se obtienen al reemplazar el valor de los parámetros u y t que aparecen en la tabla en la ecuación (3-17).
Ecuación de van der Waals:
( ) ( ) ( ) 0BAZAZB1Z MMM2
M3 =−++−
Ecuaciones de RK y SRK:
( ) ( ) 0BAZBBAZZ MM2
MMM23 =−−−+−
Ecuación de Peng-R:
( ) ( ) ( ) 0BBBAZB3B2AZB1Z 3M
2MMM
2MMM
2M
3 =−−−−−+−−
3.2.1.1. Solución analítica de la ecuación cúbica basada en el método de Cardano♥. La ecuación (3-17) se puede resolver mediante un método numérico iterativo o mediante un método analítico usando la teoría de las soluciones. El método analítico que se plantea a continuación es el método de Cardano.
En general la ecuación (3-17) es de la forma:
0RZQZPZ 112
13 =+++ (3-20)
Donde: ( )MM1 uBB1P −+−=
( )2M
2MMM1 tBuBuBAQ −−−=
( )3M
2MMM1 tBtBBAR −−−=
= Gerolano Cardano (1501-1576) junto con Nicoló Fontana (1500-1577) y Scipio Ferro, profesor de la Universidad de Bolonia fueron los primeros en dar a conocer métodos para el cálculo de las raíces del polinomio cúbico.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 30

Fundamentos de Termodinámica del Equilibrio Físico y Químico
La ecuación (3-17) se transforma en términos de la variable X cuando se hace un
cambio de variable, 3PXZ 1−= . Con la sustitución de Z se elimina el término
cuadrático y se obtiene una nueva ecuación en términos de X:
0NMXX 3 =++
donde 2
11 3
P3QM
−= ; 1
11
31 R
3PQ
3P2N +
−
=
La solución analítica de la ecuación cúbica en X da tres raíces posibles con un
discriminante D dado por:23
2N
3MD
+
=
Debido a la dificultad de encontrar un discriminante exactamente igual a cero, éste se aproxima a cero por el lado izquierdo (discriminante menor que cero) o por el lado derecho (discriminante mayor que cero). De tal manera que se usan solamente estas dos situaciones:
Si el discriminante es mayor que cero, (D > 0), hay una raíz real y dos raíces imaginarias. Este resultado significa que el sistema está en fase homogénea (frecuentemente, vapor sobrecalentado).
La raíz real se evalúa con:
3PD
2ND
2NZ 1
3/13/1
−
−−+
+−=
El volumen molar se calcula con el valor de Z:
PZRTvm = (3-21)
Si el discriminante es menor que cero (D < 0), hay tres raíces reales. Este resultado significa que el sistema se encuentra en equilibrio.
Si es una sustancia pura la raíz mayor corresponde al vapor saturado y la raíz menor al líquido saturado.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 31

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Si es una mezcla gaseosa homogénea se encuentra en su punto de rocío y se selecciona la raíz mayor, es decir Z = ZG = Zmayor y se ignoran las otras raíces; si es una mezcla en la fase líquida homogénea se selecciona la raíz menor, Z = ZL = Zmenor y el estado corresponde al punto de burbuja.
Las tres raíces se evalúan con la siguiente ecuación:
3P
32)1k(
3cos
3)M(2Z 1
k −
−+−= πθ
Donde k =1, 2, 3; θ = arccos ( φ ) y 2/3)3/M()2/N(
−−=φ .
El volumen de cada fase se evalúa con las ecuaciones:
PRTZv G
G = (3-22)
PRTZ
v LL = (3-23)
Se observa que cuando el discriminante tiende a cero el valor de M es menor que cero y el valor de φ tiende a cero. En este caso las raíces son:
3P
3)M(
3P
34cos
3)M(2Z
;3P
3)M(
3P
32cos
3)M(2Z
;3P
3)M(2Z
113
112
11
−−−=−
−=
−−−=−
−=
−−=
π
π
Se dispone de Programas en lenguaje Matlab para la determinación de propiedades termodinámicas basadas en las ecuaciones de estado cúbicas y virial.
Ejemplo 3.1. Calcule el volumen específico del vapor de agua a 1 atm (101.325 KPa) y 100º C con una calidad de 75%. Use la ecuación de SRK.Datos del agua:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 32

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Tc = 647.14 K, Pc = 22.09 MPa, w = 0.348 ; Masa molecular (PM) = 18 ;
Solución:De la Tabla 1, para SRK, u = 1, t = 0, Ωa = 0.42748; Ωb = 0.08664; fw = 0.48 + 1.57*w - 0.176*w2; Z3 - Z2 + (A - B - B2) - A*B = 0P1 = -1; Q1 = A - B - B2; R1 = -A*B;Para una sustancia pura,A = Ωa*Pr*ac/(Tr)2 donde Pr = presión reducida = P/Pc; y B = Ωb*Pr/Tr
Cálculos:Tr = 0.5763822; Pr = 4.58692*10-3 ; fw = 1.005046; ac = 1.5426055;A = 9.1048*10-3; B = 6.89492*10-4 ; Q1 = 8.41483*10-3 ; R1 = -6.277687*10-6
M = -0.3249185 ; N = -0.0712754 ; D = -4.0982*10-7 ; Ya que D < 0 entonces hay tres raíces.
φ = 0.9998385; θ = 0.017968 radianes; (-M/3)1/2 = 0.329099 ;
Z1 = 2*0.329099*cos(0.017968/3) + 1/3 = 0.99152 ; Z mayor = ZG = Z1; Z2 = 2*0.329099*cos(0.017968/3 + 2*3.1416/3) + 1/3 = 8.262*10-4 Zmenor = ZL = Z2
Z3 = 2*0.329099*cos(0.017968/3 + 4*3.1416/3) + 1/3 = 7.654*10-3
Cálculo del volumen específico del vapor húmedo: vesp = vLesp + x*(vG
esp - vLesp) ;
Vapor saturado vGesp = ZG*R*T/(P*PM) = 0.99152*8.314*373/(101.325*18) = 1.686 m3/Kg ;
Líquido saturado vLesp = ZL*R*T/(P*PM) = 8.262*10-4 *8.314*373/(101.325*18) = 1.4048*10-3 m3/Kg;
vesp = 1.4048*10-3 + 0.75*(1.686 – 1.4048*10-3) = 1.265 m3/Kg;
El volumen específico del vapor saturado de la literatura♦ es 1.6736 m3/kg con un error de 0.73% y el volumen del líquido saturado es 1.043*10-3 m3/kg con un error de 34.7%.
Ejemplo 3.2. Calcule el volumen molar de una mezcla gaseosa que consiste de 70% de metanol(1) y 30% de agua(2) a 400 kPa y 37º C. Use la ecuación de Peng Robinson.Datos:COMPONENTE yi Tci (K) Pci (KPa) wi
METANOL(1) 0,3000 512,60 8097,00 0,564AGUA(2) 0,7000 647,10 22055,00 0,345
Solución:Los cálculos obtenidos usando el programa PropSRK se muestran en la siguiente tabla:COMPONENTE fwi Tri aci αi ai zFi(ai)0.5 bi zFi*bi
METANOL 1,1586 0,6048 1,5816 1025,646 1622,124 28.193 0,0409 0,0287AGUA 0,8746 0,4791 1,6110 600,067 966,706 9.327 0,0190 0,0057
am = 1407.788; AM = 0,08477; bm = 0.03436; BM = 0.00533; P1 = -0.99467; Q1 = 0.07402; R1 = -0.0004234;
Jones J. B. & Dugan R. Ingeniería Termodinámica. Prentice Hall. 1996
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 33

Fundamentos de Termodinámica del Equilibrio Físico y Químico
M = -0.255766; N = -0.048776; D = -2.4891*10-5 < 0 Discriminante menor que cero: Hay tres raíces reales.
Z1 = 0.91421; Z2 = 0.00624; Z3 = 0.07422. Se selecciona la raíz mayor: Z = ZG = 0.91421.
El volumen molar de la mezcla es: v = (ZRT)/P = 0.91421*8.314*310/400 = 5.891 m3/kmol
Un tratamiento más complejo de las reglas de mezclado para las ecuaciones de estado cúbicas para el modelamiento de sistemas multicomponentes fue presentado por Wong-Sandler (1992) en donde los parámetros am y bm están correlacionados:
( )
∑
∑∑
=
= =
+=
−
+−+=
−=
=
nc
1i
E
i
ii
ijji
jiij
NC
1i
NC
1jijji
m
mm
RTg
RTbaxD
k1RT
aabb5.0
D1
xxb
DbRTa
σ∆
ψ
ψ
Donde kij son los parámetros de interacción binaria a partir de datos experimentales y ΔgE es la energía de Gibbs en exceso de la solución (véase más adelante el concepto de propiedades de residuales, de mezclado y en exceso).
3.3. ECUACION MODIFICADA DE BENEDICT-WEBB-RUBIN
La ecuación de Benedict-Webb-Rubin es de tipo virial y fue modificada por Starlin-Han (1972-1973) incrementando a once parámetros: Ao, Bo, Co, Do, Eo, a, b, c, d, α y γ
−
++
++
−−+
−+−−+=
22326
324o
3o
2o
oo
vexp
v1
vTc
v1
Tda
v1
TdabRT
v1
TE
TD
TCARTB
vRTP
γγα
3.4. ECUACION DE RACKET PARA LIQUIDOS SATURADOS.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 34

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Los volúmenes molares de la fase líquida obtenidos a partir de las ecuaciones de estado cúbicas presentan una desviación alrededor del 35% con respecto a los valores experimentales; por tal razón, se recomienda utilizar la ecuación de Racket para líquidos saturados.
( ) ϕRA
c
cls Z
PRTv = (3-24)
donde ϕ = (1 + (1 - Tr)2/7) y ZRA = 0.29056 - 0.08775w
Si se conoce la densidad experimental a una temperatura de referencia TREF, se recomienda usar la siguiente ecuación modificada a la temperatura de interés T:
( )ϕRATREF)T(ls Zvv = (3-25)
donde ϕ = (1 - Tr)(2/7) - (1 – (Tr)REF)(2/7) ;(Tr)REF = TREF/Tc = temperatura reducida basada en la temperatura de referencia
vls(T) = volumen molar a la temperatura T, vTref = volumen molar a la temperatura de referencia.
Ejemplo 3.3. Calcule el volumen específico del amoníaco NH3 como líquido saturado a –15º C (258 K). La presión de saturación a –15º C es 236.3 kPa.Las constantes críticas del amoníaco son: Tc = 405.7 K; Pc = 11280 kPa; w = 0.253
Solución:Cálculo de ZRA = 0.29056 – 0.08775*0.253 = 0.26836Tr = T/Tc = 258/405.7 = 0.635938; ϕ = (1 + (1 – Tr)(2/7) ) = 1.75;
VLs = (RTc/Pc)(ZRA)ϕ = (8.314*405.7/11280)(0.26836)1.75 = 0.002992 m3/kmol*(kmol/17 kg) = 1.76*10-3 m3/kg
El volumen específico reportado en la literatura es 1.519*10-3 m3/kg. El error relativo es de 16%
Ejemplo 3.4. Calcule el volumen específico del agua como líquido saturado a 25º C (298 K). La presión de saturación del agua a 25º C es 3.166 kPa. Las constantes críticas del agua son: Tc = 647.1 K; Pc = 22055 kPa; w = 0.345
Solución:Cálculo de ZRA = 0.29056 – 0.08775*0.345 = 0,2603
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 35

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Tr = T/Tc = 298/647.1 = 0,4605; ϕ = (1 + (1 – Tr)(2/7) ) = 1.8383;
VLs = (RTc/Pc)(ZRA)ϕ = (8.314*647.1/22055)(0.2603)1.8383
= 0.020545 m3/kmol*(kmol/18 kg) = 1.14*10-3 m3/kg
El volumen específico reportado en la literatura es 1.003*10-3 m3/kg. El error relativo es de 14%
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 36

Fundamentos de Termodinámica del Equilibrio Físico y Químico
4. RELACIONES TERMODINAMICAS FUNDAMENTALES
Las relaciones termodinámicas fundamentales se obtienen a partir de la combinación de la primera y segunda leyes de la termodinámica. Estas relaciones en forma diferencial son de la forma:
dzz
dyy
dxx
d);z,y,x(y,xz,xz,y
∂∂+
∂∂+
∂∂== ψψψψψψ
La combinación de la primera y segunda leyes de la termodinámica para sistemas cerrados y procesos reversibles permite obtener la siguiente relación fundamental:
Primera ley: δq – δw = du;
Segunda ley (procesos reversibles): δqrev = Tds y δwrev = Pdv.
Al reemplazar se obtiene:
du = Tds –Pdv u = u(s, v) (4-1)
Por definición, la entalpía es: h = u + Pv. Derivando, dh = (du + Pdv) + vdP, pero du + Pdv = Tds dada por la ecuación (3-1). Al reemplazar:
dh = Tds + vdP h = h(s, P) (4-2)
La energía de Gibbs está definida por: g = h – Ts. Derivando, dg = (dh - Tds) - sdT, pero dh - Tds = vdP dada por la ecuación (4-2). Al reemplazar:
dg = -sdT + vdP g = g(T, P) (4-3)
Por ultimo, la energía de Helmholtz está definida por: a = u – Ts. Derivando, da = (du - Tds) - sdT, pero du - Tds = -Pdv dada por la ecuación (4-1). Al reemplazar:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 37

