Embed Size (px)
Citation preview

k19 OFICINA ESPANOLA DEPATENTES Y MARCAS
ESPANA
k11 Numero de publicacion: 2 178 000k51 Int. Cl.7: C07K 7/08, C07K 14/00
C07K 16/44, C12N 15/11
A61K 38/16, A61K 38/10
A61K 47/48
k12 TRADUCCION DE PATENTE EUROPEA T3
k86 Numero de solicitud europea: 97941352.3k86 Fecha de presentacion: 21.08.1997k87 Numero de publicacion de la solicitud: 0 925 308k87 Fecha de publicacion de la solicitud: 30.06.1999
k54 Tıtulo: Composiciones y metodos para tratar infecciones usando analogos de indolicidina.
k30 Prioridad: 21.08.1996 US 24754 P13.01.1997 US 34949 P
k73 Titular/es: Micrologix Biotech, Inc.3650 Wesbrook MallVancouver, British Columbia V6S 2L2, CA
k45 Fecha de la publicacion de la mencion BOPI:16.12.2002
k72 Inventor/es: Fraser, Janet R.;West, Michael H., P.;Krieger, Timothy, J.;Taylor, Robert yErfle, Douglas
k45 Fecha de la publicacion del folleto de patente:16.12.2002
k74 Agente: Tavira Montes-Jovellar, Antonio
Aviso: En el plazo de nueve meses a contar desde la fecha de publicacion en el Boletın europeo de patentes,de la mencion de concesion de la patente europea, cualquier persona podra oponerse ante la OficinaEuropea de Patentes a la patente concedida. La oposicion debera formularse por escrito y estarmotivada; solo se considerara como formulada una vez que se haya realizado el pago de la tasa deoposicion (art. 99.1 del Convenio sobre concesion de Patentes Europeas).
Venta de fascıculos: Oficina Espanola de Patentes y Marcas. C/Panama, 1 – 28036 Madrid
ES
217
800
0T
3

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
DESCRIPCION
Composiciones y metodos para tratar infecciones usando analogos de indolicidina.
Campo tecnico
La presente invencion se refiere en general a composiciones para tratar infecciones causadas por mi-croorganismos, y mas especıficamente, a composiciones que comprenden analogos de indolicidina, analogosmodificados con polımeros, y a sus usos para tratar infecciones.
Antecedentes de la invencion
Para la mayorıa de los individuos sanos las infecciones son molestas, pero generalmente no son ame-nazadoras para la vida. Muchas infecciones son combatidas satisfactoriamente por el sistema inmunitariodel individuo. El tratamiento es un complemento y generalmente esta facilmente disponible en paısesdesarrollados. Sin embargo, las enfermedades infecciosas son un grave problema en paıses en desarrolloy en individuos inmunocomprometidos.
En los paıses en desarrollo, la falta de condiciones de salubridad adecuadas y la consiguiente poca hi-giene proporcionan un entorno que promueve las infecciones bacterianas, de parasitos, fungicas y vıricas.La poca higiene y las deficiencias nutricionales pueden disminuir la eficacia de las barreras naturales,tales como la piel y membranas mucosas, para la invasion de agentes infecciosos o la capacidad del sis-tema inmunitario de eliminar los agentes. Tambien, un ataque constante de patogenos puede estresarlas defensas del sistema inmunitario de la produccion de anticuerpos y celulas fagocıticas (por ejemplo,neutrofilos polimorfos) a niveles anormales. Tambien se puede producir una perdida de las defensas delhospedante debido a condiciones tales como trastornos circulatorios, obstruccion mecanica, fatiga, fumar,bebida excesiva, defectos geneticos, SIDA, trasplante de medula osea, cancer y diabetes. Un problemacorriente creciente en el mundo son las infecciones oportunistas en individuos que son VIH positivos.
Aunque puede haber vacunas disponibles para proteger frente a algunos de estos organismos, las va-cunaciones no siempre son viables, debido a factores tales como mecanismos de suministro inadecuados ypobreza economica, o eficaces debido a factores tales como suministro demasiado tarde en la infeccion, in-capacidad del paciente para montar una respuesta inmunitaria a la vacuna, o a la evolucion del patogeno.Para otros agentes patogenos no hay vacunas disponibles. Cuando no es posible la proteccion frente ala infeccion, generalmente se persigue el tratamiento de la infeccion. El arma principal en el arsenal detratamientos son los antibioticos. Aunque los antibioticos han demostrado ser eficaces frente a muchasbacterias y ası han salvado incontables vidas, no son una panacea. El uso excesivo de antibioticos enciertas situaciones ha promovido la extension de cepas bacterianas resistentes. Y muy importante, losantibacterianos no son utiles frente a infecciones vıricas.
Una variedad de organismos produce peptidos cationicos (cargados positivamente), moleculas usadascomo parte de un mecanismo de defensa no especıfico frente a microorganismos. Cuando se aıslan, estospeptidos son toxicos para una amplia variedad de microorganismos, incluyendo bacterias, hongos y ciertosvirus recubiertos. Un peptido cationico encontrado en neutrofilos es la indolicidina. Aunque la indolici-dina actua frente a muchos patogenos, existen notables excepciones y grados variables de toxicidad.
El documento WO-A-95/22338 describe en general una variedad de analogos de indolicidina, ymetodos para usar estos analogos para inhibir la supervivencia y crecimiento de microorganismos.
Uchidea et al. (Peptide Chemistry, 1995 (pp. 229-232); Protein Research Foundation, Osaka, 1996)discute la actividad antibacteriana de la indolicidina y otros peptidos.
Aunque los peptidos cationicos muestran eficacia in vitro frente a una variedad de celulas patogenasincluyendo bacterias gram positivas, bacterias gram negativas, y hongos, estos peptidos en general sontoxicos para los mamıferos cuando son inyectados, y los ındices terapeuticos normalmente son bastantepequenos. Los enfoques para reducir la toxicidad han incluido el desarrollo de un derivado o sistemade suministro que enmascara elementos estructurales implicados en la respuesta toxica o que mejora laeficacia con dosis menores. Entre otros enfoques en evaluacion se incluyen sistemas de liposomas y mi-celulares para mejorar los efectos clınicos de peptidos, proteınas, y farmacos hidrofobos, y ciclodextrinaspara aislar superficies hidrofobas durante la administracion en medio acuoso. Por ejemplo, la union apolımeros de polietilenglicol (PEG), a menudo por modificacion de grupos amino, mejora el valor me-dicinal de algunas proteınas tales como asparaginasa y adenosina-desaminasa, y aumenta la semividacirculatoria de peptidos tales como interleuquinas.
2

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Ninguno de estos enfoques ha mostrado mejorar la administracion de peptidos cationicos. Por ejemplo,se han desarrollado metodos para la sıntesis por pasos de derivados de polisorbato que pueden modificarpeptidos por reacciones de acilacion, pero la acilacion altera la carga de un peptido cationico modificadoy frecuentemente reduce o elimina la actividad antimicrobiana del compuesto. Por lo tanto, para el sumi-nistro de peptidos cationicos, ası como de otros peptidos y proteınas, es necesario un sistema que combinelas propiedades de mayor semivida circulatoria con la capacidad para formar una estructura micelular.
Aquı se describen analogos de indolicidina, disenados para ampliar su variedad y eficacia, y ademasproporcionar otras ventajas relacionadas. Tambien se describen metodos y composiciones para modificarpeptidos, proteınas, antibioticos y similares, para reducir la toxicidad, ası como para proporcionar otrasventajas.
Sumario de la invencion
Aquı se describen en general analogos de indolicidina. La invencion se refiere a un analogo de indoli-cidina que comprende hasta 25 aminoacidos y que contienen la formula I L R W P W W P W R R K. Enciertas realizaciones, los analogos de indolicidina se acoplan para formar un peptido ramificado. En otrasrealizaciones, el analogo tiene uno o mas aminoacidos cambiados por un D-aminoacido correspondiente, yen ciertas realizaciones preferidas, el aminoacido N-terminal y/o el C-terminal es un D-aminoacido. Otrasmodificaciones preferidas incluyen analogos que estan acetilados en el aminoacido N-terminal, amidadosen el aminoacido C-terminal, esterificados en el aminoacido C-terminal, modificados por incorporacionde homoserina/homoserina-lactona en el aminoacido C-terminal, y conjugados con polietilenglicol o susderivados.
En otros aspectos, se describe una molecula de acido nucleico aislada cuya secuencia comprende unao mas secuencias de codificacion de los analogos de indolicidina, vectores de expresion, y celulas hospe-dantes transfectadas o transformadas con el vector de expresion.
Otros aspectos proporcionan una composicion farmaceutica que comprende al menos un analogo deindolicidina y un tampon fisiologicamente aceptable, que opcionalmente comprende un agente antibiotico.En otras realizaciones, la composicion farmaceutica ademas comprende un agente antivırico (por ejemplo,aciclovir; hidrocloruro de amantadina; didanosina; edoxudina; famciclovir; foscarnet; ganciclovir; idoxuri-dina; interferon; lamivudina; nevirapina; penciclovir; podofilotoxina; ribavirina; rimantadina; sorivudina;stavudina; trifluridina; vidarabina; zalcitabina y zidovudina); un agente antiparasito (por ejemplo, deri-vados de 8-hidroxiquinolina; alcaloides de cinchona; derivados de nitroimidazol; derivados de piperazina;derivados de pirimidina y derivados de quinolina; albendazol; atovaquona; fosfato de cloroquina; citrato dedietilcarbamazina; eflornitina; halofantrina; iodoquinol; ivermectina; mebendazol; hidrocloruro de meflo-quina; melarsoprol B; metronidazol; niclosamida; nifurtimox; paromomicina; isetionato de pentamidina;piperazina; praziquantel; fosfato de primaquina; proguanil; pamoato de pirantel; pirimetamina; pamoatode pirivinio; gluconato de quinidina; sulfato de quinina; estibogluconato sodico; suramina y tiabendazol);un agente antifungico (por ejemplo, alilaminas; imidazoles; pirimidinas y triazoles, 5-fluorocitosina; anfo-tericina B; butoconazol; clorfenesina; ciclopirox; clioquinol; clotrimazol; econazol; fluconazol; flucitosina;griseofulvina; itraconzol; ketoconazol; miconazol; hidrocloruro de naftitfina; nistatina; sulfuro de selenio;sulconazol; hidrocloruro de terbinafina; terconazol; tioconazol; tolnaftato y undecilentato). Todavıa enotras realizaciones, la composicion se incorpora en un liposoma o un vehıculo de liberacion lenta.
Todavıa en otro aspecto, se describe una composicion para tratar una infeccion, que comprende uncompuesto de acuerdo con la invencion. La infeccion puede estar causada, por ejemplo, por un microor-ganismo, tal como una bacteria (por ejemplo, bacteria Gram negativa o Gram positiva u organismoanaerobio; son ejemplos Acinetobacter spp., Enterobacter spp., E. cofi, H. influenzae, K. pneumoniae,P. aeruginosa, S. marcescens y S. maltophilia, Bordetella pertussis; Brucella spp.; Campylobacter spp.;Haemophilus ducreyi, Helicobacter pylori; Legionella spp.; Moraxella catarrhalis; Neisseria spp.; Salmo-nella spp.; Shigella spp. y Yersinia spp.; E. faecalis, S. aureus, E. faeciurn, S. pyogenes, S. pneumoniaey estafilococos coagulasa negativos; Bacillus spp.; Corynebacterium spp.; Difetroides; Listeria spp. yEstreptococos Viridans; Clostridium spp., Bacteroides spp. y Peptostreptococcus spp.; Borrelia spp.;Chlamydia spp.; Mycobacterium spp.; Mycoplasma spp.; Propionibacterium acne, Rickettsia spp.; Tre-ponerna spp. y Ureaplasma spp.), hongos (por ejemplo, levaduras y/o mohos), parasitos (por ejemplo,protozoos, nematodos, cestodos y trematodos, tales como Babesia spp.; Balantidium coli, Blastocystis ho-minis, Cryptosporidium parvum, Encephohalitozoon spp.; Entamoeba spp.; Giardia lamblia; Leishmaniaspp.; Plasmodium spp.; Toxopiasma gondii, Trichomonas spp., Trypanosoma spp., Ascaris lumbricoides;Clonorchis sinensis, Echinococcus spp.; Fasciola hepatica, Fasciolopsis buski; Heterophyes heterophyes;
3

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Hymenolepis spp.; Schistosoma spp.; Taenia spp. y Tilchinella spiralis) o virus (por ejemplo, Alphavi-rus; Arenavirus; Bunyavirus; Coronavirus; Enterovirus; Filovirus; Flavivirus; Hantavirus; HTLV-BLV;Influenzavirus: Lentivirus; Lyssavirus; Paramyxovirus; Reovirus; Rhinovirus y Rotavirus, Adenovirus;Cytomegalovirus; Hepadnavirus; Molluscipoxvirus; Orthopoxvirus; Papillomavirus; Parvovirus; Polyo-mavirus; Simplexvirus y Varicellovirus).
En otros aspectos, se proporciona una composicion que comprende un analogo de indolicidina deacuerdo con la invencion y un antibiotico. Ademas, se proporciona un dispositivo, que puede ser undispositivo medico, que se reviste con el analogo de indolicidina y puede comprender ademas un agenteantibiotico.
En otros aspectos, se proporcionan anticuerpos que reaccionan especıficamente con cualquiera de losanalogos aquı descritos. El anticuerpo preferiblemente es un anticuerpo monoclonal o anticuerpo de ca-dena simple.
En un aspecto preferido, se describe una composicion que comprende un compuesto de acuerdo conla invencion modificado por formacion de derivado en un grupo amino con un conjugado que comprendepolioxialquilenglicol activado y un acido graso. En realizaciones preferidas, el conjugado ademas com-prende sorbitan que une el polioxialquilenglicol y el acido grado, y mas preferiblemente es polisorbato.En realizaciones preferidas, el acido graso tiene 12-18 atomos de carbono, y el polioxialquilenglicol espolioxietilenglicol, tal como con una longitud de cadena de 2 a 100. En realizaciones preferidas, el polio-xialquilenglicol se activa por irradiacion con luz ultravioleta.
Tambien se describe un metodo para preparar un compuesto de acuerdo con la invencion, modificadocon un conjugado de un polioxialquilenglicol activado y un acido graso, que comprende: (a) congelar lamezcla del conjugado de un polioxialquilenglicol activado y acido graso con el compuesto; y (b) liofilizarla mezcla congelada; en el que el compuesto tiene un grupo amino libre. En realizaciones preferidas, elcompuesto es un peptido o antibiotico. En otras realizaciones preferidas, la mezcla en la etapa (a) estaen un tampon de acetato. En un aspecto relacionado, el metodo comprende mezclar el conjugado de unpolioxialquilenglicol activado y acido graso con el compuesto; durante un tiempo suficiente para formarcompuestos modificados, en el que la mezcla esta en un tampon de carbonato que tiene un pH mayor que8,5, y el compuesto tiene un grupo amino libre. El compuesto modificado se puede aislar por HPLC defase inversa y/o precipitacion en un disolvente organico.
Tambien se describe una composicion farmaceutica que comprende al menos un compuesto modificadode acuerdo con la invencion y un tampon fisiologicamente aceptable, y en ciertas realizaciones, ademascomprende un agente antibiotico, agente antivırico, un agente antiparasito, y/o agente antifungico. Lacomposicion se puede usar para tratar una infeccion, tal como la causada por un microorganismo (porejemplo, bacterias, hongos, parasitos y virus).
Estos y otros aspectos de la descripcion seran evidentes por referencia a la siguiente descripcion de-tallada y dibujos adjuntos. Ademas, a continuacion se presentan diferentes antecedentes que describencon mas detalle ciertos procedimientos o composiciones (por ejemplo, plasmidos, etc.).
Breve descripcion de los dibujos
La Figura 1 es un SDS-PAGE que muestra el perfil de extraccion de cuerpos de inclusion (ib) de celulasenteras que contienen proteına de fusion MBI-11. La banda de la proteına de fusion se indica conla cabeza de flecha. Carril 1, patrones de proteına; carril 2, lisato total de XL1 Blue sin plasmido;carril 3, lisato total de XL1 Blue (pR2h-11, pGP1-2), cultivado a 30◦C; carril 4, lisato total deXL1 Blue (pR2h-11, pGP1-2), inducido a 42◦C; carril 5, fraccion insoluble de cuerpos de inclusiondespues de lavado con Triton X100; carril 6, extracto organico de proteına de fusion MBI-11; carril7, material concentrado no soluble en disolvente organico de extraccion.
La figura 2 es un SDS-PAGE que muestra el perfil de expresion de la proteına de fusion MBI-11 usandoplasmido pPDR2h-11. Carril 1, patrones de proteına; carril 2, MBI-11 extraıdo en disolventeorganico; carril 3, lisato total de XL1 Blue (pPDR2h-11, pGP1-2), cultivado a 30◦C; carril 4, lisatototal de XL1 Blue (pPDR2h-11, pGP1-2), inducido a 42◦C.
La figura 3 presenta los resultados de ensayo de tiempo de letalidad para MBI 11CN, MBI 11F4CN yMBI 11B7CN. Se representa el numero de unidades formadoras de colonia x 10−4 frente al tiempo.
La Figura 4 es una grafica que representa la extension de la solubilidad del peptido MBI 11CN endiferentes tampones.
4

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
La Figura 5 es un perfil de HPLC de fase inversa de MBI 11CN en la formulacion C1 (panel de la graficaizquierda) y formulacion D (panel de la grafica derecha).
La figura 6 presenta el espectro de DC de MBI 11CN y MBI 11B7CN.
La Figura 7 presenta los resultados de liberacion de colorante de ANTS/DPX de liposomas de PC dehuevo con diferentes proporciones de lıpido a proteına.
La Figura 8 presenta graficas que muestran la actividad de MBI 11B7CN frente al crecimiento de celulasen mitad de la fase logarıtmica en caldo de Terrific (TB) o caldo de Luria-Bretani (LB).
La Figura 9 muestra los resultados del tratamiento de bacterias con MBI 10CN, MBI 11CN, o un peptidotestigo solo o combinado con valinomicina.
La figura 10 es una grafica que muestra el tratamiento de bacterias con MBI 11B7CN en presencia deNaCl o Mg2+.
La figura 11 es una grafica que presenta la cantidad in vitro de MBI 11CN en el plasma frente al tiempo.Se muestran los datos para peptido en formulacion C1 y formulacion D.
La figura 12 es una grafica que presenta el cambio en los niveles de MBI 11CN in vivo en la sangre endiferentes tiempos despues de inyeccion intravenosa.
La figura 13 es una grafica que presenta el cambio en los niveles de MBI 11CN in vivo en el plasma endiferentes tiempos despues de inyeccion intraperitoneal.
La Figura 14 es una grafica que muestra el numero de animales que sobreviven a una infeccion porSASM despues de inyeccion intraperitoneal de MBI 10CN, ampicilina o vehıculo.
La Figura 15 es una grafica que muestra el numero de animales que sobreviven a una infeccion porSASM despues de inyeccion intraperitoneal de MBI 11CN, ampicilina o vehıculo.
La figura 16 es una grafica que muestra los resultados de ensayar in vivo MBI-11A1CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 17 es una grafica que muestra los resultados de ensayar in vivo MBI-11E3CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 18 es una grafica que muestra los resultados de ensayar in vivo: MBI-11F3CN frente a S.aureus (Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ipuna hora despues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 19 es una grafica que muestra los resultados de ensayar in vivo MBI-11G2CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 20 es una grafica que muestra los resultados de ensayar in vivo MBI-11CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 21 es una grafica que muestra los resultados de ensayar in vivo MBI-11B1CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 22 es una grafica que muestra los resultados de ensayar in vivo MBI-11B7CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 23 es una grafica que muestra los resultados de ensayar in vivo MBI-11B8CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
La figura 24 es una grafica que muestra los resultados de ensayar in vivo MBI-11G4CN frente a S. aureus(Smith). Se administra peptido formulado en diferentes concentraciones por inyeccion ip una horadespues de infeccion con S. aureus (Smith) por inyeccion ip.
5

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Las Figuras 25A y B presentan una grafica que muestra el numero de animales que sobreviven a unainfeccion por S. epidermidis despues de inyeccion intravenosa de MBI 10CN, gentamicina, o vehıculo.Panel A, inyeccion i.v. 15 min despues de infeccion; panel B, inyeccion i.v. 60 min despues deinfeccion.
La figura 26 es una grafica que muestra el numero de animales que sobreviven a una infeccion por SARMde ratones despues de inyeccion intravenosa de MBI 11CN, gentamicina o vehıculo.
La Figura 27 presenta senales de RP-HPLC que analizan muestras de la formacion de APS-peptidodespues de tratamiento de polisorbato activado con un agente de reduccion. Los peptidos APS-MBI-11CN se forman por liofilizacion en acido acetico-NaOH 200 mM, pH 4,6, MBI 11CN 1 mg/ml,y polisorbato 80 al 0,5 % activado. La solucion madre de polisorbato al 2,0 % activado se trata con(a) agente de no reduccion, (b) 2-mercaptoetanol 150 mM, o (c) borohidruro sodico 150 mM durante1 hora inmediatamente antes de usar.
La figura 28 presenta senales de RP-HPLC que controlan la formacion de APS-MBI 11CN frente altiempo en solucion acuosa. La reaccion se produce en tampon de carbonato sodico 200 mM pH 10,MBI 11CN 0,1 mg/ml, polisorbato 80 al 0,5 % activado. Se separan partes alıcuotas del recipientede reaccion en los tiempos indicados y se analizan inmediatamente por RP-HPLC.
Descripcion detallada de la invencion
Antes de exponer la invencion, puede ser util para entenderla exponer definiciones de ciertos terminosque se usan aquı.
Las denominaciones de aminoacidos aquı se exponen como el codigo patron de una o tres letras. Unaletra mayuscula indica un aminoacido de forma L; una letra minuscula indica un aminoacido de formaD.
Tal como se usa aquı, “indolicidina” se refiere a un peptido cationico antimicrobiano. Las indolicidinasse pueden aislar de una variedad de organismos. Una indolicidina se aısla de neutrofilos bovinos y es unpeptido de 13 aminoacidos amidado en el carboxi terminal en su forma natural (Selsted et al., J. Biol.Chem. 267:4292, 1992). Se presenta una secuencia de aminoacidos de indolicidina en SEQ ID NO: 1.
Tal como se usa aquı, un “analogo de peptido” o “analogo” de indolicidina tiene hasta 25 5 aminoacidosde longitud y tiene actividad antimicrobiana. Salvo que se indique otra cosa, un aminoacido concretose refiere a la forma L. Entre los aminoacidos basicos se incluyen arginina, lisina y derivados. Entre losrestos hidrofobos se incluyen triptofano, fenilalanina, isoleucina, leucina, valina, y derivados.
Tambien se incluyen dentro del alcance de esta descripcion derivados de aminoacidos que se han alte-rado por medios quımicos, tales como metilacion (por ejemplo, α-metilvalina), amidacion, especialmentedel aminoacido C-terminal por una alquilamina (por ejemplo, etilamina, etanolamina, y etilendiamina)y alteracion de una cadena lateral de aminoacido, tal como acilacion del grupo ε-amino de lisina. Entreotros aminoacidos que se pueden incorporar en el analogo se incluyen cualesquiera de los D-aminoacidoscorrespondientes a los 20 L-aminoacidos que se encuentran normalmente en proteınas, imino-aminoacidos,aminoacidos raros, tales como hidroxilisina, o aminoacidos no proteicos, tales como homoserina y orni-tina. Un analogo de peptido puede tener ninguno, o uno o mas de estos derivados y D-aminoacidos.
A. Analogos de indolicidina
Como se ha indicado antes, aquı se describen analogos de indolicidina. Estos analogos se puedensintetizar por metodos quımicos, especialmente usando un sintetizador de peptidos automatizado, o sepueden producir por metodos recombinantes. La eleccion de una secuencia de aminoacidos es guiada poruna formula general presentada aquı.
1. Caracterısticas del peptido
Se describen aquı analogos de indolicidina que comprenden hasta 25 aminoacidos y que contienen laformula I L R W P W W P W R R K.
Como se ha descrito antes, la modificacion de cualquiera de los restos incluyendo el N- o C-terminal,esta dentro del alcance de la descripcion. Una modificacion preferida del extremo C-terminal es ami-dacion. Otras modificaciones del extremo C-terminal incluyen esterificacion y formacion de lactona.
6

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Entre las modificaciones N-terminales se incluyen acetilacion, acilacion, alquilacion, PEG-ilacion, miris-tilacion, y similares. Adicionalmente, el peptido se puede modificar para formar un APS-peptido comose ha descrito antes. Los peptidos tambien se pueden marcar, tal como con un marcador radiactivo, unmarcador fluorescente, una marca para espectrometrıa de masas, biotina y similares.
2. Sıntesis de peptido
Los analogos de peptidos de acuerdo con la invencion se pueden sintetizar por metodos quımicospatron, incluyendo sıntesis por procedimiento automatizado. En general, los analogos de peptidos sesintetizan basandose en la estrategia patron de proteccion con Fmoc en fase solida, con HATU comoagente de acoplamiento. El peptido se escinde de la resina de la fase solida con acido trifluoroacetico quecontiene depuradores adecuados, que tambien desprotege los grupos funcionales de las cadenas laterales.Despues el peptido bruto se purifica usando cromatografıa en fase inversa preparativa. Se pueden usarotros metodos de purificacion, tales como cromatografıa de particion, filtracion en gel, electroforesis engel, o cromatografıa de intercambio ionico.
Se pueden usar otras tecnicas de sıntesis, conocidas en la tecnica, tales como estrategia de proteccioncon tBoc, o uso de diferentes reactivos de acoplamiento o similares, para preparar peptidos equivalentes.
Los peptidos se pueden sintetizar como una molecula lineal o como moleculas ramificadas. Los peptidosramificados tıpicamente contienen un nucleo peptıdico que proporciona una serie de puntos de union parapeptidos adicionales. La lisina es la mas comunmente usada para el nucleo peptıdico porque tiene ungrupo funcional carboxilo y dos grupos funcionales amina (alfa y epsilon). Tambien se pueden usar otrosdiaminoacidos. Preferiblemente, se usan dos o tres niveles de lisinas geometricamente ramificadas; estosnucleos forman una estructura de nucleo tetramero u octamero, respectivamente (Tam, Proc. Natl. Acad.Sci. USA 85:5409, 1988). Esquematicamente, se representan ejemplos de estos nucleos como se muestra:
Los puntos de union para los peptidos tıpicamente estan en su grupo funcional carboxilo a los gruposamina alfa o epsilon de las lisinas. Para sintetizar estos peptidos multımeros, se forma un derivado de laresina de la fase solida con la matriz del nucleo, y la posterior sıntesis y escision de la resina siguen losprocedimientos patron. Despues el peptido multımero tıpicamente se purifica por dialisis frente a hidro-cloruro de guanidina 4 M y despues agua, usando una membrana con un tamano de poro para retenersolo multımeros. Los peptidos multımeros se pueden usar dentro del contexto de esta descripcion comocualquiera de los peptidos lineales y se prefieren para usar para generar anticuerpos para los peptidos.
3. Produccion recombinante de peptidos
Los analogos de peptidos de acuerdo con la invencion se pueden sintetizar alternativamente por pro-duccion recombinante (vease por ejemplo, patente de EE.UU. No. 5.593.866). Son adecuados una va-riedad de sistemas hospedantes para producir analogos de peptidos, incluyendo bacterias (por ejemplo,E. coli), levaduras (por ejemplo, Saccharomyces cerevisiae), insectos (por ejemplo, Sf9), y celulas demamıferos (por ejemplo, CHO, COS-7). Se han desarrollado muchos vectores de expresion y estan dis-ponibles para cada uno de estos hospedantes. En general, en esta invencion se usan celulas bacterianasy vectores que son funcionales en bacterias. Sin embargo, a veces puede ser preferible tener vectoresque son funcionales en otros hospedantes. Aquı se discuten vectores y procedimientos para clonacion yexpresion en E. coli, y por ejemplo, en Sambrook et al. (Molecular Cloning: A Laboratory Manual, ColdSpring Harbor Laboratory Press, Cold Spring Harbor, NY, 1987) y en Ausubel et al. (Current Protocolsin Molecular Biology, Greene Publishing Co., 1995).
Se introduce una secuencia de DNA que codifica uno o mas analogos de indolicidina en un vector deexpresion adecuado para el hospedante. En realizaciones preferidas, el gen analogo se clona en un vector
7

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
para crear una proteına de fusion. La pareja de fusion se elige para que contenga una region anionica, deforma que un hospedante bacteriano este protegido del efecto toxico del peptido. La region protectoraneutraliza eficazmente los efectos antimicrobianos del peptido y tambien puede evitar la degradacion delpeptido por las proteasas del hospedante. La pareja de fusion (proteına vehıculo) aquı descrita puedefuncionar ademas para transportar el peptido de fusion a cuerpos de inclusion, el periplasma, la mem-brana exterior, o el entorno extracelular. Entre las proteınas vehıculo adecuadas en el contexto de estadescripcion se incluyen especıficamente, pero no se limitan, glutation-S-transferasa (GST), proteına Ade Staphylococcus aureus, dos dominios de union de IgG sinteticos (ZZ) de la proteına A, proteına Fde membrana exterior, β-galactosidasa (IacZ), y diferentes productos de bacteriofago λ y bacteriofagoT7. A partir de las ensenanzas que se proporciona aquı, es evidente que se pueden usar otras proteınascomo vehıculos. Ademas, no es necesario usar la proteına vehıculo entera, siempre que la region anionicaprotectora este presente. Para facilitar el aislamiento de la secuencia de peptido, se usan aminoacidossusceptibles de escision quımica (por ejemplo, CNBr) o escision enzimatica (por ejemplo, proteasa V8,tripsina) para unir el peptido y la pareja de fusion. Para expresion en E. coli, la pareja de fusion prefe-riblemente es una proteına intracelular normal que dirige la expresion hacia la formacion de cuerpo deinclusion. En dicho caso, despues de escision para liberar el producto final, no es necesario renaturalizarel peptido. En esta descripcion el casete de DNA, que comprende la pareja de fusion y el gen del peptido,se puede insertar en un vector de expresion, que puede ser un plasmido, virus u otro vehıculo conocidoen la tecnica. Preferiblemente, el vector de expresion es un plasmido que contiene un promotor inducibleo constitutivo para facilitar la transcripcion eficaz de la secuencia de DNA insertada en el hospedante.La transformacion de la celula hospedante con el DNA recombinante se puede llevar a cabo por tecnicasmediadas por Ca++, por electroporacion, u otros metodos conocidos por los expertos en la tecnica.
Brevemente, un fragmento de DNA que codifica un analogo de peptido se obtiene de un cDNA exis-tente o clon genomico, o se sintetiza. Un metodo conveniente es la amplificacion del gen a partir de unmolde de hebra simple. El molde generalmente es el producto de una sıntesis de oligonucleotido auto-matizada. Los cebadores de amplificacion se obtienen de los extremos 5’ y 3’ del molde y tıpicamenteincorporan sitios de restriccion elegidos teniendo en cuenta el sitio de clonacion del vector. Si es necesario,los codones de iniciacion y terminacion de traduccion se pueden construir en las secuencias del cebador.En la secuencia que codifica la proteına se puede optimizar el codon para expresion en el hospedanteparticular. Ası, por ejemplo, si la proteına de fusion analoga se expresa en bacterias, los codones seoptimizan para el uso bacteriano. La optimizacion de codon se lleva a cabo por sıntesis automatizadadel gen entero o region del gen, union de oligonucleotidos multiples, mutagenesis de la secuencia nativa,u otras tecnicas conocidas por los expertos en la tecnica.
Como mınimo, el vector de expresion debe contener una secuencia de promotor. Sin embargo, tambiense pueden incluir otras secuencias reguladoras. Entre dichas secuencias se incluyen un activador, sitio deunion de ribosoma, secuencia de senal de terminacion de transcripcion, secuencia de senal de secrecion,origen de replicacion, marcador seleccionable, y similares. Las secuencias reguladoras estan asociadasen funcionamiento entre sı para permitir la transcripcion y posterior traduccion. En aspectos preferi-dos, los plasmidos usados aquı para expresion incluyen un promotor disenado para expresar proteınas enbacterias. Los promotores adecuados, incluyendo tanto promotores constitutivos como inducibles, estanampliamente disponibles y son conocidos en la tecnica. Entre los promotores usados normalmente paraexpresar en bacterias se incluyen promotores de fagos T7, T3, T5 y SP6, y los operones trp, lpp y lac.Tambien se pueden usar promotores hıbridos (vease, patente de EE.UU. n◦ 4.551.433), tales como tac ytrc.
En realizaciones preferidas, el vector incluye una secuencia terminadora de transcripcion. Una “regionterminadora de transcripcion” es una secuencia que proporciona una senal que termina la transcripcionpor la polimerasa que reconoce el promotor seleccionado. El terminador de transcripcion se puede obtenerdel gen de la pareja de fusion o de otro gen, siempre que funcione en el hospedante.
En una realizacion preferida, el vector es capaz de replicacion en celulas bacterianas. Por lo tanto, elvector puede contener un origen de replicacion bacteriano. Entre los orıgenes de replicacion bacterianospreferidos se incluyen f1-ori y col E1 ori, especialmente el ori derivado de plasmidos pUC. Tambien sepueden usar vectores de bajo numero de copias (por ejemplo, pPD100), especialmente cuando el productoes perjudicial para el hospedante.
Los plasmidos preferiblemente tambien incluyen al menos un marcador seleccionable que funciona enel hospedante. Un gen marcador seleccionable confiere un fenotipo al hospedante que permite identificary/o hacer crecer selectivamente las celulas transformadas. Entre los genes de marcadores seleccionablesadecuados para hospedantes bacterianos se incluyen gen de resistencia al cloranfenicol (Cmr), gen de
8

