Embed Size (px)
Citation preview

Amino Acid Capture by Aqueous Interfaces. Implications forBiological UptakeMarilia T. C. Martins-Costa†,‡ and Manuel F. Ruiz-Lopez*,†,‡
†SRSMC, UMR 7565, University of Lorraine, BP 70239, 54506, Vandoeuvre-les-Nancy, France‡SRSMC, UMR 7565, CNRS, BP 70239, 54506, Vandoeuvre-les-Nancy, France
*S Supporting Information
ABSTRACT: The interactions of natural amino acids with water−hydrophobic interfaces arecentral to the control of key biological processes, such as passive transport, and to the overallstructure and stability of membrane proteins. We still have a very poor knowledge of theseinteractions, and our aim in this work is to investigate the thermochemistry and dynamicsproperties of simple aliphatic amino acids (glycine and valine) across a water−organicinterface. The study has been carried out by means of Born−Oppenheimer moleculardynamics simulations focusing on the role that the hydrophobicity of the side chain has on thephase transfer mechanism of the amino acid. Data for the energetics of the uptake processeshave been reported, and it is expected that the reported results will be helpful in the design offuture experiments with systems of biological relevance. We have shown that neutral tautomersexhibit a noticeable affinity for the interface that increases with increasing hydrophobicity ofthe side chain. Moreover, the zwitterionic form of valine (but not that of glycine) does alsoexhibit a significant affinity for the interface. An important finding is that the neutral and zwitterionic tautomers are roughlyisoergonic in the organic layer close to the interface. This result suggests a two-step mechanism for the water-to-organic phasetransfer that involves neutralization of a partially hydrated zwitterion in the organic layer prior to uptake into the bulk. Thoughthe mechanisms for glycine and valine are similar, the predicted energetics and dynamics for the first step display noteworthydifferences that should be measurable and may have important biological implications.
■ INTRODUCTION
The role of water−hydrophobic interfaces on atmospheric,environmental, and biological chemistry is a poorly understood,but potentially very important, topic. On the one hand, it isnow widely recognized that many polar molecules and ions arestabilized at the air−water or water−organic interfaces (see ref1 and references therein) as a result of a subtle combination ofelectrostatic-polarization and entropic effects. On the otherhand, interfacial solvation should modify the molecularproperties and the reactivity of the adsorbed solute, but incontrast to solvation in bulk phase, prediction of such effects isnot straightforward, even qualitatively.The traditional view of solvation at the interface is quite
simple: it assumes that solvation effects are roughlyintermediate between the two bulk phases. This intuitive ideahas been supported by studies on solvatochromic compoundsat the interface using second-harmonic generation spectroscopythat have led to the definition of a generalized interfacialpolarity scale.2,3 Accordingly, the polarity of a liquid interface isthe arithmetic average of the polarity of the two constituentbulk phases (for instance, the effective polarity of the air/waterinterface should be close to that of bulk butyl ether3). However,there is now compelling evidence that chemistry at the interfacecan be quite different from chemistry in the two bulk phases.4,5
Indeed, recent elaborated SFG (sum-frequency generation)experiments6 support the fact that similar molecules canundergo different polarization at the interface depending on
their relative orientation with respect to the plane of thesurface. This finding has been further supported by numericalsimulations carried out by us,5,7 according to which the reactionfield potential created by the polarized interface may besignificantly larger (in absolute value) than the potential createdby the bulk solvents and its sign depends on the H-bondingdonor or acceptor character of the solute. Hence, the activemolecular orbitals of species adsorbed at the interface (theHOMO and LUMO, in particular) may be either stabilized ordestabilized with respect to the two bulk phases, leading tointerface-enhanced or interface-inhibited chemical or photo-chemical reactions, as schematized in Figure 1.The study of interface solvation effects is, on the other hand,
a crucial step to understanding the physicochemical mecha-nisms involved in phase transfer molecular processes. Suchmechanisms have biological relevance because water−hydro-phobic interfaces represent a suitable and simple model thatallows getting useful insights on how cell membranes work.Passive diffusion of amino acids,8,9 despite its secondary rolewhen compared to assisted transport by carrier proteins, is anemblematic example. This question has stimulated experimentalresearch on the permeability properties of the lipid bilayer10,11
for establishing the background levels of amino acid transport
Received: August 21, 2013Revised: September 18, 2013Published: September 18, 2013
