Embed Size (px)
Citation preview

92
CHAPTER 5 STRUCTURE ELUCIDATION OF BACTERIAL EFFLUX
PUMPS BY IN SILICO APPROACH
5.1 Introduction
Efflux pumps are transport proteins involved in the extrusion of toxic substrates
(including virtually all classes of clinically relevant antibiotics) from within cells into
the external environment. These proteins are found in both Gram‐positive and ‐
negative bacteria as well as in eukaryotic organisms (Bambeke et al., 2000). Bacterial
multidrug efflux pumps are the major contributors of microbial resistance to several
classes of antibiotics (Webber and Piddock, 2003; Hooper, 2005) and hence are
associated with multiple drug resistance (MDR). MDR efflux is an increasingly
reported phenomenon and has been described for many organisms including
bacteria, fungi and protozoa and as a mechanism of resistance in mammalian
tumour cells. The drug efflux pumps are present in a number of clinically relevant
species like Mycobacterium tuberculosis, Staphylococcus aureus, Pseudomonas
aeruginosa, Bacillus subtilis and E. coli. Many of these efflux pumps export an
extensive range of structurally unrelated antibiotics from the cell, resulting a
reduced intracellular concentration and thus reduced susceptibility (Webber and
Piddock, 2003). The specific TetK and MRSA efflux pumps that export certain
tetracyclins, macrolides confer MDR to many strains of Staphylococcus aureus leading
to difficult management of infection, thereby increasing its chances of its becoming
fatal (Ubukata et al., 1989; Ross et al.,1990; Littlejohn et al., 1992; Guay et al.,
1993). MDR efflux pumps can be categorized into a number of families such as MF
(major facilitator) (Pao et al., 1998; Saier et al., 1999; Ward et al., 2001), MATE
(Multi drug and toxic efflux) (Paulsen et al., 1996; Chung and Saier, 2001), RND
(resistance‐nodulation‐cell division), SMR (small multi drug resistance) (Saier et al.,
1994; Paulsen et al., 1996; Tseng et al., 2003) and ABC (ATP Binding Cassette). Amid
such important characteristics, very few 3D crystal structures and mechanism of
these efflux pumps are known due to the difficulty in membrane protein
crystallization for X‐ray diffraction. In this study, we have tried to elucidate the 3D

93
structure by in silico means of three such important (as per the mandate of our
Institute) efflux pumps viz., NorA, Bmr and Rv1258c belonging to Staphylococcus
aureus, Bacillus Subtilis and Mycobacterium tuberculosis respectively.
A member of MF transporters family of efflux pumps, NorA has been
described in Staphylococcus aureus but is poorly characterized. It is a
phosphoglycoprotein (Pgp), predicted to have 388 amino acids with a molecular
mass of 42.5 kDa and is presumed to be localized in the cytoplasmic membrane.
NorA promotes the active efflux of lipophillic, monocationic compounds (ethidinium
bromide, cetrimide, benzalkonium chloride, tetraphenylphosphonium bromide [TPP}
and acriflavine) as well as hydrophilic quinolones (Yoshida et al., 1990; Ng et al.,
1994; Yu et al., 2002). The Bacillus subtilis membrane protein Bmr belongs to the
super family of major facilitator proteins and is one of the first‐discovered bacterial
multidrug‐efflux transporters (Neyfakh et al., 1991). These transporters actively
export structurally diverse organic molecules out of cells, thus making bacteria
simultaneously resistant to many toxic compounds (Lewis, 1994; Nikado, 1994;
Nikado, 1996; Paulsen et al., 1996). Bmr, causes resistance to such structurally
diverse toxins as ethidium, rhodamine, tetraphenylphosphonium (TPP), acridine
dyes, fluoroquinolone antibiotics, doxorubicin, chloramphenicol, and puromycin
(Neyfakh et al., 1991; Ahmed et al., 1995). The Bmr efflux pump is of 389 amino
acids with molecular weight of about 42.258 kDa. It is a close homolog of NorA
multidrug transporter of Staphylococcus aureus (Neyfakh, 1992). Rv1258c from
Mycobacterium tuberculosis is one of the efflux proteins, which is proteinaceous
active transporter localized in the cytoplasmic membrane of the cells. Its gene
encodes a tetracycline/aminoglycoside resistance (TAP‐2)‐like efflux pump (Ainsa et
al., 1998; Siddiqi et al., 2004). Studies have shown that the deletion of this gene
from the Mycobacterium bovis BCG chromosome increases the susceptibility of the
organism to these two drugs (De Rossi et al., 2006). This gene plays a role in
multidrug resistant (MDR) TB; the association between drug resistance and
transcription levels of this tap‐like pump prevents cytosolic accumulation of drugs.
Over expression of Rv1258c, under rifampicin pressure, has been reported

94
previously and thus explains the involvement of this clinically important efflux
protein in efflux‐mediated rifampicin resistance. It has Gene length of 1260bp and
percentage GC content 67.14%. Rv1258c M. tuberculosis has 419 amino acids and a
molecular weight of ~43287.20 Da (Siddiqi et al., 2004; Jiang et al., 2008).
5.2 Methodology
5.2.1 3D structure prediction of efflux pump NorA from Staphylococcus aureus
The prediction of transmembrane segments of NorA sequence has been carried out
using TMPred (Hofmann and Stoffel, 1993). PROSITE analysis shows that NorA
belongs to Major Facilitator Superfamily (MFS). The remote homolog of NorA has
been found by using a metaserver available at http://bioinfo.pl/meta/ (Ginalski et
al., 2003). Further, since NorA also belongs to major facilitator superfamily, the
choice of glycerol‐3‐phosphate transporter as template is appropriate being member
of same superfamily. The Structure of NorA has been modeled based on the
alignment between the NorA sequence and that of glycerol‐3‐phosphate transporter
(PDBID:1PW4) of E. coli using MODELLER 8v0 (Sali and Blundell, 1993). The modeled
structure was checked for correct stereochemistry using the program PROCHECK
(Laskowski et al., 1993). Using PROCHECK, the number of amino acids in favourable
region and outliers in Ramachandran Plot have been verified and planarity of the
planner group containing amino acids has been examined. The predicted structure
was energy minimized using the program AMBER 8 (Case et al., 2004) with ff99 force
field. Minimization was done in implicit solvent using Generalized Born model
(Hawkins et al., 1996) The detailed methodology and usage of the servers is
discussed in the material and methods chapter.
5.2.2. Identification of probable binding pockets of NorA
A grid based approach along with an “eraser” algorithm is used in order to find
probable binding pockets in the modeled structure of NorA in Cerius2 software.A
flood‐filling algorithm was employed to search unoccupied, connected grid points
with grid resolutions of 0.50 Å, radius of hydrogen atoms in protein as 2.0 Å, radius

95
of heavy atoms in protein as 2.5 Å, site opening size cut off at 5.0 Å and site cutoff as
100 site points.
Software MOE was also used for predicting the probable binding pockets of
NorA. Unlike the eraser algorithm of cerius2, the algorithm in MOE considers the
relative positions and accessibility of the receptor atoms. The parameters viz. Probe
radius1=1.4 Å, Probe radius2=1.8 Å, Isolated Donor/Acceptor = 3, Connection
distance=2.5 Å, Minimum site size = 3 and radius = 2 Å were used.
5.2.3 Docking Studies of capsaicin and known inhibitors of NorA on its predicted 3D
structure
The docking of some of the known inhibitors of NorA viz., verapamil, reserpine,
carsionic acid, isopimarine derivative, pheophorbide A and methoxyhydrocaspin
(Yoshida et al., 1990; Neyfakh, 1992; Ng et al., 1994; Saier et al., 1994) with the
predicted structure of NorA was carried out using the Ligandfit module of Cerius2
(version 4.11) and MOE. We have previously described the role of piperine, a major
constituent of Piper nigrum, as a putative bacterial (Staphylococcus aureus ) efflux
pump inhibitor (Khan et al., 2006). Further, in our pursuit to identify natural
molecules as EPIs we identified capsaicin as a novel EPI of the NorA efflux pump of S.
aureus. Docking studies of capsaicin was also carried out on the predicted structure
of NorA in order to get some insight about its mechanism of action. The docking
methodology is described in detail in the material and methods chapter.
5.2.4 Structure prediction of efflux pump Bmr from Bacillus subtilis and its model
validation
In order the select the perfect template for modelling of the efflux pump Bmr, the
sequence was submitted to various available threading servers viz., LOMETS, PHYRE,
MUSTER, SAM_T08, mGenThreader, ESyPred3d, FFAS03, PFAM‐FFAS, SAM‐T02, SP4,
FUGUE, LOOP [BIOHPC], WURST and HHpred. The structure of the Bmr protein was
modelled based on the results from these servers. The structure validation of the
modelled structure was checked using Structure analysis and validation server which

96
utilizes ProCheck, ERRAT, Verify_3D, PROVE and What_Check programmes for
verification of the 3D structure of protein. Using PROCHECK, the number of amino
acids in favourable region and outliers in Ramachandran Plot have been verified and
planarity of the planner group containing amino acids has been examined. The
details about the methodology is given in material and methods chapter.
5.2.5 Binding site identification and docking of reserpine on the predicted structure
of Bmr
The predicted structure was then analyzed using Schrödinger software and the
binding site was identified based on the literature data available on substrate
recognition residues of Bmr (Ahmed et al., 1993; Paulsen et al., 1996; Klyachko et
al., 1997; Friesner et al., 2006) and docking analysis was performed in order to study
the mechanism of reaction. The docking of the known inhibitor of Bmr viz., reserpine
(Ahmed et al., 1993; Klyachko et al., 1997) with the predicted structure of Bmr was
carried out using the Ligandfit module of Cerius2 and Glide module of Schrödinger
(Friesner et al., 2006). The detailed methodology has been described in the material
and methods chapter.
5.2.6 Structure prediction of Mycobacterium tuberculosis efflux pump Rv1258c by
comparative modeling
In order to get a reliable model, several online servers were used for modeling the
structure of Rv1258c. All the servers used for the present study is listed in the
material and methods chapter. The template for modeling was selected based on
the structure and function of the template protein. The developed protein models
were first prepared and minimized using Protein Preparation Wizard of the
Schrodinger Modeling Software Package. During minimization rmsd value was set to
0.3 A0. All the protein models were then checked for Ramachandran Core value,
disallowed residues and bad contacts using Procheck module of the SAVS server.
SAVS is Structural Analysis and Verification Server available from National Institute of
Health (NIH) for sterochemical analysis of protein models. A threshhold criteria was
set up for Ramachandran Core, bad contacts and disallowed regions in order to filter

