Embed Size (px)
Citation preview



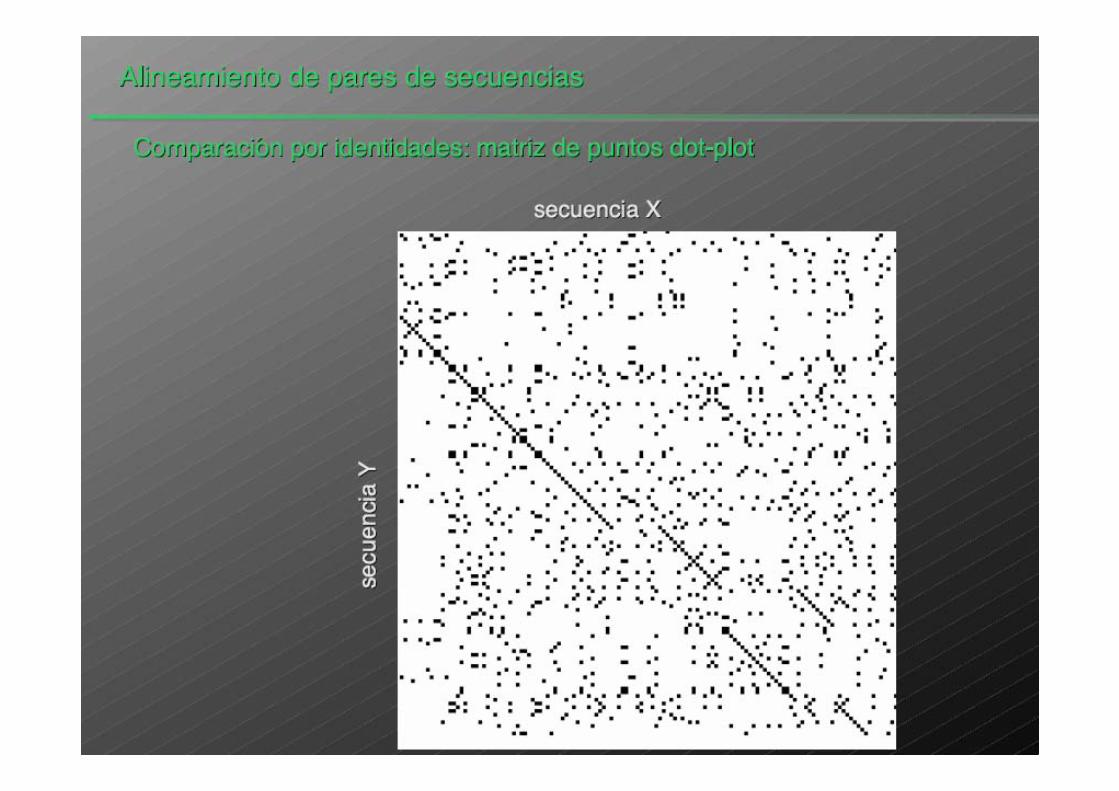

DOT PLOT: VISUALIZACIÓN DE LA SIMILITUDENTRE DOS SECUENCIAS

COMPARACION DE DOS ATPASAS DE PECES


El DOT PLOT permite una visualización rápidade la similitud entre dos secuencias
Inconvenientes:
No identifica directamente los fragmentos similares
No permite cuantificar el grado de similitud
Ventajas:
Nos muestra inversiones y estructuras repetidas
Computacionalmente eficiente


ALINEAMIENTO:
ALINEAR DOS SECUENCIAS CONSISTE EN IDENTIFICAR
LAS CORRESPODENCIAS RESIDUO-RESIDUO ENTRE
AMBAS
EL ALINEAMIENTO ES LA HERRAMIENTA BÁSICA
EN BIOINFORMATICA

