Embed Size (px)
Citation preview

ĐẠI HỌC THÁI NGUYÊN
TRƯỜNG ĐẠI HỌC NÔNG LÂM
TS. NGUYỄN TUẤN ANH (Chủ biên)
TS. ĐỖ THỊ LAN, TS. NGUYỄN THẾ HÙNG
Giáo trình PHÂN TÍCH MÔI TRƯỜNG
NHÀ XUẤT BẢN NÔNG NGHIỆP
Hà Nội - 2008

LỜI MỞ ĐẦU
Môi trường là vấn đề chung của nhân loại đang được toàn thế giới đặc biệt quan tâm. Nhiều nơi trên thế giới và ở Việt Nam môi trường đang bị suy thoái, tài nguyên thiên nhiên trở nên cạn kiệt, hệ sinh thái mất cân bằng, chất lượng cuộc sống suy giảm. Nhu cầu đào tạo các chuyên gia về nghiên cứu và bảo vệ môi trường ở nước ta hiện nay là rất cần thiết.
Môn học Phân tích môi trường là môn học nhằm cung cấp những kiến thức cơ bản nhất về cơ sở của một số phương pháp phân tích môi trường phố biến trên thế giới và một số phương pháp lấy mẫu, bảo quản mẫu, phân tích mẫu và đánh giá kết quả của các số liệu phân tích cho sinh viên chuyên ngành khoa học môi trường. Giáo trình này còn là tài liệu tham khảo cho những nhà quản lý môi trường, những kỹ thuật viên phân tích trong các phòng thí nghiệm khoa học đất, sinh học, hoá học và môi trường.
Giáo trình phân tích môi trường được tập thể tác giả của trường Đại học Nông Lâm Thái Nguyên biên soạn gồm 7 chương, được phân công như sau:
- TS. Nguyễn Tuấn Anh biên soạn chương 1, 3, 4, 5
- TS. Đỗ Thị Lan biên soạn chương 6, 7
- TS. Nguyễn Thế Hùng biên soạn chương 2
Các tác giả cám ơn sự giúp đỡ về tài liệu và đóng góp ý kiến cho việc biên soạn cuốn giáo trình này của các đồng nghiệp ở các viện nghiên cứu, trung tâm phân tích và các thầy cô giáo khoa Tài nguyên và Môi trường, trường Đại học Nông Lâm Thái Nguyên.
Trong quá trình biên soạn, chúng tôi đã tham khảo nhiều tài liệu giảng dạy và kết quả nghiên cứu có liên quan đến phân tích môi trường ở trong và ngoài nước. Tuy đã có nhiều cố gắng, song chắc chắn không tránh khỏi những thiếu sót. Tập thể tác giả mong nhận được sự góp ý của các thầy cô giáo, sinh viên và độc giả trong và ngoài nước để giáo trình này ngày càng được hoàn thiện hơn.
Các tác giả

Phần 1
NHỮNG VẤN ĐỀ CHUNG
Chương 1
MỞ ĐẦU
1.1. Môi trường
Môi trường bao gồm tất cả các yếu tố lý học, hoá học, các chất hữu cơ và vô cơ của khí quyền, thạch quyển và đại dương. Môi trường sống là tập hợp các điều kiện xung quanh có ảnh hưởng đến cơ thể sống, đặc biệt là con người. Môi trường quyết định chất lượng và sự tồn tại của cuộc sống.
Một số nhà nghiên cứu đã sử dụng thuật ngữ "vi môi trường" để chỉ rõ môi trường chức năng (functional environment), nghĩa là môi trường riêng biệt của các cá thể đặc biệt. Theo nghĩa đen, thuật ngữ này liên quan đến môi trường nhỏ, nghĩa là môi trường trực tiếp ảnh hưởng của cá thể.
1.2. Phân tích môi trường
Phân tích môi trường có thể được định nghĩa là sự đánh giá môi trường tự nhiên và những suy thoái do con người cũng như do các nguyên nhân khác gây ra. Vì vậy, phân tích môi trường bao gồm các quan trắc về các yếu tố môi trường nói chung. Đây là vấn đề rất quan trọng vì qua đó chúng ta có thể thế được yếu tố nào cần được quan trắc và biện pháp nào cần được áp dụng để quản lý, giúp chúng ta có thể tránh khỏi các thảm hoạ sinh thái có thể xẩy ra.
Trong những năm gần đây, nghiên cứu sinh thái không chỉ là sự tiếp cận về chất lượng mà còn cả về số lượng. Để có thể hiểu biết và đánh giá về một hệ sinh thái đòi hỏi phải quan trắc đầy đủ số biến động theo không gian và thời gian của cả các yếu tố môi trường, cả về số lượng và chất lượng có liên quan đến cấu trúc và chức năng của hệ. Đó là các ảnh chơi lý, hoá và sinh học của hệ sinh thái.
1.3. Sự lựa chọn phương pháp để phân tích môi trường
Việc lựa chọn phương pháp và các quy trình trong phân tích môi trường đòi hỏi phải có nhiều kinh nghiệm. Các phương pháp lựa chọn phải trả lời được những câu hỏi sau:
• Sử dụng phương pháp phân tích nào?
• Lượng mẫu có đủ cho nhiều phòng thí nghiệm không?
• Yếu tố nào hạn chế sự phát hiện, độ chính xác của các phương pháp phân tích được sử dụng?

• Người sẽ tiến hành phân tích?
• Những vấn đề gì cần chú ý để tránh làm bẩn mẫu trong quá trình bảo quản mẫu.
• Các chỉ tiêu nào cần phân tích để phản ánh thực tế khả năng độc hại của môi trường? Hàm lượng hay dạng tồn tại của các nguyên tố hoá học?
l.4. Giá trị của các số liệu trong phân tích môi trường
Công việc khó khăn đối với các nhà nghiên cứu là phải xác định được những chỉ tiêu phân tích nào là cần thiết. Việc xác định thành phần các nguyên tố là đủ hay còn cần phải phân tích các phân tử hay các nhóm chức của các chất?
Ví dụ: Khi phân tích hàm lượng tổng số các nguyên tố như: Hg, Pb, P,... có thể sẽ không đánh giá hết được tiềm năng gây hại cho sức khỏe con người. Điều này cũng tương tự như việc đánh giá mối quan hệ giữa hàm lượng tổng số của các chất ở trong đất với khả năng sử dụng của cây trồng.
Chỉ có một phần trong hàm lượng tổng số là dễ tiêu đối với thực vật. Do vậy vấn đề khó khăn là sử dụng phương pháp hoá học nào để phản ánh đúng các hoạt động của hệ rễ thực vật. Trên thực tế kết quả này thường rất hạn chế. Ví dụ đối với cây rau diếp (lettuce), hàm lượng chì trong cây có quan hệ với lượng chì chiết rút từ đất bằng HNO3 im. Trong khi với cây yến mạch (Oat), hàm lượng chì trong cây lại tương quan với chì chiết rút bằng HNO3 0,01M hoặc CH3COONH4 1M. Việc phun dung dịch CuSO4 lên là hoặc đất làm tăng hàm lượng đồng trong cây lúa mì, nhưng hàm lượng đồng trong cây lại không có tương quan với lượng đồng dễ tiêu được xác định trong dung dịch chiết rút CH3COONH4 1M, axit mạnh hoặc chất tạo phức (EDTA).
Mặc dù có những hạn chế nhất định, việc quan trắc các yếu tố riêng biệt vẫn cần được tiến hành như xác định các vùng bị ô nhiễm để ghi nhận các thay đổi về mức độ các chất ô nhiễm và các dẫn liệu của các yếu tố bên ngoài như: gió, mưa, địa hình... Để nghiên cứu xu hướng biến đổi có thể xác định một chuỗi quan trắc. Ví dụ: số liệu ở bảng 1.1 đưa ra mức độ ô nhiễm ở 4 loại chỉ thị đã được phân tích.
Bảng 1.1. Ảnh hưởng của hướng từ nguồn đối với sự tích luỹ của ion kim loại trong mẫu
Mẫu Điểm lấy mẫu Cỏ Địa y Rêu Đất
Pb (ppm) A 10 130 120 - В 49 1528 1200 - С 86 - - 270D 150 - - 230
Zn (ppm) A 102 675 1213 - В 146 1135 4870 - С 350 - - 450D 270 - - 416

Cd (ppm) A 8 68 93 - В 13 83 137 - С 9 - - 7,1D 9 - - 7,7
Số liệu bảng 1.1 cho thấy mức độ ô nhiễm thay đôi theo hưởng địa lý (hướng A,B,C,D). Tuy nhiên nếu việc lựa chọn có định hướng sẽ cho thấy mức độ nhiễm so với các vùng khác.
Những quan trắc tương tự cũng có thể được áp dụng với môi trường nước. Nhưng việc phân tích đơn thuần các mẫu nước lọc sẽ hạn chế ý nghĩa của các số liệu phân tích. Trên thực tế các chất lơ lửng và các chất lắng đọng ở các hồ nước có thể giải phóng n các chất độc hại trong các chuỗi thức ăn hoặc đời sống của các sinh vật thuỷ sinh.
Cặn lơ lửng thường là những hỗn hợp phức tạp bao gồm các chất hữu cơ, vô cơ và phức hữu cơ - vô cơ. Giữa chúng lại có sự tương tác khác nhau như các keo xét trong nước mặn có thể hấp phụ trên 2,5% axit mùn. Sự có mặt của các axit humic sẽ làm tăng khả năng hấp phụ của các chất lơ lửng. Sự thay đổi của các chất điện ly sẽ làm thay đổi qua trình này (trong nước ngọt lượng axit humic được hấp phụ là nhỏ hơn 0,4%. Do vậy tại nơi tiếp giáp giữa các vùng nước ngọt và nước mặn (vùng cửa sông) sẽ có sự biến đổi đột ngột về sự phân bố của các kim loại nặng giữa pha rắn và lỏng.
Vì các sinh vật biển có xu hướng tích luỹ các kim loại nặng khi sống trong một trường ô nhiễm nên chúng có thể được coi như các vật chỉ thị. Các số liệu này có thể so sánh với kết quả điều tra trung bình trong động vật giáp xác (tôm, cua...). Đối với thực vật hàm lượng lớn kim loại của một sinh vật có biến động lớn (hàng chục lần) so với vị trí tương đối của nó đối với nguồn ô nhiễm, nhưng hàm lượng này là tương đối ổn định trong vùng lấy mẫu và có sự khác nhau lớn so với các giá trị đã được xác định.
Tại một nơi xác định sự dao động hàm lượng của một chất có thể là 20%. Vì vậy sự khác nhau ở những nơi khác nhau phải lớn hơn để số liệu thống kê có ý nghĩa γ.
1.5. Ảnh hưởng của cân bằng
Các số liệu trong bảng 1. 1 được xem xét trên cơ sở các cân bằng như dưới đây:
Vì rêu là vật bám trên cây nên sự tích luỹ các ion kim loại có thể trước hết là từ bụi và khi xung quanh. Vì vậy, hàm lượng của một số chất có thể bi giảm do nước mưa rửa trôi. Các cây mọc trên đất có khả năng sử dụng các chất rất khác nhau và phụ thuộc vào các tính chất của đất. Khả năng hấp phụ các chất của cây cũng bị giảm sút khi có sự cạnh tranh giữa các thất hấp thu. Nếu hệ thống không quá phức tạp, quá trình hấp thu các chất có thể được biếu diễn bằng phương trình toán học như sau:
(x/m)a = k1.Ca.Sv/(1+k1Ca + k2Cb+ k3Cc+...)
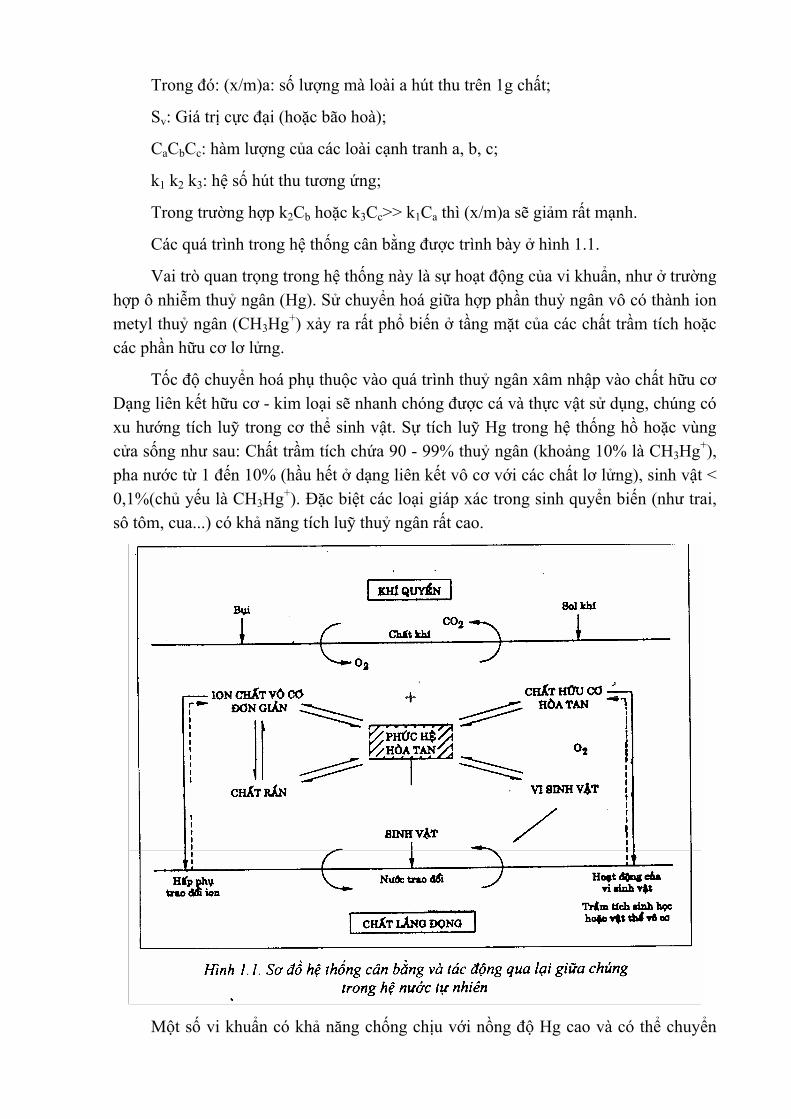
Trong đó: (x/m)a: số lượng mà loài a hút thu trên 1g chất;
Sv: Giá trị cực đại (hoặc bão hoà);
CaCbCc: hàm lượng của các loài cạnh tranh a, b, c;
k1 k2 k3: hệ số hút thu tương ứng;
Trong trường hợp k2Cb hoặc k3Cc>> k1Ca thì (x/m)a sẽ giảm rất mạnh.
Các quá trình trong hệ thống cân bằng được trình bày ở hình 1.1.
Vai trò quan trọng trong hệ thống này là sự hoạt động của vi khuẩn, như ở trường hợp ô nhiễm thuỷ ngân (Hg). Sử chuyển hoá giữa hợp phần thuỷ ngân vô có thành ion metyl thuỷ ngân (CH3Hg+) xảy ra rất phổ biến ở tầng mặt của các chất trầm tích hoặc các phần hữu cơ lơ lửng.
Tốc độ chuyển hoá phụ thuộc vào quá trình thuỷ ngân xâm nhập vào chất hữu cơ Dạng liên kết hữu cơ - kim loại sẽ nhanh chóng được cá và thực vật sử dụng, chúng có xu hướng tích luỹ trong cơ thể sinh vật. Sự tích luỹ Hg trong hệ thống hồ hoặc vùng cửa sống như sau: Chất trầm tích chứa 90 - 99% thuỷ ngân (khoảng 10% là CH3Hg+), pha nước từ 1 đến 10% (hầu hết ở dạng liên kết vô cơ với các chất lơ lửng), sinh vật < 0,1%(chủ yếu là CH3Hg+). Đặc biệt các loại giáp xác trong sinh quyển biến (như trai, sô tôm, cua...) có khả năng tích luỹ thuỷ ngân rất cao.
Một số vi khuẩn có khả năng chống chịu với nồng độ Hg cao và có thể chuyển

hoá các hợp chất hữu cơ - thuỷ ngân thống dạng thuỳ ngân tự do không hòa tan. Hơn nữa trong điều kiện kỵ khí, các vi khuẩn khử sunphat cũng có khả năng sinh ra hiđrosunfua để cố định Hg2+ dưới dạng sunfua, vi khuẩn không chuyển hoá hợp chất này thành metyl thuỷ ngân.
Xem xét các cân bằng phức tạp trong hầu hết các hệ thống tự nhiên, điều cần lưu ý không chỉ là việc lựa chọn các chỉ thị mà còn khó khăn trong công việc lấy, vận chuyển và bảo quản mẫu. Vì lý thuyết, điều cần thiết là làm ngừng trệ tất cả các quá trình hoá học cũng như sinh học bằng các phương pháp thích hợp. Các qúa trình biến đổi này sẽ được giảm tới mức tối thiểu ở nhiệt độ thấp hoặc sử dụng những phòng thí nghiệm di động. Tuy nhiên tồn tại một vấn đề là hệ thống cân bằng trong tự nhiên luôn luôn bị xáo trộn. Ví dụ như lấy một mẫu nước ở phía trên trầm tích (hoặc tách các chất lơ lửng) sẽ làm cho một hợp phần hoặc các chất hoà tan thiết lập một cân bằng mới. Tuy nhiên, về mặt tổng số vẫn không thay đổi và số là các dẫn liệu cho sự ô nhiễm.
Vấn đề tiếp theo cần chú ý để đánh giá mức độ ô nhiễm là phải lựa chọn phương pháp phân tích có độ chính xác thích hợp và cần được tiến hành trong thời gian nhất định

Chương 2
ĐỘ CHÍNH XÁC VÀ ĐỘ TIN CẬY CỦA PHÉP PHÂN TÍCH
2.1. Bảo đảm và kiểm soát chất lượng trong phân tích môi trường
Bảo đảm và kiểm soát chất lượng đòi hỏi tất cả các phòng thí nghiệm phải tuân thủ theo các hướng dìm đã được đưa ra để đảm bảo kết quả phân tích có độ tin cậy cao. Bảo đảm chất lượng thông qua hàng loạt các nguyên tắc và sự giám sát chặt chế để độ chính xác của kết quả phân tích có độ tin cậy và tính pháp lý cao. Vấn đề bảo đảm chất lượng bao gồm cả việc lấy mẫu và lo quản mẫu của các phòng thí nghiệm và trách nhiệm cũng như kỹ năng của các cá nhân phân tích và người chịu trách nhiệm. Với ý nghĩa rộng hơn thì cả kế hoạch cũng được bao gồm trong việc kiểm soát chất lượng.
Kiểm soát chất lượng phòng thí nghiệm bao gồm: các tài liệu và phương pháp tiêu chuẩn các phép thử, chuẩn bi các đường chuẩn và kiểm tra thường xuyên các thuốc thử, máy móc, xác định độ chính xác và độ tin cậy của phép phân tích, chuẩn bị sơ đồ kiểm tra.
Chuẩn bị đường chuẩn:
Các đường chuẩn được xây dụng trên cơ sở các phép đo màu hoặc sắc ký khí ở các nồng độ khác nhau và được chuẩn bi hàng ngày trước khi phân tích mẫu. Nếu kết quả đo có sai số 15% thì cần phải xây dụng lại đường chuẩn.
2.2. Sai số và độ chính xác
Sai số được thể hiện qua kết quả phân tích của các lần lặp lại. Nếu một mẫu được phân tích lặp lại nhiều lần trong cùng một điều kiện thì kết quả cũng sẽ khác nhau do sai số thí nghiệm hoặc do thao tác. Các kết quả này sẽ phân bố một cách ngẫu nhiên xung quanh một giá trị trung bình là giá trị trung bình cộng của các phép đo. Khi các kết quả đo phân bố như dáng hình quả chuông được gọi là đường cong phân bố chuẩn hoặc đường Gauss (Gaussian Curve) như hình 2.1 (trong nhiều mẫu môi trường bị nhiễm bẩn thì kết quả sẽ không theo sự phân bố chuẩn).
Giá trị trung bình (x) được tính bằng nx∑ (x: các giá trị đo; n: số lần đo)

Độ lệch chuẩn (S) sẽ xác định chiều rộng của đồ thị phân bố và được tính như sau:
Trong trường hợp hàm phân bố chuẩn thứ 68,27% diện tích nằm trong khoảng
x ± 1S; 95,45% nằm trong khoảng x ± 2S và 99,70% nằm trong khoảng x ± 3S. Giá trị 3S xung quanh giá trij trung bình là giới hạn trên và dưới trong đồ thij kiểm tra. Tất cả các giá trị nằm ngoài x ± 3S được xem độ là không bình thường. Điều này chứng tỏ rằng có vấn đề nào đó trong quá trình phân tích cần phải được xem xét ngay.
Độ lệch chuẩn cũng có thể được tính theo công thức như sau:
n: số phép đo
Mặc dù sĩ số hoặc kết quả phân tích lặp lại có thể được biểu diễn dưới dạng của độ lệch chuẩn nhưng độ lớn giá trị phân tích có thể làm thay đổi đáng kể độ lệch chuẩn lên các giá trị tương ứng. Có thể minh hoạ điều này qua 2 ví dụ sau đây:
Ví dụ 1: Hàm lượng tổng số hiđrocacbon dầu mỏ - TPH (total petroleum hiđrocacbon, TPH) trong mẫu bị nhiễm bẩn với 6 lần phân tích là 5,3 - 4,9 - 5,1 - 5,5 - 4,7 và 5,0 mg/l. Xác định độ lệch chuẩn như sau:
X x2 5,3 4,9 5,1 5,5 4,7 5,0 30,5
28,09 24,01 26,01 30,25 22,09 25,00 155,45

Ví dụ 2: Nếu kết quả phân tích TPH trong mẫu có giá trị lớn gấp 10 lần nghĩa là
53 - 49 -51- 55 - 47 và 50 mg/l thì độ lệch chuẩn sẽ là:
Nếu giá trị đo được lớn hơn, ví dụ như 530 - 490 - 510 - 550 - 470 và 500mg/1
thì S = 28,6mg/l. Như vậy độ lệch chuẩn khi biến đổi theo độ lớn các giá trị đo được là không có ý nghĩa trừ khi độ lớn của giá trị phân tích được xác định trước.
Nói một cách khác, sai số phân tích sẽ luôn có giá trị khi liên quan với giá trị của mẫu đo. Một cách biểu thì khác là độ lệch chuẩn tương đối (relative standard devlation - RSD) hoặc hệ số biến thiên (Coemcient of vanance - CV). Đây là tỷ số giữa độ lệch chuẩn và giá trị trung binh đại số:
Trong ví dụ 1 và 2 ở trên, RSD sẽ là:
Như vậy RSD ở hai ví dụ này là bằng nhau trong khi S có sự khác nhau rõ rệt
(0,29 và 2,8mg/l.)
Một cách khác biểu thị sai số là sai số chuẩn của giá trị trung bình (M), đây là tỷ số giữa S và căn bậc hai của số lần đo (n).
Trong phân tích môi trường, thông thường việc lặp lại nhiều lần là khó thực hiện
được. Vì vậy sai số của phép tính được tính toán thông qua độ khác nhau phần trăm tương đối (Relative percent difference - RPD). Tỷ số này được xác định thông qua sự phân tích lặp lại hệ lần mẫu trong một điều kiện xác định. Đó là tỷ lệ phần trăm giữa hiệu số của kết quả giữa hai lần phân tích với giá trị trung bình cộng của chúng.