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
resistencia a la ampicilina (Ampr), gen de resistencia a la tetraciclina (Tcr), gen de resistencia a la ka-namicina (Kanr), y otros conocidos en la tecnica. Para funcionar en la seleccion, algunos marcadorespueden necesitar una deficiencia complementaria en el hospedante.
En algunos aspectos, la secuencia de nucleotidos que codifica el analogo de peptido, tambien codificauna senal de secrecion, de forma que el peptido resultante se sintetiza como una proteına precursora, queposteriormente es procesada y secretada. La proteına secretada resultante se puede recuperar del espacioperiplasmico o del medio de fermentacion. Las secuencias de senales de secrecion adecuadas para usar,estan ampliamente disponibles y son conocidas (von Hejine, J. Mol. Biol. 184:99-105, 1985).
El vector tambien puede contener un gen que codifica una proteına represora, que es capaz de reprimirla transcripcion de un promotor que contiene un sitio de union del represor. La alteracion de las condi-ciones fisiologicas de la celula puede deprimir al promotor. Por ejemplo, se puede anadir una moleculaque se une competitivamente al represor, o se puede alterar la temperatura del medio de crecimiento.Entre las proteınas represoras se incluyen, pero no se limita, el represor lacl de E. coli (responsable deinduccion por IPTG), el represor λcl857 sensible a la temperatura, y similares.
Entre los ejemplos de plasmidos para expresar en bacterias se incluyen los vectores de expresion pET:pET 3a, pET 11a, pET 12a-c, y pET 15b (vease patente de EE.UU. 4.952.496; disponible en Novagen,Madison, WI). Se pueden usar vectores de bajo numero de copias (por ejemplo, pPD100) para la sobre-produccion eficaz de peptidos perjudiciales para el hospedante E. coli (Dersch et al., FEMS Micobiol.Lett.123: 19, 1994).
Los hospedantes bacterianos para los vectores de expresion T7 pueden contener copias cromosomicasde DNA que codifica la RNA-polimerasa T7 unido de forma factible a un promotor inducible (por ejem-plo, promotor lacUV; vease patente de EE.UU.. n◦ 4.952.496), tal como el encontrado en cepas de E. coliHMS174(DE3)pLysS, BL21(DE3)pLysS, HMS174(DE3) y BL21(DE3). La RNA-polimerasa T7 tambienpuede estar presente en plasmidos compatibles con el vector de expresion T7. La polimerasa puede estarcontrolada por un promotor y represor lambda (por ejemplo, pGP1-2; Tabor and Richardson, Proc. Natl.Acad. Sci. USA 82: 1074, 1985).
El peptido analogo de proteına se aısla por tecnicas patron, tales como cromatografıa de afinidad,de exclusion por tamano, o de intercambio ionico, HPLC y similares. Un peptido aislado debe mostrarpreferiblemente una banda principal por tenido con azul Coomassie del SDS-PAGE que es al menos 90 %del material.
4. Generacion de analogos por mutagenesis semialeatoria basada en amplificacion
Los analogos de indolicidina de acuerdo con la invencion, se pueden generar usando un procedimientobasado en amplificacion (por ejemplo, PCR) en el que los cebadores son designados a secuencias objetivoen los extremos 5’ y 3’ de un peptido relacionado codificado, por ejemplo indolicidina. Las condiciones deamplificacion se eligen para facilitar la mala incorporacion de nucleotidos por la polimerasa termoestabledurante la sıntesis. Por lo tanto, se introducen mutaciones aleatorias en la secuencia original, algunas delas cuales dan como resultado alteracion(es) de aminoacido(s). Los productos de amplificacion se puedenclonar en una proteına de revestimiento de un vector fago, tal como un vector fagemido, empaquetar yamplificar en un hospedante aceptable para producir una biblioteca de muestra.
Despues en estas bibliotecas se puede ensayar la actividad antibiotica de los peptidos. Brevemente,bacterias infectadas con la biblioteca se cultivan en placa, se hacen crecer, y se recubren con agarosa quecontiene una cepa bacteriana que los fagos son incapaces de infectar. Se observan las zonas de inhibicionde crecimiento en el recubrimiento de agarosa en la zona del fago que expresa un analogo con actividadantibacteriana. Estos fagos inhibidores se aıslan y se determina la secuencia de peptido clonada poranalisis de secuencia de DNA. Despues el peptido se puede sintetizar independientemente y posterior-mente investigar su actividad antibiotica.
5. Anticuerpos para analogos de indolicidina
Los anticuerpos tıpicamente se generan para un analogo de peptido especıfico usando peptidos an-tigenicos multiples (MAP) que contienen aproximadamente ocho copias del peptido unido a un pequenonucleo de peptidilo no inmunogenico para formar un inmunogen. (Vease, en general, Harlow and Lane,vease antes). Los MAP se inyectan vıa subcutanea en conejos o en ratones u otros roedores, dondepueden tener semividas suficientemente largas para facilitar la produccion de anticuerpos. Despues de
9

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
doce semanas se toman muestras de sangre, se separa el suero y se ensaya en un ensayo ELISA frente alpeptido original, indicando un resultado positivo la presencia de anticuerpos especıficos para el peptidoobjetivo. Despues este suero se puede almacenar y usar en ensayos ELISA para medir especıficamentela cantidad de analogo especıfico. Alternativamente, se pueden usar otros metodos patron de produccionde anticuerpos, por ejemplo, generacion de anticuerpos monoclonales.
En el contexto de esta descripcion, se entiende que anticuerpos incluye anticuerpos monoclonales, anti-cuerpos policlonales, anticuerpos anti-idiotıpicos, fragmentos de anticuerpo (por ejemplo, Fab, y F(ab’)2,regiones variables Fv, o regiones que determinan complementaridad). Los anticuerpos se aceptan engeneral como especıficos frente a analogos de indolicidina de acuerdo con la invencion, si se unen con unaKd mayor o igual a 10−7 M, preferiblemente mayor o igual a 10−8 M. La afinidad de un anticuerpo mono-clonal o pareja de union puede ser determinada facilmente por un experto en la tecnica (vease Scatchard,Ann. N.Y. Acad. Sci. 51: 660-672, 1949). Una vez que se han obtenido los anticuerpos adecuados, sepueden aislar y purificar por tecnicas conocidas por los expertos en la tecnica.
Los anticuerpos monoclonales tambien se pueden generar facilmente a partir de celulas de hibridomausando tecnicas convencionales (vease patente de EE.UU. n◦ RE 32.011, 4.902.614, 4.543.439, y 4.411.993;vease tambien Antibodies: A Laboratroy Manual, Harlow and Lane (eds.), Cold Spring Harbor LaboratoryPress, 1988). Brevemente, en una realizacion, se inyecta peptido en un sujeto animal tal como una ratao raton, generalmente administrado como una emulsion en un adyuvante tal como adyuvante completo oincompleto de Freunds con el fin de aumentar la respuesta inmunitaria. Generalmente el animal se vuelvea inyectar al menos una vez antes de recoger el bazo y/o nodos linfaticos e inmortalizar estas celulas. Sepueden usar diferentes tecnicas de inmortalizacion, tales como mediadas por virus Epstein-Barr o fusionpara producir un hibridoma. En una realizacion preferida, la inmortalizacion se produce por fusion conuna lınea celular de mieloma adecuada para crear un hibridoma que segrega anticuerpo monoclonal. Entrelas lıneas de mieloma adecuadas se incluyen, por ejemplo, NS-1 (ATCC n◦ TIB 18) y P3X63 - Ag 8.653(ATCC n◦ CRL 1580). Las parejas de fusion preferidas no expresan genes de anticuerpos endogenos.Despues de aproximadamente siete dıas, se puede seleccionar en los hibridomas la presencia de anticuer-pos que son reactivos frente a una proteına telomerasa. Se puede usar una gran variedad de ensayos(vease Antibodies: A Laboratory Manual, Harlow and Lane (eds.), Cold Spring Harbor Laboratory Press,1988).
Tambien se pueden usar otras tecnicas para construir anticuerpos monoclonales (vease Huse et al.,Science 246: 1275-1281, 1989; Sastry et al., Proc. Natl. Acad. Sci. USA 86:5728-5732, 1989; Alting-Meeset al., Strategies in Molecular Biology 3: 1-9, 1990; que describe tecnicas recombinantes). Estas tecnicasincluyen clonacion de la cadena pesada y ligera de cDNA de inmunoglobulina en vectores adecuados talescomo λInmunoZap(H) y λInmunoZap(L). Estos productos recombinantes se pueden seleccionar indivi-dualmente o se pueden coexpresar para formar fragmentos Fab o anticuerpos (vease Huse et al., veaseantes; Sastry et al., vease antes). Posteriormente las placas positivas se pueden convertir en un plasmidono lıtico que permite un alto nivel de expresion de fragmentos de anticuerpo monoclonal a partir de E.coli.
Igualmente, tambien se pueden construir porciones o fragmentos Fab y Fv de anticuerpos usando di-gestion enzimatica convencional o tecnicas de DNA recombinante para dar regiones variables aisladas deun anticuerpo. En una realizacion, los genes que codifican la region variable de un hibridoma que produceun anticuerpo monoclonal interesante, se amplifican usando cebadores nucleotidos para la region variable.Ademas, se pueden usar tecnicas para cambiar un anticuerpo “murino” por un anticuerpo “humano”, sinalterar la especificidad de union del anticuerpo.
B. Ensayos
Se evalua en los analogos de indolicidina de esta descripcion solos o combinados con un agente an-tibiotico u otro analogo, su potencial como agentes terapeuticos antibioticos usando una serie de ensayos.Preferiblemente, todos los peptidos se evaluan inicialmente in vitro, se seleccionan los candidatos masprometedores para la posterior evaluacion in vivo, y usando los resultados de estos ensayos, se seleccionanlos candidatos para los estudios preclınicos. Entre los ensayos in vitro se incluyen medicion de la actividadantibiotica, toxicidad, solubilidad, farmacologıa, estructura secundaria, permeabilizacion de liposoma ysimilares. Entre los ensayos in vivo se incluyen la evaluacion de la eficacia en modelos animales, antige-nicidad, toxicidad, y similares. En general, se llevan a cabo inicialmente los ensayos in vitro, seguidos delos ensayos in vivo.
10

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
1. Ensayos in vitro
Se evalua la actividad antibiotica de los analogos de indolicidina por un ensayo tal como un ensayode CIM por dilucion en agarosa o un ensayo de dilucion en caldo o de tiempo de letalidad. La actividadantibiotica se mide como la inhibicion del crecimiento o muerte de un microorganismo (por ejemplo,bacterias, hongos). Brevemente, un analogo candidato en caldo de Mueller Hinton complementado concalcio y magnesio se mezcla con agarosa fundida. Se pueden usar otras formulaciones de caldos y agarcon la condicion de que el analogo de peptido se pueda difundir libremente por el medio. La agarosa sevierte en placas petri o pocillos, se deja solidificar, y se aplica una cepa de ensayo a la placa de agarosa.La cepa de ensayo se elige, en parte, por la aplicacion pretendida del analogo. Por lo tanto, a modo deejemplo, si se desea un analogo con actividad frente a S. aureus, se usa una cepa de S. aureus. Puedeser conveniente ensayar el analogo en varias cepas y/o en aislados clınicos de las especies de ensayo. Lasplacas se incuban toda la noche, y al dıa siguiente se inspecciona visualmente el crecimiento bacteriano.La concentracion inhibidora mınima (CIM) de un analogo es la concentracion mas baja de peptido queinhibe completamente el crecimiento del organismo. Los analogos que presentan una buena actividadfrente a la cepa de ensayo, o grupo de cepas, que tıpicamente tienen una CIM menor o igual que 16µg/ml se seleccionan para posterior ensayo.
En los analogos seleccionados se puede ensayar ademas su toxicidad para celulas normales de mamıfero.Un ensayo de ejemplo es un ensayo de hemolisis de globulos rojos (GR) (eritrocitos). Brevemente, seaıslan globulos rojos de sangre entera, tıpicamente por centrifugacion, y se lavan de los componentes delplasma. Se incuba una suspension al 1 % (vol/vol) de eritrocitos en solucion salina isotonica con diferen-tes concentraciones de analogo de peptido. En general, el analogo estara en un tampon de formulacionadecuado. Despues de incubar durante aproximadamente 1 hora a 37◦C, las celulas se centrifugan, y sedetermina la absorbancia del lıquido sobrenadante a 540 nm. Se determina una medida relativa de lisispor comparacion de la absorbancia despues de la lisis completa de eritrocitos usando NH4Cl o equivalente(que establece un valor de 100 %). Un analogo que no es lıtico o solo es moderadamente lıtico, como seejemplifica en el Ejemplo 8, es conveniente y es adecuado para la posterior seleccion. Se pueden usarotros ensayos de toxicidad in vitro, por ejemplo medicion de la toxicidad frente a celulas de mamıferocultivadas, para ensayar la toxicidad in vitro.
La solubilidad del analogo de peptido en tampon de formulacion es un parametro adicional que sepuede examinar. Se pueden usar varios ensayos diferentes, tal como el aspecto en el tampon. Brevemente,el analogo de peptido se suspende en solucion, tal como caldo o tampon de formulacion. Se evalua elaspecto de acuerdo con una escala que varıa desde (a) transparente, no precipita, (b) claro, precipitadodifuso, a (c) turbio, precipitado pesado. Se pueden usar graduaciones mas finas. En general, es conve-niente menos precipitado. Sin embargo, puede ser aceptable algo de precipitado.
Se pueden llevar a cabo ensayos in vitro adicionales para evaluar el potencial del analogo como unagente terapeutico. Dichos ensayos incluyen solubilidad del peptido en formulaciones, farmacologıa en lasangre o plasma, union a proteınas del suero, analisis de estructura secundaria, por ejemplo, dicroısmocircular, permeabilizacion de liposoma, y permeabilizacion de la membrana interior bacteriana. En gene-ral, es conveniente que los analogos sean solubles y se comporten mejor que la indolicidina.
2. Ensayos in vivo
En los analogos seleccionados basandose en los resultados de los ensayos in vitro, se puede ensayar invivo la eficacia, toxicidad y similares.
La actividad antibiotica de analogos seleccionados se puede evaluar in vivo por su capacidad paramejorar infecciones microbianas usando modelos animales. En estos ensayos, un analogo es util como unagente terapeutico si la inhibicion del crecimiento de microorganismos comparada con la inhibicion convehıculo solo es estadısticamente significativa. Esta medicion se puede hacer directamente a partir decultivos aislados de fluidos o sitios corporales, o indirectamente, evaluando las tasas de supervivencia deanimales infectados. Para evaluar la actividad antibacteriana hay disponibles varios modelos animales,tales como modelos de infeccion aguda incluyendo aquellos en los que (a) ratones normales reciben unadosis letal de microorganismos, (b) ratones neutropenicos reciben una dosis letal de microorganismos o(c) conejos reciben un inoculo en el corazon, y modelos de infeccion cronica. El modelo seleccionadodependera en parte de la indicacion clınica pretendida del analogo.
A modo de ejemplo, en uno de dichos modelos de raton normal, los ratones se inoculan vıa ip o iv conuna dosis letal de bacterias. Tıpicamente, la dosis es tal que 90-100% de los animales mueren en 2 dıas.
11

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
La eleccion de la cepa de microorganismo para este ensayo depende, en parte, de la aplicacion pretendidadel analogo, y en los ejemplos que acompanan los ensayos se llevan a cabo en tres cepas diferentes deStaphylococcus. Brevemente, poco antes o despues de la inoculacion (generalmente en 60 minutos), seinyecta analogo en un tampon de formulacion adecuado. Se pueden administrar inyecciones multiples delanalogo. Los animales se observan durante hasta 8 dıas despues de infeccion y se registra la supervivenciade los animales. El tratamiento satisfactorio rescata a los animales de la muerte o retrasa la muerta aun nivel estadısticamente significativo, comparado con animales testigo sin tratamiento. Se prefieren losanalogos que muestran mejor eficacia que la propia indolicidina.
La toxicidad in vivo de un analogo se mide por administracion de un intervalo de dosis a animales,tıpicamente ratones, por una vıa definida en parte por el uso clınico pretendido. Se registra la super-vivencia de los animales, y se puede calcular la DL50, DL90−100, y dosis maxima tolerada (DMT) parapermitir la comparacion de los analogos. Se prefieren los analogos menos toxicos que la indolicidina.
Se pueden llevar a cabo ensayos in vivo adicionales para ayudar a la seleccion de analogos para el desa-rrollo clınico. Por ejemplo, se puede evaluar la inmunogenicidad de los analogos, tıpicamente por inyecciondel analogo en tampon de formulacion en animales normales, generalmente ratones, ratas o conejos. Seobtiene suero en diferentes tiempos despues de inyeccion, y se ensaya la presencia de anticuerpos que seunen al analogo. Tambien se pueden llevar a cabo ensayos despues de inyecciones multiples, protocolosde tratamiento mimetico. Los anticuerpos para los analogos se pueden identificar por ELISA, ensayos deinmunoprecipitacion, transferencias Western, y otros metodos. (Vease, Harlow and Lane, Antibodies: ALaboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1988). Se prefierenlos analogos que no provocan o provocan la produccion mınima de anticuerpos. Adicionalmente, se pue-den determinar las farmacocineticas de los analogos en animales y la histopatologıa de animales tratadoscon los analogos.
La seleccion de los analogos de indolicidina como potenciales agentes terapeuticos se basa en resulta-dos de ensayos in vitro e in vivo. En general, los analogos de peptidos que presentan baja toxicidad conaltos niveles de dosis y alta eficacia con bajos niveles de dosis son candidatos preferidos.
3. Ensayos de sinergismo
Para evaluar analogos combinados con un antibiotico u otro analogo, la combinacion se puede sometera las series de ensayos anteriores. Los antibioticos incluyen cualquier producto quımico que tienda a preve-nir, inhibir o destruir vida y como tales, los antibioticos incluyen agentes antibacterianos, antifungicidas,agentes antivıricos, y agentes antiparasitos. Simplemente a modo de ejemplo, se discuten los antibioticosantibacterianos. Los metodos para mezclar y administrar los componentes varıan dependiendo del usoclınico pretendido de la combinacion.
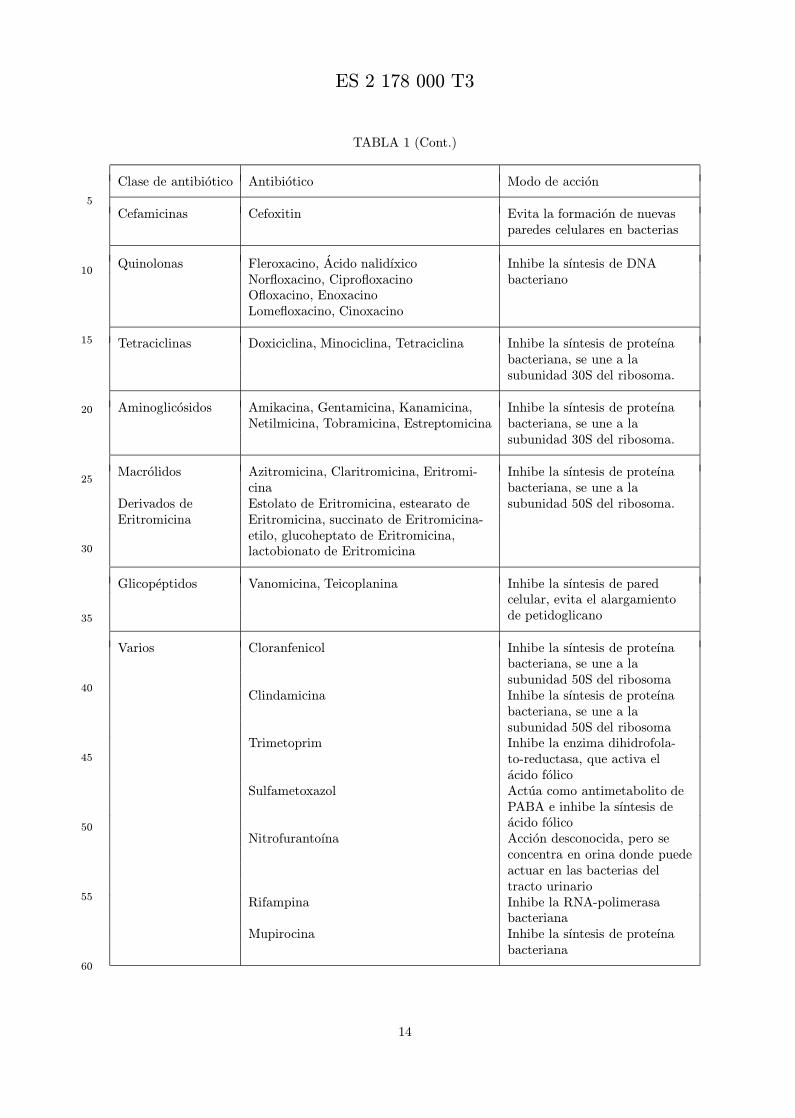
Brevemente, un ensayo de la actividad antibacteriana in vitro, el ensayo de dilucion en agarosa, seinicia con una matriz de placas que contiene cada una, una combinacion de analogo de peptido y an-tibiotico en diferentes concentraciones. Las placas se inoculan con aislados bacterianos, se incuban, yse registran las CIM de los componentes. Despues, estos resultados se usan para calcular la CIF. Entrelos antibioticos usados en el ensayo se incluyen, pero no se limitan, penicilinas, cefalosporinas, carbace-fems, cefamicinas, carbapenems, monobctamas, aminoglicosidos, glicopeptidos, quinolonas, tetraciclinas,macrolidos, y fluoroquinolonas (vease la siguiente Tabla 1).
Entre los ejemplos de antibioticos se incluyen, pero no se limitan, Penicilina G (Registro CAS N◦: 61-33-6); Meticilina (Registro CAS N◦: 61-32-5); Nafcilina (Registro CAS N◦: 147-52-4); Oxacilina (RegistroCAS N◦: 66-79-5); Cloxacilina (Registro CAS N◦: 61-72-3); Dicloxacilina (Registro CAS N◦: 3116-76-5);Ampicilina (Registro CAS N◦: 69-53-4); Amoxicilina (Registro CAS N◦: 26787-78-0); Ticarcilina (Regis-tro CAS N◦: 34787-01-4); Carbenicilina (Registro CAS N◦: 4697-36-3); Mezlocilina (Registro CAS N◦:51481-65-3); Azlocilina (Registro CAS N◦: 37091-66-0); Piperacilina (Registro CAS N◦: 61477-96-1); Imi-penem (Registro CAS N◦: 74431-23-5); Aztreonam (Registro CAS N◦: 78110-38-0); Cefalotina (RegistroCAS N◦: 153-61-7); Cefazolina (Registro CAS N◦: 25953-19-9); Cefaclor (Registro CAS N◦: 70356-03-5);Cefamandol-formiato sodico (Registro CAS N◦: 42540-40-9); Cefoxitina (Registro CAS N◦: 35607-66-0); Cefuroxima (Registro CAS N◦: 55268-75-2); Cefonicid (Registro CAS N◦: 61270-58-4); Cefmetazol(Registro CAS N◦: 56796-20-4); Cefotetan (Registro CAS N◦: 69712-56-7); Cefprozil (Registro CAS N◦:92665-29-7); Loracarbef (Registro CAS N◦: 121961-22-6); Cefetamet (Registro CAS N◦: 65052-63-3);Cefoperazona (Registro CAS N◦: 62893-19-0); Cefotaxima (Registro CAS N◦: 63527-52-6); Ceftizoxima(Registro CAS N◦: 68401-81-0); Ceftriaxona (Registro CAS N◦: 73384-59-5); Ceftazidima (Registro CASN◦: 72558-82-8); Cefepima (Registro CAS N◦: 88040-23-7); Cefixima (Registro CAS N◦: 79350-37-1);
12

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Cefpodoxima (Registro CAS N◦: 80210-62-4); Cefsulodina (Registro CAS N◦: 62587-73-9); Fleroxacino
(Registro CAS N◦: 79660-72-3); Acido nalidıxico (Registro CAS N◦: 389-08-2); Norfloxacino (RegistroCAS N◦: 70458-96-7); Ciprofloxacino (Registro CAS N◦: 85721-33-1); Ofloxacino (Registro CAS N◦:82419-36-1); Enoxacino (Registro CAS N◦: 74011-58-8); Lomefloxacino (Registro CAS N◦: 98079-51-7);Cinoxacino (Registro CAS N◦: 28657-80-9); Doxiciclina (Registro CAS N◦: 564-25-0); Minociclina (Re-gistro CAS N◦: 10118-90-8); Tetraciclina (Registro CAS N◦: 60-54-8); Amikacina (Registro CAS N◦:37517-28-5); Gentamicina (Registro CAS N◦: 1403-66-3); Kanamicina (Registro CAS N◦: 8063-07-8);Netilmicina (Registro CAS N◦: 56391-56-1); Tobramicina (Registro CAS N◦: 32986-56-4); Estreptomi-cina (Registro CAS N◦: 57-92-1); Azitromicina (Registro CAS N◦: 83905-01-5); Claritromicina (RegistroCAS N◦: 81103-11-9); Eritromicina (Registro CAS N◦: 114-07-8); estolato de Eritromicina (RegistroCAS N◦: 3521-62-8); succinato de Eritromicina y etilo (Registro CAS N◦: 41342-53-4); glucohepto-nato de Eritromicina (Registro CAS N◦: 23067-13-2); lactobionato de Eritromicina (Registro CAS N◦:3847-29-8); estearato de Eritromicina (Registro CAS N◦: 643-22-1); Vancomicina (Registro CAS N◦:1404-90-6); Teicoplanina (Registro CAS N◦: 61036-64-4); Cloranfenicol (Registro CAS N◦: 56-75-7);Clindamicina (Registro CAS N◦: 18323-44-9); Trimetoprim (Registro CAS N◦: 738-70-5); Sulfameto-xazol (Registro CAS N◦: 723-46-6); Nitrofurantoına (Registro CAS N◦: 67-20-9); Rifampina (RegistroCAS N◦: 13292-46-1); Mupirocina (Registro CAS N◦: 12650-69-0); Metronidazol (Registro CAS N◦:443-48-1); Cefalexina (Registro CAS N◦: 15686-71-2); Roxitromicina (Registro CAS N◦: 80214-83-1);Co-amoxiclavulanato; combinaciones de Piperacilina y Tazobactam; y sus diferentes sales, acidos, basesy otros derivados.
TABLA 1
Clase de antibiotico Antibiotico Modo de accion
Penicilinas Bloquea la formacion de nuevasparedes celulares en bacterias
Natural Penicilina G, Bencilpenicilina,Penicilina V, Fenoximetilpenicilina
Resistente a penicilinasa Meticilina, Nafcilina, OxacilinaCloxacilina, Dicloxacilina
Acilaminopenicilinas Ampicilina, AmoxicilinaCarboxipenicilinas Ticarcilina, CarbenicilinaUreidopenicilinas Mezlocilina, Azlocilina, Piperacilina
Carbapenems Imipenem, Meropenem Bloquea la formacion de nuevasparedes celulares en bacterias
Monobactams Aztreonam Bloquea la formacion de nuevasparedes celulares en bacterias
Cefalosporinas Evita la formacion de nuevasparedes celulares en bacterias
1a¯ Generacion Cefalotin, Cefazolina
2a¯ Generacion Cefaclor, Cefamandol
Cefuroxima, Cefonicida,Cefmetazol, Cefotetan, Cefprozil
3a¯ Generacion Cefetamet, Cefoperazona
Cefotaxima, CeftizoximaCeftriaxona, CeftazidimaCefixima, Cefpodoxima, Cefsulodina
4a¯ Generacion Cefepima
Carbacefems Loracarbef Evita la formacion de nuevasparedes celulares en bacterias
13
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 1 (Cont.)
Clase de antibiotico Antibiotico Modo de accion
Cefamicinas Cefoxitin Evita la formacion de nuevasparedes celulares en bacterias
Quinolonas Fleroxacino, Acido nalidıxico Inhibe la sıntesis de DNANorfloxacino, Ciprofloxacino bacterianoOfloxacino, EnoxacinoLomefloxacino, Cinoxacino
Tetraciclinas Doxiciclina, Minociclina, Tetraciclina Inhibe la sıntesis de proteınabacteriana, se une a lasubunidad 30S del ribosoma.
Aminoglicosidos Amikacina, Gentamicina, Kanamicina, Inhibe la sıntesis de proteınaNetilmicina, Tobramicina, Estreptomicina bacteriana, se une a la
subunidad 30S del ribosoma.
Macrolidos Azitromicina, Claritromicina, Eritromi- Inhibe la sıntesis de proteınacina bacteriana, se une a la
Derivados de Estolato de Eritromicina, estearato de subunidad 50S del ribosoma.Eritromicina Eritromicina, succinato de Eritromicina-
etilo, glucoheptato de Eritromicina,lactobionato de Eritromicina
Glicopeptidos Vanomicina, Teicoplanina Inhibe la sıntesis de paredcelular, evita el alargamientode petidoglicano
Varios Cloranfenicol Inhibe la sıntesis de proteınabacteriana, se une a lasubunidad 50S del ribosoma
Clindamicina Inhibe la sıntesis de proteınabacteriana, se une a lasubunidad 50S del ribosoma
Trimetoprim Inhibe la enzima dihidrofola-to-reductasa, que activa elacido folico
Sulfametoxazol Actua como antimetabolito dePABA e inhibe la sıntesis deacido folico
Nitrofurantoına Accion desconocida, pero seconcentra en orina donde puedeactuar en las bacterias deltracto urinario
Rifampina Inhibe la RNA-polimerasabacteriana
Mupirocina Inhibe la sıntesis de proteınabacteriana
14

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
El sinergismo se calcula de acuerdo con la siguiente formula. Una CIF de ≤ 0,5 es evidencia desinergismo, aunque pueden ser terapeuticamente utiles combinaciones con valores mayores.
CIM (peptido combinado)CIM (peptido solo)
+CIM (antibiotico combinado)
CIM (antibiotico solo)= CIF
Por ejemplo, se pueden usar antibioticos de los grupos de penicilinas, cefalosporinas, carbacefems, cefa-micinas, carbapenems, monobctams, aminoglicosidos, glicopeptidos, quinolonas, tetraciclinas, macrolidos,floroquinolonas y otros antibioticos variados, combinados con cualquiera de los peptidos aquı descritos.
C. Modificacion polimera de peptidos y proteinas
Como se indica aquı, se describen metodos y composiciones para modificar un compuesto de acuerdocon la invencion, con un ester de polisorbato activado y derivados. Las formas modificadas o derivadasse denominan aquı “peptidos modificados con APS” o “proteınas modificadas con APS”. Los compuestosmodificados con APS (por ejemplo, APS-peptidos cationicos) tienen mejores propiedades farmacologicas.
1. Caracterısticas del reactivo
Como se discute aquı, un reactivo adecuado para formar compuestos modificados con APS de acuerdocon la invencion (por ejemplo, peptidos y proteınas) comprende una region hidrofoba y una regionhidrofila, y opcionalmente un ligador. La region hidrofoba es un compuesto lipofilo con un grupo funcionaladecuado para conjugar con la region hidrofila o el ligador. La region hidrofila es un polialquilenglicol.Tal como se usa aquı, “polialquilenglicol” se refiere a polımeros de glicoles de 2 o 3 atomos de carbono.Los polialquilenos de dos atomos de carbono incluyen polietilenglicol (PEG) de diferentes pesos molecu-lares, y sus derivados, tales como polisorbatos. Los polialquilenos de tres atomos de carbono incluyenpolipropilenglicol y sus derivados.
La region hidrofoba generalmente es un acido graso, pero puede ser un alcohol graso, tiol graso, ysimilares, que tambien son compuestos lipofilos. El acido graso puede ser saturado o insaturado. Lalongitud de la cadena no parece ser importante, aunque tıpicamente se usan acidos grasos disponiblesen el comercio y tienen longitudes de cadena de C12−18. Sin embargo, la longitud puede estar limitadapor la solubilidad o solidificacion del compuesto, es decir mayores longitudes de acidos grasos son solidosa temperatura ambiente. Los acidos grasos de 12 atomos de carbono (laurilo), 14 atomos de carbono,16 atomos de carbono (palmitato) y 18 atomos de carbono (monoestearato u oleato) son longitudes decadena preferidas.
La region hidrofila es un polialquilenglicol, polietilen- o poliporopilen-glicol-monoeter. La funcion eterse forma por la union entre la cadena de polioxietileno, que preferiblemente tiene una longitud de cadenade 2 a 100 unidades monomeras, y el grupo sorbitan. El polimetilenglicol no es adecuado para adminis-trar a animales debido a la formacion de formaldehıdos, y los glicoles con una longitud de cadena de ≥4 pueden ser insolubles. Tambien son adecuadas las cadenas de polioxietileno-polioxipropileno mixtas.
No es necesario un ligador para unir las regiones hidrofila e hidrofoba, pero si se usa, debe ser unnucleofilo bifuncional capaz de reaccionar tanto con el polialquilenglicol como con la region hidrofoba. Elligador proporciona electrones para una reaccion nucleofila con el polialquilenglicol, tıpicamente formadopor reaccion con oxido de etilo u oxido de propileno. Entre los ligadores adecuados se incluyen sorbitan,alcoholes de azucares, etanolamina, etanotiol, 2-mercaptoetanol, 1,6-diaminohexano, un aminoacido (porejemplo, glutamina, lisina), otros azucares reducidos, y similares. Por ejemplo, el sorbitan forma unaunion ester con el acido graso en un polisorbato.
Entre los compuestos adecuados se incluyen polioxietilensorbitanes, tales como esteres de monolau-rato, monooleato, monopalmitato, monoestearato, trioleato, y triestearato. Estos y otros compuestosadecuados se pueden sintetizar por metodos quımicos patron o se pueden obtener en el comercio (porejemplo, Sigma Chemical Co., MO; Aldrich Chemical Co., WI; J.B. Baker, NJ).
2. Activacion del reactivo
El reactivo, generalmente un polisorbato, se activa por exposicion a la luz UV con intercambio librede aire. La activacion se logra usando una lampara que irradia a 254 nm o 302 nm. Preferiblemente, lasalida se centra en 254 nm. Longitudes de onda mayores pueden requerir tiempo de activacion mayor.Aunque existe alguna evidencia de que la luz ambiente fluorescente puede activar los polisorbatos, losexperimentos han mostrado que el uso de luz UV a 254 nm da activacion maxima antes de que la luz
15

