Embed Size (px)
Citation preview

14 April 2000
Ž .Chemical Physics Letters 320 2000 623–630www.elsevier.nlrlocatercplett
An ESR and ab-initio MO study of p –n orbital crossover inCH2 0
cycloalkylketone radical cations
Peng Wang, Kenji Komaguchi, Masaru Shiotani )
Department of Applied Chemistry, Faculty of Engineering, Hiroshima UniÕersity, Higashi 739-8527, Hiroshima, Japan
Received 4 January 2000; in final form 3 February 2000
Abstract
The cyclobutanone and cyclopentanone radical cations were generated in a halocarbon matrix by ionizing radiation at 77Ž .K. The ESR spectrum of a cyclobutanone radical cation consisted of a triple-quintet hf structure with ca. 0.9 mT 2H and
Ž .2.4 mT 4H splittings. Whereas, both cyclopentanone and 2,2,5,5-tetradeuteriocyclopentanone radical cations gave anŽ . 1identical triplet with 1.5 mT 2H splitting. Based on the H hf splittings, it was concluded that the SOMO in
Ž . Žcycloalkylketone radical cations was changed from a p oribital to a n and s orbital i.e., p –n orbitalCH2 0 C – C CH2 0.crossover when the ring size increased from a four-member to a five-member. q 2000 Elsevier Science B.V. All rights
reserved.
1. Introduction
It has been suggested based on both experimentsand theoretical calculations that for ketones such asŽ . w x w xCH CO 1,2 , CH COC H 1,2 and cC H CO3 2 3 2 5 2 4w x Ž3,4 the n -type orbital i.e., oxygen lone pair p0
.orbital can be the HOMO and either the p orCO
p -type orbital can be the next-HOMO. TheirCH2
energy sequence, however, may depend on the local: Žstructure around the C5O group i.e., substituted.group, bond angle, etc. , especially in radical cations,
because of their rather small energy difference ofless than ca. 1–2 eV. ESR can provide direct experi-
Žmental evidence on the SOMO singly occupied. wmolecular orbital of radical cations and anions 5–
x Ž .10 . Among ketones, cycloalkylketones cC H COn 2 n
) Corresponding author. Fax: q81-824-24-7736; e-mail:[email protected]
are attractive to investigate with respect to electronicstructure because they have essentially no rotationalisomers, which generally make the ESR spectralanalysis complicated, and their geometrical structurecan be easily changed using different ring sizes.
w xSymons and Boon 5 were the first to report theESR spectrum of the cyclohexanone radical cationŽ q. 1 Ž .cC H CO with a triplet H hyperfine hf split-5 10
ting of ca. 2.7 mT in a CCl F matrix at 77 K and3
attributed the triplet to two equatorial protons at theŽ .b-carbons see Scheme 1 , the rational being that
these protons are held close to the plane containingthe singly occupied p orbital on oxygen. However,immediately thereafter, their hf attribution was found
w xto be erroneous. That is, Snow and Williams 6 havereported that the ESR spectra of the non-deuteriatedcyclohexanone and 2,2,6,6-tetradeuteriocyclo-hexanone are identical, and attributed the triplet tothe protons at the g-carbons. Based on these results,
0009-2614r00r$ - see front matter q 2000 Elsevier Science B.V. All rights reserved.Ž .PII: S0009-2614 00 00207-4

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630624
Scheme 1.
