Embed Size (px)
DESCRIPTION
生命情報学入門 タンパク質立体構造予測法 20 11 年 5 月 24 日. 阿久津 達也. 京都大学 化学研究所 バイオインフォマティクスセンター. 本日の内容. 構造予測に関連する基本事項 立体構造予測法の分類 スレッディング法 3 D-1D プロファイル ポテンシャル型スコア関数を用いたスレッディング CASP まとめ. タンパク質立体構造予測. アミノ酸配列から、タンパク質の立体構造(3次元構造)をコンピュータにより推定 実験よりは、はるかに精度が悪い だいたいの形がわかれば良いのであれば、4~5割近くの予測率. アミノ酸とタンパク質. - PowerPoint PPT Presentation
Citation preview

生命情報学入門タンパク質立体構造予測法
2011 年 5 月 24 日
京都大学 化学研究所バイオインフォマティクスセンター
阿久津 達也

本日の内容 構造予測に関連する基本事項 立体構造予測法の分類 スレッディング法 3D-1D プロファイル ポテンシャル型スコア関数を用いたス
レッディング CASP まとめ

タンパク質立体構造予測 アミノ酸配列から、タ
ンパク質の立体構造(3次元構造)をコンピュータにより推定
実験よりは、はるかに精度が悪い
だいたいの形がわかれば良いのであれば、 4~5割近くの予測率
T V A C L G F S L V V G G R D
アミノ酸配列
コンピュータ
タンパク質立体構造
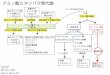
アミノ酸とタンパク質 アミノ酸:20
種類 タンパク質:ア
ミノ酸の鎖(短いものはペプチドと呼ばれる)
R
C
H N
H
R
N
O
C C C
H H O
ペプチド結合
アミノ酸
蛋白質
C
H
H N
H
R
OH
O
C アミノ基 カルボシキル基
側鎖

側鎖の例
3 CH
Ala アラニン
3 CH 3 H C
CH
Val バリン
CH
CH
HC C
HC
CH
2 CH
Phe フェニル アラニン
2 CH
C
O - O
Asp アスパラギン酸
HN
NH
2 CH
+
His ヒスチジン
2 CH
SH
Cys システイン
Gly グリシン
H

タンパク質の種類と高次構造 タンパク質の分類
球状タンパク質 繊維状タンパク質 膜タンパク質
一次構造(アミノ酸配列) 二次構造( α 、 β 、それ以外(ループ、コイ
ル)) 三次構造(三次元構造、立体構造) 四次構造(複数の鎖)

タンパク質立体構造の決定 主に X 線結晶解析か NMR 解析による
一般に X 線解析の方が精度が高い しかし、結晶中の構造しかわからない
アミノ酸配列決定より困難 半年から1年くらいかかることも珍しく無
い 既知アミノ酸配列 >> 10 万 既知立体構造 < 数万

タンパク質立体構造の特徴 基本的には鎖(ひも)
状 二種類の特徴的な構造
が頻繁に現れ、立体構造の骨格(コア)を作る α へリックス(らせん状
の部分) β シート(ひも状の部分
が並んだ部分)
α
β
ループ

構造とアミノ酸の種類の関係 ( 球状)タンパク質
内側:疎水性アミノ酸 外側:親水性アミノ酸
α へリックス 内側:疎水性 外側:親水性
β ストランド 疎水性と親水性が交互に現れる
ループ領域 親水性が高い

立体構造データベース PDB(Protein Data Bank )
タンパク質立体構造データベース 2011 年 5 月 10 日現在約 73009 データ
(ただし重複あり) SCOP
立体構造分類データベース FSSP / DALI
立体構造アライメントデータベース/アライメントサーバー

タンパク質立体構造の分類 構造分類の必要性
立体構造と機能の間には密接な関係 配列が似ていなくても構造類似のタンパク質が
多数存在 SCOP による階層的クラス分け
Class: 二次構造の組成( α 、 β 、 α+β など)に基づく分類
Fold: 構造の類似性 ← スレッディング法の対象
Superfamily: 進化的類縁性 Family: 明らかな進化的類縁性