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
ambiente de un nivel detectable de activacion.
El aire juega un papel importante en la activacion de los polisorbatos. El acceso al aire duplica lavelocidad de activacion relativa a las activaciones realizadas en recipientes hermeticamente cerrados. To-davıa no se sabe que gas es el responsable; es probable que sea un derivado de oxıgeno, aunque no hayperoxidos implicados. Se sabe que la exposicion a la luz UV de los compuestos con uniones eter generaperoxidos, que se pueden detectar y cuantificar usando tiras de ensayo de peroxido. En una reaccion, seanadio peroxido de hidrogeno en un nivel de 1 a 10 veces mayor que el encontrado en material activadopor luz UV, a una solucion de polisorbato en ausencia de luz. No se obtuvo activacion.
El reactivo se pone en un recipiente adecuado para irradiar. Una consideracion para el recipiente esla capacidad de lograr irradiacion uniforme. Por lo tanto, si el camino de onda es largo, el reactivo sepuede mezclar o agitar. La activacion requiere aire; los peroxidos no estan implicados en la activacion.El reactivo se puede activar en cualquier solucion acuosa y no es necesario tamponar.
Una activacion de ejemplo se produce en una cubeta con 1 cm de espesor de lıquido. El reactivo seirradia a una distancia menor de 9 cm a 1500 µW/cm2 (salida inicial de la fuente) durante aproximada-mente 24 horas. En estas condiciones, el reactivo activado convierte un mınimo de 85 % del peptido enAPS-peptido.
3. Modificacion de peptidos o proteınas con reactivo activado
Los peptidos o proteınas se hacen reaccionar con el reactivo APS en una fase lıquida o solida y sonmodificados por la union del derivado de APS. Los metodos aquı descritos para la union ofrecen la ven-taja de mantener la carga del peptido o proteına. Cuando la carga del peptido es crıtica para su funcion,tal como la actividad antibiotica de peptidos cationicos aquı descritos, estos metodos de union ofrecenventajas adicionales. Los metodos que unen grupos por acilacion dan como resultado la perdida de cargapositiva por conversion de los grupos amino en amido. Ademas, se sabe introducir ligador no voluminosoo potencialmente antigenico, tal como un grupo triazina, por los metodos aquı descritos.
Como se ha indicado antes, la formacion de APS-peptido se produce en fase solida o en solucionacuosa. Brevemente, en el metodo en fase solida, el peptido se suspende en un tampon adecuado, talcomo un tampon acetato. Tambien se pueden usar otros tampones adecuados que ayudan a la formacionde APS-peptido. El tampon de acetato puede ser de sodio, rubidio, litio y similares. Tambien son ade-cuadas otras soluciones de acetato, tales como HAc o HAc-NaOH. Un intervalo de pH preferido para eltampon es de 2 a 8,3, aunque se puede usar un intervalo mas amplio. Cuando el pH inicial del tamponacido acetico-NaOH se varıa, la posterior liofilizacion en tampon de acido acetico 200 mM da solo peptidomodificado de Tipo I (vease Ejemplo 14). La presencia de un componente de tampon alcalino da como re-sultado la formacion de peptidos modificados de Tipo II. Una concentracion de peptido tıpica es 1 mg/ml,que da como resultado 85-95 % de peptido modificado, sin embargo son adecuadas otras concentraciones.La principal consideracion para determinar la concentracion parece ser economica. El polımero activado(APS) se anade con exceso molar al peptido, de forma que se genera un peptido modificado con APS conuna relacion molar 1:1. Generalmente, una relacion inicial de aproximadamente 2,5:1 (APS:peptido) a5:1 (APS:peptido) da un peptido modificado con APS 1:1.
Despues la mezcla de reaccion se congela (por ejemplo, -80◦C) y se liofiliza. El acetato sodico sedesproporciona en acido acetico y NaOH durante la liofilizacion; la separacion del acido acetico volatila vacıo deja el NaOH disperso por toda la matriz solida resultante. Esta perdida de acido acetico seconfirma por un aumento del pH detectado al disolver el liofilizado. No se forma peptido modificado conAPS en tampon de acetato si las muestras solo se congelan y despues descongelan.
La reaccion de modificacion tambien puede tener lugar en solucion acuosa. Sin embargo, las modifica-ciones con APS no se producen a temperatura ambiente en ningun sistema de tampon de acetato ensayadoa pesar del pH. Las modificaciones con APS tampoco se forman en tampones de fosfato tan altos comopH 11,5. La modificacion con APS se produce en un tampon de carbonato sodico a un pH mayor queaproximadamente 8,5. Tambien se pueden usar otros tampones si soportan la formacion de derivados. Unintervalo de pH de 9-11 tambien es adecuado, y se usa mas normalmente pH 10. La reaccion se produceen dos fases: primero se forma los peptidos de Tipo I, seguido de formacion de peptidos de Tipo II.
En esta descripcion, la union se produce en un grupo amino. Para un peptido, la union se puedeproducir en el α-NH2 del aminoacido N-terminal o en el grupo ε-NH2 de la lisina. Tambien se puedenmodificar otras aminas primarias y secundarias. El bloqueo completo de todos los grupos amino por
16

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
acilacion (MBI 11CN-Y1) inhibe la formacion de APS-peptido. Por lo tanto, no se produce modificacionde los restos de arginina o triptofano. Si el unico grupo amino disponible es el grupo α-amino (porejemplo, MBI 11B9CN y MBI 11G14CN), se observa la forma de Tipo I. La inclusion de una sola lisina(por ejemplo, MBI 11B1CN, MBI 11B7CN, MBI 11B8CN), que proporciona un grupo ε-amino, da comoresultado las formas de tipo II tambien. La cantidad de Tipo II formado aumenta para peptidos con masrestos lisina.
4. Purificacion y propiedades fısicas de los peptidos modificados con APS
Los peptidos modificados con APS se pueden purificar. En casos en los que el peptido libre es toxico,la purificacion puede ser necesaria para separar peptido no modificado y/o polisorbato sin reaccionar. Sepuede usar cualquiera de una variedad de metodos de purificacion. Dichos metodos incluyen HPLC defase inversa, precipitacion con disolvente organico para separar el polisorbato, cromatografıa de exclusionpor tamanos, cromatografıa de intercambio ionico, filtracion y similares. Se prefiere el RP-HPLC. Losprocedimientos para estos metodos de separacion son conocidos.
La formacion del APS-peptido (o proteına) da como resultado la generacion de productos que con-tienen peptido que son mas hidrofobos que el peptido relacionado. Esta propiedad se puede explotarpara realizar la separacion del conjugado del peptido libre por RP-HPLC. Los conjugados se resuelven endos poblaciones basadas en su hidrofobicidad determinada por RP-HPLC; la poblacion de Tipo I eluyeligeramente antes que la poblacion de Tipo II.
Las series MBI 11 de peptidos tienen pesos moleculares entre 1600 y 2500. Cuando se hacen correr enuna columna de Superose 12, una columna de exclusion por tamanos, estos peptidos no eluyen antes queel volumen del lecho, indicando una masa molecular por debajo de 20 kDa. En contraste, los peptidosmodificados con APS eluyen a 50 kDa, demostrando ası un gran aumento de la masa molecular aparente.
Un aumento de la masa molecular aparente puede potenciar las farmacocineticas de los peptidoscationicos porque la mayor masa molecular reduce la velocidad a la que los peptidos y proteınas se elimi-nan de la sangre. La formacion de micela puede ofrecer beneficios adicionales suministrando “paquetes”de moleculas de peptido a los microorganismos mas que basandose en la union multiple de moleculasde peptido sencillas. Ademas, los peptidos modificados con APS son solubles en cloruro de metileno ocloroformo, mientras que el peptido relacionado es esencialmente insoluble. Este aumento de solubilidadorganica puede potenciar significativamente la capacidad para penetrar las barreras tisulares.
Ademas, por estudios de dicroısmo circular (DC), se observa que los peptidos modificados con APStienen una conformacion 3-dimensional alterada. Como se muestra en los ejemplos, MBI 11CN y MBI11B7CN tienen estructuras desordenadas en tampon fosfato o trifluoroetanol (TFE) acuoso al 40 % yforman una conformacion de lamina β solo por insercion en liposomas. En contraste, los espectros de DCde MBI 11CN modificado con APS y MBI 11B7CN modificado con APS indican estructura de lamina βen tampon fosfato.
D. Formulaciones y administracion
Como se ha indicado antes, se describen aquı metodos para tratar y prevenir infecciones administrandoa un paciente una cantidad terapeuticamente eficaz de un analogo de peptido de indolicidina como se hadescrito aquı. Los pacientes adecuados para dicho tratamiento se pueden identificar por caracterısticasbien establecidas de una infeccion, tales como fiebre, pus, cultivo de organismos, y similares. Las infeccio-nes que se pueden tratar con analogos de peptido incluyen las producidas o debidas a microorganismos.Entre los ejemplos de microorganismos se incluyen bacterias (por ejemplo, Gram positivas, Gram nega-tivas), hongos (por ejemplo, levaduras y mohos), parasitos (por ejemplo, protozoos, nematodos, cestodosy trematodos), virus, y priones. Los organismos especıficos en estas clases son bien conocidos (vease porejemplo, Davis et al., Microbiology, 3rd edition, Harper & Row, 1980). Entre las infecciones se incluyen,pero no se limita, sındrome de choque toxico, difteria, colera, tifus, meningitis, tos ferina, botulismo,tetano, infecciones piogenicas, disenterıa, gastroenteritis, antrax, enfermedad de Lyme, sıfilis, rubeola,septicemia y plaga.
El tratamiento eficaz de la infeccion se puede examinar de varias formas diferentes. El paciente puedepresentar menos fiebre, menor numero de organismos, menor nivel de moleculas inflamatorias (por ejem-plo, IFN-γ, IL-12, IL-1, TNF), y similares.
Los analogos de peptidos de la presente invencion, preferiblemente se administran como una compo-
17

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
sicion farmaceutica. Brevemente, las composiciones farmaceuticas aquı descritas pueden comprender unoo mas de los analogos de peptido aquı descritos, combinado con uno o mas vehıculos, diluyentes o exci-pientes fisiologicamente aceptables. Como se indica aquı el tampon de formulacion usado puede afectara la eficacia o actividad del analogo de peptido. Un tampon de formulacion adecuado contiene tampon ysolubilizador. El tampon de formulacion puede comprender tampones tales como acetato sodico, citratosodico, solucion salina tamponada neutra, solucion salina tamponada con fosfato, y similares, o sales talescomo NaCl. Se prefiere el acetato sodico. En general, se usa un tampon de acetato de 5 a 500 mM, ypreferiblemente de 100 a 200 mM. El pH de la formulacion final puede estar en el intervalo de 3 a 10, ypreferiblemente es aproximadamente neutro (aproximadamente pH 7-8). Tambien se pueden anadir so-lubilizadores tales como polioxietilensorbitanes (por ejemplo, Tween 80, Tween 20) y polioxietilen-eteres(por ejemplo, Brij 56) si el compuesto todavıa no se ha modificado con APS.
Se pueden incluir compuestos adicionales en las composiciones. Estos incluyen, por ejemplo, car-bohidratos tales como glucosa, manosa, sacarosa o dextrosa, manitol, otras proteınas, polipeptidos oaminoacidos, agentes de quelacion tales como EDTA o glutation, adyuvantes y conservantes. Como seindica aquı, las composiciones farmaceuticas de la presente invencion tambien pueden contener uno omas ingredientes activos adicionales, tales como un antibiotico (vease aquı la discusion de sinergismo) ocitoquina.
Las composiciones se pueden administrar en un vehıculo de suministro. Por ejemplo, la composicionse puede encapsular en un liposoma (vease, por ejemplo, los documentos WO 96/10585; WO 95/35094),formar complejo con lıpidos, encapsular en vehıculos de liberacion lenta o de liberacion sostenida, talescomo poligalactida, y similares. En otras realizaciones, las composiciones se pueden preparar como unliofilizado, usando excipientes adecuados para proporcionar estabilidad.
Las composiciones farmaceuticas aquı descritas se pueden administrar en diferentes formas. Por ejem-plo, se pueden administrar analogos de peptido por inyeccion intravenosa, inyeccion o implante intraperi-toneal, inyeccion o implante subcutaneo, inyeccion intradermica, lavado, inhalacion, implante, inyecciono implante intramuscular, inyeccion intratecal, lavado de vejiga, supositorios, pesarios, aplicacion topica(por ejemplo, cremas, pomadas, parches dermicos, gotas oculares, gotas para el oıdo, champus), vıaenterica, oral o nasal. El analogo se puede aplicar localmente como una inyeccion, gotas, pulverizador,comprimidos, crema, pomada, gel y similares. El analogo se puede administrar como un bolo o comodosis multiples en un periodo de tiempo.
El nivel de peptido en el suero y otros tejidos despues de administracion se puede controlar por diferen-tes tecnicas bien establecidas tales como ensayos bacterianos, cromatograficos o basados en anticuerpos,tales como ELISA.
Las composiciones farmaceuticas aquı descritas se administran de una forma adecuada para la in-feccion o enfermedad que se va a tratar. La cantidad y frecuencia de administracion se determinarapor factores tales como el estado del paciente, la causa de la infeccion, y la gravedad de la infeccion.Las dosificaciones adecuadas se pueden determinar por ensayos clınicos, pero generalmente estaran en elintervalo de aproximadamente 0,1 a 50 mg/kg.
Ademas, los analogos aquı descritos se pueden usar de la forma de los desinfectantes comunes o en cual-quier situacion en la que los microorganismos son indeseables. Por ejemplo, estos peptidos se pueden usarcomo desinfectantes de superficie, revestimientos, incluyendo union covalente, para dispositivos medicos,revestimientos para ropa, tal como para inhibir el crecimiento de bacterias o repeler mosquitos, en filtrospara purificar aire, tal como en un avion, en purificacion de agua, constituyentes de champus y jabones,conservantes alimentarios, conservantes cosmeticos, conservantes de medios, herbicidas o insecticidas,constituyentes de materiales de construccion, tales como selladores de silicona, y en el procesamiento deproductos animales, tales como curado de pieles de animales. Tal como se usa aquı, “dispositivo medico”se refiere a cualquier dispositivo para usar en un paciente, tal como un implante o protesis. Entre dichosdispositivos se incluyen stents, tubos, sondas, canulas, cateteres, injertos vasculares sinteticos, dispositi-vos controladores de la sangre, valvulas del corazon artificiales, agujas, y similares.
Para estos propositos, tıpicamente se incluyen los peptidos solos o junto con un antibiotico en com-posiciones normalmente usadas o en un aplicador adecuado, tal como para aplicar a ropa. Se puedenincorporar o impregnar en el material durante la fabricacion, tal como para un filtro de aire, o aplicadode otro modo a dispositivos. Los peptidos y antibioticos solo es necesario suspenderlos en una solucionadecuada para el dispositivo o artıculo. Los polımeros son un tipo de vehıculo que se puede usar.
18

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Los analogos, especialmente analogos marcados, se pueden usar en analisis de imagen y ensayos dediagnostico o para marcar sitios en organismos unicelulares y multicelulares eucariotas y en procariotas.Como sistema de marcaje, los analogos se puede acoplar con otros peptidos, proteınas, acidos nucleicos,antibioticos y similares.
Los siguientes ejemplos se ofrecen a modo de ilustracion, y no a modo de limitacion.
Ejemplos
Ejemplo 1
Sıntesis, purificacion y caracterizacion de analogos de peptidos
La sıntesis de peptidos se basa en la estrategia patron de proteccion con Fmoc en fase solida. Elinstrumento usado es un 9050 Plus PepSynthesiser (PerSeptive BioSystems Inc.). Se usan resinas deinjerto de polietilenglicol-poliestireno (PEG-PS) como fase solida, y se forman derivados con un ligadoraminoacido protegido con Fmoc para la sıntesis de amida C-terminal. Se usa HATU (hexafluorofosfatode O-(7-azabenzotriazol-1-il)-1,1,3,3-tetrametiluronio) como agente de acoplamiento. Durante la sıntesis,las etapas de acoplamiento se controlan continuamente para asegurar que cada aminoacido se incorporacon un alto rendimiento. El peptido se escinde de la resina de la fase solida usando acido trifluoroaceticoy depuradores adecuados, y el peptido bruto se purifica usando cromatografıa preparativa de fase inversa.
Todos los peptidos se analizan por espectrometrıa de masas para asegurar que el producto tiene lamasa molecular esperada. El producto debe tener un solo pico que da cuenta de >95 % del area de picototal cuando se somete a cromatografıa lıquida de alto rendimiento de fase inversa (RP-HPLC). Ademas,el peptido debe mostrar una sola banda que da cuenta de >90 % de la intensidad de banda total cuandose somete a electroforesis de gel de acido-urea.
El contenido de peptido, la cantidad de producto que es peptido mas que agua retenida, sal o disol-vente, se mide por analisis cuantitativo de aminoacidos, formacion de derivado de amina libre o cuanti-ficacion espectrofotometrica. El analisis de aminoacidos tambien proporciona informacion de la relacionde aminoacidos presentes en el peptido, que ayuda a confirmar la autenticidad del peptido.
Los analogos de peptidos y sus nombres estan listados en la Tabla 2. En esta tabla, y en otros sitios,los aminoacidos se indican con el codigo de aminoacidos de una letra, y las letras minusculas representanla forma D del aminoacido.
Hay que indicar, que todas las secuencias que no contienen la formula I L R W P W W P W R RK, es decir, todas las secuencias diferentes de los nombres 11B7CN, 11B16CN, 11B17CN y 11B18CN noforman parte de la presente invencion y son de naturaleza comparativa.
TABLA 2
19

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 2 (Cont.)
20

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Sufijo CN = extremo C-terminal amidado
Sufijo H = homoserina en el extremo C-terminal
Sufijo R = peptido retrosintetizado
Ejemplo 2
Sıntesis de peptidos modificados
Los analogos de indolicidina son modificados para alterar las propiedades fısicas del peptido original.Dichas modificaciones incluyen: acetilacion en el extremo N-terminal, extremo N-terminal formando de-rivado con Fmoc, polimetilacion, peracetilacion, y derivados ramificados.
Acetilacion α-N-terminal Antes de escindir el peptido de la resina y desprotegerlo, el peptido comple-tamente protegido se trata con N-acetilimidazol en DMF durante 1 hora a temperatura ambiente, que dacomo resultado la reaccion selectiva en el extremo α-N-terminal. Despues el peptido se desprotege/escindey purifica como para un peptido sin modificar.
Extremo α-N-terminal formando derivado con Fmoc Si la etapa final de desproteccion de Fmoc nose lleva a cabo, el grupo α-N-terminal-Fmoc permanece en el peptido. Despues se desprotege la cadenalateral/escinde el peptido y se purifica como para un peptido sin modificar.
Polimetilacion El peptido purificado en una solucion de metanol se trata con exceso de bicarbonatosodico, seguido de exceso de yoduro de metilo. La mezcla de reaccion se agita toda la noche a temperaturaambiente, se extrae con disolvente organico, se neutraliza y purifica como para un peptido sin modificar.Usando este procedimiento, un peptido no se metila completamente; la metilacion de MBI 11CN dio unamedia de 6 grupos metilo. Por lo tanto, el peptido modificado es una mezcla de productos metilados.
Peracetilacion Un peptido purificado en solucion de DMF se trata con N-acetilimidazol durante 1hora a temperatura ambiente. El producto bruto se concentra, se disuelve en agua, se liofiliza, se vuelvea disolver en agua y se purifica como para un peptido sin modificar. Se observa la acetilacion completade los grupos amina primarios.
Derivados de cuatro/ocho ramificaciones Los peptidos ramificados se sintetizan en un nucleo de cua-tro u ocho ramificaciones unido a la resina. La sıntesis y desproteccion/escision proceden como para elpeptido sin modificar. Estos peptidos se purifican por dialisis frente a hidrocloruro de guanidina 4 M ydespues agua, y se analizan por espectrometrıa de masas.
Los peptidos modificados usando los procedimientos anteriores estan listados en la Tabla 3.
TABLA 3
21

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 3 (Cont.)
Ejemplo 3
Produccion recombinante de analogos de peptidos
Los analogos de peptidos se preparan alternativamente por tecnica de DNA recombinante en celulashospedantes bacterianas. El peptido se prepara como una proteına de fusion, elegida para ayudar altransporte del peptido de fusion a cuerpos de inclusion, periplasma, membrana exterior o entorno extra-celular.
Construccion de plasmidos que codifican la proteına de fusion peptido MBI-11
Se usa la amplificacion por la reaccion en cadena de la polimerasa para sintetizar DNA de doble hebraque codifica los genes del peptido MBI a partir de moldes de hebra simple. Para MBI-11, se preparan 100µl de mezcla de reaccion que contiene de 50 a 100 ng de molde, 25 pm de cada cebador, MgCl2 1,5 mM,200 µM de cada dNTP, 2U de Taq-polimerasa en el tampon del suministrador. Las reacciones procedencon 25 ciclos de 94◦C durante 30 s, 55◦C durante 30 s, 74◦C durante 30 s, seguido de 74◦C durante 1min. El producto amplificado se hace digerir con BamHI y HindIII y se clona en un vector de expresionplasmido que codifica la pareja de fusion y un marcador de seleccion adecuado.
Produccion del peptido de fusion MBI-11 en E. coli
El plasmido pR2h-11 se co-electropora con pGP1-2 en la cepa XL1-Blue de E. coli, usando un promotorT7, origen de replicacion de alto numero de copias, marcador Apr y conteniendo el gen de la proteınade fusion. El plasmido pGP1-2 contiene un gen de la RNA-polimerasa T7 controlado por un promotorlambda y gen represor cl857. La expresion de la proteına de fusion es inducida por un desplazamiento dela temperatura de 30◦C a 42◦C. Los cuerpos de inclusion se lavan con solucion que contiene solubilizador yse extraen con disolvente organico de extraccion. Se analizan los perfiles de las muestras por SDS-PAGE.La Figura 1 muestra el analisis por SDS-PAGE y un perfil de extraccion del cuerpo de inclusion de lacelula entera. El contaminante principal en el disolvente organico extraıdo es β-lactamasa (Figura 1). Elnivel de expresion en estas celulas se presenta en la Tabla 4.
TABLA 4
Proteına Masa mol. % de proteına en el % en el extracto del % que es peptidode fusion (kDa) lisato de celula entera cuerpo de inclusion MBI-11
MBI-11 20,1 15 42 7,2
Ademas, se usa un vector de bajo numero de copias, pPD100, que contiene un gen de resistencia alcloranfenicol, para expresar MBI-11 con el fin de eliminar la necesidad de usar ampicilina, reduciendo asıla aparicion de β-lactamasa en el material extraıdo. El plasmido permite la expresion del gen selectiva yalto nivel de sobreproduccion de proteına en E. coli usando el bacteriofago con el sistema RNA-polimerasaT7/promotor T7 (Dersch et al., FEMS Microbiol. Lett. 123: 19-26, 1994). pPD100 contiene un gen deresistencia al cloranfenicol (CAT) como un marcador selectivo, un sitio de clonacion multiple, y unasecuencia ori obtenida del vector pSC101 de bajo numero de copias. Solo hay aproximadamente de 4 a6 copias de estos plasmidos por celula hospedante. La construccion resultante que contiene MBI-11 sellama pDR2h-11. La figura 2 presenta un analisis por electroforesis en gel de la proteına de fusion MBI-11expresada en este vector. El nivel de expresion de la proteına de fusion MBI-11 es comparable al obtenido
22
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
del plasmido pR2h-11. El producto del gen CAT no aparece, probablemente debido a la naturaleza debajo numero de copias de este plasmido, la proteına CAT no se expresa con altos niveles en pDR2h-11.
Ejemplo 4
Ensayos in vitro para medir la actividad del analogo de peptido
Ensayo de dilucion en agarosa
El ensayo de dilucion en agarosa mide la actividad antimicrobiana de peptidos y analogos de peptidos,que se expresa como la concentracion inhibidora mınima (CIM) de los peptidos.
Con el fin de mimetizar las condiciones in vivo, se usa caldo Mueller Hinton complementado concalcio y magnesio combinado con una agarosa con bajo EEO como medio de crecimiento bacteriano. Elagar mas comunmente usado se sustituye por agarosa ya que los grupos cargados en el agar evitan ladifusion del peptido por el medio. El medio se trata en autoclave y despues se enfrıa a 50 - 55◦C en unbano de agua antes de la adicion aseptica de soluciones antimicrobianas. Se anade el mismo volumen dediferentes concentraciones de solucion de peptido a la agarosa fundida enfriada que despues se vierte auna profundidad de 3 - 4 mm.
El inoculo bacteriano se ajusta a un patron de turbidez McFarland 0,5 (PML Microbiological) ydespues se diluye 1:10 antes de aplicarlo sobre la placa de agarosa. El inoculo final aplicado a la agarosaes aproximadamente 104 UFC en una mancha de 5 - 8 mm de diametro. Las placas de agarosa se incubana 35-37◦C durante de 16 a 20 horas.
Se registra la CIM como la concentracion mas baja de peptido que inhibe completamente el crecimientodel organismo, determinado por inspeccion visual. Se muestran las CIM representativas para diferentesanalogos de indolicidina en la siguiente Tabla 5.
TABLA 5
23
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
24

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
25
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
26
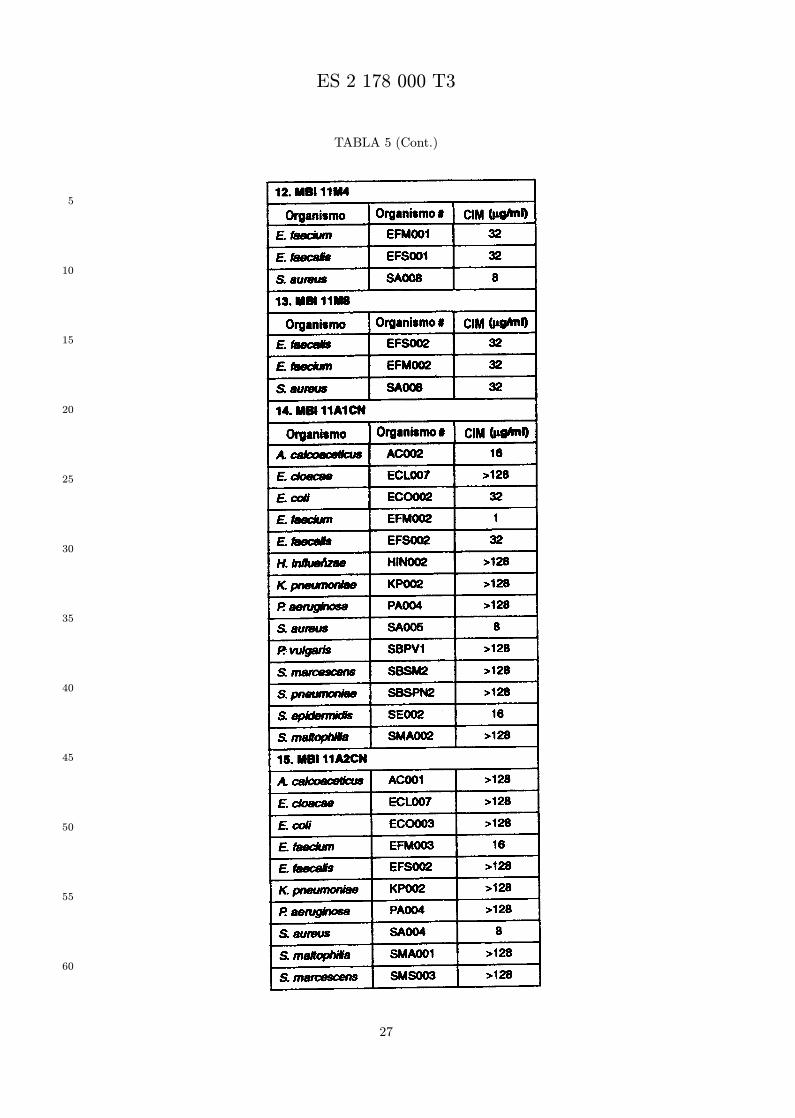
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
27
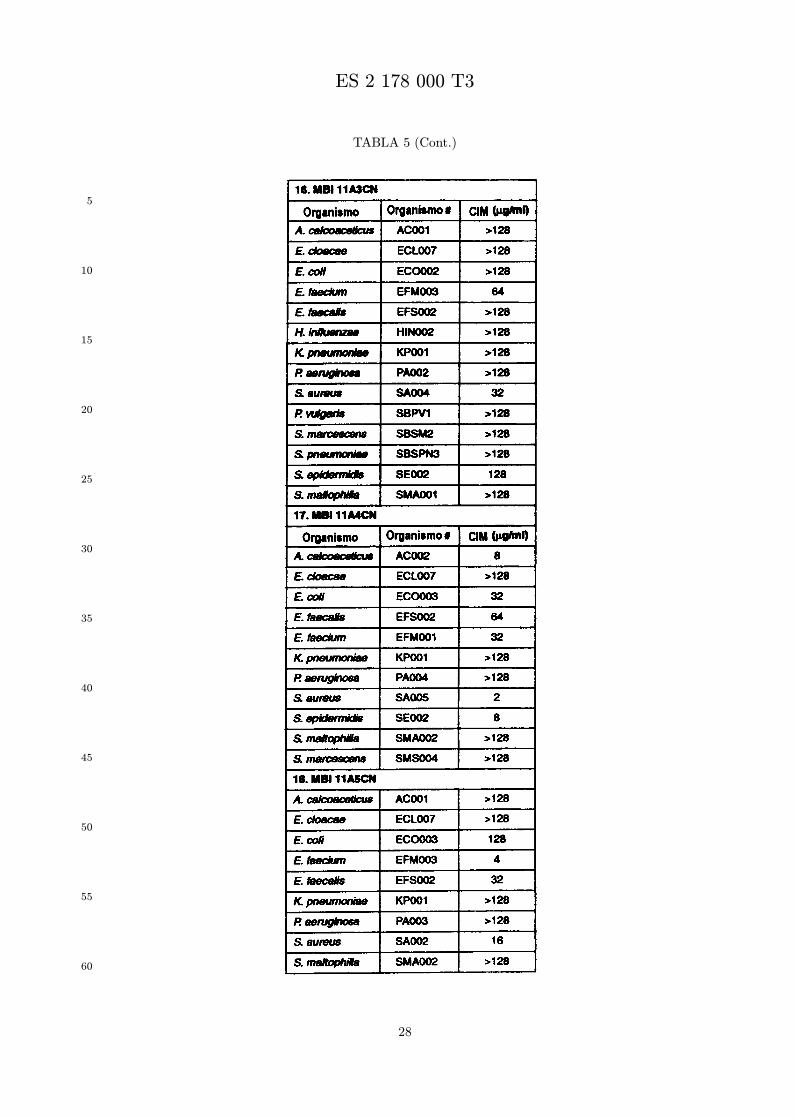
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
28
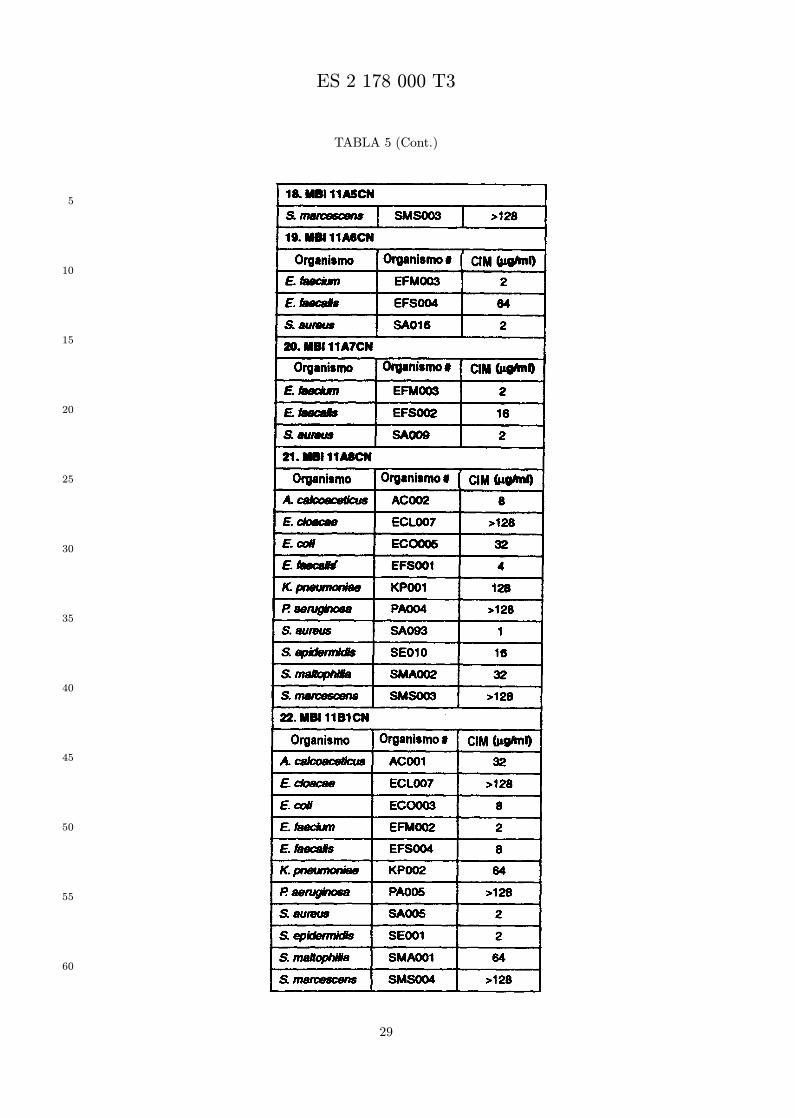
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
29
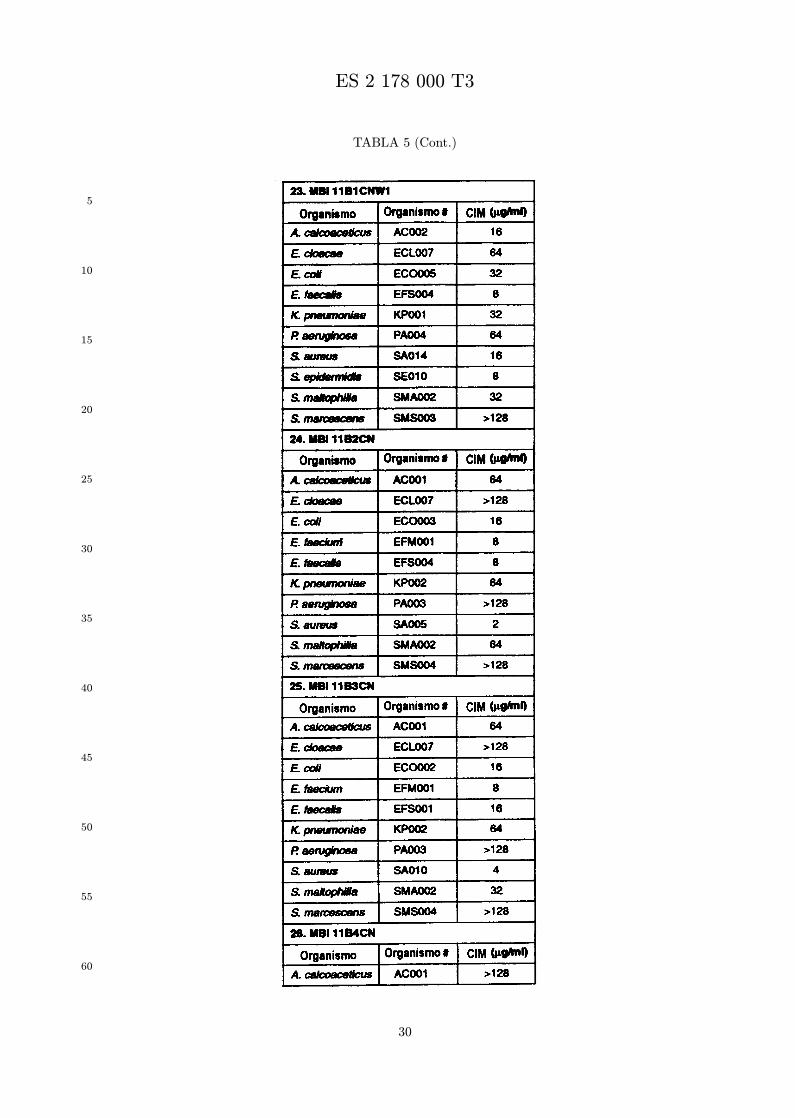
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
30

