Embed Size (px)
Citation preview

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 1/12
Biosynthesis of the lupine alkaloids I Lupinine
W.
MAREK OLEBIEWSKI'
N D
IAND. SPENSER
Department of Chemistry, McMaster University, Hamilton, Ont., Canad a
L8S
4M
Received February 5, 1985
W . MAREK OLEBIEWSKInd IAN . SPENSER.an.
J
Chem. 63, 2707 (1985).
Th e mode of incorporation into lupinine of cadaverine, intramolecularly doubly labelled with I5N and with C at the C-atom
adjacent to N, i.e.,
13C,15~- bond-labelled ,
as determined by I3C nmr spectr oscopy ; lupinine is generated from two
cadaverine-derived C5-units by a route which excludes a dimeric intermediate with C2, symm etry. The mode of incorporation
of 'H from L-(2-'H)lysine, from (R)- and (S)-(1-'H)cadaverine, and from (2-2H)-A1-piperideine nto lupinine was determined
by 'H nmr spectroscopy. T he results corroborate the conclusions from the I3C, N experiment and they establish the stereo-
chemistry of six of the steps in the biosynthetic conversion of L-lysine into lupinine.
W .
MAREK OLEBIEWSKI
t
IAN
D.
SPENSER.
an.
J
Chem. 63, 2707 (1985).
Faisant appel la spectroscopic rmn du I3C et utilisant de la cad avtr ine doublement marquee d 'une faqon intram oltculaire
par du ' 5 ~t du 13C sur le carbone voisin du ' 5 ~liaison I3C, 5N marquee), on a ttud ie le mode d'incorporation de la cadavt rine
la lupinine. Les r6sultats obtenus suggkrent que la lupinine se forme partir de deux unites en C5 dCrivtes de la cad avtr ine
par le biais d'une v oie reactionnelle qui exclut la formation d'un in termtd iaire dimkre posstdan t une symetrie C2,. Faisant
appel la rmn du 'H, on a determ int le mode d'incorporation du 'H de la L-('H-2) lysine, des ('H-2) cad avtr ines -(R ) et -(S )
et de la (ZH-2)-A1-pip~ridinela lupinine. Les rtsultats confirment les conclusions tirees sur la base des experiences conduites
avec le I3C et le I5N; de plus, elles permettent d'etablir la sterkochimie de six des Ctapes conduisant
2
la transformation
biosynthttique de la L-lys&e en lupinhe.
[Traduit par le journal]
Introduction
Speculations concerning the biosynthesis of the lupine alka-
loids go back more than 50 years. In 1931 Schopf suggested
(1, 2) that the ring skeleton of lupinine (20) might be generated
by condensation of two lysine (1) derived fragments, 5-amino-
pentanal
(3) e
'-piperideine (4) (3, 4)) and glutardialdehyde
(S),
followed by reduction (Scheme 1Bd). Schopf later aban-
doned this notion in favour of the idea (3) that the im-
inodialdehyde (7) might be the intermediate between lysine and
lupinine (Schemes 1Ba and 1Bb). This intermediate was
favoured also by Robert Robinson (4). In a further variation of
this theme it was suggested (5, 6) that lupinine arose by
rearrangement of tetrahydroanabasine (14), a dimer of
A'-piperideine (4) (3, 5) (Scheme 1Be).
The first tracer experiments to test these ideas were canied
out twenty-five years ago. Radioactivity from ~~-[2-'~C]lysine
(7) and from its decarboxylation product, [l-'4C]cadaverine
(6, 8), was indeed incorporated into lupinine. With either sub-
strate, approximately one quarter of the total activity of the
alkaloid was present within the hydroxymethyl carbon, C-11,
of lupinine. Also, the two methylene carbon atoms, C-4 and
C-6, together, accounted for one half of the total alkaloid
activity (7, 8). If the assumption is made that this activity is
distributed equally over C-4 and C-6, and that each of these two
C atoms contains one quarter of the label,' it follows that four
sites within lupinine, C- 11, C-4, C-6, and, presumably, C- 10,
each account for one quarter of the label of the intact alkaloid
(20). This conclusion was substantiated by the results of a
recent feeding experiment (9) with ~~-(4,5- '~C~)lysine.n-
'
On leave from the Department of Chemistry, University of War-
saw, 02-093 Warsaw, Poland.
'Another possible interpretation of these results is that one of the
two cen tres, C-4 or C- 6, accounts for 50% of the activity of the intact
lupinine, while the other centre is free of activity.
richment was detected at four pairs of neighbouring carbon
atoms of lupinine, C- 1 -2, C-2,-3, C-7,-8, and C-8,-9. This
labelling pattern serves as evidence that the carbon skeleton of
lupinine is generated from two lysine-derived C5-units, via an
intermediate with CZvymmetry (e.g., either cadaverine (2) or
the iminodialdehyde (7), or both). However, since the distribu-
tion of label observed in all these experiments is that which is
predicted by every one of the five hypothetical routes (Schemes
1Ba-1Be) that have been advanced to account for the deri-
vation of lupinine from lysine, available tracer evidence cannot
serve to discriminate among the biogenetic proposals.
Nor is it certain that cadaverine (2) is an intermediate
between lysine (1) and lupinine (20): Neither the observation
that incorporation of the two substrates, DL-[2-'4C]lysine nd
[l-'4C]cadaverine, leads to the same distribution of label within
lupinine, nor the labelling pattern from DL-(4,5- C,)lysine con-
stitute conclusive evidence for the obligatory intermediacy of
cadaverine. Other interpretations are possible which account
for such a distribution of label (cf. ref. 10).
The tracer experiments with doubly I3C, N- and with
'H-labelled substrates which are here reported serve as critical
tests of the hypotheses that have been advanced to account for
the origin of lupinine. 'The results, which controvert all but one
of the hypothetical sequences, provide support for the tetra-
hydroanabasine route (Scheme 1Be). Furthermore, the results
of the tracer experiments with 2H-labelled substrates clarify
stereochemical aspects of this route. Preliminary accounts of
parts of this work have appeared (1
1 ,
12).
Methods and results
In six tracer experiments, compounds, specifically labelled
with stable (Experiments 1-5) and with radioactive isotopes
(Experiment 6), were administered by the wick method to
intact plants of
upi nu s luteus
(yellow lupine) over a period of
3-6 days. Details of the experiments are summarized in Table
1.

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 2/12
27 8
CAN J CHEM.
OL
63
1985
CHO FHO CHO
C+>
HO
-3 c;3
c f
CHO
CHO
[?>
HO
NH2
CHO FHO
J
c g
- QCHO h
CHO CHO
CHO CHO CHO
cg Jsds )
rom
Bc
CHO
2 7 [ H p
lg
SCHEME
.
Five alternative hypothetical schemes o f the b iogenesis
of lupinine. A. The initial steps.
B -Be.
Variants of the intermediate
steps. CI-Ch. Variants of the final steps.
The sample of cadaverine, intramolecularly doubly labelled
( bond-labelled ) with I3C and N (Experiment 5) was pre-
pared in three steps from sodium ( C, N)cyanide and
1 bromo 4 phthalimidobutane
y the reaction sequence shown
in Scheme 2, in analogy with the preparation of a simila
labelled sample of putrescine (10).
The samples of ~~-(2-~H)lysineExperiment 3), (2-2H)-
piperideine (Experiment 4), S)- +)- l-2H)cadaverine di
drochloride (Experiment 2), and R)- -)- l-2H)cadaverine
hydrochloride (Experiment 1) were prepared by chemical a
enzymic methods, respectively, which we have previou
described (13). The samples of L-[4-3H]lysine monohyd
chloride and ~~- [6 -' ~C ]l ys in eonohydrochloride (Experim
6) were commercial products.
When the administration of tracer was complete, the pla
to which labelled substrates had been administered were mac
ated with cold methanol in a Waring blendor and the alkalo
were isolated by methanol extraction. Lupinine and sparte
were separated by chromatography on silica gel, lupinine w
converted into the hydrochloride, sparteine into the sulfate
The present paper presents a discussion of the results
tained with lupinine. The sparteine results will be dealt with
the second paper of this series.
Lupinine hydrochloride consists of a 1.7: 1 mixture of
trans and cis ring fused stereoisomers, as shown by the natu
abundance
C nmr spectrum (Fig. 1B). The C nmr spectr
(Fig. 1A) of the sample of lupinine hydrochloride from Exp
ment 5, in which (~-'~C,'~N)cadaverinen admixture w
[l-14C]cadaverine had been administered (Experiment
shows that four of the ten C-atoms, C-4, -6, -10, and -1 1, w
enriched in I3C, to an equal extent. The enrichment, abo
natural abundance, calculated for each of these four carb
atoms (Tables 2 and 3) is in agreement with the specific inc
poration of I4C (Table 1).