Article
pubs.acs.org/JPCB
© 2013 American Chemical Society 12469 dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−12474

allowed by the membrane and for discussing solute permeationin natural situations prior to the evolution of transport proteins(or where transport proteins are absent). The mechanism oftransfer is, however, difficult to unravel10,11 because amino acidsexist mainly as zwitterions in aqueous environments andbecause their side chains may contain ionizable groups.Moreover, a full understanding of the transfer process wouldneed to take into account the acido-basic properties of thewater surface, which is still a subject of controversy (see, forinstance, refs 12−22), although recent experimental work23 hassupported the original prediction12 of hydronium somewhatfavoring the water surface.Some theoretical investigations have been devoted to study
the phase transfer of natural amino acids. Most of them havefocused on their side chains and the uptake into a lipidbilayer24−26 or across the air−water interface,27 which is animportant question for understanding the thermodynamics ofmembrane protein structure and stability.28 Amino acids withN-terminal acetyl and C-terminal N-methyl amide groups at thewater−hexane interface were also studied as a model toinvestigate the effects of the interface on peptide folding.29 In arecent work, we have described the hydration shell of glycine inthe vicinity of a water−organic interface,30 whereas otherauthors have focused on the orientational ordering of asparigineand tryptophan at the air−water interface as well as the protontransfer mechanism in the case of tryptophan.31,32
However, the thermodynamics of amino acid uptakeprocesses has not been established yet, and the aim of thepresent investigation was to throw some light on this importanttopic. The present study focuses on aliphatic amino acids, theirenergetics across water−hydrophobic interfaces, and themechanism of transfer. It has been carried out using moleculardynamics (MD) simulation techniques developed in our group,which combine quantum chemistry and molecular mechanicsmethods for describing, respectively, the solute and the solvent.In this way, the possibility for proton transfer between the acidand amino groups of the amino acid induced by the chemicalenvironment is explicitly taken into account. We report andcompare free energy profiles for glycine (Gly) and valine (Val)crossing a model water−carbon tetrachloride interface. Finally,we discuss the biological relevance of the results, namely, thepossible mechanisms of transfer and the role of hydrophobicside chains.
■ METHODS
Molecular Dynamics Simulations. They were carried outin the NVT ensemble at T = 298 K using the Nose−Hooverthermostat.33,34 The system was composed of one amino acidmolecule, 1000 water molecules, and 220 CCl4 molecules. Weassumed a combined quantum mechanics and molecularmechanics (QM/MM) force field in the Born−Oppenheimerapproximation. The amino acid molecule (the solute) wasdescribed quantum mechanically at the density functionaltheory level, whereas solvent molecules were described using aclassical force field. The method allows for electrostaticembedding; that is, the Hamiltonian of the solute includesthe electrostatic interaction with the charges of the solventmolecules that were described using the force field proposedbefore for the H2O/CCl4 interface.
35 Specifically, for water, weemployed the flexible SPC force field.36 For the calculation ofsolute−solvent nonelectrostatic interactions, the van der Waalsparameters for the QM atoms were taken from the OPLS forcefield.37 The box size is 24 Å × 24 Å × 114 Å corresponding todensities of 1.00 and 1.57 for water and CCl4, respectively. Weapply periodic boundary conditions in the three directions. Acutoff of 12 Å was used for the interactions of both QM andMM systems with their environment. The time step was 0.5 fs.After equilibration, the width of the organic slab isapproximately 64 Å. We assumed a neutral pH in water(which is close to the isoelectric point of Gly and Val) so thatthe amino acids bear no net charge. The simulations were doneusing Gaussian 0338 for the QM calculations, TINKER39 for theMD simulations, and the program developed by us.40
Free Energy Calculations. Free energy has been calculatedusing the umbrella sampling41 and WHAM42,43 methodstogether with the dual level approach proposed recently.44 Inthis approach, the umbrella sampling is obtained from QM/MM MD simulations using a low-level QM method (Hartree−Fock, 3-21G basis set). Free energy perturbation theory is thenused to obtain the free energy profile at the B3LYP/6-31G(d)level. The reaction coordinate R is taken as the distancebetween the center of mass of the solute and the center of massof the organic solvent. The reaction coordinate was varied bysteps of 0.25 Å, and the bias potential force constant is k = 10kcal/mol/Å2. After thermalization, the trajectory was carriedout for 50 ps at each point of the reaction coordinate.