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
31
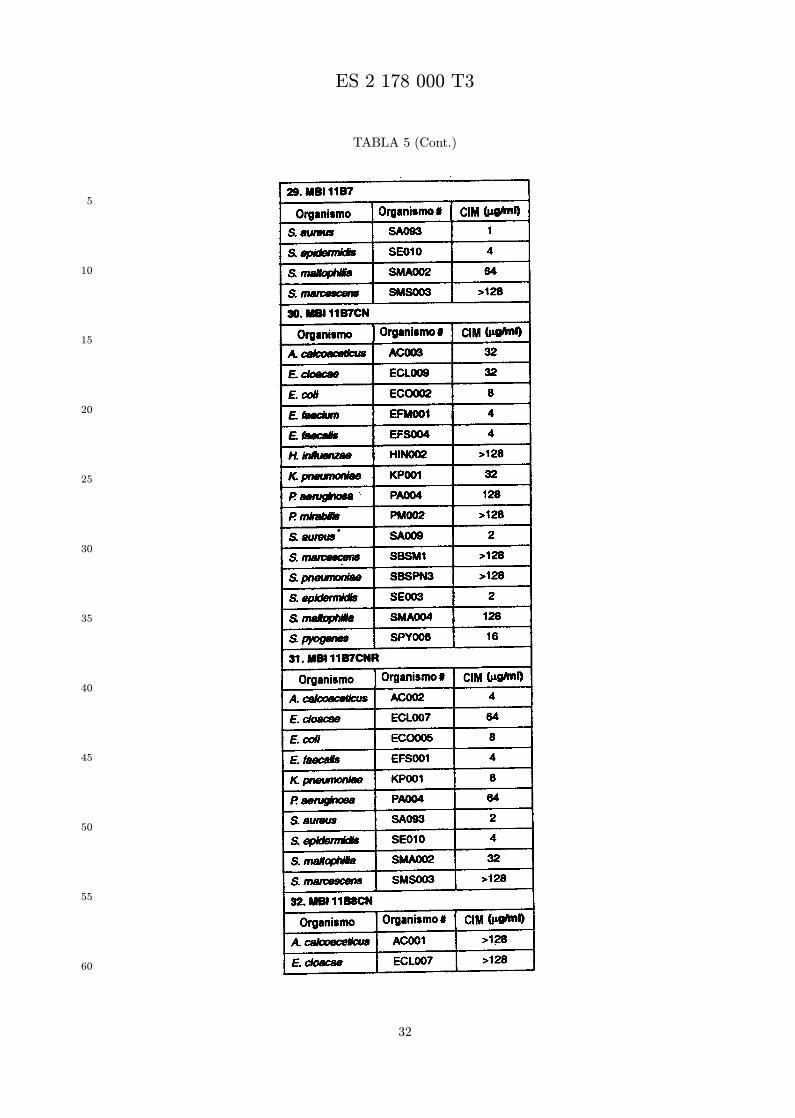
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
32
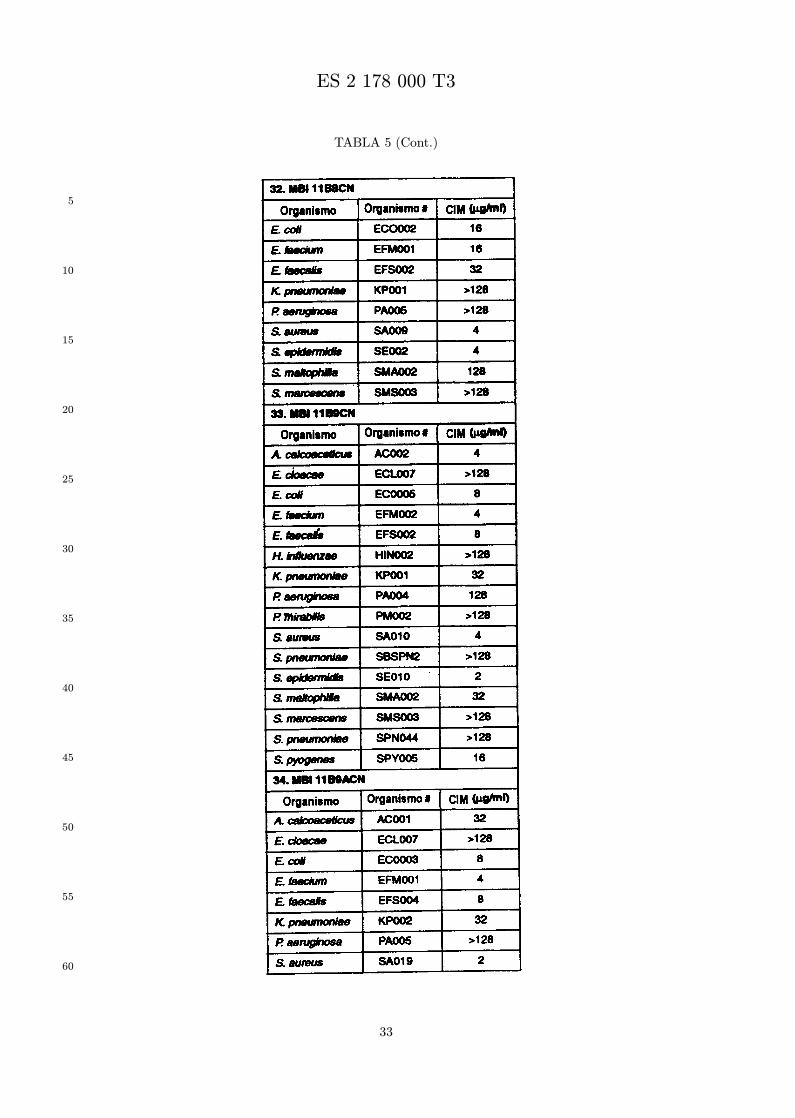
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
33

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
34
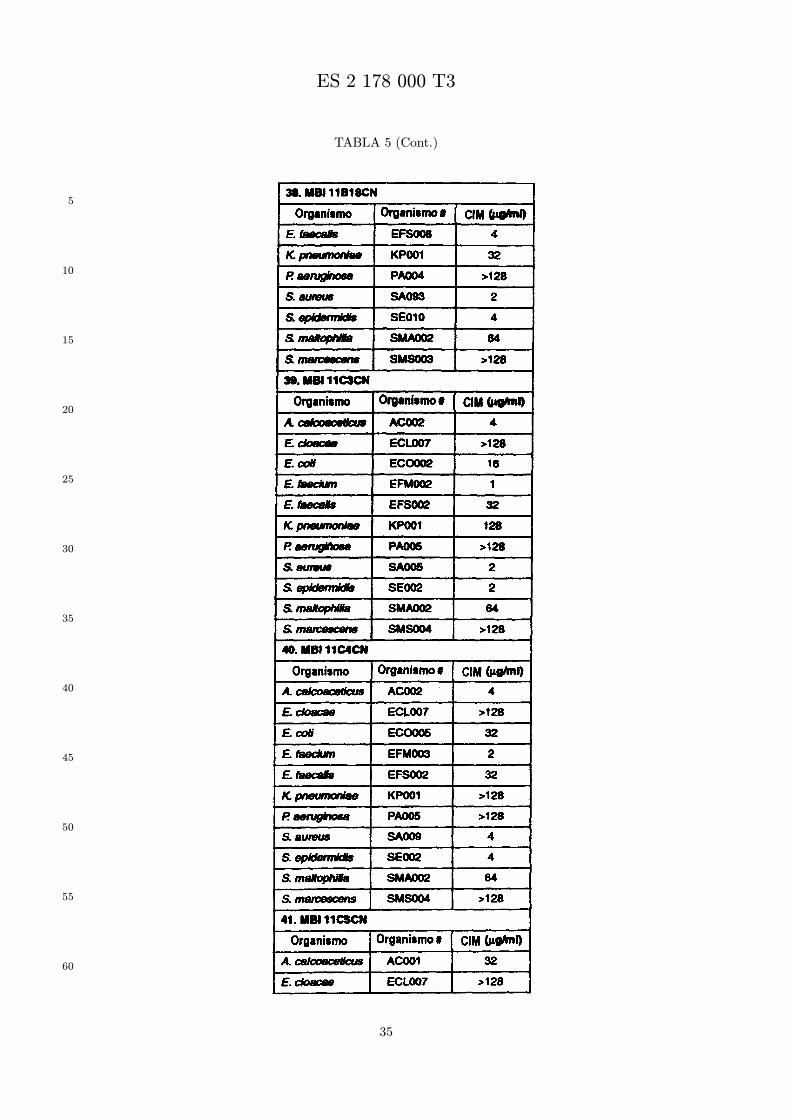
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
35
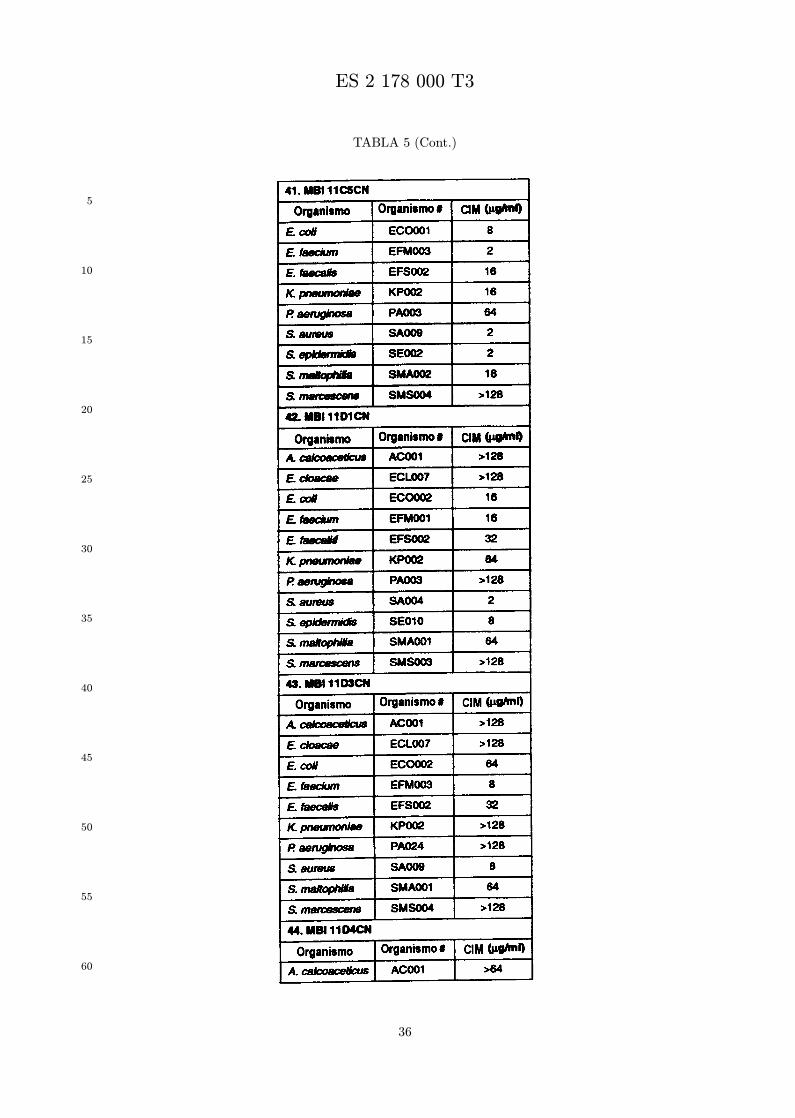
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
36
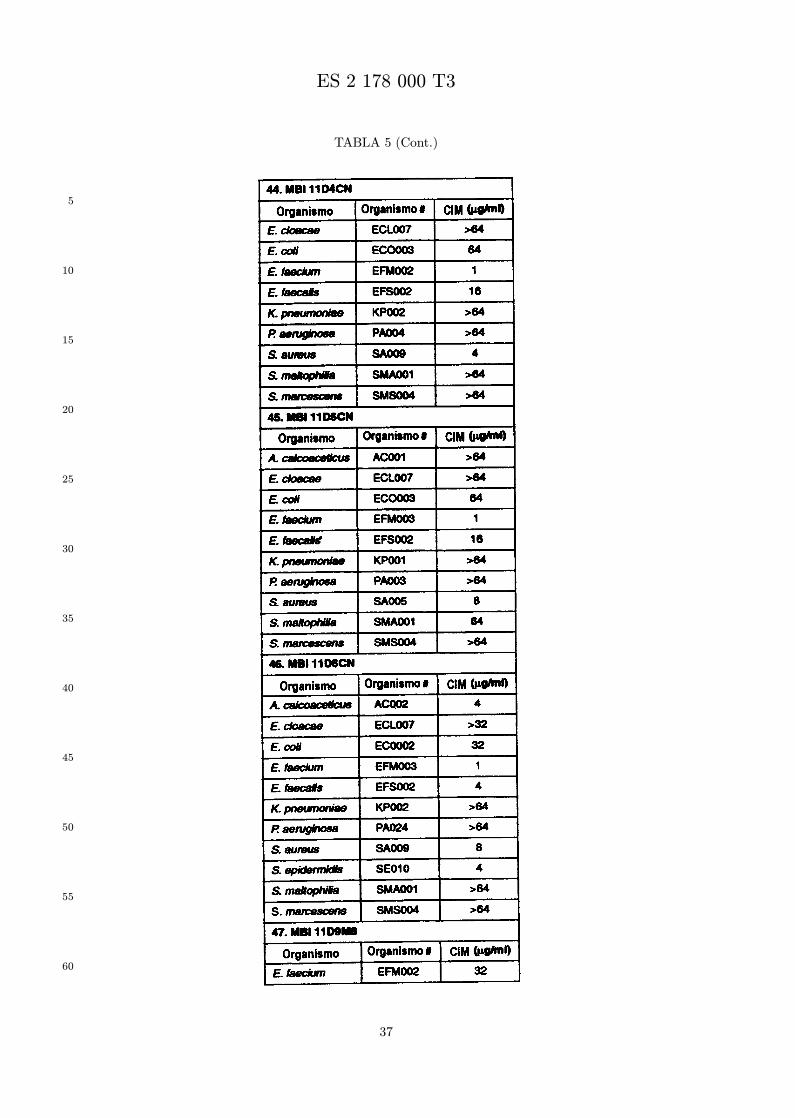
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
37
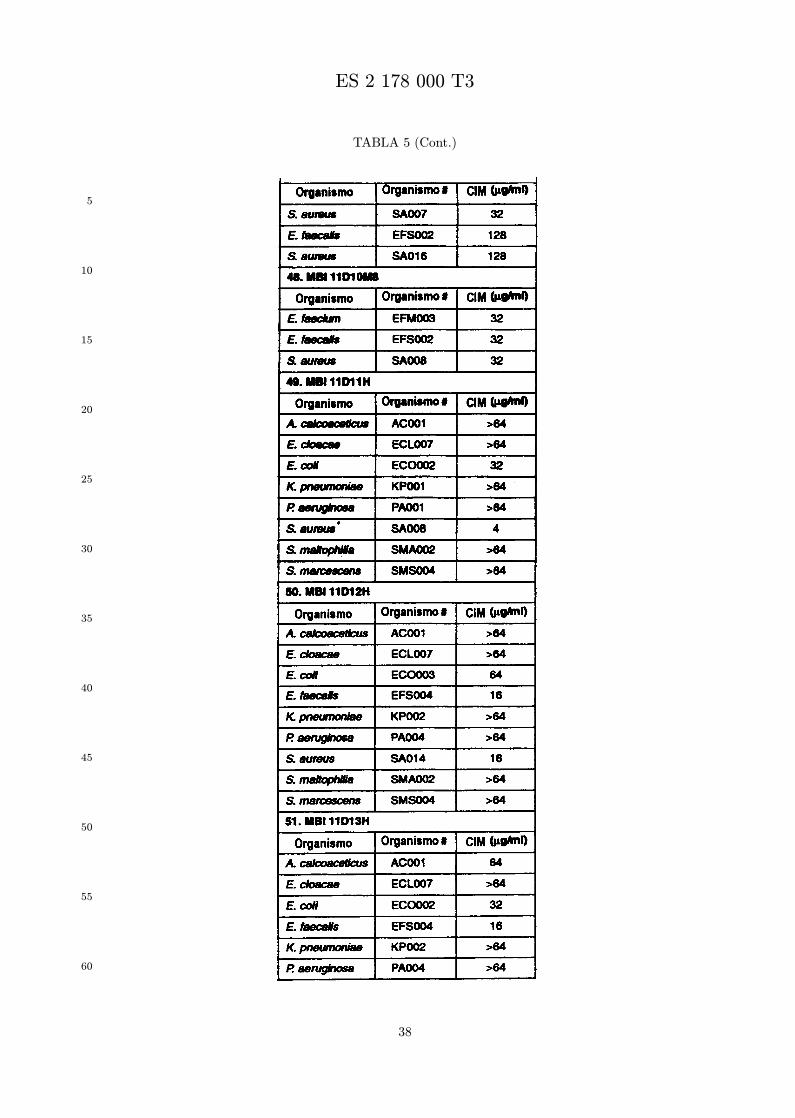
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
38
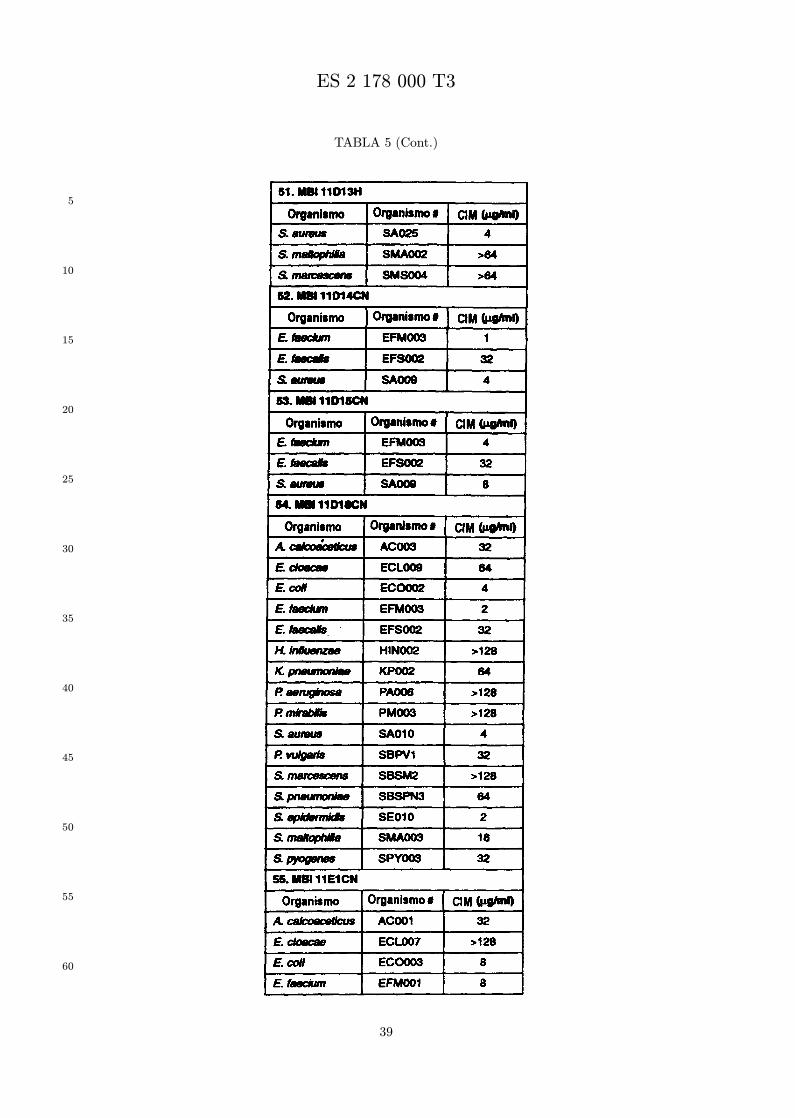
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
39
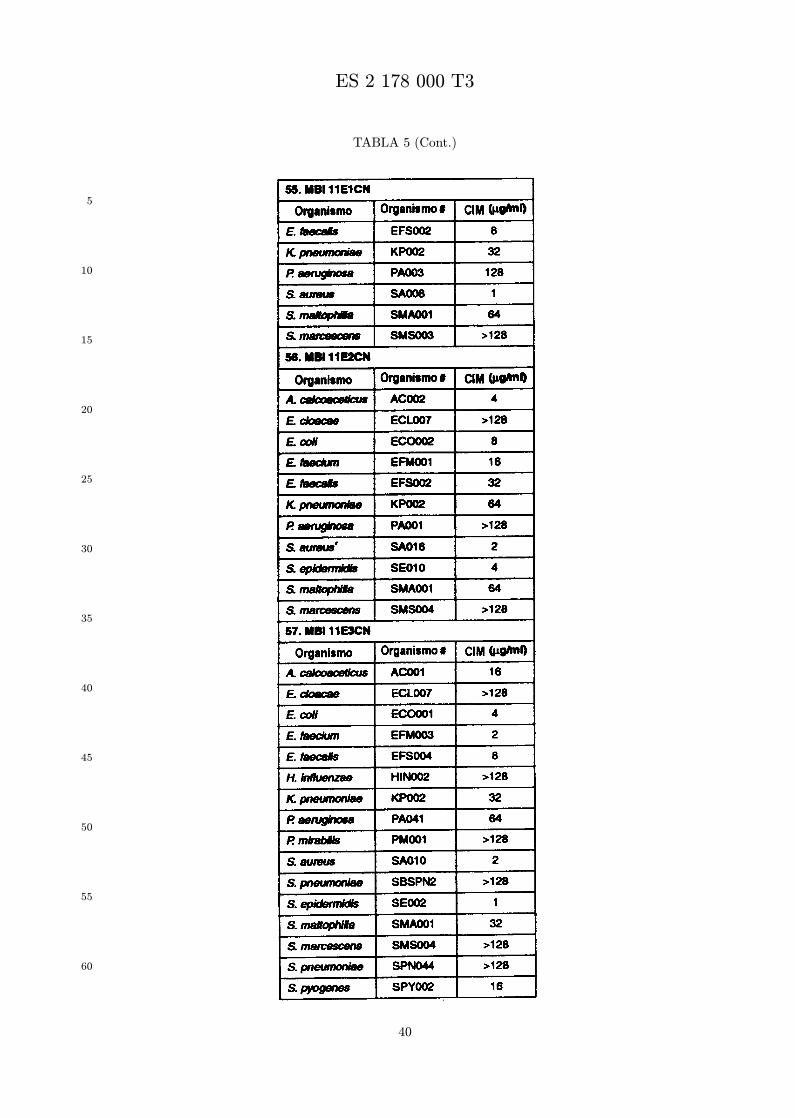
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
40

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
41
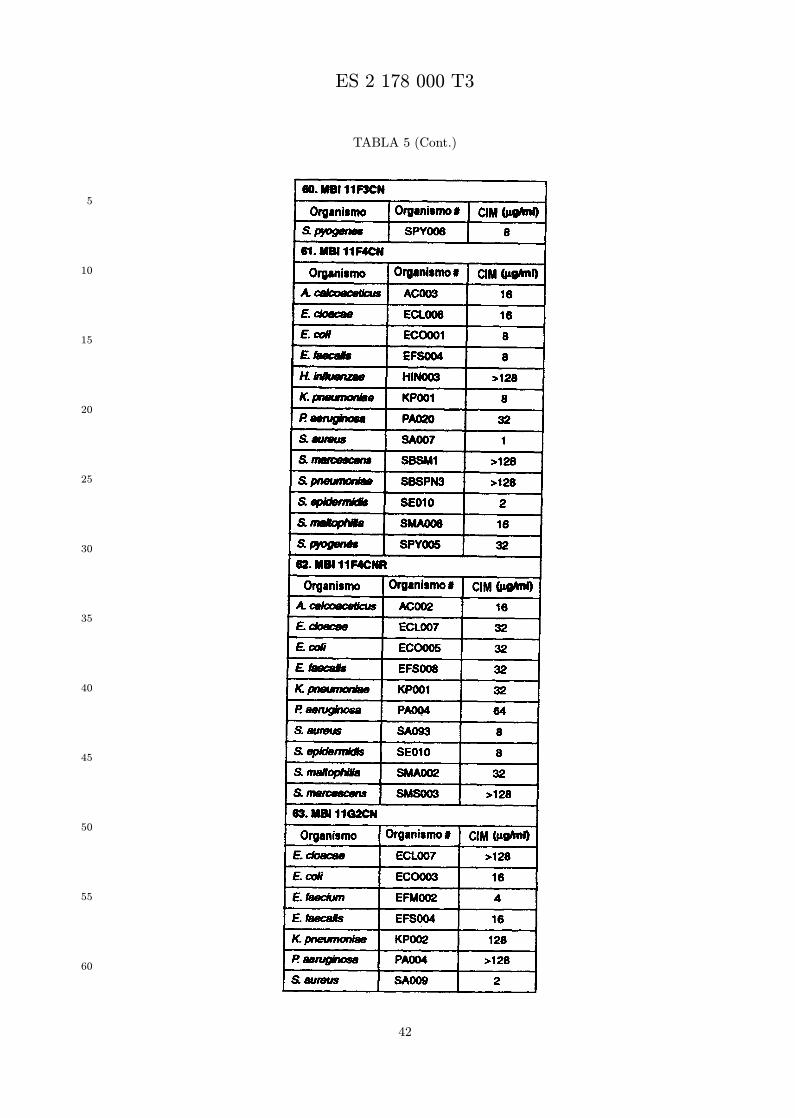
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
42
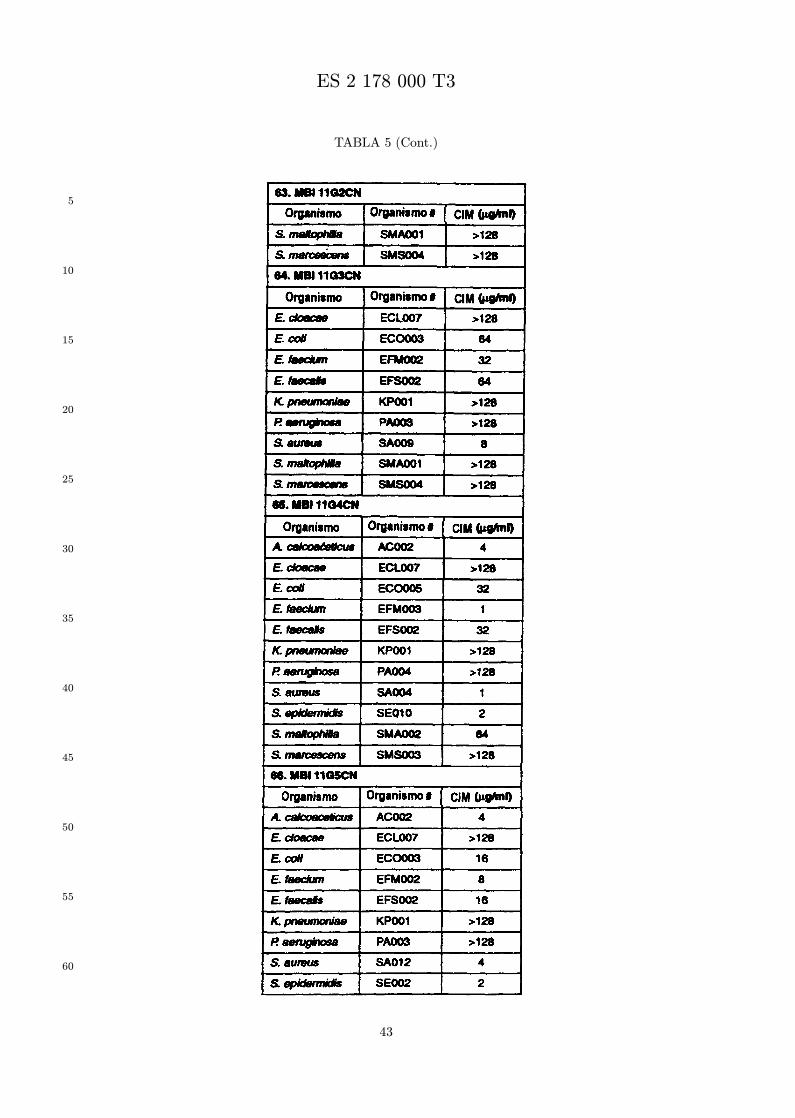
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
43
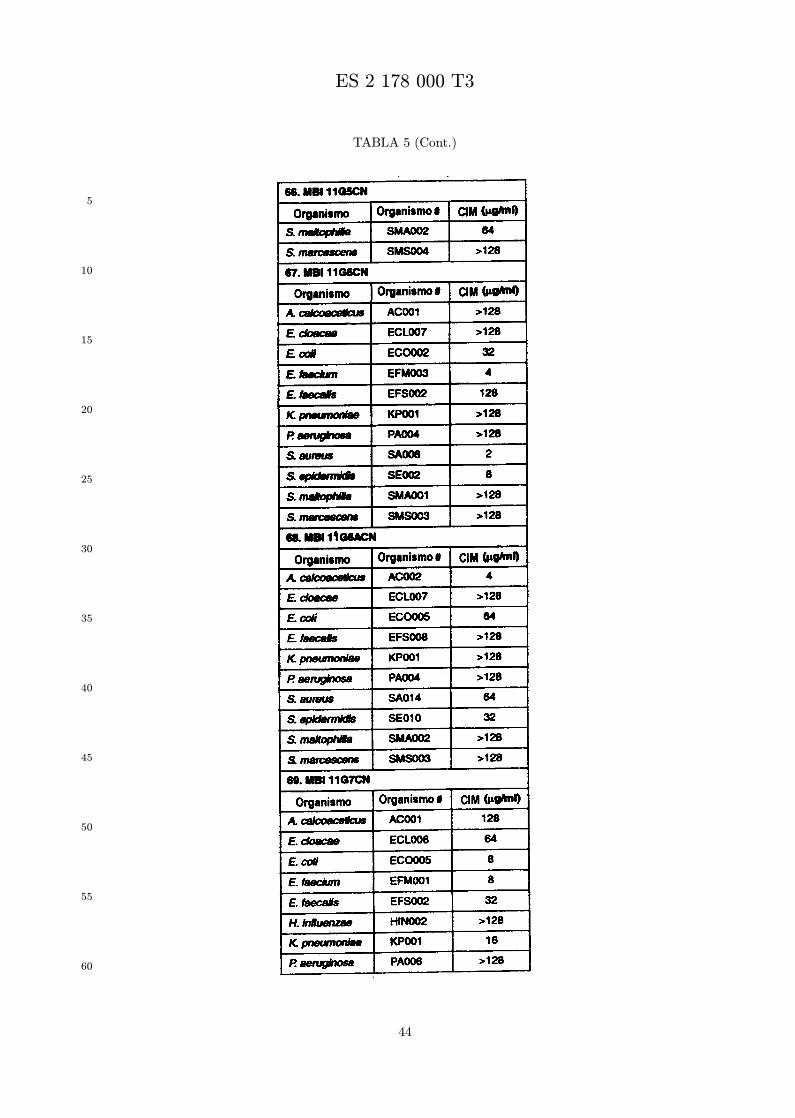
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
44