立体構造予測法の分類 物理的原理に基づく方法 ホモロジーモデリング 格子モデル 2次構造予測 スレッディング

物理的原理に基づく方法 エネルギー最小化、もしくは、微分方程式を(数値的に)解く、などの物理的原理に基づく方法
主として分子動力学法 (Molecular Dynamics) 数十残基程度であれば、実際のタンパク質やペプ
チドと似た構造を推定可能(なことがある) 構造の最適化や安定性の解析には実用的 ⇒ ホモロジーモデリング
主鎖をアラインメントで計算した後に
側鎖構造などを最適化
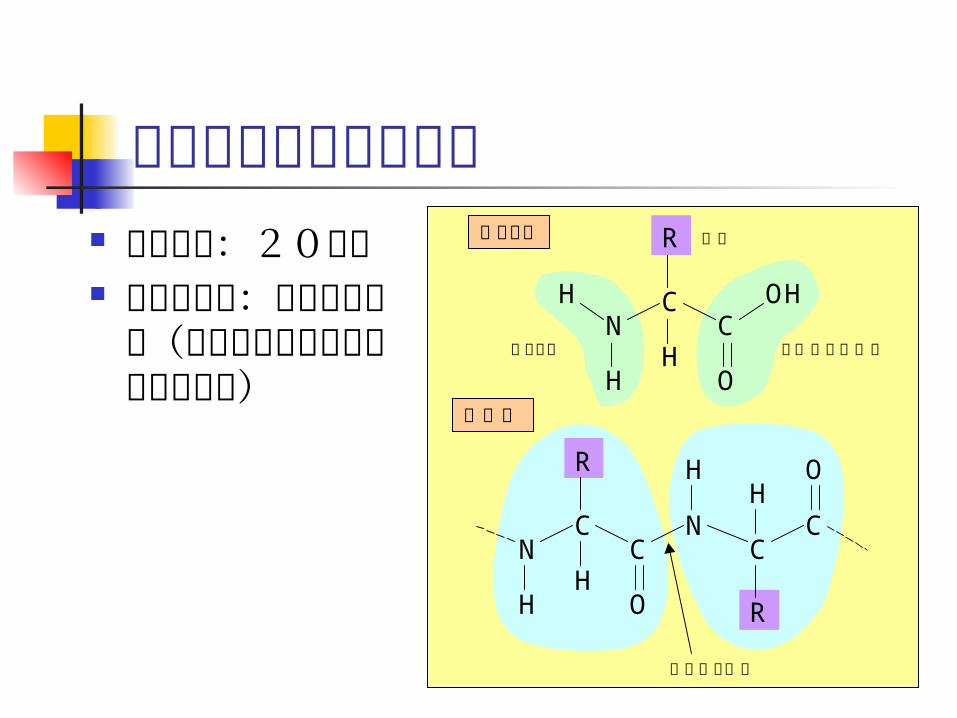
格子モデル 各残基が格子点
にあると仮定 予測よりも、
フォールディングの定性的な理解のために利用される
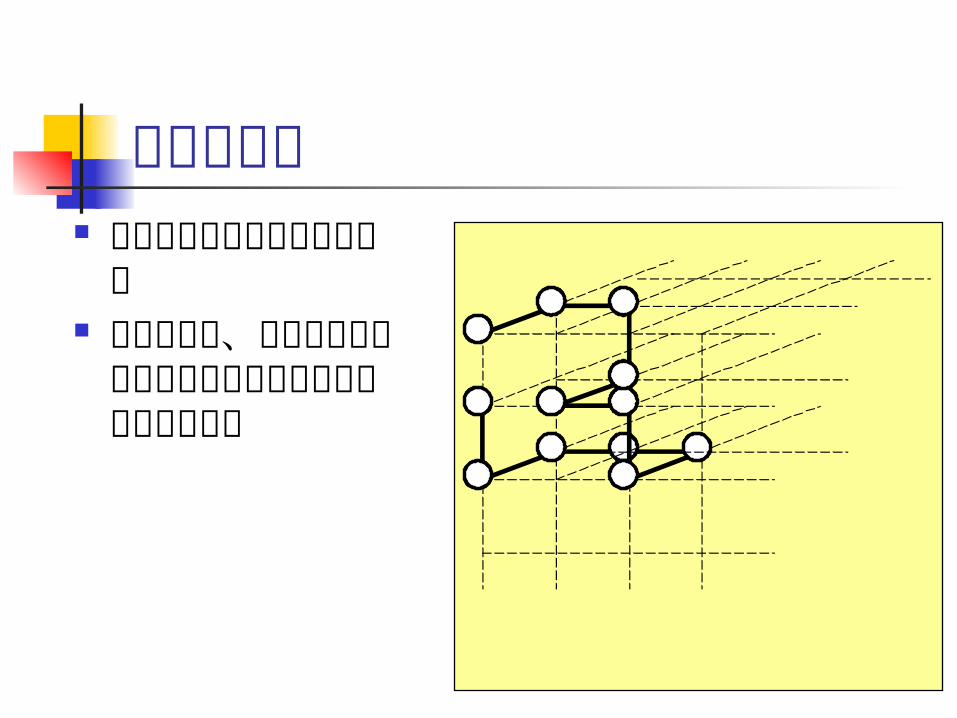
格子モデルに基づく研究 折れ畳み経路の
シミュレーションによる定性的理解 →フォールディングファンネル
エネルギー最小の構造の計算法→ NP 困難
親水性アミノ酸
疎水性アミノ酸
スコア=-9
スコア=-5
配列