97
out some poorly constructed models. Hence, all the models without any bad
contacts and for which RC core was more than 85% and disallowed region less than
0.3% were retained for further investigation. All other models were discarded.
5.2.7 Prediction of binding sites of Rv1258c
The selected protein models were prepared for docking using the Protein
preparation module of Schrodinger software. The binding pockets of the predicted
models were identified using the Sitemap module of Schrodinger. The sites present
on the opening face of the middle cavity surrounded by the transmembrane helices
were selected for docking studies. Receptor grid was generated for the protein
structures on all the identified binding sites. A grid is a 3D box that defines the region
of the receptor to be docked. It also determines the position and size of the binding
site. Further FTMap server was used to identify the small clefts formed within the
predicted binding site of Rv1258c.
5.2.8 Molecular docking studies of Rv1258c
A set of 36 known active (14 inhibitors) and inactive (22 non‐inhibitors) molecules of
Rv1258c protein were taken for the docking studies. These molecules were divided
into training set and test set. All the ligands were prepared using Ligprep module and
the conformations of all the ligands were prepared using the Confgen module of
Schrodinger software. All the conformations of the prepared ligands were docked on
to the binding pockets of the predicted protein models. SP (standard precision)
ligand docking was performed for each grid file using Glide module of the
Schrodinger software. The grid files were retained and discarded based on a
threshold value of the gilde score (which determines the binding affinity of each
ligand with the receptor grid). The final proposed models were shortlisted based on
the experimental information about the inhibitory activity of the ligands on to the
target protein and the resultant binding affinity through dock scores. The respective
binding location within the central cavity was also taken into consideration for
selecting the models. The test set molecules were then docked onto the selected
models for evaluation of their activity through docking.

98
5.3 Results and Discussion
5.3.1 Structure elucidation and docking results of NorA
The server (http//bioinfo.pl) predicted the glycerol‐3‐phosphate transporter from
E.coli [PDB ID: 1PW4] as template for the NorA sequence with the best score.
Modeled structure of NorA has been found to have 12 main helical segments
interspersed by loops of variable lengths at regular intervals (Figure 1). Analysis of
clash score and rotamer outliers using MolProbity (Davis et al., 2004) have been
found to be 5.58 and 4.38%, respectively after the energy minimization of the
modeled structure of NorA. Modeled NorA shows rectangular top and bottom with
all helices lying parallel to the long axis of the structure of NorA. The presence of
helical segments in modeled NorA structure has also been confirmed by the
prediction of secondary structure of the protein using PSIPRED (Bryson et al., 2005;
Buchan et al., 2010). The helical segments modeled using glycerol‐3‐phoaphate
transporter are found to be overlapping comparably with the transmembranous
segment as predicted by TMPred suggesting that like template, NorA is going to be a
transmembranous protein (Figure 5.1). Both N‐ and C‐termini of the modeled NorA
are going to be on the same side of membrane. The transmembranous helices‐H6
and H9 of the modeled structure are shorter but sufficient enough to transverse the
membrane. Some of the transmembrane helices are going to extend beyond the
membrane into the periplasmic and cytoplasmic sides. The arrangement of helices is
also typically similar to that of template. Most of the loops are more than five amino
acids long except L4 and L11. The central loop (L6) between H6 and H7 is the longest
one having a small helical segment (191‐198) towards the C‐terminus end of the
loop. This small helical segment has two helical turns and lies parallel to the
membrane axis.

99
Figure 5.1 : NorA amino acid sequence.Sequence of NorA showing helices (H1 to H12) regularly interspersed by lops (designated as L1 to L11) in the modeled structure and transmembrane helices predicted by TMPred are depicted as caret above the residues.

100
An opening has been observed only on the one side of the modeled structure (Figure
5.2). This opening in the modeled structure is enclosed mainly by transmembranous
segments‐ H2, H5, H10, H11 and partially by H1, H4 and H7. The latter helices (H1,
H4 and H7) form the central part of the opening and residues from these helical
segments are mainly involved in the closure the opening. The opening is funnel
shaped with cylindrical inner part. There are several amino acids protruding into the
pore with interior surface of the funnel mostly hydrophobic except at the outer
extremities. The side chains of residues Q51, S55, M109, M132 and R310 project in
the cavity of formed by the opening in the modeled structure and are available for
interaction. The aromatic amino acid residues, F140 and F341 also project their side
chains into the opening of the modeled structure. However, these lie in the closed
part of the NorA beyond the opening, thus are not available for interactions from
one side of NorA.
A total of about 17 binding pockets were identified using the software
Cerius2. Using software MOE, 22 binding pockets were identified. All these binding
pockets were compared and nine binding pockets were identified which were
common in both the above mentioned softwares that were used. The details about
these nine binding pockets are summarized in Table 5.1. All the sites have been
classified based on the number of hydrophobic contacts of the site as measured by
MOE software. Among the common binding sites, most of the binding pockets were
hydrophobic in nature with the site no. 1 being the biggest cavity and having about
354 hydrophobic contacts in comparison to 91 hydrophobic contacts of site no. 2.
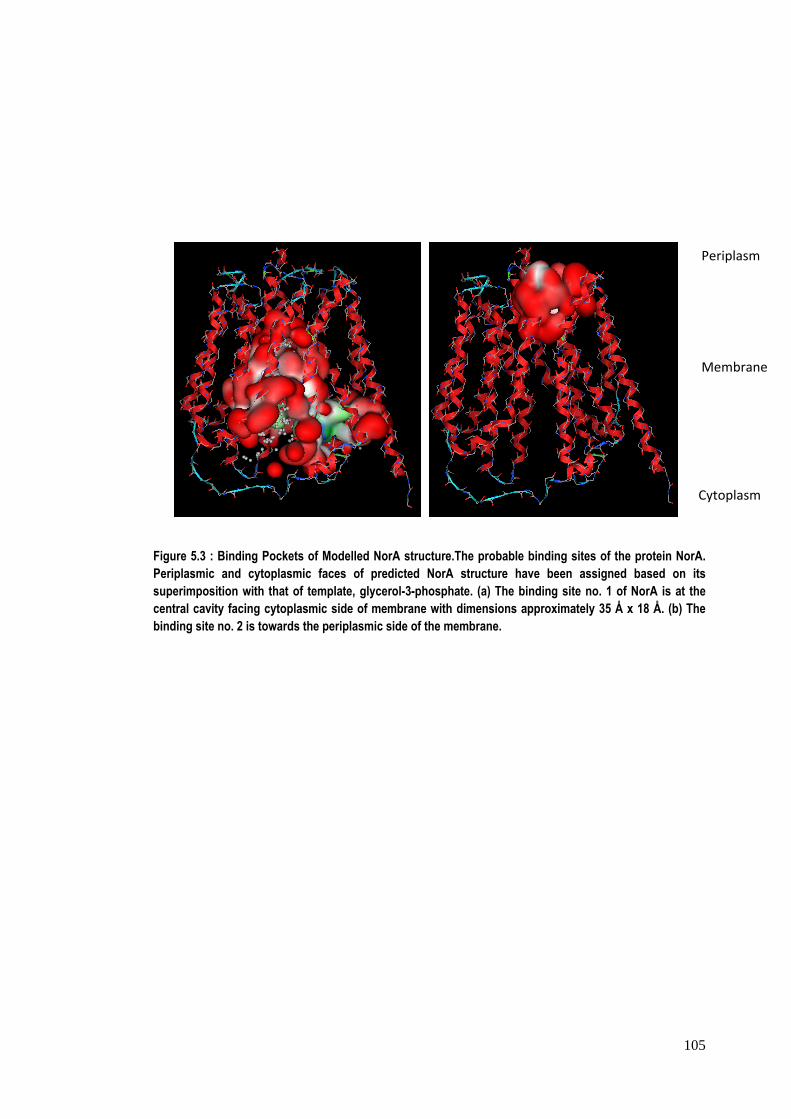
Also, all these sites, except site no. 2, are towards the cytoplasmic face of the
protein; while site no. 2 is towards the periplasmic side. The dimension of the
biggest cavity at site no. 1 is found to be approximately 35 Å x 18 Å.

Figure 5.2 : bundles. Thshows an op
Modelled Strue left panel shpening on one
ucture of NorAhows a view oe side of NorA.
A. Modeled strf modeled Nor
ructure of NorArA perpendicu
A showing tweular to membra
elve transmemane plane. The
101
mbrane helix e right panel

102
Table 5.1 Residues involved in the nine identified binding pockets of NorA.
Site No Size* Binding site residues
1
354
ile12 ile15 phe16 ile19 gly20 ile23 val44 ala45 phe47 ala48 gln51 met52 ile54
ser55 pro56 gly58 gly59 arg98 ala105 gly106 val108 met109 pro110 thr113
lys125 asn128 phe129 gly130 tyr131 met132 ser133 ile135 ile136 asn137
phe140 ile141 phe188 leu191 gln194 ser215 leu218 glu222 thr223 gly248
ile256 tyr257 phe259 asp260 lys261 met263 lys264 leu269 thr270 phe271
trp274 phe303 phe306 asp307 met308 arg310 pro311 ala312 ile313 thr314
asn315 tyr316 ser318 asn319 gln325 gly326 phe327 gly329 gly330 leu331
asn332 ser333 thr334 thr336 ser337 met338 asn340 phe341 ile342 pro344
leu374 lys377 gly378
2
91
pro24 val25 pro27 val28 tyr29 leu30 lys31 asp32 pro144 gly145 ile146 gly147
gly148 ala151 glu152 his155 arg156 pro158 phe159 ala162 ser226 leu227
thr229 ala230 asp231 asn234 tyr235 ser236 pro237 lys238 ile240 ser241
ile242 ile244
3 30 met1 met5 phe6 val7 leu8 tyr9 phe10 phe13 leu14 leu69 ile76 gly166 phe170
ile174 ile177
4 30 lys3 val7 Leu8 Leu69 ile70 ile71 lys72 leu115 ile116 ala117 asp118 ser120
pro121 leu176 asp179
5 36 gln255 ala273 leu276 leu277 val280 val301 val302 ile304 gly305 phe306
6 15 phe188 gln189 lys190 leu191 pro192 gln194 arg324 phe327
7 12 gln194 lys198 ile199 leu331 phe335
8 55 leu17 gly20 leu21 pro24 val25 glu82 gly143 pro144 ile146 gly147 pro158
phe159 tyr160 phe161 ala162
9 27 leu212 val213 phe216 gly339 asn340 ile342 ile346 ala362 ser366
* Size indicates the number of hydrophobic contacts of the site as measured by MOE [48].

