Embed Size (px)
Citation preview

INSTITUTO POLITÉCNICO NACIONAL
UNIDAD PROFESIONAL INTERDISCIPLINARIA DE
BIOTECNOLOGÍA
“CARACTERIZACIÓN GENÉTICA DE CEPAS DE LEVADURAS AISLADAS DE MEZCAL”
INFORME TÉCNICO DE LA OPCIÓN CURRICULAR EN LA MODALIDAD DE:
PROYECTO DE INVESTIGACIÓN
QUE PARA OBTENER EL TÍTULO DE INGENIERO BIOTECNÓLOGO
PRESENTA:
ANTONIO FLORES HENRY
DIRECTOR DE PROYECTO: Biol. PATRICIA LEONOR RODRÍGUEZ PASCUAL
SINODALES: Dr. GERMÁN F. GUTIÉRREZ HERNÁNDEZ Dra. MARINA OLIVIA FRANCO HERNÁNDEZ
México D.F. Mayo 2006




UPIBI
AFH RPLP
Director del Proyecto Biol. Patricia Leonor Rodríguez Pascual Sinodal 1 Dr. Germán F. Gutiérrez Hernández Sinodal 2 Dra. M. Olivia Franco Hernández Alumno Antonio Flores Henry

UPIBI
AFH RPLP
AGRADECIMIENTOS Mmmm, bueno, este como el resto del trabajo es difícil de redactar, así q solo diré gracias, jajajaja, no s cierto, mm, bueno ahí vamos.
• En primerísimo lugar agradezco a mi Ma, que fue la que me apoyo mucho durante todos mis estudios, miel gracias Ma, ja miel gracias, eso que?.
• En segundosimo lugar agradezco a mis hermanos (Nadia y Pablo) y a mi Pa, que me soportaron lo suficiente durante estos 5 años, ja, la carrera es de 4 años.
• Agradezco a mi asesora profesora Patricia, que me soporto y nunca me regaño ni me grito, jajajaja, y me defendía, por ahí se rumora que es mi madre.
• Agradezco a los profesores del departamento de bioquímica (Profa Cuca, Profa. Martha, Prof. Rodrigo, Prof. Adan, Prof Gerardo, gracias al fotodocumentador prof. y al Prof. Sergio).
• Agradezco a mis sinodales (Prof. Germán y Profa Olivia), ya que sin ellos no hubiera obtenido la calificación, jajaja, gracias al apoyo ( los primers, el termociclador y el fotodocumentador) recibido de ellos.
• Agrdezco a mi novia la Lizbetha, gracias por comprender a este ente, jajaja, TQM.
• A mis amigos las ARDILLAS (Ann, Pat, Tan, Vick, Qro 1, Qro 2, Male, Ren, Viejo, Farmer, Yom) como dice el dicho, una ardilla nace no se hace, jajaja.
• Mmm, ya es tarde, y no me recuerdo a quien mas debo de agradecer, así que haré un agradecimiento general, gracias a todos los tipos y tipas que me han ayudado.

UPIBI
AFH RPLP a
ÍNDICE TEMÁTICO
ÍNDICE TEMÁTICO--------------------------------------------------------------------------------------------a RESUMEN -------------------------------------------------------------------------------------------------------1 1. INTRODUCCIÓN--------------------------------------------------------------------------------------------2
1.1. EL MEZCAL---------------------------------------------------------------------------------------------2 1.2. PROCESO DE ELABORACIÓN DEL MEZCAL -----------------------------------------------4 1.3. LEVADURAS -------------------------------------------------------------------------------------------5 1.4. CARACTERIZACIÓN---------------------------------------------------------------------------------6 1.5. PCR (REACCIÓN EN CADENA DE LA POLIMERASA) ------------------------------------8 1.6. VENTAJAS DEL PCR-RAPD--------------------------------------------------------------------- 11 1.7 DESVENTAJAS DEL PCR-RAPD --------------------------------------------------------------- 12
2. ANTECEDENTES----------------------------------------------------------------------------------------- 13 3. JUSTIFICACIÓN ------------------------------------------------------------------------------------------ 14 4. OBJETIVOS ------------------------------------------------------------------------------------------------ 16
4.1 GENERAL---------------------------------------------------------------------------------------------- 16 4.2 ESPECÍFICOS ---------------------------------------------------------------------------------------- 16
5. ESTRATEGIA EXPERIMENTAL---------------------------------------------------------------------- 17 6. MATERIALES Y MÉTODOS --------------------------------------------------------------------------- 18 7. METODOLOGÍA------------------------------------------------------------------------------------------- 18
7.1. ELECCIÓN DE CEPAS ---------------------------------------------------------------------------- 18 7.2. OBTENCIÓN DE ADN ----------------------------------------------------------------------------- 18 7.3. DETERMINACIÓN DE CONCENTRACIÓN DE ADN-------------------------------------- 19 7.4. ESTANDARIZACIÓN DE LAS CONDICIONES DE PCR-RAPD------------------------ 19 7.5. PCR-RAPD-------------------------------------------------------------------------------------------- 20
8. RESULTADOS Y DISCUSIÓN ------------------------------------------------------------------------ 21 8.1. ELECCIÓN DE CEPAS ---------------------------------------------------------------------------- 21 8.2. OBTENCIÓN DE ADN ----------------------------------------------------------------------------- 22 8.3. DETERMINACIÓN DE CONCENTRACIÓN DE ADN-------------------------------------- 24 8.4. ESTANDARIZACIÓN DE LAS CONDICIONES DE PCR-RAPD------------------------ 24 8.5. PCR-RAPD Y ANÁLISIS DE BANDAS -------------------------------------------------------- 29
9. CONCLUSIONES----------------------------------------------------------------------------------------- 32 - ANEXO I --------------------------------------------------------------------------------------------------- 34 - ANEXO II -------------------------------------------------------------------------------------------------- 35 - ANEXO III-------------------------------------------------------------------------------------------------- 36 - ANEXO IV ------------------------------------------------------------------------------------------------- 38 - ANEXO V -------------------------------------------------------------------------------------------------- 39
11. BIBLIOGRAFÍA ------------------------------------------------------------------------------------------ 40

UPIBI
AFH RPLP b
ÍNDICE DE FIGURAS
Figura 1. Foto de la planta de Agave. ------------------------------------------------------
- - - 2
Figura 2. Entidades Federativas productoras de Mezcal. -------------------------------
- 2
Figura 3. Etapas de elaboración de mezcal. ----------------------------------------------
- - - 4
Figura 4. Fotografía de levadura. ----------------------------------------------------------
- - - - - 5
Figura 5. Proceso de desnaturalización del DNA. ---------------------------------------
- - - 7
Figura 6. Alineación de los primers al ADN. --------------------------------------------
- - - - - 7
Figura 7. Proceso de extensión del PCR. -------------------------------------------------
- - - 8
Figura 8. Amplicones. -----------------------------------------------------------------------
- - - - - - - 8
Figura 9. Amplificación exponencial del PCR. -----------------------------------------
- - - - - 9
Figura 10. Amplificación por PCR-RAPD. ----------------------------------------------
- - - - - - 9
Figura 11. Diagrama de bloques de las actividades a realizar. ------------------------- 16
Figura 12. 1er gel de electroforesis de ADN. --------------------------------------------
- - - - 21
Figura 13. 2do gel de electroforesis de ADN. --------------------------------------------
- - - - 22
Figura 14. Amplificación de ADN de cepa PJMN2 1. ----------------------------------
- - - - 25
Figura 15. Amplificación de ADN de cepa PJMN2 2. ---------------------------------- 26

UPIBI
AFH RPLP c
- - - -
Figura 16. Resultados de PCR-RAP a cepas de S. cerevisiae. --------------------- 27
Figura 17. Cámara de electroforesis. ------------------------------------------------------
- - - - 35
Figura 18. Peine de 10 dientes. -------------------------------------------------------------
- - - - 35
Figura 19. Carro de cámara de electroforesis.--------------------------------------------
- - - 35

