Embed Size (px)
Citation preview

Advanced Therapies in Europe
1

~ 500 clinical trials using ATMPs in EU
~ 270 ATMP classifications
~ 250 scientific advice requests 18 MAAs reviewed
9 ATMPs approved
3 withdrawn 1 Suspended
5 licensed ATMPs
ATMPs in Europe (2009- 2017)
Market 2

Challenges in ATMP’s availability to Patients
2009-2017 (8 years)
q 18 MAAs reviewed or
under review q 9 products approved q 3 withdrawn, 1 suspended,
1 negative opinion q 5 licensed ATMPs q Approx. 111 patients
treated
Learning from previous MAAs
ATMPs are complex products to develop, manufacture, evaluate and make available to patients
Regulatory
Quality
Non-clinical
Clinical
HTA, Price and reimbursement
Patient access
3

Challenges - areas
Quality Non-
clinical
Clinical Other
4

European Commission – EMA Action Plan on ATMPs
• Published 20 October 2017
• Builds on the issues identified at the Multi-stakeholder workshop
• Concrete actions to improve the regulatory framework for ATMPs (within the existing legal framework)
• Goal: increase the opportunity for patients to be treated with novel therapies and to promote innovation, investment and competitiveness of the EU biotech sector
5
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2017/10/WC500237029.pdf

Scientific Advice for ATMPs
• 250 SA procedures started – CAT involved (routinely) in all SA for ATMPs
• Increase in SA’s for ATMPs over period 2012 - 2016 (especially for GTMP)
SA requests until July 2017 6
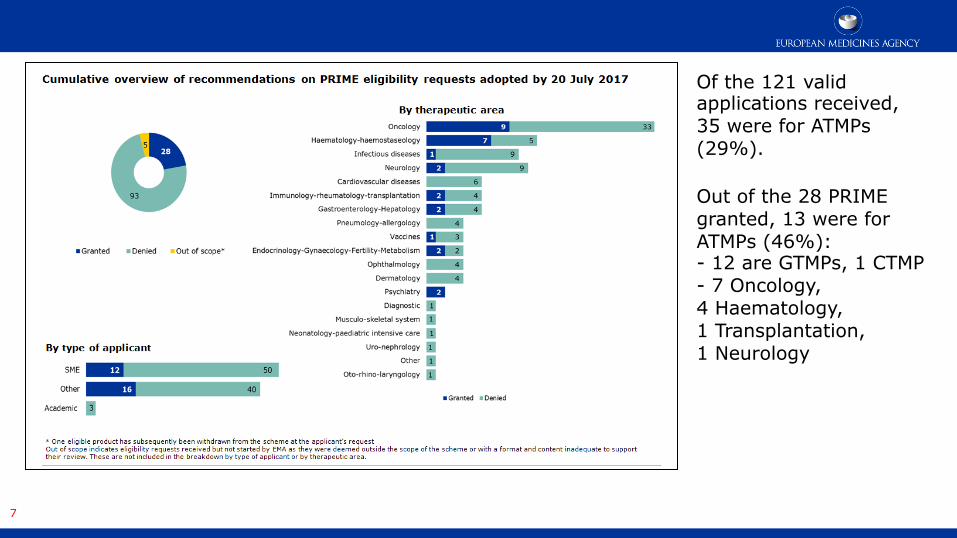
Of the 121 valid applications received, 35 were for ATMPs (29%). Out of the 28 PRIME granted, 13 were for ATMPs (46%): - 12 are GTMPs, 1 CTMP - 7 Oncology, 4 Haematology, 1 Transplantation, 1 Neurology
7

ATMPs in clinical trials
Commercial vs non-profit sponsor
Stage of development
8

EMA expert meeting on genome editing 18 October
The European Medicines Agency (EMA) convened an expert meeting on genome editing technologies. The three sessions focussed on:
1/ Genome editing and technologies: state of the art and future outlook
2/ Application of genome editing by Academia
3/ Case studies using genome editing technologies
Outcome: Discussion on scientific and regulatory opportunities and challenges of medicinal product
development using genome editing technologies. Report envisaged in 2018.
Conclusion: An active research field with promising pipeline Limited CT developments worldwide Some challenges in developing these products to be addressed
What we can and will do: • Continue to listen to developers • Support development by fostering early dialogue • Cooperation to facilitate marketing authorisation and
access to patients, at EU and international level
9