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
45
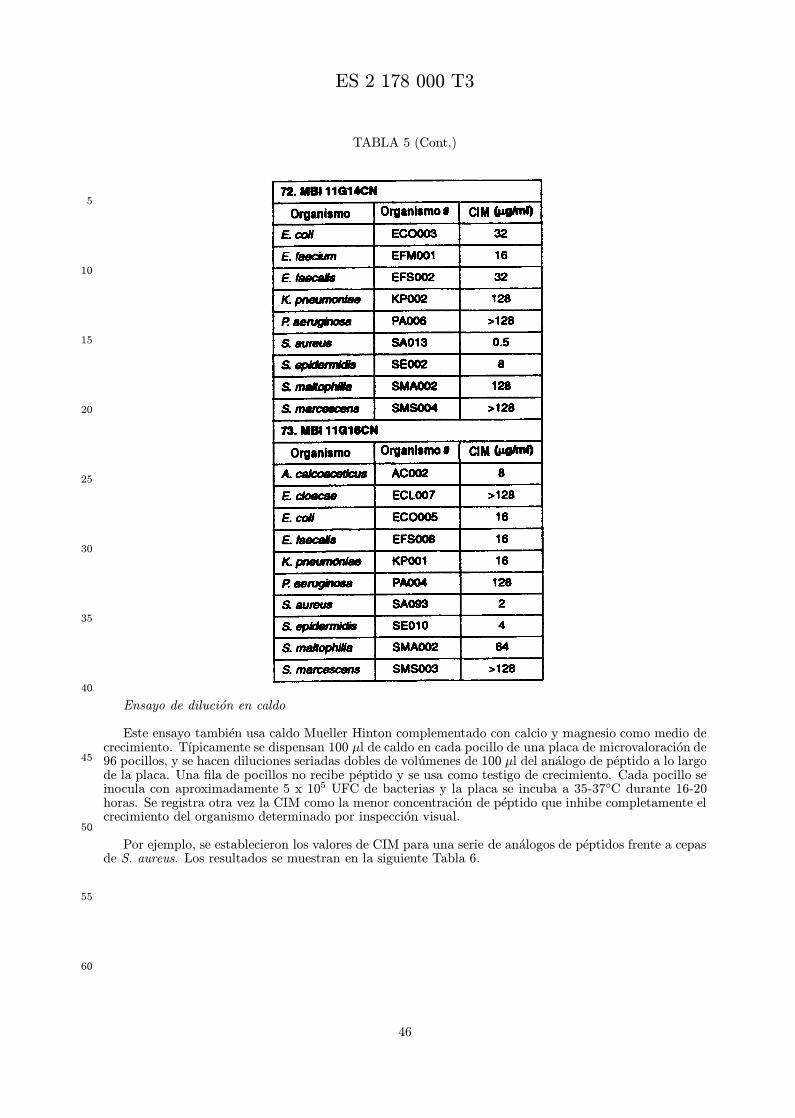
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 5 (Cont.)
Ensayo de dilucion en caldo
Este ensayo tambien usa caldo Mueller Hinton complementado con calcio y magnesio como medio decrecimiento. Tıpicamente se dispensan 100 µl de caldo en cada pocillo de una placa de microvaloracion de96 pocillos, y se hacen diluciones seriadas dobles de volumenes de 100 µl del analogo de peptido a lo largode la placa. Una fila de pocillos no recibe peptido y se usa como testigo de crecimiento. Cada pocillo seinocula con aproximadamente 5 x 105 UFC de bacterias y la placa se incuba a 35-37◦C durante 16-20horas. Se registra otra vez la CIM como la menor concentracion de peptido que inhibe completamente elcrecimiento del organismo determinado por inspeccion visual.
Por ejemplo, se establecieron los valores de CIM para una serie de analogos de peptidos frente a cepasde S. aureus. Los resultados se muestran en la siguiente Tabla 6.
46
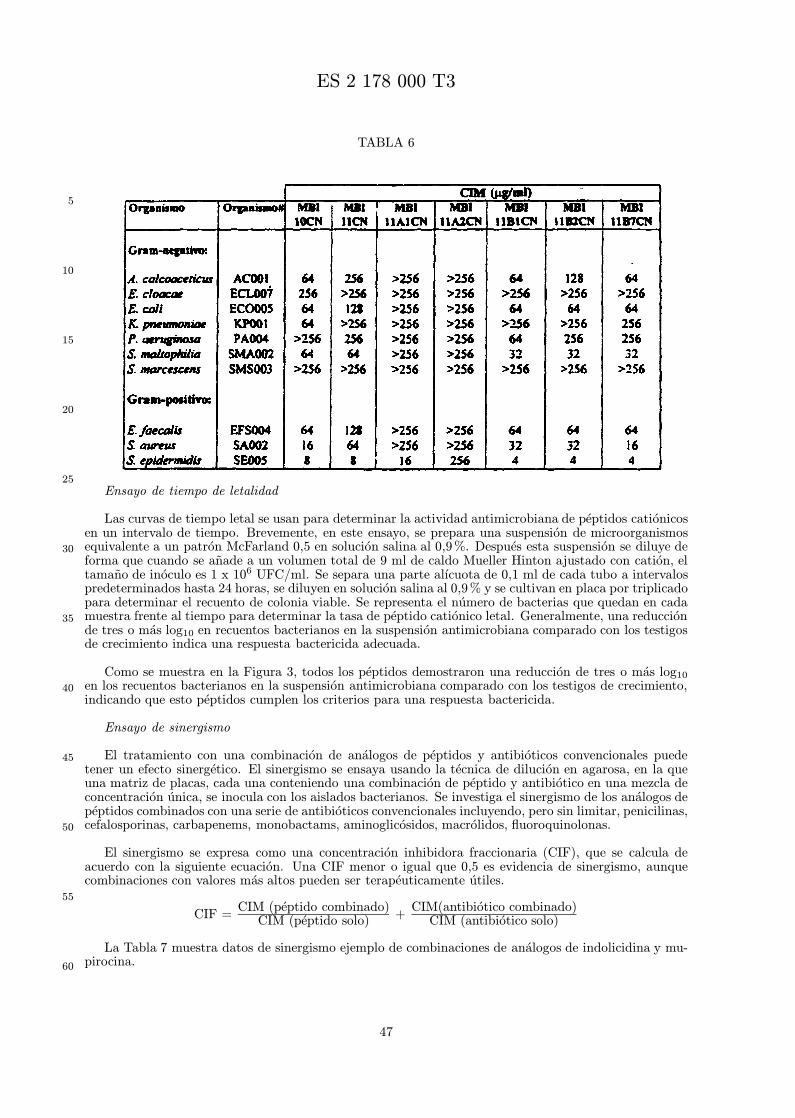
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 6
Ensayo de tiempo de letalidad
Las curvas de tiempo letal se usan para determinar la actividad antimicrobiana de peptidos cationicosen un intervalo de tiempo. Brevemente, en este ensayo, se prepara una suspension de microorganismosequivalente a un patron McFarland 0,5 en solucion salina al 0,9 %. Despues esta suspension se diluye deforma que cuando se anade a un volumen total de 9 ml de caldo Mueller Hinton ajustado con cation, eltamano de inoculo es 1 x 106 UFC/ml. Se separa una parte alıcuota de 0,1 ml de cada tubo a intervalospredeterminados hasta 24 horas, se diluyen en solucion salina al 0,9 % y se cultivan en placa por triplicadopara determinar el recuento de colonia viable. Se representa el numero de bacterias que quedan en cadamuestra frente al tiempo para determinar la tasa de peptido cationico letal. Generalmente, una reduccionde tres o mas log10 en recuentos bacterianos en la suspension antimicrobiana comparado con los testigosde crecimiento indica una respuesta bactericida adecuada.
Como se muestra en la Figura 3, todos los peptidos demostraron una reduccion de tres o mas log10
en los recuentos bacterianos en la suspension antimicrobiana comparado con los testigos de crecimiento,indicando que esto peptidos cumplen los criterios para una respuesta bactericida.
Ensayo de sinergismo
El tratamiento con una combinacion de analogos de peptidos y antibioticos convencionales puedetener un efecto sinergetico. El sinergismo se ensaya usando la tecnica de dilucion en agarosa, en la queuna matriz de placas, cada una conteniendo una combinacion de peptido y antibiotico en una mezcla deconcentracion unica, se inocula con los aislados bacterianos. Se investiga el sinergismo de los analogos depeptidos combinados con una serie de antibioticos convencionales incluyendo, pero sin limitar, penicilinas,cefalosporinas, carbapenems, monobactams, aminoglicosidos, macrolidos, fluoroquinolonas.
El sinergismo se expresa como una concentracion inhibidora fraccionaria (CIF), que se calcula deacuerdo con la siguiente ecuacion. Una CIF menor o igual que 0,5 es evidencia de sinergismo, aunquecombinaciones con valores mas altos pueden ser terapeuticamente utiles.
CIF =CIM (peptido combinado)
CIM (peptido solo) +CIM(antibiotico combinado)
CIM (antibiotico solo)
La Tabla 7 muestra datos de sinergismo ejemplo de combinaciones de analogos de indolicidina y mu-pirocina.
47

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 7
48
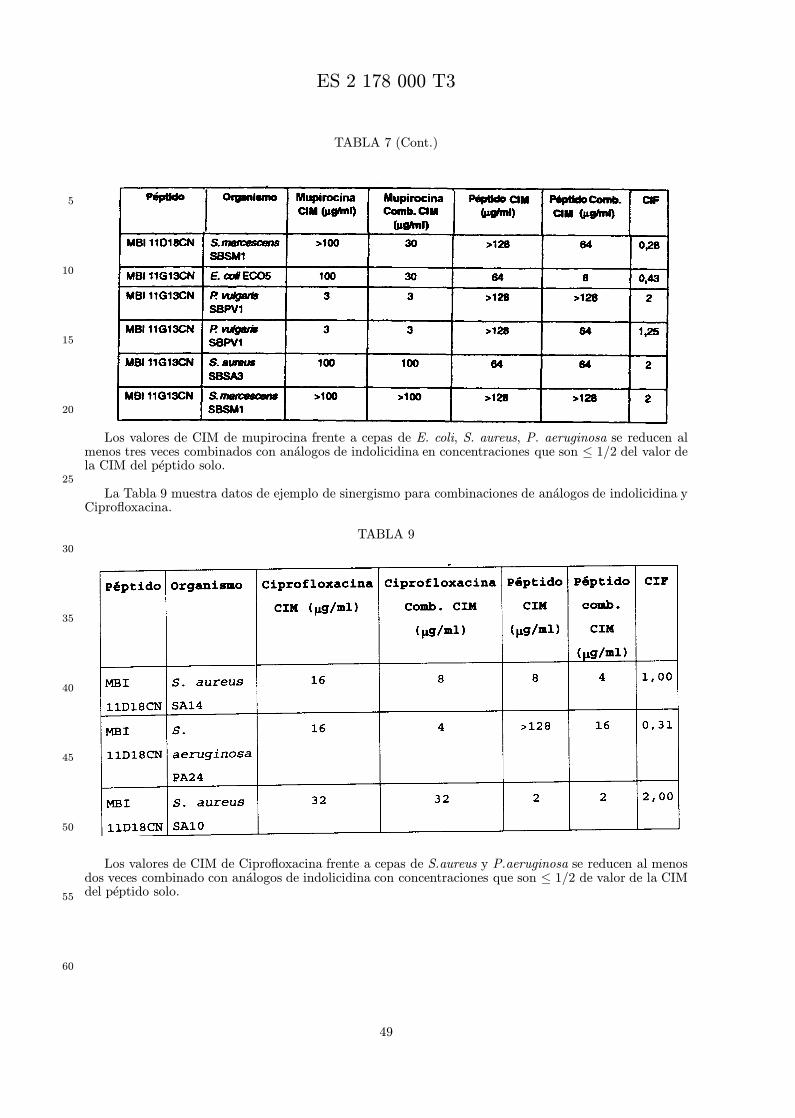
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 7 (Cont.)
Los valores de CIM de mupirocina frente a cepas de E. coli, S. aureus, P. aeruginosa se reducen almenos tres veces combinados con analogos de indolicidina en concentraciones que son ≤ 1/2 del valor dela CIM del peptido solo.
La Tabla 9 muestra datos de ejemplo de sinergismo para combinaciones de analogos de indolicidina yCiprofloxacina.
TABLA 9
Los valores de CIM de Ciprofloxacina frente a cepas de S.aureus y P.aeruginosa se reducen al menosdos veces combinado con analogos de indolicidina con concentraciones que son ≤ 1/2 de valor de la CIMdel peptido solo.
49

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Ejemplo 5
Caracterizacion bioquimica de los analogos de peptidos
Solubilidad en tampon de formulacion
El factor principal que afecta a la solubilidad de un peptido es su secuencia de aminoacidos. Lospeptidos policationicos preferiblemente son libremente solubles en soluciones acuosas, especialmente encondiciones de pH bajo. Sin embargo, en ciertas formulaciones, los peptidos policationicos pueden formarun agregado que se separa en una etapa de filtracion. Cuando las soluciones de peptido para ensayos invivo se filtran antes de administrar, se examinan la precision y reproducibilidad de los niveles de dosifi-cacion despues de filtrar.
Los peptidos disueltos en formulaciones se filtran por una membrana de filtro 0,2 µm hidrofila, ydespues se analiza el contenido total de peptido usando HPLC de fase inversa. Se prepara un patron100 % soluble para cada concentracion disolviendo el peptido en agua MilliQ. Se mide el area total depico para cada condicion y se compara con el area de pico del patron con el fin de proporcionar un valorde recuperacion relativo para cada combinacion de concentracion/formulacion.
Se preparo MBI 11CN en cuatro sistemas de tampon diferentes (A, B, C y C1) (Tabla 10, a con-tinuacion) con concentraciones de peptido de 50, 100, 200 y 400 µg/ml. Con las formulaciones A o B,ambas usadas normalmente para solvatar peptidos y proteınas, se perdio peptido por filtracion de unaforma dependiente de la concentracion (Figura 4). La recuperacion solo alcanzo un maximo de 70 %con una concentracion de 400 µg/ml. En contraste, los peptidos disueltos en las formulaciones C y C1se recuperaron completamente. Los tampones que contienen iones polianionicos parece que fomentan laagregacion, y es probable que el agregado tome la forma de una matriz que es atrapada por el filtro.Los contraiones monoanionicos son mas adecuados para mantener los peptidos de una forma soluble, noagregada, mientras que la adicion de otros agentes de solubilizacion puede mejorar mas la formulacion.
TABLA 10
Codigo Tampon de formulacion
A PBS 200 mM, pH 7,1
B Citrato sodico 100 mM, pH 5,2
C Acetato sodico 200 mM, pH 4,6
C1 Acetato sodico 200 mM/Polisorbato 80 al 0,5 %, pH 4,6
D Acetato sodico 100 mM/Polisorbato 80 activado al 0,5 %,pH 7,5: liofilizado/reconstituido
Solubilidad en caldo
La solubilidad de los analogos de peptidos se evalua en caldo de Mueller Hinton complementado concalcio y magnesio, por inspeccion visual. El procedimiento empleado es el usado para el ensayo de dilucionen caldo, excepto que no se anaden bacterias a los pocillos. El aspecto de la solucion de cada pocillo seevalua de acuerdo con la escala: (a) transparente, no precipita, (b) claro, precipitado difuso y (c) turbio,precipitado pesado. Los resultados muestran que, por ejemplo, MBI 10CN es menos soluble que MBI11CN en estas condiciones y que los analogos de MBI 11CN son menos solubles que los analogos de MBI11ACN.
Analisis por HPLC de fase inversa de formulaciones de analogos de peptidos
Se usa el HPLC de fase inversa, que proporciona un metodo analıtico para la cuantificacion de peptido,para examinar los peptidos en dos formulaciones diferentes. Se analiza una solucion de 400 µg/ml deMBI 11CN preparada en formulaciones C1 y D, usando un gradiente por pasos para resolver el peptidolibre de las otras especies. Se usan las siguientes condiciones cromatograficas patron:
50

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
- Disolvente A: acido trifluoroacetico (TFA) al 0,1 % en agua
- Disolvente B: TFA al 0,1 %/acetonitrilo al 95 % en agua
- Medio: POROS©R R2-20 (poliestireno-divinilbenceno)
Como se muestra en la Figura 5, MBI 11CN se podıa separar de dos formas, como peptido libre enformulacion C1, y como complejo de peptido-formulacion principalmente en formulacion D. Este complejosobrevive el protocolo de separacion en gradientes que contienen acetonitrilo, que se podrıa esperar querompieran la estabilidad del complejo. Tambien se observa un pico correspondiente a una pequena canti-dad (<10 %) de peptido libre en formulacion D. Si se cambia la forma del gradiente de elucion, el peptidoasociado eluye como un pico bajo ancho, que indica que los complejos de peptido en la formulacion sonheterogeneos.
Ejemplo 6
Analisis estructural de variantes de indolicidina usando espectroscopia de dicroismo circular
El dicroısmo circular (DC) es una tecnica espectroscopica que mide las estructuras secundarias depeptidos y proteınas en solucion, vease por ejemplo, R.W. Woody, (Methods in Enzymology, 246: 34,1995). El espectro de DC de peptidos con helice α se puede interpretar mas facilmente debido a losmınimos dobles caracterısticos a 208 y 222 nm. Sin embargo, para peptidos con otra estructura secun-daria, la interpretacion del espectro de DC es mas complicado y menos fiable. Los datos de DC parapeptidos se usan para relacionar estructura en solucion con actividad in vitro.
Las mediciones de DC de analogos de indolicidina se llevan a cabo en tres medios acuosos diferentes,(1) tampon de fosfato sodico 10 mM, pH 7,2, (2) tampon de fosfato y trifluoroetanol (TFE) al 40 %(vol/vol), y (3) tampon de fosfato y vesıculas grandes (liposomas) de fosfolıpidos unilamelares (100 nmde diametro) (Tabla 11). El disolvente organico TFE y los liposomas proporcionan un entorno hidrofoboque se pretende que mimetice la membrana bacteriana donde se supone que los peptidos adoptan unaconformacion activa.
Los resultados indican que los peptidos estan principalmente desordenados en tampon de fosfato (unmınimo negativo alrededor de 200 nm) con la excepcion de MBI 11F4CN, que presenta un mınimo adi-cional a 220 nm (vease a continuacion). La presencia de TFE induce estructura de lamina β en MBI 11y MBI 11G4CN, y aumenta la forma de helice α en MBI 11F4CN, aunque la mayorıa de los peptidospermanecen desordenados. En presencia de liposomas, los peptidos MBI 11CN y MBI 11B7CN, queestan desordenados en TFE, presentan estructura en lamina β (un mınimo negativo alrededor de 230nm) (Figura 6). Por lo tanto, parece que los liposomas inducen estructura secundaria mas ordenada queel TFE.
Una lamina β es la estructura secundaria predominante que aparece en un entorno hidrofobo, su-giriendo que es la conformacion principal en la forma activa asociada a membrana. En contraste, MBI11F4CN presenta mayor conformacion de helice α en presencia de TFE. El peptido MBI 11F4CN tambienes el mas insoluble y hemolıtico de los peptidos ensayados, sugiriendo que la estructura secundaria dehelice α puede introducir propiedades no deseadas en estos analogos.
Adicionalmente se registran espectros de DC para peptidos modificados con APS (Tabla 11). Los re-sultados muestran que estos compuestos tienen estructura secundaria de lamina β significativa en tamponde fosfato, que es alterada solo ligeramente en TFE.
Otra vez, los resultados de DC sugieren que una estructura de lamina β (es decir, asociada a mem-brana) es la conformacion activa preferida entre los analogos de indolicidina ensayados.
51
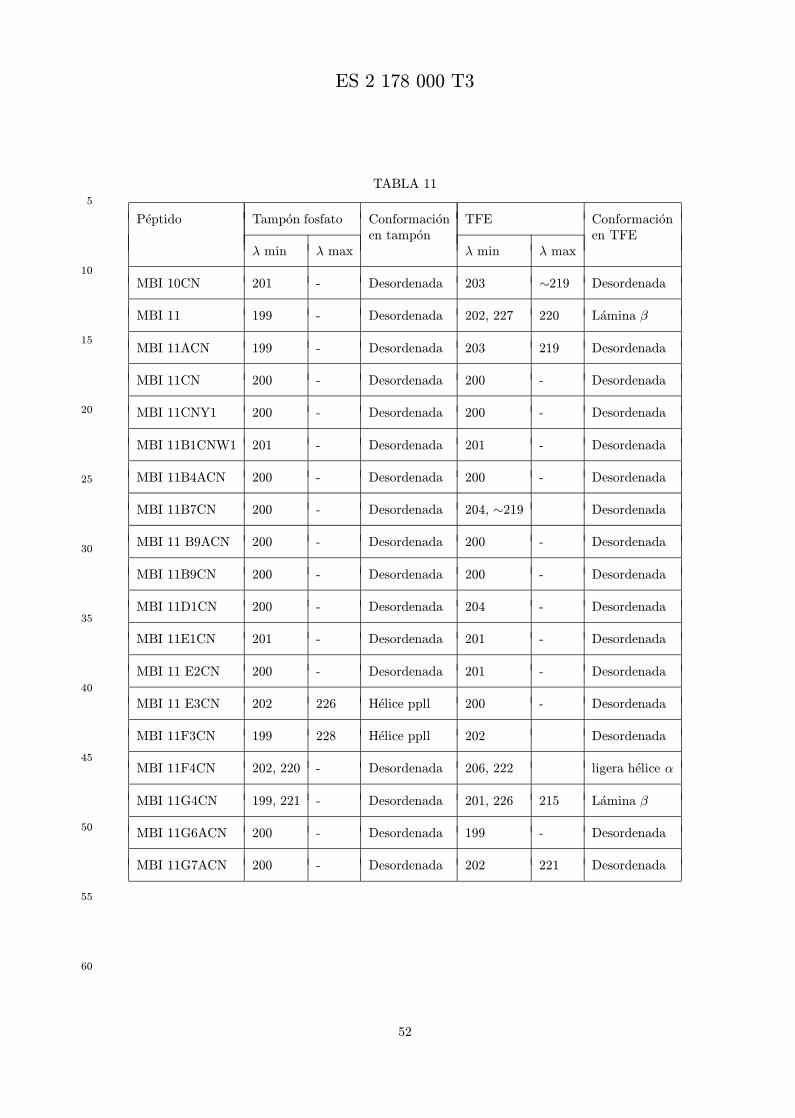
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 11
Peptido Tampon fosfato Conformacion TFE Conformacionen tampon en TFE
λ min λ max λ min λ max
MBI 10CN 201 - Desordenada 203 ∼219 Desordenada
MBI 11 199 - Desordenada 202, 227 220 Lamina β
MBI 11ACN 199 - Desordenada 203 219 Desordenada
MBI 11CN 200 - Desordenada 200 - Desordenada
MBI 11CNY1 200 - Desordenada 200 - Desordenada
MBI 11B1CNW1 201 - Desordenada 201 - Desordenada
MBI 11B4ACN 200 - Desordenada 200 - Desordenada
MBI 11B7CN 200 - Desordenada 204, ∼219 Desordenada
MBI 11 B9ACN 200 - Desordenada 200 - Desordenada
MBI 11B9CN 200 - Desordenada 200 - Desordenada
MBI 11D1CN 200 - Desordenada 204 - Desordenada
MBI 11E1CN 201 - Desordenada 201 - Desordenada
MBI 11 E2CN 200 - Desordenada 201 - Desordenada
MBI 11 E3CN 202 226 Helice ppll 200 - Desordenada
MBI 11F3CN 199 228 Helice ppll 202 Desordenada
MBI 11F4CN 202, 220 - Desordenada 206, 222 ligera helice α
MBI 11G4CN 199, 221 - Desordenada 201, 226 215 Lamina β
MBI 11G6ACN 200 - Desordenada 199 - Desordenada
MBI 11G7ACN 200 - Desordenada 202 221 Desordenada
52
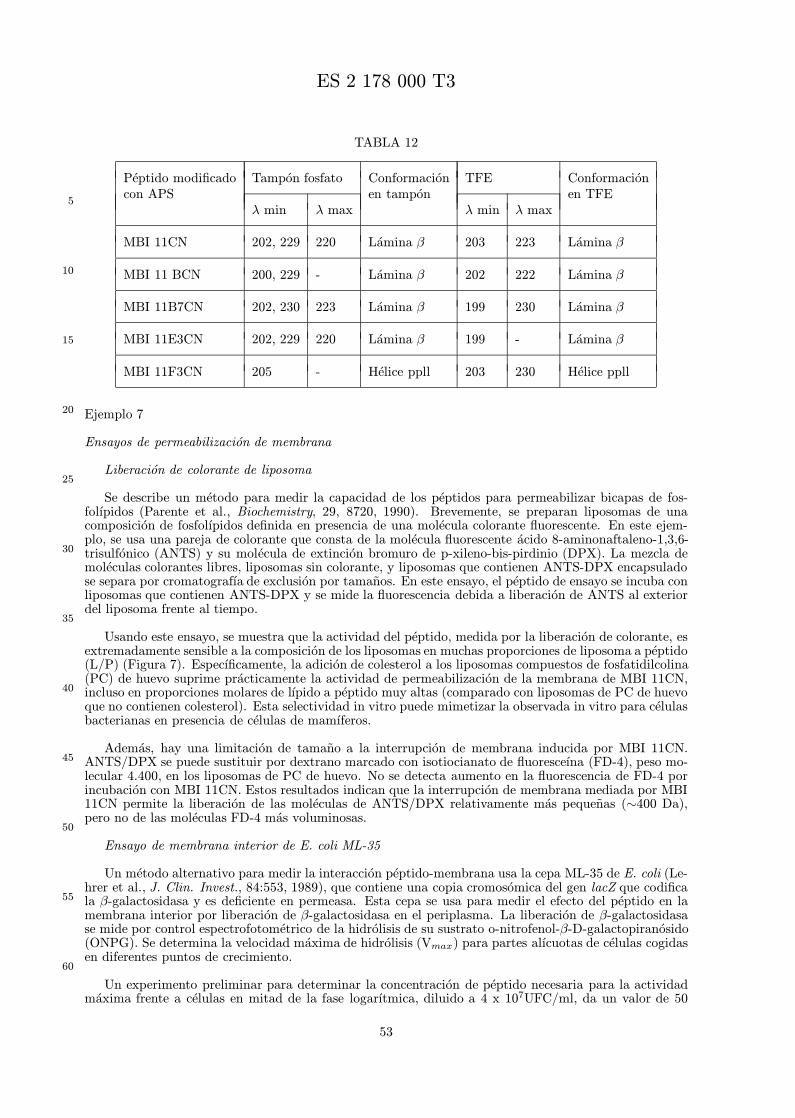
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 12
Peptido modificado Tampon fosfato Conformacion TFE Conformacioncon APS en tampon en TFE
λ min λ max λ min λ max
MBI 11CN 202, 229 220 Lamina β 203 223 Lamina β
MBI 11 BCN 200, 229 - Lamina β 202 222 Lamina β
MBI 11B7CN 202, 230 223 Lamina β 199 230 Lamina β
MBI 11E3CN 202, 229 220 Lamina β 199 - Lamina β
MBI 11F3CN 205 - Helice ppll 203 230 Helice ppll
Ejemplo 7
Ensayos de permeabilizacion de membrana
Liberacion de colorante de liposoma
Se describe un metodo para medir la capacidad de los peptidos para permeabilizar bicapas de fos-folıpidos (Parente et al., Biochemistry, 29, 8720, 1990). Brevemente, se preparan liposomas de unacomposicion de fosfolıpidos definida en presencia de una molecula colorante fluorescente. En este ejem-plo, se usa una pareja de colorante que consta de la molecula fluorescente acido 8-aminonaftaleno-1,3,6-trisulfonico (ANTS) y su molecula de extincion bromuro de p-xileno-bis-pirdinio (DPX). La mezcla demoleculas colorantes libres, liposomas sin colorante, y liposomas que contienen ANTS-DPX encapsuladose separa por cromatografıa de exclusion por tamanos. En este ensayo, el peptido de ensayo se incuba conliposomas que contienen ANTS-DPX y se mide la fluorescencia debida a liberacion de ANTS al exteriordel liposoma frente al tiempo.
Usando este ensayo, se muestra que la actividad del peptido, medida por la liberacion de colorante, esextremadamente sensible a la composicion de los liposomas en muchas proporciones de liposoma a peptido(L/P) (Figura 7). Especıficamente, la adicion de colesterol a los liposomas compuestos de fosfatidilcolina(PC) de huevo suprime practicamente la actividad de permeabilizacion de la membrana de MBI 11CN,incluso en proporciones molares de lıpido a peptido muy altas (comparado con liposomas de PC de huevoque no contienen colesterol). Esta selectividad in vitro puede mimetizar la observada in vitro para celulasbacterianas en presencia de celulas de mamıferos.
Ademas, hay una limitacion de tamano a la interrupcion de membrana inducida por MBI 11CN.ANTS/DPX se puede sustituir por dextrano marcado con isotiocianato de fluoresceına (FD-4), peso mo-lecular 4.400, en los liposomas de PC de huevo. No se detecta aumento en la fluorescencia de FD-4 porincubacion con MBI 11CN. Estos resultados indican que la interrupcion de membrana mediada por MBI11CN permite la liberacion de las moleculas de ANTS/DPX relativamente mas pequenas (∼400 Da),pero no de las moleculas FD-4 mas voluminosas.
Ensayo de membrana interior de E. coli ML-35
Un metodo alternativo para medir la interaccion peptido-membrana usa la cepa ML-35 de E. coli (Le-hrer et al., J. Clin. Invest., 84:553, 1989), que contiene una copia cromosomica del gen lacZ que codificala β-galactosidasa y es deficiente en permeasa. Esta cepa se usa para medir el efecto del peptido en lamembrana interior por liberacion de β-galactosidasa en el periplasma. La liberacion de β-galactosidasase mide por control espectrofotometrico de la hidrolisis de su sustrato o-nitrofenol-β-D-galactopiranosido(ONPG). Se determina la velocidad maxima de hidrolisis (Vmax) para partes alıcuotas de celulas cogidasen diferentes puntos de crecimiento.
Un experimento preliminar para determinar la concentracion de peptido necesaria para la actividadmaxima frente a celulas en mitad de la fase logarıtmica, diluido a 4 x 107UFC/ml, da un valor de 50
53
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
µg/ml, que se usa en todos los experimentos posteriores. Las celulas se hacen crecer en dos medios decrecimiento diferentes, caldo de Terrific (TB) y caldo Luria (LB) y se ensayan cantidades equivalentes decelulas durante sus ciclos de crecimiento. El perfil de actividad resultante de MBI 11B7CN se muestraen la figura 8. Para las celulas que se han hecho crecer en el medio TB enriquecido, se produce actividadmaxima al principio de la mitad de la fase logarıtmica (140 min), mientras que para las celulas que sehan hecho crecer en medio LB, el maximo se produce al final de la mitad de la fase logarıtmica (230min). Adicionalmente, solo en LB, se observa una bajada de actividad a 140 min. Esta disminucion deactividad puede estar relacionada con una transicion en el metabolismo, tal como un requisito para usaruna nueva fuente de energıa debido a la reduccion de la fuente original, que no se produce en el medioTB mas enriquecido. Como consecuencia de un cambio de metabolismo se producirıan cambios en elpotencial de membrana.
Para ensayar si el potencial de membrana tiene un efecto en la actividad del peptido, se examinael efecto de interrumpir el gradiente electroquımico usando el ionoforo de potasio valinomicina. Lascelulas previamente incubadas con valinomicina se tratan con peptido, y para MBI 10CN y MBI 11CNla hidrolisis de ONPG disminuye aproximadamente 50 % comparado con valinomicina no incubada pre-viamente (Figura 9). Se usa otro peptido cationico que no es sensible a la valinomicina como testigopositivo.
Se ensaya una delineacion adicional de los factores que influyen en la actividad permeabilizante dela membrana. En un ensayo de ejemplo, MBI 11B7CN se incuba previamente con tampon de HE-PES/sacarosa isotonico que contiene cloruro sodico 150 mM (NaCl) o iones magnesio 5 mM (Mg2+) y seensaya como se ha descrito antes. En la figura 10, se observa una inhibicion significativa con cualquierade las soluciones, sugiriendo la implicacion de interacciones electrostaticas en la accion permeabilizantede peptidos.
Ejemplo 8
Lisis de eritrocitos por analogos de indolcidina
Se usa un ensayo de lisis de globulos rojos (GR) para agrupar peptidos de acuerdo con su capacidadpara la lisis de GR en condiciones patron comparado con MBI 11CN y gramicidina-S. Las muestras depeptido y GR de oveja lavados se preparan en solucion salina isotonica con el pH final ajustado entre 6 y7. Las muestras de peptidos y suspension de GR se mezclan entre sı para dar soluciones que tienen 1 %(vol/vol) de GR y 5,50 o 500 µg/ml de peptido. Las mezclas de ensayo se incuban durante 1 hora a 37◦Ccon agitacion constante, se centrifugan, y se mide la absorbancia a 540 nm del lıquido sobrenadante, quedetecta la hemoglobina liberada. El porcentaje de hemoglobina liberada se determina por comparacioncon una serie de lisatos patron conocidos, en agua. Cada serie de ensayos tambien incluye MBI 11CN(500 µg/ml) y gramicidina-S (5 µg/ml) como testigos de “lisis baja” y “lisis alta”, respectivamente.
Se ensayan MBI-11B7CN-HCl, MBI-11F3CN-HCl y MBI-11F4CN-HCl usando este procedimiento ylos resultados se presentan en la siguiente Tabla 13.
TABLA 13
Peptido % de lisis con % de lisis con % de lisis con5 µg/ml 50 µg/ml 500 µg/ml
MBI 11B7CN-HCl 4 13 46
MBI 11F3CN-HCl 1 6 17
MBI 11F4CN-HCl 4 32 38
MBI 11CN-TFA N/D N/D 9
Gramicidina-S 30 N/D N/D
N/D = no hecho
54