The signals in the proton noise decoupled
C nmr spectr
due to two of the C-enriched carbon atoms, C- 10 and C-
appear as singlets, the signals due to the two others, C-6 a
C-4, as multiplets (Fig. 1A). The coupling constants of
doublet component of the multiplets are different from o
another (J13~~,15,:rans isomer 4.1 Hz, cis isomer 4.1 H
trans isomer 7.0 Hz, cis isomer 6.4 Hz; isotope s
C-4, cis +0.02, trans $0.02; C-6, cis -0.04, trans, -0.04
Each of the two sets of multiplets consists of a doublet sup
imposed on a singlet. The doublet (87 11%) (I3C-6, N)
the signals due to C-6 (trans, 6 55.7, cis, 52.9 ppm) is m
more intense than the singlet (13 2%) (I3C-6,I4N) Table
and 3), so that the latter is not apparent in the unexpan
spectrum. In the signals due to C-4 (trans, 6 54.8, cis, 6 4
ppm) the doublet (36
+
5% of signal area, Tables 2 and
(I3C-4, N) appears as a shoulder straddling the singlet (64
8%) (I3C-4,I4N).The doublet/singlet ratios at C-6 ((87
11)/(13 + 2)
=
6.7 1.3) and at C-4 ((36 + 5)/(64 + 8
0.6
+
0.1) thus differ from one another by
an
order of m
nitude (Tables 2 and 3).
Lupinine hydrochloride obtained from the feeding exp
ment with [~-4-~H/~~-6-'~C]lysineExperiment 6) was c
verted into the methiodide and crystallized to constant rad
activity and constant tritium/14C ratio. The 'H/I4C ratio of
purified lupinine methiodide was 8.4 + 0.1, approximat
double that of the intermolecularly doubly 'H/I4C labe
lysine that had served as the substrate ('H/I4C of lysine, 4.1
0.1 (Table 1)).
The deuterium nmr spectra of the samples of lupinine (f
base) from the experiments with deuterium labelled substra
(Experiments 1-4, Table 1) are shown in Fig. 2. The d
terium chemical shifts observed in the spectra of these f

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 3/12
GOLEBIEWSKI AND SPENSER
TABLE . Experiments with upinus luteus
a)
Stable isotopes
Plant material Lupinine Sparteine
Fresh Specific Specific
Expt Wt. fed Enrichment weight No. of Weight incorporation Weight incorporation
no. Substrate (mg) (at. ) (g) plants (mg) per C5 unit ( ) (mg) per C5 unit ( )
1 (R)-( l -2H)Cadaverine 150 99d 300 40 9.5 3.0 4 8
dihydrochloridea
(March, 1983)
2 (s)-(1 -2H)Cadaverine 116 93d 21 1 51 13 2.3 4 2
dihydrochloride
(Dec. 1982)
3 ~~-(2-'H)L ysine ono- 500 95d 41' 100 5 2.3 4 3
hydrochloride
(July, 1983)
4 (2-2~)-A'-Piperideine 300 9Sd 247 100 9 5.4 4 8
(Nov. 1983)
5
(~- '~C ,l- '~~ )Ca dav eri ne 40 99.99 28
dihydrochloride
105 50 10 5 47
[I '4C]Cadaverine
dihydrochlorideb
(Aug. 1982)
(b)
Radioactive isotopes
Plant material
Nominal activity
Fresh Sparteine
Expt Total Specific 3H/'4C weight No. of Lupinine 'H/14C sulfate 3 ~ / ' 4 C
no. Substrate (mCi) (mCi/mmol) ratio (g) plants (mg) ratio (mg) ratio
6 ~ ~ - [ 6 - ' ~ C ] ~ y s i n e 0.20 45
monohydrochloride'
4.1 O l h 104 36 4 8.4 0.1' 3 6. 8 0.2
L-[(Rs)-4-3H]Lysine 1.0 2 2 x 1 0 ~
monohydrochloride'
(Dec. 1983)
See Experimental.
'New England Nuclear (nominal total and specific activities, 25 pC i, 105 mCi per mmol, respectively).
'CEA France.
dMeasured by means of computer assisted peak height analysis.
'This experiment was d one during the hot summer of 1983 when the greenhouse was very hot and dry.
'Net specific activity, determined for the mixture of [I ,5-'4C]cadaverine dihydrochloride, after dilution with 1 40 mg (I- C , I-15N) cadaverin e ihydro chloride,
which was fed to the plant: 5.3 X 10' dpm per mmol.
*Specific acitivity of lupinine hydrochloride, after crystallization to constant specific activity: 3.0
X
10' dpm per mmol.
A A-mL sample of the feeding solution (25 m L) containing the doubly labelled lysine, which w as administered to the plants, was set asid e and from it lysine
hydrochloride was re-isolated by carrie r dilution and purified by ion exchange chromatography and crystallization to constant 3H /14 C atio.
'Lupinine was converted into the methiodide which was crystallized to constant 'H/I4C ratio.
N a C N
20 H9
DMSO Ac20
SCHEME . Synthesis of (1-I3C,1-I5N)cadaverine.

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 4/12
2710
CAN.
1 CHEM.
VOL.
63, 1985
TABLE. Incorporation of ( I-I3C, 1-ISN)cadaverine into trans-lupinine hydrochloridea
Natural abundance Enriched in
Chemical
13
C ,
normalized I3C, normalized Percent enrichment
Carbon
shiftb
Type of
peak areac peak areac
0.94 above natural
atom
(ppm)
signal
(ANA)
(AE)
AE/ANA~ abundancee,'
Relative percent exc
13
C above natura
abundance at
individual C atom
The sample in
D2
as contained in a 2-mm tube. Acquisition time 3.41 s, 0.293 Hz/data point; 1.6 ps pulses.
bRelative to acetone (29.8 ppm).
'Peak areas are normalized relative to C-3 100%. Estimated standard deviation *8% for singlet signals, 10% for composite signals.
dThe ratio AE/ANAs normalized so that the average value for carbon atoms 1, 2, 3, 7, 8, and 9 equals 1 O.
'((0.94 AE/ANA)
I X
1.1% for singlets; (0.94 AE/ANA) I. 1% for doublets.
'The average specific incorporation per C5 unit 1/4(15.2(? 1.6)
+
13.9(* 1.9)
+
16.4(22.0)
+
12.5(* l.5))/(99/2)
X
100% 29
rt_
2%, where 9912
I3C is the average enrichment at each terminal position of cadaverine.
TABLE.
Incorporation of (1 I3C, 1 ISN)cadaverine into cis-lupinine hydrochloridea
Natural abundance Enriched in
Chemical
13
C, normalized I3C, normalized Percent enrichment
Carbon shiftb Typ e of peak areac peak areac 1.08 above natural
atom (ppm) signal (ANA) (AE) abundancee.'
Relative percent ex
13
C above natura
abundance at
individual C atom
See footnote
a,
Table
2.
bSee footnote
b,
Table 2.
'Peak areas
are
normalized relative to C-8 100%. Estimated standard deviation 8 % or singlet signals,
k
10% for composite signals.
dThe ratio AE/ANAs normalized so that the average value for carbon atoms 1,
2,
3, 7 , 8, and 9 equals 1 O.
'((1.08 A€/ANA) )
X
1.1% for singlets; (1.08 AE/ANA)
X I . I
for doublets.
'The average specific incorporation per C5 unit 1/4(12.7(2 1.3) 12. l(f 1.5) + 15.1(21.8) 9.9(&1.2))/(99/2) X 100% 25 rt_ 2%.
lupinine samples are summarized in Table 4 and are compared
with the corresponding proton chemical shifts (Table 5). The
2H nmr spectra of two lupinine samples, that derived from
(S)-(l-2H)cadaverine (Experiment 2) and that derived from the
L-component of DL-(2-2H)lysineExperiment 3), were identi-
cal, each showing two signals, at i 1.72 (+0.01) and 1.54
ppm. The chemical shifts of the two signals (6 4.11 and 1.80
ppm) in the spectrum of the sample derived from
(2-2H)-A'-piperideine Experiment 4) corresponded to those of
two of the signals (6 4.14 and 1.77) in the spectrum of the
sample from (R)-(l-2H)cadaverine (Experiment 1). The latter
spectrum showed an additional signal at 6 2.50 ppm. The
assignment of these spectra will be discussed in the appropriate
context of the next section.
iscussion
The results of the tracer experiments with 14C-labelled y
(7) and cadaverine
6,
8) and with ~~-(4,5-'~C~)lys
9)
not discriminate among the various biogenetic hypotheses
have been advanced to account for the biosynthesis of lupin
20) (Scheme lA, B, C). The first task in a reexamination
the problem was to devise experiments capable of narrow
down the possibilities.