they proposed ‘long-range’ proton hf interactionsŽ .relayed through a trans ‘W’-plan arrangement of
s and s bonds in the radical cations of theC – C C – Hw xcarbonyl compounds. The hyperconjugation 11 is
the most probable mechanism for the unpaired elec-Ž .tron in the n and s orbital to be transferred to0 C – C
the ‘equatorial’ hydrogens at g-carbons ofcC H COq with respect to the C –C , bond, be-5 10 b a
cause the hydrogens are located at the position closeto the C –C –O plane having a large spin density,b a
w xsimilar to alkane radical cations 12,13 . When thering size decreases from a six-member to five- andfour-members, the dihedral angle between the C –Ca b
and C –H bonds may increase so as to give lessg e
effective hyperconjugation. This might cause aŽ .change in the SOMO from a n and s -type0 C – C
orbital to either a p -type or a p -type orbital,CH2 CO
which we call either a ‘p –n ’ orbital crossover orCH2 0
a ‘p –n ’ orbital crossover. However, no completeCO 0
ESR study has yet been reported for the cyclobu-Ž . Ž .tanone cC H CO and cyclopentanone cC H CO3 6 4 8
radical cations. Thus, we investigated the electronicq Žstructure of cycloalkylketones, cC H CO nsn 2 n
.3, 4 , using the low-temperature matrix ESR spec-w x 1troscopy method 12,13 . Based on the H hf split-
tings, it was found that the ‘p –n ’ orbitalCH2 0
crossover took place between the cyclobutanone andcyclopentanone radical cations. This conclusion wastheoretically supported by ab-initio MO and density
Ž .functional theory DFT calculations. Furthermore,we observed the ESR spectra of the radical anions of
Ž .cC H CO ns3–5 radiolytically generated in an 2 n
Ž .2-methyltetrahydrafuran 2MTHF matrix at 77 Kand confirmed that their SOMO is a p
U -typeC 5 O
orbital, independent of the ring size.
2. Experimental
The cycloalkylketones used in this study includedŽ .cyclobutanone cC H CO; Aldrich , cyclopentanone3 6
Ž .cC H CO; Tokyo Kasei , 2,2,5,5-tetradeuterio-4 8Ž .cylopentanone cC H CO-2,2,5,5-d , cyclohex-4 8 4
Ž .anone cC H CO; Tokyo Kasei and 2,2,6,6-tetra-5 10Ž .deuteriocyclohexanone cC H CO-2,2,6,6-d . The5 10 4
two partially deuteriated ketones were synthesizedw xafter F.E. Condon 14 . Their purities were greater
than 99% based on the 1H, 2 D and 13C-NMR spectraŽ .Bruker AMX400 . Furthermore, 95 atom% deu-terium substitution was confirmed by the NMR spec-tra. Halocarbons such as perfluorocyclohexaneŽ . Ž .cC F ; Aldrich , CCl F Tokyo Kasei and6 12 3
Ž .CFCl CF Cl Tokyo Kasei were used without fur-2 2Žther purification. 2-Methyltetrahydrofuran 2MTHF;
.Tokyo Kasei was used after drying with metallicNa.
Solid solutions containing ca. 0.5 mol% cy-cloalkylketones in either halocarbon or 2MTHF wereprepared in a Spectrosil ESR sample tube by severalcycles of freezing, degassing and thawing on a vac-uum line. They were irradiated with g-rays from60Co with a total dose of ca. 10 kGy at 77 K andthen subjected to an ESR study. These are well-established methods to generate and stabilize the

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630 625
solute radical cations or anions in a low-temperaturew xsolid matrix 12,13 . The ESR spectra were recorded
on a Bruker ESP 300E spectrometer from 4.2 K toca. 200 K. The temperature was controlled with anOxford ESR 900 continuous flow cryostat.
The geometrical structure was optimized on aCray J932r24 computer at the Information Process-
Ž .ing Center IPC , Hiroshima University, at theMP2r6-31GUU level using the GAUSSIAN 94 pro-
w x 1gram 15 . The H–hf splittings were calculated forthe optimized structure by the Density Function The-
Ž . w x UUory DFT method 16 at the B3LYPr6-31G level.
3. Results and discussion
3.1. Radical cations
Fig. 1a shows the ESR spectrum of the cyclobu-tanone radical cation in cC F at 77 K. The spec-6 12
trum consists of an isotropic triple quintet hf struc-Ž . Ž .ture with 0.9 mT 2H and 2.4 mT 4H splittings
Žwith a slight g-tensor anisotropy g s2.0002 and5
.g s2.0035 . The same triple quintet spectrum wasHobserved at 77 K using CFCl CF Cl and CFCl2 2 3
matrices instead of cC F . A possible geometrical6 12
structure of cC H COq is either planar C or3 6 2n
puckered C . Observation of the hf splittings due tos
two ‘equivalent’ and four ‘equivalent’ hydrogens
Ž .Fig. 1. ESR spectra of the cyclobutanone radical cation a andŽ .radical anion b generated in cC F and 2MTHF matrices by6 12
g-ray irradiation, respectively, at 77 K, together with the simu-Ž X . Ž X .lated ones, a and b ; the hf splittings are listed in Table 1.Ž .Note that b was obtained by subtracting the spectrum of the
Ž .matrix radical 2MTHF radical from the original one.
