Embed Size (px)
Citation preview

1
BILAG I
PRODUKTRESUMÉ

2
Dette lægemiddel er underlagt supplerende overvågning. Dermed kan nye sikkerhedsoplysninger hurtigt tilvejebringes. Læger og sundhedspersonale anmodes om at indberette alle formodede bivirkninger. Se i pkt. 4.8, hvordan bivirkninger indberettes.
1. LÆGEMIDLETS NAVN
BESPONSA 1 mg pulver til koncentrat til infusionsvæske, opløsning
2. KVALITATIV OG KVANTITATIV SAMMENSÆTNING
Hvert hætteglas indeholder 1 mg inotuzumab ozogamicin.
Efter rekonstituering (se pkt. 6.6) indeholder 1 ml opløsning 0,25 mg inotuzumab ozogamicin.
Inotuzumab ozogamicin er et antistof-lægemiddel-konjugat (ADC) sammensat af et rekombinant humaniseret IgG4 kappa CD22-rettet monoklonalt antistof (produceret i ovarieceller fra kinesisk hamster vha. rekombinant DNA-teknologi), der bindes kovalent til N-acetyl-gamma-calicheamicin dimethylhydrazid.
Alle hjælpestoffer er anført under pkt. 6.1.
3. LÆGEMIDDELFORM
Pulver til koncentrat til infusionsvæske, opløsning.
Hvid til råhvid, frysetørret kage eller pulver.
4. KLINISKE OPLYSNINGER
4.1 Terapeutiske indikationer
BESPONSA er indiceret som monoterapi til behandling af voksne med recidiverende eller refraktær CD22-positiv B-celle prækursor akut lymfoblastær leukæmi (ALL). Voksne patienter med Philadelphia-kromosompositiv (Ph+) recidiverende eller refraktær CD22-positiv B-celle prækursor ALL skal have behandlingssvigt efter mindst 1 tyrosinkinasehæmmer (TKI).
4.2 Dosering og administration
BESPONSA skal administreres under tilsyn af en læge med erfaring inden for behandling af hæmatologisk sygdom og på et sted, hvor komplet genoplivningsudstyr umiddelbart er til rådighed.Overvejes BESPONSA som behandling mod recidiverende eller refraktær B-celle ALL, skal baseline-værdien for CD22-positivitet være > 0% inden behandlingsstart, målt ved hjælp af en valideret og sensitiv test (se pkt. 5.1).
Hos patienter med cirkulerende lymfoblaster anbefales cytoreduktion med en kombination af hydroxyurea, steroid, og/eller vincristin til et perifert blasttal ≤ 10.000/mm3 inden første dosis.
Der anbefales præmedicinering med kortikosteroid, antipyretika og antihistamin inden dosering (se pkt. 4.4).
Inden behandling af patienter med høj tumorbyrde anbefales præmedicinering for at reducere urinsyreniveauet i blodet og hydrering (se pkt. 4.4).

3
Patienter skal observeres under og i mindst 1 time efter endt infusion for symptomer på infusionsrelaterede reaktioner (se pkt. 4.4).
Dosering
BESPONSA skal administreres i cyklusser på 3-4 uger.
Hos patienter, der skal fortsætte med hæmatopoietisk stamcelletransplantation (HSCT), anbefales 2 behandlingscyklusser. En tredje cyklus kan overvejes til patienter, der ikke opnår komplet remission (CR) eller komplet remission med delvis hæmatologisk restitution (CRi) og minimal residualsygdoms-(MRD) negativitet efter 2 cyklusser (se pkt. 4.4). Hos patienter, der ikke fortsætter med HSCT, kan der administreres maksimalt 6 cyklusser. Behandlingen skal seponeres hos alle de patienter, der ikke opnår CR/CRi inden for 3 cyklusser.
Skema 1 viser de anbefalede doseringsregimer.
I første cyklus anbefales en samlet dosis BESPONSA til alle patienter på 1,8 mg/m2 pr. cyklus, givet som 3 opdelte doser på dag 1 (0,8 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2). Cyklus 1 varer 3 uger, men kan forlænges til 4 uger, hvis patienten opnår CR eller CRi, og/eller for at bedre restitutionen efter toksicitet.
I de efterfølgende cyklusser er den anbefalede, samlede dosis BESPONSA 1,5 mg/m2 pr. cyklus givet som 3 opdelte doser på dag 1 (0,5 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2) til patienter, der opnår CR/CRi, eller 1,8 mg/m2 pr. forløb givet som 3 opdelte doser på dag 1 (0,8 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2) til patienter, der ikke opnår CR/CRi. Efterfølgende cyklusser varer 4 uger.
Skema 1. Doseringsregime for cyklus 1 og efterfølgende cyklusser afhængig af respons på behandlingen
Dag 1 Dag 8a Dag 15a
Doseringsregime for cyklus 1Alle patienter:Dosis (mg/m2) 0,8 0,5 0,5Længde på cyklus 21 dageb
Doseringsregime for efterfølgende cyklusser afhængig af respons på behandlingenPatienter, der har opnået CRc eller CRid:Dosis (mg/m2) 0,5 0,5 0,5Længde på cyklus 28 dagee
Patienter, der ikke har opnået CRc eller CRid:
Dosis (mg/m2) 0,8 0,5 0,5Længde på cyklus 28 dagee
Forkortelser: ANC = absolut neutrofiltal, CR = komplet remission, CRi = komplet remission med delvis hæmatologisk restitution.a +/- 2 dage (oprethold minimum 6 dage mellem doser).b For patienter, der opnår CR/CRi, og/eller for at bedre restitution efter toksicitet, kan cyklus forlænges til
28 dage (dvs. 7 dages mellemrum uden behandling med start på dag 21).c CR defineres som < 5% blaster i knoglemarven og fravær af leukæmiske blaster i perifert blod, fuld
restitution af perifere blodtal (trombocytter ≥ 100 × 109/l og ANC ≥ 1 × 109/l) og resolution af eventuel ekstramedullær sygdom.
d CRi defineres som < 5% blaster i knoglemarven og fravær af leukæmiske blaster i perifert blod, delvis restitution af perifere blodtal (trombocytter < 100 × 109/l og/eller ANC < 1 × 109/l) og resolution af eventuel ekstramedullær sygdom.
e 7 dages mellemrum uden behandling med start på dag 21.
4
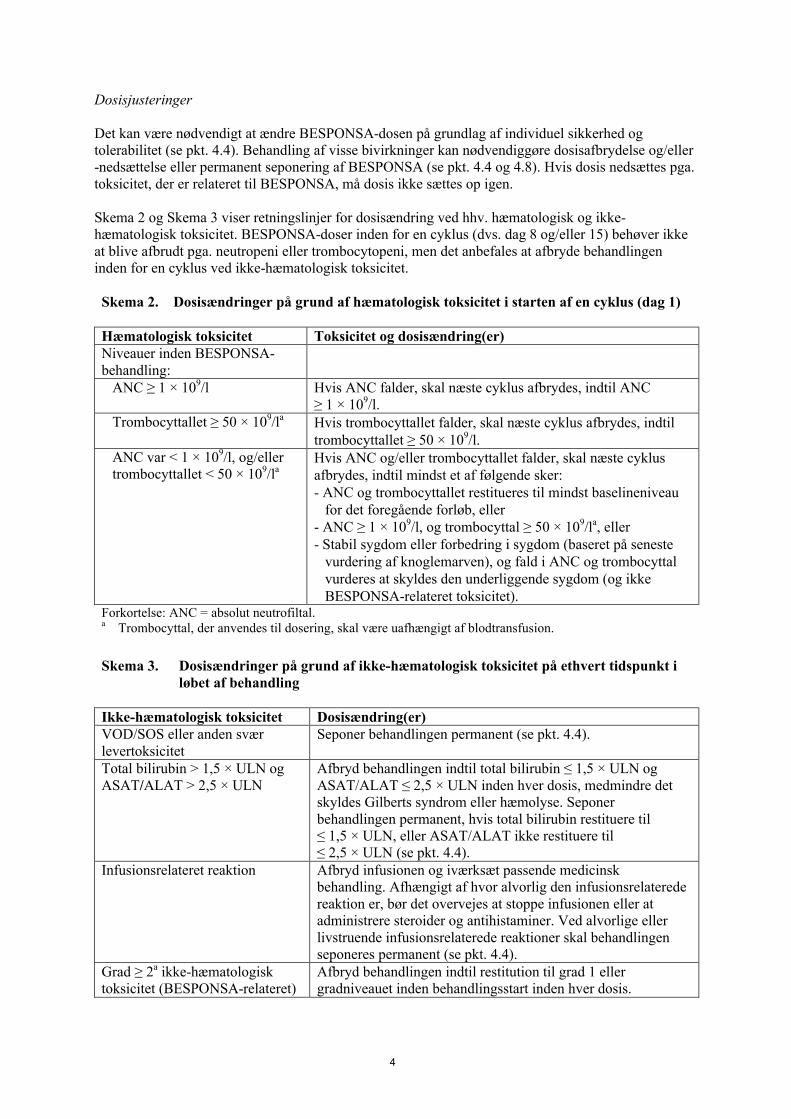
Dosisjusteringer
Det kan være nødvendigt at ændre BESPONSA-dosen på grundlag af individuel sikkerhed og tolerabilitet (se pkt. 4.4). Behandling af visse bivirkninger kan nødvendiggøre dosisafbrydelse og/eller -nedsættelse eller permanent seponering af BESPONSA (se pkt. 4.4 og 4.8). Hvis dosis nedsættes pga. toksicitet, der er relateret til BESPONSA, må dosis ikke sættes op igen.
Skema 2 og Skema 3 viser retningslinjer for dosisændring ved hhv. hæmatologisk og ikke-hæmatologisk toksicitet. BESPONSA-doser inden for en cyklus (dvs. dag 8 og/eller 15) behøver ikke at blive afbrudt pga. neutropeni eller trombocytopeni, men det anbefales at afbryde behandlingen inden for en cyklus ved ikke-hæmatologisk toksicitet.
Skema 2. Dosisændringer på grund af hæmatologisk toksicitet i starten af en cyklus (dag 1)
Hæmatologisk toksicitet Toksicitet og dosisændring(er)Niveauer inden BESPONSA-behandling:
ANC ≥ 1 × 109/l Hvis ANC falder, skal næste cyklus afbrydes, indtil ANC ≥ 1 × 109/l.
Trombocyttallet ≥ 50 × 109/la Hvis trombocyttallet falder, skal næste cyklus afbrydes, indtil trombocyttallet ≥ 50 × 109/l.
ANC var < 1 × 109/l, og/eller trombocyttallet < 50 × 109/la
Hvis ANC og/eller trombocyttallet falder, skal næste cyklus afbrydes, indtil mindst et af følgende sker:- ANC og trombocyttallet restitueres til mindst baselineniveau
for det foregående forløb, eller- ANC ≥ 1 × 109/l, og trombocyttal ≥ 50 × 109/la, eller- Stabil sygdom eller forbedring i sygdom (baseret på seneste
vurdering af knoglemarven), og fald i ANC og trombocyttal vurderes at skyldes den underliggende sygdom (og ikke BESPONSA-relateret toksicitet).
Forkortelse: ANC = absolut neutrofiltal.a Trombocyttal, der anvendes til dosering, skal være uafhængigt af blodtransfusion.
Skema 3. Dosisændringer på grund af ikke-hæmatologisk toksicitet på ethvert tidspunkt i løbet af behandling
Ikke-hæmatologisk toksicitet Dosisændring(er)VOD/SOS eller anden svær levertoksicitet
Seponer behandlingen permanent (se pkt. 4.4).
Total bilirubin > 1,5 × ULN og ASAT/ALAT > 2,5 × ULN
Afbryd behandlingen indtil total bilirubin ≤ 1,5 × ULN og ASAT/ALAT ≤ 2,5 × ULN inden hver dosis, medmindre det skyldes Gilberts syndrom eller hæmolyse. Seponer behandlingen permanent, hvis total bilirubin restituere til ≤ 1,5 × ULN, eller ASAT/ALAT ikke restituere til ≤ 2,5 × ULN (se pkt. 4.4).
Infusionsrelateret reaktion Afbryd infusionen og iværksæt passende medicinsk behandling. Afhængigt af hvor alvorlig den infusionsrelaterede reaktion er, bør det overvejes at stoppe infusionen eller at administrere steroider og antihistaminer. Ved alvorlige eller livstruende infusionsrelaterede reaktioner skal behandlingen seponeres permanent (se pkt. 4.4).
Grad ≥ 2a ikke-hæmatologisk toksicitet (BESPONSA-relateret)
Afbryd behandlingen indtil restitution til grad 1 eller gradniveauet inden behandlingsstart inden hver dosis.

5
Forkortelser: ALAT = alaninaminotransferase, ASAT = aspartataminotransferase, ULN = øvre normalgrænse, VOD/SOS = veno-okklusiv sygdom/sinusoidalt obstruktionssyndrom.a Sværhedsgrad ifølge National Cancer Institute Common Terminology Criteria for Adverse Events (NCI
CTCAE, det amerikanske cancerinstituts almindelige terminologiske kriterier for bivirkninger) version 3.0.
Skema 4 viser retningslinjerne for dosisændring afhængigt af varigheden af doseringsafbrydelser, der skyldes toksicitet.
Skema 4. Dosisændringer afhængigt af varigheden af dosisafbrydelser, der skyldes toksicitet
Varighed af dosisafbrydelser, der skyldes toksicitet
Dosisændring(er)
< 7 dage (inden for en cyklus) Afbryd næste dosis (oprethold minimum 6 dage mellem doser).≥ 7 dage Udelad næste dosis i cyklussen. ≥ 14 dage Når der opnås tilstrækkelig restitution, skal den totale dosis
nedsættes med 25 % i den efterfølgende cyklus. Hvis der er behov for yderligere dosisændring, skal antallet af doser nedsættes til 2 pr. cyklus i de efterfølgende cyklusser. Hvis en nedsættelse på 25 % af den totale dosis efterfulgt af en nedsættelse til 2 doser pr. cyklus ikke tolereres, skal behandlingen seponeres permanent.
> 28 dage Overvej permanent seponering af BESPONSA.
Særlige populationer
Ældre
Det er ikke nødvendigt at justere startdosis ud fra alder (se pkt. 5.2).
Nedsat leverfunktion
Det er ikke nødvendigt at justere startdosis til patienter med nedsat leverfunktion defineret som total bilirubin ≤ 1,5 × den øvre normalgrænse (ULN) og aspartataminotransferase(ASAT)/alaninaminotransferase (ALAT) ≤ 2,5 × ULN (se pkt. 5.2). Der er begrænset sikkerhedsinformation fra patienter med total bilirubin > 1,5 × ULN og ASAT/ALAT > 2,5 × ULN inden dosering. Afbryd behandlingen indtil restitution af total bilirubin til ≤ 1,5 × ULN og ASAT/ALAT til ≤ 2,5 × ULN inden hver dosis, medmindre det skyldes Gilberts syndrom eller hæmolyse. Seponer behandlingen permanent, hvis total bilirubin ikke restituerer til ≤ 1,5 × ULN, eller ASAT/ALAT ikke restitierer til ≤ 2,5 × ULN (se Skema 3 og pkt. 4.4).
Nedsat nyrefunktion
Det er ikke nødvendigt at justere startdosis til patienter med let, moderat eller svært nedsat nyrefunktion (kreatinin-clearance [CLcr] på hhv. 60-89 ml/min, 30-59 ml/min, eller 15-29 ml/min) (se pkt. 5.2). BESPONSAs sikkerhed og virkning er ikke undersøgt hos patienter med nyresygdom i slutstadiet.
Pædiatrisk population
BESPONSAs sikkerhed og virkning i hos børn 0-18 år er ikke klarlagt. Der foreligger ingen data.
Administration
BESPONSA er til intravenøs anvendelse. Infusionen skal administreres over 1 time.
BESPONSA må ikke administreres som intravenøst push eller bolus.