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Los peptidos que con 5 µg/ml producen la lisis de GR en una extension igual o mayor que lagramicidina-S, el testigo de “lisis alta”, se consideran que son altamente lıticos. Los peptidos que con500 µg/ml producen la lisis de los GR en una extension igual o menor que MBI 11CN, el testigo de“lisis baja”, se considera que no son lıticos. Los tres analogos ensayados son todos “moderadamentelıticos” puesto que producen mas lisis que MBI 11CN y menos que la gramicidina-S. Ademas uno delos analogos, MBI 11F3CN-HCl, es significativamente menos lıtico que las otras dos variantes en las tresconcentraciones ensayadas.
Ejemplo 9
Produccion de anticuerpos para los analogos de peptidos
Se preparan peptidos antigenicos multiples (MAP), que contienen cuatro u ocho copias del peptidoobjetivo unido a un pequeno nucleo de peptidilo no inmunogenico, como inmunogenes. Alternativamente,el peptido objetivo se conjuga con albumina de suero bovino (BSA) u ovoalbumina. Por ejemplo, se usanMBI 11CN y sus fragmentos N-terminal y C-terminal de siete aminoacidos como secuencias de peptidoobjetivo. Los inmunogenes se inyectan vıa subcutanea en conejos usando protocolos patron (vease, Har-low and Lane, Antibodies: A Laboratroy Manual, Harlow and Lane (eds.), Cold Spring Harbor LaboratoryPress, NY, 1988). Despues de repetir las inyecciones (normalmente mensualmente), se ensaya el suero deuna muestra de sangre en un ensayo ELISA frente al peptido objetivo. Un resultado positivo indica lapresencia de anticuerpos y posteriores ensayos determinan la especificidad de la union del anticuerpo alpeptido objetivo. Despues se pueden aislar los anticuerpos purificados de este suero y se pueden usar enensayos ELISA para identificar selectivamente y medir la cantidad de peptido objetivo en muestras deinvestigacion y clınicas.
Ejemplo 10
Farmacologia de analogos de peptidos en el plasma y sangre
Se determina el tiempo de vida in vitro de analogos de peptido libres en el plasma y en la sangremidiendo la cantidad de peptido presente despues de una serie de tiempos de incubacion. Se recoge sangrede ovejas, se trata con anticoagulante (no heparina) y para preparar el plasma, se centrifuga para separarlas celulas. Se anade peptido formulado a la fraccion de plasma o sangre entera y se incuba. Despues deincubar, el peptido se identifica y cuantifica directamente por HPLC de fase inversa. No es necesaria laextraccion cuando el pico de peptido libre no se superpone con ningun pico de la sangre o plasma.
Se anade una solucion de 1 mg/ml de MBI 11CN en formulaciones C1 y D a plasma de oveja recienpreparado, con una concentracion final de peptido de 100 µg/ml y se incuba a 37◦C. Se separan partesalıcuotas de plasma a diferentes tiempos y se analizan los peptidos libres por HPLC de fase inversa. Paracada cromatograma, se integra el area del pico correspondiente al peptido libre y se representa frenteal tiempo de incubacion. Como se muestra en la Figura 11, los niveles de peptido disminuyen frenteal tiempo. Ademas, cuando se administra en formulacion D, hasta 50 % del peptido se libera inmedia-tamente del complejo formulacion-peptido al anadirlo a la sangre. La curva de desintegracion para elpeptido libre da una semivida aparente en la sangre de 90 minutos tanto para la formulacion C1 como D.Estos resultados indican que en sangre de oveja, MBI 11CN es relativamente resistente a las peptidasasy proteasas del plasma. Los nuevos picos que aparecen durante la incubacion pueden ser productos deruptura del peptido.
Los niveles de peptido en el plasma in vivo se miden despues de administracion vıa iv o ip de 80-100 %de la dosis maxima tolerada de analogo de peptido en formulacion C1 o D. MBI 11CN en formulacionC1 se inyecta vıa intravenosa en la vena de la cola de ratones de cepa CD1 ICRBR. A diferentes tiemposdespues de inyeccion, los ratones se anestesian y se extrae sangre por pinchazo cardiaco. La sangre de losratones individuales se centrifuga para separar el plasma de las celulas. Despues el plasma se analiza porcolumna de HPLC de fase inversa. En los perfiles de elucion resultantes se analiza el contenido de peptidolibre por absorbancia UV a 280 nm, y estos datos se convierten en concentraciones en la sangre basandoseen un patron calibrado. Cada punto de dato representa el nivel en sangre medio de dos ratones. En esteensayo, el lımite de deteccion es aproximadamente 1 µg/ml, menos de 3 % de la dosis administrada.
El punto de tiempo mas temprano al que se puede medir el peptido es tres minutos despues deinyeccion, por lo tanto, la concentracion maxima observada (en µg/ml) se extrapola hacia atras a tiempocero (Figura 21). La concentracion inicial proyectada se corresponde bien con la concentracion esperadade entre 35 y 45 µg/ml. Sin embargo, la desintegracion es rapida, y cuando la curva se ajusta a la ecuacion
55

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
para la desintegracion exponencial, se calcula que el peptido libre que circula tiene una semivida de 2,1minutos. El peptido libre que circula no era detectable en la sangre de ratones a los que se inyecto MBI11CN en formulacion D, sugiriendo que el peptido no es liberado tan rapidamente del complejo como invitro.
Ademas, MBI 11CN tambien se administra a ratones de la cepa CD1 ICRBR por una sola inyeccionip con un nivel de dosis eficaz de 40 mg/kg. El peptido se administra tanto en formulacion C1 como Dpara determinar si la formacion de complejo del peptido tiene algun efecto en los niveles en la sangre.A diferentes tiempos despues de inyeccion, los ratones se anestesian y se extrae sangre por pinchazocardiaco. La sangre se recoge y se analiza como para la inyeccion iv.
El MBI 11CN administrado por esta vıa demostro un perfil farmacologico bastante diferente (Figura13). En formulacion C1, el peptido entro en la corriente sanguınea rapidamente, con una concentracionmaxima de casi 5 µg/ml despues de 15 minutos, que disminuyo a niveles no detectables despues de 60minutos. En contraste, el peptido en formulacion D esta presente en un nivel por encima de 2 µg/mldurante aproximadamente dos horas. Por lo tanto, la formulacion afecta a la entrada y al mantenimientode los niveles de peptido en la sangre.
Ejemplo 11
Toxicidad de analogos de peptidos in vivo
Se ensaya la toxicidad aguda de una sola dosis de diferentes analogos de indolicidina en ratones SwissCD1 usando diferentes vıas de administracion. Con el fin de determinar las toxicidades inherentes a losanalogos de peptidos en ausencia de cualesquiera efectos de la formulacion/vehıculo de suministro, todoslos peptidos se administran en solucion salina isotonica con el pH final entre 6 y 7.
Vıa intraperitoneal Se inyectan grupos de 6 ratones con dosis de peptidos entre 80 y 5 mg/kg envolumenes de dosis de 500 µl. Despues de la administracion de peptido, los ratones se observan durante unperiodo de 5 dıas, momento en el que se determinan los niveles de dosis que producen 50 % de mortalidad(DL50), dosis que produce 90-100% de mortalidad (DL90−100) y dosis maxima tolerada (DMT). Losvalores de DL50 se calculan usando el metodo de Reed y Muench (J. of Amer. Hyg. 27: 493-497, 1938).Los resultados presentados en la Tabla 14 muestran que los valores de DL50 para MBI 11CN y analogosestan en el intervalo de 21 a 52 mg/kg.
TABLA 14
Peptido DL50 DL90−100 DMT
MBI 11CN 34 mg/kg 40 mg/kg 20 mg/kg
MBI 11B7CN 52 mg/kg >80 mg/kg 30 mg/kg
MBI 11E3CN 21 mg/kg 40 mg/kg <20 mg/kg
MBI 11F3CN 52 mg/kg 80 mg/kg 20 mg/kg
Vıa intravenosa. Se inyectan grupos de 6 ratones con dosis de peptido de 20, 16, 12, 8, 4 y 0 mg/kg envolumenes de 100 µl (4 ml/kg). Despues de administracion, los ratones se observan durante un periodo de5 dıas, momento en el que se determinan los niveles de DL50, DL90−100 y DMT. Los resultados del ensayode toxicidad iv de MBI 11CN y tres analogos se muestran en la Tabla 15. Los valores de DL50, DL90−100
y DMT estan en el intervalo de 5,8 a 15 mg/kg, de 8 a 20 mg/kg y de <4 a 12 mg/kg, respectivamente.
56
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 15
Peptido DL50 DL90−100 DMT
MBI 11CN HCl 5,8 mg/kg 8,0 mg/kg <4 mg/kg
MBI 11B7CN HCl 7,5 mg/kg 16 mg/kg 4 mg/kg
MBI 11F3CN HCl 10 mg/kg 12 mg/kg 8 mg/kg
MBI 11F4CN HCl 15 mg/kg 20 mg/kg 12 mg/kg
Vıa subcutanea. La toxicidad de MBI 11CN tambien se determina despues de administracion sub-cutanea (SC). Para el ensayo de toxicidad SC, se inyectan grupos de 6 ratones con dosis de peptido de128, 96, 64, 32 y 0 mg/kg en volumenes de dosis de 300 µl (12 ml/kg). Despues de administracion, losratones se observan durante un periodo de 5 dıas. Ninguno de los animales murio con ninguno de losniveles de dosis dentro del periodo de observacion de 5 dıas. Por lo tanto, los valores de DL50, DL90−100
y DMT se ha considerado que son todos mayores que 128 mg/kg. Los ratones que recibieron niveles dedosis mayores mostraron sıntomas similares a los vistos despues de inyeccion iv, sugiriendo que el peptidoentro en la circulacion sistemica. Estos sıntomas son reversibles, desapareciendo en todos los ratones alsegundo dıa de observaciones.
Tambien se examina la toxicidad de una sola dosis de MBI 10CN y MBI 11CN en diferentes formu-laciones en ratones ICR de crıa exogenica (Tabla 16). La inyeccion intraperitoneal (grupos de 2 ratones)de MBI 10CN en formulacion D no muestra toxicidad hasta 29 mg/kg y en las mismas condiciones MBI11CN no muestra toxicidad hasta 40 mg/kg.
La inyeccion intravenosa (grupos de 10 ratones) de MBI 10CN en formulacion D muestra una dosismaxima tolerada (DMT) de 5,6 mg/kg (Tabla 16). La inyeccion de 11 mg/kg dio 40 % de toxicidad y 22mg/kg dieron como resultado 100 % de toxicidad. La inyeccion intravenosa de MBI 11CN en formulacionC (liofilizado) muestra una DMT de 3,0 mg/kg. La inyeccion de 6,1 mg/kg da como resultado 10 % detoxicidad y con 12 mg/kg 100 % de toxicidad.
TABLA 16
Peptido Vıa # Animales Formulacion DMT (mg/kg)
MBI 10CN ip 2 formulacion D >29
MBI 11CN ip 2 formulacion D >40
MBI 10CN iv 10 formulacion D 5,6
MBI 11CN iv 10 formulacion C (liofilizado) 3,0
Estos resultados se obtienen usando soluciones de peptido/tampon que se liofilizan despues de prepa-rar y se reconstituyen con agua. Si la solucion de peptido no se liofiliza antes de inyectar, si no que se usainmediatamente despues de preparar, se observa un aumento de toxicidad, y la dosis maxima toleradapuede disminuir hasta en cuatro veces. Por ejemplo, una inyeccion intravenosa de MBI 11CN como unasolucion no liofilizada, formulacion C1, de 1,5 mg/kg da como resultado 20 % de toxicidad y de 3,0 mg/kgdio 100 % de toxicidad. El analisis por HPLC de las formulaciones no liofilizada y liofilizada indica queMBI 11CN forma un complejo con polisorbato, y esta complejacion del peptido reduce su toxicidad enratones.
Ademas, los ratones se inyectan multiples veces por una vıa intravenosa con MBI 11CN (Tabla 17).En un experimento representativo, el peptido administrado en 10 inyecciones de 0,84 mg/kg en intervalos
57
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
de 5 minutos no es letal. Sin embargo, dos inyecciones de peptido de 4,1 mg/kg administradas con unintervalo de 10 minutos, dan como resultado 60 % de mortalidad.
TABLA 17
Peptido Vıa Formulacion Nivel # Intervalo Resultadode dosis∗ inyecciones de tiempo
MBI 11CN iv formulacion 0,84 10 5 min no mortalidadD
MBI 11CN iv formulacion 4,1 2 10 min 66 % deD mortalidad
∗ (mg/kg)
Para evaluar el impacto de ratones dosificados con analogo de peptido, se pueden llevar a cabo unaserie de investigaciones de histopatologıa. Se administra a grupos de ratones analogo con niveles de dosisque estan en, o por debajo de la DMT, o por encima de la DMT, o una dosis letal. Se pueden usarmultiples inyecciones para mimetizar posibles regımenes de tratamiento. Los grupos de ratones testigono se inyectan o se les inyecta solo con tampon.
Despues de inyeccion, los ratones se sacrifican en tiempos especificados y sus organos se ponen in-mediatamente en una solucion de formalina equilibrada al 10 %. Los organos de los ratones que muerencomo resultado de los efectos toxicos del analogo tambien se conservan inmediatamente. Se toman mues-tras de tejido y se preparan como microsecciones tenidas en portaobjetos que despues se examinan enel microscopio. Se evalua el dano a tejidos y esta informacion se puede usar para desarrollar analogosmejorados, metodos mejorados de administracion o regımenes de dosificacion mejorados.
Ejemplo 12
Eficacia in vivo de analogos de peptidos
Se ensaya en los analogos su capacidad de rescatar ratones de infecciones bacterianas letales. Elmodelo animal usado es una inoculacion intraperitoneal (ip) de ratones con 106-108 organismos Grampositivos con posterior administracion de peptido. Los tres patogenos investigados, S. aureus sensiblea meticilina (SASM), S. aureus resistente a meticilina (SARM), o S. epidermidis, se inyectan vıa ip enratones. Para los ratones no tratados, la muerte se produce en 12-18 horas con SASM y S. epidermis yen 6-10 horas con SARM.
El peptido se administra por dos vıas, vıa intraperitoneal, una hora despues de infeccion, o vıa intra-venosa, con una sola dosis o dosis multiples dadas en diferentes tiempos antes y despues de infeccion.
Infeccion por SASM. En un protocolo tıpico, se infectan grupos de 10 ratones vıa intraperitoneal conuna dosis DL90−100 (5,2 x 106 UFC/raton) de SASM (Smith, ATCC # 19640) inyectada en infusioncerebro-corazon que contiene 5 % de mucina. Esta cepa de S. aureus no es resistente a ningun antibioticocomun. A los 60 minutos despues de infeccion, se inyecta vıa intraperitoneal MBI 10CN o MBI 11CN enformulacion D, con los niveles de dosis establecidos. Una inyeccion de formulacion sola sirve como testigonegativo, y la administracion de ampicilina sirve como testigo positivo. La supervivencia de los ratonesse controla a las 1 2, 3 y 4 horas despues de infeccion, y despues dos veces diarias durante un total de 8dıas.
Como se muestra en la Figura 14, MBI 10CN es activo de forma maxima frente a SASM (70-80 % desupervivencia) con dosis de 14,5 a 38,0 mg/kg, aunque no se logra 100 % de supervivencia. Por debajo de14,5 mg/kg, hay una clara supervivencia dependiente de la dosis. Con estos niveles de dosis mas bajos,parece que hay un umbral dependiente del animal, de forma que los ratones mueren el dıa 2 o sobrevivenel periodo entero de ocho dıas. Como se ve en la Figura 15, por otra parte, MBI 11CN rescata 100 %de los ratones de infeccion por SASM con un nivel de dosis de 35,7 mg/kg, y por lo tanto era tan eficazcomo la ampicilina. Habıa muy poca o no habıa actividad con ninguno de los niveles de dosis mas bajos,lo cual indica que se debe lograr un nivel de peptido mınimo en la corriente sanguınea durante el tiempo
58
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
que las bacterias son un peligro para el hospedante.
Como se ha mostrado antes, los niveles de MBI 11CN se pueden mantener a un nivel mayor que 2µg/ml durante un periodo de dos horas, infiriendo que este es mayor que el nivel mınimo.
Adicionalmente, se ensayan ocho variantes basados en la secuencia de MBI 11CN frente a SASMusando el sistema experimental antes descrito. Los peptidos preparados en formulacion D se administrancon niveles de dosis en el intervalo de 12 a 24 mg/kg, y se controla la supervivencia de los ratonesinfectados durante ocho dıas (Figuras 16-24). El porcentaje de supervivencia al final del periodo deobservacion para cada variante se resume en la Tabla 18. Como se muestra en la tabla, varias de lasvariantes mostraron mayor eficacia o igual que MBI 11CN en las mismas condiciones.
TABLA 18
% de Supervivencia 24 mg/kg 18 mg/kg 12 mg/kg
100
90 11B1CN, 11F3CN
80
70 11E3CN
60 11B7CN
50 11CN
40 11G2CN
30 11B1CN
20 11G4CN
10 11CN, 11B7CN, 11G2CN11B8CN, 11F3CN
0 11A1CN 11A1CN, 11G2CN,11G4CN 11CN, 11A1CN,
11B1CN, 11B7CN,11B8CN, 11F3CN,11G4CN
Infeccion por S. epidermidis. Los analogos de peptidos generalmente tienen valores de CIM mas bajosfrente a S. epidermidis in vitro, por lo tanto, niveles mas bajos de peptido en la sangre deben ser maseficaces frente a la infeccion.
En un protocolo tıpico, se inyectan grupos de 10 ratones vıa intraperitoneal con una dosis DL90−100
(2,0 x 108 UFC/raton) de S. epidermidis (ATCC # 12228) en caldo de infusion cerebro-corazon quecontiene 5 % de mucina. Esta cepa de S. epidermidis es 90 % letal despues de 5 dıas. A los 15 min y 60min despues de infeccion, se inyectan diferentes dosis de MBI 11CN en formulacion D vıa intravenosaen la vena de la cola. Una inyeccion de formulacion sola sirve como testigo negativo, y la inyeccion degentamicina sirve como testigo positivo; ambos se inyectan a los 60 minutos despues de infeccion. Lasupervivencia de los ratones se controla a las 1, 2, 3, 4, 6 y 8 horas despues de infeccion, y despues dosveces diarias durante un total de 8 dıas.
Como se muestra en las Figuras 25A y 25B, MBI 11CN prolonga la supervivencia de los ratones. Se
59

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
observa eficacia con los tres niveles de dosis con tratamiento 15 minutos despues de inyeccion, sin em-bargo, hay menos actividad a los 30 minutos despues de infeccion y no hay efecto significativo 60 minutosdespues de infeccion. El tiempo de administracion parece ser importante en este sistema modelo, dandola mejor tasa de supervivencia una sola inyeccion de 6,1 mg/kg 15 minutos despues de inyeccion.
Infeccion por SARM La infeccion por SARM, aunque es letal en un corto periodo de tiempo, requiereuna carga bacteriana mucho mayor que SASM. En un protocolo tıpico, se inyectan grupos de 10 ratonesvıa intraperitoneal con una dosis DL90−100 (4,2 x 107 UFC/raton) de SARM (ATCC #33591) en infusioncerebro-corazon que contiene 5 % de mucina. Los protocolos del tratamiento son los siguientes, con lostiempos de tratamiento relativos al tiempo de infeccion:
- 0 mg/kg Formulacion D sola (testigo negativo), inyectada a los 0 min
- 5 mg/kg Tres inyecciones de 5,5 mg/kg a los -5, +55 y +115 min
- 1 mg/kg (2 h) Cinco inyecciones de 1,1 mg/kg a los -5, +55, +115, +175 y +235 min
- 1 mg/kg (20 min) Cinco inyecciones de 1,1 mg/kg a los -10, -5, 0, +5 y +10 min
- Vancomicina (testigo positivo) inyectada a los 0 min
Se inyecta MBI 11CN vıa intravenosa en la vena de la cola en formulacion D. Se registra la super-vivencia de los ratones a las 1, 2, 3, 4, 6, 8, 10, 12, 20, 24 y 30 horas despues de infeccion y despuesdos veces diarias durante un total de 8 dıas. No hubo cambios en el numero de ratones que sobrevivendespues de 24 h (Figura 26).
El protocolo de tratamiento de 1 mg/kg (20 min) con inyecciones separadas 5 minutos centradasen el tiempo de infeccion, retraso la muerte de los ratones en una extension significativa, quedandoun superviviente al final del estudio. Los resultados presentados en la Tabla 19 sugieren que un nivelsuficientemente alto de MBI 11CN mantenido durante un periodo de tiempo mas largo aumentarıa elnumero de ratones que sobreviven. Los resultados con 5 mg/kg y 1 mg/kg (2 h), donde no hay mejoraen la supervivencia frente al testigo negativo, indican que las inyecciones separadas 1 hora, incluso conun nivel mayor, no son eficaces frente a SARM.
TABLA 19
Porcentaje de animales que sobreviven
Tiempo de observacion Sin tratamiento Tratamiento(horas despues de infeccion)
6 50 % 70 %
8 0 40 %
10 0 30 %
12 0 20 %
Ejemplo 13
Activacion de polisorbato 80 por luz ultravioleta
Se prepara una solucion de polisorbato 80 al 2 % (peso/peso) en agua y se pone en un recipiente dereaccion adecuado, tal como una celda de cuarzo. Se pueden usar otros recipientes que son traslucidos ala luz UV o incluso opacos si se preve un camino de luz transparente o un tiempo de reaccion prolongado.Ademas, el recipiente debe permitir el intercambio de aire, pero minimizar la evaporacion.
La solucion se irradia con luz ultravioleta usando una lampara que emite a 254 nm. La irradiaciontambien se puede llevar a cabo usando una lampara que emite a 302 nm. La activacion se completa en1-14 dıas dependiendo del recipiente, la profundidad de la solucion, y de la tasa de intercambio de aire.
60

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
La reaccion se controla por un ensayo de HPLC de fase inversa, que mide la formacion de MBI 11CNmodificado con APS cuando se hace reaccionar el polisorbato activado por la luz con MBI 11CN.
Se determinan algunas propiedades del polisorbato activado. Debido a que los peroxidos son un sub-producto conocido de exponer los eteres a la luz UV, se examina la formacion de peroxido por el efectode agentes de reduccion en el polisorbato activado. Como se ve en la Figura 27A, el polisorbato activadoreacciona facilmente con MBI 11CN. El tratamiento previo con 2-mercaptoetanol (Figura 27B), un agentede reduccion suave, elimina los peroxidos detectables, pero no produce una perdida de capacidad de for-mar conjugado. El tratamiento con borohidruro sodico (Figura 27C), elimina peroxidos y eventualmenteelimina la capacidad del polisorbato activado de modificar peptidos. La hidrolisis del borohidruro enagua sube el pH y produce borato como un producto de hidrolisis. Sin embargo, ni el cambio de pH, niel borato son los responsables.
Estos datos indican que los peroxidos no estan implicados en la modificacion de peptidos por el poli-sorbato activado. El borohidruro sodico no debe afectar a epoxidos o esteres en medio acuoso, sugiriendoque el grupo reactivo es un aldehıdo o cetona. La presencia de aldehıdos en el polisorbato activado seconfirma usando un ensayo de formaldehıdo, que es especıfico para aldehıdos, incluyendo aldehıdos dis-tintos del formaldehıdo.
Ademas, el polisorbato activado se trata con 2,4-dinitrofenilhidrazina (DNPH) en un intento de cap-turar las especies reactivas. Tres componentes atrapados con DNPH se purifican y analizan por espec-troscopıa de masas. Estos componentes son derivados de polisorbato con pesos moleculares entre 1000y 1400. Esto indica que estan implicados aldehıdos de bajo peso molecular, tales como formaldehıdo oacetaldehıdo.
Ejemplo 14
Formacion de peptidos modificados con APS
Los peptidos modificados con APS se preparan en fase solida o fase lıquida. Para la preparacionen fase solida, se anaden 0,25 ml de MBI 11CN 4 mg/ml, a 0,5 ml de acido acetico-NaOH 0,4 M, pH4,6, seguido de adicion de 0,25 ml de polisorbato activado con luz UV. La mezcla de reaccion se congelaponiendola en un congelador a -80◦C. Despues de congelar, la mezcla de reaccion se liofiliza toda la noche.
Para preparar los conjugados en una fase acuosa, primero una muestra de polisorbato 80 activado porla luz UV se ajusta a pH 7,5 por adicion de NaOH 0,1 M. Esta solucion de pH ajustado (0,5 ml) se anadea 1,0 ml de carbonato sodico 100 mM, pH 10,0, seguido inmediatamente por adicion de 0,5 ml de MBI11CN 4 mg/ml. La mezcla de reaccion se incuba a temperatura ambiente durante 22 horas. El progresode la reaccion se controla por analisis en distintos puntos de tiempo usando RP-HPLC (Figura 28). Enla figura 28, el pico 2 es peptido sin reaccionar, el pico 3 es peptido modificado con APS. El tipo 1 es elque esta mas a la izquierda del pico 3 y el Tipo 2 es el de mas a la derecha del pico 3.
La Tabla 20 resume datos de varios experimentos. Salvo que se indique otra cosa en la tabla 20, lospeptidos modificados con APS se preparan por el metodo de liofilizacion en tampon de acido acetico-NaOH 200 mM, pH 4,6.
TABLA 20
Complejo
Secuencia Nombre Tipo 1 Tipo 2
ILKKWPWWPWRRKamida 11CNFase solida, pH 2,0 Sı PocoFase solida, pH 4,6 Sı SıFase solida, pH 5,0 Sı SıFase solida, pH 6,0 Sı SıFase solida, pH 8,3 Sı SıSolucion, pH 2,0 Trazas Trazas Si-Solucion, pH 10,0 Si lento
61

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 20 (Cont.)
Complejo
Secuencia Nombre Tipo 1 Tipo 2
(Ac)4-ILKKWPWWPWRRKamida 11CN-Y1 No No
ILRRWPWWPWRRKamida 11B1CN Sı Inferior
ILRWPWWPWRRKamida 11B7CN Sı Inferior
ILWPWWPWRRKamida 11B8CN Sı Inferior
ILRRWPWWPWRRRamida 11B9CN Sı Trazas
ILKKWPWWPWKKKamida 11B10CN Sı Sı
ILKKWPWWPWRRkamida 11E3CN Sı Sı
ILKKWVWWPWRRKamida 11F3CN Sı Sı
ILKKWPWWPWKamida 11G13CN Sı Sı
ILKKWPWWPWRamida 11G14CN Sı Trazas
La modificacion de los grupos amino se analiza mas determinando el numero de grupos amino pri-marios perdidos durante la union. Los peptidos sin modificar y modificados se tratan con acido 2,4,6-trinitrobencenosulfonico (TNBS) (R.L. Lundblad en Techniques in Protein Modification and Analysis pp.151-154, 1995) (Tabla 21).
Brevemente, se prepara una solucion madre de MBI 11CN 4 mg/ml y una solucion equimolar de MBI11CN modificado con APS. Una parte alıcuota de 0,225 ml de MBI 11CN o MBI 11CN modificado conAPS se mezcla con 0,225 ml de tampon de fosfato sodico 200 mM, pH 8,8. Se anade una parte alıcuotade 0,450 ml de TNBS al 1 % a cada muestra, y la reaccion se incuba a 37◦C durante 30 minutos. Se midela absorbancia a 367 nm, y se calcula el numero de grupos amino primarios modificados por moleculausando un coeficiente de extincion de 10.500 M−1 cm−1 para los derivados de trinitrofenilo (TNP).
Despues el contenido de grupos amino primarios del peptido relacionado se compara con el del co-rrespondiente peptido modificado con APS. Como se muestra a continuacion, se produce la perdida deun solo grupo amino primario durante la formacion del peptido modificado. Los peptidos que tienen unapareja 3,4 lisina dan consistentemente resultados que son 1 residuo menos de lo esperado, lo cual puedereflejar impedimento esterico despues de valoracion de un miembro del doblete.
TABLA 21
Secuencia del peptido TNP/Peptido TNP/peptido modificado Cambiocon APS
ILKKWPWWPWRRKamida 2,71 1,64 1,07
ILRRWPWWPWRRKamida 1,82 0,72 1,10
IlKKWPWWPWRRkamida 2,69 1,61 1,08
ILKKWVWWPWRRKamida 2,62 1,56 1,06
62