strongly suggests that the former can be the case.That is, assuming the C planar structure, the triplet2n
and quintet splittings are straightforwardly at-tributable to two hydrogens at the g-carbon and fourhydrogens at the b-carbons, respectively. No appre-ciable change in the spectrum was observed forcC H COq in cC F in the temperature range3 6 6 12
from 4.2 K to 175 K. This indicates that the C2n
structure is an intrinsic one, not a dynamically aver-aged one. Based on the large hf splitting due to thefour equivalent hydrogens at the b-carbons, a p -CH2
type SOMO is suggested. The possibility of either aŽ .n and s or a p -type SOMO is ruled out.0 C – C C 5 O
Boon and Symons have reported the same triple-quintet spectrum as the present one for the irradiatedsolid solution containing cC H CO in a CCl F ma-3 6 3
w xtrix at 77 K 7 . They have tentatively assigned thespectrum to a ring opening alkyl type of radical,CH CH CH COq. However, the present study re-2 2 2
vealed that their assignment of the spectrum waserroneous. It is worthy of note that the cC H COq
3 6
in CFCl CF Cl was irreversibly transformed into the2 2Ž .radical with a double triplet splitting of 2.0 mT 1H
Ž .and 3.6 mT 2H when the temperature increased to115 K. The newly formed radical was attributable tothe neutral radical which was formed by a oneproton-detachment reaction from cC H COq at the3 6
b-carbon.In order to compare the electronic structure of
cC H COq with that of other cycloalkylketone radi-3 6
cal cations, the cyclopentanone radical cations wereradiolytically generated in cC F at 77 K. Fig. 2a6 12
and b show the ESR spectra of cC H COq and4 8
cC H CO-2,2,5,5-dq, respectively. Both spectra of4 8 4
the non-deuteriated and deuteriated cyclopentanoneradical cations consist of an identical triplet with 1.5mT and gs2.0006, except for the slightly narrowerline width for the deuteriated radical cation. If theprotons responsible for the hf splitting were replacedby deuterons, the splitting should be reduced by afactor of ca. 6.5, the magnetic moment ratio of 1H to2 D. However, the observed hf splittings were inde-pendent of the deuterium substitution. Thus, that thetwo interacting protons are not located at C-2 and
Ž .C-5 i.e., two b-carbons , is conclusively demon-strated by the present experimental result. Accord-ingly, the observed triplet must be attributable to two
Ž .of the four protons at C-3 and C-4 two g-carbons .

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630626
Ž . Ž .Fig. 2. ESR spectra of the radical cations of cyclopentanone a and 2,2,5,5-tetradeuteriocylopentanone b generated in cC F , together6 12Ž . Ž .with those of the corresponding radical anions c and d generated in 2MTHF, by g-ray irradiation at 77 K. The stick diagrams show the
spectral analysis.
Ž .Then, assuming the n and s -type SOMO, the0 C – C
observed triplet is attributed to the two ‘equatorial’protons at the g-carbons, one at each g-carbon,which are located at the ‘trans’ position with respectto the C –C bond, having a large unpaired electrona b
density. The assumption is supported by the theoreti-cal calculations, as described below.
Similar to cC H COq, both spectra of4 8
cC H COq and cC H CO-2,2,6,6-dq generated5 10 5 10 4
in cC F consisted of an identical triplet, but with a6 121 Ž .larger H hf splitting of 2.6 mT 2H , at 77 K. The
splitting is in good agreement with the previouslyreported one for the normal and partially deuteriated
Žcyclohexanone radical cations 2.75 mT forq. w xcC H CO in CFCl 6,7 . Thus, it was recon-5 10 3
firmed that the cyclohexanone radical cation alsotakes the n , s -type SOMO.0 C – C
3.2. Radical anions
It was demonstrated in the above section thatwhen the ring size of cC H COq increases fromn 2 n
four-member to five- and six-members, the SOMO isŽ .changed from the p type to the n and sCH2 0 C – C
type. It is of interest to see if such orbital crossovercan be observed for the corresponding radical anions.Then, we have radiolytically generated the cy-
w xy Žcloalkylketone radical anions, cC H CO nsn 2 n.3–5 , in a 2MTHF rigid glass matrix at 77 K. The
ESR spectrum of cC H COy consisted of a triple3 6Ž . Ž .triplet with 2.7 mT 2H and 1.8 mT 2H splittings.