■ RESULTS AND DISCUSSION
Amino acids in bulk water exists mainly in ionized form orzwitterions (Zw). In the case of Gly and Val, the relativestability with respect to the nonionized or neutral form hasbeen experimentally (Gly)45 or theoretically (Val)46 deter-mined to be −7.3 kcal/mol. In gas phase and hydrophobicmedia, only the neutral form is stable. The relative stabilities atwater−hydrophobic interfaces are unknown, and estimatingthese values is one of the goals of our study.Previous works have shown that neutral amino acids may
exist in different conformations in gas phase and in solution,and Chart 1 shows the most relevant ones for Gly.47−49
Structures NI and NII are almost isoenergetic, both in the gasphase and in water solution,49 and one may anticipate similarbehavior in apolar or low polar media (see below). In watersolution, however, the lifetime of NII is too small, as itspontaneously leads to the more stable Zw tautomer (through avery small energy barrier) so that this structure should play aminor role in the measured thermodynamic properties.50
Figure 1. Scheme illustrating the interaction of two molecules AH (aproton donor) and BX (a proton acceptor) with a water−hydrophobicinterface. The molecular orbitals of the former are destabilized, whilethe molecular orbitals of the latter are stabilized. Hence, the propertiesof a hypothetical AH + BX reaction at the interface can be quitedifferent from either bulk phase.5,7
The Journal of Physical Chemistry B Article
dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−1247412470
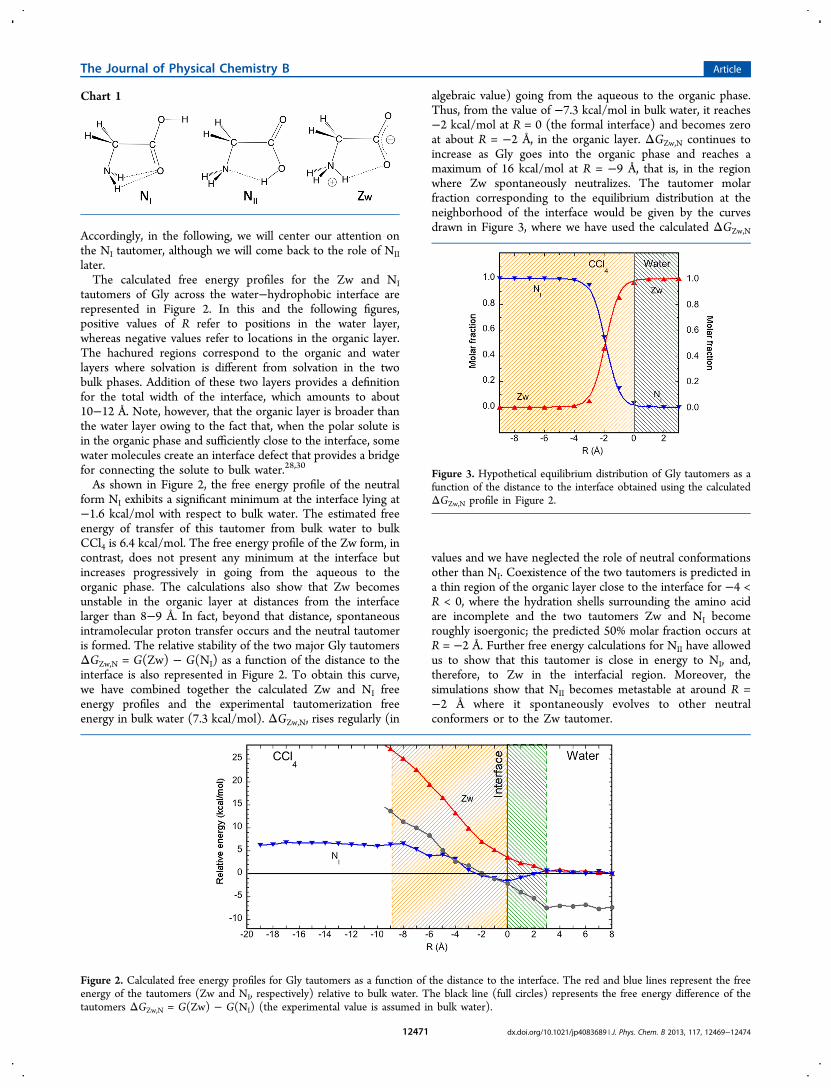
Accordingly, in the following, we will center our attention onthe NI tautomer, although we will come back to the role of NIIlater.The calculated free energy profiles for the Zw and NI
tautomers of Gly across the water−hydrophobic interface arerepresented in Figure 2. In this and the following figures,positive values of R refer to positions in the water layer,whereas negative values refer to locations in the organic layer.The hachured regions correspond to the organic and waterlayers where solvation is different from solvation in the twobulk phases. Addition of these two layers provides a definitionfor the total width of the interface, which amounts to about10−12 Å. Note, however, that the organic layer is broader thanthe water layer owing to the fact that, when the polar solute isin the organic phase and sufficiently close to the interface, somewater molecules create an interface defect that provides a bridgefor connecting the solute to bulk water.28,30
As shown in Figure 2, the free energy profile of the neutralform NI exhibits a significant minimum at the interface lying at−1.6 kcal/mol with respect to bulk water. The estimated freeenergy of transfer of this tautomer from bulk water to bulkCCl4 is 6.4 kcal/mol. The free energy profile of the Zw form, incontrast, does not present any minimum at the interface butincreases progressively in going from the aqueous to theorganic phase. The calculations also show that Zw becomesunstable in the organic layer at distances from the interfacelarger than 8−9 Å. In fact, beyond that distance, spontaneousintramolecular proton transfer occurs and the neutral tautomeris formed. The relative stability of the two major Gly tautomersΔGZw,N = G(Zw) − G(NI) as a function of the distance to theinterface is also represented in Figure 2. To obtain this curve,we have combined together the calculated Zw and NI freeenergy profiles and the experimental tautomerization freeenergy in bulk water (7.3 kcal/mol). ΔGZw,N, rises regularly (in