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
Estabilidad de los analogos de peptidos modificados con APS
Los peptidos modificados con APS demuestran un alto grado de estabilidad en condiciones que pro-mueven la disociacion de complejos ionicos o hidrofobos. Se prepara el peptido modificado con APS enformulacion D, como soluciones de 800 µg/ml en agua, solucion salina al 0,9 %, urea 8 M, guanidina-HCl8 M, 1-propanol al 67 %, HCl 1 M y NaOH 1 M, y se incuban durante 1 hora a temperatura ambiente.Se analiza en las muestras la presencia de peptido libre usando HPLC de fase inversa y las siguientescondiciones cromatograficas:
Disolvente A: acido trifluoroacetico (TFA) al 0,1 % en agua
Disolvente B: TFA al 0,1 %/acetonitrilo al 95 % en agua
Medio: POROS R2-20 (poliestireno-divinilbenceno)
Elucion: 0 % de B para 5 volumenes de columna
0-25 % de B en 3 volumenes de columna
25 % de B para 10 volumenes de columna
25-95 % de B en 3 volumenes de columna
95 % de B para 10 volumenes de columna
En estas condiciones, el peptido libre eluye exclusivamente durante la etapa de 25 % de B, y el com-plejo de formulacion-peptido durante la etapa de 95 % de B. Ninguna de las condiciones de disociacionantes mencionadas, excepto NaOH 1 M en la que se observa algo de degradacion, consiguen liberar elpeptido libre del peptido modificado con APS. Se llevan a cabo estudios adicionales con incubacion a55◦C u 85◦C durante una hora. El peptido modificado con APS es igualmente estable a 55◦C, y es sololigeramente menos estable a 85◦C. Se observa algo de hidrolisis acida, indicada por la presencia de nuevospicos en el cromatograma de HPLC, con la muestra de HCl 1 M incubada a 85◦C durante una hora.
Ejemplo 15
Purificacion de MBI 11CN modificado con APS
Se purifica una preparacion a gran escala de MBI 11CN modificado con APS. Aproximadamente 400mg de MBI 11CN se modifican con APS y se disuelven en 20 ml de agua. Se separa el MBI 11CN sinreaccionar por RP-HPLC. Despues el disolvente se evapora del conjunto de MBI 11CN modificado conAPS, y el residuo se disuelve en 10 ml de cloruro de metileno. Despues, el peptido modificado se precipitacon 10 ml de eter dietılico. Despues de 5 min a temperatura ambiente, el precipitado se recoge porcentrifugacion a 5000xg durante 10 minutos. El sedimento se lava con 5 ml de eter dietılico y se recogeotra vez por centrifugacion a 5000xg durante 10 minutos. Los lıquidos sobrenadantes se agrupan paraanalizar los subproductos de polisorbato sin reaccionar. El precipitado se disuelve en 6 ml de agua ydespues se lava por barrido con nitrogeno burbujeando durante 30 minutos para separar el eter residual.El rendimiento total a partir del MBI 11CN inicial era 43 %.
Ejemplo 16
Ensayos biologicos usando peptido modificado con APS
Todos los ensayos biologicos que comparan los peptidos modificados con APS con los peptidos sin mo-dificar se llevan a cabo en una relacion equimolar. La concentracion de peptidos modificados con APS sepuede determinar por medicion espectrofotometrica que se usa para normalizar las concentraciones paraensayos biologicas. Por ejemplo, una solucion de MBI 11CN modificado con APS de 1 mg/ml contiene lamisma cantidad de peptido que una solucion de MBI 11CN de 1 mg/ml, permitiendo ası la comparaciondirecta de datos de toxicidad y eficacia.
Los peptidos modificados con APS son al menos tan potentes como los peptidos relacionados, enensayos in vitro llevados a cabo como se describe aquı. Se presentan los valores de las CIM frente abacterias gram positivas para varios peptidos modificados con APS y se comparan con los valores obtenidosusando los peptidos relacionados (Tabla 22). Los resultados indican que los peptidos modificados son almenos tan potentes in vitro como los peptidos relacionados y pueden ser mas potentes que los peptidosrelacionados frente a cepas de E. faecalis.
63
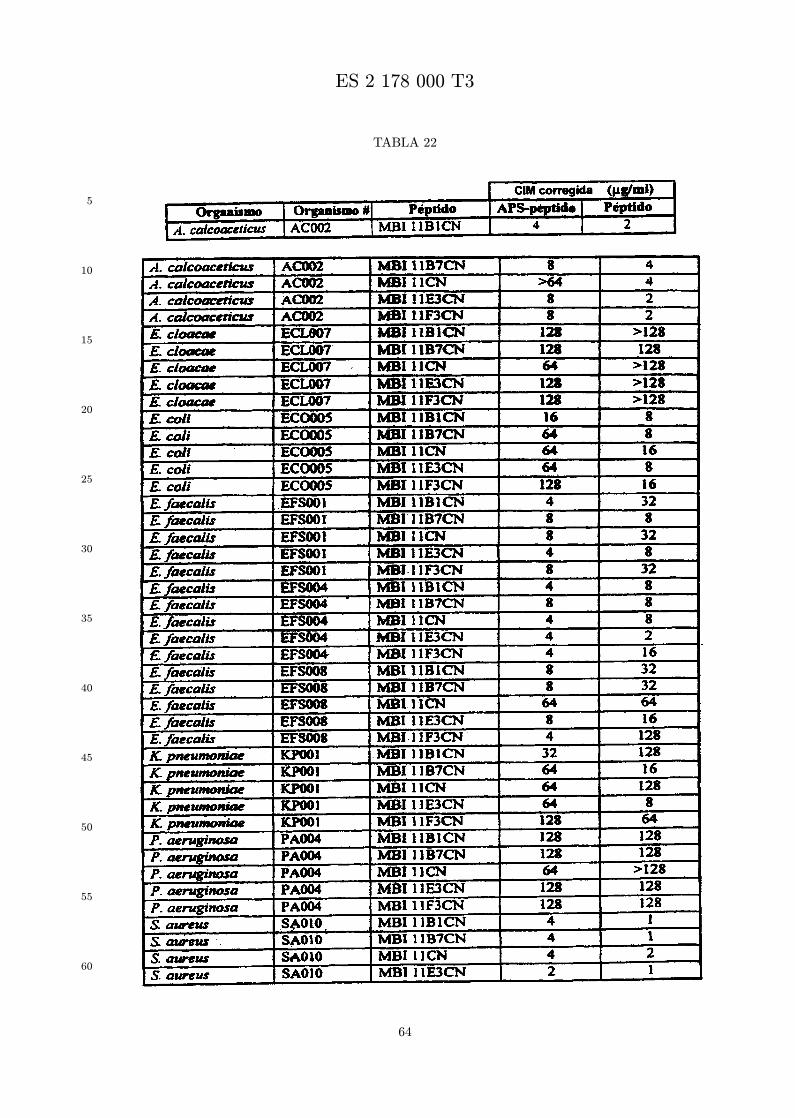
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 22
64
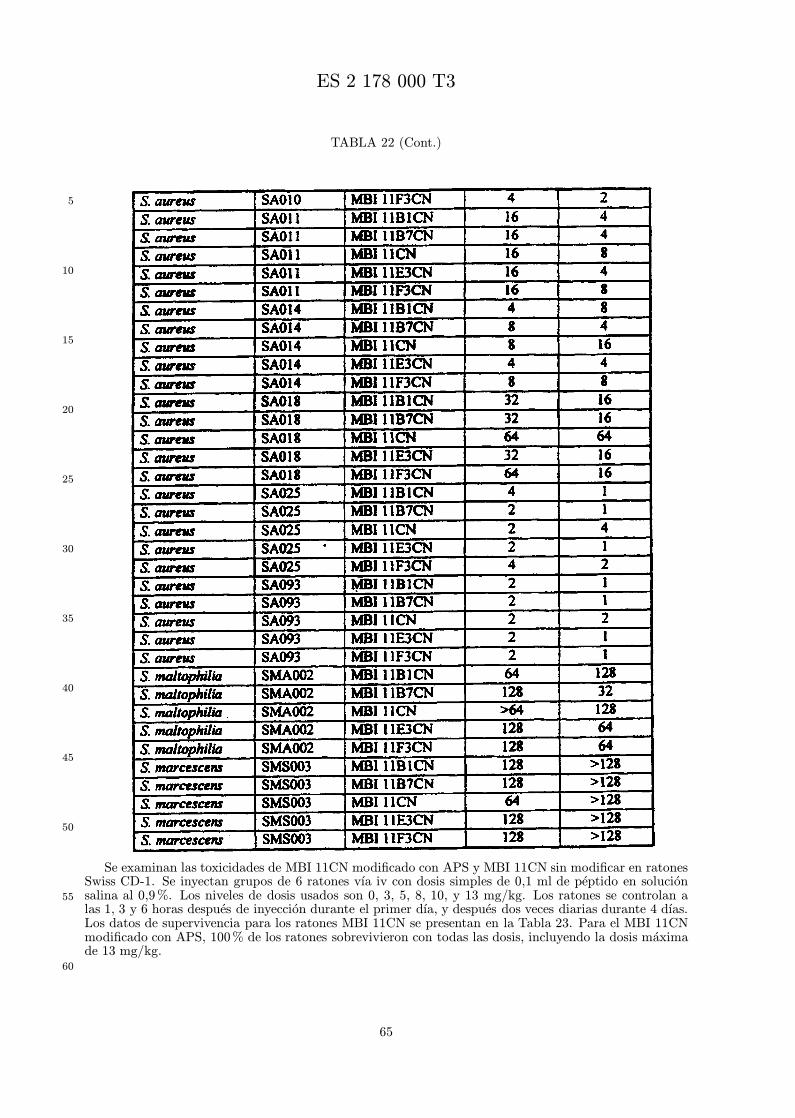
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 22 (Cont.)
Se examinan las toxicidades de MBI 11CN modificado con APS y MBI 11CN sin modificar en ratonesSwiss CD-1. Se inyectan grupos de 6 ratones vıa iv con dosis simples de 0,1 ml de peptido en solucionsalina al 0,9 %. Los niveles de dosis usados son 0, 3, 5, 8, 10, y 13 mg/kg. Los ratones se controlan alas 1, 3 y 6 horas despues de inyeccion durante el primer dıa, y despues dos veces diarias durante 4 dıas.Los datos de supervivencia para los ratones MBI 11CN se presentan en la Tabla 23. Para el MBI 11CNmodificado con APS, 100 % de los ratones sobrevivieron con todas las dosis, incluyendo la dosis maximade 13 mg/kg.
65
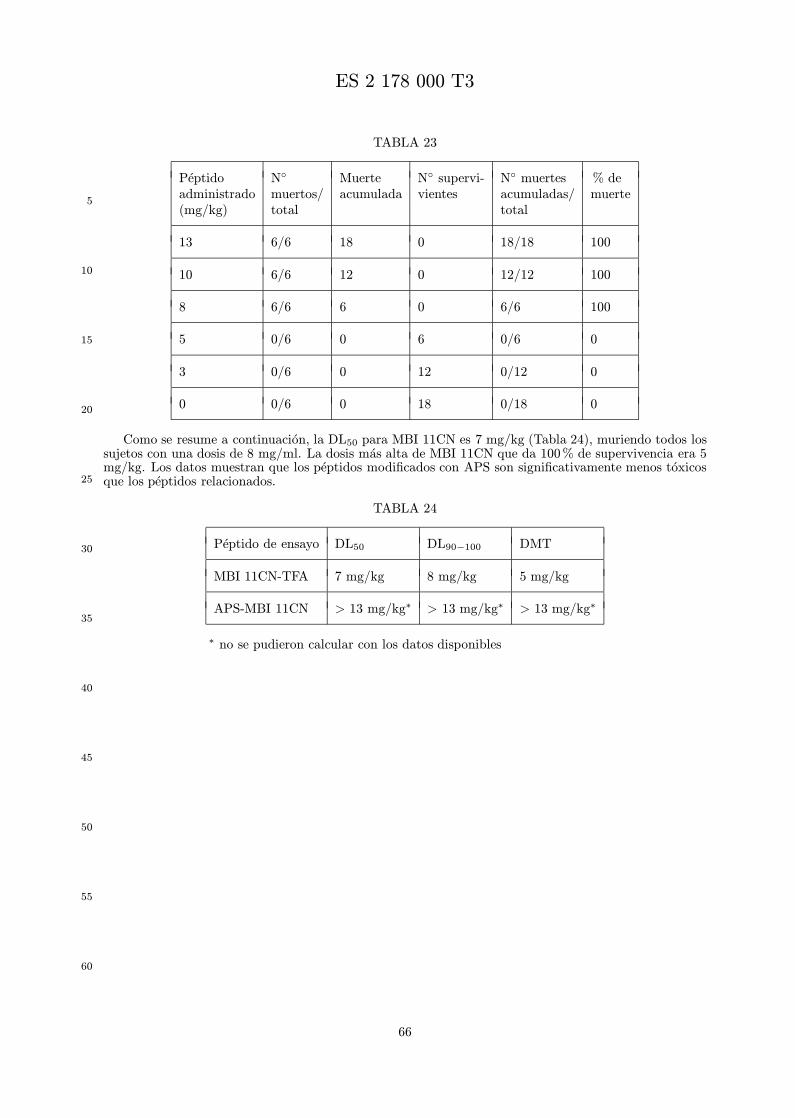
ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
TABLA 23
Peptido N◦ Muerte N◦ supervi- N◦ muertes % deadministrado muertos/ acumulada vientes acumuladas/ muerte(mg/kg) total total
13 6/6 18 0 18/18 100
10 6/6 12 0 12/12 100
8 6/6 6 0 6/6 100
5 0/6 0 6 0/6 0
3 0/6 0 12 0/12 0
0 0/6 0 18 0/18 0
Como se resume a continuacion, la DL50 para MBI 11CN es 7 mg/kg (Tabla 24), muriendo todos lossujetos con una dosis de 8 mg/ml. La dosis mas alta de MBI 11CN que da 100 % de supervivencia era 5mg/kg. Los datos muestran que los peptidos modificados con APS son significativamente menos toxicosque los peptidos relacionados.
TABLA 24
Peptido de ensayo DL50 DL90−100 DMT
MBI 11CN-TFA 7 mg/kg 8 mg/kg 5 mg/kg
APS-MBI 11CN > 13 mg/kg∗ > 13 mg/kg∗ > 13 mg/kg∗
∗ no se pudieron calcular con los datos disponibles
66

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
REIVINDICACIONES
1. Un analogo de indolicidina, que comprende hasta 25 aminoacidos y que contiene la formula: IL-RWPWWPWRRK.
2. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que dicho analogo tiene uno omas aminoacidos cambiados por un D-aminoacido correspondiente.
3. El analogo de indolicidina de acuerdo con la reivindicacion 2, en el que el aminoacido N-terminaly/o el C-terminal es un D-aminoacido.
4. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que el analogo esta acetiladoen el aminoacido N-terminal.
5. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que el analogo esta amidado enel aminoacido C-terminal.
6. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que el analogo esta esterificadoen el aminoacido C-terminal.
7. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que el analogo se modifica porincorporacion de homoserina/homoserina-lactona en el aminoacido C-terminal.
8. El analogo de indolicidina de acuerdo con la reivindicacion 1, en el que el analogo se conjuga conpolialquilenglicol o sus derivados.
9. Una molecula de acido nucleico que comprende una secuencia de acido nucleico que codifica unanalogo de indolicidina de acuerdo con la reivindicacion 1.
10. Un vector de expresion que comprende un promotor en union factible con la molecula de acidonucleico de la reivindicacion 9.
11. Una celula hospedante transfectada o transformada con el vector de expresion de la reivindicacion10.
12. Una composicion farmaceutica que comprende un analogo de indolicidina de acuerdo con unacualquiera de las reivindicaciones 1 a 8, y un tampon fisiologicamente aceptable.
13. La composicion farmaceutica de acuerdo con la reivindicacion 12, que ademas comprende unantibiotico.
14. La composicion farmaceutica de la reivindicacion 13, en la que el antibiotico se selecciona delgrupo que consta de penicilinas, cefalosporinas, carbacefems, cefamicinas, carbapenems, monobactams,quinolonas, tetraciclinas, aminoglicosidos, macrolidos, glicopeptidos, cloranfenicoles, glicilciclinas, licosa-midas y fluoroquinolonas.
15. La composicion farmaceutica de acuerdo con la reivindicacion 13, en la que el antibiotico se selec-ciona del grupo que consta de Amikacina; Amoxicilina; Ampicilina; Azitromicina; Azlocilina; Aztreonam;Carbenicilina; Cefaclor; Cefamandol-formiato sodico; Cefazolina; Cefepima; Cefetamet; Cefixima; Cefme-tazol; Cefonicid; Cefoperazona; Cefotaxima; Cefotetan; Cefoxitin; Cefpodoxima; Cefprozil; Cefsulodina;Ceftazidima; Ceftizoxima; Ceftriaxona; Cefuroxima; Cefalexina; Cefalotin; Cloranfenicol; Cinoxacino; Ci-profloxacino; Claritromicina; Clindamicina; Cloxacilina; Co-amoxiclavulanato; Dicloxacilina; Doxiciclina;Enoxacino; Eritromicina; estolato de Eritromicina; succinato de Eritromicina y etilo; glucoheptonato deEritromicina; lactobionato de Eritromicina; estearato de Eritromicina; Etambutol; Fleroxacino; Genta-micina; Imipenem; Isoniazid; Kanamicina; Lomefloxacino; Loracarbef; Meropenem; Meticilina; Metro-
nidazol; Meziocilina; hidrocloruro de Minociclina; Mupirocina; Nafcilina; Acido Nalidıxico; Netilmicina;Nitrofurantoına; Norfloxacino; Ofloxacino; Oxacilina; Penicilina G; Piperacilina; Pirazinamida; Rifabu-tin; Rifampicina; Roxitromicina; Estreptomicina; Sulfametoxazol; Sinercid; Teicoplanina; Tetraciclina;Ticarcilina; Tobramicina; Trimetoprim; Vancomicina; una combinacion de Piperacilina y Tazobactam; ysus derivados.
16. La composicion farmaceutica de acuerdo con la reivindicacion 13, en la que el antibiotico se
67

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
selecciona del grupo que consta de Amikacina; Azitromicina; Cefoxitina; Ceftriaxona; Ciprofloxacina;Co-amoxiclavulanato; Doxiciclina; Gentamicina; Mupirocina; Vancomicina; y una combinacion de Pipe-racilina y Tazobactam.
17. La composicion farmaceutica de acuerdo con una cualquiera de las reivindicaciones 12 a 16, en laque la composicion se incorpora en un liposoma.
18. Las composiciones farmaceuticas de acuerdo con una cualquiera de las reivindicaciones 12 a 16,en las que la composicion se incorpora en un vehıculo de liberacion lenta.
19. Un analogo de indolicidina de acuerdo con una cualquiera de las reivindicaciones 1 a 8, o unacomposicion farmaceutica de acuerdo con una cualquiera de las reivindicaciones 12 a 18, para uso te-rapeutico.
20. Uso de un analogo de indolicidina de acuerdo con una cualquiera de las reivindicaciones 1 a 8,o una composiciones farmaceutica de acuerdo con una cualquiera de las reivindicaciones 12 a 18, parafabricar un medicamento para tratar infecciones.
21. Uso de acuerdo con la reivindicacion 20, en el que dicha infeccion se debe a un microorganismo.
22. Uso de acuerdo con la reivindicacion 21, en el que el microorganismo se selecciona del grupo queconsta de bacterias, hongos, parasitos y virus.
23. Uso de acuerdo con la reivindicacion 22, en el que el hongo es una levadura y/o un moho.
24. Uso de acuerdo con la reivindicacion 22, en el que la bacteria es una bacteria Gram negativa.
25. Uso de acuerdo con la reivindicacion 24, en el que la bacteria Gram negativa se selecciona delgrupo que consta de Acinetobacter spp.; Enterobacter spp.; E. coli; H. influenzae; K. pneumoniae; P.aeruginosa; S.marcescens y S. maltophilia.
26. Uso de acuerdo con la reivindicacion 24, en el que la bacteria Gram negativa se selecciona delgrupo que consta de Bordetella pertussis; Brucella spp.; Campylobacter spp.; Haemophilus ducreyl; He-licobacter pylori; Legionella spp.; Moraxella catarrhalis; Neisseria spp.; Salmonella spp.; Shigella spp. yYersinia spp.
27. Uso de acuerdo con la reivindicacion 22, en el que la bacteria es una bacteria Gram positiva.
28. Uso de acuerdo con la reivindicacion 27, en el que la bacteria Gram positiva se selecciona del grupoque consta de E. faecalis; S. aureus; E. faecium; S. pyogenes; S. pneumoniae y estafilococos coagulasanegativos.
29. Uso de acuerdo con la reivindicacion 28, en el que la bacteria Gram positiva se selecciona delgrupo que consta de Bacillus spp.; Corynebacterium spp.; Difteroides: Listeria spp. y EstreptococosViridans.
30. Uso de acuerdo con la reivindicacion 22, en el que la bacteria es una bacteria anaerobia.
31. Uso de acuerdo con la reivindicacion 30, en el que la bacteria anaerobia se selecciona del grupoque consta de Clostridium spp., Bacteroides spp. y Peptostreptococcus spp.
32. Uso de acuerdo con la reivindicacion 30, en el que la bacteria se selecciona del grupo que constade Borrelia spp.; Chlamydia spp.; Mycobacterium spp.; Mycoplasma spp.; Propionibacterium acne; Ri-ckettsia spp.; Treponema spp. y Ureaplasma spp.
33. Uso de acuerdo con la reivindicacion 20, en el que el analogo de indolicidina se administra porinyeccion intravenosa, inyeccion o implante intraperitoneal, inyeccion o implante intramuscular, inyeccionintratecal, inyeccion o implante subcutaneo, inyeccion intradermica, lavado, lavado de vejiga, suposito-rios, pesarios, ingestion oral, aplicacion topica, aplicacion enterica, inhalacion, administracion en aerosolo pulverizacion o gotas nasales.
34. Un dispositivo revestido con una composicion que comprende un analogo de indolicidina de
68

ES 2 178 000 T3
5
10
15
20
25
30
35
40
45
50
55
60
acuerdo con las reivindicaciones 1-8.
35. El dispositivo de acuerdo con la reivindicacion 34, en el que la composicion ademas comprendeun agente antibiotico.
36. El dispositivo de las reivindicaciones 34 o 35, en el que el dispositivo es un dispositivo medico.
37. El dispositivo medico de acuerdo con la reivindicacion 36, en el que dicho dispositivo se seleccionadel grupo que consta de cateteres, valvulas cardiacas artificiales, canulas y stents.
NOTA INFORMATIVA: Conforme a la reserva del art. 167.2 del Convenio de Patentes Europeas (CPE)y a la Disposicion Transitoria del RD 2424/1986, de 10 de octubre, relativo a laaplicacion del Convenio de Patente Europea, las patentes europeas que designen aEspana y solicitadas antes del 7-10-1992, no produciran ningun efecto en Espana enla medida en que confieran proteccion a productos quımicos y farmaceuticos comotales.
Esta informacion no prejuzga que la patente este o no incluıda en la mencionadareserva.
69

ES 2 178 000 T3
70

ES 2 178 000 T3
71
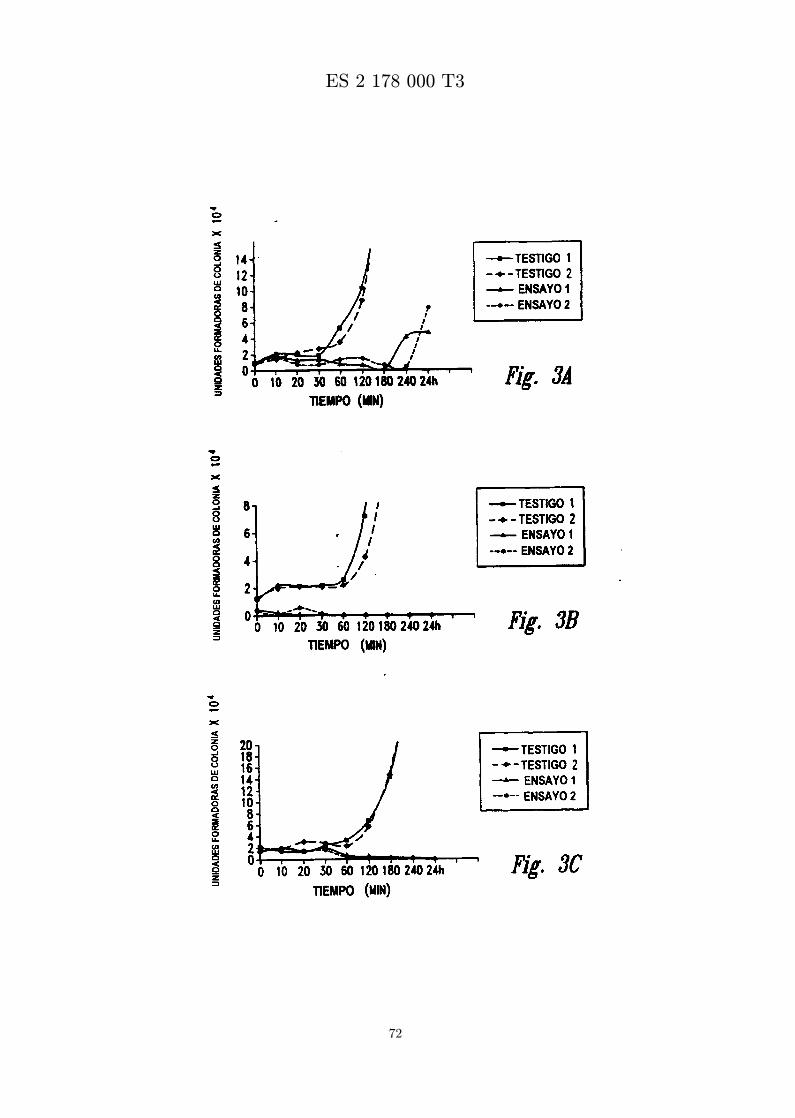
ES 2 178 000 T3
72
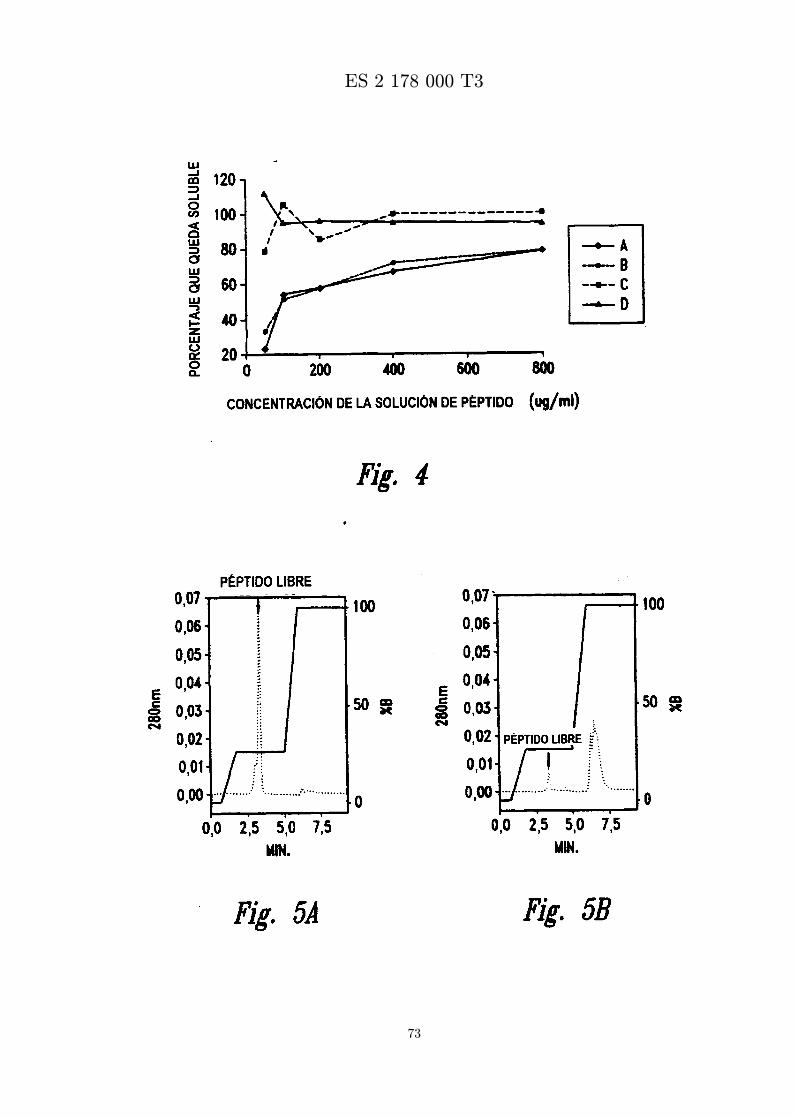
ES 2 178 000 T3
73
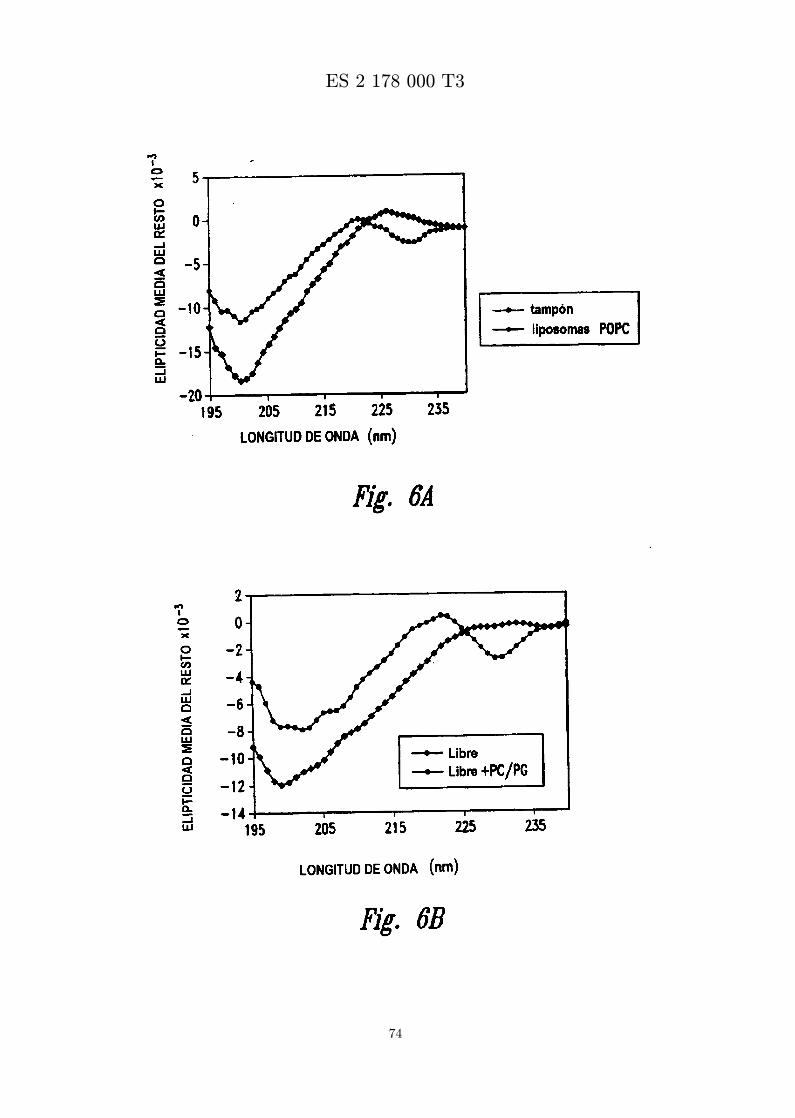
ES 2 178 000 T3
74
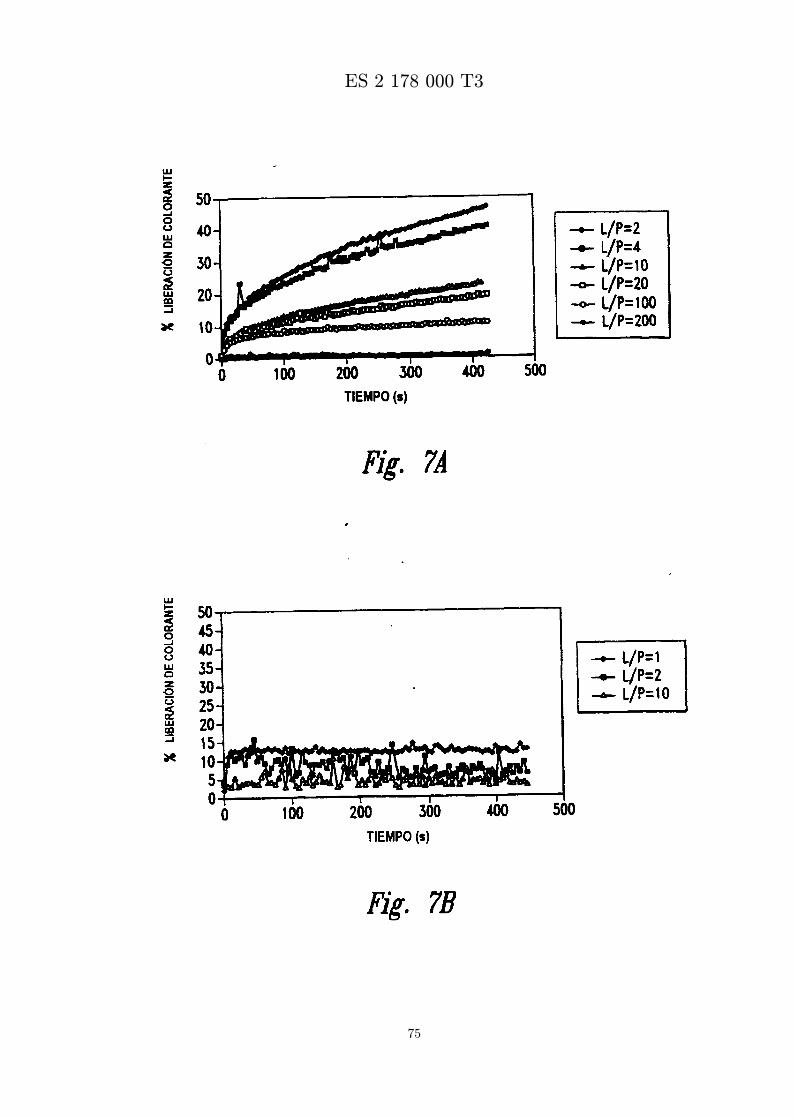
ES 2 178 000 T3
75

