Embed Size (px)
Citation preview

Saverio Santi -Scienze Chimiche- Università di Padova
1
Chimica Fisica Industriale
Proff. Armando Gennaro e Saverio Santi
Dipartimento di Scienze Chimiche
Via Marzolo 1 Padova
049 8275119 (Saverio Santi)
Ricevimento: tutti i giorni per appuntamento

Chimica Fisica Industriale
Il corso è finalizzato all’approccio alla termodinamica di non equilibrio, per acquisire le conoscenze necessarie per lo studio dei processi industriali, con riferimento sia alle trasformazioni fisiche che a quelle chimiche.
Saverio Santi -Scienze Chimiche- Università di Padova
2
Obiettivi

Chimica Fisica Industriale Modulo B
Conoscenze di base per la descrizione dei fenomeni di trasporto.
Capacità di impostare e risolvere il bilancio per le proprietà fisiche più importanti per le quali vale il principio di conservazione.
Saverio Santi -Scienze Chimiche-
Università di Padova 3

Chimica Fisica Industriale Modulo B
Bilanci dei processi industriali: bilanci di materia in presenza di reazioni chimiche.
Bilanci di energia.
Fenomeni di trasporto: trasporto di quantità di moto dei fluidi
reali; trasporto di calore: diffusività termica, conduzione, irraggiamento.
Saverio Santi -Scienze Chimiche- Università di Padova
4

Saverio Santi -Scienze Chimiche- Università di Padova
5
Chimica Fisica Industriale Modula A

Saverio Santi -Scienze Chimiche- Università di Padova
6
Cinetica Chimica
Teoria della Cinetica Chimica
Fenomeni di studio
Relazioni struttura-reattività
Effetto solvente

Saverio Santi -Scienze Chimiche- Università di Padova
7
Obiettivi
Conoscenza delle principali tipologie di reazioni complesse in fase gas e in fase condensata.
Apprendimento delle teorie fondamentali della cinetica chimica e loro interpretazione meccanicistica, delle relazioni struttura-reattività e degli effetti del mezzo di reazione.
Il corso cercherà di fornire allo studente le conoscenze necessarie per eseguire in laboratorio le misure cinetiche di base e interpretare le informazioni disponibili in pubblicazioni scientifiche e monografie

Saverio Santi -Scienze Chimiche- Università di Padova
8
Testi consigliati
K. S. Laidler, "Chemical Kinetics", Mc Graw Hill;
K. A. Connors "Chemical Kinetics", VCH.
Dispense del docente Appunti di lezioni
Saverio Santi -Scienze Chimiche- Università di Padova
9
Programma del Corso

Saverio Santi -Scienze Chimiche- Università di Padova
10
8 ore Le teorie
Teorie della velocità assoluta.
Teoria delle collisioni.
Teoria di Lindemann.
Postulato di Hammond.
Teoria di Marcus (cenni).

Saverio Santi -Scienze Chimiche- Università di Padova
11
8 ore Meccanismo
Effetto isotopico cinetico. Relazioni struttura-reattività: effetto
sostituente. Effetti del mezzo: effetto solvente, effetto
sale. NMR Dinamico

Saverio Santi -Scienze Chimiche- Università di Padova
12
8 ore Fenomemi di studio
Reazioni a catena: esempi di reazioni a catena lineare, a catena ramificata. Reazioni di polimerizzazione.
Catalisi omogenea: reazioni in soluzione, catalisi omogenea, catalisi acido-base, catalisi enzimatica.
Catalisi eterogenea: adsorbimento fisico e chimico, reazioni di superficie unimolecolari, reazioni di superficie bimolecolari.

Saverio Santi -Scienze Chimiche- Università di Padova
13
24 ore Laboratorio
Cinetica di idrolisi di un alogenuro alchilico terziario. Effetto della temperatura.
Cinetica di idrolisi di un alogenuro alchilico terziario. Effetto del solvente.
NMR dinamico: cinetica di scambio della N,N-dimetilformammide.
Teoria degli errori.
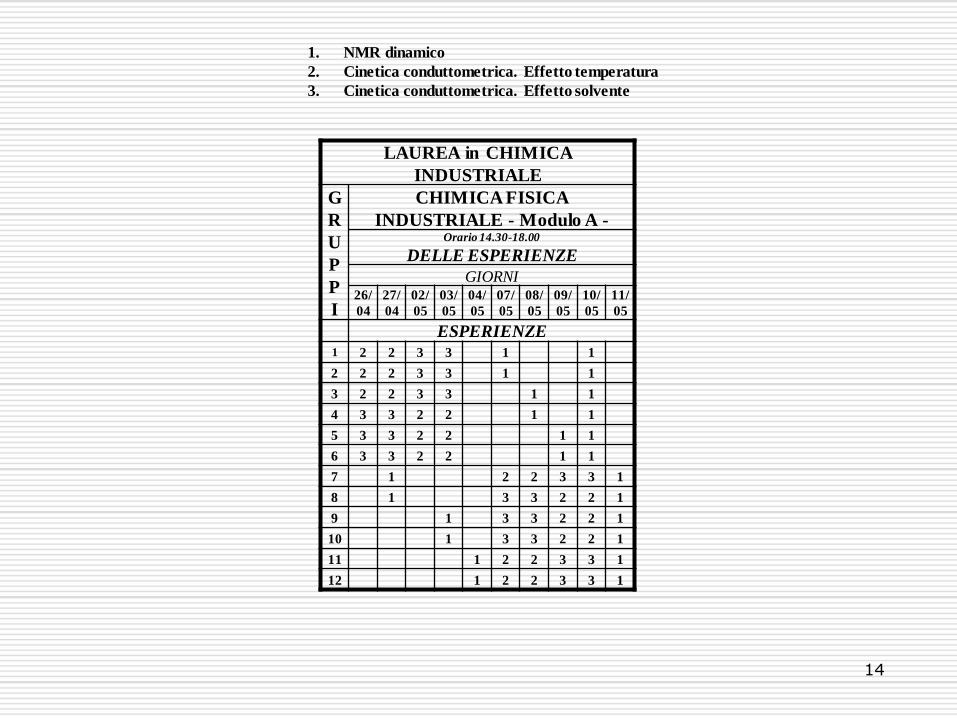
14
LAUREA in CHIMICA
INDUSTRIALE
G
R
U
P
P
I
CHIMICA FISICA
INDUSTRIALE - Modulo A - Orario 14.30-18.00
DELLE ESPERIENZE
GIORNI 26/
04
27/
04
02/
05
03/
05
04/
05
07/
05
08/
05
09/
05
10/
05
11/
05
ESPERIENZE 1 2 2 3 3 1 1
2 2 2 3 3 1 1
3 2 2 3 3 1 1
4 3 3 2 2 1 1
5 3 3 2 2 1 1
6 3 3 2 2 1 1
7 1 2 2 3 3 1
8 1 3 3 2 2 1
9 1 3 3 2 2 1
10 1 3 3 2 2 1
11 1 2 2 3 3 1
12 1 2 2 3 3 1
1. NMR dinamico
2. Cinetica conduttometrica. Effetto temperatura
3. Cinetica conduttometrica. Effetto solvente
Saverio Santi -Scienze Chimiche- Università di Padova
15
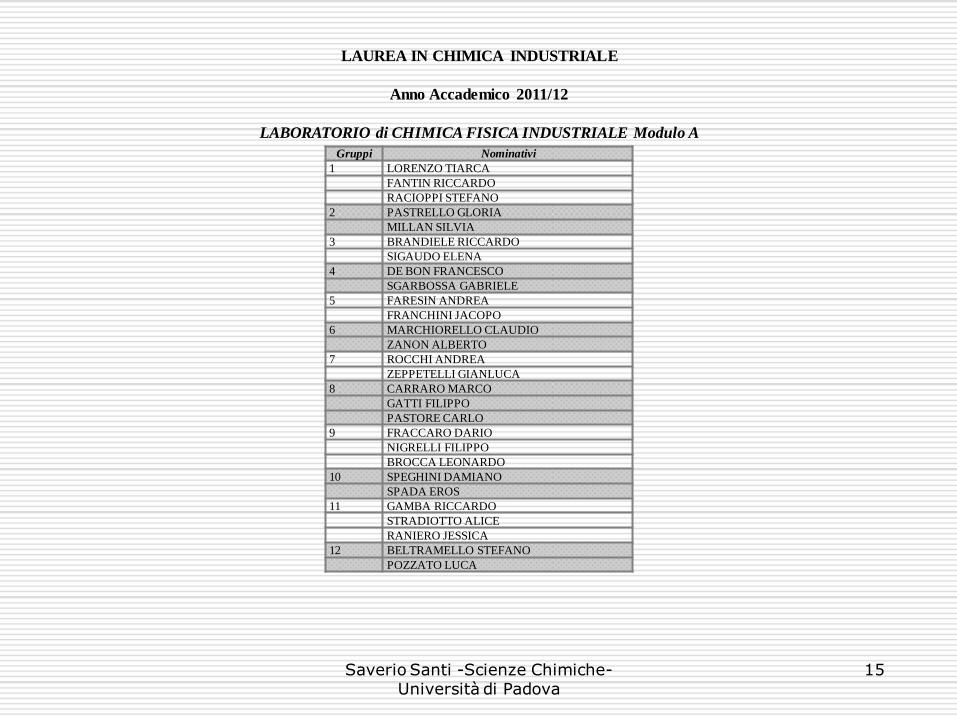
LAUREA IN CHIMICA INDUSTRIALE
Anno Accademico 2011/12
LABORATORIO di CHIMICA FISICA INDUSTRIALE Modulo A
Gruppi Nominativi
1 LORENZO TIARCA
FANTIN RICCARDO
RACIOPPI STEFANO
2 PASTRELLO GLORIA
MILLAN SILVIA
3 BRANDIELE RICCARDO
SIGAUDO ELENA
4 DE BON FRANCESCO
SGARBOSSA GABRIELE
5 FARESIN ANDREA
FRANCHINI JACOPO
6 MARCHIORELLO CLAUDIO
ZANON ALBERTO
7 ROCCHI ANDREA
ZEPPETELLI GIANLUCA
8 CARRARO MARCO
GATTI FILIPPO
PASTORE CARLO
9 FRACCARO DARIO
NIGRELLI FILIPPO
BROCCA LEONARDO
10 SPEGHINI DAMIANO
SPADA EROS
11 GAMBA RICCARDO
STRADIOTTO ALICE
RANIERO JESSICA
12 BELTRAMELLO STEFANO
POZZATO LUCA

Saverio Santi -Scienze Chimiche- Università di Padova
16
Legge Cinetica
v = k[A]a[B]b ….

Saverio Santi -Scienze Chimiche- Università di Padova
17
Effetto della temperatura

Saverio Santi -Scienze Chimiche- Università di Padova
18
Un aspetto importante della cinetica chimica riguarda l'influenza della
temperatura sulle velocità di reazione.
Le conclusioni che provengono da tali studi portano alla comprensione
degli aspetti molecolari delle reazioni chimiche.
Ci occuperemo principalmente degli effetti della temperatura sulle
reazioni elementari, che sono reazioni che avvengono in un solo stadio,
senza intermedi di reazioni identificabili.
Le reazioni elementari sono da contrapporre alle reazioni composite, che
avvengono in più stadi.
Effetto della temperatura

Molecolarità della reazione
Saverio Santi -Scienze Chimiche- Università di Padova
19
Dopo aver identificato come elementare un processo, è importante
chiedersi:
quante molecole entrano nella reazione?
Questo numero viene chiamato molecolarità della reazione.
Dalla variazione di velocità con la concentrazione possiamo di solito
determinare l'ordine di reazione.
Questo numero, puramente sperimentale, deve essere nettamente
distinto dalla molecolarità, che rappresenta una deduzione del numero
di molecole che prendono parte alla reazione.
È possibile parlare di ordine di una reazione complessa, purché la
velocità sia semplicemente proporzionale alle concentrazioni innalzate a
certe potenze.

Saverio Santi -Scienze Chimiche- Università di Padova
20
Non ha senso parlare di molecolarità se il meccanismo è di tipo composito.
Con alcune eccezioni, che vedremo in seguito, possiamo assumere che
l'ordine di una reazione elementare indichi il numero di molecole che
entrano nella reazione (ossia, che l'ordine e la molecolarità siano
uguali).
Per esempio, se una reazione elementare è di primo ordine rispetto al
reagente A e di primo ordine rispetto a un'altra sostanza B, si trova
spesso che la reazione è bimolecolare, con una molecola A e una
molecola B che entrano nella reazione.
Molecolarità della reazione
v = k1[A][B] ordine 2
A + B C + D

Saverio Santi -Scienze Chimiche- Università di Padova
21
Talvolta l'ordine cinetico non corrisponde alla molecolarità.
Si supponga, per esempio, che una reazione sia bimolecolare, ma che
un reagente sia presente in largo eccesso, in modo che la sua
concentrazione non vari apprezzabilmente mentre la reazione procede.
Inoltre (per esempio, se si tratta del solvente) la sua concentrazione può
essere la stessa in esperienze cinetiche diverse.
Se ciò si verifica, lo studio cinetico non rivelerà alcuna dipendenza della
velocità dalla concentrazione di questa sostanza.
Molecolarità della reazione

Saverio Santi -Scienze Chimiche- Università di Padova
22
Si trova frequentemente questa situazione in reazioni che avvengono in
soluzione dove il solvente può essere un reagente.
Per esempio nelle reazioni di idrolisi (in soluzione acquosa) una
molecola di acqua può reagire con una molecola di soluto.
A meno di non usare procedure speciali, che vedremo, i risultati cinetici
non rileveranno la partecipazione del solvente.
Tuttavia, se il solvente appare nell’equazione cinetica deve partecipare
alla reazione.
RX + H2O ROH + HX
v = k[RX][H2O] se [H2O] >> [RX]
v = kosservata[RX]
Molecolarità della reazione

Saverio Santi -Scienze Chimiche- Università di Padova
23
Un altro caso in cui lo studio cinetico non può rilevare se una sostanza
partecipa alla reazione si ha quando entra in gioco un catalizzatore.
Un catalizzatore è una sostanza che influenza la velocità di una reazione
senza essere consumata e può essere considerato sia un reagente che un
prodotto.
La sua concentrazione rimane costante nel tempo e l’analisi cinetica di
un singolo esperimento non ne evidenzia la partecipazione alla reazione.
La sua partecipazione può essere evidenziata misurando la velocità a
diverse concentrazioni di catalizzatore, e si trova in genere una
dipendenza lineare (ko è la costante di velocità della reazione non
catalizzata).
A P
v = k1[A][cat] v = kcat[A] + ko
cat
Molecolarità della reazione

Effetto della temperatura
Saverio Santi -Scienze Chimiche- Università di Padova
24
Sperimentalmente si trova che per un grande numero di reazioni, la
costante di velocità k è legata alla temperatura T dalla relazione
k = A e-B/T
dove A e B sono costanti. Questa relazione venne espressa da Arrhenius
e van't Hoff nella forma
k = A e-E/RT
dove R e la costante dei gas ed E è detta energia di attivazione.
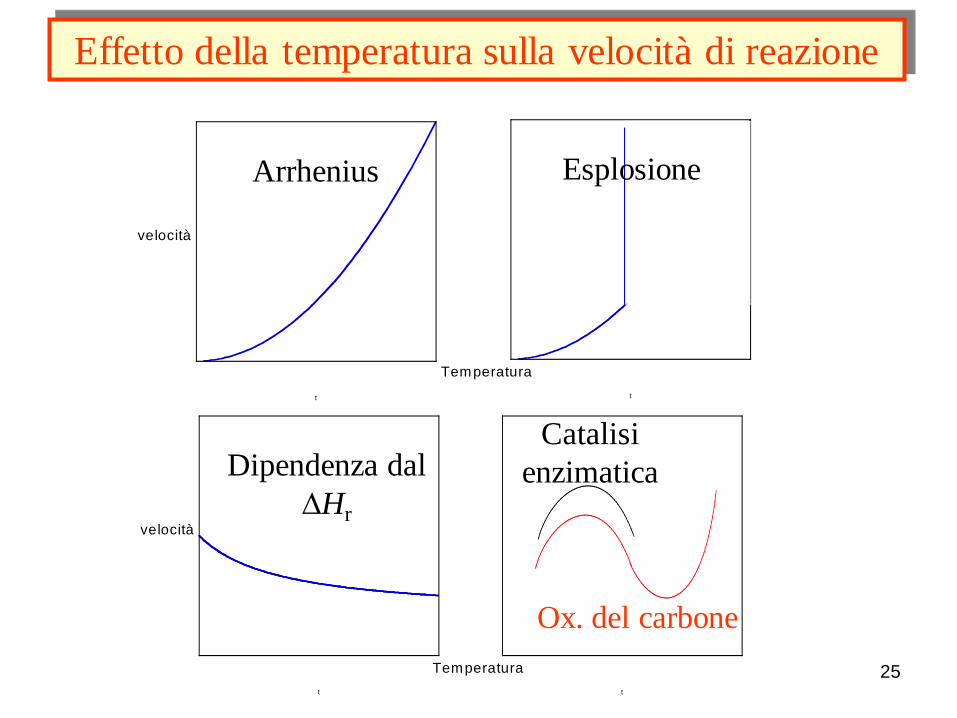
25
Effetto della temperatura sulla velocità di reazione
t
velocità
Arrhenius
t
Temperatura
Esplosione
t
velocità
Dipendenza dal
DHr
t
Temperatura
Ox. del carbone
Catalisi
enzimatica

Saverio Santi -Scienze Chimiche- Università di Padova
26
Aumento della velocità con T
k = A eDT k per T Berthelot 1862
k = A e-B/T kA per T; B = E0/R van’t Hoff 1884
k = A e-B/T kA per T; B = Ea/R Arrhenius 1889
k = ATm e-B/T dipendenza dal T del fattore pre-esponenziale Kooij 1893
k = ATm interpolazione migliore Harcourt-Esson 1895
)/TDT--(Bm 2
e AT k equazione a tre parametri van’t Hoff 1898
Effetto della temperatura

Effetto della temperatura
Saverio Santi -Scienze Chimiche- Università di Padova
27
A + B C + D k1
k -1
E 1
E -1
D U °
complesso attivato
reagenti
prodotti
van’t Hoff, sulla base della variazione della costante di equilibrio con la
temperatura, ipotizzò che una simile relazione dovesse valere anche per
la velocità di una reazione.