Trong đó: a1 và a2 là giá trị của hai lần phân tích một mẫu
Ví dụ 3: Hàm lượng Cl- trong hai lần phân tích một mau là 9,7 và 11,1 mg/l. Sai số sẽ được xác định như sau:
Độ chính xác của giá trị phân tích là mức độ chính xác của nó so với hàm lượng
thực tế có trong mẫu. Độ chính xác được đánh giá qua việc cho thêm vào mẫu một lượng nhất định dung dịch chuẩn có nồng độ đã biết. Dựa trên phần trăm của nồng độ cho thêm được phát hiện để điều chỉnh độ chính xác cho kết quả phân tích. Nhìn chung trong phân tích mẫu môi trường không yêu cầu các phép tính điều chỉnh độ sai số của kết quả phân tích. Tuy nhiên trong những phép phân tích đặc biệt nào đó, việc điều chỉnh độ sai số có thể được đặt ra. Khi phân tích một số chất hữu cơ trong nước thải, USEPA- (The Unỉted States Envừunment Protectlon Agency) đã đưa ra bốn vùng cho lượng phần trăm được xác định. Nếu giá trị của bất kỳ một phép phân tích nào đó nằm ngoài các vùng này thi các chi số QC cho phép phân tích đó sẽ không được đáp ứng.
Một ma trận lượng bổ sung phát hiện được có thể xác định bằng hữu cách: Phương pháp xác định phần trăm thu hồi từ lượng tiêu chuẩn thêm vào, chẳng hạn như phương pháp của U.S.EPA(I) và phương pháp tính phần trăm thu hồi giữa mẫu cần đo và dung dịch chuẩn(2) Lượng tìm thấy được tính toán bởi hai phương pháp sẽ cho các giá trị khác nhau.
Cách xác định 1 (U.S.EPA):
Trong đó xi: giá trị đo được cho mẫu đã hoà trộn;
xu: giá trị đo được cho mẫu không hoà trộn ở thể tích sau khi hoà trộn; k: giá trị đã biết nồng độ của mẫu chuẩn trong mẫu hoà trộn.
Cách xác định 2:
Nồng độ lý thuyết có thể tính như sau:
Trong đó: Cu: nồng độ đo được của mẫu;

Cs: nồng độ của dung dịch tiêu chuẩn;
Vu, Vs: thể tích của mẫu và dung dịch tiêu chuẩn.
Phần trăm lượng thu hồi theo các cách tính trên đây có thể được minh hoạ trong các ví dụ sau đây:
Ví dụ 4: Một mẫu nước thể được xác định có nồng độ xianua là 3,8 mg/l. Sau khi cho thêm 10 ml dung dịch tiêu chuẩn có nồng độ xianua là 50 mg/l máu vào 100 mẫu nước thải trên. Nồng độ của xianua trong hỗn hợp thu được là 8,1 mg/l .
Tính phần trăm lượng thu hồi từ dung dịch này (sau khi pha loãng) theo cách tính 1.
+ Dựa theo nồng độ xác định:
+ Dựa trên khối lượng (phương pháp chuyển đồi) khối lượng tổng số của ion
CN- trong 110 ml mẫu và dung dịch tiêu chuẩn là:
8,1 màu x 0,110 l = 0891 mg
Khối lượng ion CN- tính theo phần trăm trong 100ml mẫu cần xác định ban đầu:
3,8 mg/l x 0,100 l = 0,38 mg
Khối lượng ion CN- trong 10 ml dung dịch tiêu chuẩn là:
50 mg/l màu x 0,010 l = 0,500 mg
Tính phần trăm lượng tìm được như trong cách xác định 2.
+ Tính toán dựa trên nồng độ:
Nồng độ CN- đo được sau khi được bổ sung thêm bằng dung dịch chuẩn là 8,1 màu. Nồng độ thực tế của CN- sau khi pha trộn sẽ có giá trị bằng nồng độ CN- ban đầu trong mẫu + nồng độ CN- trong lượng dung dịch tiêu chuẩn bổ sung thêm. Hay:
+ Tính toán dựa trên khối lượng (phương pháp chuyển đổi):

Khối lượng xianua đo được trong tổng số 110 ml dung dịch (100 ml mẫu + 10 ml dung dịch chuẩn bổ sung thêm) sẽ là:
Khối lượng thực tế của xianua trong 110 ml dung dịch này là:
Ví dụ 5: Một mẫu đo được 11,7 mgl/1. Nếu bổ sung thêm 5 ml dung dịch chuẩn
có nồng độ 100 mg/l vào 50 ml dung dịch này sẽ được dung dịch có nồng độ đo được là 18,8 mg/l.
Tính lượng bổ sung tìm thấy được như trong cách tính 1 và 2 (tính trên cơ sở nồng độ).
+ Cách xác định 1:
+ Cách xác định 2:
Khi lượng tìm thấy là nhỏ hơn 100% theo U.S.EPA (cách tính 1) sẽ cho giá trị
thấp hơn khi tính theo cách 2 (ví dụ 5). Tuy nhiên nếu lượng tìm lại được mà lớn hơn 100% sẽ cho giá trị ngược lại (ví dụ 4).
Đối với việc phân tích các mẫu đất và chất thải rắn thì không cần phủ điều chinh như đối với mẫu nước vì đất và chất thải rắn phải dùng một chất lỏng để chiết rút, như ở ví dụ 6 dưới đây.
Ví dụ 6: Một mẫu đất được chết rút đề xác định hiđrocacbon dầu mỏ (PHC) bằng phương pháp đo phổ hồng ngoại. Hàm lượng PHC trong mẫu xác định được là 285 mg/kg. Thêm 2 ml dung dịch chuẩn PHC có nồng độ 1000 mg/l vào 40 g mẫu. Nồng

độ PHC mẫu này đo được là 326 mg/kg. Xác định độ tin cậy của phép phân tích theo phần trăm lượng tìm được từ lượng bổ sung của dung dịch chuẩn.
Khối lượng của PHC trong mẫu đã thêm dung dịch chuẩn là:
Khối lượng của PHC trong mẫu trước khi bổ sung dung dịch tiêu chuẩn chứa
PHC là:
Như vậy, khối lượng mẫu đã được bổ sung bằng dung dịch PHC chuẩn là 40g
chứ không phải 43g (thêm 2 ml dung dịch chuẩn có tỷ trọng khoảng 1,5 gian) theo tính toán ở trên. Thực tế là 2 ml dung môi thêm vào sẽ trộn lẫn vào chất chiết rút.
Vì vậy khối lượng của mẫu sau khi chiết rút (tức là khối lượng của phần chất rắn còn lại) hầu như không thay đôi so với trước khi chiết rút chúng.
Phần trăm phát hiện được từ lượng bổ sung thêm vào mẫu nói trên có thể được xác định theo công thức US.EPA như sau:
Không có sự điều chỉnh chính xác về khối lượng hoặc thể tích nào được đưa vào
trong cách tính toán trên. Vì vậy, xu đã được lấy là 285 mg/kg.
Phần trăm lượng tìm được trong ví dụ trên được tính theo cách 2 sẽ là:
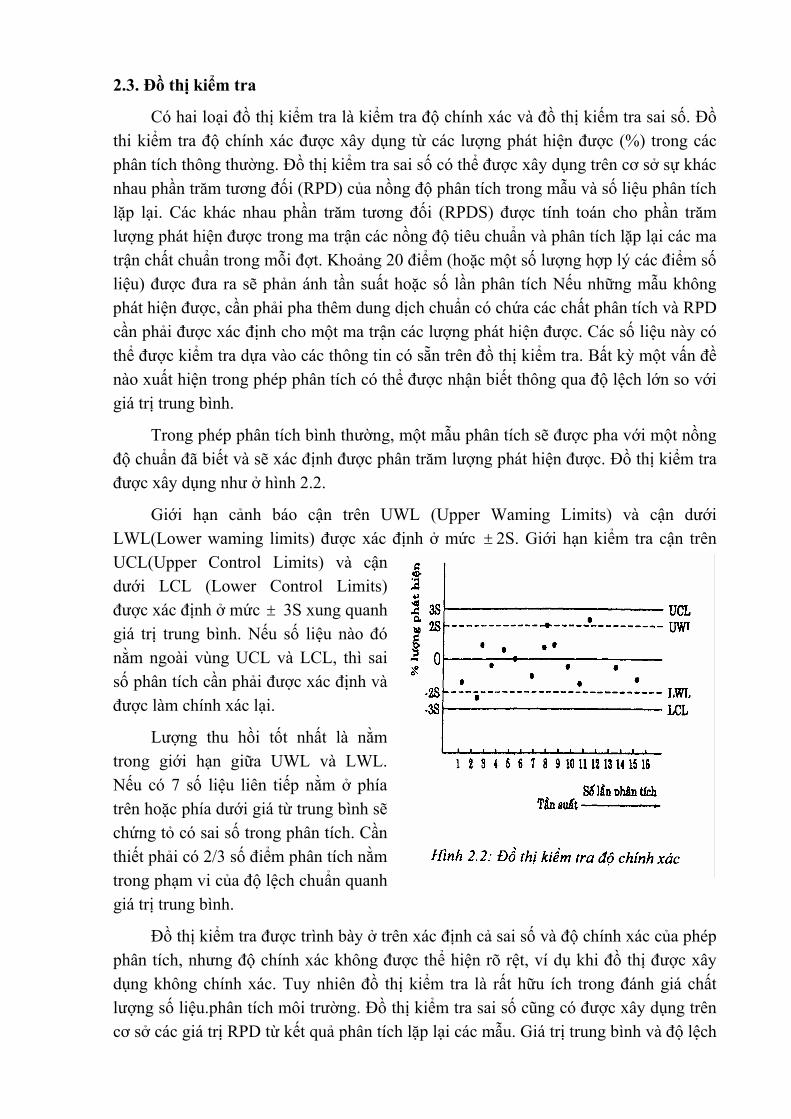
2.3. Đồ thị kiểm tra
Có hai loại đồ thị kiểm tra là kiểm tra độ chính xác và đồ thị kiếm tra sai số. Đồ thi kiểm tra độ chính xác được xây dụng từ các lượng phát hiện được (%) trong các phân tích thông thường. Đồ thị kiểm tra sai số có thể được xây dụng trên cơ sở sự khác nhau phần trăm tương đối (RPD) của nồng độ phân tích trong mẫu và số liệu phân tích lặp lại. Các khác nhau phần trăm tương đối (RPDS) được tính toán cho phần trăm lượng phát hiện được trong ma trận các nồng độ tiêu chuẩn và phân tích lặp lại các ma trận chất chuẩn trong mỗi đợt. Khoảng 20 điểm (hoặc một số lượng hợp lý các điểm số liệu) được đưa ra sẽ phản ánh tần suất hoặc số lần phân tích Nếu những mẫu không phát hiện được, cần phải pha thêm dung dịch chuẩn có chứa các chất phân tích và RPD cần phải được xác định cho một ma trận các lượng phát hiện được. Các số liệu này có thể được kiểm tra dựa vào các thông tin có sẵn trên đồ thị kiểm tra. Bất kỳ một vấn đề nào xuất hiện trong phép phân tích có thể được nhận biết thông qua độ lệch lớn so với giá trị trung bình.
Trong phép phân tích bình thường, một mẫu phân tích sẽ được pha với một nồng độ chuẩn đã biết và sẽ xác định được phân trăm lượng phát hiện được. Đồ thị kiểm tra được xây dụng như ở hình 2.2.
Giới hạn cảnh báo cận trên UWL (Upper Waming Limits) và cận dưới LWL(Lower waming limits) được xác định ở mức ± 2S. Giới hạn kiểm tra cận trên UCL(Upper Control Limits) và cận dưới LCL (Lower Control Limits) được xác định ở mức ± 3S xung quanh giá trị trung bình. Nếu số liệu nào đó nằm ngoài vùng UCL và LCL, thì sai số phân tích cần phải được xác định và được làm chính xác lại.
Lượng thu hồi tốt nhất là nằm trong giới hạn giữa UWL và LWL. Nếu có 7 số liệu liên tiếp nằm ở phía trên hoặc phía dưới giá từ trung bình sẽ chứng tỏ có sai số trong phân tích. Cần thiết phải có 2/3 số điểm phân tích nằm trong phạm vi của độ lệch chuẩn quanh giá trị trung bình.
Đồ thị kiểm tra được trình bày ở trên xác định cả sai số và độ chính xác của phép phân tích, nhưng độ chính xác không được thể hiện rõ rệt, ví dụ khi đồ thị được xây dụng không chính xác. Tuy nhiên đồ thị kiểm tra là rất hữu ích trong đánh giá chất lượng số liệu.phân tích môi trường. Đồ thị kiểm tra sai số cũng có được xây dụng trên cơ sở các giá trị RPD từ kết quả phân tích lặp lại các mẫu. Giá trị trung bình và độ lệch

chuẩn sẽ được xác định Các giới hạn UWL, LWL,UCL và LCL cũng được xác định tại ± 2S và ± 3S. Phương pháp này được coi như hình thức kiểm tra sai số bổ sung cùng với đồ thị kiểm tra lượng thu hồi ở trên. Các đồ thi kiểm tra là một phần quan trọng của chương trình kiểm tra chất lượng (QC programs) trong phân tích môi trường.

Phần 2
MỘT SỐ PHƯƠNG PHÁP
DÙNG TRONG PHÂN TÍCH MÔI TRƯỜNG
Chương 3
PHƯƠNG PHÁP TRẮC QUANG
3.1. Phương pháp so màu quang điện
Phương pháp so màu quang điện và phương pháp phân tích dựa trên sự so sánh cường độ màu của dung dịch nghiên cứu với cường độ màu của dung dịch tiêu chuẩn có nồng độ xác định.
Phương pháp này được dùng chủ yếu để xác định lượng nhỏ của các chất, tốn ít thời gian so với các phương pháp hóa học khác.
3.1.1. Định luật cơbản của phương pháp so màu
Nếu chiếu một dòng sáng (cường độ I0) vào một cuvet đúng dung dịch thì một phần của nó (cường độ Ir) bị phản xạ từ mặt cuvet, một phần khác (cường độ Ia) bị dung dịch hấp thụ, phần còn lại (cường độ It) đi qua cuvet. Ta có:
Khi sử dụng một loại cuvet có thể xem cường độ dòng ánh sáng phản xạ và
không đổi và thường không lớn nên có thể bỏ qua. Khi đó phương trình trên có dạng
I0 và It có thể đo trực tiếp còn Ia tìm được theo công thức Ia= I0 - It. Dựa trên
nghiên cứu thực nghiệm Bugơ (Bougueur) và Lămbe (Lambert) đã thiết lập định luật và phát biếu như sau: Những lớp chất có chiều dày đồng nhất trong những điều kiện khác như nhau, luôn hấp thụ một tỷ lệ bằng nhau của chùm ánh sáng chiểu vào những lớp chất đó.
Biểu thức toán học của định luật là:
Trong đó:
I0: là chiều dày lớp hấp thụ
k: hệ số tắt, hệ số này chỉ phụ thuộc vào bản chất của chất tan và bước sóng ánh sáng chiếu vào dung dịch. Do đó định luật hấp thụ ánh sáng Bugơ - Lămbê chỉ đúng cho tia đơn sắc.

Khi nghiên cứu sự hấp thụ ánh sáng của dung dịch, Bia (Beer) đã thiết lập được mối tương quan giữa hệ số tắt k với nồng độ chất hấp thụ theo phương trình:
Kết hợp những nghiên cứu của Bugơ- Lăm be- Bia thì:
Nếu nồng độ C được tính theo mol/1; Chiều dày lớp dung dịch (l) đo bằng cm thì ε được gọi là hệ số tắt phân tử hay hệ số hấp thụ phân tử; ε là một đại lượng không đổi phụ thuộc vào bước sóng ánh sáng, bản chất của chất tan, nhiệt độ dung dịch.
3.1.2. Các đại lượng thường dùng trong phương pháp so màu
Tỉ số giữa cường độ chùm sáng sau khi đi qua dung dịch (It) với cường độ chùm sáng chiếu vào dung dich(I0) gọi là độ truyền qua, kí hiệu bằng T.
Đại lượng T ứng với chiều dày lớp dung dịch bằng 1 cm gọi là hệ số truyền qua.
Logant của đại lượng nghịch đảo với độ truyền qua gọi là mật độ quang D hay độ tắt E (extinction):
Từ định nghĩa này thấy rằng mật độ quang D tỷ lệ thuận với nồng độ chất tan
trong dung dịch
3.1.3. Vùng quang phổ hấp thụ
Đặc điểm hấp thụ ánh sáng của các hợp chất màu là sự hấp thụ chọn lọc. Hệ số hấp thụ phân tử của hợp chất màu và mật độ quang của dung dịch khác nhau đối với chùm ánh sáng đi qua có bước sóng khác nhau. Vì vậy phố hấp thụ cũng là một đặc trưng điển hình của các hợp chất màu.
Khi sử dụng phương pháp so màu để định lượng một chất cần phải dùng tia đơn
sắc nào mà khi chiếu qua dung dịch, dung dịch có khả năng hấp thụ lớn nhất. Để xác

định bước sóng ánh sáng hấp thụ cực đại người ta đo giá trị mật độ quang hoặc hệ số hấp thu phân tử của dung dịch màu với những bước sóng khác nhau, cách nhau 10-20nm. Ở giá tử bước sóng nào mà mật độ quang đo được là lớn nhất thì đó là bước sóng ánh sáng thích hợp để định lượng hợp chất màu này.
3.1.4. Kính lọc màu
Để đảm bảo độ nhạy và độ chính xác của phép xác định, người ta không cho dung dịch hấp thụ một chùm ánh sáng mà chỉ cho những tia đơn sắc bị dung dịch màu hấp thụ cực đại đi qua. Muốn tách được những tia sang này người ta phải dùng kính lọc sáng (kính lọc màu).
Kính lọc sáng là tên gọi chung các môi trường như: thuỷ tinh, màng tổng hợp... chỉ cho những tia sáng thuộc một vùng xác định của quang phổ đi qua.
Kính lọc sáng trong phương pháp so màu phải đảm bảo cho ánh sang đơn sắc truyền qua đạt cực đại ở nhưng bước sóng trùng với bước sóng hấp thụ cực đại và đi qua kính lọc màu phải bị dung dịch hấp thu chọn lọc cao nhất. Muốn vậy, trước khi đo mật độ quang dung dịch một hợp chất màu chưa biết λmax (bước sóng ánh sáng bị hấp thụ cực đại) cần tiến hành phương pháp thực nghiệm: quay các kính lọc màu xem kính nào cho ánh sáng màu bị hấp thụ mạnh nhất, hoặc có thể dựa vào màu sắc của dung dịch xác định để tìm kính lọc màu thích hợp theo bảng sau:
Sáng 3. 1. Các kính lọc sử dụng cho các dung dịch màu
Màu của dung dịch Màu của kính lọc sáng Tím
Xanh Xanh lục Lục xanh
Lục Lục vàng
Vàng
Lục vàng Vàng Đỏ nâu Đỏ
Đỏ nâu Tím
Xanh h l
3.1.5. Phương pháp xác định nồng độ các chất
Khi tiến hành một loạt phép xác định, phương pháp thuận lợi nhất là phương pháp đường chuẩn. Để xây dụng đường chuẩn ta đo màu các dung dịch chuẩn của chất đó với các nồng độ hoặc hàm lượng đã biết.
Tiến hành đo giá trị mật độ quang D (hay phần trăm độ truyền qua) của dãy dung dịch chuẩn này và xây dụng đường chuẩn bằng phương pháp hồi quy tuyến tính. Tác hoành biểu diễn giá trị D, trục tung biếu diễn giá trị nồng độ hoặc hàm lượng của chất chuẩn.
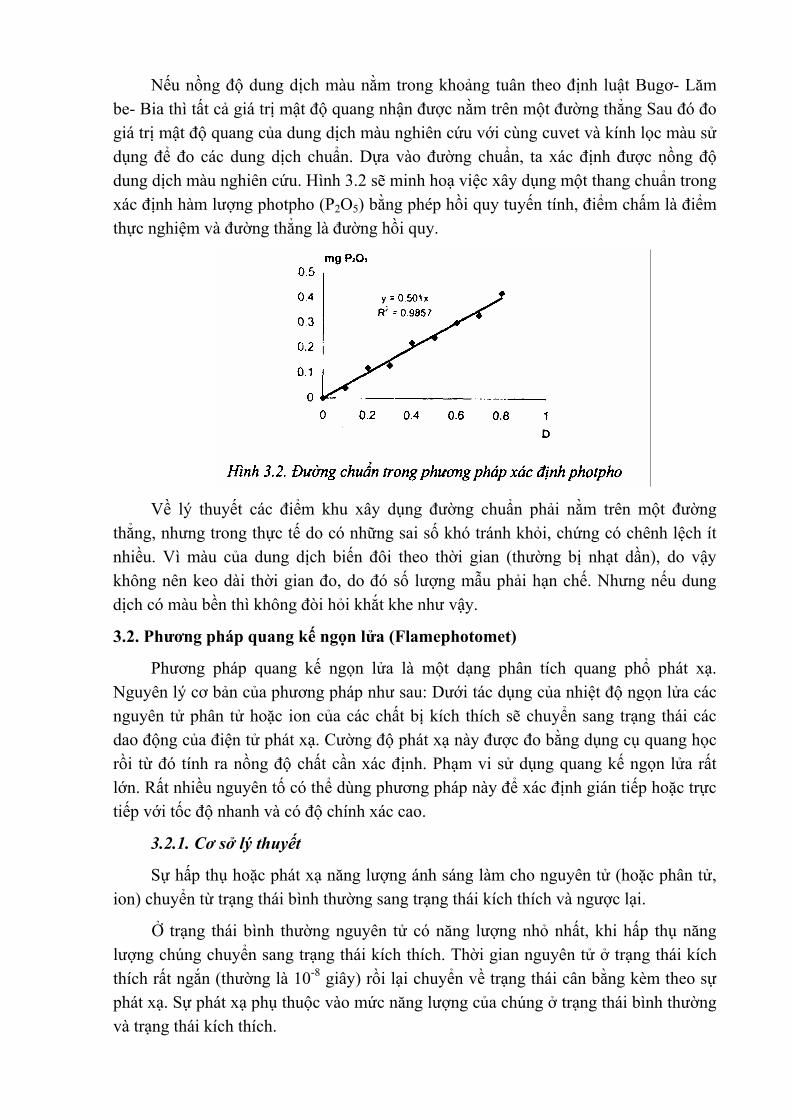
Nếu nồng độ dung dịch màu nằm trong khoảng tuân theo định luật Bugơ- Lăm be- Bia thì tất cả giá trị mật độ quang nhận được nằm trên một đường thẳng Sau đó đo giá trị mật độ quang của dung dịch màu nghiên cứu với cùng cuvet và kính lọc màu sử dụng để đo các dung dịch chuẩn. Dựa vào đường chuẩn, ta xác định được nồng độ dung dịch màu nghiên cứu. Hình 3.2 sẽ minh hoạ việc xây dụng một thang chuẩn trong xác định hàm lượng photpho (P2O5) bằng phép hồi quy tuyến tính, điểm chấm là điểm thực nghiệm và đường thẳng là đường hồi quy.
Về lý thuyết các điểm khu xây dụng đường chuẩn phải nằm trên một đường
thẳng, nhưng trong thực tế do có những sai số khó tránh khỏi, chứng có chênh lệch ít nhiều. Vì màu của dung dịch biến đôi theo thời gian (thường bị nhạt dần), do vậy không nên keo dài thời gian đo, do đó số lượng mẫu phải hạn chế. Nhưng nếu dung dịch có màu bền thì không đòi hỏi khắt khe như vậy.
3.2. Phương pháp quang kế ngọn lửa (Flamephotomet)
Phương pháp quang kế ngọn lửa là một dạng phân tích quang phổ phát xạ. Nguyên lý cơ bản của phương pháp như sau: Dưới tác dụng của nhiệt độ ngọn lửa các nguyên tử phân tử hoặc ion của các chất bị kích thích sẽ chuyển sang trạng thái các dao động của điện tử phát xạ. Cường độ phát xạ này được đo bằng dụng cụ quang học rồi từ đó tính ra nồng độ chất cần xác định. Phạm vi sử dụng quang kế ngọn lửa rất lớn. Rất nhiều nguyên tố có thể dùng phương pháp này để xác định gián tiếp hoặc trực tiếp với tốc độ nhanh và có độ chính xác cao.
3.2.1. Cơ sở lý thuyết
Sự hấp thụ hoặc phát xạ năng lượng ánh sáng làm cho nguyên tử (hoặc phân tử, ion) chuyển từ trạng thái bình thường sang trạng thái kích thích và ngược lại.
Ở trạng thái bình thường nguyên tử có năng lượng nhỏ nhất, khi hấp thụ năng lượng chúng chuyển sang trạng thái kích thích. Thời gian nguyên tử ở trạng thái kích thích rất ngắn (thường là 10-8 giây) rồi lại chuyển về trạng thái cân bằng kèm theo sự phát xạ. Sự phát xạ phụ thuộc vào mức năng lượng của chúng ở trạng thái bình thường và trạng thái kích thích.