All the hypotheses that have been advanced (Scheme
share a common initial phase, decarboxylation of lysine 1
cadaverine 2),followed by oxidative deamination of the la

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 5/12
GOLEBIEWSKI
AND
SPENSER
T R A N S IS
LUPlNlNE HCl
l r om
1- 3C,1?5~-
CADAVERINE
A
R e . 1. I3C nmr spectra (20 .15 MHz) of lupin ine hydrochloride. The spectra were recorded in the Fourier mode on a Bruker WP 80
spectrometer, in 2-mm tubes, with the natural abundance methyl signal of acetone (29.8 ppm) as internal reference. The acquisition time was
3.41 s. For further details see Tables 2 and 3. Top: Fig. ]A . Proton-noise decoupled (P.N.D.) spectrum (20 000 scans) of the 3C , 5~ -e nr ich
sample of lupinine hydrochloride (10 mg in 10
p
H20) derived from (l- 3C,1-15 N)cadaverine Experiment 5). Bottom: Fig. 1B. P.N.D .
Spectrum (18 112 scans) of a natural abundance sample of lupinine hydrochloride (17 mg in 20
p
H20).
TABLE. Incorporation of deuteriated substrates into lupinine. H nmr analysis
H nmr chemical shiftsb (ppm)
Experiment 1 Experiment 2 Experiment 3 Experiment 4
H nmr lupinine from lupinine from lupinine from lupinine from
Hydrogen chemical shiftsa R-(1- H)cadaverine S-(1- H)cadaverine ~L -( 2- ~) ly si ne (2- H)-A -piperideine
atom ( P P ~ ) 2) 3)
413 s (eq) 2.57
4 P r e ( a x ) 1 . 7 5
6 a r e ( e q ) 2.51 2.50
6P s (ax) 1.57
lo p R (ax) 1.78 1.77
11 re 3.75
11
s
4.16 4.14
The
H
nmr spectrum. Recorded in C6D6 at 250
MHz
and
400 MHz
in the Fourier mode on a Bruker WM 250 and WH 400 spectrometer,
respectively. For full assignment of the proton spectrum, see Table 5.
'The
H
nmr spectra. Recorded in
CJ-16
t 38.40
MHz
in the Fourier mode on a Bruker WM 250 spectrometer, in 10 mm tubes with
natural abundance
C6DHs (7.19
ppm) as internal reference. Acquisition time
2.048
s .
to 5-aminopentanal (3), an am inoaldehyde in equilibrium with
its intramolecular Schiff base, A -piperideine (4) (Sche me
1A).
The hypotheses also agree on the final step in the biogenetic
process, reduction of lupinal (19) to lupinine
20)
(Scheme
1C). The hypotheses differ in the proposed routes leading from
5-aminopentanal (3) SA -piperideine (4)) to lupinal (19)
(Schemes lB, 1C).
What was required for a critical examination of the bio-
genetic process was a method w hich could distinguish the dif-
ferent paths between Saminopentanal (3), the last early inter-
mediate common to all the biogenetic proposals, and lupinal
(19), the common intermediate of the final stage. One way of
differentiating among the routes would be by an experiment
cap able of tracing the fate of the intact C-N bond of
5-aminopentanal (3) into the product.
A successful experiment, leading to incorporation of such a
C-N bond into lupinine, ca n have one of three differen
outcome s. Tw o of the routes (Schemes 1Bd and 1Be) lead from
3
into 1 9, via the bicyclic compound (18) and other inter-
mediates, in such a way that the intact C-N bond of 3 is
destined to become the C-6,N bond of lupinine (Scheme 3C).
A third route (Scheme 1Bc) leads from 3 into 19, via the
bicyclic compound (17) and other intermediates, in such a way
that the intac t C-N bond of 3 is destined to become the C-4,N
bond of lupinine (Sch eme 3B ). It can be predicted that each o
these routes leads to a sample of lupinine in which only o ne o
the three C-N bonds of the product is derived from the intac
C-N bond of 3.
Th e other two routes (Schemes 1Ba and 1Bb) differ from the
above in that they give rise to a sample of lupinine which
7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 6/12
2712
CAN. J.
CHEM.
VOL. 63
1985
LUPlNlN
rom
FIG
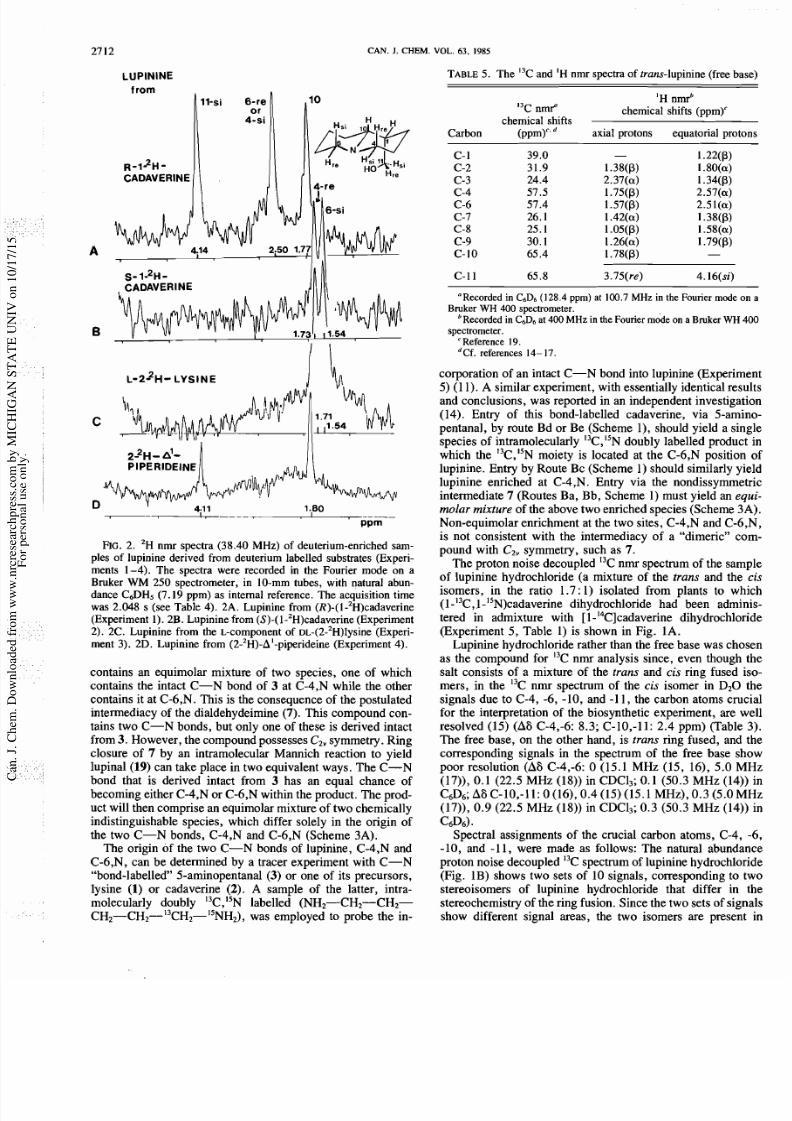
2. 'H nmr spectra (38 .40 MHz) of deuterium-enriched sam-
ples of lupinine derived from deuterium labelled substrates (Experi-
ments 1 -4). Th e spectra were recorded in the Fourier mod e on a
Bruker
WM
250 spectrometer, in 10-mm tubes, with natural abun-
dance C a H 5 (7.19 ppm) as internal reference. Th e acquisition time
was 2.048 s (see Table 4). 2A. Lupinine from (R)-(I-'~)cadaverine
(Experiment 1). 2B. Lup inine from (S)-(1 -2H)cadaverine (Experiment
2). 2C. Lupinine from the L-component of DL-(2-'H)lysine (Experi-
ment 3). 2D. Lupinine from (2-2H)-A'-piperideine (Experiment 4).
contains an equimolar mixture of two species, one of which
contain s the intact C-N bond of
3
at C-4,N while the other
contains it at C-6 ,N. This is the consequence of the postulated
intermediacy of the dialdehydeimine (7). T his comp ound con-
tains two C-N bonds, but only on e of these is derived intact
from
3.