Fundamentos de Termodinámica del Equilibrio Físico y Químico
da = -sdT - Pdv a = a(T, v) (4-4)
Las ecuaciones anteriores se aplican a un sistema cerrado sometido a cualquier proceso, bien sea para sustancia pura o mezcla homogénea a composición constante.
Para una mezcla de n moles compuesta de NC componentes, que intercambia materia y energía con los alrededores u otro sistema, las ecuaciones anteriores se modifican cuando se incluyen las derivadas parciales de cada propiedad con respeto a las moles de cada componente. De esa manera las ecuaciones de las relaciones generalizadas para sistemas abiertos son:
ii dn)nv(Pd)ns(Td)nu(d ∑+−= µ (4-5)
ii dndP)nv()ns(Td)nh(d ∑++= µ (4-6)
ii dndP)nv(dT)ns()ng(d ∑++−= µ (4-7)
ii dn)nv(PddT)ns()na(d ∑+−−= µ (4-8)
En cada ecuación µi es el potencial químico del componente i en la mezcla y se define mediante las siguientes relaciones:
jjjj n,V,Tin,T,Pin,P,Sin,V,Sii n
nanng
nnh
nnu
∂∂=
∂∂
=
∂∂=
∂∂=µ
A partir del cálculo diferencial y del criterio de la exactitud se obtienen las llamadas relaciones de Maxwell con n = 1 mol y composición constante.
vs sP
vT
−=
∂∂
∂∂
(4-9) Ps s
vPT
=
∂∂
∂∂
(4-10)
Tv v
sTP
=
∂∂
∂∂
(4.11) TP P
sTv
−=
∂∂
∂∂
(4-12)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 38

Fundamentos de Termodinámica del Equilibrio Físico y Químico
De las ecuaciones (4-5)-(4-8) se halla la dependencia directa de cada propiedad termodinámica de las propiedades independientes (variables canónicas).
u = u (s, v, ni), h = h(s, P, ni), g = g (P, T, ni), a = a(T, v, ni ).
Tomando a la temperatura y la presión como variables independientes y una ecuación de estado explícita en el volumen molar v = v (P, T, ni), las otras propiedades termodinámicas quedan finalmente, en función de estas dos propiedades intensivas cuando la composición es constante.
La entalpía y entropía molares se seleccionan como funciones básicas, las demás propiedades como la energía interna, la energía de Gibbs y la energía de Helmholtz pueden evaluarse a partir de ellas, mediante las relaciones siguientes:
u = h - Pv; g = h - Ts ; a = u - Ts
donde h = h(P,T); s = s(P,T), a composición constante. La ecuación diferencial de cada una es:
dPPhdT
Thdh
TP
+
=
∂∂
∂∂
(4-13)
dPPsdT
Tsds
TP
+
=
∂∂
∂∂
(4-14)
3.1.1. Algunas aplicaciones de las Relaciones de Maxwell.
Por definición, P
p TsTC
=
∂∂
Al derivar con respecto a la presión a T constante:
P
2
2
PTTPT
p
TvT
Ps
TT
Ts
PT
PC
∂∂−=
∂∂=
∂∂=
∂
∂∂∂
∂∂
Por integración: ∫
∂∂−=
P
0 P2
2gi
PP dPT
vTCC (4-15)
Mediante un procedimiento similar se demuestra que: ∫∞
∂∂+=
v
v2
2gi
vv dvT
PTCC
De la ecuación (4-2),
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 39

Fundamentos de Termodinámica del Equilibrio Físico y Químico
P
2
PTT TZ
PRT
TvTvv
PsT
Ph
∂∂−=
−=+
=
∂∂
∂∂
∂∂
(4-16)
Reemplazando la ecuación (4-16) en la ecuación (4-13) e integrando se obtiene el cambio de entalpía como se verá más adelante. Igualmente, reemplazando la ecuación (4-15) y la relación de Maxwell dada por la ecuación (4-12) en la ecuación (4-14) e integrando se obtiene el cambio de entropía.
∫ ∫∫∫
∂∂−=
−+= dP
TvdT
TC
;dPTvTvdTCh
P
p
Pp s ∆
∂∂∆
Con la combinación de las ecuaciones (4-11) y (4-12) se obtiene la siguiente relación:
dPZTZT
PRdP
Tvdv
TP
PPv
+
∂∂−=
−=
∂∂
∂∂
(4-17)
Esta relación es importante en la determinación de la entalpía y la entropía cuando no se dispone de una ecuación de estado explícita en el volumen, como por ejemplo, las ecuaciones cúbicas, que son explícitas en la presión.
Cálculo del coeficiente de expansión volumétrica (β). Por definición:
PTP TZ
Z1
T1
Ps
v1
Tv
v1
∂∂+=
−=
=
∂∂
∂∂β (4-18)
Para un gas ideal, Z = 1; ∂Z/∂T = 0; β = 1/T
Cálculo del coeficiente de compresibilidad isotérmica (k). Por definición:
TT P
ZZ1
P1
Pv
v1k
∂∂−=
−=
∂∂
(4-19)
Para un gas ideal Z = 1; ∂Z/∂P = 0; k = 1/P
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 40

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Los coeficientes de expansión volumétrica y de compresibilidad isotérmica tienen aplicación en la determinación de propiedades de fluidos en la región supercríticas y es un criterio para determinar la incompresibilidad de los líquidos.
Cálculo del coeficiente de Joule Thompson (μJT) o coeficiente de expansión isentálpica:
p
P
2
p
P
P
T
hJT C
TZ
PRT
C
vTvT
ThPh
PT
∂∂
=
−
∂∂
=
∂∂
∂∂
−=
∂∂=µ (4-20)
Cálculo del coeficiente de expansión isentrópica:
pp
P
P
T
ss C
ZTZT
PRT
CTvT
TsPs
PT
+
∂∂
=
∂∂
=
∂∂
∂∂
−=
∂∂=α (4-21)
El coeficiente de expansión isentrópica de un gas ideal está dado por:
Pp
P
P
T
s
gis PC
RTCTvT
TsPs
PT =
∂∂
=
∂∂
∂∂
−=
∂∂=α
Resolviendo: PCR
11P PP
TT;
PP
CR
TT
=
∂=
∂
Entalpía molar y entropía molar de gases ideales:
Las ecuaciones diferenciales (4-13) y (4-14) de la entalpía y la entropía modificadas para gases ideales son:
dPP
hdTThdh
T
id
P
idid
+
=
∂∂
∂∂
(4-22)
dPP
sdTTsds
T
id
P
idid
+
=
∂∂
∂∂
(4-23)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 41

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Para gases ideales, Pvid = RT.
De la relación de Maxwell (ec (4-12)) se tiene que PR
Tv
Ps
P
id
T
id
−=
−=
∂
∂∂
∂.
Al reemplazar este resultado en la ecuación (4-16) se obtiene:
0P
RTvP
h id
T
id
=−=
∂
∂
Reemplazando los resultados anteriores en las ecuaciones (4-22) y (4-23) se obtiene:
dTCdh idp
id = (4-22)
dPPRdT
TC
dsidpid −= (4-23)
El cambio de entalpía y entropía de un gas ideal i desde un estado de referencia a Po
y To hasta un estado a P y T se obtiene de la integración de las ecuaciones anteriores, así:
∫+=T
To
idpi
idio
idi dTChh
∫
−+=
T
To o
idpiid
ioidi P
PlnRdTTC
ss
Donde: idioh = entalpía del gas ideal i a la temperatura To
idios = entropía del gas ideal i a la temperatura To y la presión Po
Estos resultados demuestran que la entalpía de un gas ideal es independiente de la presión mientras que la entropía es función de ambas.
Ejemplo 4.1. Un cierto gas se ajusta a la siguiente ecuación de estado: bRTa
PRTv +−= donde a
y b son constantes positivas (a > b).Calcule la relación entre el coeficiente de Joule-Thompson y el coeficiente de expansión isentrópica.Solución:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 42

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Por definición: p
PJT C
vTvT
−
∂∂
=µ y p
Ps C
TvT
∂∂
=α
La relación entre estos coeficientes es la siguiente:
P
P
s
JT
TvT
vTvT
∂∂
−
∂∂
=αµ
Donde : 2P RT
apR
Tv +=
∂∂
; RTa
pRT
TvT
P+=
∂∂
; bRT
a2vTvT P −=−
∂∂
Finalmente: A1BA2
RTa
PRT
bRT
a2
PTPT
s
h
s
JT
+−=
+
−
=
∂∂
∂∂
=αµ
donde ( )2RTaPA = y RT
bPB =
4.2. PROPIEDADES TERMODINÁMICAS DE MEZCLAS DE GASES IDEALES
4.2.1. Entalpía de una mezcla gaseosa ideal. La entalpía de una mezcla de gases ideales relativa a un estado de referencia a Po y To está dado por la integración de la ecuación (4-22) aplicada a mezclas:
dTChhT
To
idpM
idoM
idM ∫+= (4-24)
Donde:id
Mh es la entalpía molar de la mezcla gaseosa ideal a T y composición yi.id
oMh es la entalpía molar de la mezcla gaseosa ideal a To y composición yi.idpMC es la capacidad calorífica molar de la mezcla.
La capacidad calorífica molar de la mezcla es una función de la temperatura y de las fracciones molares de los componentes y está dada por:
( )
+++=
= ∑∑
=
−
=
NC
1i
2i
2iiii
NC
1i
idpi
iidpM TdTcTbayR
RC
yRC
En la expresión anterior ai, bi, ci y di son las constantes de cada componente mientras que la temperatura está dada en grados absolutos.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 43

Fundamentos de Termodinámica del Equilibrio Físico y Químico
El cambio de entalpía desde el estado 1 (P1, T1) hasta el estado 2 (P2, T2), a composición constante, está dado por la ecuación (4-24) definida entre los dos estados:
( ) ( ) ( )1221
M221
M1
MM
idM TT
Td1T
3c1T
2baRh −
++++++=
ττττ∆ (4-25)
Donde 1
2
TT
=τ
;dyd;cyc;byb;ayaNC
1iiiM
NC
1iiiM
NC
1iiiM
NC
1iiiM ∑∑∑∑
====
====
La capacidad calorífica molar media de la mezcla idpMC es el término entre corchetes
de la ecuación (4-25), es decir:
( ) ( )
++++++= 2
1
M221
M1
MM
idpM T
d1T
3c
1T2b
aRCτ
τττ
4.2.2. Entropía de una mezcla gaseosa ideal. La entropía de una mezcla gaseosa ideal depende, además de la temperatura, de la presión se acuerdo con la ecuación (4-23) y de la composición.
∑∑∫
∑
==
=
−=
−=
−+=
=
NC
1iii
NC
1i
idii
idM
iidi
T
To o
iidpiid
ioidim
NC
1i
idimi
idM
)yln(yRsys
)yln(RsPPylnRdT
TC
ss
sys ;
(4-26)
Téngase en cuenta que la entropía de un componente i de una mezcla gaseosa ideal es una función de la temperatura y la presión parcial de acuerdo con el teorema de Gibbs, es decir que sim
id = F (T, pi), donde pi = yiP. Mientras que la entropía de un gas ideal i puro es una función de la temperatura y la presión, si
id = F (T, P),
El cambio de entropía para un proceso sin reacción química, desde el estado 1 (P1, T1, yi1) hasta el estado 2 (P2, T2, yi2) está dado por la siguiente ecuación:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 44