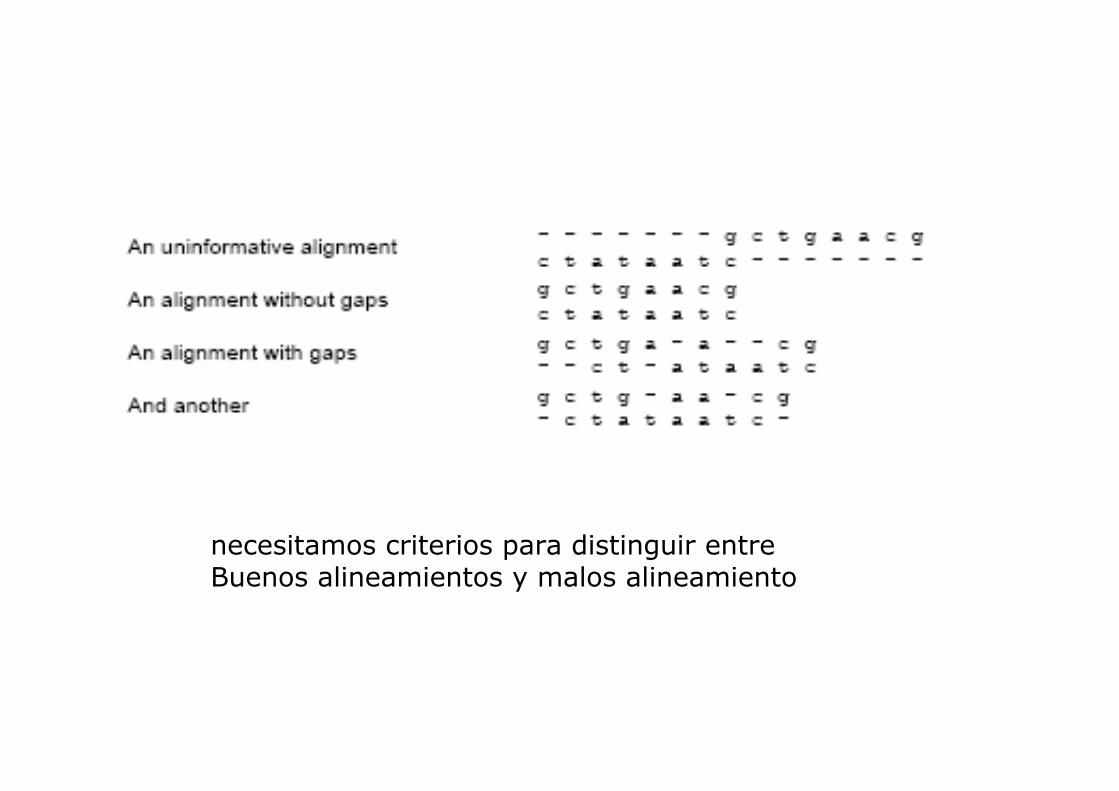
necesitamos criterios para distinguir entreBuenos alineamientos y malos alineamiento

Distancia de Hamming: dadas dos cadenas deigual longitud, se define como el número deposiciones no coincidentes (mismatches)
Distancia se Levenshtein: dadas dos cadenas, no necesariamente de la misma longitud, se define como el número de operaciones de edición(inserción, deleción, alteración) necesarias paraconvertir una en otra


Scores
Las distancias son medidas de la disimilaridadentre secuencias
El score es una medida de la similaridad entre secuencias
El score tiene introducir una penalización por introducir espacios (gap penalty)
La puntuación asignada a residuos coincidentesno tiene por qué ser igual en todos los casos, ydicha puntuación debería tener un sentido biológico








¿Cuál es la mejor ruta entre Malmo y Tromso?
Algoritmos de programacióndinámica
global
local




















Para un score x, la probabilidad de observar unscore ≥x es:P(score≥x)=1-exp(-ke–λx)
Relación distante10-5 -10-1
relacionadas10-50 -10-10
casi idénticas10-100 -10-50
Probablemente no relacionadas
P≤10-1
exactoP≤10-100
Distribución de valor extremo

Z-score = (score-media)/dev estandar
Probablemente significativo
Z≥5
Similitud observada equivalente a la aleatoria
Z=0

Indistinguible del azarE > 1
No descartar homología
0.02<E <1
Probablementehomólogas
E≤0.02
E-value de un alineamiento encontrado en una base de datos,es el número esperado de secuencias que por azar dar un scoreigual o mayor al obtenido. Resulta de multiplicar P por el tamaño de la base de datos


Zona difusa18-25%
Indistinguible del azar<18%
Alta probabilidad deEstructura y funciónsimilar
45-25%
Muy probablementeIdéntica función
<45%
Interpretación del nivel de similaridad entredos protéinas

