Embed Size (px)
Citation preview

7 - . .. -"---*-- I_-
1693282
c
EL PRESENTE TRABAJO FUE REALIZADO BAJO LA DiRECCION DEL
DR. MARCO ANTONIO QUIROZ ALFAR0 EN EL LABORATORIO DE
ELECTROQUIMICA DEL DEPTO. DE QUIMICA DE LA UNIVERSIDAD
AUTONOMA METROPOLITANA-IZTAPALAPA.
EL DESARROLLO DE LA PRESENTE TESIS CONTO CON EL APOYO
ECONOMIC0 DE PRONAES (S.E.P.) POR MEDIO DEL PROYECTO
"ELECTRODEPOSICION DE METALES Y ALEACIONES" Y DEL
CONSEJO NACIONAL DE CIENCIA Y TECNOLOGIA A TRAVES DE
SU PROGRAMA DE BECAS.

I N D I C E
INTRODUCCION
CAPITULO I
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . I Fundamentos T e ó r i c o s 4
1 . 1 . C a r a c t e r i z a c i ó n s u p e r f i c i a l d e m e t a l e s p o r procesos
d e e l e c t r o s o r c i ó n d e h i d r ó g e n o y ox igeno . . . . . . . . . . . . . 5
I . 1 . 1 . P r o c e s o s d e a d s o r c i ó n d e h i d r ó g e n o . . . . . . . . . . . . . 6
I . I . 2. D e t e r m i n a c i ó n de Q p o r V . C . . . . . . . . . . . . . . . . . 9
1 . I . 3. P r o c e s o s d e e l e c t r o s o r c i ó n de ox igeno . . . . . . . . . . 14
H , S
1 . 1 . 3 . 1 . P l a t i n o . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
1.1.3.2. P a l a d i o . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
1 . 1 . 3 . 3 . Oro 25 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2.' Depós i t os m e t á l i c o s a s u b p o t e n c i a l . . . . . . . . . . . . . . . . . . . 31
I .2. I . I n t r o d u c c i ó n . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
I .2 . 2. F o r m a c i ó n d e d e p ó s i t o s m e t á l i c o s a subpotencial
s o b r e e l e c t r o d o s P O I i c r i s t a l inos en soluciones
a c u o s a s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
I . 2 . 3. A s p e c t o s t e r m o d i n á m i c o s d e l a f o rmac i ón .de
d e p ó s i t o s m e t á l i c o s a s u b p o t e n c i a l . . . . . . . . . . . . . 39
CAPITULO I I
1 1 . 1 . Ce lda e l e c t r o q u i m i c a . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
11.2. P r e p a r a c i ó n d e e l e c t r o d o s . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
11.3. R e a c t i v o s u t i l i z a d o s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
11.4. Monta je e l e c t r ó n i c o . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
11.5. C o n d i c i o n e s e l e c t r o q u i m i c a s p a r a c a r a c t e r i z a c i ó n . . . . . 51

<.,.-.-. . ......-- ___.*_cI_-. .,. ..... .,. ... ,._I ........ --~-.*'.rr>.^il'ru~'~ru^rar------.~. .
093282
CAPITULO I11
I 1 1 . Resultados y discusión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
1 1 1 . 1 . Procesos de electrosorción de hidrógeno y oxigeno . . . . 58
1 1 1 . 1 . 1 . Platino . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
1 1 1 . 1 . 1.a. Estimación del área superficial expuesta
. a part ir de la adsorción de hidrógeno
sobre Pt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
I l l . 1 . 1.b. Estimacion del área superficial expuesta
a part ir de l a desorción de oxígeno sobre
Pt. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
I l l . 1.2. Paladio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
111.1.3. Oro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7 3
I I 1 . 2 . Depósito de adát omos de Cu a subpotencial., . . . . . . . . . . 79
111.2.1. Efecto UPD de Cu sobre Pt . . . . . . . . . . . . . . . . . . . . . . 79
111.2.2. Efecto UPD de Cu sobre Pd . . . . . . . . . . . . . . . . . . . . . . 90
I I I .2. 3 . Efecto UPD de Cu sobre Au . . . . . . . . . . . . . . . . . . . . . . 98
1 1 1 . 3 . Estimacion de A r , de Pd y Au utilizando el valor de
f obtenido por depósito de Cu a subpotencial . . . . . . . . . 106
111.3. 1 Paladio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
111.3.2. Oro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
CONCLU C I ONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
BI BLIOGRAF IA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
110
113

I N T R O D U C C I O N
Los estudios cinéticos de reacciones electroquímicas, asi como la
determinación de propiedades electrocataliticas de electrodos de
metáles nobles dependen, prioritariamenre, de las caracteristicas de
la superficie expuesta, en particular de la magnitud de su área y SU
configuración cristalográfica. Es lógico, por tanto, que una adecuada
caracterización superficial no puede ser subestimada.
A este respecto, se realizan esfuerzos cada vez más importantes a
f i n de desarrollar métodos más precisos, destacando significativamente
aquellos que se aplican "ex-situ" al sistema 187-901. Aunque estos
rnetodos dan valores muy precisos. no indican la morfología real de la
superficie. 'ya que los resultados son obtenidos. bajo condiciones
diferentes al medio ambiente normal del electrodo, y puesto que la
mayoria de ellos son de carácter destructivo, esto los hace poco
atractivos e imprácticos para determinaciones rutinarias. Por esta
razón, es prácticamente una necesidad en la investigaci6n
eiectroquimica efectuar tal caracterización mediante técnicas
sn-situ", de manera que se puedan entender la naturaleza. y origen de
las propiedades que dependen de manera directa o indirecta de la
estructura que presenta la superficie expuesta. En las últimas
décadas, los métodos electroquimicos han permitido desarrollar
tecnicas de caracterización superficial cada vez mbs adecuadas, en
particular destacan aquellas empkadas en electrodos Constituidos de
metales nobles I2.31.
,.:
1

Es sabido que este tipo de electrodos metálicos muestran gran
afinidad hacia los procesos de electrosorción de hidrógeno y oxígeno
en soluciones ácidas, lo que ha permitido su caracterización por estas
técnicas electroquímicas [1,31. Sin embargo, metales tales como Pd y
Au no han podido ser adecuadamente caracterizados por esta vía. En Pd,
el proceso de adsorción de hidrógeno es acompañado siempre por una
absorción significativa del mismo, 1 0 que impide determinar la
cantidad real adsorbida y su extención. Para Au basta decir que
presenta una adsorción casi nula de dicho eiemento, aproximadamente 2%
(37,381. Por otro lado, si bien ambos metales, Pd y Au. presentan una
adecuada electrosorción de oxígeno, la relación O/M a nivel de la
monocapa, aún no ha sido bién definida para los electrodos
constituidos de estos metales nobles.
En los últimos años, en el ámbito electroquimico se h a despertado
un enorme interés en el estudio del depósito de metales a subpotencial
(UPD : por Under Potential Deposition) sobre sustratos metálicos
ajenos. Este interés reside en ei conocimiento de la profunda
influencia que puede tener la monocapa UPD (como una fracción o
completa), sobre los electrodos sustrato usados para electrocatálisis
de reacciones electrosintéticas y celdas de combustibles, refinamiento
de superficies, estudios sobre crecimiento electroquímico de
cristales, etc. Se ha comprobado que el proceso UPD presenta una
oportunidad única para estudiar el acoplamiento de una amplia variedad
de fenómenos superficiales, tales como la adsorción, transferencia de
carga, difusión superficial, formación de películas metálicas
bidimencionales. etc. Sobre estas bases, estudios muy recientes
[6,7,121 han mostrado que el efecto UPD puede ser adecuadamente
2

utilizado para pr6positos de caracterización superficial. La
importancia del método UPD descrito, es particularmente ref lejada para
aquellas superficies metálicas cuya caracterización por las técnicas
tradicionales de electrosorción de hidrógeno y oxigeno es inoperante.
Por lo tanto, los objetivos de este trabajo van encaminados a la
aplicación del efecto UPD sobre electrodos metálicos de Pd y Au con
los siguientes propósitos:
1. Determinar las caracteristicas del depósito formado como una
funcidn del potencial aplicado al electrodo de trabajo.
2. Establecer las condiciones experimentales que permitan la
formación de una monocapa metálica sobre la superficie expuesta.
3. Determinar las caracteristicas de la superficie expuesta, en
particular la determinación de factores de rugosidad.
J. Analizar los procesos de electrosorción de oxigeno. a l a luz
de los. resultados obtenidos por aplicación del efecto UPD.
5. Establecer las relaciones O/Pt, O/Pd y OiAu correctas, cuando
una monocapa de oxígeno es formada sobre la superficie de estos
electrodos.
3

CAPITULO I
1. FUNDAMENTOS TEORICOS
;\I estudiar la cinetica de reacciones electroquimicas, es
necesario conocer la densidad de corriente que fluye a través del
electrodo donde estas ocurren. La densidad de corriente la podemos
evaluar si conocemos de antemano el valor del área real expuesta del
electrodo.
El área real del electrodo la definiremos como el área activa de la
superficie expuesta, la cual, a su vez. está relacionada e?. forma
directa al número de sitios activos accesibles en la superficie del
metal. entendiendo por sitio activo, desde el punto de vista puramente
electroquímico, .como aquel sitio capáz de efectuar una transferencia
de carea.
Para llevar a cabo la estimación del área real dei electrodo. ya
sea mediante los procesos de electrosorción de hidrógeno u oxigeno ! / o
por el depósito de metales a subpotencial (UPDI. es necesario
establecer el comportamiento electroquímico de la capa adsobida, asi
como su estequimetria con los sitios metálicos superficiales [121.
Particularmente. el efecto UPD de metales ajenos sobre sustratos de
metales nobles, parece ser el que proporciona informacion más
detallada y valiosa de las características superficiales de los
electrodos solidos l6,71.
4

1.1 CARACTERIZACION SUPERFICIAL D E METALES NOBLES POR PROCESOS
DE ELECTROSORCION D E HIDROGENO Y OXIGENO.
Los procesos de electrosorción de hidrógeno y oxígeno dependen
ampliamente' de la naturaleza del metal del electrodo. Las
características electroquímicas de dichos procesos, estan definidos y
limitados por el intervalo de electroactividad del metal en el
electrolito soporte utilizado. Así, tanto la adsorción de hidrógeno
como la formación de especies oxigenadas adsorbidas, constituyen la
erapa- intermediaria para los procesos de evolución de hidrógeno y
oxigeno moleculares respectivamente. Una de las aplicaciones prácticas
que proporciona el conocimiento de las características de los procesos
de electrosorción, es la posibilidad de realizar una adecuada
estimación del area real del electrodo para la mayoria de los
electrodos constituidos de metales nobles [ill.
Los procesos de electrosorción pueden ser estudiados de manera
directa mediante el uso de técnicas transitorias, siendo las más
adecuadas las siguientes:
a ) aquel que utiliza como perturbación una señal de corriente
alterna de amplitud pequeña, particularmente útil en el estudio de
adsorción de hidrógeno. Esta técnica permite estudiar la
pseudocapacitancia originada por la transferencia de ca.rga if51.
b) la voltamperometría cíclica [131, que consiste en aplicar un
barrido triangular repetitivo de potencial que varía de forma lineal
con el tiempo, dando como respuesta un flujo de corriente como una
función del potencial aplicado. El registro generalmente se denomina
"perfil potenciodinárnico o curva voltamperométrica".
c ) la galvanostática [13,141, se aplica una corriente constante y

se mide el potencial en función del tiempo. El registro transitorio se
conoce como "curva de carga".
1.1.1. PROCESOS DE ADCORCION DE HIDROGENO.
La técnica que utiliza c .a mide la pseudocapacitancia como una
función del potencial, relacionando la capacitancia diferencial con el
potencial y la carga mediante la relación:
C = dQ/dE (1.1)
la fracción de superficie cubierta (grado de recubrimiento1 por
electroadsorción de hidrógeno se define como Q/QH.s , siendo QH.S el
recubrimiento de la superficie por hidrógeno a la monocapa, esto
es. cada atom0 del sustrato está bloqueado por un átomo de hidrógeno,
y 0 la carga experimental obtenida asociada al proceso de
electrosorción de hidrógeno a un potencial dado. Ya que:
V dQ = Q d e ,entonces H . S
ü = Q/Q H . 5
sustituyendo dQ en (1.1) se obtiene,
(de/dE) (1.2)
De aqui que la isoterma electroquímica relaciona el grado de
recubrimiento con el potencial, por lo tanto, la técnica con c.a.
determinará la derivada de la isoterma en función del potencial. Un
ejemplo tipico de pseudocapacitancia en función del potencial se
muestra en la figura 1 . 1 . 1 . 1 , en donde además se muestra la integral
de la curva, esto es, s u isoterma elecrroquimica [I61
= 0H.S
En el caso de la voltamperometría cíclica í V.C. 1, se mide la
corriente como una función del potencial. Esta corriente se relaciona
a los parametros eléctricos de la siguiente forma:
i = dQ/dt i = dQ/dt x dE/dE i = dQ/dE x dE/dt
6

1.0
IS00
0.6
- IO00 0.5 o
E N . a a 0.4
u 500 v
0.2
O o. 1 0.2 R3 0.4 E (V/Eb")
Fig. 1 . 1 . 1 . 1 . Variación de la pseudocapacitancia en función del
potencial ( a ) , cuya integral es la curva ( b ) que representa la
isoterma electroquímica de electroabsorción de hidrógeno sobre Pt en
H SO 4 hf a 25 C 1161. 2 3
dB/dE = 0H.S i =C x dE/dt
i = (QH , s dE/dt) dWdE (1.3)
Como dE/dt = v (velocidad de barrido) es constante, - és ta técnica
deberá generar una curva similar a la curva de pseudocapacitancia. La
Fig. 1.1.1.2 muestra el perfil potenciodinámico para la electrosorción
de hidrógeno sobre P t en H SO4 I M a 25 C. En ia misma figura se
muestra también la carga de adsorción como una función del potencial
íisoterrna de adsorciónl. obtenida por integración del perfil
potenciodinámico para la adsorción de hidrógeno, después de restar la
2
7
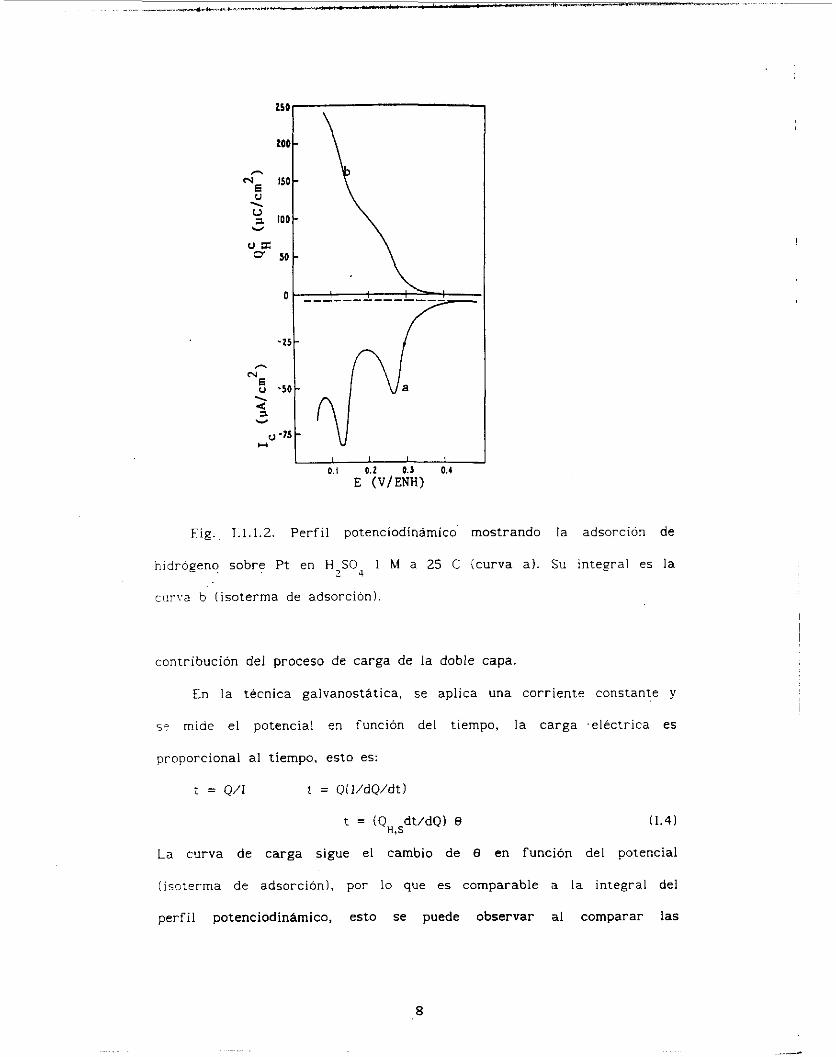
Fig. 1 ; l . l . Z . Perfil potenciodinámico mostrando l a adsorcion de
hidrogeno sobre Pt en H,S04 1 bí a 25 C (curva a) . Su integral es la
ciirva b (isoterma de adsorcion).
~
contribución del proceso de carga de la doble capa.
En la técnica galvanostática, se aplica una corriente constante y
se mide el potencial en función del tiempo, la carga .eléctrica es
proporcional a i tiempo, esto es:
t = Q/I t = O(i/dQ/dt)
t = (QH,Sdt/dQ) 6 (1.4)
La curva de carga sigue el cambio de ü en función del potencial
(isoterma de adsorción), por lo que es comparable a la integral del
perfil potenciodinámico, esto se puede observar al comparar las
8

figuras 1.1.1.1. y 1.1.1.2. Conway 118.201 apoya fuertemente el uso de
estas curvas, debido a que el procedimiento de integración de la carga
es más fácil y menos complicadas sus expresiones matemáticas, aunque
la curva integral es menos sensible a los detalles finos de ias curvas
pseudocapacitivas y a los voltamperogramas cíclicos.
El proceso de electrosorción de hidrógeno sobre Pt es
reversible, por lo tanto, el valor de la capacitancia se puede
considerar constante, excepto a altas velocidades de barrido o a
frecuencias muy elevadas. Bajo condiciones estacionarias, las
diferencias entre las características de la adsorción obtenidas por
los tres métodos previamente citados, son despreciables como se puede
apreciar en las figuras precedentes, y los diversos trabajos
publicados 117-201. Para una determinación del número de sitios
activos de la superficie del electrodo, la voltamperornetria cíclica
(\' .Cl resulta ser la técnica más ú t i l de las tres mencionadas, dada su
simplicidad y versatilidad.
1.1.2. DETERMINACION DE QH.S POR V.C.
Para llevar a cabo la determinación de QH,S es necesario saber si
l a contribución debida al proceso de adsorción de hidrógeno puede ser
separada de aquellas debidas a otros procesos que ocurren
simultaneamente, como son carga de la doble capa y evolución de
hidrógeno.
La carga de la doble capa puede ser eliminada por extrapolación
del voltamperograma en la zona no faradaica [Zl]. La exactitud de la
exrrapolación se basa en el supuesto de que la capacidad de la doble
capa es constante en el intervalo de potencial donde tiene lugar el
9

Emi n . O
IC
I I 1 I I I
Fig. 1.1.2.1. Perfil potenciodinámico catódico que muestra la
extrapolación del proceso de adsorción de hidrogeno
sobre Pt en H2C040.5 M a 25 C.
proceso de 'adsorción de hidrógeno, esto es , entre 0.0 y 0.6 Y, además
de que la contribucion de los procesos que no sean los de carga de la
doble capa, son despreciables en dicha region 131. La linea horizontal
(punteada) de la Fig. i.1.2.1 muestra l a manera de efectuar esta
correccion.
El otro problema que es aún más dif íc i l de resolver, es debido al
traslapamiento que existe entre el proceso de adsorción de. hidrógeno y
l a evolución del mismo sobre el electrodo. Por tanto, existe una
región de potencial donde ambos procesos se dan de manera simultanea,
y no se puede suprimir por un simple cambio de técnica utilizada.
Ambas contribuciones pueden ser estudiadas midiendo la
pseudocapacitancia con la frecuencia de la señal de a .c . , mediante el
uso' de un modelo de circuito equivalente apropiado. Este método no es
10

adecuado para hacer mediciones rutinarias de l a cantidad de QHS, pero
es interesante desarrollarlo ya que permite el estudio del fenómeno de
adsorción en ia región de potencial donde aparece la evolución de
hidrógeno. En la Fig. I.i.l.1 se puede observar la presencia de una
capacitancia de adsorción considerable para potenciales inferiores a
0.08 V, además de que el recubrimiento total solo es alcanzado hasta
-0.01 v.
Los trabajos hechos sobre electrodos de disco-anillo 131
condujeron a Biegler I221 a recomendar, que la cantidad QH,S
determinada por integración de la carga pasada hasta 0.08 ir (Ernin).
segundo mínimo de la curva catódica para la adsorción de hidrógeno,
fuera dividida por el grado de recubrimiento a dicho potencial,
interpolado de la isoterma derivada de los datos capacitivos (Fig.
1.1.1.11.
Para el caso del Pt. si la carga de adsorcih de hidrógeno se
determina por integración del perfil potenciodinámico de l a Fig.
1.1.2.1. con Emin=0.08 V, la división de esta carga por 0 . S 4 . valor
determinado por interpoiación de la isoterma mostrada en la Fig.
I.l.l.l.b, dará el valor de la carga correspondiente ai total de
sitios inmediatamente accesibles del electroaa, esto es, OH.S.
Una vez conocido QHS, se puede estimar ei valor del área
real del electrodo haciendo uso del estandar convencional determinado
para el metal. La carga integrada involucrada en ei proceso de
adsorción da cuenta del número de átomos de hidrógeno depositados
sobre el electrodo. Para el caso particular del Pt se ha determinado
que un átomo de hidrógeno se adsorbe en un átomo del metal 128,291
según la siguiente reacción:
11