Fundamentos de Termodinámica del Equilibrio Físico y Químico
−
−−= ∑∑∫∫
==
NC
1i o
11i1i
NC
1i o
22i2i
T
T
id1pM
T
T
id2pMid
M PPylny
PPylnyRdT
RTC
RdTRT
CRs
1
o
2
o
∆ (4-27)
Si el proceso ocurre a composición constante (yi1 = yi2), el cambio de entropía se reduce a:
( ) ( ) ( ) ( )
−−
+++++
−=
1
2123
12M
1M
M1
MidM P
PlnRTT1T2
d1T2cbln
)1TaRs τ
τττ
τ∆ (4-28)
Donde idMs = entropía molar de la mezcla gaseosa ideal a T, P y yi
Po = presión de referencia
4.2.3. Energía de Gibbs. La energía de Gibbs de una mezcla gaseosa ideal se halla a partir de la relación fundamental, g = h – Ts, usando las ecuaciones (4-24) y (4-25)
( ) ( )
( ) ( ) ( ) ( )∑ ∑∑ ∑∑
= == =
=
+=+−=
−==
NC
1i
NC
1iii
idii
NC
1i
NC
1iii
idi
idii
idM
NC
1i
idim
idii
idM
ylnyRTgyylnyRTTshyg
Tshyy,T,Pgg
(4-29)
Ejemplo 4.2. 5 kmol/h de una mezcla contiene 75% de metanol(1), 20% de etanol(2) y 5% de agua(3) se enfrían desde 250º C y 200 kPa hasta 120º C y 195 kPa cuando intercambia calor con los alrededores. Calcule el flujo de calor transferido y la entropía generada. Los alrededores están a 298 K.Datos: Constantes de Cpi/R = a + bT + cT2 + dT-2; T en K
Componente yi ai bi*103 ci*106 di*10-5 pi = yiP (kPa)METANOL(1) 0,7500 2,211 12,216 -3,450 0,000 150ETANOL(2) 0,2000 3,518 20,001 -6,002 0,000 40AGUA(3) 0,0500 3,470 1,450 0,000 0,121 10
Solución:T2 = 393 K; T1 = 523 K;aM = Σyiai = 2.535; bM = Σyibi = 13.235*10-3 ; cM = Σyici = -3.7879*10-6 ; dM = Σyidi = 0.00605*105;τ = T2/T1 = 393/523 = 0.7514; Reemplazando en la ecuación (4-25), hM2 – hM1 = -8430.27 kJ/kmolBalance de calor: Q = (5 kmol/h)(-8430.27 kJ/kmol) = -42151.37 kmol/hBalance de entropía:La composición molar no cambia. La ecuación (4-27) queda asi:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 45

Fundamentos de Termodinámica del Equilibrio Físico y Químico
K-kJ/kmol -20.125K-kJ/kmol 0.2105 20.335- =+=
−=− ∑∫
=
NC
1i 1
2i
T
T
'pM'
1M'
2M PPlnyRdT
RTC
Rss2
1
Entropía generada: Sgen = (5 kmol/h)*(-20.125 kJ/kmol-K) + (42151.37/298) = 40.82 kJ/h-K
4.3. PROPIEDADES TERMODINÁMICAS A PARTIR DEL METODO DE LAS ECUACIONES DE ESTADO
El método de las ecuaciones de estado (MEE) se basa en las ecuaciones de estado para determinar las propiedades termodinámicas de mezclas multicomponentes o de sustancias puras. La ecuación virial se usa únicamente para la fase vapor, mientras que las ecuaciones de estado cúbicas de RK, SRK y Peng-R se usan para ambas fases. Tal como se ha descrito anteriormente la solución de las ecuaciones de estado cúbicas tiene tres raíces en el factor Z o en el volumen molar, de las cuales, si son reales positivas, la mayor corresponde a la fase vapor y la menor a la fase líquida.
Antes, se deben definir los conceptos de propiedad residual, propiedad de mezclado y propiedad en exceso.
4.3.1. Propiedad Residual (ϕR(T, P, yi)). Una propiedad residual representa la diferencia entre el valor de la propiedad como gas ideal y el valor de la propiedad en su estado real, medidos a la misma temperatura T, presión P y composición.
( ) ( ) ( )y,T,Py,T,Py,T,P giR ϕϕϕ −= (4-30)
De la ecuación anterior, la propiedad en el estado real es:
( ) ( ) ( )y,T,Py,T,Py,T,P Rgi ϕϕϕ −= (4-31)
El diferencial de la propiedad ϕ(T, P, yi) con respecto a la presión a temperatura y composición constante y la integración desde el estado de gas ideal (P*→ 0, ϕ*R→ 0) hasta el estado real (T, P, yi), están dados por:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 46

Fundamentos de Termodinámica del Equilibrio Físico y Químico
dPPP
P
0 y,Ty,T
idR ∫
−
=
∂∂ϕ
∂∂ϕϕ (4-32)
Entalpía molar residual. La expresión de la entalpía molar residual se obtiene reemplazando ϕ por h en la ecuación (4-32):
;dPPh
Phh
P
0 y,Ty,T
idR ∫
−
=
∂∂
∂∂
Pero 0P
hy,T
id
=
∂
∂; por lo tanto: dP
Phh
P
0 y,T
R ∫
−=
∂∂
Finalmente, la entalpía molar residual está dada por:
dPvTvTh
P
0 y,P
R ∫
−
=
∂∂
(4-33)
La entalpía molar de una mezcla o de una sustancia pura se determina sustituyendo las ecuaciones (4-24) y (4-33) en la ecuación (4-31) aplicada a la entalpía:
∫∫
−
∂∂−+=
P
0 y,P
T
To
idpM
idoM dPv
TvTdTCh)y,T,P(h (4-34)
4.3.3. Entropía molar residual. De la misma manera, si se reemplaza ϕ por s en la ecuación (4-32), se obtiene la expresión de la entropía molar residual:
dPPs
Pss
P
0 y,Ty,T
idR ∫
−
=
∂∂
∂∂
Pero y,Py,Ty,P
id
y,T
id
Tv
Ps
PR
Tv
Ps
−=
−=
−=
∂∂
∂∂
∂∂
∂∂ y
Por lo tanto:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 47

Fundamentos de Termodinámica del Equilibrio Físico y Químico
dPPR
Tvs
P
0 y,P
R ∫
−
=
∂∂
(4-35)
Al reemplazar las ecuaciones (4-26) y (4-35) en la ecuación (4-31), se obtiene la expresión para la entropía molar:
dPPR
Tv
PPylnyR
TdTC
s)y,T,P(sT
T
NC
1i
P
0 y,P0
ii
idpMid
oM0
∫ ∑ ∫=
−
∂∂−
−+= (4-36)
A continuación se presentan las ecuaciones para la entalpía y entropía molares de mezclas basadas en la ecuación virial y las ecuaciones de estado cúbicas.
Ejemplo 4.3. Deduzca la expresión de la entalpía y la entropía residuales de una sustancia pura basada en la siguiente ecuación de estado:
BA1Z +−=Donde A = aP/(RT)2 y B = bP/(RT) con a es una función de la temperatura (tipo ecuación Peng-R)a= f(T) y b es una constante.
Solución: La ecuación de estado en términos del volumen es: bRTa
PRTv −−=
La derivada del volumen molar de esta ecuación de estado es:( )
RTdT/da
RTa
PR
Tv
2P
−+=
∂∂
. Al reemplazar en la ecuación de la entalpía residual (ecuación 4-33)
y posteriormente hacer la integración, resulta:
( )
( )B
dTda
aT2A
RTbP
dTda
aT2
RTaP
RTh
bPdTda
aT2
RTaPdPb
RTdT/daT
RTa2dPv
TvTh
2
R
P
0
P
0 y,P
R
−
−=
−
−=
−
−=
−−=
−
= ∫∫ ∂
∂
De igual manera, al reemplazar en la ecuación de la entropía residual (ecuación 4-34) y posteriormente hacer la integración, resulta:
−=
−=
−
= ∫
dTda
aT1A
Rs
dTda
aT
RTaP
RTaPdP
PR
Tvs
R
22
P
0 y,P
R
∂∂
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 48

Fundamentos de Termodinámica del Equilibrio Físico y Químico
4.3.4. Cálculo de la Entalpía molar y Entropía molar usando la ecuación virial. Debido a que la ecuación virial es explícita en el factor Z, la derivada parcial del volumen de las ecuaciones (4-33), (4-34), (4-35) y (4-36) se encuentra a partir de la definición de Z.
+=
=
y,Py,Py,P TZTZ
PR
T)ZT(
PR
Tv
∂∂
∂∂
∂∂
(4-37)
De la ecuación virial: RTBP1Z += ,
−
=
TB
dTdB
RTP
TZ mm
y,P∂∂
Al reemplazar:
m
y,P
m
y,P
y,P
m
y,P
BP
RTv
dTdB
RP1
PRT
TvT
dTdB
RP1
PR
Tv
−=
+=
+=
∂∂
∂∂
Donde:
∑∑= =
=
NC
1i
NC
1j
ijji
m
dTdB
yydTdB
(4-38)
+=
ij
)1(ij
ijij
)o(ij
ij
ij
dTrdB
wdTrdB
PcR
dTdB
(4-39)
6.2ijij
)o(ij
Tr675.0
dTrdB
= ; 2.5ijij
)1(ij
Tr722.0
dTrdB
=
Los términos dT
dB,
dTdB
,dTdB
,dTdB )1(
ij)o(
ijijm son funciones de la temperatura
únicamente.
Con la sustitución de la ecuación (4-37) en las ecuaciones (4-34) y (4-36) y la integración posterior, se obtiene como resultado las expresiones para la entalpía molar y la entropía molar de mezclas gaseosas basadas en la ecuación virial:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 49

Fundamentos de Termodinámica del Equilibrio Físico y Químico
−
=
−+= ∫
mmR
M
RM
T
To
idpM
idoMM
BdTdBTPh
hdTCh)y,T,P(h
(4-40)
=
−
−+= ∫ ∑
=
dTdBPs
sPPylnyR
TdTC
s)y,T,P(s
mRM
RM
T
To o
iNC
1ii
idpMid
oMM
(4-41)
El cambio de entalpía y entropía de un proceso determinado a composición constante, basado en la ecuación anterior, están dados por:
∫
−−
−−=−2
1 12
T
T Tm
m11
Tm
m22
idpM12 B
dTdBT*PB
dTdBT*PdTChh (4-42)
−
−
−=− ∫
12
2
1 T
m1
T
m2
T
T 1
2idpM
12 dTdBP
dTdBP
PPlnR
TdTC
ss (4-43)
Ejemplo 4.4. Una corriente a 1.5 MPa y 393.2 K que contiene 50% de dióxido de carbono y 50% de n-butano desea separarse en dos corrientes: una de 2% de CO2 y otra de 90% de CO2 a 15 MPa y 393.2 K. Use la ecuación virial para determinar el cambio de entalpía, entropía, energía de Gibbs y de exergía del proceso.Datos:
Tc (K) Pc (KPa) w Zc vc (m3/kmol)CO2 304.2 7383 0.224 0.274 0.094n-butano 425.2 3796 0.200 0.274 0.255
Solución: Base cálculo: 100 kmol de mezcla inicial.Presión y temperatura de referencia: 100 kPa y 300 K, respectivamente.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 50

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Del balance de materia: C1 = 45.44 kmol y C2 = 54.56 kmol
Del balance de energía: (Δh)proceso = (C1*hC1 + C2*hC2) -100hm = C1(hm – hC1) + C2(hm – hC2)
Donde: hm = entalpía de la mezcla con 50% de CO2; hC1 = entalpía de la corriente con 2% de CO2;hC2 = entalpía de la corriente con 90% de CO2;
Del balance de entropía: (Δs)proceso = (C1*sC1 + C2*sC2) -100sm = C1(sm – sC1) + C2(sm – sC2)
Resultados del Programa ecVIRIAL.Para P = 1.5 MPa, T = 393.2 K, y1 = 0.50: hm = hom + 6.024*103 kJ/kmol; sm = som + 1.7496 kJ/kmol-KPara P = 1.5 MPa, T = 393.2 K, y1 = 0.02: hC1 = hom + 8.277*103 kJ/kmol; sc1 = som + 4.314 kJ/kmol-KPara P = 1.5 MPa, T = 393.2 K, y1 = 0.90: hC2 = hom + 3.946*103 kJ/kmol; sC2 = som – 7.918 kJ/kmol-K
Reemplazando en las respectivas ecuaciones de balance: (Δh)proceso = -109.3 kJ/kmol; (Δs)proceso = -4.1089 kJ/kmol-K; (Δg)proceso = (Δh)proceso -T(Δs)proceso = -109.3 - 393.2(- 4.1089) = 1506.32 kJ/kmol
4.3.5. Cálculo de la entalpía molar y la entalpía molar usando las ecuaciones cúbicas. El último término integral de las ecuaciones (4-34) y (4-36) corresponde a la entalpía residual y entropía residual respectivamente. La solución de estas integrales se simplifica si se reemplaza la ecuación (4-17) en la (4-34) y en la (4-36):
∫∫ ∫ ∫
∫ ∫
∞∞
∞
−−=−=
∂∂−=
∂∂
vP
0
Pv
RT
v
P
0
v
y,vy,P
PdvRTPvPdv)Pv(dvdP
dvTPTdP
TvT
(4-44)
La integración de las ecuaciones anteriores requiere una ecuación explícita en la presión. Para las ecuaciones de estado cúbica (ecuación 3-10),
©Carlos Arturo Bello Blanco, Ing Qco M. Sc.
Separador
100 kmolCO
2 : 50%
nC4 : 50%
1500 kPa393.2 K
CO2 : 2%
nC4 : 98%
1500 kPa393.2 K
CO2 : 90%
nC4 : 10%
1500 kPa393.2 K
C1
C2
51