Như vậy, mỗi một nguyên tử hay ion biến đổi trạng thể bình thường sang trạng
thái kích thích được đặc trưng bằng một giá trị năng lượng nhất định. Vì vậy, sự phát xạ nguyên tử của bất kỳ một nguyên tố nào cũng có thành phần quang phổ đặc trưng hay cấu tạo vạch quang phổ đặc trưng.
Để ứng dụng trong phép tính định lượng người ta chọn lấy một vạch quang phổ đặc trưng tức là vạch phổ xuất hiện cuối cùng khi giảm dần nồng độ chất. Sự thay đổi cường độ của vạch này sẽ xác định lượng nguyên tử cần phân tích.
Sự kích thích quang phổ phát xạ tăng lên khi tăng nguồn năng lượng ban đầu cung cấp tức là nhiệt độ. Tuy nhiên một số nguyên tố như kim loại kiềm, kiềm thô có thể phát xạ ánh sáng ở nhiệt độ không cao lắm, vì vậy việc xác định những nguyên tố này không cần thiết phải đốt ở nhiệt độ rất cao. Nhìn chung, đề nhận được những vạch quang phổ cần có nhiệt độ thích hợp cho từng nguyên tố.
Bảng 3.2. Nhiệt độ của mã số ngọn lửa thường dùng (Theo Poluectov)
Khi tăng nồng độ nguyên tố có thể xảy ra hiện tượng hấp thụ phát xạ, tức là những nguyên tố không bị kích thích sẽ hấp thụ một phần năng lượng phát xạ. Vì thế nên bắt đầu từ một giới hạn nồng độ nào đó (đối với mỗi nguyên tố xác định) thì quan hệ giữa cường độ phát xạ 1 và nồng độ C sẽ không là tuyến tính (hình 3.3) tức là phương pháp quang kế ngọn lửa chỉ đúng và dùng được trong phạm vi nồng độ nhất định đối với từng nguyên tố.
Hỗn hợp cháy Nhiệt độ ngọn lửa (0C)
Propan- không khí Hiđro-không khí
Axetylen Hiđro - ôxy
Axetylen- ôxy Propan - N2O
1700-1 800 2000-2O45 2125-2397 2550-2660 3100-3137 2850-2900

Ngoài ra cần chú ý tới những điều kiện sau:
+ Quá trình xảy ra trong ngọn lửa: ờ điều kiện nhiệt độ cao thường có sự bay hơi lớn, phân tử các muối phân li tham gia phản ứng với các thành phần khác, kết quả là cùng một lúc trong ngọn lửa tồn tại một lượng electron tự do, ion, phân tử, nguyên tử. Cân bằng giữa dạng này phụ thuộc vào nhiệt độ của ngọn lửa, thành phần hơi của dung dịch, điều kiện ôxy hoá - khử. Vì vậy thành phần dung dịch chứa nguyên tố cần xác định có ảnh hưởng đến kết quả phân tích. Để loại trừ ảnh hưởng này, cần chuẩn bị dung dịch chuẩn có thành phần giống như thành phần dung dịch phân tích.
+ Thành phần dung dịch không những ảnh hưởng đến các quá trình xảy ra trong ngọn lửa mà còn ảnh hưởng tới quá trình bơm mẫu. Nhiệt độ, sức căng bề mặt sẽ thay đổi lượng dung dịch được hút qua vòi phần tham gia vào trong ngọn lửa, nên độ sáng của ngọn lửa cũng sẽ thay đổi. Điều đó sẽ gây nên sai số cho phép phân tích.
+ Sự có mặt của các ion lạ với lượng lớn trong dung dịch sẽ làm chuyển dịch cân bằng trong các phản ứng phân li và ion hoá dung dịch. ví dụ như cường độ phát xạ của canxi và của kim loại kiềm khác giảm xuống khi có mặt nhôm vì khi đó tạo thành một hợp chất khó phân li (nCaO.Al2O3). Lượng Canxi càng nhiều thì giá trị n càng lớn.
+ Các cation kim loại kiềm có ảnh hưởng đến sự phát xạ của nhau, thường làm táng cường độ phát xạ ảnh hưởng của các cation kim loại kiềm giảm theo thứ tự sau:
+ Ảnh hưởng của anion: Khi có mặt một vài anion trong dung dịch sẽ làm giảm
nồng độ và cường độ phát xạ của nguyên tư kim loại. Anion nitrat NO3- có ảnh hưởng
nhất đến sự phát xạ của kim loại kiềm. Mức độ ảnh hưởng tăng dần theo thứ tự sau:
+ Rất nhiều chất có mặt trong dung dịch thường tự phát xạ với những bước sóng
gần với bước sóng phân tích của nguyên tố xác định. Điều đó làm tăng kết quả xác định.
Ví dụ: Khi xác định nitrat cần chú ý vạch quang phổ của canxi, khi xác định kim

cần chú ý vạch của lối và rubiđi.
3.2.2. Quang kế ngọn lửa
- Cấu tạo: Bất kỳ một quang kế ngọn lửa nào cũng gồm những bộ phận chính sau:
+ Nguồn phát xạ: Gồm máy bơm không khí nối với ống phun mù để chuyển dung dịch sang trạng thu soi khí đưa vào ngọn lửa, bộ phận cấp hơi đốt đưa vào đèn
+ Thiết bị đơn sắc: Thường là các kính lọc phân lập, mỗi nguyên tố có kính lọc màu riêng, thường có các kính lọc của natri, kali, canxi...
+ Máy thu phát xạ: Đo cường độ phát xạ. Đa số các nước thường dùng tế bào quang điện.
+ Điện kế nhạy (đến 10-10A). Đó là kiểu điện kế gương quay
Sử dụng:
Chuẩn bị một thang dung dịch chuẩn đã biết nồng độ, có thành phần tương tự với thành phần dung dịch phân tích.
Mở bình nén không khí và khí đốt axetylen (trước khi đi vào vòi phun và đèn đốt, khí đốt và không khí cần được làm sạch bằng dung dịch H2SO4 đặc). Điều chỉnh áp suất khí đốt và không khí cho thích hợp tức là làm sao cho ngọn lửa cháy tốt (nếu ngọn lửa có màu vàng và có khỏi là thừa khí đốt, điều chỉnh sao cho ngọn lửa đều, không có màu) và dung dịch bị hút vào ổn định, vừa phải.
Quay kính lọc tương ứng.
Mở công tắc, đưa vào dung dịch chuẩn có nồng độ âm nhất. Nếu chỉ số của điện kề chỉ ngoài thang thì phải giảm màng chắn đế đọc được chỉ số trên điện kế. Tiếp tục đo thang chuẩn với nồng độ từ thấp đến cao. Số đọc phải lặp lại hai lần. Mỗi lần thay đổi dung dịch đều phải rửa đèn bằng nước cất và kim điện kế phải chỉ về 0. Vẽ đồ thị chuẩn. Thay dung dịch chuẩn bằng dung dịch phân tích. Đọc từ số trên điện kế.
Dựa vào đồ thị xác định được nồng độ chất cần phân tích.
Đóng vòi khí đốt, đóng tế bào quang điện. Rửa vòi phun bằng nước cất. Thông khí toàn bộ hệ thống.
3.3. Phương pháp quang phổ hấp thụ nguyên tử (AAS)
Như đã biết, vật chất được cấu tạo từ các nguyên tử và nguyên tử là phần tử nhỏ nhất còn giữ được tính chất của nguyên tố. Trong điều kiện bình thường, nguyên tử không thu hay phát ra năng lượng dưới dạng các bức xạ, lúc này nguyên tử tồn tại ở trạng thái cơ bản. Đó là trạng thái bền vững và có mức năng lượng thấp nhất của nguyên tử. Khi nguyên tử tự do ở trạng thái hơi, nếu chiếu một chùm tia sáng có những bước sóng nhất định vào đám hơi, nguyên tử thì các nguyên tử tự do sẽ hấp thụ

các bức xạ có bước sóng nhất định, ứng với những tia bức xạ mà nó có thể phát ra được trong quá trình phát xạ. Lúc đó nguyên tử đã nhận năng lượng dưới dạng các tia bức xạ và nó chuyền lên trạng thái kích thích, có năng lượng cao hơn trạng thái cơ bản. Đó là tính chất đặc trưng của nguyên tử ở trạng thái hơi. Quá trình đó gọi là quá trình hấp thụ năng lượng của nguyên tử Phổ sinh ra trong quá trình này được gọi là phổ hấp thụ nguyên tử. Nghiên cứu sự phụ thuộc của cường độ một vạch phổ hấp thụ của một nguyên tố và nồng độ C trong mẫu phân tích, lý thuyết và thực nghiệm cho thấy rằng. trong một vùng nồng độ C nhỏ, mối quan hệ giữa cường độ vạch phổ hấp thụ và nồng độ của nguyên tố đó trong đám hơi cũng tuân theo định luật Bugơ- Lămbe-bia.
K: Hệ số hấp thụ, phụ thuộc vào chiều dài sóng; C: Nồng độ nguyên tố cần xác
định có trong ngọn lửa; L: Chiều dày của lớp hấp thụ;
Dựa vào giá trị mật độ quang, người ta xác định nồng độ nguyên tử của nguyên
tố cần xác định trong thể tích mẫu. Biểu thức trên chứng tỏ mật độ quang của lớp hấp thụ tỷ lệ thuận với nồng độ của nguyên tử chứa trong đó tại bước sóng hấp thụ ứng với nguyên tố đó. Tính tỷ lệ này được bảo toàn trong một khoảng nồng độ nhất định, tuỳ thuộc vào tính chất của nguyên tố cần xác định và tính chất của đèn. Sự phụ thuộc trên là cơ sở thực tiễn của phương pháp phân tích hấp thụ nguyên tử định lượng.
3.3.1. Nguyên lý và thiết bị của phép đo quang phổ hấp thụ nguyên tử (AAS)
Cơ sở lý thuyết của phép đo AAS là sự hấp thụ năng lượng (bức xạ đơn sắc) của nguyên tử tự do ở trạng thái hơi (khí) khi chiếu chùm tia bức xạ qua đám hơi của nguyên tố ấy trong môi trưởng hấp thụ. Vì vậy, muốn thực hiện được phép đo phổ hấp thụ nguyên tử cần phải có các quá trình sau:
+ Chọn các điều kiện và một loạt thiết bị phù hợp đề chuyền mẫu phân tích từ trạng thái ban đầu (rắn hay dung dịch) thành trạng thái hơi của các nguyên tử tự do. Đó là quá trình nguyên tử hoá mẫu. Những thiết bị để thực hiện quá trình này gọi là hệ thống nguyên tử hoá mẫu.
+ Chiếu chùm tia sáng phát xạ của nguyên tố cần phân tích qua đám hơi nguyên tủ vừa được điều chế ở trên. Các nguyên tử của nguyên tố cần xác định trong đám hơi sẽ hấp thụ những tia bức xạ nhất định và tạo ra phổ hấp thụ của nó. Ở đây, một phần cường độ của chùm sáng đã bị một loại nguyên tử hấp thụ và phụ thuộc vào nồng độ của nguyên tố trong môi trường hấp thụ. Nguồn cung cấp nguồn tia sáng phát xạ của nguyên tố cần xác định gọi là nguồn bức xạ đơn sắc.
+ Dựa trên nguyên tắc của phép đo phổ hấp thụ nguyên tử nên dụng cụ dùng trong phương pháp này gồm những bộ phận chính như sau: Nguồn sáng, bộ hấp thụ,

thiết bị quang, thiết bị thu và ghi.
+ Nguồn sáng: Nguồn sáng chính dùng trong phương pháp phân tích quang phổ
hấp thụ nguyên tử là đèn canh rỗng. Đèn này là bình hay ống hình trụ bằng thuỷ tinh có các điện cực dùng đề đốt nóng. Bình được nạp đầy một khí trơ nào đó có áp suất thấp (agon, neon, heli, xenon...). Để có vùng bức xạ tử ngoại, cửa ra của đèn được chế tạo bằng thạch anh hoặc thuỷ tinh đặc biệt.
Catôt rỗng của đèn được chế tạo bằng một kim loại tinh khiết (hoặc bằng vật liệu khác) dưới dạng ống hình trụ mà mặt phía trong của nó được phủ bằng một chất có chứa nguyên tố cần xác định.
Tính ổn định của đèn khi làm việc là yếu tố rất quan trọng, nó ảnh hưởng đến độ chính xác và độ nhạy cảm của phương pháp phân tích. Tính ổn định đó được xác định bằng tính ổn định của nguồn duy trì khi làm việc, bằng đặc tính cấu trúc và những tỉnh chất đặc thù của đèn. Tính ổn định của đèn khi làm việc đặc biệt có ý nghĩa trong trưởng hợp sử dụng thang rộng.
Ngoài những loại đèn catôt rỗng trong thực tế phân tích người ta còn dùng các đèn không cực cao tần. Đó là các bình bằng thạch anh hay bằng thuỷ tinh có đường kính tử 10 mm đến 20mm, trong đó có đưa vào một kim loại tương ứng (hay hợp chất của nó và một khí trơ-có áp suất vài mmHg) dùng để duy trì sự phóng điện trong đèn. Sự phóng điện trong đèn được sinh ra khi đặt nó vào trong một trường điện tử của máy phát có tần số làm việc 100-2450MHz. Cũng có thể sử dụng những nguồn phù hấp thụ như là đèn hiđro, đèn xenon, đèn đơteri, nhưng các đèn này chưa được sử dụng rộng rãi trong phân tích quang phổ hấp thụ nguyên tử.
+ Bộ hấp thụ: Bộ hấp thụ dùng để chuyền chất phân tích sang trạng thái mà trong đó các chất cần xác định sẽ tồn tại dưới dạng những nguyên tử ty do, có khả năng hấp thụ ánh sáng của nguồn sáng bên ngoài. Có thể dùng các loại đèn khí làm bộ hấp thụ. Các khí thường dùng để đốt đèn ló propan, bu tan, axetylen, hiđro.

Chất oxy hoá khí đốt cháy là ôxy, được sử dụng dưới dạng tinh khiết hoặc dưới dạng hỗn hợp với không khí và một vài loại khí khác.
Đặc tính cơ bản của ngọn lửa là nhiệt độ và thành phần khí của nó, thành phần khỉ phụ thuộc vào dạng khí đốt và chất ôxy hoá. Nhiệt độ và thành phần khí của ngọn lửa xác định độ phân li của các hợp chất đưa vào và được tạo thành trong ngọn lửa. Tuỳ thuộc vào tỷ lệ giữa cacbon và oxy, ngọn lửa sẽ có tính khử hay tính ôxy hoá. Ngọn lửa có tính khử khi tỷ lệ C > 0 có tính ôxy hoá khi C < 0 và là trung hoà khi C= 0.
Đặc tính của dung môi trong mẫu được đưa vào ngọn lửa có thể ảnh hưởng đến nhiệt độ thành phần khí và làm cho ngọn lửa có tính khử hoặc tính ôxy hóa.
Trong ngọn lửa, sự nguyên tử hoá các nguyên tố hoặc các hợp chất của chúng xảy ra trong những điều kiện khác nhau, tuỳ thuộc vào hợp chất của nguyên tố ở dạng bền vững với nhiệt hoặc không bền với nhiệt.
Bộ phận phun và đèn là hai phần rất quan trọng của bộ hấp thụ. Chúng có tác dụng quyết định đến độ nhạy cảm và độ chính xác của phép phân tích. Thiết bị phun được dùng để chuyển dung dịch phân tích thành trạng thải sol khí để đưa vào ngọn lửa. Đèn cũng giữ vai trò đáng kể trong việc xác định tính ổn định và vì vậy đèn cũng xác định cả độ nhạy và độ chính xác của phép phân tích. Đèn thường có một khe liền hoặc có một số dãy các lỗ riêng biệt.
Hiện nay người ta đã nghiên cứu thành công các phương pháp nguyên tử hoá chất phân tích trong bộ phận hấp thụ, đó là phương pháp không dùng ngọn lửa và phương pháp kết hợp lò - ngọn lửa. Nhờ những phương pháp này mà độ nhạy tăng lên rất nhiều khi xác định một loạt các nguyên tố.
Kỹ thuật nguyên tử hoá không ngọn lửa
Kỹ thuật nguyên tử hoá không ngọn lửa ra đời sau kỹ thuật nguyên tử hoá trong ngọn lửa. Nhưng kĩ thuật này phát triển rất nhanh và hiện đang. được ứng dụng rất phổ biến, vì kĩ thuật này cung cấp cho phép đo AAS có độ nhạy rất cào, có khi gấp hàng trăm đến hàng nghìn lần phép đo trong ngọn lửa.
Phép đo không ngọn lửa đòi hòi một lượng mẫu tương đối nhỏ. Thông thường mỗi lần đo chi cần từ 20 đến 50 μl, do đó không cần nhiều mẫu phân tích, việc chuẩn bị mẫu cũng dễ dàng hơn.
Về nguyên tắc kỹ thuật nguyên tử hoá không ngọn lừa là quá trình nguyên tử hoá tức khắc trong thời gian rất ngắn nhờ năng lượng của dòng điện công suất lớn và trong môi trường khí trơ.
Dụng cụ để nguyên tử hoá theo kỹ thuật này gồm các nhóm chính như sau:
- Các loại cuvet graphit;
- Các loại cốc graphit;

- Các loại thuyền làm bằng kim loại chịu nhiệt như tan.
Quá trình nguyên tử hoá không ngọn lửa thường gồm 3 giai đoạn:
+ Sấy khô mẫu: Thường ở nhiệt độ 100- 1500C trong thời gian 20 đến 40 giây với lượng mẫu nhỏ hơn 100 μl - nhiệt độ và thời gian sấy phụ thuộc vào bản chất các chất ở trong mẫu và dung môi hoà tan.
+ Tro hoá: Đốt cháy các chất hữu cơ và nung luyện mẫu ở nhiệt độ thuận lợi cho gia đoạn nguyên tử hoá - nhiệt độ tro hoá phụ thuộc vào bản chất của mỗi nguyên tố và dạng hợp chất mà nguyên tố đó tồn tại - nhiệt độ tro hoá thưởng thấp hơn nhiệt độ tro hoá giới hạn của nguyên tố từ 30-500C; thời gian tro hoá thường từ 20-60 giây với lượng mẫu đưa vào cuvet nhỏ hơn 100 μl, trong đó một phần ba thời gian dùng để nâng nhiệt độ từ nhiệt độ sấy đến nhiệt độ tro hoá đã đặt; họ phần ba thời gian dùng để luyện mẫu ở nhiệt độ tro hoá đã chọn.
+ Nguyên tử hoá: Là giai đoạn quyết định cường độ của vạch phờ, thời gian rất ngắn, từ 3 đến 6 giây, đôi khi có thể đến 1 OÀ giây Tốc độ tăng nhiệt độ rất lớn (1 500- 20000C/1 giây) để đạt ngay tức khắc đến nhiệt độ nguyên tử hoá
Nhiệt độ nguyên tử hoá của mỗi nguyên tố phụ thuộc vào bản chất của nguyên tố, trạng thái và thành phần của mẫu.
+ Thiết bị quang học: Thiết bị này bao gồm, dụng cụ quang học (máy đơn sắc hay kính lọc) dùng để tách các vạch phân tích của nguồn, các thấu kinh, các màng chắn và các gương phụ đê đưa các chùm sáng từ nguồn qua bộ phận hấp thụ.
+ Thiết bị thu và ghi: Thiết bị này gồm bộ ghi ánh sáng bao gồm bộ nhân quang và các thiết bị điện để nuôi bộ khuếch đại dòng quang điện. Bộ ghi có thể là thiết bị đọc biểu kiến, thiết bị tự ghi hoặc thiết bị hiện số cùng các sơ đồ điện tương ứng của nguồn nuôi hay là thiết bị để in.
3.3.2. Những ưu điểm và nhược điểm của phép đo AAS
Cũng như các phương pháp phân tích khác, phương pháp phân tích phổ hấp thụ nguyên tử cũng có những ưu điểm và nhược điểm nhất định
Ưu điểm:
+ Phép đo phổ hấp thụ nguyên tử có độ nhạy và độ chọn lọc cao. Gần 60 nguyên tố hoá học có thể được xác định bằng phương pháp này với độ nhạy từ 1.10-4 đến 1.10-5%. Đặc biệt nếu sử dụng kĩ thuật nguyên tử hoá không ngọn lửa thì có thể đạt đến độ nhạy n.10-7%.
Chính vì có độ nhạy cao nên phương pháp phân tích này đã được sử dụng rộng rãi trong nhiều lĩnh vực để xác định lượng vết các kim loại. Đặc biệt là trong phân tích các nguyên tố vi lượng. Cũng do độ nhạy cao nên trong nhiều trường hợp không phụ làm giàu trước nguyên tố cần xác định.

+ Có thể xác định đồng thời hay liên tiếp nhiều nguyên tố trong mẫu. Các kết quả phân tích rất ổn định, sai số nhỏ. Trong trường hợp sai số không quá 15% với nồng độ ở mức ppm.
Nhược điểm:
+ Phải có hệ thống máy tương đối đắt tiền
+ Vì có độ nhạy cao nên sự nhiễm bẩn có ảnh hưởng lớn đến kết quả phân tích hàm lượng vết.
+ Phương pháp chỉ cho ta biết thành phần nguyên tố của chất ở trong mẫu phân tích mà không chỉ ra trạng thái liên kết của nguyên tố ở trong mẫu. Vì thế đây chỉ là phương pháp phân tích thành phần nguyên tố.
3.3.3. Đối tượng và phạm vi ứng dụng của phương pháp
Đối tượng chính của phương pháp là phân tích lượng nhỏ và lượng vết các nguyên tố kim loại của các chất vô cơ và hữu cơ trong các loại mẫu khác nhau như: quặng, đất, đá, nước, các sản phẩm nông nghiệp, phân bón...
Ngoài các kim loại một số phi kim như Si, P, As,Te... cũng có thể xác định chính xác bằng phương pháp này.
Trong khoảng hai chục năm trở lại đây phép đo phổ hấp phụ nguyên tử đã phát triển rất nhanh. Nó được sử dụng như là một công cụ phân tích đắc lực cho nhiều ngành khoa học và kinh tế. Hiện nay nhiều máy đo phổ hấp thụ nguyên tử đã được sản xuất với nhiều tính năng ưu việt. Vì vậy phép đo phổ hấp thụ nguyên tử là một trong những phép đo ưu việt trong hệ thống các phương pháp phân tích hiện nay.

Chương 4
PHƯƠNG PHÁP ĐIỆN HOÁ
4.1. Cực chọn lọc ion
Phương pháp phân tích điện thế là một trong những phương pháp phân tích ra đời sớm nhất. Nguyên tắc của phương pháp là đo thế cân bằng của cực nghiên cứu để xác định nồng độ cân bằng của chất phân tích hoặc theo dõi sự biến thiên nồng độ của nó khi nó tham gia vào phản ứng hóa học.
Khi nhúng cực làm bằng Pt vào dung dịch chứa chất ôxy hoá - khử liên hợp sẽ có hiện tượng trao đổi electron giữa cực và chất trong dung dịch. Sự trao đổi này sẽ gây nên phản ứng điện hoá.
Nếu như tốc độ của quá trình khử nhanh hơn quá trình ôxy hoá thì chất ôxy hoá nhận electron nhiêu hơn, Pt sẽ nhường electron cho chất ôxy hoá, lúc đó bề mặt Pt sẽ mang điện tích dương, nó sẽ hút các ion mang điện tích âm vào tạo nên lớp điện kép. Giữa hai mặt của lớp điện kép sẽ xuất hiện một thế E và thế E đó sẽ được đo trong phương pháp phân tích này.
Khi tốc độ của quá trình ôxy hoá bằng tốc độ của quá trình khử, lúc đó lượng chất ôxy hoá và lượng chất khử không thay đổi nữa. Thế E đạt tới trạng thái cân bằng. Thế cân bằng có liên quan đến nồng độ hay chính xác hơn là liên quan đến hoạt động của chất ôxy hoá và chất khử theo phương trình Nerst:
Trên thực tế không xác định được giá trị thế cân bằng của một cực riêng biệt mà
chỉ xác định thế của nó so với một cực dùng làm cực chuẩn. Trong phân tích điện thế cực này được gọi là cực so sánh (thường là các cực loại 2 như cực calomen bão hoà hoặc cực lạc clorưa). Cực cần đo thế cân bằng gọi là cực đo hoặc cực chỉ thị
Trong số các loại cực chỉ thị dùng trong phân tích điện hoá có một loại cực rất đặc biệt được chế tạo từ những loại màng đặc biệt, thế của cực phụ thuộc một cách chọn lọc vào hoạt độ chất cần xác định. Các cực này được gọi là cực chọn lọc ion.
Những công trình nghiên cứu của Nerst và của Reisenfeld về tính chất cơ bản của bề mặt màng và thế xuất hiện trên ranh giới giữa màng và dung địch đã giúp hai nhà bác học này đưa ra khái niệm về ranh giới pha của hai chất lỏng không trộn lẫn với nhau. Điện hoá học về màng được chính thức nghiên cứu từ năm 1890, trong các công trình của Ostwald- người ta đã phát hiện ra màng bán thấm là loại màng cho chuyển qua nó những loại lớn xác định.
Ngay từ thời gian đầu người ta đã nghĩ ra hai loại màng: màng đặc (gồm màng

mỏng, màng rắn) và màng lỗ xốp. Trong số các loại mảng đặc, người ta đã chế tạo được các lo màng thuỷ tinh rất mỏng và tù đó đã phát sinh ra cực màng thuỷ tinh là loại cực có thể phụ thuộc chọn lọc với hoạt độ của ion hiđro trong các dung dịch. Nhà khoa học người Nga là Nikolxki đã xây dụng thuyết trao đổi lớn của cực màng thuỷ tinh. Eisenman và các cộng sự đã xây dụng học thuyết về thế khuếch tán ở trong màng thuỷ tinh.
Tiếp theo nhà khoa học người Hugari là Pưngor và các cộng sự đã nghiên cứu thành công các loại màng rắn chọn lọc ion để chế tạo các cực. Các màng đầu tiên là các màng dị thể chứa kết tủa bạc halogenua. Sau đó người ta đã nghiên cứu và chế tạo các mạng chọn lọc ton đơn tinh thể Frant và Ross đã chế tạo được cực chọn lọc với ion florua trên cơ sở dùng đơn tinh thể lan tan florua.
Trong những năm gần đây, các nguyên lý sinh học đã được ứng dụng đê chế tạo các cực enzim trong đó màng của cực được phủ một lớp chất mang polyme chứa enzim. Chất cần xác định sẽ phản ứng trong lớp enzim và tạo nên sản phẩm gây ra tín hiệu có thể đo được
4.1.1. Lý thuyết về các thế màng của các cực chọn lọc ion
Các khái niệm cơ sở
Một cực chọn lọc lớn gồm thân cực, bên trong chứa dung dịch chất điện li có thành phần và nồng độ xác định (dung dịch 1), cuối cực là một mạng chọn lọc ion (có thể là màng rắn đồng thê hoặc dị thể, hoặc đơn tinh thê, màng thuỷ tinh hoặc màng lỏng) và bên trong thân cực có một cực so sánh nhúng vào dung dịch 1.
Thế điện động của nguyên tố điện hoá trên là:

Các cực so sánh 1 và 2 thường là các cực loại 2 có thể xác định và không đổi, do đó khi dùng cực Cll trong phân tích chỉ có thể màng ∆φM là có ý nghĩa quyết đinh Tổng số thế của cực bên trong và thế màng gọi là thế của cực Cl1, ECl1
Trong trường hợp đơn giản nhất, khi màng của cực Cll ngăn cách 2 dung dịch
trong và dung dịch nghiên cứu chỉ chứa ion cần xác định j và trong cả 2 dung dịch đó hoàn toàn không có ion khác ảnh hưởng tới thế màng được quyết định bởi ion j, vì thế màng được xác định theo hệ thức:
Trong đó: z là điện tích của ion j; aj (l) và aj (2) là hoạt độ tương ứng của j trong
các dung dịch 1, 2.
Trong trường hợp trong dung dịch phân tích có ion K là ion ngăn cản sự xác định hoạt độ của ion j, thì thế của Cll được xác định theo phương trình Nikolxki. Trong phương trình này có sự phụ thuộc của thế vào hệ số chọn lọc của cực lớn K đối với ion cần xác định j. Hệ số chọn lọc thường là hàm số của thành phần dung dịch nghiên cứu và giúp ta định hướng được ảnh hưởng của các ion cản trở tới sự xác định ion cần phân tích.
Bước nhảy thế giữa cực và môi trường trong trường hợp đơn giản tuân theo phương trình:
E0: thế điện cực trong môi trường với hoạt độ của các ion a = 1, nghĩa là điện cực
chuẩn;
R: Hằng số khí;
T: Nhiệt độ tuyệt đối của môi trường (0K);
F: Số Farađay;
n: Sự thay đổi điện tích của ion cần xác định do kết quả của phản ứng điện hoá;
ax: Hoạt độ của ion X trong môi trường nghiên cứu.
Ở 200C, phương trình có dạng:
Thế của cực phụ thuộc tuyến tính vào px cho phép chúng ta sử dụng cực đó trong
thực tiễn phân tích.