103
Docking of some of the known inhibitors of NorA viz., verapamil, reserpine,
carnosic acid, isopimarine derivative, pheophorbide A and 5’methoxyhydnocarpin
was performed on the nine common binding pockets identified using the software
cerius2 and MOE. It was found that inhibitors showed the maximum docking score at
site nos 1 and 2. At all the other binding pockets, there were significantly less scores
due to poor binding (Table 5.2). Both these binding pockets are mostly hydrophobic
in nature and one is at the cytoplasmic position while the other is towards the
periplasmic position of the protein (Figure 5.3). The table 5.2 shows the observed
docking scores of the protein and inhibitors. It is observed from this table that
verapamil and reserpine are the two best scoring molecules at both the binding sites
of NorA. Besides, hydrogen bonding and hydrophobic interactions, π‐π interactions
were observed at binding site no. 1 of protein NorA and chemical moieties of
inhibitors. In general, it has been observed that Arg 310, Ser 333 and Ser 337 of the
protein are mainly involved in hydrogen bonding with different inhibitors at binding
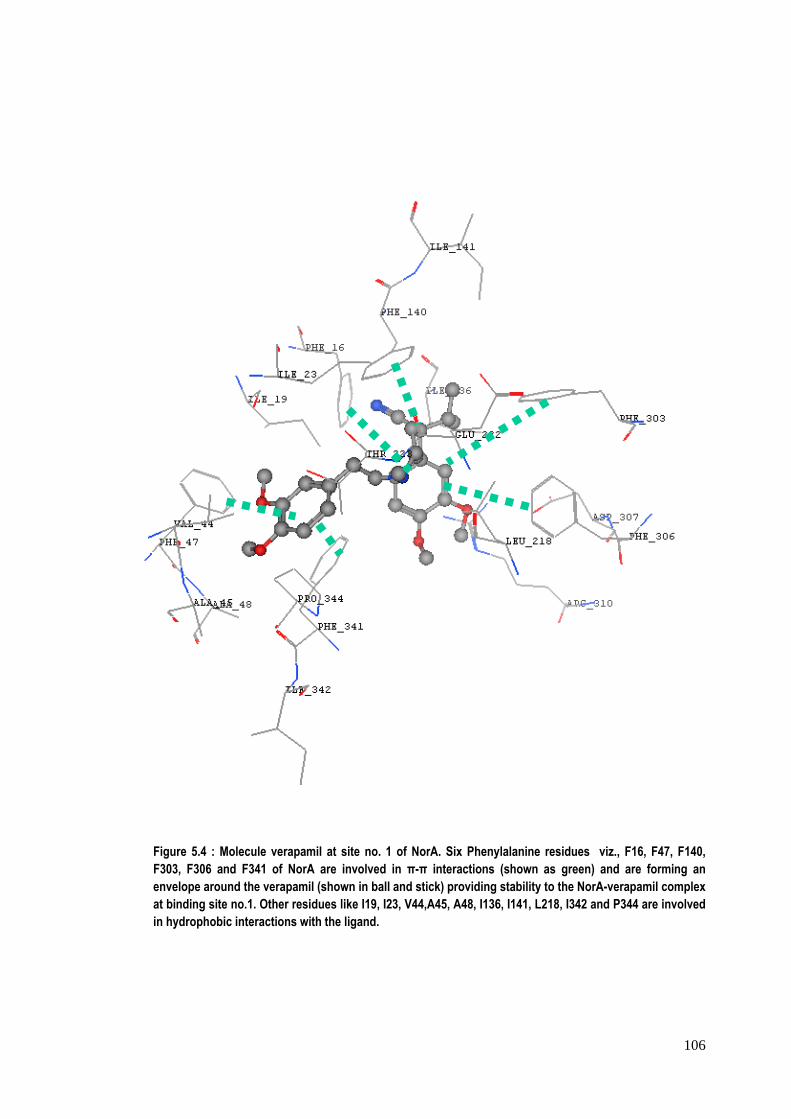
site no.1. A number of Phenylalanine residues (maximum of six with verapamil)
(predominantly at positions‐16, 47 and 341) were observed to be involved in π‐π
interactions with moieties in the inhibitors. Figure 5.4 shows such interactions
between binding site no.1 of NorA with verapamil. Both the factors‐ hydrogen
bonding and π‐π interactions contribute towards the stability of the protein‐inhibitor
complexes. Besides this, hydrophobic interactions of residues like Ala, Ile, Leu, Met
with the inhibitors were also observed and contribute towards the stability of NorA‐
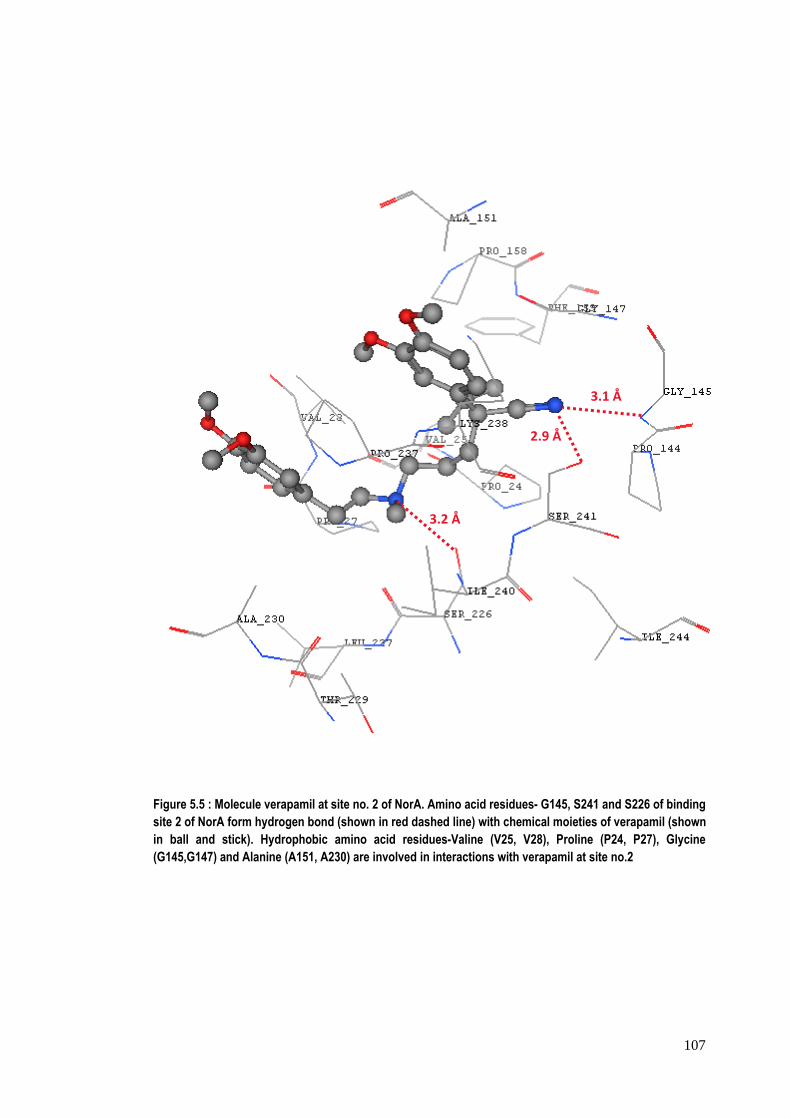
inhibitor complex., Also at binding site no. 2, three strong hydrogen bonds
contribute effectively to the stability of conformation of verapamil within the site as
shown in figure 5.5. The stable conformation of reserpine at binding site no. 1 is
attributed due to the presence of hydrogen bonding of inhibitor with serine, arginine
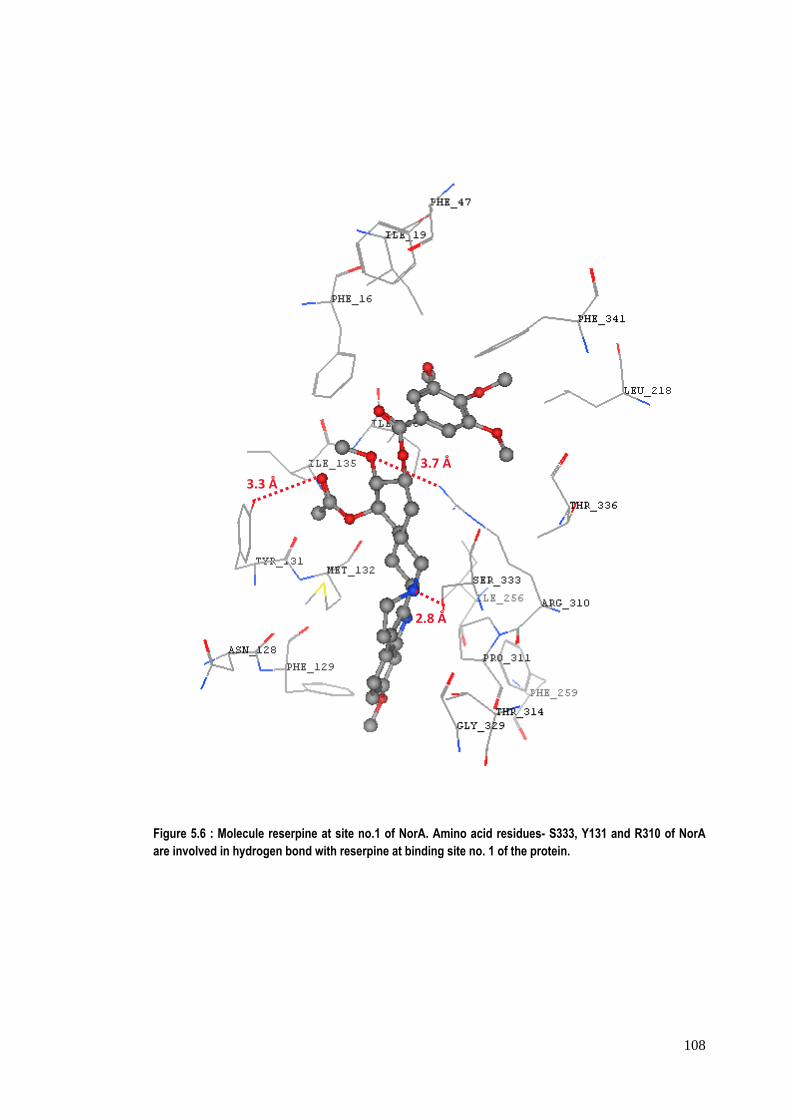
and tyrosine at site no. 1 as shown in figure 5.6. The aromatic amino acid residues‐
F16, F47, F129, F259 and F341of NorA in binding site no.1 are also involved in π‐π
interactions with phenyl rings of reserpine. The hydrophobic amino acid residues ‐
isoleucines (I19, I135, I136, I218 and I256) and M132 of NorA are involved in
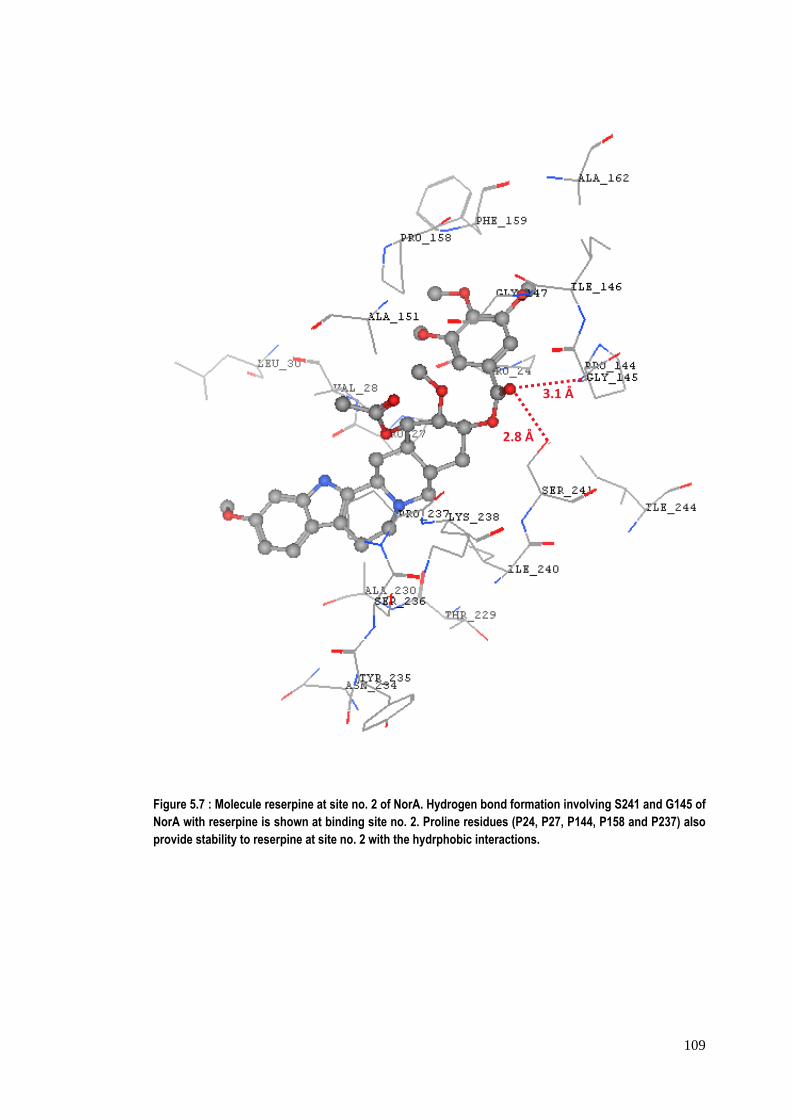
interactions with reserpine. The figure 5.7 shows serine241 & glycine145 at binding
site no. 2 of NorA are involved in hydrogen bonding with chemical moieties of
104
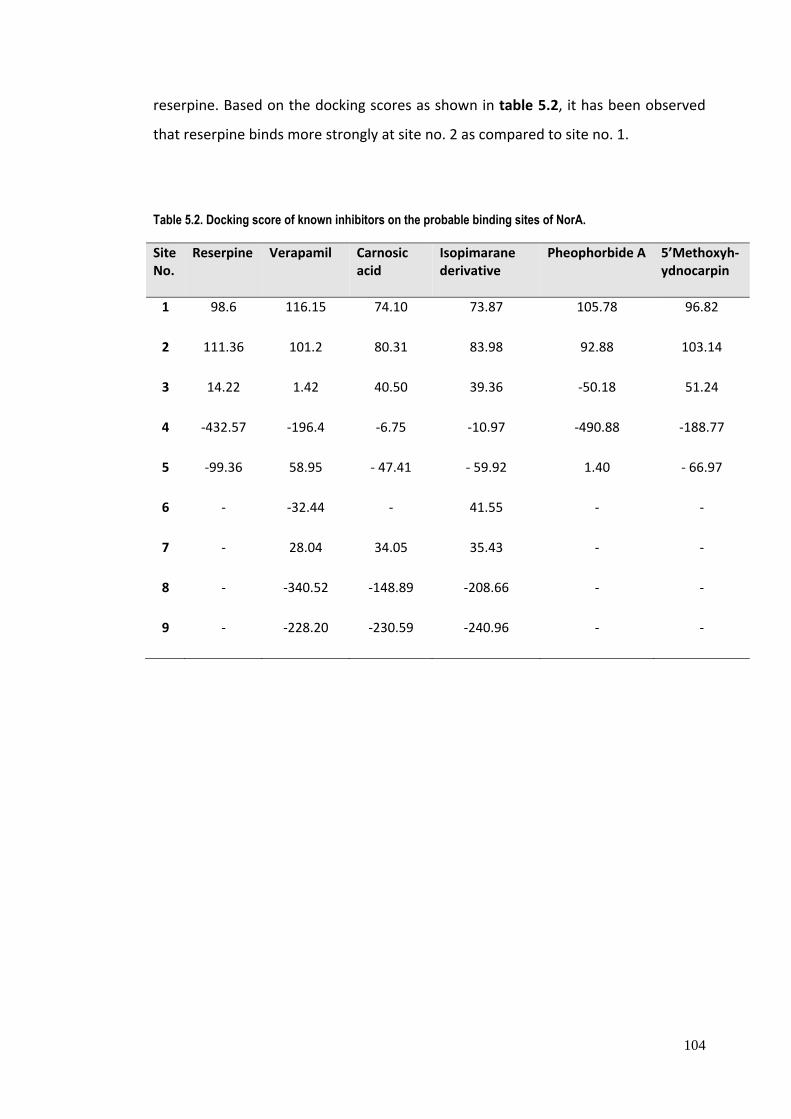
reserpine. Based on the docking scores as shown in table 5.2, it has been observed
that reserpine binds more strongly at site no. 2 as compared to site no. 1.
Table 5.2. Docking score of known inhibitors on the probable binding sites of NorA.
Site No.
Reserpine Verapamil Carnosic acid
Isopimarane derivative
Pheophorbide A
5’Methoxyh‐ydnocarpin
1 98.6 116.15 74.10 73.87 105.78 96.82
2 111.36 101.2 80.31 83.98 92.88 103.14
3 14.22 1.42 40.50 39.36 ‐50.18 51.24
4 ‐432.57 ‐196.4 ‐6.75 ‐10.97 ‐490.88 ‐188.77
5 ‐99.36 58.95 ‐ 47.41 ‐ 59.92 1.40 ‐ 66.97
6 ‐ ‐32.44 ‐ 41.55 ‐ ‐
7 ‐ 28.04 34.05 35.43 ‐ ‐
8 ‐ ‐340.52 ‐148.89 ‐208.66 ‐ ‐
9 ‐ ‐228.20 ‐230.59 ‐240.96 ‐ ‐
105
Figure 5.3 : Binding Pockets of Modelled NorA structure.The probable binding sites of the protein NorA. Periplasmic and cytoplasmic faces of predicted NorA structure have been assigned based on its superimposition with that of template, glycerol-3-phosphate. (a) The binding site no. 1 of NorA is at the central cavity facing cytoplasmic side of membrane with dimensions approximately 35 Å x 18 Å. (b) The binding site no. 2 is towards the periplasmic side of the membrane.
Cytoplasm
Membrane
Periplasm
106
Figure 5.4 : Molecule verapamil at site no. 1 of NorA. Six Phenylalanine residues viz., F16, F47, F140, F303, F306 and F341 of NorA are involved in π-π interactions (shown as green) and are forming an envelope around the verapamil (shown in ball and stick) providing stability to the NorA-verapamil complex at binding site no.1. Other residues like I19, I23, V44,A45, A48, I136, I141, L218, I342 and P344 are involved in hydrophobic interactions with the ligand.
107
Figure 5.5 : Molecule verapamil at site no. 2 of NorA. Amino acid residues- G145, S241 and S226 of binding site 2 of NorA form hydrogen bond (shown in red dashed line) with chemical moieties of verapamil (shown in ball and stick). Hydrophobic amino acid residues-Valine (V25, V28), Proline (P24, P27), Glycine (G145,G147) and Alanine (A151, A230) are involved in interactions with verapamil at site no.2
3.1 Å
2.9 Å
3.2 Å
108
Figure 5.6 : Molecule reserpine at site no.1 of NorA. Amino acid residues- S333, Y131 and R310 of NorA are involved in hydrogen bond with reserpine at binding site no. 1 of the protein.
2.8 Å
3.7 Å 3.3 Å
109
Figure 5.7 : Molecule reserpine at site no. 2 of NorA. Hydrogen bond formation involving S241 and G145 of NorA with reserpine is shown at binding site no. 2. Proline residues (P24, P27, P144, P158 and P237) also provide stability to reserpine at site no. 2 with the hydrphobic interactions.
3.1 Å
2.8 Å