Saverio Santi -Scienze Chimiche-
Università di Padova
28
k1[A][B] = k-1[C][D]
1
1
]][[
]][[
k
kK
BA
DCc
2
ln
RT
U
T
K
P
c D
2
11
RT
U
dT
kd
dT
kd D
lnln
All’equilibrio la velocità della reazione di andata deve essere
uguale alla velocità della reazione di ritorno
Allora la costante di equilibrio Kc è uguale al rapporto delle
costanti di velocità k1 e k-1
La dipendenza della Kc, dalla temperatura è e data dalla nota
equazione di van’t Hoff

Saverio Santi -Scienze Chimiche-
Università di Padova
29
Quando si usa DU ?
Per gas ideali la costante di equilibrio riferita alle
concentrazioni è:
vRTKKcP
D )( TvRvKKcP
lnlnlnln DD
T
v
T
K
T
K
PP
cPD
lnln
2
ln
RT
H
T
K
P
PD
TRT
H
T
K
P
cD
D
2
ln
V
n
V
nV
n
BA
CK
BA
C
c ]][[
][
RT
Px
RT
PxRT
Px
nRT
Pn
nRT
PnnRT
Pn
KBA
C
BA
C
c
)()()(
RTpp
pRT
pp
pK
CA
Cv
CA
Cc
RP
A + B C
)(RTKK Pc 1 RTKK cP

Saverio Santi -Scienze Chimiche-
Università di Padova
30
Quando si usa DU ?
22
ln
RT
RTH
TRT
H
T
K
P
c DD
D
D
RTHU DDD
2
ln
RT
U
T
K
P
cD

Saverio Santi -Scienze Chimiche-
Università di Padova
31
Quando si usa DU ?
2
ln
RT
U
T
K
P
cD
Per reazioni in fase gas, se la costante di equilibrio è
espressa in funzione delle concentrazioni molari, si usa
l’espressione:
L’equazione è generalmente usata anche per le reazioni
in soluzione, ma in questo caso
DD HU

Saverio Santi -Scienze Chimiche-
Università di Padova
32
2
11
RT
U
dT
kd
dT
kd D
lnln
I due processi hanno fattori di energia diversi
IRT
E
dT
kd
2
11ln
IRT
E
dT
kd
2
11ln
con E1 - E-1 = DU°
Saverio Santi -Scienze Chimiche-
Università di Padova
33
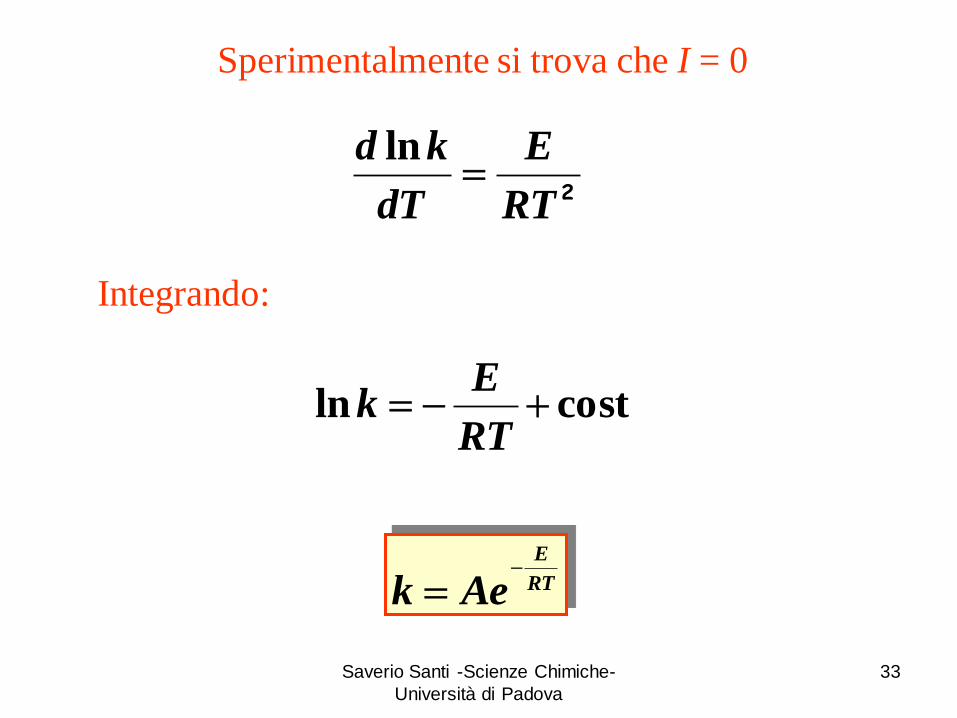
Sperimentalmente si trova che I = 0
2RT
E
dT
kd
ln
Integrando:
costln RT
Ek
RT
E
Aek

Saverio Santi -Scienze Chimiche-
Università di Padova
34
L’equazione di Arrhenius a un parametro termico è:
RT
Ea
Aek
con A, e E0 costanti indipendenti da T
L’equazione di van’t Hoff a due parametri termici è:
con A, m e E0 costanti indipendenti da T
RT
E
meTAk0
'

Saverio Santi -Scienze Chimiche-
Università di Padova
35
t
T
k
A
t
1/T
ln k
RT
E
Aek

Saverio Santi -Scienze Chimiche-
Università di Padova
36
RT
E
meTAk0
'
RT
ETmAk 0ln'lnln
differenziando:
2
0
2
0
RT
mRTE
RT
E
T
m
dT
kd
ln

Saverio Santi -Scienze Chimiche-
Università di Padova
37
2
0
2
0
RT
mRTE
RT
E
T
m
dT
kd
lnvan’t Hoff
2RT
E
dT
kda
lnArrhenius
mRTEEa
0

Saverio Santi -Scienze Chimiche-
Università di Padova
38
RT
EAk a lnln
RT
EmAk 0lnln
mRTEEa
0
RT
ETmAk 0ln'lnln
mTmAA ln'lnln
mmeTAA '
TmAmA ln'lnln

Saverio Santi -Scienze Chimiche-
Università di Padova
39
Se m non è molto grande o Ea molto piccola Ea = E0
Oscillatore armonico quantistico: En = (n + ½)ħ = (k/)
E a
E 0
coordinata di reazione
E
reagenti
complesso attivato

Saverio Santi -Scienze Chimiche-
Università di Padova
40
e
xxae
D
ka
MorsedipotenzialeeDxE e
2
1)(2)(
De
armonicopotenzialekxxE 2)(
xe
Saverio Santi -Scienze Chimiche-
Università di Padova
41
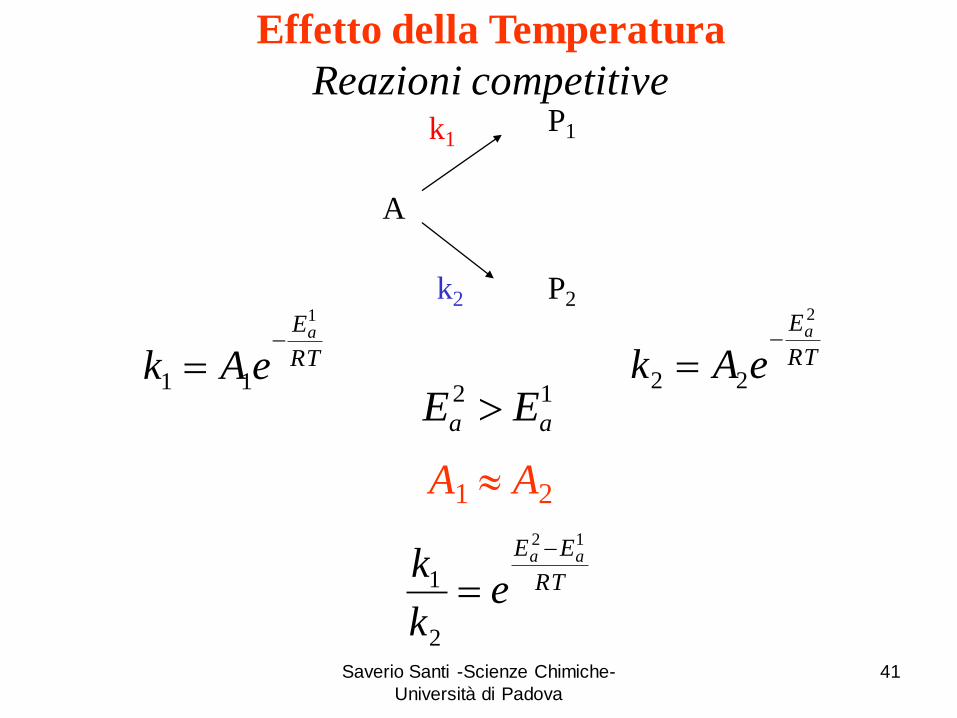
A
P1
P2
k1
k2
RT
Ea
eAk
1
11
RT
Ea
eAk
2
22
RT
EE aa
ek
k12
2
1
A1 A2
12
aa EE
Effetto della Temperatura
Reazioni competitive

Saverio Santi -Scienze Chimiche-
Università di Padova
42
RT
EE aa
ek
k12
2
1
012
RT
EEaa
Con l’aumento di T il rapporto
2
1
k
kdiminuisce, quindi k2 aumenta.
t
1/T
ln k
R
Ea
2
R
Ea
1

Saverio Santi -Scienze Chimiche-
Università di Padova
43
A B
1
1
RT
Ea
Aek
2
2
RT
Ea
Aek
1
1lnln
RT
EAk a
2
2lnln
RT
EAk a
21
12
211
211
lnTT
TT
R
E
TTR
E
k
kaa
Quanto aumenta la velocità con T ?

Saverio Santi -Scienze Chimiche-
Università di Padova
44
21
12
211
211
lnTT
TT
R
E
TTR
E
k
kaa
8.1293283
283293
98.1
10000exp
1
2
k
k
A T ambiente un aumento di 10 gradi provoca il
raddoppio della velocità
T1 = 283 K
T2 = 293 K
Ea = 10 kcal mol-1

Saverio Santi -Scienze Chimiche-
Università di Padova
45
21
12
211
211
lnTT
TT
R
E
TTR
E
k
kaa
03.112931283
12831293
98.1
10000exp
1
2
k
k
A T elevata (1000 K) un aumento di 10 gradi non ha
effetto sulla velocità.
T1 = 1283 K
T2 = 1293 K
Ea = 10 kcal mol-1
La velocità raddoppia se Ea è molto grande (200 kcal mol-1)

Saverio Santi -Scienze Chimiche-
Università di Padova
46
L’aumento di T ha effetto maggiore:
- a bassa temperatura
- a Ea elevata
Competizione tra due reazioni a differente Ea si osserva spesso
quando una stessa reazione avviene sia in fase omogenea che
eterogenea.
Ea (rx. omogenea) > Ea (rx. eterogenea)
t
1/T
ln k
)omogenea(a
E
)eterogenea(a
E
Saverio Santi -Scienze Chimiche-
Università di Padova
47
T
k
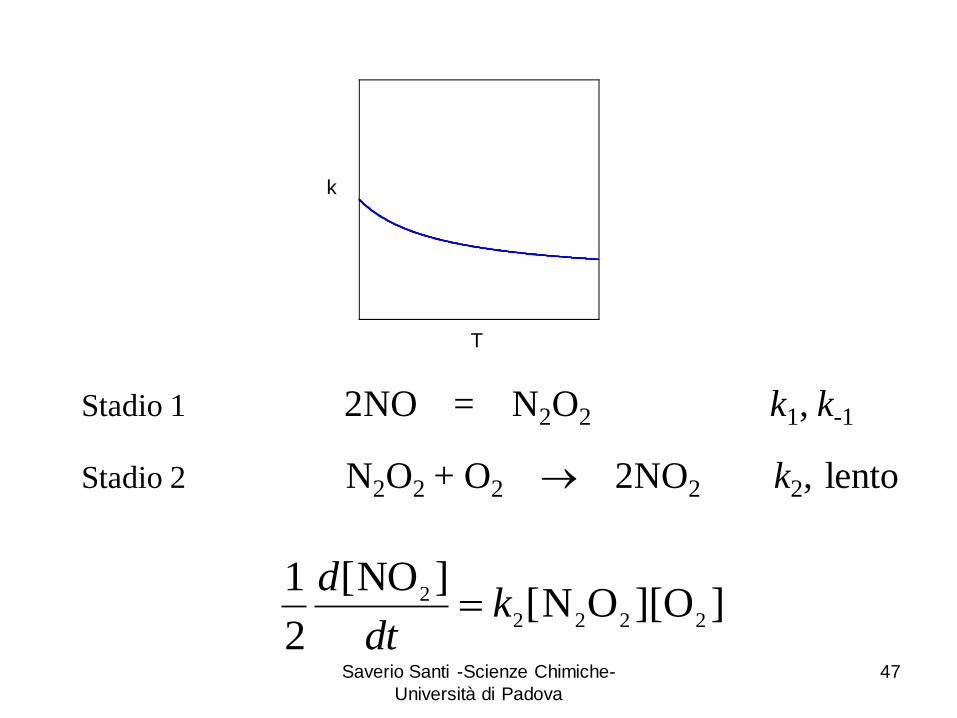
Stadio 1 2NO = N2O2 k1, k-1
Stadio 2 N2O2 + O2 2NO2 k2, lento
]O][ON[]NO[
2
12222
2 kdt
d

Saverio Santi -Scienze Chimiche-
Università di Padova
48
0]O][ON[]ON[NO][]ON[
2222221
2
1
22
kkkdt
d
]O[
NO][]ON[
221
2
1
22kk
k
]O[
]O[NO][]NO[
2
1
221
2
2
122
kk
kk
dt
d
]O][ON[]NO[
2
12222
2 kdt
d
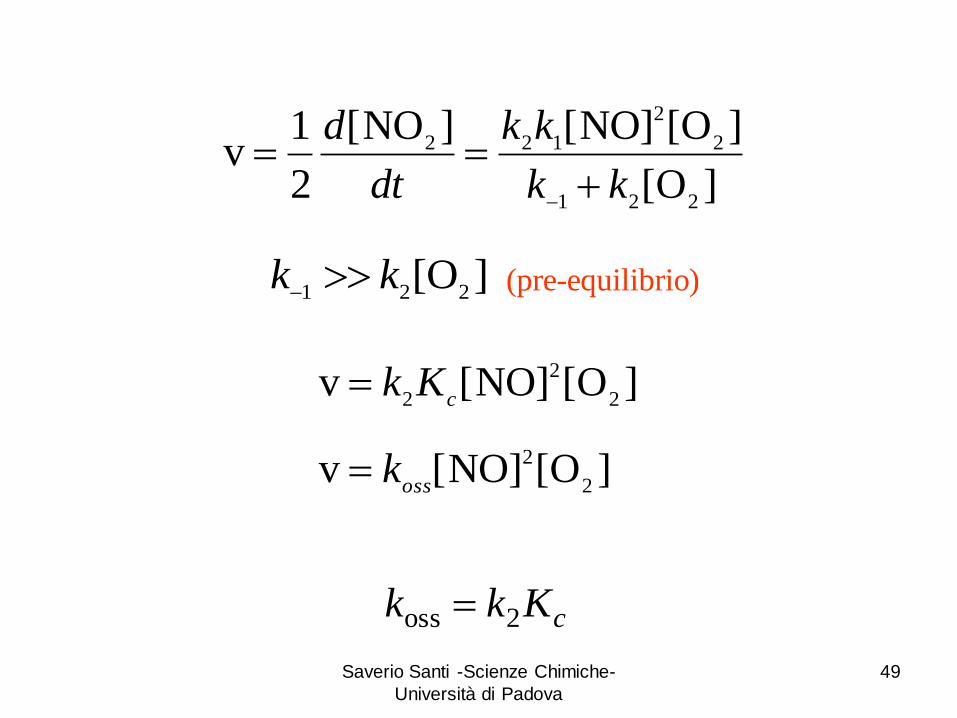
Saverio Santi -Scienze Chimiche-
Università di Padova
49
(pre-equilibrio) ]O[221
kk
]O[
]O[NO][]NO[
2
1v
221
2
2
122
kk
kk
dt
d
]O[NO][v2
2
2 cKk
cKkk 2oss
]O[NO][v2
2
ossk