However, the compound possesses CZ v ymm etry. Ring
closure of 7 by an intramolecular Mannich reaction to yield
lup inal (19 ) can take place in two equivalent ways. T he C-N
bond that is derived intact from 3 has an equal chance of
becoming either C-4,N or C -6,N within the product. Th e prod-
uct will then comprise an equimolar mixture of two chem ically
indistinguishable species, which differ solely in the o rigin of
the two C-N bonds, C-4,N and C-6,N (Scheme 3A).
The origin of the two C-N bonds of lupinine, C-4,N and
C-6,N, can be determined by a tracer experiment with C-N
bond-labelled 5-aminopentanal (3) or one of its precursors,
lysine (1) or cadaverine (2).
A sample of the latter, intra-
molecularly doubly I3C,lSN labelled (NH2-CH2-CH2-
C H ~ - C H ~ - ' ~ C H ~ - ' ~ N H ~ ) , was employed to probe the in-
TAB LE . The I3C and
H
mr spectra of trans-lup inine (free ba
'H nmrb
I3C n m f chemical shifts (ppm)'
chemical shifts
C arb on ( P P ~ a xia l p ro to ns e qu ato ria l p ro t
C-1 1 65.8 3.75(re) 4.16(si)
Recorded n C6D6 (128.4 ppm)
at
100.7 M z in the Fourier mode o
Bmker
W 400
spectrometer.
Recorded in C6D6at 400 M z in the Fourier mode on a Bmker W
spectrometer.
'Reference 19.
dCf. references 14- 17.
corporation of an intact C-N bond into lupinine (Experim
5) (1 1). A similar experiment, with essentially identical res
and conclusions, was reported in an independent investiga
(14). Entry of this bond-labelled cadaverine, via 5-am
pentanal, by route Bd or Be (Scheme l) , should yield a sin
species of intramolecularly I3C,lSNdoubly labelled produc
which the I3C,l5N moiety is located at the C-6,N position
lupin ine. Entry by Route Bc (Sche me 1 ) should similarly y
lupinine enriched at C-4,N. Entry via the nondissymme
intermediate 7 (Routes Ba, B b, Sch eme 1) must yield an e
molar mixture of the above two enriched species (Scheme 3
Non-equim olar enrichment at the two sites, C-4,N and C-6
is not consistent with the intermediacy of a dimeric c
pound with Cz, symmetry, such as 7.
The proton noise decoupled I3C nmr spectrum of the sam
of lupinine hydrochloride (a mixture of the trans and the
isomers, in the ratio 1.7 : 1) isolated from plants to wh
(1 I3C,1 15N)cadaverine dihydrochloride had been admi
tered in admixture with [l-'4 C]cad averine dihydrochlo
(Experiment 5, Table 1) is shown in Fig. 1A.
Lupinine hydrochloride rather than the fre e base was cho
as the compound for I3C nmr analysis since, even though
salt consists of a mixture of the trans and cis ring fused
mers, in the I3C nmr spectrum of the cis isomer in D 2 0
signals due to C-4, -6, -10, and -1 1, the carbon atoms cru
for the interpretation of the biosynthetic experiment, are w
resolved (15) (A6 C-4,-6: 8 .3; C-10,-1 1: 2.4 ppm ) (Table
The free base, on the other hand, is
trans
ring fused, and
corresponding signals in the spectrum of the free base s
poor resolution (A6 C-4,-6:
0 (15.1 MHz (15, 16), 5.0 M
(17)), 0.1 (22.5 M Hz (18)) in CDC13; 0. 1 (50.3 MH z (14)
C a 6 ; A6 C-10,-11: 0 (16), 0.4 (15) (15.1 MH z), 0.3 (5.0 M
(17)), 0. 9 (22.5 MHz (18)) in CDC13; 0. 3 (50.3 M Hz (14)
c a 6 ) .
Spectral assignments of the crucial carbon atoms, C-4,
-10, and -11, were made as follows: The natural abunda
proton noise decoupled I3C spectrum of lupinine hy drochlo
(Fig. 1B) shows two sets of 10 signals, corresponding to
stereoisomers of lupinine hydrochloride that differ in
stereochemistry of the ring fusion. Since the two sets of sig
show different signal areas, the two isomers are presen

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 7/12
GOLEBIEWSKl AND SPENSER
oute
1B.
C
vi
outes
106
or 10
SCHEME Incorporation of (l-'3C,1-1SN)cadaverinento lupinine. Labelling patterns predicted from the five biogenetic schemes outlined in
Scheme 1.
A
Via routes B or Bb Scheme 1.B. Via route B Scheme 1. C. Via routes BdorBe Scheme 1. Left columns: I3C,l5N ond-labelled
species. Central columns: Doubly 13C-labelled pecies. Right columns: Singly '3C-labelled pecies. = I3C; = 13C-15N.
unequal amounts (1.7 1). The less intense set of signals shows
two signals at 6 40.0 and 44.6 ppm. Signals in this spectral
region are absent from the spectra of the
trans
fused free base
(14- 18) or the trans fused salt (15) but are present in the is
fused salt (15). The signal set of lower intensity is thus due to
the is fused salt, which is the less abundant (ca. 37%), and the
signals at 6 40.0 and 44.6 pprn must be assigned (19) to C-1
and C-4, respectively, of this isomer. This assignment is con-
firmed by the off-resonance spectrum in which the signal at
6
40.0 appears as a doublet and the signal at 6 44.6 pprn as a
triplet. A second signal, which in the low intensity set appears
as a doublet, is that at 6 58.9 ppm, which must therefore be
assigned to C-10. This leaves the downfield signal at
6
61.3
pprn to be assigned to the hydroxymethyl carbon, C-11, and the
signal at
6
52.9 pprn to C-6. The corresponding resonances in
the more intense signal set, due to the
trans
fused stereoisomer,
which appear as doublets in the off-resonance spectrum, are
those at 6 36.8 pprn (C-1) and at 6 64.7 pprn (C-10). The signal
at 6 59.4 must then be assigned to C-1 1 and those at 54.8 and
55.7 pprn to the aminomethyl carbons, C-4 and C-6. Since in
the spectrum of the enriched sample of lupinine hydrochloride
(Fig. 1A) the high intensity signal at 6 55.7 appears as a doublet
and shows a similar doublet/singlet area (12521 194 = 6.4,
Table 2) as the low intensity doublet due to C-6 at 6 52.9 pprn
(10391156 = 6.7, Table 3), the signal at 6 55.7 must also be
due to C-6. Similarly, the signal at
6
54.8 pprn in the high
intensity set corresponds in doublet/singlet area (5341941 =
0.6, Table 2) to that at 6 44.6 pprn (4341786 = 0.6, Table 3)
in the low intensity set, due to C-4.
The distribution of label within the I3C,l5N nriched alkaloid
is evident from the spectrum (Fig. 1A). As expected, only four
of the ten C-atoms (C-4, C-6, C-10, C-1 1) are enriched in I3C,
to an equal extent, within experimental error (Tables 2 and 3).
The average specific incorporation of label per C5 unit (C,
unit A: C-6, -7, -8, -9, -10, C enrichment in either C-6 or
C-10 but not in both within the same molecule; C5 unit B: C-11,
-1, -2, -3, -4, I3C enrichment in either C-1 1 or in C-4, but not
in both), based on the I3C nmr data (Tables 2 and 3), was ca.
30 at.% I3C. This corresponds to an incorporation of 30199
100 = 30% of cadaverine per C5 unit, a value which is in good
agreement with that obtained from I4C measurements ((3.0
107)/(5.3 lo7)) 100 112 = 28%, Table
1

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 8/12
27 4 CAN. J CHEM.
VOL. 63 1985
The signals due to C-6 and C-4 appear as multiplets. Since
the coupling constants of the doublet component of these mul-
tiplets differ from one another, the multiplets do not arise from
I3C-6,I3C-4coupling, but are due to I3C,l5Ncoupling.
The intense I3C-6,I5Ndoublet indicates intact incorporation
of the I3C-I5N unit of the administered cadaverine into C-6,N
of lupinine. The low intensity doublet due to I3C-4,I5Narises
as a consequence of the remarkably high efficiency of incor-
poration, into the alkaloid, of the administered (l-'3C,l-15N)-
cadaverine. It can be calculated from the I3C nmr data (Tables
2 and 3) that the enriched lupinine which was biosynthesized in
the course of the experiment was diluted by natural abundance
lupinine present in the plants prior to the experiment by approx-
imately 68%. It can also be calculated that the administered
doubly enriched cadaverine was diluted by no more than 25%
of endogenous natural abundance cadaverine, prior to con-
version into lupinine. The intensity of the I3C-4,I5Ndoublet is
fully accounted for by intermolecular I3C,l5N oupling between
two monomer units, derived from this highly enriched cadav-
erine, which are incorporated into lupinine after dimerization.