110
Form the docking studies of reserpine (known inhibitor) and capsaicin in the
binding site of NorA, it was observed that Arg98 and Ile23 are involved in key
interactions and hence play an important role in the ligand binding (Figure 5.8 a &
b). Also, a hydrophobic cleft formed by Leu26, Val44 and Phe47 at one end and by
Pro24, Phe140, Ile244, Gly248 and Phe303 at the other (particularly for capsaicin)
provides some extra stability to the complex. A H‐bond formation between reserpine
and Gln51 is further supposed to provide strength to the reserpine‐NorA complex. A
strong H‐bond interaction at a distance of 1.89 Å with Ile23 of the protein NorA is
observed in case of capsaicin. The orientation of capsaicin within the binding site (as
shown in figure 5.8b) allows its aliphatic chain to extend into the hydrophobic cleft
involving residues Pro24, Phe143, Ile244, Gly248 and Phe303 which enables strong
hydrophobic interactions due to the close proximity of the ligand with these residues
(distance ranges between 1.7 Å to 3.2 Å). A weak H‐bond between Arg98 and the
hydroxyl group of the aryl moiety of capsaicin may be attributing extra stability to
the capsaicin‐NorA complex. The relative orientation of the interactive residues with
respect to the transmembrane helices is also shown in figure 5.8c. Apart from the
other key interactive residues, it was found that Val44, Phe47, Gln51, Phe140,
Ile244, Gly248 and Phe303 form the residues of the conserved domain of the MFS
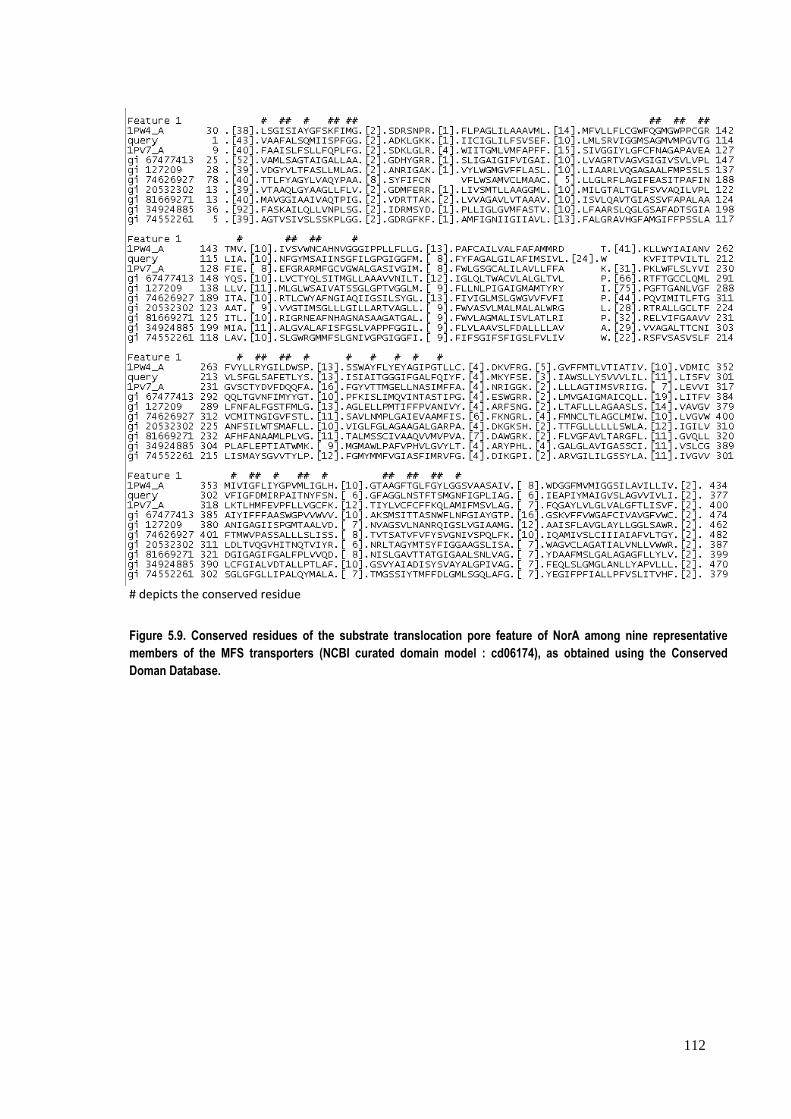
transporter family (see Figure 5.9 and Table 5.3).