Almost the same hf splittings were previously re-ported for the cC H COy generated by chemical3 6
reduction with an Na atom in the Ar matrix at 4 K:Ž . Ž . w xca. 2.7 mT 2H and ca. 1.7 mT 2H splittings 9 .
Assuming the pU -type SOMO and hyperconjuga-C 5 Ow xtion mechanism 11 , the larger and smaller triplets
are attributable to two pairs of ‘axial’ and ‘equa-torial’ hydrogens at the b-carbons with respect to theC5O bond having a large spin density. The non-equivalent two hydrogens at the one b-carbon sug-gest that cC H COy has a puckered C structure,3 6 s
contrary to the planar C structure of cC H COq.2n 3 6
The spectrum of cC H COy in 2MTHF gave a4 8Ž .triple triplet hf structure with 3.2 mT 2H and 1.7
Ž . Ž .mT 2H splittings at 77 K Fig. 2c ; these valuesbeing in good agreement with the previously re-ported ones for cC H COy generated with an alkali4 8
Ž . w xmetal in tetrahydrofuran THF . 10 Whereas,cC H CO-2,2,5,5-dy in 2MTHF gave a singlet4 8 4
which is further poorly resolved with many hf linesŽ .Fig. 2d . This indicates that the hf splittings of thenon-deuteriated radical cation were reduced by afactor of ca. 6.5, the magnetic moment ratio of 1H to2 D, in the spectrum of the partially deuteriated radi-cal cation. Thus, the triple triplet splittings must beattributable to four hydrogens at the b-carbons. Theresult is fully consistent with the p
U -type SOMO,C 5 O

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630 627
similar to cC H COq. Furthermore, we confirmed3 6
that cC H COy in 2MTHF gave a triple triplet5 10Ž . Ž .with 2.5 mT 2H and 0.6 mT 2H splittings at 77
K, whereas cC H CO-2,2,6,6-dy gave a singlet.5 10 4
These results are consistent with the pU SOMO.C 5 O
Note that the triple triplet splittings of the non-de-uteriated radical anion were almost the same as the
Ž .previously reported ones, 3.1 mT 2H and 0.7 mTŽ . w x2H , in THF with Na at 77 K 10 . Thus, it was
y Žexperimentally concluded that the cC H CO nn 2 n. Us3–5 radicals take the p -type SOMO, inde-C 5 O
pendent of the ring size. The ab-initio MO calcula-tions support this conclusion as, described below.
3.3. MO calculations
In order to obtain theoretical supports for thep –n orbital crossover in the cycloalkylketoneCH2 0
radical cations, ab-initio MO calculations were per-formed. The calculations of the cyclobutanone radi-
cal cation resulted in a geometric structure very closeto the planar C one with a dihedral angle of 179.882n
Žbetween the C2–C1–C4 and C2–C3–C4 planes see. 1the geometric parameters in Fig. 3 . The H hf
splitting calculated for the two paired hydrogens atŽ .the b-carbons was 2.21 mT 4H , which was in good
agreement with the experimental value of 2.4 mT.The calculated splitting for two hydrogens at g-
Ž .carbon C3 was 0.2 mT, this value being also ratherŽ .close to the experimental one 0.9 mT . Thus, the
C structure was supported for cC H COq by the2n 3 6
theoretical calculations. These calculations are fullyŽ .consistent with the p -type SOMO Fig. 3 , whichCH2
was proposed based on the experimental result. Fur-thermore, the calculations for the corresponding radi-
Ž y.cal anion cC H CO resulted in a puckered C3 6 s
structure which has a dihedral angle of 171.68; thecalculations were found to be consistent with theexperimental results based on the agreement betweenthe experimental and calculated 1H hf splittingsŽ .Fig. 3 .