Pt + H+ + le- - > PtíHIad (1.5)
esto permite relacionar el número de átomos adsorbidos con una
cantidad definida de electricidad. La carga por cm de área real
asociada con la adsorción de una monocapa de hidrógeno sobre los
planos de bajo índice del P t son: 208 pC para el plano (1001, 241 pC
para el plano ( 1 1 1 ) y 147 y 295 pC para el plano (1101, según si los
átomos de coordinación 7. u ambos 7 y 1 1 son los definidos como átomos
superficiales. Para superficies policristalinas, es costumbre general
1301 'asumir que la superficie consite de una distribución igual de los
tres planos de bajo índice, aunque también se ha supuesto que el plano
(1001 es el predominante 1311. Biegler [221 ha sugerido que se tomen
210 pC/cm2 como estandar convencional para el Pt. considerando el
plano (1001 como predominante y para el que corresponde una población
ae 1 . 3 ~ 1 0 ~ ~ átomosícm .
2
2
El área real expuesta para un electrodo policristalino de Pt se
puede determinar, vía electrosorción de hidrógeno, mediante ia simple
expresión:
A r = Q /210 pic cm-* 11.6)
QH,s = (Q/ept) pC (1.7)
Para electrodos constituidos de Rh, Gilman I231 ha sugerido que
l a estimación de OHS para este metal, puede ser obtenida -a partir del
perfil catódico. seleccionando adecuadamente el valor de E m i n para la
integración de la carga de adsorción. Dividiendo este resultado por el
grado de recubrimiento (e = 0.59) se obtiene la carga correspondiente
a l total de sitios activos sobre la superficie del metal [241. El
cálculo del área real es inmediato (321
H.8
A r = Q /221 pC cm-' (1.8) H S
12

Para el Ir la determinaci6n es algo más complicada, aunque los
datos capacitivos muestran que la adsorción de hidrógeno se completa
al mismo potencial que para Pt i25.261. El grado de recubrimiento
encontrado para este metal fue de 0.65, al potencial de 0.06 V.
Dividiendo la carga integrada,del perfil potenciodinámico, se
encuentra el valor de QH.S para este metal. El valor del área real
se obtiene fácilmente a partir de la siguiente expresión [3,251:
A r = QH,S/218 pC cm-* (1.91
'En el caso de electrodos constituidos de Ru, Bagotzky y Col. [331
han puesto de manifiesto que el proceso de adsorción de hidrogeno
sobre electrodos lisos, ocurre en el intervalo de 0.0 a 0.4 V, y sobre
Ru electrodepositado dicho proceso de adcorción ocurre entre 0.0 y 0.2
V. Además, estos mismos investigadores han demostrado que el proceso
de adsorción de hidrógeno procede, de manera simultanea, con una
absorción significativa del mismo, favorecida por la pronta evolución
de hidrogeno molecular sobre el electrodo. Estudios recientes 135.351
realizados por depósitos metálicos a subpotencial han conducido a una
adecuada caracterización de éste metal. Sin embargo, Woods y Col. I361
han reportado que se puede hacer el cálculo del área real mediante la
integración de la carga de adsorción de hidrógeno tomando como Emin
0.03 ir, con un grado de recubrimiento de 0.56 y un valor. de estandar
convencional de 251 pC/cm . 2
Para el Pd, el proceso de adsorción viene acompañado por el de
absorción de hidrógeno, 0.69 átomos de hidrógeno por átomo de Pd
13,241. Cuando la cantidad absorbida es pequeña, el metal conserva su
estructura. Cuando alcanza una relación atómica de H/Pd de 0.05, la
fase original a es convertida a una fase p considerablemente
!
13

expandida. Esta t ransformación , en el que ambas fases coexisten, s e
completa cuando l a re lac ión H/Pd es 0.6 . L a f a s e homogenea 6 puede
absorber más hidrógeno, y la cantidad absorbida se incrementa
linealmente con el potencial del e lectrodo L271 y por lo t a n t o con el
Log P i271. H2
La estimación del grado de recubrimiento de hidrógeno sobre é s t e
metal , ha sido uno de los problemas más d i f í c i l es de resolver , debido
funaamentaimente a la imposibilidad de separar contribuciones ajenas a
la corr iente de adsorción de hidrógeno como consecuencia propia de la
naturaleza del metal , lo que hace más especulativa la adsorción de
hidrogeno sobre Pd que p a r a los otros metaies nobles.
Para ei caso del Au. en contras te con los meta ies del grupo del
Pt . solamente adsorbe una pequeña cantidad de hidrógeno a potenciales
cercanos a la región de evolución de hidrógeno molecular. El grado de
recubrimiento de hidrógeno al potencial revers ible de hidrógeno ha
sido estimado entre 2-4 % de una monocapa [37,381.
1.1.3. PROCESOS DE ELECTROSORCION DE OXIGENO
Ei estudio de los procesos de e lectrosorción de oxígeno sobre
metales nobies, también puede rea l izarse por las técn icas ya descr i tas
en este capitulo. Aquí también emplearemos la voltamperometria cíclica
dada su simplicidad y versatilidad p a r a el estudio de t a l e s procesos
para ei oxígeno.
Los procesos de e lectrosorción de oxígeno sobre e lectrodos de
metales nobles ocurren a potenciales más positivos, que los
correspondientes a los procesos de e lectrosorción de hidrógeno, como
puede consta tarse en los procesos mostrados en l a Fig. 1.1.3.1.
14

0.2 I I
I I O 0.5 1.0 1.5
r-- PALADIO
Fig. 1.1.3.1. Perfiles potenciodinámicos para electrodos de
metales nobles a 40 mV/S en HzS04 1 M a 25 C i3.361
15

La capa de oxígeno que se forma sobre los electrodos de metales
nobles ha sido descrita en terminos de: a) quimisorción 1391, b)
incorporación bajo la superficie formando una capa dermasorbida o
aleación metal-oxígeno y c) la formación de una fase de oxido metálico
[42,431. La quimisorción se caracteriza por un incremento lineal o
casi lineal del recubrimiento con el potencial, hasta s u valor limite,
mientras que la fase de oxido se forma cuando hay un incremento agudo
en el' recubrimiento, el cual frecuentemente es irreproducible y se
acompaña de cambios significativos en la rugosidad de la superficie.
En los metales nobles, las etapas iniciales de oxidación se
pueden dar desde una pequeiia fracción de monocapa hasta la formación
de una monocapa completa, incluso hasta la formación de varias capas.
En la Fig. 1.1.3.1 se muestran los voltamperogramas
correspondientes a Pt. Rh. Ir, R u , Pd y Au. resaltando la region donde
se llevan a cabo los procesos de electrosorción de las especies
oxigenadas. Los procesos de electrosorción, aunque inician a
potenciales diferentes para cada caso, se extienden hacia potenciales
positivos, en una amplia zona, hasta alcanzar la región de evolución
de oxigeno molecular.
Para P t , Rh. Ir, Ru y Pd la región de evolución. de oxígeno
molecular se alcanza en el intervalo de potencial cercano a 1.5 V,
mientras que para el Au la evolución de oxígeno se alcanza hasta
potenciales próximos a 2.0 V. Los procesos correspondientes a la
electrodesorción de especies oxigenadas está caracterizado por un pico
de corriente catódica, situado a potenciales que son característicos
para cada metal (Pt , Rh, Pd y A d . Los otros dos metales nobles, Ir y
16

* 4c*-i---r< - I_- Ru, exhiben características electroquimicas que se alejan de este
comportamiento (para el proceso catódico). Para los metales objeto de
este estudio se hace un análisis más detallado de los procesos de
electrosorción de las especies oxigenadas, incluso para el Pt por ser
el sistema de .referencia utilizado.
1.1.3.1. PLATINO
las características de electroadsorci6n de oxigeno bajo
condiciones transitorias son muy diferentes a las exhibidas por el
hidrógeno, pués hay una considerable asimetría entre las curvas
transitorias de adsorción-desorción de oxígeno, indicativo de que ei
proceso de adsorción (de oxígeno) es irreversible, mientras que el
hidrógeno es adsorbido reversiblemente. La irreversibilidad del
proceso de adsorción depende de la cantidad de especies oxigenadas
adsorbidas. lo que puede ser observado en la Fig. 1.1.3.2, en donde
para limites superiores de potencial (EL,S) <0.9 V .el comportamiento
del proceso de adsorcion es el correspondiente a un proceso reversible
l l a s curvas san aproximadamente simetricas alrededor dei e je de
potencial).
Conforme se incrementa el limite superior de potencial hay ~ un
incremento continuo del recubrimiento. La carga que pasa antes del
comienzo de la evolución de oxigeno es aproximadamente 2 e por atom0
superficial de Pt (44-461. Estas observaciones conducen al concepto de
una monocapa de oxígeno adsorbido o PtO formado, antes del
desprendimiento de oxígeno molecular.
-
ad
Schuldiner y Co1.145,47,481 han sostenido que en u n barrido
rápido, se forma una monocapa de átomos de oxigeno adsorbidos, pero
que a velocidades lentas el oxigeno puede ser dermasorbido. A
17

50 c ! !
Fig. 1.1.3.2. Efecto del limite superior de potencial sobre el
perfil potenciodinámico de P t en H2S04 1 M a 25 C. v = 40 mV/C.
velocidades demasiado rápidas el recubrimiento a la monocapa no se
alcanza antes de que se dé la evolución de oxigeno 1481, por lo que
puede ser escogida una velocidad de barrido tal que permita alcanzar
la monocapa y que la cantidad dermasorbida sea insignificante. De esta
suposición basan el método para calcular el área superficial real de
sus electrodos.
Cuando el estudio de la capa adsorbida se efectua a potencial
constante los resultados obtenidos son diferentes de aquellos
obtenidos por técnicas transitorias. Basados en el hecho de que la
adsorción de especies oxigenadas sobre Pt es lenta, este proceso
dependerá tanto del potencial como del tiempo, por lo que a potencial
constante se pueden estudiar separadamente ambos parámetros í efectos
18

P t
.A o 1.0 1.5 2.0 2.5 3.0
E. VIER€?
Fig. 1.1.3.3. Recubrimiento de oxigeno como u n a función aei potencial
en HZSOI 1 !if a 2SC
del potencial en funcion del tiempo).
L a f igura 1.1.3.3. muestra la dependencia de la cantidad de
especies oxigenadas adsoroidas en función del potencial a tun tiempo
f i j o í - 1000 SI.
El recubrimiento de oxigeno a lcanza un valor constante a
potenciales superiores a 2 .2 V. Biegler y Woods [491 concluyeron que
e s t e recubrimiento corresponde a una monocapa que contiene aos atomos
de oxigeno por átomo superf ic ia l de P t . En la Fig. 1.1.3.3. se observa
que hay un escalón a la mitad del recubrimiento, el cual debe
corresponder a un átomo de oxigeno por átomo superf ic ia l de P t .
E x i s t e una extensa l i t e ra tura dirigida a dilucidar los mecanismos
de formación reducción de l a s especies oxigenadas sobre el P t
(50-54). Sin embargo, no hay un acuerdo general en l a interpretación
19

de los resultados. ya que los diversos autores han descrito la
naturaleza de la capa de oxígeno en términos de diferentes etapas de
la formación del óxido.
Estos estudios conducen a que el proceso más probable puede ser
representado a través de las siguientes reacciones:
2 Q:, Fc/cm
- 4Pt + H 2 0 c-- P t 4 0 H + H+ + e 55 1I.lOa)
P t 4 0 H + H20 c-- I 2PtZOH + H + e 55 í1.1Ob) - +
. 2 ~ t on + 2n20 4PtOH + ZH+ +2e- 110 (1.10c) 2
La reacción global para estas etapas es la suma de ( a ) . (bl y (cl
4Pt + 4H20 -- 4PtOH + 4H+ + 4e- 220
Se ha mostrado (521 que estas etapas sucesivas de ocupación de
gxigeno sobre Pt son las responsables de los picos anódicos observados
al inicio de la adsorción de especies oxigenadas sobre e l P t (Fig.
I . 1.3.1').
Antes de. liegar a la región de evolución de oxigeno molecular
ocrre otra etapa, que es intermedia entre estos pasos sucesivos .y la
evolución de O2 (región ancha del perfil anódico).
- + PtüH -I PtO + H + e 220 1I.IOd)
La carga total del proceso desde ( a ) hasta ( d ) es 440 pC/cm . '
Este esquema de reacción se ajusta excelentemente tanto al perfil
experimental como al cálculo de la cantidad de electricidad
involucrada por cm para la fomración de una monocapa de oxigeno
electroadsorbido en una estequiometría PtO de 1:1 , así como a los
resultados reportados por Bockris y Col. 1551 en donde se cita que a
0 = 0.27 un átomo de oxígeno comparte 2e con aproximadamente cuatro
átomos de Pt.
2
2
-
20

-- ---u-
Así el área real de electrodos constituidos de Pt se puede
determinar en base a la estequiometria PtO de ] : I , es decir:
(1.11) 2 Ar = Qa pC / (2x210 pC/cm 1
O
donde:
Qt? pC es la carga experimental integrada del perfil potenciodinámico
anódico entre los intervalos de potencial apropiados.
210 pC/cm2 correspnde al estandar convencional sugerido [301, para
un electrodo de Pt policristalino, bajo el supuesto de que la
superpicie consiste de una distribución de los tres planos cristalinos
de bajo índice de Miller, IIOO). (110) y (111) con el plano (100) como
predominante (221.
1.1.3.2. PALADIO
La electroadsorción de especies oxigenadas sobre Pd comienza
aproximadamente ai mismo potencial que sobre Pt. y se observa e l mismo
comportamiento, es decir, se incrementa la irreversibilidad a medida
que aumenta el valor del 'potencial límite superior de inversión de
barrido, tal como se muestra en la Fig. 1.1.3.4
La cantidad de especies oxigenadas adsorbidas en un barrido de
potencial aumenta en forma aproximadamente lineal con el potencial
[38,57-591.
Rand y Woods i601 investigaron la adsorción de especies
oxigenadas sobre Pd a potencial constante. En la Fig. 1.1.3.5. se
muestra la dependencia del recubrimiento del potencial aplicado a
diferentes tiempos de anodización. Se puede ver que el
recubrimiento aumenta linealmente con el potencial hasta alcanzar una
meseta definida. El recubrimiento a la meseta es independiente del
21

I : I ' 1 I
i
1.0 1.5 ,vo<P,iN#:lAl< , v "I R H L ,
0.5
Fig. 1.1.3.4. Efecto del límite superior de potencial sobre el
perfil potenciodinámico de Pd en H SO 0.5 hi a 25 C
v = 40 mV/S [31.
2 4
tiempo de anodización, cambiando a potenciales mas bajos a medida que
se incrementa el tiempo. .A aproximadamente 2 .0 \' el recubrimiento se
incrementa abruptamente y no se aproxima a un valor límite. La capa
de óxidos producido a estos potenciales elevados se reducen en una
serie de picos anchos, en vez de un solo pico bien definido del tipo
mostrado en la Fig. 1.1.3.4., que resulta de la desorción de las
especies oxigenadas cuando el recubrimiento es menor o igual que aquel
de la meseta. La reducción de la capa de altos potenciales viene
acompañado de una considerable rugosididad de la superficie
del electrodo. Este comportamiento se debe a la formación de una fase
oxida, la cual se vuelve una capa gris oscura visible bajo condiciones
más severas de polarizacibn anódica.
22

POTENCIAL (v vs RHE)
Fig. 1.1.3.5. Recubrimiento de oxígeno sobre Pd como una funcion del
potencial en H2SO4 I M a 25 C. Electrodos anodizados por
(1) 10 S , (21 100 S , (31 1000 S al potencial deseado.
La carga asociada con el recubrimiento a la meseta para un
electrodo que ha sido tratado térmicamente por calentamiento bajo
vatic para dar una superficie lisa fue 690 pC/crn* 1601, u n valor
sienificativamente menor que aquel anticipado para una relaciori 2:1 de
O/Pd, pero cercano al esperado para una capa 1:l sobre una superficie
pulida. Se concluyó, por tanto, que la meseta correspondió al escalón
observado para el Pt, es decir un recubrimiento I : ] . Esta conclusión
se confirmó por las medidas de recubrimiento de oxigeno de Burshtein y
Col. [S71, quienes estudiaron electrodos dispersos de área BET
(Brunauer, Emmet y Teller) conocida. En la Fig. 1.1.3.6. se muestran
los resultados obtenidos por ambos grupos de investigación.
"
23

O
:.f/ 0.2
I I I 1.0 1.5 2 .o
POTENOAL (V vs RHE)
Fig. 1.1.3.6. Comparación de los valores del grado de recubrimiento
de oxígeno para Pd reportado por Rand y Woods 1601 y
Burshtein y Col. [571. - , curva 3 de la Fig.
1.1.3.5.; y o, datos obtenidos de 1571 en HZS04
0 .5 M.
El trazado de la Fig. 1.1.3.6. se hace asumiendo que la meseta
corresponde a una estequiometría 1:l. La discrepancia. entre los
recubrimientos a 1.45 V puede deberse a los diferentes estados
físicos de las superficies examinadas. .
La meseta mostrada sobre la curva de recubrimiento de oxigeno en
función del potencial de anodización (Fig. 1.1.3.5.) presenta un
método para la determinación del área real de electrodos de Pd. Rand y
Woods [601 sostienen que relativamente pocas medidas de recubrimiento
24

de oxígeno en función del potencial se requieren para obtener la
meseta. Además, recomiendan tener cuidadode de evitar condiciones bajo
las cuales se desarrolla la fase óxido, ya que esto puede conducir a
cambios serios en la rugosidad de la superficie.
De la Fig. 1.1.3.6 se puede concluir que para superficies
altamente dispersas este método no puede ser aplicado, ya que la
meseta desaparece para este caso, además en la práctica es difícil
tener un electrodo bien pulido como el que utilizaron Rand y Woods en
sus investigaciones.
L Fue propuesto I601 que un cm de un plano (1001 se acepte
como estandar convencional. de acuerdo con Io propuesto para los otros
metales nobles. En este caso, un cm real es equivalente a un cambio
de 424 pC para un proceso que intercambia 2e-, es decir, el area real
es:
2
(1.12) 2
Ar = QC pC / (424 pC/cm 1 O
1.1.3.3. OR@
El oxígeno comienza a ser adsorbido sobre Au a aproximadamente
1.35 V en medio ácido 137,38,60-691 tal como se muestra en la Fig.
I . 1.3.7. No obstante. varios investigadores 170-741 han reportado
corrientes faradaicas a potenciales anódicas inferiores y han
sostenido que la carga que fluye se debe a la formación de especies
oxigenadas de Au. Bonewitz y Cchmid 1631 encontraron que el Pt cuando
es usado como contraelectrodo se disuelve y deposita sobre el
electrodo de trabajo, originando con ello la adsorción de especies
oxigenadas a potenciales inferiores, comportándose entonces el

I 1 1 100 -
0 -
-100 -
-too.
-800 -
I O 0.6 1.0 1.3
POTENCIAL (V/ERH)
Fig. 1.1.3.7. Efecto del limite anódico de barrido de potencial
sobre el perfil potenciodinámico de Au en H SO I M
a 25 C. v = 40 mV/S.
2 .
electrodo de trabajo como una aleación Pt/Au.
AI analizar el barrido anódico del Au en H SO 1 M íFip. 1.1.3.71
se observan dos picos de corriente claramente definidos a 1.47 y 1.63
\', separados por u n tercero casi imperceptible a 1.52 V . .A 1.7 V se
detecta un mínimo en la corriente que marca el comienzo de la
evolución de oxígeno. Las investigaciones hechas sobre monocristales
de Au I751 exponiendo los planos (1001, (110) ó (111) han mosrrado que
la forma del perfil potenciodinárnico depende de la orientación
cristalográf ica de la superficie del electrodo.
2 4
La desorción de especies oxigenadas la caracteriza un pico
catódico sobre el voltarnperograma cuando se usan velocidades de
26

barrido lentas, tal como se muestra en la figura precedente, mientras
que a velocidades más rápidas se alcanzan a apreciar dos picos en el
proceso catódico 1691.
La histérisis entre las curvas anódica y catódicas sobre el
voltamperograma es similar a áquel esperado para la formación de la
fase óxido [31. con solamente corrientes anódicas fluyendo a
potenciales > 1.35 V y corrientes catódicas debajo de este potencial.
Por lo tanto, el potencial metal/óxido metálico sería entonces 1.35 V;
que es un valor aigo menor a los calculados a partir de datos
termodinámicos para los pares Au/AuíOHI v Au/Au O que son de 1.46 y
1.51 V respectivamente [761. El pico de desorción cambia a potenciales
más catódicos a medida que el potencial limite superior de inversión
del barido se incrementa. lo cual es un comportamiento analog0 ai
observado para los otros metales del grupo del P t . Este cambio sugiere
una variacidn continua en la constitución de las especies oxigenadasde
la superficie en vez de una fase óxido simple.
3 - 2 3
La duda que surge es si la capa de óxido sobre Au puede ser
considerada como oxigeno quimisorbido o identificado como una fase de
óxido definida. Se ha encontrado que la formación de la pelicula sigue
la cinética de Elovich 1651 (aplicable a quimisorciónl. o que sigue
una ecuación de velocidad basado en un mecanismo de crecimiento de
óxido [771.
Rand y Woods 1601 midieron el recubrimiento de óxigeno a
potencial constante para diferentes tiempos de anodización (Fig.
1.1.3.8). Se encontró que el recubrimiento aumenta de manera continua
con el potencial, y no fue observada ninguna región donde el
recubrimiento fuera independiente del potencial, como para algunos de
27