Figure 5.8. (and Gln51 pbinding sitePhe140, Ile2proximity ofrespect to transmembrbound to the
(a) Interactionprovides stabe allows its a244, Gly248 af the ligand wthe transmem
rane helices oe central cavit
n of reserpine bility to the pr
liphatic chainand Phe303, wwith these residmbrane heliceof the proteiny of the protei
in the bindingrotein/ligand cn to extend inwhich enablesdues. (c) The s of the pred
n are colouredn.
g site of NorAcomplex. (b) Tnto the hydrops strong hydro
relative orientdicted three-dd differently a
A. H-bond formThe orientationphobic cleft iophobic interatation of the b
dimensional sand labelled. C
mation between of capsaicinnvolving residactions due tbinding site retructure of NCapsaicin is
111
en reserpine n within the dues Pro24, o the close
esidues with NorA. All 12
also shown
112
# depicts the conserved residue
Figure 5.9. Conserved residues of the substrate translocation pore feature of NorA among nine representative members of the MFS transporters (NCBI curated domain model : cd06174), as obtained using the Conserved Doman Database.

113
Table 5.3. Full name for the representative nine coded members displayed in figure 9. ID Name 1PW4_A Chain A, Crystal Structure Of The Glycerol‐3‐Phosphate Transporter From
E.Coli 1PV7_A Chain A, Crystal Structure Of Lactose Permease With Tdg (Escherichia coli) gi 67477413
RecName: Full=Probable glucose transporter rco‐3 (Neurospora crassa)
gi 127209 RecName: Full=Methylenomycin A resistance protein; AltName: Full=MMR peptide (Streptomyces coelicolor)
gi 74626927
RecName: Full=Uncharacterized transporter C757.13 (Schizosaccharomyces pombe)
gi 20532302
RecName: Full=Uncharacterized transporter ygaY (Escherichia coli)
gi 81669271
RecName: Full=Uncharacterized MFS‐type transporter Rv2456c/MT2531 (Mycobacterium tuberculosis)
gi 34924885
RecName: Full=Vesicular acetylcholine transporter; Short=VAChT; AltName: Full=Solute carrier family 18 member 3 (Homo sapiens)
gi 74552261
Quinolone resistance protein norA homolog (Pyrococcus furiosus)

114
5.3.2 In silico structure prediction and docking studies of bacterial efflux pump Bmr
from Bacillus subtilis
Since there is not good homology between the sequence of Bmr and the available
data set of sequences whose structure is known, threading approach was used in
this study for determining the 3D structure of Bmr protein using 1PV6 (lactose
permease of E. coli) as the template. The structure was modelled using FFAS and
Protmod servers (having 91% RC core value), where FFAS server was used for the
selection of the template and target template alignment and protmod server was
used for modelling the protein. The proposed 3D structure model of Bmr based on
the template 1PV6 was tested for structure validation using the Structure analysis
and validation server and the results were satisfactory.
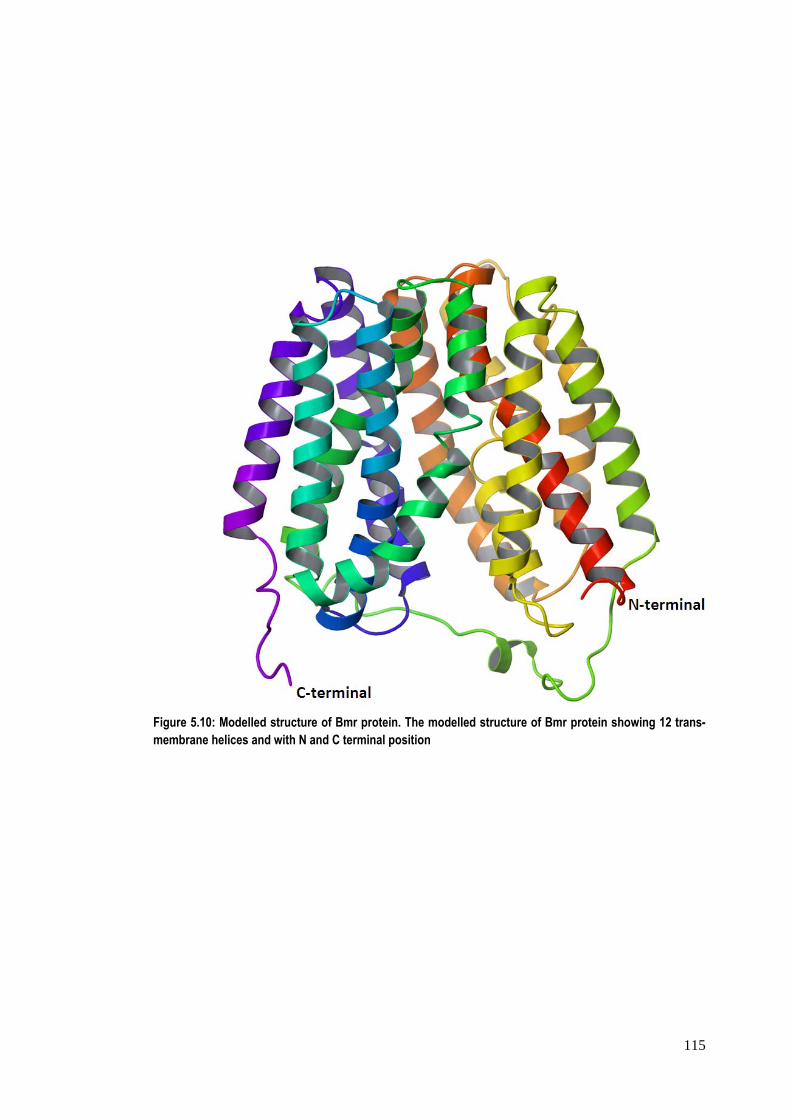
The modelled structure of Bmr has been found to have 12 main helical
segments interspersed by loops of variable lengths at regular intervals with both N‐
and C‐termini on the same side of membrane (figure 5.10), which is in corroboration
with the reported structural analysis by Paulsen et al., 1996. The transmembrane
helices of the modelled structure were also confirmed using the PSIPRED server
(Bryson et al., 2005; Buchan et al., 2010) as shown in figure 5.11. Out of the
probable binding pockets identified using the Site map module of Schrodinger, the
site comprising of the pivotal residues involved in the binding of reserpine (known
inhibitor of Bmr) (Ahmed et al., 1993; Klyachko et al., 1997) was selected for
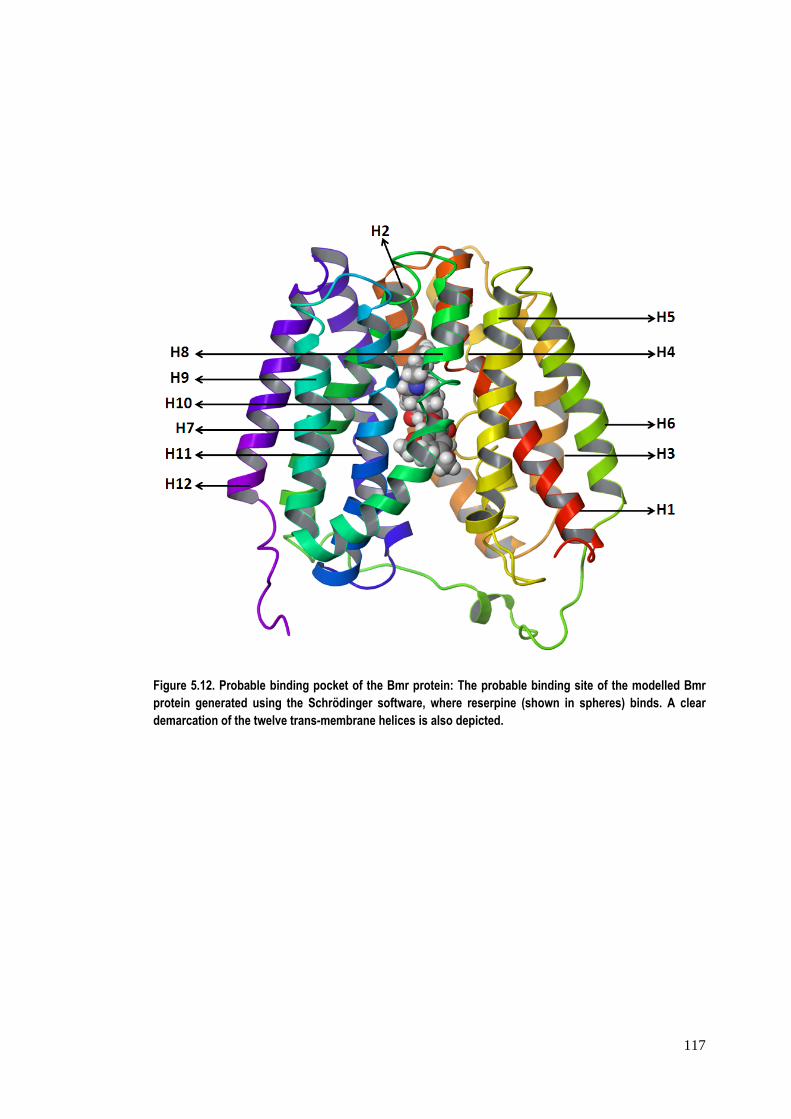
docking studies. The proposed binding pocket is depicted in figure 5.12.
115
Figure 5.10: Modelled structure of Bmr protein. The modelled structure of Bmr protein showing 12 trans-membrane helices and with N and C terminal position