algebraic value) going from the aqueous to the organic phase.Thus, from the value of −7.3 kcal/mol in bulk water, it reaches−2 kcal/mol at R = 0 (the formal interface) and becomes zeroat about R = −2 Å, in the organic layer. ΔGZw,N continues toincrease as Gly goes into the organic phase and reaches amaximum of 16 kcal/mol at R = −9 Å, that is, in the regionwhere Zw spontaneously neutralizes. The tautomer molarfraction corresponding to the equilibrium distribution at theneighborhood of the interface would be given by the curvesdrawn in Figure 3, where we have used the calculated ΔGZw,N
values and we have neglected the role of neutral conformationsother than NI. Coexistence of the two tautomers is predicted ina thin region of the organic layer close to the interface for −4 <R < 0, where the hydration shells surrounding the amino acidare incomplete and the two tautomers Zw and NI becomeroughly isoergonic; the predicted 50% molar fraction occurs atR = −2 Å. Further free energy calculations for NII have allowedus to show that this tautomer is close in energy to NI, and,therefore, to Zw in the interfacial region. Moreover, thesimulations show that NII becomes metastable at around R =−2 Å where it spontaneously evolves to other neutralconformers or to the Zw tautomer.
Chart 1
Figure 2. Calculated free energy profiles for Gly tautomers as a function of the distance to the interface. The red and blue lines represent the freeenergy of the tautomers (Zw and NI, respectively) relative to bulk water. The black line (full circles) represents the free energy difference of thetautomers ΔGZw,N = G(Zw) − G(NI) (the experimental value is assumed in bulk water).
Figure 3. Hypothetical equilibrium distribution of Gly tautomers as afunction of the distance to the interface obtained using the calculatedΔGZw,N profile in Figure 2.
The Journal of Physical Chemistry B Article
dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−1247412471
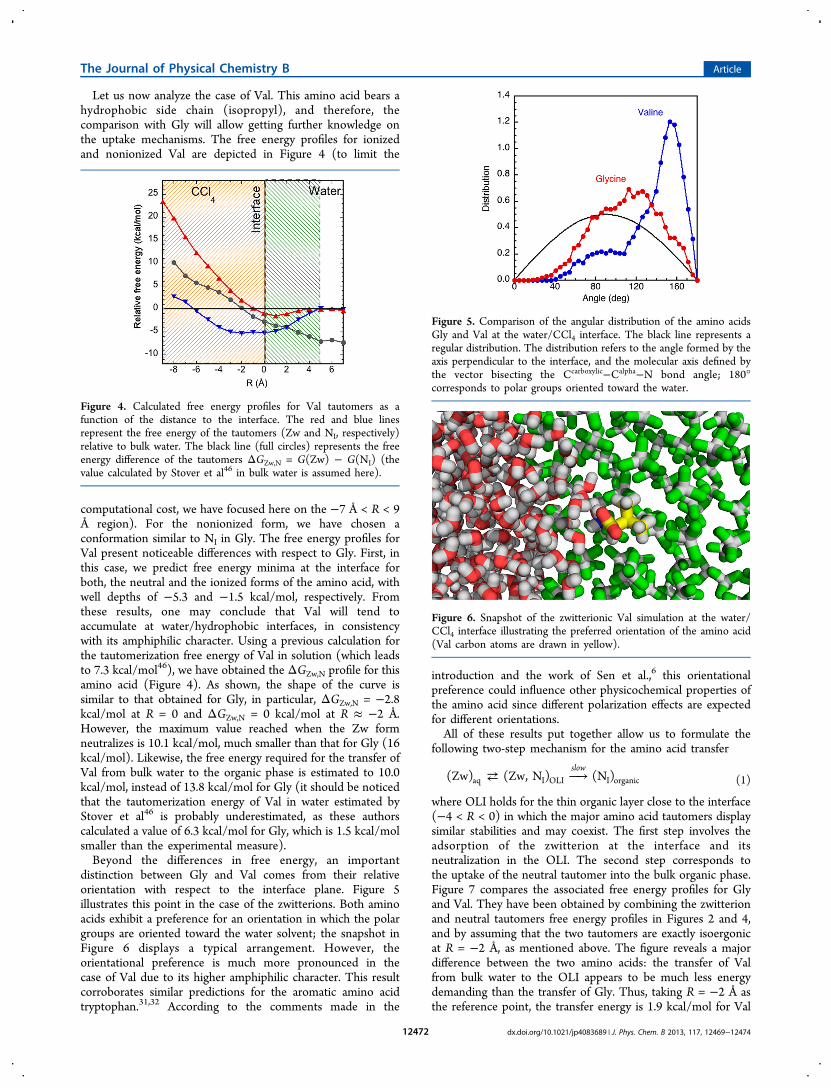
Let us now analyze the case of Val. This amino acid bears ahydrophobic side chain (isopropyl), and therefore, thecomparison with Gly will allow getting further knowledge onthe uptake mechanisms. The free energy profiles for ionizedand nonionized Val are depicted in Figure 4 (to limit the
computational cost, we have focused here on the −7 Å < R < 9Å region). For the nonionized form, we have chosen aconformation similar to NI in Gly. The free energy profiles forVal present noticeable differences with respect to Gly. First, inthis case, we predict free energy minima at the interface forboth, the neutral and the ionized forms of the amino acid, withwell depths of −5.3 and −1.5 kcal/mol, respectively. Fromthese results, one may conclude that Val will tend toaccumulate at water/hydrophobic interfaces, in consistencywith its amphiphilic character. Using a previous calculation forthe tautomerization free energy of Val in solution (which leadsto 7.3 kcal/mol46), we have obtained the ΔGZw,N profile for thisamino acid (Figure 4). As shown, the shape of the curve issimilar to that obtained for Gly, in particular, ΔGZw,N = −2.8kcal/mol at R = 0 and ΔGZw,N = 0 kcal/mol at R ≈ −2 Å.However, the maximum value