UPIBI
AFH RPLP d
ÍNDICE DE TABLAS
Tabla 1. Levaduras aisladas del proceso de elaboración del mezcal. ---------------- 12
Tabla 2. Destino de las exportaciones de mezcal. ----------------------------
- - 13
Tabla 3: Cepas seleccionadas. -------------------------------------------------------------- 20
Tabla 4. Concentraciones de ADN obtenido a partir de cepas de S. cerevisiae. --- 23
Tabla 5. Primers utilizados. -----------------------------------------------------------------
- - - - - 24
Tabla 6. Datos sobre reactivos utilizados. ------------------------------------------------
- - - - 25
Tabla 7. Datos sobre las temperaturas y ciclos del termociclador. --------------------
- 25
Tabla 8. Cepas de microorganismos seleccionadas. -------------------------------------
- 32
Tabla 9. Composición del medio de fermentación. --------------------------------------
- - - 32
Tabla 10. Composición de la solución A. -------------------------------------------------
- - - - 33

UPIBI Mayo 2006
AFH RPLP 1
RESUMEN
CARACTERIZACIÓN GENÉTICA DE CEPAS DE LEVADURAS AISLADAS DE
MEZCAL
El mezcal es una bebida alcohólica que se obtiene por la destilación de los mostos (o
jugos), obtenidos a partir de las piñas cocidas y molidas del agave. Lamentablemente esta
bebida casi no se produce a nivel industrial ya que no se tiene un control sobre los
microorganismos que intervienen durante su proceso. Es por ello el motivo de estudio. En
un trabajo anterior11a se caracterizaron cineticamente a cepas de Saccharomyces cerevisiae
que intervienen en el proceso de elaboración del mezcal, y los resultados muestran
diferencias en el comportamiento de cada cepa. Entonces se prosiguió a caracterizar desde
un punto de vista genético a 6 cepas de Saccharomyces cerevisiae utilizando la técnica de
PCR-RAPD. Para el tamizaje de primers se utilizaron 20 oligonucleotidos, a diferentes
condiciones de reacción en un volumen de 30µL, solo 8 presentaron amplificación, y el
primer que nos presento mayor bandeo fue el primer G16, este bandeo nos permitió
distinguir las cepas, ya que cada una de ellas presento diferente bandeo. Por lo que
podemos suponer que las diferencias genéticas de estas cepas podrían ser la causa del
comportamiento cinético diferente.

UPIBI Mayo 2006
AFH RPLP 2
CARACTERIZACIÓN GENÉTICA DE CEPAS DE LEVADURAS AISLADAS DE
MEZCAL
1. INTRODUCCIÓN 1.1. EL MEZCAL
A la llegada de los españoles, en 1519, el pulque era la única bebida alcohólica que se
conocía. Una vez que se introdujo en México el proceso de destilación surgieron bebidas de
alto grado alcohólico obtenidas del Agave a las que originalmente llamaron "vino de agave"
o "vino de mezcal. 1
La palabra mezcal se deriva de las palabras náhuatl Melt e Ixcalli que significan "agave
cocido al horno". De acuerdo a la Norma Oficial Mexicana (NOM-070-SCFI-1994), el
mezcal es una bebida alcohólica que se obtiene por la destilación de los mostos (o jugos),
preparados directamente con los azúcares extraídos de los agaves, mismas que son
previamente cocidas y sometidas a fermentación alcohólica. 2
Figura 1. Foto de la planta de Agave.
El mezcal se produce en casi todos los lugares de México donde hay agave, y se produce
principalmente en la región del mezcal que comprende los estados de Oaxaca, Guerrero,
Durango, San Luis Potosí y Zacatecas; y particularmente en la región que se localiza en el
Estado de Oaxaca y que comprende los distritos de Sola de Vega, Miahuatlán, Yautepec,
Tlacolula, Ocotlán, Ejutla y Zimatlán.3

UPIBI Mayo 2006
AFH RPLP 3
Figura 2. Entidades Federativas productoras de Mezcal.

UPIBI Mayo 2006
AFH RPLP 4
Para esta bebida se diferencian 2 tipos principales:
Mezcal 100% de Agave, elaborado con los mostos que únicamente contienen
azúcares provenientes del Agave.
Mezcal con otros azúcares, elaborado con un 80% de los mostos de Agave, a los
que se les adiciona un 20% de otros azúcares. 3
1.2. PROCESO DE ELABORACIÓN DEL MEZCAL
El proceso de elaboración de mezcal como ya se mencionó antes, consta de los siguientes
pasos:
Jima: en este paso se recolectan las piñas, llamada así por el aspecto que adquiere
después del corte de las hojas, del Agave. Se cava en la tierra alrededor de la planta para
sacar el tronco y las raíces, para ser despojada de las hojas (pencas), hasta dejar solo la
piña.
Cocimiento: se lleva a cabo en hornos subterráneos, se colocan trozos de piña cruda
sobre una capa de bagazo húmedo la que a su vez se encuentra encima de piedras que se
han calentado por la leña que se deposita en la parte inferior del horno, regularmente
este horneado en seco se realiza aproximadamente en 72 h a una temperatura de 82 ºC
(datos estimados), esto tiene como objetivo la hidrólisis de los polisacáridos del Agave,
(inulina), la cocción desdobla la inulina a azúcares fermentables como levulosa y
dextrosa (fructosa), en este punto también se obtiene hidroximetio-furfural, el cuál
perfila el aroma y sabor característico que determina la calidad del mezcal.
Molienda: una vez cocida la piña se cortada y se macera en molinos de piedra jalados
por caballos (empresas grandes) o son golpeados con mazos, en bateas de madera
(unidades familiares).
Fermentación: se coloca en ollas de barro grandes o en toneles de madera, se adiciona
agua corriente, “pastilla”, es decir, sulfato de amonio y se deja fermentar durante 4 días,
únicamente con los microorganismos nativos. Sin la adición de la “pastilla” la
fermentación duraba 10 días. En el municipio de Zimatlán de Álvarez, Oaxaca, la
fermentación se lleva a una temperatura media de 27 ºC. Las empresas medianas
algunas veces adicionan cultivos de levaduras y en algunos casos se suplementa el
mosto con azúcares y algunos nutrientes para aumentar el contenido alcohólico del
producto.

UPIBI Mayo 2006
AFH RPLP 5
Destilación: el producto ya fermentado, es destilado en un alambique de barro o
alambique tradicional (hecho de cobre) a fuego directo de leña. Por lo general se
realizan dos destilaciones, este proceso es heredado de padre a hijo, se dice “Que entre
más se apure el proceso, el mezcal es menos bueno” (dicho popular). Aquí se obtiene
mezcal con graduaciones de variables, que van de 40 a 50 º G.L.
Envasado: el destilado es recolectado por lo envasadores, para homogeneizarlo y
estabilizarlo dando el grado alcohólico requerido para su venta que de acuerdo con la
Norma Oficial Mexicana de Calidad del Mezcal, debe estar comprendido entre 36 y
55 % alcohol sobre volumen.
Figura 3. Etapas de elaboración de mezcal.
1.3. LEVADURAS
En el presente trabajo nos enfocaremos al proceso de fermentación y, en particular a la
caracterización genética de cepas de levaduras aisladas del proceso de elaboración de
mezcal. La fermentación es llevada a cabo por microorganismos denominados levaduras,
estos microorganismos son los encargados de transformar el azúcar en alcohol. Estos
microorganismos eucariotas son hongos del grupo de los ascomicetos cuya forma
dominante de crecimiento es unicelular. Poseen un núcleo y se multiplican por
reproducción sexual o asexual, por gemación o por fisión transversal. La reproducción
sexual, cuando ocurre, es por medio de ascosporas contenidas en un saco o asca.5

UPIBI Mayo 2006
AFH RPLP 6
Figura 4. Fotografía de levadura
En la fermentación (condiciones aeróbicas) las levaduras, utilizando el azúcar del
medio, que en este caso es inulina hidrolizada (fructosa) realizan el siguiente proceso:
Azúcar Etanol Gas carbónico Calor
En este caso, se genera gas carbónico y etanol.6
Se sabe ahora que los microorganismos que llevan acabo la fermentación de esta bebida, le
confieren propiedades organolépticas, tales como color, olor y sabor. Estas son
características necesarias que debe tener una bebida para ser atractiva al consumidor, por lo
que se trata de mejorarlas. Debido a lo anteriormente expuesto es necesario realizar una
caracterización fisiológica y genética de estos microorganismos, para poder así saber cual
es el microorganismo más indicado para la elaboración de la bebida. Se sabe que las cepas
utilizadas en enología ahora disponibles se han seleccionado a partir de la flora indígena, ya
que para este tipo de fermentación no se utilizan cepas genéticamente modificas, puesto que
resulta difícil introducir una nueva propiedad o modificar una característica sin afectar otras
propiedades cinéticas u organolépticas, que como ya se explico son esenciales para una
levadura utilizada en la fabricación del mezcal.7
1.4. CARACTERIZACIÓN
La caracterización de las levaduras se realiza desde los siguientes aspectos:
Fenotípico.
Genético.
Los caracteres fenotípicos de más fácil determinación son los estructurales y anatómicos
que pueden observarse directamente o con ayuda de lupa o microscopio, pero hay otros
como la capacidad de degradación, cinética de crecimiento, etc. los cuales son necesarios si
se les quiere dar un uso biotecnológico8.