6
BESPONSA skal rekonstitueres og fortyndes inden administration. Anvisninger i rekonstituering og fortynding af BESPONSA inden administration er anført i pkt. 6.6.
4.3 Kontraindikationer
- Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne anført i pkt. 6.1.
- Patienter, der tidligere har haft bekræftet svær eller aktiv veno-okklusiv leversygdom/sinusoidalt obstruktionssyndrom (VOD/SOS).
- Patienter med alvorlig, aktiv leversygdom (fx cirrose, nodulær regenerativ hyperplasi, aktiv hepatitis).
4.4 Særlige advarsler og forsigtighedsregler vedrørende brugen
Sporbarhed
Med henblik på forbedring af sporbarheden af biologiske lægemidler bør navn og batchnummer på det administrerede produkt angives tydeligt i patientjournalen.
Levertoksicitet, herunder veno-okklusiv leversygdom/sinusoidalt obstruktionssyndrom (VOD/SOS)
Der er rapporteret levertoksicitet, herunder svær, livstruende og nogle gange letal hepatisk VOD/SOS, hos patienter med recidiverende eller refraktær ALL, der får BESPONSA (se pkt. 4.8). BESPONSA øgede risikoen for VOD/SOS væsentligt i forhold til den risiko, der gælder for almindelige kemoterapiregimer hos denne patientgruppe. Risikoen var mest markant hos patienter, der efterfølgende fik HSCT.
I følgende undergrupper var den rapporterede hyppighed af VOD/SOS post-HSCT ≥ 50 %:- Patienter, der fik HSCT-konditionerende regimer, der indeholdt 2 alkylerende midler- Patienter ≥ 65 år og - Patienter med serumbilirubin ≥ ULN forud for HSCT
Undgå brug af HSCT-konditionerende regimer, der indeholder 2 alkylerende midler. Overvej nøje benefit/risk-forholdet inden administration af BESPONSA til patienter, hos hvem fremtidig brug af HSCT-konditionerende regimer, der indeholder 2 alkylerende midler, sandsynligvis ikke kan undgås.
Hos patienter med serumbilirubin ≥ ULN inden HSCT må der kun fortsættes med HSCT efter behandling med BESPONSA efter nøje overvejelse af benefit/risk-forholdet. Hvis disse patienter fortsætter med HSCT, skal symptomer på VOD/SOS nøje monitoreres (se pkt. 4.2).
Andre patientfaktorer, der også kan være forbundet med forhøjet risiko for VOD/SOS efter HSCT, inkluderer tidligere HSCT, alder ≥ 55 år, tidligere leversygdom og/eller hepatitis inden behandlingsstart, senere linjer af behandling (salvage) og et højere antal behandlingscyklusser.
Omhyggelig overvejelse er derfor nødvendig inden administration af BESPONSA til patienter, der tidligere har fået HSCT. Ingen patienter med recidiverende eller refraktær ALL, som blev behandlet med BESPONSA, i kliniske studier havde fået HSCT inden for de seneste 4 måneder.
Patienter med tidligere leversygdom skal nøje evalueres (fx ved ultralydsscanning, test for viral hepatitis) inden behandling med BESPONSA for at udelukke alvorlig aktiv leversygdom (se pkt. 4.3).
For patienter, der forsætter med HSCT, anbefales en behandlingsvarighed med inotuzumab ozogamicin på 2 cyklusser for at mindske risikoen for VOD/SOS. En tredje cyklus kan overvejes til de patienter, som ikke opnår en CR/CRi og MRD-negativitet efter 2 cyklusser (se pkt. 4.2).
Overvåg nøje alle patienter for symptomer på VOD/SOS, især efter HSCT. Symptomer kan inkludere forhøjede værdier for total bilirubin, hepatomegali (som kan være smertefuld), hurtig vægtstigning og

7
ascites. Alle patienter med risiko for VOD/SOS kan muligvis ikke identificeres ved målinger udelukkende af totalbilirubin. Alle patienter skal have målt deres levertal, inklusive ALAT, ASAT, total bilirubin og alkalisk fosfatase inden og efter hver dosis BESPONSA. Hos patienter, der udvikler abnorme levertal, anbefales hyppigere monitorering af levertallene og kontrol for symptomer på levertoksicitet. Hos patienter, der fortsætter med HSCT, skal der udføres hyppig monitorering af levertallene i den første måned efter HSCT, herefter mindre hyppigt i henhold til almindelig medicinsk praksis. Forhøjede leverenzymer kan nødvendiggøre dosisafbrydelse, -nedsættelse eller permanent seponering af BESPONA (se pkt. 4.2).
Behandlingen skal seponeres permanent, hvis der forekommer VOD/SOS (se pkt. 4.2). I tilfælde af svær VOD/SOS skal patienten behandles ifølge almindelig medicinsk praksis.
Myelosuppression/cytopeni
Der er indberettet neutropeni, trombocytopeni, anæmi, leukopeni, febril neutropeni, lymfopeni og pancytopeni, heraf livstruende tilfælde, hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.8).
Hos nogle patienter, der fik inotuzumab ozogamicin, er der indberettet neutropeni- og trombocytopenirelaterede komplikationer (herunder hhv. infektioner og blødning/hæmoragi) (se pkt. 4.8).
Foretag komplet blodtælling inden hver dosis BESPONA, og undersøg om der er symptomer på infektion under behandlingen og efter HSCT (se pkt. 5.1), blødning/hæmoragi og andre virkninger af myelosuppression under behandlingen. Om nødvendigt gives profylaktisk behandling mod infektion og kontroltest foretages under og efter behandlingen.
Behandling af svær infektion, blødning/hæmoragi og andre virkninger af myelosuppression, herunder svær neutropeni eller trombocytopeni, kan kræve midlertidig afbrydelse af behandlingen, nedsættelse af dosis eller seponering (se pkt. 4.2).
Infusionsrelaterede reaktioner
Der er indberettet infusionsrelaterede reaktioner hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.8).
Der anbefales præmedicinering med kortikosteroid, antipyretika og antihistamin inden dosering (se pkt. 4.2).
Patienterne skal observeres nøje under og i mindst 1 time efter endt infusion for mulig indtræden af infusionsrelaterede reaktioner, herunder symptomer såsom hypotension, hedeture og vejrtrækningsproblemer. Hvis der forekommer en infusionsrelateret reaktion, skal infusionen standses og passende medicinsk behandling iværksættes. Afhængigt af hvor alvorlig reaktionen er, bør det overvejes at stoppe infusionen eller administrere steroid og antihistamin (se pkt. 4.2). Ved alvorlige eller livstruende infusionsrelaterede reaktioner skal behandlingen seponeres permanent (se pkt. 4.2).
Tumorlysesyndrom (TLS)
Der er indberettet TLS, som kan være livstruende eller dødelig, hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.8).
For at reducere urinsyreniveauet anbefales det, at præmedicinere og hydrere patienter med høj tumorbyrde inden behandlingen påbegyndes (se pkt. 4.2).
Patienterne skal observeres for symptomer på TLS og behandles ifølge almindelig medicinsk praksis.

8
Forlængelse af QT-intervallet
Der er observeret forlængelse af QT-intervallet hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.8 og 5.2).
BESPONSA skal administreres med forsigtighed til patienter, der tidligere har haft eller er disponeret for forlænget QT-interval, som tager lægemidler kendt for at forlænge QT-intervallet (se pkt. 4.5) samt til patienter med forstyrrelser i elektrolytbalancen. Der skal tages EKG og måles elektrolytter inden behandlingsstart samt periodisk under behandlingen (se pkt. 4.8 og 5.2).
Forhøjet amylase og lipase
Der er indberettet stigninger i amylase og lipase hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.8).
Patienterne skal observeres for stigninger i amylase og lipase. Potentiel hepatobiliær sygdom skal vurderes og behandles i henhold til almindelig medicinsk praksis.
ImmuniseringSikkerheden ved vaccination med levende vira under eller efter behandling med BESPONSA er ikke blevet undersøgt. Vaccination med levende virus frarådes i mindst 2 uger inden behandlingen med BESPONSA påbegyndes, under behandlingen og indtil B-lymfocyt restitution efter sidste cyklus.
4.5 Interaktion med andre lægemidler og andre former for interaktion
Der er ikke udført interaktionsundersøgelser (se pkt. 5.2).
Baseret på in vitro-data vil samtidig administration af inotuzumab ozogamicin og hæmmere eller inducere af cytokrom P450 (CYP) eller uridindiphosphat-glucuronosyltransferase (UGT) sandsynligvis ikke ændre eksponeringen for N-acetyl-gamma-calicheamicin-dimethylhydrazid. Derudover vil inotuzumab ozogamicin og N-acetyl-gamma-calicheamicin-dimethylhydrazid sandsynligvis ikke ændre eksponeringen for substrater af CYP-enzymer, og N-acetyl-gamma-calicheamicin-dimethylhydrazid vil sandsynligvis ikke ændre eksponeringen for substrater af UGT-enzymer eller transportproteiner.
Der er observeret forlængelse af QT-intervallet hos patienter, der fik inotuzumab ozogamicin (se pkt. 4.4). Derfor bør man nøje overveje samtidig brug af inotuzumab ozogamicin og lægemidler, der er kendt for at forlænge QT-intervallet eller fremkalde Torsades de Pointes. QT-intervallet skal observeres, når disse lægemidler administreres samtidigt (se pkt. 4.4, 4.8 og 5.2).
4.6 Fertilitet, graviditet og amning
Kvinder i den fertile alder/kontraception til mænd og kvinder
Kvinder i den fertile alder skal undgå at blive gravide, mens de får BESPONSA.
Kvinder skal anvende sikker kontraception under behandlingen med BESPONSA og i mindst 8 måneder efter sidste dosis. Mænd med kvindelige partnere i den fertile alder skal anvende sikker kontraception under behandlingen med BESPONSA og i mindst 5 måneder efter sidste dosis.
Graviditet
Der foreligger ingen data fra gravide kvinder, der bruger inotuzumab ozogamicin. Baseret på ikke-kliniske sikkerhedsfund kan inotuzumab ozogamicin forårsage embryoføtal skade, når det administreres til gravide kvinder. Dyreforsøg har påvist reproduktionstoksicitet (se pkt. 5.3).

