
1
Sympathomimetika als Add-On-Therapeutika
bei Erkrankungen der oberen Atemwege:
Evidenz für Nutzen und Risiko
Diplomarbeit
zur Erreichung des akademischen Grades Magistra der Pharmazie
(Mag.a pharm.) an der Leopold-Franzens-Universität Innsbruck
Institut für Pharmazie
Fakultät für Chemie und Pharmazie
Leopold-Franzens-Universität Innsbruck
Abteilung Pharmakologie und Toxikologie
2020
Eingereicht von: ANNIKA ELLMERER
Betreuer: Univ.-Prof. Dr. med. JÖRG STRIESSNIG

2
Inhaltsverzeichnis
Abbildungsverzeichnis ....................................................................................................... 6
Tabellenverzeichnis ............................................................................................................ 7
1 Einleitung .............................................................................................................. 8
1.1 Definition: Allergische Rhinitis ............................................................................ 8
1.1.1 Klassifikation ..................................................................................................... 8
1.1.2 Mechanismus der allergischen Reaktion ........................................................... 9
1.1.3 Symptome......................................................................................................... 9
1.1.4 Ursachen und Risikofaktoren für die Entstehung .............................................. 9
1.1.5 Komorbiditäten .................................................................................................10
1.1.6 Diagnostik ........................................................................................................10
1.1.7 Häufig verwendete Scores ...............................................................................11
1.1.8 Therapie ..........................................................................................................14
1.2 Definition: Grippaler Infekt ..................................................................................21
1.2.1 Übertragung .....................................................................................................21
1.2.2 Ursachen .........................................................................................................21
1.2.3 Pathogenese und Symptome ...........................................................................22
1.2.4 Komorbiditäten .................................................................................................23
1.2.5 Epidemiologie ..................................................................................................23
1.2.6 Diagnostik ........................................................................................................23
1.2.7 Therapie ..........................................................................................................24
2 Fragestellung und Ziel der Diplomarbeit ...........................................................26
3 Methoden .............................................................................................................28
3.1 Suchstrategie .......................................................................................................28
4 Ergebnisse ...........................................................................................................29
4.1 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Antihistaminikum zur Behandlung von saisonaler allergischer Rhinitis ........29
4.1.1 Efficacy and safety of an extended-release formulation of desloratadine and
pseudoephedrine vs the individual components in the treatment of seasonal
allergic rhinitis (Pleskow et al. 2005) ................................................................29

3
4.1.2 Efficacy of once-daily desloratadine/pseudoephedrine for relief of nasal
congestion (Schenkel et al. 2002) ....................................................................33
4.1.3 Comparative efficacy and safety of a once-daily loratadine-pseudoephedrine
combination versus its components alone and placebo in the management of
seasonal allergic rhinitis (Bronsky et al. 1995) .................................................36
4.1.4 Cetirizine and pseudoephedrine retard, given alone or in combination, in patients
with seasonal allergic rhinitis (Grosclaude et al. 1997) .....................................41
4.1.5 The efficacy and safety of fexofenadine HCl and pseudoephedrine, alone and in
combination, in seasonal allergic rhinitis (Sussman et al. 1999) .......................45
4.1.6 Efficacy and safety of desloratadine/pseudoephedrine tablet, 2.5/120 mg two
times a day, versus individual components in the treatment of patients with
seasonal allergic rhinitis (Chervinsky et al. 2005) .............................................49
4.1.7 Efficacy and safety of desloratadine/pseudoephedrine combination vs its
components in seasonal allergic rhinitis (Grubbe et al. 2009) ..........................52
4.1.8 SCH 434: A new antihistamine/decongestant for seasonal allergic rhinitis (Storms
et al. 1989) .......................................................................................................56
4.1.9 Cetirizine and pseudoephedrine retard alone and in combination in the treatment
of perennial allergic rhinitis: a double-blind multicentre study (Bertrand et al 1996)
61
4.1.10 Efficacy of a leukotriene receptor antagonist in the treatment of perennial allergic
rhinitis (Jiang et al. 2006) .................................................................................65
4.1.11 Effects of terfenadine and pseudoephedrine, alone and in combination in a nasal
provocation test and in perennial rhinitis (Henauer et al. 1991) ........................67
4.1.12 Comparative Efficacy and Safety of Terfenadine with Pseudoephedrine and
Terfenadine alone in Allergic Rhinitis (Myers et al. 1998) .................................71
4.2 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Antihistaminikum zur Behandlung von Infektionen der oberen Atemwege ....74
4.2.1 Effects of pseudoephedrine and triprolidine, alone and in combination, on
symptoms of the common cold (Bye et al. 1980) ..............................................74
4.3 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Analgetikum zur Behandlung von Infektionen der oberen Atemwege ............77
4.3.1 Analgesic and Decongestant Efficacy of the Combination of Aspirin with
Pseudoephedrine in Patients With Symptoms of Upper Respiratory Tract
Infection (Eccles et al. 2014) ............................................................................77

4
4.3.2 Efficacy of a paracetamol-pseudoephedrine combination for treatment of nasal
congestion and pain-related symptoms in upper respiratory tract infection (Eccles
et al. 2006) .......................................................................................................81
5 Diskussion ...........................................................................................................85
5.1 Konklusion............................................................................................................87
6 Literaturverzeichnis ............................................................................................89
7 Abkürzungsverzeichnis ......................................................................................98
8 Anhang .................................................................................................................99
8.1 Übersichtstabellen der klinischen Studien .........................................................99
9 Eidesstattliche Erklärung ................................................................................. 136

5
Danksagung An dieser Stelle möchte ich mich zuallererst bei meinen Eltern für die jahrelange liebevolle und
geduldige Unterstützung bedanken, mit der sie mich durch mein Studium hinweg begleitet
haben. Zudem war mein Vater sowohl bei chemischen als auch pharmazeutischen Fragen
stets der beste Ansprechpartner und in gewisser Weise ein wandelndes Lexikon für mich.
Weiters möchte ich mich bei meinem Vater und bei meinem Freund Stefan für das
stundenlange Korrekturlesen dieser umfangreichen Diplomarbeit bedanken.
Mein besonderer Dank gilt jedoch Univ.-Prof. Dr. med. Jörg Striessnig, der nach Abbruch
meiner Diplomarbeit in Australien keine Sekunde zögerte und mir prompt ein neues Thema
zur Ausarbeitung Verfügung stellte. Durch seine angenehme Art und seinem stetigen Wunsch
nach neuem Wissen wurde ich während der Anfertigung dieser Arbeit bestens betreut.

6
Abbildungsverzeichnis
Abbildung 1: Aufschlüsselung des Bewertungssystems des TSS. (Schenkel et al. 2002) .....12
Abbildung 2: Mittlere Veränderungen des morgendlichen instantaneous TSS (ausgenommen
nasale Verstopfung) vom Basiswert, DL=Desloratadin, PSE=Pseudoephedrin. (Pleskow et
al. 2005) ...............................................................................................................................32
Abbildung 3: Mittlere Veränderungen des morgendlichen instantaneous NCS vom Basiswert,
DL=Desloratadin, PSE=Pseudoephedrin. (Pleskow et al. 2009) ...........................................33
Abbildung 4: Durchschnittliche Reduktion des Total Symptom Scores vom Basiswert.
(Bronsky et al. 1995) ............................................................................................................37
Abbildung 5: Durchschnittliche Reduktion des TNSS vom Basiswert. (Bronsky et al. 1995) .38
Abbildung 6: Mittelwerte für die Beurteilung der Abschwellung der Nase anhand einer
vierteiligen Skala, ASA=Acetylsalicylsäure, PSE=Pseudoephedrin. (Eccles et a. 2014) .......81

7
Tabellenverzeichnis
Tabelle 1: Klassifikationssystem der allergischen Rhinitis. (Bousquet et al. 2001) ................. 8
Tabelle 2: Jadad Score Calculation. (Jadad et al. 1996) .......................................................13
Tabelle 3: Liste der in Österreich zugelassenen oralen Antihistaminika. (Austria Codex Stand
Oktober 2020) ......................................................................................................................15
Tabelle 4: Unerwünschte Wirkungen und Kontraindikationen bei der Einnahme von α-
Sympathomimetika. (Austria Codex Stand Oktober 2020) ....................................................16
Tabelle 5: Liste der in Österreich zugelassenen abschwellenden Arzneistoffe. (Austria Codex
Stand Oktober 2020) ............................................................................................................16
Tabelle 6: Kombinationspräparate mit Phenylephrin. (Austria Codex Stand Oktober 2020) ..19
Tabelle 7: Präparate mit Pseudoephedrin. (Austria Codex Stand Oktober 2020)..................20
Tabelle 8: Virale Erreger von grippalen Infekten. (Heikkinen et al. 2003) ..............................22
Tabelle 9: Liste der in Österreich zugelassenen NSAR. (Austria Codex Stand Oktober 2020)
.............................................................................................................................................25

8
1 Einleitung 1.1 Definition: Allergische Rhinitis
Unter allergischer Rhinitis versteht man die IgE-vermittelte Entzündungsreaktion der Nase
nach Allergenexposition und die damit verbundenen nasalen und okulären Symptome.
Schätzungsweise 10 – 25% der Weltbevölkerung leiden an dieser Erkrankung, wobei die
Prävalenz weitaus höher liegen könnte, da Rhinitis von vielen Menschen nicht als echte
Erkrankung wahrgenommen wird und diese folglich keinen Arzt aufsuchen. Trotz des meist
harmlosen Krankheitsbildes, wird das Leben der Betroffenen durch die damit einhergehenden
Symptome stark beeinflusst. Zudem fallen bei der symptomatischen Behandlung von
allergischer Rhinitis erhebliche Kosten an, die sowohl die Patient*innen, als auch das
Gesundheitssystem schwer belasten. (Bousquet et al. 2001)
1.1.1 Klassifikation In vielen klinischen Studien wird zwischen der saisonalen, der ganzjährigen und der
beruflichen allergischen Rhinitis unterschieden. Die saisonale allergische Rhinitis wird meist
durch eine Vielzahl an Allergenen, welche in der freien Natur vorkommen, ausgelöst. Dazu
zählen unter anderem Pollen oder Schimmelpilze. Im Gegensatz dazu liegt die Ursache für die
Entzündungsreaktion der ganzjährigen allergischen Rhinitis vor allem bei Allergenen wie
Hausstaubmilben, Schimmelpilzen, Küchenschaben oder Tierhaaren. Die Autor*innen
Bousquet et al. verweisen in ihrem Artikel aus dem Jahr 2001 auf ein neues
Klassifikationssystem, da die Zuteilung zu einer dieser Krankheitsarten nicht in allen Fällen
eindeutig ist. Anhand der Tabelle 1 lassen sich die verschiedenen Formen und die Schwere
der allergischen Rhinitis vier Klassen zuordnen:
Klassifikationssystem der allergischen Rhinitis nach Bousquet et al. 2001
1 „Intermittierend“ Anwesenheit von Symptomen:
- An weniger als 4 Tagen pro Woche - Für weniger als 4 Wochen
2 „Persistierend“ Anwesenheit von Symptomen:
- An mehr als 4 Tagen pro Woche - Für mehr als 4 Wochen
3 „Mild“
Keiner der folgenden Punkte trifft zu: - Schlafstörungen - Beeinträchtigung der täglichen Aktivitäten bzw. beim Sport - Beeinträchtigung in der Schule oder bei der Arbeit - Besorgniserregende Symptome
4 „Moderat bis Schwer“
Einer bzw. mehrere der folgenden Punkte trifft/treffen zu: - Schlafstörungen - Beeinträchtigung der täglichen Aktivitäten bzw. beim Sport - Beeinträchtigung in der Schule oder bei der Arbeit - Besorgniserregende Symptome
Tabelle 1: Klassifikationssystem der allergischen Rhinitis. (Bousquet et al. 2001)

9
1.1.2 Mechanismus der allergischen Reaktion Allergische Rhinitis resultiert aus einer Sensibilisierung gegenüber bestimmten Allergenen und
der daraus folgenden IgE-vermittelten allergischen Reaktion, die mit einer nasalen
Entzündungsreaktion variabler Intensität verbunden ist. Die spezifischen Symptome und
nasalen Hyperreaktivitäten werden durch Zellen, Mediatoren, Zytokine, Chemokine,
Neuropeptide und Adhäsionsmoleküle ausgelöst, welche in einem komplexen Netzwerk
zusammenarbeiten. Unter einer nasalen Hyperreaktivität versteht man eine übersteigerte
Reaktion der Nase auf einen normalen Stimulus und die damit einhergehenden Symptome wie
nasale Obstruktion, Niesen oder nasale Sekretion, die sowohl getrennt voneinander, als auch
in Kombination vorkommen können. (Bousquet et al. 2008)
1.1.3 Symptome Zu den charakteristischen Symptomen von allergischer Rhinitis gehören nasale Obstruktion,
Rhinorrhoe, Niesen, Juckreiz der Nase und nasaler Ausfluss. Zusätzlich dazu leiden die
erkrankten Personen oft an okulären Symptomen, wie zum Beispiel an geröteten Augen,
tränenden Augen oder an Juckreiz der Augen. Aus all diesen Symptomen können sich zudem
Schlafstörungen sowie emotionelle Probleme entwickeln. Darüber hinaus werden die
Lebensqualität und die Arbeitsleistung der betroffenen Erwachsenen bzw. die schulische
Leistung der betroffenen Kinder zum Teil stark beeinträchtigt. (Bousquet et al. 2008)
1.1.4 Ursachen und Risikofaktoren für die Entstehung Die Ursachen für die Entstehung von allergischer Rhinitis sind bis dato noch nicht vollständig
geklärt. Allergische Rhinitis gehört zu den multifaktoriellen Erkrankungen. Dies bedeutet, dass
die Erkrankung durch ein Zusammenspiel von Genetik und exogenen Faktoren beeinflusst
wird. Ob die Ursachen für die Entstehung überdies bis in die frühe Kindheit zurückreichen ist
unklar, denn die Ergebnisse vieler Studien zu diesem Thema sind widersprüchlich und
ermöglichen keine eindeutige Aussage. Laut einer Studie von Westman et al. aus dem Jahr
2013 erhöht die Erkrankung der Eltern an allergischer Rhinits oder einer anderen allergischen
Krankheit das Risiko der Kinder an allergischer Rhinitis zu erkranken.
Zu den diskutierten Risikofaktoren für die Entstehung von allergischer Rhinitis zählen:
- Alter der Mutter - Hormonelles Gleichgewicht im Mutterleib
- Art der Geburt - Perinatale Asphyxie
- Mehrlingsschwangerschaften - Niedriges Geburtsgewicht
- Wachstumsverzögerung - Allergenexposition
- Hygiene-Hypothese - Exposition bei der Arbeit
- Ethnizität - Städtische Luftverschmutzung
(Bousquet et al. 2008)

10
1.1.5 Komorbiditäten Die Komorbiditäten von allergischer Rhinitis können in gewöhnliche bzw. kausale und
komplexe Komorbiditäten unterteilt werden. Zu den gewöhnlichen Begleiterkrankungen von
allergischer Rhinitis gehören beispielsweise Allergien, während komplexe Begleit-
erkrankungen dadurch entstehen können, dass sich eine bestehende Infektion durch
Schwellungen der Mukosa oder Stauungen des Schleimes verschlechtert. Zu den typischen
Komorbiditäten von allergischer Rhinitis gehören Asthma, Rhinosinusitis, allergische
Konjunktivitis, Nasenpolypen, Adenoide, Otitis media, Störungen der Eustachischen Röhre,
chronischer Husten, Kehlkopfentzündungen und gastroösophageale Refluxkrankheiten.
Asthma zählt zu einer der häufigsten Begleiterkrankungen von allergischer Rhinitis. Die
Unterscheidung und Diagnose gestaltet sich für praktizierende Ärzte jedoch oftmals schwierig,
da das klinische Erscheinungsbild von allergischer Rhinitis dem des Asthmas relativ ähnlich
ist. In einer Fall-Kontroll-Studie von Guerra et al. im Jahr 2002 wurde gezeigt, dass eine
bestehende Rhinitis die Wahrscheinlichkeit an Asthma zu erkranken verdreifachte. Aus
diesem Grund wird allergische Rhinitis heute zu den Risikofaktoren für Asthma gezählt.
(Bousquet et al. 2008)
1.1.6 Diagnostik Die Diagnose der Erkrankung basiert auf einer klinischen Anamnese mit den typischen
Symptomen wie Rhinorrhoe, Niesen, nasale Obstruktion und Juckreiz, sowie auf einem
diagnostischen Test. Hierbei unterscheidet man zwischen zwei Arten von Tests: Die
Anwesenheit von allergenspezifischen Antikörpern vom Typ IgE kann in der Haut mittels eines
Hauttests oder im Blut anhand einer Serumanalyse nachgewiesen werden. Der
Schnellnachweis einer IgE-vermittelten Reaktion mithilfe eines Prick-Tests (Hauttest) gehört
zu den am meisten verbreiteten Tests zur Diagnose von Allergien und wird im folgenden Teil
genauer beschrieben.
Innerhalb der Gruppe der Hauttests kann man zwischen folgenden Arten unterscheiden:
- Prick-Test
- Scratchtest
- Intradermaler Hauttest
- Prick-Prick-Test
- Atopie-Patch-Test
Für die Bestimmung der Schwere der allergischen Rhinitis, oder um Veränderungen der
Symptome aufgrund einer Therapie zu untersuchen, werden vor allem in klinischen Studien
folgende Messmethoden angewandt:
- Symptom Scores
- Visuelle Analogskalen

11
- PNIF-Messungen, akustische Rhinometrie oder Rhinomanometrie zur Ermittlung der
nasalen Obstruktion
- Inflammationsmessungen durch NO-Messungen, Analysen der Zellen und Mediatoren
in nasalen Spüllösungen und Zytologie oder Biopsien der Nase
- Reaktivitätsmessungen wie Provokationstests mit Histamin, Methacholin, Allergenen,
hypertonischer Kochsalzlösung, Capsaicin oder kalter und trockener Luft
- Messungen zur Bestimmung des Geruchssinns
(Bousquet et al. 2008)
1.1.6.1 Prick-Test Der Prick-Test ist ein Schnelltest zur Ermittlung einer allergischen Reaktion auf bestimmte
Allergene. Dazu wird die Haut des/der Patient*in auf der Innenseite des Unterarms mit Alkohol
desinfiziert und die Oberhaut mithilfe einer Lanzette in definiertem Abstand angeritzt. Die zu
untersuchenden Allergenlösungen werden anschließend in Tropfenform auf die Stellen
aufgebracht. Dabei sollte nach jeder Applikation die Nadel gewechselt werden, um die
Möglichkeit einer Kreuzkontamination zu vermeiden. Zusätzlich dazu wird eine Positivkontrolle
in Form einer Histaminlösung, sowie eine Negativkontrolle in Form einer Kochsalzlösung
aufgetragen. Nach 10 bis 20 Minuten erreicht die allergische Reaktion in Form von
Quaddelbildung ihren Höhepunkt. Aus diesem Grund sollte der Durchmesser der Quaddeln
nach 15 Minuten abgelesen werden. (Bousquet et al. 2012)
1.1.7 Häufig verwendete Scores
1.1.7.1 Total Symptom Score (TSS) Der TSS dient der Bewertung der Effektivität einer Behandlung. Er setzt sich aus acht
verschiedenen Symptomen zusammen, die anhand einer vierteiligen Skala evaluiert werden.
Dabei steht „0“ für „keine Symptome“, „1“ für „milde Symptome“, die leicht zu ertragen sind, „2“
für „moderate Symptome“, die das tägliche Leben beeinflussen, aber dennoch tolerabel sind
und „3“ für „schwere Symptome“, die das tägliche Leben stark beeinträchtigen. Die nasalen
Symptome, welche hier evaluiert werden sind folgende: Niesen, Rhinorrhoe, nasale
Obstruktion und Juckreiz. Zu den nicht-nasalen Symptomen gehören Juckreiz der Augen,
tränende Augen, gerötete Augen, Juckreiz der Ohren oder Juckreiz des Gaumens. Der TSS
wird anschließend aus der Summe aller acht individuellen Symptome errechnet und reicht
demnach von 0 bis 24 Punkten. (Storms et al. 1989)
In einer anderen Quelle wird der TSS aus der Summe von neun verschiedenen individuellen
Symptomen gebildet. Somit reicht die Skala des TSS von 0 bis 27 Punkten. Folgende nasale
Symptome werden hier evaluiert: nasaler Ausfluss, nasale Verstopfung, Juckreiz der Nase und

12
Niesen. Die nicht-nasalen Symptome sind: Juckreiz/Brennen der Augen, tränende Augen,
Rötung der Augen, Juckreiz der Ohren oder des Gaumens und Husten. (Stull et al. 2007)
In der folgenden Abbildung 1 wird die Vorgehensweise bei der Beurteilung der Schwere der
Symptome zur Berechnung des TSS genauer erläutert.
Abbildung 1: Aufschlüsselung des Bewertungssystems des TSS. (Schenkel et al. 2002)
1.1.7.2 Total Nasal Symptom Score (TNSS) Der TNSS dient der Beurteilung der Effektivität einer Behandlung im Hinblick auf die Reduktion
der nasalen Symptome. Er ist Teil des TSS, jedoch ohne alle nicht-nasalen Symptome.
Beurteilt werden hier demnach folgende Symptome: Niesen, Juckreiz der Nase, nasale
Verstopfung und Rhinorrhoe. Die Evaluierung der individuellen Symptome wird anhand einer
vierteiligen Skala durchgeführt und die Ergebnisse anschließend aufsummiert. Der TNSS
reicht somit von 0 bis 12 Punkten. (Ellis et al. 2015)
1.1.7.3 Total Nonnasal Symptom Score (TNNSS) Der TNNSS ist ebenfalls Teil des TSS und spiegelt die Effektivität einer Behandlung zur
Reduktion der nicht-nasalen Symptome wider. Auf einer vierteiligen Skala werden die
individuellen Symptome evaluiert und anschließend addiert, sodass eine Punktezahl von 0 bis
12 erreicht werden kann. Der TNNSS setzt sich aus den folgenden vier Symptomen
zusammen: Juckreiz/Brennen der Augen, tränende Augen, Rötung der Augen und Juckreiz
der Ohren/des Gaumens. (Bronsky et al. 1995, Grubbe et al. 2009)
13
1.1.7.4 Nasal Congestion Score (NCS) Der NCS, auch Nasal Stuffiness Score (NSS) genannt, ist ein Maß für die Wirksamkeit einer
Behandlung zur Reduktion der nasalen Verstopfung. Beurteilt wird hier der Grad der nasalen
Verstopfung mithilfe einer vierteiligen Skala. Dabei steht „0“ für „keine Verstopfung“, „1“ für
„milde Verstopfung“, „2“ für „moderate Verstopfung“ und „3“ steht für „schwere Verstopfung“.
Die Skala des NCS erstreckt sich somit von 0 bis 3 Punkten.
1.1.7.5 Beurteilung der Qualität von klinischen Studien
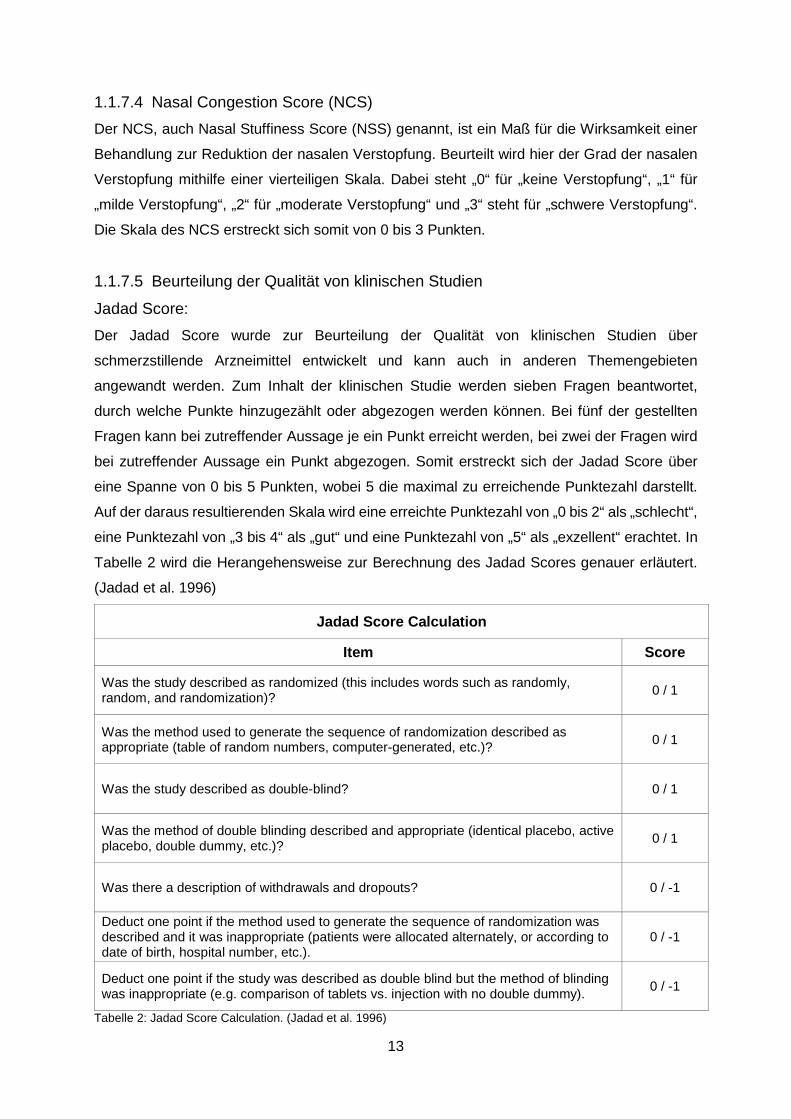
Jadad Score: Der Jadad Score wurde zur Beurteilung der Qualität von klinischen Studien über
schmerzstillende Arzneimittel entwickelt und kann auch in anderen Themengebieten
angewandt werden. Zum Inhalt der klinischen Studie werden sieben Fragen beantwortet,
durch welche Punkte hinzugezählt oder abgezogen werden können. Bei fünf der gestellten
Fragen kann bei zutreffender Aussage je ein Punkt erreicht werden, bei zwei der Fragen wird
bei zutreffender Aussage ein Punkt abgezogen. Somit erstreckt sich der Jadad Score über
eine Spanne von 0 bis 5 Punkten, wobei 5 die maximal zu erreichende Punktezahl darstellt.
Auf der daraus resultierenden Skala wird eine erreichte Punktezahl von „0 bis 2“ als „schlecht“,
eine Punktezahl von „3 bis 4“ als „gut“ und eine Punktezahl von „5“ als „exzellent“ erachtet. In
Tabelle 2 wird die Herangehensweise zur Berechnung des Jadad Scores genauer erläutert.
(Jadad et al. 1996)
Jadad Score Calculation
Item Score
Was the study described as randomized (this includes words such as randomly, random, and randomization)? 0 / 1
Was the method used to generate the sequence of randomization described as appropriate (table of random numbers, computer-generated, etc.)? 0 / 1
Was the study described as double-blind? 0 / 1
Was the method of double blinding described and appropriate (identical placebo, active placebo, double dummy, etc.)? 0 / 1
Was there a description of withdrawals and dropouts? 0 / -1
Deduct one point if the method used to generate the sequence of randomization was described and it was inappropriate (patients were allocated alternately, or according to date of birth, hospital number, etc.).
0 / -1
Deduct one point if the study was described as double blind but the method of blinding was inappropriate (e.g. comparison of tablets vs. injection with no double dummy). 0 / -1
Tabelle 2: Jadad Score Calculation. (Jadad et al. 1996)

14
Weitere wichtige Kriterien für die Beurteilung der Qualität von klinischen Studien stellen unter anderem die Registrierung der Studie, die Definition der Zielparameter bzw. der Endpunkte und etwaige Angaben zu Interessenskonflikten dar.
1.1.8 Therapie Neben der pharmakologischen Therapie spielen die Allergenimmuntherapie sowie die
Kontrolle der Umwelt durch die Reduktion der Allergenlast bei der Therapie von allergischer
Rhinitis eine wichtige Rolle. Eine Verminderung der Expositionszeit gegenüber Allergenen
durch eine Limitierung des Aufenthalts im Freien ist generell möglich, bei den Betroffenen
jedoch meist unerwünscht. Zu den wichtigsten Arzneimittelgruppen für die medikamentöse
Behandlung von allergischer Rhinitis zählen:
- Orale Antihistaminika - Topische Antihistaminika
- Intranasale Glucocorticoide - Orale Glucocorticoide
- Mastzellstabiliastoren - Leukotrienantagonisten
- Orale abschwellend wirkende Mittel - Intranasale abschwellend wirkende Mittel
- Intranasale Anticholinergika
Die Behandlung von allergischer Rhinitis sollte auf die Schwere und Dauer der Erkrankung,
die Präferenzen des/der Patient*in, auf die Effektivität der Therapie und auf die Verfügbarkeit
bzw. die Kosten des Arzneimittels abgestimmt werden. (Dykewicz et al. 2017)
1.1.8.1 Antihistaminika Zu den Antihistaminika gehören jene Arzneistoffe, die Histamin am H1-Rezeptor als
Antagonisten oder inverse Agonisten blockieren. Aufgrund vieler unerwünschten
Arzneimittelwirkungen, vor allem Sedierung, werden heutzutage hauptsächlich Antihistaminika
aus der zweiten Generation verwendet. Antihistaminika zweiter Generation lindern vor allem
die Histamin-vermittelten Symptome wie Rhinorrhoe, Niesen, Juckreiz der Nase und okuläre
Symptome. Weniger Wirkung zeigen diese jedoch bei der Reduktion der nasalen Obstruktion.
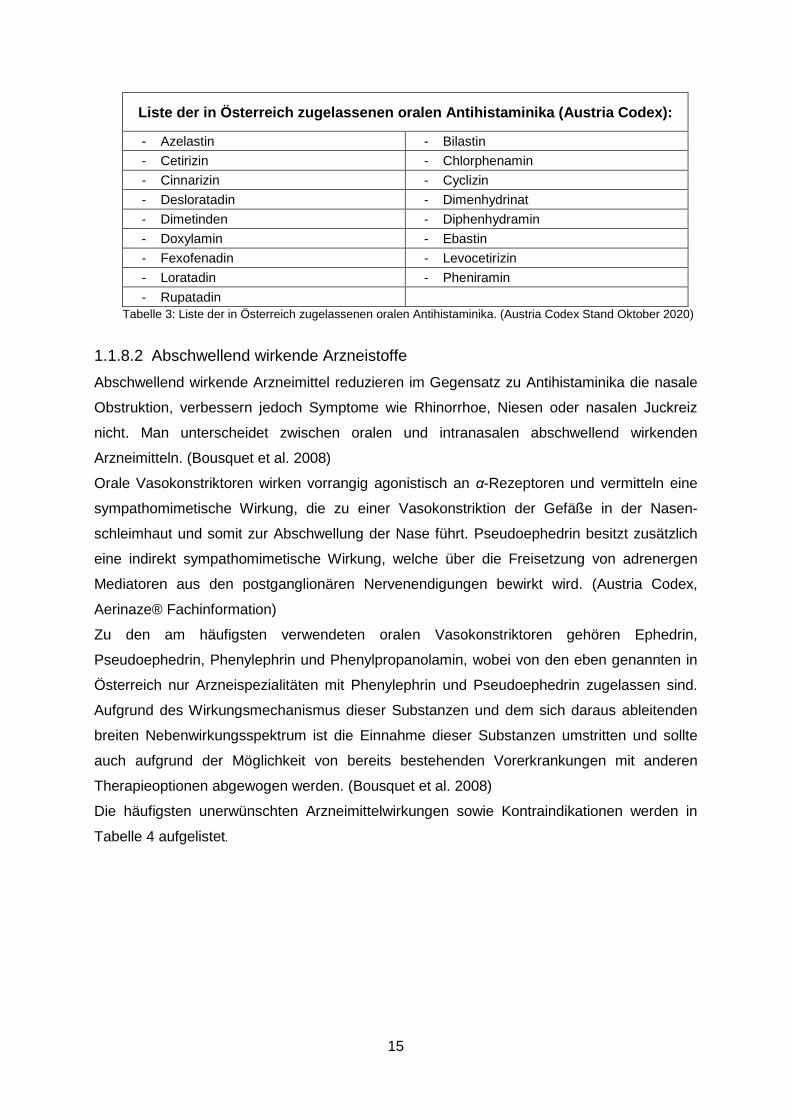
Die in Österreich zugelassenen Antihistaminika sind in Tabelle 3 zusammengefasst.
(Bousquet et al. 2008)
15
Liste der in Österreich zugelassenen oralen Antihistaminika (Austria Codex):
- Azelastin - Bilastin - Cetirizin - Chlorphenamin - Cinnarizin - Cyclizin - Desloratadin - Dimenhydrinat - Dimetinden - Diphenhydramin - Doxylamin - Ebastin - Fexofenadin - Levocetirizin - Loratadin - Pheniramin - Rupatadin
Tabelle 3: Liste der in Österreich zugelassenen oralen Antihistaminika. (Austria Codex Stand Oktober 2020)
1.1.8.2 Abschwellend wirkende Arzneistoffe Abschwellend wirkende Arzneimittel reduzieren im Gegensatz zu Antihistaminika die nasale
Obstruktion, verbessern jedoch Symptome wie Rhinorrhoe, Niesen oder nasalen Juckreiz
nicht. Man unterscheidet zwischen oralen und intranasalen abschwellend wirkenden
Arzneimitteln. (Bousquet et al. 2008)
Orale Vasokonstriktoren wirken vorrangig agonistisch an α-Rezeptoren und vermitteln eine
sympathomimetische Wirkung, die zu einer Vasokonstriktion der Gefäße in der Nasen-
schleimhaut und somit zur Abschwellung der Nase führt. Pseudoephedrin besitzt zusätzlich
eine indirekt sympathomimetische Wirkung, welche über die Freisetzung von adrenergen
Mediatoren aus den postganglionären Nervenendigungen bewirkt wird. (Austria Codex,
Aerinaze® Fachinformation)
Zu den am häufigsten verwendeten oralen Vasokonstriktoren gehören Ephedrin,
Pseudoephedrin, Phenylephrin und Phenylpropanolamin, wobei von den eben genannten in
Österreich nur Arzneispezialitäten mit Phenylephrin und Pseudoephedrin zugelassen sind.
Aufgrund des Wirkungsmechanismus dieser Substanzen und dem sich daraus ableitenden
breiten Nebenwirkungsspektrum ist die Einnahme dieser Substanzen umstritten und sollte
auch aufgrund der Möglichkeit von bereits bestehenden Vorerkrankungen mit anderen
Therapieoptionen abgewogen werden. (Bousquet et al. 2008)
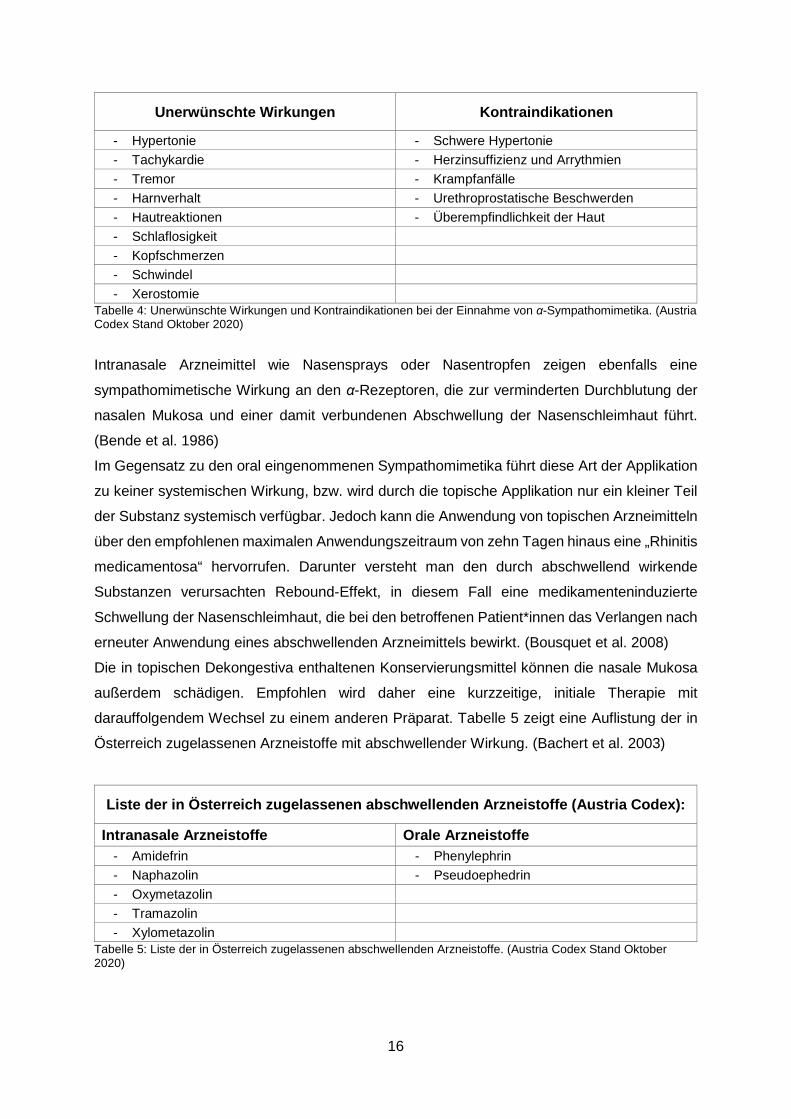
Die häufigsten unerwünschten Arzneimittelwirkungen sowie Kontraindikationen werden in
Tabelle 4 aufgelistet.
16
Unerwünschte Wirkungen Kontraindikationen
- Hypertonie - Schwere Hypertonie - Tachykardie - Herzinsuffizienz und Arrythmien - Tremor - Krampfanfälle - Harnverhalt - Urethroprostatische Beschwerden - Hautreaktionen - Überempfindlichkeit der Haut - Schlaflosigkeit - Kopfschmerzen - Schwindel - Xerostomie
Tabelle 4: Unerwünschte Wirkungen und Kontraindikationen bei der Einnahme von α-Sympathomimetika. (Austria Codex Stand Oktober 2020)
Intranasale Arzneimittel wie Nasensprays oder Nasentropfen zeigen ebenfalls eine
sympathomimetische Wirkung an den α-Rezeptoren, die zur verminderten Durchblutung der
nasalen Mukosa und einer damit verbundenen Abschwellung der Nasenschleimhaut führt.
(Bende et al. 1986)
Im Gegensatz zu den oral eingenommenen Sympathomimetika führt diese Art der Applikation
zu keiner systemischen Wirkung, bzw. wird durch die topische Applikation nur ein kleiner Teil
der Substanz systemisch verfügbar. Jedoch kann die Anwendung von topischen Arzneimitteln
über den empfohlenen maximalen Anwendungszeitraum von zehn Tagen hinaus eine „Rhinitis
medicamentosa“ hervorrufen. Darunter versteht man den durch abschwellend wirkende
Substanzen verursachten Rebound-Effekt, in diesem Fall eine medikamenteninduzierte
Schwellung der Nasenschleimhaut, die bei den betroffenen Patient*innen das Verlangen nach
erneuter Anwendung eines abschwellenden Arzneimittels bewirkt. (Bousquet et al. 2008)
Die in topischen Dekongestiva enthaltenen Konservierungsmittel können die nasale Mukosa
außerdem schädigen. Empfohlen wird daher eine kurzzeitige, initiale Therapie mit
darauffolgendem Wechsel zu einem anderen Präparat. Tabelle 5 zeigt eine Auflistung der in
Österreich zugelassenen Arzneistoffe mit abschwellender Wirkung. (Bachert et al. 2003)
Liste der in Österreich zugelassenen abschwellenden Arzneistoffe (Austria Codex):
Intranasale Arzneistoffe Orale Arzneistoffe - Amidefrin - Phenylephrin - Naphazolin - Pseudoephedrin - Oxymetazolin - Tramazolin - Xylometazolin
Tabelle 5: Liste der in Österreich zugelassenen abschwellenden Arzneistoffe. (Austria Codex Stand Oktober 2020)

17
1.1.8.3 Einnahme von Ephedra-haltigen Produkten Ephedra, die Meerträubel aus der Familie der Ephedraceae, ist ein chinesisches Heilmittel und
unter anderem als Ma-Huang, Mormonentee oder als Mexikanischer Tee bekannt. Die
sogenannten Ephedra-Alkaloide besitzen eine Amphetamin-ähnliche Struktur und weisen
daher gefäßverengende Eigenschaften auf. Zusätzlich dazu wirken sie kreislaufstimulierend,
blutdrucksteigernd, zentral erregend, stark entwässernd, appetitzügelnd und broncholytisch.
Da Produkte, die Ephedra enthalten, laut EU-Recht als Arzneimittel einzustufen sind und der
Apothekenpflicht unterliegen, wurden Ephedra-haltige Nahrungsergänzungsmittel und
Lebensmittel unter anderem als Tees zur Steigerung der Fettverbrennung oder als
Wundermittel bei der Bekämpfung von allergischer Rhinitis in den vergangenen Jahren vor
allem über das Internet verkauft. (BfR, 2002)
Zubereitungen des Ephedra-Krauts können bei allergischer Rhinitis, akutem Schnupfen,
grippalen Infekten und auch als broncholytisches Mittel bei Asthma eingesetzt werden. Laut
dem offiziellen Dokument des BfR „Risikobewertung von Pflanzen und pflanzlichen
Zubereitungen“ werden diese Anwendungen durch klinische Daten gestützt, Volksmedizinisch
werden dem Ephedra-Kraut antivirale, analgetische, antibakterielle und mukolytische
Eigenschaften zugesprochen. Traditionell wurde das Kraut der Pflanze zur Herstellung von
Tees verwendet. Nahrungsergänzungsmittel mit Ephedra-Alkaloiden werden häufig beim
Bodybuilding eingesetzt und dienen dort der Gewichtsreduktion durch Steigerung der
Fettverbrennung.
Zu den unerwünschten Nebenwirkungen von Ephedra zählen Schlaflosigkeit, Tremor,
Kopfschmerzen, Übelkeit, Erbrechen, Tachykardie und Miktionsstörungen. Bei der Einnahme
von zu hohen Dosen kann es zu Herzrhythmusstörungen, starker Hypertonie sowie zur
Entstehung einer Abhängigkeit kommen.
Aufgrund des teils stark variierenden Alkaloidgehalts in den Ephedra-haltigen Produkten, die
dem Arzneimittelgesetz nicht unterliegen und folglich kein Zulassungsverfahren durchlaufen
müssen, ist vom Verzehr dieser Produkte stark abzuraten. Aus der Einnahme von Ephedra-
haltigen Nahrungsergänzungsmitteln können schwere unerwünschte und zum Teil
lebensbedrohliche Wirkungen resultieren. Die FDA kam 2004 zu dem Schluss, dass
Nahrungsergänzungsmittel, die Ephedra-Alkaloide enthalten, ein Risiko für die Gesundheit
darstellen und aufgrund des zweifelhaften Nutzens der entsprechenden Therapie von der
Einnahme abzusehen sei (FDA, 2004). (Klenow et al. 2012) Durch die Verordnung der FDA
wurden Ephedra-haltige dem Abschnitt 402(f)(1)(A) des „Federal Food, Drug, and Cosmetic
Act (21 U.S.C. 342(f)(1)(A))“ zugeteilt. Somit ist es illegal mit Nahrungsergänzungsmitteln, die
Ephedra-Alkaloide enthalten, zu handeln. (FDA, 2004)

18
1.1.8.4 Kombination eines Antihistaminikums mit einem abschwellend wirkenden
Arzneistoff Durch die Einnahme eines Antihistaminikums werden Symptome von allergischer Rhinitis wie
Rhinorrhoe, Niesen, Juckreiz der Nase und okuläre Symptome gelindert. Keine Wirksamkeit
zeigen diese jedoch bei der Reduktion der nasalen Obstruktion. Aus diesem Grund wurden
Antihistaminika mit abschwellend wirkenden Substanzen wie Phenylephrin oder
Pseudoephedrin kombiniert. Ziel dieser Kombination ist die Erweiterung des Wirkspektrums
und die Vereinfachung des Dosierungsschemas für die Patient*innen. Durch die Einnahme
von nur einem Präparat soll sich zudem die Compliance der Betroffenen verbessern und die
Kosten der Therapie reduziert werden. (Sussman et al. 1999)
Antihistaminika an sich haben ein geringes Nebenwirkungspotential, vor allem seit der
Verbesserung der ursprünglichen ersten Generation zur zweiten, die wenig sedative
Eigenschaften besitzt. Durch die Addition eines α-Sympathomimetikums wird jedoch das
Nebenwirkungsspektrum stark erweitert. Daher ist die Verwendung einer Arzneimittel-
kombination bestehend aus einem Antihistaminikum und einem α-Sympathomimetikum mit
den Risiken und möglichen Kontraindikationen bei dem/der zu behandelnden/behandelndem
Patient*in mit dem sich bietenden Vorteil der Therapie abzuwägen. (Bousquet et al. 2008)
In den folgenden Tabellen 6 und 7 werden alle in Österreich zugelassenen Präparate, die
Phenylephrin bzw. Pseudoephedrin enthalten, angeführt.
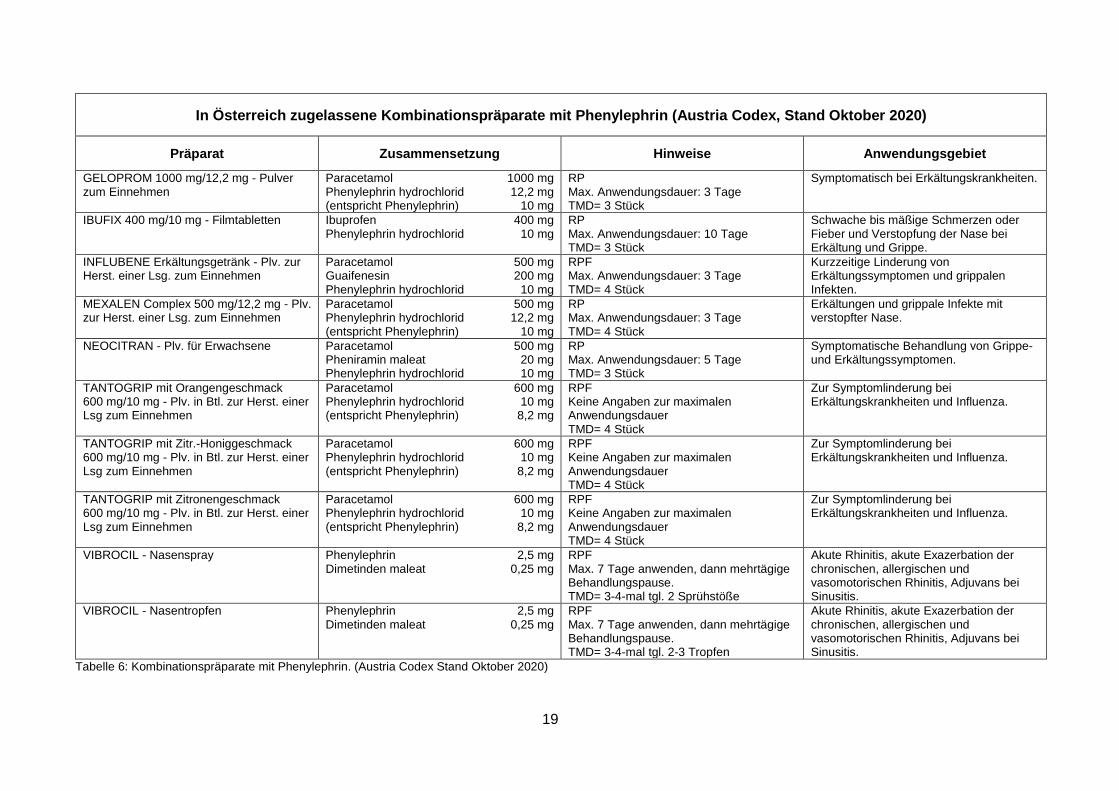
19
In Österreich zugelassene Kombinationspräparate mit Phenylephrin (Austria Codex, Stand Oktober 2020)
Präparat Zusammensetzung Hinweise Anwendungsgebiet
GELOPROM 1000 mg/12,2 mg - Pulver zum Einnehmen
Paracetamol 1000 mg Phenylephrin hydrochlorid 12,2 mg (entspricht Phenylephrin) 10 mg
RP Max. Anwendungsdauer: 3 Tage TMD= 3 Stück
Symptomatisch bei Erkältungskrankheiten.
IBUFIX 400 mg/10 mg - Filmtabletten Ibuprofen 400 mg Phenylephrin hydrochlorid 10 mg
RP Max. Anwendungsdauer: 10 Tage TMD= 3 Stück
Schwache bis mäßige Schmerzen oder Fieber und Verstopfung der Nase bei Erkältung und Grippe.
INFLUBENE Erkältungsgetränk - Plv. zur Herst. einer Lsg. zum Einnehmen
Paracetamol 500 mg Guaifenesin 200 mg Phenylephrin hydrochlorid 10 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 4 Stück
Kurzzeitige Linderung von Erkältungssymptomen und grippalen Infekten.
MEXALEN Complex 500 mg/12,2 mg - Plv. zur Herst. einer Lsg. zum Einnehmen
Paracetamol 500 mg Phenylephrin hydrochlorid 12,2 mg (entspricht Phenylephrin) 10 mg
RP Max. Anwendungsdauer: 3 Tage TMD= 4 Stück
Erkältungen und grippale Infekte mit verstopfter Nase.
NEOCITRAN - Plv. für Erwachsene Paracetamol 500 mg Pheniramin maleat 20 mg Phenylephrin hydrochlorid 10 mg
RP Max. Anwendungsdauer: 5 Tage TMD= 3 Stück
Symptomatische Behandlung von Grippe- und Erkältungssymptomen.
TANTOGRIP mit Orangengeschmack 600 mg/10 mg - Plv. in Btl. zur Herst. einer Lsg zum Einnehmen
Paracetamol 600 mg Phenylephrin hydrochlorid 10 mg (entspricht Phenylephrin) 8,2 mg
RPF Keine Angaben zur maximalen Anwendungsdauer TMD= 4 Stück
Zur Symptomlinderung bei Erkältungskrankheiten und Influenza.
TANTOGRIP mit Zitr.-Honiggeschmack 600 mg/10 mg - Plv. in Btl. zur Herst. einer Lsg zum Einnehmen
Paracetamol 600 mg Phenylephrin hydrochlorid 10 mg (entspricht Phenylephrin) 8,2 mg
RPF Keine Angaben zur maximalen Anwendungsdauer TMD= 4 Stück
Zur Symptomlinderung bei Erkältungskrankheiten und Influenza.
TANTOGRIP mit Zitronengeschmack 600 mg/10 mg - Plv. in Btl. zur Herst. einer Lsg zum Einnehmen
Paracetamol 600 mg Phenylephrin hydrochlorid 10 mg (entspricht Phenylephrin) 8,2 mg
RPF Keine Angaben zur maximalen Anwendungsdauer TMD= 4 Stück
Zur Symptomlinderung bei Erkältungskrankheiten und Influenza.
VIBROCIL - Nasenspray
Phenylephrin 2,5 mg Dimetinden maleat 0,25 mg
RPF Max. 7 Tage anwenden, dann mehrtägige Behandlungspause. TMD= 3-4-mal tgl. 2 Sprühstöße
Akute Rhinitis, akute Exazerbation der chronischen, allergischen und vasomotorischen Rhinitis, Adjuvans bei Sinusitis.
VIBROCIL - Nasentropfen Phenylephrin 2,5 mg Dimetinden maleat 0,25 mg
RPF Max. 7 Tage anwenden, dann mehrtägige Behandlungspause. TMD= 3-4-mal tgl. 2-3 Tropfen
Akute Rhinitis, akute Exazerbation der chronischen, allergischen und vasomotorischen Rhinitis, Adjuvans bei Sinusitis.
Tabelle 6: Kombinationspräparate mit Phenylephrin. (Austria Codex Stand Oktober 2020)
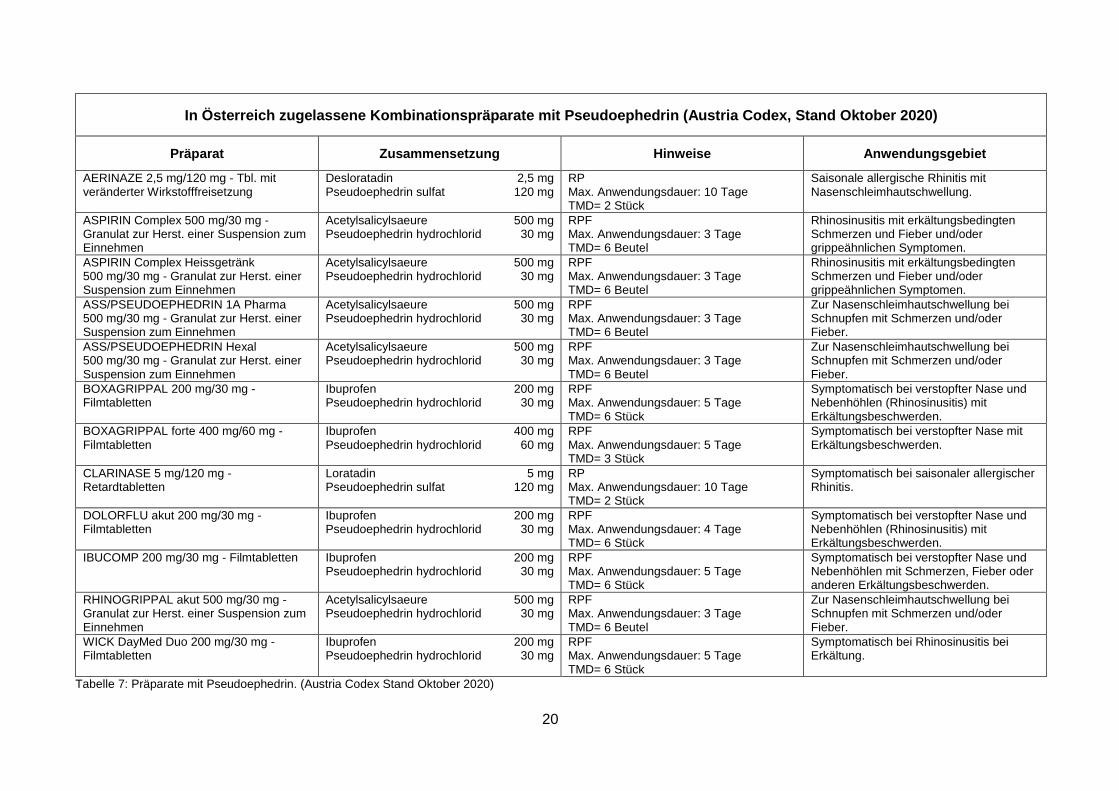
20
In Österreich zugelassene Kombinationspräparate mit Pseudoephedrin (Austria Codex, Stand Oktober 2020)
Präparat Zusammensetzung Hinweise Anwendungsgebiet
AERINAZE 2,5 mg/120 mg - Tbl. mit veränderter Wirkstofffreisetzung
Desloratadin 2,5 mg Pseudoephedrin sulfat 120 mg
RP Max. Anwendungsdauer: 10 Tage TMD= 2 Stück
Saisonale allergische Rhinitis mit Nasenschleimhautschwellung.
ASPIRIN Complex 500 mg/30 mg - Granulat zur Herst. einer Suspension zum Einnehmen
Acetylsalicylsaeure 500 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 6 Beutel
Rhinosinusitis mit erkältungsbedingten Schmerzen und Fieber und/oder grippeähnlichen Symptomen.
ASPIRIN Complex Heissgetränk 500 mg/30 mg - Granulat zur Herst. einer Suspension zum Einnehmen
Acetylsalicylsaeure 500 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 6 Beutel
Rhinosinusitis mit erkältungsbedingten Schmerzen und Fieber und/oder grippeähnlichen Symptomen.
ASS/PSEUDOEPHEDRIN 1A Pharma 500 mg/30 mg - Granulat zur Herst. einer Suspension zum Einnehmen
Acetylsalicylsaeure 500 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 6 Beutel
Zur Nasenschleimhautschwellung bei Schnupfen mit Schmerzen und/oder Fieber.
ASS/PSEUDOEPHEDRIN Hexal 500 mg/30 mg - Granulat zur Herst. einer Suspension zum Einnehmen
Acetylsalicylsaeure 500 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 6 Beutel
Zur Nasenschleimhautschwellung bei Schnupfen mit Schmerzen und/oder Fieber.
BOXAGRIPPAL 200 mg/30 mg -Filmtabletten
Ibuprofen 200 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 5 Tage TMD= 6 Stück
Symptomatisch bei verstopfter Nase und Nebenhöhlen (Rhinosinusitis) mit Erkältungsbeschwerden.
BOXAGRIPPAL forte 400 mg/60 mg - Filmtabletten
Ibuprofen 400 mg Pseudoephedrin hydrochlorid 60 mg
RPF Max. Anwendungsdauer: 5 Tage TMD= 3 Stück
Symptomatisch bei verstopfter Nase mit Erkältungsbeschwerden.
CLARINASE 5 mg/120 mg - Retardtabletten
Loratadin 5 mg Pseudoephedrin sulfat 120 mg
RP Max. Anwendungsdauer: 10 Tage TMD= 2 Stück
Symptomatisch bei saisonaler allergischer Rhinitis.
DOLORFLU akut 200 mg/30 mg -Filmtabletten
Ibuprofen 200 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 4 Tage TMD= 6 Stück
Symptomatisch bei verstopfter Nase und Nebenhöhlen (Rhinosinusitis) mit Erkältungsbeschwerden.
IBUCOMP 200 mg/30 mg - Filmtabletten
Ibuprofen 200 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 5 Tage TMD= 6 Stück
Symptomatisch bei verstopfter Nase und Nebenhöhlen mit Schmerzen, Fieber oder anderen Erkältungsbeschwerden.
RHINOGRIPPAL akut 500 mg/30 mg - Granulat zur Herst. einer Suspension zum Einnehmen
Acetylsalicylsaeure 500 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 3 Tage TMD= 6 Beutel
Zur Nasenschleimhautschwellung bei Schnupfen mit Schmerzen und/oder Fieber.
WICK DayMed Duo 200 mg/30 mg - Filmtabletten
Ibuprofen 200 mg Pseudoephedrin hydrochlorid 30 mg
RPF Max. Anwendungsdauer: 5 Tage TMD= 6 Stück
Symptomatisch bei Rhinosinusitis bei Erkältung.
Tabelle 7: Präparate mit Pseudoephedrin. (Austria Codex Stand Oktober 2020)