- 0.4
"4 I 1 I I
1.0 1.5 2.0 POTEMOAL ( V v s RHE)
Fig. 1.1.3.8. Recubrimiento de oxigeno sobre Au como una funcion
del potencial en HZCO, 1 M a 25 C. Electrodos
analizados por ( o ) 10 C, ( A ) 100 C y (0) 1000 C al
potencial deseado.
los otros metales nobles. Sin embargo, la velocidad de incremento del
recubrimiento con el potencial se hace menos marcado a medida que
aumenta el potencial. El proceso alcanza un mínimo a aproximadamente
2 .0 V antes de registrar un incremento repentino del recubrimiento,
similar al reportado para Pd. Bajo condiciones extremas de anodización
una capa visible de color naranja intenso fue observado, que en la
reducción se vuelve negro y causa severa rugosidad en la superficie
del Au. En la Fig. 1.1.3.8 se observa que a potenciales < 2 . 0 if el
recubrimiento varia muy poco cuando el tiempo de anodización fue mayor
28

que o igual a 100 C. Este mismo comportamiento fue observado por
Laitinen y Chao i561.
Brummer y Makrides 1651 consideraron que la gráfica consiste de
dos secciones lineales y que la intersección a 1.45 V marca el llenado
de una monocapa de oxígeno. Rand y Woods I601 señalaron la similaridad
entre el comportamiento del Au y los metales del grupo del Pt e
interpretaron la curva de recubrimiento en términos de una capa incial
de átomos de oxlgeno quimisorbido que se aproxima a un recubrimiento a
la monocapa alrededor de 2.0 V antes de que la nucleación y
crecimiento de la fase oxido cause una repentina elevación en l a carga
de oxígeno. La carga asociada con la capa inicial justo antes de la
formación de la fase óxido, fue 500 pC/cm , 'valor que concuerda, con
el concepto de un átomo de oxígeno por átomo de Au superficial
sobre una superficie pulida.
2
Un rapido incremento en la carga que pasa en la formación de la
capa de oxígeno cuando se desarrolla la fase de óxido fue reportado
por Laitinen y Chao 1561 y por Ogura y Col. 1731, pero la carga
correspondiente a la saturación de la capa inicial fue mucho mayor que
la reportada en la Fig.1.1.3.8, y que fueron 1.05 mC/cm para la capa
2 inicial y 1.40 mC/cm para las fase óxido.
2
Los valores reportados para la carga asociada con adsorcion de
oxígeno sobre Au bajo condiciones similares difieren
considerablemente. Por ejemplo, el recubrimiento a 1.6 V se sitúa
entre 3 0 0 pC/cmZ después de 20 C I621 y 900 pC/cm2 sobre un barrido de
108 mV/C 1671. El último valor fue para un electrodo pulido a espejo.
La película puede ser detectada por técnicas de reflectancia
[78, 67, 79-821 y elipsométricas I72, 83-851, y los cambios en los
29

parámetros ópticos ocurren cuando la carga fluye a travCs del sistema
en el proceso de adsorción. Vinnikov y Col. I851 consideran que se
forma una monocapa a 1.40 V y que éste evoluciona para dar una
monocapa de una composición diferente, que se completa a 1.80 V.
Arriba de 2.0 V se forma la fase óxido, que tiene propiedades Ópticas
diferentes de la película inicial, implicando una composición
diferente.
Las características de la capa inicial de oxígeno son totalmente
distinguibles de aquéllas de la fase óxida. En este sentido el A u se
comporta de manera similar a los metales del grupo del P t . El Au
difiere en que el oxígeno es adsorbido irreversiblemente aún a bajos
recubrimientos, sugiriendo una etapa de reconstrucción que sigue
inmediatamente a la adsorción. La adsorción pudiera procedei- via
especies OHad y Oad y que aparentemente alcanza un recubrimiento a la
monocapa de átomos de oxígeno antes de la nucleación y crecimiento de
l a fase óxido sobre la superficie. Rand y Woods [60, 861 se apoyan
en esia suposición para determinar el area de los electrodos
constituidos de Au. Estos investigadores sugieren que el recubrimiento
de oxígeno sobre un electrodo sometido a 1.80 V por 100 S en H , S 0 4 1 M
puede ser considerado próximo a una monocapa. El recubrimiento puede
ser obtenido desorbiendo la capa de óxido mediante un . barrido de
potencial catódico e integrando la carga del perfil. El plano (1001
fue escogido como el estandar convencional de área real y la carga
correspondiente a la adsorción de un átomo de oxígeno por sitio
superficial sobre dicho plano es 386 pC/cm , por lo tanto, e l área
real esta dada por la siguiente expresión:
2
Ar = Qc pC/í386 pC/cm21 (1.13) O
30

I. 2. DEPOCITOC METALICOS A SUBPOTENCIAL c93282 1.2.1. INTRODUCCION
El efecto de depósito a subpotencial (UPD). es decir, el depósito
de una submonocapa de un metal, M, sobre un sustrato metálico ajeno,
hl: a potenciales positivos repecto al potencial reversible de Nernst,
se conoce desde hace mucho tiempo. Uno de los pioneros a este respecto
fue Hevesy 1931, quién reportó en 1912 una desviación de l a ley de
Nernsi cuando depositó trazas de metales radiactivos sobre cobre. Sus
curvas, grado de recubrimiento contra potencial (ü vs E), mostraron un
residuo asimétrico a potenciales positivos, indicando que la adsorción
del metal se lleva a cabo varias decenas de mV más positivo que el
potencial reversible de Nernst. Luego de este suceso Herzfeld í941
ofreció una explicación formal a esta aparente violación a l a ley de
Nernst. al. asumir que la actividad de la fase sólida es una funcion del
grado de recubrimiento,ü, mientras el depósito no cubriera la
totalidad de la superficie del sustrato.
'4 finales de la década de 1940 varios grupos
investigaron el comportamiento del depósito y oxidación de pequeñas
cantidades de depósitos metálicos sobre electrodos de metales
nobles. Rogers y Col. 195,961 estudiaron en detalle el .deposito de
trazas de Ag y Cu radiactivos sobre diferentes sustratos metálicos
tales como Pt, Au, Pd. Rh y W en función de la concentración de iones
metálicos, electrolito soporte y agentes cornplejantes. entre otros. Su
interés principal, como el de otros grupos, fue con propósitos
analíticos.
Dos de los pioneros en la investigación de la deposición
31

electrolftica de la primera capa at6mica de un metal sobre un sustrato
metalico extraño y sus propiedades especiales fueron Mills y Willis
1971, quienes estudiaron la formación a la monocapa de Pb, TI, Sb y Ni
sobre Au y Pb sobre Ag.
A pesar de sus diferentes enfoques, todas estas investigaciones
condujeron a un número remarcable de conclusiones que fueron probadas
como completamente razonables en la década pasada. Se vi0 claramente
que la monocapa depositada a subpotencial resulta de una fuerte
interacción entre los átomos de la monocapa y el sustrato. que puede
ser descrita de una manera formal por una actividad menor que la
unidad para el depósito en la región de submonocapa. Varios resultados
experimentales conducen a la conclusión que la monocapa se distribuye
uniformemente y no crece sobre sitios activos del sustrato 197. 9S1, y
su estructura es predominantemente determinada por la del sustrato.
Se asume que la etapa inicial del electrodepósito corresponde a
la formación de la monocapa. la que a su vez, es de gran importancia
en el desarrollo de capas posteriores y por lo tanto, de las
propiedades físicas del acabado superficial. Se ha encontrado que la
segunda capa no se inicia hasta que la monocapa se completa, la cual
2 requiere de una cantidad de electricidad de aproximadamente 250 pC/cm
para un ión metálico monovalente. También fue demostrado.1971. que la
isoterma de adsorción de la monocapa puede tener una forma complicada.
Finalmente se ha señalado que existe una correlación entre el deposito
a subpotencial y el grado de desigualdad de los parámetros de red
entre sustrato y materiales depositados (961. Esto h a permitido apoyar
la idea de un desarrollo epitaxial de la monocapa, sobre la superficie
del sustrato L991.
32

1.2.2. FORMACION DE DEPOSITOS METALICOS A SUBPOTENCIAL
SOBRE ELECTRODOS POLICRISTALINOS EN SOLUCIONES ACUOSAS.
Numerosos pares metálicos han sido investigados por métodos
electroquimicos para obtener información sobre la formación de
monocapas a subpotencial. Los sistemas que han recibido mayor interés
se enruentran en esta cita bibliográfica [61.
Algunos aspectos característicos del depósito a subpotencial
(L'PD) pueden ser muy claramente observados en las curvas
voltamperométricas, como la mostrada para el sistema AgíTI en la Fig.
1.1.7.1.
+
Cuando se comienza el barrido de potencial en l a dirección
catodi.ca desde un valor anódico (0.0 V/ESC), donde la superficie del
sustrato está limpia. se genera un pico de corriente alrededor de
-0.g \ / E X debido al depósito de TI sobre el sustrato. que es oxidado
al mismo potencial al invertir el sentido del barrido . L a velocidad
de barrido (dE/dtl deberá ser suficientemente lenta, con el propósito
de evitar que se cause una polarización por concentración. Cuando el
barrido de potencial es interrupido en la región de subpotencial la
corriente decrece inmediatamente hasta cero, indicando un proceso de
adsorción reversible dependiente del potencial. Barriendo el potencial
en la región negativa del potencial reversible de Nernst, Er. el TI
será depositado en forma masiva a una velocidad limitada por la
difusión de TI del seno de la solución al electrodo. Durante el
barrido anódico, el depósito masivo será oxidado cerca del valor
+
33

LO E. I I
30
O
-10
I J O
-20 L -1,o -0.5
E (V/ESC)
Fig. 1.2.2.1. Curvas cíclicas I-E para un electrodo de Ag PT
Na2COd 0.5 M (pH 3) + TINO3 ZXIO-’~M. v = ?ú rnV/S.
termodinámico, Er, en un intervalo de potencial pequeño y
definido por la velocidad de barrido de potencial; por tanto, la
cantidad de T1 depositado dependerá del tiempo de depósito y del
potencial.
El pico de oxidación más positivo. alrededor de -0.55 V/ECC, es
llamado generalmente como “pico de la monocapa”. Se ha observado que
el pico de la monocapa solo representa una monocapa incompleta
(usualmente < 50% de una monocapa compacta), ya que una considerable
cantidad de TI se deposita a subpotencial cerca del potencial
34

termodinámico. La cantidad máxima de TI depositado a subpotencial es
constante y proporcional ai área superficial del sustrato, esto es,
aproximadamente Z X ~ O - ~ mol/cm 161. que es io esperado para un
recubrimiento a la monocapa, puesto que corresponde aproximadamente al
número de átomos superficiales por cm para un metal. Casi todos los
sistemas reportados en la literatura presentan alrededor del
mismo valor. Además, se ha encontrado que la carga requerida para
, remover ( o depositar) la cantidad máxima depositada a subpotencial es
2
2
2 del orden Zx200 pC/cm , lo cual significa que la carga transferida es
cercana al valor esperado para una descarga total de acuerdo a la
siguiente reacción:
M'+ + Ze- -- M (1.14)
Puesto que la cantidad máxima depositada a subpotencial
corresponde en muchos casos a valores muy próximos a un recubrimiento
a la .monocapa, esto sugiere muy fuertemente la .formación de una
monocapa. Una prueba directa ha sido obtenida para sustratos de
P t al medir la supresión de la adsorción de hidrógeno debido a la
deposición metálica en la región de subpotencial 1100-1041. Se sabe
que el hidrógeno se adsorbe sobre P t , antes de la evolución de
hidrógeno molecular, en dos regiones distintas de potencial, que
corresponde a átomos fuerte y débilmente enlazados 11051. .La cantidad
total de hidrógeno adsorbido sobre Pt corresponde a una carga
equivalente de 210 pC/cm , aunque este valor difiere para las
diferentes caras cristalinas del P t [1061. Esto se considera como un
recubrimiento a la monocapa que corresponde a una relación I : 1 de
Pt/Hads. Por otro lado, se sabe que el hidrógeno no se adsorbe sobre
aquellos metales que son usualmente depositados en estos estudios. Por
2
35
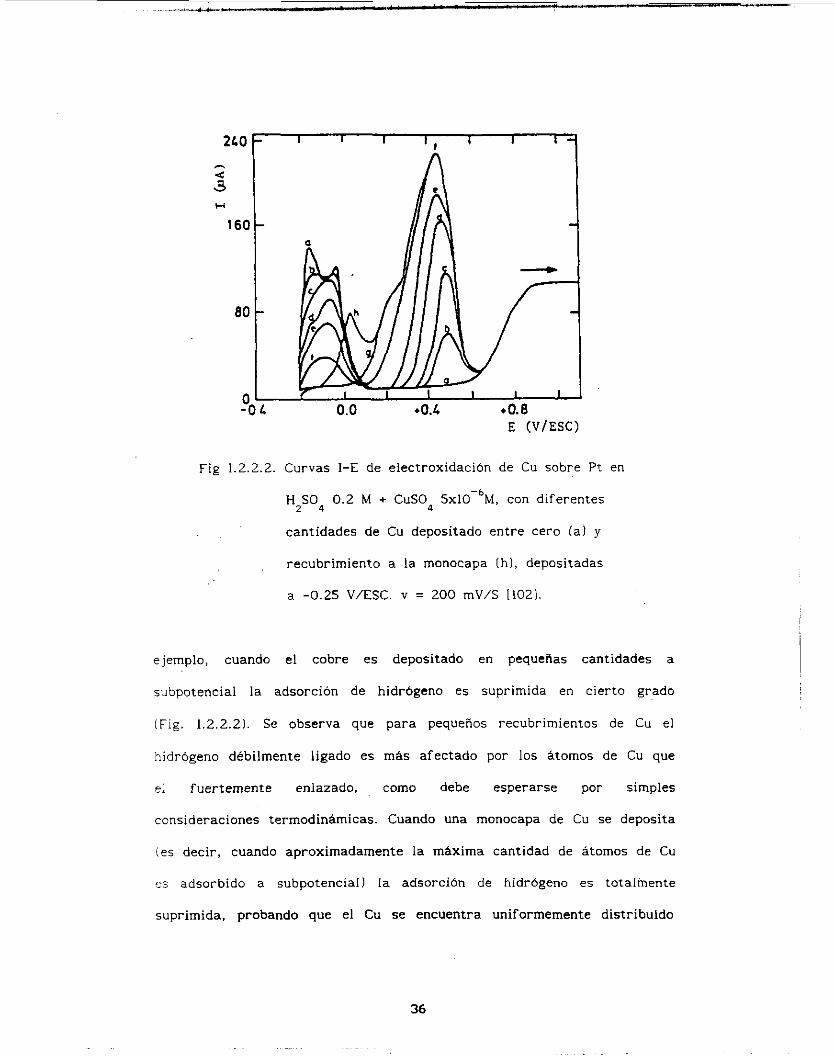
2L0 F I I I I I
E (V/ESC)
Fig 1.2.2.2. Curvas I-E de electroxidación de Cu sobre Pt en
HZS04 0.2 M + CUCO 5xlO-'M, con diferentes
cantidades de Cu depositado entre cero (a) y
recubrimiento a la monocapa íh l , depositadas
a -0.25 V/ESC. v = 200 mV/C 11021.
4
ejemplo, cuando el cobre es depositado en pequeñas cantidades a
subpotencial la adsorción de hidrógeno es suprimida en cierto grado
(Fig. 1.2.2.2). Se observa que para pequeños recubrimientos de Cu el
hidrógeno débilmente ligado es más afectado por los átomos de Cu que
ei fuertemente enlazado, como debe esperarse por simples
consideraciones termodinámicas. Cuando una monocapa de Cu se deposita
(es decir, cuando aproximadamente la máxima cantidad de átomos de Cu
p i adsorbido a subpotencial) l a adsorción de hidrógeno es totalmente
suprimida, probando que el Cu se encuentra uniformemente distribuido
36

sobre el Pt y no apilado en algunos sitios (debido a deposición
preferencial y nucleación en sitios activos), dejando algunos sitios
de la superficie de Pt sin recubrir.
Generalmente existen dos formas de definir una monocapa. El
primero, principalmente usado en física de superficie, refiere el
recubrimiento a la monocapa como el número de átomos adsorbidos.
siendo iguales al número de átomos superficiales del sustrato. El
segundo, frecuentemente usado en electroquímica, define un
recubrimiento a la monocapa como la cantidad depositada por unidad de
área superficial sobre el sustrato antes del crecimiento de la segunda
capa. Aqui el término monocapa señala un cambio importante en el
enlace del adsorbato debido a que cambia de un enlace
deposito-sustrato a un enlace puro depósito-depósito. Nos referiremos
usualmente a esta última definición cuando hablemos acerca de una
monocapa. Cuando impliquemos la primera definición, será establecida
como tal.
En la Fig. 1.2.2.3, está representada la cantidad de metal
depositado contra la cantidad de H aún adsorbido en una superficie
de P t para Cu IiOZl, Ag 11021, Bi I1041 y TI I iO i l . Existe una
relación lineal simple entre ambas cantidades, comprobando nuevamente
de manera cuantitativa la formación de una monocapa uniforme. Para los
átomos metálicos más pequeños tales como Cu y Ag la pendiente es uno
correspondiendo a una relación 1:l. esto es, un átomo de hidrógeno es
desorbido por cada átomo metálico depositado. mientras que para átomos
metálicos más grandes tales como Bi y TI la eficiencia de
despl+zamiento de Hads se incrementa, siendo aproximadamente 2: 1 para
ads
I
Bi. *Sto implica una concentración superficial de Bi, rBi, mucho
37
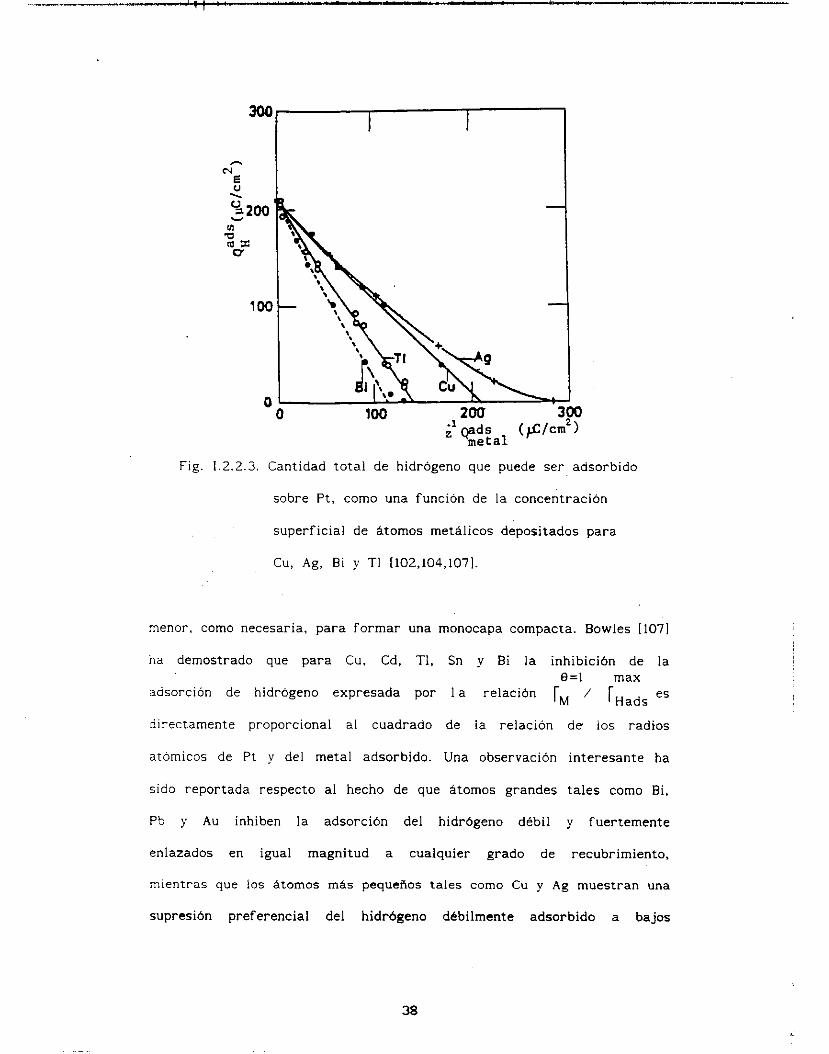
N - I ?I
Fig. 1.2.2.3. Cantidad total de hidrógeno que puede ser adsorbido
sobre P t , como una función de la concentración
superficial de átomos metálicos depositados para
C u , Ag, Bi y TI I102,104,1071.
menor, como necesaria, para formar una monocapa compacta. Bowles [I071
na demostrado que para Cu. Cd, TI. Cn y Bi la inhibición de l a
adsorción de hidrógeno expresada por I a relación rM rHads es
directamente proporcional al cuadrado de la relación d e los radios
atomicos de Pt y del metal adcorbido. Una observación interesante ha
sido reportada respecto al hecho de que átomos grandes tales como Bi,
Pb y Au inhiben la adsorción del hidrógeno débil y fuertemente
enlazados en igual magnitud a cualquier grado de recubrimiento,
mientras que los átomos más pequeños tales como Cu y Ag muestran una
supresión preferencial del hidrógeno débilmente adsorbido a bajos
ü=1 max
38

-" -7
recubrimientos del adátomo.
1.2.3. ASPECTOS TERMODINAMICOS DE LA FORMACION DE DEPOCITOS
METALICOS A SUBPOTENCIAL.
El depósito metálico a potenciales positivos respecto al
potencial reversible de Nernst ha sido descrito termodinámicamente por
una actividad menor que la unidad para la fase depositada. En base al
concepto de Herzfed [941, Roger y Stehney [lo81 derivaron una ecuación
de Nernst que fue usada en todos los trabajos en los siguientes 20
años
El potencial de equilibrio para una reacción reversible esta dada
por la siguiente expresión:
(1.15) O E = E + íRT/ZF)Ln(Aox/Ared) r r
donde:
. lox y Ared son las actividades de las especies oxidada :: reducida
respectivamente; para una reacción de deposición metálica, donde es
involucrada una fase sólida, la actividad del depósito se asume que es
constante e igual a la unidad. Sin embargo, cuando la superficie del
electrodo se recubre parcialmente por trazas de un depósito extraño,
ia actividad del depósito será menor que la unidad y variara con el
recubrimiento de la superficie [941. '
A partir de 1970, se ha venido desarrollando la descripción
termodinámica del fenómeno de formación de sub y monocapas por
depósito de metales a subpotencial a través del modelo
"pseudo-nernstiano" propuesto por Schmidt y Wüthrich IiOSl.
39