9
BESPONSA må ikke anvendes under graviditet, medmindre den mulige fordel for moderen opvejer den mulige risiko for fosteret. Gravide kvinder eller patienter, der bliver gravide, mens de får inotuzumab ozogamicin, eller behandlede mandlige patienter, der har en gravid partner, skal informeres om den potentielle risiko for fosteret.
Amning
Der foreligger ingen data om udskillelse af inotuzumab ozogamicin eller dets metabolitter i human mælk, virkningerne på det ammede barn eller virkningerne på mælkeproduktionen. På grund af risikoen for bivirkninger hos ammede børn må kvinder ikke amme under behandling med BESPONSA og i mindst 2 måneder efter sidste dosis (se pkt. 5.3).
Fertilitet
Baseret på ikke-kliniske fund kan mænds og kvinders fertilitet blive kompromitteret af behandling med inotuzumab ozogamicin (se pkt. 5.3). Der foreligger ingen information om fertiliteten hos patienter. Både mænd og kvinder skal søge rådgivning vedrørende bevarelse af fertiliteten inden behandlingen.
4.7 Virkning på evnen til at føre motorkøretøj og betjene maskiner
BESPONSA har moderat indflydelse på evnen til at føre motorkøretøj og betjene maskiner. Patienterne kan føle sig trætte under behandling med BESPONSA (se pkt. 4.8). Derfor anbefales det at være forsigtig, når man fører motorkøretøj eller betjener maskiner.
4.8 Bivirkninger
Resumé af sikkerhedsprofil
De mest almindelige bivirkninger (≥ 20 %) var trombocytopeni (51 %), neutropeni (49 %), infektion (48 %), anæmi (36 %), leukopeni (35 %), træthed (35 %), blødning (33 %), pyreksi (32 %), kvalme (31 %), hovedpine (28 %), febril neutropeni (26 %), forhøjede transaminaser (26 %), abdominalsmerter (23 %), forhøjet gamma-glutamyltransferase (21 %) og hyperbilirubinæmi (21 %).
Hos patienter, der fik BESPONSA, var de mest almindelige alvorlige bivirkninger (≥ 2 %) infektion (23 %), febril neutropeni (11 %), blødning (5 %), abdominalsmerter (3 %), pyreksi (3 %), VOD/SOS (2 %) og træthed (2 %).
Bivirkninger
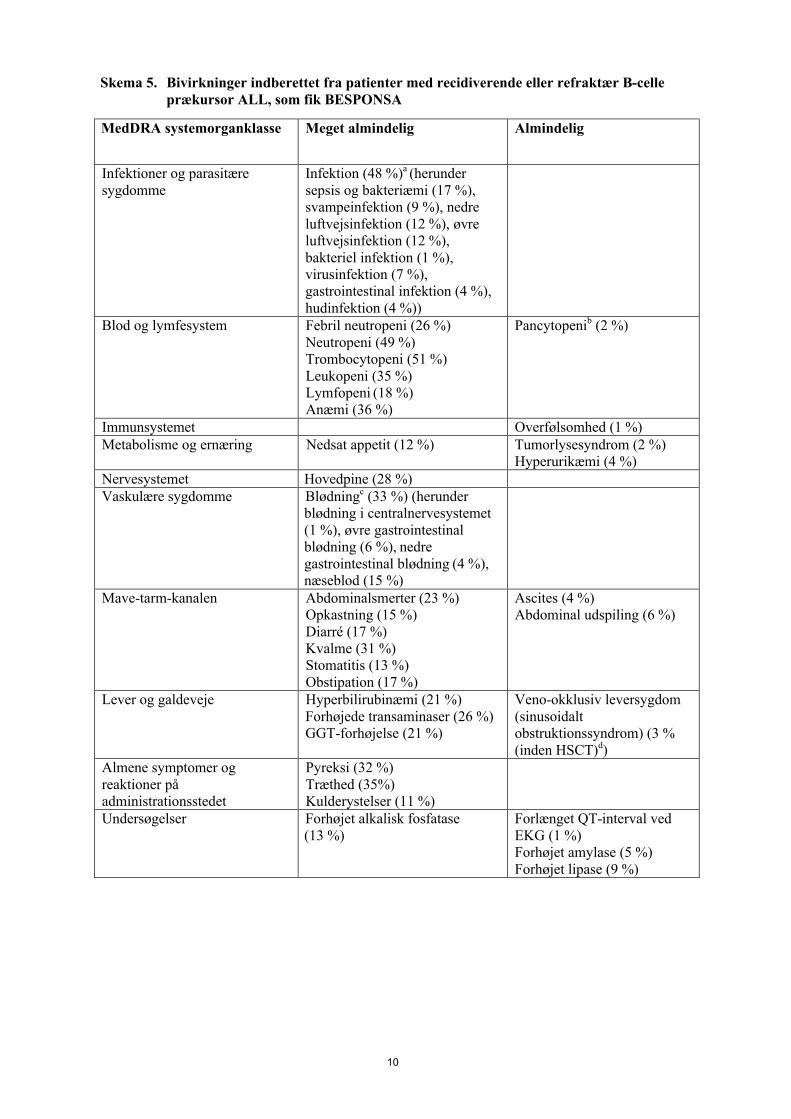
Skema 5 viser de bivirkninger, der blev indberettet fra patienter med recidiverende eller refraktær ALL, som fik BESPONSA.
Bivirkninger er opstillet efter systemorganklasse og hyppighed i følgende grupperinger: meget almindelig ( 1/10), almindelig ( 1/100 til < 1/10), ikke almindelig ( 1/1.000 til < 1/100), sjælden ( 1/10.000 til < 1/1.000), meget sjælden (< 1/10.000), ikke kendt (kan ikke estimeres ud fra forhåndenværende data). Inden for hver hyppighedsgruppe er bivirkningerne angivet efter faldende sværhedsgrad.
10
Skema 5. Bivirkninger indberettet fra patienter med recidiverende eller refraktær B-celle prækursor ALL, som fik BESPONSA
MedDRA systemorganklasse Meget almindelig Almindelig
Infektioner og parasitære sygdomme
Infektion (48 %)a (herunder sepsis og bakteriæmi (17 %), svampeinfektion (9 %), nedre luftvejsinfektion (12 %), øvre luftvejsinfektion (12 %), bakteriel infektion (1 %), virusinfektion (7 %), gastrointestinal infektion (4 %), hudinfektion (4 %))
Blod og lymfesystem Febril neutropeni (26 %)Neutropeni (49 %)Trombocytopeni (51 %)Leukopeni (35 %)Lymfopeni (18 %)Anæmi (36 %)
Pancytopenib (2 %)
Immunsystemet Overfølsomhed (1 %)Metabolisme og ernæring Nedsat appetit (12 %) Tumorlysesyndrom (2 %)
Hyperurikæmi (4 %)Nervesystemet Hovedpine (28 %)Vaskulære sygdomme Blødningc (33 %) (herunder
blødning i centralnervesystemet (1 %), øvre gastrointestinal blødning (6 %), nedre gastrointestinal blødning (4 %), næseblod (15 %)
Mave-tarm-kanalen Abdominalsmerter (23 %)Opkastning (15 %)Diarré (17 %)Kvalme (31 %)Stomatitis (13 %)Obstipation (17 %)
Ascites (4 %)Abdominal udspiling (6 %)
Lever og galdeveje Hyperbilirubinæmi (21 %)Forhøjede transaminaser (26 %)GGT-forhøjelse (21 %)
Veno-okklusiv leversygdom (sinusoidalt obstruktionssyndrom) (3 % (inden HSCT)d)
Almene symptomer og reaktioner på administrationsstedet
Pyreksi (32 %)Træthed (35%)Kulderystelser (11 %)
Undersøgelser Forhøjet alkalisk fosfatase (13 %)
Forlænget QT-interval ved EKG (1 %)Forhøjet amylase (5 %)Forhøjet lipase (9 %)

11
Skema 5. Bivirkninger indberettet fra patienter med recidiverende eller refraktær B-celle prækursor ALL, som fik BESPONSA
MedDRA systemorganklasse Meget almindelig Almindelig
Traumer, forgiftninger og behandlingskomplikationer
Infusionsrelateret reaktion (10 %)
Bivirkninger inkluderede behandlingskrævende hændelser af alle årsager, der indtraf ved eller efter cyklus 1, dag 1, inden for 42 dage efter sidste dosis BESPONSA, men inden start på ny kræftbehandling (inklusive HSCT).Foretrukne termer er anvendt i henhold til MedDRA (Medical Dictionary for Regulatory Activities) version 19.1.
Forkortelser: ALL = akut lymfoblastær leukæmi, EKG = elektrokardiogram, GGT = gamma-glutamyltransferase, HSCT = hæmatopoietisk stamcelletransplantation.a Infektion omfatter også andre typer infektion (11 %). Bemærk, at patienter kan have haft > 1 type infektion.b Pancytopeni omfatter følgende indberettede foretrukne termer: Knoglemarvssvigt, febril knoglemarvsaplasi
og pancytopeni.c Blødning omfatter også andre typer af blødning (17 %). Bemærk, at patienter kan have haft > 1 type
blødning.d VOD/SOS inkluderer 1 yderligere patient med veno-okklusiv leversygdom, der indtraf på dag 56 uden
intervenerende HSCT. VOD/SOS blev også indberettet hos 18 patienter efter en efterfølgende HSCT.
Beskrivelse af udvalgte bivirkninger
Levertoksicitet, herunder veno-okklusiv leversygdom/sinusoidalt obstruktionssyndrom (VOD/SOS)
I det kliniske pivotalstudie (N = 164) blev der indberettet VOD/SOS hos 23 (14 %) patienter herunder5 (3 %) patienter under studiebehandlingen eller under opfølgning uden intervenerende HSCT. Blandt de 79 patienter, som fortsatte med HSCT (hvoraf 8 fik yderligere behandling (salvage) efter behandling med BESPONSA inden fortsættelse med HSCT), blev der indberettet VOD/SOS hos18 (23 %) patienter. Fem ud af de 18 VOD/SOS-hændelser, der indtraf efter HSCT, var letale (se pkt. 5.1).
Der blev indberettet VOD/SOS op til 56 dage efter sidste dosis med inotuzumab ozogamicin uden intervenerende HSCT. Den gennemsnitlige tid fra HSCT til indtræden af VOD/SOS var 15 dage (interval: 3-57 dage). Ud af de 5 patienter, der fik VOD/SOS under behandling med inotuzumab ozogamicin, men uden intervenerende HSCT, fik 2 patienter også HSCT inden BESPONSA-behandlingen.
Ud af de patienter, der fortsatte med HSCT efter behandling med BESPONSA, blev der indberettet VOD/SOS hos 5/11 (46 %) patienter, der fik HSCT både inden og efter behandling med BESPONSA, 13/68 (19 %) patienter, der kun fik HSCT efter behandling med BESPONSA.
I forhold til andre risikofaktorer blev VOD/SOS rapporteret hos 6/11 (55 %) patienter, der fik et HSCT-konditionerende regime, der indeholdt 2 alkylerende midler og 9/53 (17 %) patienter, der fik et HSCT-konditionerende regime, der indeholdt 1 alkylerende middel, 7/17 (41 %) patienter, der var ≥ 55 år og 11/62 (18 %) patienter, der var < 55 år, og 7/12 (58 %) patienter med serumbilirubin ≥ ULN inden HSCT og hos 11/67 (16 %) patienter med serumbilirubin < ULN inden HSCT.
I pivotalstudiet (N = 164) blev der indberettet hyperbilirubinæmi og forhøjede transaminaser hos hhv. 35 (21 %) og 43 (26 %) patienter. Der blev indberettet grad ≥ 3 hyperbilirubinæmi og forhøjede transaminaser hos hhv. 9 (6 %) og 11 (7 %) patienter. Mediantiden til indtræden af hyperbilirubinæmi og forhøjede transaminaser var hhv. 73 dage og 29 dage.
Se pkt. 4.4 vedrørende klinisk behandling af levertoksicitet, herunder VOD/SOS.

12
Myelosuppression/cytopeni
I pivotalstudiet (N = 164) blev der indberettet trombocytopeni og neutropeni hos hhv. 83 (51 %) og 81 (49 %) patienter. Der blev indberettet grad 3 trombocytopeni og neutropeni hos hhv. 23 (14 %) og 33 (20 %) patienter. Der blev indberettet grad 4 trombocytopeni og neutropeni hos hhv. 46 (28 %) og 45 (27 %) patienter. Der blev indberettet febril neutropeni, som kan være livstruende, hos 43 (26 %) patienter.
Se pkt. 4.4 vedrørende klinisk behandling af myelosuppression/cytopeni.
Infektioner
I pivotalstudiet (N = 164) blev der indberettet infektioner, herunder alvorlige infektioner, hvoraf nogle var livstruende eller letale, hos 79 (48 %) patienter. Hyppigheden af følgende typer af specifikke infektioner var: sepsis og bakteriæmi (17 %), nedre luftvejsinfektion (12 %), øvre luftvejsinfektion (12 %), svampeinfektion (9 %), virusinfektion (7 %), gastrointestinal infektion (4 %), hudinfektion (4 %) og bakteriel infektion (1 %). Der blev indberettet letale infektioner, herunder lungebetændelse, neutropenisk sepsis, sepsis, septisk shock og sepsis pseudomonas hos 8 (5 %) patienter.
Se pkt. 4.4 vedrørende klinisk behandling af infektioner.
Blødning/hæmoragi
I pivotalstudiet (N = 164) blev der indberettet blødning/hæmoragiske hændelser, de fleste af mild sværhedsgrad, hos 54 (33 %) patienter. Hyppigheden af følgende typer af specifikke blødning/hæmoragiske hændelser var: næseblod (15 %), øvre gastrointestinal blødning (6 %), nedre gastrointestinal blødning (4 %), blødning i centralnervesystemet (CNS) (1 %). Der blev indberettet grad 3/4 blødning/hæmoragiske hændelser hos 8/164 (5 %) patienter. Der blev indberettet én grad 5 blødning/hæmoragisk hændelse (intra-abdominal blødning).
Se pkt. 4.4 vedrørende klinisk behandling af blødning/hæmoragiske hændelser.
Infusionsrelaterede reaktioner
I pivotalstudiet (N = 164) blev der indberettet infusionsrelaterede reaktioner hos 17 (10 %) patienter. Alle hændelser havde en sværhedsgrad på ≤ 2. Infusionsrelaterede reaktioner forekom generelt i cyklus 1 og kort efter endt infusion af inotuzumab ozogamicin, og de forsvandt spontant eller med medicinsk behandling.
Se pkt. 4.4 vedrørende klinisk behandling af infusionsrelaterede hændelser.
Tumorlysesyndrom (TLS)
I pivotalstudiet (N = 164) blev der indberettet TLS, som kan være livstruende eller letalt, hos 4/164 (2 %) patienter. Der blev indberettet grad 3/4 TLS hos 3 (2 %) patienter. TLS forekom generelt kort efter endt infusion af inotuzumab ozogamicin og forsvandt med medicinsk behandling.
Se pkt. 4.4 vedrørende klinisk behandling af TLS.
Forlængelse af QT-intervallet
I pivotalstudiet (N = 164) blev der målt maksimale stigninger i QT-interval korrigeret for hjertefrekvens vha. Fridericia-formlen (QTcF) ≥ 30 msek og ≥ 60 msek fra baseline hos henholdsvis30/162 (19 %) og 4/162 (3 %) patienter. En stigning i QTcF intervallet på > 450 msek blev observeret hos 26/162 (16 %) patienter. Ingen patienter havde en stigning i QTcF-intervallet på > 500 msek. Der blev indberettet forlænget QT-interval af grad 2 hos 2/164 (1 %) patienter. Der blev ikke indberettet forlænget QT-interval af grad ≥ 3 eller Torsades de Pointes.

13
Se pkt. 4.4 vedrørende regelmæssig måling af EKG og elektrolytter.
Forhøjet amylase og lipase
I pivotalstudiet (N = 164) blev der indberettet forhøjet amylase og lipase hos hhv. 8 (5 %) og 15 (9 %) patienter. Der blev indberettet forhøjet amylase og lipase af grad ≥ 3 hos hhv. 3 (2 %) og 7 (4 %) patienter.
Se pkt. 4.4. vedrørende regelmæssig monitorering af forhøjet amylase og lipase.
Immunogenicitet
I kliniske studier af BESPONSA til patienter med recidiverende eller refraktær ALL blev 7/236 (3 %) patienter testet positive for antistoffer mod inotuzumab ozogamicin. Ingen patienter blev testet positiv for neutraliserende antistoffer mod inotuzumab ozogamicin. Hos patienter, der blev testet positive for antistoffer mod inotuzumab ozogamicin, blev der ikke detekteret nogen indvirkning på clearance af BESPONSA baseret på populationsfarmakokinetisk analyse. Antallet af patienter var for lille til at vurdere virkningen af antistoffer mod inotuzumab ozogamicin på virkning og sikkerhed.
Indberetning af formodede bivirkninger
Når lægemidlet er godkendt, er indberetning af formodede bivirkninger vigtig. Det muliggør løbende overvågning af benefit/risk-forholdet for lægemidlet. Læger og sundhedspersonale anmodes om at indberette alle formodede bivirkninger via det nationale rapporteringssystem anført i Appendiks V.
4.9 Overdosering
I kliniske studier hos patienter med recidiverende eller refraktær ALL var den maksimale enkeltdosis og gentagne doser inotuzumab ozogamicin hhv. 0,8 mg/m2 og 1,8 mg/m2 pr. cyklus, givet som 3 opdelte doser på dag 1 (0,8 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2) (se pkt. 4.2). Overdosering kan give de samme bivirkninger som dem, der ses ved den anbefalede terapeutiske dosis (se pkt. 4.8).
I tilfælde af overdosering skal infusionen standses midlertidigt, og patienten skal kontrolleres for levertoksicitet og hæmatologisk toksicitet (se pkt. 4.2). Det bør overvejes at genoptage BESPONSA ved den korrekte, terapeutiske dosis, når al toksicitet er forsvundet.
5. FARMAKOLOGISKE EGENSKABER
5.1 Farmakodynamiske egenskaber
Farmakoterapeutisk klassifikation: Antineoplastiske midler, andre antineoplastiske stoffer, monoklonale antistoffer, ATC-kode: L01XC26.
Virkningsmekanisme
Inotuzumab ozogamicin er et ADC sammensat af et CD22-rettet monoklonalt antistof, der bindes kovalent til N-acetyl-gamma-calicheamicin dimethylhydrazid. Inotuzumab er et humaniseret immunoglobulin klasse G subtype 4 (IgG4) antistof, der specifikt genkender humant CD22. Det lille molekyle, N-acetyl-gamma-calicheamicin, er et cytotoksisk produkt.
N-acetyl-gamma-calicheamicin er kovalent forbundet med antistoffet med en syrelabil binding. Ikke-kliniske data tyder på, at BESPONSAs aktivitet mod cancer skyldes bindingen af ADC til tumorceller, med CD22-ekspression, efterfulgt af internalisering af ADC-CD22-komplekset og den intracellulære frigivelse af N-acetyl-gamma-calicheamicin dimethylhydrazid via hydrolytisk spaltning af bindingen.