The observed difference in the doublet/singlet ratios of the
signals due to C-6 and due to C-4 serves as evidence that an
intermediate with C2, symmetry, such as
7
cannot be impli-
cated in lupinine biosynthesis. The biogenetic proposals shown
in Schemes 1Ba and 1Bb are thus eliminated.
Furthermore, since the intact
l3C-I5N unit of the adminis-
tered cadaverine is maintained at C-6,N and not at C-4,N of
lupinine, the sequence shown in Scheme lBc, according to
which the precursor C-N bond enters C-4,N of lupinine, is no
longer in contention.
The biogenetic schemes which need to be examined further
are those shown in Schemes 1Bd and 1Be. These two schemes
differ in the timing of the loss of the nitrogen atom of one of
the two cadaverine-derived aminopentanal units. Scheme 1Bd
postulates that this nitrogen atom is lost at an early stage, so
that the aminopentanal-derived intermediate which serves as
the precursor of the C5 chain, C-4, -3, -2, -1,
-
11, of lupinine
is glutardialdehyde (5), a compound wih C2 symmetry.
Scheme lBe, on the other hand, postulates that this nitrogen
atom is eliminated at a late stage of biosynthesis, following
formation of the CI dimers, 14 and 15.
An experiment with 5-aminopentanal or A'-piperideine, la-
belled at the sp2 carbon atoms with I3C or I4C, would serve as
a critical test of these two suggested routes. In one instance
(Scheme lBd), three of the carbon atoms of lupinine, C-10,
C-11, and C-4, should carry label. In the other instance
(Scheme
lBe), label should be present at two of these carbons,
C-10 and C-11, but not at the third, C-4. Similarly, an experi-
ment with 5-aminopentanal or A'-piperideine labelled with
deuterium at the sp2 carbon atom would distinguish these two
routes, provided that it can be demonstrated that the integrity
of the C-D bond is maintained in the course of the biogenetic
process. The results of an experiment with
(2-2H)-A'-piperi-
deine (Experiment
4
that fulfils this condition will be dis-
cussed later. On the basis of this experiment and of other
experiments with deuteriated substrates (Experiments 1-3)
(see later) the hypothesis based on the intermediacy of glu-
tardialdehyde (Scheme 1Bd) can be discounted.
The biogenetic sequence shown in Scheme 1Be remains. It
is consistent with all available experimental evidence.
The initial step in the biosynthetic process leading to lupinine
from primary metabolites is the decarboxylation of lysine
1)
to
yield cadaverine
2).
Two stereochemical questions concerning
this step must be answered. First, it must be establ
whether L-(i.e., (S)-)lysine or D-(i.e., (R)-)lysine serves a
substrate. Secondly, it must be established whether the d
boxylation process which converts this lysine into cadav
takes place with net retention of configuration or with
inversion of configuration. We have answered both
questions.
The result of the experiment with intermolecularly do
labelled [~- ~H /~ ~- '~ C] ly s i neExperiment 6) demonstrate
L-lysine, rather than D-lysineor DL-lysine, erves as the pr
sor of lupinine in L luteus. The 3H/14C atio of the do
labelled lysine that was administered to the plants was 4
0.1. The 3H/14C atio of the lupinine methiodide that
obtained was 8.4 0.1 (Table 1). From these results it c
calculated that, within experimental error, lupinine is de
entirely (102 3%) from L-lysine( of product derived
L-substrate 50 (3H/14C atio of pr ~d uc t) /( ~H /' ~Cat
substrate)) (20).
Demonstration that the decarboxylation of
L-lysine o ca
erine takes place with net retention of configuration c
from two of the experiments with deuteriated substrates.
These experiments (Experiments 1-4), which solve
only this stereochemical problem, but which also answer
other stereochemical questions concerning the biosynt
steps of the route from lysine into lupinine that involve t
formations at those carbon atoms of lupinine which orig
from the terminal carbon atoms of cadaverine, will no
discussed.
In the first two of these experiments, samples of cadave
chirally deuteriated at C- I, were used as substrates. The
ples of lupinine obtained from these experiments were e
ined by 'H n'mr. The 'H nmr spectra are shown in Figs. 2A
2B. Chemical shifts were assigned by comparison with
corresponding 'H nmr chemical shifts (Table 4).
Unambiguous assignment of the resonances due to th
protons of lupinine was not possible with one-dimensiona
spectroscopy, even at 400 MHz, because of substantial ov
of some of the signals. Homo- and heteroscalar corre
two-dimensional 'H and 'H,I3C nmr spectroscopy (COSY
J-resolved 2D 'H nmr spectroscopy were employed for a
plete assignment of the spectrum. Assignment of the down
signals, due to the 11 re and 1 si protons, had been rep
(21). A full discussion of the 'H nmr spectrum of lupinine
appear elsewhere (19). Assignments of the signals due t
protons of importance in this study are given in Table 4. T
assignments were originally made on the basis of a spec
determined at 250 MHz and subsequently confirmed o
basis of 400-MHz two-dimensional spectra (Table 5). But
at this frequency the signals for two pairs of protons, 4 a
6a, and 4P and lop, which appeared as two unresolved si
at 250 MHz, were insufficiently separated for unequi
assignment of the deuterium signals at chemical shifts c
sponding to these positions:
4a (eq) 2.57 ,6a (eq) 2.51; 4P
1.75, 10P (ax) 1.78 ppm. Assignment of the other three
tons of interest, 6P (ax), 1.57; 11-re, 3.75; 1-si, 4.16
did not pose a problem.
On the basis of the proton resonances, the deuterium si
in the spectrum of lupinine derived from (R)-(I-'H)cadav
(Fig. 2A) can be assigned as follows:
6
4.14, H- 11 si (c
sponding to the signal at
6
4.16 ppm in the 'H spectru
2.50, either H-6a (2.51) or H-4a (2.57); 6 1.77, either
(1.75) or H-1OP (1.78 ppm). Similarly, the signals in the
trum of lupinine from (S)-(1-'H)cadaverine (Fig. 2B) ar

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 9/12
GOLEBIEWSKl
AND
SPENSER
2715
to the following protons: 8 1.73, either H-4P (1.75) or H-1OP
(1.78); 1.54, H-6P (1.57 ppm).
These spectral assignments lead to four possible sets of deu-
teriated positions in the sample of lupinine from (R)-(1-'H)-
cadaverine, and to two possible sets in the sample of lupinine
from (S)-(1-'H)cadaverine, a total of eight possib le com bina-
tions for the assignment of the two spectra:
Spectrum 2A:
Spectrum
2B:
lupinine from
lupinine from
R) -
1-'H)cadaverine S) - 1 'H)cadaverine
6 4.14 6 2.50 6 1.77 6 1.54 6 1.73
A num ber of these combinations of assignments can be elim-
inated on the basis of considerations w hich follow from the fact
that the samples of lupinine from (R)- and from
(S)-(l-2H )cadave rine give different 'H nmr spectra and that
incorporation is thus stereospecific.
Firstly, it can be assumed that, since incorporation is stereo-
specific, the and protons at any one carbon atom cannot
both be deuteriated in a given lupinine sample. Thus, assign-
ment 3 for spectrum 2A, in which H-4a and H-4P both carry
deuterium, can be discounted.
Secondly, it can be assumed that since incorporation is
I
stereospecific, the two samples of lupinine derived from the
two enantiomers of (1-'H)cadaverine cannot both bear deu-
terium at the sam e site. Thus, the pair (1 a) for the assign -
ment of the spectra of the lupinine samples derived from (R)-
and (S)-(l-2H)cadaverine, respectively, can
be
discounted,
since assignment 1 and assignment a both have a resonance
assigned to H-4P. Similarly, the pairs (2 b) and (4 b) are
eliminated, since in each pair both assignments have a reso-
nance due to H-1OP.
A further restriction follows from the mode of incorporation
of I3C- and I4C-labelled substrates into cad averine . F rom these
experim ents it was concluded that the two C5 chains of lu -
pinine, C-6, -7, -8, -9 -10 and C -4, -3 , -2, -1, -1 1, ultimately
originate from one and the same C5 precursor. The o bserved
localization of label in all experiments with I4C-labelled racers
was consistent with equimolar distribution of radioactivity be-
tween the two C 5 chains , and the mode of incorpo ration of I3C
from l-13C,l-15N)cadaverine,ssdem onstrated by I3C nmr,
proved this equimolar distribution vide upra). It is quite un-
likely, therefore, that a sample of lupinine formed bio-
synthetically from a specifically deuteriated C5-substrate would
contain two d euteriated sites in one of the two C5 units, while
the other C 5 unit is free of deuterium . Yet this is the situation
presented by assignment b for spectrum 2B. According to this
assignment, both deuterium sites are located on the C5-unit,
C-6, -7, -8, -9 -10, while the other C,-unit, C-4, -3, -2, -1,
-1 1, is devoid of deu terium. In the light of the results of earlier
biosynthetic experiments such a distribution is improbable.