1 . . - .
Este modelo describe el proceso de electrosorci6n mediante el
siguiente equilibrio:
M'Mads M'+ + Ze- -I (1.16)
cuyo potencial de equilibrio se expresa mediante la siguiente
ecuación:
Z+ = Eo Z+ (RT/ZF) Ln lAMz+/A I (1.17) EM/M M/M
donde:'
,A Z+ es la actividad de los iones metálicos en solución, MZ+
A es la actividad de las especies metálicas depositadas a
subpotencial para un grado de recubrimiento entre cero y uno.
hf
Los demás parámetros tienen su significado usual.
Las objeciones para este modelo I l iOl , radican en el hecho de que
el potencia1 ' aplicado para lograr un depósito a submonocapa involucra
l a activi.dad de las especies M depositadas a subpotencial. Sin
embargo, implícitamente se selecciona la actividad de la especie M
como su estado de referencia, tomando A = 1, io que lleva a considerar
un proceso de multicapas como estado de referemncia. Esto dificulta la
interpretación de los resultados obtenidos por depósito a
subpotencial.
Bruckenstein ill01 ha propuesto una descripción termodinámica
formal del efecto UPD. Este formalismo se fundamenta en la definición
del "potencial de equilibrio termodinámico de submonocapa", que
conduce a una expresión similar a la ecuación de Nernst.
Con la finalidad de poder relacionar O, l a concentración de los
iones metálicos en solución y el potencial aplicado al electrodo,
40

Bruckenstein propone el siguiente proceso de depósito metálico "3 (1.18) - I+ M'+ + aupoe is) +- M''-'UPD (SI
cuyo potencial de equilibrio está definido por la siguiente expresión:
A E ~ ~ = A E M ~ + ( R T / T , , ~ ~ F )Ln( AMz+/AML I (1.19)
donde:
AEhlL es el potencial de equilibrio de submonocapa
es ei potencial estandar de submonocapa AE:lL
z- es la actividad de los iones metálicos MZi en solución .Ah,
Z -
i es la actividad de la especie M depositada a subpotencial a grado
es la Valencia del ión metálico en solución
\:>
de recubrimiento e
Y es la Valencia de electrosorción
5 es el sustrato.
:,F?
El potencial estandar de monocapa está relacionado con la energía
l ibro estandar de formación de submonocapa de acuerdo a la siguiente
expresión:
La Valencia de electrosorción es un número fraccionario que
describe el comportamiento de aquellos adátomos capaces de retener una
carga parcial, como resultado del proceso de depósito a subpotencial,
i n cambio la Valencia de Nernst (Z) es un número entero.' La Valencia
de electrosorción, que es la resultante entre la diferencia de
funciones trabajo del sustrato y del adátomo, está definida por la
siguiente ecuación:
(1.21)
Esta ecuación determina el desplazamiento del potencial de
equilibrio de la monocapa con el cambio en la composición de la

solución a grado de recubrimiento constante.
El potencial reversible para la formación del depósito en
multicapas
Mz+ + Ze- (MI <I M(M1 (1.22)
está dado por la siguiente expresión:
A E ~ = A$ + (RTIZF) Ln A ~ = + (1.23)
Se establece el subpotencial A U como la diferencia del potencial
de equilibrio de submonocapa y la diferencia de potencial de
equilibrio del depósito masivo, es decir:
A U = AE - AEr (1.24) ML
sustituyendo las expresiones (1.19) y (1.23) en (1.241 tendremos:
A V = AEo - AE: + RT/F [VaupD - 1/Z 1 Ln A Z+ - (RT/a Fl Ln AML
(1.25)
ML !,1 U P D
donde:
AEzL - AEO= AU' y AEo es el potencial estandar de. equilibrio para el
deposito masivo del par M/MZ'
además:
arreglando:
AU' = AG'IZF - A C ; ~ / ~ , , , F (1.26)
La ecuación (1.251 establece un nuevo estado de referencia que
tiene la contribución debida al depósito masivo y la contribución
debida a la Valencia de electrosorción. '
Kolb y Col. [lill, han definido el subpotencial A U como la
diferencia entre el potencial del pico de oxidación del depósito
masivo y el potencial del pico de oxidación de submonocapa (pico más
positivo), observados sobre el perfil potenciodinámico. Para procesos
P
42

de electrosorción con 8 < 0.2, la isoterma de adsorción de Langmuir
describe apropiadamente el proceso, y el desplazamiento del
subpotencial se puede expresar por la siguiente ecuación:
AU =(PG'/ZF-AG~~/'",~F+RT/F [ i / ~upD- l /~~ Ln A ~ = + - RT/a,,,F Ln 8/14 P
(1.27)
Esta ecuación solo puede ser usada para experimentos
potenciodinámicos, si la reacción de electroxidación es totalmente
reversible i1101. AGiL es el cambio de energía libre estandar para el
efecto UPD a e = 0.5.
La energía libre de monocapa depende del grado de recubrimiento,
de aqui que muchos sustratos sean energéticamente heterogeneos. y las
especies de la monocapa experimenten interacciones laterales [ iE1.
De las' heterogeneidades intrinsecas debidas a las caracreristicas
estructurales de la superficie, quizás el factor más importante en üPD
es ei cambio en la función trabajo del sustrato que acompaña la
formación de una capa superficial a UPD. Asumiendo que la energía
libre de la monocapa decrece linealmente con el grado de
recubrimiento,e, tendremos:
donde AG;;" es el cambio de energía libre estandar en el limite de
e -> O y f es el parametro de Temkin que es una medida de las
interacciones particula-sustrato. Por lo tanto, el subpotencial está
dado por:
A G O RT RT A U = [ z r - -F]+i 'UPD [& - ]Ln AMz+ - - 'UPOF
43

e AGe=' se puede obtener de la intercepción de la gráfica AU vs fe+Lnl_e
usando el valor previamente determinado de T~~~ í1131.
ML
Si existe una considerable diferencia en eiectronegatividades
entre el sustrato y las especies de la monocapa, puede ocurrir alguna
polarización por carga, y esto conduciría a la existencia de una
pequeña carga positiva sobre las especies de la monocapa [6,1111.
Esta polarización por carga es el resultado de la naturaleza del
depósito y no implica un mecanismo UPD que involucre una reacción de
transferencia parcial de carga en el sentido convencional faradáico.
Conway y Coi. [1141 han tratado este caso (polarización por carga) en
términos de un dipolo imagen inducido, es decir, ei complejo
metal-adátomo puede ser considerado como un dipolo superficial. Estas
interacciones dipolo-dipolo conducirían a una disminución adicional en
la energía .libre de la monocapa con el recubrimiento, la cual puede
ser identificad,a por un
obtiene:
A G M ~ =
donde g es el parámetro
antes señaladas. Ahora
2/3 término e . A s í , para esta interaccion se
hGe" + fRTü + gRTü2'3 (1.30)
de Frumkin que da cuenta de las interacciones
podemos escribir una nueva expresión del
ML
subpotencial que incluye los parámetros f y g previamente señalados
RT f e
(1.31)
La diferencia en las energías libres entre los procesos de
submonocapa y multicapas se puede relacionar a otras propiedades
termodinámicas. AU ha sido relacionado con las diferencias de función
44

(1.321
trabajo [ill1 mediante la sigüiente expresión:
AU =uA@ P
donde:
AU corresponde a la ecuación (1.271 y a = 0.5 V/eV. P
La Fig. 1.2.2.4 muestra el perfil anódico de seis diferentes
sistemas, en los que se puede aspreciar el AU (AE - AE I para cada
uno de tales sistemas.
p .ML r
Los picos de corriente y su potencial, que definen el AU , están
indicados en cada figura con una flecha. Algunos perfiles de los
sistemas indicados, exhiben un tercer pico de pequeña magnitud entre
los picos señalados. La cantidad de electricidad relacionada con este
pico, es mucho menor que para el pico registrado a valores más
positivos de potencial. Conway y Col. 11141 explican la presencia de
multipicos a través de la existencia de estados de adsorción
multiples..
P
La .relacion serniempirica de Kolb [I111 se ajusta bastante bien
para depósitos a subpotencial sobre sustratos poiicristalinos. Sin
embargo, para sustratos monocristalinos esta relación no se satisface.
Para monocristales 11151 se ha sugerido que la dependencia de la
función trabajo del sustrato con la orientación cristalográfica. no es
reflejada en los valores de AU , puesto que las energías de adsorcion
del depósito a subpotencial no dependen únicamente de A+, sino también
de las interacciones laterales y verticales.
P
El concepto de AEML, potencial de equilibrio de monocapa. conduce
a la definición de un sobrepotencial para el proceso de depósito a
subpotencial. Así, cuando el potencial aplicado (AE ) difiere del
potencial de equilibrio de submonocapa, AEMLe, a recubrimiento 6. el
aPP
45

LO
O
-. 80
o LO 4
O
N . Y U
20
10
O
-1.0 -as O -0.5 O E ( V/ESC )
Fig. 1.2.2.4. Curvas de electroxidación para varios metales
depositados sobre Ag y Au en NaZSO4 I M ípH 31
(a y cl y NaC104 I M (pH 31 (b, d. e y f ) .
[Mz+l = ZXIO-~. v = 20 mV/S. Las flechas
indican los picos de corriente de electro-
oxidacion del depósito masivo y de la
monocapa Iiiil.
46

-- e
sobrepotenciai es Ill31
I
ne = AE app - AEMLe (1.33) I
Así, ocurre un proceso faradáico, dando origen a la formacion de
una monocapa hasta que el sobrepotencial decae a cero cuando 9 alcanza
= AEMLe. En la referencia I61 están el equilibrio
reportados valores de AU para diversos sistemas con sus respectivas
citas bibliográficas.
y AEapp
P
47

CAPITULO I1
En este capitulo se describen las condiciones experimentales de
trabajo, pureza de reactivos, preparación de electrodos, montaje
electrónico y las caracteristicas de la celda empleada en el
desarrollo de esta investigación.
11.1. CELDA ELECTROQUIMICA
Tanto para los estudios de electrosorción de hidrógeno y oxígeno,
como para el depósito de Cu a subpotencial, se utilizó una celda de
vidrio Pyrex del tipo convencional para tres electrodos. La celda, con
capacidad de 50 mi , presenta cinco entradas y una salida (para
realizar cambio de eiectrolito). De las entradas de la celda, tres
fueron asignadas a los electrodos de trabajo, referencia y
contra-electrodo, ésta última también funciona como evacuacion de N
del sistema a través de una trampa de agua. Las dos entradas restantes
2
son empleadas para el llenado de la celda con electrolito soporte, y
entrada de flujo de N al sistema para mantener una atmósfera inerte
durante 10s experimentos. La entrada que sirve para el llenado de la
celda tiene acoplado un embudo de adición, el cual permite desoxigenar
dosis nuevas de electrolito soporte para experiencias subsiguientes.
La salida de la celda tiene adaptado un vidrio poroso que, cuando el
experimento así lo requiere, permite agitar la soiución mediante flujo
de N . El diagrama esquemático de la celda se muestra en la Fig.
1 1 . 1 . 1 .
2
2
48

¿
Fig.II.1.1 Celda electroquímica para el estudio de electrodos
de metales nobles en H2SO4 0.5 M. Se utilizó
también para el depósito de adátomos de Cu.
49

11.2. PREPARACION DE ELECTRODOS
Para los electrodos de trabajo se emplearon alambres de 0.5 mm,
$, de diversas longitudes, de cada uno de los metales estudiados. En
el caso de electrodos de Pd, se usaron también presentaciones en
placa.
Para los electrodos de Pt y Pd el contacto eléctrico con el
circuito exterior, se hizo mediante un alambre de cobre, mismo que fue
unido al metal con soldadura de estaño. Posterioremente se selló en
u n tubo de vidrio Pyrex con resina epóxica y siliflex, tal como se
muestra en la Fig. 11.2.1.
En los electrodos de Au, el contacto eléctrico también se efectuó
a través de un alambre de cobre, pero en este caso ambos metales fueron
fundidos a la flama, luego de lo cual se selló ai vidrio Pyrex en la
forma ya descrita.
Los electrodos de Pd y Au, placas y alambres respectivamente, se
pulieron con lija de carburo de silicio grado 600 y se enjuagaron con
agua de'sionizada (R > 1s MR ) antes de ser introducidos a la celda.
En el caso de electrodos de P t una limpieza con agua regia por 30 C
seguida por enjuagues con agua desionizada, fue requerida antes de ser
usados. Como contra electrodo se usó una barra de grafito vidriado,
cuya construcción fue similar al descrito para los electrodos de
trabajo, salvo que se usa mercurio para realizar el contacto eléctrico
entre el cobre (conductor externo1 y el grafito.
El electrodo de referencia empleado fue Hg/HgZCO4 ,HZC04 0.5 M de
Tacussel Tipo C 8 No. 329.562. pero todos los potenciales reportados
en este trabajo estan referidos al potencial reversible de hidrógeno
(ERH).
50

_LII_ A - - . ~. . . . ,. -
11.3. REACTIVOS UTILIZADOS
Los procesos de electrosorción de hidrógeno y oxígeno, así como
el depósito de Cu a subpotencial, realizados sobre los tres metales
nobles utilizados en el curso de ésta investigación fueron llevados a
cabo en medio H SO4 0.5 M. Esta solución se preparó a partir de HZS04
(98% g.r. Baker Analyzed) y agua desionizada (R > 18 MR). El H O
deionizada fue obtenida en columnas de MILLIPORE Modelo AllLLI-Q
(Continental Motor Systems)
2
2
Alambre de Cobre a Tubo de v i d r i o Pyrex
Soldadura do Estaño &Res ina E pox i t a
Fig. 11.2.1. Diagrama esquemático de la fabricación de 10s
electrodos utilizados en el desarrollo
experimental.
51

Para los experimentos de depósitos de Cu a subpotencial la
concentración de iones Cu2' empleada se f i jó en l ~ l O - ~ M usando como
fuente de iones Cu CuS04.5H20 grado analítico (Técnica Química.
C.A.).
Z+
La electroxidación de los adátomos de cobre, previamente
depositados, se efectuó en soluciones de H SO 0.5 M libre de iones
Cu . Los metales nobles empleados como electrodos de trabajo fueron
de alta pureza, 99.9997. (Jonhson Mafley,Co. ).
2 4
2+
2 1 NZ utilizado, para desoxigenar el electrolito y mantener
atmósfera inerte, fue proporcionado por INFRA de México. Previo a su
paso por el sistema, el gas N fue tratado pasándolo por tres trampas:
la primera conteniendo una solución de pirogalol, la segunda agua
desionizada y la última H SO concentrado. Este tratamiento permitió
retirar el oxígeno residual del gas N comercial, evitando ademss. el
arrastre de'pirogalol al sistema.
2
2 4
2
11.4. MONTAJE ELECTRONIC0
El diagrama esquemático del montaje electrónico acoplado a la
celda electroquímica se muestra en la Fig. 11.4.1. Los componentes del
mismo son los siguientes:
1. Potenciostatoígalvanostato PAR Modelo 273 con programador de
señales triangulares de pendiente variable integrado. Su funcion es
proporcionar el voltaje necesario entre los electrodos de trabajo, W.
! contra-electrodo, C-E; y
mantener la diferencia de potencial entre los electrodos de
referencia, R, y de trabajo, W , según el programa de potencial
seleccionado. Visto desde otro ángulo, el potenciostato es un elemento
ajustar dicho voltaje con el propósito de
52

Fig.Il.4.1. Diagrama esquemático del montaje electr6nico utilizado
53

activo, cuya funci6n es forzar a través del electrodo de trabajo la
corriente necesaria para alcanzar el potencial deseado.
2. Graficador x-y-t HP Modelo 7090A. Este instrumento nos
permitió registrar los perfiles potenciodinámicos de nuestro sistema
de manera directa e inmediata.
11.5. CONDICIONES ELECTROQUIMICAS PARA CARACTERIZACION
Ya que tanto los procesos de electrosorción de hidrógeno y
oxigeno. asi como los procesos de deposición y oxidación de adátomos
de Cu. fueron estudiados por métodos electroquímicos, es necesario
señalar aquí las condiciones electroquímicas bajo las cuales estos
estudios fueron desarrollados.
1. Voltamperornetria Cíclica (VC). Mediante esta tecnica. el
potencial impuesto al electrodo de trabajo se hace variar, entre dos
valores límites y de manera repetitiva. como una función dei tiempo.
LGS límites de potencial, potenciales intermedios, amplitudes de
potencial y velocidades de barrido, dE/dt, constituyen el programa de
potencial. el cual es desarrollado por el generador de señales. El
programa de potencial debe ser seleccionado para cada tipo de electrodo
de trabajo usado, y establecido de acuerdo al tipo de anáiisis a
desarrollar. Un programa de potencial típico es mostrado en la
Fig.(II.5.1)
Esta técnica fue usada para estudiar los procesos de
electrosorción de hidrógeno y oxígeno en P t , Pd y A u , así como para
estudiar los procesos de oxidación de adátomos de Cu. Las condiciones
establecidas para cada caso, son las siguiente:

TABLA 11. 5.1 Condiciones experimentales para el estudio
de los procesos de electrosorción de hidró-
geno y oxigeno.
P t Pd Au
E i n f . 0.08 0.31 0 . 0 3 V
Esup. 1.66 1.56 1.86 V
Ex 0.05 0.05 0 . 0 5 v
dE/dt 0.050 o . O50 O . 050 mV/S
TABLA 11.5.2. Condiciones experimentales para el depósito y
electroxidación de adátomos de Cu.
P t /cu Pd/Cu Au/Cu
E . : 0.51- 0.217 0.41- 0.21 0.53- 0 .21 i.
t : 180 1 80 600 S
d
d
C.Patrón: 0.08- 1.51 0.31- 1.56 0.03- 1.81 \
dE/dt: 0.100 0.05 O . 05 mV/S
La respuesta del sistema es seguida por la variación .de corriente
como una función del programa de potencial impuesto. Las curvas
corriente-potencial correspondientes son obtenidas por medio de un
registrador x-y, o bien a través de un osciloscopio, dependiendo
básicamente de la velocidad del barrido. P i r a los electrodos de P t , Pd
y Au , utilizados en este trabajo, estas curvas 1-E son representadas
en las figuras que se muestran en el Capitulo 111.
55

Ei
Fig.II.5.1. Programa de potencial aplicado al sistema para
estudiar los procesos de electrosorci6n de
especies oxigenadas en H SO 0.5 M a una
velocidad de barrido de 50 mV/S.
7 . 4
56

2. MCtodo Potenciostático. Esta técnica se basa en someter ai
electrodo de trabajo a la imposición de un potencial de valor
constante. El potencial aplicado y su duración se pueden establecer
manualmente, a través del potenciostato, o siguiendo un programa
preestablecido en el genarador de señales. La respuesta del sistema
puede ser analizada en términos de la corriente y/o carga asociadas
ai valor de potencial impuesto.
Este tipo de técnica electroquímica fue la seleccionada para
1levar.a cabo el depósito de Cu a subpotencial. Los diferentes valores
de potencial usados, fueron establecidos a partir del valor
correspondiente al potencial de equilibrio de Nernst para la
concentración de iones Cu empleada: 2.
E 2+ = Eo 2+ + 0.03 log [Cu"] C" /C" C" /C"
Eo 2+ = 0.34 V, C" /C"
como [c~'+I = 1x10.~ M
entonces:
y
E Z+ = 0.22 VIE" cu /cu
Todos los depósitos efectuados a E > E 2+ + IO mV son a w C" /C"
denominados como "depósito a subpotencial", mientras que a condiciones
de E < E 2+ se denominan como "depósito a sobrepotencial". La
primera condición fue la mantenida durante todos los experimentos de
deposición de Cu. Siguiendo la carga involucrada en el depósito de Cu
como una función del tiempo a un potencial dado, es posible determinar
l a cantidad máxima depositada a ese potencial.
a w C" /cu
El tiempo necesario para alcanzar el equilibrio de
adsorción-desorción de los adátomos de Cu, es función del valor de
potencial impuesto. Así, para los valores de potencial más positivos
(respecto a ECu~+/Cu) el tiempo al cual Q es máxima es el más largo, cu

tien d e a E z+ Por tanto, es cu /CU' reduciéndose conforme E
recomendable determinar t
aPP
a l mayor valor posible de subpotencial. max
Considerando ambas técnicas, es posible seguir el recubrimiento
de la superficie con adátomos de Cu en función del potencial de
deposición. La representaci 6 n de Qcu vs Ed , la isoterma
electroquímica, permite caracterizar la superficie metálica expuesta.
2 6
58