˚ q y q yŽ .Fig. 3. Geometrical structures bond length in A and angle in degree of cC H CO , cC H CO , cC H CO and cC H CO3 6 3 6 4 8 4 8
optimized by ab-initio MO calculations using Gaussian 94rMP2r6-31GUU, together with the illustration of SOMOs. The experimental 1HŽ . UUhf splittings are compared with the theoretical ones in parentheses calculated at DFT B3LYPr6-31G level for the optimized structures.
( )P. Wang et al.rChemical Physics Letters 320 2000 623–630628
The calculations of the cyclopentanone radicalŽ q.cation cC H CO resulted in the C structure4 8 2
with a twist angle of 11.98 between the C2–C1–C5Žplane and the C3–C4 bond see the bond distances in
.Fig. 3 . The C structure was calculated to be 243s
kJrmol higher than the C one. The C structure is2 2
fully consistent with the experimental results. Thatis, the 1H hf splitting calculated for two equatorialhydrogens at the g-carbons was 1.48 mT, which is inexcellent agreement with the experimental value of1.5 mT. Thus, as expected, the SOMO ofcC H COq was concluded to consist of the ‘in-4 8
Ž .plane’ n and s orbital as depicted in Fig. 3.0 C – C
Furthermore, the experimental triplet hf splitting ofŽ q.the cyclohexanone radical cation cC H CO was5 10
very well reproduced by the calculations: 2.6 mTŽ . Ž .exp. vs. 2.86 mT calc. for the two equatorialhydrogens at the g-carbons. The dihedral angle be-tween the C –C and C –H bonds was calculateda b g e
to be 1.58 for cC H COq, this angle being much5 10q Ž .smaller than that for cC H CO 46.38 . This could4 8
be the reason why the experimental 1H hf splitting is
q Ž .larger for cC H CO 2.6 mT than for5 10q Ž .cC H CO 1.5 mT , because of more effective4 8
hyperconjugation in the former radical cation. Thus,Ž .the calculations supported the n and s SOMO0 C – C
of both cC H COq and cC H COq.4 8 5 10
The calculated 1H hf splittings for cyclopen-tanone and cyclohexanone radical anions were alsonicely compared with the experimental ones, asshown in Table 1. Thus, the p
U -type SOMO wasC 5 O
both experimentally and theoretically confirmed fory Ž .cC H CO ns3–5 .n 2 n
In order to demonstrate the p –n orbitalCH2 0
crossover more clearly, ab-initio calculations werecarried out at the ROHFr6-31GUU level. The resultsfor cyclobutanone and cyclopentanone are schemati-cally shown in Fig. 4. It can be seen from the figure,that the energy level sequence of the higher threeoccupied molecular orbitals is the n , s –p –0 C – C CH2