Saverio Santi -Scienze Chimiche-
Università di Padova
50
cKkk 2oss
22
2ln
RT
U
RT
E
dT
kdaoss
D
dT
Kd
dT
kd
dT
kdc
lnlnln 2oss
2
ln
RT
U
T
K
P
c D
2
22ln
RT
E
dT
kda

Saverio Santi -Scienze Chimiche-
Università di Padova
51
22
2ln
RT
U
RT
E
dT
kdaoss
D
Se la reazione di equilibrio è esotermica:
DU° DH° < 0
2aEU D
0ln
dT
kdoss
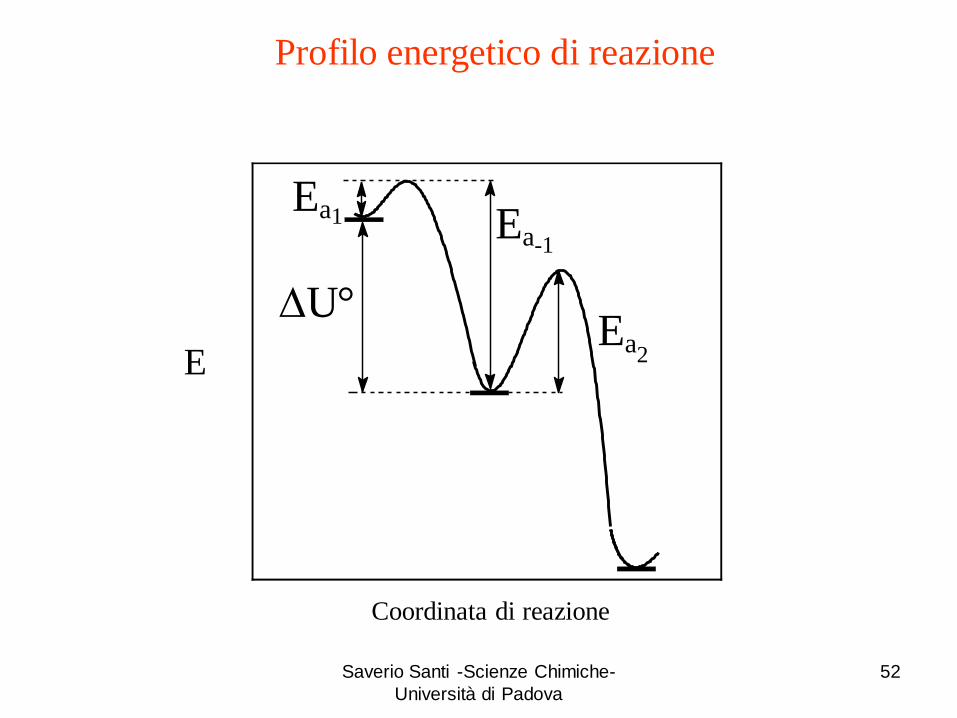
Saverio Santi -Scienze Chimiche-
Università di Padova
52
Coordinata di reazione
E
DU°
Ea1
Ea2
-1Ea
Profilo energetico di reazione

Saverio Santi -Scienze Chimiche-
Università di Padova
53
Legge di Distribuzione di Boltzmann
La dipendenza esponenziale della k da E/RT
è il punto cruciale di tutte le teorie cinetiche
RT
Ea
Aek
RT
E
meTAk0
'
dipendenza che si trova nella
Legge di Distribuzione di Boltzmann
T
ε
i
i
λen Bk

Saverio Santi -Scienze Chimiche-
Università di Padova
54
T
ε
i
i
λen Bk
ni numero di molecole nello stato i-esimo a energia ei
kB = R/L costante di Boltzmann
l attività assoluta T
μ
e Bk
kBT ln l potenziale chimico per molecola

55
Analoga espressione si ottiene considerando una
distribuzione Gaussiana della velocità di una molecola
di gas:
2
2
22/12 )2(
1)(
u
eup
velocità a una dimensione
u è la componente della velocità lungo l’asse x
p(u
)
+u 0 -u
Distribuzione Gaussiana della velocità

Saverio Santi -Scienze Chimiche-
Università di Padova
56
kTum2
1
2
1 2 e
Secondo la teoria cinetica dei gas l’energia cinetica media
è:
Poiché la velocità può essere positiva o negativa
allora
0u
m
kTu 22
kT
um
ekT
mup 2
2/1
2/12
)2()(
kT
x
ekT
mup
e
2/1
2/1
)2()(

Saverio Santi -Scienze Chimiche-
Università di Padova
57
duekT
mudup kT
xe
2/1
2/1
)2()()(
La probabilità che una molecola abbia una componente
della velocità compresa tra u e u + d(u) è :
p(u
)
u
e
ee
e
ee d
md
m
m
dum
uum2/12/1
2/1
2
2
12
2
1
2
12
2
1
ovvero l’area sottesa dalla curva:
0

Saverio Santi -Scienze Chimiche-
Università di Padova
58
e
e
e
demkT
mudup kT
x
2/12/1
2/1
2
1
)2()()(
)(
2
1)(
)2()()(
2/12/1
2/1
ee
e
dm
udduekT
mudup kT
x
ee
e
dekT
udup kT
x
2/1)(2
1)()(
p(u
)
+u 0 -u
xkT
x
x dekT
udupN
dN x
ee
e
2/1)(
1)()(2
frazione di molecole aventi energie
tra ex e ex + dex in una dimensione

Saverio Santi -Scienze Chimiche-
Università di Padova
59
yxkTkT
yx
ddeekTN
dN yx
eeee
ee
2/1)(
1
In due dimensioni la frazione di molecole aventi energie
tra ex + ey e ex + ey + d(ex + ey) è:
Ponendo e = ex + ey e integrando tra 0 e e si ottiene:
ee
dekTN
dNkT
)(
1
La frazione f* di molecole aventi energie maggiore di e* si
ottiene integrando tra e* e ∞ e fornisce l’interpretazione del
termine esponenziale dell’equazione di Arrhenius:
kTef*
*e

Saverio Santi -Scienze Chimiche-
Università di Padova
60
n*/l 0 1
e*/kBT
4
3
2
1
Il numero di molecole a energia e* aumenta con la T
T2 > T1
e*
e
ε
ε)(1
d
dN
N
T1
T2

Saverio Santi -Scienze Chimiche-
Università di Padova
61
T
ε
i
i
λen Bk
T
εε
en
nB
k
1
2
12
T
E
en
nR
1
2a
e* = e2 - e1
Ea = Le*
Ea1
2 Se e* è la differenza tra l’energia
dello stato attivato 2 (e2) e l’energia
dello stato fondamentale 1 (e1),
dalla legge di distribuzione di
Boltzmann si ricava il rapporto tra
il numero di molecole che hanno
energia e2 e il numero di molecole
che hanno energia e1:

Saverio Santi -Scienze Chimiche-
Università di Padova
62
Teoria delle collisioni
A + B P
Reazioni in fase gas bimolecolari
Calcola il numero di collisioni ZAB sulla base
della Meccanica Statistica (Maxwell):
Teoria cinetica dei gas
• A e B sono sfere rigide di diametro dA e dB
• la sezione d’urto non dipende dall’energia
(stati quantici) di A e B
• la velocità media di una molecola è
• una collisione dà P quando er e*
2/1
π
8
m
Tku B

Saverio Santi -Scienze Chimiche-
Università di Padova
63
B
B
BBA
BdB
AdAdAB
Sezione d’urto = dAB2
Si ha un urto quando il
parametro d’urto è p dAB
La velocità relativa media di una
molecola è ur
dAB=(dA + dB)/2

Saverio Santi -Scienze Chimiche-
Università di Padova
64
B
B
BBA
urt
Il numero medio di urti di una molecola di A (ZA )
che si muove con velocità media ur con le
molecole di B è dato dal volume del cilindro per il
numero di molecole di B per unità di volume NB
per la velocità stessa:
r2AB BA ud πNZ
Frequenza collisionale
1s
2ABd π
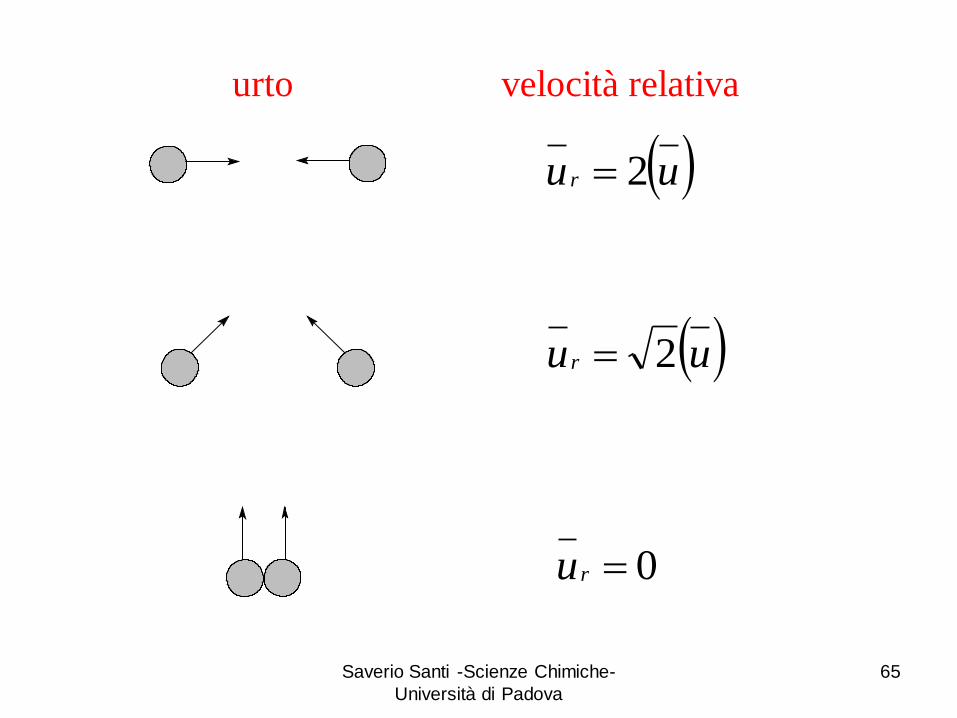
Saverio Santi -Scienze Chimiche-
Università di Padova
65
uur 2
uu r 2
0ru
urto velocità relativa

Saverio Santi -Scienze Chimiche-
Università di Padova
66
Per NA molecole di A il numero totale di collisioni è:
ruNNZ 2
AB BA ABd π
2/1
π
8
m
Tku B
2/12/1
π
)(8
π
8
π
8
BA
BAB
B
B
A
Br
mm
mmTk
m
Tk
m
Tku
2/1
πμ
8
Tku B
r
2/122
BAr uuu
2/1
2
AB BA ABμ
π8d
TkNNZ B
Densità Collisionale m-3 s-1

Saverio Santi -Scienze Chimiche-
Università di Padova
67
2A P
ruNZ 2
AA Ad π
uuuuu AAAr 22222
uNZ 2d π 2
AA A
2
AA
AA
ZNZ
2/1
2
A
2
AA
πd 2
A
B
Am
TkNZ m-3 s-1
2/1
π
8
m
Tku B
Il fattore 2 evita di commettere l’errore di
contare due volte un urto tra 2 molecole di A

Saverio Santi -Scienze Chimiche-
Università di Padova
68
Solo le molecole che possiedono energia e e*
danno luogo a collisioni che portano a prodotti:
T
E
en
nR
1
2a
e* = e2 - e1
Ea = Le*
Ea
1
2

Saverio Santi -Scienze Chimiche-
Università di Padova
69
2/1
2
A
2
AA
πd2
A
BA
m
TkNZ
La velocità di scomparsa di A per la reazione:
si ottiene moltiplicando ZAA per il fattore esponenziale
2A P
TR
E
AAA eZ
dt
dN
TR
E
A
B2AA
A em
Tkπd N
dt
dN
2/1
22

Saverio Santi -Scienze Chimiche-
Università di Padova
70
22 ][
][Ak
dt
Ad
TR
E
A
B2AA
A em
Tkπd N
dt
dN
2/1
22
L
NA
AL
Z
N
LZz AAA
A
AAAA ][
][ 22
T
A
B em
TkLk R
E2/1
2
A2
πd 2
Dividendo per otteniamo la costante di velocità in m3 s-1
Moltiplicando per L otteniamo la costante in unità molari m3 mol-1 s-1
2
AN
TAAezk R
E
2

Saverio Santi -Scienze Chimiche-
Università di Padova
71
22 ][
][Ak
dt
Ad
TR
E
A
B2AA
A em
Tkπd N
dt
dN
2/1
22
Dividendo per L otteniamo:
TR
E
A
B2AAA e
m
Tkπ
L
d N
dt
Ad
Ldt
dN
2/122][
TR
E
A
B2AA e
m
Tkπ
L
d NAk
2/122
2
2][
L
NAe
m
Tkπ
AL
d Nk ATR
E
A
B2AA
][][
22/1
2
2
2
T
A
B em
TkLk R
E2/1
2
A2
πd 2
Poiché si ottiene
da cui si ricava che , con
TAAezk R
E
2
Dimostrazione alternativa
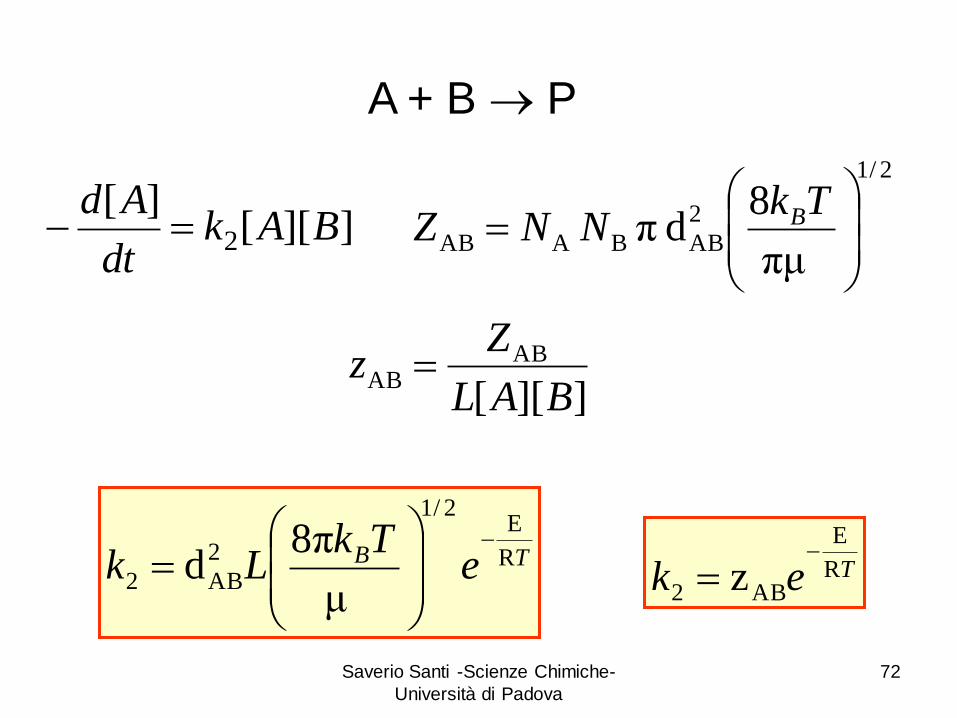
Saverio Santi -Scienze Chimiche-
Università di Padova
72
A + B P
2/1
2
AB BA ABπμ
8d π
TkNNZ B]][[
][2 BAk
dt
Ad
]][[
ABAB
BAL
Zz
TB eTk
Lk R
E2/1
2
AB2μ
π8d
Tek R
E
AB2 z

Saverio Santi -Scienze Chimiche-
Università di Padova
73
TR
E
ABezk
2TR
E
meTAk
'
2/1
2/1
2
ABμ
π8d T
kLz B
AB
2/1'TAA
2/1
2
ABμ
π8d'
Bk
LA

Saverio Santi -Scienze Chimiche-
Università di Padova
74
2/1
2
AB
3
μ
π8d10
Tk
LA Blitri mol-1 s-1
Per le razioni in fase gas tra reagenti semplici l’accordo è molto
buono e A è dell’ordine di 7 * 1010 - 7 * 1011 litri mol-1 s-1
La stima del valore di A ponendo dab = 5 Å, μ = 2 * 10-22 g, T = 300 K
è 4 * 1011 litri mol-1 s-1.
calcolato
osservato
A
AP
I risultati in soluzione sono controversi e si possono trovare valori di
P ≈ 1, << 1, ma anche >> 1.