UPIBI Mayo 2006
AFH RPLP 7
Los caracteres genotípicos son más difíciles de determinar, puesto que se requiere algún
método de extracción de ADN y así poder aplicar alguna técnica de marcadores
moleculares para su descripción, en la mayoría de los casos ésta caracterización es
fragmentaria.
En la caracterización genotípica de las levaduras involucradas en la fermentación del
mezcal se determinaron los parámetros cinéticos de estas, como se encontraron diferencias
importantes procedimos a realizar una caracterización genética que ayude a explicar el
comportamiento de éstos microorganismos. 10
Las técnicas moleculares están siendo utilizadas para la caracterización genotípica. Estas
proporcionan una base para la determinación de especies microbiológicas. 9
Los marcadores moleculares se pueden relacionar con un rasgo genético y DNA. Cuando
un marcador molecular es invariable en todos los organismos estudiados se dice que es
monomórfico, pero cuando presenta diferencias en el peso molecular, actividad enzimática,
estructura, o sitios de restricción, o sea variación se dice que es variable. A veces el grado
de variación es tal que se denominan hipervariable.10
Los marcadores moleculares sirven de señal, la cual ayuda a revelar la ubicación relativa
de genes en el genoma. Los genes polimorfitos del genoma constituyen los mejores
marcadores moleculares debido a que los individuos pueden diferir en múltiples maneras.10a
Los primeros marcadores se basaron en la identificación de proteínas e isoenzimas por
electroforesis en geles de almidón o poliacrilamida, pero esta técnica no era capaz de
detectar suficientes polimorfismos entre variedades o especies próximas debido a que las
proteínas son el resultado de la expresión génica, que puede ser distinta de unos tejidos a
otros, de una etapa de desarrollo a otra, de un medio ambiente a otro, y de una época del
año a otra.
Los avances de la tecnología del DNA recombinante han permitido el desarrollo de los
marcadores moleculares basados en el DNA, consiguiendo estabilidad en la identificación
de especies y variedades. El número de técnicas descritas es cada vez más numeroso, pero
se pueden reunir en 3 categorías: RFLP, STS y MAAP10, en este trabajo solo nos
enfocaremos a la categoría MAAP, ya que la técnica de PCR-RAPD se engloba en esta.
- MAAP (Múltiples perfiles arbitrarios de amplificación): Término genérico con el que se
designan las técnicas que emplean primers arbitrarios para generar huellas dactilares
complejas. Entre las cuales caben destacar: PCR (Reacción en Cadena de la Polimerasa),

UPIBI Mayo 2006
AFH RPLP 8
AFLP (Polimorfismo de la longitud de los fragmentos amplificados) y AP-PCR (PCR con
primers arbitrarios).
1.5. PCR (REACCIÓN EN CADENA DE LA POLIMERASA)
La PCR (Polymerase Chain Reaction) es una técnica "in vitro" la cual se basa en la
amplificación enzimática de una región de ADN que corresponden a un gen o parte de él, lo
amplifica más de un millón de veces, siempre y cuando se conozca una parte de su
secuencia de nucleótidos. Esta técnica utiliza dos oligonucleótidos (primers) sintéticos de
10-20 nucleótidos que son complementarios a los extremos de la región que se quiere
amplificar. Estos, actúan como cebadores para la síntesis del ADN la cual es habitualmente
catalizada por una enzima llamada Taq polimerasa (aislada de la bacteria Thermus
aquáticus) cuya temperatura optima esta entre 79 y 85 ºC. A esta temperatura la enzima es
capaz de realizar una extensión de más de 60 nucleótidos por segundo en regiones ricas en
uniones G-C. Esta temperatura permite la unión específica de los primers y la extensión, así
aumenta el nivel de exigencia de la reacción. La reacción se lleva a cabo en una serie de
ciclos cada uno de los cuales incluye tres fases o pasos:
Desnaturalización: Se usan temperaturas de 90 a 95 ºC que producen la rotura de los
puentes de hidrógeno intercatenarios (separa las cadenas). La temperatura se mantiene unos
minutos para conseguir la separación de las hebras, esto evita que se renaturalizarse y no
permita hibridación de los primers.
Figura 5. Proceso de desnaturalización del DNA.
Hibridación: Llamada también annealing o de emparejamiento, se disminuye la
temperatura en un rango comprendido entre los 40 y los 60ºC para producir la unión de los
primers a los extremos del fragmento que se va a amplificar.

UPIBI Mayo 2006
AFH RPLP 9
Figura 6. Alineación de los primers al ADN.
La temperatura de fusión o annealing (Tm, “melting temperature) depende de varios factores
y es relativamente específica para cada primer. La longitud de los primers y la secuencia
son críticas en la designación de los parámetros de una amplificación. Una fórmula simple
para calcular la Tm es la siguiente:
)(2)(4 TACGTm +++= ……………..ec 1.
Extensión: También llamada amplificación, durante este paso la Taq polimerasa incorpora
nucleótidos en el extremo 3' del primer utilizando como molde la cadena de ADN
previamente desnaturalizada. Se lleva a una temperatura entre 72 a 75ºC ya que es la
temperatura a la que la Taq polimerasa alcanza su máxima actividad. Al final hay que dejar
un minuto más para que pueda amplificar lo máximo.
Figura 7. Proceso de extensión del PCR.
Al final del primer ciclo de la PCR, hay dos dobles hebras de DNA idénticas a la original;
cada nueva hebra producida se denomina “amplicón”.
Figura 8. Amplicones.
Este ciclo de tres pasos (desnaturalización, alineamiento y extensión) se repite varias veces.
Así se crean primero dos, cuatro, ocho, .... 2n copias (donde n = número de ciclos), después
de 30 ciclos se obtienen un millón de copias. Este proceso es llamado amplificación
exponencial9 debido a que la secuencia objetivo es copiada una y otra vez con el objeto de
hacer las copias necesarias para que haya el suficiente material genético para realizar las
respectivas pruebas.
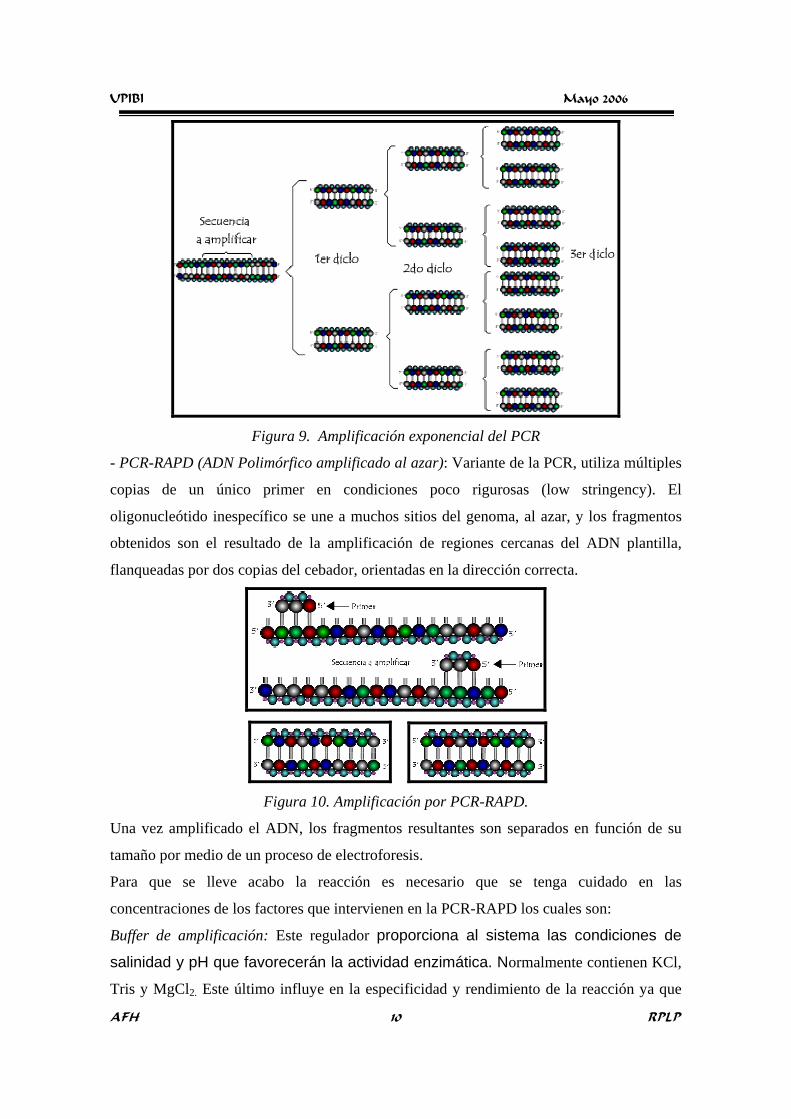
UPIBI Mayo 2006
AFH RPLP 10
Figura 9. Amplificación exponencial del PCR
- PCR-RAPD (ADN Polimórfico amplificado al azar): Variante de la PCR, utiliza múltiples
copias de un único primer en condiciones poco rigurosas (low stringency). El
oligonucleótido inespecífico se une a muchos sitios del genoma, al azar, y los fragmentos
obtenidos son el resultado de la amplificación de regiones cercanas del ADN plantilla,
flanqueadas por dos copias del cebador, orientadas en la dirección correcta.
Figura 10. Amplificación por PCR-RAPD.
Una vez amplificado el ADN, los fragmentos resultantes son separados en función de su
tamaño por medio de un proceso de electroforesis.
Para que se lleve acabo la reacción es necesario que se tenga cuidado en las
concentraciones de los factores que intervienen en la PCR-RAPD los cuales son:
Buffer de amplificación: Este regulador proporciona al sistema las condiciones de
salinidad y pH que favorecerán la actividad enzimática. Normalmente contienen KCl,
Tris y MgCl2. Este último influye en la especificidad y rendimiento de la reacción ya que