p for neutral cC H CO, but is changed to theC 5 O 3 6
n , s –p –p for neutral cC H CO. When0 C – C C 5 O CH2 4 8
the one-electron oxidation reaction takes place toform cC H COq, the energy level of the original3 6
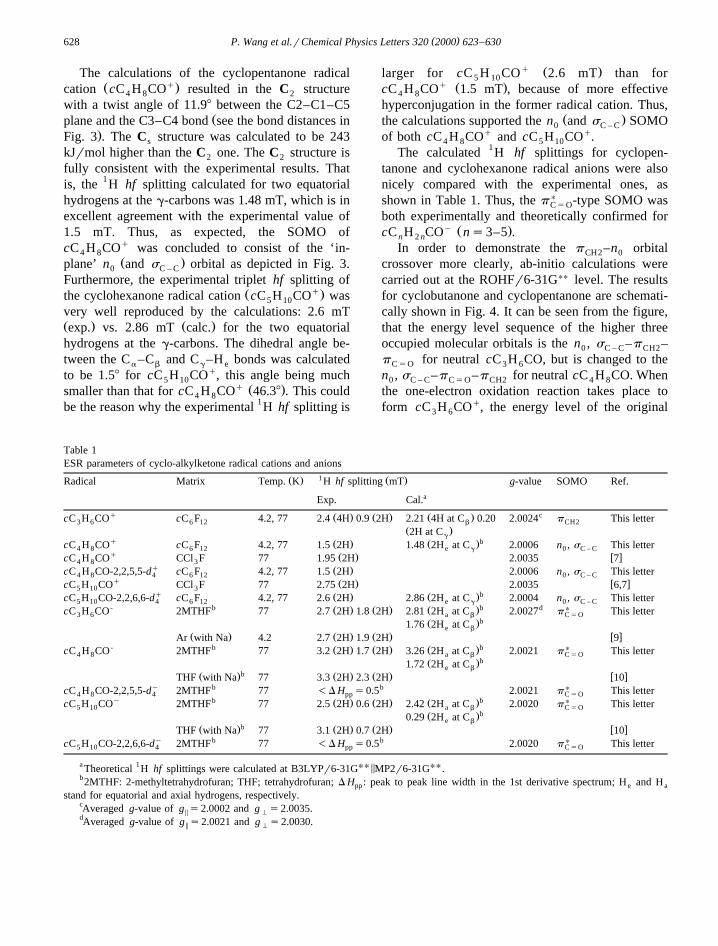
Table 1ESR parameters of cyclo-alkylketone radical cations and anions
1Ž . Ž .Radical Matrix Temp. K H hf splitting mT g-value SOMO Ref.aExp. Cal.
q cŽ . Ž . Ž .cC H CO cC F 4.2, 77 2.4 4H 0.9 2H 2.21 4H at C 0.20 2.0024 p This letter3 6 6 12 b CH2Ž .2H at Cg
q bŽ . Ž .cC H CO cC F 4.2, 77 1.5 2H 1.48 2H at C 2.0006 n , s This letter4 8 6 12 e g 0 C – Cq Ž . w xcC H CO CCl F 77 1.95 2H 2.0035 74 8 3
q Ž .cC H CO-2,2,5,5-d cC F 4.2, 77 1.5 2H 2.0006 n , s This letter4 8 4 6 12 0 C – Cq Ž . w xcC H CO CCl F 77 2.75 2H 2.0035 6,75 10 3
q bŽ . Ž .cC H CO-2,2,6,6-d cC F 4.2, 77 2.6 2H 2.86 2H at C 2.0004 n , s This letter5 10 4 6 12 e g 0 C – CU- b b dŽ . Ž . Ž .cC H CO 2MTHF 77 2.7 2H 1.8 2H 2.81 2H at C 2.0027 p This letter3 6 a b C 5 O
bŽ .1.76 2H at Ce b
Ž . Ž . Ž . w xAr with Na 4.2 2.7 2H 1.9 2H 9U- b bŽ . Ž . Ž .cC H CO 2MTHF 77 3.2 2H 1.7 2H 3.26 2H at C 2.0021 p This letter4 8 a b C 5 O
bŽ .1.72 2H at Ce bbŽ . Ž . Ž . w xTHF with Na 77 3.3 2H 2.3 2H 10
Uy b bcC H CO-2,2,5,5-d 2MTHF 77 -D H s0.5 2.0021 p This letter4 8 4 pp C 5 Oy b Ž . Ž . Ž .b UcC H CO 2MTHF 77 2.5 2H 0.6 2H 2.42 2H at C 2.0020 p This letter5 10 a b C 5 O
bŽ .0.29 2H at Ce bbŽ . Ž . Ž . w xTHF with Na 77 3.1 2H 0.7 2H 10
Uy b bcC H CO-2,2,6,6-d 2MTHF 77 -D H s0.5 2.0020 p This letter5 10 4 pp C 5 O
a 1 UU 5 UUTheoretical H hf splittings were calculated at B3LYPr6-31G MP2r6-31G .b2MTHF: 2-methyltetrahydrofuran; THF; tetrahydrofuran; D H : peak to peak line width in the 1st derivative spectrum; H and Hpp e a
stand for equatorial and axial hydrogens, respectively.cAveraged g-value of g s2.0002 and g s2.0035.5 HdAveraged g-value of g s2.0021 and g s2.0030.5 H

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630 629
Fig. 4. Energy level diagrams of cC H CO and cC H CO showing how each MO level of the neutral molecules is shifted when they3 6 4 8
suffered one electron loss or addition to form their radical cations or anions, respectively. The energy levels were calculated by the ab-initioROHFr6-31GUU method.