21
1.2 Definition: Grippaler Infekt
Unter einem grippalen Infekt versteht man eine milde Infektion der oberen Atemwege, die mit
den für die Krankheit typischen Symptomen wie nasaler Verstopfung, nasalem Ausfluss,
Niesen, Halsschmerzen und Husten einhergeht. Es handelt sich hierbei um einen Überbegriff
für eine heterogene Gruppe an Erkrankungen, die von zahlreichen viralen Erregern aus
verschiedenen Gruppen ausgelöst werden können. Obwohl sich der Großteil dieser viralen
Infektionen auf den Bereich der oberen Atemwege beschränkt und von selbstlimitierendem
Charakter ist, können sich die Infektionen auf benachbarte Organe ausbreiten und zu
bakteriellen Komplikationen führen. Grippale Infekte stellen aufgrund der vielen Arztbesuche,
Therapiekosten und Fehlstunden in der Arbeit oder Schule eine enorme ökonomische
Belastung für die Gesellschaft dar. (Heikkinen et al. 2003)
1.2.1 Übertragung Bei der Übertragung der Krankheit unterscheidet man zwischen drei Mechanismen:
- Handkontakt von Sekreten, die das Virus enthalten, entweder durch direkten Kontakt
mit der infizierten Person oder indirekten Kontakt mit einer Oberfläche
- Kleine Aerosolteilchen, die über einen längeren Zeitraum in der Luft schweben und
eingeatmet werden
- Große Aerosolteilchen, die direkt von einer infizierten Person unter anderem durch
Niesen oder Husten übertragen werden
(Heikkinen et al. 2003)
1.2.2 Ursachen Rhinoviren gehören zu den häufigsten Erregern für grippale Infekte in allen Altersgruppen und
verursachen jährlich 30 – 50% aller Infektionen der oberen Atemwege. Im Herbst, wenn die
Infektionszahlen ihren Höhepunkt erreichen, sind Rhinoviren hier sogar für 80% der
Infektionen verantwortlich. Neben den Rhinoviren können endemische Coronaviren,
Influenzaviren, Adenoviren u. v. a. m. für die Entstehung eines grippalen Infektes verant-
wortlich sein. In der folgenden Tabelle 8 werden die Häufigkeiten verschiedener Erreger
angeführt. Als Grundlage für diese Schätzung diente eine Studie von Mäkelä et al. aus dem
Jahr 1998. (Heikkinen et al. 2003)
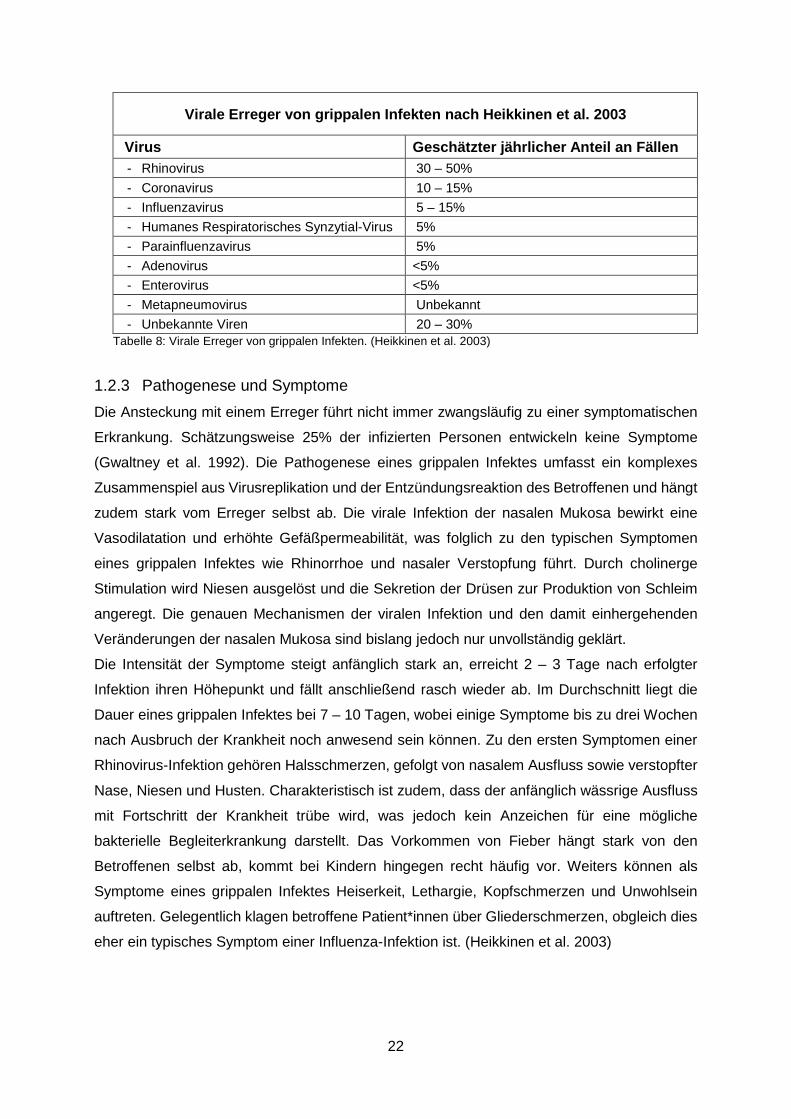
22
Virale Erreger von grippalen Infekten nach Heikkinen et al. 2003
Virus Geschätzter jährlicher Anteil an Fällen - Rhinovirus 30 – 50% - Coronavirus 10 – 15% - Influenzavirus 5 – 15% - Humanes Respiratorisches Synzytial-Virus 5% - Parainfluenzavirus 5% - Adenovirus <5% - Enterovirus <5% - Metapneumovirus Unbekannt - Unbekannte Viren 20 – 30%
Tabelle 8: Virale Erreger von grippalen Infekten. (Heikkinen et al. 2003)
1.2.3 Pathogenese und Symptome Die Ansteckung mit einem Erreger führt nicht immer zwangsläufig zu einer symptomatischen
Erkrankung. Schätzungsweise 25% der infizierten Personen entwickeln keine Symptome
(Gwaltney et al. 1992). Die Pathogenese eines grippalen Infektes umfasst ein komplexes
Zusammenspiel aus Virusreplikation und der Entzündungsreaktion des Betroffenen und hängt
zudem stark vom Erreger selbst ab. Die virale Infektion der nasalen Mukosa bewirkt eine
Vasodilatation und erhöhte Gefäßpermeabilität, was folglich zu den typischen Symptomen
eines grippalen Infektes wie Rhinorrhoe und nasaler Verstopfung führt. Durch cholinerge
Stimulation wird Niesen ausgelöst und die Sekretion der Drüsen zur Produktion von Schleim
angeregt. Die genauen Mechanismen der viralen Infektion und den damit einhergehenden
Veränderungen der nasalen Mukosa sind bislang jedoch nur unvollständig geklärt.
Die Intensität der Symptome steigt anfänglich stark an, erreicht 2 – 3 Tage nach erfolgter
Infektion ihren Höhepunkt und fällt anschließend rasch wieder ab. Im Durchschnitt liegt die
Dauer eines grippalen Infektes bei 7 – 10 Tagen, wobei einige Symptome bis zu drei Wochen
nach Ausbruch der Krankheit noch anwesend sein können. Zu den ersten Symptomen einer
Rhinovirus-Infektion gehören Halsschmerzen, gefolgt von nasalem Ausfluss sowie verstopfter
Nase, Niesen und Husten. Charakteristisch ist zudem, dass der anfänglich wässrige Ausfluss
mit Fortschritt der Krankheit trübe wird, was jedoch kein Anzeichen für eine mögliche
bakterielle Begleiterkrankung darstellt. Das Vorkommen von Fieber hängt stark von den
Betroffenen selbst ab, kommt bei Kindern hingegen recht häufig vor. Weiters können als
Symptome eines grippalen Infektes Heiserkeit, Lethargie, Kopfschmerzen und Unwohlsein
auftreten. Gelegentlich klagen betroffene Patient*innen über Gliederschmerzen, obgleich dies
eher ein typisches Symptom einer Influenza-Infektion ist. (Heikkinen et al. 2003)

23
1.2.4 Komorbiditäten Zu den typischen Begleiterkrankungen von grippalen Infekten gehören Nasennebenhöhlen-
entzündungen, Lungenentzündungen und akute Otitis media vor allem bei Kindern. Eine
Lungenentzündung kann sowohl die Folge einer viralen Infektion sein, die sich in den
Lungenbereich ausgebreitet hat, als auch aufgrund einer bakteriellen Superinfektion
entstanden sein. Die Ergebnisse vieler Studien deuten des Weiteren drauf hin, dass akute
Exazerbationen bei Asthmapatient*innen in klarem Zusammenhang mit Infektionen der oberen
Atemwege stehen. (Heikkinen et al. 2003)
1.2.5 Epidemiologie Die Häufigkeit von grippalen Infekten nimmt in den Ländern der gemäßigten Zone auf der
Nordhalbkugel im Herbst stark zu, bleibt über den Winter hoch und nimmt mit Frühlingsbeginn
ab, während grippale Infekte in tropischen Regionen vorwiegend zur Regenzeit vorkommen.
Zudem nimmt die Zahl der grippalen Infekte, an denen eine Person pro Jahr erkrankt, mit dem
Alter ab. So erkranken im Durchschnitt jüngere Kinder an 6 – 8 grippalen Infekten pro Jahr,
Erwachsene hingegen nur an 2 – 4.
Verschiedene diskutierte Risikofaktoren, die eine Rolle bei der Entstehung von grippalen
Infekten spielen könnten, sind bislang nicht bestätigt. Vermutet wird jedoch, dass die
Exposition gegenüber kleinen Kindern ebenso wie psychischer Stress das Risiko selbst an
einem grippalen Infekt zu erkranken erhöht. Vermutet wird zudem, dass körperliche Betätigung
in normalem Ausmaß die Entstehung verhindern kann, während körperliche Verausgabung
das Gegenteil bewirkt. (Heikkinen et al. 2003)
1.2.6 Diagnostik Die Diagnose von grippalen Infekten stellen erwachsene Patient*innen meist selbst.
Problematisch kann dies jedoch bei Kleinkindern sein, die ihre Symptome nicht selbst
schildern können. Schwierig ist hier die Unterscheidung zwischen einer meist harmlosen
grippalen Infektion und einer invasiven bakteriellen Erkrankung, vor allem da Fieber häufig das
dominanteste Symptom zu Erkrankungsbeginn darstellt. Halsschmerzen, die mit einer
Streptokokken-Infektion einhergehen, können teilweise auf eine gewöhnliche virale Infektion
hindeuten, die Unterscheidung ist jedoch meist einfach, da die primären Symptome eines
grippalen Infektes - die Verstopfung der Nase und der nasale Ausfluss - bei einer
Streptokokken-Infektion sehr selten vorkommen.
Folgende Methoden werden zur Identifikation von Viren herangezogen:
- Isolation des Virus in einer Zellkultur
- Antigen-Detektion
- PCR (Heikkinen et al. 2003)

24
1.2.7 Therapie Aufgrund der Vielschichtigkeit von grippalen Infekten gibt es bis dato keine allgemein effektive
Therapie, jedoch kann die Erkrankung durch eine Vielzahl von rezeptfreien Medikamenten
symptomatisch behandelt werden. Aufgrund der unterschiedlichen Bedürfnisse der Erkrankten
und der als unterschiedlich unangenehm empfundenen Symptome, sollte die Therapie speziell
auf die Beschwerden jedes/jeder einzelnen Patient*in angepasst werden.
Zu den wichtigsten Arzneimittelgruppen für die symptomatische Behandlung von grippalen
Infekten gehören:
- Orale abschwellend wirkende Mittel - Intranasale abschwellend wirkende Mittel - Orale Antihistaminika - NSAR - Antitussiva - Mukolytika - Zink
(Heikkinen et al. 2003)
1.2.7.1 Abschwellend wirkende Arzneistoffe Symptome wie nasale Verstopfung und nasaler Ausfluss, die mit grippalen Infekten
einhergehen, werden von vielen Erkrankten als besonders störend erachtet. Durch orale bzw.
intranasale abschwellend wirkende Arzneistoffe kann die nasale Obstruktion stark reduziert
werden. Genauere Informationen bezüglich des Wirkmechanismus und der verschiedenen in
Österreich erhältlichen Arzneistoffe sind dem Kapitel „Allergische Rhinitis“ unter dem Punkt
„Therapie“ zu entnehmen. (Heikkinen et al. 2003)
1.2.7.2 Antihistaminika Antihistaminika der ersten Generation können aufgrund ihrer anticholinergen Wirkungen bei
der Bekämpfung von Niesen und Rhinorrhoe eingesetzt werden. Antihistaminika der zweiten
Generation mit vorrangig peripherer Histamin-antagonistischer Wirkung scheinen hingegen
bei der Behandlung von grippalen Infekten nicht effektiv zu sein. (Gwaltney et al. 1997)
Bezüglich der genaueren Beschreibung des Wirkmechanismus von Antihistaminika und den
in Österreich zugelassenen Arzneistoffen wird auf das Kapitel „Allergische Rhinitis“ und deren
Therapie verwiesen. (Heikkinen et al. 2003)
1.2.7.3 NSAR und Paracetamol Nichtsteroidale Antirheumatika hemmen die Cyclooxygenase und bewirken so eine
verminderte Produktion von Prostaglandinen und Thromboxanen. Paracetamol wird aufgrund
seiner stark antipyretischen Wirkung oft bei der Behandlung von grippalen Infekten eingesetzt.
Anders als die COX-Hemmer gehört es jedoch zur Gruppe der nicht-sauren, schwachen
Analgetika mit fehlender COX-Inhibition. (Gelbe Liste)

25
Laut einem Cochrane Review aus dem Jahr 2015 mildern NSAR die Schmerzen, welche durch
grippale Infekte verursacht werden, es gibt jedoch keine eindeutige Evidenz, dass die
respiratorischen Symptome gelindert werden. Daher sollten die Vorteile dieser Therapie mit
den Risiken und Nebenwirkungen abgewogen werden. (Kim et al. 2015)
Die in Österreich zugelassenen NSAR sind in Tabelle 9 zusammengefasst.
Liste der in Österreich zugelassenen NSAR (Austria Codex):
- Acetylsalicylsäure - Calciumcarbasalat - Dexibuprofen - Dexketoprofen - Diclofenac - Ibuprofen - Indometacin - Ketoprofen - Lornoxicam - Mefenaminsäure - Meloxicam - Naproxen - Phenazon - Piroxicam
Tabelle 9: Liste der in Österreich zugelassenen NSAR. (Austria Codex Stand Oktober 2020)

26
2 Fragestellung und Ziel der Diplomarbeit
Allergische Rhinitis gehört mit einer Lebenszeitprävalenz von 20% zu den häufigsten
allergischen Erkrankungen weltweit. Das alltägliche Leben der Betroffenen wird durch
allergische Rhinitis stark beeinträchtigt, Komorbiditäten können entstehen, zahlreiche
Fehltage in der Schule bzw. der Arbeit können die Folge sein und das Gesundheitssystem
wird durch die verursachten Kosten stark belastet. In Deutschland betrugen die direkten und
indirekten Kosten der allergischen Rhinitis für das Gesundheitswesen und die Wirtschaft im
Jahr 2000 schätzungsweise 240 Millionen Euro. (Bachert et al. 2003)
Ebenso wie allergische Rhinitis zählen Erkrankungen der oberen Atemwege zu den häufigsten
Erkrankungen weltweit. Erwachsene leiden im Schnitt unter 2 – 4 grippalen Infekten jährlich,
während dieser Wert bei Schulkindern bei 6 – 8 liegt. (Heikkinen et al. 2003) Bei einer
Annahme von zwei Erkrankungen der oberen Atemwege, wobei hier die Fälle von Influenza
ebenfalls miteinberechnet werden, käme man allein in den USA auf 600 Millionen Fälle jährlich.
Aufgrund des meist harmlosen Verlaufs der Krankheit ist eine Selbstdiagnose der Erkrankung
üblich und die Behandlung der Symptome erfolgt durch rezeptfrei erhältliche Präparate meist
eigenständig durch die Betroffenen. (Eccles et al. 2014)
Arzneimittelkombinationen bestehend aus einem Antihistaminikum oder Analgetikum und
einem α-Sympathomimetikum werden häufig für die Therapie von solchen Erkrankungen
empfohlen. Die Addition eines α-Sympathomimetikums soll die nasale Obstruktion, welche
eine häufige Begleiterscheinung von allergischer Rhinitis oder grippalen Infekten darstellt,
reduzieren und somit dazu führen, dass Betroffene nur ein Präparat einzunehmen haben. Es
besteht die Annahme, dass sich dadurch die Compliance der Patient*innen erhöhen könnte
und die Kosten für die Therapie gesenkt werden könnten. Zudem wird der regelmäßige
Gebrauch von topischen, intranasal abschwellend wirkenden Arzneimittel nicht empfohlen, da
sich daraus eine Rhinitis medicamentosa entwickeln könnte.
Die Sinnhaftigkeit des Einsatzes dieser Kombinationspräparate wird jedoch immer wieder
hinterfragt, da sich das Nebenwirkungsspektrum durch die Addition eines α-Sympatho-
mimetikums deutlich erweitert. Durch die Aktivierung der α-Rezeptoren kann es zu
kardiovaskulären Veränderungen wie einer Erhöhung des Blutdruckes oder Tachykardie
kommen. Gegenanzeigen von Kombinationspräparaten mit Pseudoephedrin sprechen unter
anderem gegen die Anwendung dieser Präparate bei bestehenden kardiovaskulären
Vorerkrankungen, wie z.B. schweren Hypertonien, Tachyarrhythmien oder ischämischen
Herzerkrankungen.
Aufgrund der großen Anzahl an veröffentlichten Studien zu diesem Thema mit teils stark
divergierenden Ergebnissen und dem Mangel von eindeutigen Aussagen in bereits
vorhandenen Reviews fällt eine objektive Meinungsbildung zur Abwägung von Nutzen und

27
Risiko schwer. Daher war es das Hauptziel dieser Diplomarbeit die Evidenz für Nutzen und
Risiko von diesen spezifischen Arzneimittelkombinationen, welche zur Therapie von
allergischer Rhinitis und grippalen Infekten in Österreich zugelassen sind, anhand einer
systematischen Literaturrecherche von randomisierten und kontrollierten klinischen Studien zu
erfassen.

28
3 Methoden 3.1 Suchstrategie
Für die Beschaffung der Literatur zu den klinischen Studien wurde die Plattform „PubMed“
(https://pubmed.ncbi.nlm.nih.gov/) verwendet. Im Zuge dessen wurde nach geeigneten
klinischen Studien zur Beurteilung der Evidenz und Sicherheit von Arzneimittelkombinationen
für die Behandlung von grippalen Infekten und allergischer Rhinitis recherchiert. Dabei wurde
unter folgenden Stichworten gesucht:
- [common cold AND combination therapy] 293 Treffer
- [phenylephrine AND (clinical trial OR clinical study)] 1159 Treffer mit dem Filter:
„Humans”
- [pseudoephedrine AND (clinical trial OR clinical study)] 246 Treffer mit dem Filter:
„Humans”
- [phenylephrine AND oral AND decongestant] 59 Treffer mit den Filtern: „Clinical Trial“
und „Meta-Analysis“
Die Ergebnisse dieser Recherche wurden anschließend ausgewertet und all jene Studien, die
für die Bearbeitung dieser Arbeit interessant erschienen, in eine Tabelle übertragen (siehe
Anhang). Aufgrund verschiedener Ausschlusskriterien, welche im folgenden Absatz taxativ
aufgelistet werden, wurden nur 15 dieser Studien in die finale Auswahl für eine genauere
Analyse aufgenommen.
Ein Ausschlussgrund war, wenn der Komponentenbeweis nicht erbracht wurde.
Beispielsweise wurde in einer Vielzahl von Studien die Effektivität und Sicherheit einer
Arzneimittelkombination, bestehend aus einem Antihistaminikum und Pseudoephedrin,
gegenüber der Monotherapie mit Pseudoephedrin untersucht. Um jedoch den Vorteil der
Kombination gegenüber der Monotherapie mit einem Antihistaminikum zu ermitteln, wäre eine
weitere Untersuchungsgruppe mit dem Antihistaminikum allein nötig. War dies nicht der Fall,
so wurde die Studie für die Ausarbeitung der Fragestellung dieser Arbeit nicht herangezogen.
Des Weiteren wurden jene Studien ausgeschlossen, bei denen die Teilnehmer*innenzahl
unter 30 lag, oder ein Arzneistoff untersucht wurde, welcher in Österreich nicht zugelassen ist.
Folgende drei Studien wurden allerdings in die Analyse miteinbezogen, obwohl die
untersuchten Wirkstoffe Triprolidin und Terfenadin in Österreich mittlerweile nicht mehr
zugelassen sind: Bye et al. 1980, Henauer et al. 1991 und Myers et al. 1998. Zu einer Studie,
die ansonsten die Kriterien zur Teilnahme an der Analyse erfüllte (Backhouse et al. 1990),
waren die Originaldaten im vorgegebenen Zeitrahmen nicht zugänglich. Außerdem wurden all
jene Studien ausgeschlossen, bei denen die allergische Reaktion auf bestimmte Allergene
durch einen Provokationstest, und somit nicht auf klinisch relevante Weise erzeugt wurde,
bzw. ein grippaler Infekt durch die gezielte Infektion mit einem Erreger ausgelöst wurde.

29
4 Ergebnisse
In den folgenden Kapiteln werden die Ergebnisse der systematischen Literaturrecherche der
Plattform „PubMed“ genauer beschrieben. Nach anfänglichem Ausschlussverfahren wurden
für die detaillierte Analyse und Ausarbeitung dieser Diplomarbeit 15 Studien ausgewählt. Die
entsprechenden Auswahlkriterien wurden im vorherigen Kapitel genauer erläutert.
4.1 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Antihistaminikum zur Behandlung von saisonaler allergischer Rhinitis
4.1.1 Efficacy and safety of an extended-release formulation of desloratadine and
pseudoephedrine vs the individual components in the treatment of seasonal
allergic rhinitis (Pleskow et al. 2005) Ziel dieser Studie war es, die Wirksamkeit und Sicherheit einer Arzneimittelkombination
bestehend aus Desloratadin und Pseudoephedrin zur Anwendung bei Patient*innen mit
saisonaler allergischer Rhinitis im Vergleich zu Desloratadin oder Pseudoephedrin als
Einzelsubstanzen zu ermitteln. Die Studie wurde im Herbst des Jahres 2000 an 47
verschiedenen Untersuchungszentren in den USA durchgeführt und vom „Schering-Plough
Research Institute“ gesponsert.
Voraussetzung für die Teilnahme an der Studie war ein allgemein guter Gesundheitszustand,
ein Alter von mindestens 12 Jahren und eine Vorerkrankung mit allergischer Rhinitis seit
mindestens zwei Jahren. Zudem wurde die Erkrankung mittels eines positiv ausfallenden
Prick-Tests oder intradermaler Injektion und Anwesenheit von Symptomen während der
Screening-Phase bestätigt.
1121 Studienteilnehmer*innen nahmen an dieser randomisierten, doppelblinden Studie mit
zwei Kontrollgruppen teil. Nach einer zweiwöchigen Screening-Phase wurden die
Studienteilnehmer*innen randomisiert und erhielten entweder die Kombination aus 5 mg
Desloratadin mit 240 mg Pseudoephedrin, die Monotherapie mit 5 mg Desloratadin, oder die
Monotherapie mit 240 mg Pseudoephedrin. Die Arzneimittelkombination bestand aus einem
Tablettenkern, der Pseudoephedrin kontrolliert freisetzte, und einer Tablettenhülle mit
Desloratadin. Die Pseudoephedrin-Monotherapie wurde ebenfalls als Tablette mit
kontrollierter Freisetzung verabreicht. Die Proband*innen wurden wie folgt eingeteilt: 372
Personen erhielten Desloratadin-Pseudoephedrin, 372 Personen Desloratadin und 377
Pseudoephedrin. Alle Behandlungsgruppen nahmen die entsprechenden Medikamente
einmal täglich morgens über einen Zeitraum von zwei Wochen ein. Die Teilnehmer*innen
mussten an folgenden Tagen zum Untersuchungszentrum: Screening, Tag 1, Tag 8, und Tag

30
15. Von den 1121 randomisierten Studienteilnehmer*innen beendeten 1047 die zweiwöchige
Studie. Das entspricht einem Follow-Up von 93%.
Die Studienteilnehmer*innen zeichneten die Intensität verschiedener nasaler Symptome
(Rhinorrhoe, verstopfte Nase, Niesen, juckende Nase) sowie nicht-nasaler Symptome
(juckende Augen, tränende Augen, juckende Ohren, juckender Gaumen) anhand eines
Tagebuchs auf. Die Evaluation der Symptome erfolgte zweimal täglich, jeweils für den
Zeitraum der vergangenen 12 h (reflective score), sowie zum Evaluierungszeitpunkt
(instantaneous score). Zur Bewertung der Intensität der Symptome wurde eine vierteilige
Skala verwendet aus welcher anschließend der TSS errechnet wurde. Der Basiswert des TSS
(ausgenommen nasale Verstopfung) lag zu Beginn bei 15,0 in der Desloratadin-
Pseudoephedrin-Gruppe, bei 14,7 in der Desloratadin-Gruppe sowie bei 15,8 in der
Pseudoephedrin-Gruppe. Der Basiswert des NCS lag zu Beginn bei 2,6 in allen Behandlungs-
gruppen.
Zur Einschätzung der Wirksamkeit wurden zwei primäre Endpunkte festgelegt. Der primäre
Endpunkt des antiallergisch wirksamen Bestandteils Desloratadin war die Veränderung des
Mittelwerts aus dem morning and evening reflective TSS (ausgenommen nasale Verstopfung)
vom Basiswert bis Studienende. Verglichen wurden hierbei primär Desloratadin-
Pseudoephedrin vs. Pseudoephedrin. Der primäre Endpunkt des abschwellend wirkenden
Bestandteils Pseudoephedrin war die Veränderung des Mittelwerts aus dem morning and
evening reflective NCS vom Basiswert bis Studienende. Verglichen wurden hier primär
Desloratadin-Pseudoephedrin vs. Desloratadin.
Als Grundlage zur Analyse der Ergebnisse diente die ITT-Population. Hierzu wurden all jene
Proband*innen gerechnet, die mindestens eine Dosis des Studienmedikaments eingenommen
hatten. Die Effektivitätsparameter wurden mittels der Zwei-Wege-Varianzanalyse (two-way
ANOVA) ausgewertet.
Die Arzneimittelkombination bestehend aus Desloratadin-Pseudoephedrin war der
Monotherapie mit Pseudoephedrin sowie der Monotherapie mit Desloratadin zur Reduktion
des TSS (ausgenommen nasaler Verstopfung) während der gesamten Dauer der Studie
statistisch signifikant (p=0,001 und p≤0,02) überlegen. Durch die Arzneimittel wurde eine
Reduktion vom Basiswert des mean morning and evening reflective TSS in folgender Höhe
erreicht: -6,09 (39%) mit Desloratadin-Pseudoephedrin, -5,08 (32%) mit Pseudoephedrin, und
-5,10 (34%) mit Desloratadin.
Die Arzneimittelkombination bestehend aus Desloratadin-Pseudoephedrin war der
Monotherapie mit Desloratadin zur Reduktion des NCS an den Tagen 2,3,4 sowie während
der Intervalle von Tag 1-8 und Tag 9-15 statistisch signifikant (p≤0,001) überlegen. Ebenfalls
statistisch signifikant (p≤0,02) überlegen war die Arzneimittelkombination der Monotherapie
mit Pseudoephedrin an den Tagen 3,4 sowie während der Intervalle von Tag 1-8 und Tag 9-

31
15. Durch die Arzneimittel wurde eine Reduktion vom Basiswert (2,6) in folgender Höhe
erreicht: -0,90 (33%) mit Desloratadin-Pseudoephedrin, -0,74 (28%) mit Desloratadin, und -
0,78 (29%) mit Pseudoephedrin. Allgemein war die Reduktion des NCS durch die
Arzneimittelkombination statistisch signifikant (p=0,009) größer im Vergleich zu den
Monotherapien. Es gab während der gesamten Studiendauer keine statistisch signifikanten
(p=0,53) Unterschiede im Hinblick auf die Reduktion des NCS zwischen den Monotherapie-
Gruppen mit Desloratadin oder Pseudoephedrin.
Desloratadin-Pseudoephedrin reduzierte den morgendlichen TSS (instantaneous) statistisch
signifikant höher als Desloratadin (p≤0,007) bzw. Pseudoephedrin (p≤0,008) allein zu allen
Zeitpunkten und Intervallen. Die Kombination war des Weiteren der Monotherapie mit
Desloratadin zur Reduktion des morgendlichen NCS (instantaneous) statistisch signifikant
(p≤0,005) überlegen. Beim Vergleich der Desloratadin-Pseudoephedrin-Gruppe mit der
Pseudoephedrin-Gruppe zur Reduktion des morgendlichen NCS konnte jedoch kein
anhaltender statistisch signifikanter Unterschied festgestellt werden.
Bei der Bewertung des therapeutischen Ansprechens auf die unterschiedlichen Medikamente
zum Endpunkt war die Arzneimittelkombination bestehend aus Desloratadin-Pseudoephedrin
der Monotherapie mit Pseudoephedrin statistisch signifikant (p=0,005) und der Monotherapie
mit Desloratadin numerisch überlegen. Hier gibt es jedoch keine genaueren Angaben, ob die
Bewertung des therapeutischen Anprechens durch die Prüfärzt*innen oder die Patient*innen
selbst erfolgte.
Es gab während der Studie keine unerwarteten oder unerwünschten Arzneimittelwirkungen,
die mit der Verabreichung der Substanzen in Verbindung gebracht werden konnten. Die
häufigsten Nebenwirkungen waren Kopfschmerzen, Xerostomie und Schlaflosigkeit. Es gab
mehr Fälle von Xerostomie und Schlaflosigkeit in der Pseudoephedrin-Gruppe im Vergleich
zur Arzneimittelkombination von Desloratadin mit Pseudoephedrin. Außerdem führte die
Einnahme von Desloratadin-Pseudoephedrin sowie Pseudoephedrin allein zu einer Erhöhung
der Herzfrequenz. Klinische Laborparameter, Elektrokardiogramme sowie Vitalparameter
wurden durch die Einnahme der Arzneimittel in nicht klinisch relevantem Ausmaß verändert.
Die Autor*innen schließen aus dieser Studie, dass die Ergebnisse früherer Studien
untermauert wurden. Die Kombination aus einem Antihistaminikum und Pseudoephedrin biete
zusätzliche therapeutische Vorteile bei der Behandlung von saisonaler allergischer Rhinitis
gegenüber den entsprechenden Monotherapien. Die Annahme, dass Desloratadin intrinsische
abschwellende Eigenschaften aufweise, welche durch die Kombination mit Pseudoephedrin
verstärkt würden, werde laut den Autor*innen durch die Ergebnisse dieser Studie bekräftigt.
Das Nebenwirkungsprofil der Arzneimittelkombination sei mit dem der Pseudoephedrin
Monotherapie zu vergleichen.
32
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Bei dieser Studie ist
anzumerken, dass keine Angaben über eine Registrierung gemacht wurden. Auf der
Registrierungsplattform für klinische Studien „Clinical Trials“ (https://clinicaltrials.gov) konnte
die Studie nicht gefunden werden. Des Weiteren gab es keine genauere Beschreibung, wie
die Randomisierung der Studienteilnehmer*innen erfolgte, und ob die aktiven Kontrollen, in
diesem Fall die Monotherapie mit Desloratadin bzw. Pseudoephedrin, ununterscheidbar von
der Arzneimittelkombination mit Desloratadin-Pseudoephedrin waren. Zudem fehlen die
Angaben zu den Messwerten des morgendlichen TSS bzw. des morgendlichen NCS. Hier wird
nur erwähnt, dass die Unterschiede signifikant seien, die Originaldaten der Studie sind jedoch
unbedingt erforderlich. Auch bei der Veränderung der Herzfrequenz werden keine genaueren
Werte genannt.
Die Schlussfolgerung der Autor*innen, dass in dieser Studie die abschwellenden
Eigenschaften der Monotherapie mit Desloratadin bewiesen wurden, ist an dieser Stelle
kritisch zu hinterfragen. Da die Studie über keine Placebokontrolle verfügt, kann keine
allgemeine Aussage der Wirkung der Monotherapien getroffen werden, denn dazu müsste
man untersuchen, welcher Anteil des positiven Effektes allein durch Placebo erreicht werden
könnte.
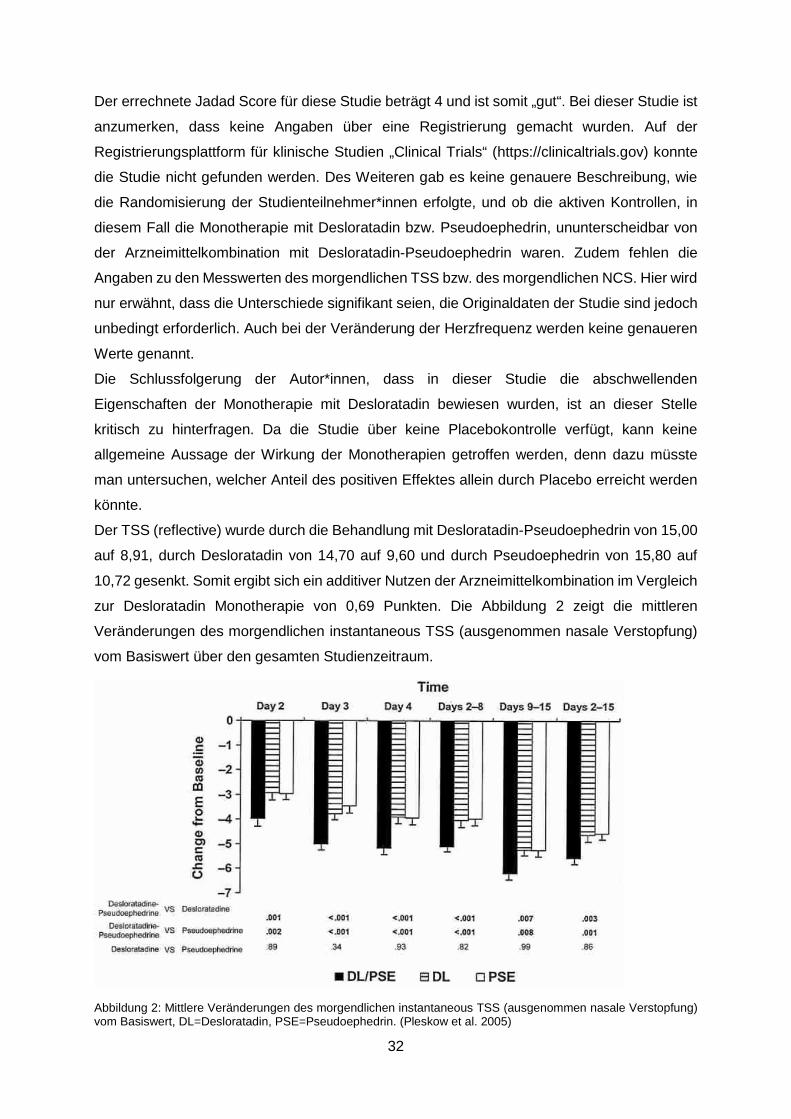
Der TSS (reflective) wurde durch die Behandlung mit Desloratadin-Pseudoephedrin von 15,00
auf 8,91, durch Desloratadin von 14,70 auf 9,60 und durch Pseudoephedrin von 15,80 auf
10,72 gesenkt. Somit ergibt sich ein additiver Nutzen der Arzneimittelkombination im Vergleich
zur Desloratadin Monotherapie von 0,69 Punkten. Die Abbildung 2 zeigt die mittleren
Veränderungen des morgendlichen instantaneous TSS (ausgenommen nasale Verstopfung)
vom Basiswert über den gesamten Studienzeitraum.
Abbildung 2: Mittlere Veränderungen des morgendlichen instantaneous TSS (ausgenommen nasale Verstopfung) vom Basiswert, DL=Desloratadin, PSE=Pseudoephedrin. (Pleskow et al. 2005)
33
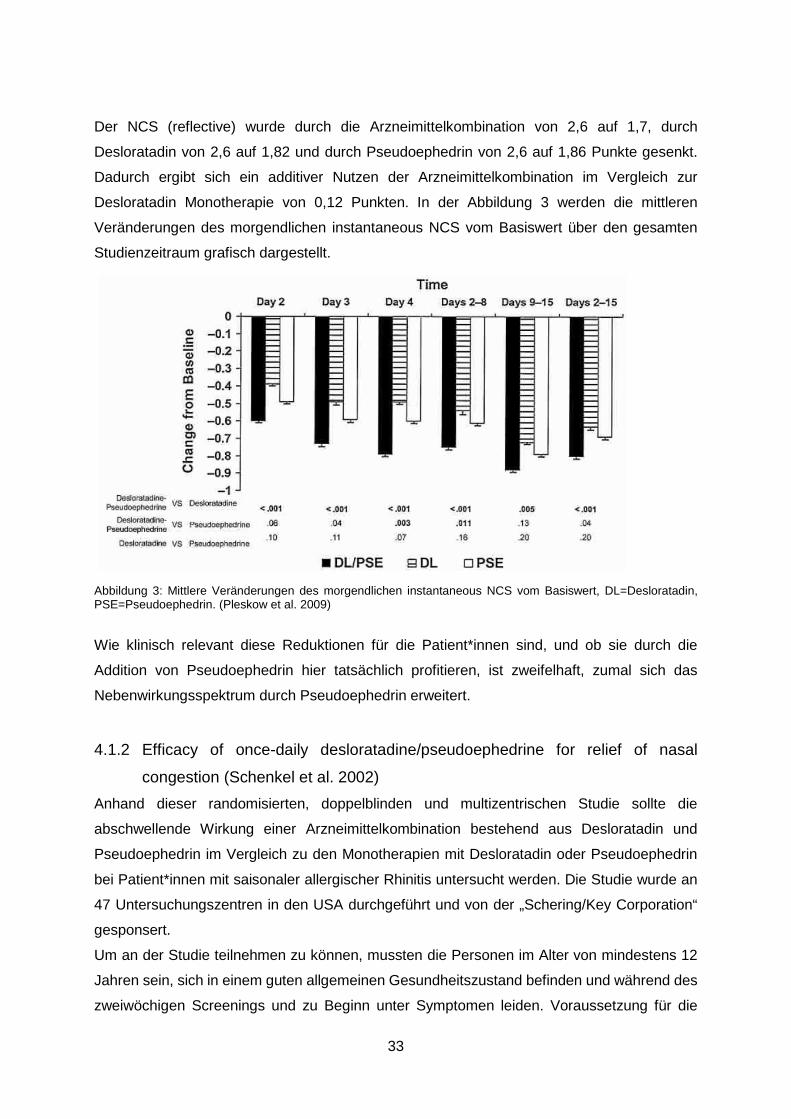
Der NCS (reflective) wurde durch die Arzneimittelkombination von 2,6 auf 1,7, durch
Desloratadin von 2,6 auf 1,82 und durch Pseudoephedrin von 2,6 auf 1,86 Punkte gesenkt.
Dadurch ergibt sich ein additiver Nutzen der Arzneimittelkombination im Vergleich zur
Desloratadin Monotherapie von 0,12 Punkten. In der Abbildung 3 werden die mittleren
Veränderungen des morgendlichen instantaneous NCS vom Basiswert über den gesamten
Studienzeitraum grafisch dargestellt.
Abbildung 3: Mittlere Veränderungen des morgendlichen instantaneous NCS vom Basiswert, DL=Desloratadin, PSE=Pseudoephedrin. (Pleskow et al. 2009)
Wie klinisch relevant diese Reduktionen für die Patient*innen sind, und ob sie durch die
Addition von Pseudoephedrin hier tatsächlich profitieren, ist zweifelhaft, zumal sich das
Nebenwirkungsspektrum durch Pseudoephedrin erweitert.
4.1.2 Efficacy of once-daily desloratadine/pseudoephedrine for relief of nasal
congestion (Schenkel et al. 2002) Anhand dieser randomisierten, doppelblinden und multizentrischen Studie sollte die
abschwellende Wirkung einer Arzneimittelkombination bestehend aus Desloratadin und
Pseudoephedrin im Vergleich zu den Monotherapien mit Desloratadin oder Pseudoephedrin
bei Patient*innen mit saisonaler allergischer Rhinitis untersucht werden. Die Studie wurde an
47 Untersuchungszentren in den USA durchgeführt und von der „Schering/Key Corporation“
gesponsert.
Um an der Studie teilnehmen zu können, mussten die Personen im Alter von mindestens 12
Jahren sein, sich in einem guten allgemeinen Gesundheitszustand befinden und während des
zweiwöchigen Screenings und zu Beginn unter Symptomen leiden. Voraussetzung für die

34
Teilnahme war des Weiteren eine Erkrankung an saisonaler allergischer Rhinitis seit
mindestens zwei Jahren, die durch einen positiven Hauttest auf saisonale Allergene, der nicht
älter als ein Jahr war, bestätigt wurde.
1018 Proband*innen wurden nach der Eingangsphase einer der drei Untersuchungsgruppen
nach zufälligem Prinzip zugeteilt. 336 Patient*innen erhielten die Kombination aus 5 mg
Desloratadin und 240 mg Pseudoephedrin, 340 erhielten 5 mg Desloratadin und 342 erhielten
240 mg Pseudoephedrin. Die Kombinationstablette war aus einem Tablettenkern mit
Pseudoephedrin in kontrollierter Freisetzung und einer Hülle aus Desloratadin zusammen-
gesetzt. Alle Medikamente wurden einmal täglich für einen Zeitraum von 15 Tagen verabreicht.
955 (94%) Studienteilnehmer*innen beendeten die Studie.
Die Teilnehmer*innen zeichneten die Intensität ihrer nasalen Symptome (Niesen, Schnupfen,
nasale Verstopfung, juckende Nase), sowie ihrer nicht-nasalen Symptome (juckende Augen,
tränende Augen, juckende Ohren oder Gaumen, gerötete Augen) anhand einer vierteiligen
Skala zweimal täglich morgens und abends auf. Hierbei wurde festgehalten, wie sich die
Proband*innen in den vergangenen 12 Stunden (reflective score) und zum Zeitpunkt der
Aufzeichnung (instantaneous score) fühlten.
Der primäre Endpunkt wurde als die Veränderungen des mittleren NCS (reflective score) vom
Basiswert während des gesamten 15-tägigen Behandlungszeitraums festgelegt. Die
Basiswerte des NCS waren zu Beginn der Studie wie folgt: 2,56 in der Untersuchungsgruppe
mit Desloratadin-Pseudoephedrin, 2,57 in der mit Desloratadin, sowie 2,54 in der mit
Pseudoephedrin.
Alle Studienteilnehmer*innen, die mindestens eine Dosis eines Arzneimittels erhalten hatten,
wurden auf Basis der ITT-Methode in die Effektivitäts- und Sicherheitsanalyse miteinbezogen.
Während des gesamten Behandlungszeitraums war die Arzneimittelkombination bestehend
aus Desloratadin und Pseudoephedrin im Hinblick auf die Reduktion des mittleren NCS
(reflective score) der Monotherapie mit Desloratadin, sowie der Monotherapie mit
Pseudoephedrin statistisch signifikant (p<0,01) überlegen. Des Weiteren war die Kombination
Desloratadin-Pseudoephedrin zu allen Untersuchungszeitpunkten statistisch signifikant
besser, ausgenommen an Tag 1, als die Kombination nur numerisch (p=0,052) besser als
Pseudoephedrin abschnitt. Die Reduktion des mittleren NCS vom Basiswert lag während der
15-tägigen Untersuchung in der Desloratadin-Pseudoephedrin-Gruppe bei 0,85 (32%), in der
Pseudoephedrin-Gruppe bei 0,70 (27%) und in der Desloratadin-Gruppe bei 0,65 (25%).
Desloratadin-Pseudoephedrin war außerdem effektiver als die Einzelsubstanzen zur
Reduktion der nasalen Verstopfung am Morgen. Die Kombination bestehend aus Desloratadin
und Pseudoephedrin erreichte eine Reduktion von 0,75 (28%) verglichen mit 0,61 (22%) in der
Pseudoephedrin-Gruppe (p<0,01) und 0,59 (21%) in der Desloratadin-Gruppe (p<0,01) über
den gesamten Behandlungszeitraum. Es gab keine statistisch signifikanten Unterschiede im

35
Hinblick auf die Reduktion der verstopften Nase zwischen der Monotherapie Gruppe mit
Desloratadin und der mit Pseudoephedrin.
Während des gesamten Studienzeitraums traten keine unerwarteten oder unüblichen
Nebenwirkungen durch die Medikamenteneinnahme auf. Die häufigsten unerwünschten
Arzneimittelwirkungen in den Gruppen Desloratadin-Pseudoephedrin, Desloratadin sowie
Pseudoephedrin waren Xerostomie (jeweils 7%, 2% und 7%) und Schlaflosigkeit (jeweils 5%,
1% und 8%). Die vermehrte Anhäufung unerwünschter Wirkungen in den beiden Gruppen mit
Desloratadin-Pseudoephedrin und Pseudoephedrin ist vermutlich auf die pharmakologischen
Eigenschaften von Pseudoephedrin zurückzuführen. Während der Studie traten keine klinisch
relevanten Veränderungen von Vitalparametern, Elektrokardiogrammen oder klinischen
Laborparametern auf.
Aus diesen Ergebnissen schließen die Autor*innen, dass die Arzneimittelkombination
bestehend aus Desloratadin und Pseudoephedrin Vorteile gegenüber der Monotherapie mit
Pseudoephedrin im Hinblick auf ihre abschwellenden Eigenschaften hat. Dies liefere weitere
Beweise für die angeblich abschwellende Wirkung, die der Monotherapie mit Desloratadin
zugesprochen wird. Des Weiteren sei der additive Effekt auf die Abschwellung der
Nasenschleimhaut, welcher durch die Arzneimittelkombination erreicht wurde, ein Hinweis
dafür, dass Desloratadin und Pseudoephedrin über unterschiedliche Mechanismen
abschwellend wirken. Das Nebenwirkungsspektrum der Arzneimittelkombination und
Pseudoephedrin sei des Weiteren sehr ähnlich.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Zu dieser Studie ist
anzumerken, dass keine Angaben über eine Registrierung gemacht wurden. Auf der
Registrierungsplattform für klinische Studien „Clinical Trials“ (https://clinicaltrials.gov) war die
Studie nicht zu finden. Über die Art der Randomisierung gibt es ebenfalls keine Angaben.
Außerdem wäre es wichtig zu wissen, ob die aktive Kontrolle, in diesem Fall die Monotherapie
mit Desloratadin oder Pseudoephedrin, unterscheidbar von der Arzneimittelkombination
bestehend aus Desloratadin und Pseudoephedrin war. Ein weiterer wichtiger Punkt, welcher
in dieser Studie fehlt, ist die Angabe über das statistische Modell, denn es wird an keiner Stelle
erwähnt nach welchem Prinzip die p-Werte berechnet wurden.
Der mittlere NCS (reflective) wurde in der Untersuchungsgruppe mit Desloratadin-
Pseudoephedrin von 2,56 auf 1,71, in der mit Desloratadin von 2,57 auf 1,92 und in der mit
Pseudoephedrin von 2,54 auf 1,84 gesenkt. Der Unterschied des NCS zwischen der
Kombination aus Desloratadin und Pseudoephedrin bzw. der Pseudoephedrin Monotherapie,
der durch die Addition von Pseudoephedrin erreicht wurde, beträgt hier demnach 0,21 Punkte.
Somit ist fraglich, ob die Patient*innen von diesem Unterschied profitieren, bzw. ob dieser nicht
nur statistisch signifikant, sondern auch klinisch relevant ist.

36
4.1.3 Comparative efficacy and safety of a once-daily loratadine-pseudoephedrine
combination versus its components alone and placebo in the management of
seasonal allergic rhinitis (Bronsky et al. 1995) Ziel dieser Studie war es, eine Arzneimittelkombination namens „SCH 434 QD“, die aus
Loratadin und Pseudoephedrin besteht, mit den Einzelkomponenten als Monotherapie oder
Placebo im Hinblick auf die Effektivität und Sicherheit zu vergleichen. Diese randomisierte,
doppelblinde und placebokontrollierte Studie wurde an 14 Untersuchungszentren in den USA
im Herbst 1989 durchgeführt und vom „Schering-Plough Research Institute“ gesponsert.
Personen im Alter von 12 bis 60 Jahren und einer mindestens einjährigen Geschichte mit
saisonaler allergischer Rhinitis konnten an der Studie teilnehmen. Voraussetzung für die
Teilnahme war außerdem ein allgemein guter Gesundheitszustand und ein positiv ausfallender
Prick-Test, der die bestehende Allergie bestätigte.
Nach der Eingangsphase wurden 879 Studienteilnehmer*innen für einen Zeitraum von vier bis
sieben Tagen mit Placebo behandelt, um die Basiswerte des TSS festzulegen und
Patient*innen mit zu geringen Symptomen auszuschließen. Anschließend wurden die
verbliebenen 874 Teilnehmer*innen vier verschiedenen Behandlungsgruppen zugeteilt und
erhielten: 10 mg Loratadin und 240 mg Pseudoephedrin 1xtgl., 10 mg Loratadin 1xtgl., 120 mg
Pseudoephedrin 2xtgl., oder Placebo. Die Arzneimittelkombination war aus einem
Tablettenkern mit Pseudoephedrin mit kontrollierter Freisetzung und einer Hülle aus Loratadin
zusammengesetzt. Die Pseudoephedrin Monotherapie wurde als Tablette mit kontrollierter
Freisetzung verabreicht. Um die vollständige Verblindung der Medikamente zu gewährleisten,
wurden alle Behandlungen zweimal täglich morgens und abends für einen Zeitraum von zwei
Wochen verabreicht. 221 Patient*innen erhielten Loratadin-Pseudoephedrin, 217 erhielten
Loratadin, 220 Pseudoephedrin und 216 Placebo. Von den anfänglichen 874 Studienteil-
nehmer*innen beendeten 820 die Studie. Das entspricht einer Drop-Out Rate von 6%.
Die Studienteilnehmer*innen zeichneten die Intensität der Symptome für den gesamten
Studienzeitraum einschließlich der Placebo-Phase mithilfe eines Tagebuchs auf. Die
Wirksamkeit der Arzneimittel wurde durch die Beurteilung von acht verschiedenen nasalen
und nicht-nasalen Symptomen durch die Studienteilnehmer*innen sowie durch Prüfärzt*innen
ermittelt. Dazu wurde die Intensität der Symptome auf einer Skala von 0 bis 3 bewertet und
anschließend der TSS errechnet. Eine globale Bewertung der therapeutischen Effekte auf die
Symptome der Proband*innen wurde durch die Teilnehmer*innen und durch Prüfärzt*innen an
den Tagen 4, 8 und 15 während der aktiven Behandlungsphase durchgeführt.
Der für den primären Endpunkt herangezogene Symptom Score war nicht klar definiert.
Vergleiche wurden für durchschnittliche Veränderungen der Scores vom Basiswert an Tag 4
und am Endpunkt durchgeführt. Der TSS war zu Beginn der Behandlung wie folgt: 16,3 in der
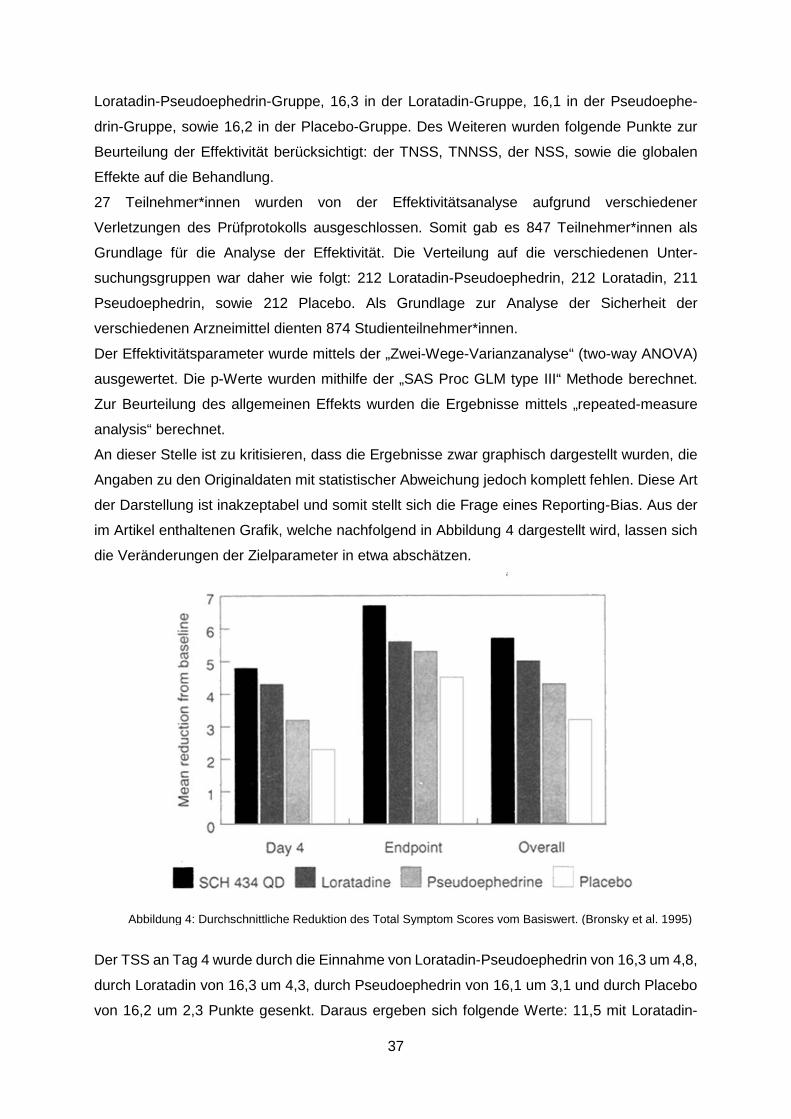
37
Loratadin-Pseudoephedrin-Gruppe, 16,3 in der Loratadin-Gruppe, 16,1 in der Pseudoephe-
drin-Gruppe, sowie 16,2 in der Placebo-Gruppe. Des Weiteren wurden folgende Punkte zur
Beurteilung der Effektivität berücksichtigt: der TNSS, TNNSS, der NSS, sowie die globalen
Effekte auf die Behandlung.
27 Teilnehmer*innen wurden von der Effektivitätsanalyse aufgrund verschiedener
Verletzungen des Prüfprotokolls ausgeschlossen. Somit gab es 847 Teilnehmer*innen als
Grundlage für die Analyse der Effektivität. Die Verteilung auf die verschiedenen Unter-
suchungsgruppen war daher wie folgt: 212 Loratadin-Pseudoephedrin, 212 Loratadin, 211
Pseudoephedrin, sowie 212 Placebo. Als Grundlage zur Analyse der Sicherheit der
verschiedenen Arzneimittel dienten 874 Studienteilnehmer*innen.
Der Effektivitätsparameter wurde mittels der „Zwei-Wege-Varianzanalyse“ (two-way ANOVA)
ausgewertet. Die p-Werte wurden mithilfe der „SAS Proc GLM type III“ Methode berechnet.
Zur Beurteilung des allgemeinen Effekts wurden die Ergebnisse mittels „repeated-measure
analysis“ berechnet.
An dieser Stelle ist zu kritisieren, dass die Ergebnisse zwar graphisch dargestellt wurden, die
Angaben zu den Originaldaten mit statistischer Abweichung jedoch komplett fehlen. Diese Art
der Darstellung ist inakzeptabel und somit stellt sich die Frage eines Reporting-Bias. Aus der
im Artikel enthaltenen Grafik, welche nachfolgend in Abbildung 4 dargestellt wird, lassen sich
die Veränderungen der Zielparameter in etwa abschätzen.
Der TSS an Tag 4 wurde durch die Einnahme von Loratadin-Pseudoephedrin von 16,3 um 4,8,
durch Loratadin von 16,3 um 4,3, durch Pseudoephedrin von 16,1 um 3,1 und durch Placebo
von 16,2 um 2,3 Punkte gesenkt. Daraus ergeben sich folgende Werte: 11,5 mit Loratadin-
Abbildung 4: Durchschnittliche Reduktion des Total Symptom Scores vom Basiswert. (Bronsky et al. 1995)
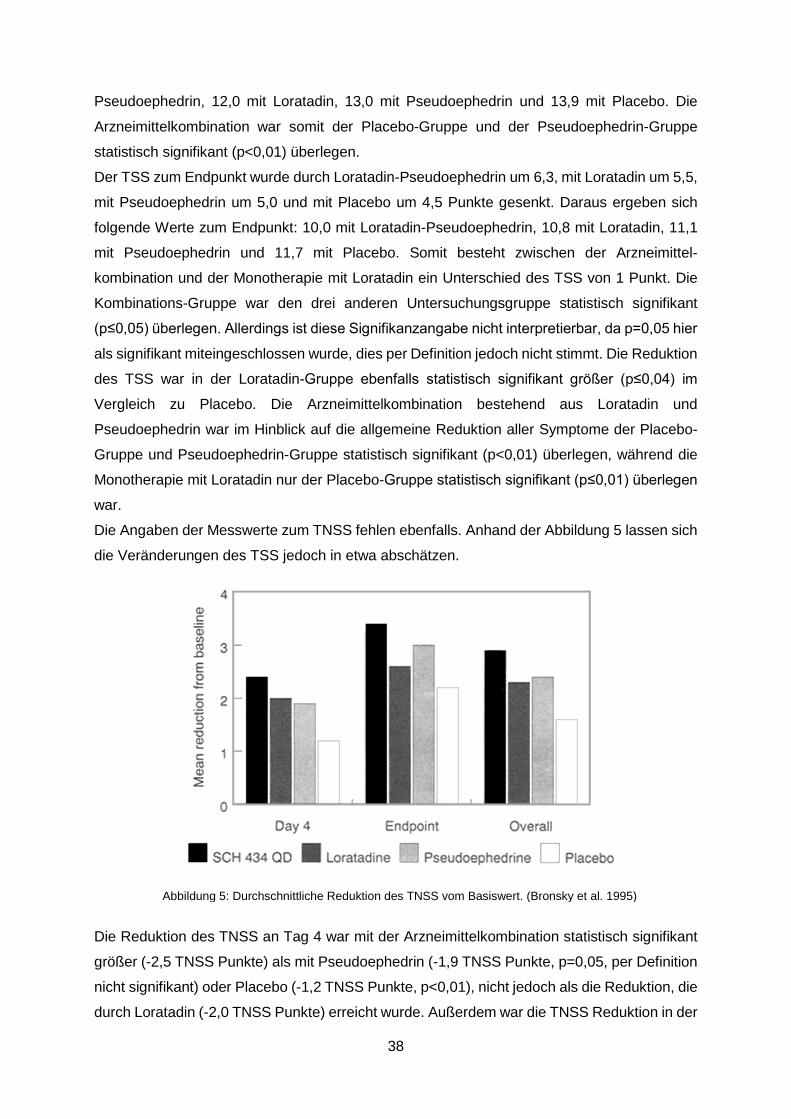
38
Pseudoephedrin, 12,0 mit Loratadin, 13,0 mit Pseudoephedrin und 13,9 mit Placebo. Die
Arzneimittelkombination war somit der Placebo-Gruppe und der Pseudoephedrin-Gruppe
statistisch signifikant (p<0,01) überlegen.
Der TSS zum Endpunkt wurde durch Loratadin-Pseudoephedrin um 6,3, mit Loratadin um 5,5,
mit Pseudoephedrin um 5,0 und mit Placebo um 4,5 Punkte gesenkt. Daraus ergeben sich
folgende Werte zum Endpunkt: 10,0 mit Loratadin-Pseudoephedrin, 10,8 mit Loratadin, 11,1
mit Pseudoephedrin und 11,7 mit Placebo. Somit besteht zwischen der Arzneimittel-
kombination und der Monotherapie mit Loratadin ein Unterschied des TSS von 1 Punkt. Die
Kombinations-Gruppe war den drei anderen Untersuchungsgruppe statistisch signifikant
(p≤0,05) überlegen. Allerdings ist diese Signifikanzangabe nicht interpretierbar, da p=0,05 hier
als signifikant miteingeschlossen wurde, dies per Definition jedoch nicht stimmt. Die Reduktion
des TSS war in der Loratadin-Gruppe ebenfalls statistisch signifikant größer (p≤0,04) im
Vergleich zu Placebo. Die Arzneimittelkombination bestehend aus Loratadin und
Pseudoephedrin war im Hinblick auf die allgemeine Reduktion aller Symptome der Placebo-
Gruppe und Pseudoephedrin-Gruppe statistisch signifikant (p<0,01) überlegen, während die
Monotherapie mit Loratadin nur der Placebo-Gruppe statistisch signifikant (p≤0,01) überlegen
war.
Die Angaben der Messwerte zum TNSS fehlen ebenfalls. Anhand der Abbildung 5 lassen sich
die Veränderungen des TSS jedoch in etwa abschätzen.
Die Reduktion des TNSS an Tag 4 war mit der Arzneimittelkombination statistisch signifikant
größer (-2,5 TNSS Punkte) als mit Pseudoephedrin (-1,9 TNSS Punkte, p=0,05, per Definition
nicht signifikant) oder Placebo (-1,2 TNSS Punkte, p<0,01), nicht jedoch als die Reduktion, die
durch Loratadin (-2,0 TNSS Punkte) erreicht wurde. Außerdem war die TNSS Reduktion in der
Abbildung 5: Durchschnittliche Reduktion des TNSS vom Basiswert. (Bronsky et al. 1995)

39
Loratadin- und Pseudoephedrin-Gruppe statistisch signifikant (p<0,01) größer als in der
Placebo-Gruppe. Zum Endpunkt der Aufzeichnung war die Reduktion des TNSS in der
Untersuchungsgruppe der Kombination von Loratadin und Pseudoephedrin statistisch
signifikant (-3,5 TNSS Punkte, p<0.01) größer als in der mit Loratadin (-2,6 TNSS Punkte) und
Placebo (-2,2 TNSS Punkte). Pseudoephedrin war ebenfalls statistisch signifikant (3,0 TNSS
Punkte, p<0,01) der Placebo-Gruppe überlegen. Die globale Reduktion des TNSS war in allen
drei Arzneimittelgruppen statistisch signifikant (p<0,01) größer als mit Placebo.
Sowohl an Tag 4 als auch zum Zeitpunkt des letzten Kontrolltermins waren die
Arzneimittelkombination und die Untersuchungsgruppe mit Loratadin im Hinblick auf die
Reduktion des TNNSS der Gruppe mit Pseudoephedrin und Placebo statistisch signifikant
(p≤0,03) überlegen. Der Unterschied der Reduktion des TNNSS zwischen Pseudoephedrin
und Placebo war nicht statistisch signifikant. Bei der globalen Bewertung der Reduktion des
TNNSS waren die Arzneimittelkombination, sowie die Loratadin-Gruppe der Pseudoephedrin
und Placebo-Gruppe statistisch signifikant (p<0,01) überlegen.
Die Reduktion des NSS war an Tag 4 und am Endpunkt in der Untersuchungsgruppe mit
Loratadin-Pseudoephedrin statistisch signifikant (p<0.03) größer als in der mit Placebo. Der
Unterschied zwischen der Arzneimittelkombination und Pseudoephedrin an Tag 4 war nicht
statistisch signifikant. Zum Zeitpunkt der letzten Kontrolle war die Reduktion durch die
Arzneimittelkombination statistisch signifikant (p<0,01) größer als mit Loratadin. Die erreichte
Reduktion des NSS war mit Pseudoephedrin statistisch signifikant (p≤0,05) größer als mit
Placebo an Tag 4 und zum Endpunkt und ebenfalls statistisch signifikant größer (p<0,01) als
mit Loratadin zum Endpunkt. Es gab keine Unterschiede im Hinblick auf die Reduktion des
NSS zwischen Loratadin und Placebo. Die allgemeine Reduktion des NSS war in der
Untersuchungsgruppe mit Loratadin und Pseudoephedrin statistisch signifikant (p≤0,02)
größer als in der mit Loratadin und Placebo. Die Untersuchungsgruppe mit Pseudoephedrin
war der mit Placebo ebenfalls statistisch signifikant (p=0,04) überlegen.
In der Arzneimittelgruppe mit Loratadin-Pseudoephedrin sprachen auf Basis einer Evaluierung
durch Prüfärzt*innen mit 58% die meisten Teilnehmer*innen „exzellent“ oder „gut“ auf das
Arzneimittel an und es gab am wenigsten Fälle von Therapieversagen. Verglichen dazu waren
die Zahlen in den übrigen Untersuchungsgruppen wie folgt: 49% in der Loratadin-Gruppe, 49%
in der Pseudoephedrin-Gruppe und 39% in der Placebo-Gruppe. Das allgemeine Ansprechen
auf die verschiedenen Medikamente war in der Gruppe mit Loratadin-Pseudoephedrin an Tag
8, 15 und zum Endpunkt statistisch signifikant (p≤0,02) besser im Vergleich zu Loratadin und
Pseudephedrin als Monotherapie und zu allen Zeitpunkten statistisch signifikant (p<0,01)
besser als Placebo. Zu den annähernd gleichen Ergebnissen kamen auch die Studien-
teilnehmer*innen bei der Bewertung des therapeutischen Ansprechens auf die Medikamente.