reached when the Zw formneutralizes is 10.1 kcal/mol, much smaller than that for Gly (16kcal/mol). Likewise, the free energy required for the transfer ofVal from bulk water to the organic phase is estimated to 10.0kcal/mol, instead of 13.8 kcal/mol for Gly (it should be noticedthat the tautomerization energy of Val in water estimated byStover et al46 is probably underestimated, as these authorscalculated a value of 6.3 kcal/mol for Gly, which is 1.5 kcal/molsmaller than the experimental measure).Beyond the differences in free energy, an important
distinction between Gly and Val comes from their relativeorientation with respect to the interface plane. Figure 5illustrates this point in the case of the zwitterions. Both aminoacids exhibit a preference for an orientation in which the polargroups are oriented toward the water solvent; the snapshot inFigure 6 displays a typical arrangement. However, theorientational preference is much more pronounced in thecase of Val due to its higher amphiphilic character. This resultcorroborates similar predictions for the aromatic amino acidtryptophan.31,32 According to the comments made in the
introduction and the work of Sen et al.,6 this orientationalpreference could influence other physicochemical properties ofthe amino acid since different polarization effects are expectedfor different orientations.All of these results put together allow us to formulate the
following two-step mechanism for the amino acid transfer
⇄ ⎯ →⎯⎯(Zw) (Zw, N ) (N )slow
aq I OLI I organic (1)
where OLI holds for the thin organic layer close to the interface(−4 < R < 0) in which the major amino acid tautomers displaysimilar stabilities and may coexist. The first step involves theadsorption of the zwitterion at the interface and itsneutralization in the OLI. The second step corresponds tothe uptake of the neutral tautomer into the bulk organic phase.Figure 7 compares the associated free energy profiles for Glyand Val. They have been obtained by combining the zwitterionand neutral tautomers free energy profiles in Figures 2 and 4,and by assuming that the two tautomers are exactly isoergonicat R = −2 Å, as mentioned above. The figure reveals a majordifference between the two amino acids: the transfer of Valfrom bulk water to the OLI appears to be much less energydemanding than the transfer of Gly. Thus, taking R = −2 Å asthe reference point, the transfer energy is 1.9 kcal/mol for Val
Figure 4. Calculated free energy profiles for Val tautomers as afunction of the distance to the interface. The red and blue linesrepresent the free energy of the tautomers (Zw and NI, respectively)relative to bulk water. The black line (full circles) represents the freeenergy difference of the tautomers ΔGZw,N = G(Zw) − G(NI) (thevalue calculated by Stover et al46 in bulk water is assumed here).
Figure 5. Comparison of the angular distribution of the amino acidsGly and Val at the water/CCl4 interface. The black line represents aregular distribution. The distribution refers to the angle formed by theaxis perpendicular to the interface, and the molecular axis defined bythe vector bisecting the Ccarboxylic−Calpha−N bond angle; 180°corresponds to polar groups oriented toward the water.
Figure 6. Snapshot of the zwitterionic Val simulation at the water/CCl4 interface illustrating the preferred orientation of the amino acid(Val carbon atoms are drawn in yellow).