14
Aktivering af N-acetyl-gamma-calicheamicin dimethylhydrazid inducerer dobbeltstrengede brud på DNA, hvorefter stop af cellecyklus og apoptotisk celledød induceres.
Klinisk virkning og sikkerhed
Patienter med recidiverende eller refraktær ALL, der havde fået 1 eller 2 tidligere behandlingsregimer for ALL - Studie 1
BESPONSAs sikkerhed og virkning hos patienter med recidiverende eller refraktær CD22-positivALL blev evalueret i et åbent, internationalt, multicenter fase 3-studie (Studie 1), hvor patienter blev randomiseret til at få BESPONSA (N = 164 (164 fik behandling)) eller investigators valg af kemoterapi (N = 162 (143 fik behandling)) specifikt fludarabin + cytarabin + granulocyt kolonistimulerendefaktor (FLAG) (N=102 (93 fik behandling)), mitoxantron/cytarabin (MXN/Ara-C) (N=38 (33 fik behandling)) eller højdosis cytarabin (HIDAC) (N=22 (17 fik behandling)).
Egnede patienter var ≥ 18 år og havde Philadelphia-kromosomnegativ (Ph-) eller Ph+ recidiverende eller refraktær B-celle CD-22 prækursor ALL.
CD22-ekspression blev vurderet ved hjælp af flowcytometri baseret på knoglemarvsaspirat. Hos patienter med utilstrækkeligt knoglemarvaspirat blev der testet på en perifer blodprøve. Hos patienter med både utilstrækkeligt knoglemarvsaspirat og utilstrækkelige cirkulerende blaster blev CD22-ekspression vurderet ved hjælp af immunhistokemi.
I det kliniske studie var sensitiviteten på visse lokale test lavere end den centrale laboratorietest. Der skal derfor anvendes validerede test, som har demonstreret høj sensitivitet.
Alle patienter skulle have ≥ 5% knoglemarvsblaster og tidligere have fået 1 eller 2 induktionskemoterapiregimer for ALL. Patienter med Ph+ B celle prækursor ALL skulle haft behandlingssvigt med 1 anden- eller tredjegenerations TKI og standardkemoterapi. Skema 1 (se pkt. 4.2) viser de doseringsregimer, der blev brugt til at behandle patienterne.
De to primære endepunkter var CR/CRi, som blev vurderet af en blindet uafhængig bedømmelseskomite for endepunkter, og samlet overlevelse (OS). De sekundære endepunkter inkluderede MRD-negativitet, varighed af remission (DoR), HSCT-rate og progressionsfri overlevelse (PFS). Den primære analyse af CR/CRi og MRD-negativitet blev foretaget hos de første 218 randomiserede patienter, og analyse for OS, PFS, DoR, og HSCT-rate blev foretaget hos alle 326 randomiserede patienter.
Ud af 326 randomiserede patienter (ITT-population) havde 215 (66%) af patienterne fået et tidligere behandlingsregime og 108 (33%) af patienterne havde fået 2 tidligere behandlingsregimer for ALL. Gennemsnitsalder var 47 år (interval: 18-79 år), varigheden af første remission var < 12 måneder for 206 (63%) af patienterne og 55 (17%) af patienterne havde fået HSCT før behandling med BESPONSA eller investigators valg af kemoterapi. De 2 behandlingsgrupper var generelt i overensstemmelse med hensyn til demografi og sygdomskarakteristika ved baseline. I alt havde 276 (85%) af patienterne Ph- ALL. Ud af 49 (15%) patienter med Ph+ havde 4 ikke tidligere fået en TKI, 28 patienter havde fået 1 TKI tidligere og 17 patienter havde fået 2 TKI’ere tidligere. Den hyppigste anvendte TKI (42 patienter) var dasatinib efterfulgt af imatinib (24 patienter).
Baseline-karakteristika var ens hos de 218 først randomiserede patienter.
Af de 326 patienter (ITT-populationen) havde 253 patienter prøver, der kunne testes både lokalt og centralt for CD22. Centrale og lokale laboratorietest viste ≥70% CD22-positive leukæmiske blaster ved baseline hos henholdsvis 231/253 (91,3%) og 130/253 (51,4%) af patienterne.
15
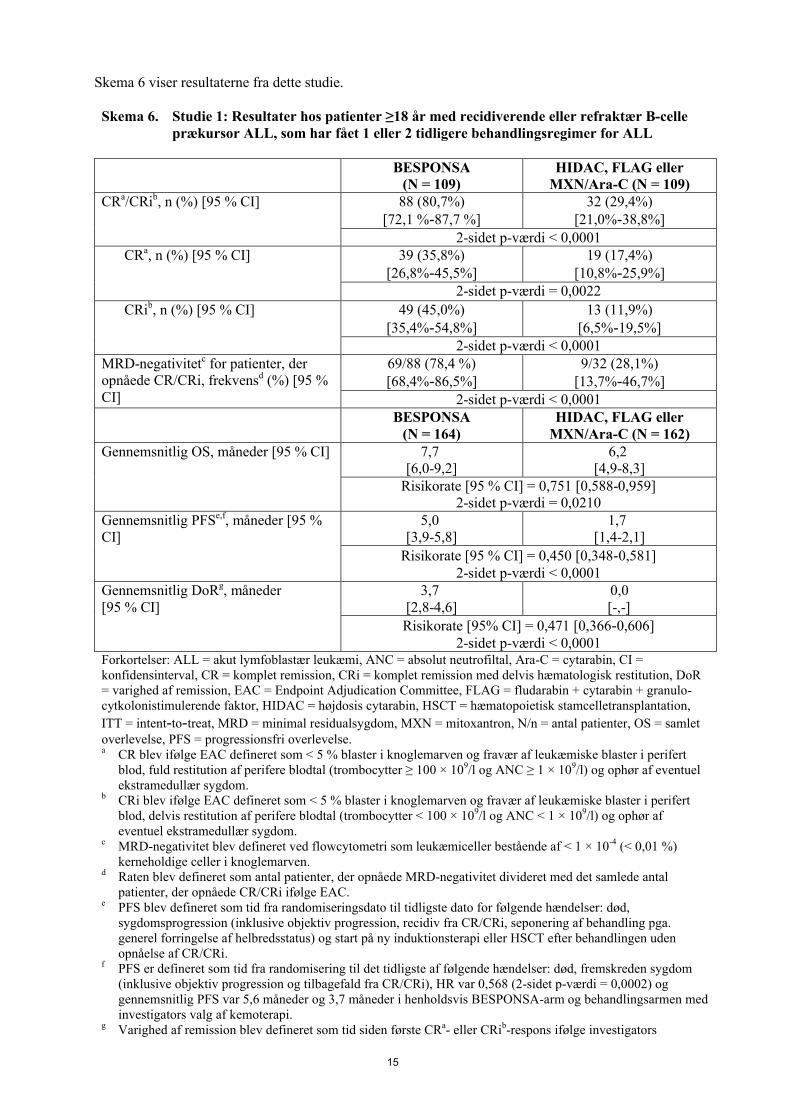
Skema 6 viser resultaterne fra dette studie.
Skema 6. Studie 1: Resultater hos patienter ≥18 år med recidiverende eller refraktær B-celle prækursor ALL, som har fået 1 eller 2 tidligere behandlingsregimer for ALL
BESPONSA(N = 109)
HIDAC, FLAG eller MXN/Ara-C (N = 109)
CRa/CRib, n (%) [95 % CI] 88 (80,7%)[72,1 %-87,7 %]
32 (29,4%)[21,0%-38,8%]
2-sidet p-værdi < 0,0001CRa, n (%) [95 % CI] 39 (35,8%)
[26,8%-45,5%]19 (17,4%)
[10,8%-25,9%]2-sidet p-værdi = 0,0022
CRib, n (%) [95 % CI] 49 (45,0%)[35,4%-54,8%]
13 (11,9%)[6,5%-19,5%]
2-sidet p-værdi < 0,0001MRD-negativitetc for patienter, der opnåede CR/CRi, frekvensd (%) [95 % CI]
69/88 (78,4 %)[68,4%-86,5%]
9/32 (28,1%)[13,7%-46,7%]
2-sidet p-værdi < 0,0001BESPONSA
(N = 164)HIDAC, FLAG eller
MXN/Ara-C (N = 162)Gennemsnitlig OS, måneder [95 % CI] 7,7
[6,0-9,2]6,2
[4,9-8,3]Risikorate [95 % CI] = 0,751 [0,588-0,959]
2-sidet p-værdi = 0,0210Gennemsnitlig PFSe,f, måneder [95 % CI]
5,0[3,9-5,8]
1,7[1,4-2,1]
Risikorate [95 % CI] = 0,450 [0,348-0,581]2-sidet p-værdi < 0,0001
Gennemsnitlig DoRg, måneder [95 % CI]
3,7[2,8-4,6]
0,0 [-,-]
Risikorate [95% CI] = 0,471 [0,366-0,606]2-sidet p-værdi < 0,0001
Forkortelser: ALL = akut lymfoblastær leukæmi, ANC = absolut neutrofiltal, Ara-C = cytarabin, CI = konfidensinterval, CR = komplet remission, CRi = komplet remission med delvis hæmatologisk restitution, DoR = varighed af remission, EAC = Endpoint Adjudication Committee, FLAG = fludarabin + cytarabin + granulo-cytkolonistimulerende faktor, HIDAC = højdosis cytarabin, HSCT = hæmatopoietisk stamcelletransplantation,
ITT = intent-to-treat, MRD = minimal residualsygdom, MXN = mitoxantron, N/n = antal patienter, OS = samlet overlevelse, PFS = progressionsfri overlevelse.a CR blev ifølge EAC defineret som < 5 % blaster i knoglemarven og fravær af leukæmiske blaster i perifert
blod, fuld restitution af perifere blodtal (trombocytter ≥ 100 × 109/l og ANC ≥ 1 × 109/l) og ophør af eventuel ekstramedullær sygdom.
b CRi blev ifølge EAC defineret som < 5 % blaster i knoglemarven og fravær af leukæmiske blaster i perifert blod, delvis restitution af perifere blodtal (trombocytter < 100 × 109/l og ANC < 1 × 109/l) og ophør af eventuel ekstramedullær sygdom.
c MRD-negativitet blev defineret ved flowcytometri som leukæmiceller bestående af < 1 × 10-4 (< 0,01 %) kerneholdige celler i knoglemarven.
d Raten blev defineret som antal patienter, der opnåede MRD-negativitet divideret med det samlede antal patienter, der opnåede CR/CRi ifølge EAC.
e PFS blev defineret som tid fra randomiseringsdato til tidligste dato for følgende hændelser: død, sygdomsprogression (inklusive objektiv progression, recidiv fra CR/CRi, seponering af behandling pga. generel forringelse af helbredsstatus) og start på ny induktionsterapi eller HSCT efter behandlingen uden opnåelse af CR/CRi.
f PFS er defineret som tid fra randomisering til det tidligste af følgende hændelser: død, fremskreden sygdom (inklusive objektiv progression og tilbagefald fra CR/CRi), HR var 0,568 (2-sidet p-værdi = 0,0002) og gennemsnitlig PFS var 5,6 måneder og 3,7 måneder i henholdsvis BESPONSA-arm og behandlingsarmen med investigators valg af kemoterapi.
g Varighed af remission blev defineret som tid siden første CRa- eller CRib-respons ifølge investigators