40
Hier wurde das therapeutische Ansprechen auf die Arzneimittel mit „exzellent“ oder „gut“ von
61% in der Loratadin-Pseudoephedrin-Gruppe, 52% in der Pseudoephedrin-Gruppe, 47% in
der Loratadin-Gruppe sowie 33% in der Placebo-Gruppe bewertet.
Es gab statistisch signifikant (p≤0,05) mehr Fälle von unerwünschten Arzneimittelwirkungen in
der Gruppe mit Loratadin-Pseudoephedrin und in der mit Pseudoephedrin im Vergleich zu
Placebo. Die häufigsten unerwünschten Nebenwirkungen in der Loratadin-Pseudoephedrin-
Gruppe waren Kopfschmerzen, Xerostomie, Schläfrigkeit, Pharyngitis, Nervosität und
Schlaflosigkeit. Es gab keine Unterschiede bezüglich der Häufigkeit des Auftretens von
Schläfrigkeit in den verschiedenen Untersuchungsgruppen. In den Untersuchungsgruppen mit
Loratadin-Pseudoephedrin und Pseudoephedrin gab es statistisch signifikant (p≤0,04) mehr
Fälle von Schlaflosigkeit und Nervosität als in denen mit Loratadin oder Placebo. Außerdem
war die Häufigkeit von Xerostomie in den beiden Gruppen ebenfalls statistisch signifikant
(p≤0,05) höher im Vergleich zu Loratadin oder Placebo. Während der Studie kam es zu einer
geringen Veränderung der Laborparameter, diese waren jedoch nicht klinisch relevant.
Die Herzfrequenz wurde durch die Gabe der Arzneimittelkombination bestehend aus
Loratadin-Pseudoephedrin um 3,3-4,5 Schläge pro Minute erhöht und erreichte somit
statistische Signifikanz (p≤0,02) gegenüber der Loratadin- oder Placebo-Gruppe mit einer
Erhöhung von 0,1-0,6 und 0,2-0,9 Schlägen pro Minute. In der Pseudoephedrin-Gruppe kam
es ebenfalls zu einer Erhöhung der Herzfrequenz um 2,5-3,0 Schläge pro Minute, dieser
Unterschied war jedoch nicht statistisch signifikant.
Aus diesen Ergebnissen schließen die Autor*innen, dass „SCH 434 QD“ ein sicheres
Arzneimittel zur kurzzeitigen Behandlung von saisonaler allergischer Rhinitis ist und sowohl
gegenüber Placebo als auch den Monotherapien mit Loratadin oder Pseudoephedrin
statistisch signifikant überlegen ist.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Zu dieser Studie ist
anzumerken, dass keine Angaben über eine Registrierung gemacht wurden und auch auf der
internationalen Registrierungsplattform „Clinical Trials“ (https://clinicaltrials.gov) war die Studie
nicht zu finden. Anfangs wird zwar erwähnt, dass ein institutionelles Komitee das
Studienprotokoll sowie die Einwilligungserklärung, die von jeder/jedem Studienteilnehmer*in
unterschrieben werden musste, bewilligt hat. Eine Registrierung ist dennoch notwendig. Es
gab des Weiteren keine genaue Beschreibung des Verfahrens zur Randomisierung der
Studienteilnehmer*innen und die Angaben der Originaldaten bzw. der Absolutwerte der
Ergebnisse fehlen ebenfalls. Ein weiterer Kritikpunkt dieser Studie ist mit Sicherheit, dass der
primäre Endpunkt nicht beschrieben wird. Die Tabellen führen lediglich die errechneten p-
Werte an und auch die Angaben von Standardabweichungen fehlen hier komplett. Eine

41
objektive Beurteilung der Ergebnisse fällt daher schwer. Aufgrund der eindeutig mangelhaften
Durchführung sind die Ergebnisse dieser Studie nur bedingt aussagekräftig.
4.1.4 Cetirizine and pseudoephedrine retard, given alone or in combination, in
patients with seasonal allergic rhinitis (Grosclaude et al. 1997) In dieser randomisierten, doppelblinden Studie wurde die Wirksamkeit und Sicherheit einer
Arzneimittelkombination bestehend aus Cetirizin und Pseudoephedrin zur Behandlung von
allergischer Rhinitis im Vergleich zu den Einzelsubstanzen untersucht. Die Studie wurde im
Zeitraum von März bis September des Jahres 1992 an 30 Untersuchungszentren in Frankreich
und 13 Untersuchungszentren in Deutschland durchgeführt.
Voraussetzung für die Teilnahme an der Studie war ein Alter von 12 bis 65 Jahren, eine
Erkrankung mit allergischer Rhinitis aufgrund von Pollen seit mindestens einem Jahr und ein
positiver Haut- bzw. RAST-Test.
687 Patient*innen im Alter von 9 bis 66 Jahren nahmen an dieser Studie teil und wurden
anhand einer computergenerierten Randomisierungsliste einer der drei Untersuchungs-
gruppen zugeteilt. Die Studienteilnehmer*innen erhielten demnach entweder eine
Arzneimittelkombination bestehend aus 5 mg Cetirizin und 120 mg Pseudoephedrin retard
oder eine Monotherapie mit 5 mg Cetirizin oder 120 mg Pseudoephedrin retard. In der
Untersuchungsgruppe mit Cetirizin-Pseudoephedrin befanden sich nach der Eingangsphase
230 Patient*innen, in der mit Cetirizin 231 und in der mit Pseudoephedrin 226. Alle Arzneimittel
wurden zweimal täglich über einen Zeitraum von zwei Wochen verabreicht. Um sowohl die
Verblindung der Patient*innen als auch die der Prüfärzt*innen zu garantieren, wurden pro
Dosis je zwei optisch ununterscheidbare Kapseln verabreicht: eine Dosis der
Arzneimittelkombination bestand aus einer Kapsel Cetirizin und einer Kapsel Pseudoephedrin
retard, die Monotherapie mit Cetirizin bestand aus einer Kapsel Cetirizin und einer
Placebokapsel und die Monotherapie mit Pseudoephedrin bestand aus einer Kapsel
Pseudoephedrin retard und einer Placebokapsel. Zusätzlich zur Untersuchungsmedikation
war es den Studienteilnehmer*innen erlaubt, die bereits zuvor begonnene Einnahme von
Arzneimitteln (z.B. inhalative Corticosteroide ≤400 µg pro Tag) zur Behandlung von Asthma
fortzusetzen. Die Teilnehmer*innen mussten an insgesamt drei Tagen zu einem der
Untersuchungszentren: Zu Studienbeginn, eine Woche nach Beginn der Studie erfolgte die
Kontrolluntersuchung und nach zwei Wochen die Abschlussuntersuchung. 616 (90%)
Studienteilnehmer*innen beendeten die zweiwöchige Studie. Die Verteilung in den Gruppen
war zum Endpunkt wie folgt: 210 (91%) in der Kombinations-Gruppe, 208 (90%) in der
Cetirizin-Gruppe, und 198 (88%) in der Pseudoephedrin-Gruppe.
An den Kontrollterminen beurteilten die Prüfärzt*innen folgende Symptome mittels einer
vierteiligen Skala: nasale Obstruktion, Niesen, Rhinorrhoe, nasaler Juckreiz sowie Juckreiz bei
den Augen. Die vierteilige Skala wertete die Intensität der Symptome wie folgt: „0“ „abwesend“,

42
„1“ „mild“, „2“ „moderat“ und „3“ „schwer“. Die Patient*innen zeichneten dieselben Symptome
täglich anhand derselben vierteiligen Skala auf. Diese Aufzeichnungen wurden in ein
Tagebuch eingetragen und stellten die Grundlage für den primären Effektivitätsparameter dar.
Am letzten Tag der Evaluierung beurteilten die Prüfärzt*innen außerdem den allgemeinen
Behandlungseffekt mithilfe einer fünfteiligen Skala. Darin stellte „0“ „schlechter“, „1“ „keine
Veränderungen“, „2“ „leichte Verbesserung“, „3“ „deutliche Verbesserung“, sowie „4“
„Symptomfreiheit“ dar.
Der primäre Endpunkt war der Prozentsatz an „angenehmen Tagen“ mit einem MSS von 0 bis
1 und wurde mithilfe der täglich aufgezeichneten fünf Symptomen errechnet. Der MSS setzte
sich zusammen aus dem Mittelwert der am höchsten bewerteten Symptomen pro Patient pro
Tag. Der Zeitraum für die Berechnung der Anzahl an „angenehmen Tagen“ war Tag 2 bis 13.
Zusätzlich wurden mehrere sekundäre Endpunkte zur Beurteilung der Effektivität festgelegt:
Der mittlere „5-Symptom-Score“, der mittlere „4-Symptom-Score“ (ausgenommen nasale
Obstruktion) und die mittleren individuellen Symptome über den gesamten
Behandlungszeitraum.
Die Werte für den „5-Symptom-Score“ waren zu Beginn wie folgt: 1,92 Punkte in der
Kombinations-Gruppe, 1,99 Punkte in der Cetirizin-Gruppe, sowie 1,96 Punkte in der
Pseudoephedrin-Gruppe. Die Basiswerte für den „4-Symptom-Score“ lagen in der
Kombinations-Gruppe bei 1,83 Punkten, in der Cetirizin-Gruppe bei 1,92 Punkten und in der
Pseudoephedrin-Gruppe bei 1,88 Punkten.
Als Grundlage für die Analyse der Daten diente die ITT-Population. Dazu zählten all jene
Studienteilnehmer*innen, welche ein Arzneimittel erhalten hatten. Um die Verteilung der
„angenehmen Tage“ zwischen den Untersuchungsgruppen zu vergleichen wurde der „Kruskal-
Wallis Test“ angewandt. Die Unterschiede zwischen Cetirizin oder Pseudoephedrin und der
Arzneimittelkombination wurde mittels „Wilcoxon-Mann-Whitney-Test“ berechnet. Allgemeine
Vergleiche zwischen den Gruppen wurden mittels „One-way ANOVA“ und „Two-by-two Com-
parisons“ unter zu Hilfenahme des „t-Test“ gemacht.
In der Untersuchungsgruppe mit Cetirizin-Pseudoephedrin gab es nach einem Einnahme-
zeitraum von zwei Wochen statistisch signifikant (p<0001) mehr „angenehme Tage“ als in den
Untersuchungsgruppen mit Cetirizin oder Pseudoephedrin. Der Median lag hier bei 53% in der
Kombinations-Gruppe, bei 31% in der Cetirizin-Gruppe und bei 33% in der Pseudoephedrin-
Gruppe. Die Mittelwerte waren wie folgt: 51% „angenehme Tage“ mit der Einnahme von
Cetirizin-Pseudoephedrin, 40% mit Cetirizin, sowie 37% mit Pseudoephedrin. Die Mittelwerte
des „5-Symptom-Scores“ lagen in der Arzneimittelkombinations-Gruppe bei 0,85, in der
Cetirizin-Gruppe bei 1,03 (p<0,001) und in der Pseudoephedrin-Gruppe bei 1,14 (p<0,001)
Punkten.

43
Die Reduktion des “4-Symptom-Scores” (ausgenommen nasale Obstruktion), welche mit der
Arzneimittelkombination bestehend aus Cetirizin und Pseudoephedrin erreicht wurde, war
statistisch signifikant (p<0,001) größer im Vergleich zu Cetirizin oder Pseudoephedrin allein.
Die Mittelwerte dafür lagen bei 0,77 Punkten in der Untersuchungsgruppe mit Cetirizin-
Pseudoephedrin, bei 0,93 Punkten mit Cetirizin und bei 1,12 Punkten mit Pseudoephedrin.
Die einzelnen Symptome wurden durch die Arzneimittelkombination im Vergleich zu Cetirizin
mit Ausnahme der juckenden Augen statistisch signifikant (p≤0,01) verbessert. Außerdem war
Cetirizin-Pseudoephedrin statistisch signifikant (p≤0,01) besser im Vergleich zu
Pseudoephedrin zur Reduktion aller Symptome mit Ausnahme der verstopften Nase.
Bei der Beurteilung des MSS durch die Prüfärzt*innen schnitt die Kombination von Cetirizin
und Pseudoephedrin statistisch signifikant (p<0,01) besser ab als die Monotherapie mit
Cetirizin oder Pseudoephedrin. Die Mittelwerte hierfür waren nach der ersten Woche 1,53 für
Cetirizin-Pseudoephedrin, 1,76 für Cetirizin und 1,82 Punkten für Pseudoephedrin. Zum
Endpunkt der Studie lagen die Mittelwerte für Cetirizin-Pseudoephedrin bei 1,29, für Cetirizin
bei 1,63 (p<0,001) und bei Pseudoephedrin bei 1,53 (p=0,031) Punkten.
Die allgemeine Beurteilung des Therapieeffekts durch die Prüfärzt*innen fiel zugunsten der
Arzneimittelkombination aus. Diese war statistisch signifikant besser zur Behandlung
saisonaler allergischer Rhinitis als Cetirizin (p=0,001) oder Pseudoephedrin (p=0,007),
welches jeweils als Monotherapeutikum verabreicht wurde. Der Therapieeffekt wurde von 69%
der Studienteilnehmer*innen in der Untersuchungsgruppe mit Cetirizin-Pseudoephedrin, von
56% in der mit Cetirizin und von 58% in der mit Pseudoephedrin als „gut“ bzw. „exzellent“
beurteilt.
Während des gesamten Studienzeitraums kam es zu keinerlei schwerwiegenden
unerwünschten Arzneimittelwirkungen. Nebenwirkungen wurde von 68 Patient*innen (30%) in
der Kombinations-Gruppe, von 54 (23%) in der Cetirizin-Gruppe und von 68 (30%) in der
Pseudoephedrin-Gruppe berichtet. Dieser Unterschied war jedoch nicht statistisch signifikant.
Die häufigsten unerwünschten Arzneimittelwirkungen in der Cetirizin-Gruppe waren
Schläfrigkeit (6%) und Kopfschmerzen (4%), während in der Kombinations- bzw.
Pseudoephedrin-Gruppe vor allem über Kopfschmerzen (Kombination: 4%, Pseudoephedrin:
7%), Schlaflosigkeit (Kombination: 7%, Pseudoephedrin: 11%) und Xerostomie (Kombination:
7%, Pseudoephedrin: 4%) berichtet wurde. Minimale Veränderungen der Blutwerte bei sieben
Patient*innen wurden als nicht klinisch relevant gewertet. Die Herzfrequenz erhöhte sich durch
die Arzneimitteleinnahme um 3,2 Schläge pro Minute in der Kombinations- und um 2,2 Schläge
pro Minute in der Pseudoephedrin-Gruppe.
Aus den Ergebnissen dieser Studie schließen die Autor*innen, dass die
Arzneimittelkombination bestehend aus Cetirizin und Pseudoephedrin die Symptome von
saisonaler allergischer Rhinitis stärker als die jeweiligen Einzelsubstanzen abschwächt.

44
„Histamin-induzierte“ Reaktionen wurden durch die Kombination von Cetirizin und
Pseudoephedrin im Vergleich zu Pseudoephedrin allein stärker reduziert. Im Vergleich zu
Cetirizin alleine erwies sich die Kombination sowohl effektiver zur Reduktion der verstopften
Nase als auch zur Reduktion von Rhinorrhoe, Niese und Juckreiz der Nase. Die Autor*innen
erklären sich diesen Effekt dadurch, dass die Reduktion der nasalen Obstruktion zur
Verbesserung anderer nasaler Symptome führe. Unerwünschte Arzneimittelwirkungen kamen
in der Untersuchungsgruppe mit Cetirizin-Pseudoephedrin doppelt so oft wie in der
Untersuchungsgruppe mit Cetirizin vor. Die Anmerkung der Autor*innen war hier, dass dies
der Preis für die höhere Effektivität sei. Die abschließende Aussage der Autor*innen war daher,
dass die Kombination von 5 mg Cetirizin und 120 mg Pseudoephedrin, bei zweimal täglicher
Gabe über einen Zeitraum von zwei Wochen eine effektive und sichere Behandlungsmethode
für saisonale allergische Rhinitis sei.
Der errechnete Jadad Score für diese Studie beträgt 5 und ist somit „exzellent“. Zu dieser
Studie ist anzumerken, dass keine Angaben über eine Registrierung gemacht wurden. Sie war
sowohl auf der Registrierungsplattform für klinische Studien „Clinical Trials“
(https://clinicaltrials.gov) als auch auf der europäischen Seite „EU Clinical Trials Register“
(https://www.clinicaltrialsregister.eu/) nicht auffindbar. Die Studie wurde jedoch gemäß der
Deklaration von Helsinki und den Europäischen Richtlinien der „Good Clinical Practice“
durchgeführt. Das Studienprotokoll wurde vom Ethikkomitee in Frankreich und Deutschland
bewilligt.
Im Gegensatz zu anderen Studien wurde hier nicht der TSS als primärer Endpunkt festgelegt,
sondern die Anzahl an Tagen, die von den Patient*innen als „angenehm“ (ohne bzw. mit
milden Symptomen) empfunden wurde. Diese Art der Evaluierung ist anspruchsvoller, da hier
jeweils das am schlechtesten bewertete Symptom für die Analyse berücksichtigt wird.
Ergänzend dazu wurde in dieser Studie auch die Gesamtheit aller Symptome anhand des
„mean scores“ bewertet.
Die Durchführung der Studie scheint professionell geplant gewesen zu sein und es gibt genaue
Angaben zur statistischen Analyse.
Der prozentuelle Anteil an „angenehmen Tagen“ lag in der Kombinations-Gruppe mit Cetirizin
und Pseudoephedrin bei 51% und in der Cetirizin-Gruppe bei 40%. Der Unterschied, der durch
die Addition von Pseudoephedrin erreicht wird liegt hier demnach bei 11%. Rechnet man den
prozentuellen Anteil auf die Tage um, ergibt sich der Gewinn von 1,2 „angenehmen Tagen“
bei einem Zeitraum von 12 Tagen. Zusätzlich wäre es bei dieser Studie interessant gewesen,
welcher Anteil dieses Effekts allein durch die Gabe von Placebo erreicht worden wäre.
Zusammenfassend zeigt auch diese Studie zwar einen signifikanten, allerdings auch nur
klinisch geringen Nutzen für die Kombination.

45
4.1.5 The efficacy and safety of fexofenadine HCl and pseudoephedrine, alone and
in combination, in seasonal allergic rhinitis (Sussman et al. 1999) Ziel dieser Studie war es, die Effektivität und Sicherheit einer Arzneimittelkombination
bestehend aus Fexofenadin und Pseudoephedrin zur Behandlung saisonaler allergischer
Rhinitis im Vergleich zu den Einzelsubstanzen zu untersuchen. Diese randomisierte,
doppelblinde Studie wurde an verschiedenen Untersuchungszentren in Kanada durchgeführt
und erhielt finanzielle Unterstützung von „Hoechst Marion Roussel“.
Voraussetzung für die Teilnahme an dieser Studie war ein Alter von 12 bis 65 Jahren, ein
positiv ausfallender Prick-Test, der die bestehende Erkrankung an saisonaler allergischer
Rhinitis bestätigte sowie Evidenz für das Ansprechen von Antihistaminika zur Behandlung der
durch saisonale allergische Rhinitis verursachten Symptome.
Nach anfänglichem Screening folgte die Placebo-Phase, in welcher die Patient*innen für drei
bis fünf Tage Placebo-Tabletten einnahmen. Dies hatte die Ausscheidung von „Placebo-
Respondern“ und die Bestimmung der Basiswerte des TSS zum Zweck. 651 Teilnehmer*innen
erfüllten beim zweiten Screening die Voraussetzungen für die Teilnahme und wurden
daraufhin einer der drei Untersuchungsgruppen zugeteilt. Sieben bis zehn Tage nach der
Einnahme der Arzneimittel mussten die Teilnehmer*innen für einen dritten Besuch zum
Untersuchungszentrum und nach weiteren sieben bis zehn Tagen erneut zur Vollendung der
Studie erscheinen. Insgesamt nahmen die Patient*innen die jeweiligen Arzneimittel somit für
einen Zeitraum von 14 bis 20 Tagen ein. Die Einteilung in die verschiedenen
Untersuchungsgruppen war wie folgt: 215 Teilnehmer*innen erhielten die Kombination
bestehend aus 60 mg Fexofenadin und 120 mg Pseudoephedrin mit verzögerter Freisetzung,
218 erhielten die Monotherapie mit 60 mg Fexofenadin und 218 erhielten die Monotherapie
mit Pseudoephedrin mit kontrollierter Freisetzung. Die Arzneimittel wurden jeweils morgens
und abends um 7 Uhr bzw. 19 Uhr eingenommen. Fexofenadin bzw. Pseudoephedrin wurde
in Form von Kapseln verabreicht, wohingegen die Arzneimittelkombination als Tablette
verabreicht wurde. Um die vollständige Verblindung beiderseits zu garantieren, wurden daher
jeweils zusätzlich zum Arzneimittel eine Placebo-Tablette bzw. eine Placebo-Kapsel
eingenommen. Von den anfänglichen 651 Studienteilnehmer*innen beendeten 588 die Studie.
Das entspricht einem Follow-Up von 90%. Der prozentuelle Anteil an Studienteilnehmer*innen,
welche die Studie beendeten, war wie folgt: 94% in der Kombinations-Gruppe, 93% in der
Fexofenadin-Gruppe sowie 84% in der Pseudoephedrin-Gruppe.
Zur Bewertung der Effektivität der Arzneimittel zeichneten die Teilnehmer*innen ihre
Symptome einmal täglich um 19 Uhr direkt vor der Einnahme des jeweiligen Arzneimittels auf.
Bewertet wurden hier folgende fünf Symptome: nasale Obstruktion, Niesen, Rhinorrhoe,
juckende Nase, juckender Gaumen bzw. Rachen, sowie juckende oder tränende Augen. Die

46
Proband*innen evaluierten ihre Symptome über den Zeitraum der vergangenen 12 h (reflective
score), sowie zum Evaluierungszeitpunkt (instantaneous score). Außerdem wurden die
Symptome zur Schlafenszeit (bedtime TSS) einmalig nach Einnahme der ersten Placebo
Tablette bzw. der ersten Arzneimitteldosis aufgezeichnet. Zur Bewertung der Stärke der
Symptome wurde eine fünfteilige Skala verwendet und aus dieser wurde der TSS aus der
Summe der fünf Symptome errechnet. Der Basiswert des reflective TSS – NCS war zu Beginn
wie folgt: 7,84 in der Arzneimittelkombinations-Gruppe, 8,19 in der Fexofenadin-Gruppe und
7,97 in der Pseudoephedrin-Gruppe. Der reflective NCS lag zu Beginn in der Kombinations-
Gruppe bei 2,32, in der Fexofenadin-Gruppe bei 2,36 und in der Pseudoephedrin-Gruppe bei
2,34.
Zusätzlich zu den oben angeführten Angaben wurden die Studienteilnehmer*innen gebeten,
die Auswirkungen der Arzneimitteleinnahme auf ihr tägliches Leben und auf die Arbeit vor
Beginn der Studie und zum Zeitpunkt der Vollendung zu bewerten. Folgende Parameter
wurden hierfür evaluiert: verpasste Schul-/Arbeitszeit, Produktivität bei der Arbeit bzw. in der
Schule, generelle Produktivität und Beeinträchtigung des täglichen Lebens.
Es gab in dieser Studie zwei primäre Endpunkte: Die Veränderung vom Basiswert des
reflective TSS ausgenommen der nasalen Obstruktion (TSS – NCS) und die Veränderung vom
Basiswert des reflective NCS. Die sekundären Endpunkte wurden in Form der Veränderungen
des instantaneous TSS – NCS, der Veränderungen des instantaneous NCS, der Werte für die
individuellen Symptome, der bedtime TSS – NCS sowie der bedtime NCS festgelegt.
Als Grundlage zur Analyse der primären und sekundären Endpunkte diente die ITT-
Population. Die Basiswerte wurden mittels ANOVA und die Effektivitätsparameter mittels
Kovarianzanalyse (ANCOVA) berechnet.
Die Arzneimittelkombination bestehend aus Fexofenadin und Pseudoephedrin führte zu einer
statistisch signifikant (p<0,0001) höheren Reduktion des reflective TSS (ausgenommen nasale
Obstruktion) im Vergleich zu Pseudoephedrin allein. Der Unterschied zur Monotherapie mit
Fexofenadin war nicht statistisch signifikant (p=0,1579). Die Reduktion des reflective TSS -
NCS lag in der Arzneimittelkombinations-Gruppe bei 2,32, in der Fexofenadin-Gruppe bei 2,05
und in der Pseudoephedrin-Gruppe bei 1,42 Punkten.
Die Reduktion des reflective NCS, die durch die Arzneimittelkombination erreicht wurde, lag
bei 0,56 Punkten und war statistisch signifikant (p<0,0005) größer als die Reduktion durch die
Therapie mit Fexofenadin allein (0,36). Der Unterschied der Kombination beider Arzneistoffe
zur Monotherapie mit Pseudoephedrin war nicht statistisch signifikant (0,45, p=0,0590).
Die Kombinationstherapie bestehend aus Fexofenadin und Pseudoephedrin führte zu einer
statistisch signifikant größeren Reduktion der folgenden Symptome im Vergleich zur
Monotherapie mit Pseudoephedrin: Niesen (p<0,0001), Rhinorrhoe (p=0,0002), juckende
Nase, juckender Gaumen bzw. Rachen (p=0.0002), und juckende oder tränende Augen

47
(p=0.0006). Außerdem war die Kombinationstherapie der Monotherapie mit Fexofenadin zur
Reduktion der nasalen Verstopfung statistisch signifikant überlegen (p=0,0005).
Die Veränderungen des instantaneous TSS – NCS und des instantaneous NCS fielen ähnlich
zu den oben genannten Ergebnissen aus. Die Reduktion des instantaneous TSS
(ausgenommen nasale Verstopfung) und des bedtime TSS (ausgenommen nasale
Verstopfung), die durch die Arzneimittelkombination erreicht wurde, war statistisch signifikant
(p=0,0001 und p=0,0341) größer als die Reduktion durch Pseudoephedrin allein. Der
Unterschied zu Fexofenadin allein war nicht statistisch signifikant. Die Kombination von
Fexofenadin und Pseudoephedrin war der Monotherapie mit Fexofenadin, nicht jedoch der
Monotherapie mit Pseudoephedrin, im Hinblick auf die Reduktion des instantaneous NCS
statistisch signifikant (p=0,0007) überlegen. Der bedtime NCS wurde durch die Kombination
ebenfalls stärker reduziert als durch Fexofenadin allein. Hierbei war der Unterschied jedoch
nicht statistisch signifikant (p=0,1939).
Zu Beginn der Studie lag der Wert für die Beeinträchtigung des täglichen Lebens in allen
Untersuchungsgruppen bei 44%. Nach Abschluss der Studie sank dieser Wert in der
Kombinations-Gruppe um 13%, in der Fexofenadin-Gruppe um 9,8% und in der
Pseudoephedrin-Gruppe um 7,9%. Die Reduktion, welche durch die Kombination der beiden
Arzneistoffe erreicht wurde, war der Monotherapie mit Pseudoephedrin statistisch signifikant
(p=0,006) überlegen. Der Unterschied zur Monotherapie mit Fexofenadin war nicht statistisch
signifikant.
Während des Studienzeitraums kam es in der Untersuchungsgruppe mit Fexofenadin-
Pseudoephedrin bzw. Pseudoephedrin allein zu statistisch signifikant (p<0,001) mehr Fällen
von unerwünschten Arzneimittelwirkungen im Vergleich zur Untersuchungsgruppe mit
Fexofenadin. Die häufigsten unerwünschten Arzneimittelwirkungen waren Kopfschmerzen
(Kombinations-Gruppe 9,3%, Fexofenadin-Gruppe 7,3%, Pseudoephedrin-Gruppe 12,4%)
und Schlaflosigkeit (Kombinations-Gruppe 11,2%, Fexofenadin-Gruppe 1,8%, Pseudo-
ephedrin-Gruppe 12,8%). Während der gesamten Studiendauer veränderten sich die
klinischen Laborparameter, Elektrokardiogramme sowie Vitalparameter in nicht klinisch
relevantem Ausmaß.
Die Autor*innen schließen aus den Ergebnissen dieser Studie, dass die Histamin-vermittelten
Symptome durch die Arzneimittelkombination im Vergleich mit Pseudoephedrin allein stärker
reduziert werden konnten. Des Weiteren sei die Arzneimittelkombination der Monotherapie mit
Fexofenadin zur Reduktion des NCS überlegen. Die Veränderungen der Werte, die am Ende
des Dosierungsintervalls aufgezeichnet wurden (instantaneous TSS – NCS und instantaneous
NCS), lieferten ähnliche Ergebnisse und würden das empfohlene zweimal tägliche
Dosierungsschema bestärken. Auch bei der Bewertung der einzelnen Symptome kamen die
Autor*innen zu dem Schluss, dass die Kombinationstherapie der Monotherapie mit Ausnahme

48
der nasalen Obstruktion in allen Aspekten statistisch signifikant überlegen sei. Beim Vergleich
der Kombinationstherapie mit Fexofenadin sei diese im Hinblick auf die Reduktion der
verstopften Nase ebenfalls statistisch signifikant überlegen. Die vermehrte Anhäufung von
Fällen von Schlaflosigkeit in der Kombinations-Gruppe sowie in der Pseudoephedrin-Gruppe
sei auf die Wirkung auf das zentrale Nervensystem von Pseudoephedrin zurückzuführen.
Außerdem traten Kopfschmerzen in der Pseudoephedrin-Gruppe vermehrt auf. Dies sei
jedoch in früheren Studien ebenfalls bemerkt worden. Abgesehen von diesen Nebenwirkungen
sei Pseudoephedrin generell als ein sicheres Arzneimittel anzusehen und in vielen Ländern
der Welt rezeptfrei zu erhalten. Die Autor*innen vertreten die Meinung, dass es möglich sei
durch die Kombination von Fexofenadin und Pseudoephedrin eine höhere Compliance bei den
Patient*innen zu erreichen, da diese nur ein Medikament einnehmen müssten. Außerdem
könne sich dies positiv auf die Kosten für die Patient*innen auswirken, weil nur ein Präparat
gekauft werden müsste. Die Verbesserung der Lebensqualität und die Verminderung der
Arbeitsbeeinträchtigungen seien mit der Arzneimittelkombination ebenfalls größer, was zum
Schluss führe, dass die Kombination der Monotherapie vorzuziehen sei.
Der Jadad-Score für diese Studie beträgt 4 und ist somit „gut“. In dem Text zu dieser Studie
gab es keine Angaben über eine etwaige Registrierung. Auf der Registrierungsplattform für
klinische Studien „Clinical Trials“ (https://clinicaltrials.gov) war die Studie nicht auffindbar.
Anfangs wird erwähnt, dass die Studie in Übereinstimmung mit den „Canadian Guidelines“
durchgeführt wurde. Die Prüfer*innen erhielten die Zustimmung von einem institutionellen
Prüfungsausschuss und jede/jeder Studienteilnehmer*in musste eine schriftliche Einver-
ständniserklärung vor Beginn der Studie ablegen. Darüber hinaus wurde in der Studie auf die
Art der Randomisierung nicht genauer eingegangen.
In dieser Studie gibt es sowohl genaue Angaben über die statistische Berechnung und die
Modelle, die hierfür verwendet wurden, als auch die Absolutwerte der primären Endpunkte
inklusive Abweichungen. Die expliziten Absolutwerte der sekundären Endpunkte sind in
diesem Text nicht angeführt, diese lassen sich jedoch aus der enthaltenen Grafik herauslesen.
Anfangs lag der reflective TSS – NCS in der Kombinations-Gruppe bei 7,84, in der
Fexofenadin-Gruppe bei 8,19 und in der Pseudoephedrin-Gruppe bei 7,97 Punkten. Dieser
wurde durch die Einnahme der Arzneimittel um 2,32, 2,05 bzw. 1,42 Punkte gesenkt. Die
Reduktion des TSS – NCS, die durch die Addition von Pseudoephedrin erreicht wurde, beträgt
demnach 0,27 Punkte.
Der reflective NCS lag zu Beginn in der Kombinations-Gruppe bei 2,32, in der Fexofenadin-
Gruppe bei 2,36 und in der Pseudoephedrin-Gruppe bei 2,34. Durch die Arzneimitteleinnahme
wurde der reflective NCS um 0,56 (auf 1,76), 0,36 (auf 2,00) bzw. 0,45 (auf 1,89) Punkte

49
gesenkt. Durch die Addition von Pseudoephedrin zu Fexofenadin wurde der reflective NCS
somit um zusätzlich 0,2 Punkte gesenkt.
Ob dies für die Patient*innen spürbare Effekte mit sich bringt (d.h. ein um 0,24 Punkte geringer
NCS durch Kombination im Vergleich zu Fexofenadin alleine) und sich daher die Addition eines
Wirkstoffes, welcher ein deutliches Nebenwirkungsspektrum aufweist, lohnt, ist auch in dieser
Studie daher fraglich.
4.1.6 Efficacy and safety of desloratadine/pseudoephedrine tablet, 2.5/120 mg two
times a day, versus individual components in the treatment of patients with
seasonal allergic rhinitis (Chervinsky et al. 2005) Ziel dieser randomisierten, doppelblinden Studie war es, die Effektivität und Sicherheit einer
Kombination von Desloratadin und Pseudoephedrin zur Behandlung saisonaler allergischer
Rhinitis im Vergleich zur Monotherapie mit Desloratadin bzw. Pseudoephedrin zu untersuchen.
Die Studie wurde an 20 Untersuchungszentren in den USA durchgeführt.
Einschlusskriterien für die Teilnahme an der Studie waren eine Erkrankung an saisonaler
allergischer Rhinitis seit mindestens zwei Jahren, ein positiv ausfallender Prick-Test, der nicht
älter als ein Jahr war und die bestehende Erkrankung bestätigte sowie ein Alter von
mindestens 12 Jahren.
650 Studienteilnehmer*innen wurden nach anfänglicher Screening-Phase einer der drei
Untersuchungsgruppen zugeteilt und erhielten eines der folgenden Arzneimittel über einen
Zeitraum von zwei Wochen: 2,5 mg Desloratadin und 120 mg Pseudoephedrin zweimal
täglich, 5 mg Desloratadin einmal täglich, oder 120 mg Pseudoephedrin zweimal täglich. Die
Arzneimittelkombination bestand aus einer Matrixtablette, die Pseudoephedrin kontrolliert
freisetzte. Die Verteilung in den drei Gruppen war wie folgt: 214 Teilnehmer*innen erhielten
das Kombinations-Präparat, 214 erhielten Desloratadin und 222 erhielten Pseudoephedrin.
604 (93%) Studienteilnehmer*innen beendeten die zweiwöchige Studie. 200 (94%) davon
wurden mit Desloratadin-Pseudoephedrin behandelt, 200 (94%) erhielten Desloratadin und
204 (92%) nahmen Pseudoephedrin ein.
Am ersten Screening-Tag bewerteten alle Studienteilnehmer*innen die Intensität ihrer
Symptome anhand einer vierteiligen Skala. Hierbei wurden nicht-nasale (juckende bzw.
brennende Augen, tränende Augen, juckender Gaumen, juckende Ohren) sowie nasale
Symptome (Rhinorrhoe, Niesen, juckende Nase, nasale Verstopfung) bewertet. Während der
Studie zeichneten alle Teilnehmer*innen ihre Symptome zweimal täglich mithilfe eines
Tagebuchs auf. Beurteilt wurden hier die vergangenen 12 Stunden (reflective) und der Zustand
zum Evaluierungszeitpunkt (instantaneous). Diese Aufzeichnungen dienten zur Berechnung
des TSS.

50
Die primären Endpunkte dieser Studie wurden als die mittleren Veränderungen des reflective
TSS (ausgenommen nasale Verstopfung) vom Basiswert im Vergleich zu Pseudoephedrin,
sowie die mittleren Veränderungen des NSS vom Basiswert im Vergleich zu Desloratadin
festgelegt. Die sekundären Endpunkte waren der TSS (inklusive nasale Verstopfung), der
TNSS und der TNNSS.
Die Basiswerte für den TSS (ausgenommen nasale Verstopfung) lagen in der Kombinations-
Gruppe bei 15,2, in der Desloratadin-Gruppe bei 14,7 und in der Pseudoephedrin-Gruppe bei
14,9 Punkten. Die Werte für den TNSS lagen anfangs bei 6,9 in der Untersuchungsgruppe mit
Desloratadin-Pseudoephedrin, 6,8 in der mit Desloratadin und 6,8 Punkten in der mit
Pseudoephedrin. Der TNNSS lag bei 8,3 (Desloratadin-Pseudoephedrin), 7,9 (Desloratadin)
und 8,1 (Pseudoephedrin) Punkten.
Die Effektivitätsparameter wurden mittels der Zwei-Wege-Varianzanalyse (two-way ANOVA)
ausgewertet.
Nach zweiwöchiger Einnahme der Arzneimittel war die Kombination bestehend aus
Desloratadin und Pseudoephedrin der Monotherapie mit Desloratadin bzw. der Monotherapie
mit Pseudoephedrin zur Reduktion des reflective TSS (ausgenommen nasale Verstopfung)
statistisch signifikant (p≤0,001) überlegen. Die Reduktion des reflective TSS lag in der
Kombinations-Gruppe bei 6,7 (43%), in der Desloratadin-Gruppe bei 5,4 (36%) und in der
Pseudoephedrin-Gruppe bei 5,3 (35%) Punkten.
Die Einnahme der Kombination von Desloratadin und Pseudoephedrin führte zu einer
statistisch signifikant (p=0,005) größeren Reduktion des reflective NSS im Vergleich zu
Desloratadin, jedoch nicht im Vergleich zu Pseudoephedrin als Monotherapie. Die Reduktion,
welche durch die Kombination erreicht wurde, lag bei 0,92 (36%) Punkten. Durch die
Monotherapie mit Desloratadin wurde eine Reduktion des reflective NSS von 0,73 (29%)
Punkten und mit Pseudoephedrin von 0,83 (32%) Punkten erreicht.
Der reflective TNSS (ausgenommen nasale Verstopfung) wurde durch die Einnahme von
Desloratadin-Pseudoephedrin um 3,0 (42%), durch Desloratadin um 2,4 (35%) und durch
Pseudoephedrin um 2,3 (34%) Punkte gesenkt. Somit war die Kombinationstherapie den
Monotherapien statistisch signifikant (p≤0,001) überlegen.
Während der Studie wurde der reflective TNNSS durch die Einnahme der Kombinations-
therapie um 3,7 (44%) Punkte gesenkt. Diese Reduktion war statistisch signifikant größer als
jene, die durch die Einnahme von Desloratadin allein (3,0, 37%, p=0,003) und durch die
Einnahme von Pseudoephedrin allein (3,0, 37%, p=0,001) erreicht wurde.
Beim Vergleich des reflective TSS (inklusive nasale Verstopfung) zeigte sich eine statistisch
signifikante (p=0,001) Überlegenheit der Arzneimittelkombination bestehend aus Desloratadin
und Pseudoephedrin im Vergleich zur Monotherapie mit Desloratadin oder Pseudoephedrin.
Der reflective TSS wurde durch die Kombinationstherapie um 7,6 (42%) Punkte gesenkt,

51
während die Monotherapie mit Desloratadin sowie die Monotherapie mit Pseudoephedrin nur
zu einer Reduktion von 6,1 (35%) Punkten führten.
Während der Studie kam es in der Untersuchungsgruppe mit Desloratadin-Pseudoephedrin
zu unerwünschten Arzneimittelwirkungen bei 41% der Teilnehmer*innen. In der Desloratadin-
Gruppe wurden von 37% und in der Pseudoephedrin-Gruppe von 42% der Teilnehmer*innen
über Nebenwirkungen berichtet. Zu den häufigsten unerwünschten Arzneimittelwirkungen
zählten Schlaflosigkeit (10%), Kopfschmerzen (8%) und Xerostomie (6%). Durch die
Einnahme der Medikamente kam es zu keinen klinisch relevanten Veränderungen der
Vitalparameter, Elektrokardiogramme oder Laborparametern.
Aus den Ergebnissen dieser Studie schließen die Autor*innen, dass die Kombination eines
Antihistaminikums und eines abschwellenden Arzneistoffes eine effektive und gut verträgliche
Therapiemöglichkeit bei saisonaler allergischer Rhinitis sei. Durch die Kombination reduziere
man die Gesamtheit der Symptome einer saisonalen allergischen Rhinitis bei vergleichbarem
Nebenwirkungsspektrum wie bei Einnahme der Einzelsubstanzen. Zusammengefasst sei der
Gebrauch der Arzneimittelkombination von Desloratadin (2,5 mg) und Pseudoephedrin
(120 mg) bei Patient*innen mit saisonaler allergischer Rhinitis zu empfehlen.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Das
Prüfungsprotokoll der Studie wurde in jedem Untersuchungszentrum vom dort zuständigen
Prüfungsausschuss bewilligt und jede/jeder Studienteilnehmer*in stimmte dem Ablauf der
Studie durch eine schriftliche Einwilligungserklärung zu. Auf der internationalen
Registrierungsplattform für klinische Studien „Clinical Trials“ (https://clinicaltrials.gov) war die
Studie nicht zu finden und es gibt im Studienprotokoll keine Angaben zu einer Registrierung.
Des Weiteren gab es keine genauere Beschreibung, wie die Randomisierung der
Studienteilnehmer*innen in der Studie erfolgte.
Es gab in der Studie keine Angaben dazu, ob die Monotherapie mit Pseudoephedrin ebenfalls
als Tablette mit kontrollierter Freisetzung verabreicht wurde. Außerdem wurde nicht
beschrieben, wie die vollständige Verblindung beider Seiten sichergestellt wurde. Die
Monotherapie mit Desloratadin wurde nur einmal täglich verabreicht, wohingegen die
Monotherapie mit Pseudoephedrin als auch das Kombinationspräparat zweimal täglich
eingenommen wurde. Wichtig wäre, zu wissen, ob die Untersuchungsgruppe, die Desloratadin
einnahm, abends eine Placebo-Tablette erhielt. Wäre dies nicht der Fall, wäre keine
vollständige Verblindung garantiert.
Der TSS wurde durch die Einnahme der Medikamente in der Kombinations-Gruppe von 15,2
auf 8,5, in der Desloratadin-Gruppe von 14,7 auf 9,3 und in der Pseudoephedrin-Gruppe von
14,9 auf 9,6 Punkte gesenkt. Durch die Zugabe von Pseudoephedrin zu Desloratadin ergibt

52
sich somit ein additiver Nutzen von 0,8 Punkten bei der Reduktion des reflective TSS
(ausgenommen nasale Verstopfung).
In der Studie gibt es keinerlei Angaben zu den Basiswerten des NSS. Anhand der angeführten
Prozentangaben lässt sich jedoch der Basiswert ermitteln. Der NSS wurde folglich in der
Gruppe mit Desloratadin-Pseudoephedrin von 2,6 auf 1,68, in der Gruppe mit Desloratadin
von 2,5 auf 1,77 und in der Gruppe mit Pseudoephedrin von 2,6 auf 1,77 Punkte reduziert.
Hier ergibt sich durch die Addition von Pseudoephedrin zur Monotherapie mit Desloratadin ein
zusätzlicher Nutzen von 0,09 Punkten.
Das Nebenwirkungsspektrum wurde durch die Addition von Pseudoephedrin kaum merklich
vergrößert. Dies würde die Anwendung einer Kombinationstherapie bekräftigen, jedoch ist
fraglich, ob der Unterschied von 0,8 Punkten, bzw. 0,09 Punkten für die
Studienteilnehmer*innen als klinisch relevant wahrnehmbar ist.
4.1.7 Efficacy and safety of desloratadine/pseudoephedrine combination vs its
components in seasonal allergic rhinitis (Grubbe et al. 2009) Ziel dieser Studie war es, zu untersuchen, ob eine Arzneimittelkombination bestehend aus
Desloratadin und Pseudoephedrin zu einer effektiveren Symptomreduktion bei saisonaler
allergischer Rhinitis führte, als die Einzelsubstanzen. Zusätzlich dazu wurde die Sicherheit
dieser Arzneimittelkombination auf Grundlage der von den Teilnehmer*innen berichteten
unerwünschten Arzneimittelwirkungen und von Vitalparametern, Elektrokardiogrammen und
Laborparametern bewertet. Die Studie wurde zur Pollenzeit im Herbst in mehreren
Untersuchungszentren in den USA durchgeführt und erhielt redaktionelle sowie finanzielle
Unterstützung von „Schering Plough“.
Voraussetzung für die Teilnahme an dieser randomisierten, doppelblinden Studie war eine
vorliegende Erkrankung an saisonaler allergischer Rhinitis seit mindestens zwei Jahren, die
durch einen positiven Prick-Test bestätigt werden musste, ein ansonsten guter
Gesundheitszustand sowie ein Alter von mindestens 12 Jahren.
Nach 3- bis 14-tägiger Eingangsphase wurden 598 Patient*innen randomisiert und erhielten
die Kombination aus 2,5 mg Desloratadin und 120 mg Pseudoephedrin zweimal täglich. Die
Monotherapiegruppen erhielten einmal täglich 5 mg Desloratadin bzw. zweimal täglich 120 mg
Pseudoephedrin. Die entsprechenden Arzneimittel wurden morgens (2 Tabletten) und abends
(1 Tablette) für einen Zeitraum von 15 Tagen eingenommen. Um die vollständige Verblindung
beider Seiten zu gewährleisten, wurden fehlende Tabletten im Dosierungsschema durch
Placebo-Tabletten ergänzt. Die Arzneimittelkombination war aus einem Tablettenkern, der
Pseudoephedrin kontrolliert freisetzt, und einer Tablettenhülle mit Desloratadin
zusammengesetzt. Die Proband*innen wurden wie folgt eingeteilt: 200 Personen erhielten
Desloratadin-Pseudoephedrin, 198 Personen Desloratadin, und 200 Pseudoephedrin. Die

53
Teilnehmer*innen mussten an folgenden Tagen zum Untersuchungszentrum: Screening, Tag
1, Tag 8 und Tag 15. Von den 598 randomisierten Studienteilnehmer*innen beendeten 561
die zweiwöchige Studie. Das entspricht einem Follow-Up von 93,8%. Davon erhielten 189
Personen Desloratadin-Pseudoephedrin, 191 Personen Desloratadin, und 181
Pseudoephedrin.
Die Studienteilnehmer*innen zeichneten die Intensität verschiedener nasaler Symptome
(Rhinorrhoe, verstopfte Nase, Niesen, juckende Nase) sowie nicht-nasaler Symptome
(juckende oder brennende Augen, tränende Augen, juckende Ohren oder juckender Gaumen)
anhand einer vierteiligen Skala zweimal täglich auf. Die Proband*innen evaluierten ihre
Symptome für den Zeitraum der vergangenen 12 h (reflective score) sowie zum
Evaluierungszeitpunkt (instantaneous score). Aus den Aufzeichnungen der Patient*innen
wurde der reflective TSS (ausgenommen nasale Verstopfung) errechnet. Dieser lag zu Beginn
der Studie bei 14,18 in der Desloratadin-Pseudoephedrin-Gruppe, bei 14,83 in der
Desloratadin-Gruppe und bei 14,06 Punkten in der Pseudoephedrin-Gruppe. Die Werte des
reflective NCS lagen bei 2,47 in der Untersuchungsgruppe mit Desloratadin-Pseudoephedrin,
bei 2,50 in der mit Desloratadin und bei 2,45 Punkten in der mit Pseudoephedrin.
Zur Beurteilung der Effektivität der Arzneimittel wurden zwei primäre Endpunkte festgelegt.
Der primäre Endpunkt des antiallergisch wirksamen Bestandteils in der
Arzneimittelkombination Desloratadin-Pseudoephedrin war die Veränderung des mittleren
reflective TSS (ausgenommen nasale Verstopfung) vom Basiswert im Vergleich zu
Pseudoephedrin. Der primäre Endpunkt des abschwellend wirkenden Bestandteils in der
Arzneimittelkombination Desloratadin-Pseudoephedrin war die Veränderung des mittleren
reflective NCS vom Basiswert im Vergleich zu Desloratadin.
Die sekundären Endpunkte in dieser Studie waren der instantaneous TSS, der TNSS, der
TNNSS, die Intensität der individuellen Symptome, der generelle Gesundheitszustand und das
Ansprechen auf die Therapie.
Die Effektivitätsparameter wurden mittels der Zwei-Wege-Varianzanalyse (two-way ANOVA)
ausgewertet.
Während der 15-tägigen Untersuchungsphase war die Arzneimittelkombination bestehend aus
Desloratadin und Pseudoephedrin der Monotherapie mit Desloratadin bzw. jener mit
Pseudoephedrin zur Reduktion des reflective TSS (ausgenommen nasale Obstruktion)
statistisch signifikant (p<0,001) überlegen. Die Reduktion, welche durch die Arzneimittel-
einnahme erreicht wurde, lag in der Kombinations-Gruppe bei 6,54 (46%), in der Desloratadin-
Gruppe bei 5,09 (34%) und in der Pseudoephedrin-Gruppe bei 5,07 (36%) Punkten.
Der reflective NCS wurde durch die Einnahme von Desloratadin-Pseudoephedrin um 0,93
(37%), durch Desloratadin um 0,66 (27%) und durch Pseudoephedrin um 0,75 (31%) Punkte

54
gesenkt. Somit war die Kombination der Monotherapie mit Desloratadin (p<0,001), aber nicht
der Monotherapie mit Pseudoephedrin (p=0,006) statistisch signifikant überlegen.
Durch die Einnahme der Arzneimittelkombination wurde eine größere Reduktion des
instantaneous TSS (ausgenommen nasale Obstruktion) erreicht als durch die Einnahme von
Desloratadin bzw. Pseudoephedrin allein. Durch die Einnahme von Desloratadin in
Kombination mit Pseudoephedrin wurde der instantaneous TSS um 6,27 (45%), durch
Desloratadin um 4,92 (36%: p<0,001) und durch Pseudoephedrin um 5,19 (35%, p=0,011)
Punkte reduziert.
Bei Beurteilung der Reduktion des instantaneous NCS wurde eine Überlegenheit der
Arzneimittelkombination gegenüber den Einzelsubstanzen beobachtet. Die Reduktionen lagen
bei 0,81 (33%) durch Desloratadin-Pseudoephedrin, bei 0,60 (23%, p=0,002) durch
Desloratadin und 0,66 (28%, p=0,032) Punkten durch Pseudoephedrin.
Bei der Bewertung des generellen Gesundheitszustandes durch die Patient*innen schnitt
Desloratadin-Pseudoephedrin mit einer Reduktion der Symptome von 1,06 (40%) besser ab
als Desloratadin mit einer Reduktion von 0,95 (34%) oder als Pseudoephedrin mit einer
Reduktion von 0,91 (34%) Punkten. Nur beim Vergleich von Desloratadin-Pseudoephedrin mit
Pseudoephedrin wurde statistische Signifikanz (p=0,062) beobachtet.
Während der gesamten Dauer der Studie wurde über keine schwerwiegenden oder
unerwarteten unerwünschten Arzneimittelwirkungen berichtet. Die häufigsten unerwünschten
Arzneimittelwirkungen waren Xerostomie (10%), Schlaflosigkeit (10%) und Kopfschmerzen
(7%) in der Untersuchungsgruppe, die Desloratadin in Kombination mit Pseudoephedrin
einnahm. Auf ähnliche Ergebnisse kam man in der Gruppe mit Pseudoephedrin: Xerostomie
(8%), Schlaflosigkeit (14%) und Kopfschmerzen (12%). In der Gruppe, die Desloratadin
einnahm, kam es zu weniger Fällen von Nebenwirkungen mit einer Häufigkeit von 2% für
Xerostomie, 3% für Schlaflosigkeit und 7% für Kopfschmerzen. Durch die Einnahme der
Arzneimittel kam es zu einem Anstieg der Herzfrequenz in der Desloratadin-Pseudoephedrin-
Gruppe von 3,9 Schlägen pro Minute und in der Pseudoephedrin-Gruppe von 3,0 Schlägen
pro Minute. Über die gesamte Studiendauer hinweg wurden keine klinisch relevanten
Veränderungen der Vitalparameter, Elektrokardiogramme sowie der klinischen Labor-
parameter beobachtet.
Die antihistaminische Wirkung der Kombination war während der gesamten Studie der
Monotherapie mit Pseudoephedrin überlegen. Die abschwellende Wirkung, die durch die
Kombination erreicht wurde, war größer als jene, welche durch die Monotherapie mit
Desloratadin erreicht werden konnte. Zu ähnlichen Ergebnissen kam man bei der Beurteilung
der individuellen Symptome. Die Kombination der Arzneistoffe wurde generell gut vertragen
und zeigte sich als ein sicheres Arzneimittel zur Behandlung von saisonaler allergischer
Rhinitis. Die Autor*innen schließen daher aus den Ergebnissen dieser Studie, dass die

55
Kombinationstherapie aus Desloratadin und Pseudoephedrin eine zusätzlich anti-
histaminische bzw. abschwellende Wirkung im Vergleich zu den Einzelsubstanzen besitzt und
aus diesem Grund die Resultate von früheren Studien bekräftigen würde. Schlussfolgernd
halten die Autor*innen fest, dass die Kombination bestehend aus Desloratadin und
Pseudoephedrin eine wichtige Alternative für Patient*innen mit saisonaler allergischer Rhinitis
sei, die ihre nasale Obstruktion durch die Einnahme von Desloratadin allein nicht ausreichend
reduzieren könnten.
Der Jadad-Score für diese Studie beträgt „4“ und ist somit „gut“. Es gab in der Studie keine
Angaben zu einer Bewilligung des Studienprotokolls durch einen Prüfungsausschuss sowie zu
einer erfolgten schriftlichen Einwilligungserklärung seitens der Teilnehmer*innen. Des
Weiteren war die Studie auf der internationalen Registrierungsplattform für klinische Studien
„Clinical Trials“ (https://clinicaltrials.gov) nicht auffindbar. Auf die Details, wie die
Randomisierungsliste in dieser Studie generiert wurde wird hier ebenfalls nicht eingegangen.
Obwohl die Bewertung der Veränderungen des TNSS und des TNNSS zu den sekundären
Endpunkten in dieser Studie gehören, werden diese bei den Ergebnissen nicht angeführt.
Abgesehen davon gibt es in dieser Studie ausreichend Informationen zu den Absolutwerten
und den statistischen Modellen, die zur Berechnung der Ergebnisse angewandt wurden. Die
Studie scheint in Summe von guter Qualität zu sein und gewissenhaft durchgeführt worden zu
sein.
Wichtig für die Beurteilung der Wirksamkeit eines Arzneimittels ist die Symptomreduktion, die
von den Patient*innen beobachtet wurde. In dieser Studie wurde die Verbesserung des
Gesundheitszustandes bzw. der Symptomatik der saisonalen allergischen Rhinitis durch die
Studienteilnehmer*innen selbst evaluiert. Hierbei zeigte sich im Vergleich zu Pseudoephedrin
jedoch nur eine geringe, wenngleich statistisch signifikante, Überlegenheit der Arzneimittel-
kombination. Daher ist fraglich, ob die Patient*innen tatsächlich von der Addition von
Pseudoephedrin bei der Behandlung von saisonaler allergischer Rhinitis profitieren.