Ž .HOMO n , s sharply drops, but the next0 C – CŽ .HOMO p remains at almost the same level soCH2
that the latter orbital becomes the SOMO. ForcC H CO, the energy level sequence is maintained4 8
even after a one-electron oxidation so as to give theŽ .n and s SOMO. The energy separation be-0 C – C
tween pU and p
U orbitals is so large that noCH2 C 5 O
orbital crossover takes place for the cyclobutanoneand cyclopentanone radical anions.
4. Conclusion
The present ESR study of the cycloalkylketoneradical cations, cC H COq, revealed that then 2 n
SOMO was changed from the p orbital to the nCH2 0Ž .and s orbital when the ring size increased fromC – C
Ž . Ž .four n s 3 to five and six n s 4, 5 . ThecC H COq and cC H COq radicals have the4 8 5 10
‘twist’ C structure and the ‘chair-like’ C structure,2 s
respectively. The unpaired electron in these radicalcations can be effectively transferred to the ‘trans’
Ž .C –H hydrogen from the s C –C orbital whichg a b
is conjugated with the n orbital having a large spin0
density. Whereas, the cC H COq radical has the3 6ŽC planar with the smaller C –C –C and C –2n b a b b
.C –C bond angles close to 908. In this structureg b
the unpaired electron in cC H COq is less effec-n 2 n
tively transferred to the ‘trans’ C –H hydrogens andg
the SOMO turned out to be the p orbital.CH2
Acknowledgements
The present study was partially supported by asubsidy for Science Research from the Japanese
Ž .Ministry of Education Grant No. 08240105 .
References
w x1 K. Kimura, S. Katsumata, T. Yamazaki, H. Wakabayashi, J.Ž .Electron Spectrosc. 6 1975 41.
w x2 K. Kimura, S. Katsumata, Y. Achiba, T. Yamazaki, S. Iwata,Handbook of HeI Photoelectron Spectra of FundamentalOrganic Molecules, Japan Scientific Societies Press,TokyorHalsted Press, New York, 1980, p. 156.
w x Ž .3 R. Hoffmann, R.B. Davidson, J. Am. Chem. Soc. 93 19715699.
w x4 W.L. Jorgensen, L. Salem, The Organic Chemist’s Book ofOrbitals, Academic Press, New YorkrLondon, 1973, p. 197.
w x Ž .5 M.C.R. Symons, P.J. Boon, Chem. Phys. Lett. 89 1982516.
w x6 L.D. Snow, F. Williams, J. Chem. Soc. Chem. Commun.Ž .1983 1090.

( )P. Wang et al.rChemical Physics Letters 320 2000 623–630630
w x7 P.J. Boon, M.C.R. Symons, K. Ushida, T. Shida, J. Chem.Ž .Soc. Perkin Trans. II 1984 1213.
w x8 L.B. Knight Jr., B.W. Gregory, S.T. Cobranchi, F. Williams,Ž .X.Z. Qin, J. Am. Chem. Soc. 110 1988 327.
w x Ž .9 R. Koppe, P.H. Kasai, J. Phys. Chem. 98 1994 12904.w x Ž .10 J.E. Bennett, B. Milee, A. Thomas, J. Chem. Soc. A 1968
298.w x11 N.M. Atherton, Electron Spin Resonance, New Yorkr
LondonrSydneyrToronto, 1973, p. 103.
w x12 M. Shiotani, ESR studies of radical cations in solid matrices,Ž .Magn. Res. Rev. 12 1987 333.
w x Ž .13 A. Lund, M. Shiotani Eds. , Radical Ionic System, Kluwer,Dordrecht, 1991, p. 99, p. 127.
w x Ž .14 F.E. Condon, J. Am. Chem. Soc. 73 1951 4675.w x15 M.J. Frisch et al., Gaussian 94, Gaussian, Pittsburgh, PA,
1995.w x Ž .16 A.D. Becke, Phys. Rev. A 38 1988 3098.