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( )( )2
mmm2m
m
mmy,v tbvubvT/aT
)bv(RT
TPT
−+∂∂
−−
=
∂∂
(4-45)
La sustitución de las ecuaciones (4-44) y (4-45) en las ecuaciones (4-34) y (4-36) permite calcular la entalpía y entropía residuales con las siguientes ecuaciones:
−+
−
+
−−= ∫∞
mv
m2mmm
2m
m
mm
mRM dv
tbvubvdTa
Tda1a
RT11
RTPvRTh
( )
−+−
−
−= ∫∞
mv
m2mmm
2m
m
mmRM dv
tbvubvdTda
R1
RTbvPlnRs
La solución particular de cada ecuación de estado estará en función de sus parámetros específicos u, t, am y bm.
4.3.5.1. Ecuación de Redlich-Kwong (RK) y Soave-Redlich-Kwong (SRK). La expresión para la entalpía molar de ambas ecuaciones de estado es similar y es la siguiente:
+
−−−−=
−+= ∫
m
M
m
m
M
Mm
RM
T
T
RM
idpM
idoMFM
ZB
1lndTa
Tda1BA
1ZRTh
hdTCh)z,T,P(h0
(4-46)
Para la entropía molar es la siguiente ecuación:
+
+−−=
−
−+= ∫ ∑
=
m
M
m
m
M
MMm
RM
RM
T
To o
FiNC
1iFi
idpMid
oMFM
ZB
1lndTa
TdaBA
)BZln(Rs
sP
PzlnzR
TdTC
s)z,T,P(s
(4-47)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 52

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Para la ecuación (RK), 5.0dTa
Tdam
m −= y para las ecuaciones (SRK) y (Peng-R):
m
NC
1iriiwiFi
m
m
a
Tfz
dTaTda ∑
=−=α
(4-48)
4.3.5.2. Ecuación de Peng-Robinson. La entalpía molar y la entropía molar de mezclas se evalúan con las siguientes ecuaciones:
−+
−−−−=
−+= ∫
M
M
m
m
M
MRM
T
To
RM
idpM
idoMFM
B4142.0ZB4142.2Z
lndTa
Tda1B22
A1ZRTh
hdTCh)z,T,P(h
(4-49)
( )
−+
+−−=
−
−+= ∫ ∑
=
M
M
m
m
M
MM
RM
T
To
RM
o
FiNC
1iFi
idpMid
oMFM
B4142.0ZB4142.2Z
lndTa
TdaB22
ABZlnRs
sP
PzlnzR
TdTC
s)z,T,P(s
(4-50)
Como se mencionó anteriormente, las ecuaciones cúbicas tienen la capacidad de predecir las propiedades termodinámicas de ambas fases. Para evaluar la entalpía y la entropía se debe resolver previamente la ecuación cúbica de Z (ecuación (3-17) para la ecuación de estado especificada.
Ejemplo 4.5. Una mezcla gaseosa formada por 74% de nitrógeno(1), 13% de agua(2), 6.5% de CO2(3) y 6.5% de O2(4) a 1.0 MPa y 1000 K fluye a través de una turbina adiabática donde se expande hasta 110 kPa y 570 K. Por cada kmol/s de flujo molar, calcule: a) la potencia generada por la turbina, en kW; b) la irreversibilidad, en kW, si los alrededores están a 298 K. Use la ecuación de Peng-Robinson con el Programa PropPENGR©
Datos de las constantes de Cp/R = a + bT + cT2 + d/T2:Componente a b*103 c*106 d*10-5
N2 3.280 0.593 0.000 0.040H2O 3.470 1.450 0.000 0.121
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 53

Fundamentos de Termodinámica del Equilibrio Físico y Químico
CO2 5.457 1.045 0.000 -1.157O2 3.639 0.506 0.000 -0.227
Solucion:Balance de energía en la turbina: ws = (h1 –h2) = (hid)1 – (hid)2 - (hR
1 – hR2),
Donde (hid)1 – (hid)2 es la diferencia de entalpía de la mezcla gaseosa como gas ideal y se evalúa con la
ecuación (4-25) y (hR1 – hR
2) es la diferencia de las entalpías residuales.
Balance de entropía: Sgen = (s1 –s2) = (sid)1 – (sid)2 - (sR1 – sR
2)Donde (sid)1 – (sid)2 es la diferencia de entropía de la mezcla gaseosa como gas ideal y se evalúa con la ecuación (4-27) y (sR
1 – sR2) es la diferencia de las entropías residuales.
Usando el Programa PropPENGR© con las especificaciones y datos necesarios, los resultados son los siguientes:am = 0.74*3.28 + 0.13*3.47 + 0.065*5.457 + 0.065*3.639 = 3.4695; Igualmente, bm = 0.728*10-3; cm = 0; dm = -0.045*105;
Para las condiciones de entrada:
P1 = 1000 kPa, T1 = 1000 K, (hid)1 = (hid)oM + 22920.371 kJ/kmol; (sid)1 = (sid)oM + 26.85 kJ/kmol-K;AM = 4.0367*10-4; BM = 2.797*10-3; Z = 1.0024; (T/a)(da/dT) = -3.2026;
hR1 = -8.314*1000*[1.0024 – 1 –
(4.0367*10-4/(21.5*2.797*10-3))*(4.2036)*ln[(1.0024 + 2.4142*2.797*10-3)/(1.0024 - 0.4142*2.797*10-3)]] = -5.903
sR1 = -8.314*[ln(1.0024 – 2.797*10-3) +
(4.0367*10-4/(21.5*2.797*10-3))*(-3.2036)*ln[(1.0024 + 2.4142*2.797*10-3)/(1.0024 - 0.4142*2.797*10-3)]] = 0.014
Para las condiciones de salida:P2 = 110 kPa, T2 = 570 K, (hid)2 = (hid)oM + 8823.4 kJ/kmol; (sid)2 = (sid)oM + 26.9822 kJ/kmol-K;AM = 4.1053*10-4; BM = 5.3048*10-4; Z = 1.00012; (T/a)(da/dT) = -1.383; hR
2 = 4.133 kJ/kmol; sR2 = 8.125*10-3 kJ/kmol-K;
(hid)1 – (hid)2 = (22920.371 – 8823.4) = 14096.97 kJ/kmol; (hR
1 – hR2) = (-5.903 – 4.133) = -10.036 kJ/kmol;
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 54
N2 : 74,0%
H2O : 13,0%
CO2 : 6.5%
O2 : 6.5%
1 kmol/s1000 kPa1000 K
1 2N
2 : 74,0%
H2O : 13,0%
CO2 : 6.5%
O2 : 6.5%
1 kmol/s110 kPa570 K
Wneto

Fundamentos de Termodinámica del Equilibrio Físico y Químico
h1 – h2 = (hid)1 – (hid)2 - (hR1 – hR
2) = 14096.97 – (-10.036) = 14107.006 kJ/kmol;
Potencia de la turbina: Ws = (1 kmol/s)(14107.006 kJ/kmol) = 14107 kW
(sid)1 – (sid)2 = (26.9822 – 26.85) = 0.1322 kJ/kmol-K; (sR2 – sR
1) = (8.125*10-3 – 0.014) = -5.875*10-3;
s2 – s1 = (sid)1 – (sid)2 - (sR2 – sR
1) = 0.1322 – (-5.875*10-3) = 0.1381 kJ/kmol-K
Entropía generada: Sgen = (1 kmol/s)(0.1381 kJ/kmol-K) = 0.1381 kW/K;
Irreversibilidad = I = ToSgen = 298*0.1381 = 41.15 kW
Eficiencia de la turbina según la segunda ley: Wrev = Ws + I = 14107 + 41.15 = 14148.15 kW
ηII = Ws/Wrev = 14107/14148.15 = 0.997 (99.7%)
4.4. PROPIEDAD PARCIAL MOLAR DE UN COMPONENTE EN UNA MEZCLA
Hasta ahora las propiedades termodinámicas se han determinado en procesos a composición constante. Sin embargo, muchas aplicaciones de la termodinámica en ingeniería química se basan en sistemas donde la composición de los componentes cambia ya sea por proceso de mezclado, de separación, procesos de transferencia de masa entre fases o procesos donde ocurren reacciones químicas.En esta sección se estudiarán los sistemas de composición variable en los cuales el cambio en las moles de cada componente afecta las propiedades termodinámicas; esto quiere decir que cualquiera propiedad termodinámica (Θ = nθ) será función de la presión, la temperatura y las moles de cada componente ni y tendrá la forma:
)n.....n,n,n,n,T,P(n NC4321θθ =
El diferencial total de esta propiedad será:
( ) ∑=
∂∂+
∂∂+
∂∂=
NC
1kk
n,T,Pkn,Pn,Tdn
nndT
TndP
Pnnd
j
θθθθ (4-51)
Por definición, una propiedad parcial molar de un componente i en una mezcla es el cambio de la propiedad total de la mezcla con respecto al cambio de las moles del componente i, manteniendo constante la presión, la temperatura y la composición de los demás componentes.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 55

Fundamentos de Termodinámica del Equilibrio Físico y Químico
jn,T,Pii n
)n(
=
∂θ∂θ (4-52)
Una propiedad fundamental en la termodinámica de las soluciones es la energía de Gibbs. El diferencial total de la energía de Gibbs es:
( )
( ) ∑
∑
=
=
∂∂
+−=
∂∂
+
∂∂
+
∂∂
=
NC
1kk
n,T,Pk
NC
1kk
n,T,Pkn,Pn,T
dnnngdT)ns(dP)nv(ngd
dnnngdT
TngdP
Pngngd
j
j
(4-53)
Usando la definición de propiedad parcial molar, la derivada parcial de ng con respecto a las moles de la especie k es la energía de Gibbs parcial molar y se define como el potencial químico de la especie k:
kn,T,Pk
k gnng
j
=
∂∂
=µ (4-54)
Sustituyendo el potencial químico de cada especie, el diferencial de la energía de Gibbs está dado por:
( ) ∑=
+−=NC
1kkkdndT)ns(dP)nv(ngd µ (4-55)
Donde n son las moles totales de la mezcla.
Para una propiedad θ :
( ) ∑=
+
∂∂+
∂∂=
NC
1kkk
x,Px,TdndT
TndP
Pnnd θθθθ (4-56)
Dado que nk = xkn, dnk = xkdnk + nkdxk y d(nθ) = ndθ + θdn. La sustitución de estas ecuaciones en (4-56) resulta:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 56

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( )∑=
++
∂∂+
∂∂=+
NC
1kkkkkk
x,Px,TdxndnxdT
TndP
Pndnnd θθθθθ (4-57)
Al reagrupar en los términos comunes a n y a dn se obtiene:
0dnxndxdTT
dPP
dNC
1kkk
NC
1kkk
x,Px,T=
−+
−
∂∂−
∂∂− ∑∑
==
θθθθθθ (4-58)
Cada término entre corchetes de la derecha de la ecuación anterior debe ser cero, por lo tanto se encuentran dos relaciones importantes:
∑=
=NC
1kkkx θθ ó ∑
=
=NC
1k
kknn θθ (4-59)
∑=
+
∂∂+
∂∂=
NC
1kkk
x,Px,TdxdT
TdP
Pd θθθθ (4-60)
La derivada de θ dada por la ecuación (4-59) proporciona una expresión para dθ :
∑∑∑===
+
∂∂+
∂∂=+=
NC
1kkk
x,Px,T
NC
1kkk
NC
1kkk dxdT
TdP
Pdxdxd θθθθθθ
(4-61)
El arreglo de la ecuación anterior es la ecuación de Gibbs/Dühem:
0dxdTT
dPP
NC
1kkk
x,Px,T=−
∂∂+
∂∂ ∑
=
θθθ(4-62)
En el caso particular en que la temperatura y la presión son constantes, la ecuación Gibbs/Dühem se reduce a:
0dxNC
1kkk =∑
=
θ (4-63)
Las ecuaciones (4-62), (4-63) para la energía de Gibbs quedan transformadas en:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 57

Fundamentos de Termodinámica del Equilibrio Físico y Químico
0gdxdTTgdP
Pg NC
1kkk
x,Px,T=−
∂∂
+
∂∂ ∑
=
(4-64)
0dxgdxNC
1kkk
NC
1kkk == ∑∑
==
µ (4-65)
4.4.1. Fugacidad y coeficiente de fugacidad.
4.4.1.1. Fugacidad y coeficiente de fugacidad de una sustancia pura. Para un fluido puro a temperatura constante, la ecuación (4-3) se reduce a dgi = vidP; si es un gas ideal el volumen molar se halla con la ecuación de estado (vid)i = R*T/P, entonces
( )
( )P
dPPlnd
PlnRTddPP
RTdg idi
=
==
Por integración:
( )PlnRT)T(g iidi += ϕ (4-66)
Si el fluido es un gas no ideal, se espera que
( )
( )( )iii
i
ii
flnRT)T(g
dPPZflnd
flnRTddPP
ZRTdg
+=
=
==
ϕ
(4-67)
Donde fi = fugacidad del gas puro i.
La energía de Gibbs molar residual de la sustancia i se encuentra restando las dos ecuaciones diferenciales anteriores:
−=−=PflnRTggg i
iidi
Ri (4-68)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 58