56
4.1.8 SCH 434: A new antihistamine/decongestant for seasonal allergic rhinitis
(Storms et al. 1989) Ziel dieser Studie war es, die Effekte einer Arzneimittelkombination namens „SCH 434“, die
aus Loratadin und Pseudoephedrin besteht, und von Loratadin zur Behandlung von
Patient*innen mit saisonaler allergischer Rhinitis zu untersuchen. Dazu wurde die Effektivität
und Sicherheit dieser Arzneimittelkombination mit der Monotherapie mit Loratadin oder
Pseudoephedrin, sowie mit Placebo verglichen. Diese randomisierte, doppelblinde und
placebokontrollierte Studie wurde an sieben Untersuchungszentren in den USA zur Pollenzeit
im August bis Oktober durchgeführt und von der „Schering-Plough Corporation“ gesponsert.
Um an dieser Studie teilnehmen zu können, mussten die Patient*innen seit mindestens zwei
Jahren an saisonaler allergischer Rhinitis erkrankt sein und einen positiven Hauttest
vorweisen, der diese Erkrankung bestätigte.
444 Proband *innen wurden nach anfänglicher Screening-Phase zur Teilnahme ausgewählt
und anhand einer computergenerierten Randomisierungsliste einer der vier Untersuchungs-
gruppen zugewiesen. Die erste Untersuchungsgruppe erhielt „SCH 434“ und somit 5 mg
Loratadin kombiniert mit 120 mg Pseudoephedrin, die zweite erhielt 5 mg Loratadin, die dritte
erhielt 120 mg Pseudoephedrin und die vierte Gruppe erhielt Placebo. Alle Arzneimittel wurden
zweimal täglich über einen Zeitraum von 14 Tagen eingenommen. „SCH 434“ und die
Monotherapie mit Pseudoephedrin wurden in Tablettenform verabreicht, Loratadin wurde in
Kapselform verabreicht und Placebo in Tabletten- oder Kapselform. Um die vollständige
Verblindung beider Seiten zu garantieren, erhielten alle Studienteilnehmer*innen daher
zweimal täglich eine Tablette und eine Kapsel. Alle Teilnehmer*innen wurden an den Tagen
4, 8 und 15 von einer/einem Prüfärztin/Prüfarzt untersucht. 435 Studienteilnehmer*innen
beendeten die zweiwöchige Studie. Das entspricht einem Follow-Up von 98%. 111
Studienteilnehmer*innen erhielten die Arzneimittelkombination bestehend aus Loratadin und
Pseudoephedrin, 109 erhielten die Monotherapie mit Loratadin, 109 erhielten die
Monotherapie mit Pseudoephedrin und 106 erhielten Placebo. 442 Teilnehmer*innen wurden
für die Sicherheitsanalyse eingeschlossen und 435 für die Effektivitätsanalyse.
Die Studienteilnehmer*innen zeichneten die Intensität ihrer Symptome vor Studienbeginn und
während des gesamten Behandlungszeitraums auf. Dazu wurde eine vierteilige Skala
verwendet, aus welcher anschließend der TSS errechnet wurde. Ergänzend dazu wurde das
allgemeine therapeutische Ansprechen auf die Behandlung durch die Patient*innen selbst und
durch die Prüfärzt*innen anhand einer fünfteiligen Skala an den Follow-Up-Terminen
durchgeführt. Dabei stand „1“ für „exzellent“, „2“ für „gut“, „3“ für „mittelmäßig“, „4“ für „schlecht“
und „5“ für „Therapieversagen“. Die Bewertung der Sicherheit der Arzneimittel basierte auf den
beobachteten Veränderungen relativ zu den Basiswerten während der Studiendauer.

57
Der primäre Endpunkt dieser Studie wurde nicht definiert. Die Validität der Studie ist daher
zweifelhaft.
Die Analyse der Effektivitätsparameter wurde mittels der „Zwei-Wege-Varianzanalyse“ (two-
way ANOVA) durchgeführt. Die individuellen Symptome zur Beurteilung des generellen
Ansprechens auf die Therapie wurden zusätzlich mithilfe von linearer Regression auswertet,
um die Ergebnisse der Varianzanalyse zu bestätigen.
Bei der Beurteilung des TSS schnitt die Arzneimittelkombination an Tag 4 statistisch signifikant
(p≤0,03) besser ab als alle anderen Untersuchungsgruppen. Die beiden Monotherapien mit
Loratadin oder Pseudoephedrin waren der Placebo-Gruppe ebenfalls statistisch signifikant
überlegen (p≤0,02). Die Reduktion, welche durch die Einnahme der Arzneimittel erreicht
wurde, lag bei 51% in der Kombinations-Gruppe, bei 42% in der Loratadin-Gruppe, bei 38% in
der Pseudoephedrin-Gruppe und bei 28% in der Placebo-Gruppe. Zum Endpunkt (Tag 15) lag
die Reduktion bei 55% mit Loratadin-Pseudoephedrin, 50% mit Loratadin, 46% mit
Pseudoephedrin und bei 38% mit Placebo. Die Verbesserung des TSS, welche durch die
kombinierte Einnahme von Loratadin-Pseudoephedrin erreicht wurde, war statistisch
signifikant (p≤0,05) besser als jene durch eine alleinige Pseudoephedrin- bzw.
Placebobehandlung. Die Monotherapie mit Loratadin war der Behandlung mit Placebo zum
Endpunkt ebenfalls statistisch signifikant (p≤0,01) überlegen.
Die Gesamtheit aller nasalen Symptome wurde durch die Einnahme von Loratadin-
Pseudoephedrin an Tag 4 um 48%, durch Loratadin um 34%, durch Pseudoephedrin um 35%
und durch Placebo um 24% reduziert. Somit war die Kombination allen anderen Therapien
statistisch signifikant überlegen (p≤0,01). Verglichen mit Placebo waren auch die Loratadin-
und Pseudoephedrin-Gruppe statistisch signifikant (p≤0,02) besser. Zum Endpunkt lagen die
Werte in der Kombinations-Gruppe bei 49%, in der Loratadin-Gruppe bei 43%, in der
Pseudoephedrin-Gruppe bei 43% und in der Placebo-Gruppe bei 33%. Die Therapie mit der
Arzneimittelkombination (p≤0,01) sowie die Monotherapien mit Loratadin (p≤0,05) oder
Pseudoephedrin (p≤0,05) waren der Placebo-Gruppe zur Reduktion aller nasalen Symptome
zum Endpunkt statistisch signifikant überlegen. Der Unterschied zwischen der
Arzneimittelkombination zu den Monotherapien war nicht statistisch signifikant.
Die nicht-nasalen Symptome (gerötete Augen, juckende Augen, tränende Augen und
juckende/r Gaumen/Ohren) wurden durch die Einnahme von Loratadin-Pseudoephedrin an
Tag 4 um 53%, durch Loratadin um 53%, durch Pseudoephedrin um 41% und durch Placebo
um 34% gesenkt. Zum Endpunkt lag die Reduktion, die durch die Einnahme der Medikamente
erreicht wurde, bei 62% in der Kombinations-Gruppe, bei 60% in der Loratadin-Gruppe, bei
49% in der Pseudoephedrin-Gruppe und bei 46% in der Placebo-Gruppe. Sowohl an Tag 4
als auch zum Endpunkt war der Unterschied zwischen den Untersuchungsgruppen mit

58
Loratadin-Pseudoephedrin und Loratadin zur Untersuchungsgruppe mit Pseudoephedrin
sowie zur Untersuchungsgruppe mit Placebo statistisch signifikant (p≤0,01).
Der NSS wurde durch die Einnahme der Kombination an Tag 4 um 42% reduziert. In der
Loratadin-Gruppe lag die Reduktion bei 22%, in der Pseudoephedrin-Gruppe bei 38% und in
der Placebo-Gruppe bei 28%. Die durch die Arzneimittelkombination erreichte Reduktion war
statistisch signifikant (p≤0,01) größer als durch Loratadin oder Placebo, nicht jedoch als jene
durch Pseudoephedrin. Pseudoephedrin war statistisch signifikant besser als Loratadin
(p≤0,05) und geringfügig besser als Placebo (p=0,07). Zum Endpunkt reduzierte die
Arzneimittelkombination den NSS um 42%, die Loratadin-Gruppe um 35%, die
Pseudoephedrin-Gruppe um 42% und die Placebo-Gruppe um 28%. Die Verbesserung des
NSS war durch die Einnahme von Loratadin-Pseudoephedrin sowie durch die Einnahme von
Pseudoephedrin allein statistisch signifikant (p=0,05) besser als durch Placebo. Zwischen
Loratadin und Placebo gab es keinen Unterschied im Hinblick auf die Reduktion des NSS.
Die Autoren geben keine Absolutwerte, sondern nur die Prozentpunkte der Veränderung der
SS an. Somit ist es nicht möglich den Ausgangsschweregrad der Symptome zu Beginn und
damit den absoluten klinischen Nutzen zu beurteilen.
Bei der Beurteilung des allgemeinen Ansprechens auf die Therapie durch die Prüfärzt*innen
schnitt die Arzneimittelkombination an Tag 4 statistisch signifikant (p<0,01) besser ab als alle
anderen Untersuchungsgruppen. Die Werte an Tag 4 waren wie folgt: 2,4 für die Kombination,
2,8 für Loratadin, 2,9 für Pseudoephedrin und 3,2 für Placebo. Zum Endpunkt gab es zwischen
den aktiven Arzneimittelgruppen keine Unterschiede mehr, alle drei waren jedoch der Placebo-
Gruppe statistisch signifikant (p≤0,03) überlegen. Die Werte zum Endpunkt lagen bei 2,5 für
die Kombination, bei 2,7 für Loratadin, bei 2,7 für Pseudoephedrin und bei 3,0 Punkten für
Placebo.
Eine Auswertung des allgemeinen Therapieansprechens durch die Patient*innen selbst ergab
ähnliche Ergebnisse. An Tag 4 war die Arzneimittelkombination allen anderen Untersuchungs-
gruppen statistisch signifikant überlegen. Bei der Analyse zum Endpunkt wurden die Gruppen
Loratadin-Pseudoephedrin, Loratadin und Pseudoephedrin allein besser als die Placebo-
Gruppe bewertet.
Während der Studiendauer kam es zu statistisch signifikant (p=0,01) mehr Fällen von
unerwünschten Arzneimittelwirkungen in der Untersuchungsgruppe mit Loratadin und
Pseudoephedrin im Vergleich zur Placebo- und Loratadin-Gruppe. In der Arzneimittel-
kombinations-Gruppe berichteten 66 (59%) Teilnehmer*innen von unerwünschten Arznei-
mittelwirkungen. In der Loratadin-Gruppe taten dies 42 (38%), in der Pseudoephedrin-Gruppe
55 (50%) und in der Placebo-Gruppe 39 (36%) der Teilnehmer*innen. Die häufigsten
Nebenwirkungen waren Sedierung, Schwindel, Schlaflosigkeit, Nervosität, Xerostomie,
Müdigkeit und Kopfschmerzen. Fälle von Sedierung kamen in allen Untersuchungsgruppen in

59
gleichem Maße vor. In der Kombinations-Gruppe berichteten 8% der Teilnehmer*innen von
derartigen Beschwerden, in der Loratadin-Gruppe 7%, in der Pseudoephedrin-Gruppe 8% und
in der Placebo-Gruppe 5%. Fälle von Nervosität kamen vermehrt mit der Monotherapie von
Pseudoephedrin (7%) vor. In der Kombinations-Gruppe gaben von 5% der Teilnehmer*innen
das Auftreten von Nervosität an, in der Loratadin-Gruppe 4% und in der Placebo-Gruppe 2%.
Schlaflosigkeit kam zu einem statistisch signifikant (p=0,01) häufigeren Anteil in der
Kombinations- (19%) und der Pseudoephedrin-Gruppe (17%) vor. Der Anteil in der Loratadin-
Gruppe lag bei 4% und in der Placebo-Gruppe bei 3%. Über Xerostomie wurde von 24% der
Teilnehmer*innen berichtet, welche die Kombination einnahmen, von 5% in der Loratadin-
Gruppe, von 12% in der Pseudoephedrin-Gruppe und von 4% in der Placebo-Gruppe. Somit
verursachte die Arzneimittelkombination und die Monotherapie mit Pseudoephedrin statistisch
signifikant (p=0,01) mehr Fälle von Xerostomie als Loratadin oder Placebo. Während der
Studie kam es zu einer geringen Veränderung der Laborparameter, diese waren jedoch nicht
klinisch relevant.
Die Autor*innen schließen aus den Ergebnissen dieser Studie, dass das Präparat namens
„SCH 434“ die Symptome von saisonaler allergischer Rhinitis stärker reduziert, als Loratadin
oder Pseudoephedrin allein und besser als Placebo wirkte. Bei der Beurteilung der Ergebnisse
an Tag 4 war die Arzneimittelkombination den anderen Untersuchungsgruppen im Hinblick auf
die Reduktion des TSS, der nasalen Symptome, des NSS, und des nasalen Ausflusses
statistisch signifikant überlegen. Loratadin und Pseudoephedrin allein schnitten ebenfalls
besser als Placebo ab. Die restlichen Daten dieser Studie würden den Vorteil der Kombination
gegenüber den Einzelkomponenten jedoch nicht untermauern. Die Autor*innen nennen als
Beispiel den TSS (nasal und nicht-nasal), der nur an Tag 4 durch die Kombination stärker
reduziert wurde als durch Loratadin allein. Ebenso schnitt die Kombination nur an Tag 4 bei
der Beurteilung des allgemeinen Therapieansprechens besser als die Einzelsubstanzen ab.
Die Ergebnisse der Studie seien jedoch laut den Autor*innen ein Grund für die Anwendung
der Arzneimittelkombination, da diese eine bessere Therapieoption als die Einzelsubstanzen
darstelle. Der Einsatz von Kombinationspräparaten habe den Vorteil, dass die Patient*innen
nur ein Medikament einnehmen müssten und dies zusätzlich billiger als zwei separate
Verschreibungen sei. Darüber hinaus sei „SCH 434“ effektiver als Placebo. Außerdem sei das
Kombinationspräparat effektiver als Loratadin oder Pseudoephedrin zur Reduktion vieler
nasaler Symptome an Tag 4. Abschließend halten die Autor*innen fest, dass das Präparat
„SCH 434“ eine effektive und sichere Therapieoption bei saisonaler allergischer Rhinitis sei.
Der errechnete Jadad Score für diese Studie beträgt 5 und ist somit „exzellent“. Allerdings fehlt
die Definition des primären Zielparameters. Die Einwilligungserklärung und das Studien-

60
protokoll wurden von der für das jeweilige Untersuchungszentrum zuständigen Ethik-
kommission geprüft. Jede/jeder Teilnehmer*in gab vor Beginn der Studie eine schriftliche
Einwilligungserklärung ab. Angaben zu einer Studienregistrierung gibt es jedoch keine. Es gibt
in dieser Studie ausreichend genaue Angaben über die Art der Randomisierung sowie über
die statistischen Modelle, die zur Berechnung der Ergebnisse verwendet wurden. Die zu
Beginn der Studie aufgezeichneten Basiswerte und die Endwerte (TSS, NSS) wurden jedoch
nicht angegeben. Bei der Analyse der Ergebnisse wird nur die prozentuelle Reduktion der
verschiedenen Parameter genannt, allerdings fehlen die Absolutwerte. Außerdem gibt es
keine Angaben über den primären bzw. sekundären Endpunkt dieser Studie. Folgende
Effektivitätsparameter wurden analysiert: Der TSS, die nasalen Symptome, die nicht-nasalen
Symptome, der NSS, der Juckreiz der Nase, der nasale Ausfluss und der Juckreiz der Augen.
Obwohl die Autor*innen selbst bei Evaluierung der Ergebnisse an Tag 4 nur bei wenigen
Parametern einen Vorteil der Kombination gegenüber den Einzelsubstanzen feststellen
konnten, gaben sie dennoch eine Empfehlung zur Anwendung der Arzneimittelkombination
ab. Auf Grundlage der durch die Studie gelieferten Daten erscheint dies jedoch
widersprüchlich.
Der TSS wurde durch die Einnahme der Kombination um 55%, mit Loratadin um 50%, mit
Pseudoephedrin um 46% und mit Placebo um 38% gesenkt. Durch die Addition von
Pseudoephedrin zu Loratadin ergibt sich somit ein additiver Nutzen von 5%.
Der NSS wurde durch Loratadin-Pseudoephedrin um 42%, durch Pseudoephedrin ebenfalls
um 42%, durch Loratadin um 35% und durch Placebo um 28% reduziert. Hier ergibt sich ein
additiver Nutzen von 7%.
Wichtig für die Beurteilung der Effektivität ist außerdem die Evaluierung durch die
Patient*innen selbst. Die Angaben der Patient*innen selbst fehlen hier allerdings vollständig.
Die Ergebnisse der Evaluierung durch die Prüfärzt*innen verhielten sich jedoch laut den
Autor*innen sehr ähnlich. Bei der Bewertung des allgemeinen Therapieansprechens wurde die
Arzneimittelkombination mit 2,5 Punkten bewertet, Loratadin mit 2,7, Pseudoephedrin mit 2,7
und Placebo mit 3,0. Allein durch die Gabe einer Placebo-Tablette wurde die Therapie somit
mit „mittelmäßig“ bewertet. Durch die Gabe der Arzneimittelkombination ergibt sich ein
Unterschied von 0,5 Punkten. Vergleicht man die Kombination mit den Monotherapien, ergibt
sich ein Unterschied von nur 0,3 Punkten.
Schlaflosigkeit kommt in der Kombinations-Gruppe bei 19% der Teilnehmer*innen vor, und in
der Pseudoephedrin-Gruppe zu 17%. Die Loratadin-Gruppe kommt hier nur auf 4% und die
Placebo-Gruppe auf 2%. Fälle von Xerostomie treten in der Kombinations-Gruppe bei 24%
der Patient*innen auf, in der Pseudoephedrin-Gruppe bei 12%, in der Loratadin-Gruppe bei
5% und in der Placebo-Gruppe bei 4%.

61
Ob die Patient*innen daher tatsächlich von der Addition von Pseudoephedrin profitieren, ist in
dieser klinischen Studie sehr kritisch zu hinterfragen, da durch die Addition das
Nebenwirkungsspektrum beträchtlich erweitert wird. Allerdings ist durch Mängel in
Studiendesign und Präsentation der Daten die Validität der Studie insgesamt als gering zu
bewerten.
4.1.9 Cetirizine and pseudoephedrine retard alone and in combination in the
treatment of perennial allergic rhinitis: a double-blind multicentre study
(Bertrand et al 1996) Ziel dieser Studie war es, die Effektivität einer Arzneimittelkombination bestehend aus Cetirizin
und Pseudoephedrin mit jener der Einzelsubstanzen zur Behandlung von ganzjähriger
allergischer Rhinitis zu vergleichen. Die Studie wurde an acht verschiedenen
Untersuchungszentren in Belgien und Luxemburg durchgeführt.
Voraussetzung für die Teilnahme an der Studie war eine seit mindestens einem Jahr
bestehende Erkrankung an ganzjähriger allergischer Rhinitis, die mittels eines Prick-Tests
oder RAST bestätigt wurde, sowie ein Alter von 12 bis 65 Jahren. Zusätzlich mussten die
Patient*innen unter folgenden Symptomen leiden: nasale Obstruktion, Rhinorrhoe und Niesen.
Der Gebrauch von Medikamenten abseits der Studie war nicht gestattet, jedoch durften
Personen, die an Asthma litten, ihre bestehende Behandlung mit bis zu 400 µg inhalativen
Corticosteroiden pro Tag fortsetzen.
Nach anfänglicher Screening-Phase und Bestimmung der Basiswerte der Symptome wurden
210 Proband*innen zur Teilnahme ausgewählt, randomisiert und einer der drei
Untersuchungsgruppen zugeteilt, sodass es pro Untersuchungsgruppe 70 Teilnehmer*innen
gab. Die Arzneimittelkombination bestehend aus 5 mg Cetirizin und 120 mg Pseudoephedrin
wurde in Kapselform verabreicht, die Monotherapie mit 5 mg Cetirizin als Tablette und die
Monotherapie mit 120 mg Pseudoephedrin wiederum als Kapsel. Um die vollständige
Verblindung der Teilnehmer*innen und der Prüfärzt*innen zu garantieren wurde bei jeder
Einnahme die fehlende Kapsel bzw. Tablette durch eine Placebo-Kapsel bzw. -Tablette
ersetzt. Alle Arzneimittel wurden zweimal täglich über einen Zeitraum von drei Wochen
verabreicht.
Von den anfänglichen 210 Studienteilnehmer*innen beendeten 171 (81,4%) die Studie. 19
Teilnehmer*innen aus der Kombinations-Gruppe, 7 aus der Cetirizin-Gruppe und 13 aus der
Pseudoephedrin-Gruppe brachen die Studie vorzeitig ab.
Die Studienteilnehmer*innen mussten nach Beginn der Behandlung zu zwei weiteren
Terminen ins Untersuchungszentrum. Der zweite Besuch fand zwischen dem fünften und
neunten Tag der Behandlung statt und der letzte Besuch diente dem Abschluss der Studie.
Die Teilnehmer*innen zeichneten die Intensität ihrer Symptome täglich am Abend anhand

62
einer vierteiligen Skala auf. Beim letzten Besuch der Teilnehmer*innen führten die
Prüfärzt*innen eine allgemeine Evaluation mittels einer fünfteiligen Skala durch.
Zur Beurteilung der Wirksamkeit der untersuchten Arzneimittel wurde die Methode der
höchsten SS verwendet. Pro Tag wurde für jede/jeden Patient*in das am schlechtesten
bewertete Symptom ausgewählt und daraus der höchste „daily score“ berechnet.
Anschließend wurde durch den „daily score“ der Prozentsatz an Tagen bestimmt, an denen
die Symptome abwesend bzw. mild, also mit einem SS von 0 bzw. ≤1 waren. Ein ähnliches
Prinzip wurde zur Bestimmung der nasalen Obstruktion angewandt, um den prozentuellen
Anteil an Tagen zu ermitteln, an denen die nasale Obstruktion mild bzw. abwesend war. Der
Basiswert wurde festgelegt als das am schlechtesten bewertete Symptom an den Tagen 0 (ein
Tag vor Beginn der Studie) und 1. Dieser lag zu Beginn der Studie in der Kombinations-Gruppe
bei 2,3, in der Cetirizin-Gruppe bei 2,4 und in der Pseudoephedrin-Gruppe bei 2,4 Punkten.
Die Werte für den NCS lagen in der Kombinations-Gruppe anfänglich bei 2,1, in der Cetirizin-
Gruppe bei 2,2 und in der Pseudoephedrin-Gruppe bei 2,2 Punkten.
Der primäre Endpunkt dieser Studie war zwar nicht eindeutig definiert, kann aber als der
Prozentsatz an Tagen, an denen die Symptome von ganzjähriger allergischer Rhinitis mild
oder abwesend waren, angenommen werden. Dennoch sollte in einer Studie der primäre
Endpunkt eindeutig definiert werden. Andernfalls ist es als Mangel bei der Beurteilung der
Qualität einer klinischen Studie anzusehen.
Als Grundlage für die Analyse der Effektivitätsparameter diente die ITT-Population, zu der all
jene gezählt wurden, die randomisiert wurden. Wurden bei der globalen Analyse der
Ergebnisse statistisch signifikante Unterschiede gefunden, so wurde ein Paarvergleich mit
einer der folgenden Methoden durchgeführt: CMH-Test, Fisher-Test und Wilcoxon-Mann-
Whitney-Test.
Nach dreiwöchiger Behandlung mit einem der drei Arzneimittel lag der Prozentsatz für die
Tage, an denen die Symptome abwesend waren, in der Untersuchungsgruppe mit Cetirizin-
Pseudoephedrin bei 12%, in der mit Cetirizin allein bei 7% und in der mit Pseudoephedrin
allein bei 5%. Keiner dieser Unterschiede war statistisch signifikant. Der prozentuelle Anteil an
Tagen, an denen die Symptome mild waren, lag in der Kombinations-Gruppe bei 65%, in der
Cetirizin-Gruppe bei 46% und in der Pseudoephedrin-Gruppe bei 41%. Hier war die
Kombinations-Gruppe der Gruppe mit Cetirizin (p=0,003) und der Gruppe mit Pseudoephedrin
(p=0,0001) statistisch signifikant überlegen.
Der prozentuelle Anteil an Tagen ohne nasale Obstruktion war in der Kombinations-Gruppe
mit 30% statistisch signifikant höher als in der Cetirizin-Gruppe mit 17% (p=0,005) und als in
der Pseudoephedrin-Gruppe mit 18% (p=0,025). Der Unterschied zwischen der Monotherapie
mit Cetirizin und Pseudoephedrin war nicht statistisch signifikant (p=0,477). Zu denselben
Ergebnissen kam man bei Betrachtung des Prozentsatzes an Tagen, an denen die nasale

63
Obstruktion mild war. Hier lagen die Werte in der Gruppe mit Cetirizin-Pseudoephedrin bei
79%, in der mit Cetirizin bei 53% und in der mit Pseudoephedrin bei 62%. Der Unterschied
zwischen Cetirizin und Pseudoephedrin war hier ebenfalls nicht statistisch signifikant
(p=0,139). Die Kombination war jedoch den Monotherapien statistisch signifikant (p=0,0001
Cetirizin, p=0,003 Pseudoephedrin) überlegen.
Die Arzneimittelkombination war des Weiteren gegenüber den Einzelsubstanzen zur
Reduktion der nasalen Versstopfung überlegen. Zusätzlich dazu verbesserte die Kombination
Symptome wie Rhinorrhoe und Niesen mehr als Pseudoephedrin allein. Die Monotherapie mit
Cetirizin war der mit Pseudoephedrin zur Reduktion von den Symptomen Niesen und Juckreiz
der Nase oder der Augen überlegen.
Der Wert für das am schlechtesten bewertete Symptom wurde durch die Einnahme von
Cetirizin-Pseudoephedrin zum Zeitpunkt des zweiten Besuches auf 1,38, durch Cetirizin allein
auf 1,76 und durch Pseudoephedrin allein auf 1,85 Punkte reduziert. Der Unterschied
zwischen der Arzneimittelkombination und den Monotherapien war hier statistisch signifikant
p=0,001). Zum Zeitpunkt des dritten Besuches war jedoch kein Unterschied zwischen den
Untersuchungsgruppen festzustellen.
Bei der allgemeinen Evaluation des Therapieeffekts durch die Prüfärzt*innen war die
Arzneimittelkombination der Monotherapie mit Cetirizin (p=0,028) sowie der Monotherapie mit
Pseudoephedrin (p=0,018) statistisch signifikant überlegen. In der Kombinations-Gruppe
wurde der Therapieeffekt zu 66% als „gut“ bzw. „exzellent“ bewertet, in der Cetirizin-Gruppe
zu 53% und in der Pseudoephedrin-Gruppe zu 42%.
Während der Studiendauer kam es in der Pseudoephedrin-Gruppe vermehrt zu Berichten über
unerwünschte Arzneimittelwirkungen. Der Unterschied zwischen den Gruppen war jedoch
nicht statistisch signifikant. In der Cetirizin-Gruppe waren die häufigsten Nebenwirkungen
Schläfrigkeit (9%) und Bronchitis (6%), in der Pseudoephedrin-Gruppe war es Schlaflosigkeit
(10%). Andere unerwünschte Arzneimittelwirkungen waren Übelkeit (9%), Kopfschmerzen
(7%) und Asthenie (6%). In der Arzneimittelkombinations-Gruppe wurde vermehrt über
Schläfrigkeit (13%), Kopfschmerzen (11%), Pharyngitis (7%), Nervosität (6%) und Xerostomie
geklagt. Durch die Einnahme der Arzneimittel kam es während der gesamten Studiendauer zu
keinen klinisch relevanten Veränderungen der Laborparameter, der Herzfrequenz oder des
Blutdrucks.
Die Autor*innen schließen aus den Ergebnissen dieser Studie, dass die
Arzneimittelkombination bestehend aus Cetirizin und Pseudoephedrin die Symptome von
ganzjähriger allergischer Rhinitis stärker reduzierte, als Cetirizin oder Pseudoephedrin allein.
Die allgemeine Evaluation und die Beurteilung der einzelnen Symptome würden diese
Ergebnisse untermauern. Die Methode der höchsten SS sei des Weiteren eine besonders
genaue Art, die Effektivität von Arzneimitteln zu untersuchen, da verschiedene Symptome das

64
Leben der Patient*innen unterschiedlich stark beeinträchtigen. Für die/den eine/einen
Patient*in sei nasale Obstruktion das am schwersten zu ertragende Symptom, für die/den
andere/anderen das Niesen. Daher könne man auf diese Art und Weise den Nutzen eines
Arzneimittels zur Reduktion der individuellen Beschwerden der Patient*innen besser
untersuchen. Es bestehe laut den Autor*innen dieser Studie ein klinischer Vorteil der
Kombination gegenüber den Monotherapien, da die Kombination die Symptome von
ganzjähriger allergischer Rhinitis besser reduziere, als die Einzelsubstanzen. Besonders
ausgeprägt sei dieser zusätzliche Effekt, wenn nasale Obstruktion vorliege. Cetirizin sei zwar
von den Teilnehmer*innen am besten vertragen worden und habe das geringste
Nebenwirkungsprofil, das sei jedoch kein klinisch relevanter Vorteil, da die Kombination
effektiver sei. Abschließend halten die Autor*innen fest, dass die Kombination bestehend aus
5 mg Cetirizin und 120 mg Pseudoephedrin eine effektive und gut verträgliche Therapieoption
bei ganzjähriger allergischer Rhinitis sei, vor allem wenn nasale Verstopfung stark vertreten
sei.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Anfangs wird
erwähnt, dass die Studie in Einklang mit der Deklaration von Helsinki durchgeführt wurde und
das Studienprotokoll von der zuständigen Ethikkommission geprüft wurde. Alle
Teilnehmer*innen gaben zu Beginn der Studie eine schriftliche Einwilligungserklärung ab. In
der Studie fehlen allerdings die Angaben zu einer Registrierung. Die Studie war weder auf der
internationalen Registrierungsplattform für klinische Studien „Clinical Trials“
(https://clinicaltrials.gov) noch auf der europäischen Seite „EU Clinical Trials Register“
(https://clinicaltrials.gov/) zu finden. Des Weiteren gibt es keine genauere Beschreibung, wie
die Randomisierung der Studienteilnehmer*innen in der Studie erfolgte. Die Definition des
primären Zielparameters war nicht eindeutig. Daher kann nicht beurteilt werden, ob das
Studienziel erreicht wurde.
Darüber hinaus scheint die Studie scheint von guter Qualität zu sein. Es gibt genaue Angaben
über die statistischen Modelle und auch die Absolutwerte der Ergebnisse des primären
Endpunktes wurden angegeben. Zwar fehlen die Absolutwerte zu den einzelnen Symptomen,
allerdings lassen sich diese aus den diversen Grafiken in etwa herauslesen. Die Autor*innen
sprechen von nicht-klinisch relevanten Auswirkungen der Arzneimittel auf die Herzfrequenz
und auf den Blutdruck. Interessant wäre allerdings zu wissen, wo hier die Grenze zu „nicht-
klinisch relevant“ gezogen wurde, also wie sehr die Herzfrequenz durch die Gabe der
Medikamente verändert wurde.
Der prozentuelle Anteil an angenehmen Tagen lag in der Kombinations-Gruppe bei 65%, in
der Cetirizin-Gruppe bei 46% und in der Pseudoephedrin-Gruppe bei 41%. Durch die Addition

65
von Pseudoephedrin erreichte man demnach einen größeren prozentuellen Anteil an
angenehmen Tagen in einem Ausmaß von 19%.
Die Anzahl an Tagen, an denen die nasale Verstopfung mild war, lag in der Gruppe mit
Cetirizin-Pseudoephedrin bei 79%, mit Cetirizin bei 53% und mit Pseudoephedrin bei 62%.
Durch die Zugabe von Pseudoephedrin zum Antihistaminikum wird folglich ein Gewinn von
26% verzeichnet.
Die allgemeine Evaluation durch die Prüfärzt*innen zeigte ebenfalls einen statistisch
signifikanten Vorteil der Kombination gegenüber der Einzelsubstanzen. Die Unterschiede der
am schlechtesten bewerteten Symptome war jedoch nur zum Zeitpunkt des zweiten Besuchs
statistisch signifikant. Gegen Ende der Studie gab es zwischen den Untersuchungsgruppen
keinen Unterschied mehr.
Insgesamt kann diese Studie einen Vorteil der Kombination im Vergleich zu Cetirizin in Hinblick
auf die Anzahl symptomarmer Tage belegen. Allerdings scheint diese Wirkung lediglich durch
die Reduktion der nasalen Obstruktion zustande zu kommen, da sich im Zeitverlauf die Scores
für alle anderen Symptome nicht unterscheiden.
4.1.10 Efficacy of a leukotriene receptor antagonist in the treatment of perennial
allergic rhinitis (Jiang et al. 2006) Ziel dieser Studie war es, die Effektivität des Leukotrien-Rezeptor-Antagonisten Zafirlukast zur
Behandlung ganzjähriger allergischer Rhinitis im Vergleich zu Loratadin und der Kombination
von Loratadin-Pseudoephedrin zu untersuchen. Die randomisierte, einfachblinde Studie wurde
von Januar 2002 bis April 2003 am Taichung Veterans General Hospital in Taiwan
durchgeführt und erhielt finanzielle Unterstützung vom Taichung Veterans General Hospital.
Einschlusskriterien für die Teilnahme an der Studie waren ein Alter von 15 bis 70 Jahren, eine
Erkrankung an ganzjähriger allergischer Rhinitis seit mindestens zwei Jahren und ein positiv
ausfallender Haut-Test, der die bestehende Erkrankung bestätigte.
93 Patient*innen erfüllten die Voraussetzungen zur Teilnahme an der Studie und wurden
anhand einer computergenerierten Randomisierungsliste einer der drei Untersuchungs-
gruppen zugeteilt. 32 Personen erhielten 20 mg Zafirlukast zweimal täglich, 30 Personen
erhielten 10 mg Loratadin einmal täglich und 31 Personen erhielten zweimal täglich eine
Kombination aus 5 mg Loratadin und 120 mg Pseudoephedrin. Alle Arzneimittel wurden über
einen Zeitraum von 14 Tagen eingenommen. Weder die Teilnehmer*innen, noch die
Techniker, welche für die Rhinomanometrie zuständig waren, wussten welcher der drei
Untersuchungsgruppen sie zugeteilt waren.
Vor Beginn der Behandlung und zwei Tage nach Einnahme der letzten Dosis evaluierten die
Studienteilnehmer*innen ihre nasalen Symptome mithilfe einer vierteiligen Skala. Zu
denselben Zeitpunkten wurde mittels Rhinomanometrie die MCA bestimmt. Der Basiswert für

66
den TSS lag zu Beginn der Studie in der Zafirlukast-Gruppe bei 8,1, in der Loratadin-Gruppe
bei 7,8 und in der Loratadin-Pseudoephedrin-Gruppe bei 7,8 Punkten.
In dieser Studie gab es keine Angaben darüber, wie der primäre bzw. sekundäre Endpunkt
festgelegt wurde.
Patient*innen, die das entsprechende Arzneimittel weniger als elf Tage lang einnahmen,
wurden von der Analyse ausgeschlossen. Die Werte der SS, die MCA sowie der
inspiratorische und exspiratorische Druck, welcher mittels Rhinomanometrie gemessen
wurde, wurden mithilfe des Wilcoxon-Mann-Whitney-Tests verglichen. Die Ergebnisse wurden
zwischen den Gruppen durch den Kruskal-Wallis-Test analysiert.
Der TSS wurde nach zweiwöchiger Behandlung durch Zafirlukast auf 5,9 (p<0,0001), durch
Loratadin auf 6,0 (p<0,0001) und durch Loratadin-Pseudoephedrin auf 5,9 (p=0,0003) Punkte
reduziert. Durch die Einnahme von Loratadin und der Kombination von Loratadin mit
Pseudoephedrin wurden sowohl die individuellen Symptome als auch der TSS statistisch
signifikant reduziert. Durch Zafirlukast wurden der TSS und alle individuellen Symptome bis
auf Niesen ebenfalls statistisch signifikant reduziert.
Bei der Reduktion der nasalen Obstruktion war die Gruppe mit Zafirlukast den anderen beiden
Untersuchungsgruppen statistisch signifikant (p=0,014) überlegen. Die Werte lagen nach
Behandlung mit Zafirlukast bei 1,9, mit Loratadin bei 2,4 und mit Loratadin-Pseudoephedrin
bei 2,3 Punkten.
Die MCA erhöhte sich nach Abschluss der Studie in allen drei Untersuchungsgruppen, die
Unterschiede zu den Werten zu Beginn der Studie und zwischen den drei Gruppen waren
jedoch nicht statistisch signifikant. Der inspiratorische und der exspiratorische Druck
verringerten sich in allen Untersuchungsgruppen, der jeweilige Unterschied war jedoch nicht
statistisch signifikant.
Zu den unerwünschten Arzneimittelwirkungen, die während der Studie beobachtet wurden,
gab es keine Angaben.
Die Autor*innen dieser Studie schließen aus den Ergebnissen, dass Zafirlukast eine
ausgeprägtere Reduktion der nasalen Verstopfung bewirke als Loratadin oder die Kombination
bestehend aus Loratadin und Pseudoephedrin. Ein Vorteil der Kombination von Loratadin und
Pseudoephedrin gegenüber der Monotherapie von Loratadin alleine werde hier nicht
beobachtet. Letzteres könne allerdings an der geringen Teilnehmer*innenzahl liegen. Obwohl
in früheren Studien die Überlegenheit der Kombination eines Antihistaminikums mit
Pseudoephedrin gezeigt wurde, könne dies durch die Ergebnisse dieser Studie nicht bewiesen
werden. Die Kombination war der Monotherapie mit Loratadin zur Reduktion der nasalen
Obstruktion im Zuge dieser Untersuchung nicht überlegen. Zafirlukast hingegen sei bei der
Reduktion der nasalen Verstopfung den anderen Arzneimitteln überlegen und habe im Hinblick
auf die Symptome der Rhinorrhoe und des Juckreizes der Nase dieselben Ergebnisse wie

67
Loratadin geliefert. Daher sei Zafirlukast eine effektive Therapieoption der ganzjährigen
allergischen Rhinitis.
Der errechnete Jadad Score für diese Studie beträgt 2 und ist somit „schlecht“. Es gibt keine
Angaben zu einer Registrierung der Studie. Das Studienprotokoll wurde von der
Ethikkommission des Taichung Veterans General Hospital geprüft und vor Beginn der Studie
wurde von jede/jeder Teilnehmer*in eine schriftliche Einwilligungserklärung abgegeben.
Ein Minuspunkt dieser Studie ist mit Sicherheit, dass es sich hierbei um keine
Doppelblindstudie gehandelt hat, sondern lediglich die Teilnehmer*innen selbst nicht wussten,
welches Medikament ihnen verabreicht wurde. Außerdem wurde nicht genauer beschrieben,
ob diejenigen, die Loratadin erhielten, abends zusätzlich eine Placebo-Tablette einnahmen.
Wäre dieser Punkt nicht eingehalten worden, wäre keine vollständige Verblindung seitens der
Patient*innen garantiert gewesen. Der primäre bzw. sekundäre Endpunkt wurde nicht definiert
und es gibt des Weiteren keine Angaben, wie viele Patient*innen die Studie vorzeitig
beendeten und aus welchen Gründen dies geschah. Die Studie scheint von unzureichender
Qualität zu sein weshalb ihre Aussagekraft nur sehr begrenzt ist.
4.1.11 Effects of terfenadine and pseudoephedrine, alone and in combination in a
nasal provocation test and in perennial rhinitis (Henauer et al. 1991)
Studie 1 – Nasaler Provokationstest
Ziel dieser randomisierten und doppelblinden Studie war es, mittels Provokationstests die
allergische Reaktion auf Graspollen nach Einnahme von Placebo, Terfenadin,
Pseudoephedrin oder einer Kombination der beiden Arzneimittel zu untersuchen. Die Angaben
zum Land, in dem die Studie durchgeführt wurde, sowie zu etwaigen Sponsoren dieser Studie
fehlen hier.
Voraussetzung für die Teilnahme an dieser Studie war eine bestehende Erkrankung an
allergischer Rhinitis, welche durch einen positiven Prick-Test bestätigt wurde sowie ein
positiver RAST für Graspollen.
13 Patient*innen nahmen an dieser Studie teil und waren zu Beginn symptomfrei. Die Studie
wurde im cross-over Design so geführt, dass alle Teilnehmer*innen vier verschiedene
Behandlungen erhielten, wobei zwischen den unterschiedlichen Arzneimitteln je eine Woche
als Auswaschphase diente. Aufgrund einer computergenerierten Randomisierungsliste
erfolgte die Zuteilung zu folgenden Untersuchungsgruppen: Gruppe 1 erhielt 60 mg Terfenadin
und 120 mg Pseudoephedrin mit verzögerter Freisetzung, Gruppe 2 erhielt 60 mg Terfenadin,
Gruppe 3 erhielt 120 mg Pseudoephedrin mit verzögerter Freisetzung und Gruppe 4 erhielt

68
Placebo. Die Teilnehmer*innen erhielten fünf der zugeteilten Tabletten und wurden
angewiesen zwei Tage vor Beginn der Studie pro Tag zwei Stück der Tabletten einzunehmen.
Die letzte Tablette sollte zwei Stunden vor Testbeginn eingenommen werden. Um die
vollständige Verblindung beider Seiten zu garantieren, wurden optisch ununterscheidbare
Tabletten verwendet. 12 Teilnehmer*innen beendeten die Studie. Das entspricht einem
Follow-Up von 92%.
Der Ablauf des Provokationstests war wie folgt: Zuerst wurden 0,2 ml bestehend aus
0,3 mg/ml menschlichem Albumin und 4 mg/ml Phenol in die beiden Nasenlöcher mittels eines
Zerstäubers gesprüht. Anschließend wurde alle zehn Minuten ein standardisiertes Allergen in
steigender Konzentration von 100 bis 100.000 BU/ml eingesprüht. Der Schwellenwert der
allergischen Reaktion wurde überschritten, wenn zwei der folgenden Kriterien innerhalb von
zehn Minuten nach Verabreichung der Dosis erfüllt waren: nasaler Ausfluss >0,5 g, Niesen ≥4
oder die nasale IPFR sank um ≥40%.
Der primäre Endpunkt dieser Studie war der Schwellenwert der allergischen Reaktion. Die
Daten wurden nach logarithmischer Transformation durch das allgemeine lineare Modell
analysiert.
Der Schwellenwert der allergischen Reaktion lag in der Placebo-Gruppe bei 196 BU/ml, in der
Pseudoephedrin-Gruppe bei 757 BU/ml, in der Terfenadin-Gruppe bei 1790 BU/ml und in der
Kombinations-Gruppe bei 3860 BU/ml. Sowohl die Terfenadin- (p=0,039) als auch die
Pseudoephedrin-Gruppe (p=0,026) waren der Placebo-Gruppe statistisch signifikant
überlegen. Die mittlere nasale IPFR, gemessen zum Zeitpunkt der höchsten
Allergenkonzentration, wurde durch Terfenadin allein um 46% und durch Terfenadin-
Pseudoephedrin um 56% gesenkt. Die Monotherapie mit Terfenadin war hier der
Kombinationstherapie überlegen, da die nasale IPFR durch die Kombination stärker reduziert
wurde.
Während der Studie kam es zu sieben Berichten von unerwünschten Arzneimittelwirkungen,
unter anderem über Xerostomie, Schläfrigkeit, Nervosität und Asthenie.
Studie 2 – Ganzjährige allergische Rhinitis
In dieser randomisierten und doppelblinden Studie sollte die Effektivität und Sicherheit einer
Arzneimittelkombination bestehend aus Terfenadin und Pseudoephedrin mit der Monotherapie
mit Terfenadin bei Patient*innen mit ganzjähriger allergischer Rhinitis verglichen werden. Die
Angaben zum Land, in dem die Studie durchgeführt wurde, sowie zu etwaigen Sponsoren
dieser Studie fehlen hier.
50 Patient*innen mit bestehender Erkrankung an ganzjähriger allergischer Rhinitis wurden zur
Teilnahme an der Studie ausgewählt und aufgrund eines computergenerierten

69
Randomisierungscodes einer der zwei Untersuchungsgruppen zugeteilt. 25 Teilnehmer*innen
der ersten Gruppe erhielten 60 mg Terfenadin und 25 Teilnehmer*innen der zweiten Gruppe
erhielten 60 mg Terfenadin kombiniert mit 120 mg Pseudoephedrin. Alle Arzneimittel wurden
in Tablettenform verabreicht und über einen Zeitraum von 14 Tagen zweimal täglich
eingenommen. Drei Teilnehmer*innen (94%) brachen die Studie aufgrund unerwünschter
Arzneimittelwirkungen vorzeitig ab.
Die Studienteilnehmer*innen zeichneten die Intensität ihrer Symptome täglich anhand einer
vierteiligen Skala auf. Bewertet wurden hierbei folgende Symptome: nasale Verstopfung,
Niesen, Rhinorrhoe, Juckreiz der Nase und/oder des Rachens, Juckreiz der Augen, tränende
Augen und rötliche Augen. Dieselben Symptome wurden von den Prüfärzt*innen wöchentlich
mithilfe einer visuellen Analogskala evaluiert, auf der 0 mm für „abwesend“ und 100 mm für
„schwer“ stand. Zu Beginn der Studie und an den Tagen 7 und 14 wurden die Durchblutung,
Schwellung, Sekretion oder Obstruktion der Nase mittels Rhinoskopie unter Zuhilfenahme
einer vierteiligen Skala evaluiert. Zusätzlich wurde die Funktion des Geruchssinns zu
Studienbeginn und an Tag 14 durch verschiedene aromatische Geruchsstoffe überprüft.
Der primäre Endpunkt dieser Studie war die allgemeine Bewertung der Verbesserung der
Symptome, der durch die Prüfärzt*innen evaluiert wurde. Zu den sekundären Endpunkten
gehörten die Ergebnisse der rhinoskopischen Untersuchung sowie die Evaluierung der
Symptome, die durch die Prüfärzt*innen beurteilt wurde. Die Unterschiede zwischen den
Gruppen wurden durch den CMH-Test oder den Wilcoxon-Mann-Whitney-Test analysiert.
Die deutliche Verbesserung der Symptome wurde häufiger in der Kombinations-Gruppe
bestehend aus Terfenadin und Pseudoephedrin beobachtet, der Unterschied zur Terfenadin-
Gruppe war jedoch nicht statistisch signifikant (Tag 7 p=0,27, Tag 14 p=0,69). In der
Untersuchungsgruppe mit Terfenadin in Kombination mit Pseudoephedrin gaben 10
Teilnehmer*innen an, dass ihre Symptome deutlich verbessert wurden bzw. abwesend waren.
In der Untersuchungsgruppe mit Terfenadin taten dies 5 Personen. Die Ergebnisse der
Rhinoskopie zeigten eine Überlegenheit der Kombination gegenüber der Monotherapie,
jedoch wurde nur im Hinblick auf die Schwellung statistische Signifikanz gezeigt (p=0,01). Der
Geruchssinn wurde durch die Einnahme der Medikamente nur minimal verbessert.
Während der Studie kam es in der Terfenadin-Pseudoephedrin-Gruppe häufiger zu Berichten
von unerwünschten Arzneimittelwirkungen. 20 der 25 Teilnehmer*innen klagten hier über
Nebenwirkungen, während es in der Terfenadin-Gruppe nur 9 Teilnehmer*innen waren. Dieser
Unterschied war statistisch signifikant (p=0,004). Zu den berichteten Nebenwirkungen
gehörten unter anderem Schläfrigkeit, Xerostomie, Kopfschmerzen, Nervosität und Übelkeit.
Der Blutdruck veränderte sich in der Kombinations-Gruppe von 122/81 mmHg auf 120/79
mmHg während er in der Terfenadin-Gruppe unverändert blieb. Die Herzfrequenz stieg in der
Kombinations-Gruppe von 67 auf 72 und sank in der Terfenadin-Gruppe von 69 auf 67 Schläge

70
pro Minute. 15 (65%) Patient*innen der Kombinations-Gruppe und 18 (78%) der Terfenadin-
Gruppe gaben an das Medikament, sofern verfügbar, wieder zu nehmen.
Die Autor*innen schließen aus den Ergebnissen der zwei Studien, dass die Kombination
bestehend aus Terfenadin und Pseudoephedrin effektiver zur Reduktion der Symptome von
allergischer Rhinitis als Terfenadin alleine sei, der Unterschied sei jedoch gering. Die
Annahme, dass die nasale Verstopfung durch die Kombination mit Pseudoephedrin stärker
verbessert werde, konnte in dieser Studie nicht bewiesen werden.
Im nasalen Provokationstest schnitt Terfenadin besser als Pseudoephedrin ab, ein Ergebnis
welches die Autor*innen der Studie verwunderte. Da die Schwelle der allergischen Reaktion
vor allem von der Rhinorrhoe und der nasalen Obstruktion abhängig war, vermutete man, dass
Pseudoephedrin als abschwellendes Arzneimittel besser als das Antihistaminikum Terfenadin
abschneiden würde. Die Autor*innen schlussfolgern, dass die Kombination effektiver als die
Einzelsubstanzen wirke, das Nebenwirkungsspektrum jedoch erweitert würde.
Der errechnete Jadad Score für beide Studien beträgt 5 und ist somit „exzellent“. Beide Studien
wurden in Einklang mit der Deklaration von Helsinki durchgeführt und das Studienprotokoll
wurde von der dort zuständigen Ethikkommission geprüft. Alle Patient*innen wurden vor
Beginn der Studie über den Nutzen als auch über die Risiken der Studie informiert und gaben
eine mündliche Einwilligungserklärung ab. Angaben zu einer Registrierung der Studien fehlen
allerdings in beiden Fällen.
Da es sich bei der ersten Studie um einen Provokationstest handelt, sind die Ergebnisse für
die Bearbeitung dieser Diplomarbeit von geringerem Interesse. Der Fokus in dieser Arbeit liegt
auf Studien, bei denen die Patient*innen auf natürlichem Wege den Allergenen ausgesetzt
waren. Des Weiteren wurde die Studie über einen sehr kurzen Zeitraum und mit einer kleinen
Studienpopulation durchgeführt. Der Effekt der Arzneimittel lässt sich nach derart kurzer Zeit
nur schwer abschätzen, ebenso die unerwünschten Arzneimittelwirkungen.
Die zweite Studie scheint von guter Qualität zu sein. Die Originaldaten der Evaluation durch
die Patient*innen selbst fehlen jedoch. Der primäre Endpunkt spiegelt die Ergebnisse der
Evaluation der Prüfärzt*innen wider, interessant wäre jedoch auch die Bewertung durch die
Studienteilnehmer*innen. Die Teilnehmer*innenzahl bei der zweiten Studie ist ebenfalls sehr
gering. Dies könnte eine mögliche Ursache für die minimalen Unterschiede zwischen den
Untersuchungsgruppen sein. Die Ergebnisse dieser Studie favorisieren jedoch die
Verwendung der Arzneimittelkombination nicht, da durch die Monotherapie mit Terfenadin ein
ähnlicher therapeutischer Effekt erreicht wurde und das bei einem geringeren
Nebenwirkungsprofil.