CAPITULO 111
111. RESULTADOS Y DISCUSION
Se presentan en este capítulo los resultados concernientes a los
estudios electroquímicos desarrollados sobre los electrodos de Pt , Pd
Au. En la primera parte del capítulo se analizaran las
características electroquimicas de cada uno de los electrodos de
:rabajo utilizados. hacia los procesos de electrosorción de hidrógeno
:: oxígeno. En la segunda parte del mismo, se presentarán los
resultados concernientes al depósito de Cu a subpotencial sobre los
mismos electrodos de trabajo.
En ambos casos, los resultados obtenidos con los electrodos de P t
i e r a n los primeros en presentarse a f i n de que éstos sean tomaaos como
referencia para el análisis de los otros dos metales estudiados.
1 1 1 . 1 . PROCESOS DE ELECTROCORCION DE HlDROGENO Y OSIGESO
1 1 1 . 1 . 1 . Plat ino
La figura III.1.1.l. muestra un voltamperograma tipico para un
electrodo policristalino de Pt . El voltamperograma exhibe tres
regiones de potencial bien definidas. las cuales son caracteristicas
para este t ipo de electrodos metálicos. En la región A se eiectúan
los procesos de adsorción-desorción de hidrógeno. El proceso de
adsorcion de hidrógeno es caracterizado por la aparición de dos picos
de corriente catódica. los cuales, para propósitos de identificación.
se han etiquetado como Ha y Ha aduciendo a la naturaleza del enlace
Pt-H . Ya que el pico Ha es el primero en aparecer durante la
adsorción y el último en desaparecer durante la desorcion, éste ha
f d
ads f
59

I
C
m
z O -
o. 2 0.4
O
Z O W w
-
1.4 E, V/ERH O. 6 O. 8 1.0 I. 2
2 Fig. 111.1.1.1. Perfil potenciodinárnico para Pt (Ageom=0.25 crn 1,
obtenido en H SO 0.5 M entre los límites de
0.08 hasta 1.51 V. v = 50 mV/S.
2 4
60

energéticos de la superficie de P t , generando, por tanto, el enlace
Pt-Hads más fuerte. Una vez cubiertos los sitios de mayor energía, los
restantes comenzarán a ser ocupados, lo cual se manifiesta por la
aparición del segundo pico de corriente catódica, Ha. La inversión del
sentido del barrido de potencial, hacia la dirección de valores
positivos, da lugar el proceso de desorción de hidrógeno, siendo
caracterizado éste por la porción anódica del voltamperograma en la
región. A. Los picos de corriente anódica Hd y H son los equivalentes
anodicos a los picos de corriente catódica H l y H: respectivamente. El
pico de corriente anódico intermedio, H i , es el asociado a la
desorcion de hidrogeno desde los sitios de P t correspondientes al
piano de bajo índice de Miller (110) y cuya energía de enlace es
intermedia con respecto a los sitios de planos preferenciales. El pico
de corriente catódica equivalente no es observado en el espectro
eiectroquímico, debido al traslape entre los picos de adsorción
preferenciales.
d
d d
r
d
En la región B del voltamperograma II1.1.1.1, se observan
fundamentalmente los procesos de carga y descarga de la doble capa
eléctrica, la cual, para fines prácticos, se puede considerar que
permanece inalterable en todo el intervalo de potencial estudiado.
Finalmente, en la región C del voltamperograma I l l . l . l .1 se
dan los procesos característicos de adsorción-deserción de oxigeno y,
eventualmente, el proceso de evolución del mismo. El aumento de
corriente anódica a partir de 0.8 V. se origina por el inicio del
proceso de oxidación de la superficie del electrodo de Pt . el cual
durante el barrido de potencial y hasta un valor de potencial
61

I
C
1 .4 E,V/ERH o. 2 0.4 O. 6 O. 8 1.0 I. 2
Fig. III.l.l.1. Perfil potenciodinárnico para Pt (A =0.25 cm-I. geom
obtenido en H SO4 0.5 bi entre los límites de
0.08 hasta 1.51 V. v = 50 mV/S.
2
60

sido asociado a la adsorci6n de hidr6geno sobre los sitios más
energéticos de la superficie de P t , generando, por tanto, el enlace
Pt-Hadp más fuerte. Una vez cubiertos los sitios de mayor energía. los
restantes comenzarán a ser ocupados, io cual se manifiesta por la
aparición del segundo pico de corriente catódica, H:. La inversion del
sentido del barrido de potencial, hacia la dirección de valores
positivos. da lugar el proceso de desorción de hidrógeno, siendo
caracterizado éste por la porción anódica del voltamperograma en la
region AI Los picos de corriente anódica Hd y H)' son los equivalentes
anódicos a los picos de corriente catódica H i y H i respectivamente. El
pico de corriente anódico intermedio, Hd, es el asociado a la
desorcion de hidrogeno desde los sitios de P t correspondientes al
plano de bajo índice de Miller (110) y cuya energía de enlare es
intermedia con respecto a los sitios de planos preferenciales. El pico
de corriente catódica equivalente no es observado en el espectro
electroquimico. debido a l traslape entre los picos de adsorción
pref erenciales.
d
En la región B del voltamperograma III.l.I.1, se observan
fundamentalmente los procesos de carga y descarga de la doble capa
electrica, la cual, para fines prácticos, se puede considera:. que
permanece inalterable en todo el intervalo de potencial estudiado.
Finalmente, en la región C del voltamperograma 1 i l . l . l . l se
dan 10s procesos caracterlsticos de adsorción-desorción de oxígeno y?
eventualmente, el proceso de evolución del mismo. Ei aumenta de
corriente anódica a partir de 0.8 V, se origina por el inicio del
proceso de oxidación de la superficie del electrodo de Pt . e l cual
durante el barrido de potencial y hasta un valor de potencial
61
. .. . . . : .;.. ,<<I. +: . I.

alrededor de 1.5 V, recubre la superficie de Pt con una monocapa
monomolecular de oxígeno adsorbido. Si el límite superior de potencial
fuese incrementado más allá de 1.5 V, los procesos de formación de
óxidos y evolución de oxigeno tomarían lugar. Debe tomarse en cuenta,
sin embargo, que el valor de potencial de 1.5 V no define con
precisión la demarcación entre los procesos de adsorción y evolución
de oxigeno en Pt . ya que ésta es dependiente de varios factores tales
como electrolito soporte, factor de rugosidad. velocidad de barrido de
porencial. etc. Debido a esto, la evaluación de parámetros de
superficies, a partir de la carga asociada a esta porción del
voltarnperograma, no es confiable.
Cuando la dirección del barrido de potencial es invertida,
partiendo del limite superior de potencial programado, l a porción
catocica del voltarnperograma mostrado en la Fig. 1 1 1 . 1 . 1 . 1 exhibe un
pico de corriente ancho y centrado alrededor de 0.78 V. En esle caso,
y a diferencia de lo que ocurre en la porción anódica. la unica
conrrihucion al pico de corriente catódica observado es el proceso de
desorcion de oxigeno de la superficie de Pt. Esto es debido a que
cualquier contribución por evolución de oxigeno y/o formacion de
óxiaos. sólo es observada a valores altos de potencial I > 2 . 0 \ ' I y
ba!o condiciones de anodización extrema. Por tanto, es lógico esperar
que la altura, y en alguna medida la forma, del pico de corriente
catódica sea sólo función del grado de recubrimiento de la superficie
alcanzando una altura máxima de P t por el oxigeno adsorbido,
cuando 9 = I . La dependencia en la altura del pico de corriente
catódica con el grado de recubrimiento por oxigeno, puede ser
adecuadamente analizada variando el límite superior de potencial. Los
e P i
Pt
62

~
0.2 0 . 4 O. 6 0.8 1.0 I .2 1.4 1.6
EL.S(V/ERH)
2 Fig. III.1.1.2. Curvas I-E de Pt ( A = 0.25 cm ) obtenidas geom
en ti SO 0.5 M. Potencial limite superior de
barrido desde 0.91 hasta 1.66 V con incrementos
de 0.1 V . v = 50 mV/S.
2 4
63

voltamperogramas obtenidos a diferentes valores del limite superior de
potencial, son mostrados en la Fig.III.1.1.2. Dos aspectos relevantes
pueden ser observados de esta figura. En primer lugar se puede ver que
conforme el limite superior de potencial se incrementa, los perfiles
potenciodinámicos anódico y catódico se vuelven más irreversibles, lo
cual es una consecuencia de la diferencia de energía invoiucrada en
los procesos de adsorción y desorción correspondientes. Sin embargo,
ia relación entre ia carga involucrada en la adsorción. Oa, :i üquella
invoiucrada en el proceso de desorcion, O', es siempre aproximadamente
la unidad, esto es, O"/O" - 1. Por lo tanto, podemos asumir que la
cantidad de oxigeno adsorbida es la misma que se desorbe en el barrido
inverso de potencial. A valores del limite superior de potencial
mayores de 1.15 Y , la relacion Qa/O' > I reflejando inmediaramenre la
contribucion de un proceso adicional al de adsorción u? oxigeno
durante el barrido directo de potencial. Esto confirma et manera
categoricü la imposibilidad de utilizar la carga involucraou en el
proceso de adsorción de oxieeno para calculos estimativos de
parámetros superficiales.
O 0
0 0
El segundo aspecto a considerar, es el hecho de que l a posición
del pico de corriente catódica es prácticamente indepentiiente del
limite superior de potencial. Hay una variación no mayor de 7ü mY en
un intervalo del limite superior de potencial mayor a 600 rn\. lo cual
permite suponer que en el proceso de reducción, caracterizado por este
pico de corriente catódica, solo un tipo de especie oxigenada está
siendo involucrada. La obtención de una linea recta en la
representación gráfica de O vs ELS (Fig. I 11.1.1.3.) confirma esta
suposición. A vaiores de potencial mayores a 1.15 V, la representación
O
64

gráfica de Q de
la participación de procesos adicionales a la adsorción de oxigeno,
probablemente la formación de óxidos superiores de P t y/o evolución de
oxígeno, impidiendo la evaluación correcta de 00 a partir de estos
valores de potencial.
vs ELS muestra que Q:/Qk > 1, lo cual es indicativo O
Aún cuando la gráfica de QL vs E trazada entre 0 .9 1.5 V no
presenta cambios significativos de pendientes en este intervalo de
potencial, se ha establecido en la literatura [50-541 que IX probable
mecanismo para la adsorción de oxígeno es el siguiente:
LS
- Pt + H o - Pt (OH) + H+ + e I
2 - ads -
- p t n + ~ n * + e I I ads ?
Pt (OH) + H20 ~
ads
siendo la etapa I una reacción de equilibrio rápida. en nonde el
primer electrón es intercambiado. Esta etapa solo es susce.tible de
ser evidenciada a traves de metodos rapidos de perturbacion, por
ejemplo un barrido de potencial a velocidades no menores a 5 \' S . La
especie oxigenada inicialmente formada, PtiOHi continua S'J proceso
do oxidación, 11, formando la especie oxigenada Ptü Is cual se
conserva hasta que un grado de recubrimiento 0 - 1 es alcanzado.
Esta es base para considerar la adsorción de oxigeno sobre Pt. un
- 1
ads'
ads'
Pt
metodo de estimación de areas superficiales. Sin embarp, estas
consideraciones deben ser tomadas con reserva debidi a las
controversias encontradas en la literatura respecto al t ipo final de
especie oxigenada formada 147,491.
1II.l.l.a. Estimación del área superficial expuesra a partir
de la adsorción de hidrógeno sobre Pt
La estimación de áreas reales de electrodos de P t , se basa en e1
65 " , '7 ..._ *. .,

O. 9 1.0 1.1 I . 2 1.3 I. 4 I. 5
EL.s( V/ ER H 1
Fig. III.1.1.3. Representación gráfica de las cargas anódicas
y catódicas para P t en función del
superior de potencial (0 vs E
a 25 C. A
limite
1 en H SOJ U.5 hl 0 LS 2 2 = 0.25 cm . Y = 50 mi!/%
geom
66

De aqui que si uno:es capaz de determinar el número de átomos de
hidrógeno adsorbidog entonces será posible determinar ei número de o.
sitios activos sobr% la superficie expuesta y, por consiguiente, el
valor del área real. 2 t *.
Sabemos que el! proceso de electroadsorción de hidrógeno involucra
la transferencia de -un electrón, por tanto, determinando la carga de
electroadsorción de : hidrógeno a máximo recubrimiento es posible
determinar la cant idd total de hidrógeno adsorbido. Sin embargo. como !?
f u e mencionado al ' inicio del capitulo 111.1.1. y observado en Fig
111.1.1.1. antes de que la superficie expuesta de Pt sea totalmente
saturada por hidrógeno adsorbido, da inicio el proceso suplementario
de evolución de hidhgeno, esto es, ambos procesos, electroadsorción y
evolucion, .se dan dmultáneamente a partir de 0.08 \:. .As] para el
caso particular de Pt , si la carga de hidrógeno es determinada por
integración del pe#il potenciodinamico catódico de la Fig. 111.1.1.1,
con E = 0.08 entonces la división de esta integral por 0.77,
dara la carga correspondiente al total de sitios, mientras que si es
dividida por 0.84. 117,221. la carga corresponderá a. los sitios
inmediatamente accmibles. Del valor calculado y tomando como estandar
convencional de carga QE, = 210 pCícrn (para el intercambio de
le-] 1221. el área- real expuesta puede ser estimada mediante la
. m i n
2

. . . . . . .. . . . . . ~ ~~. . - ~
De la Fig. 111.1.1.1 se tiene que Q" = 61.4 pC, por lo que
= 0.34 cm . Dado que el área geométrica del electrodo de Pt es :
H 2
*pt
2 = 0.25 cm , el factor de rugosidad correspondiente es: A
f = Ar/A = 1.36 Este último valor es una medida de la rugosidad de
la superficie expuesta, que para muchos propósitos es más útil que el
mismo valor del área real. Desde un punto de vista más estricto, es
este factor de rugosidad lo que caracteriza una superficie rnetalica
expuesta.
gc o rn
g c o m
1ll.l.l.b. Estimación del área superficial expuesta a partir
de la desorción de oxígeno sobre Pt
Sobre una base similar a la adsorción de hidrógeno. e l aren real
expuesta de electrodos de Pt puede ser evaluada partiendo UP ¡a carga
de electrodesorción de oxigeno. En este caso es la desorción rio ia
adsorción el proceso considerado, ya que como mencionanlos
anteriormente. la evaluación de la cantidad exacta de ,?sieeno
abbsorbido es incierta tanto por la ocurrencia a; procesos
suplementarios. como por el desacuerdo existente en el t i pc Sinal de
especie oxigenada formada. No obstante lo anterior, tomanao como
estandar interno la adsorción de hidrógeno, asi como el hecr,n de que
los procesos suplementarios anódicos en la region de 'oxieenn se
manifiestan en otras regiones catódicas diferentes al pico de
corriente catódica, caracterisitico para la desorción de oxigeno. se
pueden hacer evaluaciones de área más o menos confiables. En este caso
partimos del supuesto de que a valores de potencial alrededor de 1.5 V
la superficie de Pt es totalmente cubierta por una capa monoatomica de
oxígeno en una estequiometría I:1, es decir, formando la especie PtO.
68

Así, la carga catódica asociada al pico de corriente catódica,
observado en la región C del voltamperograma de la Fig. 1 1 1 . 1 . 1 . 1 ,
deberá corresponder a la desorción de una monocapa de oxígeno
adsorbida. En este caso, ninguna corrección por recubrimiento es
necesaria, pero el estandar convencional de carga deberá ser ahora de
420 pC/cm debido a que dos electrones son involucrados en el proceso
de adsorción-desorclón de oxígeno. De aquí que:
2
QC,G A r = 2íQ;t ,pC/cm 2
Del pico de corriente catódica (Fig 111.1.1.1) se ha calculado que
= 141.4 p ~ , y como Q P ~ = 210 piC/cmZ entonces:
2 A r = 0.34 cm f = 1.36
Los valores de .4r obtenidos por ambos procesos son ipiaies. lo
que implica que a 1.51 V la especie oxigenada adsorbida en realidad
esta formando' una monocapa de O sobre la superficie de¡ electrodo
de P:. es decir, ,la especie adsorbida es PtQ.
ads
111.1.2. Paladio
La Fig. II1.1.2.1 muestra el perfil potenciodinamico de uno de
= 0.31 cm ) utilizados en el curs'? de esta los electrodos de Pd (A
investigación. Esta curva exhibe tres regiones de potencial bien
definidas, las cuales son caracteristicas de los electrodos
constituidos por este metal. En la región A se llevan a cabo 10s
procesos de adsorción-desorción de hidrógeno, pero también tiene lugar
la absorción del mismo. El proceso de adsorción ocurre al efectuar el
barrido de potencial hacia valores menores de potencial (partiendo de
la región de la doble capa eléctrca). observándose un incremento
2
geom
69

continuo en la corriente pico a medida que se avanza hacia valores
menores de potencial, no detectándose los picos de corriente de
adsorción para los diferentes planos cristalinos del metal, como es el
caso para los metales nobles como son Pt, Rh e Ir 131. A l invertir ia
aireccion del barrido de potencial se observa un sólo pico de
corriente de desorción de hidrógeno a 0.27 \', sin ninguna estructura
de picos caracteristica de los tres planos de bajo indice de híiller.
Debido al problema de absorción de hidrogeno que presenta el metal no
se traba-io a potenciales menores de 0.31 V.
En la región B del voltamperograma (Fig. 111.1.2.1). se presentan
IC' procesos de carga y descarga de la doble capa eléctrica. Para
f i n o s practicos, ia carga debida a estos procesos se considera que
D o x i l a n x e constante en el intervalo de potenciai estudiado.
Finalmente. en la región C ocurren los procesos de
aisnrcinn-desorción de oxigeno y. posiblemente. se present- la
e..?i,Jci:ri del mismo. Como en el P t , el proceso de adsorcion de oxi,geno
ccrnienza I 0.S i y se extiende a valores de potencial alrededor de
! , 5 's., a partir del cuai se pueden presentar tanto la formacion de
oxiacs. como la evolución de oxígeno molecular. De la Fig. 111.1.2.1
se GueOe observar que a partir de 1.0 \' la corriente anódica se
incrementa paulatinamente en el perfil potenciodinámico, io que
di:^iculta el estudio superficial del electrodo por adsorcioii de
esFecies oxigenadas. A l invertir el sentido del barrido de potencial,
so liera a cabo la desorción de las especies oxigenadas. Este proceso
se manifiesta en el voltamperograma con la aparicion de un pico de
ccF!-:ente catódica agudo a 0.68 V. La altura del pico catódico varia
con el valor del potencial limite superior (E 1 de inversion del LS
70

I
I
o
Z O (3 W
-
a
I 1
a2 0.4 O. 6 0.0. 1.0 1.2 1.4 E, ( V
Fig. 111.1.2.1. Perfi l potenciodinámico de un electrodo de Pd
2 = 0.31 crn I en H SO4 0.5 M, desde 0.21
gcom 2 ( A
hasta 1.56 V. v = 50 rnV/S.
71
O. 2 0.4 0.6 o. a 1.0 1.2 I .4 E. V/ERW
Fig. 111.1.2.2. Curvas I-E mostrando el efecto del E de
inversión del barrido de potencial para Pd
(A
con incrementos de 0.1 V. v = 50 mV/S.
L,S
2 = 0.31 cm 1, desde 0.86 hasta 1.56 \
gcom
12

barrido de potencial. por lo que se puede hacer un estudio más
detallado del proceso de desorción de oxígeno modificando este
parámetro, tal como se observa con la Fig. 111.1.2.2.
La variación de la altura del pico de corriente catódica viene
acompañado de un desplazamiento del potencial pico E , y es más
marcado a medida que aumenta la altura del pico de corriente. Para un
intervalo de potencial de 0.6 V (PELS = 0.86 - 1.46 V) el E se
modifica en 0.09 V , lo que es indicativo que la especie adsorbida
sobre el' Pd posee mayor energía que la adsorbida sobre P r . poi- lo
tanto, l a caracterización del Pd por desorción de la especie adsorbida
deberá hacerse aún con más cuidado que para el caso del Pt.
P
P
Por otro lado, la relación Oa/QC es aproximadamente izual a la
unidad en un estrecho intervalo de potencial, 0.86 - 0.96 \ ' , :; para
valores superiores de potencial la relación crecen en forma continua,
Oa/Oo ?. 1. 'Este pequeño intervalo de potencial donde C1''/9~-1
(proceso reversible) hace suponer que a potenciales mayores e);? 0.96 V
O 0
r , o
existe u n proceso paralelo ai de adsorción de oxígeno, adema; a? la
posible evolución prematura de oxígeno molecular en los sltios más
activos de la superficie (el electrodo posee alto valor de rugosidad,
p w su método de preparación). Woods y Col. 12.361 han reportado que
la razón por la cual (O'/Qc) > I es debido a la disolución c i i l Pd a
potenciales mayores que y/o iguales a 1.0 V.
O 0
L a Fig. 111.1.2.3. muestra la representación gráfica de las
cargas anódica y catódica en función del potencial limite superior.
Por tanto, es evidente que una estimación del área real del electrodo
a partir de la curvas de la Fig 111. 1.2.3 sería imprecisa. por 10
que, este cálculo se efecturá en el siguiente subcapitulo mediante el
73