Saverio Santi -Scienze Chimiche-
Università di Padova
75
CONFRONTO TEORIA-ESPERIMENTO:
Reazioni Elementari in Fase Gas
Reazioni Unimolecolari
Reazioni Bimolecolari
Reazioni Trimolecolari
Reazioni elementari in fase gas non sono comuni: spesso il meccanismo è complesso.
Un analisi attenta permette in alcuni casi di ottenere dati accurati su un singolo stadio elementare.
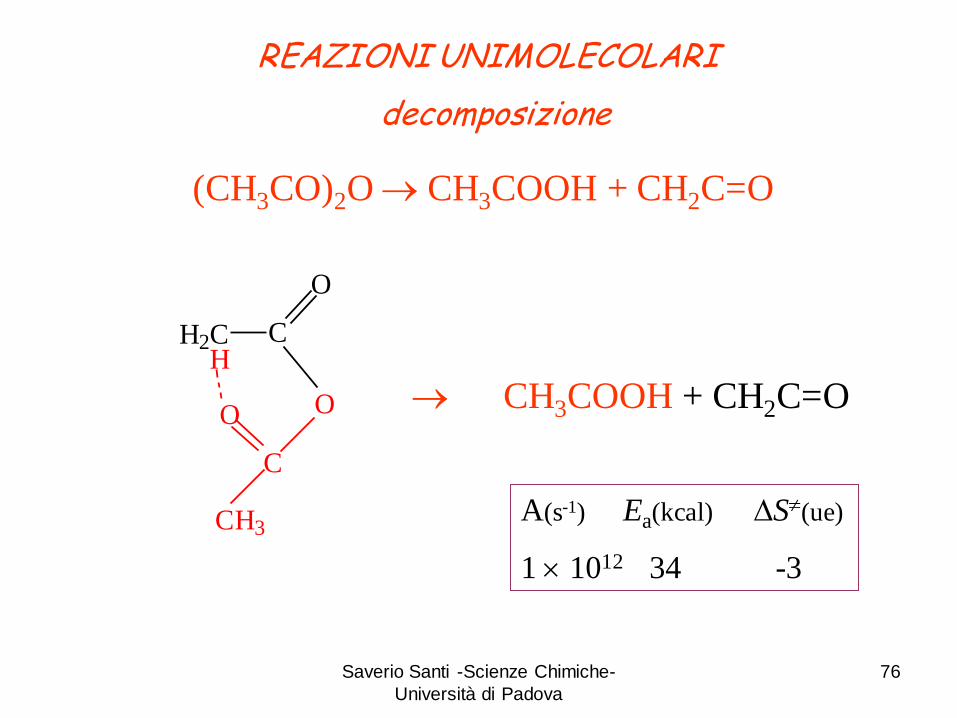
Saverio Santi -Scienze Chimiche-
Università di Padova
76
REAZIONI UNIMOLECOLARI
decomposizione
(CH3CO)2O CH3COOH + CH2C=O
H2C C
O
H
OO
C
CH3
CH3COOH + CH2C=O
A(s-1) Ea(kcal) DS(ue)
1 1012 34 -3

Saverio Santi -Scienze Chimiche-
Università di Padova
77
REAZIONI UNIMOLECOLARI
isomerizzazione
cis-stilbene trans-stilbene 6 1012 43 -1
estere dimetilmaleico estere dimetilfumarico 7 105 27 -32
ciclopropano propene 1.5 1015 65 11
A(s-1) Ea(kcal) DS(ue)

Saverio Santi -Scienze Chimiche-
Università di Padova
78
A P
Collisione con le pareti?
Radiazione esterna ?
Collisione tra molecole ?

Saverio Santi -Scienze Chimiche-
Università di Padova
79
Ipotesi di Lindemann-Christiansen
A + A A + A* k1
k -1
k2
A* P

Saverio Santi -Scienze Chimiche-
Università di Padova
80
2
11][Akv velocità di “energizzazione”
*]][[11
AAkv
velocità di “de-energizzazione”
*][22
Akv velocità di conversione

Saverio Santi -Scienze Chimiche-
Università di Padova
81
*][2 Akv
a pressione di A sufficientemente elevata
*]][[][1
2
1AAkAk
211vvv
][*][1
1 Ak
kA
pre-equilibrio
A + A A + A* k1
k -1
k2
A* P

Saverio Santi -Scienze Chimiche-
Università di Padova
82
*][2
Akv
][*][1
1 Ak
kA
][1
1
2A
k
kkv
a pressione di A sufficientemente elevata
la reazione ha una cinetica del I ordine

Saverio Santi -Scienze Chimiche-
Università di Padova
83
a pressione di A molto bassa
12 vv 2
1][Akv
a pressione di A sufficientemente bassa la
reazione ha una cinetica del II ordine
A + A A + A* k1
k -1
k2
A* P

Saverio Santi -Scienze Chimiche-
Università di Padova
84
A + A A + A* k1
k -1
k2
A* P
Ipotesi dello stato stazionario (Hinshelwood)
0*][
dt
Ad
0*][*]][[][*][
21
2
1
AkAAkAk
dt
Ad

Saverio Santi -Scienze Chimiche-
Università di Padova
85
0*][*]][[][21
2
1
AkAAkAk
0)][*]([][21
2
1
kAkAAk
21
2
1
][
][*][
kAk
AkA
*][2
Akv
21
2
1
2][
][
kAk
Akkv

Saverio Santi -Scienze Chimiche-
Università di Padova
86
21
2
1
2][
][
kAk
Akkv
1
1
2
][
k
Akkv
[A] sufficientemente elevata
21][ kAk
2
1][Akv
[A] sufficientemente bassa
][12
Akk

Saverio Santi -Scienze Chimiche-
Università di Padova
87
21
2
1
2][
][
kAk
Akkv
21
1
2][
]['
kAk
Akkk
][
1
'
1
121
1
Akkk
k
k
1/k'
1/[A]
1/k1
k-1
/k1k
2
k'
[A]
1
21
k
kk
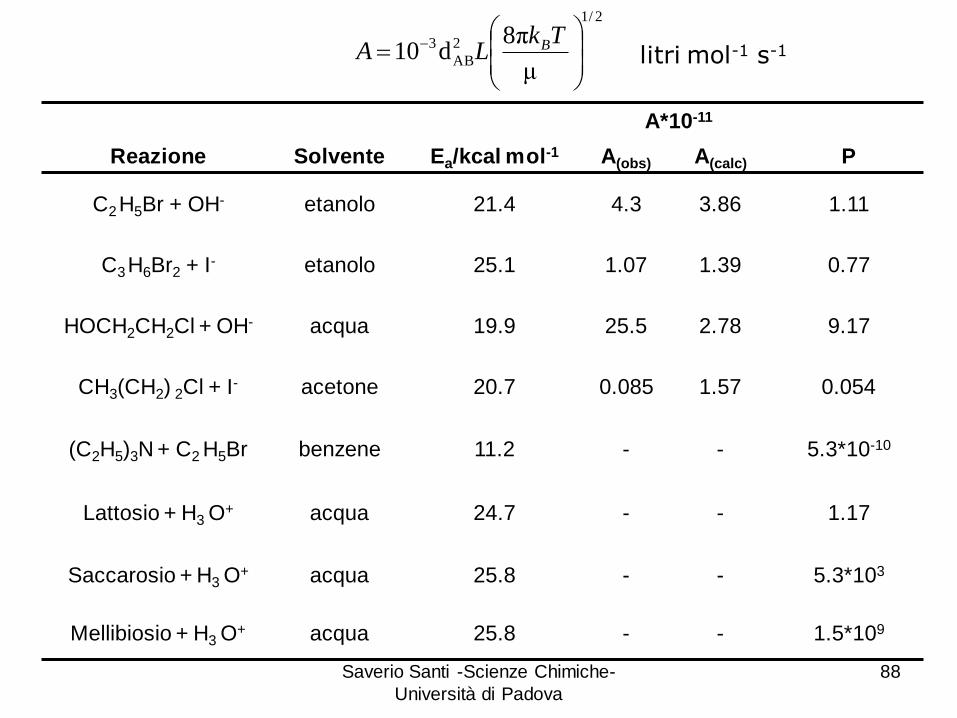
Saverio Santi -Scienze Chimiche-
Università di Padova
88
A*10-11
Reazione Solvente Ea/kcal mol-1 A(obs) A(calc) P
C2 H5Br + OH- etanolo 21.4 4.3 3.86 1.11
C3 H6Br2 + I- etanolo 25.1 1.07 1.39 0.77
HOCH2CH2Cl + OH- acqua 19.9 25.5 2.78 9.17
CH3(CH2) 2Cl + I- acetone 20.7 0.085 1.57 0.054
(C2H5)3N + C2 H5Br benzene 11.2 - - 5.3*10-10
Lattosio + H3 O+ acqua 24.7 - - 1.17
Saccarosio + H3 O+ acqua 25.8 - - 5.3*103
Mellibiosio + H3 O+ acqua 25.8 - - 1.5*109
2/1
2
AB
3
μ
π8d10
Tk
LA Blitri mol-1 s-1

Saverio Santi -Scienze Chimiche-
Università di Padova
89
Molti fattori contribuiscono alla deviazione dalla teoria:
1. Le molecole non sono sferiche: la teoria non tiene conto le
richieste geometriche della reazione
+
+
prodotti
prodotti
+ prodotti
Forti impedimenti sterici portano a valori di P << 1
+ prodotti

Saverio Santi -Scienze Chimiche-
Università di Padova
90
2. La collisione può non provocare la liberazione di energia
cinetica al momento giusto e nel luogo giusto.
3. La presenza di contributi entropici molto favorevoli può
provocare valori di P >> 1.
4. Il calcolo per le reazioni in soluzione è complicato dalla
presenza del solvente che può partecipare alla formazione
dello stato di transizione.
5. La teoria delle collisioni non fornisce una accurata
interpretazione della barriera di energia né un metodo per
calcolare l’energia di attivazione o ipotizzare la struttura
dello stato di transizione.
Tuttavia la teoria delle collisioni ha il merito di definire un
modello per la comprensione del comportamento cinetico di
reazioni “tipiche” o “normali”

Saverio Santi -Scienze Chimiche-
Università di Padova
91
Dalla teoria delle collisione risulta che il fattore pre-
esponenziale dipende da T:
2/1'TAA
Sperimentalmente non è apprezzabile poiché si determina
per estrapolazione a 01
T
T
1
kln
Aln
e l’incertezza è in genere elevata.

Saverio Santi -Scienze Chimiche-
Università di Padova
92
La teoria delle collisioni considera efficace ogni collisione. Tuttavia,
se le molecole sono complesse, solo in una piccola frazione delle
collisioni (per P << 1) le molecole si avvicineranno nel corretto modo
perché la reazione avvenga.
Per mantenere valida la teoria è stata proposta l’introduzione nel
fattore pre-esponenziale di un fattore sterico P (o fattore di
probabilità) che rappresenta la frazione delle collisioni efficaci:
TABePzk R
Ea
2
Il calcolo di P non può essere del tutto soddisfacente ed è
inconsistente con il fatto che all’equilibrio il rapporto tra le costanti
cinetiche delle reazioni opposte è una costante di equilibrio e deve
contenere un termine entropico.
E’ necessario un approccio alternativo al problema.

Saverio Santi -Scienze Chimiche-
Università di Padova
93
Consideriamo la reazione bimolecolare:
A2 + B2 = 2AB
T
E
ezPk R111
1
T
E
ezPk R111
-1
T
EE
P ezP
zP
k
kK R
11
11
1
11-1
Sappiamo dalla termodinamica che
T
HS
T
G
P eeek
kK RRR
1
1
D
DD
da cui risulta che e
R
11
11
S
ezP
zPD
T
H
T
EE
ee RR
1-1D

L’orientazione delle molecole e gli altri effetti non possono essere
introdotti in modo soddisfacente modificando la teoria delle
collisioni tra sfere rigide. Non è sufficiente collegare i fattori di
probabilità con la probabilità che i reagenti collidano. Devono
essere interpretati in termini di entropia di attivazione.
RT
Gklnkln
D 11
lnRT
Gkln
D
1
1 lnRT
Gkln
D
2
1
D
D
T
HS
ekRR
1
11
Teoria della velocità assoluta
Una trattazione molto più soddisfacente venne proposta da
Henry Eyring nel 1935 (e indipendentemente da M. G. Evans e
M. Polanyi). Il punto di partenza è la teoria di van’t Hoff:
La Teoria della Velocità Assoluta ha definito in modo rigoroso.

Eyring, Evans e Polanyi, svilupparono teorie
quantitative della velocità di reazione basate
sull’idea del ruolo critico dello stato di
transizione nel controllo della velocità.
Nel passaggio dallo stato iniziale allo stato
finale il sistema reagente deve attraversare
una regione del cammino di reazione, detta
stato di transizione, la cui energia potenziale
è la più alta del cammino.
Teoria della Velocità Assoluta
(o dello Stato di Transizione)

La Teoria della Velocità Assoluta si basa su alcuni postulati:
• I sistemi molecolari che raggiungono il passo non tornano
più indietro (se provengono dai reagenti diventano prodotti
e viceversa).
• Si assume la legge di Maxwell-Boltzmann per la
distribuzione dell’energia delle molecole reagenti e la
concentrazione del complesso attivato può essere sempre
calcolata rispetto ai reagenti con la teoria dell’equilibrio
chimico.
• Si può separare il moto del sistema per il superamento del
passo dagli altri moti associati al complesso attivato.
• La velocità di reazione è uguale al prodotto delle
concentrazioni delle specie dello stato di transizione e della
frequenza con la quale queste specie diventano prodotti.

Queste assunzioni si riferiscono ad una reazione elementare che
da questo punto di vista può essere definita come una reazione
con unico stato di transizione:
A + B Y + Z
v = [X≠ ]
La meccanica statistica ha permesso la derivazione della teoria
A + B
X≠
Y + Z
reazionedicoordinata
pE

A + B Y + Z
A + B XS≠ + XD
≠ Y + Z
Consideriamo la reazione all’equilibrio:
Esistono due tipi di complessi attivati, quelli che nell’immediato
passato erano reagenti, XS≠, e quelli che nell’immediato passato
erano prodotti, XD≠.
Tuttavia, Eyring fa l’assunzione che anche quando il sistema è
lontano dall’equilibrio la concentrazione di XS≠ rimane invariata
poiché la loro formazione non può essere influenzata dalla
presenza o meno di molecole di prodotto.
A + B XS≠ Y + Z
Immaginiamo ora di rimuovere in qualche modo i prodotti dal
sistema: i complessi di tipo XD≠ non ci saranno più.

A + B X≠ Y + Z
La velocità di flusso delle molecole di X≠ provenienti dai
reagenti è la stessa all’equilibrio e lontana dall’equilibrio.
KBA
X
]][[
][
Si tratta di un “quasi-equilibrio” nel senso che non vale il
Principio di Le Chatelier e ci si riferisce solo alle molecole di X≠ provenienti dai reagenti.
La termodinamica statistica permette di calcolare le
concentrazione delle specie che partecipano ad un equilibrio
utilizzando le funzioni di partizione.