71
4.1.12 Comparative Efficacy and Safety of Terfenadine with Pseudoephedrine and
Terfenadine alone in Allergic Rhinitis (Myers et al. 1998) Ziel dieser Studie war es, eine Arzneimittelkombination, die aus Terfenadin und
Pseudoephedrin besteht, mit den Einzelkomponenten im Hinblick auf die Effektivität und
Sicherheit bei Patient*innen mit allergischer Rhinitis zu vergleichen. Diese randomisierte und
doppelblinde Studie wurde an einem Untersuchungszentrum in Indien durchgeführt und erhielt
finanzielle Unterstützung von Torrent Pharmaceuticals.
41 Patient*innen im Alter von 15 bis 56 Jahren wurden nach anfänglicher Screening-Phase
zur Teilnahme an der Studie ausgewählt und nach zufälligem Prinzip einer der zwei
Untersuchungsgruppen zugeteilt. Die erste Gruppe erhielt die Arzneimittelkombination
bestehend aus 60 mg Terfenadin und 120 mg Pseudoephedrin und die zweite Gruppe erhielt
die Monotherapie mit 60 mg Terfenadin. Die Kombination der beiden Arzneimittel bestand aus
60 mg Terfenadin und 10 mg Pseudoephedrin mit sofortiger Freisetzung sowie 110 mg
Pseudoephedrin mit verzögerter Freisetzung. Beide Arzneimittel wurden als optisch
ununterscheidbare Tabletten zweimal täglich über einen Zeitraum von 14 Tagen
eingenommen. Die Teilnehmer*innen erhielten zu Beginn der Studie den Tablettenvorrat für
eine Woche. Nach dieser Woche kamen die Studienteilnehmer*innen erneut zum
Untersuchungszentrum, um die Medikamente für die folgende Woche abzuholen. 22
Patient*innen wurden der Kombinations-Gruppe zugeteilt und 19 erhielten die Monotherapie
mit Terfenadin.
32 der anfänglichen 41 Teilnehmer*innen beendeten die zweiwöchige Studie. Das entspricht
einem Follow-Up von 78%. 4 Patient*innen beendeten die Studie aufgrund unerwünschter
Arzneimittelwirkungen vorzeitig und 5 Teilnehmer*innen erschienen nicht zum ersten Follow-
Up-Termin.
Die Evaluation der Symptome sowie der unerwünschten Arzneimittelwirkungen fand zu Beginn
der Studie, eine Woche nach Studienbeginn und zum Abschluss der Studie statt. Im Zuge
dessen wurden folgende Symptome beurteilt: nasale Verstopfung, Niesen, laufende Nase,
nasaler Ausfluss, Juckreiz der Nase, tränende Augen sowie gereizte Augen. Zusätzlich wurde
die Intensität der generellen Anzeichen einer allergischen Rhinitis evaluiert: die nasale
Obstruktion, die Rhinorrhoe, die Schwellung der Schleimhaut, die nasale Durchblutung und
die Rötung der Augen.
Nach Abschluss der Studie wurde der generelle Therapieerfolg durch die Patient*innen und
die Prüfärzt*innen bewertet. Hier stand „exzellent“ für eine Reduktion der Symptome im
Ausmaß von 90%, die Symptome waren somit weitestgehend abwesend. „Gut“ stand für eine
Reduktion der Symptome im Ausmaß von 25% bis 89%. Demnach verbesserten sich einige
Symptome, während andere noch präsent blieben. „Mittelmäßig“ bedeutete eine Reduktion

72
von 25% oder weniger und somit, dass die meisten Symptome unverändert blieben. „Schlecht“
stand für kein Ansprechen auf die Therapie bzw. eine Verschlechterung des Zustandes.
Wie der primäre bzw. sekundäre dieser Studie definiert wurde, wurde an keiner Stelle erwähnt.
Die Ergebnisse der Studie wurden mittels Binomialtest analysiert.
Die beobachtete Symptomreduktion, welche durch die Einnahme der Kombination von
Terfenadin und Pseudoephedrin erreicht wurde, war der von Terfenadin alleine statistisch
signifikant überlegen. Bei der Evaluation des allgemeinen Therapieeffekts durch die
Teilnehmer*innen selbst und durch die Prüfärzt*innen wurde die Arzneimittelkombination bei
46% der Patient*innen mit „exzellent“ bewertet, während dieser Wert in der Terfenadin-Gruppe
bei 11% lag. Dieser Unterschied war statistisch signifikant (p=0,0485). Der Therapieeffekt
wurde in der Kombinations-Gruppe zu 27% und in der Terfenadin-Gruppe zu 37% mit „gut“
bewertet. Somit wurde der Therapieeffekt von 73% der Kombinations-Gruppe und von 47%
der Terfenadin-Gruppe mit „exzellent“ bzw. „gut“ bewertet. Die Unterschiede zwischen der
Effektivität beider Arzneimittel war statistisch signifikant (p<0,05). Bei Befragung der
Patient*innen favorisierten 16 der 22 Teilnehmer*innen in der Untersuchungsgruppe mit
Terfenadin-Pseudoephedrin die Behandlung, während es in der Untersuchungsgruppe mit
Terfenadin nur 8 von 19 waren. Dieser Unterschied war statistisch signifikant.
Während der Studie kam es in keiner der beiden Untersuchungsgruppen zu schwerwiegenden
unerwünschten Arzneimittelwirkungen. In den beiden Untersuchungsgruppen wurde unter
anderem über Schlaflosigkeit, Schwindel und Sedierung berichtet. Durch die Einnahme der
Arzneimittel kam es zu keinen Veränderungen der routinemäßig untersuchten
Laborparameter.
Die Autor*innen schließen aus den Ergebnissen ihrer Studie, dass die Überlegenheit der
Arzneimittelkombination gegenüber der Monotherapie mit Terfenadin zur Behandlung von
allergischer Rhinitis gegeben ist. Die Evaluation durch die Patent*innen und durch die
Prüfärzt*innen habe einen zusätzlichen Nutzen der Kombination gezeigt. Beide Arzneimittel
wurden von den Proband*innen dieser Studie gut vertragen und seien daher eine sichere
Therapieoption bei allergischer Rhinitis, wobei die Kombination die Symptome schneller und
effektiver verbessere als die Monotherapie.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. In der Studie gibt es
keine Angaben über eine Registrierung, sie wurde jedoch von der dort zuständigen
Ethikkommission geprüft. Es gibt weder Angaben zu den Einschlusskriterien zur Teilnahme an
der Studie, noch wird genauer erläutert, wie die Teilnehmer*innen randomisiert wurden. Die
Absolutwerte der Symptomreduktion fehlen ebenfalls. Es wurde lediglich erwähnt, dass die
beschriebenen Symptome statistisch signifikant reduziert wurden, der p-Wert zu dieser
Aussage fehlt jedoch gänzlich.

73
Aufgrund der geringen Teilnehmer*innenzahl ist die Aussagekraft dieser Studie gering, da sich
viele unerwünschte Arzneimittelwirkungen erst bei einer größeren Studienpopulation
manifestieren. Darüber hinaus fällt eine objektive Bewertung der Ergebnisse schwer, da einige
Absolutwerte fehlen und keine krankheitsspezifischen Symptome analysiert wurden. Auch
aufgrund der kleinen Studienpopulation lässt sich keine Aussage zur Wirksamkeit der
Kombination gegenüber der Monotherapie aussprechen.

74
4.2 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Antihistaminikum zur Behandlung von Infektionen der oberen Atemwege
4.2.1 Effects of pseudoephedrine and triprolidine, alone and in combination, on
symptoms of the common cold (Bye et al. 1980) Ziel dieser Studie war es, die Effektivität und Sicherheit einer Arzneimittelkombination
zusammengesetzt aus Triprolidin und Pseudoephedrin zur symptomatischen Behandlung von
grippalen Infekten im Vergleich zu den Einzelsubstanzen und zu Placebo zu untersuchen.
Diese randomisierte, doppelblinde und placebokontrollierte Studie fand von Oktober 1976 bis
März 1977 an vier verschiedenen Untersuchungszentren in England statt.
466 gesunde Proband*innen wurden vorab zur Teilnahme ausgewählt, wovon 199 Personen
an grippalen Infekten erkrankten. Insgesamt berücksichtigte man für die Studie die Daten von
243 solcher registrierten Infekte. Erfüllten die erkrankten Patient*innen die Voraussetzungen
zur Teilnahme, wurden diese von Krankenpfleger*innen untersucht und nach zufälligem
Prinzip einer der vier Untersuchungsgruppen zugeteilt. Pro Untersuchungszentrum wurden 4
verschiedene Randomisierungslisten erstellt: Eine für weibliche Teilnehmerinnen unter 40
Jahren, eine für weibliche Teilnehmerinnen über 40 Jahren, eine für männliche
Teilnehmer*innen unter 40 Jahren und eine für männliche Teilnehmer*innen über 40 Jahren.
56 Teilnehmer*innen erhielten die Arzneimittelkombination bestehend aus 2,5 mg Triprolidin
und 60 mg Pseudoephedrin, 61 erhielten die Monotherapie mit 2,5 mg Triprolidin, 63 erhielten
die Monotherapie mit 60 mg Pseudoephedrin und 63 Teilnehmer*innen erhielten Placebo. Die
entsprechenden Medikamente wurden den Teilnehmer*innen in optisch ununterscheidbarer
Tablettenform ausgehändigt. Die Studienteilnehmer*innen wurden angewiesen, pro Tag
maximal drei der Tabletten so lange wie notwendig (jedoch maximal zehn Tage lang)
einzunehmen. 235 Patient*innen beendeten die Studie. Das entspricht einem Follow-Up von
97%. In der Kombinations-Gruppe beendeten 55 Teilnehmer*innen die Studie, in der
Triprolidin-Gruppe waren es 59, in der Pseudoephedrin-Gruppe 61 und in der Placebo-Gruppe
60.
Die Effektivität der Behandlung wurde anhand der Aufzeichnungen der Patient*innen beurteilt.
Dazu protokollierten diese täglich die Intensität von 12 verschiedenen Symptomen anhand
einer vierteiligen Skala. Die allgemeine Evaluierung im Hinblick auf die Verbesserung der
Symptome fand an den Tagen 8 – 10 durch die Krankenpfleger*innen und Teilnehmer*innen
statt. Den Proband*innen wurde zu diesem Anlass die Frage gestellt, ob sie glaubten, während
der Studie Placebo erhalten zu haben. Die Häufigkeit der 12 evaluierten Symptome war zu
Beginn wie folgt: „schwerer Kopf“ 95%, „rinnende Nase“ 88%, „Niesen“ 84%, „verstopfte Nase“
80%, „Halsschmerzen“ 77%, „Kopfschmerzen“ 73%, „Husten“ 72%, „Krankheitsgefühl“ 69%,

75
„Sputum“ 63%, „Heiserkeit“ 59%, „Gliederschmerzen“ 47% und „Fieber“ 40%. Es gab jedoch
keinerlei Information über die Homogenität der Studienarme in Hinblick auf die üblichen
Baseline-Parameter bzw. mögliche Unterschiede im Schweregrad der Erkrankung.
Es gibt in dieser Studie keine Angaben zu den primären oder sekundären Endpunkten. Die
Analyse der Werte für die Symptome wurde mithilfe des Chi-Quadrat-Tests durchgeführt.
Durch die Einnahme der Arzneimittelkombination wurde die Häufigkeit des Niesens an den
Tagen 2,3 und 4 im Vergleich zu Placebo statistisch signifikant reduziert. Pseudoephedrin
reduzierte die Häufigkeit an den Tagen 2 und 3 statistisch signifikant und Triprolidin an Tag 2.
Die nasale Obstruktion war zu Beginn des grippalen Infektes am stärksten und wurde durch
die Einnahme der Kombination oder durch Pseudoephedrin allein statistisch signifikant
reduziert. Die übrigen Symptome wurden durch die Einnahme der Medikamente nicht
statistisch signifikant verbessert. Da kein primärer Endpunkt definiert wurde, beschränkt sich
die Bewertung der Wirksamkeit auf die Beurteilung einzelner statistisch signifikant
unterschiedlicher Symptome.
Die Ergebnisse der Evaluierung im Hinblick auf die Verbesserung der Symptome zeigten, dass
die Kombination bestehend aus Triprolidin und Pseudoephedrin und die Monotherapie mit
Pseudoephedrin die Symptome des grippalen Infektes statistisch signifikant (p<0,01) bei mehr
Proband*innen verbessert hatten als Placebo. In der Kombinations-Gruppe gaben 28 (58%)
Teilnehmer*innen an, dass ihre Symptome durch die Einnahme der Arzneimittel verbessert
wurden. In der Triprolidin-Gruppe waren das 27 (48%), in der Pseudoephedrin-Gruppe 29
(53%) und in der Placebo-Gruppe 14 (25%) Personen. Ein statistisch signifikanter Nutzen der
Kombination gegenüber Triprolidin ist somit für diesen Endpunkt nicht belegbar.
All jene Teilnehmer*innen, die von der Einnahme der Arzneimittel nicht profitierten, gaben bei
der Frage, ob sie glaubten, dass sie Placebo eingenommen hätten, an, dass sie zur Placebo-
Gruppe gehörten. Im Gegensatz dazu gaben all jene Proband*innen, die von der
Arzneimitteleinnahme profitierten, an, dass sie kein Placebo erhalten hatten, sogar wenn sie
tatsächlich zur Placebo-Gruppe gehörten.
Während der Studie kam es in der Placebo-Gruppe zu den meisten Fällen von unerwünschten
Arzneimittelwirkungen. In der Pseudoephedrin-Gruppe wurde von 25 der Teilnehmer*innen
über Schlaflosigkeit berichtet, während in der Placebo-Gruppe hingegen lediglich neun dieser
Fälle vorkamen. Dieser Unterschied war statistisch signifikant.
Die Autor*innen schließen aus den Ergebnissen dieser Studie, dass signifikant mehr
Teilnehmer*innen von der Therapie mit Pseudoephedrin allein bzw. mit der Kombination aus
Triprolidin und Pseudoephedrin im Gegensatz zu Placebo profitierten. Der Unterschied
zwischen Triprolidin und Placebo war nicht statistisch signifikant.

76
Der errechnete Jadad-Score für diese Studie beträgt 4 und ist somit „gut“. In der Studie gibt
es keine genaueren Angaben dazu, wie die Teilnehmer*innen randomisiert wurden. Des
Weiteren wird nicht erwähnt, ob für die Studie Pseudoephedrin mit verzögerter Freisetzung
verwendet wurden. Die Studie wurde vermutlich nicht registriert, da es hierzu ebenfalls keine
Angaben gibt und die Studie auf der internationalen Registrierungsplattform für klinische
Studien „Clinical Trials“ (https://clinicaltrials.gov) sowie auf der europäischen Seite „EU Clinical
Trials Register“ (https://www.clinicaltrialsregister.eu/) nicht zu finden ist.
In dieser Studie fehlen sowohl die Angaben des primären Endpunktes als auch die
Absolutwerte der Ergebnisse. Die p-Werte wurden ebenfalls nicht angegeben. Des Weiteren
gab es keine Angaben über die Homogenität der Studienarme oder über die demografischen
Daten der Teilnehmer*innen. Diese Punkte erschweren die Bewertung der Qualität dieser
Studie sowie die Evaluation der Ergebnisse.
Betrachtet man die Ergebnisse der generellen Symptomreduktion, so wurde durch die
Einnahme der Kombination eine Verbesserung bei 58% der Teilnehmer*innen erzielt, in der
Triprolidin-Gruppe bei 48%, in der Pseudoephedrin-Gruppe bei 53% und in der Placebo-
Gruppe bei 25%. Durch die Kombination von Pseudoephedrin und Triprolidin ergibt sich somit
ein additiver Nutzen von 10% im Vergleich zu Triprolidin allein.
Aufgrund der schlechten Qualität der Studie und der mangelhaften Studiendaten lässt sich
kein Nutzen für die Kombination gegenüber Triprolidin alleine ableiten.

77
4.3 Wirksamkeit und Sicherheit eines α-Sympathomimetikums zusätzlich zu einem Analgetikum zur Behandlung von Infektionen der oberen Atemwege
4.3.1 Analgesic and Decongestant Efficacy of the Combination of Aspirin with
Pseudoephedrine in Patients With Symptoms of Upper Respiratory Tract
Infection (Eccles et al. 2014) Ziel dieser Studie war es, die Wirksamkeit und Sicherheit einer Arzneimittelkombination von
Acetylsalicylsäure und Pseudoephedrin zur symptomatischen Behandlung von Infektionen der
oberen Atemwege im Vergleich zu den Einzelsubstanzen oder Placebo zu untersuchen. Die
Studie wurde am „Common Cold Centre“ in Cardiff, Großbritannien im Zeitraum von
September 2009 bis März 2012 durchgeführt und von „Bayer HealthCare“ gesponsert. Die
Studie wurde im Juni 2009 im „EU trials register“ registriert und ist unter der EudraCT-Nummer
2009-011355-46 zu finden.
Voraussetzung zur Teilnahme an der Studie war eine Infektion der oberen Atemwege seit
maximal drei Tagen und damit verbundenen Schmerzen, sowie eine verstopfte Nase.
Während des Screenings wurden nur jene Patient*innen eingeschlossen, deren
Schmerzsymptome zumindest den Wert 2 (moderat) einer vierteiligen Skala erreichten, sowie
all jene mit einer NAR >0,25 PA/cm³/s.
833 Proband*innen nahmen an dieser randomisierten, doppelblinden und placebo-
kontrollierten Studie teil. Nach anfänglichem Screening und der Bestimmung der Basiswerte
wurden die Studienteilnehmer*innen nach zufälligem Prinzip in folgende Untersuchungs-
gruppen eingeteilt: Die erste Gruppe erhielt zwei Sachets mit je 500 mg Acetylsalicylsäure und
30 mg Pseudoephedrin, die zweite Gruppe erhielt zwei Sachets mit je 500 mg
Acetylsalicylsäure, die dritte Gruppe erhielt zwei Sachets mit je 30 mg Pseudoephedrin und
die vierte Gruppe erhielt zwei Placebo-Sachets. Der Inhalt der Sachets wurden zur Einnahme
in einem Glas Wasser aufgelöst und getrunken. Da es nicht möglich war die Sachets optisch
voneinander zu unterscheiden, wurde so die Verblindung der Studienteilnehmer*innen und
Prüfärzt*innen sichergestellt. Um auch eine geschmackliche Differenzierung auszuschließen,
wurden dem Placebo-Präparat dieselben Hilfsstoffe, die in den anderen Arzneimitteln
enthalten waren, zugesetzt. 829 Patient*innen (99,5%) beendeten die einwöchige Studie und
wurden somit in der ITT-Population analysiert. Für die Analyse der Sicherheit der Arzneimittel
wurden alle 833 Studienteilnehmer*innen berücksichtigt.
Die Teilnehmer*innen besuchten das Untersuchungszentrum im Rahmen der Studie
insgesamt an zwei Tagen. An Tag 1 wurden die Patient*innen ausgewählt und alle benötigten
Basiswerte evaluiert. Anschließend erhielten alle geeigneten Proband*innen eine Dosis des
entsprechenden Arzneimittels und folgende Parameter wurden im Abstand von je einer Stunde

78
über einen Zeitraum von vier Stunden beurteilt: NAR, schmerzstillende Wirkung, Intensität der
Schmerzen, Linderung der verstopften Nase und Intensität der nasalen Verstopfung. Die
Einschätzung der eben genannten Parameter erfolgte anhand einer vier- bzw. fünfteiligen
Skala.
Nach Ende des vierstündigen Beobachtungszeitraums wurden die Studienteilnehmer*innen
beauftragt, noch ein bis zwei weitere Dosen des entsprechenden Arzneimittels im Abstand von
mindestens vier Stunden einzunehmen sowie drei Dosen täglich im Abstand von mindestens
vier Stunden an den Tagen 2 und 3.
An den Tagen 1-3 zeichneten die Proband*innen jeweils am Abend folgende Parameter mittels
einer Skala auf: schmerzstillende Wirkung, Intensität der Schmerzen, Linderung der
verstopften Nase und Intensität der nasalen Verstopfung. An Tag 3 wurden zudem die
allgemeine schmerzstillende Wirkung sowie die allgemeine Linderung der verstopften Nase
evaluiert. Die Studienteilnehmer*innen wurden gebeten, innerhalb von sieben Tagen nach
Einnahme der ersten Dosis zum abschließenden Follow-Up-Termin im Untersuchungszentrum
zu erscheinen.
Zur Einschätzung der Effektivität der verschiedenen Arzneimittel wurden zwei primäre
Endpunkte festgelegt: Zum einen die Linderung der nasalen Verstopfung, gemessen als die
AUC der NAC für den Zeitraum von 0-4 Stunden, und zum anderen die schmerzstillende
Wirkung, gemessen anhand des „total pain relief scores“ für den Zeitraum von 0-4 Stunden.
Im Hinblick auf die nasale Verstopfung wurden die Arzneimittelkombination bestehend aus
Acetylsalicylsäure und Pseudoephedrin mit der Monotherapie aus Acetylsalicylsäure
verglichen. Das Ausmaß der Schmerzlinderung wurde primär zwischen der Arzneimittel-
kombination und der Monotherapie aus Pseudoephedrin verglichen.
Sekundäre Endpunkte waren die NAC zu den Zeitpunkten 0-1,0-2 und 0-3 Stunden nach
Verabreichung der initialen Dosis, die Veränderungen der Intensität der Verstopfung der Nase
zu den Zeitpunkten 0-4 Stunden sowie an den Tagen 0-3, die Linderung der nasalen
Verstopfung zu den Zeitpunkten 0-4 und an den Tagen 0-3 und zuletzt die allgemeine
Linderung der verstopften Nase. Außerdem zählten zu den sekundären Endpunkten die
Veränderungen der Schmerzintensität zu den Zeitpunkten 0-4 Stunden und an den Tagen 0-
3, die schmerzstillende Wirkung an den Tagen 0-3 und die allgemeine Linderung der
Schmerzen.
Als Grundlage für die Analyse der Effektivität diente die ITT-Population mit 829
Teilnehmer*innen. Unterschiede zwischen den verschiedenen Behandlungsgruppen wurden
mittels „ANOVA“ ausgewertet. Um die Unterschiede zwischen den Arzneimittelgruppen
darzustellen, wurde die „Methode der kleinsten Quadrate“ angewandt. Da die Erwartungen an
die „ANOVA“ der primären und sekundären Endpunkte nicht erfüllt wurden, wurden die
Endpunkte daraufhin mittels eines nichtparametrischen „Mann-Whitney U-test“ analysiert.

79
Die NAC wurde durch die Arzneimittelkombination bestehend aus Acetylsalicylsäure und
Pseudoephedrin im Vergleich zur Monotherapie mit Acetylsalicylsäure (p<0,001) oder Placebo
(p<0,001), aber nicht im Vergleich zur Monotherapie mit Pseudoephedrin (p=0,59) statistisch
signifikant erhöht.
Die Kombination von Acetylsalicylsäure mit Pseudoephedrin war der Acetylsalicylsäure- und
Placebo-Gruppe zur Linderung der verstopften Nase nach vier Stunden statistisch signifikant
(p<0,001) überlegen, nicht jedoch der Pseudoephedrin-Gruppe (p=0,9). Der Unterschied
entsprach ca. 60 cm3s über den gesamten Zeitraum. Die Angaben, die diesen Unterschied mit
der subjektiven Symptomatik in Korrelation stellen, fehlen jedoch. Nach drei Tagen war die
Arzneimittelkombination der Acetylsalicylsäure- (p=0,016) und Placebo-Gruppe (p<0,001)
immer noch statistisch signifikant überlegen. Im Vergleich zur Pseudoephedrin-Gruppe gab es
weiterhin keinen statistisch relevanten Unterschied.
Die Veränderungen der subjektiven Intensität der verstopften Nase nach vier Stunden zeigten
eine statistische Überlegenheit (p=0,008) der Kombinationstherapie gegenüber Placebo, aber
nicht gegenüber den Monotherapien mit Acetylsalicylsäure oder Pseudoephedrin. Nach drei
Tagen war die Kombination der Placebo-Gruppe weiterhin statistisch signifikant (p=0,048)
überlegen, nicht jedoch gegenüber den anderen Untersuchungsgruppen.
Bei der allgemeinen subjektiven Beurteilung der Linderung der verstopften Nase war die
Arzneimittelkombination der Acetylsalicylsäure- (p=0,04) und Placebo-Gruppe (p<0,001), nicht
jedoch der Pseudoephedrin-Gruppe (p=0,2) statistisch signifikant überlegen.
Da die Analyse der schmerzstillenden Wirkung der verschiedenen Arzneimittel nicht Thema
dieser Diplomarbeit ist, wird hier nicht weiter darauf eingegangen und auf den Originalartikel
verwiesen.
Während der gesamten Studiendauer kam es zu keinerlei unerwarteten unerwünschten
Ereignissen. Die häufigsten unerwünschten Arzneimittelwirkungen betrafen das gastro-
intestinale System mit einer Verteilung von 8% in der Acetylsalicylsäure-Pseudoephedrin-
Gruppe, 8% in der Acetylsalicylsäure-Gruppe, 5% in der Pseudoephedrin-Gruppe und 4% in
der Placebo-Gruppe. Des Weiteren kamen Störungen des Nervensystems mit einer Häufigkeit
von 4% Acetylsalicylsäure-Pseudoephedrin-Gruppe, 1% in der Acetylsalicylsäure-Gruppe, 2%
in der Pseudoephedrin-Gruppe und 5% in der Placebo-Gruppe vor.
Aus den Ergebnissen dieser Studie schließen die Autor*innen, dass durch die Verabreichung
der Arzneimittelkombination bestehend aus Acetylsalicylsäure und Pseudoephedrin eine
schmerzstillende sowie abschwellende Wirkung erreicht wird. Der abschwellende Effekt, der
durch die Arzneimittelkombination erreicht wird, sei größer als durch Placebo oder
Acetylsalicylsäure, nicht jedoch größer als durch Pseudoephedrin erzielte Effekt. Der
schmerzstillende Effekt der Kombinationstherapie sei größer als der mit Placebo,
Pseudoephedrin oder Acetylsalicylsäure.

80
Die Intensität der Schmerzen bzw. die Intensität der Verstopfung der Nase wurden anhand
einer 4-teiligen Skala bewertet, während die Schmerzlinderung bzw. Abschwellung der Nase
anhand einer 5-teiligen Skala bewertet wurden. Bei genauerer Betrachtung fällt auf, dass nur
bei der 4-teiligen Skala statistische Signifikanz erreicht wurde. Eine mögliche Ursache dafür
sei laut den Autor*innen, dass die vierteilige Skala ungenauer als die fünfteilige Skala sei und
daher die Unterschiede in der fünfteiligen Skala nicht statistisch relevant seien.
Zusammengefasst sei die Arzneimittelkombination den Monotherapien überlegen und die
Anwendung einer fixen Kombination von Acetylsalicylsäure und Pseudoephedrin zur
Behandlung von Erkrankungen der oberen Atemwege, die mit verstopfter Nase und
Schmerzen einhergehen, sei gerechtfertigt.
Der errechnete Jadad Score für diese Studie beträgt 4 und ist somit „gut“. Zu dieser Studie ist
anzumerken, dass es keine genauen Angaben über die Art und Weise der Randomisierung
gibt, nach der die Studienteilnehmer*innen den verschiedenen Untersuchungsgruppen
zugeteilt wurden. In der Studie wurden außerdem nicht nur ein, sondern zwei primäre
Endpunkte festgelegt.
Die Beurteilung der Ergebnisse fällt in dieser Studie schwer, da es keine Angaben der
Originaldaten bzw. der Absolutwerte der Ergebnisse gibt. In den Tabellen werden
ausschließlich die p-Werte angeführt.
Anhand der im Artikel enthaltenen Grafik, welche nachfolgend als Abbildung 6 zu finden ist,
kann man die Werte für den „congestion relief score“ in etwa ablesen. Der „congestion relief
score“ wurde durch die Arzneimittelkombination mit Acetylsalicylsäure und Pseudoephedrin
um 1,5 Punkte, durch Pseudoephedrin um 1,5 Punkte, durch Acetylsalicylsäure um 1,3 Punkte
und durch Placebo um 1,3 Punkte erhöht. Die Addition von Pseudoephedrin zu
Acetylsalicylsäure zur Behandlung grippaler Infekte führt somit zu einer Verbesserung des
„congestion relief scores“ um 0,2 Punkte. Aufgrund des selbstlimitierenden Charakters dieser
Erkrankung ist die klinische Relevanz dieses Unterschieds fraglich. Die Einnahme
sympathomimetisch wirkender Substanzen ist in Bezug auf Nutzen und Risiko abzuwägen.
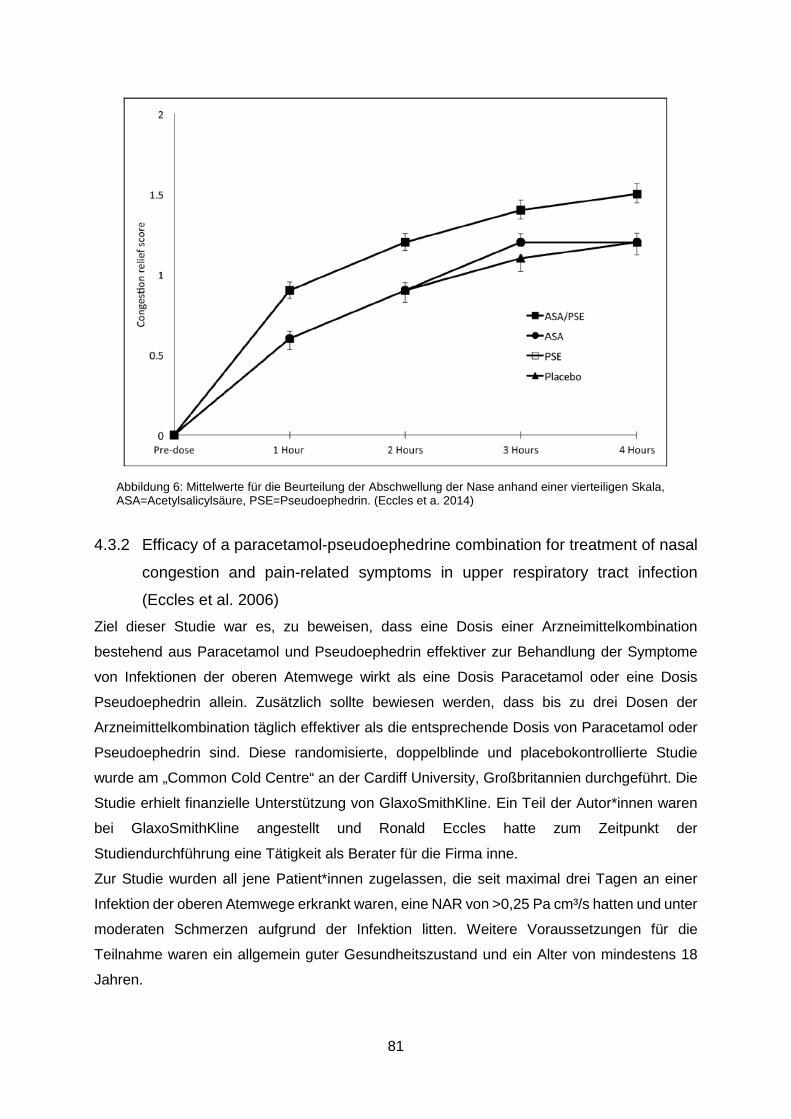
81
4.3.2 Efficacy of a paracetamol-pseudoephedrine combination for treatment of nasal
congestion and pain-related symptoms in upper respiratory tract infection
(Eccles et al. 2006) Ziel dieser Studie war es, zu beweisen, dass eine Dosis einer Arzneimittelkombination
bestehend aus Paracetamol und Pseudoephedrin effektiver zur Behandlung der Symptome
von Infektionen der oberen Atemwege wirkt als eine Dosis Paracetamol oder eine Dosis
Pseudoephedrin allein. Zusätzlich sollte bewiesen werden, dass bis zu drei Dosen der
Arzneimittelkombination täglich effektiver als die entsprechende Dosis von Paracetamol oder
Pseudoephedrin sind. Diese randomisierte, doppelblinde und placebokontrollierte Studie
wurde am „Common Cold Centre“ an der Cardiff University, Großbritannien durchgeführt. Die
Studie erhielt finanzielle Unterstützung von GlaxoSmithKline. Ein Teil der Autor*innen waren
bei GlaxoSmithKline angestellt und Ronald Eccles hatte zum Zeitpunkt der
Studiendurchführung eine Tätigkeit als Berater für die Firma inne.
Zur Studie wurden all jene Patient*innen zugelassen, die seit maximal drei Tagen an einer
Infektion der oberen Atemwege erkrankt waren, eine NAR von >0,25 Pa cm³/s hatten und unter
moderaten Schmerzen aufgrund der Infektion litten. Weitere Voraussetzungen für die
Teilnahme waren ein allgemein guter Gesundheitszustand und ein Alter von mindestens 18
Jahren.
Abbildung 6: Mittelwerte für die Beurteilung der Abschwellung der Nase anhand einer vierteiligen Skala, ASA=Acetylsalicylsäure, PSE=Pseudoephedrin. (Eccles et a. 2014)

82
384 Patient*innen wurden anfangs gescreent, wovon 305 die Voraussetzungen für die
Teilnahme erfüllten und anschließend nach zufälligem Prinzip einer der folgenden
Untersuchungsgruppen zugeordnet wurden: Die erste Gruppe mit 76 Teilnehmer*innen erhielt
die Kombination aus 1000 mg Paracetamol und 60 mg Pseudoephedrin, die zweite Gruppe
mit 76 Teilnehmer*innen erhielt die Monotherapie mit 1000 mg Paracetamol, die dritte Gruppe
mit 76 Teilnehmer*innen erhielt die Monotherapie mit 60 mg Pseudoephedrin und die vierte
Gruppe mit 77 Teilnehmer*innen erhielt Placebo. Die Medikamente wurden in Tablettenform
verabreicht und maximal dreimal täglich mit einem Dosierungsabstand von vier Stunden
eingenommen.
Jede/jeder Teilnehmer*in musste an zwei Tagen ins Untersuchungszentrum: An Tag 1 erfolgte
das Screening sowie die Zuteilung zu einer der Untersuchungsgruppen und an Tag 4 wurde
die abschließende Untersuchung der Teilnehmer*innen durchgeführt. Die Studienteil-
nehmer*innen erhielten an Tag 1 die erste Dosis des zugeteilten Medikaments im
Untersuchungszentrum und wurden gebeten, die folgenden drei Tage die Einnahme des
Medikaments mit maximal drei Tabletten pro Tag fortzusetzen. Die NAR und die Intensität der
Schmerzen wurden 1, 2, 3 sowie 4 Stunden nach Einnahme der ersten Dosis im
Untersuchungszentrum bestimmt. Die NAR wurde mittels Rhinomanometrie bestimmt und
daraus wurde die NAC berechnet. Zusätzlich dazu zeichneten die Teilnehmer*innen täglich
gegen Abend die Intensität der Schmerzen und die Verstopfung der Nase anhand eines
Tagebuchs auf. Der NCS wurde unter Zuhilfenahme einer vierteiligen Skala durch die
Patient*innen selbst beurteilt. An Tag 4 wurden die Tagebücher der Patient*innen den
Prüfärzt*innen übergeben und die allgemeine Effektivität der jeweiligen Arzneimittel bestimmt.
Zur Beurteilung der allgemeinen Verbesserung der nasalen Obstruktion wurde eine fünfteilige
Skala verwendet.
Die primären Endpunkte dieser Studie waren die AUC der NAC 0 – 4 Stunden nach
Verabreichung der ersten Dosis und die Schmerzlinderung 0 – 4 Stunden nach Verabreichung
der ersten Dosis. Zu Beginn lagen die Werte für die mittlere NAC in der Kombinations-Gruppe
bei 206,2 cm³/s, in der Paracetamol-Gruppe bei 206,9 cm³/s, in der Pseudoephedrin-Gruppe
bei 210,8 cm³/s und in der Placebo-Gruppe bei 217,6 cm³/s. Auf die Ergebnisse im Hinblick
auf die Schmerzlinderung, welche durch die Arzneimitteleinnahme erreicht wurde, wird hier
nicht weiter eingegangen, da dies außerhalb des thematischen Rahmens dieser Diplomarbeit
liegt.
Die sekundären Endpunkte waren die NAC nach 1 Stunde, die Veränderungen der Schmerzen
nach 4 Stunden, die Veränderungen der Schmerzen nach 3 Tagen und die Veränderungen
der nasalen Obstruktion nach 3 Tagen. Der NCS lag zu Beginn der Studie bei 1,9 in der
Kombinations-Gruppe, bei 2,0 in der Paracetamol-Gruppe, bei 1,8 in der Pseudoephedrin-
Gruppe und bei 1,8 Punkten in der Placebo-Gruppe.

83
Die primären und sekundären Endpunkte wurden mittels der Kovarianz-Analyse bestimmt.
Die AUC der NAC 0 – 4 Stunden nach Verabreichung der ersten Dosis lag in der
Kombinations-Gruppe bei 864,8, in der Paracetamol-Gruppe bei 713,2, in der
Pseudoephedrin-Gruppe bei 915,1 und in der Placebo-Gruppe bei 779,0. Die
Arzneimittelkombination war somit der Monotherapie mit Paracetamol und der Placebo-
Gruppe im Hinblick auf die NAC statistisch signifikant (p=0,0001) überlegen. Der mittlere
Unterschied der AUC der NAC (0 – 4 h) lag hier bei 153,9 im Vergleich zu Paracetamol und
bei 120,3 im Vergleich zu Placebo.
Eine Stunde nach Verabreichung der ersten Dosis lag die NAC in der Kombinations-Gruppe
bei 206,3 cm³/s, in der Paracetamol-Gruppe bei 167,9 cm³/s, in der Pseudoephedrin-Gruppe
bei 219,2 cm³/s und in der Placebo-Gruppe bei 176,3 cm³/s. Eine Stunde nach Verabreichung
der Arzneimittel war die Kombinations-Gruppe der Placebo-Gruppe im Hinblick auf die NAC
statistisch signifikant (p<0,0001) überlegen.
Die Summe aller nasalen Veränderungen während den Tagen 0 – 3 lag in der Gruppe von
Paracetamol-Pseudoephedrin bei 1,7, in der Gruppe von Paracetamol alleine bei 1,3, in der
Gruppe von Pseudoephedrin alleine bei 1,2 und in der Placebo-Gruppe bei 0,9 Punkten. Die
Angaben der Endwerte fehlen hier. Somit kann man die klinische Relevanz dieser Reduktion
nicht beurteilen, da man keinen Vergleich zu den Ausgangswerten herstellen kann. Die p-
Werte lagen hier bei 0,0209 beim Vergleich der Arzneimittelkombination mit Paracetamol allein
und bei 0,0042 beim Vergleich der Arzneimittelkombination mit Placebo. Die Arzneimittel-
kombination war der Paracetamol- und Placebo-Gruppe somit im Hinblick auf die Reduktion
der nasalen Obstruktion statistisch signifikant überlegen.
Durch die Einnahme der Arzneimittel kam es zu einer allgemeinen Verbesserung der nasalen
Obstruktion (globale Bewertung) von 1,7 Punkten in der Kombinations-Gruppe, von 1,3 in der
Paracetamol-Gruppe, von 1,4 in der Pseudoephedrin-Gruppe und von 1,0 in der Placebo-
Gruppe. Die Kombination reduzierte die nasale Verstopfung statistisch signifikant (p=0,0004)
stärker als Placebo.
Während der Studie kam es bei 7% der Teilnehmer*innen in der Kombinations-Gruppe zu
unerwünschten Arzneimittelwirkungen. In der Paracetamol-Gruppe wurde von 3% der
Teilnehmer*innen über Nebenwirkungen berichtet, in der Pseudoephedrin-Gruppe von 5% und
in der Placebo-Gruppe von 7%. Es kam während der gesamten Studiendauer zu keinen
schweren unerwünschten Arzneimittelwirkungen.
Die mittleren Werte der nasalen Obstruktion wurden durch die Arzneimittelkombination um
58% gesenkt. Im Vergleich dazu lag die Reduktion, welche durch die Gabe von Placebo-
Tabletten erreicht wurde bei 39%. Die Unterschiede zwischen der Arzneimittelkombination und
den Einzelsubstanzen waren während der Studie gering, aber dennoch war die Kombination
beiden Monotherapien überlegen. Alle Arzneimittel wurden von den Patient*innen gut

84
vertragen und zeigten ein gutes Sicherheitsprofil. Aus den Ergebnissen dieser Studie
schließen die Autor*innen daher, dass die Arzneimittelkombination eine sichere und effektive
Therapieoption für Infektionen der oberen Atemwege darstelle.
Der Jadad Score für diese Studie beträgt 3 und ist somit „gut“. Alle Patient*innen stimmten
dem Ablauf der Studie vorab durch eine schriftliche Einwilligungserklärung zu. Das
Studienprotokoll und die Einwilligungserklärung wurden vom „Bro Taf Ethics Commitee“
bewilligt. Die Studie wurde in Einklang mit der Deklaration von Helsinki und Good Clinical
Practice durchgeführt. Die Angaben zur Registrierung der Studie fehlen jedoch, und auch auf
der internationalen Registrierungsplattform für klinische Studien „Clinical Trials“
(https://clinicaltrials.gov) sowie auf der europäischen Seite „EU Clinical Trials Register“
(https://www.clinicaltrialsregister.eu/) ist die Studie nicht auffindbar. Über die Beschaffenheit
der Tabletten und die Zusammensetzung gibt es ebenfalls keine genauen Angaben. Welche
Arzneistoffe die verschiedenen Untersuchungsgruppen einnehmen und in welcher Dosierung
diese verabreicht werden, wird nur kurz in der Einleitung und im Abstract erwähnt. Des
Weiteren wird über die Art und Weise, wie die Randomisierung erfolgte, nicht berichtet.
Bei dieser Studie fehlen zudem die Absolutwerte der Vergleiche der Arzneimittelkombination
mit Pseudoephedrin. Die Kombination wird hier nur mit Paracetamol und Placebo verglichen.
Gerade die Werte im Vergleich zu Pseudoephedrin wären allerdings zur Einschätzung der
allgemeinen Wirksamkeit durchaus von Interesse. Allgemein sind die Ergebnisse der Studie
unübersichtlich formuliert. Die Autor*innen liefern des Weiteren keine Informationen zur
Korrelation der NAC oder der AUC der NAC mit den für die/den Patient*in spürbaren
Auswirkungen. Aus diesem Grund ist eine Beurteilung der tatsächlichen klinischen Relevanz
für die Patient*innen nicht möglich.
Durch die Kombination der Arzneistoffe Paracetamol und Pseudoephedrin wurden die Werte
für die allgemeinen Veränderungen der nasalen Obstruktion um 58% und durch die Einnahme
von Placebo um 39% gesenkt. Durch die Einnahme der Kombination ergibt sich hier demnach
ein Unterschied von 19%. Da die Unterschiede zu Paracetamol alleine nur sehr klein waren,
ist der Nutzen der Kombination als gering einzuschätzen.

85
5 Diskussion
Essenziell für die Beurteilung der Effektivität einer Arzneitherapie mit Kombinationspräparaten
sind Gegenüberstellungen mit Placebo und den entsprechenden Einzelsubstanzen sowie
Vergleiche mit ähnlichen Studien, um die Reproduzierbarkeit der Ergebnisse zu validieren.
Aufgrund der heterogenen Arten, die Symptome in den Untersuchungen zu evaluieren, stellt
sich die allgemeine Beurteilung der Effektivität und der Vergleich zwischen den Studien als
sehr komplex dar. Bei einem Großteil der hier untersuchten Studien wurde der TSS und der
NCS mit den Werten zu Beginn der Studie und nach Studienabschluss verglichen. Die
Basiswerte für den TSS lagen hier anfänglich meist bei 15,0-16,0 Punkten und wurden durch
die Gabe der Kombination um ca. 6,0 Punkte reduziert. Durch die Gabe eines
Antihistaminikums konnte dieser Wert um ca. 5,0 Punkte verringert werden. Die Basiswerte
für den NCS lagen oftmals bei ca. 2,5 Punkten und wurden durch die Einnahme eines
Kombinationspräparates um 0,8-1,0 Punkte bzw. durch die Einnahme eines Antihistaminikums
um 0,6-0,7 Punkte reduziert. Der daraus errechnete additive Nutzen einer Therapie mit
Kombinationspräparaten lag meist bei ca. 0,7 Punkten bei der Reduktion des TSS und bei 0,1-
0,2 Punkten bei der Reduktion des NCS. Somit zeichnete sich beim Vergleich der Studien
durchaus eine gewisse Reproduzierbarkeit der Ergebnisse ab.
Die Resultate der meisten dieser Studien deuten auf eine Überlegenheit der Kombinations-
präparate gegenüber den Einzelsubstanzen hin. Dennoch ist der tatsächlich klinisch relevante
Nutzen gering, denn die Überlegenheit bei zusätzlicher Gabe von Pseudoephedrin war zwar
meist statistisch signifikant, eine klinische Relevanz ist daraus allerdings nicht ableitbar. So
profitiert ein/eine Patient*in zwar objektiv von der Reduktion der nasalen Obstruktion, wenn
man die NAC misst, subjektiv ist dieser Unterschied hingegen kaum merklich. Somit steht
diesem fraglichen Nutzen eine Vergrößerung des Nebenwirkungsspektrums gegenüber.
Eine wichtige Grundlage für die Beurteilung der Sicherheit, der in dieser Diplomarbeit
untersuchten Arzneimittelkombinationen, stellen die Berichte über unerwünschte
Arzneimittelwirkungen dar. Besonders die kardiovaskulären Veränderungen, die nach
Einnahme der Präparate beobachtet wurden, sind bei der Einschätzung des Nutzens dieser
Medikamente von Bedeutung. Pleskow et al. berichteten im Jahr 2005 über eine geringe
Steigerung der Herzfrequenz nach Einnahme einer Arzneimittelkombination bestehend aus
Desloratadin und Pseudoephedrin sowie nach Einnahme der Monotherapie mit
Pseudoephedrin. In der Studie von Bronsky et al. 1995 wurde die Herzfrequenz in der
Kombinations-Gruppe um 3,3-4,5 Schläge pro Minute statistisch signifikant (p≤0,02)
gegenüber der Loratadin- und Placebo-Gruppe erhöht. In der Pseudoephedrin-Gruppe wurde
die Herzfrequenz ebenfalls um 2,5-3,0 Schläge pro Minute erhöht. Ebenso berichteten
Grosclaude et al. 1997 über eine Erhöhung der Herzfrequenz in der Kombinations-Gruppe um

86
3,2 und in der Pseudoephedrin-Gruppe um 2,2 Schläge pro Minute. Auch von Grubbe et al.
wurde 2009 über dieselbe unerwünschte Arzneimittelwirkung berichtet. Im Zuge dieser Studie
wurde die mittlere Herzfrequenz durch die Einnahme der Arzneimittelkombination um 3,9 und
durch die Einnahme von Pseudoephedrin alleine um 3,0 Schläge pro Minute erhöht. Bertrand
et al. erwähnten 1996 in ihrer Studie ebenfalls eine Veränderung der Herzfrequenz, nannten
jedoch keine genaueren Werte. In den restlichen Studien wurde nicht erwähnt, ob die
Herzfrequenz während der Studie gemessen wurde und ob Veränderungen feststellbar waren.
Die Herzfrequenz erhöhte sich somit in einigen, der in dieser Arbeit untersuchten, Studien um
durchschnittlich 3 Schläge pro Minute.
Hierbei ist allerdings zu beachten, dass ausschließlich Patient*innen mit allgemein gutem
Gesundheitszustand und keinen kardiovaskulären Vorerkrankungen in die Studien einge-
schlossen wurden. Die Veränderungen kardiovaskulärer Parameter, die bei Patient*Innen mit
dementsprechenden Vorerkrankungen und Begleittherapien (z.B. mit β- oder α-Adrenozeptor-
Antagonisten) auftreten können, sind daher weitgehend unklar. In einer randomisierten,
doppelblinden und placebokontrollierten Studie von Chua et al. aus dem Jahr 1989 wurden
die kardiovaskulären Effekte einer Dosis von Pseudoephedrin bei 20 hypertensiven
Patient*innen untersucht, mit dem Ergebnis, dass die Herzfrequenz sowie der systolische
Blutdruck durch die Einnahme von Pseudoephedrin statistisch signifikant (p>0,01 und p<0,03)
erhöht wurden. Der diastolische Blutdruck und der mittlere arterielle Druck blieben jedoch
beinahe unverändert. (Chua et al. 1989)
1992 wurden in einer randomisierten, doppelblinden und placebokontrollierten Studie die
Auswirkungen von Pseudoephedrin auf den Blutdruck und die Herzfrequenz bei 28
hypertensiven Patient*innen untersucht. Obwohl die Veränderungen des Blutdrucks und der
Herzfrequenz, die während der Studie beobachtet wurden, nicht statistisch signifikant waren,
zeichnete sich eine deutliche Korrelation zwischen der Einnahme von Pseudoephedrin und
der Steigerung der beiden Parameter ab. Die Autor*innen schließen aus den Ergebnissen
dieser Studie, dass die Einnahme von Pseudoephedrin bei hypertensiven Patient*innen sicher
sei, es jedoch bei einer größeren Studienpopulation durchaus zu klinisch relevanten
kardiovaskulären Veränderungen kommen könnte. Es sei daher ratsam eine gute
medizinische Kontrolle der hypertensiven Patient*innen vor Anwendung eines Präparates mit
Pseudoephedrin vorauszusetzen. (Beck et al. 1992)
Bei der Therapie von allergischer Rhinitis müsste das entsprechende Kombinationspräparat
über mehrere Monate hinweg eingenommen werden, was folglich zu einer Steigerung der
Herzfrequenz über einen längeren Zeitraum führen würde. Wie sich eine derartige
Beeinflussung des Herzens auf die Gesundheit der Patient*innen mittel- und langfristig
auswirken kann, ist schwer abzuschätzen. Es sollten daher bei der Auswahl des Präparates

87
bestehende Vorerkrankungen und mögliche Risiken der Behandlung unbedingt berücksichtigt
werden.
Neben den kardiovaskulären unerwünschten Arzneimittelwirkungen gibt es in den analysierten
Studien zahlreiche Berichte über Nebenwirkungen, die eindeutig auf die Einnahme von
Pseudoephedrin-haltigen Arzneimitteln zurückzuführen sind.
Schenkel et al. 2002 schrieb über Nebenwirkungen wie Xerostomie und Schlaflosigkeit, die
vermehrt in den Untersuchungsgruppen mit Pseudoephedrin vorkamen. So gab es sowohl in
der Kombinations-Gruppe als auch in der Pseudoephedrin-Gruppe bei 7% der
Teilnehmer*innen Fälle von Xerostomie, während in der Desloratadin-Gruppe nur 2% der
Teilnehmer*innen unter diesem Symptom litten. 5% der Patient*innen in der Kombinations-
Gruppe und 8% der Patient*innen in der Pseudoephedrin-Gruppe berichteten über
Schlaflosigkeit. Demgegenüber gab es in der Desloratadin-Gruppe nur bei 1% der
Patient*innen Fälle von Schlaflosigkeit.
Weiters gab es Berichte über Nervosität und Kopfschmerzen in den Untersuchungsgruppen,
die ein α-Sympathomimetikum einnahmen. In der Studie von Storms et al. aus dem Jahr 1989
litten 7% der Teilnehmer*innen, die Pseudoephedrin allein einnahmen, an Nervosität, während
dies von 5% in der Kombinations-Gruppe, von 4% in der Loratadin-Gruppe und von 2% in der
Placebo-Gruppe berichtet wurde. Schlaflosigkeit wurde von 19% in der Kombinations-Gruppe
und von 17% in der Pseudoephedrin-Gruppe angegeben, wohingegen nur 4% der Loratadin-
Gruppe und 2% der Placebo-Gruppe darunter litten. Ebenfalls gab es eindeutig mehr Fälle von
Xerostomie in der Gruppe mit Pseudoephedrin alleine (24%) im Vergleich zur Kombinations-
Gruppe (12%), der Loratadin-Gruppe (5%) und der Placebo-Gruppe (4%).
In beinahe allen in dieser Arbeit analysierten Studien wird über eine Korrelation zwischen der
Einnahme von Pseudoephedrin und dem Auftreten von Störungen des Nervensystems
berichtet.
5.1 Konklusion
Die Einschätzung des Nutzens von Arzneimittelkombinationen, welche aus einem
Antihistaminikum und einem α-Sympathomimetikum bestehen und zur Behandlung von
allergischer Rhinitis eingesetzt werden, fällt aufgrund der geringen Überlegenheit der
Kombination gegenüber den Einzelsubstanzen schwer. Bei Patient*innen, die aufgrund einer
Erkrankung an allergischer Rhinitis sehr unter nasaler Obstruktion leiden, kann die Therapie
mit einer Kombination, welche ein α-Sympathomimetikum enthält, in Betracht gezogen
werden, vor allem da die Anwendung von topischen intranasal abschwellend wirkenden
Arzneimitteln für einen Zeitraum von maximal zehn Tagen vorgesehen ist und die Betroffenen
oft monatelang unter nasalen Symptomen leiden. Jedoch kann es bei der Langzeitanwendung
von oralen Vasokonstriktoren zu einer Verringerung der Aktivität von Pseudoephedrin

88
kommen, einer sogenannten Tachyphylaxie. Daher ist es ratsam, die Anwendung auf einen
kurzen Zeitraum zu beschränken und nach Besserung des kongestiven Zustandes der Mukosa
auf die Monotherapie mit einem Antihistaminikum umzusteigen. Die entsprechenden
Fachinformationen spiegeln die Ergebnisse der in dieser Arbeit untersuchten Studien ebenfalls
wider und empfehlen einen maximalen Anwendungszeitraum von zehn Tagen. (Austria Codex,
Fachinformation zu Aerinaze® und Clarinase®)
Präparate, die Pseudoephedrin und ein Antihistaminikum enthalten und zur Anwendung bei
allergischer Rhinitis vorgesehen sind, sind in Österreich rezeptpflichtig. Die Abgabe in der
Apotheke sollte daher nur nach Vorlage eines Rezeptes erfolgen. Angesichts der
kardiovaskulären Nebenwirkungen dieser Arzneimittel sollte die Behandlung bei allen
Patient*innen mit den möglicherweise bestehenden Vorerkrankungen und den damit
einhergehenden Risiken abgewogen werden. Vorranging sind daher die behandelnden Ärzte
für die Beurteilung des individuellen kardiovaskulären Risikos zuständig. Zusätzlich dazu
sollten die Patient*innen von Apotheker*innen hinsichtlich der Risiken und Nebenwirkungen
informiert werden. Beim Auftreten von unerwünschten Arzneimittelwirkungen wie Hypertonien,
Tachykardien, Herzrhythmusstörungen, Übelkeit oder neurologischen Symptomen wie Kopf-
schmerzen ist die Behandlung umgehend abzubrechen.
Für die Therapie von grippalen Infekten zeigt sich bei der Anwendung von
Arzneimittelkombinationen bestehend aus einem Analgetikum und einem α-Sympatho-
mimetikum kein klinisch relevanter Vorteil für die Patient*innen. Aufgrund des
selbstlimitierenden Charakters dieser Krankheit und einer durchschnittlichen Dauer der
Symptome von zehn Tagen sowie des fraglichen klinischen Nutzens der systemischen
Vasokonstriktion ist hier, wenngleich vergleichende Studiendaten fehlen (PMID 27748955) die
Anwendung eines intranasal abschwellend wirkenden Arzneimittels dem Einsatz von
Kombinationspräparaten vorzuziehen. Zusätzlich kann ein Analgetikum zur allgemeinen
Verbesserung des Wohlbefindens und zur Linderung von Kopfschmerzen verwendet werden.
Da jedoch viele Kombinationspräparate mit Pseudoephedrin oder Phenylephrin in der
Apotheke rezeptfrei erhältlich sind, liegt die Aufgabe hier bei den Apotheker*innen, die
Kund*innen entsprechend zu informieren und zu beraten.

89
6 Literaturverzeichnis
Akerlund A, Klint T, Olén L, Rundcrantz H (1989) Nasal decongestant effect of oxymetazoline
in the common cold: an objective dose-response study in 106 patients. J Laryngol Otol
103:743–746.
Aschan G (1974) Decongestion of nasal mucous membranes by oral medication in acute
rhinitis. A rhinomanometric study to demonstrate synergism between antihistamines and
adrenergic substance. Acta Otolaryngol 77:433–438.
Austria Codex, Stand Oktober 2020, http://msweb1.uibk.ac.at/acfi/Default.aspx.
Bachert C, Borchard U, Wedi B, Klimek L, Rasp G, Riechelmann H, Schultze-Werninghaus G,
Wahn U, Ring J (2003) Allergische Rhinokonjunktivitis - Leitlinie der Deutschen
Gesellschaft für Allergologie und klinische Immunologie.
Backhouse CI, Rosenberg RM, Fidler C (1990) Treatment of seasonal allergic rhinitis: a
comparison of a combination tablet of terfenadine and pseudoephedrine with the individual
ingredients. Br J Clin Pract 44:274–279.
Badorrek P, Dick M, Schauerte A, Hecker H, Murdoch R, Luettig B, Hohlfeld JM, Krug N (2009)
A combination of cetirizine and pseudoephedrine has therapeutic benefits when compared
to single drug treatment in allergic rhinitis. Int J Clin Pharmacol Ther 47:71–77.
Badorrek P, Dick M, Hecker H, Schaumann F, Sousa AR, Murdoch R, Hohlfeld JM, Krug N
(2011) Anti-allergic drug testing in an environmental challenge chamber is suitable both in
and out of the relevant pollen season. Ann Allergy Asthma Immunol 106:336–341.
Barchuk WT, Salapatek AM, Ge T, D'Angelo P, Liu X (2013) A proof-of-concept study of the
effect of a novel H3-receptor antagonist in allergen-induced nasal congestion. J Allergy
Clin Immunol 132:838-46.e1-6.
Beck RA, Mercado DL, Seguin SM, Andrade WP, Cushner HM (1992) Cardiovascular effects
of pseudoephedrine in medically controlled hypertensive patients. Arch Intern Med
152:1242–1245.
Bende M, Löth S (1986) Vascular effects of topical oxymetazoline on human nasal mucosa.
The Journal of Laryngology & Otology 100:285–288.
Berkowitz RB, Connell JT, Dietz AJ, Greenstein SM, Tinkelman DG (1989) The effectiveness
of the nonsedating antihistamine loratadine plus pseudoephedrine in the symptomatic
management of the common cold. Ann Allergy 63:336–339.
Berkowitz RB, McCafferty F, Lutz C, Bazelmans D, Godfrey P, Meeves S, Liao Y, Georges G
(2004) Fexofenadine HCl 60 mg/ pseudoephedrine HCl 120 mg has a 60-minute onset of
action in the treatment of seasonal allergic rhinitis symptoms, as assessed in an allergen
exposure unit. Allergy Asthma Proc 25:335–343.