16
vurdering til datoen for en PFS-hændelse eller censureringsdatoen, hvis der ikke blev dokumenteret en PFS-hændelse. Analyse var baseret på ITT-populationen med patienter uden remission, der blev givet en varighed på nul og blev anset som en hændelse.
Blandt de første 218 randomiserede patienter opnåede 64/88 (73 %) og 21/88 (24 %) af responderende patienter ifølge EAC CR/CRi i hhv. cyklus 1 og 2 i BESPONSA-armen. Ingen yderligere patienter opnåede CR/CRi efter cyklus 3 i BESPONSA-armen.
Fund vedrørende CR/CRi- og MRD-negativitet fra de første 218 randomiserede patienter stemte overens med de resultater, der sås hos alle 326 randomiserede patienter.
Blandt alle 326 randomiserede patienter var sandsynligheden for overlevelse efter 24 måneder 22,8 % i inotuzumab BESPONSA-armen og 10 % i behandlingsarmen med investigators valg af kemoterapi.
I alt fik 79/164 (48,2 %) af patienterne i BESPONSA-armen og 36/162 (22,2 %) af patienterne i behandlingsarmen med investigators valg af kemoterapi opfølgende HSCT. Dette inkluderer 70 og 18 patienter i henholdsvis BESPONSA-armen og i behandlingsarmen med investigators valgs af kemoterapi som fortsatte direkte til HSCT. Hos de patienter, som fortsatte direkte til HSCT, var mediantiden mellem sidste dosis af inotuzomab ozogamicin og HSCT 4,8 uger (interval: 1-19 uger). Der sås en forbedring i OS for BESPONSA versus investigators valg af kemoterapiarm, hos patienter som fik HSCT. På trods af større hyppighed af tidlig død post-HSCT (på dag 100) i BESPONSA-armen, sås en sen-overlevelsesfordel ved BESPONSA. Hos patienter, som fik en opfølgende HSCT-behandling, var den mediane OS 11,9 måneder (95% CI: 9,2-20,6) for BESPONSA versus 19,8måneder (95% CI: 14,6-26,7) for investigators valg af kemoterapi. Sandsynligheden for overlevelse ved 24 måneder var 38,0 % (95% CI: 27,4-48,5) versus 35,5 % (95 % CI: 20,1-51,3) for henholdsvis BESPONSA og investigators valg af kemoterapi. I tilføjelse hertil, var sandsynligheden for overlevelse ved 24 måneder i BESPONSA-armen 38,0 % (95% CI: 27,4-48,5) for patienter som fik opfølgende HSCT sammenlignet med 8,0 % (95 % CI: 3,3-15,3) for patienter, som ikke fik en opfølgende HSCT.
BESPONSA forbedrede OS i forhold til investigators valg af kemoterapi for alle stratificeringsfaktorer, herunder varighed af første remission ≥ 12 måneder, status af salvage-behandling 1 og alder < 55 år ved randomisering. Der fandtes også en tendens til forbedret OS med BESPONSA for patienter med andre prognostiske faktorer (Ph-, ingen tidligere HSCT, 90 % positivitet for CD22 i leukæmiske blaster ved baseline, ingen perifere blaster ved baseline og baseline hæmoglobin ≥ 10 g/dl, baseret på eksplorativ analyse. Patienter med MLL-genrearrangement (mixed lineage leukemia), inklusive t(4;11), der generelt havde lavere CD22 ekspression inden behandlingen, havde et dårligere OS-resultat efter behandling med BESPONSA eller investigators valg af kemoterapi.
For patientrapporterede resultater var de fleste scorer for funktionsdygtighed og symptomer til fordel for BESPONSA sammenlignet med investigators valg af kemoterapi. For patientrapporterede resultater, som blev målt vha. spørgeskemaet om livskvalitet fra European Organisation for Research and Treatment of Cancer (EORTC QLQ-C30), var BESPONSA signifikant bedre estimeret gennemsnitlig efter-baseline-scorer (hhv. BESPONSA og investigators valg af kemoterapi) for funktionsdygtighed ift. social rolle (64,7 versus 53,4: forbedringsgrad lille), fysisk funktionsdygtighed (75,0 versus 68,1: forbedringsgrad lille), social funktionsdygtighed (68,1 versus 59,8: forbedringsgrad medium) og appetittab (17,6 versus 26,3: forbedringsgrad lille) sammenlignet med investigators valg af kemoterapi. Der var en tendens til fordel for BESPONSA, forbedringsgrad lille, for estimerede gennemsnitlige efter-baseline-scorer (hhv. BESPONSA og investigators valg) for overordnet helbredsstatus/livskvalitet (QoL) (62,1 versus 57,8), kognitiv funktion (85,3 versus 82,5), dyspnø (14,7 versus 19,4), diarré (5,9 versus 8,9), træthed (35,0 versus 39,4). Der var en tendens til fordel for RESPONSA for estimerede gennemsnitlige efter-baseline-scorer, som blev målt vha. spørgeskemaet EuroQoL 5 Dimension (EQ-5D) (hhv. BESPONSA og investigators valg af kemoterapi) for EQ-5D-indekset (0,80 versus 0,76, minimal betydende forskel for kræft = 0,06).
17
Patienter med recidiverende eller refraktær ALL, der havde fået 2 eller flere tidligere behandlingsregimer for ALL - Studie 2
BESPONSAs sikkerhed og virkning blev evalueret i et enkeltarmet, åbent, multicenter fase 1/2-studie (Studie 2). Egnede patienter var ≥ 18 år med recidiverende eller refraktær B-celle prækursor ALL.
Af de 93 screenede patienter blev 72 allokeret til studiemedicin og behandlet med BESPONSA. Gennemsnitsalder var 45 år (interval: 20-79), 76,4% havde fået ≥2 tidligere behandlinger, 31,9% havde tidligere fået HSCT og 22,2% var pH+. De hyppigste årsager til ophør af behandlingen var: sygdomsprogression/recidiv (30 (41,7%)), resistent sygdom (4 (5,6%)), HSCT (18 (25,0%)) og bivirkninger (13 (18,1%)).
I studiets fase 1 fik 37 patienter BESPONSA ved en total dosis på 1,2 mg/m2 (n = 3) 1,6 mg/m2
(n = 12) eller 1,8 mg/m2 (n = 22). Den anbefalede BESPONSA-dosis blev fastlagt til 1,8 mg/m2/cyklus administreret ved en dosis på 0,8 mg/m2 på dag 1 og 0,5 mg/m2 på dag 8 og 15 i en cyklus på 28 dage med dosisnedsættelse efter opnåelse af CR/CRi.
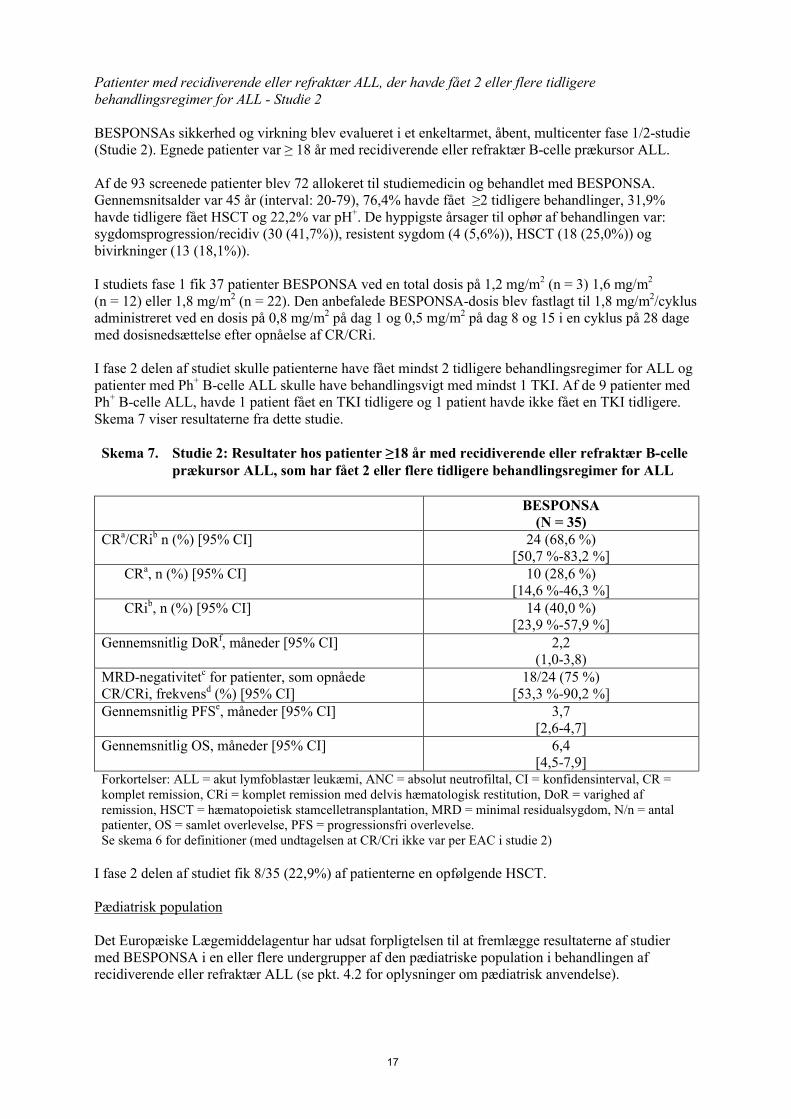
I fase 2 delen af studiet skulle patienterne have fået mindst 2 tidligere behandlingsregimer for ALL og patienter med Ph+ B-celle ALL skulle have behandlingsvigt med mindst 1 TKI. Af de 9 patienter med Ph+ B-celle ALL, havde 1 patient fået en TKI tidligere og 1 patient havde ikke fået en TKI tidligere. Skema 7 viser resultaterne fra dette studie.
Skema 7. Studie 2: Resultater hos patienter ≥18 år med recidiverende eller refraktær B-celle prækursor ALL, som har fået 2 eller flere tidligere behandlingsregimer for ALL
BESPONSA(N = 35)
CRa/CRib n (%) [95% CI] 24 (68,6 %)[50,7 %-83,2 %]
CRa, n (%) [95% CI] 10 (28,6 %)[14,6 %-46,3 %]
CRib, n (%) [95% CI] 14 (40,0 %)[23,9 %-57,9 %]
Gennemsnitlig DoRf, måneder [95% CI] 2,2(1,0-3,8)
MRD-negativitetc for patienter, som opnåede CR/CRi, frekvensd (%) [95% CI]
18/24 (75 %)[53,3 %-90,2 %]
Gennemsnitlig PFSe, måneder [95% CI] 3,7[2,6-4,7]
Gennemsnitlig OS, måneder [95% CI] 6,4[4,5-7,9]
Forkortelser: ALL = akut lymfoblastær leukæmi, ANC = absolut neutrofiltal, CI = konfidensinterval, CR = komplet remission, CRi = komplet remission med delvis hæmatologisk restitution, DoR = varighed af remission, HSCT = hæmatopoietisk stamcelletransplantation, MRD = minimal residualsygdom, N/n = antal patienter, OS = samlet overlevelse, PFS = progressionsfri overlevelse.Se skema 6 for definitioner (med undtagelsen at CR/Cri ikke var per EAC i studie 2)
I fase 2 delen af studiet fik 8/35 (22,9%) af patienterne en opfølgende HSCT.
Pædiatrisk population
Det Europæiske Lægemiddelagentur har udsat forpligtelsen til at fremlægge resultaterne af studier med BESPONSA i en eller flere undergrupper af den pædiatriske population i behandlingen af recidiverende eller refraktær ALL (se pkt. 4.2 for oplysninger om pædiatrisk anvendelse).

18
5.2 Farmakokinetiske egenskaber
Hos patienter med recidiverende eller refraktær ALL behandlet med inotuzumab ozogamicin ved den anbefalede startdosis på 1,8 mg/m2/cyklus (se pkt. 4.2) blev steady state-eksponering opnået i behandlingscyklus 4. Den gennemsnitlige (SD) maksimale serumkoncentration (Cmax) af inotuzumab ozogamicin var 308 ng/ml (362). Det gennemsnitlige (SD) simulerede totale område under koncentrations-tidskurven (AUC) pr. cyklus ved steady state var 100 mkgt/ml (32,9).
Fordeling
Bindingen in vitro af N-acetyl-gamma-calicheamicin dimethylhydrazid til humane plasmaproteiner er ca. 97 %. N-acetyl-gamma-calicheamicin dimethylhydrazid in vitro er et substrat af P-glykoprotein (P-gp). Hos mennesker var det totale fordelingsvolumen af inotuzumab ozogamicin ca. 12 liter.
Biotransformation
N-acetyl-gamma-calicheamicin dimethylhydrazid in vitro blev primært metaboliseret via non-enzymatisk reduktion. Hos mennesker var serumniveauerne af N-acetyl-gamma-calicheamicin dimethylhydrazid typisk under kvantificeringsgrænsen (50 pg/ml), men sporadisk målbare niveauer af ikke-konjungeret calicheamicin på op til 276 pg/ml fandtes hos nogle patienter.
Elimination
Inotuzumab ozogamicins farmakokinetik er velkarakteriseret af en 2-kompartmentmodel med lineære og tidsbestemte clearance-komponenter. Clearance af inotuzumab ozogamicin ved steady state hos 234 patienter med recidiverende eller refraktær ALL var 0,0333 l/t, og den endelige halveringstid (t½) ved afslutningen af cyklus 4 var ca. 12,3 dage. Efter administration af multiple doser sås en 5,3 gange akkumulering af inotuzumab ozogamicin mellem cyklus 1 og 4.
Populationsfarmakokinetisk analyse af 765 patienter indikerer, at legemsoverfladearealet påvirkede disposition af inotuzumab ozogamicin i betydelig grad. Dosis af inotuzumab ozogamicin administreres baseret på legemsoverfladearealet (se pkt. 4.2).
Alder, race og køn
Baseret på populationsfarmakokinetisk analyse påvirker alder, race og køn ikke i betydelig grad fordelingen af inotuzumab ozogamicin.
Nedsat leverfunktion
Der er ikke udført studier af inotuzumab ozogamicin hos patienter med nedsat leverfunktion.
Baseret på en populationsfarmakokinetisk analyse af 765 patienter var clearance af inotuzumab ozogamicin hos patienter med nedsat leverfunktion, defineret af National Cancer Institute Organ Dysfunction Working Group (NCI ODWG) kategori B1 (total bilirubin ≤ ULN og ASAT > ULN, n = 133) eller B2 (total bilirubin > 1,0-1,5 × ULN og ASAT af alle niveauer, n = 17) den samme som hos patienter med normal leverfunktion (total bilirubin/ASAT ≤ ULN, n = 611) (se pkt. 4.2). Hos3 patienter med nedsat leverfunktion, defineret af NCI ODWG kategori C (total bilirubin > 1,5-3 × ULN og ASAT på alle niveauer) og 1 patient med nedsat leverfunktion, defineret af NCI ODWG kategory D (total bilirubin > 3 × ULN og ASAT af alle niveauer) lod inotuzumab ozogamicin-clearance ikke til at være reduceret.
Nedsat nyrefunktion
Der er ikke udført studier af inotuzumab ozogamicin hos patienter med nedsat nyrefunktion.

19
Baseret på populationsfarmakokinetiske analyser af 765 patienter var clearance af inotuzumab ozogamicin hos patienter med let nedsat nyrefunktion (CLcr 60-89 ml/min, n = 237), moderat nedsat nyrefunktion (CLcr 30-59 ml/min, n = 122) eller svært nedsat nyrefunktion (CLcr 15-29 ml/min, n = 4) den samme som hos patienter med normal nyrefunktion (CLcr ≥ 90 ml/min, n = 402) (se pkt. 4.2). Inotuzumab ozogamicin er ikke undersøgt hos patienter med nyresygdom i slutstadiet (se pkt. 4.2).
Hjerte-elektrofysiologi
Farmakokinetisk/farmakodynamisk populationsanalyse indikerede, at der var en korrelation mellem forøgede serumkoncentrationer af inotuzumab ozogamicin og forlængelse af QTc-intervaller hos patienter med ALL og patienter med non-Hodgkin lymfom (NHL). Medianen (øvre grænse af 95% CI) for ændring i QTcF ved en subterapeutisk Cmax koncentration var 3,87 msek (7,54 msek).
I et randomiseret klinisk studie af patienter med recidiverende eller refraktær ALL (Studie 1) blev der målt maksimale stigninger i QTcF interval på ≥ 30 msek og ≥ 60 msek fra baseline hos henholdsvis 30/162 (19 %) og 4/162 (3 %) patienter i inotuzumab ozogamicin-armen henholdsvis 18/124 (15 %) og 3/124 (2 %) i behandlingsarmen med investigators valg af kemoterapi. Stigninger i QTcF interval på > 450 msek og > 500 msek blev observeret hos henholdsvis 26/162 (16 %) og ingen af patienterne i inotuzumab ozogamicin-armen versus 12/124 (10 %) og 1/124 (1 %) af patienterne i behandlingsarmen med investigators valg af kemoterapi (se pkt. 4.8).
5.3 Prækliniske sikkerhedsdata
Toksicitet ved gentagne doser
De primære målorganer hos dyr inkluderede leveren, knoglemarven og lymfoide organer med associerede hæmatologiske ændringer, nyrerne og nervesystemet. Andre observerede ændringer inkluderede påvirkning af reproduktionsorganerne hos hanner og hunner (se nedenfor) og præ-neoplastiske og neoplastiske leverlæsioner (se nedenfor). De fleste påvirkninger var reversible eller delvist reversible, undtagen påvirkningerne i leveren og nervesystemet. Relevansen for mennesker, af de irreversible fund hos dyr, kendes ikke.
Genotoksicitet
Inotuzumab ozogamicin var klastogent in vivo i knoglemarven hos hanmus. Dette stemmer overens med den kendte induktion af brud på DNA af calicheamicin. N-acetyl-gamma-calicheamicin dimethylhydrazid (det cytotoksiske stof, der frigives fra inotuzumab ozogamicin) var mutagent i en in vitro bakteriel mutationstest (Ames).
Carcinogenicitet
Der er ikke udført carcinogenicitetsstudier med inotuzumab ozogamicin. I toksicitetsstudier udviklede rotter ovalcellehyperplasi, ændrede hepatocellulære foci og hepatocellulære adenomer i leveren ved ca. 0,3 gange den kliniske eksponering hos mennesker baseret på AUC. Hos 1 abe blev der detekteret ændring af et hepatocellulært focus ved ca. 3,1 gange den kliniske eksponering hos mennesker baseret på AUC ved afslutningen af den 26 uger lange doseringsperiode. Relevansen for mennesker af disse fund hos dyr kendes ikke.
Reproduktionstoksicitet
Administration af inotuzumab ozogamicin til hunrotter ved den maternelle toksiske dosis (ca. 2,3 gange den kliniske eksponering hos mennesker baseret på AUC) inden parring og i løbet af den første gestationssuge resulterede i embryoføtal toksicitet, inklusive øget resorption og færre levedygtige embryoner. Den maternelle toksiske dosis (ca. 2,3 gange den kliniske eksponering hos mennesker baseret på AUC) resulterede også i væksthæmning hos fosteret, herunder lavere fostervægt og forsinket ossifikation af skelettet. Der forekom også let væksthæmning hos rottefostre ved ca.