二次構造予測 アミノ酸配列中の各残基
が、 α 、 β 、それ以外のどれに属するかを予測
でたらめに推定しても、33.3%の的中率
最も高精度なソフトを使えば、 70%~ 80%の的中率
ニューラルネット、 HMM 、サポートベクタマシンなどの利用
α β それ以外
L K I A P

ニューラルネットによる二次構造予測
出力層
中間層 (隠れ層)
入力層
Leu Asn Lys Ala Val Thr Gly

膜タンパク質の膜貫通領域予測
膜貫通領域 α へリック
ス 7~17残基程度の疎水性指標の平均値をプロット
平均値が高い部分が膜貫通領域と推定
D G A I L V P R V K Q
A 1.8 C: 2.5 D: - 3.5 E: -3.5 F: 2.8 ...
疎水性指標
膜タンパク
細胞膜

フォールド予測( Fold Recognition)
精密な3次元構造ではなく、だいたいの形( fold) を予測
立体構造は 1000種類程度の形に分類される、との予測 (Chotia, 1992) に基づく
T V A C L G F S L V V G G R D
1000個のテンプレート構造
アミノ酸配列

タンパク質スレッディング
T V A C L G F S L V V G R D
アミノ酸配列
K
立体構造
立体構造(テンプレート)とアミノ酸配列の間のアライメント

スレッディングとアライメント
T V A C L G F S L V V G R D
入力アミノ酸配列
K
立体構造 A G L F S G L G Y
A G L G S V L G
A G L F G S L G Y
A G L G S V L G

スレディング法の分類 プロファイルによるスレッディング
PSI-BLAST 3D-1D 法 構造アライメント結果に基づくスレッディング
残基間ポテンシャルによるスレッディング コンタクトポテンシャル 距離依存ポテンシャル その他のポテンシャル

プロファイル アライメント
におけるスコア行列と類似
スレッディングの場合、残基位置ごとにスコア (位置依存スコア )
A
C
D
E
残基1 残基2 残基3 残基4 3.8
1.5
-1.5
0.2
-3.5
1.3
-2.9
2.1
-0.3
3.7
2.3
-4.6
3.1
-1.3
1.2
4.2
立体構造
残基1
残基2
残基3
残基4

プロファイルによるアライメント
動的計画法(DP) により最適解を計算
スコア行列のかわりにプロファイルを使う
AED ...... アミノ酸配列:
A
C
D
E
残基1 残基2 残基4 3.8
1.5
-1.5
0.2
-3.5
1.3
-2.9
-4.1
-0.3
3.7
2.3
-4.6
3.1
-1.3
1.2
4.2
残基3
プロファイル:
123 .....AED .....
アライメント 1234 .....A-ED .....
1- 23 .....AEDC ...
スコア 3.8-4.1+4.2 =3.9
3.8-2.0+3.7+ 3.1=8.7
3.8-2.0-2.9+ -0.3=-1.4
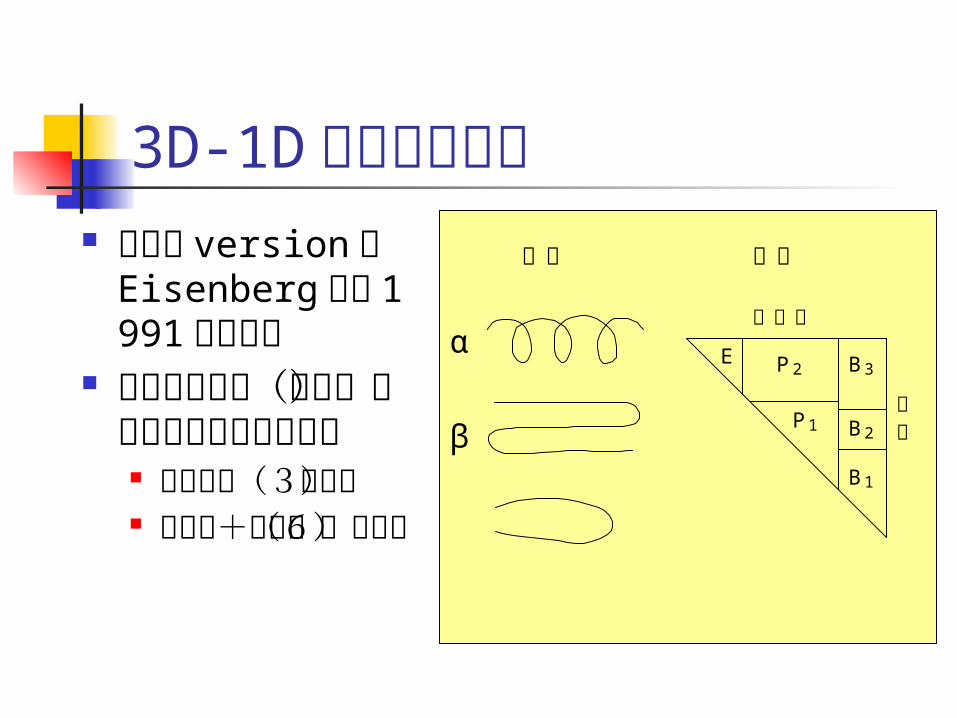
3D-1D プロファイル 最初の version
は Eisenberg らが 1991 年に提案
構造中の残基(位置)を18種類の環境に分類 二次構造(3種
類) 内外性+極性
(6種類)
E P 2
P 1 B 2
B 1
B 3
内外性
極性
α
β
主鎖 側鎖