90
Berkowitz RB, Woodworth GG, Lutz C, Weiler K, Weiler J, Moss M, Meeves S (2002) Onset
of action, efficacy, and safety of fexofenadine 60 mg/pseudoephedrine 120 mg versus
placebo in the Atlanta allergen exposure unit. Annals of Allergy, Asthma & Immunology
89:38–45.
Bertrand B, Jamart J, Marchal JL, Arendt C (1996) Cetirizine and pseudoephedrine retard
alone and in combination in the treatment of perennial allergic rhinitis: a double-blind
multicentre study. Rhinology 34:91–96.
BfR (2020) BgVV und BfArM warnen: Schwere Gesundheitsschäden durch Ephedra-Kraut.
Bousquet J et al. (2012) Practical guide to skin prick tests in allergy to aeroallergens. Allergy
67:18–24.
Bousquet J et al. (2008) Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in
collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy 63
Suppl 86:8–160.
Bousquet J, van Cauwenberge P, Khaltaev N (2001) Allergic rhinitis and its impact on asthma.
Journal of Allergy and Clinical Immunology 108:S147-334.
Britton MG, Empey DW, John GC, McDonnell KA, Hughes DT (1978) Histamine challenge and
anterior nasal rhinometry: their use in the assessment of pseudoephedrine and triprolidine
as nasal decongestants in subjects with hayfever. Br J Clin Pharmacol 6:51–58.
BRONSKY E, BOGGS P, FINDLAY S, GAWCHIK S, GEORGITIS J, MANSMANN H,
SHOLLER L, WOLFE J, MELTZER E, MORRIS R (1995) Comparative efficacy and safety
of a once-daily loratadine-pseudoephedrine combination versus its components alone and
placebo in the management of seasonal allergic rhinitis. Journal of Allergy and Clinical
Immunology 96:139–147.
Bye CE, Cooper J, Empey DW, Fowle AS, Hughes DT, Letley E, O'Grady J (1980) Effects of
pseudoephedrine and triprolidine, alone and in combination, on symptoms of the common
cold. Br Med J 281:189–190.
Caenen M, Hamels K, Deron P, Clement P (2005) Comparison of decongestive capacity of
xylometazoline and pseudoephedrine with rhinomanometry and MRI. Rhinology 43:205–
209.
Chervinsky P, Nayak A, Rooklin A, Danzig M (2005) Efficacy and safety of
desloratadine/pseudoephedrine tablet, 2.5/120 mg two times a day, versus individual
components in the treatment of patients with seasonal allergic rhinitis. Allergy Asthma Proc
26:391–396.
Chiang Y-C, Shyur S-D, Chen T-L, Huang L-H, Wen T-C, Lin M-T, Yang H-C, Liang P-H, Kao
Y-H, Wang T-C (2006) A randomized controlled trial of cetirizine plus pseudoephedrine
versus loratadine plus pseudoephedrine for perennial allergic rhinitis. Asian Pac J Allergy
Immunol 24:97–103.

91
Chua SS, Benrimoj SI, Gordon RD, Williams G (1989) A controlled clinical trial on the
cardiovascular effects of single doses of pseudoephedrine in hypertensive patients. Br J
Clin Pharmacol 28:369–372.
Cohen BM (1972) Clinical and physiologic "significance" of drug-induced changes in nasal
flow/resistance. Eur J Clin Pharmacol 5:81–86.
Connell JT (1969) Effectiveness of topical nasal decongestants. Ann Allergy 27:541–546.
Connell JT (1979) A novel method to assess antihistamine and decongestant efficacy. Ann
Allergy 42:278–285.
Connell JT, Gold AJ, Zola EM, Paule CL (1984) Efficacy of a timed-release
antihistamine/decongestant tablet for symptoms of nasal allergy. Drug Intell Clin Pharm
18:244–247.
Connell JT, Williams BO, Allen S, Cato A, Perkins JG (1982) A double-blind controlled
evaluation of Actifed and its individual constituents in allergic rhinitis. J Int Med Res 10:341–
347.
CORREN J, HARRIS A, AARONSON D, BEAUCHER W, BERKOWITZ R, BRONSKY E,
CHEN R, CHERVINSKY P, COHEN R, FOURRE J (1997) Efficacy and safety of loratadine
plus pseudoephedrine in patients with seasonal allergic rhinitis and mild asthma. Journal
of Allergy and Clinical Immunology 100:781–788.
Day JH, Briscoe MP, Ratz JD, Danzig M, Yao R (2009) Efficacy of loratadine-montelukast on
nasal congestion in patients with seasonal allergic rhinitis in an environmental exposure
unit. Annals of Allergy, Asthma & Immunology 102:328–338.
Debelic M, Radielovic P (1973) Zur Therapie der banalen Rhinitis. Doppelblind-kontrollierte
klinische Prüfung. Schweiz Rundsch Med Prax 62:1074–1077.
Diamond L, Gerson K, Cato A, Peace K, Perkins JG (1981) An evaluation of triprolidine and
pseudoephedrine in the treatment of allergic rhinitis. Ann Allergy 47:87–91.
Doyle WJ, Riker DK, McBride TP, Hayden FG, Hendley JO, Swarts JD, Gwaltney JM (1993)
Therapeutic effects of an anticholinergic-sympathomimetic combination in induced
rhinovirus colds. Ann Otol Rhinol Laryngol 102:521–527.
Dressler WE, Myers T, Rankell AS, London SJ, Poetsch CE (1977) A system of
rhinomanometry in the clinical evaluation of nasal decongestants. Ann Otol Rhinol Laryngol
86:310–317.
Dykewicz MS et al. (2017) Treatment of seasonal allergic rhinitis: An evidence-based focused
2017 guideline update. Ann Allergy Asthma Immunol 119:489-511.e41.
Eccles R, Jawad M, Jawad S, Ridge D, North M, Jones E, Burnett I (2006) Efficacy of a
paracetamol-pseudoephedrine combination for treatment of nasal congestion and pain-
related symptoms in upper respiratory tract infection. Curr Med Res Opin 22:2411–2418.

92
Eccles R, Eriksson M, Garreffa S, Chen SC (2008) The nasal decongestant effect of
xylometazoline in the common cold. Am J Rhinol 22:491–496.
Eccles R, Jawad MSM, Jawad SSM, Angello JT, Druce HM (2005) Efficacy and safety of single
and multiple doses of pseudoephedrine in the treatment of nasal congestion associated
with common cold. Am J Rhinol 19:25–31.
Eccles R, Voelker M (2014) Analgesic and Decongestant Efficacy of the Combination of Aspirin
with Pseudoephedrine in Patients With Symptoms of Upper Respiratory Tract Infection.
Clin Pharmacol Drug Dev 3:118–125.
Edwards N, Elcock HW, Pyke R, Griffiths MV, Resouly A (1973) An antihistamine-
sympathomimetic amine combination in vasomotor rhinitis. Practitioner 211:220–223.
Ellis AK, Soliman M, Steacy L, Boulay M-È, Boulet L-P, Keith PK, Vliagoftis H, Waserman S,
Neighbour H (2015) The Allergic Rhinitis - Clinical Investigator Collaborative (AR-CIC):
nasal allergen challenge protocol optimization for studying AR pathophysiology and
evaluating novel therapies. Allergy Asthma Clin Immunol 11:16.
Empey DW, Bye C, Hodder M, Hughes DT (1975) A Double-blind crossover trial of
pseudoephedrine and triprolidine, alone and in combination, for the treatment of allergenic
rhinitis. Ann Allergy 34:41–46.
Empey DW, Frosolono MF, Hughes DT, Perkins JG (1984) Comparison of pseudoephedrine
and triprolidine, alone and in combination in preventing nasal congestion in subjects with
allergic rhinitis using nasal histamine challenge. Br J Clin Pharmacol 18:86–89.
Erffmeyer JE, McKenna WR, Lieberman PL, Yoo TJ, Taylor WW (1982) Efficacy of
phenylephrine-phenylpropanolamine in the treatment of rhinitis. Southern medical journal
75:562–564.
European Medicines Agency (1998) Terfenadine | European Medicines Agency.
https://www.ema.europa.eu/en/medicines/human/referrals/terfenadine. Accessed Sep 17,
2020.
Falliers CJ, Redding MA (1980) Controlled comparison of anew antihistamine-decongestant
combination to its individual components. Ann Allergy 45:75–80.
FDA (2004) Final Rule Declaring Dietary Supplements Containing Ephedrine Alkaloids
Adulterated Because They Present an Unreasonable Risk; Final Rule.
https://www.govinfo.gov/content/pkg/FR-2004-02-11/html/04-2912.htm. Accessed Oct 12,
2020.
Ferguson EA, Eccles R (1997) Changes in nasal nitric oxide concentration associated with
symptoms of common cold and treatment with a topical nasal decongestant. Acta
Otolaryngol 117:614–617.

93
Gelbe Liste (2019) Nichtsteroidale Antirheumatika/Antiphlogistika (NSAR). https://www.gelbe-
liste.de/wirkstoffgruppen/nichtsteroidale-antiphlogistika-antirheumatika. Accessed Sep 10,
2020.
Grosclaude M, Mees K, Pinelli ME, Lucas M, van de Venne H (1997) Cetirizine and
pseudoephedrine retard, given alone or in combination, in patients with seasonal allergic
rhinitis. Rhinology 35:67–73.
Grossman J, Bronsky EA, Lanier BQ, Linzmayer MI, Moss BA, Schenkel EJ, Selner JC (1989)
Loratadine-pseudoephedrine combination versus placebo in patients with seasonal allergic
rhinitis. Ann Allergy 63:317–321.
Grubbe RE, Lumry WR, Anolik R (2009) Efficacy and safety of desloratadine/pseudoephedrine
combination vs its components in seasonal allergic rhinitis. J Investig Allergol Clin Immunol
19:117–124.
Guerra S, Sherrill DL, Martinez FD, Barbee RA (2002) Rhinitis as an independent risk factor
for adult-onset asthma. Journal of Allergy and Clinical Immunology 109:419–425.
Gwaltney JM, Druce HM (1997) Efficacy of brompheniramine maleate for the treatment of
rhinovirus colds. Clin Infect Dis 25:1188–1194.
Gwaltney JM, Hayden FG (1992) Psychological stress and the common cold. N Engl J Med
326:644-5; author reply 645-6.
Hamilton LH, Chobanian SL, Cato A, Perkins JG (1982) A study of sustained action
pseudoephedrine in allergic rhinitis. Ann Allergy 48:87–92.
Heikkinen T, Järvinen A (2003) The common cold. The Lancet 361:51–59.
Henauer S, Seppey M, Huguenot C, Pécoud A (1991) Effects of terfenadine and
pseudoephedrine, alone and in combination in a nasal provocation test and in perennial
rhinitis. Eur J Clin Pharmacol 41:321–324.
Hilding DA (1994) Literature review: the common cold. Ear Nose Throat J 73:639-43, 46-7.
Horak F, Toth J, Marks B, Stübner UP, Berger UE, Jäger S, Burtin B, Duby C (1998) Efficacy
and safety relative to placebo of an oral formulation of cetirizine and sustained-release
pseudoephedrine in the management of nasal congestion. Allergy 53:849–856.
Horak F, Zieglmayer P, Zieglmayer R, Lemell P, Yao R, Staudinger H, Danzig M (2009) A
placebo-controlled study of the nasal decongestant effect of phenylephrine and
pseudoephedrine in the Vienna Challenge Chamber. Annals of Allergy, Asthma &
Immunology 102:116–120.
Howarth PH, Harrison K, Smith S (1993) The influence of terfenadine and pseudo-ephedrine
alone and in combination on allergen-induced rhinitis. Int Arch Allergy Immunol 101:318–
321.
Jackson RH, Garrido R, Lynes TE (1975) Ru-Tuss in the symptomatic treatment of allergic
rhinitis. Ann Allergy 35:172–174.

94
Jadad AR, Moore R, Carroll D, Jenkinson C, Reynolds DM, Gavaghan DJ, McQuay HJ (1996)
Assessing the quality of reports of randomized clinical trials: Is blinding necessary?
Controlled Clinical Trials 17:1–12.
Jawad SS, Eccles R (1998) Effect of pseudoephedrine on nasal airflow in patients with nasal
congestion associated with common cold. Rhinology 36:73–76.
Jiang R-S (2006) Efficacy of a leukotriene receptor antagonist in the treatment of perennial
allergic rhinitis. J Otolaryngol 35:117–121.
Kaminszczik I, Barbon L (1983) Relieving symptoms of upper respiratory allergies and the
common cold: azatadine maleate/pseudoephedrine sulfate syrup versus placebo. J Int Med
Res 11:101–107.
Kim SY, Chang Y-J, Cho HM, Hwang Y-W, Moon YS (2015) Non-steroidal anti-inflammatory
drugs for the common cold. Cochrane Database Syst Rev:CD006362.
Klenow S, Latté K, Wegewitz U, Dusemund P, Pöting A, Appel K, Großklaus R, Schumann R,
Lampen A (2012) Risikobewertung von Pflanzen und pflanzlichen Zubereitungen. Berlin:
BfR.
Klimek L, Schumacher H, Schütt T, Gräter H, Mueck T, Michel MC (2017) Factors associated
with efficacy of an ibuprofen/pseudoephedrine combination drug in pharmacy customers
with common cold symptoms. Int J Clin Pract 71.
LaForce C, Gentile DA, Skoner DP (2008) A randomized, double-blind, parallel-group,
multicenter, placebo-controlled study of the safety and efficacy of extended-release
guaifenesin/pseudoephedrine hydrochloride for symptom relief as an adjunctive therapy to
antibiotic treatment of acute respiratory infections. Postgrad Med 120:53–59.
Latte J, Taverner D (2007) Clinical trial of 3 days of treatment with oral pseudoephedrine for
the common cold in the southern hemisphere. Am J Rhinol 21:452–455.
Latte J, Taverner D, Slobodian P, Shakib S (2004) A randomized, double-blind, placebo-
controlled trial of pseudoephedrine in coryza. Clin Exp Pharmacol Physiol 31:429–432.
Lebacq E, Gline JP, Joue P, Stockis A, Calderon P, van Schoor O, Deroubaix X, Schwegler F
(1994) Exploratory study of the decongestive effect of Rhinopront syrup in adults and in
children with acute rhinitis. Clin Trials Metaanal 29:113–124.
Loose I, Winkel M (2004) Clinical, double-blind, placebo-controlled study investigating the
combination of acetylsalicylic acid and pseudoephedrine for the symptomatic treatment of
nasal congestion associated with common cold. Arzneimittelforschung 54:513–521.
Mäkelä MJ, Puhakka T, Ruuskanen O, Leinonen M, Saikku P, Kimpimäki M, Blomqvist S,
Hyypiä T, Arstila P (1998) Viruses and bacteria in the etiology of the common cold. J Clin
Microbiol 36:539–542.

95
McFadden EA, Gungor A, Ng B, Mamikoglu B, Moinuddin R, Corey J (2000)
Loratadine/pseudoephedrine for nasal symptoms in seasonal allergic rhinitis: a double-
blind, placebo-controlled study. Ear Nose Throat J 79:254, 257-8, 260 passim.
Meltzer EO, Berman GD, Corren J, Pedinoff AJ, Doyle G, Waksman JA, Butkerait P, Cooper
SA, Berlin RG, Wason S (2004) Addition of ibuprofen to pseudoephedrine and
chlorpheniramine in the treatment of seasonal allergic rhinitis. Annals of Allergy, Asthma &
Immunology 93:452–459.
Meltzer EO, Casale TB, Gold MS, O'Connor R, Reitberg D, del Rio E, Weiler JM, Weiler K
(2003) Efficacy and safety of clemastine-pseudoephedrine-acetaminophen versus
pseudoephedrine-acetaminophen in the treatment of seasonal allergic rhinitis in a 1-day,
placebo-controlled park study. Annals of Allergy, Asthma & Immunology 90:79–86.
Meltzer EO, Ratner PH, McGraw T (2015) Oral Phenylephrine HCl for Nasal Congestion in
Seasonal Allergic Rhinitis: A Randomized, Open-label, Placebo-controlled Study. J Allergy
Clin Immunol Pract 3:702–708.
Meltzer EO, Ratner PH, McGraw T (2016) Phenylephrine hydrochloride modified-release
tablets for nasal congestion: a randomized, placebo-controlled trial in allergic rhinitis
patients. Ann Allergy Asthma Immunol 116:66–71.
Meran A, Morse J, Gibbs TG (1990) A cross-over comparison of acrivastine, pseudoephedrine
and their combination in seasonal allergic rhinitis. Rhinology 28:33–40.
Moinuddin R, deTineo M, Maleckar B, Naclerio RM, Baroody FM (2004) Comparison of the
combinations of fexofenadine-pseudoephedrine and loratadine-montelukast in the
treatment of seasonal allergic rhinitis. Annals of Allergy, Asthma & Immunology 92:73–79.
Mucha SM, deTineo M, Naclerio RM, Baroody FM (2006) Comparison of montelukast and
pseudoephedrine in the treatment of allergic rhinitis. Arch Otolaryngol Head Neck Surg
132:164–172.
Myers EN, Panda NK, Mann SBS (1998) Comparative Efficacy and Safety of Terfenadine with
Pseudoephedrine and Terfenadine alone in Allergic Rhinitis. Otolaryngol Head Neck Surg
118:253–255.
Nathan RA, Finn AF, LaForce C, Ratner P, Chapman D, Guia EC de, Hewlett D, Kramer B
(2006) Comparison of cetirizine-pseudoephedrine and placebo in patients with seasonal
allergic rhinitis and concomitant mild-to-moderate asthma: randomized, double-blind study.
Annals of Allergy, Asthma & Immunology 97:389–396.
Picon PD, Costa MB, da Veiga Picon R, Fendt LCC, Suksteris ML, Saccilotto IC, Dornelles
AD, Schmidt LFC (2013) Symptomatic treatment of the common cold with a fixed-dose
combination of paracetamol, chlorphenamine and phenylephrine: a randomized, placebo-
controlled trial. BMC Infect Dis 13:556.

96
Pleskow W, Grubbe R, Weiss S, Lutsky B (2005) Efficacy and safety of an extended-release
formulation of desloratadine and pseudoephedrine vs the individual components in the
treatment of seasonal allergic rhinitis. Annals of Allergy, Asthma & Immunology 94:348–
354.
Prenner B, Anolik R, Danzig M, Yao R (2009) Efficacy and safety of fixed-dose
loratadine/montelukast in seasonal allergic rhinitis: effects on nasal congestion. Allergy
Asthma Proc 30:263–269.
Reinecke S, Tschaikin M (2005) Untersuchung der Wirksamkeit von Oxymetazolin auf die
Rhinitisdauer. Ergebnisse einer plazebokontrollierten Doppelblindstudie bei akuter Rhinitis.
MMW Fortschr Med 147 Suppl 3:113–118.
Robert M, Llorens M, García E, Luria X (2004) Efficacy and tolerability of ebastine 10 mg plus
pseudoephedrine 120 mg in the symptomatic relief of the common cold. Eur J Intern Med
15:242–247.
Roth RP, Cantekin EI, Bluestone CD, Welch RM, Cho YW (1977) Nasal decongestant activity
of pseudoephedrine. Ann Otol Rhinol Laryngol 86:235–242.
Schachtel BP, Voelker M, Sanner KM, Gagney D, Bey M, Schachtel EJ, Becka M (2010)
Demonstration of the analgesic efficacy and dose-response of acetylsalicylic acid with
pseudoephedrine. J Clin Pharmacol 50:1429–1437.
Schenkel E, Corren J, Murray JJ (2002) Efficacy of once-daily desloratadine/pseudoephedrine
for relief of nasal congestion. Allergy Asthma Proc 23:325–330.
Selner JC, Koepke JW, Staudenmayer H, Dolen WK, Glover GC, Linzmayer MI, Mooney JJ,
Wiener MB (1991) Assessment of nasal patency by rhinoscopic measurement of cross
sectional nasal airway area: correlation with subjective nasal symptoms. Ann Allergy
66:43–47.
Sperber SJ, Sorrentino JV, Riker DK, Hayden FG (1989) Evaluation of an alpha agonist alone
and in combination with a nonsteroidal antiinflammatory agent in the treatment of
experimental rhinovirus colds. Bull N Y Acad Med 65:145–160.
Sperber SJ, Turner RB, Sorrentino JV, O'Connor RR, Rogers J, Gwaltney JM (2000)
Effectiveness of pseudoephedrine plus acetaminophen for treatment of symptoms
attributed to the paranasal sinuses associated with the common cold. Arch Fam Med
9:979–985.
Stokes JR, Romero FA, Allan RJ, Phillips PG, Hackman F, Misfeldt J, Casale TB (2012) The
effects of an H3 receptor antagonist (PF-03654746) with fexofenadine on reducing allergic
rhinitis symptoms. J Allergy Clin Immunol 129:409-12, 412.e1-2.
Storms WW, Bodman SF, Nathan RA, Chervinsky P, Banov CH, Dockhorn RJ, Jarmoszuk I,
Zeitz HJ, McGeady SJ, Pinnas JL, Greenstein S (1989) SCH 434: A new