With these restrictions, the assignment of spectrum 2B is
unambiguous. The signals at 1.7 3 and 1.54 ppm can be
assigned to deuterium at H-4P and H-6P, respectively. Two
possible assignments remain for spectrum 2A. While the sig-
nals at 4.14 and 1.77 ppm can be unequivocally assigned to
deuterium at H-11-si and H-lOP, respectively, the signal at S
2.50 ppm is due either to H-6a or to H-4 a.
Notwithstanding this remaining ambiguity in the assignment
of one of the 'H nI tr spectra of the 'H-labelled samp les of
lupinine, the stereochemistry of every step of the biosynthetic
process from cadaverine into lupinine can now be deduced
from the available evidence.
As sho wn by the I3C nmr spectrum (Fig. 1A) of lupinine
derived from (1-I3C,1-I5N)cadaverine Experiment 5), four car-
bon atom s of lupinine, C-6 and C -10 of one of the C5 units and
C-4 and C-11 of the other, are derived from an a-carbo n atom
of cadaverine. As shown further by this spectrum, only on e of
the three C-N bonds of lupinin e, N, C- 6, represents an intact
C-N bond of cadav erine, whereas the three carbon atoms,
C-4, C -10, and C-1 1 of lupinine represent cadaverine carbon
atoms from which a nitrogen atom had been detached in the
course of the biosynthetic process.
Biochemical separation of a primary amino group from a
carbon atom is invariably accompanied by loss of hydrogen
atom from the a-carbon, either by oxidation (catalyzed by a
dehydrogenase, E .C. 1.4.1 ., or by an oxidase, E.C. 1.4 .3 or
by transamination (catalyzed by an aminotransferase, e.g.,
E.C.2.6.1.). These processes are stereospecific, i.e., the
a-hyd rogen atom must be in the correct steric environment in
order to be removed.
If, in the course of lupinine biosynthesis, removal of a hy-
drogen atom from the cadaverine carbon that is destined to
become C-4, C-10, and C-1 1 of lupinine indeed takes place
stereospecifically, then only one or the other, but not both, of
the samples of lupinine derived from the two enantiomers of
(l-2H)cad averine should carry d euterium at these positions.
The carbon atom destined to become C-6 of lupinine, on the
other hand, is shown by the experiment with '3C,15N-labelle
cadaverine to remain attached to the original nitrogen atom
throughout the biosynthetic process. Deuterium should then be
found at C-6 in both samples of lupinine. It follows that the
signal at
8
2.5 0 ppm in the spectrum of the sample of lupinine
derived from (S)-(l-2H )cadave rine (the S-lupinine ) is due to
a 6 proton rather than to a 4 proton.
The stereospecificity of incorporation of deuterium from
(R)- and from (S)-(l-2H)cadaverine into the two proton sites at
C- 6 of lup inine serves as clear evidence that the integrity of the
C-D bond of cadaverine is maintained on route into lupinin e.
Deu terium from (R)-(1-'H)cadaverine enters the re-site (a ) at
C-6, whereas deuterium from (S)-(1-'H)cadaverine enters the
si-site (P).
With the complete assignment of the signals in the 2H nmr
spectra of the deuteriated sam ples of lupinine (Figs. 2A and B ),
the stereochemical course of the biosynthetic steps from cadav-
erine into lupinine becomes clear.
The carbon atoms destined to become C-10 and C-11 of
lupinine are derived, according to Schemes 1Bd and 1B e (and
also lBc), from the aldehyde group of the intermediate
5-aminopentanal (3), a compound that is formed, early in the
biosynthetic sequence, directly from cadaverine by loss of an
amino group. Deuterium is present at C-10 and C-1 1 in the
lupin ine sam ple derive d from (R)-(1-'H)cadaverine ( R-
lupinine ) (Scheme 4) but not in that derived from
(S)-(l-2H)c adave rine ( S-lupinine ) (Scheme 5). It follow s that
removal of the amino group in the course of conversion of
cadaverine 2)
into 5-aminopentanal (3) is accompanied by
stereospecific loss of the si-proton from the a-carbon atom of
cadaverine (step a, Schemes 4 and 5).

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 10/12
2716
CAN. J CHEM.
VOL.
63
1985
attack from
C a f a c e
C D O
entry of H-lrom
- r c ; Fee
h N y l
Ndsintry of n
H ~ , D ~
rCHsi
H r e from C-re f
SCHEME
The biosynthetic route from cadaverine into lupinine
and its stereochemistry: incorporation of (R)-(1-'H)cadaverine.
The observation that a signal due to deuterium at C-4 is
foun d in the spectrum of S-lupinine (Fig . 2B) but not in that
of R-lupinine (Fig. 2A) proves that glutardialdeh yde (5)
(Schem e 1B d) cannot be implicated in the biosynth etic process:
Loss of the amino group of to yield 5 can, in principle, be
accompanied by loss of the si proton or the re proton from the
carbon to nitrogen. If the si proton were lost, then the sample
of S-lupinine should show only one deuterium signal, due to
deuterium at carbon-6 , while the sample of R-lupinine should
show four signals, due to deuterium at carbons 6, lO ,4, and 11.
If the re-proton were lost then, as a consequence of the C2
symmetry of glutardialdeh yde, the sample of S-lupinine
should show signals due to deuterium at carbons 6, 4, and 1 1,
in the ratio 2: 1 : 1, while the sample of R-lupinine should
show signals due to deuterium at carbons 6, 10, 4, and 11, in
the ratio 2: 2: 1 1. This is not observed. Scheme 1Bd is thereby
disproven . The amin o group is thus shed at a later stage of the
biosynthetic process (step c, Schemes 4 and 5). Since deu-
terium at C-4 is preserved in S-lupinine (Scheme 5) and not
in R-lupinine (Scheme 4), it is the re-proton which is lost,
together with the amino group, in this step.
Since the deuterium at C-4 of the S-lupinine is found in the
re position, reduction of the iminium bond (step d, Scheme 5)
must take place by entry of the reducing hydride ion from the
si-face at C-4. Finally, since deuterium at C-11 of the
R-lupinine is found in the si position, reduction of the carbo-
nyl carbon (step e, Sch eme 4), must take place by entry of th e
hydride ion from the re-face. The two experiments with en-
antiotopically deuteriated (l-2H)cadaverin e thus clarify the hid-
den stereoch emistry of fou r steps in the bio synthetic sequence.
The ov ert stereochemistry at C-1 and C-10 of lupinine is deter-
mined in the dimerization step (step b, Schemes 4 and 5) that
leads from A'-piperideine (4) into tetrahydroanabasine (14).
The proposed reaction sequence (Schemes lBe, 4, and 5)
postulates that A'-piperideine serves as an intermediate be-
tween cadaverine and lupinine. The validity of the proposed
sequence and its stereochemistry can be further tested by means
of an experim ent with deuterium -labelled A'-piperidein e (Ex-
attack from
C a f a c e
enlry of H-froni
Ci/e pf face
from C-re or
-
C-si face
SCHEME. The biosynthetic route from cadaverine into lupi
and its stereochemistry: incorporation of (S)-(I-'H)cadaverine.
periment 4). If the conclusions drawn from the experim
with chirally deuteriated cadaverine (Experim ents 1 and 2)
correct, it can be predicted that deuterium from (2-
A'-piperideine must enter at C-10 and at C-11-si of lupin
The 2H nrnr spectrum of a sam ple of lup inine derived f
(2-2H)-A l-piperideine s shown in Fig. 2D . The predictions
fully substantiated.
The final stereoch emical question that was answered in
investigation concerns the prochirality of the decarboxyla
of lysine in L. luteus. The evidence which shows that L-ly
is the precursor of lupinine in
L.
luteus (Experiment 6)
already been discussed (vide supra). Decarboxylation
L-lysine yields cadaverine. In princip le, this decarboxyla
can take place either with net retention or with net inversio
configuration. Decarboxylation of
L-(i.e., (S)-)(2-2H)ly
with net retention of configuratio n yields (S)-(l-2H )cad aver
whereas decarboxylation with net inversion yields
(I-'H)cadaverine. Incorporation into lupinine of the L-comp
ent of ~ ~ - ( 2 - ~ H ) l y s i n eields a sample of lupinine whose
nmr spectrum (Fig. 2C) is identical with that of the lupin
sample obtained when (S)-(l-2H )cadaverine served as the
strate (Fig. 2B).