QW 600
5 0 0
400
300
200
I O 0
, I5 I. 2 I. 3 1.4 1.1 EL,sW/ERH) 0.9 1.0 0.9 1.0
, I ,5 I. 2 I. 3 1.4 1.1 EL,sW/ERH)
Fig. III.1.2.3. Representación gráfica de las cargas anódicas
y catódicas en función del potencial limite
superior (Oo vs E
Y = 50 mV/S.
1, para Pd en HZSOJ 0.5 M L,S
74

depósito de Cu a subpotencial.
111.1.3. Oro
La Fig. 111.1.3.1 muestra un perfil potenciodinámico tipico para
= 0.19 cm'l en H SO4 0.5 M. un electrodo policristalino de Au ( A
El voltamperograma muestra dos regiones de potencial bien definidos.
La región A exhibe una amplia zona de potencial donde únicamente se
llevan a cabo procesos de carga y descarga de la doble capa electrica,
que para fines prácticos se puede considerar que permanece constante
en ei 'intervalo de potencial estudiado. La región B del perfil
potenciodinaico (Fig. 111.1.3.1) muestra los procesos de
adsorcion-desorcion de especies oxigenadas naturalmente. la
formacion de óxidos y evolución de oxizeno molecular. Cuando se
efecttx el barrido de potencial en la direccion positiva se observa un
fluio de corriente anódica a partir de 1.37 Y, debido a la adsorción
de especies oxigenadas sobre la superficie del electrodo. con tres
picos de corriente muy proximos entre S I (1.41, 1.1s y . 1.5'' i',
respectivamente] tal como se aprecia en el perfil potenciodinaniico de
la Fig. 111.1.3.1. Estos picos están relacionados con la adsoi-cion de
especies oxigenadas por los diferentes pianos cristalinos del metal,
correspondiendo al pico I la adsorción de especies por los sitios mas
energéticos de la superficie, es decir, los sitios de menoi' eiiergia
seran los últimos en registrar especies adsorbidas. A 1.7 Y el perfil
anódico registra una corriente minima y posteriormente sufre un
incremento abrupto al aproximarse a la región de formación de oxidos y
evoiucion de oxigeno molecular. Arvia y Col. 11161 han postuiado que
la especie adsorbida a potenciales cercanos a 1.38 V reacciona con el
medio, dando como resultado una serie de picos de corriente al
2 geom

R E G I O N A
I
R E G I O N 6
, I
0.2 O. 4 0.6 0.8 1.0 1.2 I .4 1.6 E, V/ERH
Fig. 111.1.3.1. Perf i l potenciodinámico para Au 1.4 =0.19 crn'.l.
obtenido en H SO 0.5 M a 25 C entre los limites
0.03 hasta 1.86 V. Y = 50 mV/S.
geom
2 4
76

. 2. . . . .
invertir el sentido del barrido de potencial y que además. cuya carga
es mayor que la correspondiente al proceso de adsorción ( Q L / Q z < I).
Cuando la dirección del barrido de potencial es invertido,
partiendo del potencial límite superior (E ) programado, se registra
un pico de corriente agudo en el perfil catódico alrededor de 1.15 V
(Fig, 111.1.3.1). cuya altura. depende del valor de ELS de inversión
del barrido de potencial. Coni0 en el caso de los otros dos metaies
nobles aquí tratados, la carga obtenida del perfil potenciodinámico
catódico' es una información directa de la naturaleza de la especie
desorbida ( o reducida). Es por tanto evidente, que la altura del pico
de corriente catódica será diferente para cada valor de ELS de
inversión del barrido de potencial tal como se muestra m l a Fig.
111.1.3.2. A medida que el valor de E se incrementa la desorcioii de
la especie adsorbida se vuelve más difícil indicando con ello que el
proceso es más irreversible.
LS
LS
La Fig 111.1.3.3 muestra la representación gráfica de las cargas
involucradas en los procesos de adsorcion y desorción c e especies
oxigenadas. En esta Fig. 111.1.3.3 se observa que ambos valores de
carga se incrementan de manera continua hasta alcanzar una meseta
sobre la isoterma electroquimica donde las cargas poseen tl mismo
valor. Finalmente vuelven a incrementarse, haciéndolo más .rápidamente
la carga anódica. A bajos grados de recubrimiento la relacion de ia
carga anódica y catódica (Qa/Q 1 es menor que la unidad, siendo
iguales en la región de la meseta y mayores que la unidad
apotencciaies mayores que 1.8 V. apoya I O
observado por Arvía y Col. [1161, mientras que QYQO' - I apoya un
proceso reversible y Qa/Qc > I sugiere la formac6n de óxidos o los
. .
C
O 0
La relación Q : / Q ~ < I
O 0
O 0

I 1 I
o. 2 0.4 0.6 O. 8 I .o 1.2 I .4 1.6 E, V/EAh
Fig. 111.1.3.2. Perfiles potenciodinárnicos mostrando el efecto
del ‘ELS de inversión del barrido de potencial
para Au ( A = 0.19 cm I , desde 1.48 hasta
1.86 V con incrementos de potencial variables.
2
geom
78

procesos de evolución de oxígeno molecular.
Se puede realizar una estimación del área real del electrodo
(región de meseta), considerando que ü = 1 a E = 1.7 y asumiendo que
la especie adsorbida es A u 2 0 3 ill 6 I . Usando la misma expresión
previamente utilizada para Pt. tenemos:
LS
C Qo K
A r = n pC/cm *I
donde: '
cia es el estandar convencional calculado para un eiectrodo
policristalino de A u [1171. cuyo valor es 184 pC/cm .
Au
2
De la Fig. 111.1.3.2 se determina el valor de 0' v considerando 0 -
un proceso que intercambia tres electrones por atomo superficid, de hu
tendremos finalmente el valor del área real del electrodo
2 00 ='358 pic Ar = 0.65 cm f = 3.42
En el siguiente subcapitulo se hará una estimación mas ronfiable
del valor del área real de éste electrodo a la luz de los rostiltados
obtenidos por depósito de Cu a subpotencial.
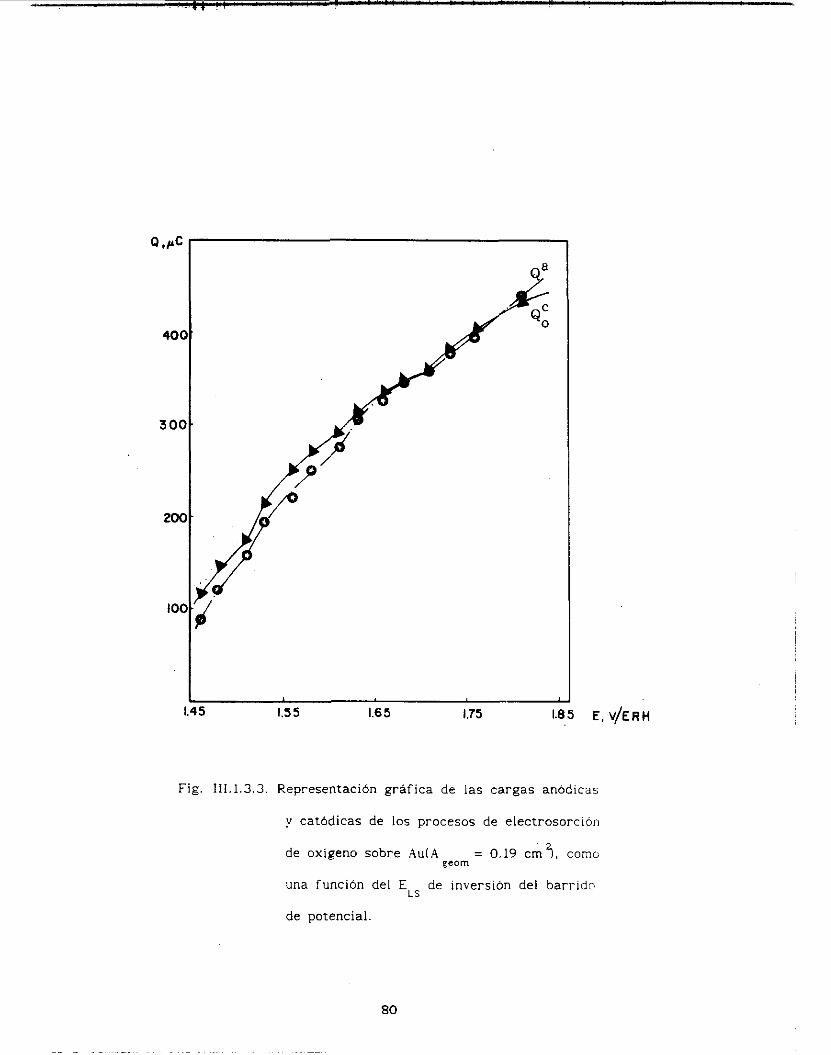
1.45 1.5 5 1.65 1.75 1.85 E , V/ERH
Fig. 111.1.3.3. Representación gráfica de las c a r g a s anódicas
y catódicas de los procesos de electrosorción
de oxigeno sobre Au(A = 0.19 c m 5 , corno
u n a función del E de inversión del barrido
de potencial.
geom
LS
80

111.2. DEPOSITO DE ADATOMOC DE Cu A SUBPOTENCIAL
En este subcapitulo se presenta el estudio electroquimico
correspondiente al depósito de Cu a subpotencial sobre los electrodos
de P t , Pd y Au utilizados. Los resultados experimentales se reportan
en la misma secuencia presentada previamente para los procesos de
electrosorción de hidrógeno y oxigeno. Los electrodos de Pt y .Au son
los mismos que Aquellos usados en los procesos de electrosorción de
hidrógeno, y oxigeno. mientras que para Pd un electrodo recién
preparado fue utilizado. Esto último es debido al hecho de que. si el
electrodo e s dejado por tiempos prolongados en la solución IHqSCf4 0.5
M) y i o es retirado de la misma, se observan modificaciones permanentes
en ICE correspondientes voitamperogramas de referencia.
Antes de efectuar la caracterización superficial de estos
electrodos, via .e fecto UPD de Cu. se lleva a cabo uii estudio
preliminar del sistema con e l objeto de encontrar el tiempo oprimo de
deposito ít ):- d
III.2.l.Efecto UPD de Cu sobre Pt
La Fig. 111.2.1.1 muestra el perfil potenciodinamico p a r a la
electroxidacion de
durante t = 5 min, obtenido en H2C0
Cu . El pico de corriente anódica más negativo (pico IVI corresponde
a la oxidación del Cu depositado en multicapas, ya que La cantidad
depositada se presenta como función de t
corriente anódica observados a potenciales más positivos, [ I , 1 1 y
Ill), se atribuyen al Cu depositado a subpotencial. La carga total de
Cu, previamente depositado sobre Pt a E . = 0.24 \' d
0.5 M en presencia de iones d 4
2+
mientras que los picos de d *

O. 6 O. 8 1.0 1.2 1.4 E,V 0.2 O. 4
Fig. 111.2.1.1. Perfil potenciodinámico de electroxidación de Cu
sobre Pt en H SO4 0.5 M en presencia de iones Cu".
v = 100 mV/S, E = 0.24 V, t = 5 min y ICu"1 =
2
d <I
I x M.
82 v ... ,

oxidaci6n de Cu involucrada en estos tres picos an6dicos, alcanza un
valor límite con t d' Este perfil potenciodináico de electroxidación de
Cu, obtenido en presencia de iones Cu , tiene un carácter más bien
cualitativo, ya que suobjetivo es poder diferenciar la oxidacion del
CU depositado a UPD de áquel depositado en forma masiva.
2+
Una vez comprobada la existencia del efecto UPD (por la presencia
de los picos anódicos I. I1 y 111, Fig. iii.2.1.1) se determina el
tiempo óptimo de depósito, esto es, el tiempo necesario para alcanzar
las condiciones de equilibrio de adsorción-desorcion de los adátomos
de Cu sobre el sustrato de Pt.
La Fig. I11.2.1.2 muestra los perfiles potenciodinaniicos de
electroxidación de Cu. los cuales fueron depositados a Ed = 0.51 L'
en función del tiempo de depósito y obtenidos en H SO 0 . 5 \I libre de
iones Cu . Se puede observar que la altura del pico anodico I se
incrementa con el aumento de t hasta alcanzar una altura maxima a
t = 3 min. As¡ .mismo, dos efectos siinuitáneos fueron observados: 11)
disminución progresiva de la region de adsorción-desorcioii de
hidrógeno liO21, y (2) un desplazamiento en el potencial de inicio del
proceso de oxidacion de la superficie del Pt .
2 4
2+
d
d
La inhibición de los procesos de electrosorción del hidrogeiio se
debe al incremento en la cantidad de Cu adsorbido sobre el Pt .
bloqueando con ello los sitios activos para la adsorción de hidrogrno.
La carga de oxidación de Cu. calculada por integracion de los
perfiles potenciodinámicos correspondientes. está representada en
función de t en la Fig 111.2.1.3. d
AI principio, la carga se incrementa rápidamente hasta alcanzar
un valor máximo, luego de lo cual parece apreciarse una ligera
83

I I I I
0.2 0.4 0.6 0.8 1.0 I. 2 1.4 E ,V
Fig. 111.2.1.2. P e r f i l e s potenciodinárnicos de electroxidacion dc
adátomos de Cu sobre P t , como u n a función del
tiempo de depósito a un potencial constante
E = 0.51 v, [CU'+I = I x 10.~ y = so rnv..'c d
Las curvas I-E s e obtuvieron en ausencia de
iones Cu . 21
84

Fig. 111.2.1.3. Variación de l a carga asociada a la eiectroxidación
de adátomos de Cu sobre Pt. como una funcion del
tiempo de depósito (cargas obtenidas por integración
del área bajo la curva I-E de la Fig. 111.2.1.2).
85
_ _ ~ _ . ~. ~
. . . ..< - . ,. . .

disminución en la misma. Esta disminución es fundamentalmente debida a
la impresición de la integración, a mayores valores de t va que el
perfil de oxidación de Cu se traslapa con el inicio de la oxidación de
la superficie de Pt.
d ' .
Una vez conocido el tiempo óptimo para la adsorción de adátomos
de Cu sobre Pt , se efectúan los depósitos a subpotencial a diversos
valores de E . La Fig. 111.2.1.4. muestra el perfil potenciodinámico
de la electroxidación de Cu (en H SO 0.5 M libre de iones ¿uZ'1,
depositado a 0 .22 V durante 3 min. la otra curva corresponde al
perfil de la curva patron del P t en ei mismo medio. Se observa rariibien
ia inhibición total de los procesos de electrosorción para el
hidrogeno y la existencia de los cuatro picos de corriente anodica de
d
2 4
eiecrroxidacion de Cu previamente citados (Fig. 111.2.1.11. La Fig.
111.7.1.5 muestra las curvas I-E de la electroxidacion de adaionios de
i3:i obtenidos a otros potenciales de depósito para el mismo sistema
IPt/cu*'). L a representacion grafica de las cargas asociadas a estos
perfiles potenciodinamicos es la curva mostrada en la Fig. 111.2.1.6.
Se observa que la cantidad de Cu adsorbida aumenta gradualniente a
medida que se incrementa el potencial de depósito. A 0.46 \' la
isoterma electroquimica (Fig. 111.2.1.6) exhibe un punto de inflexión,
debida posiblemente a un rearreglo en la estrucutra de los adátomos
sobre la superficie del PI. permitiendo con ello la adsorciori de una
mayor cantidad de Cu por el sustrato. Este punto de inflexion parece
estar asociado a la aparición del pico I1 en el perfil
potenciodinámico de la curva de electroxidacion de Cu. Continuando
hacia potenciales negativos se encuentra una meseta, justo antes de
alcanzar el depósito masivo, donde la cantidad de Cu adsorbido no es
86
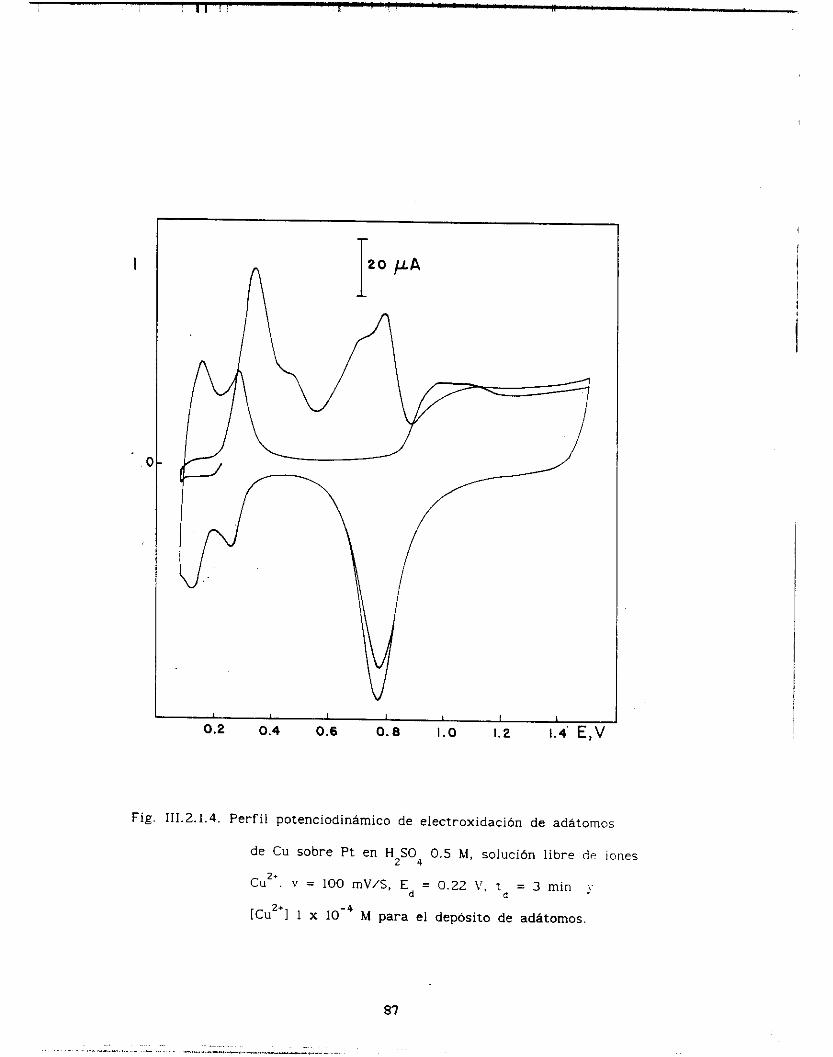
I
I
0.2 0.4 0.6 O. 0 1.0 1.2 1.4' E,V
Fig. 111.2.1.4. Perfil potenciodinámico de electroxidación de adátomos
de Cu sobre P t en H SO
CU . v = 100 mVíS, E = 0.22 \', t = 3 min
[Cu*'I 1 x
0.5 M, solución libre de ¡ones 2 4
2+
d d
M para el dep6sito de adátomos.
I
I
87
.. ~ . . ..~ ~. . . ,. .

'C
Ed : 0.65, 0.61, 0.56. 0.51
0.46, 0.41, 0.36, 0.26. 0.218, 0.217 \
I
0 . 4 0.6 0.8 I . o I. 2 1.4 E.V 0.2
Fig. 111.2.1.5. Perfiles potenciodinámicos de electroxidación de
adátomos de Cu sobre Pt en H SO 0.5 M, libre de
iones Cu , obtenidos a varios Ed. t = 3 min.
v = 100 mV/S y [cuZ+i = i x M.
2 4 2r
d
88

función del potencial. Finalmente se observa un incremento brusco de
la carga, que corresponde al depósito de cobre en multicapas..
La aparición de esta meseta indica que a pesar de proporcionar
mayor energia al sistema, ya no es posible depositar más Cu sobre el
sustrato. lo que significa que ya no hay más sitios disponibles para
la adsorción de Cu y , por lo tanto, se ha alcanzado el recubrimiento a
la monocapa (61. En esta region de la meseta, cada átomo de Cu se
adsorbe sobre un átomo de Pt intercambiando en el proceso 2e- segun la
siguiente' reacción: -
> cuo ( 1 1 1 . 1 1 cuZ* + 2e - Ahora estamos en posibilidades de poder esrimar el valor del área
real del electrodo empleando el mismo criterio que para la adsorción
de hidrógeno mostrado previamente en este mismo capitulo ílil.l.l.a1.
Tendremos que el área real estara dada por l a siguiente expresion:
(111.2)
donde:
OCU es la carga requerida para formar una monocapa de Cu sobre el
sustrato íPt).
el número de electrones intercambiados por átomo de Cu adsorbido.
estandar convencional de carga para un proceso que
m
n
O Q P t
intercambia un electrón.
Midiendo la carga de adsorción de Cu a la monocapa (región de la
meseta Fig. 111.2.1.6.) y conociendo los otros parámetros de la
ecuación (111.2) se puede obtener el valor del área real y el factor
de rugosidad para este metal (Pt).
89 .i: .

cFu:r,c 200
15c
100
5a
- 0.4 0.5 O. 6 E, v o. I 0.2 O. 3
Fig. 111.2.1.6. Isoterma de adsorción de adátomos de Cu
depositados sobre Pt íA = 0.25 cm 2 ) geom
en función del potencial de depósito.
td = 3 min.
90

Q C U = 141 fiC m
2 Ar = 0 . 3 4 cm f = 1.36
Otra manera de determinar el
en cuenta los siguientes puntos [71:
área con adátomos es la que tiene
1. Conocer el area promedio ocupada por átomo del sustrato
metálico.
2. Conocida la relación entre los radios atómicos (R / RH,).
Esto nos va a permitir suponer una estequiometria entre ambos metales.
Para el caso del Pt/CuZt la relación es de. 1.08. lo que induce a
suponer una estequiometría I:1, esto es, todos los átomos de Cu
adquieren la geometrla superficial del Pt l1021.
M
3. Todos los sitios superficiales han sido cubiertos.
5 QC" rn NT = N =
C" M - (111.3) n e
donde:
o=* es la carga requerida para formar una monocapa de Cu sobre el
sustrato.
m
n es el número de electrones intercambiados en el proceso de
descarga del adátomo.
e es La carga elemental del electron 11.602 x C). '
NT es el número de átomos de Cu que forman la monocapa. cu
es el número de sitios activos del metal para el depósito "M adátomos de Cu.
Sustituyendo los valores numéricos correspondientes en la
ecuación (111.3) se obtiene que el número de átomos de Cu descargados
sobre el Pt son 4.4 x
91