The Journal of Physical Chemistry B Article
dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−1247412472

and 7.0 kcal/mol for Gly. In contrast, in the second step of theprocess, the uptake into the bulk organic phase from the OLI,the energetics are similar for the two amino acids: about 8 kcal/mol for Val and 7 kcal/mol for Gly (see Figure 7; the curves forVal and Gly are roughly parallel in the region −8 Å < R < −2Å). Overall, a lower energy is required for the water-to-organicphase transfer in the case of the amphiphilic amino acid Val byroughly 4 kcal/mol.Finally, one might wonder how the mechanism in eq 1 would
compare with the alternative process: neutralization in the bulkaqueous phase, followed by phase transfer of the neutral form.Actually, the experimentally determined free energy ofactivation for the neutralization process of glycine in bulkwater at 298 K is quite large, 14.6 kcal/mol,51 and the activationenergy for the corresponding backward process can be deducedto be about only 7 kcal/mol.50 In other words, the neutraltautomer should not survive long enough to reach the interfaceand the kinetics of the overall phase transfer process should becontrolled by diffusion of the amino acid across the interface.The intramolecular proton transfer will readily take place in theOLI induced by the lower polarity of the environment.
■ CONCLUSIONSIn the past, the understanding of amino acid uptakemechanisms has been limited by the lack of experimentalthermochemical data. In this study, an elaborated theoreticalapproach has allowed us to obtain, for the first time, a detailedfree energy scheme for the water-to-organic phase transfer ofthe aliphatic amino acids Gly and Val and to analyze the role ofthe hydrophobicity of the side chain. In the energeticscalculations, two major results have been obtained. First, ithas been shown that the neutral and zwitterionic tautomers ofboth Gly and Val become almost isoergonic in the organic layerclose to the interface (OLI), where solute hydration is stronglyreduced with respect to bulk water. Second, the simulationsconfirm the existence of free energy minima at the interface forthe neutral forms, and also for the zwitterionic form in the caseof Val. Using these data, a two-step uptake mechanism has beenproposed consisting of the adsorption−neutralization of thezwitterion at the OLI, followed by the uptake of the nonionizedform into the organic phase. Overall, the calculated freeenergies of water-to-organic phase transfer amount to 13.8 and
10.0 kcal/mol for Gly and Val, respectively. The easier transferof valine with respect to glycine is obviously due to the higherhydrophobicity of its side chain, but the simulationsdemonstrate its decisive role in facilitating the adsorption atthe interface during the first step of the uptake process.
■ ASSOCIATED CONTENT
*S Supporting InformationComplete ref 19. This material is available free of charge via theInternet at http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
This work was supported by the French ANR (09-BLAN-0180-01), the CNRS, and the University of Lorraine. The authorsthank the French computational facility CINES (projectlct2550).
■ ABBREVIATIONS
Gly, glycine; Val, valine; MD, molecular dynamics; QM/MM,quantum mechanics and molecular mechanics; OLI, organiclayer close to the interface
■ REFERENCES(1) Petersen, P. B.; Saykally, R. J. On the Nature of Ions at the LiquidWater Surface. Annu. Rev. Phys. Chem. 2006, 57, 333−364.(2) Wang, H. F.; Borguet, E.; Eisenthal, K. B. Polarity of LiquidInterfaces by Second Harmonic Generation Spectroscopy. J. Phys.Chem. A 1997, 101, 713−718.(3) Wang, H. F.; Borguet, E.; Eisenthal, K. B. Generalized InterfacePolarity Scale Based on Second Harmonic Spectroscopy. J. Phys. Chem.B 1998, 102, 4927−4932.(4) Donaldson, D. J.; Valsaraj, K. T. Adsorption and Reaction ofTrace Gas-Phase Organic Compounds on Atmospheric Water FilmSurfaces: A Critical Review. Environ. Sci. Technol. 2010, 44, 865−873.(5) Martins-Costa, M. T. C.; Anglada, J. M.; Francisco, J. S.; Ruiz-Lopez, M. F. Reactivity of Volatile Organic Compounds at the Surfaceof a Water Droplet. J. Am. Chem. Soc. 2012, 134, 11821−11827.(6) Sen, S.; Yamaguchi, S.; Tahara, T. Different Molecules ExperienceDifferent Polarities at the Air/Water Interface. Angew. Chem., Int. Ed.2009, 48, 6439−6442.(7) Martins-Costa, M. T. C.; Anglada, J. M.; Francisco, J. S.; Ruiz-Lopez, M. Reactivity of Atmospherically Relevant Small Radicals at theAir−Water Interface. Angew. Chem., Int. Ed. 2012, 51, 5413−5417.(8) Ellory, J. C.; Jones, S. E. M.; Young, J. D. Glycine Transport inHuman Erythrocytes. J. Physiol. 1981, 320, 403−422.(9) Tunnicliff, G. Amino Acid Transport by Human ErythrocyteMembranes. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 1994,108, 471−478.(10) Chakrabarti, A. C.; Deamer, D. W. Permeation of Membranesby The Neutral Form of Amino Acids and Peptides: Relevance to theOrigin of Peptide Translocation. J. Mol. Evol. 1994, 39, 1−5.(11) Chakrabarti, A. C. Permeability of Membranes to Amino Acidsand Modified Amino Acids: Mechanisms Involved in Translocation.Amino Acids 1994, 6, 213−229.(12) Petersen, M. K.; Iyengar, S. S.; Day, T. J. F.; Voth, G. A. TheHydrated Proton at the Water Liquid/Vapor Interface. J. Phys. Chem. B2004, 108, 14804−14806.