116
Pred: CCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCH AA: MEKKNITLTILLTNLFIAFLGIGLVIPVTPTIMNELHLSG Pred: HHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHH AA: TAVGYMVACFAITQLIVSPIAGRWVDRFGRKIMIVIGLLF Pred: HHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHHHH AA: FSVSEFLFGIGKTVEMLFISRMLGGISAPFIMPGVTAFIA Pred: HHCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCC AA: DITTIKTRPKALGYMSAAISTGFIIGPGIGGFLAEVHSRL Pred: CCCCCCCHHHHHHHEEEEECCCCCCCCCHHHHCCCCCHHH AA: PXXXXXXXXXXXXILSILTLREPERNPENQEIKGQKTGFK Pred: HHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCH AA: RIFAPMYFIAFLIILISSFGLASFESLFALFVDHKFGFTA Pred: HHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHH AA: SDIAIMITGGAIVGAITQVVLFDRFTRWFGEIHLIRYSLI Pred: HHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHHH AA: LSTSLVFLLTTVHSYVAILLVTVTVFVGFDLMRPAVTTYL Pred: HCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH AA: SKIAGNEQGFAGGMNSMFTSIGNVFGPIIGGMLFDIDVNY Pred: HHHHHHHHHHHHHHHHHHHHCHHHCCCCC AA: PFYFATVTLAIGIALTIAWKAPAHLKAST
Figure 5.11. PSIPRED results: The figure depicts the PSIPRED results for the Bmr protein sequence. Pred: Predicted secondary structure; AA: target sequence
117
Figure 5.12. Probable binding pocket of the Bmr protein: The probable binding site of the modelled Bmr protein generated using the Schrödinger software, where reserpine (shown in spheres) binds. A clear demarcation of the twelve trans-membrane helices is also depicted.

118
In order to confirm the accuracy of the modelled structure of Bmr and the
proposed binding pocket, the molecule reserpine was docked on to the binding
pocket of the prepared protein (Bmr_model) using the Glide module of Schrodinger
software suite. The Val286 residue of the protein was mutated first by Leucine
(Bmr_V286L) and then by Glycine (Bmr_V286G) residues and in both the cases,
reserpine was docked on to the identified binding pocket. Further, in order to verify
the built model, the Phe143 residue was mutated by isoleucine (Bmr_F143I) and
then by leucine. Subsequently, the Phe306 residue was also mutated by leucine
(Bmr_F306L) and serine (Bmr_F306S). The prepared proteins were also taken for
similar docking studies on the ligandfit module of Cerius2. Since a similar trend was
observed in Cerius2 also for first three cases, the docking studies for rest of the four
cases was done only in Glide in order to save time. The binding affinities
(MMGBSA_DGbind: results of Glide docking) and dock score (results of ligandfit) of
reserpine onto the modelled structure of Bmr and the mutated structures are shown
in Table 5.4.
The interactive residues of the protein with reserpine revealed that there are direct
hydrogen bonding and π‐π interactions between reserpine and Arg313 and Phe309
residues of the modelled structure of Bmr. Besides this, Phe143 also plays an
important role in providing extra stability to the protein‐ligand complex by means of
hydrophobic interactions, as also reported in the literature (Klyachko et al., 1997). It
is observed that the distance of Val286 from the binding site is more than 10Ǻ.
Although Val286 is not directly involved in binding but it does play an important role
in determining the substrate specificity, which may be due to intra‐molecular
interactions.
The importance of these residues for reserpine accommodation in the binding site
was further confirmed by in silico mutations. The in silico mutations of val286,
phe143 and phe306 of Bmr binding site leads to the reduced binding affinity of the
reserpine which imply that mutation of any one of these residues leads to the
conformation alteration of reserpine within the binding site. It is also observed that
the due to the conformational change, accommodation of reserpine is not as