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Por definición el coeficiente de fugacidad de una sustancia pura es la relación entre la fugacidad y la presión
Pfi
i =φ (4-69)
De acuerdo con la ecuación (4-67) la energía de Gibbs molar residual del gas puro i se relaciona con el coeficiente de fugacidad de la siguiente manera:
( )RTgln
Ri
i −=φ (4-70)
Se observa que en la condición de gas ideal cuando P → 0, 1 y →→ iRi 0g φ .
4.4.1.2. Fugacidad y coeficiente de fugacidad de un componente i en una mezcla. La energía de Gibbs parcial molar de un componente i en una mezcla gaseosa ideal se halla al reemplazar las ecuaciones (4-29) y (4-66) en la relación fundamental de la energía de Gibbs:
( )PylnRT)T(ylnRTgsThg iiiidi
idi
idim
idi
idim +=+==−= ϕµ (4-71)
Para un componente i en una mezcla no ideal, su energía de Gibbs parcial molar está dada por la ecuación anterior modificada:
+== iiii flnRT)T(g ϕµ (4-72)
Donde if = fugacidad del componente i en la solución o mezcla.
La energía de Gibbs parcial molar residual del componente i se halla restando las dos ecuaciones anteriores:
−=−=
PyflnRTgggi
ii
idim
Ri (4-73)
En efecto si la mezcla es ideal Pyf ii = donde yiP es la presión parcial del componente i en la fase vapor y además su energía de Gibbs parcial molar residual es cero.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 59

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Se define como coeficiente de fugacidad del componente i en una mezcla a la relación entre su fugacidad y su presión parcial:
Py
fˆi
ii =φ (4-74)
La ecuación (4-73) se reacomoda para dar:
( )RTgˆln
Ri
i −=φ (4-73)
4.4.1.3. Cálculo del coeficiente de fugacidad de una sustancia pura. La combinación de las ecuaciones (4-66) y (4-67) permite encontrar la siguiente expresión para determinar el coeficiente de fugacidad de un gas puro:
( )P
dP1Z)Pln(d)fln(d)ln(d ii −=−=φ
Por integración,
( ) ∫∫ −=−=P
0
Ri
P
0ii dPv
RT1
PdP1Z)ln( φ (4-75)
El coeficiente de fugacidad de sustancias puras se evalúa usando la ecuación anterior a partir de datos PvT o una ecuación de estado.
A partir de la definición de propiedades residuales, el coeficiente de fugacidad se relaciona con la energía de Gibbs molar residual y ésta a su vez con la entalpía y entropía residuales de la siguiente manera:
( ) Ri
Riii
idi
Ri TshlnRTggg −=−=−= φ
( )RTh
Rsln
Ri
Ri
i −=φ (4-76)
Ejemplo 4.6. A partir de la expresión del coeficiente de fugacidad dada por la ecuación (4-76) encuentre el coeficiente de fugacidad de una sustancia pura que se basa en la ecuación de estado y usando las propiedades residuales del ejemplo 4.3.Solución:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 60

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Al reemplazar las propiedades residuales del ejemplo 4.3, el coeficiente de fugacidad de una sustancia pura basada en la misma ecuación de estado está dado por:
( )
( ) ABln
BdTda
aT2A
dTda
aT1A
RTh
Rsln
i
Ri
Ri
i
−=
+
−−
−=−=
φ
φ
En forma similar, los coeficientes de fugacidad de una sustancia pura a partir de las propiedades residuales dadas por las ecuaciones (4-40), (4-41), (4-46), (4-47), (4-49, (4-50) se evalúan con las siguientes expresiones:
• Ecuación virial:
( )RTPBB
dTdBT
RTP
dTdB
RP
RTh
Rsln ii
iiiiii
Ri
Ri
i =
−
−
=−=φ (4-77)
El resultado será el mismo si se resuelve la ecuación (4-75):
( ) ∫∫ =
=−=
P
0
iiP
0ii RT
PBP
dPRT
PBP
dP1Z)ln( φ
• Ecuaciones de Redlich Kwong y Soave Redlich Kwong:
+−−−−=
ZB1ln
BA)BZln()1Z()ln( iφ (4-78)
• Ecuación de Peng Robinson:
−−++−−−−=
B)12(ZB)12(Zln
B2A)BZln()1Z()ln( 5.1iφ (4-79)
4.4.1.4. Fugacidad de una sustancia pura en el equilibrio líquido-vapor. Una sustancia pura en equilibrio líquido-vapor se encuentra en su punto de saturación, es decir que a la temperatura de equilibrio le corresponde una presión de saturación PS
i y debe cumplirse que
)fln(RT)fln(RTdgdg Li
Gi
Li
Gi === (4-80)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 61

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Donde Si
Li
Gi fff == es la fugacidad del fluido i puro en el punto de saturación a la
temperatura T.
Bajo estas condiciones existe un coeficiente de fugacidad de saturación que es la relación entre la fugacidad de saturación y la presión de saturación:
Si
SiS
i Pf
=φ (4-81)
En el equilibrio líquido vapor también se debe cumplir que: ;0dgdg Li
Gi =− pero
dPvdTsdg;dPvdTsdg Li
Li
Li
Gi
Gi
Gi +−=+−=
Al reemplazar en la expresión anterior se obtiene la ecuación de Clapeyron:
( )( )
( )( )L
iGi
Li
Gi
Li
Gi
Li
Gi
sati
vvThh
vvss
dTdP
−−
=−−
=
Si se considera que Gi
Li
Gi vvv ≈− y que el vapor saturado se comporta como un gas
ideal, P/RTv Gi = , al reemplazar en la ecuación de Clapeyron se obtiene la ecuación
de Clausius- Clapeyron:( ) ( )
2
Li
Gi
sati
RThh
dTPlnd −
= (4-82)
Con la integración de la ecuación anterior y asumiendo que la entalpía de vaporización es aproximadamente constante, se obtiene el siguiente resultado:
( ) ( ) CRT
hhPlnL
iG
isati +
−−=
Donde C es una constante de integración.
Se han propuesto muchas correlaciones empíricas entre la presión de saturación y la temperatura. Una de ellas es la ecuación de Antoine dada por:
( ) ( )iiisat
i CT/BAPln +−=
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 62

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Donde Ai, Bi y Ci son constantes de cada sustancia i. Por ejemplo para el agua las constantes de Antoine son: A = 16.2887, B = 3816.44 y C = -46.13. La presión se saturación del agua a 100º C (373.15 K) es, según la ecuación de Antoine 101.326 kPa. Si se compara con 101.325 kPa el resultado es excelente.
4.4.1.5. Efecto de la presión sobre la fugacidad. Combinando las ecuaciones (4-3) y (4-67) se encuentra la siguiente ecuación que muestra el efecto de la presión sobre la fugacidad.
RTv
P)fln( i
x,T
i =
∂∂
(4-83)
Integrando desde la presión de saturación hasta la presión del sistema se obtiene:
∫=
=
P
PiS
iSi
iS
i
iS
i
dPvRT1
Pfln
ffln
φ
La fugacidad de i se halla despejando fi :
= ∫
P
Pi
Si
Sii
Si
dPvRT1expPf φ (4-84)
Si el fluido es un líquido puro incompresible el volumen molar es constante y la ecuación anterior se puede integrar para obtener la fugacidad, así:
−=
RT)PP(vexpPf
Si
SiS
iSi
Li φ (4-85)
Donde fiL = fugacidad de la sustancia como líquido puro a la presión P y a la
temperatura T. vi
S = volumen molar del líquido saturado a la temperatura de saturación
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 63

Fundamentos de Termodinámica del Equilibrio Físico y Químico
El término exponencial representa el efecto de la presión sobre la fugacidad del líquido y se denomina Efecto Poynting. El efecto Poynting tiende a la unidad cuando la presión es baja; en este caso la fugacidad del líquido estará dada por:
Si
Si
Li Pf φ= (4-86)
4.4.1.6. Efecto de la temperatura sobre la fugacidad. La relación entre la fugacidad y la temperatura se encuentra a partir de la definición de la energía de Gibbs. Derivando a (ng/(RT) con respecto a la temperatura a presión y composición constante:
( ) dTRTngngd
RT1
RTngd 2−=
Remplazando a d(ng) dada por la ecuación (4-7):
∑=
+−=NC
1iiidngdT)ns(dP)nv()ng(d (4-7)
y )ns(T)nh()ng( −=
Se obtiene la relación fundamental de la termodinámica de las soluciones:
∑=
+−=
NC
1ii
i2 dn
RTgdT
RTnhdP
RTnv
RTngd (4-87)
La relación anterior aplicada a las condiciones residuales se transforma en:
( )∑
∑
=
=
−−=
+−=
NC
1iii2
RRR
NC
1ii
Ri
2
RRR
dnˆlndTRTnhdP
RTnv
RTngd
dnRTgdT
RTnhdP
RTnv
RTngd
φ
(4-88)
A partir de la definición de propiedad parcial molar y usando la ecuación (4-59), el coeficiente de fugacidad se puede obtener a partir de la ecuación (4-88):
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 64

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( ) ( )( )
( )i
NC
1ii
NC
1i
Ri
i
R
Ri
n,T,Pi
R
i
ˆlnyRTgy
RTg
RTg
nRT/ngˆln
j
φ
φ
∑∑==
−=−=
−=
∂
∂−=
(4-89)
Esta ecuación demuestra que ( )iˆln φ es una propiedad parcial molar de ngR/(RT)
A presión y temperatura constantes, la ecuación de Gibbs/Dühem para la energía de Gibbs es:
( )∑=
=NC
1iii 0ˆlndy φ (4-90)
A presión y composición constantes, la ecuación (4-88) se transforma en la expresión que relaciona el efecto de la temperatura sobre el coeficiente de fugacidad de un fluido puro:
( )P
i
P
i
P
Ri
2
Ri
Tfln
Tln
T)RT/(g
RTh
=
=
∂
∂−=
∂∂
∂φ∂
(4-91)
A partir de la ecuación anterior se puede determinar la entalpía del líquido puro en términos de la fugacidad dada por la ecuación (4-85) o (4-86):
P
Li2id
iiL TflnRThh
−=
∂∂
(4-92)
Donde P
sati
P
sati
P
Li
TPln
Tln
Tfln
∂
∂+
∂
∂=
φ∂
∂según la ecuación (4-86)
La ecuación (4-87) muestra las relaciones entre las propiedades parciales molares.
Ejemplo 4.7. Calcule la entalpía latente de vaporización del agua a 120º C (393.15 K) si el vapor saturado es gas ideal, es decir su entalpía residual es cero). Use las constantes de Antoine del agua. Solución: La condición de idealidad del vapor saturado implica que el coeficiente de fugacidad en el punto de saturación es igual a uno, por lo tanto, de la ecuación (4-92) se desprende:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 65

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( )22
P
si2
P
Li2
fgiLiGiLid
i CTBRT
TPlnRT
TflnRThhhhh
+=
=
==−=−
∂∂
∂∂
Reemplazando los valores:
hfg = 8.314*393.152*3816.44/(393.15 – 46.13)2 = 40726.4 kJ/kmol = 2262.58 kJ/kg
El valor experimental en las tablas de vapor es 2202.3 con un error relativo de 2.73%
La aplicación del criterio de la exactitud a la ecuación (4-87) da las relaciones entre propiedades residuales molares:
( )( ) ( )( )2
i
ij,n,T,Pi
2
n,P
i
RTh
nRT/nh
TRT/g
jj
−=
∂
∂−=
∂
∂
≠ (4-93)
( )( ) ( )( )RTv
nRT/nv
PRT/g i
ij,n,T,Pin,T
i
jj
=
∂
∂=
∂∂
≠ (4-94)
La entalpía parcial residual del componente i se encuentra a partir de la ecuación (4-93) en función de la energía de Gibbs parcial residual y de acuerdo con la ecuación (4-89), con el coeficiente de fugacidad:
( )y,P
i
y,P
i
y,P
Ri
2i
idi
2
Ri
Tfln
Tˆln
T)RT/(g
RT)hh(
RTh
=
=
−=
−=
∂∂
∂φ∂
∂∂
(4-95)
La entalpía parcial molar del componente i en la fase líquida se puede determinar a partir de la ecuación anterior en términos de la fugacidad:
y,P
Li2id
i
y,P
i2idiiL T
flnRThTˆlnRThh
−=
−=
∂∂
∂φ∂
(4-96)
4.4.2. Cálculo del coeficiente de fugacidad de un componente en una mezcla usando las ecuaciones de estado.
El coeficiente de fugacidad de un componente i en una mezcla se obtiene al desarrollar la ecuación (4.89) para una ecuación de estado dada.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 66