Saverio Santi -Scienze Chimiche-
Università di Padova
100
T
i
i
en Bk
ε
λ
Le funzioni di partizione
Distribuzione di Boltzmann
La frazione di molecole rispetto al numero totale N aventi energia εi è
i
T
Tj
ji
j
e
e
N
nf
B
B
k
ε
k
ε
fi rappresenta la probabilità che una molecola si trovi nello stato a
energia εj.
Il denominatore di questa espressione ha un significato speciale, si
indica con il simbolo q se riferito ad una singola molecola ed è
denominato funzione di partizione molecolare, o Q se riferito ad
un sistema di molecole ed è denominato funzione di partizione
canonica.
In tedesco è detta Zustandsumme, somma degli stati.
Infatti, rappresenta la somma di tutti gli stati energetici.
i
T
i
eQ Bk
ε

Saverio Santi -Scienze Chimiche-
Università di Padova
101
Una molecola possiede diversi tipi di energia, traslazionale,
rotazionale, vibrazionale ed elettronica, :
εT = εt + εr + εv + εe εt < εr < εv < εe
I contributi sono indipendenti e la funzione di partizione è
fattorizzabile:
Consideriamo due stati energetici εr e εv, ciascuno a due livelli,
abbiamo quattro stati:
εv1 + εr1, εv1 + εr2, εv2 + εr1, εv2 + εr2,
TTTT
rvrrvrv
eeeeq B
22
B
1
B
21
B
11
k
εε
k
εε
k
εε
k
εε 2

TT
r
TT
v
rrvv
eeqeeq B
2
B
1
B
2
B
1
k
ε
k
ε
k
ε
k
ε
Definiamo qv e qr funzioni di partizione degli stati vibrazionali
e rotazionali
rvqqq
TTTT
rrvv
eeeeq B
2
B
1
B
2
B
1
k
ε
k
ε
k
ε
k
ε
Raccogliendo:
In generale:
trve qqqqq
La meccanica statistica permette di ottenere le espressioni per le
differenti funzioni di partizione e ne consente il calcolo rigoroso.
qt > qr > qv > qe

Saverio Santi -Scienze Chimiche-
Università di Padova
103
Energia elettronica
La separazione dei livelli elettronici è elevata, con ε0 = 0. Il
primo termine è uguale a 1, già il secondo sarà molto piccolo.
TTTTeeeeq B
3
B
2
B
1
B
0
k
ε
k
ε
k
ε
k
ε
1eq
La funzione di partizione per ogni tipo di energia è data la somma
delle funzioni di partizione dell’energia corrispondenti a ciascun
livello a partire da ε0 che è assunto come zero:
ε0
ε1
ε2
ε3
ε4
ε5

Per una gas ideale in una dimensione di lunghezza l si ha:
l
h
Tmkq B
t
2/12
Poiché ci sono tre gradi di libertà traslazionali lungo le tre
coordinate si ha:
33231
3
2/3
10102 m
V
qV
h
Tmkq t
t
B
Energia traslazionale

Saverio Santi -Scienze Chimiche-
Università di Padova
105
2
2
2
10108
h
TIkq B
r
Per molecole lineari si hanno due gradi di libertà rotazionali:
Per molecole non lineari si hanno tre gradi di libertà rotazionali :
Energia rotazionale
Per una libera rotazione interna (un grado di libertà):
101
82/12
h
TIkq B
r
32
3
2/32/132
101088
h
TkIIIq BCBA
r
è il numero di simmetria: assicura che configurazioni identiche
non siano contate più di una volta
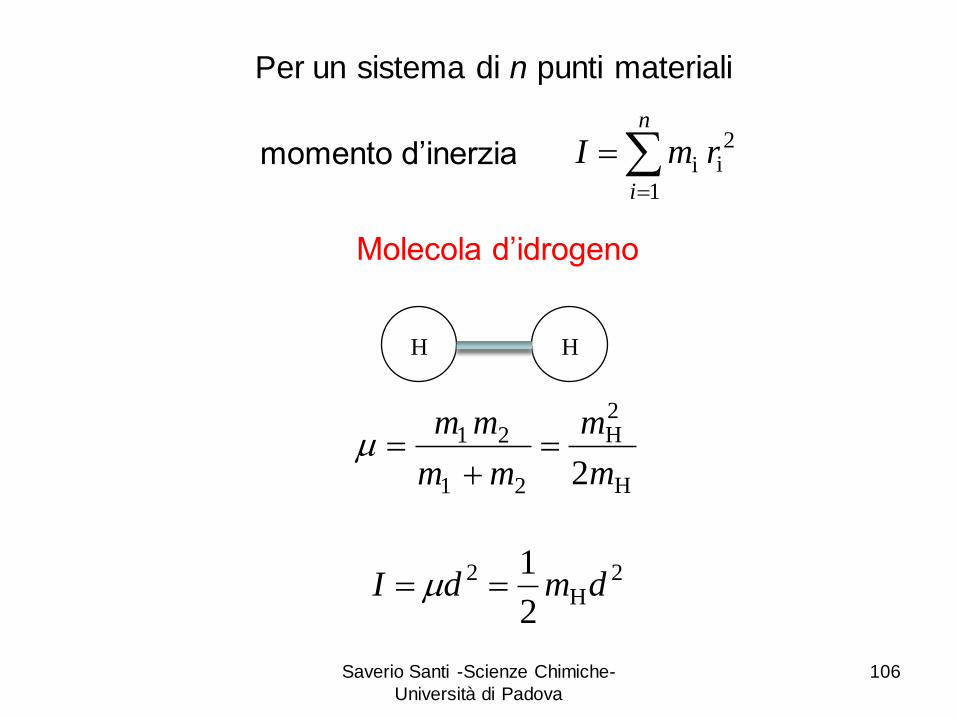
Saverio Santi -Scienze Chimiche-
Università di Padova
106
2H
2
2
1dmdI
Molecola d’idrogeno
n
i
rmI1
2ii
H H
H
2H
21
21
2m
m
mm
mm
Per un sistema di n punti materiali
momento d’inerzia

Saverio Santi -Scienze Chimiche-
Università di Padova
107
Il numero di simmetria
H Cl 1
Per molecole lineari non simmetriche = 1:
H1 H2 H1 H2 2
Si ottiene contando il numero di possibili orientazioni equivalenti
della molecola ottenute per rotazione ma non per riflessione:
O1 C 2 O2 O2 C O1
H1
O 2 H2 H2
O H1
H1
N H3
H2
H1
N H2
H3
H2
N H1
H3
3
H D
per molecole lineari simmetriche = 2:

Gradi di libertà molecolari
Saverio Santi -Scienze Chimiche-
Università di Padova
108
Per una molecola costituita da N atomi 3 N gradi di libertà
3 traslazionali
2 rotazionali in molecole lineari
3 rotazionali in molecole non lineari

Molecole lineari
Saverio Santi -Scienze Chimiche-
Università di Padova
109
Due gradi di libertà rotazionali (a) descrivono la rotazione della molecola attorno
al suo centro di massa.
La rotazione attorno all’asse molecolare (b) non cambia la posizione della
molecola.

Saverio Santi -Scienze Chimiche-
Università di Padova
110
Il numero totale di gradi di libertà è 3N (N numero di atomi).
Sottraendo i gradi di libertà traslazionale (3) e rotazionali (2 o 3)
si ottiene il numero di gradi di libertà vibrazionali di una
molecola:
3N – 6 gradi di libertà per una molecola non lineare;
3N – 5 gradi di libertà per una molecola lineare
Per una molecola biatomica (N = 2) c’è un solo grado di libertà
vibrazionale la cui funzione di partizione è
101
1
1
kT
hv
e
q

Saverio Santi -Scienze Chimiche-
Università di Padova
111
trv qqqq
La funzione di partizione molecolare è data dalla relazione:
Ogni energia può essere rappresentata come la differenza tra
l’energia del livello iesimo e l’energia del livello zero ε0. Ponendo
εi = (εi + ε0)- ε0 si ottiene: )εε(εε 00
ii eee
La funzione di partizione può essere riscritta come:
i
kT
εε
kT
ε i
eeq
)( 00
'0
qeq kT
ε

Saverio Santi -Scienze Chimiche-
Università di Padova
112
A + B X≠ Y + Z
X≠ ≡ A B≠
La costante di equilibrio classica per la reazione:
A + B Z
Vnn
n
CC
CK
BA
Z
BA
Z
Il numero di molecole n di ogni reagente e prodotto contenuto
nel volume V può essere espresso in funzione della
distribuzione di Boltzmann:

T
i
i
en Bk
ε
λ
distribuzione di Boltzmann
i
T
i
eQ Bk
ε
funzione di partizione di n molecole nel volume V
A
T
A Qeλn Bk
ε
A
A
0
Infatti
B
T
B Qeλn Bk
ε
B
B0
Z
T
Z Qeλn Bk
ε
Z
Z
0
BAZ
0
k
ε
000
0
εεεεconB D
D
T
BA
Z
BA
Z eQQ
VQK
ll
l
Sostituendo nella costante di equilibrio:
'0ε
QeQTkB

Saverio Santi -Scienze Chimiche-
Università di Padova
114
l attività assoluta T
e Bk
μ
kBT ln l potenziale chimico per molecole
Ricordando che:
e che all’equilibrio d =0, nel nostro caso Z = A + B ,
otteniamo lZ = lAlB
T
BA
Z eQQ
VQK Bk
ε0D

Saverio Santi -Scienze Chimiche-
Università di Padova
115
Abbiamo visto che la funzione di partizione per l’energia
traslazionale in tre dimensioni di un gas di volume V è:
V
h
Tmkq B
t
3
2/32
T
BA
Z eQQ
QK Bk
ε0D
Quindi nel calcolo della costante di equilibrio il termine V si
semplifica T
BA
Z eQQ
QK Bk
ε0D
In quantità molari (E0 = LDe0 e kBL = R)
T
E
BA
Z eQQ
QK R
0

Saverio Santi -Scienze Chimiche-
Università di Padova
116
Con lo stesso procedimento otteniamo la costante di quasi-
equilibrio
T
E
BA
eQQ
QK R
0
A + B X≠
Se A e B contengono NA e NB atomi lo stato attivato X≠ contiene
3(NA e NB ) – 6 gradi di libertà vibrazionali se la molecola non
è lineare, 3(NA e NB ) – 5 se è lineare.
Una di queste ha caratteristiche diverse dalle altre, è una
vibrazione a bassa frequenza priva di forza di richiamo che
trasforma X≠ in prodotti.

Saverio Santi -Scienze Chimiche-
Università di Padova
117
Se X≠ è una molecola biatomica (N = 2, 3N – 5 gradi di libertà)
questo modo normale di vibrazione è l’unico possibile e tende a 0
per dare i prodotti:
h
Tk
Tk
he
B
B
Tk
h
B
11
1
1
1lim
0
Applicando uno dei postulati della teoria dello stato di transizione:
“Si può separare il moto del sistema per il superamento del
passo dagli altri moti associati al complesso attivato"
T
E
BA
B eQQ
Q
h
TkK R
0
h
TkQQ B

Saverio Santi -Scienze Chimiche-
Università di Padova
118
Se X≠ è una molecola triatomica lineare (N = 3, 3N – 5 gradi di
libertà) ci sono quattro modi normali di vibrazioni:
A + BC X≠ AB + C
X≠ ≡ [A B C]≠
Due bending
Due stiramenti
+ + -

Saverio Santi -Scienze Chimiche-
Università di Padova
119
Superfici di energia potenziale
energia potenziale
A-B B-C
X≠

Saverio Santi -Scienze Chimiche-
Università di Padova
120
T
E
BA
B eQQ
Q
h
Tk
BA
XR
0
]][[
][
Uno dei postulati della teoria dello stato di transizione è:
“La velocità di reazione è uguale al prodotto delle
concentrazioni delle specie dello stato di transizione e della
frequenza con la quale queste specie diventano prodotti"
T
E
BA
B eQQ
Q
h
TkBAX R
0
]][[][
][ Xv
T
E
BA
B eQQ
Q
h
TkBA R
0
]][[
v ]][[ BAkv T
E
BA
B eQQ
Q
h
Tkk R
0
Kh
Tkk B

Saverio Santi -Scienze Chimiche-
Università di Padova
121
Derivazione alternativa
Il moto che permette il passaggio del passo non è un moto
vibrazionale ma un moto traslazionale.
A + B
Y + Z
reazionedicoordinata
pEd
Tutti i complessi che transitano nel tratto del passo di lunghezza
arbitraria d sono complessi attivati.

Saverio Santi -Scienze Chimiche-
Università di Padova
122
La funzione di partizione traslazione speciale che porta a prodotti
per la particella di massa m≠ in un sistema in una dimensione di
lunghezza d:
d
h
kTmqt
2/12
Perciò la funzione di partizione che descrive lo stato di transizione
X≠ ricordando che Q≠ è la funzione di partizione di tutti i moti
eccetto quello del superamento del passo) è:
Qh
kTmQ d
2/1
2
e deve essere introdotta nell’equazione:
T
E
BA
eQQ
Q
BA
XR
0
]][[
][

Saverio Santi -Scienze Chimiche-
Università di Padova
123
Ricavando la concentrazione del complesso attivato si ottiene:
T
E
BA
eQQ
Q
h
kTmBAX R
2/1 02]][[][
d
La velocità media di una particella che si muove in una
direzione, secondo la teoria cinetica dei gas, è data
dall’espressione: 2/1
2
m
kTu
e quindi la frequenza di attraversamento del passo, , è:
d
1
2
2/1
m
kT
Abbiamo visto che la velocità di reazione è v = [X≠], per cui

Saverio Santi -Scienze Chimiche-
Università di Padova
124
T
E
BA
BB eQQ
Q
m
Tk
h
TkmBAX R
2/12/1 01
2
2]][[][v
dd
T
E
BA
eQQ
Q
h
kTBA R
0
]][[v
La distanza viene eliminata e quindi la definizione del suo lavoro è
irrilevante.
Il risultato è identico a quello ottenuto con la derivazione
precedente.
Concettualmente l’equivalenza delle due derivazioni si spiega
considerando che una vibrazione a bassa frequenza priva di forza
di richiamo la cui frequenza tende a zero diventa una traslazione.

Postulato di Hammond
Lo stato di transizione di una reazione elementare si trova più
vicino alla coordinata di reazione dello stato, iniziale o finale, che
ha un’energia maggiore.
La struttura di uno stato di transizione assomiglia a quella della
specie che gli è più vicina in energia. In una reazione endotermica,
la struttura dello stato di transizione assomiglia a quella dei
prodotti, mentre in una reazione esotermica assomiglia a quella dei
reagenti.
reazionedicoordinata
pE
reazionedicoordinata
pE
reazionedicoordinata
pE

Saverio Santi -Scienze Chimiche-
Università di Padova
126
La teoria dello stato di transizione ha fornito un'interpretazione
del fattore pre-esponenziale A.
Equazione di Eyring:
K
h
Tkk B
Formulazione termodinamica della teoria dello stato di transizione

Saverio Santi -Scienze Chimiche-
Università di Padova
127
K
h
Tkk B
RT
G
eK
D
DDD STHG
si ottiene la formulazione termodinamica
della teoria della stato di transizione:
RT
H
R
S
eeh
Tkk
D
D
B

RTaEAek
/
Confrontiamo le due equazioni:
2
ln
RT
E
dT
kda
dT
Kd
TdT
kd
ln1ln
Passando al logaritmo naturale
Applicando l'equazione di van'Hoff alla costante di
equilibrio K si ricava:
2
ln
RT
U
dT
Kd
D
K
h
Tkk B

Saverio Santi -Scienze Chimiche-
Università di Padova
129
2
ln
RT
U
T
K
P
cD
Per reazioni in fase gas si usa l’espressione:
L’equazione è generalmente usata anche per le reazioni
in soluzione, ma in questo caso
DD HU
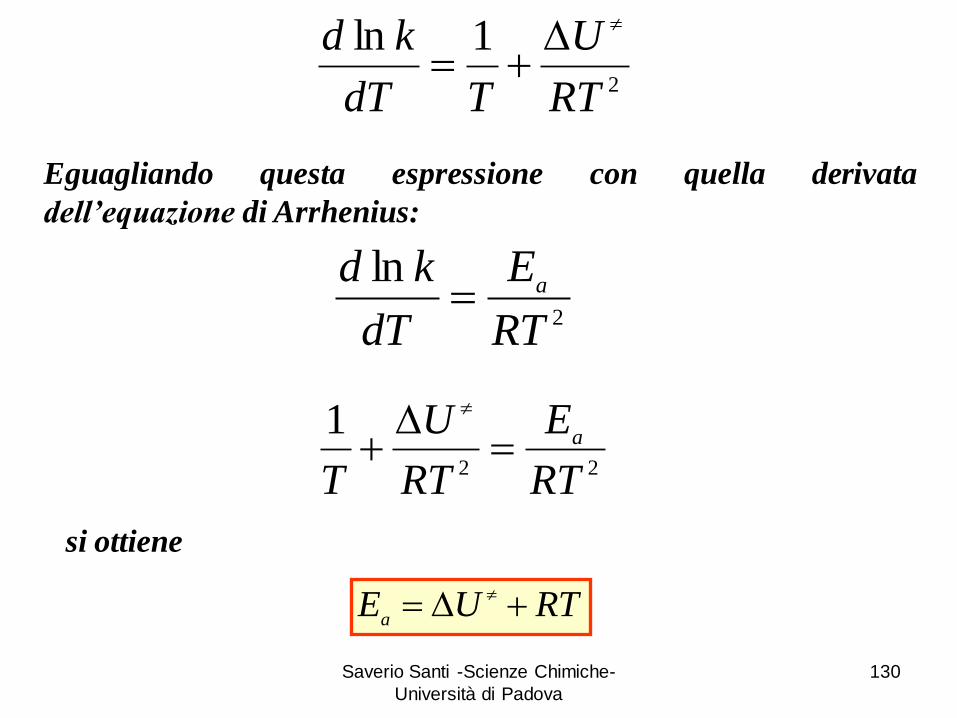
Saverio Santi -Scienze Chimiche-
Università di Padova
130
2
1ln
RT
U
TdT
kd D
2
ln
RT
E
dT
kda
Eguagliando questa espressione con quella derivata
dell’equazione di Arrhenius:
22
1
RT
E
RT
U
T
aD
si ottiene
RTUEa
D