It follows that, en rou te to lupinine, L-(2 -2H)lys ine s c
verted into (S)-(l-2H)cadaverine, i.e., in the process lead
from L -lysine to cadaverine the carboxyl gro up of ly sin
replaced by a proton w ith net retention of configuration . T
stereochem ical course is consisten t with th e prochirality of
reactions catalyzed by o ther L-amino acid decarbo xylases
(E.C.4.1.1.).
Th e experimental evidence here presented serves as a crit
test of the biogenetic hypotheses of lu pinine biosynthesis
have been proposed. All but one of the hypotheses are
proven on the basis of the results of the six biosy nthetic exp
ments which are here reported. The remaining hypoth
(Scheme 1Be) which is presented in full detail in Schem
and 5 , is entirely consistent with all the experimental evide
This evidence throws light on the steps from lysine into
pinine and on the stereochem istry of several of these step

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 11/12
GOLEBIEWSKI AND SPENSER 2717
xperimental
Extraction of lupinine and sp arteine from Lupinus luteus
The fresh plant m aterial was imm ediately ground in a blendor with
methanol and extracted in a Soxh let apparatus for 8 h. T he extract was
concentrated in vacuo and sulfuric acid (1 M , 1 mL) was added to the
residue. The mixture was extracted with ether (4
x
10 mL) and the
aqueous phase filtered through Celite and extracted with chloroform
(3 10 mL). The aqueous phase was neutralized with solid potassium
carbonate, basified with potassium hydroxide (50% w/v, 2 mL), and
extracted with methylene chloride (4 x 15 mL). The solvent was
evaporated in vacuo and the residue once again taken through an
acidlb ase cycle (sulfuric acid, potassium hydroxide, as above), yield-
ing a basic fraction (25 mg) which contained lupinine (20) and spar-
teine as the major components.
The alk aloid fraction (25 mg) was applied to a silica gel column (10
185 mm, 4 g, 230-400 mesh, BDH). The column was washed, in
turn, with methylene chloride (10 mL), 3% methanol in methylene
chloride/0.880 ammonia, 50 0: 1 (20 mL) and 5% methanol in meth-
ylene chloride/0.880 ammonia, 333 : 1 (40 mL). The lupinine fraction
was eluted with 8% methanol in methylene chloride/0.880 amm onia,
166: 1 (120 m L), the spa rteine fraction with 12% methanol in methyl-
ene chloride/0.880 ammonia, 100 : 1 (80 mL). Yield of crude alka-
loids: lupinine, 10 mg; sparteine, 4.5 mg. Lupinine was dissolved in
hydrochloric acid (0.1 M, 0.7 mL), water and excess of acid were
removed in vacuo, and the residue dried in vacuo. Recrystallization
from a mixture of methanol-acetone gave lupinine hydrochloride, mp
208-209 °C (lit. (23) mp 207-209 °C). A small sample of lupinine was
converted into lupinine methiodide, lit. (23) mp 295-296°C.
Sparteine was dissolved in acetone (2 drops) and a solution of
sulfuric acid in absolute ethanol (I M, 60 FL freshly prepared) was
added. Crystals of the sulfate derivative formed immediately, were
filtered off, and washed with a mixture of acetone and absolute ethanol
(3 , 3 drops), mp 265-266°C (dec.) (lit. (24) mp 264.5-265.5 C).
Radioactive materials
~ ~ - [ 6 - ' ~ C ] L y s i n end ~ - [ 4- ,H ] l~s in e ere obtained from commer-
cial sources (see Table 1).
Administration of radioactive tracers to L. luteus
Plants were grown from seed and used for tracer experiments 6
weeks after germination. The exp eriment was carried out in the growth
chambers (Experiment 6). Tracer solution was administered to 36
plants by wick in a single dose. The plants were allowed to grow 3
days in contact with tracer.
Materials labelled with stable isotopes
(1 13C,1 N)Cadav erine dihy drochloride (24)
5-Phthalimido pentano(1 -'3C,1 -ISN)nitrile 22)
A solution of
1-bromo-4-phthalimidobutane
21) (0.85 g) in di-
methyl sulfoxide (1.6 m L) was added dropwise over 2 min to a stirred
solution of sodium (I3C,l5N)cyanide 99 at.% I3C, 99 at.% I5N, MSD
Isotopes, M ontreal) (0.1 5 g) in aqueous dimethyl sulfoxide (1.6 mL
DM SO, 0 .2 mL H2 0) . The solution was stirred 4 h at 65°C and left
overnight at room temperature. Water (12 mL) was added and the
mixture extracted with benzene (4 5 mL). The benzene extract was
reextracted with water, dried, and evaporated to dryness in vacuo,
yielding crystalline product (680 mg). A small sample was re-
crystallized from absolute ethanol, mp 69-70°C; 'H nmr (CDC1,) 6:
8.13-7.87 (4H, m), 3.83 (2H, t,
J
6 Hz, H-5) 2.65-2.35 (2H, m,
H-4), 2.1-1.9 (4H, m); I3C nmr (CDCI,) 6: 119 .2 (d, J 1 3 ~ l s 16.7
HZ), 36.7 (C-5), 27.6 (C-4), 22.7 (C -3), 16 .6 (d, J 55.2 (HZ, C-2);
ms m z : 230 (6.5%), 188 (31, M CH 2- 13 C'5 ~)) ,60 (100, M
(C3H5- CHN)), 149 (21), 104 (98), 77 (12), 76 (13).
l-N-Acetyl-5-N-phthaloyl-l,5-diamino(l-13~,11 5 ~ )entane (23)
Crude
5-phthalimid0pentano(l-~~~,l- ~~)nitrile
22) (0.67 g) in
acetic anhyd ride (3 mL) was ad ded to a suspension of freshly prepared
Raney n ickel catalyst (0.5 g) in acetic anhydride (7 mL). (The cataly st
had been freshly prepared imm ediately before use by gradual addition
of sodium hydroxid e pellets to a suspension of nickel aluminum alloy
(1 1 w/ w, BDH ) in water, without cooling. This mixture was left for
30 min and heated on the steam bath for 1 h. The solid was then
washed with water, 95% alcohol, absolute alcohol, and acetic anhy-
dride.) Hyd rogenation was carried out at 80-90°C and 1 atm for 3 h.
The mixture was centrifuged, the supernatant solution was decanted,
and the metallic residue washed with acetic anhydride. The solvent
was evaporated in vacuo to yield the acetyl derivative as a solid (yield
0.8 g, m p 134-136 C, after recrystallization from absolute ethanol);
'H nmr (CDCI,) 6: 7.9-7.7 (4H, m), 6.0 (I H, dtd,
J I ~ N H
9.5 Hz,
J C H ~ - I N H
1 .2 H Z , J I ~ C . N H.8 HZ ), 3.67 (2H , t, J 6.6 HZ, H-5), 3.23
(2H, dm, J I ~ ~ . ~ .39 HZ), 1.94 (3H, d , J 12 HZ), 1.8--1.3 (6H, m);
I3C nmr (CDCl,) 6: 170.2 (1 5 ~ C 0 - ), 68.6 (NCO-), 134.1 (C-3',-
4'), 132.3 (C-1',-6'), 123 .3 (C-2',-5'), 39.5 (d,
J l3 C .1 .1 5 ~
10.2 HZ,
C-I ) , 37.7 (C-5), 29.0 (d,
J
33 HZ, C-2), 28.2 (d ,
J
7.1 HZ, C-4), 24.1
(CH,), 23.3 (d, J 8.5 HZ, C-3).
(1 C,
1
~)Ca dave rine dihydrochloride (24)
Hydroch loric acid (6 M , 14 mL) was added to the crude acetyl
derivative (23) and the mixture was stirred for 18 h at 100°C. Crys-
tallization of phthalic acid started on cooling and was complete after
1 h at 0°C. The phthalic acid was removed by centrifugation and the
supernatant solution was extracted with ethyl acetate (3
x
10 mL).