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Para la ecuación de (RK) por ejemplo, la energía de Gibbs residual se halla a partir de la entropía residual y la entalpía residual:
++−−
−=
nZnBnZln
nBAn)nnZ(
nnBnZlnn
RTng 2R
Ejemplo 4.8. Deduzca la expresión del coeficiente de fugacidad del componente k en una mezcla cuya energía de Gibbs residual está dada por la entropía y entalpía residuales encontradas en el ejemplo 4.3:
Solución:La energía de Gibbs residual de una mezcla se halla a partir de la entalpía y entropía residuales y los coeficientes de fugacidad se obtienen a partir de la ecuación (4-89):
( ) ( )( )jn,T,Pi
R
i nRT/ngˆln
∂
∂−=φ (4-89)
Donde:
( ) ( )
∑∑∑
∑∑
==
=
=
==
ii
iiim
2
iii
m
iiim
2
kkkm
m2
mR
nnbnnb;n
anna
byb;ayaRT
PnbRT
PnaRTng
donde -
La solución de la ecuación (4-89) para encontrar los coeficientes de fugacidad es:
( )( )( ) ( )kk2
2
i
NC
iiki
NC
ii
2
kin,T,Pk
Rˆlnb
RTP
n
anaann2
RTP
nRT/ng
i
φ−=−
−
=
∂
∂ ∑∑≠
( )( )
−−
=
−−
= 1
aa2A
bbB1
aa2
RTPa
bb
RTPbˆln
m
k
m
k
m
k2
m
m
kmkφ
Para una sustancia pura el resultado es el mismo obtenido en el ejemplo 4.5 con ak = am, bk = bm
4.4.2.1. Ecuación virial. La expresión del coeficiente de fugacidad del componente i en una mezcla a partir de la ecuación virial está dada por:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 67

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( ) ( )( )
−+= ∑∑
= =
NC
1j
NC
1kjkjikjiii 2yy
21B
RTP)ˆln δδφ (4-97)
Donde δji = 2Bji - Bjj - Bii; δii = δjj = δkk = 0; δji = δij
Los coeficientes de fugacidad de los componentes de una mezcla gaseosa binaria se evalúan con la ecuación anterior reducida a NC = 2 mediante las siguientes expresiones:
[ ]1222111 yB
RTP)ˆln( δφ += [ ]21
21222 yB
RTP)ˆln( δφ += (4-98)
Donde δ12 = 2B12 – B11 – B22
Para una sustancia pura la ecuación (4-98) se convierte en la ecuación (4-77):
=RTPBexp ii
iφ (4-77)
En el punto de saturación, el coeficiente de fugacidad está en función de la temperatura:
=
RTBPexp ii
SiS
iφ (4-99)
4.4.2.2. Ecuaciones Cúbicas. Ecuaciones de (RK) y de (SRK). La expresión del coeficiente de fugacidad del
componente i en una mezcla a partir de las ecuaciones de Redlich-Kwong (RK) y Soave (SRK) está dada por:
+−−−−−=
m
M'i
'i
M
MMm
'imi Z
B1ln)BA(
BA
)BZln(B)1Z()ˆln( φ (4-100)
Para una sustancia pura la ecuación anterior se convierte en la ecuación (4-78):
+−−−−=
ZB1ln
BA)BZln()1Z()ln( iφ (4-78)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 68

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Ecuación de (Peng-R). El coeficiente de fugacidad del componente i en una mezcla se determina con la siguiente ecuación basada en la ecuación de Peng-Robinson::
−−
++−−−−−=
Mm
Mm'i
'i
M5.1M
Mm'imi B)12(Z
B)12(Zln)BA(
B2A
)BZln(B)1Z()ˆln( φ (4-101)
Para una sustancia pura la ecuación anterior se transforma en la ecuación (4-79):
−−++−−−−=
B)12(ZB)12(Zln
B2A)BZln()1Z()ln( 5.1iφ (4-79)
En las ecuaciones (4-100) y (4-101), m
i'i a
a2A = y m
i'i b
bB =
Ejemplo 4.9. Calcular la entalpía y entropía residuales, los coeficientes de fugacidad de los componentes de una mezcla gaseosa formada por 10% (molar) de NH3 y 90% de agua (H2O) a 180º C (453 K) y 500 KPa. Use la ecuación de Peng-Robinson (Peng-R).
Datos:Componente yi Tci(ºK) Pci (KPa) wi
NH3 0.10 406.0 11280 0.250H2O 0.90 647.3 22090 0.348
Solución:Ecuación de PENG-R. De la Tabla 1, u = 2, t = 1 ;
Z3 - (1-BM)Z2 + (AM -2BM -3BM2) - (AMBM - B2
M - B3M) = 0
fwi = 0.37464 + 1.54226*wi - 0.26992*wi2 ;
aci = (1 + fwi(1-Tri1/2))2 ; Ωa = 0.45724; Ωb = 0.0778
Cálculos:Tri fwi aci αi ai bi
NH3 0.69983 0.878658 1.307842 599.487 784.034 0.0189539H2O 1.11576 0.743335 0.918060 461.857 424.012 0.0232812
am = 743.09 (KPa)(m3/(Kmol)2 ; bm = 0.0193866 m3 /Kmol. AM =0.0261936; BM = 2.5737*10-3 P1 = -0.9974262 ; Q1 =0.0210263 ; R1 = -6.07735*10-5 ; M = -0.3105933 ; N = -0.0665736;
Cálculo del discriminante: D < 0 (Existen tres raíces. La mezcla gaseosa está en su punto de rocío)φ = 0.9992307; θ /3 = 0.013075 radianes;
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 69
Fundamentos de Termodinámica del Equilibrio Físico y Químico
Cálculo de las raíces: Z1 = 0.976; Z2 = 3.4539*10-3 ; Z3 = 0.018027
Ya que la mezcla está en fase gaseosa, el valor de Zm = 0.976Masa molecular promedio: PM = 17.9 Cálculo de la entalpía residual: hR
M = 257.56 KJ/Kmol = 14.39 KJ/KgCálculo de la entropía residual: sR
M = 0.3704 KJ/(Kmol-K) = 0.0207 KJ/(Kg-K)
Cálculo de los coeficientes de fugacidad:Componente A’
i B’i
iφNH3 2.054361 0.97768 0.98H2O 1.51077 1.20089 0.99
Ejemplo 4.10. Una mezcla que contiene 90% molar de metano (1), 5% de propano (2), y 5% de n-pentano (3) a 100º C (373 K) y 20 atm (2026.5 KPa) entra a una turbina. Calcular el volumen específico, los coeficientes de fugacidad, la entalpía y la entropía residuales si se usa la ecuación de SRK.DATOS: 1 atm = 101.325 KPa. R = 0.08206 atm-m3/(Kmol-ºK) = 8.314 KJ/(Kmol-K)Componente Tc (K) Pc (atm) w
Metano 190.6 45.4 0.007Propano 369.8 41.9 0.145
n-pentano 469.6 33.3 0.251
Solución: Cálculos:
Tri fwi aci αi ai bi
CH4 1.956978 0.4909813 0.6466372 2.303397 1.4894621 0.0298481C3H8 1.008653 0.7039496 0.9939308 9.395096 9.3380192 0.0625748C5H12 0.794293 0.8629818 1.1965418 22.809651 22.809651 0.1002614De acuerdo con la ecuación de SRK,am = 2.2200398 atm(m3/Kmol)2; AM = 0.0473925 ; bm = 0.0350137 m3/Kmol; BM = 0.0228785; P1 = -1.0 ; Q1 = 0.239905 ; R1 = -1.0842693*10-3; M = -0.3093428; N = 0.0671615 ; Discriminante: D = 3.1298969*10-5 > 0; Existe una raíz real; D1/2 = 5.5945482*10-3; D1 = 0.3396284 ; D2 = 0.303609; Z = 0.97657;
PMmezcla = 20.2 Kg/Kmol; vmolar = 1.4946 m3/Kmol ; v = 0.074 m3/Kg
Tdam/(amdT) = -0.815065; hR = 16.96 KJ/Kg; sR = 0.00356 KJ/(Kg-K)
Coeficientes de fugacidad:Componente A’
i B’i
iφCH4 1.6381903 0.8524691 0.99
C3 H8 4.1018238 1.7921099 0.90C5H12 6.4107514 2.8634906 0.83
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 70

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Ejemplo 4.11. Encontrar la expresión del coeficiente de expansión volumétrica β de una sustancia pura usando las ecuaciones cúbicas. Solución:
Por definición,PT
vv1
=∂∂β
Ya que RTPvZ = ;
+=
PP TZTZ
PR
Tv
∂∂
∂∂
La ecuación de estado cúbica general es: 0RZQZPZ 112
13 =+++
112
P
1
P
1
P
12
P QZP2Z3TR
TQZ
TPZ
TZ
++
+
+
−=
∂
∂∂∂
∂∂
∂∂
Sus derivadas parciales están dadas por las siguientes ecuaciones:
−=
TB
)1u(TP M
P
1
∂∂
∂∂ ( )
++−
=
TB
B)tu(2uTA
TQ M
MM
P
1
∂∂
∂∂
∂∂
( )( )
+−+
−=
TBtB3B2AB
TA
TR M2
MMMMM
P
1
∂∂
∂∂
∂∂ ;
Ta
aT2
TA
TA m
m
MM
−−=
∂∂
∂∂
La expresión
Ta
aT m
m ∂∂
está dada por las ecuaciones (4-48). Finalmente, TB
TB MM −=
∂∂
Finalmente, la expresión para el coeficiente de expansión volumétrica queda en función del cambio de Z con respecto a la temperatura encontrado anteriormente, así:
PTZ
Z1
T1
+=
∂∂β
Ejemplo 4.12. Deduzca la ecuación que permita determinar el coeficiente de compresibilidad isotérmica k usando la ecuación de Peng-Robinson (Peng-R).Solución:
Por definición, TP
vv1k
−=∂∂
. Ya que PZRTv = ;
−
=
PZ
PZ
PRT
Pv
TT ∂∂
∂∂
Reemplazando en la ecuación anterior se obtiene,
TP
ZZ1
P1k
−=
∂∂
Donde: 11
2T
1
T
1
T
12
T QZP2Z3PR
PQZ
PPZ
PZ
++
+
+
−=
∂
∂∂
∂∂∂
∂∂
Para la ecuación de Peng-Robinson, P1 = (BM - 1); Q1 = AM - 2BM - 3B2M ; R1 = -(AM BM - B2
M - B3M)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 71

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Derivando con respecto a la presión PB
PP M
T
1 =
∂∂
; ( )2MMM
T
1 B6B2AP1
PQ
−−=
∂∂
;
( )3M
2MBM
T
1 B3B2BA2P1
PR
−−−=
∂∂
Reemplazando,
++−−−−−+
+=)QZP2Z3(Z
)B3B2BA2(Z)B6B2A(ZB1
P1k
112
3M
2MMM
2MMM
2M
4.5. PROPIEDADES TERMODINAMICAS A PARTIR DE MODELOS DE ACTIVIDAD
Las ecuaciones de estado cúbicas no tienen en cuenta las interacciones entre las moléculas y la polaridad de una solución líquida, por lo tanto, su aplicación en la fase líquida puede causar errores apreciables. Debido a esto su uso no se recomienda en soluciones o mezclas de componentes polares.
En este método las propiedades termodinámicas de la fase vapor se evalúan con una ecuación de estado como las ecuaciones cúbicas o con más frecuencia, con la ecuación virial (a presiones bajas y moderadas), mientras que las de la fase líquida se evalúan con modelos de coeficientes de actividad.
Para entender el significado de coeficiente de actividad se debe explicar algunos conceptos y definiciones básicas.
4.5.1. Fugacidad en el estado estándar. El estado estándar se define como el estado de una sustancia líquida pura a las condiciones de presión P y de temperatura T del sistema y se denota con el símbolo º.
Por lo tanto, la fugacidad en el estado estándar de un líquido puro, (f ºi), según la
ecuación (4-85) corresponde a:
−=
RT)PP(vexpPf
Si
SiS
iSi
Lºi φ (4-102)
4.5.2. Fugacidades en soluciones ideales. El cambio de energía de Gibbs de una sustancia pura a temperatura constante está por: dgi = vidP = RTdln(fi)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 72