4.1.2. Một vài loại điện cực chỉ thị thông thường
Điện cực màng kali: Điện cực màng kali để xác định hoạt độ của ion K+, được cấu tạo từ một ống thuỷ tinh hoặc ống nhựa, phần cuối của ống là màng mỏng chọn lọc ion. Bên trong ống được đổ đầy dung dịch KCl 0,1 M, điện cực so sánh bạc clorua được những trong dung dịch này.
Điện cực màng kali nhạy với ion kém trong phạm vi nồng độ xác định với điện cực EM.K-01 của Liên Xô (cũ) giới hạn này là từ 10-1 đến l.10-4. Khi có mặt Na+ với tỉ lệ K+:Na+ là 1:20 và NH4+ với tỉ lệ K+:NH4+ là 1:200 thì các ion Na+ và NH4+ sẽ cản trở phép xác định K+ bằng điện cực EM.K-01.
Trước khi bắt đầu làm việc, cần rửa khoang bên trong của điện cực hai lần bằng nước cất tại nhiệt độ phòng và hai lần bằng dung dịch KCl 0,1M. Sau đó rót vào điện cực 1,5-2,5 ml KCl 0,1 M rồi nhúng điện cực so sánh bên trong vào.
Để điện cực vào dung dịch KCl 0,1 M ít nhất là 1 ngày 1 đêm và sau đó vẫn giữ trong dung dịch này. Dùng dung dịch KCl chuẩn để kiểm tra điện cực sau khi đã chuẩn bị xong.
Dung dịch KCl, mol/1 γ± aK+ pK
+
0,2000 0,1000 0,0500 0,0100 0,0050 0,0010 0,0005 0,0001
0,718 0,770 0,816 0,902 0,927 0,965
0,436 0,O770 0,O408 0,0090 0,0O46 0,0010 0,0005 0,000 1
0,84 1,11 1,39 2,O4 2,34 3,02 3,30 4,00
Để kiểm tra điện cực cần thiết lập một mạng đo, điện cực K được nhúng vào trong dung dịch KCl chuẩn, điện cực so sánh được nhúng trong dung dịch KCl bão hoà. Giữa hai dung dịch này được nối bằng cầu dẫn làm đầy bằng nga đã trộn với lãi axetat (CH3COOLi). Đo trị số sau khi nhúng điện cực vào dung dịch sau 5 phút.
Nếu điện cực hoạt động tốt, tiến hành đo thế của dung dịch thí nghiệm. Dựa vào thang chuẩn giữa thế và pK+ tính ra được hoạt độ của K+ trong dung dịch nghiên cứu.

* Điện cực màng nitrat:
Điện cực màng nitrat EM - NO3-01 do Liên Xô (cũ) chế tạo có cấu tạo tương tự như điện cực kali, chỉ khác là màng được bão hoà bằng ion NO3
- Khoang bên trong của điện cực chứa dung dịch KNO3 0,1 M và dung dịch KCl 0,005M, sau đó nhúng điện cực so sánh bên trong vào. Điện cực so sánh bên ngoài thường dùng là điện cực calomen (thường dùng là điện cực bạc clorua).
Thế của điện cực màng nitrat trong giá trị pa từ 2 - 9 và hoạt độ NO3- từ 0,001-
1M tuân theo phương trình:
E = E0 - 0,058.PNO3-
Các anion khác như Cl- HCO3-, SO4
2- với tỷ lệ là NO3-: Cl- = 1: 1; NO3
-: HCO3-
= 1: 5; NO3-: SO4
2-: 1:5 sẽ cản trở việc xác định SO42-.
Trước khi dùng điện cực nitrat mới, cần phải rửa khoang bên trong cực hai lần bằng nước cất và hai lần bằng dung dịch KNO3 và KCl với nồng độ như trên, sau đó rót vào 1,5 - 2,5 ml dung dịch này rồi nhúng điện cực so sánh bên trong vào. Điện cực sau khi chuẩn bị xong, được giữ qua một ngày đêm trong dung dịch KNO3 0,1 M và sau này vẫn giữ trong dung dịch đó
Sau khi chuẩn bị điện cực xong, tiến hành kiểm tra sự phụ thuộc thế của cực vào giá trị PNO3
-. Thiết lập mạch đo gồm điện cực so sánh và điện cực nitrat đều nhúng trong dung dịch NaNO3 chuẩn. Sau 5 phút, ghi số trên máy đo.
Nồng độ NaNO3(mol/1) γ± aNO3- p NO3
-
0,1000 0,0500 0,0100 0,0050 0,0010 0,0005 0, 0001
0,765 0,812 0,900 0,926 0,964 0,975 1,000
0,O765 0,O406 0,0090 0,0064 0,0010 0,0005 0,0001
1,11 1,30 2,O4 2,34 3,02 3,30 4,00

Xây dụng đồ thị theo kết quả đo thế trong 3 - 4 dung dịch chuẩn. Khi thay đổi giá trị pNO3
- theo đơn vị độ dốc của đồ thị là 58mV.
Các điện cực nhạy với khí
Điện cực nhạy với khí đầu tiên được xem như là bộ thu để xác định thế đã được sử dụng là điện cực dùng đề xác định CO2 trong không khí. Hiện nay đã có các điện cực nhạy với khí để xác định NH3, SO2, H2... và các chất khí khác. Nguyên tắc làm việc của các điện cực này dựa trên sự chỉ thị của ion được tạo thành do phân tử khí phản ứng với nước.
Thế của điện cực nhạy với khí được xác định bằng phương trình:
Do vậy cũng có khả năng xác định hàm lượng khí trong mẫu thông qua việc xác
định thế.
Khó khăn cơ bản đối với các điện cực nhạy với khí là việc xây dụng những điều kiện đề thiết lập nhanh cân bằng giữa chất khí và chất lỏng, và việc chọn điện cực bên trong để khí khuếch tán và tham gia thiết lập cân bằng tạo nên những ion được xác định bằng điện cực chọn lọc ton.
Cấu tạo của các điện cực nhạy với khí:
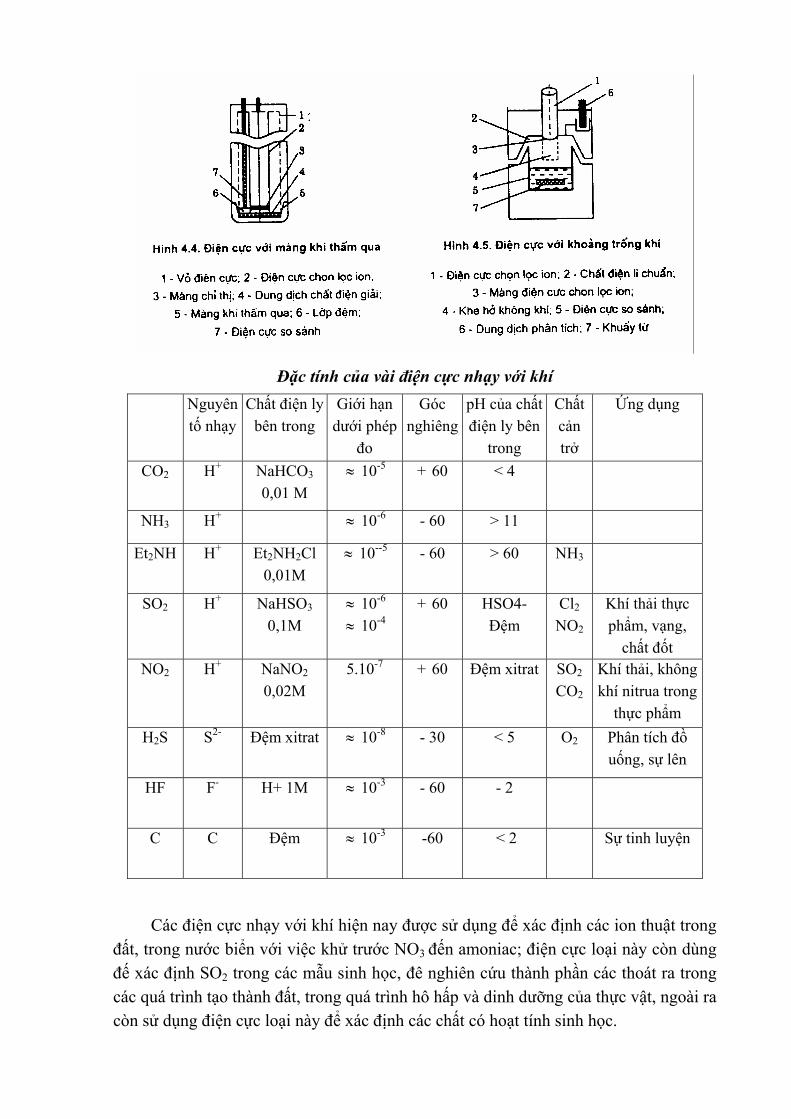
Đặc tính của vài điện cực nhạy với khí
Nguyên tố nhạy
Chất điện ly bên trong
Giới hạn dưới phép
đo
Góc nghiêng
pH của chất điện ly bên
trong
Chất cản trở
Ứng dụng
CO2 H+ NaHCO3 0,01 M
≈ 10-5 + 60 < 4
NH3 H+ ≈ 10-6 - 60 > 11
Et2NH H+ Et2NH2Cl 0,01M
≈ 10--5 - 60 > 60 NH3
SO2 H+ NaHSO3 0,1M
≈ 10-6
≈ 10-4 + 60 HSO4-
Đệm Cl2 NO2
Khí thải thực phẩm, vạng,
chất đốt NO2 H+ NaNO2
0,02M 5.10-7 + 60 Đệm xitrat SO2
CO2 Khí thải, không khí nitrua trong
thực phẩm H2S S2- Đệm xitrat ≈ 10-8 - 30 < 5 O2 Phân tích đồ
uống, sự lên
HF F- H+ 1M ≈ 10-3 - 60 - 2
C C Đệm ≈ 10-3 -60 < 2 Sự tinh luyện
Các điện cực nhạy với khí hiện nay được sử dụng để xác định các ion thuật trong đất, trong nước biển với việc khử trước NO3 đến amoniac; điện cực loại này còn dùng đế xác định SO2 trong các mẫu sinh học, đê nghiên cứu thành phần các thoát ra trong các quá trình tạo thành đất, trong quá trình hô hấp và dinh dưỡng của thực vật, ngoài ra còn sử dụng điện cực loại này để xác định các chất có hoạt tính sinh học.

Các điện cực so sánh
Điện cực so sánh hiện nay thường được sử dụng cùng với các điện cực chọn lọc ton là điện cực bạc clorua. Đây là điện cực thuận tiện, đơn giản, bền và không đòi hỏi phải bảo quản ngoài việc làm đầy bên trong cực bằng dung dịch liệt. Điện cực bạc clorua có vỏ làm bằng thuỷ tinh, cuối cực có sợi amiăng để tiếp xúc giữa điện cực với môi trưởng bên ngoài. Bên trong ống có dung dịch kali clorua, dây dẫn bạc có bề mặt xốp đã được phủ bằng kết tủa bạc clorua.
Thế của điện cực được xác định theo nồng độ dung dịch KCl. Khi nồng độ KCl không thay đổi thì giá trị thế của điện cực cũng không thay đổi
Khi phân tích mẫu ngoài thực địa nên dùng dung dịch KCl bão hoà.
Thế của điện cực bạc clorua:
Dung dịch KCl E(V) ở 250C
Dung dịch KCl bão hoà + 0,202(200C)
KCl 1,0 M + 0,238
KCl 0,1M + 0,290
HCl 0,1N + 0,289
HCl 1,0 M + 0,218
Cầu dẫn điện
Cầu dẫn điện dùng để trực lập mạng điện với các cực chọn lọc ion là một ống thủy tinh hình chữ U, đường kính bên trong 3-5 mm; dài khoảng 5 - 6 cm; được nạp đầy aga trong dung dịch một muối bão hoà (KCl, KNO3, CH3COOLi... khi dùng cực chọn lọc ion kali, nạp vào trong cầu do 1 điện dung dịch liti axetat, còn khi dùng cực bạc clorua người ta nạp dung dịch kali nitrat.
Để chuẩn bị cầu dẫn điện lấy 3g aga đã nghiền nhỏ cho vào cốc, thêm vào 100 ml nước cất giữ qua đêm. Sau đó đặt lên bếp, đun cẩn thận và thêm từ từ 10 g muối, khuấy đều bằng thìa thủy tinh đến khi tan hoàn toàn.
Đun hỗn hợp trên ở nhiệt độ 50-600C rồi rót cẩn thận vào ống chữ U; giữ cho đến khi hỗn hợp nguội đi. Cần chú ý rằng trong quá trình cho hỗn hợp vào và giữ hỗn hợp nguội không để tạo bọt khí trong ống.
Giữ cầu dẫn điện trong dung dịch muối bão hoà (muối nạp vào trong ống). Trước khi dùng, dùng nước cất rửa qua cầu dẫn điện, dùng giấy lọc lau khô. Một đầu cầu được để trong dung dịch nghiên cứu còn đầu kia nhúng vào trong dung dịch KCl bão hoà có cắm điện cựu so sánh.
Sử dụng các điện cực chọn lọc ion trong điều kiện ngoài thực địa
Các điện cực chỉ thị làm bằng thuỷ tinh và các điện cực chọn lọc bằng màng có

thể dùng để xác định hoạt độ của các ion trong các điều kiện ngoài thực địa, nghĩa là xác định trực tiếp trong đất nếu độ ẩm của đất không nhỏ hơn 15% trong nước thiên nhiên, nước thải, nước công nghiệp, nước tưới. Khi làm việc ngoài thực địa, nguồn điện cho máy là phi.
Trước khi ra thực địa cần kiểm tra các điện cực chỉ thị theo các dung dịch chuẩn. Tất cả cả các điện cực chỉ thị, trừ điện cực chọn lọc ion bạc clorua, đều phải nhúng trong dung dịch bởi vì nếu màng bị khô, thậm chí chỉ trong thời gian ngắn cũng có tác hại đến cực, làm rối loạn chức năng của cực.
Cần xây dụng đồ thị chuẩn trước tại nhiệt độ tiến hành phép đo ở ngoài địa thực.
Phân tích nước trong điều kiện thực địa, tiến hành tương tự như phân tích dung dịch trong phòng thí nghiệm.
Khi đo hoạt độ của các ion trong đất, điện cực chỉ thị làm bằng thủy tinh và điện cực so sánh được cắm vào đất với khoảng cách chính xác 5 - 6cm. Khi dùng điện cực màng cần đào lỗ trước với độ sâu bằng độ sâu của phép đo và đường kính lỗ tương ứng với đường kính của cực. Sau khi nhúng các điện cực vào trong đất cần ấn nhẹ để làm chặt xung quanh cực. Sau 15 - 30 phút, tuỳ thuộc vào độ ẩm của đất, tiến hành đo hoạt độ lớn cần xác định
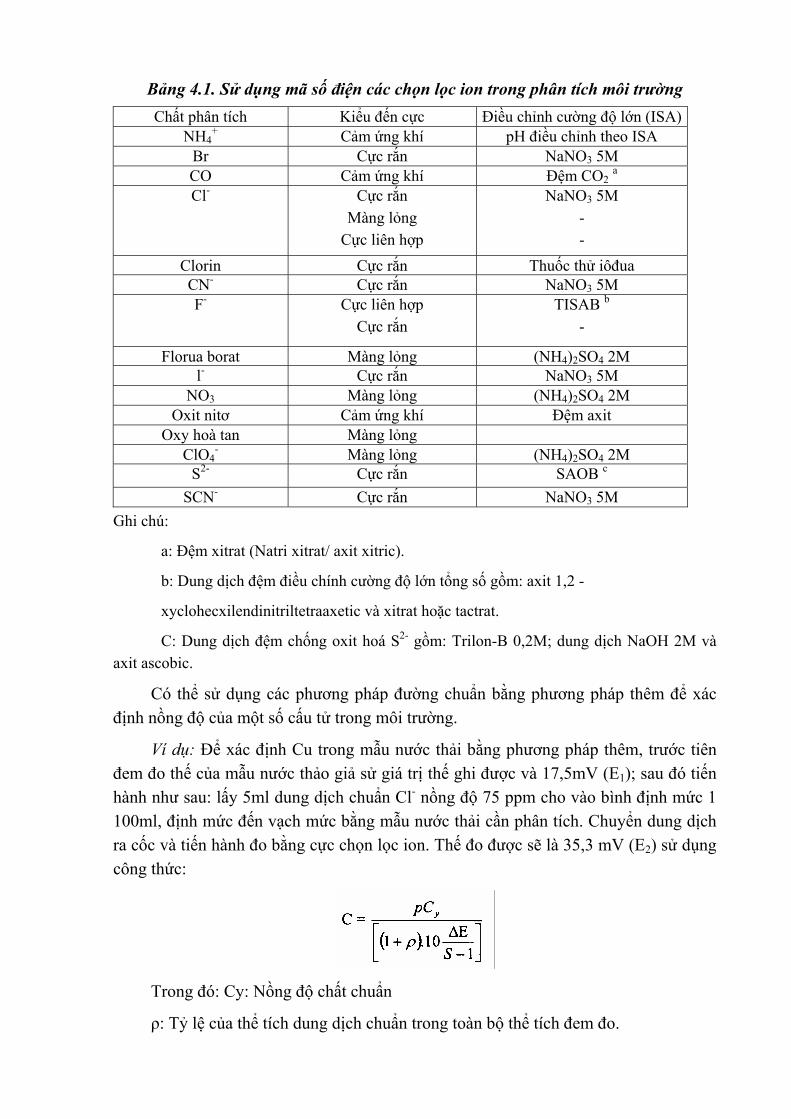
Bảng 4.1. Sử dụng mã số điện các chọn lọc ion trong phân tích môi trường Chất phân tích Kiểu đến cực Điều chỉnh cường độ lớn (ISA)
NH4+ Cảm ứng khí pH điều chỉnh theo ISA
Br Cực rắn NaNO3 5M CO Cảm ứng khí Đệm CO2 a Cl- Cực rắn
Màng lỏng Cực liên hợp
NaNO3 5M - -
Clorin Cực rắn Thuốc thử iôđua CN- Cực rắn NaNO3 5M F- Cực liên hợp
Cực rắn TISAB b
-
Florua borat Màng lỏng (NH4)2SO4 2M l- Cực rắn NaNO3 5M
NO3 Màng lỏng (NH4)2SO4 2M Oxit nitơ Cảm ứng khí Đệm axit
Oxy hoà tan Màng lỏng ClO4
- Màng lỏng (NH4)2SO4 2M S2- Cực rắn SAOB c
SCN- Cực rắn NaNO3 5M Ghi chú:
a: Đệm xitrat (Natri xitrat/ axit xitric).
b: Dung dịch đệm điều chính cường độ lớn tổng số gồm: axit 1,2 -
xyclohecxilendinitriltetraaxetic và xitrat hoặc tactrat.
C: Dung dịch đệm chống oxit hoá S2- gồm: Trilon-B 0,2M; dung dịch NaOH 2M và axit ascobic.
Có thể sử dụng các phương pháp đường chuẩn bằng phương pháp thêm để xác định nồng độ của một số cấu tử trong môi trường.
Ví dụ: Để xác định Cu trong mẫu nước thải bằng phương pháp thêm, trước tiên đem đo thế của mẫu nước thảo giả sử giá trị thế ghi được và 17,5mV (E1); sau đó tiến hành như sau: lấy 5ml dung dịch chuẩn Cl- nồng độ 75 ppm cho vào bình định mức 1 100ml, định mức đến vạch mức bằng mẫu nước thải cần phân tích. Chuyển dung dịch ra cốc và tiến hành đo bằng cực chọn lọc ion. Thế đo được sẽ là 35,3 mV (E2) sử dụng công thức:
Trong đó: Cy: Nồng độ chất chuẩn
ρ: Tỷ lệ của thể tích dung dịch chuẩn trong toàn bộ thể tích đem đo.

S: Độ dốc của điện cực.
∆E = E2 - E1
Áp dụng công thức cho ví dụ trên Cy = 75 ppm; ρ = 5ml/100ml = 0,05
∆E = 35,3 - 17,5 = 17,8 mV; S = 58,1 mV.
Do đó nồng độ của Cl- trong mẫu phân tích là 3,34 ppm.
4.2. Phương pháp cực phổ
Phương pháp là phương pháp phân tích do nhà bác học Tiệp Khắc (cũ) Iaroslap Hayropski phát minh năm 1922. Phương pháp này đã được sử dụng rộng rãi để định lượng các chất vô cơ và hữu cơ trong các đối tượng khác nhau. Nhờ sự hoàn thiện của kỹ thuật cực phổ nên ngày nay đã có hàng loạt các phương pháp cực phổ khác nhau: cực phô một chiều dòng khuếch tán, cực phổ xoay chiều, cực phổ hỗn hống, cực phổ xung vi phân...
4.2.1. Cực phổ một chiều dòng khuếch tán (cực phổ cổ điển)
Nguyên lý:
Cơ sở của phương pháp là nghiên cứu đường biểu diễn sự phụ thuộc của cường độ dòng điện vào thế của bình điện phân có cấu tạo đặc biệt chứa ton can khảo sát
I = f(E)
Đường biểu diễn sự phụ thuộc đó được gọi là đường Von-Ampe hay còn gọi là đường cực phổ hoặc sóng cực phổ.
Để có đường Von-Ampe ta dùng máy cực phổ. Sơ đồ đơn giản của máy như sau:
Catôt hay dùng là điện cực giọt thuỷ ngân là điện cực chỉ thị còn nuốt là đáy thuỷ
ngân hay điện cực calomen bão hoà làm điện cực so sánh.
Thế E đặt vào sẽ tạo nên phân cực catôt (φk) và anôt (φA).
Cân bằng tổng quát của thế bên ngoài đặt vào có thể biểu thị như sau:

Ở đây iR là sự giảm thế khi dòng đi qua dung dịch. Các thành phần (pH và iR
trong cực phổ có khuynh hướng tiến tới cực tiều do bề mặt lớn của điện cực so sánh và trong dung dịch so sánh có chất điện ly trơ (nền), làm giảm điện trở R của dung dịch. Do đó thế bên ngoài đặt vào chi gây nên sự phân cực của điện cực giọt thuỷ ngân. Nếu dùng điện cực giọt thuỷ ngân làm điện cực chỉ thị và điện cực so sánh là cực Calomen bão hoà (thế của cực này trong cực phổ được quy ước bằng không) thì:
Sự phụ thuộc giữa thế của điện cực giọt thuỷ ngân và cường độ dòng điện biểu
thị bằng đồ thị dưới dạng đường cong cực phổ (cực phổ đồ). Dạng tổng quát của cực phổ đầu được biểu diễn ở hình 4.8.
Khi đạt được thế khử (hoặc ôxy hoá) của ion nào đó có trong dung dịch, quá trình tích phân được bắt đầu, cường độ dòng điện tăng lên rõ rệt. Sự tăng lên của cường độ dòng chỉ khi trong dung dịch còn lớp khuếch tán sát điện cực có chứa ion gây nên dòng điện phân khi nồng độ lớn bằng không thì quá trình tăng cường độ dòng sẽ ngừng lại. Khi đó bắt đầu hiện tượng phân cực nồng độ. Khi không có các ion được khử hay được oxy hoá ở lớp sát điện cực, cường độ dòng lúc này được gọi là dòng giới hạn khuếch tán, nó có giá trị không đôi do có sự khuếch tán chất từ dung dịch đến lớp sát điện cực.
Cường độ dòng khuếch tán giới hạn tỷ lệ với tốc độ khuếch tán chất đến điện cực, tức là tỷ lệ với nồng độ của chất có trong dung dịch.
id = K.C
Trong đó K là hệ số tỷ lệ
Phương trình trên chính là phương trình dòng khuếch tán giới hạn.
D.Incovic đã đưa ra phương trinh chi tiết:
Trong đó: n: số electron tham gia phản ứng;
D: hệ số khuếch tán;
m: khối lượng thủy ngân thoát ra từ mao quản trong một đơn vị thời gian,
τ: chu kỳ giọt thuỷ ngân;
C: nồng độ của chất.
Giá trị hệ số tỷ lệ K phụ thuộc vào đặc tính của điện cực sử dụng, đặc tính của ion được khử hoặc ôxy hoá, đặc tính của chất điện li trơ và nhiệt độ của dung dịch cực phổ. Trong trường hợp khử cation, giá trị dòng khuếch tán giới hạn có liên quan đến

nồng độ của nguyên tố phân tích chỉ là một phần của dòng tổng số ít.
Trong đó:
ik: dòng tụ điện;
ig: dòng điện phân yếu do các tạp chất trong dung dịch gây nên;
ih: dòng di chuyển;
Dòng di chuyển xuất hiện là do sự di chuyển của các ion dưới tác dụng của lực điện trường, để loại trừ cần thêm vào trong dung dịch phân tích chất điện li trơ.
Dòng điện phân yếu được loại trừ bằng cách tinh chế cẩn thận chất điện li trơ.
Dòng tụ điện sinh ra do lớp điện kép của dòng khuếch tán ở sát điện cực tích điện, dòng này nói chung cũng khó loại trừ. Dòng tụ điện và dòng điện phân cực gộp lại thành dòng dư.
Dòng dư trong nhiều trường hợp lớn đến mức không thể đo được sóng cực phổ, đặc biệt khi xác định các kim loại nặng trong đất, trong nước và trong thực vật đòi hỏi tiến hành ở độ nhạy cao.
Để làm giảm dòng dư một cách đáng kể, hiện nay trong tất cả các máy cực phổ đều có thiết bị bổ chính. Sử dụng thiết bị bổ chính cho phép nhận được dạng sóng cực phổ tốt hơn rất nhiều và như thế sẽ nâng cao hơn độ nhạy và độ lặp lại của kết quả.
Khi sử dụng cột thuỷ ngân sẽ thu được những kết quả tốt vì khi đó sự tăng của dòng bổ chính nhỏ hơn nhiều so với sự tăng của dòng khuếch tán.
Nguyên nhân làm tăng dòng khuếch tán là do điện cực chuyên động tiếp tuyến của giọt thuỷ ngân, chuyển động này gây nên việc khuấy trộn lớp dung dịch ở cạnh giọt, trên đường cong cực phô có xuất hiện cực đại. Để loại trừ cực đại người ta thêm vào dung dịch những chất hoạt động bề mặt như aga- aga, gelatin... khi đó chuyển động tiếp tuyến của thuỷ ngân sẽ giảm đi.
Sóng cực phổ của ion xác định thường bị sai lệch do có xuất hiện sóng của ôxy, ôxy chứa trong dung dịch của ion xác định được trên điện cực giọt thuỷ ngân.
Để loại oxy trước khi phân tích cực phô người ta cho khí H2 hay N2 sục qua dung
dịch. Trong trường hợp những dung dịch có tính chất axit, đề loại ôxy có thể dùng CO2. Còn trong môi trường kiềm thường natri sunfit, thêm 0,1 g Na2SO3 trong 100ml

dung dịch có thể khử hoàn toàn được ôxy trong thời gian khoảng 5 phút.
Phân tích cực phổ định tính và định lượng
Khi xác định định tính ion được khử (hoặc ôxy hoá) người ta dựa vào giá trị thế bán sóng E1/2 Giá trị thế bán sóng bằng giá trị thế của điện cực chỉ thị khi cường độ dòng bằng 1/2 giá trị của dòng giới hạn.
Giá trị thế bán sóng không phụ thuộc vào nồng độ của chất phản ứng, không phụ thuộc vào vận tốc chảy của giọt thuỷ ngân qua mao quản và các yếu tố khác. Đây là một hằng số đặc trưng cho ion trong một nền nhất định. Vì thế việc lựa chọn chất điện ly trơ (chất nền) có ý nghĩa to lớn trong phân tích cực phố. Hiện nay, người ta dùng các chất điện ly có thành phần khác nhau (các axit, các muối trung tính, các dung dịch kiềm, các chất tạo phức khác nhau) làm chất nền. Đối với đa số chất nền, người ta đã thành lập được các nguyên tố có khả năng được khử hoặc được ôxy hoá và các giá trị thế bán sóng. Các bản này có ghi trong các sách chuyên khảo hoặc các sổ tay.
Khi phân tích một nguyên tố hay một vài nguyên tố phải chọn chất điện ly trơ (chất nền) để loại trừ ảnh hưởng của các ion kèm theo.
Giá trị của dòng giới hạn hay chiều cao của sóng cực phổ trong những điều kiện nhất định tỷ lệ với nồng độ lớn cần khảo sát. Để xác định chiều cao của sóng, người ta thường dùng phương pháp của Hon.
Theo phương pháp này người ta kẻ 3 đường thẳng ở trên sóng, từ các giao điểm lại kẻ các đường song song với trục hoành.
Khoảng cách giữa các đường thẳng này là chiều cao sóng cực phổ. Từ điểm giữa của chiều cao sóng cực phổ kẻ đường thẳng song song với trục hoành. Từ giao điểm của đường thẳng này với sóng cực phổ, kẻ đường thẳng song song với trục tung. Đường thẳng này cắt trục hoành tại điểm là giá trị thế bán sóng. Giá trị thế bán sóng là giá trị tính đối với điện cực calome bão hoà.