UPIBI Mayo 2006
AFH RPLP 11
los iones Mg+2 actúan como cofactores de la enzima. Es necesario probar diferentes
cantidades de MgCl2 ya que un exceso origina una acumulación de productos inespecíficos
y una cantidad pobre disminuye el rendimiento de la amplificación.
Primers: Para estos hay una serie de reglas a seguir:
La longitud debe estar comprendida entre 10 y 20 bases ya que se ha comprobado
que primers de mayor longitud (30-35 bases) no aumentan el rendimiento y los
primers cortos carecen de suficiente especificidad.
El contenido en G-C se recomienda que debe ser entre 40-60 %.
La secuencia de los primers debe comenzar y terminar con 1-2 bases púricas.
Para evitar la formación de dímeros de primers es necesario comprobar que los
primers no contengan secuencias complementarias entre sí.
Desoxinucleótidos Trifosfato: Las concentraciones de dNTPs y de MgCl2 se encuentran
relacionadas ya que el Mg+2 se une a los dNTPs con lo que concentraciones elevadas de
dNTPs inhibirían la reacción al no tener la Taq polimerasa suficiente Mg como para
incorporar dNTPs.
Taq-Polimerasa: La actividad de este enzima se ve influenciada por la concentración de
dNTPs, de Mg y de algunos iones monovalentes de manera que concentraciones elevadas
de los mismos inhiben dicha actividad.
ADN Molde o “Template": El ADN que la Taq polimerasa utiliza como molde para la
síntesis de nuevas cadenas polinucleotídicas. Depende de los siguientes factores:
Marcador Que Se Va a Amplificar: Hay marcadores cuyos primers son más
específicos o cuyas condiciones de amplificación están mejor optimizadas que las
de otros. Por eso puede darse el caso de que cierta cantidad de ADN amplifique
para unos marcadores pero no para otros.
Calidad del ADN: Cuando se trabaja con ADN cuya calidad es óptima no suele
haber problemas en la amplificación y cantidades del mismo por encima e incluso
por debajo de los 5 ηg rinden buenos resultados.
1.6. VENTAJAS DEL PCR-RAPD
Esta técnica no requiere una gran cantidad de ADN, además puede realizarse a
partir de una preparación de ADN cruda
Es rápido y mucho más fácil de realizar, aunque susceptible a contaminación.
Tiene un gran poder discriminativo entre especies no relacionadas.

UPIBI Mayo 2006
AFH RPLP 12
1.7 DESVENTAJAS DEL PCR-RAPD
Esta técnica genera un perfil de cada aislamiento bacteriano, pero con reducida
reproducibilidad.
Las técnicas basadas en marcadores moleculares esta siendo muy utilizadas para
discriminar entre distintos tipos de organismos, ya que nos proporcionan patrones que son
únicos de una especie o de una población. Las técnicas mas utilizadas para la
discriminación genotípica son la PCR-RAPD y los RFLPs. En algunos casos se utilizan
combinaciones de estas técnicas, las cuales nos proporcionan una mejor discriminacion13,14,
y la información obtenida nos proporciona características genotípicas particulares que
pueden ser útiles en biotecnología. Un organismo muy importante en biotecnología es la
levadura S. cerevisiae, ya que participa en procesos de producción de bebidas alcohólicas
de importancia comercial ( vino, mezcal, etc), en la producción de compuestos
recombinantes (proteínas, vacunas) y en la producción de biomasa (panificación). Como
ejemplo podemos decir que mediante la técnica de PCR-RAPD se han podido discriminar a
cepas S. cerevisiae de la fabricación de cerveza Ale y Lager, la técnica demostró ser rápida
y confiable para distinguir entre cepas cercanamente relacionadas18.
En los últimos años ha tomado importancia el estudio de las cepas industriales de levadura,
con el fin de tener un control sobre el proceso de fermentación y sobre la calidad del
producto16.Derivado de ello, se ha tenido mucho interés en estudiar sus características
genotípicas, y con ello discriminar entre las diferentes cepas para seleccionar a las mejores.

UPIBI Mayo 2006
AFH RPLP 13
2. ANTECEDENTES Se tiene reportado el aislamiento de diferentes especies de microorganismos a partir de
ollas de fermentación del municipio de Zimatlán11, Oaxaca, los cuales ya se identificaron y
caracterizaron cinéticamente11a. De ellos se seleccionaron los correspondientes a las
levaduras del género Saccharomyces, las cuáles se pueden observar en la tabla 1.
Tabla 1. Levaduras aisladas del proceso de elaboración del mezcal.
Clave Grupo microbiano Especie microbiana Temporada de aislamiento
M3-A01-l Levadura Saccharomyces cerevisiae Verano
M3-A20-l Levadura Saccharomyces cerevisiae Verano PJAG1 Levadura Saccharomyces cerevisiae Verano PJAG2 Levadura Saccharomyces cerevisiae Verano PJMN2 Levadura Saccharomyces cerevisiae Verano PJMN32 Levadura Saccharomyces cerevisiae Verano PJMV1 Levadura Saccharomyces cerevisiae Verano PJMV2 Levadura Saccharomyces cerevisiae Verano