97
antihistamine/decongestant for seasonal allergic rhinitis. Journal of Allergy and Clinical
Immunology 83:1083–1090.
Stroh JE, Ayars GH, Bernstein IL, Kemp JP, Podleski WK, Prenner BM, Schoenwetter WF,
Salzmann JK (1988) A comparative tolerance study of terfenadine-pseudoephedrine
combination tablets and pseudoephedrine tablets in patients with allergic or vasomotor
rhinitis. J Int Med Res 16:420–427.
Stübner UP, Toth J, Marks B, Berger UE, Burtin B, Horak F (2001) Efficacy and safety of an
oral formulation of cetirizine and prolonged-release pseudoephedrine versus
xylometazoline nasal spray in nasal congestion. Arzneimittelforschung 51:904–910.
Stull DE, Krouse J, Meltzer EO, Roberts L, Kim S, Frank L, Naclerio R, Lund V, Long A (2007)
Development and validation of the Congestion Quantifier seven-item test (CQ7): a
screening tool for nasal congestion.
Sussman GL, Mason J, Compton D, Stewart J, Ricard N (1999) The efficacy and safety of
fexofenadine HCl and pseudoephedrine, alone and in combination, in seasonal allergic
rhinitis. J Allergy Clin Immunol 104:100–106.
Tarasido JC (1980) Azatadine maleate/pseudoephedrine sulfate repetabs versus placebo in
the treatment of severe seasonal allergic rhinitis. J Int Med Res 8:391–394.
Taverner D, Danz C, Economos D (1999) The effects of oral pseudoephedrine on nasal
patency in the common cold: a double-blind single-dose placebo-controlled trial. Clin
Otolaryngol Allied Sci 24:47–51.
Virtanen H (1983) A slow release combined preparation (dexchlorpheniramine +
pseudoephedrine) for symptomatic treatment of the common cold. J Laryngol Otol 97:159–
163.
Weiler JM, Gellhaus M, Donnelly A, Weiler K (1990) Randomized, double-blind, parallel
groups, placebo-controlled study of efficacy and safety of Rynatan in the treatment of
allergic rhinitis using an acute model. Ann Allergy 64:63–67.
Westman M, Kull I, Lind T, Melén E, Stjärne P, Toskala E, Wickman M, Bergström A (2013)
The link between parental allergy and offspring allergic and nonallergic rhinitis. Allergy
68:1571–1578.
Wheatley LM, Togias A (2015) Clinical practice. Allergic rhinitis. N Engl J Med 372:456–463.
Zhang Y, Mallefet P (2018) Time-to-onset of cold and flu symptom relief: A randomized,
double-blind, placebo-controlled pilot study for a multi-symptom combination product. Int J
Clin Pharmacol Ther 56:604–611.
Zubizaretta J (1980) Azatadine maleate/pseudoephedrine sulfate repetabs versus placebo in
the treatment of severe perennial allergic rhinitis. J Int Med Res 8:395–399.
Zumpft CW (1975) Double-blind comparison of metizoline hydrochloride (Ellsyl) and
phenylephrine in allergic and vasomotor rhinitis. Curr Ther Res Clin Exp 17:40–46.
98
7 Abkürzungsverzeichnis
AEU = allergen exposure unit AUC = area under the curve BU/ml = Bethesda Unit/ml CHM = Cochran-Mantel-Haenszel COX = Cyclooxgygenase ECC = environmental challenge chamber IPFR = inspiratory peak flow rate ITT = intention-to-treat MCA = minimal cross-sectional area MSS = maximal symptom score NAC = nasal airflow conductance NAR = nasal air flow resistance NCS = nasal congestion score NSAR = nichtsteroidales Antirheumatikum NSS = nasal stuffiness score PCR = Polymerase-Kettenreaktion PE = Phenylephrin PNIF = peak nasal inspiratory flow PP = per protocol PSE = Pseudoephedrin RAST = Radio-Allergo-Sorbent-Test RP = Rezeptpflichtig RPF = Rezeptfrei SS = symptom score TMD = Tagesmaximaldosis TNNSS = total nonnasal symptom score TNSS = total nasal symptom score TSS = total symptom score UAW = unerwünschte Arzneimittelwirkung
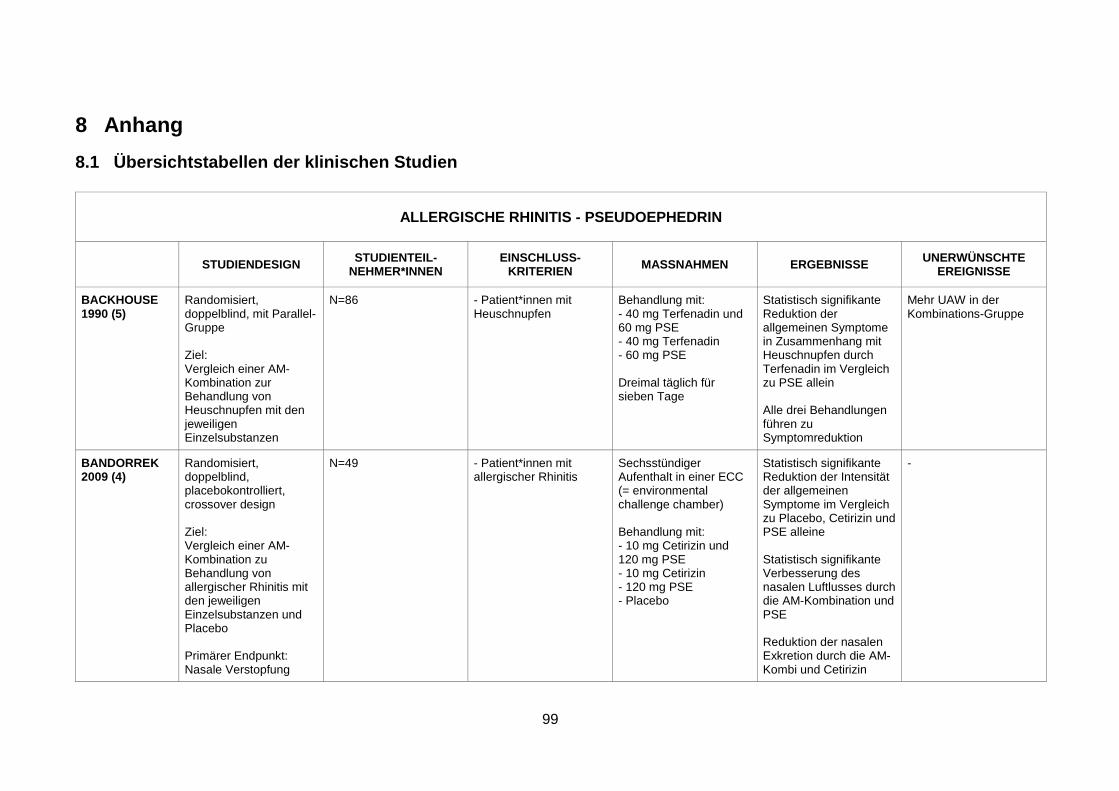
99
8 Anhang 8.1 Übersichtstabellen der klinischen Studien
ALLERGISCHE RHINITIS - PSEUDOEPHEDRIN
STUDIENDESIGN STUDIENTEIL-NEHMER*INNEN
EINSCHLUSS- KRITERIEN MASSNAHMEN ERGEBNISSE UNERWÜNSCHTE
EREIGNISSE
BACKHOUSE 1990 (5)
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich einer AM-Kombination zur Behandlung von Heuschnupfen mit den jeweiligen Einzelsubstanzen
N=86 - Patient*innen mit Heuschnupfen
Behandlung mit: - 40 mg Terfenadin und 60 mg PSE - 40 mg Terfenadin - 60 mg PSE Dreimal täglich für sieben Tage
Statistisch signifikante Reduktion der allgemeinen Symptome in Zusammenhang mit Heuschnupfen durch Terfenadin im Vergleich zu PSE allein Alle drei Behandlungen führen zu Symptomreduktion
Mehr UAW in der Kombinations-Gruppe
BANDORREK 2009 (4)
Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Vergleich einer AM-Kombination zu Behandlung von allergischer Rhinitis mit den jeweiligen Einzelsubstanzen und Placebo Primärer Endpunkt: Nasale Verstopfung
N=49 - Patient*innen mit allergischer Rhinitis
Sechsstündiger Aufenthalt in einer ECC (= environmental challenge chamber) Behandlung mit: - 10 mg Cetirizin und 120 mg PSE - 10 mg Cetirizin - 120 mg PSE - Placebo
Statistisch signifikante Reduktion der Intensität der allgemeinen Symptome im Vergleich zu Placebo, Cetirizin und PSE alleine Statistisch signifikante Verbesserung des nasalen Luftlusses durch die AM-Kombination und PSE Reduktion der nasalen Exkretion durch die AM-Kombi und Cetirizin
-
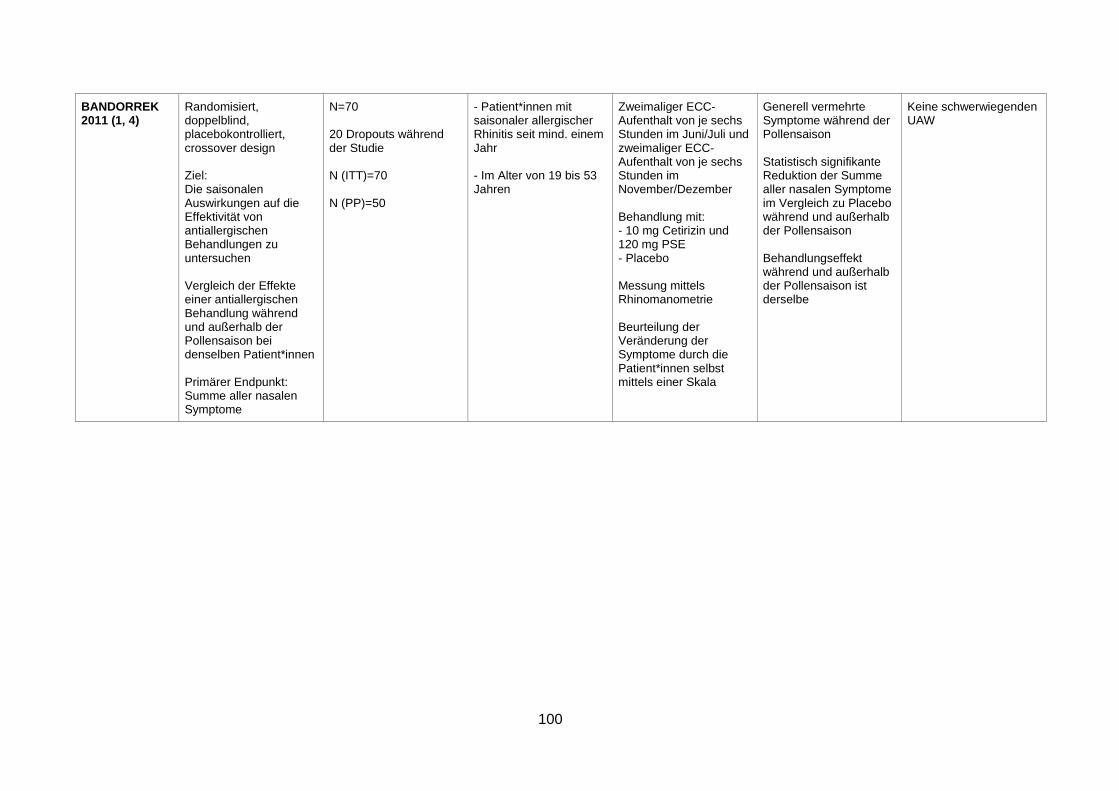
100
BANDORREK 2011 (1, 4)
Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Die saisonalen Auswirkungen auf die Effektivität von antiallergischen Behandlungen zu untersuchen Vergleich der Effekte einer antiallergischen Behandlung während und außerhalb der Pollensaison bei denselben Patient*innen Primärer Endpunkt: Summe aller nasalen Symptome
N=70 20 Dropouts während der Studie N (ITT)=70 N (PP)=50
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. einem Jahr - Im Alter von 19 bis 53 Jahren
Zweimaliger ECC- Aufenthalt von je sechs Stunden im Juni/Juli und zweimaliger ECC-Aufenthalt von je sechs Stunden im November/Dezember Behandlung mit: - 10 mg Cetirizin und 120 mg PSE - Placebo Messung mittels Rhinomanometrie Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Generell vermehrte Symptome während der Pollensaison Statistisch signifikante Reduktion der Summe aller nasalen Symptome im Vergleich zu Placebo während und außerhalb der Pollensaison Behandlungseffekt während und außerhalb der Pollensaison ist derselbe
Keine schwerwiegenden UAW
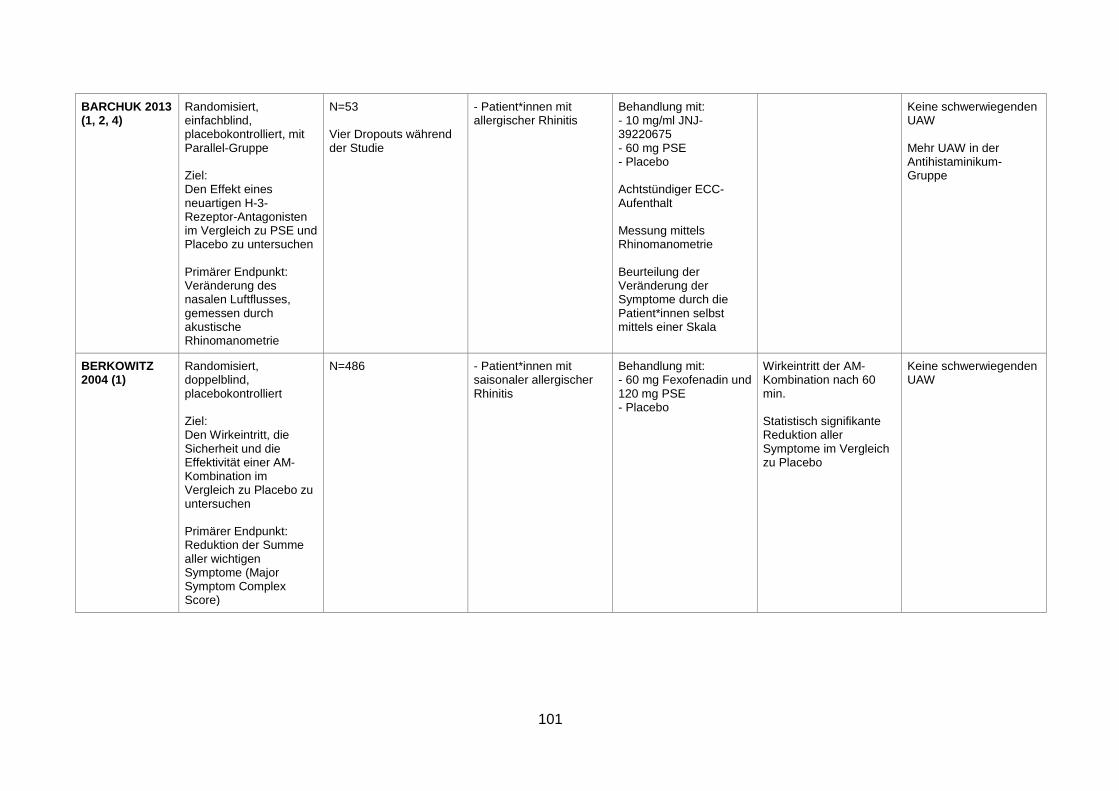
101
BARCHUK 2013 (1, 2, 4)
Randomisiert, einfachblind, placebokontrolliert, mit Parallel-Gruppe Ziel: Den Effekt eines neuartigen H-3-Rezeptor-Antagonisten im Vergleich zu PSE und Placebo zu untersuchen Primärer Endpunkt: Veränderung des nasalen Luftflusses, gemessen durch akustische Rhinomanometrie
N=53 Vier Dropouts während der Studie
- Patient*innen mit allergischer Rhinitis
Behandlung mit: - 10 mg/ml JNJ-39220675 - 60 mg PSE - Placebo Achtstündiger ECC-Aufenthalt Messung mittels Rhinomanometrie Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Keine schwerwiegenden UAW Mehr UAW in der Antihistaminikum-Gruppe
BERKOWITZ 2004 (1)
Randomisiert, doppelblind, placebokontrolliert Ziel: Den Wirkeintritt, die Sicherheit und die Effektivität einer AM-Kombination im Vergleich zu Placebo zu untersuchen Primärer Endpunkt: Reduktion der Summe aller wichtigen Symptome (Major Symptom Complex Score)
N=486 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 60 mg Fexofenadin und 120 mg PSE - Placebo
Wirkeintritt der AM-Kombination nach 60 min. Statistisch signifikante Reduktion aller Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW
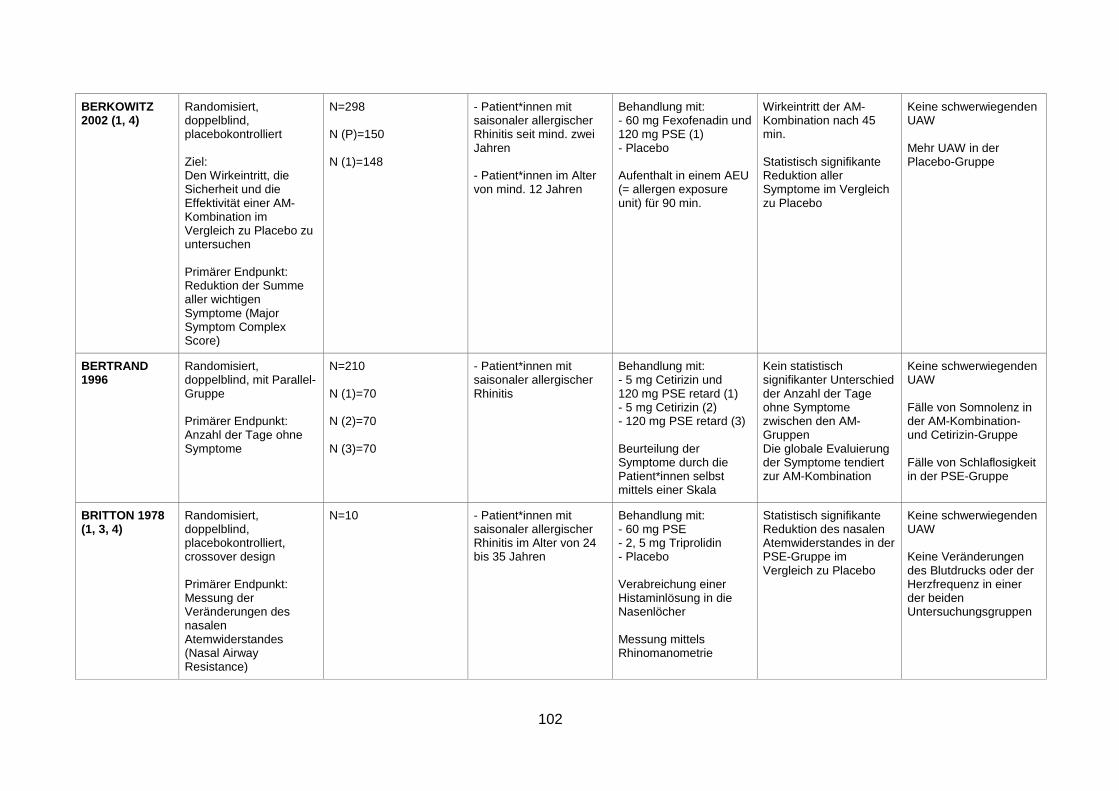
102
BERKOWITZ 2002 (1, 4)
Randomisiert, doppelblind, placebokontrolliert Ziel: Den Wirkeintritt, die Sicherheit und die Effektivität einer AM-Kombination im Vergleich zu Placebo zu untersuchen Primärer Endpunkt: Reduktion der Summe aller wichtigen Symptome (Major Symptom Complex Score)
N=298 N (P)=150 N (1)=148
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren - Patient*innen im Alter von mind. 12 Jahren
Behandlung mit: - 60 mg Fexofenadin und 120 mg PSE (1) - Placebo Aufenthalt in einem AEU (= allergen exposure unit) für 90 min.
Wirkeintritt der AM-Kombination nach 45 min. Statistisch signifikante Reduktion aller Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Mehr UAW in der Placebo-Gruppe
BERTRAND 1996
Randomisiert, doppelblind, mit Parallel-Gruppe Primärer Endpunkt: Anzahl der Tage ohne Symptome
N=210 N (1)=70 N (2)=70 N (3)=70
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 5 mg Cetirizin und 120 mg PSE retard (1) - 5 mg Cetirizin (2) - 120 mg PSE retard (3) Beurteilung der Symptome durch die Patient*innen selbst mittels einer Skala
Kein statistisch signifikanter Unterschied der Anzahl der Tage ohne Symptome zwischen den AM-Gruppen Die globale Evaluierung der Symptome tendiert zur AM-Kombination
Keine schwerwiegenden UAW Fälle von Somnolenz in der AM-Kombination- und Cetirizin-Gruppe Fälle von Schlaflosigkeit in der PSE-Gruppe
BRITTON 1978 (1, 3, 4)
Randomisiert, doppelblind, placebokontrolliert, crossover design Primärer Endpunkt: Messung der Veränderungen des nasalen Atemwiderstandes (Nasal Airway Resistance)
N=10 - Patient*innen mit saisonaler allergischer Rhinitis im Alter von 24 bis 35 Jahren
Behandlung mit: - 60 mg PSE - 2, 5 mg Triprolidin - Placebo Verabreichung einer Histaminlösung in die Nasenlöcher Messung mittels Rhinomanometrie
Statistisch signifikante Reduktion des nasalen Atemwiderstandes in der PSE-Gruppe im Vergleich zu Placebo
Keine schwerwiegenden UAW Keine Veränderungen des Blutdrucks oder der Herzfrequenz in einer der beiden Untersuchungsgruppen
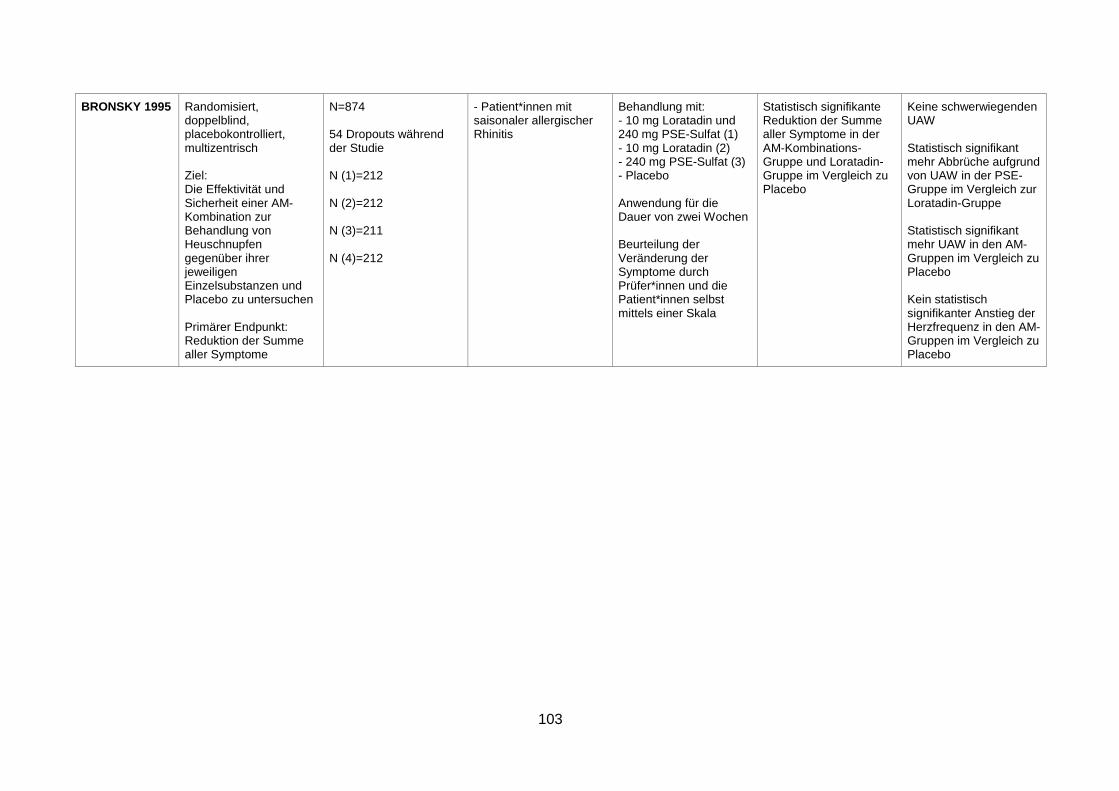
103
BRONSKY 1995 Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Die Effektivität und Sicherheit einer AM-Kombination zur Behandlung von Heuschnupfen gegenüber ihrer jeweiligen Einzelsubstanzen und Placebo zu untersuchen Primärer Endpunkt: Reduktion der Summe aller Symptome
N=874 54 Dropouts während der Studie N (1)=212 N (2)=212 N (3)=211 N (4)=212
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 10 mg Loratadin und 240 mg PSE-Sulfat (1) - 10 mg Loratadin (2) - 240 mg PSE-Sulfat (3) - Placebo Anwendung für die Dauer von zwei Wochen Beurteilung der Veränderung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Summe aller Symptome in der AM-Kombinations-Gruppe und Loratadin-Gruppe im Vergleich zu Placebo
Keine schwerwiegenden UAW Statistisch signifikant mehr Abbrüche aufgrund von UAW in der PSE-Gruppe im Vergleich zur Loratadin-Gruppe Statistisch signifikant mehr UAW in den AM-Gruppen im Vergleich zu Placebo Kein statistisch signifikanter Anstieg der Herzfrequenz in den AM-Gruppen im Vergleich zu Placebo
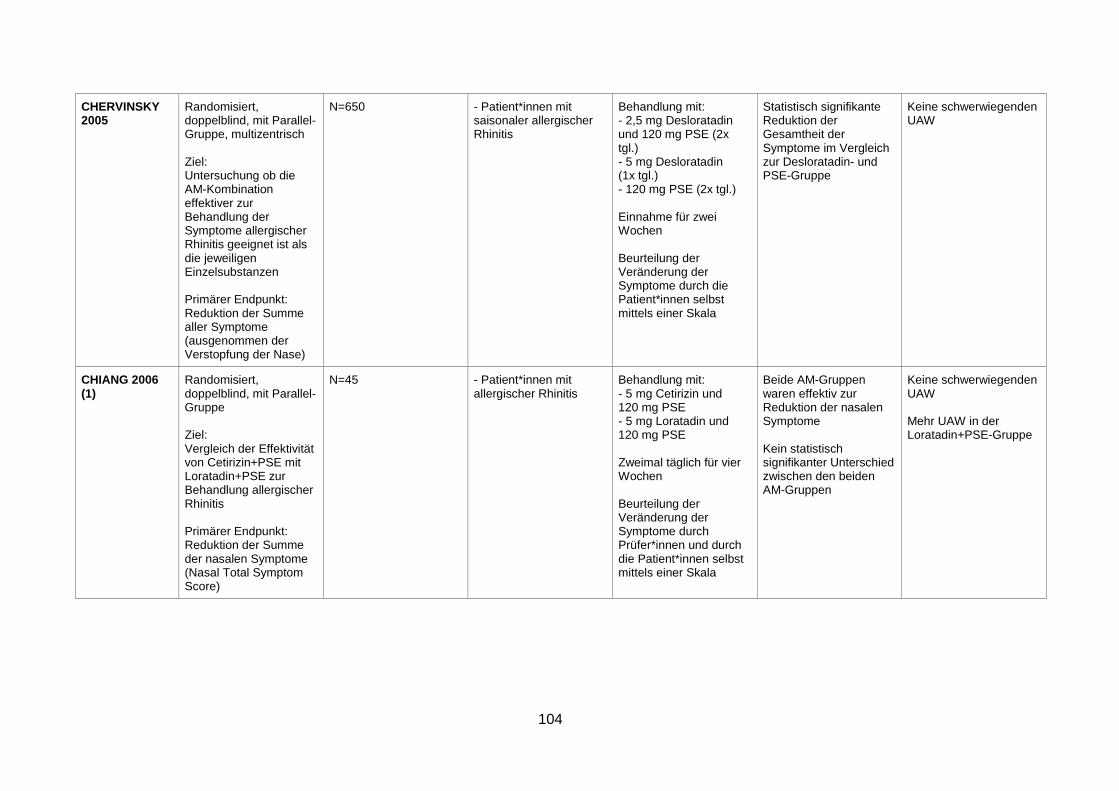
104
CHERVINSKY 2005
Randomisiert, doppelblind, mit Parallel-Gruppe, multizentrisch Ziel: Untersuchung ob die AM-Kombination effektiver zur Behandlung der Symptome allergischer Rhinitis geeignet ist als die jeweiligen Einzelsubstanzen Primärer Endpunkt: Reduktion der Summe aller Symptome (ausgenommen der Verstopfung der Nase)
N=650 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 2,5 mg Desloratadin und 120 mg PSE (2x tgl.) - 5 mg Desloratadin (1x tgl.) - 120 mg PSE (2x tgl.) Einnahme für zwei Wochen Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Gesamtheit der Symptome im Vergleich zur Desloratadin- und PSE-Gruppe
Keine schwerwiegenden UAW
CHIANG 2006 (1)
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich der Effektivität von Cetirizin+PSE mit Loratadin+PSE zur Behandlung allergischer Rhinitis Primärer Endpunkt: Reduktion der Summe der nasalen Symptome (Nasal Total Symptom Score)
N=45 - Patient*innen mit allergischer Rhinitis
Behandlung mit: - 5 mg Cetirizin und 120 mg PSE - 5 mg Loratadin und 120 mg PSE Zweimal täglich für vier Wochen Beurteilung der Veränderung der Symptome durch Prüfer*innen und durch die Patient*innen selbst mittels einer Skala
Beide AM-Gruppen waren effektiv zur Reduktion der nasalen Symptome Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen
Keine schwerwiegenden UAW Mehr UAW in der Loratadin+PSE-Gruppe
105
CONNELL 1979 (1, 2)
- Ziel: Vergleich der Effekte einer AM-Kombination zur Behandlung von Heuschnupfen mit den jeweiligen Einzelsubstanzen
- - Behandlung mit: - Azatadin maleat und PSE-Sulfat - Azatadin maleat - PSE-Sulfat
Kombination der beiden besser als die Einzelsubstanzen
-
CONNELL 1984 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Vergleich der Effektivität einer AM-Kombination zur Behandlung der Symptome von allergischer Rhinitis mit Placebo
N=94 N (P)=48 N (1)=46
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren
Behandlung mit: - 5 mg Carbinoxamin und 120 mg PSE (1) - Placebo Zweimal täglich für zwei Tage Messung des nasalen Luftflusses mittels Rhinomanometrie
Statistisch signifikante Reduktion der Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW
CONNELL 1982 (2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Vergleich einer AM-Kombination zur Behandlung der Symptome von Heuschnupfen mit den entsprechenden Einzelsubstanzen Primärer Endpunkt: Nasaler Luftfluss (Total Airflow Rates)
N=135 - Patient*innen mit Heuschnupfen seit mind. zwei Jahren
Behandlung mit: - 2,5 mg Triprolidin und 60 mg PSE - 2,5 mg Triprolidin - 60 mg PSE - Placebo Messung mittels Rhinomanometrie Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Verstopfung der Nase durch die Kombination im Vergleich zu Triprolidin und Placebo Statistisch signifikante Reduktion der Heuschnupfen-Symptome durch die Kombination im Vergleich zu PSE und Placebo
Keine schwerwiegenden UAW Vermehrt Fälle von Schläfrigkeit in der Kombinations- und Triprolidin-Gruppe
106
CORREN 1997 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Die Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis und mildem Asthma zu untersuchen
N=193 N (P)=96 N (1)=97 N (analysiert)=184
- Patient*innen mit allergischer Rhinitis und damit einhergehendem Asthma seit mind. zwei Jahren - Im Alter von 12 bis 70 Jahren
Behandlung mit: - 5 mg Loratadin und 20 mg PSE (1) - Placebo Zweimal tägliche Einnahme für sechs Wochen Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels eines Tagebuchs Messung der PEFR (=Peak Expiratory Flow Rates)
Statistisch signifikante Verbesserung der Summe aller Schnupfen-Symptome im Vergleich zu Placebo Statistisch signifikante Verbesserung der Summe aller Asthma-Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Vermehrt Fälle von Schlaflosigkeit in der AM-Gruppe
DIAMOND 1981 (1)
Doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination zur Behandlung allergischer Rhinitis zu untersuchen
- - Patient*innen mit allergischer Rhinitis
Behandlung mit: - Triprolidin und PSE - Placebo
- -
EMPEY 1975 (2) Doppelblind, placebokontrolliert, crossover design Ziel: Vergleich der Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis mit den entsprechenden Einzelsubstanzen und Placebo
N=40 - Patient*innen mit allergischer Rhinitis
Behandlung mit: - 2,5 mg Triprolidin und 60 mg PSE - 2,5 mg Triprolidin - 60 mg PSE - Placebo
Triprolidin und PSE sind statistisch signifikant besser als Placebo zur Reduktion der Symptome Die Kombination beider AM ist den Einzelsubstanzen überlegen
Keine schwerwiegenden UAW
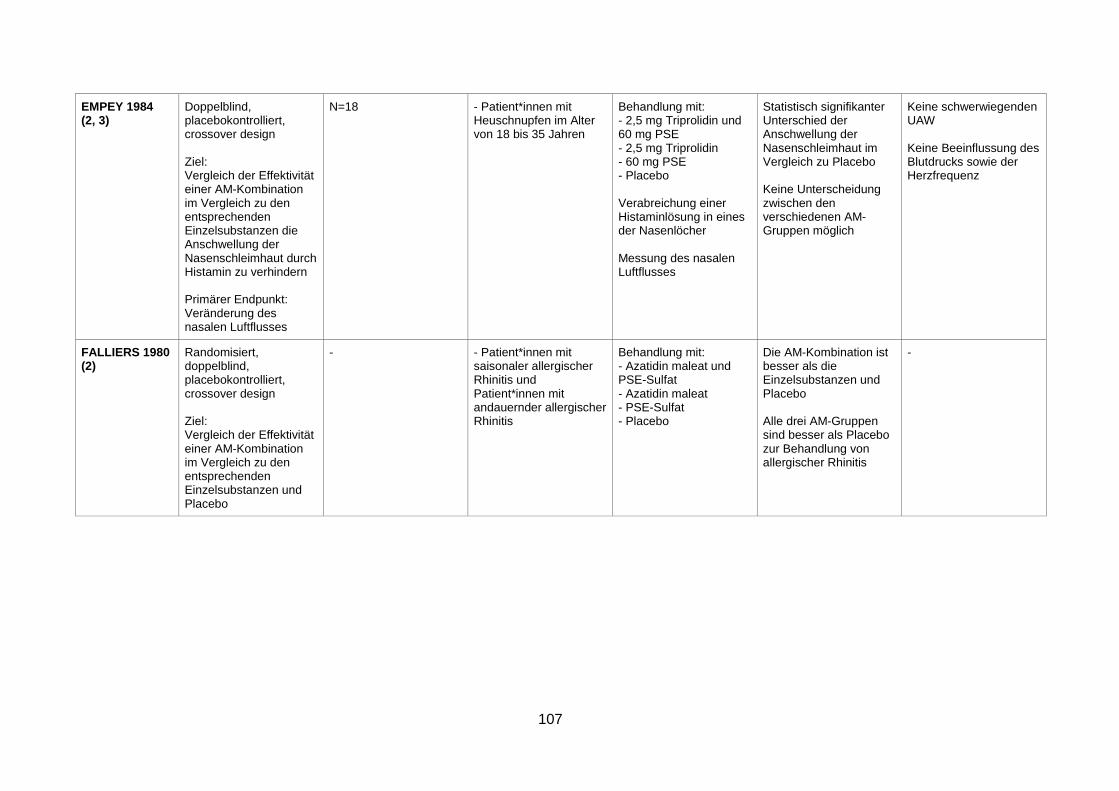
107
EMPEY 1984 (2, 3)
Doppelblind, placebokontrolliert, crossover design Ziel: Vergleich der Effektivität einer AM-Kombination im Vergleich zu den entsprechenden Einzelsubstanzen die Anschwellung der Nasenschleimhaut durch Histamin zu verhindern Primärer Endpunkt: Veränderung des nasalen Luftflusses
N=18 - Patient*innen mit Heuschnupfen im Alter von 18 bis 35 Jahren
Behandlung mit: - 2,5 mg Triprolidin und 60 mg PSE - 2,5 mg Triprolidin - 60 mg PSE - Placebo Verabreichung einer Histaminlösung in eines der Nasenlöcher Messung des nasalen Luftflusses
Statistisch signifikanter Unterschied der Anschwellung der Nasenschleimhaut im Vergleich zu Placebo Keine Unterscheidung zwischen den verschiedenen AM-Gruppen möglich
Keine schwerwiegenden UAW Keine Beeinflussung des Blutdrucks sowie der Herzfrequenz
FALLIERS 1980 (2)
Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Vergleich der Effektivität einer AM-Kombination im Vergleich zu den entsprechenden Einzelsubstanzen und Placebo
- - Patient*innen mit saisonaler allergischer Rhinitis und Patient*innen mit andauernder allergischer Rhinitis
Behandlung mit: - Azatidin maleat und PSE-Sulfat - Azatidin maleat - PSE-Sulfat - Placebo
Die AM-Kombination ist besser als die Einzelsubstanzen und Placebo Alle drei AM-Gruppen sind besser als Placebo zur Behandlung von allergischer Rhinitis
-
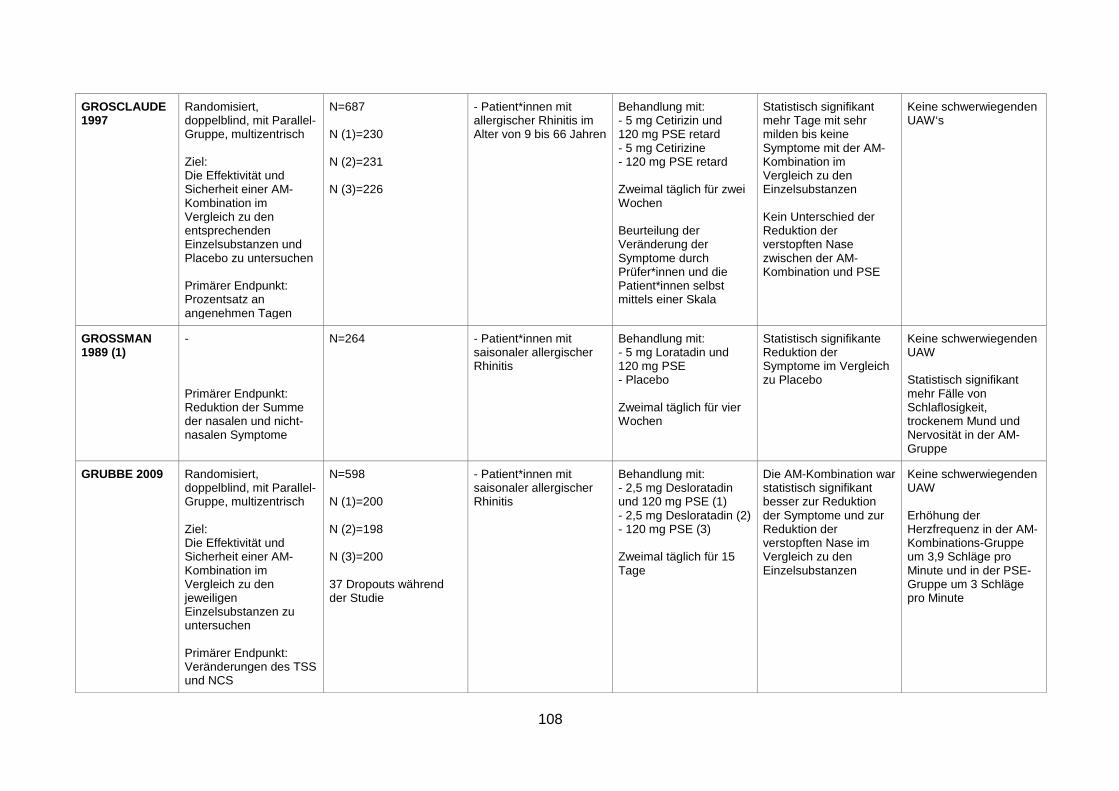
108
GROSCLAUDE 1997
Randomisiert, doppelblind, mit Parallel-Gruppe, multizentrisch Ziel: Die Effektivität und Sicherheit einer AM-Kombination im Vergleich zu den entsprechenden Einzelsubstanzen und Placebo zu untersuchen Primärer Endpunkt: Prozentsatz an angenehmen Tagen
N=687 N (1)=230 N (2)=231 N (3)=226
- Patient*innen mit allergischer Rhinitis im Alter von 9 bis 66 Jahren
Behandlung mit: - 5 mg Cetirizin und 120 mg PSE retard - 5 mg Cetirizine - 120 mg PSE retard Zweimal täglich für zwei Wochen Beurteilung der Veränderung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Statistisch signifikant mehr Tage mit sehr milden bis keine Symptome mit der AM-Kombination im Vergleich zu den Einzelsubstanzen Kein Unterschied der Reduktion der verstopften Nase zwischen der AM-Kombination und PSE
Keine schwerwiegenden UAW‘s
GROSSMAN 1989 (1)
- Primärer Endpunkt: Reduktion der Summe der nasalen und nicht-nasalen Symptome
N=264 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 5 mg Loratadin und 120 mg PSE - Placebo Zweimal täglich für vier Wochen
Statistisch signifikante Reduktion der Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Statistisch signifikant mehr Fälle von Schlaflosigkeit, trockenem Mund und Nervosität in der AM-Gruppe
GRUBBE 2009 Randomisiert, doppelblind, mit Parallel-Gruppe, multizentrisch Ziel: Die Effektivität und Sicherheit einer AM-Kombination im Vergleich zu den jeweiligen Einzelsubstanzen zu untersuchen Primärer Endpunkt: Veränderungen des TSS und NCS
N=598 N (1)=200 N (2)=198 N (3)=200 37 Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 2,5 mg Desloratadin und 120 mg PSE (1) - 2,5 mg Desloratadin (2) - 120 mg PSE (3) Zweimal täglich für 15 Tage
Die AM-Kombination war statistisch signifikant besser zur Reduktion der Symptome und zur Reduktion der verstopften Nase im Vergleich zu den Einzelsubstanzen
Keine schwerwiegenden UAW Erhöhung der Herzfrequenz in der AM-Kombinations-Gruppe um 3,9 Schläge pro Minute und in der PSE-Gruppe um 3 Schläge pro Minute
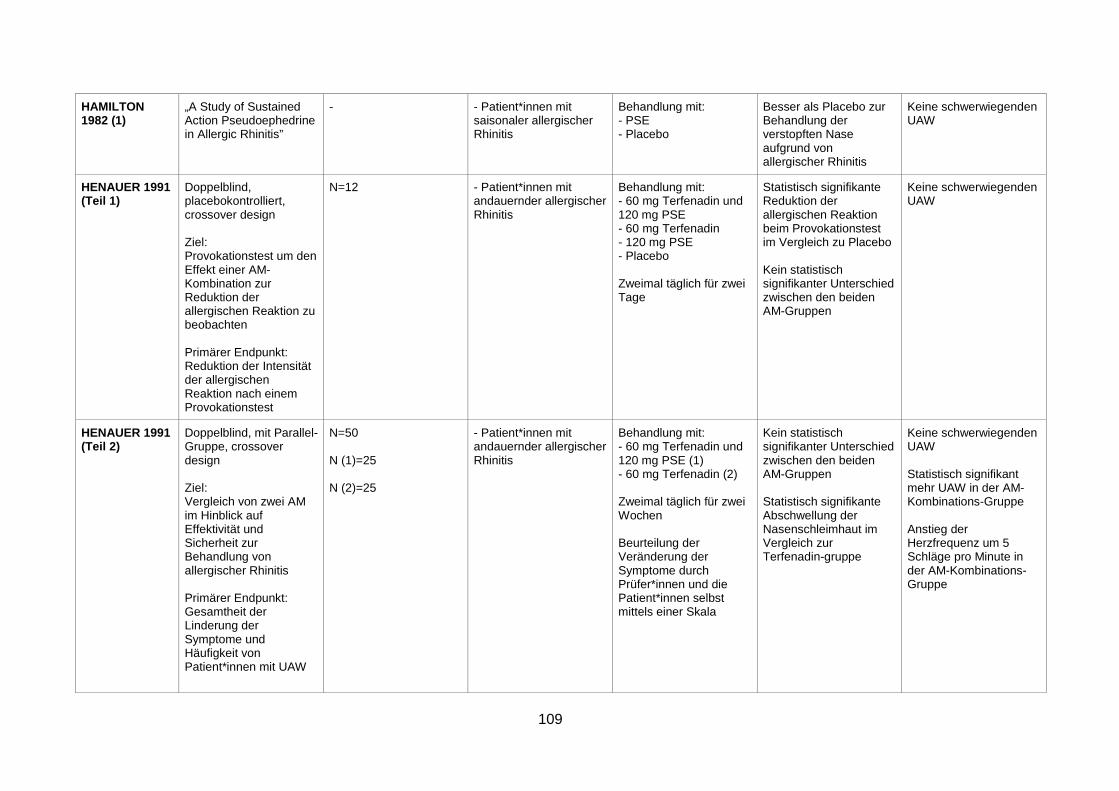
109
HAMILTON 1982 (1)
„A Study of Sustained Action Pseudoephedrine in Allergic Rhinitis”
- - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - PSE - Placebo
Besser als Placebo zur Behandlung der verstopften Nase aufgrund von allergischer Rhinitis
Keine schwerwiegenden UAW
HENAUER 1991 (Teil 1)
Doppelblind, placebokontrolliert, crossover design Ziel: Provokationstest um den Effekt einer AM-Kombination zur Reduktion der allergischen Reaktion zu beobachten Primärer Endpunkt: Reduktion der Intensität der allergischen Reaktion nach einem Provokationstest
N=12 - Patient*innen mit andauernder allergischer Rhinitis
Behandlung mit: - 60 mg Terfenadin und 120 mg PSE - 60 mg Terfenadin - 120 mg PSE - Placebo Zweimal täglich für zwei Tage
Statistisch signifikante Reduktion der allergischen Reaktion beim Provokationstest im Vergleich zu Placebo Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen
Keine schwerwiegenden UAW
HENAUER 1991 (Teil 2)
Doppelblind, mit Parallel-Gruppe, crossover design Ziel: Vergleich von zwei AM im Hinblick auf Effektivität und Sicherheit zur Behandlung von allergischer Rhinitis Primärer Endpunkt: Gesamtheit der Linderung der Symptome und Häufigkeit von Patient*innen mit UAW
N=50 N (1)=25 N (2)=25
- Patient*innen mit andauernder allergischer Rhinitis
Behandlung mit: - 60 mg Terfenadin und 120 mg PSE (1) - 60 mg Terfenadin (2) Zweimal täglich für zwei Wochen Beurteilung der Veränderung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen Statistisch signifikante Abschwellung der Nasenschleimhaut im Vergleich zur Terfenadin-gruppe
Keine schwerwiegenden UAW Statistisch signifikant mehr UAW in der AM-Kombinations-Gruppe Anstieg der Herzfrequenz um 5 Schläge pro Minute in der AM-Kombinations-Gruppe
110
HORAK 1998 (1, 3, 4)
Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Die Effektivität einer AM-Kombination zur Behandlung der verstopften Nase zu untersuchen Primärer Endpunkt: Reduktion der Summer aller Symptome
N=24 - Patient*innen mit andauernder allergischer Rhinitis aufgrund von Hausstaubmilben
Behandlung mit: - 5 mg Cetirizin und 120 mg PSE - Placebo Zweimal täglich für eine Woche Zweimaliger Aufenthalt in der Wiener Provokationskammer für mehrere Stunden Auswaschphase von mind. 2 Wochen und anschließend erneute Behandlung mit dem alternativen Präparat
Statistisch signifikante Reduktion der Verstopfung der Nase im Vergleich zu Placebo
Ein Fall von moderaten UAW
HOWARTH 1993 (1, 3, 4)
Doppelblind, placebokontrolliert, crossover design Ziel: Den Effekt von PSE auf die Intensität der allergischen Reaktion nach Konfrontation mit einem Allergen zu untersuchen
N=12 - Patient*innen mit andauernder allergischer Rhinitis
Behandlung mit: - 60 mg PSE - Placebo Dreimal täglich, zwei Tage vor dem Provokationstest Messung mittels Rhinomanometrie
Statistisch signifikante Reduktion des nasalen Widerstandes im Vergleich zu Placebo
-
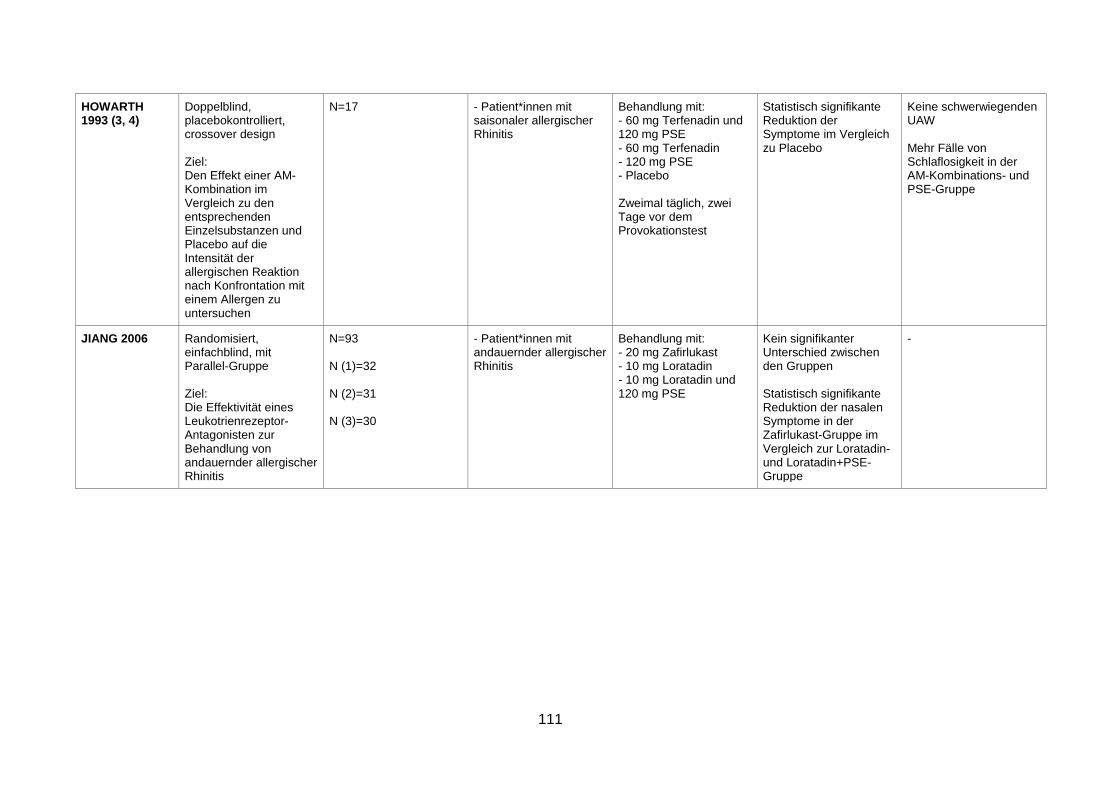
111
HOWARTH 1993 (3, 4)
Doppelblind, placebokontrolliert, crossover design Ziel: Den Effekt einer AM-Kombination im Vergleich zu den entsprechenden Einzelsubstanzen und Placebo auf die Intensität der allergischen Reaktion nach Konfrontation mit einem Allergen zu untersuchen
N=17 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 60 mg Terfenadin und 120 mg PSE - 60 mg Terfenadin - 120 mg PSE - Placebo Zweimal täglich, zwei Tage vor dem Provokationstest
Statistisch signifikante Reduktion der Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Mehr Fälle von Schlaflosigkeit in der AM-Kombinations- und PSE-Gruppe
JIANG 2006 Randomisiert, einfachblind, mit Parallel-Gruppe Ziel: Die Effektivität eines Leukotrienrezeptor-Antagonisten zur Behandlung von andauernder allergischer Rhinitis
N=93 N (1)=32 N (2)=31 N (3)=30
- Patient*innen mit andauernder allergischer Rhinitis
Behandlung mit: - 20 mg Zafirlukast - 10 mg Loratadin - 10 mg Loratadin und 120 mg PSE
Kein signifikanter Unterschied zwischen den Gruppen Statistisch signifikante Reduktion der nasalen Symptome in der Zafirlukast-Gruppe im Vergleich zur Loratadin- und Loratadin+PSE-Gruppe
-
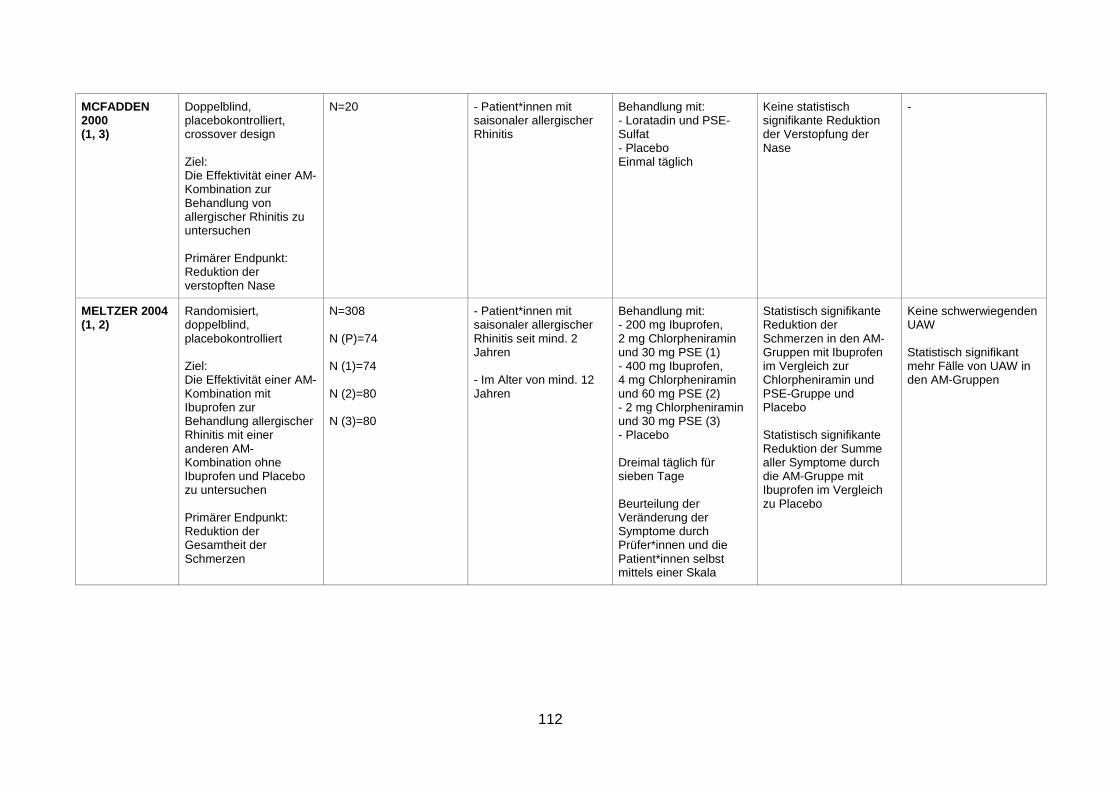
112
MCFADDEN 2000 (1, 3)
Doppelblind, placebokontrolliert, crossover design Ziel: Die Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis zu untersuchen Primärer Endpunkt: Reduktion der verstopften Nase
N=20 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - Loratadin und PSE-Sulfat - Placebo Einmal täglich
Keine statistisch signifikante Reduktion der Verstopfung der Nase
-
MELTZER 2004 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination mit Ibuprofen zur Behandlung allergischer Rhinitis mit einer anderen AM-Kombination ohne Ibuprofen und Placebo zu untersuchen Primärer Endpunkt: Reduktion der Gesamtheit der Schmerzen
N=308 N (P)=74 N (1)=74 N (2)=80 N (3)=80
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. 2 Jahren - Im Alter von mind. 12 Jahren
Behandlung mit: - 200 mg Ibuprofen, 2 mg Chlorpheniramin und 30 mg PSE (1) - 400 mg Ibuprofen, 4 mg Chlorpheniramin und 60 mg PSE (2) - 2 mg Chlorpheniramin und 30 mg PSE (3) - Placebo Dreimal täglich für sieben Tage Beurteilung der Veränderung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Schmerzen in den AM-Gruppen mit Ibuprofen im Vergleich zur Chlorpheniramin und PSE-Gruppe und Placebo Statistisch signifikante Reduktion der Summe aller Symptome durch die AM-Gruppe mit Ibuprofen im Vergleich zu Placebo
Keine schwerwiegenden UAW Statistisch signifikant mehr Fälle von UAW in den AM-Gruppen
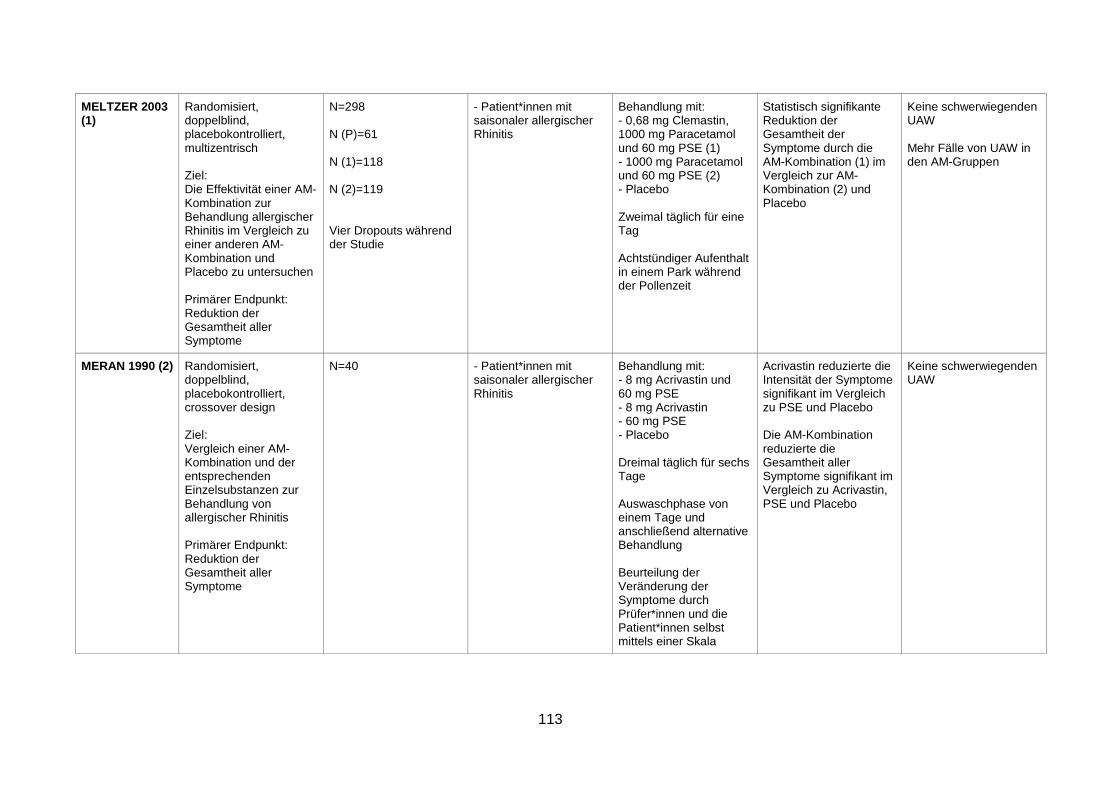
113
MELTZER 2003 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Die Effektivität einer AM-Kombination zur Behandlung allergischer Rhinitis im Vergleich zu einer anderen AM-Kombination und Placebo zu untersuchen Primärer Endpunkt: Reduktion der Gesamtheit aller Symptome
N=298 N (P)=61 N (1)=118 N (2)=119 Vier Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 0,68 mg Clemastin, 1000 mg Paracetamol und 60 mg PSE (1) - 1000 mg Paracetamol und 60 mg PSE (2) - Placebo Zweimal täglich für eine Tag Achtstündiger Aufenthalt in einem Park während der Pollenzeit
Statistisch signifikante Reduktion der Gesamtheit der Symptome durch die AM-Kombination (1) im Vergleich zur AM-Kombination (2) und Placebo
Keine schwerwiegenden UAW Mehr Fälle von UAW in den AM-Gruppen
MERAN 1990 (2) Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Vergleich einer AM-Kombination und der entsprechenden Einzelsubstanzen zur Behandlung von allergischer Rhinitis Primärer Endpunkt: Reduktion der Gesamtheit aller Symptome
N=40 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 8 mg Acrivastin und 60 mg PSE - 8 mg Acrivastin - 60 mg PSE - Placebo Dreimal täglich für sechs Tage Auswaschphase von einem Tage und anschließend alternative Behandlung Beurteilung der Veränderung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Acrivastin reduzierte die Intensität der Symptome signifikant im Vergleich zu PSE und Placebo Die AM-Kombination reduzierte die Gesamtheit aller Symptome signifikant im Vergleich zu Acrivastin, PSE und Placebo
Keine schwerwiegenden UAW
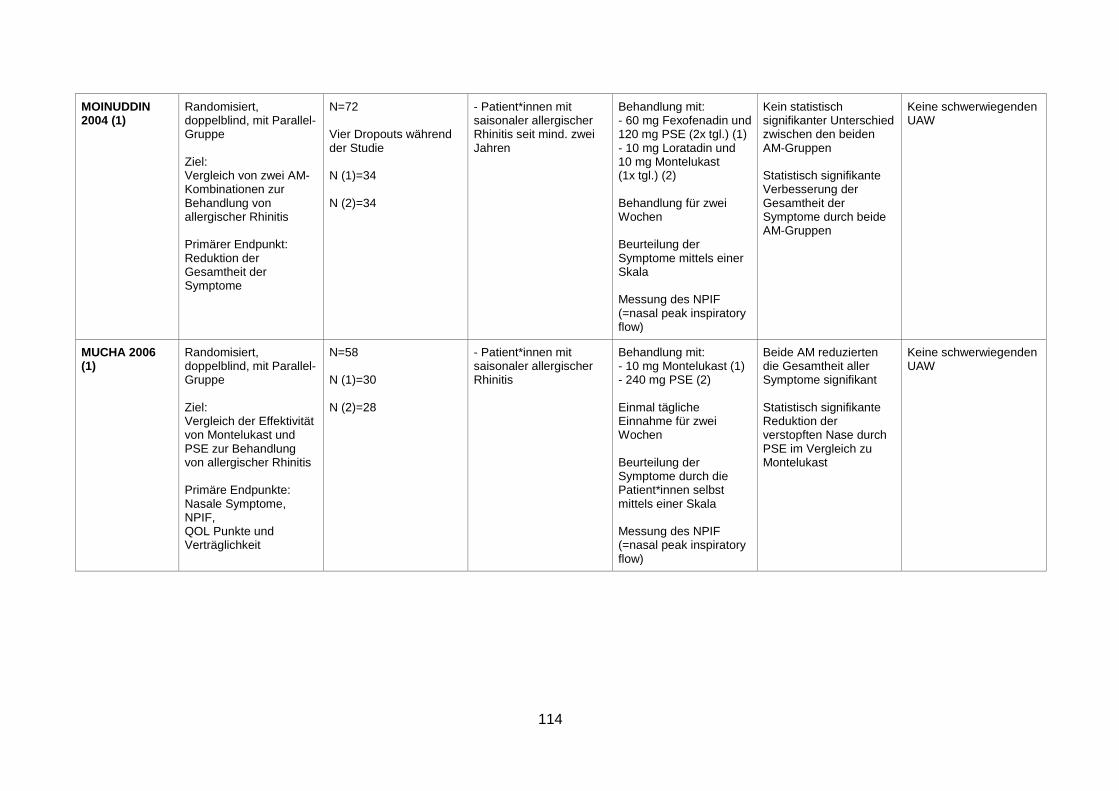
114
MOINUDDIN 2004 (1)
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich von zwei AM-Kombinationen zur Behandlung von allergischer Rhinitis Primärer Endpunkt: Reduktion der Gesamtheit der Symptome
N=72 Vier Dropouts während der Studie N (1)=34 N (2)=34
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren
Behandlung mit: - 60 mg Fexofenadin und 120 mg PSE (2x tgl.) (1) - 10 mg Loratadin und 10 mg Montelukast (1x tgl.) (2) Behandlung für zwei Wochen Beurteilung der Symptome mittels einer Skala Messung des NPIF (=nasal peak inspiratory flow)
Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen Statistisch signifikante Verbesserung der Gesamtheit der Symptome durch beide AM-Gruppen
Keine schwerwiegenden UAW
MUCHA 2006 (1)
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich der Effektivität von Montelukast und PSE zur Behandlung von allergischer Rhinitis Primäre Endpunkte: Nasale Symptome, NPIF, QOL Punkte und Verträglichkeit
N=58 N (1)=30 N (2)=28
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 10 mg Montelukast (1) - 240 mg PSE (2) Einmal tägliche Einnahme für zwei Wochen Beurteilung der Symptome durch die Patient*innen selbst mittels einer Skala Messung des NPIF (=nasal peak inspiratory flow)
Beide AM reduzierten die Gesamtheit aller Symptome signifikant Statistisch signifikante Reduktion der verstopften Nase durch PSE im Vergleich zu Montelukast
Keine schwerwiegenden UAW
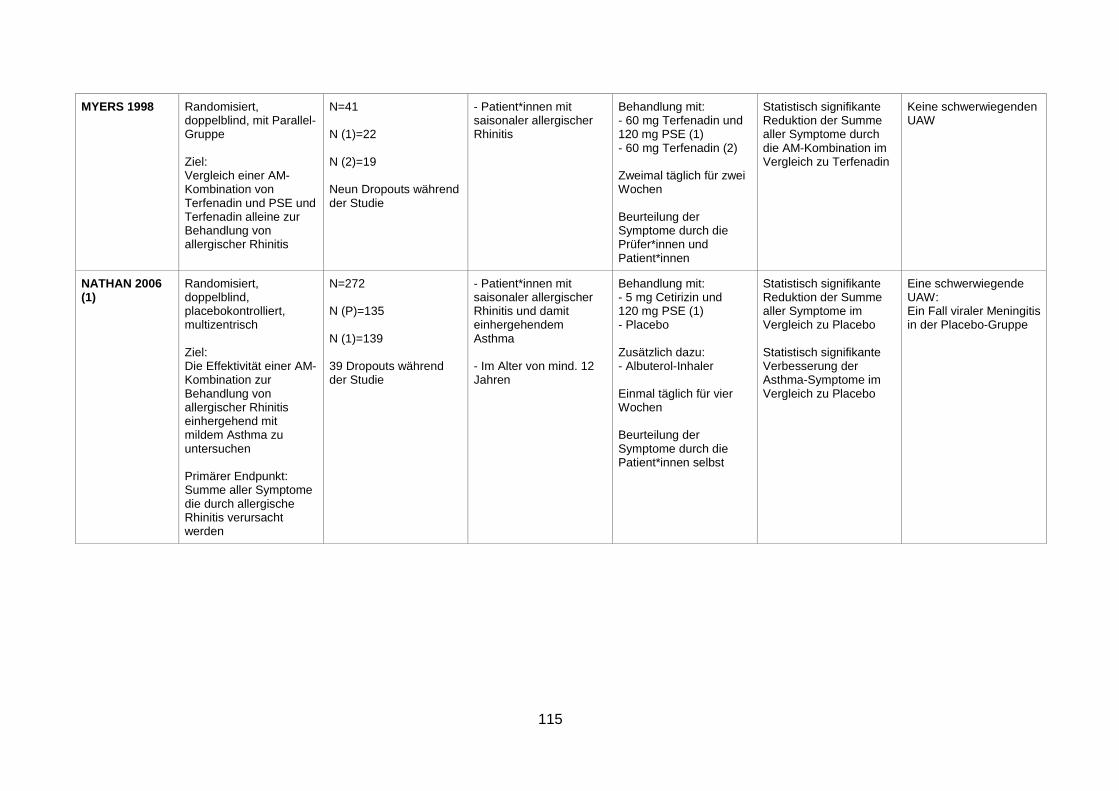
115
MYERS 1998 Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich einer AM-Kombination von Terfenadin und PSE und Terfenadin alleine zur Behandlung von allergischer Rhinitis
N=41 N (1)=22 N (2)=19 Neun Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 60 mg Terfenadin und 120 mg PSE (1) - 60 mg Terfenadin (2) Zweimal täglich für zwei Wochen Beurteilung der Symptome durch die Prüfer*innen und Patient*innen
Statistisch signifikante Reduktion der Summe aller Symptome durch die AM-Kombination im Vergleich zu Terfenadin
Keine schwerwiegenden UAW
NATHAN 2006 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Die Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis einhergehend mit mildem Asthma zu untersuchen Primärer Endpunkt: Summe aller Symptome die durch allergische Rhinitis verursacht werden
N=272 N (P)=135 N (1)=139 39 Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis und damit einhergehendem Asthma - Im Alter von mind. 12 Jahren
Behandlung mit: - 5 mg Cetirizin und 120 mg PSE (1) - Placebo Zusätzlich dazu: - Albuterol-Inhaler Einmal täglich für vier Wochen Beurteilung der Symptome durch die Patient*innen selbst
Statistisch signifikante Reduktion der Summe aller Symptome im Vergleich zu Placebo Statistisch signifikante Verbesserung der Asthma-Symptome im Vergleich zu Placebo
Eine schwerwiegende UAW: Ein Fall viraler Meningitis in der Placebo-Gruppe
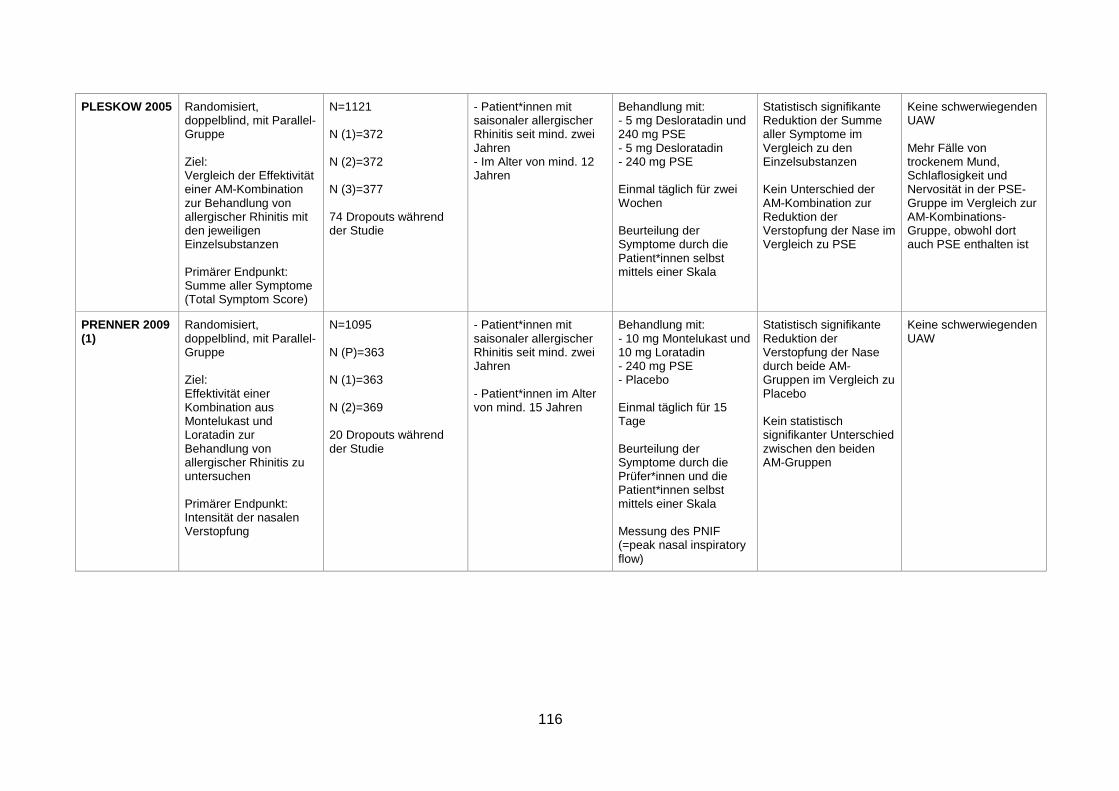
116
PLESKOW 2005 Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich der Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis mit den jeweiligen Einzelsubstanzen Primärer Endpunkt: Summe aller Symptome (Total Symptom Score)
N=1121 N (1)=372 N (2)=372 N (3)=377 74 Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren - Im Alter von mind. 12 Jahren
Behandlung mit: - 5 mg Desloratadin und 240 mg PSE - 5 mg Desloratadin - 240 mg PSE Einmal täglich für zwei Wochen Beurteilung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Summe aller Symptome im Vergleich zu den Einzelsubstanzen Kein Unterschied der AM-Kombination zur Reduktion der Verstopfung der Nase im Vergleich zu PSE
Keine schwerwiegenden UAW Mehr Fälle von trockenem Mund, Schlaflosigkeit und Nervosität in der PSE-Gruppe im Vergleich zur AM-Kombinations-Gruppe, obwohl dort auch PSE enthalten ist
PRENNER 2009 (1)
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Effektivität einer Kombination aus Montelukast und Loratadin zur Behandlung von allergischer Rhinitis zu untersuchen Primärer Endpunkt: Intensität der nasalen Verstopfung
N=1095 N (P)=363 N (1)=363 N (2)=369 20 Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren - Patient*innen im Alter von mind. 15 Jahren
Behandlung mit: - 10 mg Montelukast und 10 mg Loratadin - 240 mg PSE - Placebo Einmal täglich für 15 Tage Beurteilung der Symptome durch die Prüfer*innen und die Patient*innen selbst mittels einer Skala Messung des PNIF (=peak nasal inspiratory flow)
Statistisch signifikante Reduktion der Verstopfung der Nase durch beide AM-Gruppen im Vergleich zu Placebo Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen
Keine schwerwiegenden UAW
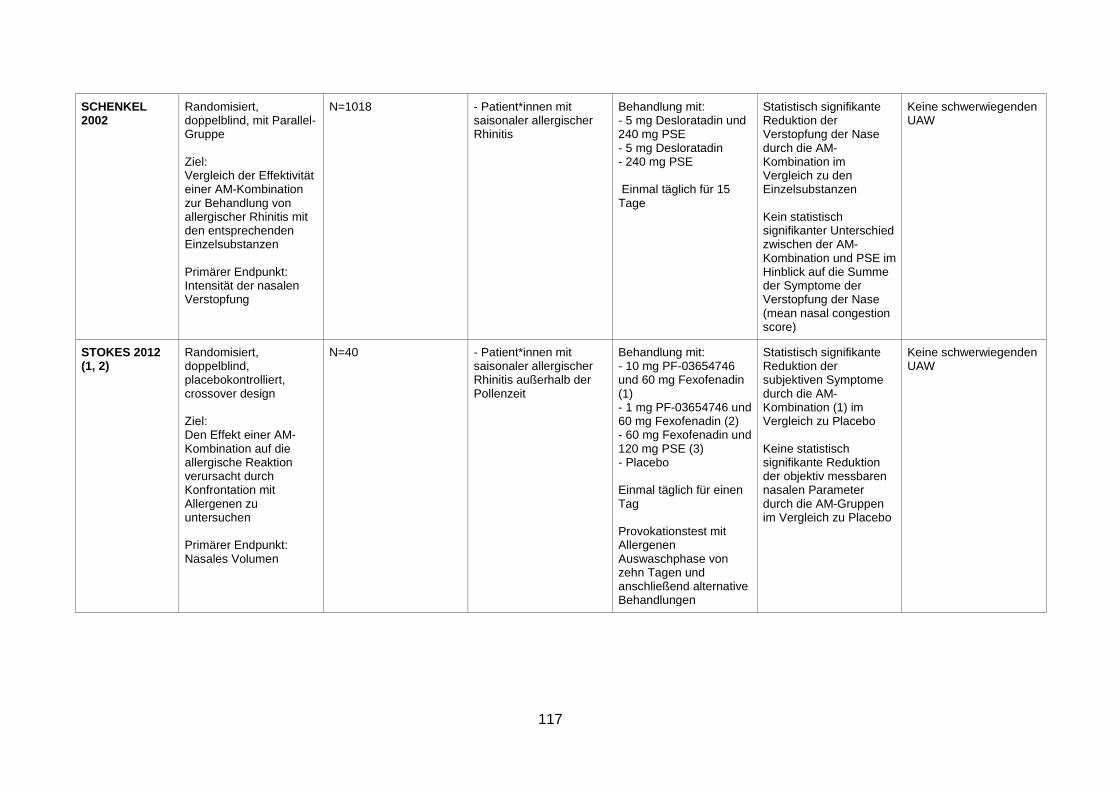