Saverio Santi -Scienze Chimiche-
Università di Padova
131
RTUEa
D DDD VPUH
RTVPHEa
DD
la variazione di volume tra lo stato attivato e il reagente è nullo
RTnVP DD
non vi è variazione di n nella formazione dello stato di transizione
0D n
Per una reazione unimolecolare in fase gas
A P

Saverio Santi -Scienze Chimiche-
Università di Padova
132
RTHEa
D
RT
H
R
S
eeh
Tkk
D
D
B RT
E
R
S a
eeh
Tkek
D
B
RTaEAek
/
Confrontando con l’equazione di Arrhenius:
si ricava
R
S
eh
TkeA
D
B

Saverio Santi -Scienze Chimiche-
Università di Padova
133
RTVPHEa
DD
RTnVP DD
vi è variazione di n nella formazione dello stato di transizione 1D n
Per una reazione unimolecolare in fase gas
A + B P
RTHEa
2D
si ottiene RT
E
R
S a
eeh
Tkek
D
B2
da cui si ricava R
S
eh
TkeA
D
B2

Saverio Santi -Scienze Chimiche-
Università di Padova
134
RT
aE
R
S
eeh
Tkek
D
B2RT
aE
R
S
eeh
Tkek
D
B
Le equazione ricavate per reazioni in fase gas
vanno bene anche in soluzione

Saverio Santi -Scienze Chimiche-
Università di Padova
135
Plot di Arrhenius
1/T
lnk
lnA
R
Ea
RTaEAek
/
1/T
R
H D
RT
H
R
S
eeh
Tkk
D
D
B
ln(k/T)
Plot di Eyring
R
S
h
kBD
ln

Saverio Santi -Scienze Chimiche-
Università di Padova
136
Significato dei parametri termodinamici di attivazione
RX + 2H2O ROH + H3O+ + X-
Stechiometria
R-X R Xd+ d-
R+ X-+
Meccanismo di tipo SN1
Nello stato di transizione è presente una parziale rottura
del legame C-X e una parziale separazione di carica

DH dipende:
- dal grado di rottura del legame R-X (l'energia di
legame C-Cl è DH° = 81 kcal mol-1)
- dal contributo negativo dell'energia di solvatazione
DS risentirà:
- dell'aumento di disordine dovuto alla parziale
dissociazione
- dell'aumento di ordine nello stato di transizione
R X
+-+ -+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
R Xd+ d-
+-
+
-
+-
+
-
+-
+
-
+-
+
-+
-
+
-
+-
+
-
+-
+
-
+-
+
-+
-
+ -

Saverio Santi -Scienze Chimiche-
Università di Padova
138
Per la reazione di idrolisi del cloruro di t-butile a
30 °C in una miscela al 30% di acqua/etanolo si
trovano i seguenti valori:
1-1-
-1
molK cal20
mol kcal 17
D
D
S
H
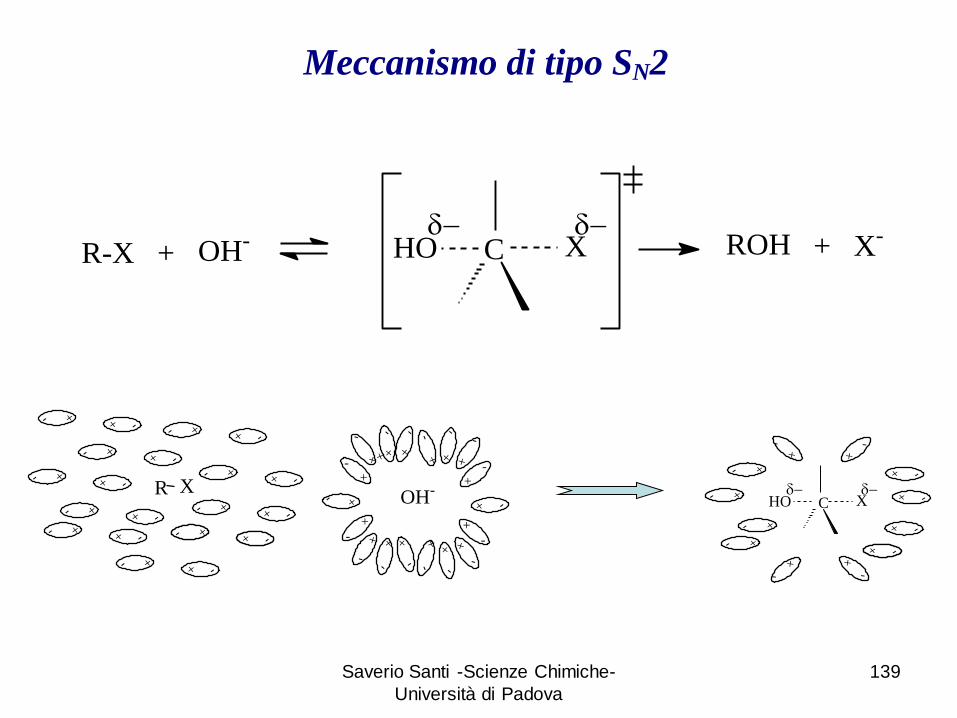
Saverio Santi -Scienze Chimiche-
Università di Padova
139
R-X + C XHO ROH + X-OH-
d d
Meccanismo di tipo SN2
+ -
+
-
+-
+
-
+-
+
-
+-
+
-
+-
OH-+-
+
-+
-
+
-
+-
+
-
+-
+
-
+-
+
-
-
+
R X
+-+ -+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
XHOd d
C
+ -+ -
+-
+-
+ -
+ -
+ -
+-
+-
+-
+-
+-
+-

Saverio Santi -Scienze Chimiche-
Università di Padova
140
DH
è minore di quello relativo ad un meccanismo
dissociativo:
l'energia di parziale rottura del legame C-X è in parte
compensata dall'energia di formazione del legame C-O
(l'energia di legame C-O è DH° = 85.5 kcal mol-1)
DS
- positivo poiché la carica relativa è maggiormente dispersa
nello stato di transizione che è quindi meno solvatato dei
reagenti
- contributo negativo dovuto alla diminuzione di disordine
dovuto al minor numero di molecole nello stato attivato.

Saverio Santi -Scienze Chimiche-
Università di Padova
141
R-X + C XH2O ROH + HXH2Od d
Meccanismo di tipo SN2
R X
+-+ -+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-+ -
+-
+
-
+-
+
-
+-
+
-
+-
+
-+
-
+
-
+-
+
-
+-
+
-
+-
+
-+
-
+ - XH2Od d
C
+-
+
-
maggiore solvatazione rispetto ai reagenti
DH minore DS negativo

Saverio Santi -Scienze Chimiche- Università di Padova
142
Effetto del mezzo

Saverio Santi -Scienze Chimiche-
Università di Padova
143
REAZIONI IN SOLUZIONE
Parametri che influenzano la costante di velocità:
Solvente (costante dielettrica)
Pressione idrostatica
Forza ionica
Sostituenti

Saverio Santi -Scienze Chimiche-
Università di Padova
144
1. correlare la velocità in soluzione con la velocità in fase gas;
2. considerare le velocità in solventi diversi;
3. esaminare una reazione in un solvente particolare
variando la costante dielettrica o la forza ionica del mezzo.
L'effetto del mezzo di reazione sulla velocità delle reazioni
chimiche è una problematica molto complessa e può essere
affrontata da differenti prospettive e livelli di comprensione.
Vi sono infatti diversi aspetti dell'influenza dell'ambiente di
reazione sulla velocità che permettono di verificare se è
possibile:
Effetto solvente

Saverio Santi -Scienze Chimiche-
Università di Padova
145
La teoria dello stato di transizione ha fornito
un'interpretazione del fattore pre-esponenziale A.
Equazione di Eyring:
K
h
Tkk B
kB = costante di Boltzman
h = costante di Plank
K = costante di equilibrio
tra stato di transizione X e reagenti.
A + B X≠

Saverio Santi -Scienze Chimiche-
Università di Padova
146
K non è la vera costante di equilibrio termodinamica,
bensì la corrispondente quantità che coinvolge le
concentrazioni:
]][[
][
BA
XK

Saverio Santi -Scienze Chimiche-
Università di Padova
147
La vera costante di equilibrio termodinamica 0
K è
espressa in funzione delle concentrazioni
termodinamiche, ovvero le attività:
BAaa
aK
0
BA
KK
0

dipende dalle caratteristiche di una specifica reazione e
non può essere determinato con i metodi fisici più comuni.
Può essere altresì stimato noti i coefficienti di
attività dei reagenti A e B e la costante di velocità.
ricavando l'espressione di K dall'equazione
K
h
Tkk B
sostituendola nell'equazione di Eyring si ricava:
BAkk 00B
0 Kh
Tkk

Saverio Santi -Scienze Chimiche-
Università di Padova
149
A + B AB P
Kh
Tkk B
AB
BA0γ
γγK
h
Tkk B
]][[
][
BA
ABK
AB
BA0
γ
γγlogloglog kk
Equazione di Brönsted

Saverio Santi -Scienze Chimiche-
Università di Padova
150
Reagenti e complesso attivato apolari
decomposizione del pentossido di azoto:
I reagenti hanno struttura simile a quella del complesso attivato
la velocità della reazione in fase gas
è molto prossima alla velocità in soluzione
NO + N2O5 3NO2
Stadio 1 N2O5 NO2 + NO3
Stadio 2 NO + NO3 2NO2 k2 >> k1

Saverio Santi -Scienze Chimiche-
Università di Padova
151
Solvente k 105, dm
-3 mol
-1 s
-1 Ea, kcal
Fase gas 1.65 24.7
Nitrometano 1.53 24.5
Bromo 2.15 24.0
Pentacloroetano 2.2 25.0
Tetracloruro di carbonio 2.35 24.1
Dicloro etilene 2.38 24.4
cloroformio 2.74 24.6
il rapporto tra i coefficienti di attività è circa unitario.
52
0
ONkk
Saverio Santi -Scienze Chimiche-
Università di Padova
152
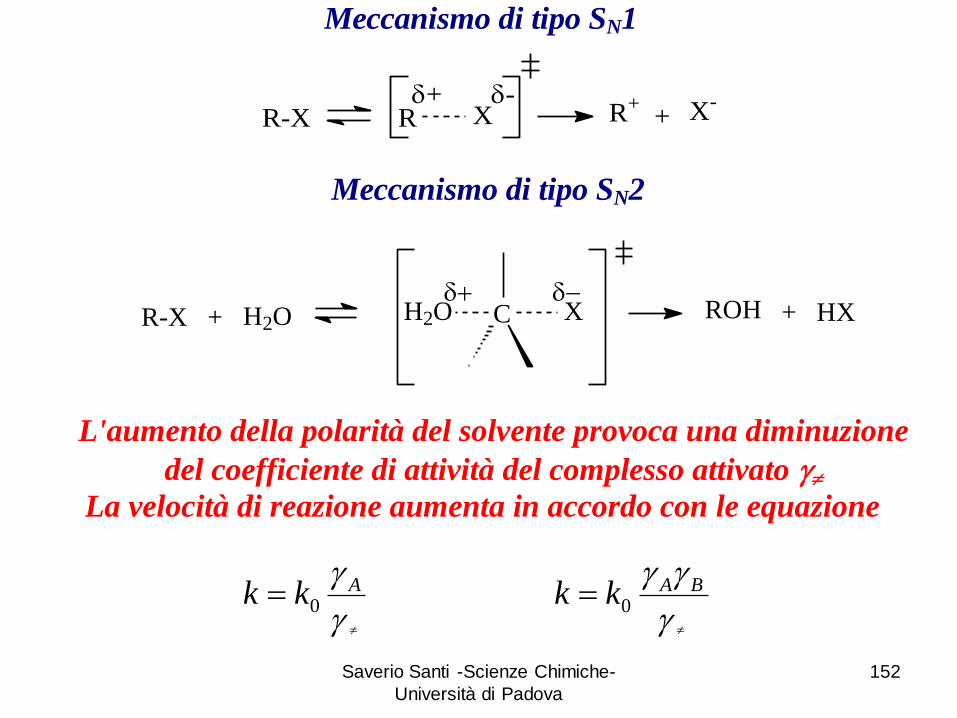
R-X R Xd+ d-
R+ X-+
Meccanismo di tipo SN1
R-X + C XH2O ROH + HXH2Od d
Meccanismo di tipo SN2
L'aumento della polarità del solvente provoca una diminuzione
del coefficiente di attività del complesso attivato
La velocità di reazione aumenta in accordo con le equazione
Akk 0
BAkk 0

Saverio Santi -Scienze Chimiche-
Università di Padova
153
Kh
Tkk BDall’equazione
RT
G
h
Tkk
3.2loglog B
Dsi ricava (1)
BAkk 0Nell’equazione 0B0 K
h
Tkk si ha che
RT
G
h
Tkk B
3.2loglog 0
0
Dda cui si ricava (2)
DD
BA
RT
GG
k
klog
3.2
)(log 0
0
Sottraendo l’equazione 2 all’equazione 1 si ricava

Saverio Santi -Scienze Chimiche-
Università di Padova
154
0
log3.2k
kRTG D d
variazione di energia libera di attivazione
)( 0
DDD GGGd
RT
GG
k
k
3.2
)(log 0
0
DD
La grandezza DGd è definita come
effetto solvente

Saverio Santi -Scienze Chimiche-
Università di Padova
155
Effetto solvente
A X P
Supponiamo che le uniche forze interagenti tra
soluto e solvente siano di tipo elettrostatico.
é il momento di dipolo del reagente, é il momento di dipolo del complesso attivato
DG = - DGl + DG° + DGl
g g
l l
DGl
DG
DGl
DG°
DG: variazione di energia libera di un forte dipolo da un mezzo a
costante dielettrica unitaria ad un altro a costante dielettrica e

Saverio Santi -Scienze Chimiche-
Università di Padova
156
DG = - DGl + DG° + DGl
Dalla teoria elettrostatica di Kirkwood (J. Chem. Phys. 1934, 2, 351):
- trascura le forze di van der Waals
- solo forze elettrostatiche dipolo-dopolo
- distribuzione di carica simmetrica nella molecola
- l’effetto del solvente è descritto in funzione della sola costante dielettrica e
D
12ε
1εμln
3
2
rRTG
DD
1
13
2
3
2
02ε
ε
r
μ
r
μGG

157
DD
1
13
2
3
2
02ε
ε
r
μ
r
μGG
1
11lnln
3
2
3
2
02ε
ε
r
μ
r
μ
RTkk
kln
12ε
1ε
RT
GG
k
k )(ln 0
0
DDRicordando che

158
meccanismo reagenti complesso attivato pendenza
RX (a)d+
R---Xd- + +
SN1 RX
+ (b) d+
R---Xd+ -
Y- + RX (c) d-
Y---R---Xd- -
Y + RX (d) d+Y---R---X
d- + +
Y- + RX
+ (e) Y---R---X - -
SN2
Y + RX+ (f) d+
Y---R---Xd+ -
kln
12ε
1ε
a,d
b,c,f
e