UPIBI Mayo 2006
AFH RPLP 14
3. JUSTIFICACIÓN
El mezcal una bebida conocida internacionalmente como el "Coñac mexicano"3 y se busca
que se pueda posicionar y alcanzar un status como el que tiene hoy en día el Tequila.
La industria del mezcal es una de las pocas con las que cuenta el estado de Oaxaca, y este
junto con Guerrero y Chiapas son una de las tres entidades federativas más pobres de
México que se localiza al sureste de la República Mexicana. La pobreza y la marginación
son bastante agudas en estas entidades del territorio mexicano. Ante la escasez de fuentes
de empleo, los oaxaqueños optan por emigrar a otras zonas del país pero, sobre todo, a los
Estados Unidos de Norteamérica.
La entidad se sostiene del comercio y el turismo, pero muy poco de la industria. En este
contexto, la industria del mezcal es la más importante y dinámica por la cantidad de
empleos y divisas que genera a través de las exportaciones a algunos países de América,
Europa y Asia como se puede apreciar en la tabla 2.
Tabla 2. Destino de las exportaciones de mezcal12.
América Europa Asia Argentina Bolivia Canadá Colombia Chile Ecuador El Salvador EUA Guatemala Honduras Panamá Paraguay Perú Uruguay Venezuela
Alemania España Francia Grecia Italia Países Bajos Portugal Reino Unido Suecia Suiza
Hong Kong Japón Taiwán Turquía
La identificación y caracterización de cepas de levaduras fermentadoras es una tecnología
esencial en la enología. Desde el punto de vista biotecnológico, resulta de gran utilidad
conocer las características de los diferentes microorganismos y así poder elegir a los
mejores para la producción7. Esto es particularmente cierto en el caso del mezcal, puesto
que existe una gran diversidad de las especies que están involucradas en su proceso de
elaboración y se estarían aportando datos valiosos sobre este tipo de microorganismos y con
los resultados obtenidos se puede tener un mayor control sobre los microorganismos y por
ende sobre este tipo de procesos (fermentación). Lamentablemente no hay información

UPIBI Mayo 2006
AFH RPLP 15
suficiente hasta ahora sobre esta caracterización de los microorganismos (levaduras) que
participan en este proceso fermentativo de la producción de mezcal. Se sabe también, que
las modificaciones genéticas a estos microorganismos no son una buena opción, al menos
para la fermentación, ya que cuando se pretende realizar una modificación en solo un gen,
el mezcal se ve afectado, ya que no cumple con las características deseadas por el
consumidor (sabor, olor, color, etc.). Por ello se ha preferido seleccionar a las mejores
cepas, previa caracterización7.
En la mayoría de los grupos de seres vivos la caracterización del genotipo no existe o es
incompleta. Las aplicaciones de la técnicas basadas en los marcadores moleculares son muy
diversas y estas se vienen empleando en la diferenciación de individuos13 , análisis
filogenéticos y taxonómicos, mapeo de genomas, cuantificación de variabilidad génica intra
e interespecífica, o propensión a infecciones, localización de resistencia a enfermedades y
dispersión de especies. Es por esto que se decidió utilizar una técnica de este tipo para
realizar el estudio. La técnica a utilizar es la PCR-RAPD, puesto que no requiere una
gran cantidad de ADN, además puede realizarse a partir de una preparación de
ADN cruda, es decir, no se tiene que hacer un tratamiento al DNA antes de ser
utilizado, es rápida y mucho más fácil de realizar, tiene un gran poder
discriminativo entre especies no relacionadas, puesto que solo utiliza iniciadores
de 10 nucleótidos y es reproducible bajo ciertas circunstancias.
Por los motivos ya expuestos se hace necesario realizar una caracterización genotípica de
dichos microorganismos, además que una vez concluido el proyecto se pretende ofrecer al
Consejo Regulador De Mezcal, así como una asesoría sobre como pueden obtener mejores
resultados para que puedan producir el mezcal de manera industrial y de este modo poder
incrementar tanto las fuentes de empleo como la economía del estado.
Se eligieron estas cepas puesto que hay muchos datos reportados en bibliografía acerca de
estas, además que son fáciles de manejar, ya que tienen una velocidad de crecimiento
relativamente alta.

UPIBI Mayo 2006
AFH RPLP 16
4. OBJETIVOS
4.1 GENERAL
Caracterizar genéticamente mediante PCR-RAPD a 6 cepas de levaduras aisladas
del proceso de fermentación del mezcal
4.2 ESPECÍFICOS
Establecer las condiciones, de la técnica PCR-RAPD, óptimas para la
caracterización genotípica de las cepas de S. cerevisiae.
Detectar los marcadores moleculares específicos de las 6 cepas de S. cerevisiae con
ayuda de la PCR-RAPD.
Encontrar los marcadores moleculares que permitan diferenciar cepas de la misma
especie relacionadas industrialmente (en el mismo proceso)

UPIBI Mayo 2006
AFH RPLP 17
5. ESTRATEGIA EXPERIMENTAL La organización de las actividades que se realizaron se muestra a continuación:
Revisión bibliográfica
Elección de cepas
Estandarización de las condiciones de
PCR-RAPD
Tamizaje de oligonucleótidos
PCR-RAPD a Saccharomyces
Análisis de patron de bandas
Elaboración de informe final
Obtención de DNA
Revisión bibliográfica
Figura 11. Diagrama de bloques de las actividades a realizar.
Revisión bibliográfica
Revisión bibliográfica
Revisión bibliográfica
Revisión bibliográfica
Revisión bibliográfica

UPIBI Mayo 2006
AFH RPLP 18
6. MATERIALES Y MÉTODOS Se utilizaron los siguientes materiales y equipos: Termociclador Brinkman,
Espectrofotometro UV-vis Perkin Helmer, Microcentrifuga Micro Hettech, centrifuga
Hermle, dNTPs Invitrogen, Taq Polimerasa Invitrogen, Buffer de reacción Invitrogen,
Cloruro de magnesio Invitrogen, Fotodocumentador Kodack, Primers Carl Roth serie C y
G, ADN S. cerevisiae.
7. METODOLOGÍA
7.1. ELECCIÓN DE CEPAS
Se seleccionaron del banco de cepas de levaduras aisladas de los mostos de mezcal, del
departamento de Biología de la Unidad Profesional Interdisciplinaria de Biotecnología. 6
cepas de S. serevisiae asilados del proceso de elaboración de mezcal.
7.2. OBTENCIÓN DE ADN
Se utilizó la técnica de rompimiento celular, el cual se describe a continuación:
1. Resembrar las cepas en sus respectivos medios sólidos, ver tabla 10 (anexo I).
2. Una vez que se obtuvo la suficiente biomasa se realizaron observaciones al microscopio
para verificar que no hay contaminación.
3. Se preparó 100 mL de medio para el crecimiento de los microorganismos (descrito en el
anexo I).
4. Se incubó a 28 °C en agitación (150 rmp), durante 48 h en promedio (con algunas
variaciones, dependiendo de la cepa), hasta obtener una absorbancia(A, 540 nm) de 0.9
a 1.0.
5. Se retiró de la incubación y se verificó que no hubiera contaminación con una
observación en fresco al microscopio.
6. El resto de la metodología descrita se llevó a cabo en baño de hielo.
7. Se pasó el medio a tubos Falcon de 15 mL.
8. Se centrifugó a 5000 rpm por 15 minutos.
9. Se desechó el sobrenadante y se resuspendió el sedimento con 3 mL de agua destilada.
10. Se pasó a tubos Eppendorf y se añadió 0.3 g de perlas de vidrio, 200 µL de cloroformo-
fenol y 200 µL de solución A (ver composición en anexo II).
11. Se agitó en Vortex cada tubo Eppendorf por 25 minutos en intervalos de 5 minutos.

UPIBI Mayo 2006
AFH RPLP 19
12. Adicionar 200 µL de TE 1X.
13. Se centrifugó a 13000 rpm.
14. Se recuperó el sobrenadante, el cual contenía el ADN, con ayuda de una pipeta Pasteur,
y se transfirió a un tubo Falcon.
15. Se adicionaron dos volúmenes de etanol absoluto al tubo y se mezcló por inversión,
para precipitar el ADN.
16. Se dejo en refrigeración a una temperatura de -28 °C toda la noche para ayudar a la
precipitación del ADN.
17. Se centrifugo a 13000 rpm.
18. Se desecho el sobrenadante.
19. Se resuspendió el sedimento en 400 µL de TE 1X.
20. Se corrió una electroforesis en gel para verificar la presencia de ADN (ver protocolo en
anexo III).
21. El vial se mantuvo en refrigeración.
7.3. DETERMINACIÓN DE CONCENTRACIÓN DE ADN
1. Se realizaron diluciones 1:50 del ADN con TE1X
2. Se leyeron las muestras que solo contiene TE 1X (blanco) al espectro (Perkin Elmer
DU-60.) UV-visible a las longitudes de onda 260 y 280.
3. Se determinaron las concentraciones de ADN de cada muestra como conforme a los
cálculos mostrados en el anexo IV.
7.4. ESTANDARIZACIÓN DE LAS CONDICIONES DE PCR-RAPD
La estandarización de condiciones se realizo en el Laboratorio de Proyectos e
Investigación, departamento de Biología de la Unidad Profesional Interdisciplinaria de
Biotecnología.
1. Se reviso en bibliografía las condiciones y temperaturas necesarias para llevar a cabo
esta reacción.
2. Se realizo la técnica en condiciones de esterilidad con un volumen final de reacción de
30 µL.
3. Se observaron los amplificados con una electroforesis en gel de agarosa al 2 % (ver
método de preparación en anexo III).
4. Se documento el gel y se imprimió.