3D-1D プロファイル
3D- 1Dスコア 3D- プロファイル
E
P
2 P
1
B
3
内外性
極性
B
2 B
1
残基1
残基3
残基2
残基4
タンパク質立体構造
A
R
Y
W
-0.66
0.18
-1.67
1.00
-0.79
-1.16
0.07
1.17
-0.91
-2.16
0.17
1.05
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・ B 1 B 1 α B 1 β
・ ・ ・ ・・・
環境クラス
アミノ酸
A
R
Y
W
-0.79
-1.16
0.07
1.17
-0.91
-2.16
0.17
1.05
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・
・ ・ ・ ・・・
残基1 残基2 残基3
-0.79
-1.16
0.07
1.17

その他のプロファイル 配列のマルチプルアライメントに基づ
くプロファイル PSI-BLAST 、 HMM
立体構造のマルチプルアライメントに基づくプロファイル作成
角度情報なども考慮したプロファイル プロファイル vs プロファイルによる
アライメント

ポテンシャル型スコア関数を用いたスレッディング
全体のポテンシャルエネルギーを最小化( Σfd(X,Y) が最小となるようなスレッディングを計算)
精度向上が期待できる
でも計算時間が問題
T V A C L G F S L V V G R D
アミノ酸配列
K
立体構造
d
f (T, F) d

プロファイル型スコア関数とポテンシャル型スコア関数
プロファイル型スコア関数 (Eisenberg et al. 1991)
ポテンシャル型スコア関数 (Miyazawa, Sippl, . . .)
A
C
D
E
Pos1 Pos2 Pos3 Pos4 3.8
1.5
-1.5
0.2
-3.5
1.3
-2.9
2.1
-0.3
3.7
2.3
-4.6
3.1
-1.3
1.2
4.2
d
d
score
A
L

フラグメント・アセンブリ法
Univ. Washington の Baker らが開発 現時点では最強の方法と考えられている方法 数残基から十数残基の断片構造をプロファイル比較法などを用いて既知構造データベースから取得 => 各断片配列ごとにいくつかの候補を選ぶ
フラグメントをつなぎ合わせることにより全体構造を予測。つなぎ合わせる際には分子動力学法などによるエネルギー最適化などを行う

立体構造予測におけるブレークスルー
スレッディング法の発明 (Eisenberg et al., 1991) 構造既知の配列と類似性が無い配列の構造予測
PSI-BLAST の開発 (Altschul et al, 1997) プロファイルに基づくマルチプルアライメント
の繰り返し実行によるスレッディング David Baker による ab initio 予測 (1997)
統計情報+シミュレーション

立体構造予測コンテスト:CASP
CASP (Critical Assessment of Techniques for Protein Structure Prediction)
ブラインドテストにより予測法を評価① 半年以内に立体構造が実験により決定する見込みの配列(数十種類)をインターネット上で公開
② 参加者は予測結果を送付③ 構造決定後、正解とのずれなどを評価、順位づ
け

CASP の経過と結果の公表 CASP1 (1994), CASP2(1996),
CASP3(1998), CASP4(2000), CASP5(2002), CASP6(2004), CASP7(2006), CASP8(2008), CASP9(2010)
CAFASP(1998,2000,2002,2004,2006) 完全自動予測法の評価
結果の公表 会議 ホームページ
http://predictioncenter.gc.ucdavis.edu/ 学術専門誌 (Proteins)

まとめ 立体構造予測
正確な座標は予測できない だいたいの形の予測であれば4~5割近く
タンパク質スレッディング法が有力 近年では、フラグメントアセンブリー法が有力
二次構造予測であれば、 70%-80%程度

参考文献 阿久津達也:バイオインフォマティクスの数理と
アルゴリズム、共立出版、2007. 丸山修、阿久津達也:バイオインフォマティクス
–配列データ解析と構造予測、朝倉書店、2007.
藤博幸:タンパク質機能解析のためのバイオインフォマティクス、共立出版、2004.