Figure 7. Schematic mechanism and calculated free energy profiles forGly and Val uptake processes. OLI stands for the organic layer close tothe interface (−4 < R < 0) where neutralization of the zwitterionsoccurs.
The Journal of Physical Chemistry B Article
dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−1247412473

(13) Petersen, P. B.; Saykally, R. J. Evidence for an EnhancedHydronium Concentration at the Liquid Water Surface. J. Phys. Chem.B 2005, 109, 7976−7980.(14) Tarbuck, T. L.; Ota, S. T.; Richmond, G. L. SpectroscopicStudies of Solvated Hydrogen and Hydroxide Ions at AqueousSurfaces. J. Am. Chem. Soc. 2006, 128, 14519−14527.(15) Iuchi, S.; Chen, H. N.; Paesani, F.; Voth, G. A. Hydrated ExcessProton at Water-Hydrophobic Interfaces. J. Phys. Chem. B 2009, 113,4017−4030.(16) Takahashi, H.; Maruyama, K.; Karino, Y.; Morita, A.; Nakano,M.; Jungwirth, P.; Matubayasi, N. Energetic Origin of Proton Affinityto the Air/Water Interface. J. Phys. Chem. B 2011, 115, 4745−4751.(17) Buch, V.; Milet, A.; Vacha, R.; Jungwirth, P.; Devlin, J. P. WaterSurface is Acidic. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7342−7347.(18) Beattie, J. K.; Djerdjev, A. M.; Warr, G. G. The Surface of NeatWater is Basic. Faraday Discuss. 2008, 141, 31−39.(19) Petersen, P. B.; Saykally, R. J. Is the Liquid Water Surface Basicor Acidic? Macroscopic vs. Molecular-Scale Investigations. Chem. Phys.Lett. 2008, 458, 255−261.(20) Winter, B.; Faubel, M.; Vacha, R.; Jungwirth, P. Behavior ofHydroxide at the Water/Vapor Interface. Chem. Phys. Lett. 2009, 474,241−247.(21) Mundy, C. J.; Kuo, I. F. W.; Tuckerman, M. E.; Lee, H. S.;Tobias, D. J. Hydroxide Anion at the Air−Water Interface. Chem. Phys.Lett. 2009, 481, 2−8.(22) Mishra, H.; Enami, S.; Nielsen, R. J.; Stewart, L. A.; Hoffmann,M. R.; Goddard, W. A.; Colussi, A. J. Bronsted Basicity of the Air−Water Interface. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 18679−18683.(23) Yamaguchi, S.; Kundu, A.; Sen, P.; Tahara, T. QuantitativeEstimate of the Water Surface pH Using Heterodyne-DetectedElectronic Sum Frequency Generation. J. Chem. Phys. 2012, 137,151101.(24) MacCallum, J. L.; Drew Bennett, W. F.; Peter Tieleman, D.Distribution of Amino Acids in a Lipid Bilayer from ComputerSimulations. Biophys. J. 2008, 94, 3393−3404.(25) Johansson, A. C. V.; Lindahl, E. Position-Resolved Free Energyof Solvation for Amino Acids in Lipid Membranes from MolecularDynamics Simulations. Proteins: Struct., Funct., Bioinf. 2008, 70, 1332−1344.(26) Johansson, A. C. V.; Lindahl, E. Titratable Amino Acid Solvationin Lipid Membranes as a Function of Protonation State. J. Phys. Chem.B 2009, 113, 245−253.(27) Shaytan, A. K.; Ivanov, V. A.; Shaltan, K. V.; Khokhlov, A. R.Free Energy Profiles of Amino Acid Side Chain Analogs Near Water-Vapor Interface Obtained via MD Simulations. J. Comput. Chem. 2010,31, 204−216.(28) White, S. H.; Wimley, W. C. Membrane Protein Folding andStability: Physical Principles. Annu. Rev. Biophys. Biomol. Struct. 1999,28, 319−365.(29) Chipot, C.; Pohorille, A. Conformational Equilibria ofTerminally Blocked Single Amino Acids at the Water−HexaneInterface. A Molecular Dynamics Study. J. Phys. Chem. B 1998, 102,281−290.(30) Martins-Costa, M. T. C.; Ruiz-Lopez, M. F. Simulation ofAmino Acid Diffusion Across Water/Hydrophobic Interfaces. Phys.Chem. Chem. Phys. 2011, 13, 11579−11582.(31) Vohringer-Martinez, E.; Toro-Labbe, A. Amino Acids at Water−Vapor Interfaces: Surface Activity and Orientational Ordering. J. Phys.Chem. B 2010, 114, 13005−13010.(32) Vohringer-Martinez, E.; Toro-Labbe, A. The Mean ReactionForce: A Method to Study the Influence of the Environment onReaction Mechanisms. J. Chem. Phys. 2011, 135, 064505.(33) Nose, S. A Unified Formulation of the Constant TemperatureMolecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511−519.(34) Hoover, W. G. Canonical Dynamics: Equilibrium Phase-SpaceDistributions. Phys. Rev. A 1985, 31, 1695−1697.(35) Moreira, N. H.; Skaf, M. S. Structural Characterization of theH2O/CCl4 Liquid Interface Using Molecular Dynamics Simulations.Prog. Colloid Polym. Sci. 2004, 128, 81−85.