20
0,4 gange den kliniske eksponering hos mennesker baseret på AUC.
Baseret på ikke-kliniske fund anses inotuzumab ozogamicin for at have potentiale til at påvirke reproduktion og fertilitet hos mænd og kvinder (se pkt. 4.6). I toksicitetsstudier med gentagne doser hos rotter og aber omfattede de fund, der vedrørte reproduktionsevnen hos hunner, atrofi af ovarier, uterus, vagina og mælkekirtler. Niveauet for ingen observeret bivirkning (NOAEL) for påvirkningen af reproduktionsorganer hos hunrotter og -aber var hhv. ca. 2,2 og 3,1 gange den kliniske eksponering hos mennesker baseret på AUC. I toksicitetsstudier med gentagne doser hos rotter inkluderede fund vedrørende reproduktion hos hanner degeneration af testiklerne associeret med hypospermi og atrofi af prostata og sædblæren. NOAEL blev ikke identificeret for påvirkning af reproduktionsorganerne hos handyr, som blev observeret ved ca. 0,3 gange den kliniske eksponering hos mennesker baseret på AUC (se pk t. 4.6).
6. FARMACEUTISKE OPLYSNINGER
6.1 Hjælpestoffer
SukrosePolysorbat 80NatriumchloridTrometamol
6.2 Uforligeligheder
Da der ikke foreligger studier af eventuelle uforligeligheder, må dette lægemiddel ikke blandes med andre lægemidler end dem, der er anført under pkt. 6.6.
6.3 Opbevaringstid
Uåbnet hætteglas
4 år
Rekonstitueret opløsning
BESPONSA indeholder ikke bakteriostatiske konserveringsmidler. Den rekonstituerede opløsning skal bruges med det samme. Hvis den rekonstituerede opløsning ikke kan bruges med det samme, kan den opbevares i op til 4 timer i køleskab (2-8 °C). Beskyttes mod lys. Må ikke nedfryses.
Fortyndet opløsning
Den fortyndede opløsning skal bruges straks eller opbevares ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.
6.4 Særlige opbevaringsforhold
Opbevares i køleskab (2 °C-8 °C). Må ikke nedfryses. Opbevares i den originale karton for at beskytte mod lys.
Opbevaringsforhold efter rekonstitution og fortynding, se pkt. 6.3.

21
6.5 Emballagetype og pakningsstørrelser
Type I, ravfarvet hætteglas med en prop af chlorobutylgummi og flip off-hætte med krympeforsegling indeholdende 1 mg pulver.
Hver karton indeholder 1 hætteglas.
6.6 Regler for bortskaffelse og anden håndtering
Vejledning i rekonstituering, fortynding og administration
Anvend aseptisk teknik under rekonstituering og fortynding. Inotuzumab ozogamicin (som har en densitet på 1,02 g/ml ved 20 °C/68 °F) er lysfølsomt og skal beskyttes mod ultraviolet lys under rekonstituering, fortynding og administration.
Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding.
Rekonstituering
Beregn dosis (mg) og antallet af nødvendige hætteglas BESPONSA. Rekonstituér hvert 1 mg hætteglas med 4 ml vand til injektionsvæske for at få en
engangsopløsning på 0,25 mg/ml BESPONSA. Hvirvl forsigtigt glasset for at fremme opløsning. Må ikke rystes. Se efter, om den rekonstituerede opløsning indeholder partikler eller er misfarvet. Den
rekonstituerede opløsning skal være klar til let uklar, farveløs og i det væsentlige fri for synlige fremmedlegemer. Hvis der ses partikler eller misfarvning, må opløsningen ikke anvendes.
BESPONSA indeholder ikke bakteriostatiske konserveringsmidler. Den rekonstituerede opløsning skal bruges med det samme. Hvis den rekonstituerede opløsning ikke kan bruges med det samme, kan den opbevares i køleskab (2-8 °C) i op til 4 timer. Beskyttes mod lys. Må ikke nedfryses.
Fortynding
Beregn det volumen af rekonstitueret opløsning, der er nødvendig for at opnå den korrekte dosis i forhold til patientens legemsoverfladeareal. Træk denne mængde op af hætteglasset/-glassene vha. en sprøjte. Beskyttes mod lys. Eventuelt ubrugt, resterende rekonstitueret opløsning i hætteglasset skal kasseres.
Kom den rekonstituerede opløsning i en infusionsbeholder med natriumchloridopløsning 9 mg/ml (0,9 %) til injektion, så det totale nominelle volumen er 50 ml. Slutkoncentrationen skal være mellem 0,01-0,1 mg/ml. Beskyttes mod lys. Det anbefales at bruge en infusionsbeholder af polyvinylchlorid (PVC) (med eller uden di(2-ethylhexyl)phthalat (DEHP)), polyolefin (polypropylen og/eller polyethylen) eller ethylenvinylacetat (EVA).
Vend forsigtigt infusionsbeholderen om for at blande den fortyndede opløsning. Må ikke rystes.
Den fortyndede opløsning skal bruges straks eller opbevares ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.
Administration
Hvis den fortyndede opløsning opbevares i køleskab (2-8 °C), skal den stå ved stuetemperatur(20-25 °C) i ca. 1 time inden administration.

22
Filtration af den fortyndede opløsning er ikke nødvendigt. Hvis den fortyndede opløsning imidlertid filtreres, anbefales det at anvende polyethersulfon (PES)-, polyvinylidenfluorid (PVDF)- eller hydrofile polysulfon (HPS)-baserede filtre. Der må ikke anvendes filtre fremstillet af nylon eller blandede celluloseestere (MCE).
Infusionsposen skal beskyttes mod lys ved hjælp af afskærmning, der beskytter mod ultraviolet lys (dvs. ravgule, mørkebrune eller grønne poser eller aluminiumsfolie) under infusion. Infusionsslangen behøver ingen beskyttelse mod lys.
Den fortyndede opløsning infunderes over 1 time ved en hastighed på 50 ml/time ved stuetemperatur (20-25 °C). Beskyttes mod lys. Det anbefales at bruge infusionsslanger fremstillet af PVC (med eller uden DEHP), polyolefin (polypropylen og/eller polyethylen) eller polybutadien.
BESPONSA må ikke blandes eller administreres som infusion med andre lægemidler.
Skema 8 viser opbevaringstider og -forhold for rekonstituering, fortynding og administration af BESPONSA.
8. Opbevaringstider og -forhold for rekonstitueret og fortyndet BESPONSA-opløsning
Maksimal tid fra rekonstitution til fuldendt administration ≤ 8 timera
Rekonstitueret opløsning
Rekonstitueret opløsningEfter start på fortynding Administration
Den rekonstituerede opløsning skal bruges straks eller efter opbevaring i køleskab (2-8 °C) i op til 4 timer. Beskyttes mod lys. Må ikke nedfryses.
Den fortyndede opløsning skal bruges straks eller efter opbevaring ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.
Hvis den fortyndede opløsning opbevares i køleskab (2-8 °C), skal den stå ved
stuetemperatur (20-25 °C) i ca. 1 time inden administration. Administrer den fortyndede opløsning som infusion over 1 time ved en hastighed på 50 ml/time ved stuetemperatur (20-25 °C). Beskyttes mod lys.
a Med ≤ 4 timer mellem rekonstituering og fortynding.
Bortskaffelse
BESPONSA er kun til engangsbrug.
Ikke anvendt lægemiddel samt affald heraf skal bortskaffes i henhold til lokale retningslinjer.
7. INDEHAVER AF MARKEDSFØRINGSTILLADELSEN
Pfizer Europe MA EEIGBoulevard de la Plaine 171050 BruxellesBelgien
8. MARKEDSFØRINGSTILLADELSESNUMMER (-NUMRE)
EU/1/17/1200/001

23
9. DATO FOR FØRSTE MARKEDSFØRINGSTILLADELSE/FORNYELSE AFTILLADELSEN
Dato for første markedsføringstilladelse: 29. juni 2017
10. DATO FOR ÆNDRING AF TEKSTEN
Yderligere oplysninger om dette lægemiddel findes på Det Europæiske Lægemiddelagenturs hjemmeside http://www.ema.europa.eu.

24
BILAG II
A. FREMSTILLER AF DET BIOLOGISK AKTIVE STOF OG FREMSTILLER ANSVARLIG FOR BATCHFRIGIVELSE
B. BETINGELSER ELLER BEGRÆNSNINGER VEDRØRENDE UDLEVERING OG ANVENDELSE
C. ANDRE FORHOLD OG BETINGELSER FOR MARKEDSFØRINGSTILLADELSEN
D. BETINGELSER ELLER BEGRÆNSNINGER MED HENSYN TIL SIKKER OG EFFEKTIV ANVENDELSE AF LÆGEMIDLET

25
A. FREMSTILLER AF DET BIOLOGISK AKTIVE STOF OG FREMSTILLER ANSVARLIG FOR BATCHFRIGIVELSE
Navn og adresse på fremstilleren af det biologisk aktive stof
Wyeth Pharmaceutical Division of Wyeth Holdings LLC, 401 North Middletown Road, Pearl River, New York (NY) 10965USA
Navn og adresse på den fremstiller, der er ansvarlig for batchfrigivelse
Pfizer Ireland PharmaceuticalsGrange Castle Business ParkClondalkinDublin 22Irland
B. BETINGELSER ELLER BEGRÆNSNINGER VEDRØRENDE UDLEVERING OG ANVENDELSE
Lægemidlet må kun udleveres efter ordination på en recept udstedt af en begrænset lægegruppe (se bilag I: Produktresumé, pkt. 4.2).
C. ANDRE FORHOLD OG BETINGELSER FOR MARKEDSFØRINGSTILLADELSEN
Periodiske, opdaterede sikkerhedsindberetninger (PSUR’er)
Kravene for fremsendelse af periodiske, opdaterede sikkerhedsindberetninger for dette lægemiddel fremgår af listen over EU-referencedatoer (EURD list), som fastsat i artikel 107c, stk. 7, i direktiv 2001/83/EF, og alle efterfølgende opdateringer offentliggjort på den europæiske webportal for lægemidler.
Indehaveren af markedsføringstilladelsen skal fremsende den første PSUR for dette præparat inden for 6 måneder efter godkendelsen.
D. BETINGELSER ELLER BEGRÆNSNINGER MED HENSYN TIL SIKKER OG EFFEKTIV ANVENDELSE AF LÆGEMIDLET
Risikostyringsplan (RMP)
Indehaveren af markedsføringstilladelsen skal udføre de påkrævede aktiviteter og foranstaltningervedrørende lægemiddelovervågning, som er beskrevet i den godkendte RMP, der fremgår af modul 1.8.2 i markedsføringstilladelsen, og enhver efterfølgende godkendt opdatering af RMP.
En opdateret RMP skal fremsendes: på anmodning fra Det Europæiske Lægemiddelagentur når risikostyringssystemet ændres, særlig som følge af, at der er modtaget nye oplysninger, der
kan medføre en væsentlig ændring i benefit/risk-forholdet, eller som følge af, at en vigtig milepæl (lægemiddelovervågning eller risikominimering) er nået.

26
BILAG III
ETIKETTERING OG INDLÆGSSEDDEL

27
A. ETIKETTERING

28
MÆRKNING, DER SKAL ANFØRES PÅ DEN YDRE EMBALLAGE
YDRE KARTON
1. LÆGEMIDLETS NAVN
BESPONSA 1 mg pulver til koncentrat til infusionsvæske, opløsning inotuzumab ozogamicin
2. ANGIVELSE AF AKTIVT STOF/AKTIVE STOFFER
Hvert hætteglas indeholder 1 mg inotuzumab ozogamicin.Efter rekonstituering indeholder hvert hætteglas 0,25 mg/ml inotuzumab ozogamicin.
3. LISTE OVER HJÆLPESTOFFER
SukrosePolysorbat 80NatriumchloridTrometamol
4. LÆGEMIDDELFORM OG INDHOLD (PAKNINGSSTØRRELSE)
Pulver til koncentrat til infusionsvæske, opløsning1 hætteglas1 mg
5. ANVENDELSESMÅDE OG ADMINISTRATIONSVEJ(E)
Læs indlægssedlen inden brug.i.v. infusion efter rekonstituering og fortynding.Kun til engangsbrug.
6. SÆRLIG ADVARSEL OM, AT LÆGEMIDLET SKAL OPBEVARES UTILGÆNGELIGT FOR BØRN
Opbevares utilgængeligt for børn.
7. EVENTUELLE ANDRE SÆRLIGE ADVARSLER
8. UDLØBSDATO
EXP

29
9. SÆRLIGE OPBEVARINGSBETINGELSER
Opbevares i køleskab. Må ikke nedfryses. Opbevares i den originale pakning for at beskytte mod lys.
10. EVENTUELLE SÆRLIGE FORHOLDSREGLER VED BORTSKAFFELSE AF IKKE ANVENDT LÆGEMIDDEL SAMT AFFALD HERAF
11. NAVN OG ADRESSE PÅ INDEHAVEREN AF MARKEDSFØRINGSTILLADELSEN
Pfizer Europe MA EEIGBoulevard de la Plaine 171050 BruxellesBelgien
12. MARKEDSFØRINGSTILLADELSESNUMMER (-NUMRE)
EU/1/17/1200/001
13. FREMSTILLERENS BATCHNUMMER
Lot
14. GENEREL KLASSIFIKATION FOR UDLEVERING
15. INSTRUKTIONER VEDRØRENDE ANVENDELSEN
16. INFORMATION I BRAILLESKRIFT
Fritaget fra krav om brailleskrift.
17. ENTYDIG IDENTIFIKATOR – 2D-STREGKODE
Der er anført en 2D-stregkode, som indeholder en entydig identifikator.
18. ENTYDIG IDENTIFIKATOR - MENNESKELIGT LÆSBARE DATA
PC:SN:NN:

30
MINDSTEKRAV TIL MÆRKNING PÅ SMÅ INDRE EMBALLAGER
HÆTTEGLAS
1. LÆGEMIDLETS NAVN, STYRKE OG/ELLER ADMINISTRATIONSVEJ(E)
BESPONSA 1 mg pulver til koncentrat inotuzumab ozogamicini.v. infusion efter rekonstituering og fortynding
2. ADMINISTRATIONSMETODE
Kun til engangsbrug.
3. UDLØBSDATO
EXP
4. BATCHNUMMER
Lot
5. INDHOLD ANGIVET SOM VÆGT, VOLUMEN ELLER ANTAL DOSER
6. ANDET

31
B. INDLÆGSSEDDEL

32
Indlægsseddel: Information til patienten
BESPONSA 1 mg pulver til koncentrat til infusionsvæske, opløsninginotuzumab ozogamicin
Dette lægemiddel er underlagt supplerende overvågning. Dermed kan der hurtigt tilvejebringes nye oplysninger om sikkerheden. Du kan hjælpe ved at indberette alle de bivirkninger, du får. Se sidst i afsnit 4, hvordan du indberetter bivirkninger.
Læs denne indlægsseddel grundigt, inden du får dette lægemiddel, da den indeholder vigtige oplysninger.
- Gem indlægssedlen. Du kan få brug for at læse den igen. - Spørg lægen, apotekspersonalet eller sundhedspersonalet, hvis der er mere, du vil vide.- Kontakt lægen, apotekspersonalet eller sundhedspersonalet, hvis du får bivirkninger, herunder
bivirkninger, som ikke er nævnt her. Se afsnit 4.
Se den nyeste indlægsseddel på www.indlaegsseddel.dk
Oversigt over indlægssedlen
1. Virkning og anvendelse2. Det skal du vide, før du får BESPONSA3. Sådan får du BESPONSA4. Bivirkninger5. Opbevaring6. Pakningsstørrelser og yderligere oplysninger
1. Virkning og anvendelse
Det virksomme stof i BESPONSA er inotuzumab ozogamicin. Det tilhører en gruppe lægemidler, som er målrettet kræftceller. Disse lægemidler kaldes antineoplastiske midler.
BESPONSA bruges til at behandle voksne med akut lymfoblastær leukæmi. Akut lymfoblastær leukæmi er kræft i blodet, hvor der er for mange hvide blodlegemer. BESPONSA er beregnet til behandling af akut lymfoblastær leukæmi hos voksne patienter, der tidligere har prøvet andre behandlinger, men hvor disse behandlinger ikke har virket.
BESPONSA virker ved at binde sig til celler med et protein, der kaldes CD22. Lymfoblastære leukæmiceller har dette protein. Efter at have bundet sig til de lymfoblastære leukæmiceller overfører lægemidlet et stof til cellerne, som forstyrrer cellernes DNA og til slut dræber dem.
2. Det skal du vide, før du får BESPONSA
Brug ikke BESPONSA: hvis du er allergisk over for inotuzumab ozogamicin eller et af de øvrige indholdsstoffer i
BESPONSA (angivet i punkt 6). hvis du tidligere har haft en sygdom, hvor blodkarrene i leveren var beskadiget og tilstoppet af
blodpropper (veno-okklusiv sygdom), eller du har det nu. hvis du har en alvorlig leversygdom, fx skrumpelever (hvor leveren ikke fungerer ordentligt
på grund af langvarige skader), eller sygdom med symptomer på grund af øget modstand ved blodets gennemstrømning gennem leveren (nodulær regenerativ hyperplasi), som kan være forårsaget af kronisk brug af lægemidler, eller aktiv leverbetændelse (en sygdom, hvor der er betændelsestilstand i leveren).

33
Advarsler og forsigtighedsregler
Kontakt lægen, apotekspersonalet eller sundhedspersonalet, hvis du:
tidligere har haft leverproblemer eller leversygdom, eller hvis du har symptomer på en alvorlig tilstand, der kaldes hepatisk veno-okklusiv sygdom, hvor blodkarrene i leveren bliver beskadiget og tilstoppet af blodpropper. Veno-okklusiv sygdom kan være dødelig og er forbundet med hurtig vægtstigning, smerter i den øverste højre side af maven, forstørret lever, væskeophobning som giver udspiling af maven, og blodprøver som viser forhøjet bilirubin og/eller leverenzymer (kan give gulfarvning af hud og øjne). Denne sygdom kan forekomme under behandling med BESPONSA eller efter efterfølgende stamcelletransplantation. En stamcelletransplantation er et indgreb, hvor man transplanterer en anden persons stamceller (celler, som udvikler sig til nye blodlegemer) ind i din blodbane. Du kan få dette indgreb udført, hvis din sygdom reagerer optimalt på behandlingen.
har symptomer på et lavt antal blodlegemer, der kaldes for neutrofile (nogen gange ledsaget af feber), røde blodlegemer, hvide blodlegemer, lymfocytter eller et lavt antal blodplader. Disse symptomer omfatter betændelse eller feber, eller at man let får blå mærker eller ofte får næseblod.
har haft symptomer på en reaktion i forbindelse med infusion, såsom feber og kulderystelser eller åndedrætsbesvær under eller kort tid efter infusionen af BESPONSA.
har haft symptomer på tumorlysesyndrom, der kan være forbundet med symptomer i maven og tarmene (fx kvalme, opkastning, diarré), hjertet (fx ændringer i hjerterytmen), nyrerne (fx mindre mængde urin, blod i urinen), nerverne og musklerne (fx muskelspasmer, muskelsvækkelse, muskelkramper) under eller kort efter infusionen af BESPONSA.
tidligere har haft eller er disponeret for ændring i hjertets elektriske aktivitet (forlænget QT-interval), som kan forårsage alvorlig uregelmæssig hjerterytme, tager lægemidler kendt for at forlænge QT-intervallet, og/eller har forstyrrelser i elektrolytbalancen (fx calcium, magnesium, kalium).
har forhøjet indhold af amylase eller lipaseenzymer, hvilket kan være et tegn på problemer med bugspytkirtlen eller leveren og galdeblæren eller galdegangene.
Fortæl det straks til lægen, apotekspersonalet eller sundhedspersonalet, hvis du bliver gravid under behandlingen med BESPONSA og i op til 8 måneder efter afslutning af behandlingen.
Lægen vil regelmæssigt tage blodprøver for at måle dine blodtal under behandlingen med BESPONSA, se også afsnit 4.
Under behandlingen, især i de første par dage efter behandlingsstart, kan du risikere et alvorligt fald i antallet af hvide blodlegemer (neutropeni), som kan være ledsaget af feber (febril neutropeni).
Du vil muligvis, især i de første par dage efter start af behandlingen, have forhøjede leverenzymer. Lægen vil regelmæssigt tage blodprøver for at måle leverenzymer under behandlingen med BESPONSA.
Behandling med BESPONSA kan muligvis forårsage en alvorlig uregelmæssig hjerterytme (ændring i de elektriske impulser i hjertet, som kaldes forlængelse af QT-intervallet). Lægen tager et EKG (elektrokardiogram) og blodprøver for at måle elektrolytterne i blodet (fx calcium, magnesium, kalium) inden den første dosis BESPONSA, og regelmæssigt under behandlingen.
Lægen vil overvåge, om du får symptomer på tumorlysesyndrom, efter du har fået BESPONSSA, se afsnit 4.
Børn og unge
BESPONSA må ikke bruges til børn og unge under 18 år, da der ikke foreligger nogen oplysninger om anvendelse til børn og unge.

34
Brug af anden medicin sammen med BESPONSA
Fortæl altid lægen eller apotekspersonalet, hvis du tager anden medicin eller har gjort det for nylig. Dette gælder også håndkøbsmedicin og naturlægemidler.
Graviditet, amning og frugtbarhed
Hvis du er gravid eller ammer, har mistanke om, at du er gravid, eller planlægger at blive gravid, skal du spørge din læge eller apotekspersonalet til råds, før du bruger dette lægemiddel.
Prævention
Du skal undgå at blive gravid eller blive far til et barn. Kvinder skal bruge sikker prævention under behandlingen og i mindst 8 måneder efter sidste dosis. Mænd skal bruge sikker prævention under behandlingen og i mindst 5 måneder efter sidste dosis.
Graviditet
Virkningen af BESPONSA på gravide kvinder kendes ikke, men baseret BESPONSAs virkningsmekanisme kan det måske skade det ufødte barn. Du må ikke få BESPONSA under graviditet, medmindre din læge mener, at det er det bedste lægemiddel for dig.
Kontakt straks lægen, hvis du eller din partner bliver gravid under behandlingen med dette lægemiddel.
Frugtbarhed
Mænd og kvinder bør søge rådgivning om bevarelse af frugtbarheden inden behandling.
Amning
Hvis du skal i behandling med BESPONSA, skal du stoppe med at amme under behandlingen og i mindst 2 måneder efter behandlingen. Tal med lægen.
Trafik- og arbejdssikkerhed
Hvis du føler dig usædvanligt træt (dette er en meget almindelig bivirkning ved BESPONSA), bør du ikke køre eller betjene maskiner.
3. Sådan får du BESPONSA
Brug altid lægemidlet nøjagtigt efter lægens, apotekspersonalets eller sundhedspersonalets anvisning. Er du i tvivl, så spørg lægen, apotekspersonalet eller sundhedspersonalet.
Sådan gives BESPONSA Lægen vil fastsætte den korrekte dosis. En læge eller sygeplejerske giver dig BESPONSA gennem et drop i en vene (intravenøs
infusion). Infusionen gives over 1 time. Doserne gives en gang om ugen. Hvert behandlingsforløb består af 3 doser.
Hvis behandlingen fungerer godt, og du skal have en stamcelletransplantation (se afsnit 2), får du 2 eller højst 3 behandlingsforløb.
Hvis behandlingen fungerer godt, men du ikke skal have en stamcelletransplantation, får du op
til højst 6 behandlingsforløb.
Hvis du ikke reagerer på behandlingen inden for 3 behandlingsforløb, vil behandlingen blive
stoppet.

35
Lægen vil muligvis ændre din dosis, afbryde eller helt stoppe behandlingen med BESPONSA,
hvis du får visse bivirkninger.
Lægen vil muligvis nedsætte din dosis afhængigt af hvordan du reagerer på behandlingen.
Lægen vil tage blodprøver under behandlingen for at undersøge, om du har bivirkninger, og
om du reagerer på behandlingen.
Spørg lægen, apotekspersonalet eller sundhedspersonalet, hvis der er noget, du er i tvivl om.
Lægemidler, der gives inden behandling med BESPONSA
Inden behandlingen med BESPONSA får du andre lægemidler (præmedicinering) for at mindskereaktioner forårsaget af infusionen og andre mulige bivirkninger. Lægemidlerne kan omfatte binyrebarkhormoner (fx dexamethason), febernedsættende midler (antipyretika) og antihistaminer (mod allergiske reaktioner).
Du vil muligvis få medicin og ekstra væske, inden du bliver behandlet med BESPONSA, for at forhindre, at der opstår tumorlysesyndrom. Tumorlysesyndrom er forbundet med forskellige symptomer i maven og tarmene (fx kvalme, opkastning, diarré), hjertet (fx ændringer i hjerterytmen), nyrerne (fx mindre mængde urin, blod i urinen), nerverne og musklerne (fx muskelspasmer, muskelsvækkelse, muskelkramper).
4. Bivirkninger
Dette lægemiddel kan som al anden medicin give bivirkninger, men ikke alle får bivirkninger. Nogle af disse bivirkninger kan være alvorlige.
Fortæl det straks til lægen, hvis du bemærker symptomer på en eller flere af følgende alvorlige bivirkninger:
Reaktion i forbindelse med infusionen (se afsnit 2), symptomerne kan være feber og kulderystelser eller vejtrækningsbesvær under eller kort tid efter infusionen af BESPONSA.
Veno-okklusiv leversygdom (se afsnit 2), symptomerne kan være hurtig vægtstigning, smerter i den øverste højre side af maven, forstørret lever, væskeophobning som giver hævelse af maven, og blodprøver som viser forhøjet bilirubin og/eller leverenzymer (kan give gulfarvning af hud og øjne).
Lavt antal blodlegemer (neutrofile) sommetider ledsaget af feber, røde blodlegemer, hvide blodlegemer, lymfocytter eller et lavt antal blodplader (se afsnit 2), symptomerne kan være betændelse eller feber, eller at man let får blå mærker eller ofte får næseblod.
Tumorlysesyndrom (se afsnit 2), dette kan give forskellige symptomer i mave-tarmkanalen (fx kvalme, opkastning og diarre), hjertet (fx ændringer i hjerterytmen), nyrerne (fx nedsat mængde urin, blod i urinen), nerver og muskler (muskelspasmer, -svaghed og -kramper)
Ændre hjertets elektriske aktivitet (forlænge QT-intervallet) (se afsnit 2), der kan give alvorlig uregelmæssig hjerterytme. Fortæl lægen, hvis du bliver svimmel, ør eller besvimer.
Andre bivirkninger kan omfatte:
Meget almindelige bivirkninger (kan påvirke flere end 1 ud af 10 personer): Infektioner Et lavere antal hvide blodlegemer, hvilket kan medføre generel svækkelse og en tendens til at
udvikle infektion Et lavere antal blodlegemer, der kaldes for lymfocytter (en slags hvide blodlegemer), hvilket
kan medføre en tendens til at udvikle infektion Et lavere antal røde blodlegemer, hvilket kan medføre træthed og stakåndethed Nedsat appetit Hovedpine

36
Blødning Smerter i mave/underliv Opkastning Diarré Kvalme Mundbetændelse Forstoppelse Forhøjet indhold af bilirubin, hvilket kan medføre gulfarvning af huden, øjnene og andet væv Feber Kulderystelser Træthed Højt indhold af leverenzymer i blodet (der kan være tegn på leverskade)
Almindelige bivirkninger (kan forekomme hos op til 1 ud af 10 personer): Et lavere antal af forskellige typer blodlegemer For højt indhold af urinsyre i blodet Omfattende væskeansamling i underlivet Udspiling af underlivet/maven Ændringer i hjerterytmen (kan ses på EKG) Unormalt højt indhold af amylase i blodet (et enzym, der er nødvendigt for fordøjelsen og
omdannelse af stivelse til sukkerstoffer) Unormalt højt indhold af lipase i blodet (et enzym, der er nødvendigt for, at kroppen kan
fordøje fedt fra maden) Overfølsomhed
Indberetning af bivirkninger
Hvis du oplever bivirkninger, bør du tale med din læge, sygeplejerske eller apoteket. Dette gælder også mulige bivirkninger, som ikke er medtaget i denne indlægsseddel. Du eller dine pårørende kan også indberette bivirkninger direkte til Lægemiddelstyrelsen via det nationale rapporteringssystem anført i Appendiks V. Ved at indrapportere bivirkninger kan du hjælpe med at fremskaffe mere information om sikkerheden af dette lægemiddel.
5. Opbevaring
Opbevar lægemidlet utilgængeligt for børn.
Brug ikke lægemidlet efter den udløbsdato, der står på hætteglassets etiket og kartonen efter Exp. Udløbsdatoen er den sidste dag i den nævnte måned.
Uåbnet hætteglas
- Opbevares i køleskab (2-8 °C). - Opbevares i den originale karton for at beskytte mod lys. - Må ikke nedfryses.
Rekonstitueret opløsning
- Bruges straks eller opbevares i køleskab (2-8 °C) i op til 4 timer. - Beskyttes mod lys.- Må ikke nedfryses.