Để phân tích cực phổ, người ta thường lấy những lượng thể tích nhỏ (3 -5ml) để rút ngắn được thời gian giải phóng oxy khỏi dung dịch.
Trong phân tích định lượng, để xác định nồng độ người ta sử dụng các phương pháp: phương pháp dung dịch chuẩn, phương pháp thêm và phương pháp đường chuẩn. Thuận lợi nhất khi phân tích hàng loạt mẫu là phương pháp đường chuẩn.
Đồ thị đường chuẩn: trục tung ghi giá trị chiều cao của sóng (h, mm), trục hoành ghi giá trị nồng độ C. Đường chuẩn thường đi qua gốc toạ độ. Đôi khi đồ thị không đi qua gốc toạ độ mà cắt trục tung hoặc trục hoành ở một điểm nào đấy. Trong trường hợp cắt trục tung có nghĩa chưa hoàn toàn triệt tiêu dòng dư hay dung dịch nền bị nhiễm nguyên tố cần xác định; khi cắt trục hoành có nghĩa là dòng khuếch tán giới hạn được tính chưa hoàn toàn.
4.2.2.Cực phổ hỗn hống (hay Von-Ampe hoà tan)
Cơ sở lý thuyết của phương pháp.
Để tiến hành cực phổ hỗn hống, người ta thực hiện qua 2 giai đoạn:
+ Điện phân đế làm giàu chất cần phân tích nên bề mặt cực đo dưới dạng một kết tủa (kim loại, hợp chất khó tan). Cực đo thường là cực thuỷ ngân treo có kích thước nhỏ như giọt thuỷ ngân trong cực phô cổ điển, cực đã quay bằng vật liệu trơ (như than thuỷ tinh, than nhão tinh kết, than ngâm tẩm platin...), cực màng thuỷ ngân trên bề mật cực rắn trơ. Quá trình điện phân thường tiến hành trên các máy cực phổ thông thưởng tại thế không đổi khi khuấy dung dịch với vận tốc đều. Nếu dùng cực rắn đã thì dùng các cực quay quanh trục của nó, nếu dùng cực thuỷ tinh ngân tĩnh thì khuấy dung dịch bằng máy khuấy từ.
+ Hoà tan kết tủa, làm giàu và ghi đo giá trị cường độ dòng tại thế thay đổi không gián đoạn (sự tan nuốt).
Đường cong biểu diễn sự phụ thuộc của cường độ trong quá trình hoà tan nuốt có dạng pic. Theo chiều sâu pic người ta tìm được nồng độ của ion kim loại trong dung dịch và theo thế của pic sẽ biết được bản chất của kim loại.
Hình 10a biểu diễn sóng cực phổ của ion Pb2+ nồng độ 2.10-4 M trong dung dịch KCNS 0,1 M. Hình 10b là đường cực phổ hỗn hống (von - ampe hoà tan) của dung dịch chứa ion Pb2+ nồng độ 10-6, KCNS 0,1 M khi điện phân làm giàu trong 5 phút tại thế (- 0,7 V). Dùng cực thuỷ tinh thuỷ ngân treo có kích thước tương tụ giọt thuỷ tinh ngân khi đo cực phổ, tốc độ phân cực hoà tan nuốt là 25mV/s cả hai đường cong được ghi cùng một độ nhạy của điện kế.
Qua đó ta thấy phương pháp cực phổ hỗn hống có tốc độ nhạy cao hơn phương pháp cực phổ điên rất nhiều.
Những yếu tố chủ yếu để xác định cường độ dòng nuốt là: nồng độ kim loại trong dung dịch, bán kính giọt thuỷ ngân, thể tích dung dịch, thời gian điện phân, thế điện
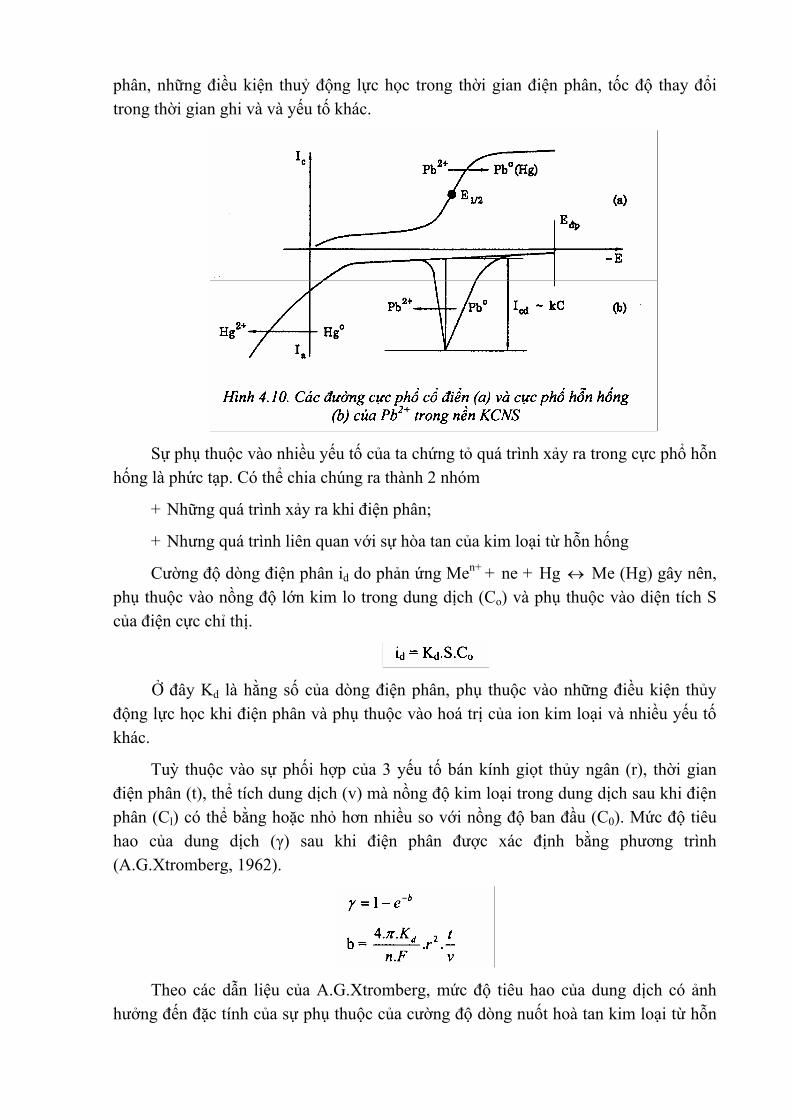
phân, những điều kiện thuỷ động lực học trong thời gian điện phân, tốc độ thay đổi trong thời gian ghi và và yếu tố khác.
Sự phụ thuộc vào nhiều yếu tố của ta chứng tỏ quá trình xảy ra trong cực phổ hỗn
hống là phức tạp. Có thể chia chúng ra thành 2 nhóm
+ Những quá trình xảy ra khi điện phân;
+ Nhưng quá trình liên quan với sự hòa tan của kim loại từ hỗn hống
Cường độ dòng điện phân id do phản ứng Men+ + ne + Hg ↔ Me (Hg) gây nên, phụ thuộc vào nồng độ lớn kim lo trong dung dịch (Co) và phụ thuộc vào diện tích S của điện cực chỉ thị.
Ở đây Kd là hằng số của dòng điện phân, phụ thuộc vào những điều kiện thủy
động lực học khi điện phân và phụ thuộc vào hoá trị của ion kim loại và nhiều yếu tố khác.
Tuỳ thuộc vào sự phối hợp của 3 yếu tố bán kính giọt thủy ngân (r), thời gian điện phân (t), thể tích dung dịch (v) mà nồng độ kim loại trong dung dịch sau khi điện phân (Cl) có thể bằng hoặc nhỏ hơn nhiều so với nồng độ ban đầu (C0). Mức độ tiêu hao của dung dịch (γ) sau khi điện phân được xác định bằng phương trình (A.G.Xtromberg, 1962).
Theo các dẫn liệu của A.G.Xtromberg, mức độ tiêu hao của dung dịch có ảnh
hưởng đến đặc tính của sự phụ thuộc của cường độ dòng nuốt hoà tan kim loại từ hỗn

hống và nồng độ của ion kim loại trong dung dịch:
KA là hằng số anot, KA phụ thuộc vào hệ số khuếch tán của kim loại trong hỗn
hống, tốc độ thay đổi thế khi ghi cực phổ, bán kính giọt thuỷ ngân...
Nếu b < 0, 1 thì, ≈γ b ở và phương trình trên có dạng
Phương trình này đúng khi tiến hành phân tích với thể tích dung dịch lớn, thời
gian điện phân ngắn và bán kính giọt thuỷ tinh nhỏ. Nồng độ các ion kim loại còn lại trong dung dịch trong trường hợp này thực tế không đổi.
Giá trị anốt tỷ lệ của ion kim loại trong dung dịch, thời gian điện phân và bán kính của thuỷ ngân.
Nếu b > 3 thì ≈γ 1, phương trình trên sẽ có dạng:
Khi thể tích dung dịch nhỏ, thời gian điện phân lớn, bán kính giọt thuỷ ngân lớn
thì cường độ dòng nuốt tỷ lệ thuận với nồng độ kim loại trong dung dịch, thể tích dung dịch, tỷ lệ nghịch với bán kính giọt thuỷ ngân và không phụ thuộc vào thời gian điện phân. Nồng độ ion trong dung dịch khi kết thúc quá trình điện phân thường nhỏ hơn 5% so với nồng độ ban đầu.
Phương trình (a) được tuân theo trong khoảng giá trị 0, 1 < b < 3
Kĩ thuật tiến hành xác định
Để xác định một hay một vài nguyên tố, người ta đặt vào bình điện một thế có giá trị cao hơn thế bán sóng của nguyên tố bị khử âm nhất là 0,2 - 0,3 V. Tại thế này trong thời gian đã cho, tiến hành điện phân dung dịch thí nghiệm và thường xuyên khuấy đều dung dịch. Sau khi dung dịch không bị xáo trộn, người ta bắt đầu hoà tan dượt kim loại từ hỗn thống khi thế giảm đều.

(Nồng độ của Zn2+, Cd2+, Bi3+, Cu2+ là 10-8m; Pb2+ và Te+ là 10-7 M điện phân 5
phút tại e = - 1,4V so với cực calomen bão hòa, cực làm việc là giọt thuỷ ngân treo)
Trong phương pháp Von - Ampe hoà tan thường nhúng vào bình điện phân chứa dung dịch phân tích 3 cực:
Cực làm việc là cực trên đó xảy ra phản ứng kết tủa chất cần phân tích dưới dạng kim loại hoặc hợp chất khó tan.
Cực so sánh, thường là một lon cực loại II như cực calomen hoặc cực bạc clorua.
Cực so sánh có thể không đổi và phải giữ thế này không đổi trong suốt quá trình làm việc đặc biệt khi tiến hành liên tiếp các thực nghiệm trong thời gian điện phân dài. Đề bào đảm được điều đó, cần chế tạo cực so sánh có diện tích bề mặt đủ lớn đề mật độ dòng qua cực đủ nhỏ.
Cực phù trợ thường là một cực platin
Có 3 cực làm việc chính:
Cực giọt thuỷ ngân tĩnh dưới dạng giọt treo hoặc giọt ngồi
Cực rắn hình đĩa.
Cực màng thuỷ ngân được điều chế tại chỗ trên bề mặt cực rắn đĩa
Trong cực phổ hỗn hống người ta hay dùng nhất là cực giọt thuỷ ngân tĩnh. Đó là một giọt thuỳ ngân có kích thước nhỏ (đường kính 0,8 - 1,5 mm) và bất động được treo trên một mao quản bằng thuỷ tinh hoặc đặt trên một mặt thuỷ tinh hơi lõm, ở giữa có một đoạn nhỏ và gắn platin để dẫn điện. Để đảm bảo tính chính xác và độ lặp lại của các phép xác định, yêu cầu chủ yếu của cực giọt thuỷ ngân tĩnh là phải có kích thước không đổi và có độ lặp đủ cao, vì sau mỗi một lần đo phải tạo một giọt khác có kích thước như giọt đã dùng trong vẫn đo trước. Thông thường người làm phân tích không thể chế tạo được cực giọt treo anh có chất lượng cao và vì vậy chỉ có các hãng mới sản xuất được cực treo tốt. Cực giọt thuỷ ngân tĩnh của hăng PAR (Mĩ) và cực giọt thuỷ ngân treo kiểu Kemula của hãng Birkmann (Mĩ) là cực tốt nhất.

Nếu có được một giọt Hg tĩnh có chất lượng tốt thì việc phân tích sẽ rất thuận lợi vì:
+ Khoảng thể cho phép dùng cực thuỷ ngân rất rộng, xác định được một số lớn kim loại. Trong môi trường axit khoảng thế dừng được tốt nhất là -0,15 đến -1,2V; trong môi trường trung tính và kiềm khoảng thế được mở rộng nhiều: từ -0,15 đến gần -2V.
+ Thuận lợi cho việc chọn điều kiện phân tích như chọn thành phần dung dịch nền, chọn thế điện phân, đặc biệt khi phân tích các kim loại trong mẫu có thành phần phức tạp vì có thể tham khảo, nghiên cứu những tài liệu khác về phân tích cực phổ để biết tính chất cực phổ của các chất khử cực khác nhau trong các nền cực phổ khác nhau. Xác định nồng độ chất phân tích: Nồng độ của kim loại trong dung dịch phân tích thường xác định bằng phương pháp thêm. Khi phân tích nhiều mẫu có cùng một thành phần có thể sử dụng phương pháp đường chuẩn. Cũng có những trường hợp để giảm ảnh hưởng do sự biến đổi kích thước của hạt, do sự thay đổi nhiệt độ, thời gian điện phân và các yếu tố khác đến kết quả phân tích, có tác giả đã sử dụng chất chuẩn trong Khi sử dụng chất chuẩn trong đê tính nồng độ người ta không dùng chiều cao lực của quá trình hoà tan nuốt kim loại từ hỗn hống mà dùng tỷ lệ chiều sâu giữa lực của nguyên tố cần xác định và lực của nguyên tồ đưa vào trong dung dịch làm chất chuẩn trong.
4.2.3. Cực phổ sung
Những phương pháp phân cực điện cực hoạt động bằng những xung điện áp gián đoạn có biên độ và bề rộng (thời gian tồn tại) xác định gọi là những phương pháp cực phổ xung. Có hai phương pháp cực phổ xung cơ bản hiện đang được sử dụng phố biến:
Phương pháp cực phổ xưng biến đổi đều (Normal pulse polarography -NNP)
Phương pháp cực phổ xung vi phân (Dìfferential pulse polarography - DPP)
4. 2. 3.1. Phương pháp cực phổ sung biến đổi đều (NPP)
Trong phương pháp này điện cực giọt thuỷ ngân được phân cực bằng một điện áp một chiều chọn trước và được giữ không đôi trong suốt quá trình đó, điện áp này được gọi là điện áp khởi điểm, tương ứng với chất sóng trong phương pháp cực phổ cổ điển, trong đỗ mỗi một chu kỳ giọt, điện cực được phân cực bổ sung bằng một xung vuông góc có khoảng tồn tại rất ngắn (40 đến 100 ms) được đưa vào sát nút trước khi giọt rơi (hoặc chu kì kết thúc). Sau thời gian đó xung bị ngắt và thế điện cực trở về điện áp khởi điểm. Biên độ xung tăng dẫn theo thời gian với một tốc độ đều giồng như tốc độ quyết thế tuyến tính trong cực phổ cổ điển. Cường độ dòng cực phổ được ghi theo một trong hai cách sau:
Ghi cường độ dòng cực phổ tại một thời điểm xác định sau khi đặt xung (thường là 17ms trước khi ngắt xung), phần lớn các máy hiện đang sử dụng thực hiện theo cách

này.
Ghi cường độ dòng cực phổ hai lần, lần thứ nhất trước khi đặt xung và lần thứ hai sau khi đặt xung (trong 2 khoảng thời gian giống nhau: 17 ms trước khi đặt và 17 ms trước khi ngắt xung). Cách này cho hiệu quả tốt hơn nhưng khá phức tạp.
Phương pháp NPP có thể đạt độ nhạy 2.10-7M cho cả hai quá trình thuận nghịch và bất thuận nghịch.
4.2.3.2. Phương pháp cực phổ sung vi phân (DPP)
Trong trường hợp này, điện cực được phân cực bằng một điện áp một chiều biến thiên tuyến tính với một tốc độ chậm, nhưng cuối mỗi một chu kì giọt (giọt rơi cưỡng bức nhờ một bộ gõ) trên khung điện áp biến đổi một chiều, người ta đặt thêm một xung vuông góc với biên độ trong khoảng 10 - 100mV và độ dải xung từ 40- 100 ms. Cường độ dòng cực phổ được ghi hai lần, lần một tại thời điểm τ1, thường là trong 17ms trước khi nạp xung là lần thứ hai tại thời điểm τ2, thường là 17ms trước khi ngắt xung. Hai giá trị này được đưa vào bộ so sánh và kết quả chuyển ra bộ ghi là hiệu sồ của hai giá trị đo. Đường cực phổ có dạng pic. Ưu điểm nổi bật của phương pháp này và có độ nhạy cao với các hợp chất vô cơ và hữu cơ ở các quá trình thuận nghịch và không thuận nghịch và quan trọng hơn là đường I-E có dạng định cực đại, sau mỗi một đỉnh dòng điện tại trở về trạng thái nền, do đó phương pháp có độ phân giải rất cao, có thể đạt đến 50.000 (ví dụ có thể ghi 10-7M Cd2+ khi có mặt 5.10-3 Cu2+ mà không cần tách). Điều này cho phép phân tích trực tiếp nhiều chất trong cùng một dung dịch. Một hướng phân tích có nhiều triển vọng là sử dụng kết hợp DPP với Von -Ampe hoà tan, ví dụ có thể xác định 10-9 M Pb2+ và thời gian không quá 3 phút. Có thể nói, hiện nay DPP là phương pháp cực phô hoàn chỉnh nhất vì phương pháp này có độ nhạy cao, độ chọn lọc cao và có tính vạn năng đối với hầu hết các đối tượng phân tích.

Chương 5
CÁC PHƯƠNG PHÁP PHÂN TÍCH SẮC KÝ
5.1. Mở đầu
Năm 1906, nhà bác học Nga Michael Tvest đã cho dung dịch các sắc tố thực vật trong ete dầu hoả lên cột nhồi bột mịn canxi cacbonat, ông thấy các sắc tố bị hấp phụ lên trên đầu cột. Khi cho ete dầu hoả lên cột, các sắc tố di chuyển trong cột từ trên xuống dưới, mỗi sắc tố có một tốc độ riêng, tách thành những vùng hay vòng màu xếp chồng lên nhau, hình thành một hệ mà Tvest gọi đó là sắc đồ. Ông đặt tên cho phương pháp tách này là sắc ký (Chromatography). Trong tiếng Hy Lạp chroma là màu, graphein là viết. Tên gọi này ngày nay vẫn được sử dụng mặc dù phương pháp này còn được dùng tách các chất không màu.
5.2. Một số khái niệm
5.2.1. Quá trình sắc ký
Sắc ký là một nhóm các phương pháp hoá lý dừng để tách các thành phần của một hỗn hợp. Sự tách sắc ký được dựa trên sự phân chia khác nhau của các chất khác nhau vào hai pha luôn tiếp xúc và không hoà lẫn vào nhau: một pha tĩnh và một pha động (trong thí nghiệm của Tvest: pha tĩnh là canxi cacbonat, pha động là ete dầu hoả).
Quá trình sắc ký gồm 3 giai đoạn chính
a) Đưa hỗn hợp lên pha tĩnh (ví dụ: đưa dung dịch các sắc tố lên đầu cột canxi cacbonat). Các chất được giữ trên pha tĩnh.
b) Cho pha động chạy qua pha tĩnh (dung môi đe dầu hoả qua cột), pha động sẽ kéo theo các chất di chuyển trên pha tĩnh với tốc độ khác nhau, tác khỏi nhau và có vị trí khác nhau trên pha tĩnh tạo thành sắc đồ. Giai đoạn này gọi là khai triển sắc ký.
Nếu tiếp tục cho pha động chạy qua thì các chất có thể lần lượt bị kéo ra ngoài pha tĩnh (ví dụ: ra khỏi cột). Đó là quá trình rửa giải và dung môi dùng được và dung môi rửa giải (eluent), dịch hứng được ờ cuối cột gọi là dịch rửa giải (eluate).
Nếu các chất được tách trên pha tĩnh (sắc ký khai thêm ta có thể lấy từng phần pha tĩnh có mang chất (phân đoạn bột trên cột) đem chiết lấy chất.
Nếu các chất được tách ra ngoài pha tĩnh (sắc ký rửa giải) ta có thể hứng thu lấy các phân đoạn dịch rửa giải có các chất cần phân tích.
c) Phát hiện các chất: Các chất màu có thể phát hiện dễ dàng, các chất không màu có thể phát hiện bằng đèn tử ngoại hay bằng các thuốc thử. Trong sắc ký rửa giải có thể phát hiện các chất khi chúng đi ra khỏi cột bằng cách cho dung dịch rửa giải đi qua một bộ phận phát hiện gọi là đetectơ đặt sau cột.