UPIBI Mayo 2006
AFH RPLP 20
5. Se analizaron los resultados obtenidos y se eligió el primer que nos proporciono un
mayor bandeo.
6. Se variaron las temperaturas, concentraciones y las condiciones hasta conseguir el
mayor bandeo posible.
7.5. PCR-RAPD
1. La técnica se llevó a cabo con los resultados obtenidos durante la estandarizacion de
condiciones.
2. Se hizo un tubo como testigo negativo (sin DNA), con el cual se verifico que no hubiera
contaminación que interfiera con la técnica.
3. Los amplificados se revelaron con una electroforesis en gel de agarosa al 2 % (ver
método de preparación en anexo III).
4. Se documento el gel y se imprimió.

UPIBI Mayo 2006
AFH RPLP 21
8. RESULTADOS Y DISCUSIÓN
8.1. ELECCIÓN DE CEPAS
Se seleccionaron las siguientes cepas.
Tabla 3: Cepas seleccionadas.
Clave Grupo Especie
M3-A20-l Levadura Saccharomyces cerevisiae
PJAG1 Levadura Saccharomyces cerevisiae
PJAG2 Levadura Saccharomyces cerevisiae
PJMN2 Levadura Saccharomyces cerevisiae
PJMV1 Levadura Saccharomyces cerevisiae
PJMV2 Levadura Saccharomyces cerevisiae
Las cepas seleccionadas corresponden a la especie Saccharomyces cerevisiae, siendo todas
estas aisladas del mosto de fermentación del mezcal en las temporadas de primavera y
verano, a partir de 2 palenques diferentes del municipio de Zimatlán de Álvarez en el
estado de Oaxaca.
Como ya se menciono, la intención de este trabajo es encontrar diferencias genéticas en las
levaduras, puesto que son del género y especie pero su comportamiento durante la
fermentación es diferente.

UPIBI Mayo 2006
AFH RPLP 22
8.2. OBTENCIÓN DE ADN
El ADN obtenido a partir de las diferentes cepas de Saccharomyces se muestra a
continuación en la figura 12.
Figura 12. Gel de azarosa al 1% para electroforesis de ADN.
Los carriles corresponden a:
M Marcador Lamba HindIII
4 M3-A20-l
5 PJAG1
6 PJAG2
8 PJMV1
9 PJMV2
Como se puede apreciar en la figura 12 todas las muestras, de interes (carriles 4, 5, 6, 8, 9),
en el gel tienen presencia de ADN. Se observa que la muestra M3-A20-l (carril 4) tiene una
mayor cantidad de ADN con respecto a las otras. Las muestras de los carriles 1, 2, 3 y 7
corresponden a muestras de un ADN que fue utilizado para estandarizar la técnica de
extracción. En la foto podemos apreciar que el tamaño del ADN de levadura es de alto peso
molecular, alrededor de 23,130 pares de bases.
Se observa en el gel la presencia de RNA, el cual deberá eliminarse, para la reacción de
PCR se lleve a cabo de una forma mas eficiente. Para ello se probó la purificación de las
muestras por columnas con gel, pero no se obtuvieron resultados satisfactorios, al parecer el
DNA se quedaba atrapado en el gel junto con las impurezas. Por ello se utilizó la hidrólisis
por RNAsa. (Anexo V).

UPIBI Mayo 2006
AFH RPLP 23
Figura 13. 2do gel de electroforesis de ADN.
La figura 13 nos muestra la presencia de ADN de las siguientes cepas, y se marca el carril
correspondiente:
1 M3-A20-l (2)
2 M3-A20-l (3)
4 PJMN2
7 PJAG2
9 PJAG1
11 PJMV2
12 PJMV1
Como puede observarse en ambos geles, la concentración de ADN obtenida a partir de las
diferentes cepas es variable a pesar de que se obtuvo un crecimiento similar en todas, para
ello es necesaria determinar la concentración exacta mediante espectrofotometría, con el fin
de ajustar la concentración en los ensayos de PCR. Sin embargo puede observarse que se
obtuvo ADN en todas las cepas probadas.
Es importante resaltar que algunas de las cepas tuvieron un tiempo de crecimiento
demasiado lento, por lo que este paso llevo mas tiempo de los programado.

UPIBI Mayo 2006
AFH RPLP 24
8.3. DETERMINACIÓN DE CONCENTRACIÓN DE ADN
Los resultados obtenidos durante este paso se muestran en la tabla 4.
Tabla 4. Concentraciones de ADN obtenido a partir de cepas de S. cerevisiae.
Cepa de
Saccharomyces As260 As280 Concentración(µg/mL)
M3-A20-l 1.9513 0.9382 4.87825
PJAG1 1.5249 0.7463 3.81225
PJAG2 1.9310 0.9274 4.8275
PJMN2 0.7691 0.3723 1.92275
PJMV1 2.5227 1.2027 6.432885
PJMV2 0.0919 0.0435 0.22975
Candida 2.3833 1.0641 5.95825
Como se observa en la tabla anterior hay una buena concentración de ADN obtenida para
poder realizar la PCR de manera adecuada, excepto en la muestra PJMV2 que observa un
valor de 0.22975 µg/mL. Se observa en las muestras PJMN1 y Candida un valor alto en la
columna de As280 que es la correspondiente a la absorbancia de las proteínas lo que indica
una mayor cantidad que en las otras muestras, probablemente por una ineficiente
desproteinización. Sin embargo, esto no afectó la PCR hay una gran cantidad en, lo cual
puede interferir al momento de realizar dicha técnica. La memoria de cálculo de
concentraciones de ADN se puede revisar en el anexo IV.
8.4. ESTANDARIZACIÓN DE LAS CONDICIONES DE PCR-RAPD
Los primers utilizados durante el tamizaje de oligonucleotidos están mostrados en la
tabla 5, cabe mencionar que en los resultados de las amplificaciones solo se muestran los
resultados más representativos, ya que la mayoría de los primers utilizados no mostraban
resultados de amplificación o mostraban solo una banda, Esto se comprobó in silico ya que
se consulto la genoteca de la CNBI15, la mayoría de los primers de la serie R no tenían
sitios de unión al genoma de la levadura, por lo cual no fueron aptos para esta técnica.

UPIBI Mayo 2006
AFH RPLP 25
Tabla 5. Primers utilizados.
No. Primer Secuencia (5’-3’) % G-C
1 C11 AAA GCT GCG G 60
2 G16 AGC GTC CTC C 70
3 G10 CTA CGG AGG A 60
4 C15 GAC GGA TCA G 60
5 G03 GAG CCC TCC A 70
6 R16 GAT CAA GTC C 50
7 R21 GAT CAT AGC C 50
8 R28 GAT CAT AGC G 50
9 R27 GAT CAT GGT C 50
10 R25 GAT CGC ATT G 50
11 R24 GAT CTA ACC G 50
12 R20 GAT CTC AGA C 50
13 R19 GAT CTG ACA C 50
14 R04 GGA ACC AAT C 50
15 R01 GGT ACT AAG A 40
16 C07 GTC CCG ACG A 70
17 R15 GTT TTC GCA G 50
18 C14 TGC GTG CTT C 60
19 R10 TGG TAA AGG G 50
20 C12 TGT CAT CCC C 60
Las condiciones a las que se obtuvieron mejores resultados fueron las siguientes, el
volumen final de reacción es de 30 µL:

UPIBI Mayo 2006
AFH RPLP 26
Tabla 6. Datos sobre reactivos utilizados.
Reactivo Concentración
dNTPs 300 µM
Taq Polimerasa 1.5 U
Buffer de reacción 3 µL
Cloruro de magnesio 15 mM
Primers 1µM
ADN 50 ηg
ddH2O estéril cpb (30 µL)
Tabla 7. Datos sobre las temperaturas y ciclos del termociclador.
Paso Temperatura (˚ C) Tiempo (min) No. de ciclos
Desnaturalización 93 1 1
Desnaturalización 92 1 55
Alineamiento 35 1 55
Extensión 71 1 55
Extensión 71 5 1
Tapa 110 Todo el tiempo Todo el tiempo
Guardar 4 - Desde que termine el PCR-RAPD
Los resultados obtenidos de este paso son satisfactorios, podemos observar que
necesitamos una temperatura de alineamiento baja, lo cual nos permite obtener gran
polimorfismo, necesario para poder encontrar diferencias entre las cepas. Aparte notamos
que los tiempos de desnaturalización, alineamiento y extensión son cortos y se necesitan
muchos ciclos (55) para que se aprecie una buena amplificación.