117
SCHENKEL 2002
Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich der Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis mit den entsprechenden Einzelsubstanzen Primärer Endpunkt: Intensität der nasalen Verstopfung
N=1018 - Patient*innen mit saisonaler allergischer Rhinitis
Behandlung mit: - 5 mg Desloratadin und 240 mg PSE - 5 mg Desloratadin - 240 mg PSE Einmal täglich für 15 Tage
Statistisch signifikante Reduktion der Verstopfung der Nase durch die AM-Kombination im Vergleich zu den Einzelsubstanzen Kein statistisch signifikanter Unterschied zwischen der AM-Kombination und PSE im Hinblick auf die Summe der Symptome der Verstopfung der Nase (mean nasal congestion score)
Keine schwerwiegenden UAW
STOKES 2012 (1, 2)
Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Den Effekt einer AM-Kombination auf die allergische Reaktion verursacht durch Konfrontation mit Allergenen zu untersuchen Primärer Endpunkt: Nasales Volumen
N=40
- Patient*innen mit saisonaler allergischer Rhinitis außerhalb der Pollenzeit
Behandlung mit: - 10 mg PF-03654746 und 60 mg Fexofenadin (1) - 1 mg PF-03654746 und 60 mg Fexofenadin (2) - 60 mg Fexofenadin und 120 mg PSE (3) - Placebo Einmal täglich für einen Tag Provokationstest mit Allergenen Auswaschphase von zehn Tagen und anschließend alternative Behandlungen
Statistisch signifikante Reduktion der subjektiven Symptome durch die AM-Kombination (1) im Vergleich zu Placebo Keine statistisch signifikante Reduktion der objektiv messbaren nasalen Parameter durch die AM-Gruppen im Vergleich zu Placebo
Keine schwerwiegenden UAW
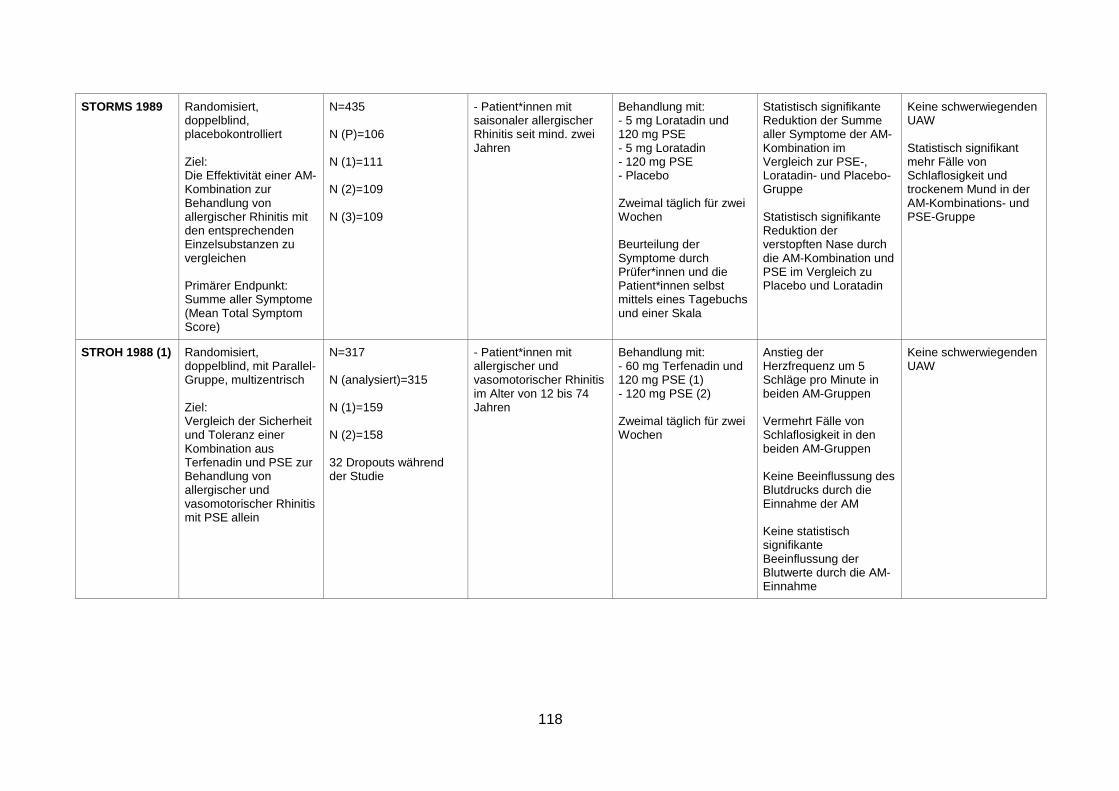
118
STORMS 1989 Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination zur Behandlung von allergischer Rhinitis mit den entsprechenden Einzelsubstanzen zu vergleichen Primärer Endpunkt: Summe aller Symptome (Mean Total Symptom Score)
N=435 N (P)=106 N (1)=111 N (2)=109 N (3)=109
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren
Behandlung mit: - 5 mg Loratadin und 120 mg PSE - 5 mg Loratadin - 120 mg PSE - Placebo Zweimal täglich für zwei Wochen Beurteilung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels eines Tagebuchs und einer Skala
Statistisch signifikante Reduktion der Summe aller Symptome der AM-Kombination im Vergleich zur PSE-, Loratadin- und Placebo-Gruppe Statistisch signifikante Reduktion der verstopften Nase durch die AM-Kombination und PSE im Vergleich zu Placebo und Loratadin
Keine schwerwiegenden UAW Statistisch signifikant mehr Fälle von Schlaflosigkeit und trockenem Mund in der AM-Kombinations- und PSE-Gruppe
STROH 1988 (1) Randomisiert, doppelblind, mit Parallel-Gruppe, multizentrisch Ziel: Vergleich der Sicherheit und Toleranz einer Kombination aus Terfenadin und PSE zur Behandlung von allergischer und vasomotorischer Rhinitis mit PSE allein
N=317 N (analysiert)=315 N (1)=159 N (2)=158 32 Dropouts während der Studie
- Patient*innen mit allergischer und vasomotorischer Rhinitis im Alter von 12 bis 74 Jahren
Behandlung mit: - 60 mg Terfenadin und 120 mg PSE (1) - 120 mg PSE (2) Zweimal täglich für zwei Wochen
Anstieg der Herzfrequenz um 5 Schläge pro Minute in beiden AM-Gruppen Vermehrt Fälle von Schlaflosigkeit in den beiden AM-Gruppen Keine Beeinflussung des Blutdrucks durch die Einnahme der AM Keine statistisch signifikante Beeinflussung der Blutwerte durch die AM-Einnahme
Keine schwerwiegenden UAW
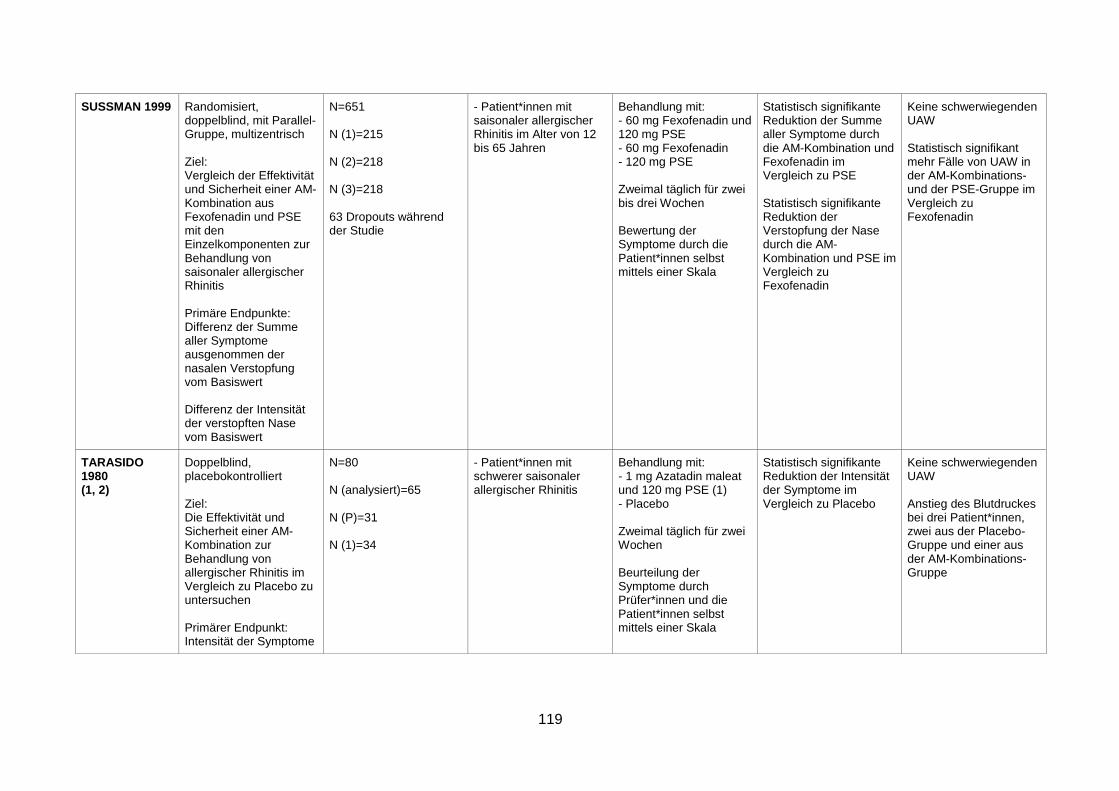
119
SUSSMAN 1999 Randomisiert, doppelblind, mit Parallel-Gruppe, multizentrisch Ziel: Vergleich der Effektivität und Sicherheit einer AM-Kombination aus Fexofenadin und PSE mit den Einzelkomponenten zur Behandlung von saisonaler allergischer Rhinitis Primäre Endpunkte: Differenz der Summe aller Symptome ausgenommen der nasalen Verstopfung vom Basiswert Differenz der Intensität der verstopften Nase vom Basiswert
N=651 N (1)=215 N (2)=218 N (3)=218 63 Dropouts während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis im Alter von 12 bis 65 Jahren
Behandlung mit: - 60 mg Fexofenadin und 120 mg PSE - 60 mg Fexofenadin - 120 mg PSE Zweimal täglich für zwei bis drei Wochen Bewertung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Summe aller Symptome durch die AM-Kombination und Fexofenadin im Vergleich zu PSE Statistisch signifikante Reduktion der Verstopfung der Nase durch die AM-Kombination und PSE im Vergleich zu Fexofenadin
Keine schwerwiegenden UAW Statistisch signifikant mehr Fälle von UAW in der AM-Kombinations- und der PSE-Gruppe im Vergleich zu Fexofenadin
TARASIDO 1980 (1, 2)
Doppelblind, placebokontrolliert Ziel: Die Effektivität und Sicherheit einer AM-Kombination zur Behandlung von allergischer Rhinitis im Vergleich zu Placebo zu untersuchen Primärer Endpunkt: Intensität der Symptome
N=80 N (analysiert)=65 N (P)=31 N (1)=34
- Patient*innen mit schwerer saisonaler allergischer Rhinitis
Behandlung mit: - 1 mg Azatadin maleat und 120 mg PSE (1) - Placebo Zweimal täglich für zwei Wochen Beurteilung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Intensität der Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Anstieg des Blutdruckes bei drei Patient*innen, zwei aus der Placebo-Gruppe und einer aus der AM-Kombinations-Gruppe
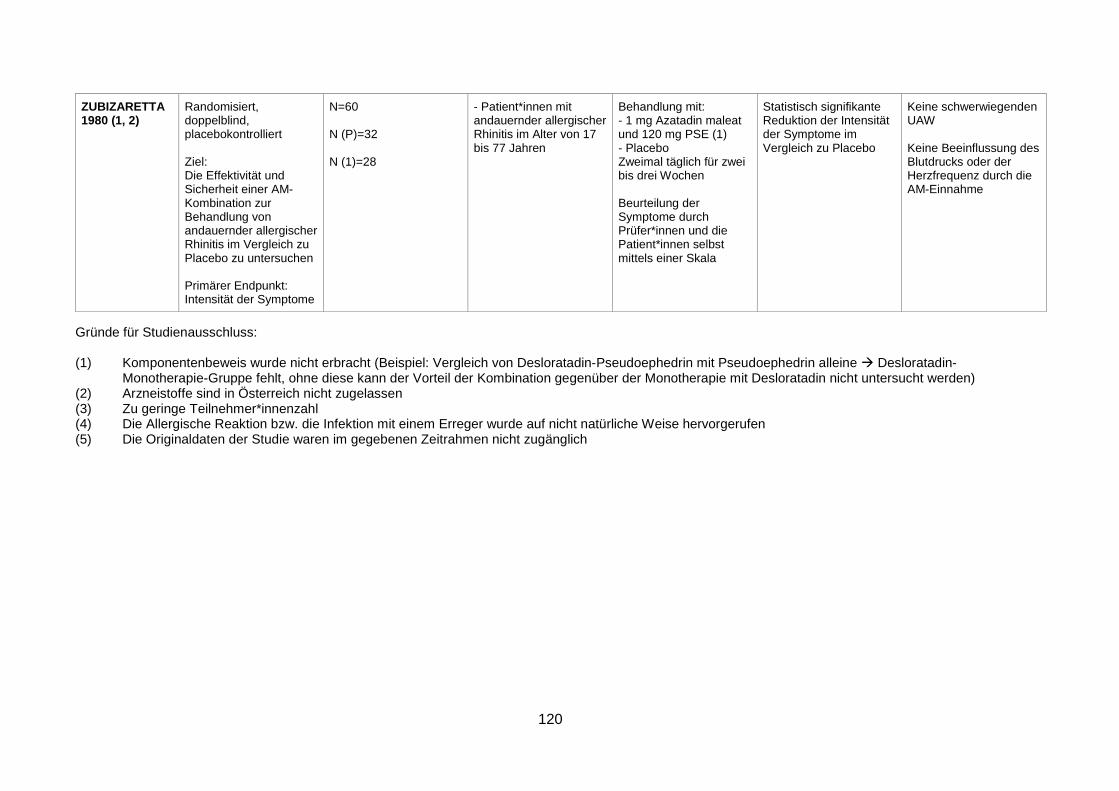
120
ZUBIZARETTA 1980 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität und Sicherheit einer AM-Kombination zur Behandlung von andauernder allergischer Rhinitis im Vergleich zu Placebo zu untersuchen Primärer Endpunkt: Intensität der Symptome
N=60 N (P)=32 N (1)=28
- Patient*innen mit andauernder allergischer Rhinitis im Alter von 17 bis 77 Jahren
Behandlung mit: - 1 mg Azatadin maleat und 120 mg PSE (1) - Placebo Zweimal täglich für zwei bis drei Wochen Beurteilung der Symptome durch Prüfer*innen und die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Intensität der Symptome im Vergleich zu Placebo
Keine schwerwiegenden UAW Keine Beeinflussung des Blutdrucks oder der Herzfrequenz durch die AM-Einnahme
Gründe für Studienausschluss: (1) Komponentenbeweis wurde nicht erbracht (Beispiel: Vergleich von Desloratadin-Pseudoephedrin mit Pseudoephedrin alleine Desloratadin-
Monotherapie-Gruppe fehlt, ohne diese kann der Vorteil der Kombination gegenüber der Monotherapie mit Desloratadin nicht untersucht werden) (2) Arzneistoffe sind in Österreich nicht zugelassen (3) Zu geringe Teilnehmer*innenzahl (4) Die Allergische Reaktion bzw. die Infektion mit einem Erreger wurde auf nicht natürliche Weise hervorgerufen (5) Die Originaldaten der Studie waren im gegebenen Zeitrahmen nicht zugänglich
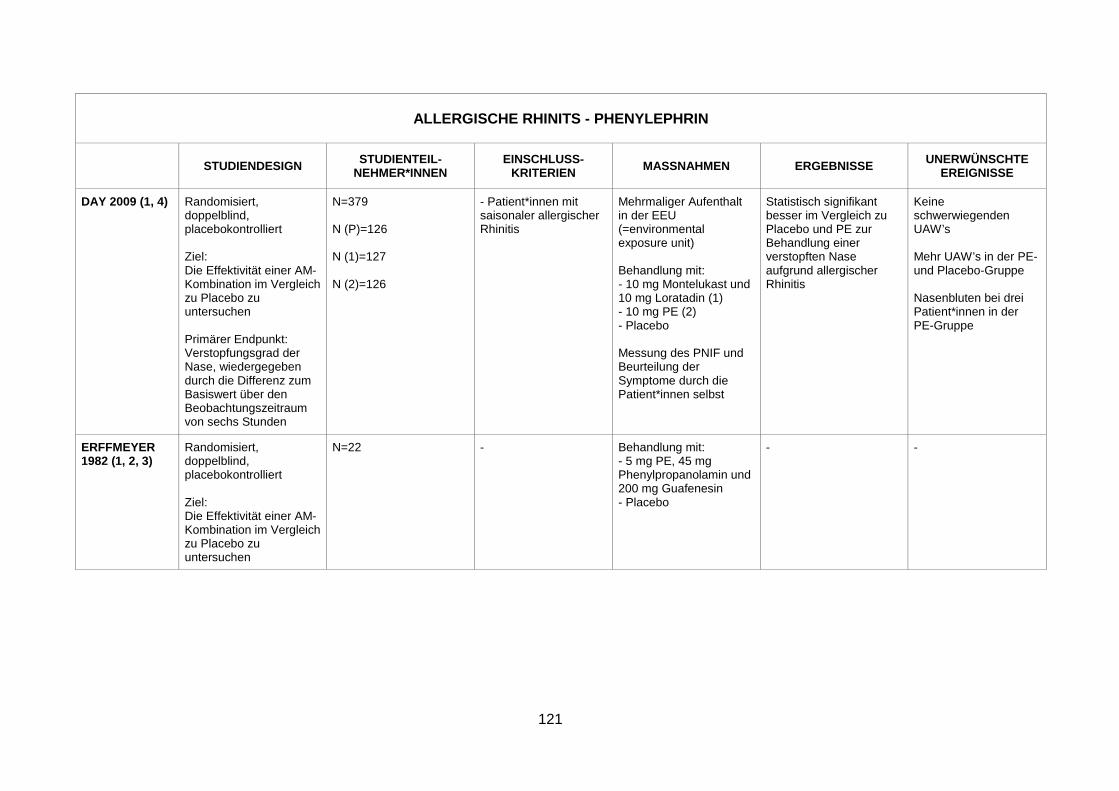
121
ALLERGISCHE RHINITS - PHENYLEPHRIN
STUDIENDESIGN STUDIENTEIL-NEHMER*INNEN
EINSCHLUSS-KRITERIEN MASSNAHMEN ERGEBNISSE UNERWÜNSCHTE
EREIGNISSE
DAY 2009 (1, 4) Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination im Vergleich zu Placebo zu untersuchen Primärer Endpunkt: Verstopfungsgrad der Nase, wiedergegeben durch die Differenz zum Basiswert über den Beobachtungszeitraum von sechs Stunden
N=379 N (P)=126 N (1)=127 N (2)=126
- Patient*innen mit saisonaler allergischer Rhinitis
Mehrmaliger Aufenthalt in der EEU (=environmental exposure unit) Behandlung mit: - 10 mg Montelukast und 10 mg Loratadin (1) - 10 mg PE (2) - Placebo Messung des PNIF und Beurteilung der Symptome durch die Patient*innen selbst
Statistisch signifikant besser im Vergleich zu Placebo und PE zur Behandlung einer verstopften Nase aufgrund allergischer Rhinitis
Keine schwerwiegenden UAW’s Mehr UAW’s in der PE- und Placebo-Gruppe Nasenbluten bei drei Patient*innen in der PE-Gruppe
ERFFMEYER 1982 (1, 2, 3)
Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination im Vergleich zu Placebo zu untersuchen
N=22 - Behandlung mit: - 5 mg PE, 45 mg Phenylpropanolamin und 200 mg Guafenesin - Placebo
- -
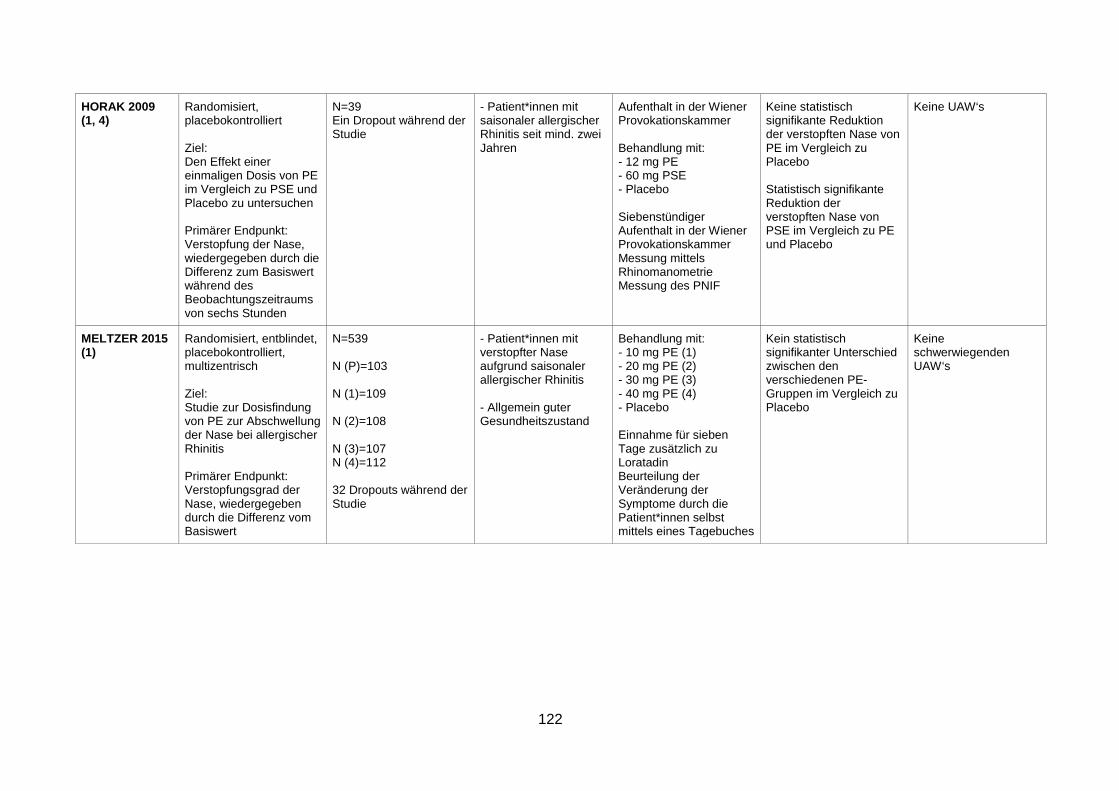
122
HORAK 2009 (1, 4)
Randomisiert, placebokontrolliert Ziel: Den Effekt einer einmaligen Dosis von PE im Vergleich zu PSE und Placebo zu untersuchen Primärer Endpunkt: Verstopfung der Nase, wiedergegeben durch die Differenz zum Basiswert während des Beobachtungszeitraums von sechs Stunden
N=39 Ein Dropout während der Studie
- Patient*innen mit saisonaler allergischer Rhinitis seit mind. zwei Jahren
Aufenthalt in der Wiener Provokationskammer Behandlung mit: - 12 mg PE - 60 mg PSE - Placebo Siebenstündiger Aufenthalt in der Wiener Provokationskammer Messung mittels Rhinomanometrie Messung des PNIF
Keine statistisch signifikante Reduktion der verstopften Nase von PE im Vergleich zu Placebo Statistisch signifikante Reduktion der verstopften Nase von PSE im Vergleich zu PE und Placebo
Keine UAW‘s
MELTZER 2015 (1)
Randomisiert, entblindet, placebokontrolliert, multizentrisch Ziel: Studie zur Dosisfindung von PE zur Abschwellung der Nase bei allergischer Rhinitis Primärer Endpunkt: Verstopfungsgrad der Nase, wiedergegeben durch die Differenz vom Basiswert
N=539 N (P)=103 N (1)=109 N (2)=108 N (3)=107 N (4)=112 32 Dropouts während der Studie
- Patient*innen mit verstopfter Nase aufgrund saisonaler allergischer Rhinitis - Allgemein guter Gesundheitszustand
Behandlung mit: - 10 mg PE (1) - 20 mg PE (2) - 30 mg PE (3) - 40 mg PE (4) - Placebo Einnahme für sieben Tage zusätzlich zu Loratadin Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels eines Tagebuches
Kein statistisch signifikanter Unterschied zwischen den verschiedenen PE-Gruppen im Vergleich zu Placebo
Keine schwerwiegenden UAW‘s
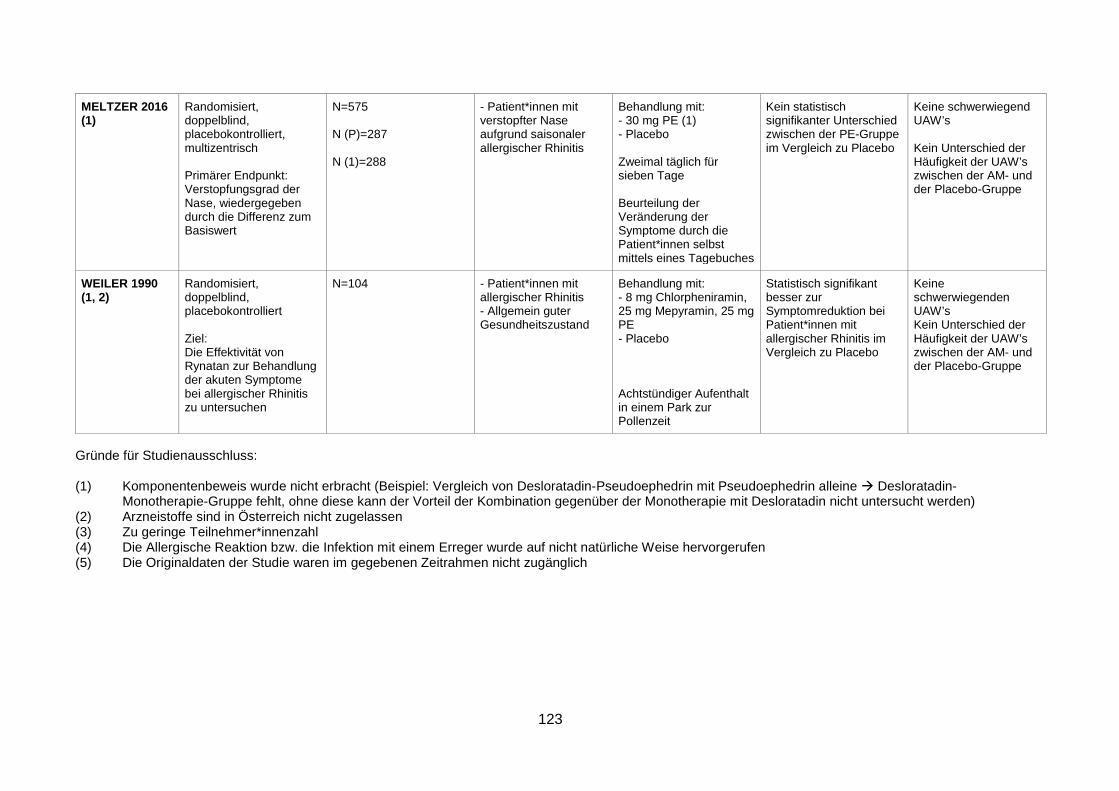
123
MELTZER 2016 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Primärer Endpunkt: Verstopfungsgrad der Nase, wiedergegeben durch die Differenz zum Basiswert
N=575 N (P)=287 N (1)=288
- Patient*innen mit verstopfter Nase aufgrund saisonaler allergischer Rhinitis
Behandlung mit: - 30 mg PE (1) - Placebo Zweimal täglich für sieben Tage Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels eines Tagebuches
Kein statistisch signifikanter Unterschied zwischen der PE-Gruppe im Vergleich zu Placebo
Keine schwerwiegend UAW’s Kein Unterschied der Häufigkeit der UAW’s zwischen der AM- und der Placebo-Gruppe
WEILER 1990 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität von Rynatan zur Behandlung der akuten Symptome bei allergischer Rhinitis zu untersuchen
N=104 - Patient*innen mit allergischer Rhinitis - Allgemein guter Gesundheitszustand
Behandlung mit: - 8 mg Chlorpheniramin, 25 mg Mepyramin, 25 mg PE - Placebo Achtstündiger Aufenthalt in einem Park zur Pollenzeit
Statistisch signifikant besser zur Symptomreduktion bei Patient*innen mit allergischer Rhinitis im Vergleich zu Placebo
Keine schwerwiegenden UAW’s Kein Unterschied der Häufigkeit der UAW’s zwischen der AM- und der Placebo-Gruppe
Gründe für Studienausschluss: (1) Komponentenbeweis wurde nicht erbracht (Beispiel: Vergleich von Desloratadin-Pseudoephedrin mit Pseudoephedrin alleine Desloratadin-
Monotherapie-Gruppe fehlt, ohne diese kann der Vorteil der Kombination gegenüber der Monotherapie mit Desloratadin nicht untersucht werden) (2) Arzneistoffe sind in Österreich nicht zugelassen (3) Zu geringe Teilnehmer*innenzahl (4) Die Allergische Reaktion bzw. die Infektion mit einem Erreger wurde auf nicht natürliche Weise hervorgerufen (5) Die Originaldaten der Studie waren im gegebenen Zeitrahmen nicht zugänglich
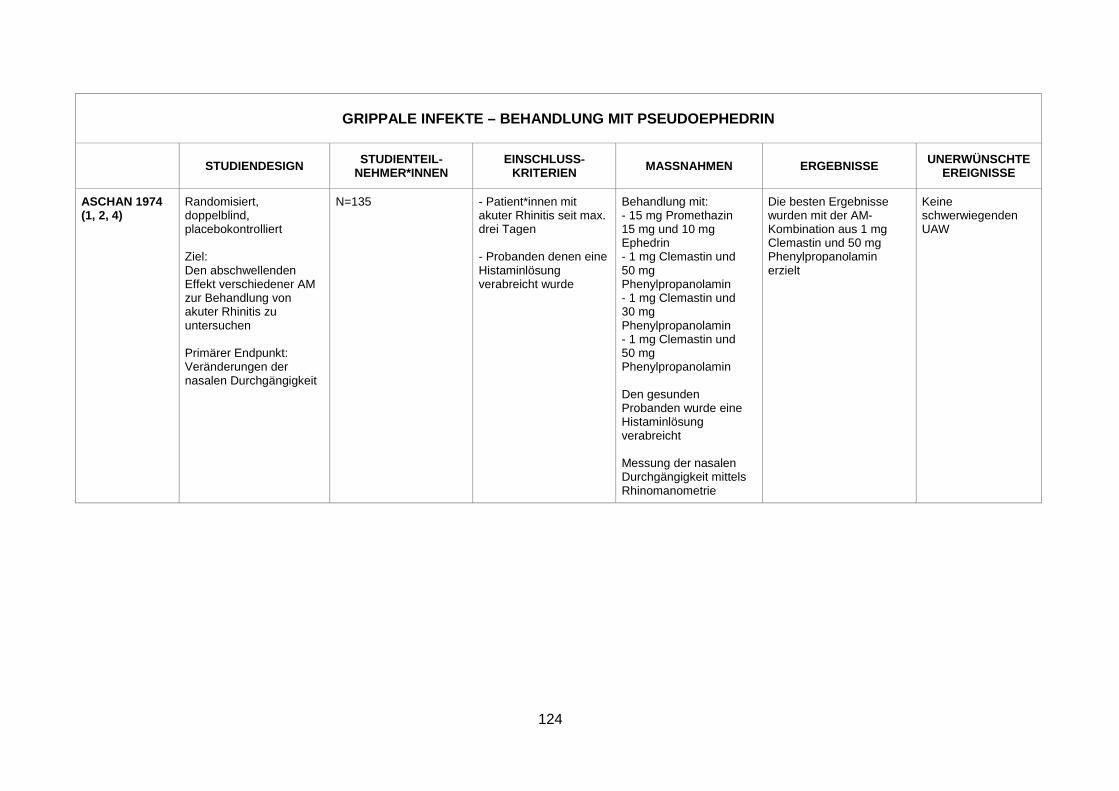
124
GRIPPALE INFEKTE – BEHANDLUNG MIT PSEUDOEPHEDRIN
STUDIENDESIGN STUDIENTEIL-NEHMER*INNEN
EINSCHLUSS-KRITERIEN MASSNAHMEN ERGEBNISSE UNERWÜNSCHTE
EREIGNISSE
ASCHAN 1974 (1, 2, 4)
Randomisiert, doppelblind, placebokontrolliert Ziel: Den abschwellenden Effekt verschiedener AM zur Behandlung von akuter Rhinitis zu untersuchen Primärer Endpunkt: Veränderungen der nasalen Durchgängigkeit
N=135 - Patient*innen mit akuter Rhinitis seit max. drei Tagen - Probanden denen eine Histaminlösung verabreicht wurde
Behandlung mit: - 15 mg Promethazin 15 mg und 10 mg Ephedrin - 1 mg Clemastin und 50 mg Phenylpropanolamin - 1 mg Clemastin und 30 mg Phenylpropanolamin - 1 mg Clemastin und 50 mg Phenylpropanolamin Den gesunden Probanden wurde eine Histaminlösung verabreicht Messung der nasalen Durchgängigkeit mittels Rhinomanometrie
Die besten Ergebnisse wurden mit der AM-Kombination aus 1 mg Clemastin und 50 mg Phenylpropanolamin erzielt
Keine schwerwiegenden UAW
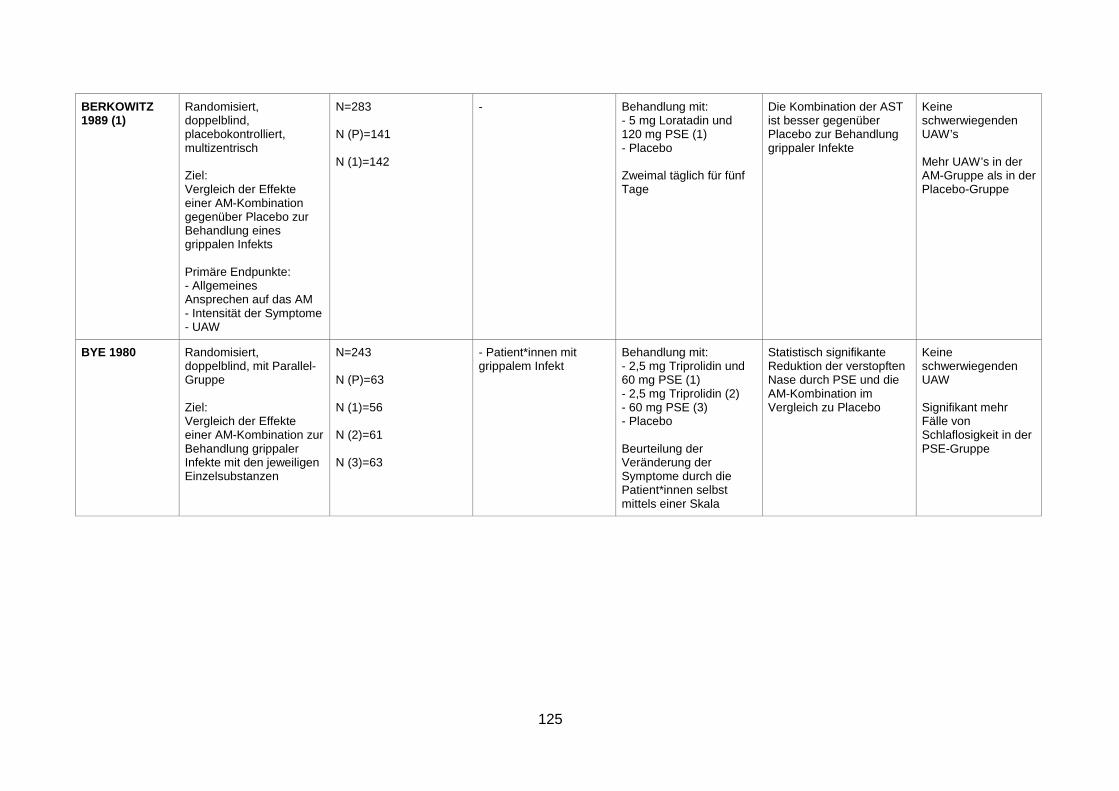
125
BERKOWITZ 1989 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Vergleich der Effekte einer AM-Kombination gegenüber Placebo zur Behandlung eines grippalen Infekts Primäre Endpunkte: - Allgemeines Ansprechen auf das AM - Intensität der Symptome - UAW
N=283 N (P)=141 N (1)=142
- Behandlung mit: - 5 mg Loratadin und 120 mg PSE (1) - Placebo Zweimal täglich für fünf Tage
Die Kombination der AST ist besser gegenüber Placebo zur Behandlung grippaler Infekte
Keine schwerwiegenden UAW’s Mehr UAW’s in der AM-Gruppe als in der Placebo-Gruppe
BYE 1980 Randomisiert, doppelblind, mit Parallel-Gruppe Ziel: Vergleich der Effekte einer AM-Kombination zur Behandlung grippaler Infekte mit den jeweiligen Einzelsubstanzen
N=243 N (P)=63 N (1)=56 N (2)=61 N (3)=63
- Patient*innen mit grippalem Infekt
Behandlung mit: - 2,5 mg Triprolidin und 60 mg PSE (1) - 2,5 mg Triprolidin (2) - 60 mg PSE (3) - Placebo Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der verstopften Nase durch PSE und die AM-Kombination im Vergleich zu Placebo
Keine schwerwiegenden UAW Signifikant mehr Fälle von Schlaflosigkeit in der PSE-Gruppe
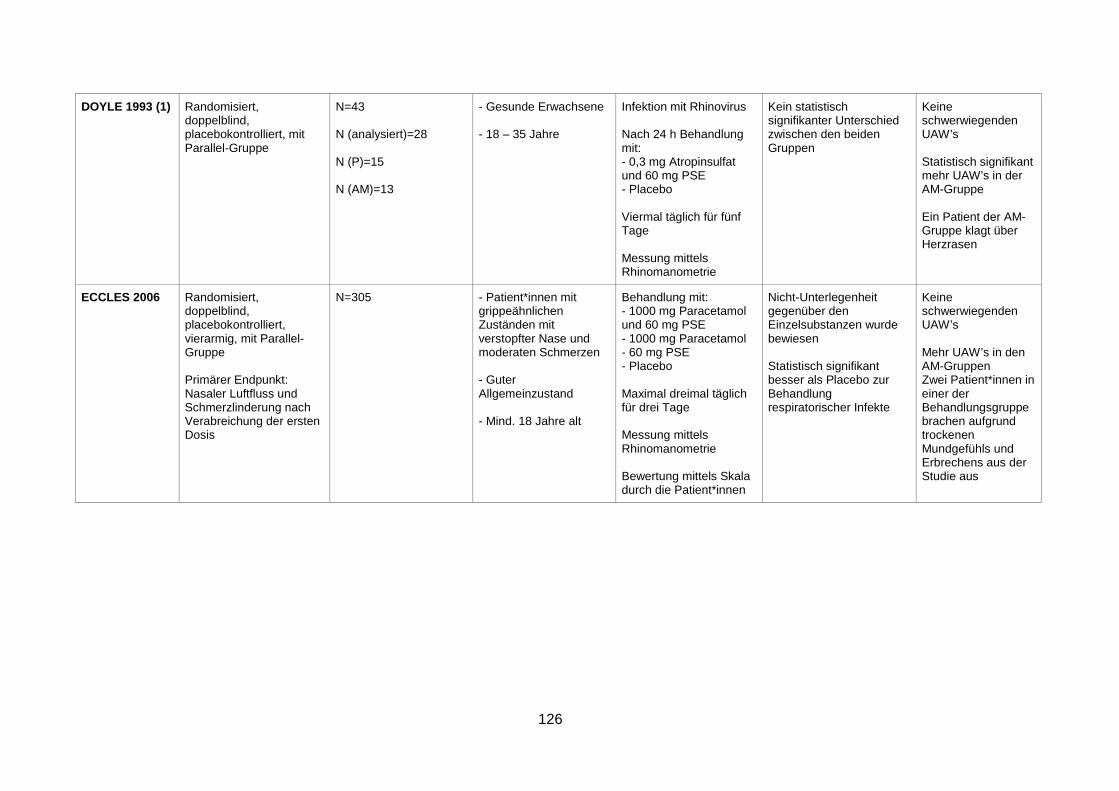
126
DOYLE 1993 (1) Randomisiert, doppelblind, placebokontrolliert, mit Parallel-Gruppe
N=43 N (analysiert)=28 N (P)=15 N (AM)=13
- Gesunde Erwachsene - 18 – 35 Jahre
Infektion mit Rhinovirus Nach 24 h Behandlung mit: - 0,3 mg Atropinsulfat und 60 mg PSE - Placebo Viermal täglich für fünf Tage Messung mittels Rhinomanometrie
Kein statistisch signifikanter Unterschied zwischen den beiden Gruppen
Keine schwerwiegenden UAW’s Statistisch signifikant mehr UAW’s in der AM-Gruppe Ein Patient der AM-Gruppe klagt über Herzrasen
ECCLES 2006 Randomisiert, doppelblind, placebokontrolliert, vierarmig, mit Parallel-Gruppe Primärer Endpunkt: Nasaler Luftfluss und Schmerzlinderung nach Verabreichung der ersten Dosis
N=305
- Patient*innen mit grippeähnlichen Zuständen mit verstopfter Nase und moderaten Schmerzen - Guter Allgemeinzustand - Mind. 18 Jahre alt
Behandlung mit: - 1000 mg Paracetamol und 60 mg PSE - 1000 mg Paracetamol - 60 mg PSE - Placebo Maximal dreimal täglich für drei Tage Messung mittels Rhinomanometrie Bewertung mittels Skala durch die Patient*innen
Nicht-Unterlegenheit gegenüber den Einzelsubstanzen wurde bewiesen Statistisch signifikant besser als Placebo zur Behandlung respiratorischer Infekte
Keine schwerwiegenden UAW’s Mehr UAW’s in den AM-Gruppen Zwei Patient*innen in einer der Behandlungsgruppe brachen aufgrund trockenen Mundgefühls und Erbrechens aus der Studie aus
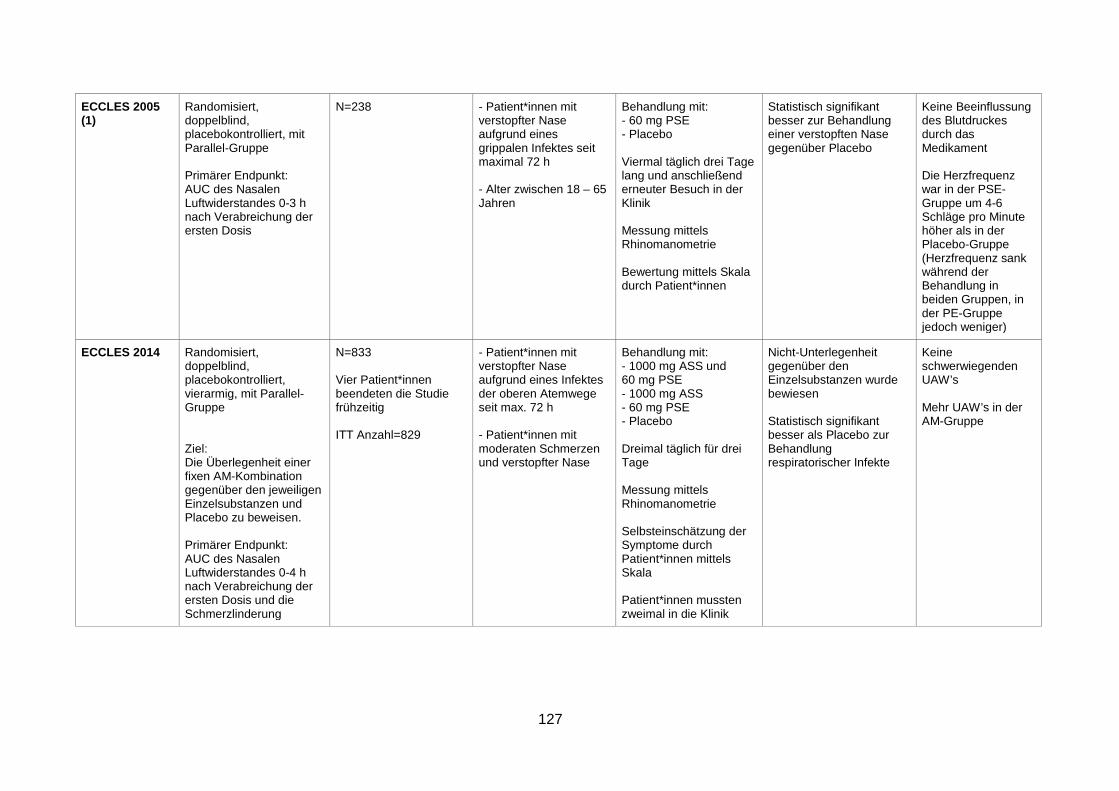
127
ECCLES 2005 (1)
Randomisiert, doppelblind, placebokontrolliert, mit Parallel-Gruppe Primärer Endpunkt: AUC des Nasalen Luftwiderstandes 0-3 h nach Verabreichung der ersten Dosis
N=238
- Patient*innen mit verstopfter Nase aufgrund eines grippalen Infektes seit maximal 72 h - Alter zwischen 18 – 65 Jahren
Behandlung mit: - 60 mg PSE - Placebo Viermal täglich drei Tage lang und anschließend erneuter Besuch in der Klinik Messung mittels Rhinomanometrie Bewertung mittels Skala durch Patient*innen
Statistisch signifikant besser zur Behandlung einer verstopften Nase gegenüber Placebo
Keine Beeinflussung des Blutdruckes durch das Medikament Die Herzfrequenz war in der PSE-Gruppe um 4-6 Schläge pro Minute höher als in der Placebo-Gruppe (Herzfrequenz sank während der Behandlung in beiden Gruppen, in der PE-Gruppe jedoch weniger)
ECCLES 2014 Randomisiert, doppelblind, placebokontrolliert, vierarmig, mit Parallel-Gruppe Ziel: Die Überlegenheit einer fixen AM-Kombination gegenüber den jeweiligen Einzelsubstanzen und Placebo zu beweisen. Primärer Endpunkt: AUC des Nasalen Luftwiderstandes 0-4 h nach Verabreichung der ersten Dosis und die Schmerzlinderung
N=833 Vier Patient*innen beendeten die Studie frühzeitig ITT Anzahl=829
- Patient*innen mit verstopfter Nase aufgrund eines Infektes der oberen Atemwege seit max. 72 h - Patient*innen mit moderaten Schmerzen und verstopfter Nase
Behandlung mit: - 1000 mg ASS und 60 mg PSE - 1000 mg ASS - 60 mg PSE - Placebo Dreimal täglich für drei Tage Messung mittels Rhinomanometrie Selbsteinschätzung der Symptome durch Patient*innen mittels Skala Patient*innen mussten zweimal in die Klinik
Nicht-Unterlegenheit gegenüber den Einzelsubstanzen wurde bewiesen Statistisch signifikant besser als Placebo zur Behandlung respiratorischer Infekte
Keine schwerwiegenden UAW’s Mehr UAW’s in der AM-Gruppe
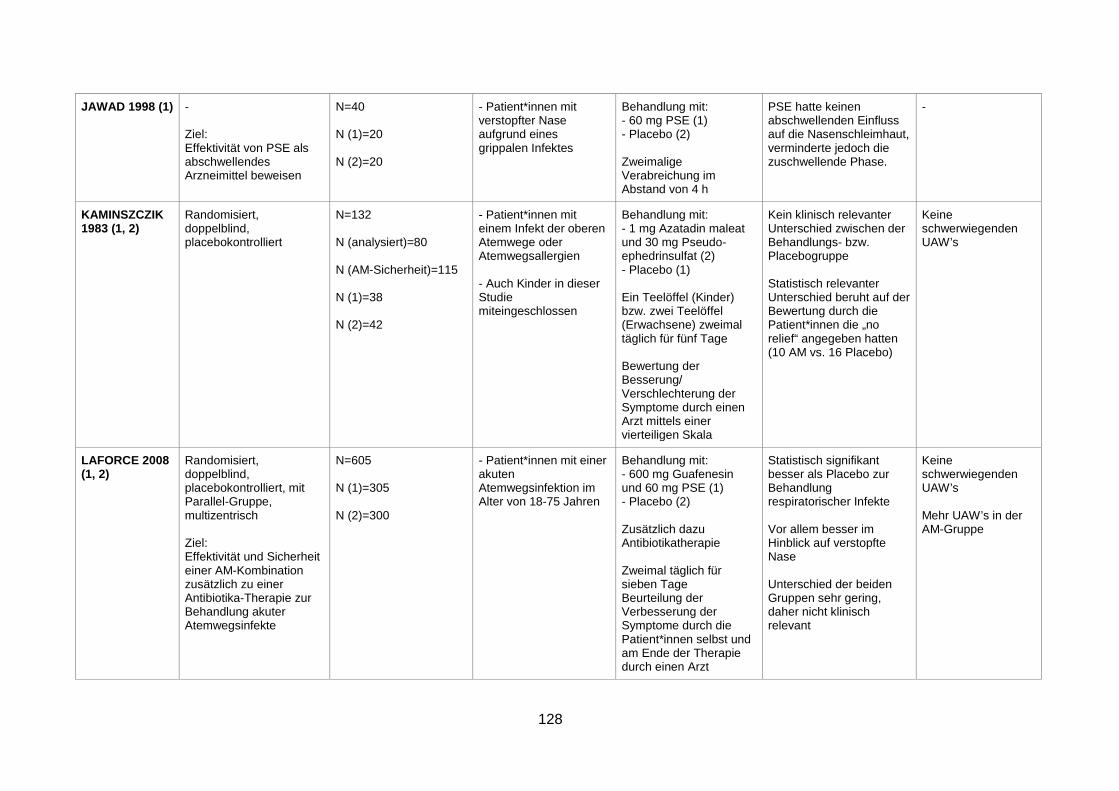
128
JAWAD 1998 (1) - Ziel: Effektivität von PSE als abschwellendes Arzneimittel beweisen
N=40 N (1)=20 N (2)=20
- Patient*innen mit verstopfter Nase aufgrund eines grippalen Infektes
Behandlung mit: - 60 mg PSE (1) - Placebo (2) Zweimalige Verabreichung im Abstand von 4 h
PSE hatte keinen abschwellenden Einfluss auf die Nasenschleimhaut, verminderte jedoch die zuschwellende Phase.
-
KAMINSZCZIK 1983 (1, 2)
Randomisiert, doppelblind, placebokontrolliert
N=132 N (analysiert)=80 N (AM-Sicherheit)=115 N (1)=38 N (2)=42
- Patient*innen mit einem Infekt der oberen Atemwege oder Atemwegsallergien - Auch Kinder in dieser Studie miteingeschlossen
Behandlung mit: - 1 mg Azatadin maleat und 30 mg Pseudo-ephedrinsulfat (2) - Placebo (1) Ein Teelöffel (Kinder) bzw. zwei Teelöffel (Erwachsene) zweimal täglich für fünf Tage Bewertung der Besserung/ Verschlechterung der Symptome durch einen Arzt mittels einer vierteiligen Skala
Kein klinisch relevanter Unterschied zwischen der Behandlungs- bzw. Placebogruppe Statistisch relevanter Unterschied beruht auf der Bewertung durch die Patient*innen die „no relief“ angegeben hatten (10 AM vs. 16 Placebo)
Keine schwerwiegenden UAW’s
LAFORCE 2008 (1, 2)
Randomisiert, doppelblind, placebokontrolliert, mit Parallel-Gruppe, multizentrisch Ziel: Effektivität und Sicherheit einer AM-Kombination zusätzlich zu einer Antibiotika-Therapie zur Behandlung akuter Atemwegsinfekte
N=605 N (1)=305 N (2)=300
- Patient*innen mit einer akuten Atemwegsinfektion im Alter von 18-75 Jahren
Behandlung mit: - 600 mg Guafenesin und 60 mg PSE (1) - Placebo (2) Zusätzlich dazu Antibiotikatherapie Zweimal täglich für sieben Tage Beurteilung der Verbesserung der Symptome durch die Patient*innen selbst und am Ende der Therapie durch einen Arzt
Statistisch signifikant besser als Placebo zur Behandlung respiratorischer Infekte Vor allem besser im Hinblick auf verstopfte Nase Unterschied der beiden Gruppen sehr gering, daher nicht klinisch relevant
Keine schwerwiegenden UAW’s Mehr UAW’s in der AM-Gruppe
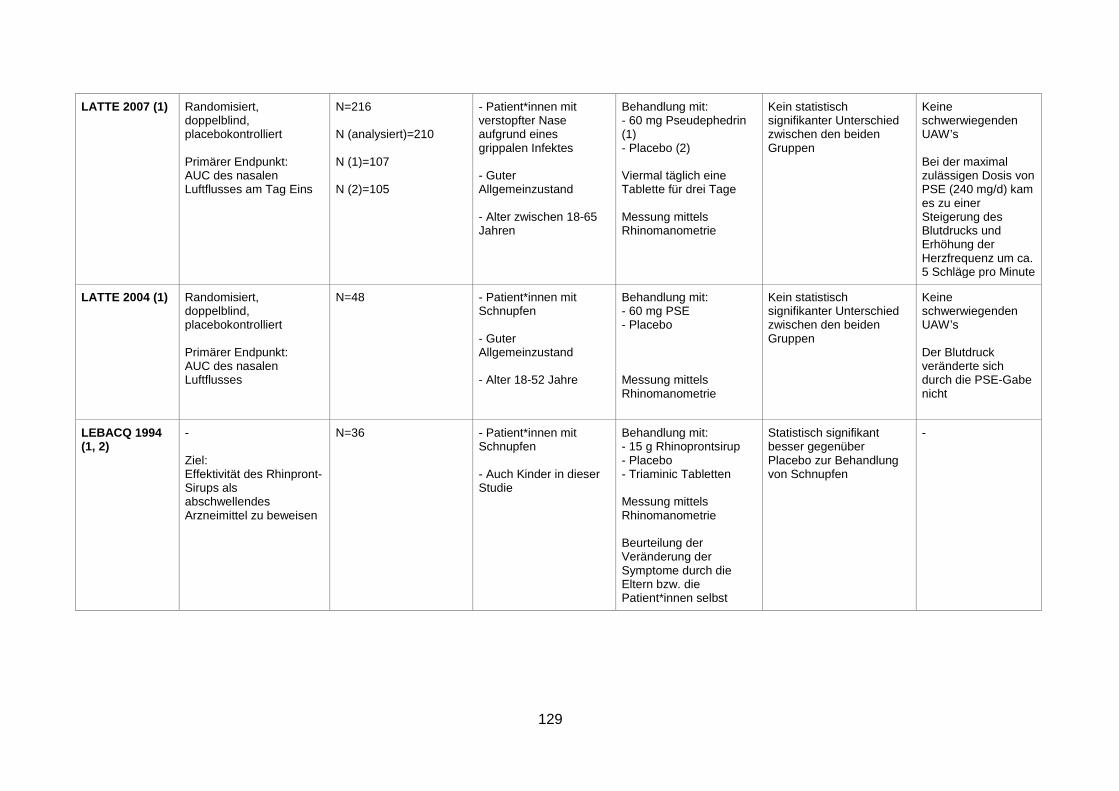
129
LATTE 2007 (1) Randomisiert, doppelblind, placebokontrolliert Primärer Endpunkt: AUC des nasalen Luftflusses am Tag Eins
N=216 N (analysiert)=210 N (1)=107 N (2)=105
- Patient*innen mit verstopfter Nase aufgrund eines grippalen Infektes - Guter Allgemeinzustand - Alter zwischen 18-65 Jahren
Behandlung mit: - 60 mg Pseudephedrin (1) - Placebo (2) Viermal täglich eine Tablette für drei Tage Messung mittels Rhinomanometrie
Kein statistisch signifikanter Unterschied zwischen den beiden Gruppen
Keine schwerwiegenden UAW’s Bei der maximal zulässigen Dosis von PSE (240 mg/d) kam es zu einer Steigerung des Blutdrucks und Erhöhung der Herzfrequenz um ca. 5 Schläge pro Minute
LATTE 2004 (1) Randomisiert, doppelblind, placebokontrolliert Primärer Endpunkt: AUC des nasalen Luftflusses
N=48 - Patient*innen mit Schnupfen - Guter Allgemeinzustand - Alter 18-52 Jahre
Behandlung mit: - 60 mg PSE - Placebo Messung mittels Rhinomanometrie
Kein statistisch signifikanter Unterschied zwischen den beiden Gruppen
Keine schwerwiegenden UAW’s Der Blutdruck veränderte sich durch die PSE-Gabe nicht
LEBACQ 1994 (1, 2)
- Ziel: Effektivität des Rhinpront-Sirups als abschwellendes Arzneimittel zu beweisen
N=36 - Patient*innen mit Schnupfen - Auch Kinder in dieser Studie
Behandlung mit: - 15 g Rhinoprontsirup - Placebo - Triaminic Tabletten Messung mittels Rhinomanometrie Beurteilung der Veränderung der Symptome durch die Eltern bzw. die Patient*innen selbst
Statistisch signifikant besser gegenüber Placebo zur Behandlung von Schnupfen
-
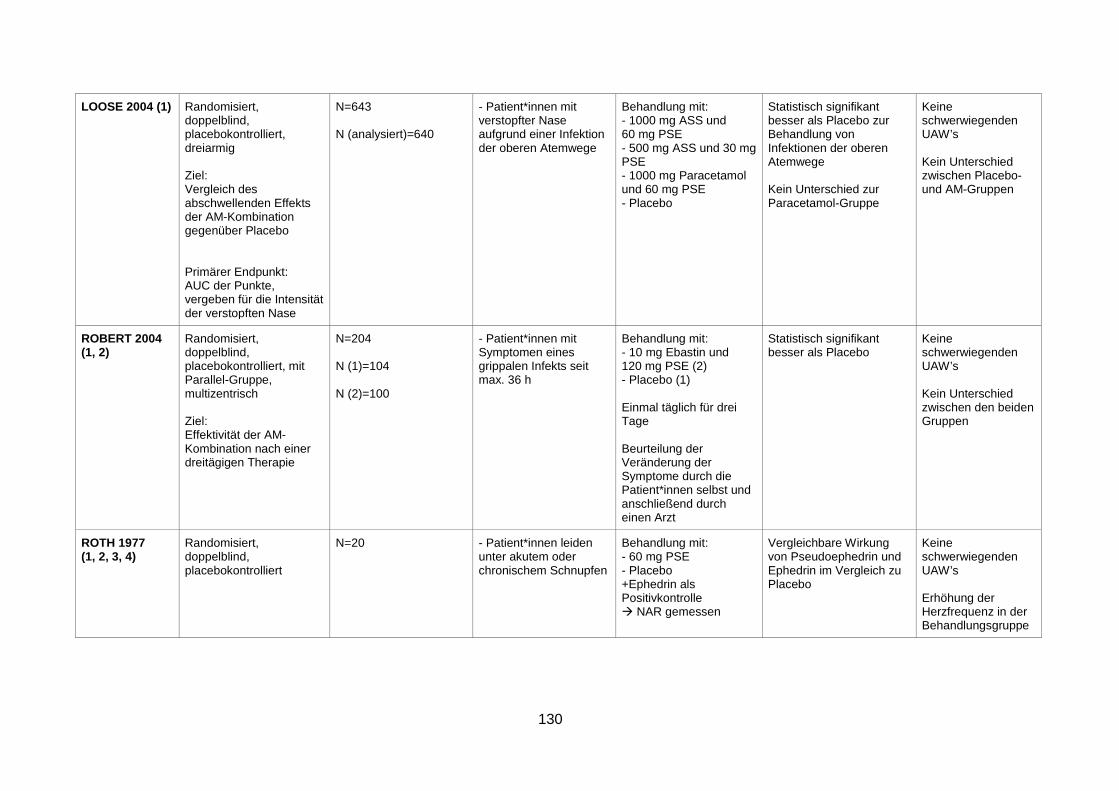
130
LOOSE 2004 (1) Randomisiert, doppelblind, placebokontrolliert, dreiarmig Ziel: Vergleich des abschwellenden Effekts der AM-Kombination gegenüber Placebo Primärer Endpunkt: AUC der Punkte, vergeben für die Intensität der verstopften Nase
N=643 N (analysiert)=640
- Patient*innen mit verstopfter Nase aufgrund einer Infektion der oberen Atemwege
Behandlung mit: - 1000 mg ASS und 60 mg PSE - 500 mg ASS und 30 mg PSE - 1000 mg Paracetamol und 60 mg PSE - Placebo
Statistisch signifikant besser als Placebo zur Behandlung von Infektionen der oberen Atemwege Kein Unterschied zur Paracetamol-Gruppe
Keine schwerwiegenden UAW’s Kein Unterschied zwischen Placebo- und AM-Gruppen
ROBERT 2004 (1, 2)
Randomisiert, doppelblind, placebokontrolliert, mit Parallel-Gruppe, multizentrisch Ziel: Effektivität der AM-Kombination nach einer dreitägigen Therapie
N=204 N (1)=104 N (2)=100
- Patient*innen mit Symptomen eines grippalen Infekts seit max. 36 h
Behandlung mit: - 10 mg Ebastin und 120 mg PSE (2) - Placebo (1) Einmal täglich für drei Tage Beurteilung der Veränderung der Symptome durch die Patient*innen selbst und anschließend durch einen Arzt
Statistisch signifikant besser als Placebo
Keine schwerwiegenden UAW’s Kein Unterschied zwischen den beiden Gruppen
ROTH 1977 (1, 2, 3, 4)
Randomisiert, doppelblind, placebokontrolliert
N=20 - Patient*innen leiden unter akutem oder chronischem Schnupfen
Behandlung mit: - 60 mg PSE - Placebo +Ephedrin als Positivkontrolle NAR gemessen
Vergleichbare Wirkung von Pseudoephedrin und Ephedrin im Vergleich zu Placebo
Keine schwerwiegenden UAW’s Erhöhung der Herzfrequenz in der Behandlungsgruppe
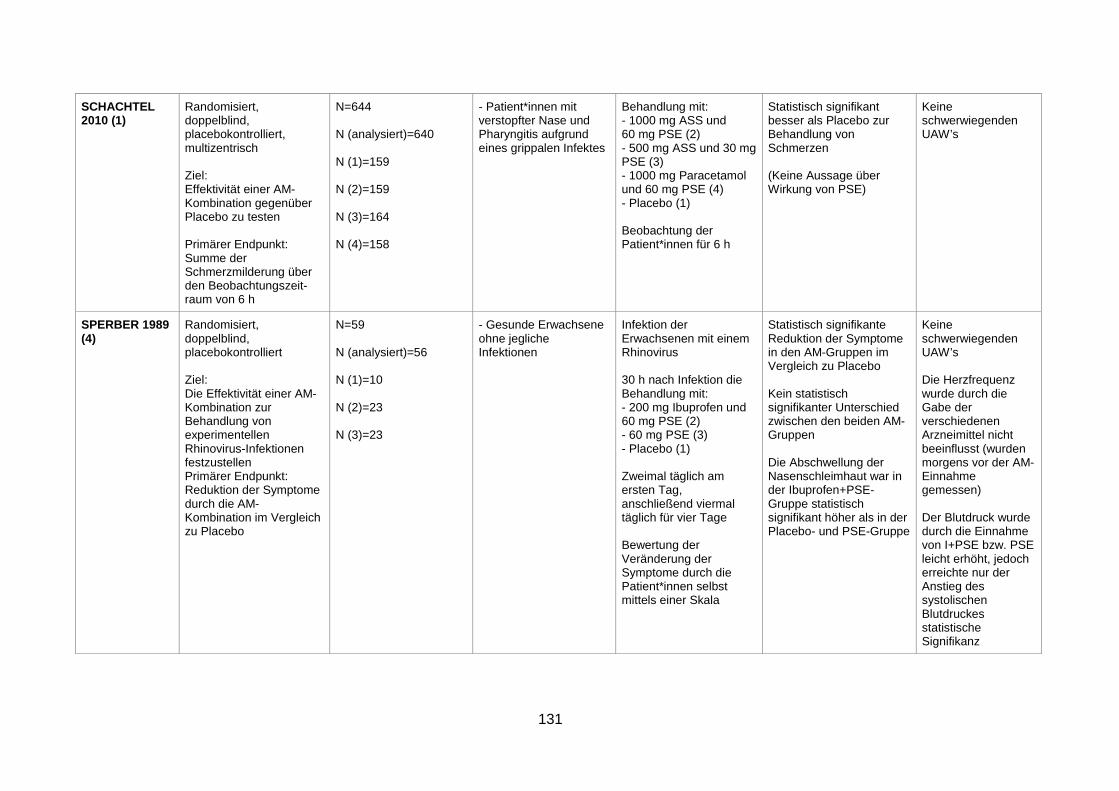
131
SCHACHTEL 2010 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Ziel: Effektivität einer AM-Kombination gegenüber Placebo zu testen Primärer Endpunkt: Summe der Schmerzmilderung über den Beobachtungszeit-raum von 6 h
N=644 N (analysiert)=640 N (1)=159 N (2)=159 N (3)=164 N (4)=158
- Patient*innen mit verstopfter Nase und Pharyngitis aufgrund eines grippalen Infektes
Behandlung mit: - 1000 mg ASS und 60 mg PSE (2) - 500 mg ASS und 30 mg PSE (3) - 1000 mg Paracetamol und 60 mg PSE (4) - Placebo (1) Beobachtung der Patient*innen für 6 h
Statistisch signifikant besser als Placebo zur Behandlung von Schmerzen (Keine Aussage über Wirkung von PSE)
Keine schwerwiegenden UAW’s
SPERBER 1989 (4)
Randomisiert, doppelblind, placebokontrolliert Ziel: Die Effektivität einer AM-Kombination zur Behandlung von experimentellen Rhinovirus-Infektionen festzustellen Primärer Endpunkt: Reduktion der Symptome durch die AM-Kombination im Vergleich zu Placebo
N=59 N (analysiert)=56 N (1)=10 N (2)=23 N (3)=23
- Gesunde Erwachsene ohne jegliche Infektionen
Infektion der Erwachsenen mit einem Rhinovirus 30 h nach Infektion die Behandlung mit: - 200 mg Ibuprofen und 60 mg PSE (2) - 60 mg PSE (3) - Placebo (1) Zweimal täglich am ersten Tag, anschließend viermal täglich für vier Tage Bewertung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der Symptome in den AM-Gruppen im Vergleich zu Placebo Kein statistisch signifikanter Unterschied zwischen den beiden AM-Gruppen Die Abschwellung der Nasenschleimhaut war in der Ibuprofen+PSE-Gruppe statistisch signifikant höher als in der Placebo- und PSE-Gruppe
Keine schwerwiegenden UAW’s Die Herzfrequenz wurde durch die Gabe der verschiedenen Arzneimittel nicht beeinflusst (wurden morgens vor der AM-Einnahme gemessen) Der Blutdruck wurde durch die Einnahme von I+PSE bzw. PSE leicht erhöht, jedoch erreichte nur der Anstieg des systolischen Blutdruckes statistische Signifikanz
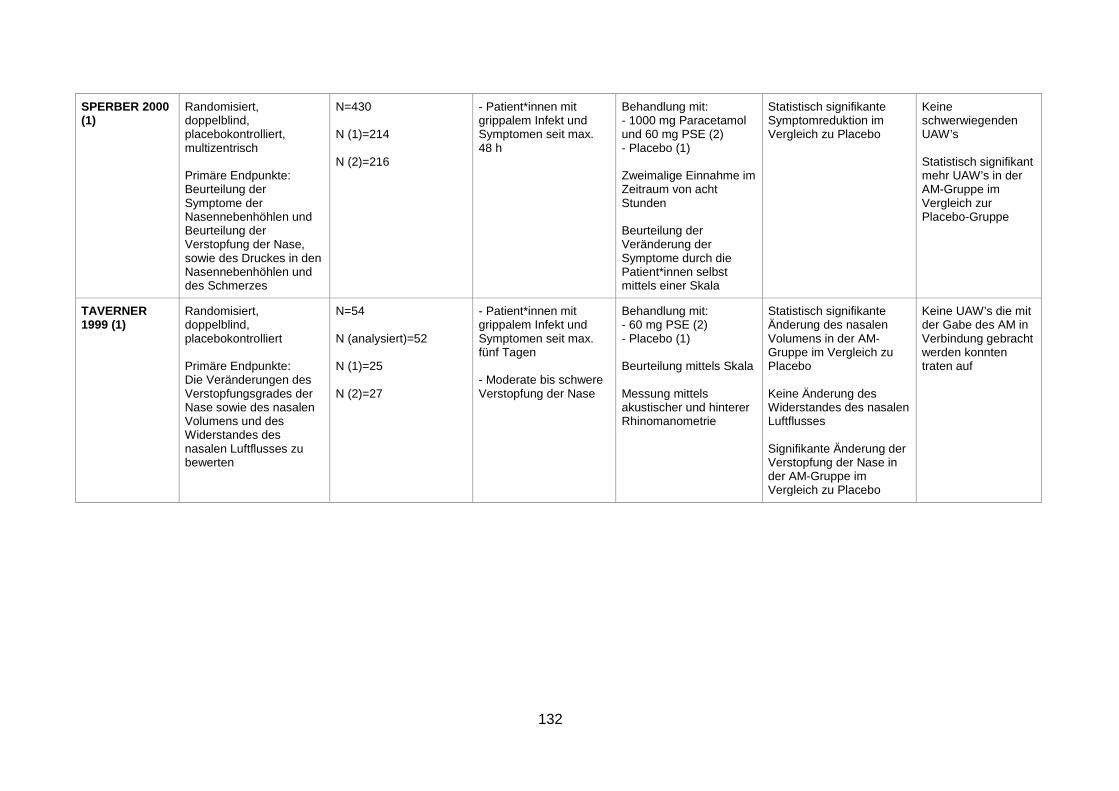
132
SPERBER 2000 (1)
Randomisiert, doppelblind, placebokontrolliert, multizentrisch Primäre Endpunkte: Beurteilung der Symptome der Nasennebenhöhlen und Beurteilung der Verstopfung der Nase, sowie des Druckes in den Nasennebenhöhlen und des Schmerzes
N=430 N (1)=214 N (2)=216
- Patient*innen mit grippalem Infekt und Symptomen seit max. 48 h
Behandlung mit: - 1000 mg Paracetamol und 60 mg PSE (2) - Placebo (1) Zweimalige Einnahme im Zeitraum von acht Stunden Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Symptomreduktion im Vergleich zu Placebo
Keine schwerwiegenden UAW’s Statistisch signifikant mehr UAW’s in der AM-Gruppe im Vergleich zur Placebo-Gruppe
TAVERNER 1999 (1)
Randomisiert, doppelblind, placebokontrolliert Primäre Endpunkte: Die Veränderungen des Verstopfungsgrades der Nase sowie des nasalen Volumens und des Widerstandes des nasalen Luftflusses zu bewerten
N=54 N (analysiert)=52 N (1)=25 N (2)=27
- Patient*innen mit grippalem Infekt und Symptomen seit max. fünf Tagen - Moderate bis schwere Verstopfung der Nase
Behandlung mit: - 60 mg PSE (2) - Placebo (1) Beurteilung mittels Skala Messung mittels akustischer und hinterer Rhinomanometrie
Statistisch signifikante Änderung des nasalen Volumens in der AM-Gruppe im Vergleich zu Placebo Keine Änderung des Widerstandes des nasalen Luftflusses Signifikante Änderung der Verstopfung der Nase in der AM-Gruppe im Vergleich zu Placebo
Keine UAW’s die mit der Gabe des AM in Verbindung gebracht werden konnten traten auf
133
VIRTANEN 1983 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Effektivität einer AM-Kombination zur Linderung von nasalen und das Ohr betreffenden Symptomen festzustellen Primäre Endpunkte: Symptomreduktion der Nase und der Ohren in den beiden Behandlungs-Gruppen
N=93 N (analysiert)=80 N (1)=41 N (2)=39
- Patient*innen mit grippalem Infekt und Symptomen seit max. 48 h
Behandlung mit: - 6 mg Dexchlorphenir-amin maleat und 120 mg PSE-Sulfat (2) - Placebo (1) Zweimal tägliche für fünf Tage Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Statistisch signifikante Reduktion der nasalen Symptome in der AM-Gruppe im Vergleich zu Placebo Statistisch signifikante Symptomreduktion der Ohrenbeschwerden am dritten Tag
Keine schwerwiegenden UAW’s Mehr UAW’s in der AM-Gruppe, dieser Unterschied war jedoch nicht statistisch signifikant
ZHANG 2019 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Primärer Endpunkt: Reduktion der Summer aller Symptome (im Verlauf von 4 h)
N=53 N (1)=27 N (2)=25
- Patient*innen mit grippeähnlichen Symptomen und grippalen Infekten seit max. 48 h
Behandlung mit: - Paracetamol, Dextrometorphan hydrobromid und Chlorpheniramin maleat, PSE (2) - Placebo (1) Beurteilung der Veränderung der Symptome durch die Patient*innen selbst mittels einer Skala
Kein Unterschied zwischen den beiden Behandlungs-Gruppen
Keine schwerwiegenden UAW‘s
134
GRIPPALE INFEKTE - PHENYLEPHRIN
STUDIENDESIGN STUDIENTEIL-NEHMER*INNEN
EINSCHLUSS- KRITERIEN MASSNAHMEN ERGEBNISSE UNERWÜNSCHTE
EREIGNISSE
COHEN 1972 (1) Randomisiert, doppelblind, placebokontrolliert, crossover design Ziel: Messung der Nasendurchgängigkeit nach Behandlung mit PE im Vergleich zu Placebo
N=48 Keine Studien-abbrecher
- Patient*innen leiden aufgrund eines grippalen Infektes unter verstopfter Nase - Symptome seit 24 bis 48 h
Behandlung mit: - 10 mg PE - 15 mg PE - 25 mg PE - Placebo Messung mittels Rhinomanometrie Kontrolle des Blutdrucks und der Herzfrequenz
Signifikante Verbesserung der Nasendurchgängigkeit im Vergleich zu Placebo Leichter Anstieg der Herzfrequenz bei Behandlung mit PE Keine Beeinflussung des Blutdrucks
Keine schwerwiegenden UAW Diese korrelieren mit der verabreichten PE-Dosis
PICON 2013 (1, 2)
Randomisiert, doppelblind, placebokontrolliert Ziel: Effektivität und Sicherheit einer Kombination von Paracetamol, Chlorpheniramin und PE zu bewerten Primärer Endpunkt: Die Summe der erreichten Punkte auf einer 4-teiligen Likert-Skala
N=146 N (1)=73 N (2)=73 Alter: 18 -60 Jahre 8 Dropouts N (analysiert)=146
- Patient*innen leiden unter grippeähnlichen Zustanden oder einem grippalen Infekt
Behandlung mit: - 400 mg Paracetamol, 4 mg Chlorpheniramin und 4 mg PE (2) - Placebo (1) 5-mal täglich für 48 – 72 h Patient*innen mussten an drei Tagen in die Klinik Bewertung durch 4-teilige Skala
Statistisch signifikanter Unterschied der Reduktion der Symptome im Vergleich zu Placebo Keine statistische Signifikanz im Bezug auf die Tage mit anhaltenden Symptomen
Keine schwerwiegenden UAW Kein statistisch signifikanter Unterschied der unerwünschten Ereignisse zwischen Behandlungs- und Placebo-Gruppe Keine Beeinflussung der Herzfrequenz
135
DEBELIC 1973 (1, 2)
Randomisiert, doppelblind „Zur Therapie der banalen Rhinitis.“ Primärer Endpunkt: Intensität der verstopften Nase
- - Behandlung mit: - 1 mg Clemastine und 50 mg Phenylpropanolamine 50 mg (2x tgl.) - 0,2 mg Belladonna und 4 mg Chlorphenamin - 50 mg Phenylpropanolamin (2x tgl.) Einnahme für acht Tage
- -
Gründe für Studienausschluss: (1) Komponentenbeweis wurde nicht erbracht (Beispiel: Vergleich von Desloratadin-Pseudoephedrin mit Pseudoephedrin alleine Desloratadin-
Monotherapie-Gruppe fehlt, ohne diese kann der Vorteil der Kombination gegenüber der Monotherapie mit Desloratadin nicht untersucht werden) (2) Arzneistoffe sind in Österreich nicht zugelassen (3) Zu geringe Teilnehmer*innenzahl (4) Die Allergische Reaktion bzw. die Infektion mit einem Erreger wurde auf nicht natürliche Weise hervorgerufen (5) Die Originaldaten der Studie waren im gegebenen Zeitrahmen nicht zugänglich

136
9 Eidesstattliche Erklärung
Ich erkläre hiermit an Eides statt durch meine eigenhändige Unterschrift, dass ich die
vorliegende Arbeit selbständig verfasst und keine anderen als die angegebenen Quellen und
Hilfsmittel verwendet habe. Alle Stellen, die wörtlich oder inhaltlich den angegebenen Quellen
entnommen wurden, sind als solche kenntlich gemacht.
Die vorliegende Arbeit wurde bisher in gleicher oder ähnlicher Form noch nicht als Magister-
/Master-/Diplomarbeit/Dissertation eingereicht.
Datum Unterschrift
Recommended

