119
favoured as was in wild type, except in the case of Bmr_V286G where the strong
interaction and probably less steric clashes favours the strong binding of reserpine
within the binding pocket.
The change of orientation of reserpine within the binding pocket due to
mutation at Val286 plays a significant role in the binding affinity of the ligand with
the receptor. The interaction figures of reserpine with Bmr_model, Bmr_V286L and
Bmr_V286G are shown in Figures 5.13, 5.14 and 5.15 respectively. From the
interaction figures, it is observed that Arg313 of wild type Bmr is involved in H‐
bonding and π‐cation interaction with reserpine and Phe309 is involved in π‐π
interaction with the six‐membered ring of trimethoxy benzoic acid moiety of
reserpine. Docking studies with the V286L mutation reveals that there is a complete
reorientation of reserpine within the binding pocket, with the indole moiety being in
the vicinity of Phe309 providing a possibility of π‐π interaction, whereas Arg313 is
involved in H‐bonding with oxygen of methyl ester moiety of reserpine. But, the
complex energy increases significantly to ‐65.497 Kcal/mol thereby indicating some
steric clashes due to this orientation. A very stable complex is observed for reserpine
and Bmr_V286G wherein the role of Phe143 residue in substrate recognition is also
signified, as also reported in literature. Phe143 and Phe309 are involved in π‐π
interactions with the five‐membered and six‐membered rings respectively of the
indole moiety of reserpine. The energy value of 172.7 Kcal/mol indicates the high
stability of the complex. The mutations of Phe143 and Phe306 with isoleucine,
leucine and serine also decreases the complex binding affinity as is indicated in the
energy values in Table 5.4. Moreover, the orientation remains similar to the one in
Bmr_V286L and Bmr_V286G (as shown in figures 4‐6 and S1‐S4) thereby, indicating
that the mutation not only changes the approach of the ligand towards the protein,
but also the orientation of the ligand within the binding pocket. The role of Phe143
and Phe306 in substrate recognition seems to be significant as any mutation to these
residues lowers the binding affinity of reserpine towards Bmr.
120
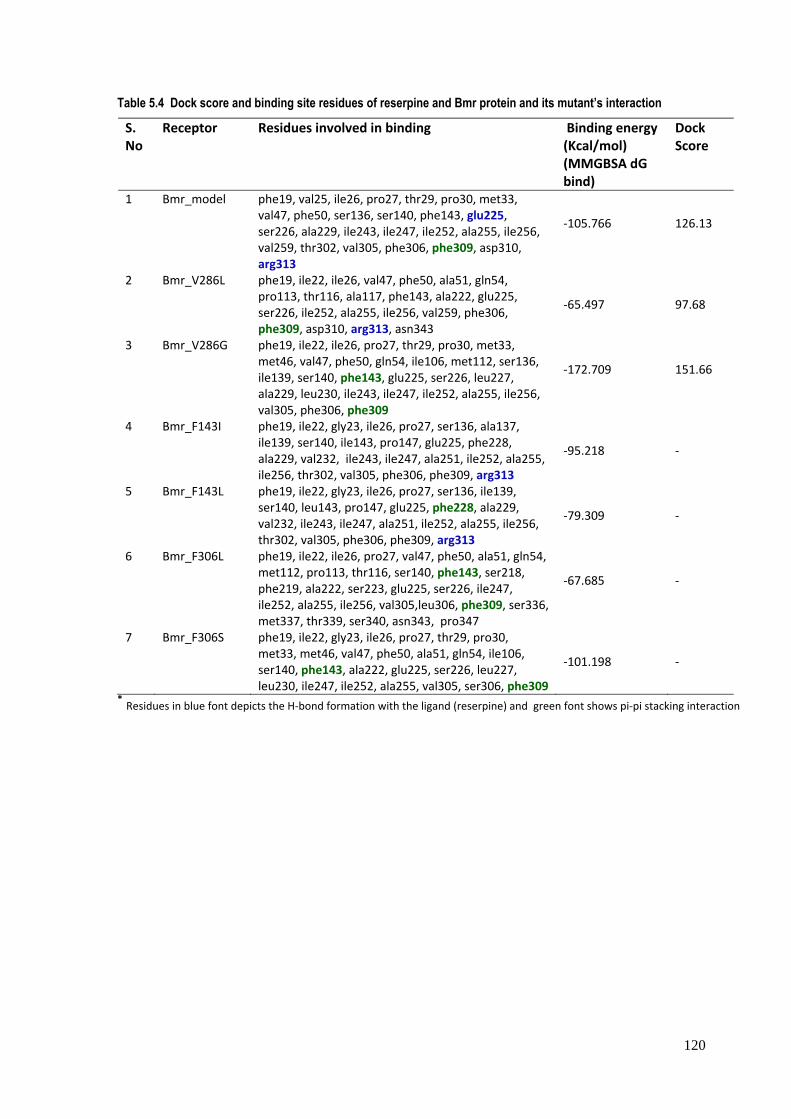
Table 5.4 Dock score and binding site residues of reserpine and Bmr protein and its mutant’s interaction S. No
Receptor Residues involved in binding Binding energy (Kcal/mol) (MMGBSA dG bind)
Dock Score
1 Bmr_model phe19, val25, ile26, pro27, thr29, pro30, met33, val47, phe50, ser136, ser140, phe143, glu225, ser226, ala229, ile243, ile247, ile252, ala255, ile256, val259, thr302, val305, phe306, phe309, asp310, arg313
‐105.766 126.13
2 Bmr_V286L phe19, ile22, ile26, val47, phe50, ala51, gln54, pro113, thr116, ala117, phe143, ala222, glu225, ser226, ile252, ala255, ile256, val259, phe306, phe309, asp310, arg313, asn343
‐65.497 97.68
3 Bmr_V286G phe19, ile22, ile26, pro27, thr29, pro30, met33, met46, val47, phe50, gln54, ile106, met112, ser136, ile139, ser140, phe143, glu225, ser226, leu227, ala229, leu230, ile243, ile247, ile252, ala255, ile256, val305, phe306, phe309
‐172.709 151.66
4 Bmr_F143I phe19, ile22, gly23, ile26, pro27, ser136, ala137, ile139, ser140, ile143, pro147, glu225, phe228, ala229, val232, ile243, ile247, ala251, ile252, ala255, ile256, thr302, val305, phe306, phe309, arg313
‐95.218 ‐
5 Bmr_F143L phe19, ile22, gly23, ile26, pro27, ser136, ile139, ser140, leu143, pro147, glu225, phe228, ala229, val232, ile243, ile247, ala251, ile252, ala255, ile256, thr302, val305, phe306, phe309, arg313
‐79.309 ‐
6 Bmr_F306L phe19, ile22, ile26, pro27, val47, phe50, ala51, gln54, met112, pro113, thr116, ser140, phe143, ser218, phe219, ala222, ser223, glu225, ser226, ile247, ile252, ala255, ile256, val305,leu306, phe309, ser336, met337, thr339, ser340, asn343, pro347
‐67.685 ‐
7 Bmr_F306S phe19, ile22, gly23, ile26, pro27, thr29, pro30, met33, met46, val47, phe50, ala51, gln54, ile106, ser140, phe143, ala222, glu225, ser226, leu227, leu230, ile247, ile252, ala255, val305, ser306, phe309
‐101.198 ‐
* Residues in blue font depicts the H‐bond formation with the ligand (reserpine) and green font shows pi‐pi stacking interaction

Figure 5.13:reserpine winteraction r
: Interaction fiwhere Arg313 respectively. G
gure of Bmr_mand Phe309 a
Glu225 is also
model with resare involved iinvolved in H-
serpine: Interan H-bonding (bonding with
active residue(2.254Å), π-careserpine at a
s of the Bmr ation interactio distance of 2.
121
protein with on and π-π 316 Å.

Figure 5.14interaction bonding witindole moiet
. Interactive of the mutateth reserpine aty.
residues of Bed Bmr modelat a idstance o
Bmr_V286L m (where Val28
of 2.091Å, whe
mutant type w86 is mutated ereas Phe309
with reserpine: by Leucine). is involved in
: The figure Arg313 is inv
n π-π interacti
122
depicts the volved in H-ion with the
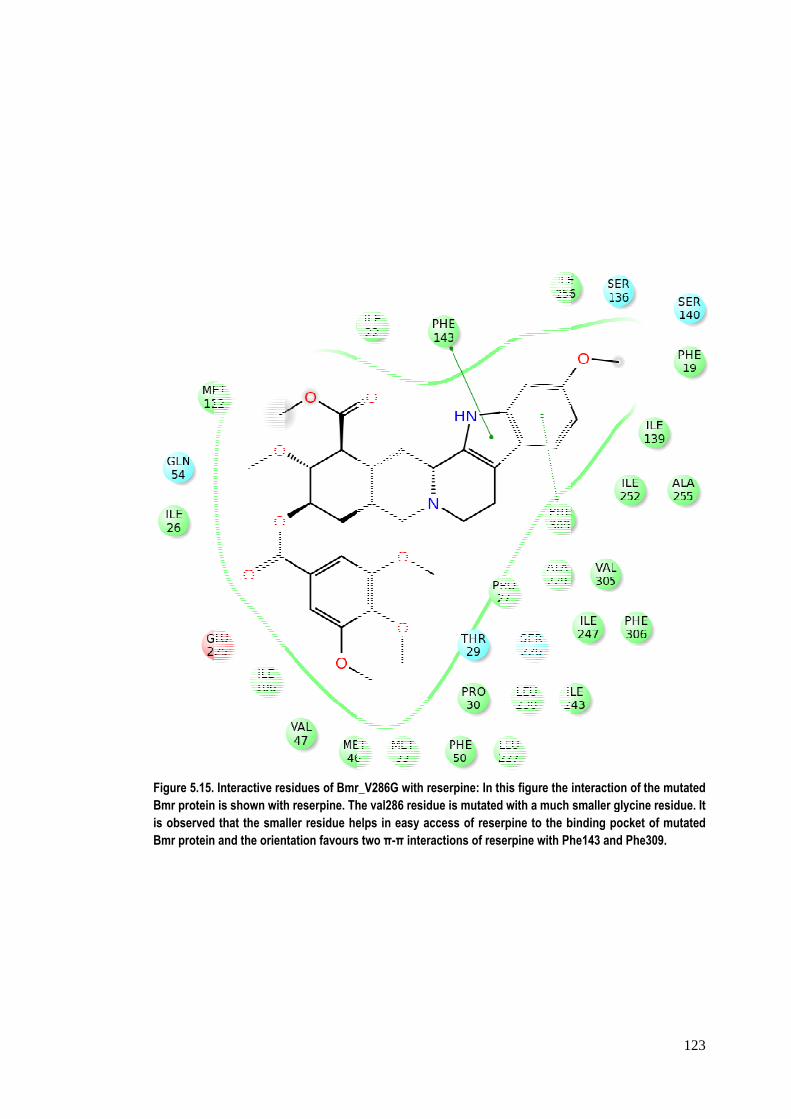
Figure 5.15.Bmr proteinis observedBmr protein
Interactive res is shown with that the sma and the orien
sidues of Bmrh reserpine. Thller residue hetation favours
r_V286G with rhe val286 residelps in easy as two π-π inter
reserpine: In thdue is mutatedaccess of reseractions of res
his figure the id with a much rpine to the b
serpine with Ph
interaction of smaller glycin
binding pockethe143 and Phe
123
the mutated ne residue. It t of mutated e309.

124
From the interaction figures it is clear that Phe143 and Phe306 are directly involved
in reserpine binding through strong hydrophobic interactions, whereas Val286 is not
directly involved in the interaction, as also reported in the literature.
5.3.3 In silico 3D structure prediction and docking studies of Mycobacterium
tuberculosis efflux pump Rv1258c
A total of 48 models were built from the various selected servers. In order to filter
out some poorly constructed model a filter criterion was set where the value for bad
contacts was set at zero and RC core value was set more than 85%. Based on the set
threshold value of ‘bad contacts’ and ‘RC core’, a total of 17 models were selected.
The templates selected for modeling were 1PW4, 1PV6, 2GFP, 2CFQ, 2VCH, 2CFP,
307P, 2V8N, 1PV7. Out of these 1PW4, 1PV6, 2CFQ and 2GFP are the 3D structures
of the transporters from the Multi Facilitator Superfamily (MFS), which gives more
robustness to our predicted models based on the functional aspect. The final model
consists of 12 transmembrane helices which is common in all MDR efflux pumps. The
model alongwith the description of helices and loops is shown in figure 5.16. There
are two 6‐α‐helical bundles that are connected by a large cytoplasmic loop and this
structural feature is conserved in MFS transporters (Huang et al., 2003). Top five
binding sites were selected for consideration of docking studies from each model.
Finally, a total of 41 binding pockets, only those which lie within the central cavity of
the transmembrane helices, were considered for docking analysis. Docking of 7
active and 11 inactive molecules were carried out onto the selected binding pockets,
for further pruning of the obtained models for accuracy. Based upon the information
from the template structures, it was observed that the binding location of the
substrate lies within the centre of the main hydrophobic cavity at a similar distance
from the central helices. Accordingly, this information about the location of binding
site was utilized in order to segregate and filter the most probable proposed model
with binding site location and a total of five binding pockets were selectively chosen
out of fourty‐one. The interaction studies and glide score revealed that modeller‐
multi‐4‐mini2 as the best model among all. Further docking studies of the test set (7
active and 11 inactive molecules) was carried out on the binding pocket of modeller‐

125
multi‐4‐mini2. The results show that almost all the active molecule occupies the
central location within the binding cavity thereby completely blocking the efflux
passage. And most of the inactive compounds bind towards the peripheral of the
central pocket thereby rendering the passage open for efflux. Hence it signifies that
even if the ligand binds with good affinity but does not cover the central passage, it
may not act as a potent inhibitor. Hence the location of the ligand binding site within
the large central cavity plays an important role in determining the potency of the
inhibitor and must be taken into account while designing inhibitors for Rv1258c. The
identified binding site was further explored in order to identify the smaller binding
clefts that are formed within the pocket. Interestingly, it was observed that there are
eight small binding clefts of different size and property that are formed within the
identified binding pocket. This also supports the fact that this efflux pump is a
multidrug transporter. Further, the docking of some first line and second line
antibiotics such as amikacin, rifampicin, ciprofloxacin, ofloxacin, moxifloxacin and
gatifloxacin was carried out on the predicted model of Rv1258c.