10

Wide engagement with stakeholders
Regulatory and Scientific Process
Streamline EMA internal regulatory processes + Promote use of early access tools + Harmonise and guide on application documents & national requirements +
Raise awareness of regulatory process, early engagement and collaboration + Develop different models of collaboration and engagement with the wider community of ATMP stakeholders
Take home messages
Committee Advanced Therapies (CAT)
Academia
Patients
Payers
INVESTORS
11

Thank you for your attention
Ana Hidalgo-Simon [email protected] European Medicines Agency 30 Churchill Place • Canary Wharf • London E14 5EU • United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact
Follow us on @EMA_News
With thanks to: Veronika Jekerle, Patrick Celis and Falk Ehmann

Back up slides

• Donation, procurement and testing according to Dir. 2004/23/EC (2006/86/EC)
• Variability of the starting material (autologous & allogeneic)
• GMP requirements for early manufacturing steps (e.g. cell extraction from
biopsies)
• Excipients/reagents qualified for clinical use (e.g. growth factors, cytokines):
EDQM General monograph 5.2.12 Raw materials for the production of cell-
based and gene therapy medicinal products
• Characterisation methods and potency
• Comparability
• Medical device or structural component & administration device
Quality challenges
14

Non-clinical challenges
• Suitability of animal models (species specificity)
• Predictability for safety (i.e. long-term safety)
• Homologous model / disease model (efficacy/ proof of concept)
• Large animal model necessary? (surgical procedure, tissue regeneration)
• GLP environment (for toxicity studies)
• Biodistribution & methods for tracking cells (systemic applications)
15

Clinical challenges
• Long-term efficacy
• Safety
Ø Clinical control available/feasible/ethical
Ø Standardised collection & delivery procedure
Ø Dose available & justified
Ø Duration of follow-up (efficacy/safety)
Ø Traceability
16

Additional elements
• Growing innovation ecosystems in the EU
• Need to raise awareness of regulatory process among a diverse set of stakeholders, sometimes outside the classic sphere of influence of regulators
• Early engagement and collaboration needed: with developers and users
• Patient access: develop different models for reimbursement and payment
17

Purpose Aims to formalise and structure the collaboration between the Agency and Academia in the wider context of the European Regulatory System for Medicines
Implementation Action plan, monitoring and reporting
Scope Framework will cover collaboration between Agency and academia, covering areas of common interest in relation to medicines for human and veterinary use. Queries relating to a specific product and/or regulatory procedure fall outside the scope of this framework
§ Mapping of academic entities with an interest in the regulatory activities
§ Evolution of available expertise to keep pace with advances in scientific knowledge
§ Identifying opportunities to promote research and knowledge generation
§ Promoting and reinforcing dialogue through effective communication
§ Monitoring progress and output of the cooperation with academia
Key elements
Framework of collaboration with academia
18

International dimension
Until recently:
• Product development was very regional, although the need for alignment of the scientific and regulatory requirements was identified early on (ATMP cluster started in 2008, IPRF cell therapy and gene therapy groups since 2011 / 2012 respectively)
Situation today:
• Most Regulatory Authorities have by now a clear legal framework for the approval of Gene and Cell-based Medicines
• Limited approvals of ATMPs (worldwide), but active pipeline (development of new products, clinical trials)
Already here:
• CAR-T’s: worldwide development, approvals in US, ongoing evaluations in EU, submissions to other authorities
• Gene therapy products: (ophthalmologic indication, orphan) approved US • Tissue engineered product: e.g. MACI approved in US and Spherox approved in the EU • IND / CTA (Scientific advice) for same products
Direction of travel: • Strengthening of the EMA-FDA interactions 19

Several elements under construction
l GMP for ATMP –published in November 2017 https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf
l Application of GLP through development – clarification published in CTFA and EMA websites in 2017
l ATIMP Guideline - drafting ongoing l Revised S&E follow-up / Risk Management Plan guideline –
drafting ongoing l Q&A for minimally manipulated ATMPs - published l Revised definition for orphan designation – published l Revised GM Cell Guideline – Concept paper consultation closed l Comparability of ATMPs (Q&A) – to start 2018
20





