Por último multiplicando el número de átomos activos del sustrato
por el área promedio ocupada por átomo para un electrodo
policristalino de P t [I171 tenemos:
Ar = 4.4 x I O l 4 átomos Pt (8.33 10~'6cmz/átomos P t )
A r = 0 .37 cm f = 1.48 2
111.2.2. Efecto UPD de Cu sobre Pd
La Fig. 111.2.2.1 representa el perfil potenciodinámico para la
electroxidación de átomos de Cu. los cuales han sido previamente
depositados sobre Pd a partir de una solución 1 ~ 1 0 . ~ M en iones Cu
usando como electrolito soporte H SO 0.5 M. El depósito se obtuvo
aplicando un potencial de 0.21 V al electrodo de Pd durante 3 min. En
Zt
2 4
la Fig. I11.2.2.1 se observan tres picos asociados al proceso de
eiectroxidacion del Cu. Los picos I y I I corresponden a la
electroxidación de Cu depositado a subpotencial, mientras que el pico
111 se relaciona a la electroxidacion del Cu depositado en multicapas.
La Fig. 111.2.2.2 muestra las curvas ¡-E de la electroxidacion de
Cu depositados a 0.41 V en función del tiempo de depósito. En estos
perfiles se observa que ei pico de corriente anódica se incrementa con
el tiempo hasta los 3 m i n , luego sufre un decremento ( t > 3 rninl
acompañado de la aparición de un pico de corriente a potenciales más
positivos (1.17 VI. Este pico de corriente anódica más positiva
aparece en la misma región de potencial que en el Pt. La explicación a
la disminución de la corriente anódica y la aparicion de este pico de
corriente a potenciales más positivos es la misma que se di6
previamente para el Pt.
d
92

T
I
O. 6 O. 8 1.0 1.2 1.4 E ( V ) 02 O. 4
Fig. 111.2.2.1. Perfil potenciodinámico de electroxidación de
adátornos de Cu sobre Pd (A
H SO 0.5 M , en presencia de iones Cu ,
v = 40 mV/C, E = 0.21 V, t = 3 min.
= 0.18 cm2) en geom
2i
2 4
d d
[CU'+I = M O - ~ M.
93
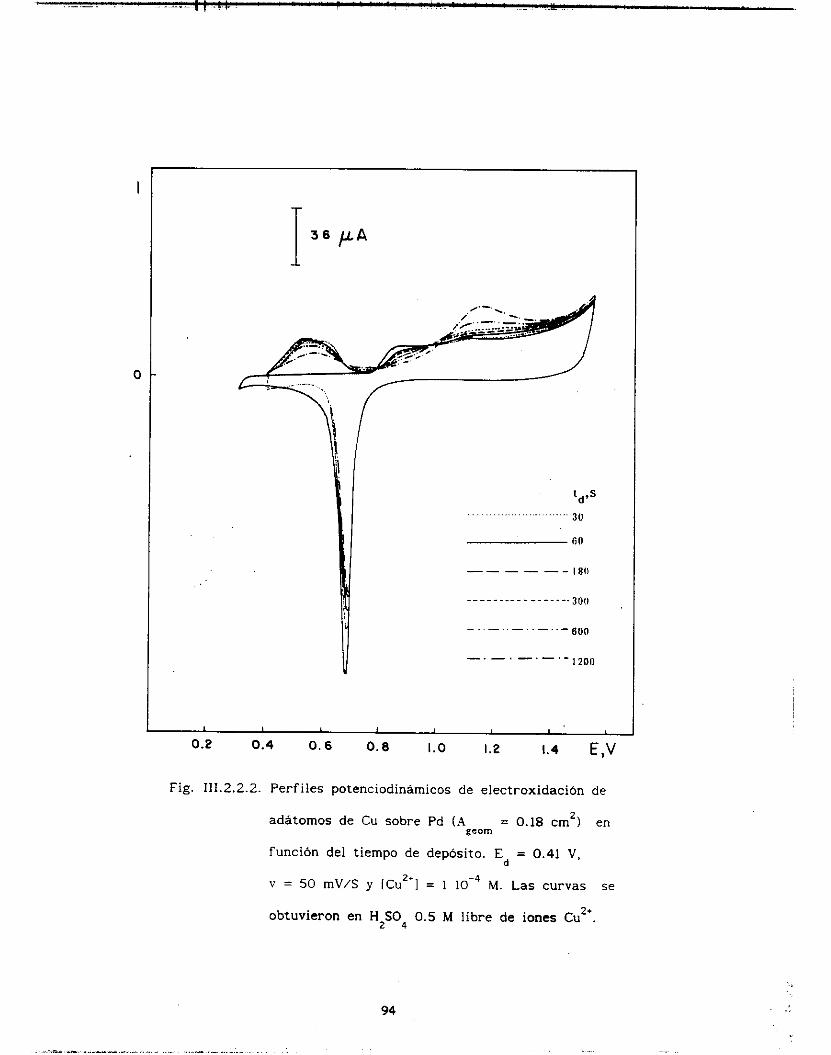
3u
(in
. . . . . . . . . . . . . . . . .. . . . . . . . . .
I811
3011
600 - . . - . . - . . - . . -
12on
0.2 0.4 O. 6 O. 8 1.0 1.2 1.4 E,V
Fig. 111.2.2.2. Perf ilec potenciodinámicos de electroxidación de
adátornos de Cu sobre Pd ( A = 0.18 crn ) en
función del tiempo de depósito. E = 0.41 V,
v = 50 mV/C y [Cu2'l = 1 M. Las curvas se
obtuvieron en H2S04 0.5 M libre de iones Cu".
2 gcom
d
94

i La Fig. 111.2.2.3 muestra la representación grdfica de las cargas
de electroxidación de Cu como una función del tiempo de depósito a un
potencial f i j o (0.41 VI. Al inicio, la carga se incrementa
rápidamente, alcanza un valor máximo (donde permanece casi constante
durante 2 min aproximadamente) y finalmente decrece paulatinamente. De
esta Fig. 111.2.2.3 se
min, se ha alcanzado
sistema.
Ahora se puede
concluye que para un tiempo de depósito de 3
el equilibrio adsorción-desorción del Cu para el
calcular la cantidad adsorbida en función del
potencial de depósito, la Fig. 111.2.2.4 muestra los perfiles
potenciodinárnicos de electroxidación de los adátomos de Cu en medio
= 0.18 cm ) . H SO4 0.5 hi (solución libre de iones Cuz') sobre PdIA
A diferencia del P t , los perfiles de la electroxidacion de Cu sobre Pd
no muestran una estructura de picos claramente definida, sino hasta
que se ha alcanzado un cierto grado de recubrimiento (E > 0.31 VI o
hasta que aparece el depósito masivo.
2
2 geom
d
La representación gráfica de las cargas asociadas a los perfiles
potenciodinámicos (Fig. 111.2.2.4) es la isoterma electroquímica
mostrada en la Fig. 111.2.2.5.
Se puede observar que la cantidad adsorbida se incrementa de
forma permanente a medida que disminuye el potencial de. depósito. A
diferencia del P t , la isoterma electroquimica mostrada por el Pd no
presenta punto de inflexión alguno, sin embargo alcanza una meseta y
continúa s u crecimiento a medida que se disminuye el valor del
potencial de depósito, mostrando un crecimiento abrupto en ia carga de
electroxidación al iniciarse el depósito de Cu en multicapas.
Considerando que en la región de la meseta se ha alcanzado una

Fig. 111.2.2.3. Representación gráfica de l a carga de electroxiaacion
de Cu en funcion del tiempo de depósito para Pd
(.A 2 = 0.18 cm 1. E = 0.41 V y v = 50 rnViS.
geom d
96
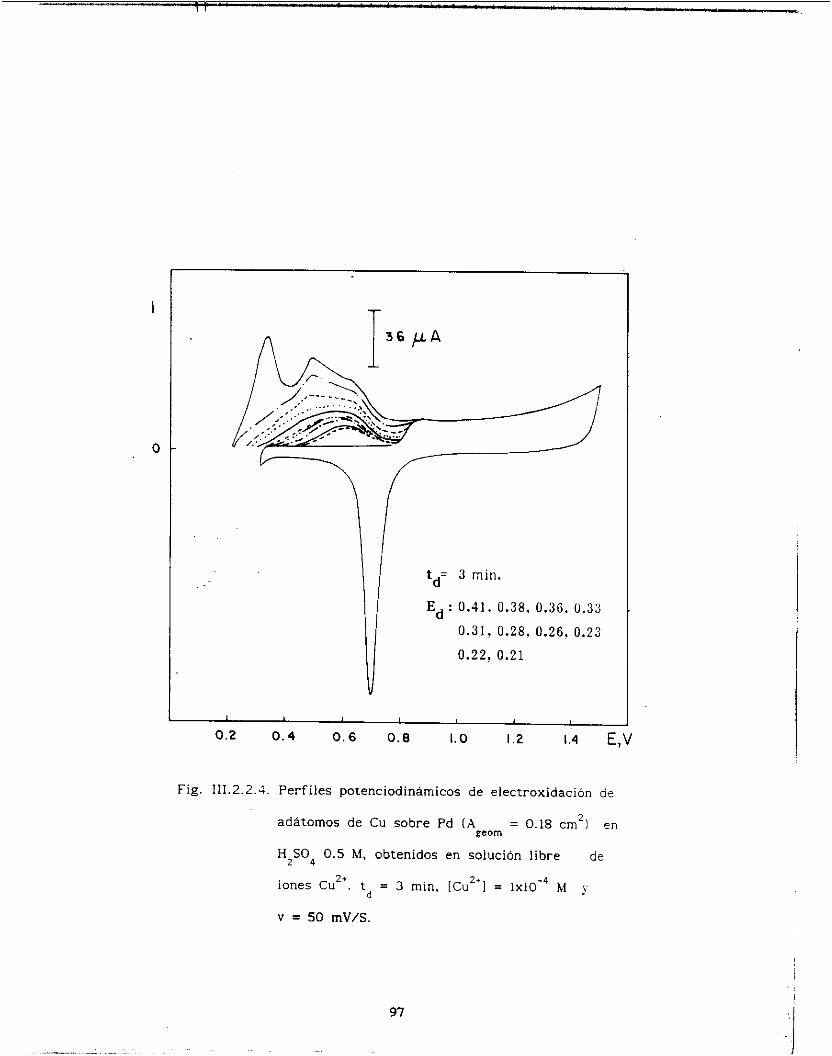
T
t = 3 min.
Ed : 0.41. 0.38. 0.36. 0.33
d
0.31, 0.28, 0.26. 0.23
0.22, 0.21
O. 4 0 .6 0.0 I. o I .2 1.4 E,V 0.2
Fig. 111.2.2.4. P e r f i l e s potenciodinámicos de electroxidación a,
= 0.18 cm I en 2 adátomos de Cu sobre Pd (A
H2SO4 0.5 M, obtenidos en solución l ibre
iones cu . t = 3 min, i ~ u * + ~ = 1x10'~ M
Y = 50 mV/C.
geom
de
2+ d
97

0.2 0.3
Fig. 111.2.2.5. lsoterma de adsorción de adátornos de Cu sobre
= 0.18 cm 2 ) en función del potencial geom
Pd (A
de depósito td = 3 rnin.

monocapa completa (relación Pd/Cu 1:l) podemos estimar el valor del
área real del electrodo a partir de este dato mediante el uso de la
siguiente expresión:
C "
Q m P C
(111.4)
donde:
Q C " es la carga a la monocapa (158 pC). rn
n es ei número de electrones intercambiados en el proceso (2e-1.
es el estandar determinado para un electrodo de Pd ';d
2 policristalino 11171. Su valor calculado es 204 pC/cm ,
Sustituyendo el valor numérico de cada uno de los parámetros tenemos:
A r = 0.39 cm f = 2 . 2 . 2
y de acuerdo con Quiroz y Coi. I71 que consideraron, área promedio por
átomo, .relación de radios atómicos y sitios superficiales totales
bloqueados, tenemos:
R /R = 1.07. lo que implica que la relación Pd/Cu es 1:I. Pd Cu
IIII.5)
Todos los parámetros conservan su significado (previamente descritos)
NT = N = 4.9 x 10 átomos de Cu descargados sobre Pd. S 14
C" M
El área real se obtiene multiplicando el número de átomos de Pd,
bloqueados por Cu, por el área promedio ocupada por un átomo
policristalino de Pd 11171, es decir:
14 . A r = 4.9 x 10 atornos Pd (8.21 x 10.'' cm2/átomos Pdl
2 A r = 0.4 cm f = 2.2
99

111.2.3. Efecto UPD de Cu sobre Au
La Fig. 111.2.3.1 muestra el perfil potenciodinámico ciclico del
depósito y eiectroxidación de Cu sobre un electrodo de Au en H-SO
0 . 5 M con presencia de iones Cu (zona de potencial < 0.6 i'!. En la
misma Fig. 111.2.3.1 se observa también la formación y reduccion de
especies oxigenadas sobre dicho electrodo (región de potencial ' 0.6V)
Esta curia se obtuvo con el propósito de observar la existencia del
efecto UPD de Cu sobre Au. Los picos I y I I corresponden áI depósito
de Cu a subpotencial, mientras que el pico I l l es el correspondiente
al Cu depositado en multicapas. El pico I\' está relacionado con el
depósito de Cu sobre el electrodo de .4u.
i 3
2+
La Fig. 111.2.3.2 muestra los perfiles potenciodinámicos, para la
electroxidación de adátomos de Cu. previamente depositados sobre ,\u, a
diferentes tiempos de depósito ( E = 0.46 \!l. A tiempos de deposito
superiores a 10 m i n . se observan disminuciones en la altura ur,l pico
de corriente de la curva de electroxidación de (:u. Este iFnónieno
parece estar relacionado a la difusión de Cu hacia el interior del
electrodo, ya que lo acompaña u n pico de corriente anodica que se
observa a potenciales más positivos (1.2 \ . I , y que crece con e : tiempo
de depósito.
d
Las cargas asociadas a la electroxidación de adatomos ae Cu en
función de\ tiempo de depósito, a un potencial f i j o , están mostradas
en la Fig. 111.2.3.3. AI inicio, la carga se incrementa rápidamente
(hasta los 60 SI. alcanza un valor máximo a los 3 min y permanece
constante durante u n tiempo aproximado de 7 min. En este intervaio oe
tiempo la cantidad adsorbida no es función del tiempo de depósito,
100

1 I
0.8 1.0 1.2 1.4 . 1.6 E,1 0.2 0.4 0.6
Fig. 111.2.3.1. Perfil potenciodinárnico mostrando el depósito y
electroxidación de adátomos de Cu sobre
policristalino (A
1 x
Au
= 0.19 c m 2 I . ICu 2. 1 = geom
M en HZS04 0.5 M y v = 50 mV/S.
101

I
4u
I I
0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 E,V
Fig. 111.2.3.2. Perfiles potenciodinámicos de electroxidación de
2 adátomos de Cu sobre Au (A = 0.19 cm 1, como
una función del tiempo de deposito.
fueron obtenidas en H SO 0.5 M libre de iones
Cu2*. v = 50 mV/S.
geom
Las curvas
2 4
102 ..u ...

2 0 40 6 0
b. S
Fig. 111.2.3.3. Representación g r á f i c a de la carga de
e lectroxidación de Cu sobre .4u en
función del tiempo de deposito.
E = 0.46 V. d
103

esto es, la adsorci6n-desorci6n de adatornos se encuentra en
equilibrio. Para potenciales inferiores este equilibrio deberá
alcanzarse en un tiempo menor, sin embargo, el tiempo de depósito
aplicado al electrodo de trabajo, para obtener la carga máxima para
los potenciales escogidos, fue siempre de 10 min; justo antes de
iniciarse el abatimiento de la carga (Fig. 111.2.3.31, evitando con
ello la aparición del segundo pico anódico en el perfil de
electroxidación de los adátomos de Cu.
La' Fig. 111.2.3.4 muestra los perfiles de electroxidación de Cu
sobre Au a diversos potenciales de depósito, desde 0 .53 V
hasta 0.21 \', a un mismo tiempo de depósito, t = 10 min. d
Las cargas asociadas a la electroxidación de adatomos d e Cu
depositados a diferentes potenciales, están representados graf icamente
en la isoterma electroquimica mostrada en l a Fig. 111.2.3.5.
La .¡soterma electroquimica de adsorción muestra que la carga se
incrementa . a medida que se disminuye el valor del potencial de
C " depósito, alcanza una meseta ( Q = 206 pCl y finalmente presenta un
incremento brusco de la carga (depósito de Cu en multicapas). .A 0.35 V
la isoterma electroquimica registra un punto de inflexion y está
localizado a 213 de Ocu . Este punt o está relacionado con la
aparición del pico 11 de la curva de electroxidación (Fig. 111.2.3.1).
correspondiente a la región de subpotencial para adátomos de Cu sobre
Au.
rn
rn
2+ Considerando que la descarga de los iones Cu es total y que
cada ión Cu se adsorbe sobre un átomo del sustrato intercambiando
2e ,en el proceso, tendremos que el área real del electrodo estará
dada por la siguiente expresión :
z+
-
104
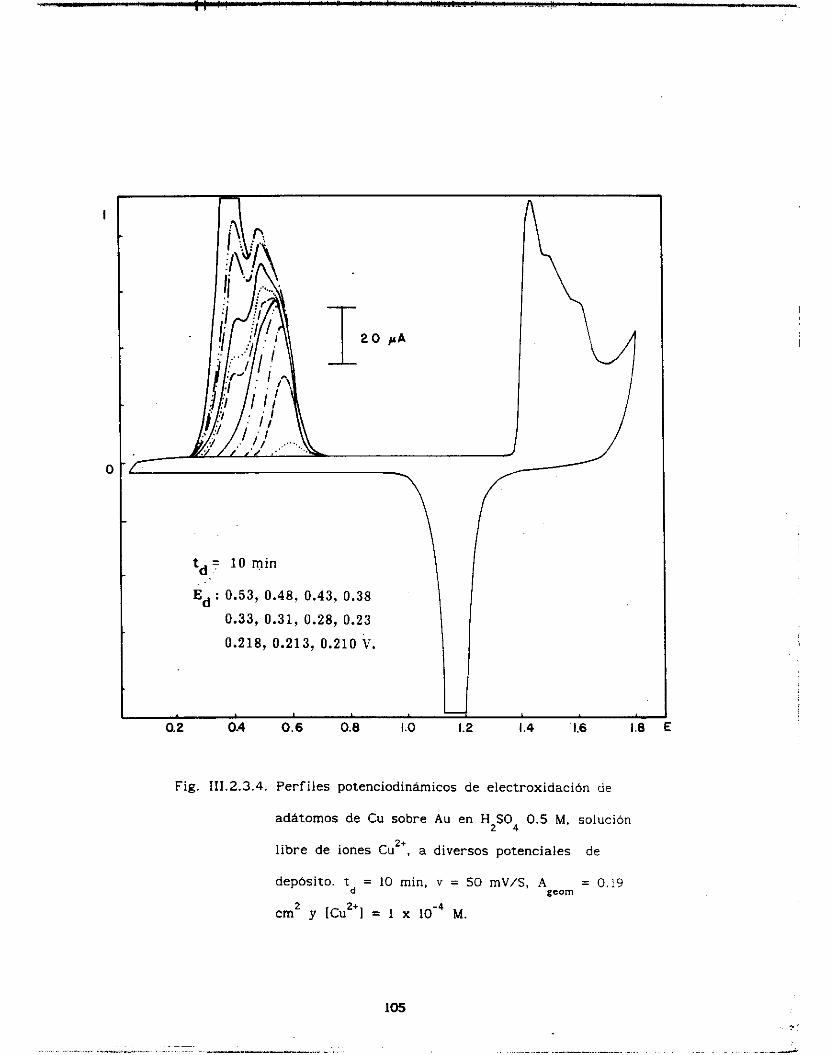
I
O
o. 2 0.4 , 0.6 , 0.8 , 1.0 ,:I, 1.2 1.4 1.6 , 18 ,
t = 10 rnin
Ed : 0.53, 0.48. 0 .43, 0.38
0.33, 0.31, 0.28, 0.23
0.218, 0.213, 0.210 V.
d
Fig. 111.2.3.4. Perf ¡les potenciodinárnicos de electroxidación de
adátomos de Cu sobre Au en H SO4 0.5 M, solución
libre de iones Cuz*. a diversos potenciales
depósito. t = 10 rnin, v = 50 rnV/S, A = 0.19 d gcom
2
de
2 cm y ic2'1 = I x M.