126
Figure 5.16. Predicted 3D structure model of Rv1258c. (a) the transmembrane helices numbered from 1 - 12 and (b) the loops interspersed by these helices.
(a)
(b)

127
The sequence identity of Rv1258c and the template sequence [a multidrug
transporter EmrD from E. coli (PDB ID: 2GFP)] was found to be 36%. Using the site
map module of the Schrodinger suite, the predicted structure of Rv1258c revealed
five probable binding pockets. The docking studies of reserpine, a known inhibitor,
and piperine on these binding pockets revealed one of the binding sites showing
better binding affinity for these inhibitors. Reserpine and piperine had a Glide score
of 23.75 Kcal/mol and 25.97 Kcal/mol, respectively, showing better binding affinity
of piperine for the receptor than reserpine. The figure showing the interaction of
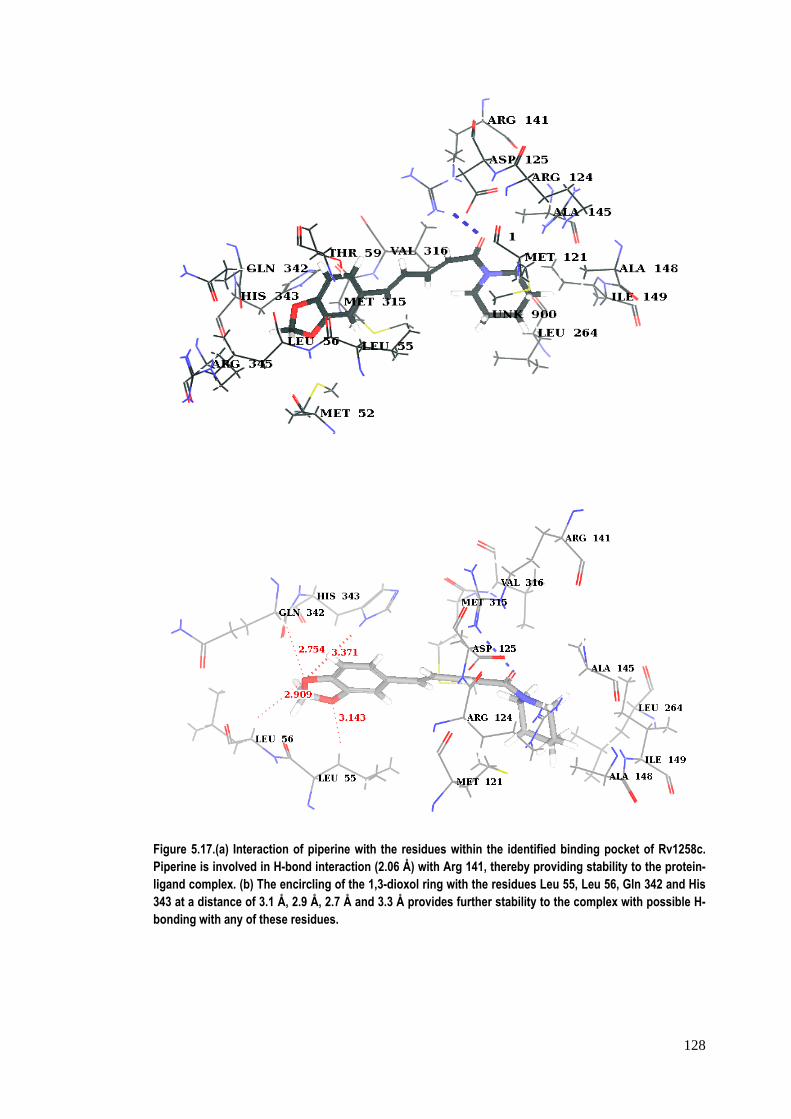
piperine with Rv1258c revealed that it is involved in H‐bond interaction (2.06A°) with
Arg141 to form a protein–ligand complex (Figure 5.17a). Furthermore, the 1,3‐dioxol
ring of the piperine molecule is surrounded by Leu55, Leu56, Gln342 and His343
residues at a favourable distance (all ≤3 A° ) for H‐bonding, thereby providing more
stability to the ligand in the pocket (Figure 5.17b). Additionally, piperine and the
heterocyclic ring of His343 are seen to be favourably oriented to interact with a π‐π
interaction between the two.
128
Figure 5.17.(a) Interaction of piperine with the residues within the identified binding pocket of Rv1258c. Piperine is involved in H-bond interaction (2.06 Å) with Arg 141, thereby providing stability to the protein-ligand complex. (b) The encircling of the 1,3-dioxol ring with the residues Leu 55, Leu 56, Gln 342 and His 343 at a distance of 3.1 Å, 2.9 Å, 2.7 Å and 3.3 Å provides further stability to the complex with possible H-bonding with any of these residues.
129
Further, the docking of some first line and second line antibiotics such as
amikacin, rifampicin, ciprofloxacin, ofloxacin, moxifloxacin and gatifloxacin was
carried out on the predicted model of Rv1258c. Based on the dock scores (as shown
in Table 5) of these drugs on Rv1258c, it was found that apart from rifampicin (which
is a known binder of Rv1258c), amikacin showed a good binding affinity with this
efflux pump.
Table 5.5. Results of docking of several known drugs on the predicted model of Rv1258c
Drugs Rifampicin Ciprofloxacin Amikacin Ofloxacin Moxifloxacin Gatifloxacin
Dock score
‐8.950 ‐4.841 ‐9.718 ‐4.728 ‐4.722 ‐4.526
5.4 Conclusion
A strong structural commonality is observed between the efflux pumps of the MFS
family. All the predicted structures shows the similar formation of 12
transmembrane helices with a large central cavity for diverse substrate binding. A
ligand that binds to the central core of the cavity acts as an inhibitor to these
proteins by blocking the central passage, as also is reported in the template
structures Lactose permease (1PV6) and Emrd (2GFP) which are multi‐drug
transporters in E coli. The helices involved in the formation of the internal cavilty are
observed to be similar as in the template. The interactive residues are different
which might be the determining factor for the substrate selectivity of these efflux
pumps. The docking results and the residual interaction figures show the importance
of the binding location for a molecule to be the inhibitor of these efflux pumps.
Further the formation of large number of binding clefts within the central binding
pocket supports the multidrug binding on this efflux pump. Amid so many similarities
there still exists some specificity among the 3D structures of these pumps, which is
discussed below.

130
5.4.1 NorA from Staphylococcus aureus
Two main binding sites‐ central hydrophobic pocket and periplamic pocket have
been identified for NorA The amino acids of diverse chemical nature have been
found to be present in these sites. Further, known inhibitors of NorA have been
found to have maximum docking scores for these sites. The type of interactions
which have been found to be responsible stabilization of NorA‐inhibitor complexes
include π‐π interactions, hydrogen bonding and hydrophobic interactions. This will
provide clues for elucidation of the reaction mechanism of an efflux pump like NorA
and aid in designing of novel and potent inhibitors of efflux pump, like NorA.
5.4.2 Bmr from Bacillus subtilis
The in silico mutations performed on the modelled structure confirmed the
robustness of the predicted structure. The effect of mutation was assessed by
change in the binding affinity of reserpine, based on the dock score and binding
affinity. On comparing the affinity of wild type with the mutants, it was clear that
any mutation to these residues leads to significant change in reserpine binding, as
reported in the literature (Ahmed et al., 1993; Klyachko et al., 1997). Besides this,
the study also revealed the importance of the residues Arg313 and Phe309 in
reserpine binding, where these amino acids are directly involved in H‐bonding and
significant π‐π interactions with the reserpine molecule.
It is also inferred from the study that Val286 is not directly involved in the
binding of the substrate, but do play a role in substrate recognition due to intra‐
molecular interactions. Any change in this residue significantly affects the reserpine
binding. Substitutions for Val286, which is located in the IX putative transmembrane
domain of Bmr change the sensitivity of the transporter to inhibitory action of
reserpine. Substitution of leucine for Val286 leads lowering of reserpine sensitivity,
substitution of glycine for Val286 increases the sensitivity. The results further
suggest that the 286th residue of Bmr is not a critical residue directly involved in the
inhibitor recognition. This residue is just a component of the “wall” of the reserpine‐
binding “pocket.” It is also very clear in the modelled structure that these significant

131
residues are located in different regions of the Bmr transporter, the 5th (F143), 9th
(V286) and 10th (F306) transmembrane domains of the proposed structure, thus
validating that the reserpine‐binding site of Bmr may represent a spatial structure
formed by interacting transmembrane domains. The proposed structure of this
important efflux pump of Bacillus subtilis would help significantly in understanding
the mechanism of action of the drug efflux and hence would be instrumental in the
design of specific efflux pump inhibitor in order to increase the efficacy of the drugs.
5.4.3 Rv1258c from Mycobacterium tuberculosis
The binding site of the predicted model was determined using the experimental
inhibitory values of some active and inactive molecules from our institute’s
molecular repository. The robustness of the model was determined using the test set
molecules having experimental inhibitory activity data. Also the model was used for
docking with known antibiotics and it was found that amikacin and refampicin shows
better binding, and other drugs (ciprofloxacin, ofloxacin, moxifloxacin and
gatifloxacin) showed very weak binding to the predicted structure of Rv1258c. The
information about the interactive residues, helices and binding modes of inhibitors
within the protein would be very useful for designing more potent inhibitors of this
protein even in the absence of a 3D crystal structure.





