37
Fortyndet opløsning
- Bruges straks eller opbevares ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding.
- Beskyttes mod lys.- Må ikke nedfryses.
Dette lægemiddel skal efterses visuelt for partikler og misfarvning før det gives. Hvis der ses partikler eller misfarvning, må det ikke bruges.
Spørg på apoteket, hvordan du skal bortskaffe medicinrester. Af hensyn til miljøet må du ikke smide medicinrester i afløbet, toilettet eller skraldespanden.
6. Pakningsstørrelser og yderligere oplysninger
BESPONSA indeholder:
Aktivt stof: inotuzumab ozogamicin. Hvert hætteglas indeholder 1 mg inotuzumab ozogamicin. Efter rekonstituering indeholder 1 ml opløsning 0,25 mg inotuzumab ozogamicin.
Øvrige indholdsstoffer: sucrose, polysorbat 80, natriumchlorid og trometamol.
Udseende og pakningsstørrelser
BESPONSA er et pulver til koncentrat til infusionsvæske, opløsning. En pakke BESPONSA indeholder:
1 hætteglas indeholdende en hvid til råhvid frysetørret kage eller pulver.
Indehaver af markedsføringstilladelsen
Pfizer Europe MA EEIGBoulevard de la Plaine 171050 BruxellesBelgien
Fremstiller
Pfizer Ireland PharmaceuticlasGrange Castle Business ParkClondalkinDublin 22Irland
Hvis du ønsker yderligere oplysninger om dette lægemiddel, skal du henvende dig til den lokale repræsentant for indehaveren af markedsføringstilladelsen:
Belgique/België/BelgienPfizer S.A. / N.V.Tél/Tel: +32 (0)2 554 62 11
LietuvaPfizer Luxembourg SARL filialas LietuvojeTel: + 370 52 51 4000
БългарияПфайзер Люксембург САРЛ, Клон БългарияТел.: +359 2 970 4333
Luxembourg/LuxemburgPfizer S.A.Tél/Tel: +32 (0)2 554 62 11

38
Česká republikaPfizer PFE, spol. s r.o.Tel: +420 283 004 111
MagyarországPfizer Kft.Tel: +36-1-488-37-00
DanmarkPfizer ApSTlf: +45 44 20 11 00
MaltaV.J. Salomone Pharma Ltd.Tel: +356 21220174
DeutschlandPfizer Pharma GmbHTel: +49 (0)30 550055 51000
NederlandPfizer bvTel: +31 (0)10 406 43 01
EestiPfizer Luxembourg SARL Eesti filiaalTel: +372 666 7500
NorgePfizer Norge ASTlf: +47 67 52 61 00
ΕλλάδαPfizer Ελλάς A.E.Τηλ: +30 210 6785 800
ÖsterreichPfizer Corporation Austria Ges.m.b.H.Tel: +43 (0)1 521 15-0
EspañaPfizer, S.L.Tel: +34 91 490 99 00
PolskaPfizer Polska Sp. z o.o.Tel: +48 22 335 61 00
FrancePfizerTel: +33 (0)1 58 07 34 40
PortugalPfizer Biofarmacêutica, Sociedade Unipessoal LdaTel: +351 21 423 5500
HrvatskaPfizer Croatia d.o.o.Tel: + 385 1 3908 777
RomâniaPfizer Romania S.R.L.Tel: +40 (0) 21 207 28 00
IrelandPfizer Healthcare IrelandTel: 1800 633 363 (toll free)+44 (0)1304 616161
SlovenijaPfizer Luxembourg SARLPfizer, podružnica za svetovanje s področjafarmacevtske dejavnosti, LjubljanaTel: + 386 (0)1 52 11 400
ÍslandIcepharma hf.Sími: +354 540 8000
Slovenská republikaPfizer Luxembourg SARL, organizačná zložkaTel: + 421 2 3355 5500
ItaliaPfizer S.r.l.Tel: +39 06 33 18 21
Suomi/FinlandPfizer OyPuh/Tel: +358 (0)9 43 00 40
ΚύπροςPfizer Ελλάς Α.Ε. (Cyprus Branch)Τηλ: +357 22 817690
SverigePfizer Innovations ABTel: +46 (0)8 550-520 00
LatvijaPfizer Luxembourg SARL filiāle LatvijāTel: + 371 670 35 775
United KingdomPfizer LimitedTel: +44 (0) 1304 616161

39
Denne indlægsseddel blev senest ændret
Andre informationskilder
Du kan finde yderligere oplysninger om dette lægemiddel på Det Europæiske Lægemiddelagenturs hjemmeside: http://www.ema.europa.eu. Der er også links til andre websteder om sjældne sygdomme og om, hvordan de behandles.
Denne indlægsseddel findes på alle EU-/EØS-sprog på Det Europæiske Lægemiddelagenturs hjemmeside.
Nedenstående oplysninger er til læger og sundhedspersonale. Se venligst produktresumeet for den fulde information om dosering og dosisjustering.
Administration
BESPONSA er til intravenøs anvendelse. Infusionen skal administreres over 1 time.
BESPONSA må ikke administreres som intravenøst push eller bolus.
BESPONSA skal rekonstitueres og fortyndes inden administration.
BESPONSA skal administreres i behandlingsforløb på 3-4 uger.
Hos patienter, der skal fortsætte med hæmatopoietisk stamcelletransplantation (HSCT), anbefales 2 behandlingscyklusser. En tredje cyklus kan overvejes til patienter, der ikke opnår CR/CRi og MRD-negativitet efter 2 cyklusser. Hos patienter, der ikke fortsætter med HSCT, kan der administreres op til maksimalt 6 cyklusser. Alle patienter, der ikke opnår CR/CRi inden for 3 cyklusser, skal have seponeret behandlingen (se pkt. 4.2 i produktresuméet).
Nedenstående skema viser de anbefalede doseringsregimer.
I første cyklus anbefales en samlet dosis til alle patienter på 1,8 mg/m2 pr. forløb, givet som 3 opdelte doser på dag 1 (0,8 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2). Cyklus 1 varer 3 uger, men kan forlænges til 4 uger, hvis patienten opnår CR eller CRi, og/eller for at bedre restitutionen efter toksicitet.
I de efterfølgende cyklusser er den anbefalede, samlede dosis 1,5 mg/m2 pr. cyklus givet som 3 opdelte doser på dag 1 (0,5 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2) til patienter, der opnår CR/CRi, eller 1,8 mg/m2 pr. forløb givet som 3 opdelte doser på dag 1 (0,8 mg/m2), 8 (0,5 mg/m2) og 15 (0,5 mg/m2) til patienter, der ikke opnår CR/CRi. Efterfølgende cyklusser er af 4 ugers varighed.
Doseringsregime for cyklus 1 og efterfølgende cyklusser ifølge respons på behandlingen
Dag 1 Dag 8a Dag 15a
Doseringsregime for cyklus 1Alle patienter:
Dosis (mg/m2) 0,8 0,5 0,5Længde på cyklus 21 dageb
Doseringsregime for efterfølgende cyklusser ifølge respons på behandlingenPatienter, der har opnået CRc eller CRid:
Dosis (mg/m2) 0,5 0,5 0,5Længde på cyklus 28 dagee

40
Doseringsregime for cyklus 1 og efterfølgende cyklusser ifølge respons på behandlingen
Dag 1 Dag 8a Dag 15a
Patienter, der ikke har opnået CRc eller CRid:
Dosis (mg/m2) 0,8 0,5 0,5Længde på cyklus 28 dagee
Forkortelser: ANC = absolut neutrofiltal, CR = komplet remission, CRi = komplet remission med delvis hæmatologisk restitution.a +/- 2 dage (oprethold 6 dage mellem doser).b For patienter, der opnår CR/CRi, og/eller for at bedre restitution efter toksicitet, kan cyklus forlænges til
28 dage (dvs. 7 dages mellemrum uden behandling med start på dag 21).c CR defineres som < 5% blaster i knoglemarven og fravær af leukæmiske blaster i perifert blod, fuld
restitution af perifere blodtal (trombocytter ≥ 100 × 109/l og ANC ≥ 1 × 109/l) og resolution af eventuel ekstramedullær sygdom.
d CRi defineres som < 5% blaster i knoglemarven og fravær af leukæmiske blaster i perifert blod, delvis restitution af perifere blodtal (trombocytter < 100 × 109/l og/eller ANC < 1 × 109/l) og resolution af eventuel ekstramedullær sygdom.
e 7 dages mellemrum uden behandling med start på dag 21.
Vejledning i rekonstituering, fortynding og administration
Anvend aseptisk teknik under rekonstituering og fortynding. Inotuzumab ozogamicin (som har en densitet på 1,02 g/ml ved 20 °C/68 °F) er lysfølsomt og skal beskyttes mod ultraviolet lys under rekonstituering, fortynding og administration.
Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding.
Rekonstituering
Beregn dosis (mg) og antallet af nødvendige hætteglas BESPONSA. Rekonstituér hvert 1 mg hætteglas med 4 ml vand til injektionsvæske for at få en
engangsopløsning på 0,25 mg/ml BESPONSA. Hvirvl forsigtigt glasset for at fremme opløsning. Må ikke rystes. Se efter, om den rekonstituerede opløsning indeholder partikler eller er misfarvet. Den
rekonstituerede opløsning skal være klar til let uklar, farveløs og i det væsentlige fri for synlige fremmedlegemer. Hvis der ses partikler eller misfarvning, må opløsningen ikke anvendes.
BESPONSA indeholder ikke bakteriostatiske konserveringsmidler. Den rekonstituerede opløsning skal bruges med det samme. Hvis den rekonstituerede opløsning ikke kan bruges med det samme, kan den opbevares i køleskab (2-8 °C) i op til 4 timer. Beskyttes mod lys. Må ikke nedfryses.
Fortynding
Beregn det volumen rekonstitueret opløsning, der er nødvendig for at opnå den korrekte dosis i forhold til patientens legemsoverfladeareal. Træk denne mængde op af hætteglasset/-glassene vha. en sprøjte. Beskyttes mod lys. Eventuelt ubrugt, resterende rekonstitueret opløsning i hætteglasset skal kasseres.
Kom den rekonstituerede opløsning i en infusionsbeholder med natriumchloridopløsning 9 mg/ml (0,9 %) til injektion, så det totale nominelle volumen er 50 ml. Slutkoncentrationen skal være på mellem 0,01-0,1 mg/ml. Beskyttes mod lys. Det anbefales at bruge en infusionsbeholder af polyvinylchlorid (PVC) (med eller uden di(2-ethylhexyl)phthalat [DEHP]), polyolefin (polypropylen og/eller polyethylen) eller ethylenvinylacetat (EVA).
Vend forsigtigt infusionsbeholderen om for at blande den fortyndede opløsning. Må ikke rystes.

41
Den fortyndede opløsning skal bruges straks, opbevares ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.
Administration
Hvis den fortyndede opløsning opbevares i køleskab (2-8 °C), skal den stå ved stuetemperatur
(20-25 °C) i ca. 1 time inden administration. Filtration af den fortyndede opløsning er ikke nødvendigt. Hvis den fortyndede opløsning
imidlertid filtreres, anbefales det at anvende polyethersulfon (PES)-, polyvinylidenfluorid (PVDF)- eller hydrofile polysulfon (HPS)-baserede filtre. Der må ikke anvendes filtre fremstillet af nylon eller blandede celluloseestere (MCE).
Infusionsposen skal beskyttes mod lys ved hjælp af afskærmning, der beskytter mod ultraviolet lys (dvs. ravgule, mørkebrune eller grønne poser eller aluminiumsfolie) under infusion. Infusionsslangen behøver ingen beskyttelse mod lys.
Den fortyndede opløsning infunderes over 1 time ved en hastighed på 50 ml/time ved stuetemperatur (20-25 °C). Beskyttes mod lys. Det anbefales at bruge infusionsslanger fremstillet af PVC (med eller uden DEHP), polyolefin (polypropylen og/eller polyethylen) eller polybutadien.
BESPONSA må ikke blandes eller administreres som infusion med andre lægemidler.
Opbevaringstider og -forhold for rekonstitueret og fortyndet BESPONSA opløsning.
Maksimal tid fra rekonstitution til fuldendt administration ≤ 8 timera
Rekonstitueret opløsning
Fortyndet opløsningEfter start på fortynding Administration
Den rekonstituerede opløsning skal bruges straks eller efter opbevaring i køleskab (2-8 °C) i op til 4 timer. Beskyttes mod lys. Må ikke nedfryses.
Den fortyndede opløsning skal bruges straks eller efter opbevaring ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.
Hvis den fortyndede opløsning opbevares i køleskab (2-8 °C), skal den stå ved stuetemperatur (20-25 °C) i ca. 1 time inden administration. Administrer den fortyndede opløsning som infusion over 1 time ved en hastighed på 50 ml/time ved stuetemperatur (20-25 °C). Beskyttes mod lys.
a med ≤4 timer i mellem rekonstituering og fortynding.
Opbevaringsforhold og holdbarhed
Uåbnede hætteglas
4 år
Rekonstitueret opløsning
BESPONSA indeholder ikke bakteriostatiske konserveringsmidler. Den rekonstituerede opløsning skal bruges med det samme. Hvis den rekonstituerede opløsning ikke kan bruges med det samme, kan den opbevares i køleskab (2-8 °C) i op til 4 timer. Beskyttes mod lys. Må ikke nedfryses.

42
Fortyndet opløsning
Den fortyndede opløsning skal bruges straks eller opbevares ved stuetemperatur (20-25 °C) eller i køleskab (2-8 °C). Den maksimale tid fra rekonstituering til fuldendt administration skal være ≤ 8 timer, og der skal være ≤ 4 timer mellem rekonstituering og fortynding. Beskyttes mod lys. Må ikke nedfryses.





