5.2.2. Phân loại các phương pháp sắc ký
- Theo bản chất vật lý các pha
Pha động có thể là một chất lỏng hay chất khí, pha tĩnh có thể là một chất rắn (hạt xốp hay bột mắn) hay một chất lỏng (được giữ trên một chất mang rắn). Do đó, dựa vào bản chất các pha ta phân biệt các phương pháp (trong tên của phương pháp, pha động được nêu trước pha tĩnh):
Sắc ký lỏng - lỏng (liquid - liquid chromatography LLC)
Sắc ký lỏng - rắn (liquid - song chromatography LSC)
Sắc ký khí - lỏng (gas - liquid chromatography GLC)
Sắc ký khí - rắn (gas - solid chromatography GSC)
Hai phương pháp đầu gọi chung là sắc ký lỏng (LC).
Hai phương pháp cuối gọi chung là sắc ký khí (GC).
- Theo hiện tượng sắc ký
Sắc ký hấp phụ (absorption chromatography): pha tĩnh là một chất rắn có khả năng hấp phụ, đó là các phương pháp sắc ký lỏng - rắn và khí - rắn.
Sắc ký phân bố (partition chromatography): pha tĩnh là chất lỏng không hoà lẫn được với pha động, chất lỏng này được bao trên bề mặt của một chất rắn gọi là giá hay chất mang và phải là chất trơ, không tham gia vào sắc ký. Sắc ký phân bố bao gồm sắc ký lỏng - lỏng và sắc ký khí - lỏng.
Sắc ký trao đổi ion (ion - exchange chromatography): pha tĩnh là chất nhựa trao đổi ion chớp chất cao phân tử có mang những ion có khả năng trao đổi với các ion cùng dấu của dung dịch hỗn hợp sắc ký).
Sắc ký theo loại cỡ (size - exclusion chromatography): còn gọi là sắc ký trên gel. Các phân tử cỡ lớn sẽ được loạt các phân tử nhỏ hơn sẽ được tách theo kích thước do các phân tử nhỏ di chuyển chậm hơn.
- Theo kỹ thuật và phương tiện sắc ký
a) Theo phương pháp giữ pha tĩnh:
Sắc ký trên cột (column chromatography: CC) pha tĩnh được chứa trong một cột bằng kim loại hay thuỷ tinh.
Sắc ký lớp mỏng (thin layer chromatography: TLC): pha tĩnh được tráng đều và giữ trên mặt phẳng của bản thuỷ tinh, nhựa hay nhôm. Lớp mỏng pha tĩnh thường là: silicagel, nhôm oxit, xenlulozơ, chất nhựa trao đổi lớn và có chiều dày khoảng 0,25 - 0,5mm.
Sắc ký giấy (paper chromatography - PC) pha tĩnh (lỏng) được thấm trên một loại

giấy lọc đặc biệt gọi là giấy sắc ký
b) Theo cách cho pha động chạy ta có:
Sắc ký khai triển (development chromatography) cho pha động kép các chất chạy và tách trên pha tĩnh (Sắc đồ nằm trên pha tĩnh). Sắc ký rửa giải (elution chromatoglaphy) cho pha động chạy và kép các chất lần lượt ra ngoài pha tính (ra khỏi cột, ra khỏi giấy).
Ghi chú: Sắc ký lớp mỏng và sắc ký lớp giấy được gọi chung là sắc ký mặt phẳng (planar chromatography).
Sắc ký lỏng có thể tiến hành trên cột, trên mặt phẳng (lớp mỏng, giấy).
Sắc ký được tiến hành trên cột
Sắc ký được khai triển thưởng được thực hiện trên mặt phẳng (giấy và lớp mỏng).
Sắc ký rửa giải thường tiến hành trên cột
- Phân loại sắc ký theo bản chất các pha
Ghi chú:
GSC: sắc ký khí - rắn GLC: sắc ký khí - lỏng
LSC: sắc ký lỏng - rắn IEC: sắc ký trao đổi lớn
BPC: sắc ký pha liên kết. IC: sắc ký ion
LLC: sắc ký lỏng - lỏng SEC: sắc ký loại cổ.
AC: sắc ký ái lực PC: sắc ký giấy
TLC: sắc ký lớp mỏng
5.2.3. Sự tách sắc ký và sắc đồ
- Sự tách sắc ký và sắc đồ
Khi cho một dung dịch chứa chất A và B chảy qua một cột chứa chất hấp thu Nồng độ của chất A và B trong dung dịch sẽ giảm đi do một phần các chất này phân bố chuyển vào chất hấp thu. Nồng độ của các chất tan A, B trên chất hấp thu sẽ tăng dần và A, B bị chất tách ra khỏi dung dịch. Đồng thời cũng xảy ra một quá trình ngược lại, các chất A, B trên cột hấp thụ lại phân bố tan trở lại vào dung dịch. Quá trình này phụ thuộc vào hệ số phân bố của A, B trong dung dịch và chất hấp thu cho đến khi đạt cân bằng.
Tốc độ hấp thu = tốc độ giải hấp
Tuy nhiên vì pha động (dung dịch) luôn di chuyên nên cân bằng trên lại bị phá vỡ, và một cân bằng mới lại được thiết lập. Cứ như vậy qua rất nhiều cân bằng (có khi hàng nghìn cân bằng pha) các chất A, B được tách ra khỏi nhau do khả năng phân bố

của A, B vào chất hấp thu hay khả năng bị giữ trên cột của A và B là khác nhau.
Ví dụ: cho một dung dịch chứa chứa ion đồng và ion coban lên trên một cột có chất hấp thu (1), như thế đồng và coban bị hấp thu trên cột (2). Sau đó tiếp tục cho dung môi qua cột, đồng và co ban sẽ kéo dần xuống và dần dần chúng tách ra thành các lớp (3), dung môi tiếp tục được cho qua cột và 2 lớp Cu2+ và Co2+ sẽ được tách ra khỏi nhau (4,5), tại thời điểm t2 sẽ thấy được 2 dải tách riêng ra khỏi nhau trên cột: ta có sắc đồ ngay trên cột. Nếu dừng ở đây ta có sắc ký khai triển
Nếu tiếp tục thêm dung môi, tới thời điểm t3, dải Co2+ sẽ tách ra khỏi cột, nhờ
thiết bị đetectơ ta sẽ thu được tín hiệu dưới dạng một đỉnh hay một lúc, ở thời điểm t4
dải Cu2+ ra khỏi cột lại và cho ta thêm một tức tương ứng. Ta thu được một sắc đồ gồm hai lúc riêng biệt ứng với 2 chất trên. Đây là phương pháp sắc ký rửa giải và sắc đồ thu được gọi là sắc đồ rửa giải. Vị trí của lực trên trục thời gian là cơ sở để định tính chất tương ứng và diện tích dưới tức là đặc trưng định lượng của chất.
Trong quá trình này ta thấy một sự khác biệt về khả năng phân bố trong chất hấp thu trên cột: Cu2+ được hấp thu mạnh hơn nên sẽ đẩy Co2+ ra, sở dĩ có hiện tượng đó là vì:
Độ tan của đồng trong dung môi nhỏ hơn so với coban
Ái lực của chất hấp thu đối với Cu2+ mạnh hơn so với Co2+
Vì hai lý do trên nên đồng sẽ bị giải hấp thu ít hơn. Nồng độ lớn Co2+ trong dung môi từ nhỏ đến cực đại rồi giảm đến 0.
Tốc độ di chuyên của dải phụ thuộc vào tốc độ di chuyển của pha lỏng (pha động) và mức độ hấp thu của chất hấp thu đối với chất tan. Chất nào bị hấp thu mạnh nhất sẽ di chuyển chậm nhất và nó bị dung môi giải hấp ít nhất.
Cứ như vậy trong điều kiện thích hợp ta sẽ tách được hoàn toàn các chất ra khỏi nhau: Do tác dụng làm chậm (retardation), do sự phân bố hấp thu khác nhau của pha
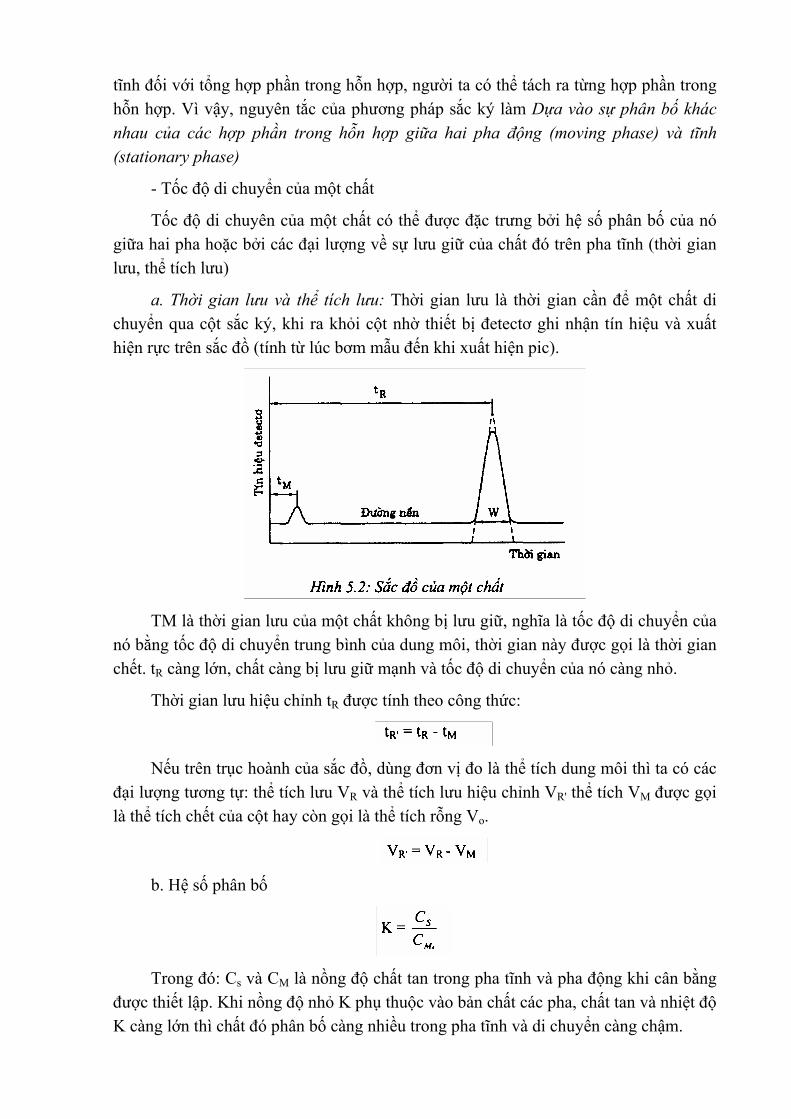
tĩnh đối với tổng hợp phần trong hỗn hợp, người ta có thể tách ra từng hợp phần trong hỗn hợp. Vì vậy, nguyên tắc của phương pháp sắc ký làm Dựa vào sự phân bố khác nhau của các hợp phần trong hỗn hợp giữa hai pha động (moving phase) và tĩnh (stationary phase)
- Tốc độ di chuyển của một chất
Tốc độ di chuyên của một chất có thể được đặc trưng bởi hệ số phân bố của nó giữa hai pha hoặc bởi các đại lượng về sự lưu giữ của chất đó trên pha tĩnh (thời gian lưu, thể tích lưu)
a. Thời gian lưu và thể tích lưu: Thời gian lưu là thời gian cần để một chất di chuyển qua cột sắc ký, khi ra khỏi cột nhờ thiết bị đetectơ ghi nhận tín hiệu và xuất hiện rực trên sắc đồ (tính từ lúc bơm mẫu đến khi xuất hiện pic).
TM là thời gian lưu của một chất không bị lưu giữ, nghĩa là tốc độ di chuyển của
nó bằng tốc độ di chuyển trung bình của dung môi, thời gian này được gọi là thời gian chết. tR càng lớn, chất càng bị lưu giữ mạnh và tốc độ di chuyển của nó càng nhỏ.
Thời gian lưu hiệu chỉnh tR được tính theo công thức:
Nếu trên trục hoành của sắc đồ, dùng đơn vị đo là thể tích dung môi thì ta có các
đại lượng tương tự: thể tích lưu VR và thể tích lưu hiệu chỉnh VR' thể tích VM được gọi là thể tích chết của cột hay còn gọi là thể tích rỗng Vo.
b. Hệ số phân bố
Trong đó: Cs và CM là nồng độ chất tan trong pha tĩnh và pha động khi cân bằng
được thiết lập. Khi nồng độ nhỏ K phụ thuộc vào bản chất các pha, chất tan và nhiệt độ K càng lớn thì chất đó phân bố càng nhiều trong pha tĩnh và di chuyển càng chậm.

c. Hệ số dung lượng K' (hệ số phân bố khối lượng)
Trong đó: Qs và QM là lượng chất tan phân bố trong pha anh và pha động, K' phụ
thuộc vào bản chất các pha, bản chất chất tan, nhiệt độ và đặc điểm của cột. Để tách một hỗn hợp chất, người ta thường chọn cột, pha động và các điều kiện phân tích khác sao cho K' nằm trong khoảng từ 1 đến 8.
- Sự doãng pic và hình dáng pic
a. Hình dáng pic: Hình dáng lý tưởng của tức là lực đối xứng, nhưng thực tế lúc sắc ký chỉ gần đối xứng. Chiều rộng của tục được đo ở 1/10 chiều cao của pic.
b. Sự doãng pic: Sự doãng pic là kết quả của sự di chuyển nhanh, chậm khác nhau của các phân tử của cùng một chất khi đi qua cột sắc khí. Các lực ra chậm bao giờ cũng tù hơn.
c. Hiệu lực của cột: Hiệu lực của cột thường được đo bằng hai thông số: số đĩa lý thuyết N và chiều cao đĩa lý thuyết H. Ta có thể coi như cột sắc khí được chia thành N lớp hay N tầng mỏng, ở mỗi một lớp sự phân bố chất tan vào hai pha đạt trạng thái cân bằng. Những tầng mỏng giả định này được gọi là địa lý thuyết. Nếu cột sắc khí có
chiều dài là L thì: H = NL . Cột có N lớn là cột có hiệu lực cao.
d. Độ phân giải: Độ phân giải Rs là tỉ số khoảng cách giữa hai pic và độ rộng trung bình của pic.
Rs = 0,75 hai pic tách không tốt, còn xen phủ nhau nhiều;
Rs = 1,0 hai pic tách khá tốt, còn xen phủ nhau 4%;
Rs = l,5 hai pic tách hoàn toàn (chi xen phủ 0,3%).
5.3. Sắc ký lỏng hiệu năng cao
Sắc ký lỏng hiệu năng cao (high perfonnance liquid chromatography-HPLC) còn được gọi là sắc ký lỏng cao áp hay sắc ký lỏng hiện đại. Trong phương pháp này pha tĩnh được dùng là những hạt có kích thước rất nhỏ, khoảng 10 micromet, có hiệu suất tách rất cao, có hạt nhỏ đòi hỏi phải dùng bơm để nén dung môi qua cột.
5 3.1. Máy sắc ký lỏng hiệu năng cao
Sơ đồ nguyên tắc của một máy sắc ký lỏng hiệu năng cao như sau:

- Các thành phần chính của máy sắc ký lỏng hiệu năng cao
a) Bình chứa dung môi
Thường làm bằng thuỷ tinh, đôi khi làm bằng thép không rỉ, trong phương pháp rửa giải thông thường chỉ cần một bình chứa dung môi, trong phương pháp rửa giải gradient thường dùng 2, 3, 4 bình chứa các dung môi khác nhau và hệ dung môi rửa giải là hỗn hợp của các loại dung môi trên trộn lẫn với nhau theo ti lệ đã được xác định. Cần loại các hạt và các khí hoà tan trong dung môi.
b. Hệ thống bơm
Bơm dùng trong phương pháp phải tạo được áp suất cao (3000 - 6000 psi hay khoảng 250 - 500 at), lưu lượng bơm khoảng 0,1 đến 10 ml/ph, phải trơ với các dung môi. Hệ thống bơm này phải có khả năng bơm pha động qua cột ở áp suất cao mà không bị ngắt quãng hoặc tạo nhịp sóng có thể làm sai lệch các lực thu được. Có nhiều kiểu bơm đã dùng trong các máy HPLC; nhưng kiểu phông xoay chiều hiện nay được dùng phổ biến nhất.
c) Hệ bơm mẫu (hệ tiêm mẫu)
Mẫu được bơm vào cột nhờ hệ thống van mẫu. Người ta bơm mẫu vào vòng mẫu với thể tích thường là từ 10 đến 20 μl.
d. Cột sắc ký
Kiểu cột thường phụ thuộc vào phương pháp dùng để tách, nhưng thường là những cột bằng thép không gỉ, có cỡ lỗ chính xác đường kính từ 3 đến 4 túm và dài từ 10 đến 30cm (những cột dùng cho SEC đường rộng hơn và dài đến 100cm). Loại cột này có hiệu lực rất cao, có số (ra lý thuyết lên đến 100.000 đĩa cho 1m chiều dài cột)

e) Đetectơ
Là bộ phận phát hiện các chất khi chứng ra khỏi cột và ghi nhận các tín hiệu ghi trên sắc đồ. Hiện đang sử dụng các loại đetectơ sau:
Đetectơ tử ngoại (UV): Dùng đèn thuỷ ngân cho vạch 254nm. Nhiều chất hấp thụ ở bước sóng này.
Đetectơ tử ngoại và khả kiến (UV-VIS): Dùng phổ quang kế lựa chọn bước sóng từ 195 đến 750 nm. Loại này hiện nay được dùng nhiều nhất.
Đetectơ điện hoá và đetectơ huỳnh quang có độ nhạy và độ chọn lọc cao dùng trong phân tích vết. Dùng đetectơ điện hoá có thể phát hiện được những lượng picrogam (10-12g)
5.3.2. Các phương pháp sắc ký hiệu năng cao
a) Sắc ký phân bố hiệu năng cao
Gồm hai loại: Sắc khí lỏng - lỏng và sắc ký pha liên kết.
- Sắc ký lỏng - lỏng (LLC): Pha tĩnh là chất lỏng được bao trên bề mặt của các hạt chất mang, tức là được hấp phụ trên chất mang.
- Sắc ký pha liên kết (BPC): Pha tĩnh được gắn hoá học với chất mang tạo ra liên kết siloxan nối nhóm cơ - silic với silicagen.
b. Sắc ký hấp phụ hiệu năng cao (sắc ký lỏng - rắn LSC)
Pha tĩnh là chất rắn mà trên bề mặt có chứa các nhóm hiđroxil phân cực, pha động là một dung môi không phân cực.
c) Sắc ký trao đối ion hiệu năng cao (IEC)
Pha tĩnh rắn chứa các nhựa trao đổi lớn dưới dạng bột mịn, pha động thường là dung dịch nước.
d. Sắc ký lỏng hiệu năng cao trên gel (sốc ký loại cỡ SEC)
Phương pháp này được ứng dụng chủ yếu cho các chất có phân tử lượng lớn. Chất nhồi cho SEC là những hạt xốp của silicagent hay các polyme có kích thước nhỏ ( 10 μm). Pha tĩnh là dung môi nằm trong các lỗ xốp của hạt, pha động là dung môi chảy giữa các hạt. Chỉ những phân từ nhỏ mới khuếch tán vào lớp xốp, khi rửa giải các phân tử sẽ lần lượt ra theo cỡ từ lớn đến nhỏ.
5.4. Sắc ký khí (GC - Gas chromatography)
Sắc ký khí là phương pháp được dùng để tách các chất ở thể khí bay hơi, với pha độ là chất khí, gọi là khí mang (carrier gas).
Như đã nêu ở trên, có thể chứa phép sắc ký khí thành sắc ký khí rắn (GSC - gas solid chromatography) trong đó pha tĩnh rắn là một chất hấp phụ, đây là sắc ký khí hấp
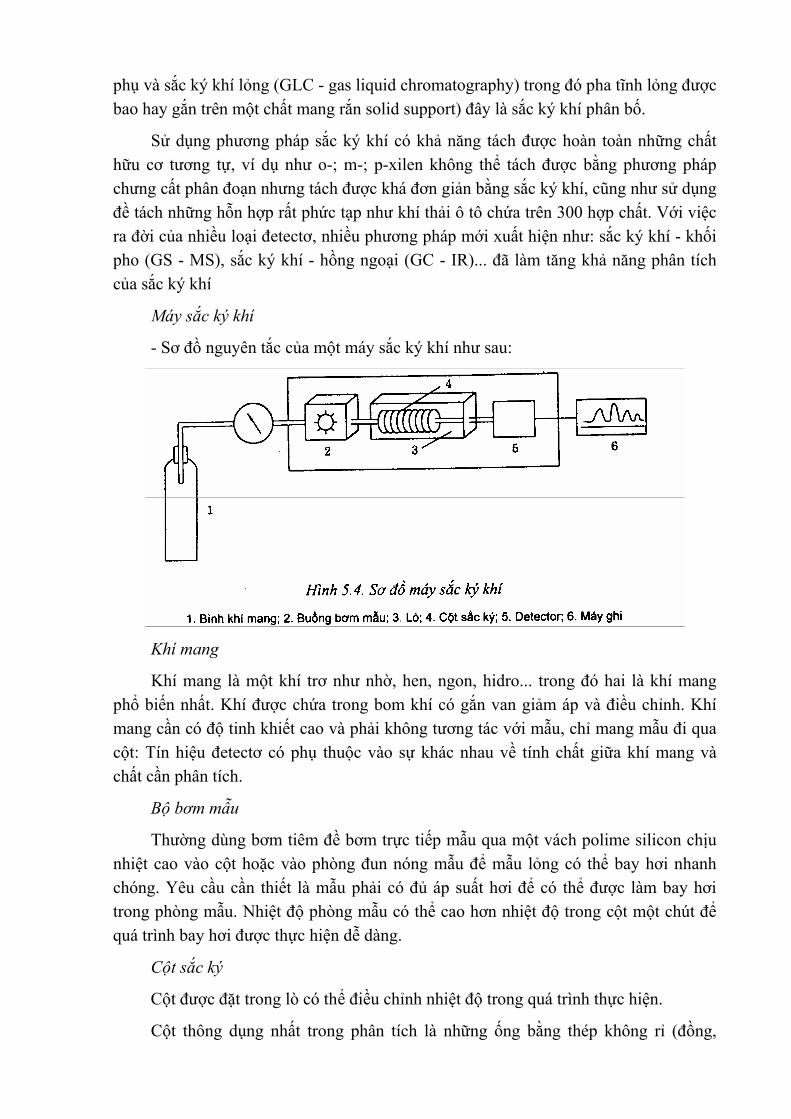
phụ và sắc ký khí lỏng (GLC - gas liquid chromatography) trong đó pha tĩnh lỏng được bao hay gắn trên một chất mang rắn solid support) đây là sắc ký khí phân bố.
Sử dụng phương pháp sắc ký khí có khả năng tách được hoàn toàn những chất hữu cơ tương tự, ví dụ như o-; m-; p-xilen không thể tách được bằng phương pháp chưng cất phân đoạn nhưng tách được khá đơn giản bằng sắc ký khí, cũng như sử dụng đề tách những hỗn hợp rất phức tạp như khí thải ô tô chứa trên 300 hợp chất. Với việc ra đời của nhiều loại đetectơ, nhiều phương pháp mới xuất hiện như: sắc ký khí - khối pho (GS - MS), sắc ký khí - hồng ngoại (GC - IR)... đã làm tăng khả năng phân tích của sắc ký khí
Máy sắc ký khí
- Sơ đồ nguyên tắc của một máy sắc ký khí như sau:
Khí mang
Khí mang là một khí trơ như nhờ, hen, ngon, hidro... trong đó hai là khí mang phổ biến nhất. Khí được chứa trong bom khí có gắn van giảm áp và điều chỉnh. Khí mang cần có độ tinh khiết cao và phải không tương tác với mẫu, chỉ mang mẫu đi qua cột: Tín hiệu đetectơ có phụ thuộc vào sự khác nhau về tính chất giữa khí mang và chất cần phân tích.
Bộ bơm mẫu
Thường dùng bơm tiêm đề bơm trực tiếp mẫu qua một vách polime silicon chịu nhiệt cao vào cột hoặc vào phòng đun nóng mẫu để mẫu lỏng có thể bay hơi nhanh chóng. Yêu cầu cần thiết là mẫu phải có đủ áp suất hơi để có thể được làm bay hơi trong phòng mẫu. Nhiệt độ phòng mẫu có thể cao hơn nhiệt độ trong cột một chút để quá trình bay hơi được thực hiện dễ dàng.
Cột sắc ký
Cột được đặt trong lò có thể điều chỉnh nhiệt độ trong quá trình thực hiện.
Cột thông dụng nhất trong phân tích là những ống bằng thép không rỉ (đồng,