The aqueous layer was evaporated in vacuo. The residue was dried
(0.45 g) and recrystallized from 95% ethanol. After the first crop of
crystals (0.17 g), mp 255-257 C, was filtered off, the mother liquors
were chromatographed on an ion exchange column (Dowex 50-X4,
H + form, 2.1 mequiv./mL, 1.3
x
cm). The column was washed
with water (20 mL) and the produ ct was eluted with hyd rochloric acid
(1.5 M, 200 mL), yielding a further crop (0.13 g) of
(1-'3C,I-15N)cadaverine ihydrochloride. Total yield 57% (relative to
NaI3Cl5N); H nmr (DzO) 6: 3.76 (I H, t, J 7.2 Hz), 2.97 (2H , t,
J
7.2
Hz) , 2.18 ( lH , t ,
J
7. 2 Hz), 1.45- 1.75 (6H, m); J13C.H.I144 HZ; he
signals at 6 3.76 and 2.18 ppm are doublets of triplets, due to I3C,H-1
coupling of the 1-I3CH2 roup. The signal at 6 2.97 ppm is due to the
5-CH2 group; I3C nmr (D,O) 6: 40.0 (d,
J I ~ ~ . ~ . ~ ~ N
.4 HZ), 27.0 (s, d,
J13c.2 13c.l 35 .4 HZ,), 23.4 (s, C-3).
D L - ( Z - ' H ) L ~ S ~ ~ ~onohyd rochloride was prepared (13) in two steps
from diethyl 2-acetamidomalonate and
1-bromo-4-phthalimidobutan
(21). Condensation yielded diethyl 2-acetamido-2- 4-phthalimido
butyl)malonate, whose hydrolysis in D CI/ D2 0 yielded the desired
product. D~-(2 -'H)L ~sin e onohydrochloride: IH nmr (D2 0) 6: 3.7
(ca. 0.05 H, H-2), 3.05 (2H, t,
J
7.2 Hz, H-6), 1.3-2.0 (m, 6H).
( 2 - ' ~ ) - A ' - ~ i ~ e r i d e i n end (S)-(+)-(I -'H)cadaverine hydrochlo ride
were prepared from the above DL-(2-'H)lysine (vide infra).
(2-'H)-A1-Piperideine (cf. ref. 13)
Freshly crystallized N-bromosuccinimide (0.32 g) was added to a
so lu t ion of D ~- ( 2- ' ~ ) l~s ineonohydrochloride (0.64 g) in water (90
mL) and the solution was heated at 50°C in a nitrogen atmosphere on
a rotary evaporator under mild suction until colorless. Three more
portions of N-bromo succinimide (3 0.3 2 g) were added and reaction
continued in the same way. T he total reaction time was 55 min. The
solution was used for feeding without further purification.
A small portion of the solution was co ncentrated and the 'H and 'H
nmr spectra of the mixture were determined; 'H nmr 6: 7.5 (s), 6. 9 (s),
6.3 (s) , 4.5 (H 20 ), 3.5 (bm), 3.2 (s) , 3.0 (m), 2.6 (s) ,
(CH2-CO),NH, succinimide);
'H
nmr 6: 8.7 (rel. area I ), 4. 9 (rel.
area 2.4), 4.5 (DHO, natural abund ance) ppm . When the solution was
evaporated and redissolved in water, the 'H nmr spectrum changed
considerably: 'H
n r
6: 8.3 (rel. area 5.7), 8.1 (1.3), 6.7 (1.0), 4.9
(1.8), 3 .8 (4 .3). When the solution was basified (K2C0 3) and ex-
tracted six times with chloroform , the chloroform evaporated, and the
residue redissolved in water, a further spectral change was observed:
'H nmr 6: 7.9 (rel. area 0. I), 3. 1 (0.9).
(R)-(-)- and (S)-(+)-(l-ZH)Cadaverineydrochloride
(R)-(-)-(l- ~)Cadaverine
hydrochloride was prepared by decar-
he
value, 18.4 Hz, for
J13c.z.13c.l
that was reported in our pre-
liminary com municatio n (I 1) was in error. We thank D r.
E
Leete for
pointing out this mistake.

7/24/2019 BIOSINTESIS LUPINin
http://slidepdf.com/reader/full/biosintesis-lupinin 12/12
2718 CAN.
J
CHEM.
VOL.
63 1985
boxylation of the L-com ponent of DL-lysine in deuterium oxid e, cata-
lyzed by L-lysine decarboxylase (13). 'H nmr ( D2 0) : 2.95 (3.01 H,
t, J 7.2 Hz, H-1,5)
1.45-1.75 (m, 6H); ca. 1 nondeuteriated
product; ms mlz: 89 (M'
I ,
9 ) 88 (M ', 100 +NHsCHD (CH2)4),
87 (M' 1, 14).
s)- +)- l-2~)~adaverine ydrochloride was obtained (13) by
decarboxylation, catalyzed by L-lysine decarboxylase, of the
L -co mp on ent of ~ ~ - ( 2 - ~ H ) l ~ s i n e95 at. 2-'H). (S)-(+)-(1-2H )-
Cadaverine dihydrochloride (93 at. L2H): 'H nmr (D 20 ) similar to
that of
(R)-(-)-(1- H)cadaverine
hydrochloride, above; ms mlz: 89
(M' 1 , 10.6 ), 88 (M ', loo ), 87 (M' 1, 16), 7 non-deuteriated
product, based on computer assisted peak height analysis.
Administration of enriched substrates to L. luteus
The plants used in these experiments were grown from seed and
were used in feeding experiments 3-6 weeks after germination. The
feeding experiments were carried out in the greenhouse (summer
months, Experiments 3 , 5) or in the growth chambers (winter mon ths,
Experiments 1, 2, 4). Tracer solution was administered by wick over
a period of 5-6 days; the plants were then grown for a further period
of 3-4 days before being harvested.
Acknowledgements
We are grateful to Professor Dr. M. W iewiorow ski, Univer-
sity of Pozn an, for a gift of seeds of
Lupinus luteus
o Thelma
Leech, Greenhouse Supervisor, McMaster University, for
providing facilities for our experiments, and to
J.
Ian A.
Thompson and Brian G. Sayer, Department of Chemistry, for
recording nmr spectra. We are greatly indebted to Dr. R. E.
Lenkinsk i, South Western Ontario NMR Facility, Un iversity of
Guelph , for determinin g COSY and 2-D resolved spectra.
This investigation was supported by a grant from the Natural
Sciences and Engineering Research Council of Canada.
1. C. SCHOPF, . SCHMIDT,nd W. B RAU N. er. 64 683 (1931).
2. C. SCHOPF.n IX Co ngress of the International Union of Pu re and
Applied Chemistry, Madrid. Vol. 5. 1934 . pp. 1 97- 198.
3. C. SCHOPF.Angew. Chem. 61, 31 (1949).
4. R. ROBINSO N. tmctura l relations of natural products. Oxfo
1955. p. 74.
5. C. SCHOPF.Angew. Chem. 69, 69 (1957).
6 . H . R . SCH U T~E.rch. Pharrn. (Weinheim), 293, 1006 (196
7. H. R . SCHUITE nd H. HINDORF. . Naturforsch. Teil B , 19 ,
(1 964).
8. M. SOUCEKnd H. R. SCHUITE.Angew. Chem. Int. Ed. En
1, 597 (1962).
9. J. R AN A nd D. J. ROBINS. . Chem. Research (S), 16 4 (198
10. G. GRUE-SBRENSENnd I. D. SPENSER. an. J. Chem. 60,
(1982).
11. W. M. GOLEBIEWSKInd I. D. SPENSER.. Chem. Soc. Ch
Commun. 1509 (1983).
12. W. M. GOLEBIEWSKInd I. D. SPENSER.. Am. Chem. Soc. 1
1441 (1984).
13. J. C. RICHARDSnd I. D. SPE NSER. etrahedron, 39, 3
(1983).
14. J. R AN A nd D. J. ROBINS. . Chem. Soc. Chem. C ommun.
(1984).
15. K. SHIM A nd U . KURASH IGE. ippon Kagaku Kaishi,
(1983); Chem. Abstr. 99, 54040a (1983).
16. E. WENKERT, . CHAUNC Y,. G. DA VE , . R. JEFFCOAT,
SCHELL,nd H. P. SCHENK.. Am. Chem. Soc. 95 ,842 7 (19
17. F. BOHLMANNnd R. ZEISBERG. hem. Ber. 108, 1043 (19
18. H. PODKOWINSKAnd J. SKOLIK.Org. Magn. Reson. 22,
(1984).
19. W. M . GOLEBIEWSKI.O be published.
20. E. LEISTNER, . N. GUPTA nd I. D. S PENSER.. Am. Ch
SOC.95, 4040 (1973).
21. H. PODKOWINSKA.esz. Nauk. Akad. Ekon. Poznaniu, Ser
109, 107 (1983); Chem. Abstr. 100, 51872b (1984).
22. J. C. RICHARDSnd I. D. SPENSER. an. J . Chem. 60, 2
(1982).
23. J. F COUCH. . Am. Chem. Soc. 56, 2434 (1934).
24. J. F COUCH. . Am. Chem. Soc. 59, 1469 (1937).