Saverio Santi -Scienze Chimiche-
Università di Padova
159
DD
1
1333
2
02ε
ε
r
μ
r
μ
r
μGG
B
2B
A
2A
A + B X P
1
11lnln
333
2
02ε
ε
r
μ
r
μ
r
μ
RTkk
B
2B
A
2A
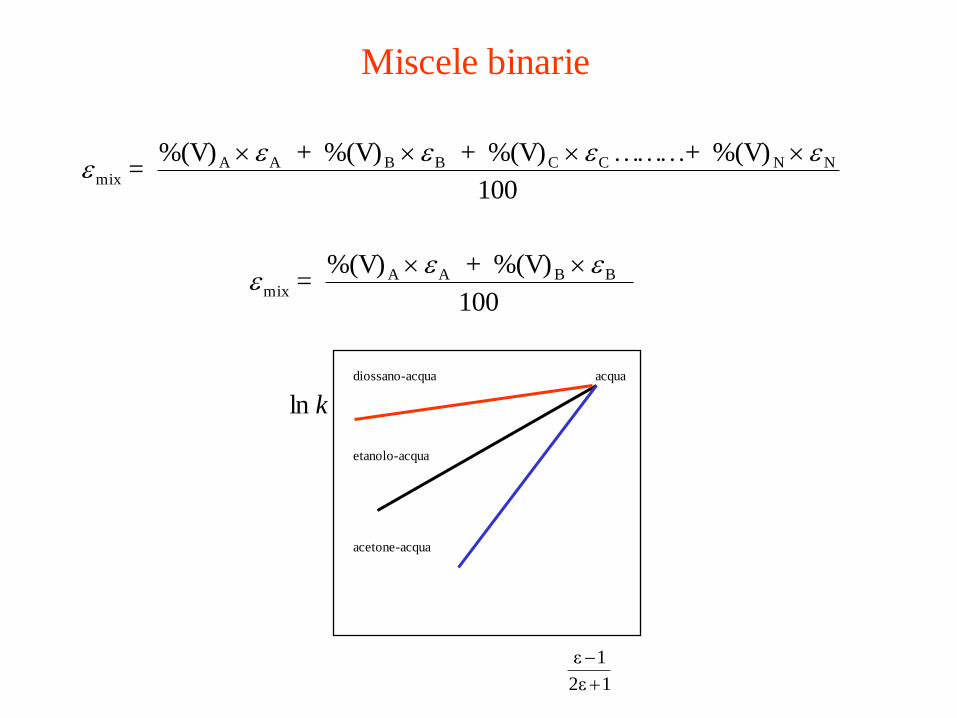
Miscele binarie
100
%(V) +………%(V) + %(V) + %(V) = NNCCBBAA
mix
eeeee
100
%(V) + %(V) = BBAA
mix
eee
12ε
1ε
kln
diossano-acqua
etanolo-acqua
acetone-acqua
acqua

Saverio Santi -Scienze Chimiche-
Università di Padova
161
-12
-11
-10
-9
-8
0.472 0.473 0.474 0.475 0.476 0.477 0.478 0.479
30° C25° C
y = -92.587 + 174.94x R= 0.98576
y = -96.523 + 181.82x R= 0.8473

Ioni in soluzione
AB
BA0
γ
γγlogloglog kk
IBd1
IAzγlog
i
2
i
i
Introducendo l’equazione di Debye-Hückel per ogni i
e nell’ipotesi che
ABBAi dddd
IBd1
I]Az-z[zkk ABBA
222
0loglog
2
2
1i
i
i zcI forza ionica

Saverio Santi -Scienze Chimiche-
Università di Padova
163
2BAAB )z(zz
2poichè
IBd1
IAz2zloglog 0
BAkk
si ottiene
IBd1
I]Az-z[zkk ABBA
222
0loglog
IBd1
I]A)z(z-z[zkk
2BABA
22
0loglog

Saverio Santi -Scienze Chimiche-
Università di Padova
164
IAz2zloglog 0 BAkk
Per soluzioni a forza ionica molto bassa
Iz2z0.1loglog 0 BAkk
0
lnk
k
I
(a) [Co(NH3)5 Br]2+
+ Hg2+
[Co(NH3)5 H2O]3+
+ HgBr2 (zAzB = 4)
(b) S2O82-
+ 2I- 2SO4
2- + I2 (ordine 1 rispetto a I
-) (zAzB = 2)
(c) CH3COOC2H5 + OH-
CH3COO- + C2H5OH (zAzB = 0)
(d) H+ + Br
- + 1/2H2O2 1/2Br2 + H2O (zAzB = -1)
(e) [Co(NH3)5Br]2+
+ OH- [Co(NH3)5 OH]
2+ + Br
-(zAzB = -2)
Equazione di Brönsted-Bjerrum
In soluzione acquosa A = 0.509
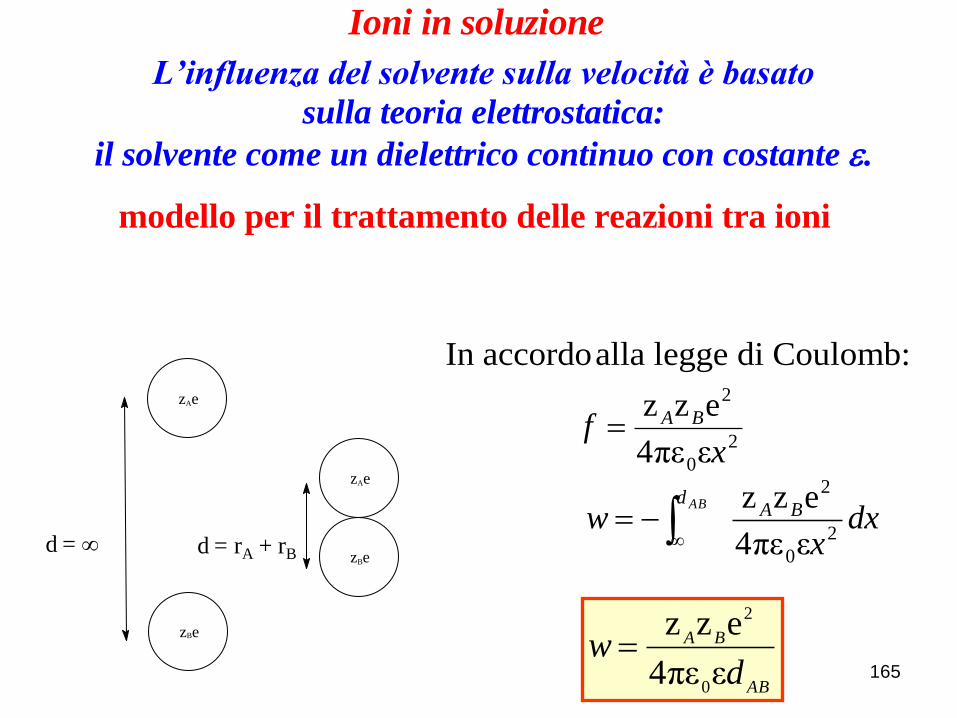
165
zAe
zBe
zAe
zBed = d = rA + rB
Ioni in soluzione
modello per il trattamento delle reazioni tra ioni
L’influenza del solvente sulla velocità è basato
sulla teoria elettrostatica:
il solvente come un dielettrico continuo con costante e.
dxx
w BAd AB
2
0
2
επε4
ezz
AB
BA
dw
επε4
ezz
0
2
2
0
2
επε4
ezz
xf BA
In accordo alla legge di Coulomb:

Saverio Santi -Scienze Chimiche-
Università di Padova
166
AB
BA
dGG
επε4
ezz
0
2
0 DD
Kh
Tkk B D GKRT ln
Tkdkk
BAB
BA
επε4
ezzlnln
0
2
0
0ε
ln)(lnlim kk
lavoro elettrostatico
lavoro non elettrostatico
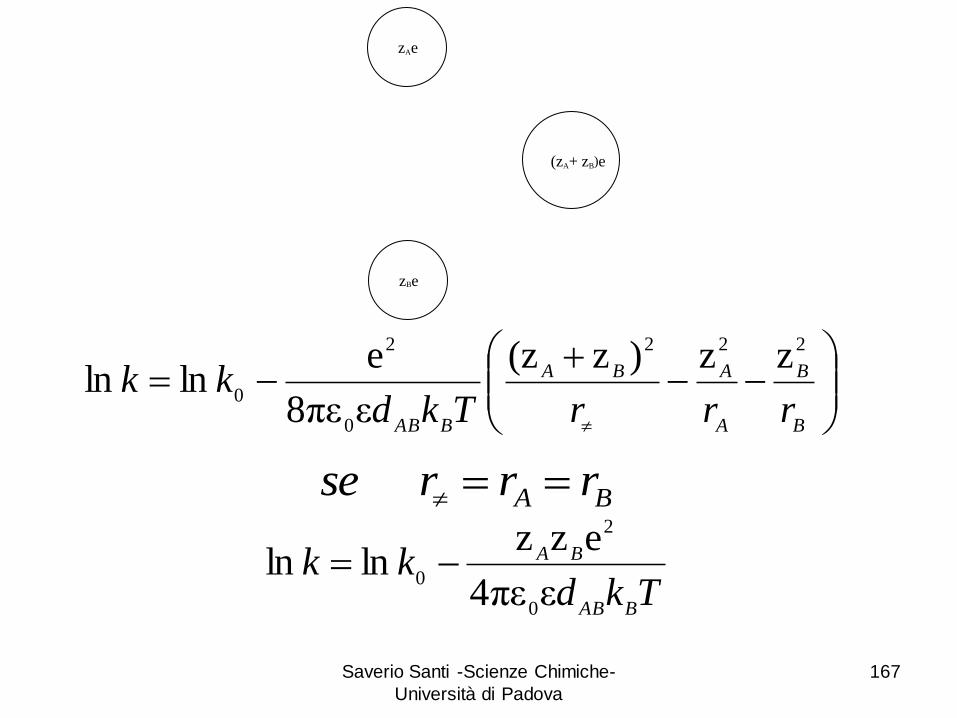
Saverio Santi -Scienze Chimiche-
Università di Padova
167
zB)e(zA+
zBe
zAe
B
B
A
ABA
BABrrrTkd
kk222
0
2
0
zz)z(z
επε8
elnln
Tkdkk
BAB
BA
επε4
ezzlnln
0
2
0
BA rrrse

Saverio Santi -Scienze Chimiche-
Università di Padova
168
Effetto della pressione idrostatica
RT
V
dP
K
T
aD
ln
RT
V
dP
K
T
D
ln
equilibrio in soluzione
equilibrio complesso attivato
K
h
Tkk BPoiché
RT
V
dP
k
T
D
ln
la costante di velocità dipende dalla pressione

Saverio Santi -Scienze Chimiche-
Università di Padova
169
RT
Vkk
D
0lnln
Sperimentalmente si trova che a temperatura costante il lnk è lineare
rispetto alla pressione, quindi DV≠ non dipende dalla pressione.
L’equazione differenziale può essere integrata:
dove k0 è la costante di velocità a pressione zero (solitamente molto
simile al valore a pressione atmosferica)
0
0.5
-0.5
Tipo II: DV≠ e DS≠ < 0
Tipo I: DV≠ e DS≠ ≈ 0
Tipo III: DV≠ e DS≠ > 0
ln(k/k0) 0.5 1
Pressione/108 Pa

Saverio Santi -Scienze Chimiche-
Università di Padova
170
Si trova una sostanziale linearità tra DV≠ e DS≠ per le reazioni tra ioni che
possono essere suddivise in tre categorie:
Tipo I. Reazioni che dipendono poco dalla pressione e che hanno
fattori pre-esponenziali “normali” (109-1011 M-1 s-1).
Tipo II. Reazioni con valori di k che aumentano molto all’aumento della
pressione e che hanno fattori pre-esponenziali piccoli.
Tipo III. Reazioni che diminuiscono molto in k all’aumento della
pressione e che hanno fattori pre-esponenziali grandi.
In soluzione DV≠ dipende da due contributi:
(DV≠)r la variazione del volume delle molecole di reagenti quando
formano il complesso attivato;
(DV≠)s la variazione del volume delle molecole di solvente
(DV≠)s è particolarmente importante nelle reazione tra ioni.

Saverio Santi -Scienze Chimiche-
Università di Padova
171
Elettrostrizione
+
+
+
+
+
+
–
–
maggiore elettrostrizione
diminuzione di entropia
minore elettrostrizione
aumento di entropia
DS≠ < 0 DS≠ > 0
DVs≠ < 0
k aumenta con la pressione k diminuisce con la pressione
DVs≠ > 0

Saverio Santi -Scienze Chimiche- Università di Padova
172
Effetto isotopico

Saverio Santi -Scienze Chimiche-
Università di Padova
173
EFFETTO ISOTOPICO
Reazioni di equilibrio: effetto isotopico termodinamico
Reazioni non di equilibrio:
effetto isotopico cinetico
primario
secondario
H2 2H
D2 2D
HD H + D
RH R + H
RD R + D
CR2CHD CR2CH + D

Saverio Santi -Scienze Chimiche-
Università di Padova
174
Effetto isotopico termodinamico
H2 2H
RT
molkcal
H
HH e
q
qK
1
2
5.1022
kT
hv
H
HvHrHtH
eh
IkT
h
kTmqqqq
1
1822
2
3
2/3
2
2222
3
2/32
h
kTmqq H
HtH
D2 2D
2
H
2
H
2
H
2
1
2dmd
m
mI
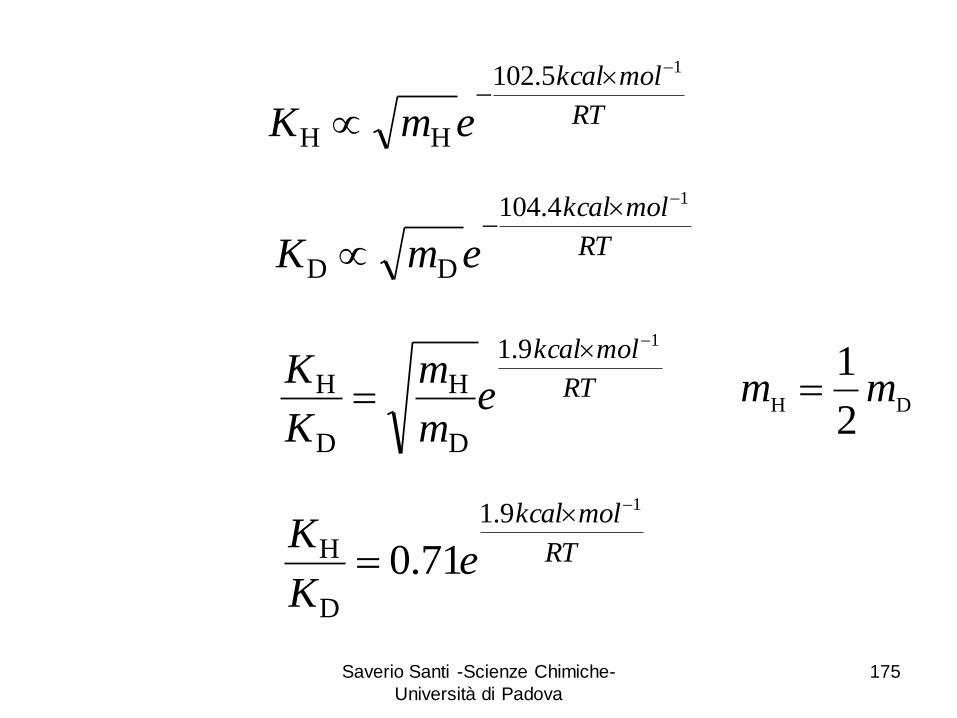
Saverio Santi -Scienze Chimiche-
Università di Padova
175
RT
molkcal
emK
15.102
HH
RT
molkcal
emK
14.104
DD
RT
molkcal
em
m
K
K19.1
D
H
D
H
DH2
1mm
RT
molkcal
eK
K19.1
D
H 71.0

Saverio Santi -Scienze Chimiche-
Università di Padova
176
RT
molkcal
eK
K19.1
D
H 71.0
T = 298 K 18D
H K
K
HD H + D
9D
H K
K
Saverio Santi -Scienze Chimiche-
Università di Padova
177
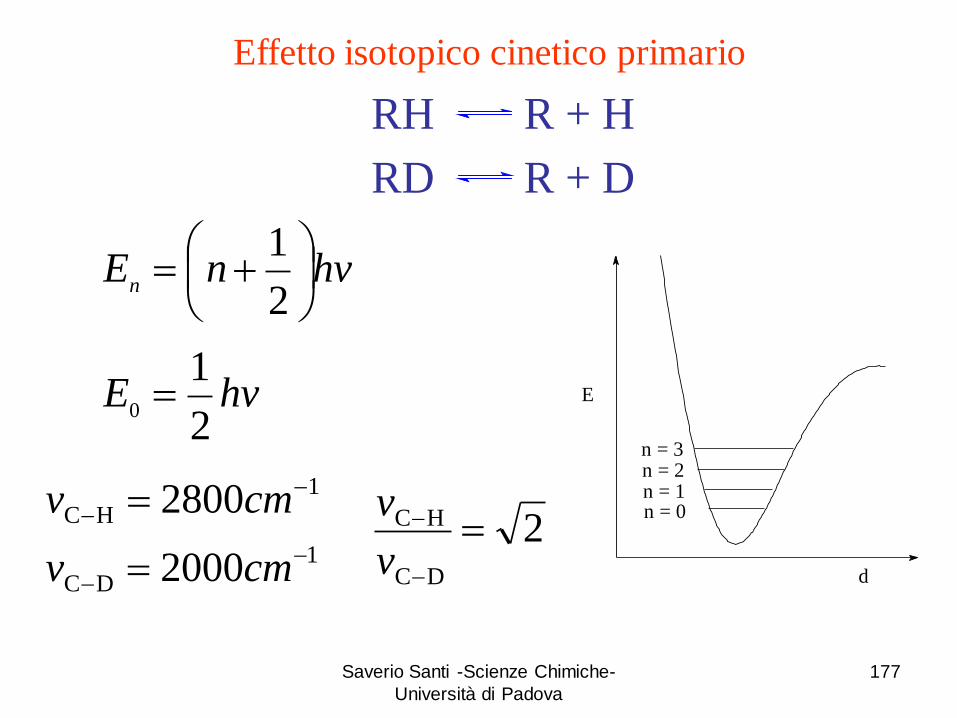
Effetto isotopico cinetico primario
RH R + H
RD R + D
hvnEn
2
1
hvE2
10
1
DC
1
HC
2000
2800
cmv
cmv
E
d
n = 3n = 2
n = 0n = 1
2DC
HC
v
v