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Integrando desde (P = 0, fi = P) hasta (P, fºLi) para un líquido puro:
( )i
oLi
P
0i lnRT
PflnRTdPv φ=
=∫ ;
Para un componente i en una solución:
== iii flnRTddPvgd
El resultado de la integración de la ecuación anterior desde (P = 0, Pxf iL
i = ) hasta
(P, Lif ) es el siguiente:
( )ii
Li
P
0i
ˆlnRTPx
flnRTdPv φ=
=∫
La diferencia de las dos integrales arroja el siguiente resultado:
∫ −−=
=
=
P
0ii
i
iº
ii
Li
ºii
Li dP)vv(
RT1ˆ
lnfx
flnPfx
Pflnφφ
Por definición, una solución se considera ideal cuando el volumen molar de cada componente en la solución es igual al volumen molar de dicho componente puro, medidos a la misma presión y temperatura, es decir:
)T,P(v)x,T,P(v ºii =
Si la solución es ideal se cumple la igualdad de los volúmenes molares y la fugacidad del componente i en una solución ideal está dado por:
Lºii
ideali fxf =
Una solución es ideal en los límites cuando xi→1 para sistemas completamente miscibles y cuando xi→0 para sistemas parcialmente miscibles. Es decir:
Lºi
i
ideali
i
fx
flim
1x=
→Lº
iiideal
i fxf = (4-103)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 73

Fundamentos de Termodinámica del Equilibrio Físico y Químico
La ecuación anterior se conoce como la regla de Lewis-Randall y se aplica a soluciones de componentes completamente miscibles.
Para soluciones de componentes parcialmente miscibles (solubilidad parcial) se cumple la ley de Henry:
ii
ideali
i
Hx
flim
0x=
→
iiideal
i Hxf = (4-104)
Donde Hi es la constante experimental de Henry.
4.5.3. PROPIEDAD DE MEZCLADO (∆w(M)).
Una propiedad de mezclado se defina como la diferencia entre el valor de la propiedad como solución líquida en su estado real y su valor a partir del estado estándar de sus componentes puros, medidos a las mismas condiciones de presión, temperatura y composición molar.
∑ ∑∑= ==
−=−=−=NC
1i
NC
1i
ºiii
ºiii
NC
1ii
)M( )ww(x)wx()wx(www ∆ (4-105)
La propiedad de mezclado representa la diferencia de la propiedad en un proceso a presión y temperatura constantes cuando se forma una solución a partir de sus componentes líquidos puros.
Si la propiedad es la entalpía se tiene la entalpía de mezclado o calor de solución; y si es la energía de Gibbs se tiene la energía de Gibbs de mezclado o energía de Gibbs de solución.
La expresión para la entalpía de mezclado se puede encontrar modificando la ecuación (4-93) y reemplazándola en la ecuación (4-105). En la ecuación (4-93) a la fugacidad de un componente i le corresponde su entalpía parcial molar; de la misma
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 74

Fundamentos de Termodinámica del Equilibrio Físico y Químico
manera a la fugacidad de un líquido puro en el estado estándar le corresponde una entalpía en ese estado (hi
º). Por lo tanto,
x,P
ºii2º
ii T)ffln(RT)hh(
−=−
∂∂
(4-106)
Reemplazando en (4-93) la entalpía de mezclado es:
∑ ∑= =
−=−=
NC
1i
NC
1ix,P
ºii
i2º
iii)M(
T
fflnxRT)hh(xh
∂
∂∆
Si la solución es ideal se cumple la regla de Lewis-Randall, es decir,
iLº
iideal
i xff = .
Reemplazando en la ecuación anterior el término diferencial es cero:
∑ ∑= =
=
−=−=
NC
1i
NC
1i x,P
ii
2ºiii
ideal)M( 0T
xlnxRT)hh(xh∂
∂∆
El resultado anterior quiere decir que significa que el calor de mezclado de una solución ideal es cero.
De acuerdo con la ecuación (4-93), la expresión de la energía de Gibbs de mezclado es:
∑=
−=NC
1i
ºiii
M
)gg(xRT1
RTg∆
(4-107)
La expresión de la entropía de mezclado se obtiene usando la (4-7):
iidngdP)nv(dT)ns()ng(d ∑++−=
La aplicación del criterio de la exactitud a esta ecuación, da la relación de Maxwell,
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 75

Fundamentos de Termodinámica del Equilibrio Físico y Químico
( )i
n,T,Pin,P
i snns
Tg
j
−=
∂
∂−=
∂∂
De la ecuación (4-7) para n = 1 se obtiene la entropía del líquido puro:ºi
n,P
ºi s
Tg
−=
∂∂
Al reemplazar las ecuaciones anteriores en la ecuación (4-93) se encuentra la entropía de mezclado:
( )x,P
ºii
NC
1ii
ºii
NC
1ii
M
T)gg(x
R1ssx
R1
Rs
−−=−= ∑∑
==∂
∂∆ (4-108)
Similarmente, la del volumen de mezclado es:
( )x,T
NC
1i
ºii
i
NC
1i
ºiii
M
P)gg(x
RTPvvx
RTP
RTvP ∑∑
==
−=−=
∂∂∆
(4-109)
La energía de Gibbs de mezclado se puede evaluar integrando la ecuación (4-67) desde un estado como líquido en el estado estándar hasta un estado en la solución, es decir,
=−⇒= ∫∫ º
i
iºi
f
fii
g
gi f
flnRT)gg()fln(dRTgdi
ºi
i
ºi
(4-110)
El argumento del término logarítmico se define como actividad del componente i en la solución,
ºi
ii f
fa = (4-111)
La ecuación anterior está de acuerdo con la regla de Lewis-Randall. Según la ley de Henry la actividad está definida como:
i
iºi H
fa = (4-112)
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 76

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Igualmente las demás propiedades de mezclado resultan ser funciones de la actividad.
== ∏∑
==
NC
1i
xi
NC
1iii
Mi)a(ln)aln(x
RTg∆
(4-113)
( )x,P
NC
1i
ii
M
Tlnalnx
RTh ∑
=
−=
∂∂∆
(4-114)
x,T
NC
1i
ii
M
Pln)aln(x
RTvP ∑
=
=
∂∂∆
(4-115)
∑=
+
∂
∂−=
NC
1ii
x,P
ii
M
alnTalnTx
Rs∆
(4-116)
Las ecuaciones anteriores están en función de la actividad. Los modelos de actividad relacionados en este trabajo permiten determinar la actividad y por consiguiente, las propiedades de mezclado. 4.5.4. ACTIVIDAD DE SOLUCIONES IDEALES
Según regla de Lewis-Randall o la ley de Henry, la actividad de un componente en una solución ideal es igual a su composición (xi), ii xa = . Con este resultado las propiedades de mezclado de soluciones ideales se determinan así:
( ) 0xln*xRTgNC
1iii
)ideal(M ≤= ∑=
∆ ;
0h )ideal(M =∆ ;
0v )ideal(M =∆ ;
0)xln(*xRsNC
1iii
)ideal(M ≥−= ∑=
∆
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 77

Fundamentos de Termodinámica del Equilibrio Físico y Químico
De estas relaciones nuevamente se concluye que una solución ideal tiene una entalpía de mezclado y un volumen de mezclado iguales a cero. También se concluye que un proceso de mezclado genera entropía o sea que el proceso es irreversible.
Las propiedades de soluciones ideales están dadas por las siguientes expresiones:
∑∑==
+=NC
1iii
NC
1i
ºii
ideal )xln(xRTgxg∆ (4-117)
∑=
=NC
1i
ºii
ideal hxh∆ (4-118)
∑=
=NC
1i
ºii
ideal vxv∆ (4-119)
∑∑==
−=NC
1iii
NC
1i
ºii
ideal )xln(xRsxs∆ (4-120)
4.5.5. PROPIEDAD EN EXCESO (ϕE).
Se define como una propiedad en exceso a la diferencia entre la propiedad de una solución en su estado real y su valor si fuera una solución ideal a las mismas condiciones de presión, temperatura y composición. Es decir,
ϕE(P,T,zFi) = ϕ (P,T,zFi) - ϕ ideal(P,T,zFi) (4-121)
ϕE = (ϕ - ϕ º) - (ϕ ideal - ϕ º)
De acuerdo a la ecuación (4-118) la entalpía de mezclado es igual a la entalpía en exceso; de la misma manera se cumple para el volumen, según la ecuación (4-119). Mientras que la energía de Gibbs en exceso es:
gE = gM - gM(ideal)
Reemplazando las ecuaciones (4-113) y (4-117):
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 78

Fundamentos de Termodinámica del Equilibrio Físico y Químico
=
=−= ∏∑ ∑ ∑
== = =
NC
1i i
iNC
1i
NC
1i
NC
1i i
iiiiii
E
xaln
xalnx)xln(x)aln(x
RTg
(4-122)
El argumento del logaritmo natural de la ecuación anterior se define como coeficiente de actividad de un componente i en una solución:
i
ii x
a=γ (4-123)
i
ºiº
i xa
=γ (4-124)
Dondeγi = coeficiente de actividad del componente i según la regla de Lewis-Randall,γi
º = coeficiente de actividad del componente i según la ley de Henry.
Por tanto,
∑∑==
=
=
NC
1iii
NC
1i
Ei
i
E
)ln(xRTg
xRTg
γ (4-125)
Según la regla de Lewis-Randall, el coeficiente de actividad de un componente en una solución tiende a uno cuando la fracción molar de dicho componente también tiende a uno.
11x
lim ii
=→
γ
La condición anterior se cumple para sistemas completamente miscibles y que se ajustan a la ley de Raoult y además, establece la convención simétrica:
γi → 1, xi → 1.
Según la ley de Henry, el coeficiente de actividad del componente i tiende a uno cuando su fracción molar tiende a cero (dilución infinita).
10x
lim ºi
i
=→
γ
La condición anterior se cumple en soluciones parcialmente miscibles que se ajustan a la ley de Henry y establece la convención asimétrica:
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 79

Fundamentos de Termodinámica del Equilibrio Físico y Químico
γiº → 1, xi → 0.
La expresión de la fugacidad del componente i en una solución líquida está dada para cada convención:
- Según la regla de Lewis-Randall:
Lºiii
Li fxf γ= (4-126)
- Según la ley de Henry:
iºii
Li Hxf γ= (4-127)
Usando la definición de propiedad parcial molar, de la ecuación (4-125) se deduce que:
=
=
RTg
n)RT/(ng()ln(
Ei
n,T,Pi
E
i
j∂
∂γ (4-128)
Esta ecuación demuestra que ( )iln γ es una propiedad parcial molar de ngE/(RT)
4.5.6. MODELOS DE COEFICIENTES DE ACTIVIDAD.
Se han desarrollado muchos modelos empíricos de la energía de Gibbs en exceso en función de la temperatura y las moles de los componentes que tienen la forma:
( )NC321
E
n,.......n,n,n,TfRTng
=
Para sistemas binarios la función de la energía de Gibbs en exceso más conveniente es de la forma
( ) ( ) .......xxCxxBARTxx
g 22121
21
E
+−+−+=
Donde A, B, C son constantes para una temperatura dada.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 80

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Si B = C = 0, el modelo anterior se convierte en ARTxx
g21
E
= y los coeficientes de
actividad quedan expresados por: ( ) 221 Axln =γ y ( ) 2
12 Axln =γ
Los modelos de Margules, van Laar, Wilson, NRTL, UNIQUAC, UNIFAC, y sus modificaciones recientes, permiten encontrar las expresiones para los coeficientes de actividad.
Ejemplo 4.13. La energía de Gibbs en exceso de una solución binaria de las especies 1 y 2 basada
en el modelo de Margules de dos parámetros A12 y A21 es: ( ) 21121212
ExxxAxA
RTg
+=
Deduzca la expresión del coeficiente de actividad para cada especie.
Solución:La expresión de los coeficientes de actividad se encuentra usando la ecuación (4.27) en términos de n1 y n2. La energía de Gibbs en exceso para n moles es:
( )2
12212
12212
21121
212
E
nnnA
nnnA
nnn
nnA
nnA
RTng
+
=
+=
Para la especie 1: ( ) ( )( ) ( )
−
+
−=
∂∂
= 211
22141
2
122
2n,T,P1
E
1 nnn
nn2nA
nn2nnAn
nRT/ngln
2
γ
Simplificando y teniendo en cuenta que x1 = n1/n, x2 = n2/n y x1 + x2 = 1, la expresión del coeficiente de actividad de la especie 1 es:
( ) ( ) ( ) 211221123
21 xxAA2Axln −+=γ
Similarmente: ( ) ( ) ( ) 212112213
12 xxAA2Axln −+=γ
En este trabajo se presentan los modelos de coeficientes de actividad basado en la teoría de las soluciones regulares propuesto por van Laar y por Scatchard-Hildebrand, el modelo de composición local de Wilson y los modelos de contribución de grupos UNIQUAC y UNIFAC para mezclas de componentes completamente miscibles. Estos modelos dependen del tipo de solución, de la naturaleza de los componentes y de la temperatura y en menor grado de la presión.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 81