thép) hoặc bằng thuỷ tinh dài từ 1 đến 10 m và có đường kính từ 2 đến 4 mm. Chúng được uốn cuộn tròn cho khớp với phòng lò. Cột được nhồi bằng những phần tử rắn hoạt động như một pha tĩnh (GSC): đó là những hạt nhỏ xếp gắn trên mặt trong cột hoặc pha tĩnh được tẩm trên các hạt nhỏ đó. Trong sắc ký khí lỏng (GLC), pha tĩnh được giữ trên chất mang, đó là những hạt chất rắn nhỏ, bền nhiệt, trơ về mặt hoá học, có lỗ cỡ 1 - 5 μm, bề mặt riêng lớn từ 1 - 10 m2/g.
Pha tĩnh phải chịu nhiệt, hoá lỏng ở nhiệt độ phân tích, trơ về hoá học. Thường hay sử dụng các polyme xốp hoặc gel aluminosilicat khử nước.
Đeteclơ
Yêu cầu về đetectơ rất nghiêm ngặt: Mọi hợp phần có trong 0,1 μl mẫu phải được phát hiện ở mức 1% cho nên phải nhanh chóng phát hiện được 0,002 μl (có khối lượng cỡ 10-6 g) mẫu. Các đetectơ hiện nay đo được những lượng nhỏ hơn đến mấy bậc. Hiện nay trong phương pháp GC người ta sử dụng những loại đetectơ sau:
Đetectơ dẫn nhiệt (TCD): Việc vận hành của đetectơ dựa trên sự cân bằng nhiệt cửa một dây dẫn nung nóng. Khi có chất cần phân tích đi qua thì độ dẫn điện của hỗn hợp khí sẽ thấp đi, dây sẽ nóng lên, xuất hiện một tín hiệu điện. Đetectơ dẫn nhiệt thích hợp với nhiều chất cần phân tích.
Đetectơ ion hoá ngọn lửa (FID): Khi đốt cháy các hợp chất hữu cơ trong ngọn lửa, chúng sẽ tạo ra các ion. Các điện cực ở gần ngọn lửa sẽ phát hiện ra sự có mặt của các ion bằng sự xuất hiện một dòng điện nhỏ chạy qua mạch điện. Có thể đo được những dòng ngay cả khi chứng chỉ xấp xi bằng 10-12A. Đây là đetectơ được dùng nhiều nhất, phát hiện được đến 10-9g.
Đetectơ hấp thụ electron (đetectơ bắt electron - ECD): Nhờ có nguồn phóng xạ như 3H hoặc 63Ni, khí mang bị ion hoá tạo nên một "dòng nhất định" trong mạch đetectơ. Khi một chất phân tích hấp thụ electron được giải hấp nó sẽ làm giảm dòng này, đặc biệt khi hợp chất có chứa nguyên tố có độ âm điện cao. Dòng sẽ giảm đi, tạo ra tín hiệu ghi để. Đetectơ đặc biệt nhạy cảm với những hợp chất chứa halogen. Đetectơ hấp thụ electron rất thuận lợi cho việc phân tích môi trường. Độ nhạy cao, đạt tới 10-12g.
Thông tin GC
Sắc đồ thông thường biểu thị trên hai trục x và y, trong đó tín hiệu đetectơ ghi trên trục y và thời gian ghi trên trục x.
Khi một hợp phần của mẫu được giải hấp và phát hiện nhờ đetectơ thì tín hiệu đó sẽ được ghi lại. Thời gian ghi được ở lúc tín hiệu là hàm của KD và vì thế nó cung cấp thông tin định tính về mẫu giống như thế bán sóng trong cực phổ, và sẽ nhận biết được một hợp phần trong mẫu.
Thông tin định lượng từ sắc đồ được rút ra từ chỗ tín hiệu đetectơ là hàm của thời
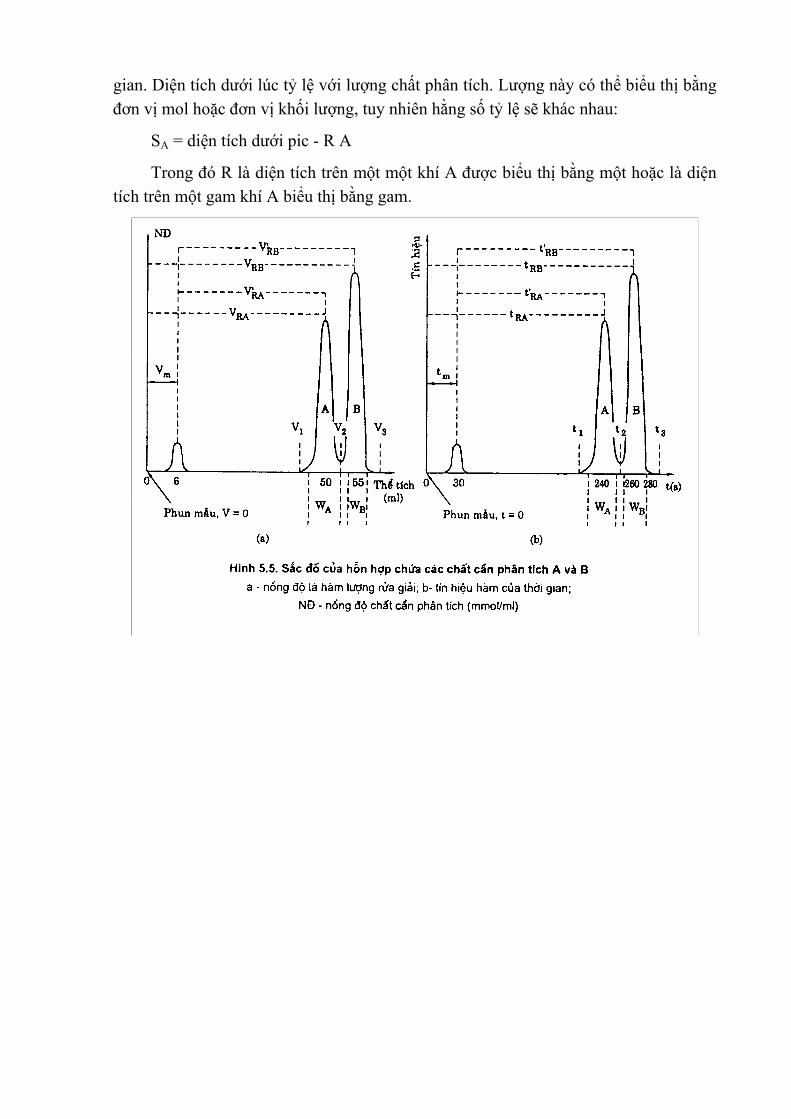
gian. Diện tích dưới lúc tỷ lệ với lượng chất phân tích. Lượng này có thể biểu thị bằng đơn vị mol hoặc đơn vị khối lượng, tuy nhiên hằng số tỷ lệ sẽ khác nhau:
SA = diện tích dưới pic - R A
Trong đó R là diện tích trên một một khí A được biểu thị bằng một hoặc là diện tích trên một gam khí A biểu thị bằng gam.

Chương 6
CÁC LOẠI NƯỚC VÀ CÁC PHƯƠNG PHÁP PHÂN TÍCH NƯỚC
6.1. Đại cương về các loại nước
6.1.1. Nước thiên nhiên
Nước thiên nhiên bao gồm các loại nước có nguồn thiên nhiên như sông ngòi, hồ, ao, suối, mạch ngầm, biển, đại dương. Có thể nói nước thiên nhiên là một hệ dị thể nhiều thành phần, vì nước thiên nhiên luôn chứa những lượng nào đó các chất tan và không tan có nguồn gốc vô cơ cũng như hữu cơ. Các chất đó được đưa vào từ khí quyển, từ đất và các nguồn chứa hoặc các mạch nước chảy qua. Các chất đó còn được tạo thành do hoạt động sống và sự phân huỷ của các cơ thể động vật, thực vật sống trong nước hoặc đáy các nguồn nước. Các hoạt động của con người trong các lĩnh vực khác nhau của sản xuất và đời sống hàng ngày cũng ảnh hưởng tới thành phần nước trong thiên nhiên. Việc bón phân và sự cải tạo đất bằng phương pháp hóa học, các phế thải của các nhà máy, các khu công nghiệp đặc biệt là nước thải công nghiệp và hoạt động khác của con người cũng đưa vào nước thiên nhiên những chất vô cơ và hữu cơ. Có nhiều cách phân loại nước thiên nhiên, nguyên tắc của sự phân loại đó phụ thuộc vào mục đích phân loại và nghiên cứu nước.
Dựa vào nguồn gốc của nước, có thể chia thành các loại; nước khí quyển (nước mưa, tuyết), nước ngầm, nước bề mặt (nước sông, ngòi, hồ ao, biển...).
Dựa vào nguyên tắc và mục đích sử dụng nước có các loại như: nước ăn, nước sinh hoạt, nước kỹ thuật, nước chữa bệnh,...
Dựa vào hàm lượng và đặc tính của các chất có trong nước, chia thành: nước ngọt (chứa hàm lượng chất tan < 0,1%), nước khoáng (hàm lượng chất tan từ 0,1 -2,5%) nước biển (hàm lượng chất tan từ 2,5-5,0%), nước muối (hàm lượng chất tan >5%). Ngoài ra người ta còn phân chia thành các loại như nước cứng, nước mềm, nước trong, nước đục, nước có màu, nước có mùi,...
Nước thiên nhiên được đặc trưng bằng một loạt các tính chất như độ đục, mùi vi, độ kiềm, độ cung, cặn không tan,... sự tồn tại của các loại vi sinh vật khác nhau, các quần thể phù du, các loại rong rêu cũng có thể làm cho nước bị nhiễm bẩn.
Dựa trên các tính chất hoá lý có thể chia các hợp phần hóa học thành 3 nhóm lớn như sau:
a. Các chất hoà tan trong nước dưới dạng các ion và phân tử: Các con vô cơ trong nước với hàm lượng lớn nhất là Na+, K+, Ca2+, Mg2+, Cl-, HCO3
2-, CO32-, SO4
2-. Các ion có hàm lượng nhỏ hơn là Fe2+, Fe3+, Mn2+, Br-, I-, F-, BO2
-, HPO-24, HSO3
-, HS-, HSiO3-. Các ion vô cơ còn lại có hàm lượng rất nhỏ trong nước được gọi là các

ion của các nguyên tố vi lượng và siêu vi lượng. Các khí có hàm lượng lớn là O2, CO2, N2, H2S. Các khí như CH4, Ar, RN,... có hàm lượng nhỏ. Các chất tan hữu cơ thường gặp trong nước tự nhiên là fomanđehit, phenol.
b. Các hợp phần tạo nên những hệ keo trong nước bao gồm các hệ keo vô cơ như SiO2, Al(OH)3, Fe(OH)3. Các hệ keo hữu cơ như các chất mùn. Các hệ keo thường làm cho nước có màu.
c. Các chất tạo với nước các phần tử như cát, sét và các hợp chất hữu cơ khác nhau.
Các hợp phần sinh học của nước thiên nhiên bao gồm các loại vi khuẩn, vinh động vật có kích thước lớn, rong rêu, tảo... trong nước thiên nhiên có chứa một lượng thức ăn cho các thuỷ sinh vật. Vì vậy, thành phần định lượng và định tính của chúng phụ thuộc nhiều vào thành phần hoá học của nước. Đối với các vi sinh vật trong nước, bên cạnh những vi sinh vật có lợi còn có những vi sinh vật có hạn. Vì vậy trong việc phân tích nước thì xác định chỉ tiêu vi sinh vật cũng có ý nghĩa hết sức quan trọng
6.1.2. Nước thải
Cùng với sự phát triển của văn minh nhân loại, nhu cầu về nước ngày càng tăng, lượng nước công nghiệp cũng như nước sinh hoạt thải ra ngày càng nhiều, gây ô nhiễm đáng kề đến nước mặt và môi trường. Do đó, nhiều vùng nước mặt đã bị ô nhiễm các loại hợp chất hóa học và các loại vi sinh vật độc hại.
Trong những năm gần đây, nguy cơ làm ô nhiễm môi trường do các chất hữu cơ, đặc biệt là các sản phẩm công nghiệp và khai thác chế biến dầu mỏ gây ra ngày càng tăng. Việc nhiễm các loại thuốc trừ sâu, diệt cỏ, các chất tây rửa tổng hợp cũng là mối đe doạ đáng kể đến sự tồn tại của các loài thuỷ sinh và con người. Ngoài ra còn phải kể tới các chất thải phóng xạ thải ra từ các phòng thí nghiệm.
Có thể nói nước thải là một hệ dị thể phức tạp, bao gồm rất nhiều chất tồn tại dưới các trạng thái khác nhau. Nếu như nước thải công nghiệp chứa rất nhiều các hoá chất vô cơ và hữu cơ thì nước thải sinh hoạt lại chứa rất nhiều các chất dưới dạng prôtein, hiđratcacbon, mỡ, các chất thải ra từ người và động vật, ngoài ra còn phải kê đến các loại rác rưởi như giấy, gỗ. các chất hoạt động bề mặt,... Các hợp chất vô cơ thường gặp ở đây là các ion như: Na+, K+, Mg2+, Ca2+, Cl-, CO3
2-, SO42-. Ngoài ra nước
thải sinh hoạt còn chứa các vi khuẩn, virut, rong, rêu.
Với những ngành sản xuất khác nhau thì trong các nước thải sẽ có những loại hoá chất khác nhau như:
- Khai khoáng: Các kim loại, các axit vô cơ.
- Gia công đồ gỗ flo, kẽm.
- Đồ gốm: Ba, Cd, Li, Mg, Se

- Đồ da: Ca, H2S, Na2S, CR, Zn, Ni...
- Luyện cốc: NH3, H2S, Các kiềm,...
- Công nghiệp sơn: Ba, ClO3-, Cd, Co, Pb, Zn, Mn...
- Hoá dược: B, Br, NH4+, K, các axit, kiềm, các chất hữu cơ,...
- Thuỷ tinh: H3BO3, As...
Trong số các hoá chất gây nhiễm bẩn nguồn nước thì Hg, Be, Cd, Pb, As, Se có độc tính rất cao.
Nước thải sinh hoạt có thành phần hóa học đơn giản hơn, chủ yếu bao gồm: K, Na, Fe, Pb, Zn, Ni, Mn, Hg, Ag, Co.
Việc phân tích nước thải rất khó khăn và phức tạp. Khi phân tích cần phải tiến hành các quá trình tách, làm giàu, lựa chọn các phương pháp có độ nhạy, độ chọn lọc cao. Một trong những đặc dính gây khó khăn cho việc phân tích nước thải là tính chất không bền vững của nói
Đối với nước ăn, việc phân tích hóa học đơn giản vì loại nước này đã được xử lý về mặt cơ, hoá và sinh học.
6.2. Phân tích nước
6.2.1. Phân tích nước thiên nhiên
Việc phân tích đề đánh giá chất lượng nước thiên nhiên phụ thuộc vào mục đích sử dụng loại nước đó, nhưng nói chung để phân tích đẩy đủ một mẫu nước tài nguyên cần xác định khảo sát một số loại chỉ tiêu sau đây:
- Các thông số về vật lý và cảm quan như nhiệt độ, vị, độ đục, tỷ trọng
- Các thông số về thành phần hóa học như hàm lượng các cation, anion và các chất hữu cơ.
- Đối với nước sinh hoạt, ngoài các chỉ tiêu trên còn phải xác định và đánh giá cả về chỉ tiêu vi sinh vật.
6.2.2. Phân tích nước thải
Thành phần nước thải khóc nhiều so với nước thiên nhiên, vì thế phân tích nước thải lại theo một sơ đồ khác. Mẫu nước thải thường phân tích ngay trong ngày lấy mẫu. Sau đây là các chỉ tiêu cần phân tích đối với nước thải:
- Nhiệt độ
- Màu sắc.
- Mùi vị
- Độ đục

- pH.
- Lượng kết tủa
- Độ bền tương đối của nước.
- Các chất lơ lửng.
- Tổng số muối tan
- Các chất màu, chất hữu cơ độc hại.
- Kim loại nặng
6.2.3. Những điều cần lưu ý khi phân tích nước
Phải lấy và bảo quản mẫu đúng quy cách và tuân theo một cách nghiêm ngặt các quy tắc lấy mẫu. Thông thường mẫu phải đựng trong bình polietylen.
Phải chọn phương pháp phân tích thích hợp.
Phải sử dụng đúng quy cách các dụng cụ và hoá chất.
6.3. Lấy và bảo quản mẫu nước
6.3.1. Xác định vị trí lấy mẫu
Mẫu lấy để xác định tính chất thông thường của nước có thể lay ngưu nhiên từ bề mặt vào thời gian buổi sáng. Tuy nhiên, khi nghiên cứu một cách chỉ tiết hơn, cần phải đặt kế hoạch và xác định vi trí lấy mẫu trước
Đối với vùng nước rộng như: ao hồ, mẫu lấy phải đại diện cho nước toàn bộ vùng cần phải lấy nước ở nguồn thải vào và ra khỏi hồ cũng như tác động của con người đến nguồn nước.
Tuỳ theo vùng nước nông hay sâu mà có thể lấy mẫu ở một tầng, ở hai tầng (tầng mặt và tầng đáy), ba tầng hoặc nhiều hơn nữa ở các sông, suối có thể lấy dọc theo dòng nước và theo chiều ngang nếu đủ rộng ờ những nơi có nguồn nước thải, cần lấy các điểm trước và sau điểm hoà nhập của nguồn nước thải.
6.3.2. Lấy mẫu nước
Vị trí lấy mẫu phụ thuộc vào mục đích của nghiên cứu. Thời gian và tần suất lấy mẫu phụ thuộc vào mức độ biến động của các chất, có thể theo các thời gian trong ngày. Trong các ngày nắng, pa của nước có thể tăng, đặc biệt là nước có khả năng đệm thấp do khả năng trao đổi CO2 của thực Vật thuỷ sinh và nồng độ P có thể sẽ bị giảm đi.
Trong nhiều trường hợp lấy mẫu vào buổi sáng là thích hợp vì lúc đó sự biến động các chất diễn ra chậm. Trong trường hợp chung, có thể lấy mẫu 3 tháng một lần, nhưng khi nghiên cứu chi tiết có thể lấy mẫu theo chu kì 7 hoặc 14 ngày từ mùa xuân đến mùa thu.

Trong trường hợp nước đồng nhất, có thể chỉ cần lấy một mẫu đơn lẻ cũng đủ đề đại diện cho môi trường nước thời điềm lấy mẫu. Ví dụ như ở dòng suối chảy và không khác biệt nhiều thì có thể lấy ở độ sâu trung bình ở giữa suối. Tương tự như vậy khi lấy mẫu trong hồ chứa nước hoặc trong bề chứa nước. Tuy nhiên, tính đồng nhất giữa các tầng nước cũng cần phải khẳng định bằng việc lấy một loạt các mẫu ở các độ sâu khác nhau để kiểm tra sơ bộ trước. Nếu lấy nước qua máy bơm, ống dẫn hoặc vòi nước thì cần phải để nước chảy khoảng 5-6 lít trước khi lấy mẫu.
Trong trường hợp chất lượng nước bị biến đôi theo thời gian thì mẫu cũng cần phải được lấy theo các thời gian thích hợp. Như trong trường hợp có sự thay đổi của dòng chảy, của các nguồn nước thải,... mẫu có thể lấy ở nhiều nguồn khác nhau phụ thuộc vào nguồn: nước. Cần phải tránh lấy mẫu ở những khu vực đặc biệt như vùng nước đọng, cỏ dại mọc nhiều hoặc có nước ngầm xâm nhập vào.
Lượng nước cần lấy tối thiểu để đảm bảo cho các phép phân tích, thường từ 1 đến 2 lít. Dụng cụ đựng mẫu cần phải được làm sạch bằng các biện pháp cần thiết như rửa bằng các chất tẩy rửa hoặc dung dịch axit. Đối với phân tích vi sinh vật thì dụng cụ đựng mẫu cần phải được vô trùng.
Có thể làm giảm các tác động của các quá trình xảy ra trong mẫu bằng việc thêm các hoá chất cần thiết, điều chỉnh pH hoặc giữ mẫu ở nhiệt độ thấp (40C).
6.3.3. Lấy và bảo quản mẫu
Mục tiêu chính của công việc này là nhàm chuyển mẫu đến các nơi phân tích và đảm bảo sự biến đổi của mẫu là tối thiểu, chất phân tích sẽ không có sự thay đổi đáng kể về hàm lượng. Tuy nhiên, trong nhiều trường hợp, tính chất của nước có thể bị thay đổi, ví dụ như Na+ hay Ca2+ có sự biến động rất ít trong khi Mo và các vitamin thì bị biến đổi rất nhiều.
Vì vậy, dụng cụ lấy mẫu và dụng cụ đựng mẫu phải đảm bảo sạch và phải áp dụng các biện pháp cần thiết để giảm sự biến đổi của mẫu đến mức tối thiểu.
- Lấy mẫu nước
Khi lấy mẫu nước cần phải đảm bảo không làm xáo trộn các tầng nước. Người ta thường dùng 4 loại dụng cụ lấy mẫu sau:
Dụng cụ lấy mẫu hình trụ mở.
Loại Runner và Kemmerer: có dạng hình trụ mở, dung tích từ 1 đến 3 lít có nắp đậy ở mỗi đầu. Các nắp này có thể được mở ra hoặc đóng vào nhờ một hệ thống dây ống Ruttner làm bằng nhựa còn ống Kemmerer làm bằng đồng. Khi nắp ống được mở ra, nước sẽ để qua, đến độ sâu cần lấy mẫu, người ta thường kẻo, hạ ống lên xuống vài lần (dao động trong khoảng 25cm) trước khi đóng nắp lại để lấy mẫu.
Dụng cụ lấy mẫu Friedinger: dụng cụ này có dạng gần giống với loại Runner và Kemmerer, nhưng nắp được mở ra 900. Tất cả các phần bên trong đều không làm bằng

kim loại. Dung tích dao động từ 3,5-5 lít, dụng cụ này có khung đê gắn nhiệt kế.
- Dụng cụ lấy mẫu có thoát khí, cổ hẹp
Dạng bình Dussart: dụng cụ này rất đơn giản, có thể tự làm lấy được. Dùng một bình có cổ hẹp (dạng cổ chai) có nút cao su với hai ống nhỏ. Một ống được kéo dài xuống tận đáy bình, còn một ống chỉ cần xuyên qua nắp cao su. Đầu trên của hai ống nhỏ này được nối với nhau bằng một ống nhỏ hình U hoặc nối bằng ống cao su mềm. Một vật đủ nặng buộc phía dưới bình đê kẻo bình chìm xuống nước óng chữ U được buộc chặt vào cổ bình và nối với dây kéo đề điều khiển khi lấy mẫu. Thả bình đến độ sâu cần thiết, kéo ống chữ U, nước sẽ tràn vào bình, đầy không khí thoát ra.
Dụng cụ lấy mẫu Valas: Tương tự như dụng cụ Dussart nhưng phần đóng mở có từ tỉnh và cấu tạo phức tạp hơn. Sau khi mở đề lấy mẫu, nắp sẽ được đậy kín lại. Dụng cụ này thường sử dụng để lấy mẫu phân tích vi sinh vật.
- Dụng cụ lấy mẫu bằng bơm
Dùng một ống bằng cao su hoặc nhựa (đủ nặng) để thả xuống độ sâu cần thiết. Dùng bơm để hút tạo dòng nước liên tục và mẫu nước được lấy. Nếu cần phân tích các chất khí thì đầu ống cần đặt sát đáy bình đựng mẫu và để lượng nước chảy qua ít nhất khoảng gấp 3 lần thể tích bình đựng mẫu. Để lấy mẫu nước ở một tang nhất định, ống cao su cần đặt song song với nhau ở khoảng giữa của hai đĩa nhựa có đường kính khoảng 10cm.
Dụng cụ lấy mẫu này sử dụng các loại bơm tay để tránh sự tiếp xúc của kim loại với nước. Nó có ưu điểm là ít làm xáo trộn mẫu nước. Thể tích của ống dài 10m, thiết diện lcm2 chỉ là 1 lít. Dụng cụ này thích hợp để lấy các mẫu hỗn hợp với các độ sâu khác nhau.
- Dụng cụ lấy mẫu nước có nhiều chức năng khác nhau.
Dụng cụ lấy mẫu Vondorn: đây là dụng cụ được dùng rất phổ biến để lấy mẫu nước. Gồm một ống hình trụ bằng chất dẻo, 2 van cao su nối với nhau bằng ống cao su ở phía trên và phía dưới ống. Hai van cao su này có thể đóng mở nhờ một chiếc khoá ở phía ngoài ống trụ. Khi thả dụng cụ xuống nước, 2 van cao su đều mở, đến độ sâu cần thiết sẽ được đóng lại nhờ hệ thống điều khiển (messenger). Mẫu được chuyên sang bình chứa nhờ lỗ nhỏ ở thành hình trụ.
6.3.4. Một số chỉ dẫn khi lấy mẫu nước.
Khi lấy mẫu đối với nguồn nước chảy như sông, suối thì nên lấy mẫu ở chỗ chảy mạnh nhất.
Mẫu lấy ở các hồ thiên nhiên, hồ chứa các ao đầm thì lấy mẫu ở các vị trí khác nhau và những độ sâu và những độ sâu khác nhau. Không nên lấy mẫu trung bình đối với loại mẫu này.