Saverio Santi -Scienze Chimiche-
Università di Padova
178
2/1
μπ2
1
κv
H
HR
HRμ mmm
mm
E0H
E0D
DED DEH
2H
D
DC
HC
m
m
v
v
HDvv D
o
H
oEE

Saverio Santi -Scienze Chimiche-
Università di Padova
179
RT
E
eAk
Da
DD
RT
E
eAk
Ha
HH
E0H
E0D
D
aE
H
aE
RT
-EE
eA
A
k
kDa
Ha
D
H
D
H
D
0
H
0
D
a
H
a -EE-EE
RT
-EE
ek
kD0
H0
D
H DH AA
D
0
H
0
H
a
D
a -EEEE

Saverio Santi -Scienze Chimiche-
Università di Padova
180
2H
D
DC
HC
m
m
v
v
RT
-EE
ek
kD0
H0
D
H hvE2
10
RT
-vvh
ek
k2
D
H
D0
H0
1DC
1HC
2000
2800
cmv
cmv
RT
Lcmh
ek
k2
800
D
H
1

Saverio Santi -Scienze Chimiche-
Università di Padova
181
(cm-1) (s-1) (s-1) (m-1) c (m s-1)
(s-1) = 8 104 (m-1) 2.998108 (m s-1) =
= 2.4 1013 (s-1)
7)K(15.298)molJK(314.82
)mol(10022.6)s(104.2Js10626.6
D
H11
12311334
ek
k
RT
Lcmh
ek
k2
800
D
H
1

RH + R’ [R…H…R’] R + R’H
RH------R'
R + R'H
RH + R'
R---H---R'
R + R'H
RH + R'
C-DC-H
R----H-R'
R + R'H
RH + R'
legame RH rottoquasi completamente
legame RH rafforzato
H equidistante da R e R'
effetto isotopico primario massimo
effetto isotopico primario ridotto
effetto isotopico primario negativo
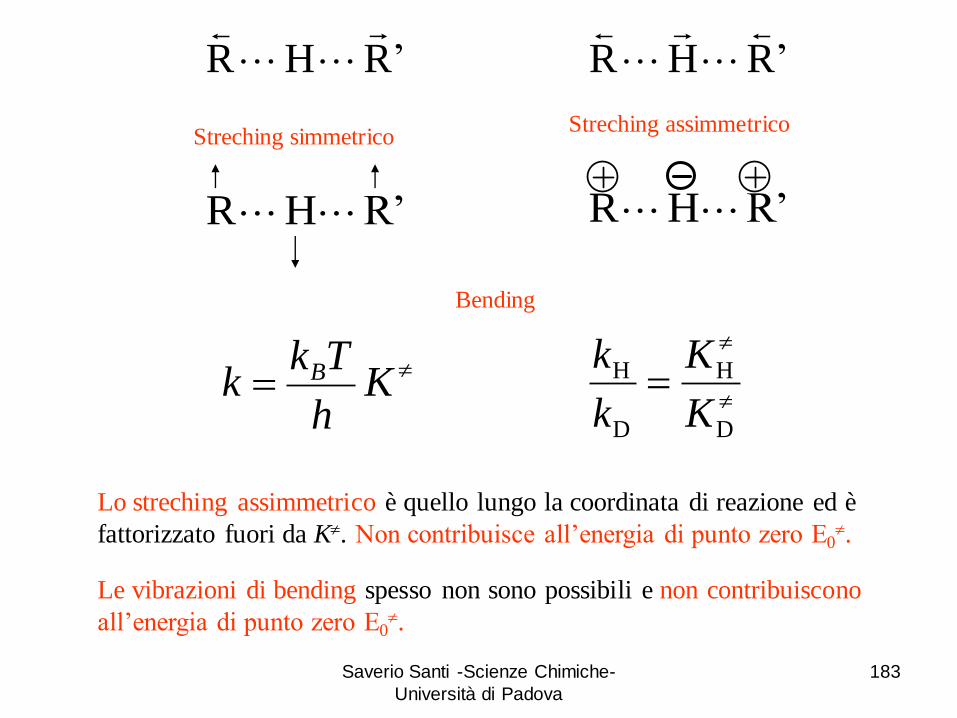
Saverio Santi -Scienze Chimiche-
Università di Padova
183
’RHR
Streching simmetrico
’RHR
Streching assimmetrico
R’HR R’HR
Bending
Kh
Tkk B
D
H
D
H
K
K
k
k
Lo streching assimmetrico è quello lungo la coordinata di reazione ed è
fattorizzato fuori da K. Non contribuisce all’energia di punto zero E0.
Le vibrazioni di bending spesso non sono possibili e non contribuiscono
all’energia di punto zero E0.

Saverio Santi -Scienze Chimiche-
Università di Padova
184
Lo streching simmetrico non contribuisce all’energia di
punto zero E0 solo se H è equidistante da R e R’ (e non si
muove se mR = mR’): effetto isotopico massimo
’RHR
Streching simmetrico
R’HR R’HR
Lo streching simmetrico contribuisce all’energia di punto
zero E0 se H non è equidistante da R e R’: effetto
isotopico ridotto
R’RH
Se il legame RH nello stato di transizione è rafforzato la
sostituzione isotopica accelera la reazione: effetto isotopico
negativo
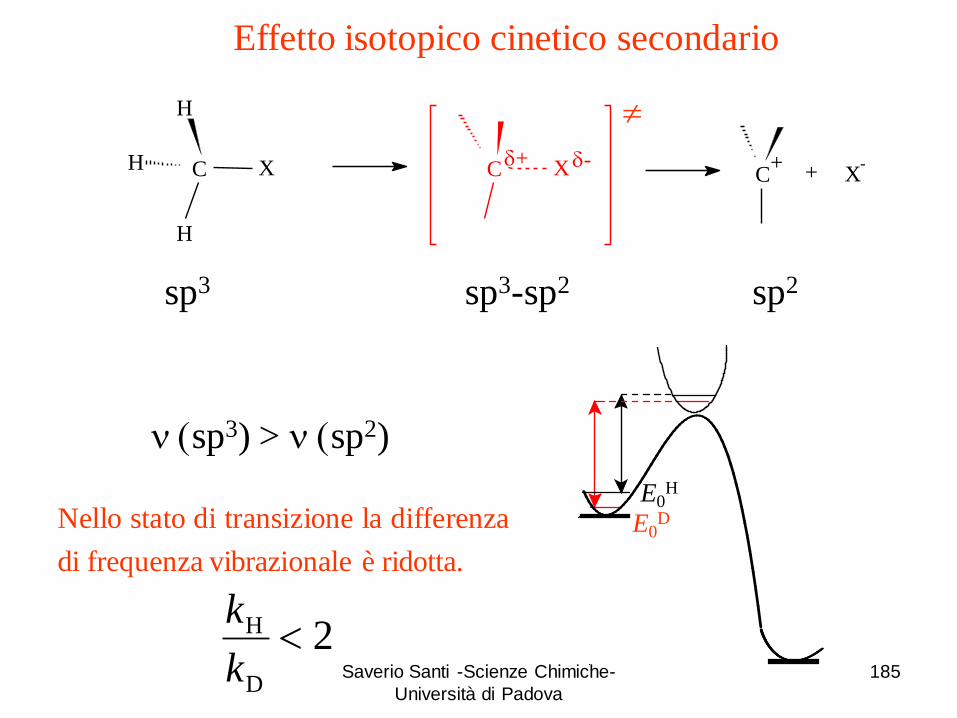
Saverio Santi -Scienze Chimiche-
Università di Padova
185
Effetto isotopico cinetico secondario
sp3 sp3-sp2 sp2
sp3) > sp2)
Nello stato di transizione la differenza
di frequenza vibrazionale è ridotta. E0
D
E0H
2D
H k
k
+C X XCd+ d-
C X-
+
H
H
H

Saverio Santi -Scienze Chimiche-
Università di Padova
186
Effetto sostituente:
-induttivo -sterico
rH > rD
R-H
R-D
rD rH
H sostituente con maggiore ingombro sterico
D sostituente più elettron-donatore
oscillatore
anarmonico

Saverio Santi -Scienze Chimiche- Università di Padova
187
Reazioni a catena
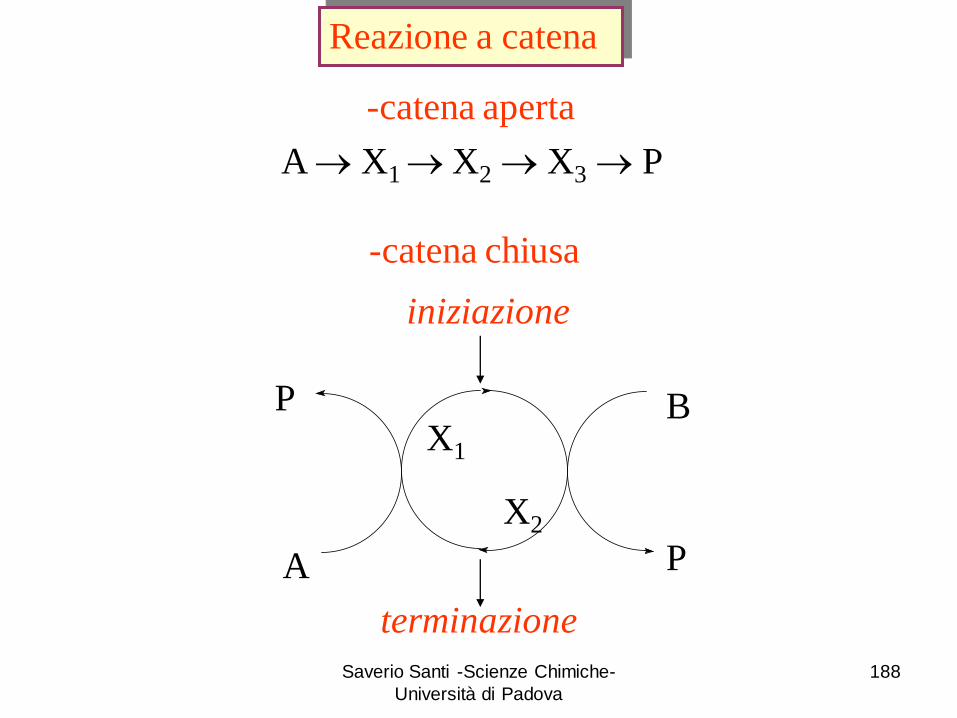
Saverio Santi -Scienze Chimiche-
Università di Padova
188
iniziazione
terminazione
A
P X1
B
P
X2
Reazione a catena
A X1 X2 X3 P
-catena aperta
-catena chiusa

Saverio Santi -Scienze Chimiche-
Università di Padova
189
Lunghezza di catena: numero medio di cicli
einiziazion reazione velocità
reazione globale velocitàγ
A X1 iniziazione
X1 + B X2 + P propagazione
X2 + A X1 + P propagazione
X2 + P X1 + B inibizione
X1 A terminazione

Saverio Santi -Scienze Chimiche-
Università di Padova
190
1 Br2 2Br iniziazione
2 Br + H2 HBr + H propagazione
3 H + Br2 HBr + Br propagazione
-2 H + HBr Br + H2 inibizione
-1 2Br Br2 terminazione
H2 + Br2 2HBr
Sperimentalmente: ]Br[[HBr]'1
]][BrH[
2
21
22
k
kv

1 Br2 2Br
2 Br + H2 HBr + H
3 H + Br2 HBr + Br
-2 H + HBr Br + H2
-1 2Br Br2
Si trascurano le reazioni:
H2 2H legame più forte di Br2v
-3 HBr + Br H + Br2 legame HBr più forte di Br2
H + Br HBr [H] molto bassa
2H H2 [H] molto bassa
HBr H + Br legame HBr più forte di Br2

Saverio Santi -Scienze Chimiche-
Università di Padova
192
0[H][HBr]][H][Br][Br][H]H[
22322 kkkdt
d
0[Br]2][H][Br[H][HBr]][Br][H-][Br2B 2
12322221 kkkkkdt
]r[d
1 Br2 2Br
2 Br + H2 HBr + H
3 H + Br2 HBr + Br
-2 H + HBr Br + H2
-1 2Br Br2
0[Br]][Br 2
121
kk
2/1
1
21][Br
[Br]
k
k
sommando le due equazioni
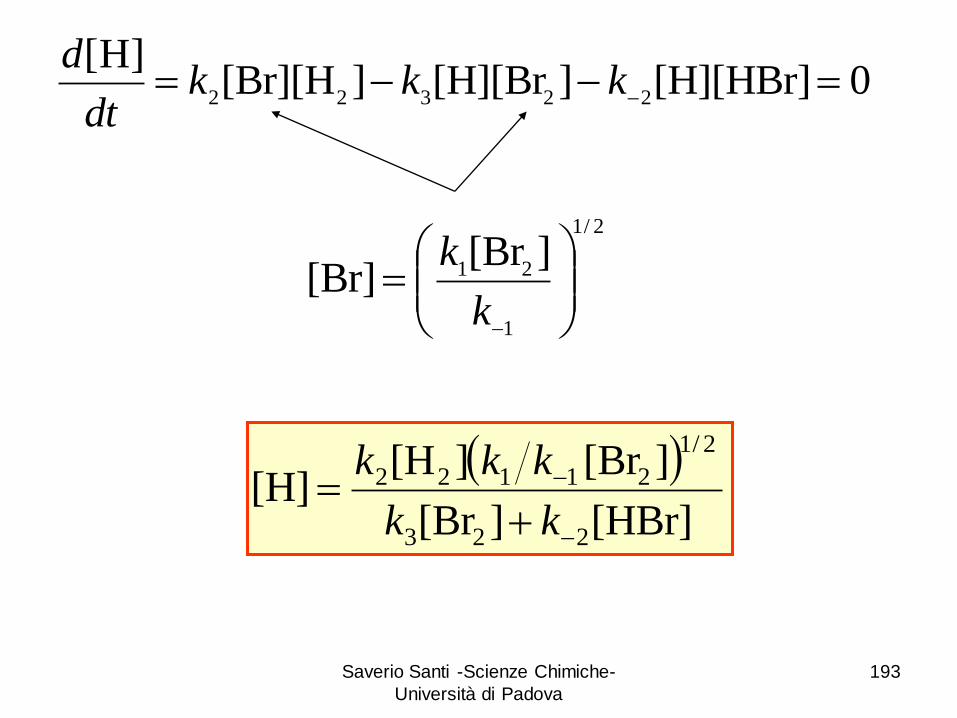
Saverio Santi -Scienze Chimiche-
Università di Padova
193
0[H][HBr]][H][Br][Br][H]H[
22322
kkk
dt
d
2/1
1
21][Br
[Br]
k
k
[HBr]][Br
][Br][H[H]
223
2/1
21122
kk
kkk

0[H][HBr]][H][Br][Br][H]H[
22322 kkkdt
d
0[Br]2][H][Br[H][HBr]][Br][H-][Br2B 2
12322221 kkkkkdt
]r[d
1 Br2 2Br
2 Br + H2 HBr + H
3 H + Br2 HBr + Br
-2 H + HBr Br + H2
-1 2Br Br2
0[Br]][Br 2
121
kk
2/1
1
21][Br
[Br]
k
k

Saverio Santi -Scienze Chimiche-
Università di Padova
195
][H][Br2]HB[
23kdt
rd
][Br
[HBr]1
][Br][H2]HB[
23
2
2/1
21122
k
k
kkk
dt
rd
[HBr]][Br
][Br][H[H]
223
2/1
21122
kk
kkk
][Br
[HBr]][Br
][Br][H2
]HB[2
223
2/1
211223
kk
kkkk
dt
rd
][Br
[HBr]][Br
23
223
k
kk
Moltiplicando e dividendo per k3[Br2]:

Saverio Santi -Scienze Chimiche-
Università di Padova
196
][Br
[HBr]1
][Br][H2]HB[
23
2
2/1
21122
k
k
kkk
dt
rd
]Br[[HBr]'1
]][BrH[
2
21
22
k
kv
2/1
1122 kkkk3
2'k
kk
Sperimentalmente:





