Significación de alineamientos
Técnica de Montecarlo:Barajar secuencias>Repetir alineamiento>Apuntar score

Interpretación de la homología
• Regla de Doolitle para proteínas(Doolitle’s rule of thumb)
Si dos secuencias son >100 aa y su identidad es:
– > 25%, probablemente están relacionadas
– 15-25 % (twilight zone), podrían estar relacionadas, pero habría que demostrarlo con otras pruebas
– <15 %, probablemente no están relacionadas

Interpretación de la homología
• Los alineamientos son fáciles con secuencias muy parecidas
• Por debajo de un umbral, los alineamientos pueden no tener un significado biológico
– Twilight zone
• Por debajo de este umbral hay que disponer de más información (estructura, etc.)

Filtros

Se aplican filtros para evitar el efecto de:
Regiones de baja complejidad (filtros SEG y DUST)
Secuencias repetidas (filtros específicos)
Regiones coil-coil (filtro COILS)

Conceptos básicos• Homología (homology):
• Concepto cualitativo
• Implica la existencia de una relación evolutiva
• Similitud (similarity):• Concepto cuantitativo
• Concepto o cantidad que expresa cómo de parecida es una cosa a otra:
• Identidad (DNA y proteínas)
• Conservación funcional (prot.)
Reeck et al. 1987 "Homology" in proteins and nucleic acids : a terminology muddle and a way out of it. Cell 50: 667



Principales algoritmos para alineamientos• Para alineamientos múltiples
• Métodos progresivos– Alinean las secuencias más parecidas y luego van incorporando el
resto poco a poco – Más rápidos, aunque menos precisos que en prog. dinámica– Producen un buen alineamiento, pero que no es necesariamente el
óptimo
– Ej. Clustal, PILEUP, T-Coffee
• Métodos iterativos– Rápidos y precisos– Construyen un alineamiento inicial que se revisa progresivamente para
conseguir mejorar la puntuación
– Ej. Multalin, Dialign

Clustal
• Dos versiones:– Clustal W: en servidor web– Clustal X: con interfaz gráfica para Windows
• Método:– Se alinean separadamente todos los pares de secuencias y calcula una
matriz de distancias que indica la divergencia entre cada par de secuencias
– A partir de la matriz de distancias se calcula un “arbol guía” (Neighbor-Joining)
– Las secuencias se alinean progresivamente siguiendo el orden de lasramas del arbol guía
• Características:– Fortalezas:
• Alta velocidad– Problemas:
• Alineamiento depende de las secuencias iniciales• No se puede corregir en el alineamiento la adición inicial de gaps

Operational options
Output options
Input options, matrix choice, gap opening penalty
Gap information,output tree type
File input in GCG, FASTA, EMBL, GenBank, Phylip, or several other formats

Secuencias no alineadas

Misma región, después del alineamiento

Multalin
• Disponible en servidores Web; también versión local, pero sin interfaz gráfica
• Método:– Alinea progresivamente, primero por pares y luego grupos de
secuencias, y calcula sus puntuaciones– Recalcula las puntuaciones de los alineamientos de cada dos
secuencias durante la producción del alineamiento– Los alineamientos recalculados se incorporan al alineamiento global– El programa para cuando nos e mejoran las puntuaciones
• Características:– Puede producir mejores alineamientos que algoritmos progresivos– Problemas:
• Requiere mucha computación

Editores de alineamientos
• Sirven para
– Editar manualmente un alineamiento
– importar o exportar datos
– Mejorar la presentación visual de los alineamientos




AGTVATVSCAGTSATHACIGRCARGSCIGEMARLACIGDYARWSC
.........
IGTVARVSC <= Ejemplo de secuencia consenso
Secuencias consenso

Ejemplo:AGTVATVSC AGTSATHAC IGRCARGSC IGEMARLAC IGDYARWSC
......... IGTVARVSC <= Ejemplo de secuencia consenso
podríamos generar el siguiente patrón:[AI]-G-X-X-A-[RT]-[SA]-C
Expresiones regulares
prosite


Perfiles
Permiten mejorar alinen secuencias distantes
Position-specific scoring matrix

Dominios yFamilias de proteínas

REDES NEURONALES





Modelos ocultos de MarkovHMM


PSI-BLAST






