(36) Toukan, K.; Rahman, A. Molecular-Dynamics Study of AtomicMotions in Water. Phys. Rev. B 1985, 31, 2643−2648.(37) Jorgensen, W. L.; Maxwell, D. S.; Tirado-Rives, J. Developmentand Testing of the OPLS All-Atom Force Field on ConformationalEnergetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996,118, 11225−11236.(38) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Montgomery, J. J. A.; Vreven, T.;Kudin, K. N.; Burant, J. C.; et al. Gaussian 03, Revision C.02; GaussianInc.: Wallingford, CT, 2004.(39) Ponder, J. W. TINKER: Software Tools for Molecular Design 4.2;Washington University School of Medicine: Saint Louis, MO, 2004.(40) Martins-Costa, M. T. C. A Gaussian 03/TINKER 4.2 Interface forBorn-Oppenheimer Ab Initio and Hybrid QM/MM Simulations;University of Lorraine - CNRS: Vandoeuvre-les-Nancy, France,2006. Contact: [email protected].(41) Torrie, G. M.; Valleau, J. P. Nonphysical Sampling Distributionsin Monte Carlo Free-Energy Estimation: Umbrella Sampling. J.Comput. Phys. 1977, 23, 187−199.(42) Kumar, S.; Bouzida, D.; Swendsen, R. H.; Kollman, P. A.;Rosenberg, J. M. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput.Chem. 1992, 13, 1011−1021.(43) Roux, B. The Calculation of the Potential of Mean Force UsingComputer Simulations. Comput. Phys. Commun. 1995, 91, 275−282.(44) Retegan, M.; Martins-Costa, M.; Ruiz-Lopez, M. F. Free EnergyCalculations Using Dual-Level Born−Oppenheimer Molecular Dy-namics. J. Chem. Phys. 2010, 133, 064103.(45) Wada, G.; Tamura, E.; Okina, M.; Nakamura, M. On the Ratioof Zwitterion Form to Uncharged Form of Glycine at Equilibrum inVarious Media. Bull. Chem. Soc. Jpn. 1982, 55, 3064−3067.(46) Stover, M. L.; Jackson, V. E.; Matus, M. H.; Adams, M. A.;Cassady, C. J.; Dixon, D. A. Fundamental Thermochemical Propertiesof Amino Acids: Gas-Phase and Aqueous Acidities and Gas-PhaseHeats of Formation. J. Phys. Chem. B 2012, 116, 2905−2916.(47) Hu, C. H.; Shen, M. Z.; Schaefer, H. F. Glycine ConformationalAnalysis. J. Am. Chem. Soc. 1993, 115, 2923−2929.(48) Tortonda, F. R.; Pascual-Ahuir, J. L.; Silla, E.; Tunon, I.;Theoretical, A. Study of Solvent Effects on the ConformationalEquilibria of Neutral Glycine in Aqueous Solution. J. Mol. Struct.:THEOCHEM 2003, 623, 203−210.(49) Benbrahim, N.; Rahmouni, A.; Ruiz-Lopez, M. F.; Theoretical,A. Study of Medium Effects on the Structure of the Glycine AnalogueAminomethylphosphonic Acid. Phys. Chem. Chem. Phys. 2008, 10,5624−5632.(50) Tunon, I.; Silla, E.; Ruiz-Lopez, M. F. On the TautomerizationProcess of Glycine in Aqueous Solution. Chem. Phys. Lett. 2000, 321,433−437.(51) Slifkin, M. A.; Ali, S. M. Thermodynamic Parameters of theActivation of Glycine Zwitterion Protonation Reactions. J. Mol. Liq.1984, 28, 215−221.
The Journal of Physical Chemistry B Article
dx.doi.org/10.1021/jp4083689 | J. Phys. Chem. B 2013, 117, 12469−1247412474





