UPIBI Mayo 2006
AFH RPLP 27
Figura 14. Gel de amplificación de ADN de cepa PJMN2 1.
Los carriles corresponden a la amplificación de ADN con 8 primers diferentes, y se
presentan a continuación.
1 Marcador Lamba HindIII
2 Testigo negativo
3 R28
4 R25
5 R01
6 R32
7 R10
8 R19
9 R21
10 R15
Como se puede observar los primers utilizados no son los adecuados, debido a que en la
mayoria de los carriles (3, 4, 6, 8 y 9) no hay amplificación, en los carriles 7 y 10 solo
presentan una banda, solo en el carril 5 se aprecia que hay 2 bandas, por lo cual se
probaron otros primers, y los que presentan mayor polimorfismo se muestran en la
figura 15.

UPIBI Mayo 2006
AFH RPLP 28
Figura 15. Amplificación de ADN de cepa PJMN2 2.
Los carriles corresponden a la amplificación de ADN con 8 primers diferentes, y se
presentan a continuación.

UPIBI Mayo 2006
AFH RPLP 29
1 Marcador φx174/HaeIII
2 G10
3 C12
4 C07
5 G15
6 C15
7 C11
8 G16
9 C13
10 Testigo negativo
Como se observa en la figura 15 el primer correspondiente al carril número 8, primer G16,
es el que presenta mayor numero de bandas, por lo cual, es el oligonucleotido que se
selecciona para realizar la técnica de PCR-RAPD. Se observa que los amplificados son
todos de bajo peso molecular (entre 1,353 y 310 pares d bases). En el carril 9,
correspondiente al primer C13, se observa solo una banda de alto peso molecular,
aproximadamente de 1,353 bases, también se puede apreciar que las bandas están por
arriba de las 300 bases.
8.5. PCR-RAPD Y ANÁLISIS DE BANDAS
Los resultados obtenidos de la PCR-RAPD se muestran en la figura 16.
Figura 16. Resultados de PCR-RAP con primer G16 a cepas de S. cerevisiae.

UPIBI Mayo 2006
AFH RPLP 30
Las muestras se encuentran dispuestas de la siguiente manera:
No. de
bandas
Tamaño aproximado
(pb)
1 Marcador φx174/HaeIII - -----
2 Testigo negativo 0 -----
3 PJMV1 1 491
4 PJMV2 4 975, 834, 731 y 491
5 PJAG2 2 975 y 491
6 PJAG1 4 1822, 1250, 1147 y 491
7 M3-A20 1 731
8 PJMN2 4 1027, 834, 650 y 491
9 M3-A10 2 731 y 667
Se observan los amplificados obtenidos a partir del ADN de las diferentes cepas de S.
cerevisiae en los carriles 3 al 8 y en el carril número 9 el amplificado a partir de una cepa
de Candida, esto con el fin de observar las diferencias de bandeo en S. cerevisiae. Los
fragmentos son de entre 1822 y 491 pares de bases aproximadamente. Se puede observar en
los carriles 3, 4, 5, 6 y 8 una banda común de 491 bases, la cuál podría considerarse una
banda característica de especie. Se aprecia que la muestra M3-A20, correspondiente al
carril 7, comparte una banda con la muestra PJMV2, correspondiente al carril número 4,
esta banda es aproximadamente de 731 bases. En la muestra control (carril 9) se observan 2
bandas de 731 y 667 bases.
Es interesante resaltar que a pesar de que todas las muestras corresponden a la misma
especie y que la amplificación se desarrolló con el mismo oligonucleótido y bajo las
mismas condiciones, el resultado en el número de bandas y el tamaño de las mismas es
diferente en cada caso. Esto es significativo en el sentido de que indica que si bien algunas
comparten secuencias comunes (ver carriles 3, 4, 5, 6 y 8), también muestran diferencias
genotípicas. Estas diferencias podrían traducirse también en diferencias fenotípicas, lo que
explicaría su comportamiento diferente en las cinéticas de crecimiento desarrolladas
anteriormente11a, así como las diferencias en cuanto a la producción de etanol8a. También es
indicativo de que se trata efectivamente de diferentes cepas (diferentes aislados) y no de un
artificio en el aislamiento de las cepas. Las cepas que presentan mayor polimorfismo fueron
las cepas PJMV2, PJAG1 y PJMN2, mostrando 4 bandas cada una. Este número de bandas

UPIBI Mayo 2006
AFH RPLP 31
que se obtuvieron son relativamente pocas, lo que indica que estos primers tienen pocos
sitios de unión al ADN de S. cerevisiae, y tal como se anotó anteriormente los primers al
azar no tienen muchos sitios de unión en el genoma de S. cerevisiae.
Estos resultados son similares a los reportados anteriormente17, ya que la mayoría de los
individuos comparten al menos una banda y solo unos pcoos individuos tienen un patrón de
bandeo único.
En la parte inferior del gel se puede se observan una mancha que corresponde a las
proteínas, que no se pudieron eliminar en el aislamiento del ADN.

UPIBI Mayo 2006
AFH RPLP 32
9. CONCLUSIONES
La técnica de rompimiento mecánico fue adecuada para la obtención de ADN a partir
de levaduras.
La estandarización de condiciones de la PCR-RAPD para DNA de S. cerevisiae nos
proporciona un patrón de bandeo característico una caracterización de nuestras cepas.
Se obtuvo una banda que posiblemente pertenezca a un marcador de población, la
cual nos indica que las cepas de S. cerevisiae son de la misma especie pero que
poseen diferencias genéticas.
El primer G16 nos permite diferenciar entre cada cepa de S. cerevisiae.
Se obtuvieron marcadores moleculares que nos permiten diferenciar entre cepas de
S. cerevisiae relacionadas industrialmente.
se encontró variabilidad genética entre las cepas de S. cerevisiae aisladas del proceso
de fermentación de mezcal.

UPIBI Mayo 2006
AFH RPLP 33
10. RECOMENDACIONES PARA EL TRABAJO FUTURO
Se recomienda realizar el análisis de patrón de bandeo, así como realizar una prueba de
PCR-RAPD con los primers C12 y C07, ya que estos muestran un bandeo mas definido y
que suponemos que nos puede ayudar a discriminar de una mejor forma, y posteriormente
realizar una comparación de los resultados que se obtengan con los resultados obtenidos en
este trabajo para poder tener bases mas sólidas y fundamentadas sobre las diferencias
genotípicas de estas levaduras.
PPaarraa TTooddoo MMaall MMeezzccaall YY PPaarraa TTooddoo BBiieenn TTaammbbiiéénn
UPIBI Mayo 2006
AFH RPLP 34
- ANEXO I
Especificación de medios para los diferentes microorganismos
A continuación se listan los medios sólidos para el mantenimiento de cada una de las cepas
y preparación de tubos inóculo.
Tabla 8. Cepas de microorganismos seleccionadas.
Clave Grupo microbiano Especie microbiana Medio sólido
M3-A01-l Levadura Saccharomyces cerevisiae ASD
M3-A09-l Levadura Saccharomyces cerevisiae ASD
M3-A20-l Levadura Saccharomyces cerevisiae ASD
PJAG1 Levadura Saccharomyces cerevisiae PDA
PJAG2 Levadura Saccharomyces cerevisiae PDA
PJMN2 Levadura Saccharomyces cerevisiae PDA
PJMN32 Levadura Saccharomyces cerevisiae PDA
PJMV1 Levadura Saccharomyces cerevisiae ASD
PJMV2 Levadura Saccharomyces cerevisiae ASD
ASD= Agar Sabouraud Dextrosa
PDA= Agar papa dextrosa
El medio empleado para la obtención de biomasa para la purificación de ADN se muestra a
continuación:
Tabla 9. Composición del medio líquido para la obtención de biomasa.
Compuesto Cantidad (g/L)
Fructosa 70
Extracto de levadura 4
K2HPO4 0.5
KH2PO4 0.5
(NH)4SO4 10
MgSO4 0.01
pH final 4.5 - 5