ES 2 178 000 T3
76
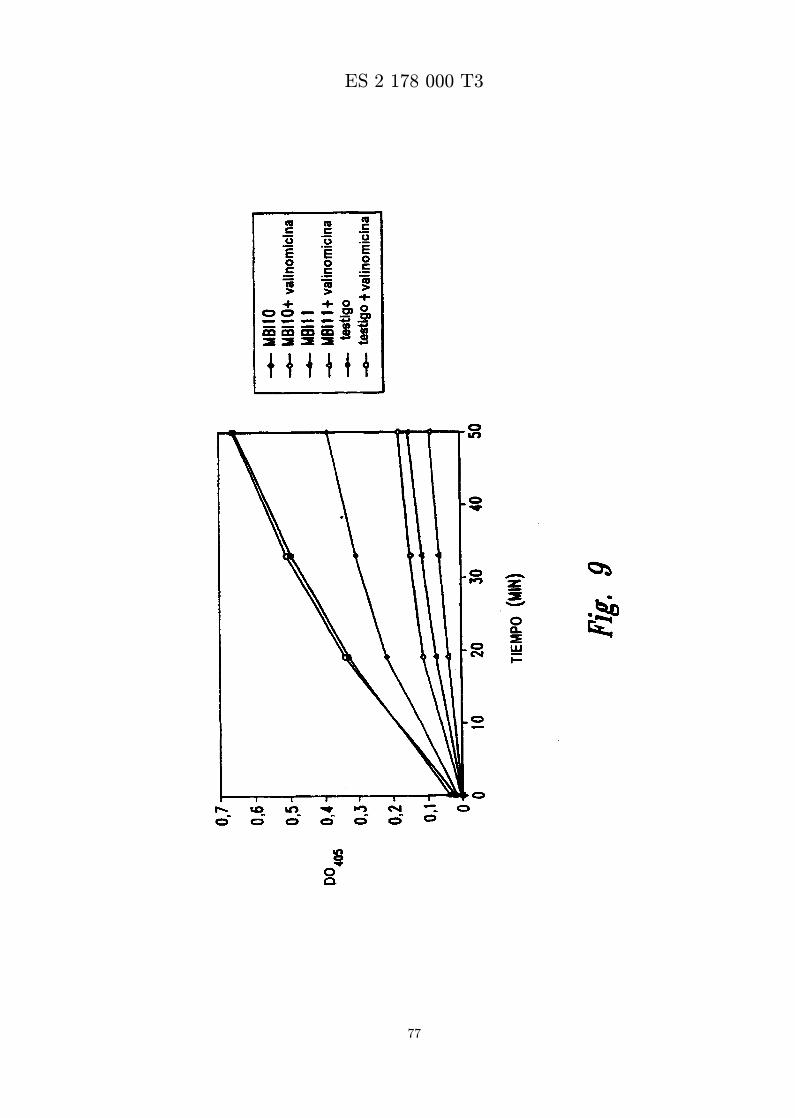
ES 2 178 000 T3
77
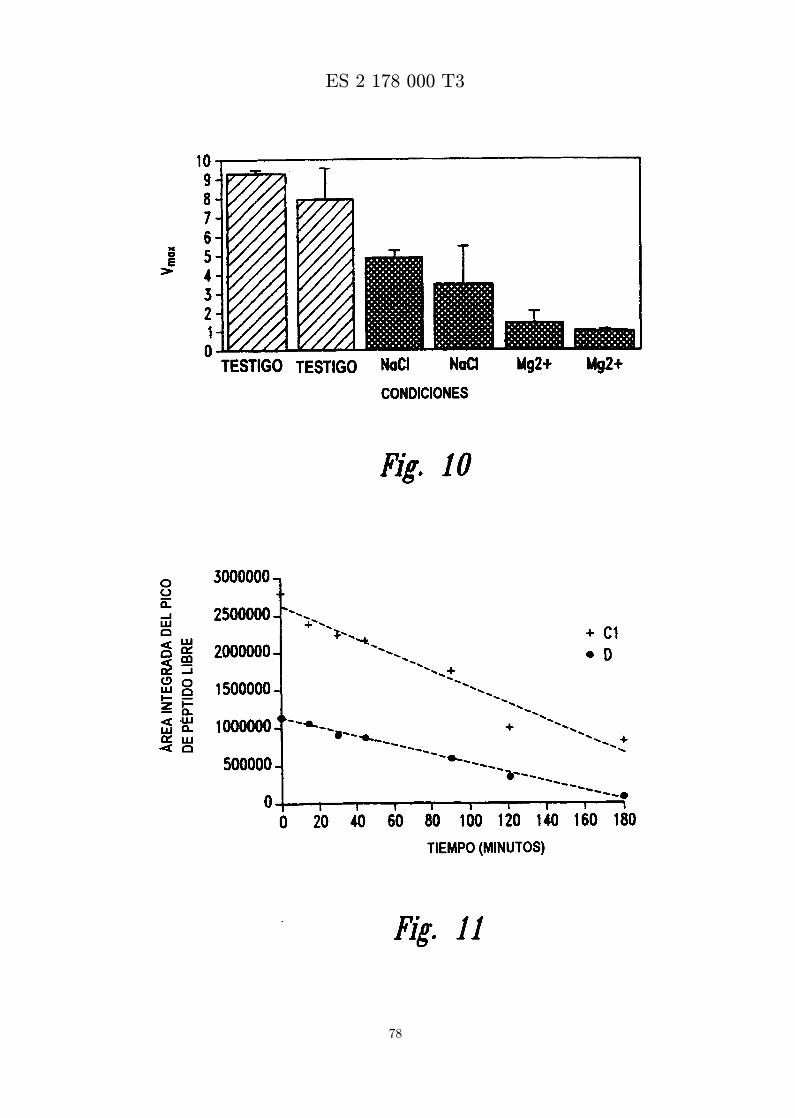
ES 2 178 000 T3
78
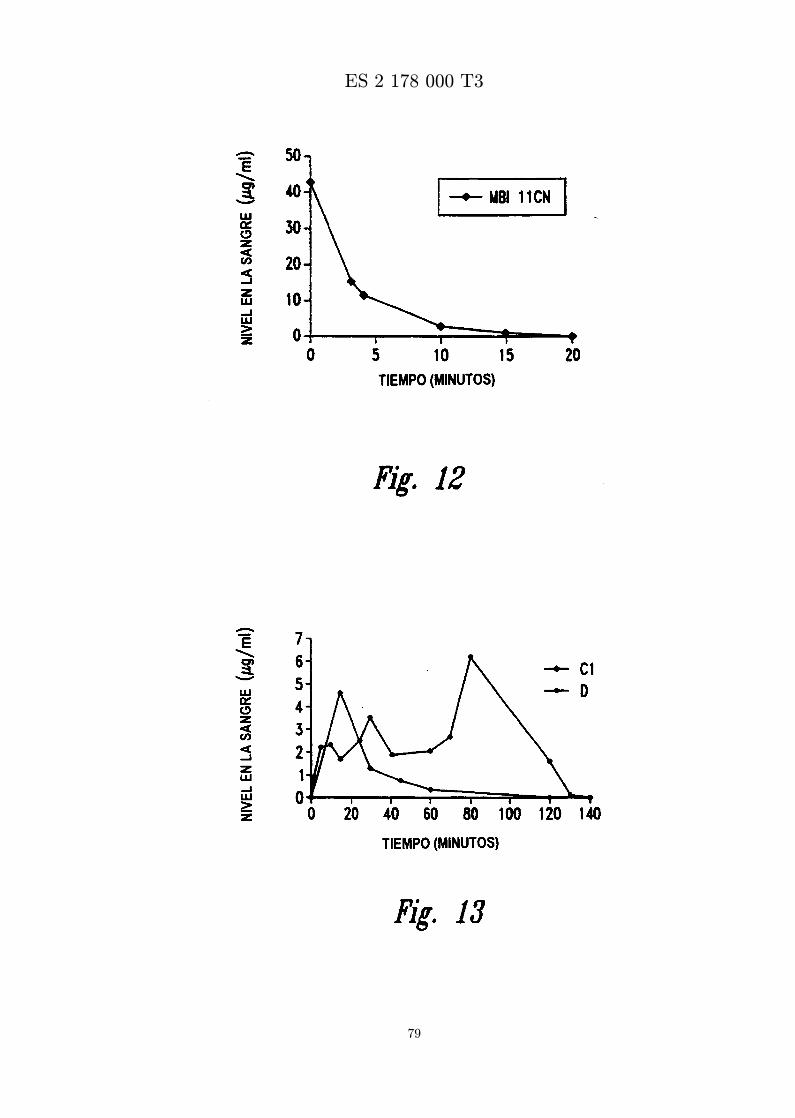
ES 2 178 000 T3
79
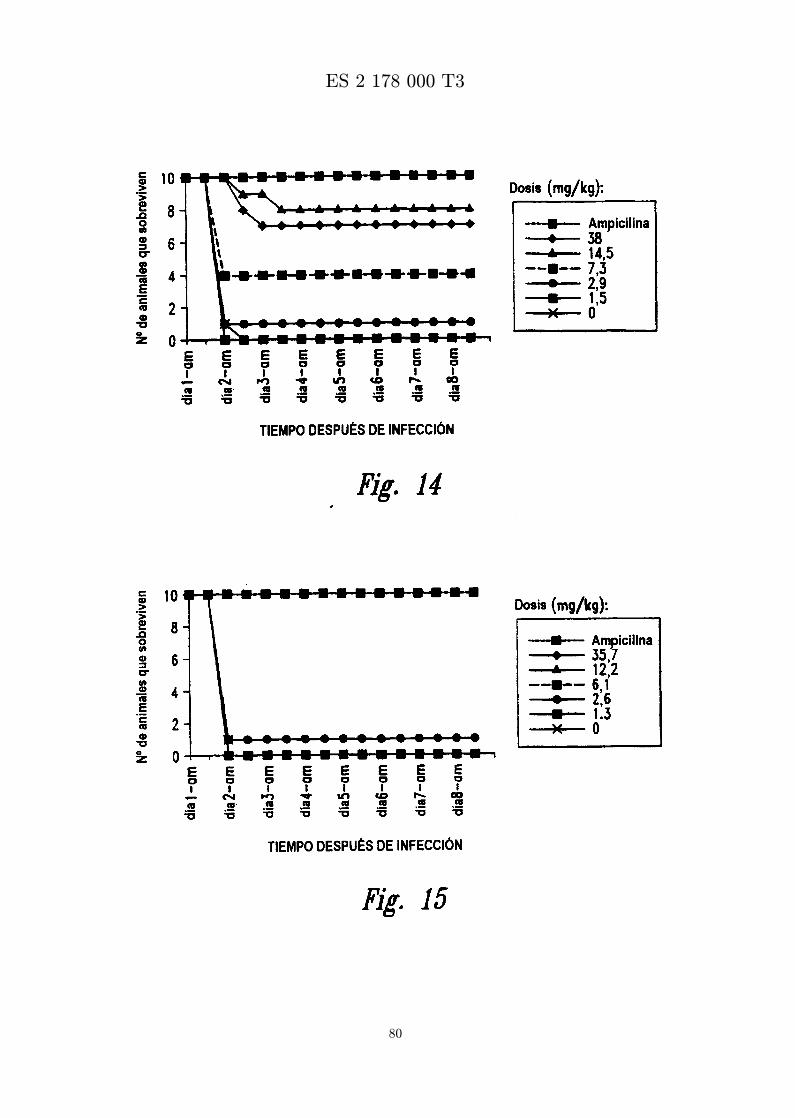
ES 2 178 000 T3
80
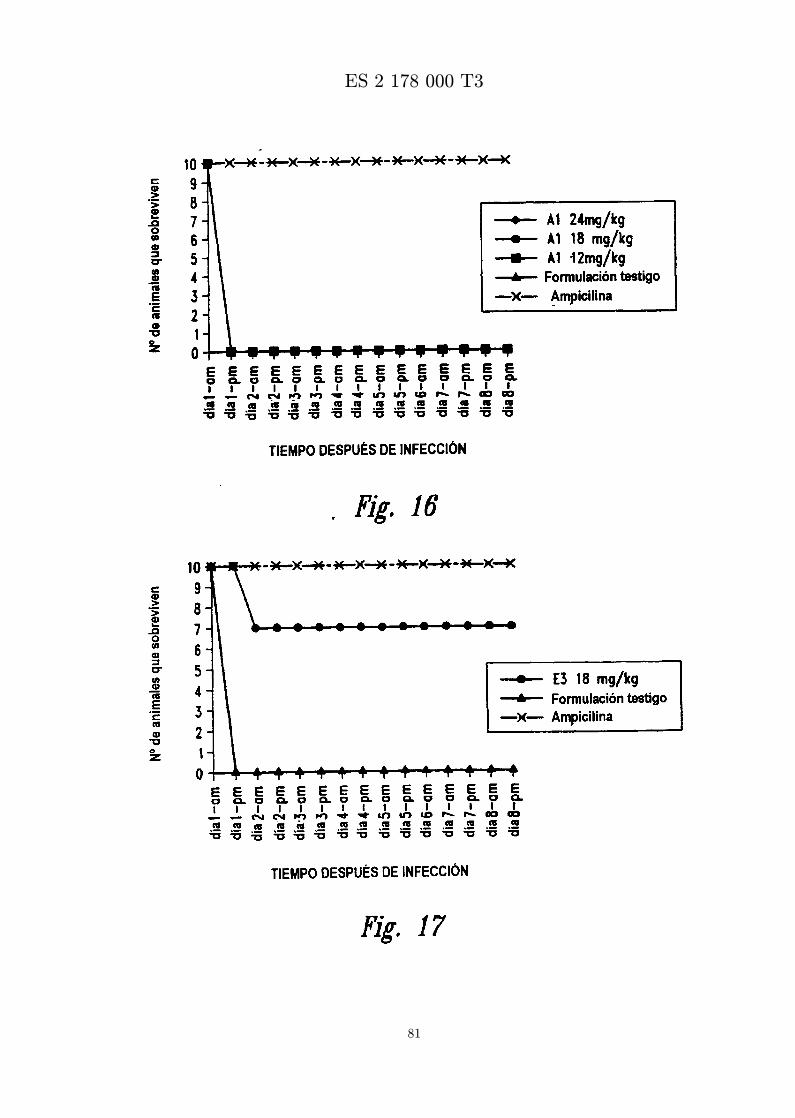
ES 2 178 000 T3
81
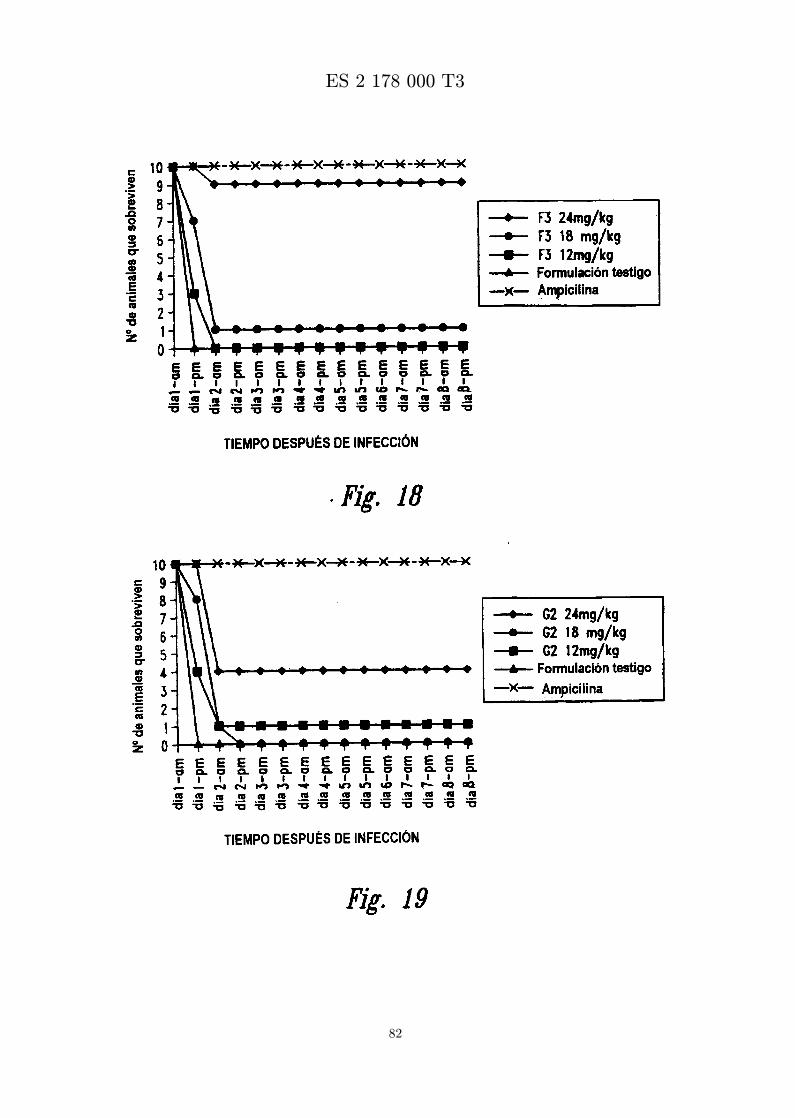
ES 2 178 000 T3
82
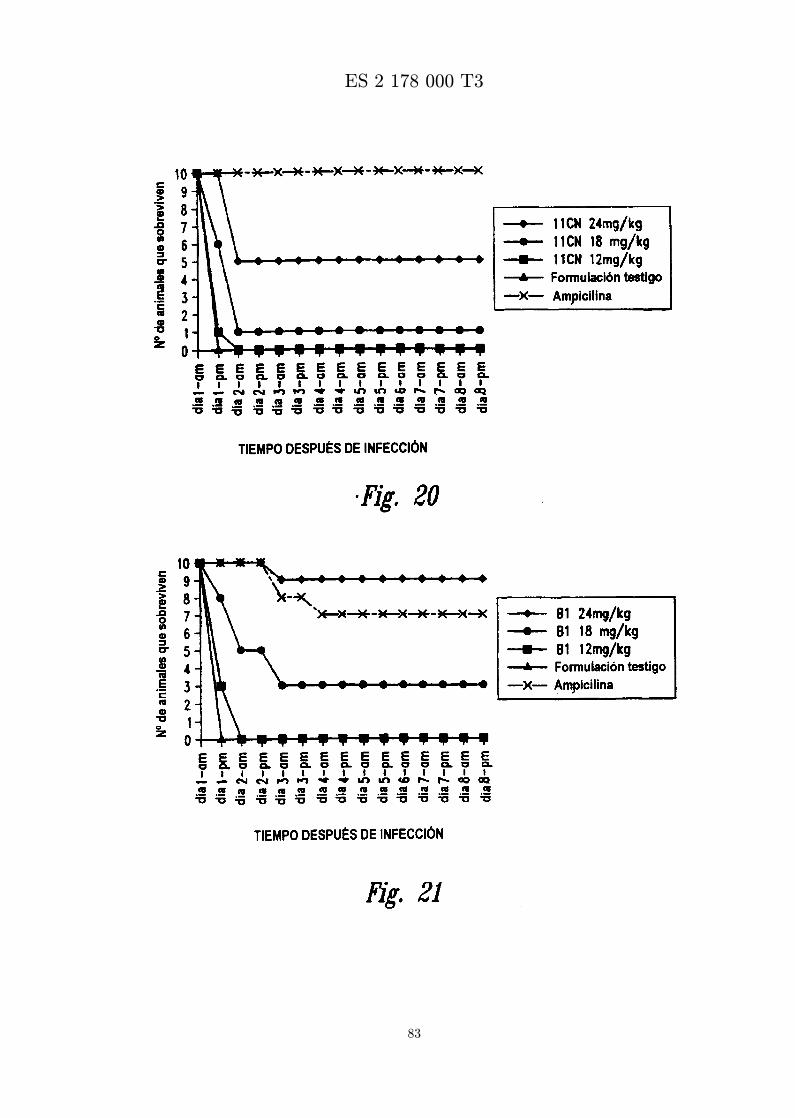
ES 2 178 000 T3
83
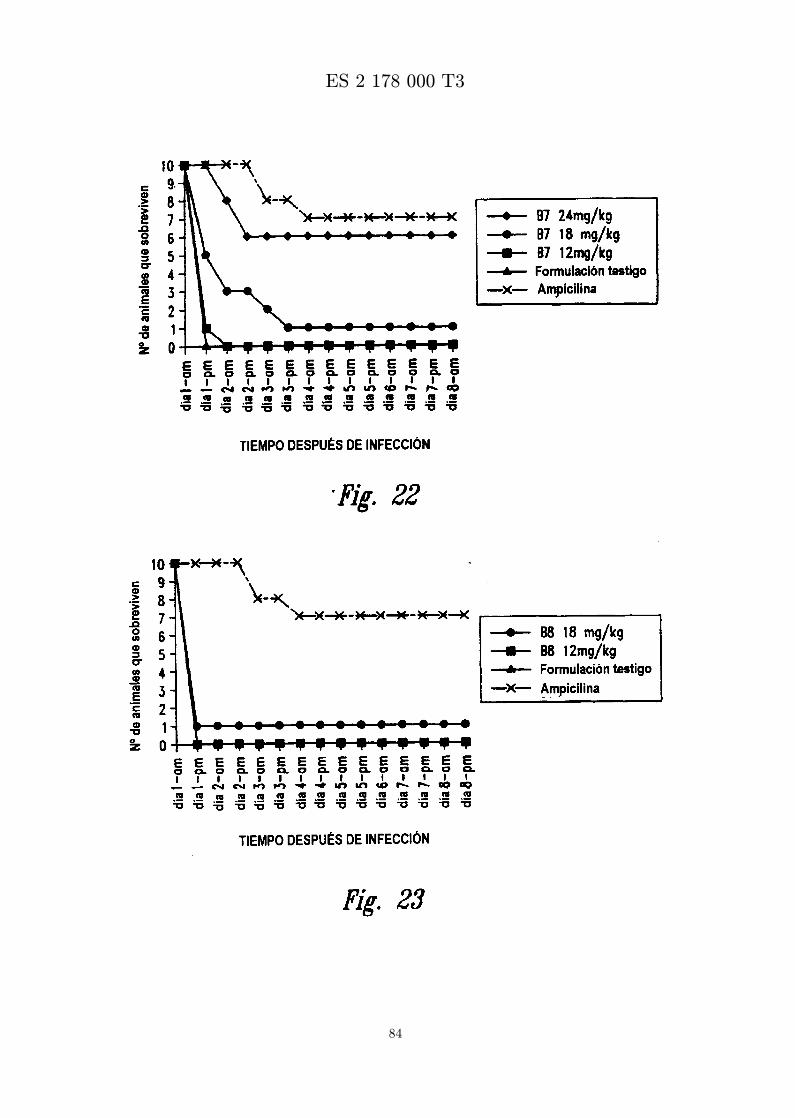
ES 2 178 000 T3
84
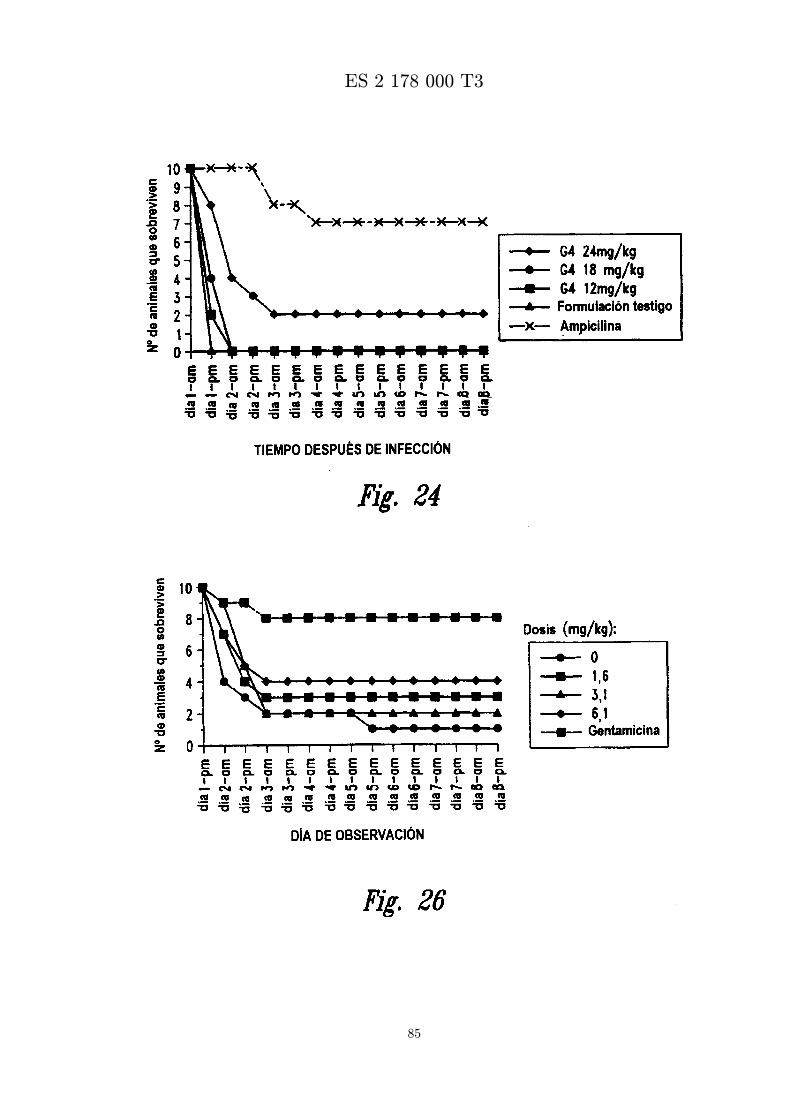
ES 2 178 000 T3
85
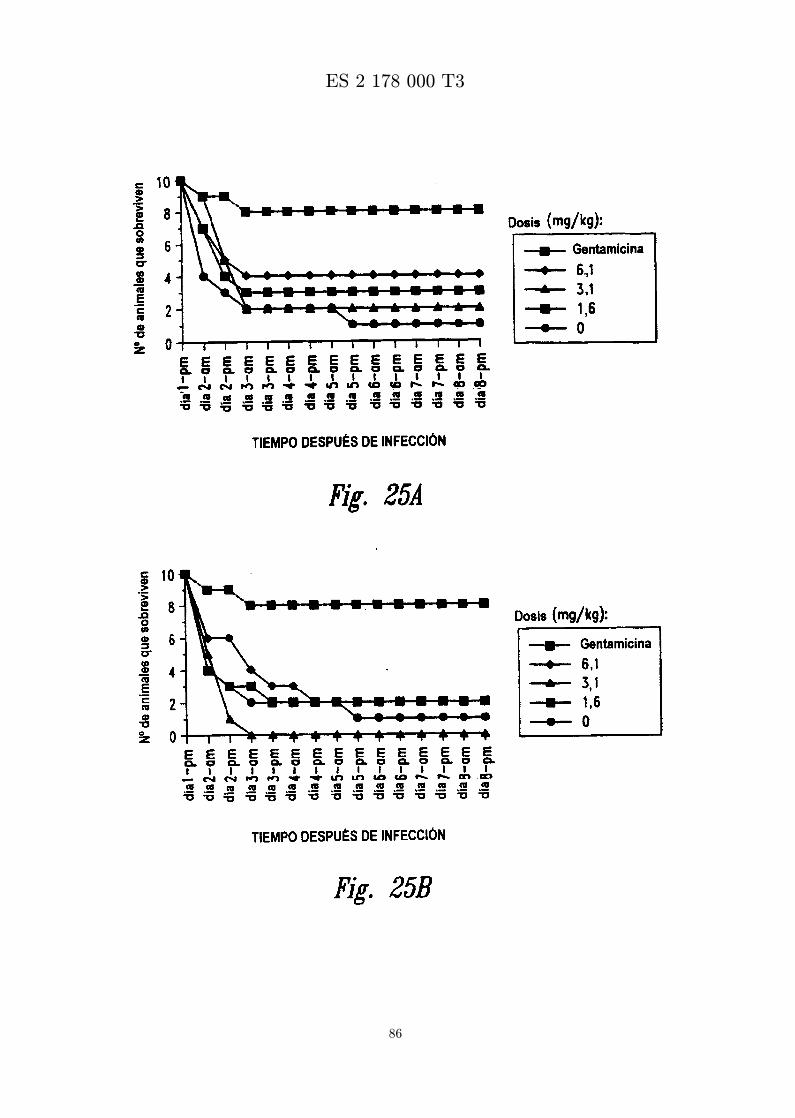
ES 2 178 000 T3
86
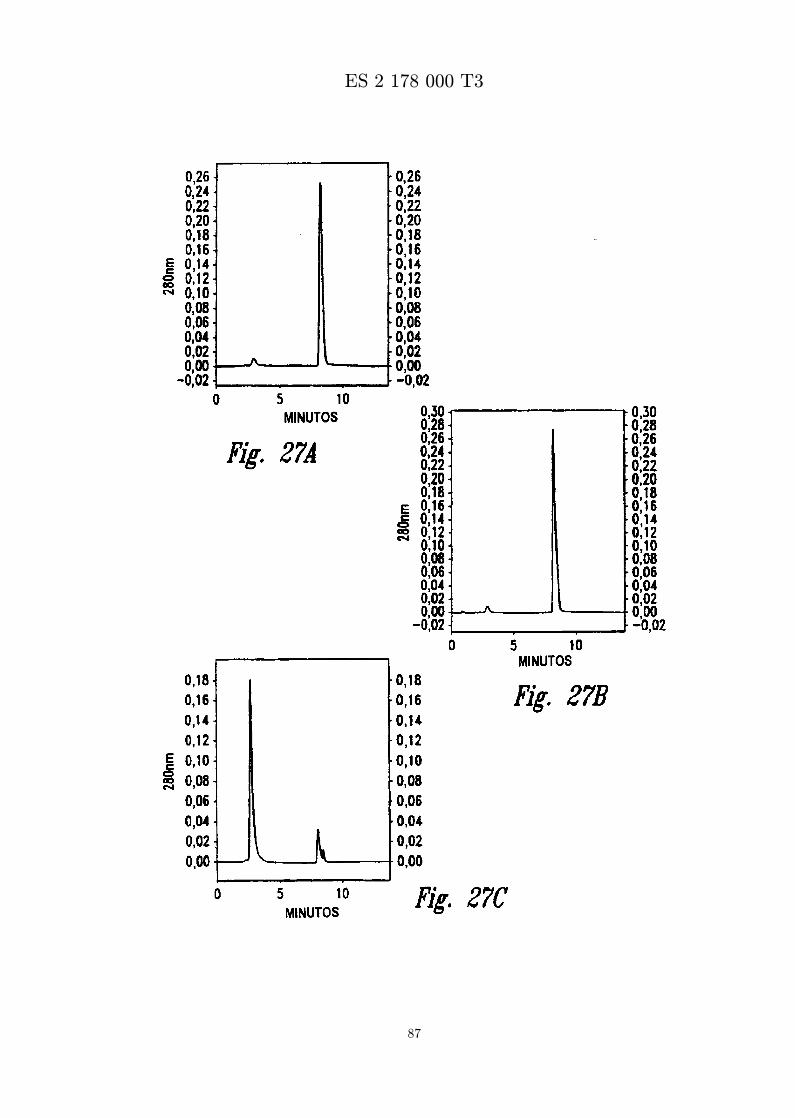
ES 2 178 000 T3
87
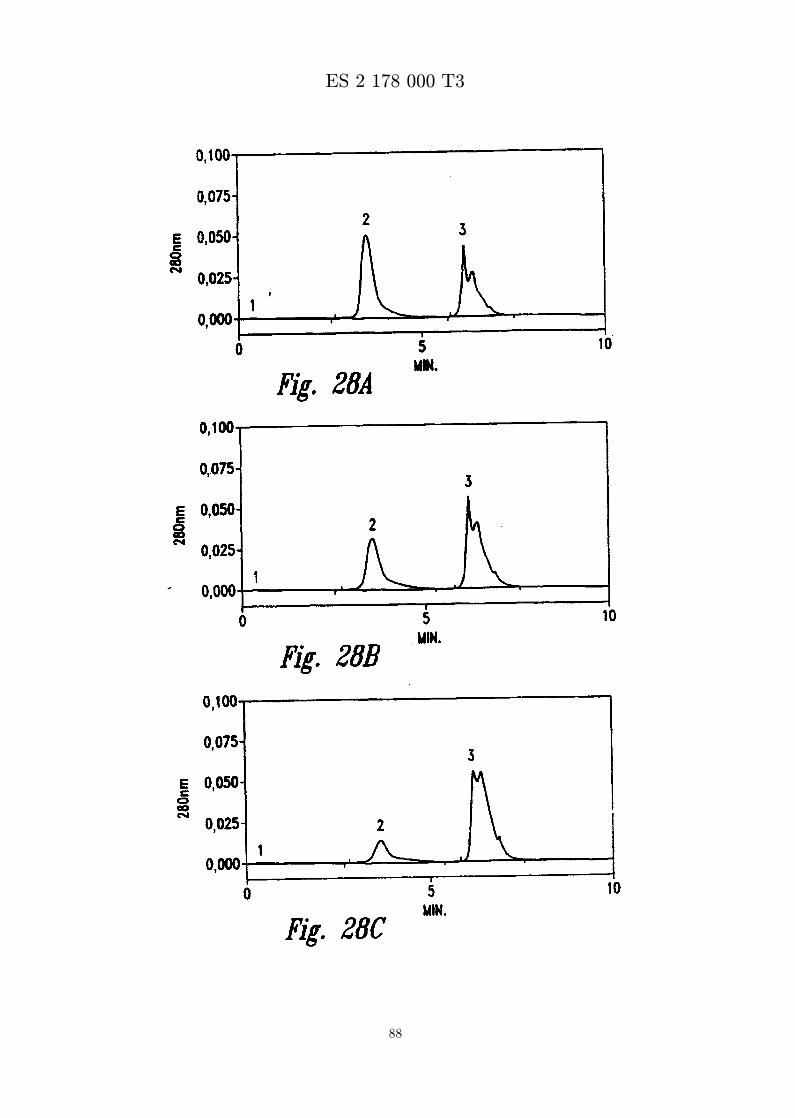
ES 2 178 000 T3
88




