Fundamentos de Termodinámica del Equilibrio Físico y Químico
Debido a la complejidad matemática de estos modelos para sistemas multicomponentes, algunos cálculos de propiedades se han reducido a mezclas binarias (x1, x2). Para sistemas multicomponentes se recomienda implementar procedimientos por computadoras debido a su complejidad de cálculo.
4.5.6.1. Ecuación de van Laar*. Van Laar consideró una mezcla de dos líquidos y supuso que se mezclan a temperatura y presión constantes de manera que: 1) no hay cambio de volumen, es decir, el volumen en exceso es cero, vE = 0; 2) la entropía en exceso es cero, sE = 0. En este caso gE = hE.
La expresión para el coeficiente de actividad de un componente i en una mezcla multicomponente es la siguiente:
∑∑==
−
==
−+
−=
NC
1jjijji
NC
1jijjij
2
jii
iji
i
iji
Ax;Ax
)x1(x
1)x1(
ln
ΨΨ
ΨΨΨ
γ
(4-129)
Donde Aij es el parámetro de interacción binaria entre el componente i y el componente j y es función de la temperatura según la siguiente ecuación:
RTA
A'ij
ij = (4-130)
Aij’ es la energía de interacción binaria entre i y j que se puede tomar constante.
Aii = Ajj = 0; Aij ≠ Aji. Si Aij = 0, γi = 1; si Aij > 0, γi > 1; si Aij < 0, γi < 1
Para mezclas binarias la ecuación de van Laar para cada componente se reduce a:
2
212
121
121
AxAx1
Aln
+
=γ (4-131)
* van Laar, J. J. Phys. Chem. Vol 83 (1913), p. 599-608
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 82

Fundamentos de Termodinámica del Equilibrio Físico y Químico
2
121
212
212
AxAx1
Aln
+
=γ (4-132)
4.5.6.1.1. Determinación de los parámetros de interacción binaria.
• Determinación de los parámetros a partir de datos experimentales.Cuando se conocen los coeficientes de actividad de una mezcla binaria a una temperatura dada (TEXP), los parámetros binarios a la temperatura de interés pueden evaluarse usando la ecuación (4-130):
=
TTAA EXP
)T(12)T(12 EXP (4-133)
=
TTAA EXP
)T(21)T(21 EXP (4-134)
• A partir de coeficientes de actividad a dilución infinita. Los parámetros binarios se pueden estimar a partir de datos experimentales de coeficientes de actividad a dilución infinita a una temperatura experimental.
Cuando x1 →0; x2 → 1, la ecuación (4-131) se reduce a: A12 = ln(γ1∝)
Cuando x2 →0; x1 → 1, la ecuación (4-131) se reduce a: A21 = ln(γ2∝)
Donde γ1∝ y γ2
∝ son los coeficientes de actividad a dilución infinita de los componentes 1 y 2, respectivamente, evaluados a una temperatura experimental. Los parámetros binarios quedan expresados en función de la temperatura de acuerdo con las ecuaciones (4-133) y (4-134).
Ejemplo 4.14. El sistema binario acetato de metilo (1) – benceno (2) se ajusta a la ecuación de van Laar con los parámetros de interacción binaria A12= 183.97/T y A21= 65.23/T, donde T está en K. Calcule el efecto de la temperatura sobre los coeficientes de actividad de cada componente de una solución que contiene 30% de acetato de metilo a 101.325 kPa desde 30 hasta 80º C. Tome intervalos de 10º C.Solución: A T = 303 K; A12= 183.97/303 = 0.6072; A21= 65.23/303 = 0.2153;
1245.0
2153.0*7.06072.0*3.01
6072.0
AxAx1
Aln 22
212
121
121 =
+
=
+
=γ
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 83
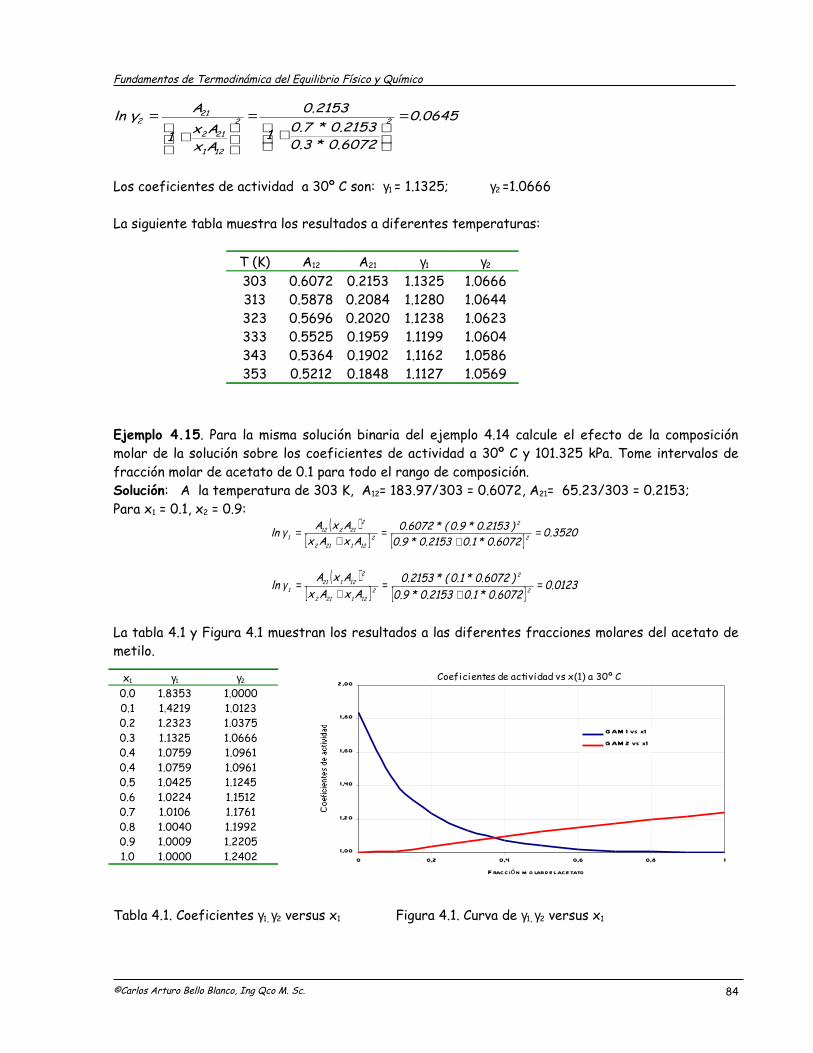
Fundamentos de Termodinámica del Equilibrio Físico y Químico
0645.0
6072.0*3.02153.0*7.01
2153.0
AxAx1
Aln 22
121
212
212 =
+
=
+
=γ
Los coeficientes de actividad a 30º C son: γ1 = 1.1325; γ2 =1.0666
La siguiente tabla muestra los resultados a diferentes temperaturas:
T (K) A12 A21 γ1 γ2
303 0.6072 0.2153 1.1325 1.0666313 0.5878 0.2084 1.1280 1.0644323 0.5696 0.2020 1.1238 1.0623333 0.5525 0.1959 1.1199 1.0604343 0.5364 0.1902 1.1162 1.0586353 0.5212 0.1848 1.1127 1.0569
Ejemplo 4.15. Para la misma solución binaria del ejemplo 4.14 calcule el efecto de la composición molar de la solución sobre los coeficientes de actividad a 30º C y 101.325 kPa. Tome intervalos de fracción molar de acetato de 0.1 para todo el rango de composición.Solución: A la temperatura de 303 K, A12= 183.97/303 = 0.6072, A21= 65.23/303 = 0.2153; Para x1 = 0.1, x2 = 0.9:
( )[ ] [ ] 3520.0
6072.0*1.02153.0*9.0)2153.0*9.0(*6072.0
AxAxAxAln 2
2
2121212
221212
1 =+
=+
=γ
( )[ ] [ ] 0123.0
6072.0*1.02153.0*9.0)6072.0*1.0(*2153.0
AxAxAxAln 2
2
2121212
212121
1 =+
=+
=γ
La tabla 4.1 y Figura 4.1 muestran los resultados a las diferentes fracciones molares del acetato de metilo.
Tabla 4.1. Coeficientes γ1, γ2 versus x1 Figura 4.1. Curva de γ1, γ2 versus x1
©Carlos Arturo Bello Blanco, Ing Qco M. Sc.
x1 γ1 γ2
0.0 1.8353 1.00000.1 1.4219 1.01230.2 1.2323 1.03750.3 1.1325 1.06660.4 1.0759 1.09610.4 1.0759 1.09610.5 1.0425 1.12450.6 1.0224 1.15120.7 1.0106 1.17610.8 1.0040 1.19920.9 1.0009 1.22051.0 1.0000 1.2402
84
Coeficientes de actividad vs x(1) a 30º C
1,00
1,2 0
1,40
1,60
1,80
2 ,00
0 0,2 0,4 0,6 0,8 1
F racc i n m o lar d e l ace tatoó
G AM 1 vs x1
G AM 2 vs x1

Fundamentos de Termodinámica del Equilibrio Físico y Químico
PROBLEMAS PROPUESTOS
Los siguientes problemas se pueden resolver usando las ecuaciones de estado cúbicas para ambas fases y la ecuación virial para la fase gaseosa. Compare los resultados con los valores reportados en la literatura cuando sea posible. Puede usar los programas ecRK©, ecSRK©, ecPENGR© y ecVIRIAL©.
1. Una corriente de vapor de agua a 1000 kPa y 250º C se mezcla adiabáticamente con otra corriente de agua líquida a 1000 kPa y 150º C. La mezcla que sale se encuentra como líquido saturado a 970 kPa. Calcule: a) el flujo másico de vapor a 250º C por cada kmol/h de agua líquida a 150º C; b) la eficiencia termodinámica del proceso basada en la segunda ley. La presión de saturación del agua, en kPa, en función de la temperatura absoluta en K, está dada por: ln(Psat) = 16.5362 – 3985.44/(T – 38.9974).
2. Una turbina de vapor recibe vapor a 25 MPa y 600º C donde se expande reversible e isotérmicamente hasta 1.4 MPa. Calcule la potencia por cada kmol/s de vapor. Si el flujo de calor proviene de un depósito térmico que está a 620º C, calcule la potencia máxima. El entorno se encuentra a 300 K.
3. A un reactor continuo entra 1 kmol/h de CO y 10 kmol/h de vapor de agua donde reaccionan a 10 atm y 550º C según la siguiente reacción:
CO(g) + H2O(g) ―――› H2(g) + CO2(g)
La conversión del CO es de 25%. Calcule el flujo de calor transferido y la irreversibilidad del proceso. To = 300 K y Po = 100 kPa.
4. Resuelva el problema 3 si los productos salen a 700º C.
5. Calcule el flujo de calor para relaciones molares CO/H2O de 10, 15, 20, 25, 30, 35, 40, 45 y 50 si la reacción ocurre a 550º C. Construya la curva Q vs relación CO/H2O. Analice el efecto de CO/H2O sobre Q.
6. Analice el efecto de la temperatura sobre el flujo de calor para una relación CO/H2O de 25. La temperatura varía desde 300 hasta 1000 K. Construya la curva Q vs T.
7. Una mezcla de 95% de metano, 3% de etano y 2% de dióxido de carbono entran a una cámara de combustión a 300º C donde se quema con aire estequiométrico a 300º C (79% de nitrógeno y 21% de oxígeno). La combustión es completa y los gases de combustión salen 1200º C. Todo el proceso ocurre a 2 bares (200 kPa). Calcule la entalpía de combustión en kJ/kmol de mezcla.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 85

Fundamentos de Termodinámica del Equilibrio Físico y Químico
8. Deduzca las expresiones de la entalpía y entropía residuales de una mezcla basada en la ecuación de van der Waals:
( )( )
j
jj
j
2j
jj
jjmj
jjm2m
m
m PcRTc
81b;
PcRTc
6427a;byb;aya
va
bvRTP ===
=−
−= ∑∑ donde
9. A partir de la energía de Gibbs residual: Rns
RTnh
RTng RRR
−= , deduzca las expresiones para el
coeficiente de fugacidad de cada componente.
10. Si una mezcla de 90% de metano y 10% de etanol se encuentra a 600º C y 2000 kPa, calcule la entalpía residual y la entropía residual y los coeficientes de fugacidad de cada componente. Compare con los resultados arrojados por la ecuación de Peng Robinson.
©Carlos Arturo Bello Blanco, Ing Qco M. Sc. 86
Recommended

![[Aviónica – Tercer Año] 2 CURSO ÁREA ASIGNATURA MODULO HORAS EVALUACIÓN ABAST. TEC. AVANZADA LOGÍSTICA I PRESENCIALES 10 Al Finalizar el Modulo APLICACIÓN OBJETIVO Adquiri](https://img.pdfslide.tips/doc/110x75/6027d7e183f1495899018a6b/-avinica-a-tercer-ao-2-curso-rea-asignatura-modulo-horas-evaluacin.jpg)



![201015 Termodinamica Modulo[1]](https://img.pdfslide.tips/doc/110x75/5572003f49795991699f1423/201015-termodinamica-modulo1.jpg)