UPIBI Mayo 2006
AFH RPLP 35
- ANEXO II
Composición de la solución A
La composición de la solución A, empleada en la purificación de ADN, es como sigue.
Tabla 10. Composición de la solución A.
Componente Cantidad
Triton X-100 al 10% 200 µL
SDS al 10% 1mL
NaCl 5M 200 µL
Tris pH 8 1M 100µL
EDTA 0.5M 20 µL
Aforar con agua destilada a 10mL

UPIBI Mayo 2006
AFH RPLP 36
- ANEXO III
Electroforesis
En esta parte se explican los pasos a seguir para realizar la técnica de electroforesis en gel
de agarosa (0.5% y 2%).
1. Colocar dentro del carro para electroforesis el peine, mas adecuado, según el
número de pozos que se requieran.
2. Pesar 0.5 o 2 gramos de agarosa y adicionar 100 mL de regulador TE 1X. Calentar a
ebullición 1 minuto para disolver. (Con cuidado para evitar el derrame de la agarosa
sobrecalentada)
3. Adicionar 8 µL de bromuro de etidio de 10 mg/mL y homogeneizar sin formar
burbujas o espuma, (como el bromuro de etidio es un potente mutageno y
cancerigeno se debe de manejar con guantes y no respirar los vapores de la agarosa
adicionada de este compuesto).
4. Vaciar la agarosa en el carro de electroforesis a modo de cubrir por lo menos 5 mm
de los dientes del peine colocado.
5. Dejar que gelifique.
6. Sacar con sumo cuidado el peine, evitando romper los pozos formados.
7. Colocar el carro dentro de la cámara de electroforesis y llenarla con regulador
TE 1X, a modo de cubrir el gel. Cuidar que no queden burbujas en los pozos.
8. Sobre una superficie lisa (un trozo de parafilm) colocar muestras de 3 µL del
colorante para corrida, tantas, como muestras de DNA se tengan.
9. Sobre esa superficie mezclar 15 µL de la solución de DNA con los 3 µL del
colorante, homogeneizando perfectamente.
10. Tomar esta mezcla con la micropipeta y vaciarla cuidadosamente en cada uno de los
pozos del gel, sin derramarlas. Anotar el orden en que se fueron colocando las
muestras en el gel.
11. Mezclar así mismo 2 µL del marcador de peso molecular de 250 µg/µL con 2 µL de
colorante y vaciarlos en uno de los pozos.

UPIBI Mayo 2006
AFH RPLP 37
12. Cerrar la cámara de electroforesis y conectar los cables. Desarrollar la electroforesis
a 80-90 mV (para geles de 8 x 10 cm aprox.), durante 60 minutos, o hasta que el
primer colorante llegue a las tres cuartas partes del gel.
13. Transcurrido el tiempo, sacar el gel con guantes y observar las bandas de DNA
cromosómico con una lámpara de luz U.V.
Figura 17. Cámara de electroforesis. Figura 18. Peine de 10 dientes.
Figura 19. Carro de cámara de electroforesis.

UPIBI Mayo 2006
AFH RPLP 38
- ANEXO IV
Memoria de cálculo para determinar la concentración de ADN
Para determinar la molaridad de ADN se realiza la siguiente secuencia de cálculo.
Como se conoce la absorbancia, la cual es de As260 = 0.2847, del ADN se puede
determinar la concentración de esta de la siguiente manera:
con esta concentración se puede calcular la Molaridad con la siguiente ecuación
mLgADN µ]][[ =
1000
50][
260 mLgFDAs
ADN
µ⋅⋅
= ……………..ec 2.
Donde:
[ADN] = concentración de ADN.
As260 = Absorbancia de la muestra de ADN a 260 ηm.
FD = Factor de dilución.
mLgmL
g
ADN µµ
87825.41000
50509513.1][ =
⋅⋅=

UPIBI Mayo 2006
AFH RPLP 39
- ANEXO V
Hidrólisis de RNA
1. A una muestra de ADN con RNA de 400 µL adicionar 0.4 µL de RNAsa.
2. Incubar a 37 °C durante 1 h.
3. Sacar de la incubación y adicionar 100 µL de acetato de sodio 2.5 M.
4. Adicionar dos volúmenes de etanol absoluto.
5. Dejar precipitar toda la noche a -28°C.
6. Centrifugar a 5000 rpm por 15 minutos.
7. Desechar el sobrenadante.
8. Resuspender la pastilla en 150 µL de TE1X.
9. Correr una electroforesis en gel de azarosa al 1% para verificar que se haya eliminado el
RNA.

UPIBI Mayo 2006
AFH RPLP 40
11. BIBLIOGRAFÍA
1 Marín, G. 2005. Orígenes del mezcal. www.aquioaxaca.com/mezcal.htm.
2 Ramales, M. 2005. La industria del mezcal y la economía oaxaqueña.
www.eumed.net/cursecon/ecolat/mx/ramales-mezcal-a.htm.
3 Estado de Oaxaca. 2005. Mezcal de Oaxaca. http://oaxaca.gob.mx/mezcal/.
4 Perez, R. 2006. Mezcal, bebida que tiene un puente con los dioses.
www.macroeconomia.com.mx/articulos.php?id_sec=30&id_art=735&id_ejemplar=51.
5 Collado, Q. 2005. Levaduras y la fermentación alcohólica (II).
http://www.verema.com/opinamos/tribuna/articulos/levaduras02.htm.
6 Collado, Q. 2005. Levaduras y la fermentación alcohólica (I).
Verema.com/opinamos//tribuna/articulos/levadura01.asp.
7 Claude Flanz y Coordinador. Enología: Fundamentos científicos y tecnológicos. 2da Ed.
2003.AMV ediciones - Ediciones Mundi Prensa. Madrid, España
8 Quezada, F. 2005. Biotecnología para el uso sostenible de la biodiversidad.
Capacidades locales y mercados potenciales. Unidad de publicaciones CAF.
9 M.E. Gilles-González 1997.
10 2005. www.monografias.com/trabajos19/musaceas/ musaceas.shtml.
10ª Karp, G. 2001. Biología celular y molecular. 1ra edición. McGraw-Hill
Interamericana. México. 416.
11 Duran, D; Ruiz, A. 2003. Aislamiento, identificación y caracterización de especies
microbianas del mezcal. UPIBI. 2003. 15.
11a Bautista, M; Cerrillo, M. 2005. Caracterización de cepas de levaduras productoras de
mezcal. Upibi. 2005. 18-62.
12 Banco Nacional de Comercio Exterior (BANCOMEXT), 2002.
13 Mitterdofer, G. Mayer, H. Viernstein, H. 2002. Clustering of Saccharomyces boulardii
strains within the species S. cerevisiae using molecular typing techniques. Journal of
Applied Microbiology. Volumen 93: 521-530.
14 Williams John G. K., et al, Genetic Analysis using random polymorphic DNA markers,
Methods in enzymology, Vol. 218, 1993
15 2006. www.ncbi.nih.gov/.
16 Gschaedler, Téllez, Fallad, Caracterización molecular y comparación de cepas de
levaduras aisladas de los procesos de elaboración de mezcal y sotol. CIATEJ. (2001).

UPIBI Mayo 2006
AFH RPLP 41
Memorias electrónicas del IX Congreso nacional de Biotecnología y Bioingeniería.
Veracruz, México.
17 Guerra, J. Pataro, C. Moreira, E. 2001. Genetic diversity of Saccharomyces cerevisiae
strains during 24 h fermentative cycle for the production of the artisanal Brazilian cachaca.
Letters in Applied Microbiology. Volumen 33: 106-111.
18 Tonari-Lehoczki, J. Dlauchy, D. 2000. Delimitation of brewing yeast strains using
different molecular techniques. International Journal of Food Microbiology.
Volumen 62: 37-45.





