30 (
2 O(
IO'
t t
I I '. O. 3 , 0.4 , \ 4 0.5 Edep. V/EI
Fig. 111.2.3.5. Isoterma de adsorción de adátomos de Cu sobre
2 = 0.19 cm ) en función del potencial peom
Au ( A
de dep6sito. td = 10 min.
106

Todos los parámetros conservan su significado (previamente
definidos), excepto el valor de carga por cm para una superficie
policristalina de Au 11171,
2
2 = 184 pC/cm QZ"
C " Es necesario hacer una corrección al valor d e Qm por la
diferencia de radios entre el sustrato y el adsorbato [1071, según la
siguiente expresión:
Q C U = 206 IRS /RAdIZ pic lII1.71 rn
sustituyendo los valores de los radios atómicos del sustrato y del
adsorbato 11.41 y 1.25 A respectivamente) tendremos que Q = 260.8
pC y los valores de Ar y f obtenidos son:
o C "
rn
2 Ar = 0.71 cm f = 3.14
Finalmente. por el método alternativo utilizado por Quiroz y Col.
I71 tenemos:
íII1.81
cuyos parámetros ya fueron definidos anteriormente. El número de
sitios activos disponibles del metal son:
NM = 8.14 x IOL4 átomos de Au
y sabiendo que el área ocupada por un átomo poiicristalino de Au
I1171 es 9.06 x IO"b cm , tendremos que el Ar y f estimados son: 2
2 A r = 0.74 cm f = 3.89

111.3 ESTIMACION DE A r DE Pd Y Au UTILIZANDO EL VALOR DE f
OBTENIDO POR DEPOSITO DE Cu A SUBPOTENCIAL.
111.3.1. Paladio
Bajo la consideración de que un electrodo estabilizado (perfil
potenciodinámico invariante) es utilizado para efectuar el estudio de
los procesos de electrosorción de oxígeno o el depósito de adátomos de
Cu a subpotencial, y suponiendo que el factor de rugosidad no cambia,
se puede utilizar la Ec. reportada por Dickertmann y Col. [IISI para
evaluar el grado de recubrimiento de la especie adsorbida.
C
‘o ’ Ageom e =
O
M n . f . Q
l111.9)
donde:
0 es el grado de recubrimiento de la especie adsorbida.
QO
Ageom
n es el número de electrones intercambiados en el proceso.
f es el factor de rugosidad del electrodo, determinado por
es la carga involucrada en el proceso de electrodesorción.
es el área geométrica del electrodo utilizado.
depósito de Cu a subpoencial.
es el estandar convencional de carga para el electrodo utilizado Q:
Lli71.
Una vez conocido el valor de éste parámetro, se puede hacer una
adecuada estimación del área real del electrodo a partir de l a carga
determinada del perfil potenciodinámico catódico de ia curva patrón
del Pd, mediante la siguiente expresión:
108

c Q, / e A r =
n . Q i , (111.101
Para el electrodo de Pd utilizado en los procesos de
= 0.31 cm , se efectúa la estimación electrosorción de oxígeno, A
de e al mismo valor de E que el de la curva patrón utilizada en el
depósito de adátomos de Cu d subpotencial (A = 0.18 cm 1. Asi a
= 1.56 V la carga asociada al proceso de electrodesorción de
especies' oxigenadas es de 442.2 pC (para el electrodo de mayor área).
Además se cuenta con los siguientes datos:
n = 2e
f = 2.2
2 geom
LS 2
georn
- (Considerando que la especie formada es PdO).
(evaluado por efecto UPDI.
O 2 = 204 pC / cm (estandar convencional para Pd policristalinol.
QPd
A s i el valor de I3 calculado es 1.59 y. por lo tanto, el valor
2 estimado- de. Ar es 0.39 cm .
Volviendo a . la Fig. I11.1.2.3 y considerando que la carga del
perfil catódico refle ja realmente la especie adsorbida sobre el
electrodo, se tiene lo siguiente:
AI primer cambio de pendiente (punto 6 de la curva anódica) el
valor de la carga catódica es 146.8 pC. Si la especie formada es PdOH,
entonces se puede escribir el siguiente equilibrio:
Pd + H20 <I PdOH + H' + le-
146.8 pC
1 ( 204 rrC/crn'l A r =
2 A r = 0.72 cm f = 2.32
AI segundo cambio de pendiente (punto 11 de la curva
lII1.11)
nódic I el
109

valor de la carga cat6dica es 299.2 IC. Si consideramos ahora que la
especie formada es PdO, tendremos:
PdOH ,- PdO + Hi + le- (111.12)
o bién la reacción global
Pd + HZO +- PdO + 2H' + 2e- (111.13)
2 9 9 . 2 pC
24204 pC/cmz 1 A r =
f = 2.35 2 A r = 0.73 cm
Los valores de f así determinados son muy similares al ootenido
por depósito de Cu a subpotencial y por tanto, la especie que se forma
sobre el Pd es Pdo. en una relación estequiométrica 1:l.
II1.3.2. Oro
Del mismo modo, se puede estimar el valor de Ar para el electrodo
de A u utilizado. Considerando que la especie adsorbida en la region de
¡a meseta observada en la isoterma elctroquimica es A u 2 O j como ya fue
mencionado con anterioridad 11161, y que el valor de la carga catodica
involucrada en dicha región es 358 pC, se puede evaluar 0 utilizando
la Ec.111.9, esto es:
358 pC í 0 . 1 9 cmz
3 ( 3 . 7 4 ) 1 8 4 pC / cmZ) = 0.91 _-- e =
Este valor implica que a E = 1.71 V la superficie del electrodo
aun no se encuentra saturada por la esprcie adsorbida (Au O 1, por lo
tanto habrá que corregir el valor de Qo al efectuar la estimacion del
valor del área real para aquellos electrodos constituidos de este
LS
2 3 C
110

metal. Así para el electrodo de Au utilizado en el curso de esta
investigación el valor estimado de A r por adsorción de especies
oxigenadas, fue:
358 FC / 0.91
3(184 FC / cm 7 A r =
2 A r = 0.71 cm
111
f = 3.74

C O N C L U S I O N E S
a) Platino
PARAMETROS DE REFERENCIA. -
= 0.25 cmL
= 210 pC/cm lie-)
Ageom
QP t
1. Area geométrica:
2. Estandar convencioval de carga: O 2
ELECTROADSORCION DE HIDROGENO.
C 1. Carga experimental: Q, = 61.4 pic
C 2. Carga correg ida a l a saturación:
3. Area real expuesta:
4. Factor de rugosidad:
QH,s= 7 3 . 1 p C
A r l P t H ) = 0 .35 cmz
f = 1 . 4 P t
ELECTRODESORCION DE OXIGENO.
C
O I , Carga experimental : Q = 141.4 pl3
2. Area real expuesta: A r ( P t 0 ) = 0 . 3 4 crn'.
3. Factor de rugosidad: f p t = 1 . 4
DEPOSITO DE COBRE A SUBPOTENCIAL.
1 . Carga experimental a la meseta:
2. Area real expuesta: .Ar(PtCuJ = 0 . 3 4 c m ' ~
Qcu = 141 pic rn
3. Factor de rugosidad: f = 1 . 4 P t
CORRELACION O/Pt
Estos resutados muestran, de manera contundente, que el depósito
de metales a subpotencial. Cu en este caso en particular, se presenta
como un método alternativo adecuado para la estimación de areas reales
de electrodos de metales nobles. Los cálculos efectuados tanto para la
electrodesorción de oxígeno como para la electroxidación de Cu,
confirman plenamente que la estequiometria Pt:O a nivel de la monocapa
es 1:l. es decir, bajo la formación de la especie oxigenada PtO.
112

b) Paladio
PARAMETROS DE REFERENCIA.
I . Area geométrica:
2. Estandar convencional de carga:
ELECTRODESORCION DE OXIGENO.
i . Carga experimental :
2. Carga c o r r e g i d a a 8 = 1:
3. Area real expuesta :
4. Factor de rugosidad:
DEPOSITO DE COBRE A SUBPOTENCIAL.
1. Carga experimental a la meseta:
2. Area real expuesta :
= 0.18 cm'
= 204 pC/cm
Ageom
QP d
O 2
C Qo = 253.1 pC
C Qo,s = 159.2 p C
Ar(Pd0) = 0 . 3 9 cm'
f P d = 2.17
Q c U = 158 pC rn
AríPdCu) = 0 . 3 9 cmz
= 2.17 P d
3. Factor de rugosidad:
CORRELACION O/Pd
Estos resultados confirman plenamente que la especie oxigenada
que se. forma sobre la superficie del Pd, a nivel de la monocapa es
Pdo. Por lo tanto, el depósito de Cu a subpotencial puede servir como
referencia interna en la estimación del área real de electrodos
constituidos de Pd policristalino.

c) Oro
PARAMETROS DE REFERENCIA.
2 = 0.19 c m Ageom
QA u
1. Area geométrica:
2. Estandar convencinal de carga:
ELECTRODESORCION DE OXIGENO.
O = 184 pC/cm'íle-)
1. Carga experimental :
2. Carga correg i d a a e = 1:
3. Area real expuesta : Ar(Au O = 0.71 cm
QZ = 358 pC
Q , , = 393.4 pC C
2 2 3
4. Factor de rugosidad: f =3.74 Aiu
DEPOSITO DE COBRE A SUBPOTENCIAL.
1. Carga experimental a la meseta: Qc" = 205 pC
2. Carga c o r r e g i d a ( D i f . r a d i o s ) : Q c " = 261 pC
3. Area . real expues ta : A r ( A u C u ) . = 0.71 cm
m
m . c 2
4. Factor de rugosidad: f = 3 . 7 4 A U
CORRELACION OíAu.
Los resultados obtenidos para Au policristalino, muestran que la
especie oxigenada que se forma a nivel de la monocapa es AuZ03, y no
AuO como ha sido reportado en la literatura. Por consiguiente, el
depósito de Cu a subpotencial puede ser utilizado como referencia
interna en la estimación del área real de electrodos constituidos de
Au policristalino.
114
. .

B I B L I O G R A F I A
1. R. Woods, J. Electroanal. Chem., 49, 217 (19741.
2. D.A.J. Rand and R. Woods, J. Electroanal. Chem.. 35, 209 (1972).
3. R. Woods, Chemisorption at electrodes, in Electroanalytical
Chemistry (A.J. Bard, Ed.), vol. 9. Marcel Dekker, N.Y. 1976.
4. R. Parsons, Surf. Sci.. 101, 316 (1980).
5. C.M. Ferro, A.J. Calandra and A.J. Arvía, J. Electroanal. Chem.,
65, 953 (19751.
5. D.M. Kolb, Physical and Electrochemical Properties of Metallic
Substrates, in Advances in Electrochemistry and Electrochemical
Engineering IH. Gerischer and C.W. Tobias, Eds.), vol. 11, P.
125. John Wiley, N . i . 1978.
7. M.A. Quiroz, Contribucion al Estudio Electroqulmico de Rutenio
y Platino/Rutenio, Tesis Doctoral, Universidad Autónoma
Metropolitana-lztapalapa 1985.
S. M.A. . Ouiroz, Y, Meas, E. Larny-Pitara and J . Barbier. J .
Electroanal. Chern.. 157, 65 (19831.
9. R.R. Adzic, Israel J . Chem, 18, 166 (1979).
10. G. Lafranconi. F. Mazza, E. Sivieri and S. Torchio. Corros. Sci..
18, 517 (1978):
11. M. Breiter, J . Electrochem. Soc., 114, 1261 (1967). '
12. hi. Peuckert. F.P. Coenen and H.P. Bonze], Electrochirn. Acta, 29,
1305 (1989).
13. D. Gilroy and B.E. Conway, Can. J. Chem., 46, 875 (1975).
14. M Breiter. in Trans. Symp. Electrode Processes. 1959 (E.
Yeager, Ed.), J. Wiley. N.Y. P. 307 (1961).
115

15. M. Brei te r , H. Karnrnermaier and C.A. Knorr , Z. Elektrochern.. 60, 37
(1956 1.
16. J . Honz and L. Nernec, Collect. Czech. Chem Commun.. 34, 2030
( 1969).
17. S. Gilrnan, J . Electroanal. Chem., 7, 382 (1964).
18. B.E. Conway, J. Electroanal. Chem., 8, 486 (1964).
19. T. HepeI and F.H. Pollak, J. Electrochem. Soc.. 131. 2094 (1984).
20. S. Hadzi-Jordanov, H. Angerstein-Kozlowska, hi. Vokovic and B.E.
Conway, J . Phys. Chem., 81, 2271 (1977).
21. R. Payne, J . Electroanal. Chem., 19, 1 (1968).
22. T. Biegler. D.A.J. Rand and R. Woods, J . Electroanal. Chem.. 29,
269 (19711.
23. S. Gilman. J . Phys. Chem., 71, 4330 119671.
24. D.A.J . Rand and R. Woods, J . Electroanal . Chem. 31, 29 (19711.
25. S. Gilman, J . Phys. Chem.. 71, 4339 (1967).
26. A. Capon and R. Parsons, J. Electroanal . Chem.. 39, 275 11972).
27. A.N. Frumkin. in Advances in Electrochemistry and Electrochemical
Engineering. (P. Delahay, Ed.) . vol. 3, cap. 5. Interscience.
N . Y . (1963).
28. J.E. Benson and Boudart. J . Catal . ,1 . 701 119651.
29. J.F. Connolly, R.J. Flannery and C. Aronovits, J . Electrochem.
Soc., 113. 577 (1966).
30. S. GiIrnan, Electrochim. Acta, 9, 1025 11964).
31. S.B. Brurnrner, J . Phys. Chem.. 69, 562 11965).
32. S. Gilman, in Electroanal. Chern., vol. 2, P. 111, A.J. Bard, Ed.
Arnold Press . London 11967).
116

33. V.S. Bagotzky, A.M. Sukundin and E.K. Tesseva. Electrochim. Acta,
21, 29 (1976).
34. M.A. Quiroz, I. Gonzalez. H. Vargas and Y. Meas, Electrochim.
Acta, 31. 503 (1986).
35. M.A. Quiroz. L. Calgado and Y. Meas, Electrochim. Acta, 33. 435
(1988).
36. D.M. Michell. D.A.J. Rarid and R. Woods, J. Electroanal. Chem.,
89, I1 (1978).
37. M.. Breiter, C.A. Knorr and V. Volkl, Z. Elektrochem., 59, 681
(19551.
38. F.G. Will and C.A. Knorr, 2. Elektrochern., 64, 270 (1960).
39. H.A. Laitinen and M.S. Chao, J . Electrochem. Soc., 105, 726
( 1961 I .
40. S. Cchuldiner and T.B. Warner, J . Electrochem. Soc., 112, 212
(1965).
41. J.P. Hoare,. in The Electrochemistry of Oxigen, Interscience,
N.Y.. chap. 11, (1968).
12. J.P. Hoare, Advan. Electrochem. Electrochem. Eng.. 6, 201 11967).
43. M. Becker and M. Breiter. 2. Elektrochem., 60, 1080 (19561.
44. J.S. Mayell and S.H. Langer. J. Electrochem. Soc., 111, 438
( 1964 I .
45. C. Cchuldiner and R.M. Roe, J. Electrochem. Coc., 110, 332
( 1963).
46. H.A. Laitinen and C.G. Enke, J. Electrochem. Soc., 107. 773
( 1960 1.
47. M. Rosen and Schuldiner. J. Electrochem. Soc., 117, 35 (19701.
117

~. .. , , - ~ . .
48. T.B. Warner and S. Schuldiner, J. Electrochem. Soc., 112, 853
(1965).
49. T. Biegler and R. Woods,J. Electroanal . Chem., 20. 73 (1969).
50. A. Damjanovic, A. Dey and J.O'M. Bockris, Electrochim Acta, 11,
791 (1966).
51. K.J. Vetter and J.W. Schultze. J. Electroanal. Chem., 34, 131
(19721.
s 52. H. Angerstein-Kozlowska, B.E.- Conway and W.B.A. Sharp, J .
Electroanal. Chem., 43, 9 (1973).
53. V.S. Bagotzky, i u . B . Vassiliev, 0.A Khazova and S.S. Sedova.
Electrochim. Acta, 16, 913 (1971).
54. B.V. Tilak, H. Angerstein-Kozlowska and B.E. Conway, J.
Electroanal. Chem., 48. 1 (1973).
55. M.L.B. Rao, A . Damjanovic and J.O'M Bockris, J. Phys. Chern.. 67,
2508 (1963).
56. N .R . d e Tacconi, A.J. Calandra and A.J. Arvía, J . Electroanal.
Chern.. 51, 25 (1974).
57. R.Kh. Burshtein, M.R. Tarasevich and V.S. Vilinskaya,
Eiektrokhim.. 6, 1497 11970).
58. R . K h . Burshtein, V.S. Vilinskaya and M.R. Tarasevich,
Elektrokhim., 6, 1861 (1970).
59. V.A. Gromyko, Elektrokhim., 7 , 885 (1971).
60. G.M. Schnid and N. Hackerman; J . Electrochem. Soc., 108. 310
(1961 1.
61. C.M. Ferro, A.J. Calandra and A.J Arvia, J . Electroanal . Chem.,
65, 963 (1975).

62. D.E. Icenhower, H.B. Urbach and J.H. Harrison, J. Electrochem.
Soc.. 117, 1500 (1970).
63. R.A. Bonewitz and G.M. Schmid, J. Electrochem. Soc., 117, 1367
(1970).
64. G. Deborin and B.V. Ershler, Acta Physicochim., 59, 347 (1940).
65. S.B. Brummer and A.C. Makrides, J. Electrochem. Soc.. 111. 1122
11964).
66. G. Gruneberg, Electrochim. Acta, 10, 339 (1965).
67. T. Takamura. K. Takamura, W . Nippe and E. Yeager, J. Electrochem.
Soc., 117. 626 (19701.
68. A.A. Michri, A.G. Pshenichnikov and R.Kh. Burshtein, Elektrokhim,
S. 364 11972).
69. M.D. Gol’dshtein. Ts.1. Zalkind and V.1. Veseiovskii,
Elektrokhim, S. 606 11972).
70. C.E.S. El’ Wakkad and A.M. Shams El Din, J. Chem. Coc.. 3098
(1954).
71. G.hl. Schrnid and R.N. O’Brien. J . Electrochem. Coc., 11, 832
(1964).
72. R.C. Sirohi and M.A. Genshaw, J. Electrochem. Soc., 116. 910
(1969).
73. K. Ogura. C . Haruyama and K. Nagasaki, J . Electrochem. Soc., 11S,
531 11971).
71. D.M. McArthur, J. Electrochem. Soc., 119, 672 (1972).
75. A. Hamelin and M. Sotto, C.R. Acad, Sci, Ser. C., 271, 609 (1970).
76. M. Pourbaix, Atlas d’Equilibres Electrochimiques, Gauthiers
Villars. Paris 11963).
119

77. J.W. Schuitze and K.J. Vetter, Ber Bunsenges Phys. Chem. 75. 470
(19711.
78. J.D.E. Mclntyre and D.M. Kolb, Syrnp. Fa raday Soc., 4, 99 (19701
79. K.J. Cathro and D.F.A. Koch., J. Electrochem. Soc.. 111, 1416
(19641.
SO. B.D. Cahan, J . Horkans and E. Yeager, Syrnp. Faraday Soc., J. 36
( 1970).
S1. D.M. Kolb and J.D.E. Mclntyre, Sur f . Sci., 28, 321 (19711.
S2. R.M. Lazorenko-Manevich and T.N. Ctoyanovskaya, Elektrokhirn., S,
982 (19721.
83. W.E. Reid and J. Kruger, Nature , 203, 402 (19641.
84. J.L. Ord and D.L. De Smet, J.Electrochern. Soc., 118, 206 11971).
S5. i u .Ya . Vinnikov,V.A. Chepelin and V.1. Veselovskii, Elektrokhirn.,
8, 1229 (19721.
86. D.A.J. Rand and R. Woods, J . Electroanal . Chern., 44, S3 í!9731.
S7. R . h l . . lshikawa and A.T. Hubbard. J . Electroanal . Cheni.. 6 2 . 317
(19761.
SS. E. Yeager , W.E. O’Grady, M.Y.C. Woo and P. Hagans, J
Electrochem. Soc.. 125, 348 (1978).
S9. P.N. Ross, J r . . J . Electroanal Chern., 76, 139 (19771.
90. C.M. Elliot and R.W. hlurray, Anal. Chem.. 4s. 1247 (19761.
91. K . Kinoshita and P. S tonehar t , in Modern Aspects of
Electrochemistry IJ.O’h!. Bockris and B.E. Conway, Eds. J, No. 12,
183,Plenurn, N.Y. (19771.
92. C.L. Scortichini and C.N. Reylley, J . Cat . , 79, 138 (1983).
93. G.V. Hevesy. Physik. Z., 13, 715 (19121.
94. K.F. Herzfeld, Physik, Z., 14. 29 (1913).
120
_. . ~

95. L.B. Rogers, D.P. Krause, J.C. Griess. Jr. and D.B. Ehrlinger, J.
Electrochem. Soc., 95, 33 (1949).
96. R.C. De Geiso and L.B. Rogers, J. Electrochem. Soc.. 106, 433
( 1959).
97. T. Mills and G.C. Willis, J . Electrochem. Soc., 100, 452 (1953).
98. M.M. Nicholson, J . Am. Chem. Soc.. 79, 7 (1957).
99. G.I. Finch, H. Wilman and L. Yang, Disc. Faraday Soc., 1 , 144
(1947).
100. B.J: Bowles, Electrochim. Acta, 15, 589 (1970).
IO!. B.J. Bowles, Electrochim. Acta, 15, 737 (19701.
107. C.H. Cadle and S. Bruckenstein, Anal. Chem.. 43, 1858 (1971;.
103. M.Z. Hassan, D.F. Untereker and Bruckenstein, J . Eiectroanal.
Chem.. 42. 161 (1973).
104. C . H . Cadle and C. Bruckenstein, Anal. Chem., 41. 1993 (1972,.
105. !d Breiter, Electrochim. Acta, 7. 25 (1962).
106. F.C. W~ i l l , J . Electrochem. Coc., 112. 451 í1965).
107. B.J. Bowley, Nature, 212, 1456 (1966).
10s. L.B. Rogers and A.F. Ctehney. J. Electrochem. Soc.. 95, 25
(1949).
100. E. Schmidt and N. Wuthrich, J . Electroanal. Chem.. Z S , 349
í 19701.
110. C. Cwathirajan and C. Bruckenstein, Elctrochim. Acta. 2S, 865
(1983).
11;. D.M. Kolb, hi. Przasnyski and H. Gerischer, J . Electroanal. Chem.,
5-1, 25 (1974).
113. B.E. Conway, Theory and Principles of Electrode Processes, Ronald
Press, N.Y. (1965).
i
121

113. S. Cwathirajan, H. Mizota and S. Bruckenstein, J. Phys. Chem..
86, 2480 (19821.
114. B.E. Conway and H. Angerstein-Kozlowzka, J . Electroanal. Chem.,
113, 63 (19801.
115. H. Bort, K. Juttner. W.J. Lorenz and E. Schmidt, J. Electroanal.
Chem., 9 0 , 143 (19781.
116. C.M. Ferro, A.J. Calandra and A.J. Arvía. J . Electroanal. Chern..
59, 239 (1975).
117. H. Kubicka, J . Catal. 12, 223 [1968).
115. D. Dickertrnann. J .W. Schultze and K.J. Vetter, J Electroanal.
Chern., 55, 429 (19741.
119. K. Kinoshita and P.N. Ross, J . Electroanal. Chem., 7s . 313
I19771





