Mẫu lấy từ các mạch, các giếng, các hồ chứa nước nhân tạo thì nên lấy ở những độ sâu cần thiết.
Mẫu nước mưa chỉ cần hứng vào lúc trời mưa và cần ghi rõ thời điểm lấy mẫu.
Mẫu từ các trạm và các vòi nước sinh hoạt, thông thường được lấy trực tiếp tại các ống nước dẫn ra.
Mẫu nước thải, nôn lấy mau trung bình, trước khi lấy mẫu cần nêu cứu kỹ yêu cầu, mục đích sử dụng nước và các điều kiện sinh hoạt khác.
6.3.5. Bảo quản mẫu nước trước khi phân tích
Trong khoảng thời gian từ lúc lấy mau nước đến khi phân tích, hàm lượng của các phần có thể bị thay đổi với mức độ khác nhau
Nhiệt độ và pH của nước là đại lượng biến đổi nhanh nhất, vì vậy hai chỉ tiêu này cần xác định ngay tại nơi lấy mẫu. Hàm lượng một số khí cũng cần phải được xác định ngay như O2, CO2, H2S, Cl2.
Đối với việc xác định các kim loại nặng trong nước thì cần tuân thủ theo những quy trình sau đây để bảo quản mẫu thêm các hoá chất tinh khiết vào trong một lít nước.
Ag : Thêm 5 ml HNO3 đặc,
Ai : Thêm 5 ml HCl đặc,
As : Thêm 5 ml HCl
Ca : Thường không phải xử lý,
Cd : Thêm 5ml HNO3 đặc,
Co : Thêm 5ml HNO3 đặc,
Cr : Thêm 5ml HNO3 đặc,
Cu: Thêm 5ml HNO3 khi xác định tổng hàm lượng sắt. Thêm 25 ml CH3COONa 1M dùng để xác định các dạng khác nhau của sắt,
Hg : Thêm vào 1 ml HNO3 đặc,
Mg : Không cần phải xử lý,
Mg : Thêm 5 ml HNO3 đặc,
NH3 : Thêm 1 ml H2SO4 đặc và 2 - 3 ml CHCl3, bảo quản ở nhiệt độ 3 - 40C,
Ni : Thêm 5 ml HNO3
PO43- : Thêm vào 1 - 2 ml CHCl3,
Pb : Thêm 3 ml NHO3 đặc và 2 ml CH3COOH đặc,

Zn : Thêm 1 ml H2SO4 đặc.
SO42- : Thêm 2 - 4 CHCL3.
CN- : Giữ mẫu ở nhiệt độ 3 -40C, tăng ph, cần xác định ngay,
SIO32- : Đựng trong bình polietylen, khi hàm lượng SiO2 cao cần thêm 1
ml H2SO4 (1:3),
F- : phải đựng trong bình polietylen,
Cr : Thêm 2 - 4 ml CHCl3.
Borat: Đựng trong bình polietylen, hoặc bình thuỷ tinh không có bo (nên xác định ngay khi lấy mẫu),
SCN-: Nên xác định ngay, không nên để quá một đêm,
Phenol: Nếu hàm lượng vượt quá 100 mg/l thì không câng xử lý mẫu và không được lưu giữ quá 5 đêm, nếu hàm lượng < 100 mg/l có thể thêm vào 4 g NaOH/11, nếu hàm lượng < 0,5 mg/l cần phải phân tích ngay sau khi lấy mẫu,
HCHO: Không cần xử lý, cần phân tích ngay
Đường và tinh bột: không nên xác định chậm hơn 1 ngày đêm sau khi lấy mẫu.
6.4. Xác định thành phần hoá học của nước
6.4 1. Xác định pH
Ngày nay, việc xác định pH trở nên đơn giản do sự phát triển của khoa học công nghệ. Giá trị pH được xác định bằng phép đo trên máy đo pH (pH meter). Điểm cần lưu ý khi đo pH là việc bảo quản điện cực do các đặc điểm về khí hậu của vùng nhiệt đới. Trước khi tiến hành đo cần phải kiêm tra máy bằng cách điều chỉnh máy với các dung dịch đệm có giá trị pH = 4,00; 7,00; 10,00. Điện cực phải luôn được bảo quản trong dung dịch bảo quản.
6.4.2. Độ cứng
Độ cứng của nước do các kim loại kiềm thổ hoá trị hai, chủ yếu là Ca và Mg gây nên. Người ta thường phân biệt độ cứng cacbonat và độ cứng phi cacbonnat. Lượng lớn Ca và Mg tương ứng với các muốn của các axit vô cơ mạnh như HCl, H2SO4, HNO3 gọi là độ cứng phi Cacbonat. Độ cứng toàn phần là tổng của hai loại độ cứng trên.
Độ cứng thường được biểu thị bằng số mặt đương lượng gam của các ion Ca, Mg trong 1 lít nước hoặc đôi khi bằng thang độ cứng của Đức, ký hiệu là OH (l đơn vị độ cứng của Đức tương ứng với 10mg/l CaO/l).
a. Xác định độ cứng của nước bằngphươngpháp chuẩn độ Comptexon
Phương pháp này dựa trên phản ứng tạo phức của các ion Ca2+, Mg2+ với anion

etylenđiamintettraaxetat (ký hiệu là Y4-) các phức đó có độ bền không cao, phức CaY2- có hằng số bền là 1010,7, bền hơn phức MnY2- có hằng số bền là 108,7 vì nồng độ của các ion Ca2+ và Mg2+ trong nước thường không cao, nên để phản ứng chuẩn độ xảy ra hoàn toàn, người ta thường tiến hành chuẩn độ trong môi trường đệm NH4OH + NH4Cl có pH = 10,00. Dung dịch chuẩn là Complexon III. Để xác định điểm tương đương người ta dùng chỉ thị ET_OO (Eriocrom T đen). Trong môi trường chuẩn độ, chất chỉ thị sẽ phản ứng với ion Ca2+ và Mg2+ để tạo thành phức có màu đỏ.
Trình tự phân tích
Lấy 100 ml nước cần xác định độ cứng, thêm vào 10 ml dung dịch đệm (nếu độ kiềm của mẫu nước lớn thì phải dùng dung dịch HCI 0,1 M để điều chỉnh cho pH dung dịch bằng 7-8 theo giấy chỉ thị ứng hợp trước khi thêm dung dịch đệm), thêm một lượng nhỏ chất chỉ thị (khoảng bằng một hạt đỗ xanh), dung dịch có màu đỏ. Chuẩn dung dịch bằng dung dịch chuẩn Complexon III cho tới khi dung chuyển từ màu đỏ sang màu xanh. Nếu trong dung dịch có mặt các ion cản trở như Fe3+, Cu2+ thì cần chuẩn độ lại với mẫu nước khác và cách tiến hành như sau: Sau khi cho 10 ml dung dịch đệm, ta thêm vài giọt KCN 5% để che ion cản trở rồi mới thêm chất chỉ thị và tiến hành chuẩn độ như trên.
Tính kết quả
Độ cứng toàn phần của nước được tính như sau:
Trong đó: a: thể tích dung dịch Complexon III tiêu tốn;
V: thể tích mẫu nước lấy để phân tích (ml);
0,025: nồng độ dung dịch Complexon III (mol/l);
2: số đương lượng gam độ cứng tương ứng với 1 mol Complexon III.
Hoá chất
- Dung dịch Complexon III 0,025M: hoà tan 9,306g Complexon III trong 1 lít nước cất hai lần
- KCN 50%: cân 5g KCN hoà tan trong 100 ml nước cất hai lần.
- Eriocrom T đen cân 0,05g chất chỉ thị trộn với 10g NaCl.
- Đệm NH4OH + NH4Cl (pH = 10), hoà tan 25g NH4Cl trong 100 ml nước thêm 200ml NH4OH 20%, sau đó thêm nước cất đến 1 1ít.
b. Xác định Ca và Mg
Để xác định hàm lượng Ca và Mg trong nước sinh hoạt, nước uống, các loại

nước bề mặt, nước thải, người ta thường dừng phương pháp chuẩn độ Complexon III (Trilon B).
Phức EDTA - Ca bền hơn EDTA - Mg. Khi pH của dung dịch nằm trong khoảng 12- 13, EDTA - Ca bền vững trong khi đó EDTA - Mg bị phân huỷ vì tạo ra hiđroxit - Mg. Do đó có thể chuẩn độ Ca trong môi trường của NaOH có pH = 12- 13 với chỉ thị Murexit.
Xác định Ca.
Trình tự phân tích
Cho mẫu nước vào bình tam giác có V: 250ml, thể tích mẫu nước được lấy để không chứa quá 15mg Ca. Dùng nước cất pha loãng thành 100 ml, nếu thấy cần thiết. Nếu mẫu nước có tính axit thì cần trung hoà bằng dung dịch NaOH. Nếu mẫu nước có độ kiềm cao hơn 6 mđlg/l thì cần thêm vào lượng tương đương dung dịch HCl 0,1N, đun sôi dung dịch và để nguội. Sau đó thêm vào 2ml NaOH 2M và một ít chất chỉ thị murexit. Chuẩn độ từ từ bằng dung dịch EDTA 0,025M cho đến khi dung dịch chuyển từ màu hồng sang tím.
Tính kết qủa:
Trong đó a: thể tích dung dịch EDTA 0,025M;
V: thể tích mẫu nước chuẩn độ (ml);
1000: 2 x 0,025 x 20 x 1000.
Hoá chất
- EDTA 0,025M. Xem phần trên
- NaOH 2M: hoà tan 8g NaOH trong 100 ml nước cất hai lần.
- HCl 0,1 N: pha 8,2ml HCl đặc trong 1 lít nước cất hai lần
Xác định Mg.
Sau khi đã xác định Ca như trên, tiến hành xác định tổng số Ca + Mg tại các giả trị pH = 10 với chỉ thị Enocrom T đen.
Trình tự phân tích
Lấy vào bình tam giác có V = 250ml lượng thể tích như trong xác định Ca. Xử lý mẫu như đã làm đối với Ca. Thêm vào 5ml Hiđroxilamin 10%, vài giọt KCN 10% và một lượng nhỏ chỉ thị Eriocrom T đen rồi tiến hành chuẩn độ bằng EDTA 0,025M.
Tính kết qủa

Trong đó:
V1: thể tích EDTA chuẩn Ca + Mg (ml);
V2: thể tích Ca (ml);
V: thể tích mẫu nước (ml).
Hoá chất
Hidroximalin 10%: hoà tan 10g NH2OH.HCl trong 100 ml nước cất hai lần
6.4.3. Xác định Cu
Hàm lượng đồng trong các loại nước thiên nhiên và trong các nguồn nước sinh hoạt thường không lớn lắm, dao động trong khoảng từ 0,001 - 1 mg/1. Trong nước, Cu thường tồn tại ở dạng ion Cu2+, hoặc dưới dạng phức xianua, tactrat...
Để xác định Cu người ta thường dùng phương pháp trắc quang với đietylđithiocacbonat (DDC).
Phản ứng giữa Cu2+ với Pb-DDC xảy ra tại giá trị pH = 1 - 1,5. Trong trường hợp này cùng với Cu còn có Bi, Hg, Ag tham gia vào phản ứng. Các nguyên tố còn lại không tham gia phản ứng nên phương pháp này có độ chọn lọc cao. Hg và Ag không gây cản trở vì phức của chúng không có màu. Bitmut (Bi) chỉ gây cản trở khô nồng độ của nó vượt quá 30 μg/l. Nếu nồng độ Bi vượt quá giới hạn thì cần phải lắc phức DDC- kim loại với 25ml HCl 5M trong 5 phút, khi đó phức Bi-DDC bị phân huỷ còn Cu-DDC không bị phân huỷ.
Khi trong mẫu có chứa phức Cu-xianua, thì cần phải phá huỷ phức đó bằng cách thêm 0,5ml H2SO4 (1 : l); 5mL HNO3 đặc Vào 200-250ml mẫu, cho mẫu vào cốc chịu nhiệt rồi đặt lên bếp, làm bay hơi dung dịch cho đến khi gần cạn khô (quá trình phá huỷ này nên tiến hành trong tủ hút). Để nguội thêm vào 1 ml HCl đặc và làm bay hơi đến khô. Để nguội, thêm vào nước cất 2 lần, đun nóng dung dịch đến sôi để hoà tan hết lượng muối rắn, lọc và chuyền toàn bộ dung dịch thu được vào bình định mức thích hợp, đinh mức bằng nước cất hai lần và giữ lấy để phân tích.
Trình tự phân tích
Lấy một thể tích nước cần phân tích để trong mẫu chứa khoảng 0,2-0,6 μg Cu/l, cho vào phễu chiết dung tích 200-500 ml. Pha loãng mẫu nước nếu cần đến thể tích 100 ml rồi thêm vào 5 giọt HCl (1:1), từ buret thêm vào một cách chính xác 10 ml Pb- DDC trong toluen. Cẩn thận đậy nút phễu chiết tại và lắc trong hai phút. Giữ yên phễu chiết để cho hai tướng phân lớp, tách bỏ phần tướng nước phía dưới, phần tướng hữu cơ chứa Cu-DDC có màu vàng được chuyển vào cuvet và tiến hành so màu tại bước

sóng λ = 430nm.
Lập đường chuẩn
Lấy vào các phễu chiết 0,00; 0,2; 0,5; 1,0; 2,0; 3,0; 4,0; 5,0; 6,0 ml dung dịch chuẩn Cu2+ có nồng độ 1μg Cu/ml, pha loãng bằng nước cất đến 100 ml và tiến hành như với mẫu.
Tính kết quả
Dựa vào đường chuẩn xây dụng mối tương quan y = a.x + b
Trong đó y: hàm lượng Cu2+
x: mật độ quang đo được
Hoá chất
- Pb-DDC trong toluen: Chuẩn bi một phễu chiết sạch có V =1000 ml, thêm vào đó 50 -100 ml nước cất hai lần, 0,1g Pb(NO3)2 loại tinh khiết hoá học, tắc kĩ để muối đó tan hết. Hoà tan 0,1 g Na-DDC trong lượng nước tối thiểu rồi thêm vào phễu chiết đề kết tủa hết Pb-DDC. Thêm vào phễu chiết 250ml toluen, đậy nút phễu chiết và lắc mạnh, toàn bộ kết tủa Pb-DDC sẽ tan hết vào trong toluen, tách bỏ tướng.nước, phần tướng hữu cơ được lọc qua giấy lọc vào bình màu nâu, dung dịch này bền trong khoảng 3 tháng
HCl loãng: Pha dung dịch HCl 0,01N.
- Dung dịch Cu2+ nồng độ 1 μg Cu/ml: Hoà tan 3,906 mg CuSO4.5H2O trong 1 lít nước cất hai lần.
6.4.4. Xác định Pb bằng phương pháp trắc quan đithizon
Hàm lượng Pb trong nước thiên nhiên rất nhỏ, nằm trong khoảng 0,001-0,02mg/1. Trong nước sinh hoạt cũng có lượng vết Pb do ống nước được làm bằng Pb. Trong nước thải đặc biệt là nước thải của các nhà máy sản xuất Pb, Zn, hàm lượng Pb có thể lên tới 6 -7 màu. Chỉ trong nước thải có thể ở dạng tan hoặc dạng muối khó tan như sunfat, cacbonat, sunfua.
Đithizon-điphenylthiocacbonat là thuốc thử hữu cơ có khả năng tạo phức càng của với Pb2+. Phức Pb-đithiozonat có màu đỏ tan trong toluen, phúc này được so màu tại bước sóng λ =520 nm.
Pb-đithiozonat được chiết chọn lọc và định lượng từ dung dịch có pH= 8 - 9, có chứa lượng dư CN- là chất dùng để che nhiều kim loại khác cùng bị chiết với Pb. Trong môi trường trên cùng bị chiết với Pb chỉ có Ta, Bi,Sn. Ở đây Tali không cản trở, nhưng Bi và Sn gây cản trở, do vậy chúng cần được tách trước từ môi trường axit. Thêm hiđrazin vào dung dịguội thêm dung địch mẫu nước, đun nóng để khử Sn (IV) xuống Sn (VI) và khử các chất khác. Sau khi để nguội thêm dung dịch kalinatritactrat vào, đưa dung dịch đến pH = 2,5 - 3 bằng axit tactric, rồi tiến hành triết nhiều lần, mỗi

lần bằng 5/ ml đithizon trong toluen cho tới khi màu xanh của đithizon không đổi. Tách lấy tướng nước. Tiến hành chiết như vậy không những tách được thiếc mà còn tách được cả Hg, Ag, Cu.
Nếu trong mẫu có chứa lượng đáng kể các chất hữu cơ thì cần phải vô cơ hoá chúng bằng hỗn hợp axit mạnh như H2SO4 và HClO4. Các chất ôxy hoá cần khử trước bằng hiđrazin.
Trình tự phân tích
Thêm vào cốc 100 ml mẫu nước đã được pha loãng hoặc cô nước để trong đó chứa từ 0,01 - 0,1 mg Pb. Sử dụng bếp cách thuỷ, làm bay hơi dung dịch đến khi còn 30 ml, chỉnh pH tới 7 theo giấy chỉ thị tổng hợp. Thêm vào 10 ml hiđroxilamin 10% và đun nóng hỗn hợp ở to = 90 - 950C trong khoảng 12 phút. Để nguội dung (tích, dùng axit HCl loãng hoặc axetec loãng chỉnh pH dung dịch tới 3. Chuyển toàn bộ dung dịch trên vào phễu chiết, từ buret thêm cẩn thận 10 ml dung dịch đithizon và tiến hành chiết cho đến khi màu xanh của đithizon không thay đổi. Tách lấy tướng nước, rồi chỉnh pH của dung dịch tới 8-9 bằng dung dịch amoniac, thêm vào 5 - 1 0 ml dung dịch KCN 10%. Sau đó chuyển toàn bộ dung dịch vào phễu chiết, thêm vào chính xác 10 ml dung dịch đithizon và tiến hành chiết cho tới khi màu xanh của đithizon không thay đổi. Gộp tất cả các phần đithizon chiết được lại, chuyên vào một phễu chiết sạch rồi tiến hành rửa bằng dung dịch amoniac loãng, khi đó phức của Pb-đithizonat có màu đỏ.
Lập đường chuẩn
Thêm vào một loạt cốc 0; 2,5; 5,0; 10,0; 20,0; 30,0; 40,0; 50,0 ml dung dịch chuẩn chứa 0,002 mgPb/ml và thêm nước cất vào mỗi cốc đến thể tích 100 ml. Như vậy ta được các dung dịch có hàm lượng tương ứng 0; 0,05; 0,1... mg Pb/lít. Rồi tiến mậu tương tự như đối với mẫu.
Tính kết quả
Dựa vào đường chuẩn xây dụng mối tương quan y = a.x + b
y: hàm lượng Pb trong mẫu
x: mật độ quang đo được. Từ đó thay giá trị mật độ quang đo được của mẫu vào phương trình trên ta sẽ tính được hàm lượng Pb có trong mẫu.
Hoá chất
Đithizon: Hoà tan 50 mg đithizon trong 100 ml toluen trong phễu chiết thể tích 1000ml. Thêm vào đó 200ml nước cất và 5 - 10 ml dung dịch NH3 đặc. Đậy nút phễu chiết cẩn thận và lắc trong hai phút. Lúc này đithizon đã chuyển sang tướng nước, sau khi hai tướng đã phân lớp, tách bỏ phần dung môi phía trên, lại thêm vào tướng nước 20ml toluen, lắc hỗn hợp, tách bỏ phần toluen. Cẩn thận thêm vào phễu chiết 200ml toluen và dung dịch HCl loãng cho đến khi tướng nước có phản ứng axit rõ thử bằng giấy chỉ thị pH. Đậy nút phễu lại và lắc hỗn hợp để chuyển hoàn toàn đithizon sang

tướng hữu cơ. Chuyển tướng hữu cơ sang một phễu chiết khác, rửa nó vài lần, mỗi lần bằng 50 ml nước cất họ lần cho đến khi tướng nước hết phản ứng axit. Tách bỏ phần tướng nước, tướng hữu cơ chứa đithizon được chuyển vào bình màu nâu sạch và khô, bảo quản dung dịch ở nhiệt độ thấp và tránh ánh sáng. Dung dịch bền trong vài tháng.
- Dung dịch làm việc: Lấy một thể tích dung dịch đithizon gốc pha cùng với một thể tích toluen, động trong bình thuỷ tinh màu nâu sẫm. Dung dịch bền trong một tuần.
- Dung dịch rửa amoniac loãng Pha 175 ml NH3 đặc bằng nước cất hai lần tới thể tích 1 lít.
- Kali natri tactrat 10%: Hoà tan 10 gam KNaC4H4O6.2H2O trong 100 ml nước cất hai lần.
- KCN 10%: Hoà tan 10 gam KCN trong 100 ml nước cất hai lần đã được kiềm hóa.
- Dung dịch 0,002 mg Pb/ml: Hoà tan 3,197 mg Pb(NO3)2 trong nước cất hai lần thành 1 lít
6. 4.5. Xác định Zn bằng phương pháp trắc quan đithizon
Kẽm trong nước tự nhiên chủ yếu từ các nguồn nước thải, đặc biệt nước thải của các nhà máy luyện kim, công nghiệp hoá chất, các nhà máy sợi tống hợp. Các dạng tồn tại của Zn trong nước và các ion đơn, ton phức với xianua, cacbonat, sunfua...
Nói chung khi lấy mẫu nước để phân tích cần thêm 1 ml axit H2SO4 đặc vào 1 lít nước, trừ trường hợp mẫu có chứa xianua.
Để xác định Zn trong nước người ta dùng phương pháp trắc quan đithizon là phương pháp cho phép xác định đến với phần trăm miligam kẽm trong 1 lít nước.
Ion Zn tạo phức với đithizon tạo thành Kẽm-đithizonat có màu đỏ hồng. Phản ứng giữa kẽm và đithizon xảy ra tại pH = 5 và dung dịch có chứa lượng dư S2O3
2-, CN- dùng để che Cd, Ni, Pb... Do đó nếu trong mẫu nước không có các ion này hoặc có nhưng với hàm lượng nhỏ hơn kem thì không cần dùng xianua. Thiếc (II) cần được ôxy hoá trước thành Thiếc (IV) bằng H2O2 và đun sôi kỹ để phân huỷ H2O2 còn dư. Các chất ôxy hoá được đithizon cần được loại trừ trước khi xác định. Fe (III) cần được loại trừ trước bằng cách kết tủa nó dưới dạng hiđroxit và lọc bỏ.
Trình tự phân tích
Lấy 100 - 200 ml nước cần phân tích cho vào phễu chiết, mẫu nước cần được pha loãng trước hoặc làm bay hơi để chứa khoảng 0,005 - 0,03 mg Zn. Dùng axit axetic hoặc NaOH loãng chỉnh pH của dung dịch tới 7 rồi thêm vào chính xác 20ml dung dịch đệm, có pH:5 và 20ml dung dịch che. Từ buret thêm vào chính xác loạn dung dịch đithizin, đậy nút phễu, lắc cẩn thận trong khoảng 2 phút. Quá trình chiết kết thúc khi màu của đithizon giữ nguyên màu xanh. Gộp tất cả các phần đithizon được chiết lý

chuyên vào một phễu chiết sạch khô khác rồi tiến hành rửa đithizon dư bằng dung dịch amoniac loãng như đối với chì. Khi đó phức Zn - đithizonat có màu hồng xuất hiện. Phức màu này được đem so màu tại bước sóng λ = 530 nm.
Lập đường chuẩn
Chuẩn bị 7 phễu chiết sạch, thêm vào lần lượt từng phễu 0; 10;20;30;40;50;60 mà dung dịch có nồng độ 0,0005 mg Zn/ml. Thêm nước cất 2 lần vào để được thể tích là 100ml rồi tiến hành như đối với mẫu phân tích.
Tính kết quả
Dựa trên việc lập thang chuẩn sẽ xác định được mối tương quan hàm số y = a.x + b.
Trong đó: y: hàm lượng Zn trong mẫu.
x: mật độ quang
Hoá chất
Dung dịch che: Hoà tan 120 g Na2S2O3.5H2O; 0,15g (NH4)2C2O4.2H2O; l,5g KCN (nếu cần), 24 g CH3COONa.3H2O, 10 ml NH3 2N, 70 ml HCl là trong nước cất hai lần rồi chuyển vào bình định mức 1 lít và định mức đến vạch mức.
Dung dịch đệm pH = 5. Lấy 73,4 ml CH3COOH 1N và 50 ml NaOH lN thêm nước cất đến 500 ml và lắc đều.
- Đithizon: Sử dụng như trong xác định Pb.
- Dung dịch chuẩn Zn: Xem phần xác định Hg trong đất.
6.4.6. Xác định Hg bằng phương pháp trắc quan đithizon
Thuỷ ngân đôi khi có trong nước chảy ra từ các mỏ và trong nước thải sản xuất các chất màu, dược phẩm và chất nồ. Việc xác định Hg là rất quan trọng và cần thiết, vì các hợp chất của nó thường rất độc.
Để xác định Hg thường dùng phương pháp chiết trắc quang đithizon.
Phương pháp này rất đặc trưng và chọn lọc đối với Hg, vì nó được chiết hoàn toàn từ môi trường có độ axit rất cao, trong môi trường này tuyệt đại đa số các kim loại khác không bị chiết. Phương pháp cho phép xác định từ vài phần trăm miligam tới hàng chục miligam trong 1 lít nước.
Nếu để mẫu nước lâu mới tiến hành phân tích thì cần thêm vào 1 ml HNO3 đặc cho 1 lít mẫu.
Trong môi trường axit chỉ có Au, Cu cùng bị chiết với Hg. Để che hai nguyên tố này phải dùng Complexon III và Thioxianat (SCN-).
Trong môi trường đệm axetat có chứa Complexon III và Thioxianat (SCN-) chỉ có Au (m) và có thể cả Pt (II) có ảnh hưởng đến việc xác định, nhưng hai nguyên tố đó

thường ít khi có mặt trong nước.
Chất hữu cơ có màu thường tách nước bằng toluen. Nếu mẫu có chứa nhiều chất hữu cơ thì cần vô vơ hoá trước khi xác định.
Trình tự phân tích
Cho vào phễu chiết một thể tích nước thích hợp để trong đó có chứa 0,005 - 0,1 mg Hg, pha loãng đến 100 ml bằng nước cất hai lần. Thêm vào 20ml dung dịch đệm, 10 ml Complexon (m), 10 ml KSCN. Từ buret thêm chính xác 10 ml đithizon, đậy nút phễu chiết, lắc cẩn thận trong khoảng 2 phút. Quá trình chiết kết thúc khi màu xanh của đithizon không thay đổi. Gộp tất cả các phần đithizon đã chiết được lại, chuyển vào một phễu chiết sạch khô. Dùng dung dịch amoniac loãng 5% để rửa đithizon dư. Tách bỏ phần tướng nước, khi đó phức Hg - đithizonat có màu đỏ da cam xuất hiện Phức màu này được so màu tại bước sóng λ = 490 nm.
Lập đường chuẩn
Phép tập đường chuẩn tương tự như khi xác định các nguyên tố khác. Đường chuẩn ở đây được lập với các dung dịch ứng với 0; 0,05; 0,1 mg Hg/l. Tiến hành tương tự như đối với mẫu phân tích.
Tính kết quả
Tương tự như đối với các nguyên tố khác ta xây dụng hàm tương quan; y = a.x + b
Trong đó: y: hàm lượng Hg trong mẫu;
x: mật độ quang
Hoá chất
Dung dịch đệm axetat (pH = 4): Hoà tan 57 ml CH3COOH đặc và 82g CH3COONa.3H2O trong nước cát thành 1 lít.
Complexon III 0,05N. Hòa tan 9,306 g EDTA trong nước cất thành 1 lít.
KSCN: Pha dung dịch 0,1 M
Dung dịch chuẩn Mg: Xem phần xác định Hg trong đất.
6.4.7. Xác định Fe
Hàm lượng của Fe có trong nước thiên nhiên phụ thuộc rất nhiều vào nguồn nước và những vùng mà nguồn nước chảy qua. Ngoài ra còn tuỳ thuộc vào pH và sự có mật của một số chất như CO3
2-, O2, S2 và các chất hữu cơ tan trong nước, chúng sẽ oxy hoá hay khử Fe và có thể làm cho Fe nằm ở dạng tan hay kết tủa.
Để xác định Fe trong nước, thường dùng phương pháp so màu với các thuốc thử như axit sunfoxalixilic hoặc o-phenalthrolin.
Muốn xác định sắt nằm ở các dạng khác nhau, một điều quan trọng cần phải chú

ý là cách ấy mẫu. Để xác định tổng lượng sắt có trong mẫu cần phải xử lý mỗi mẫu nước với 25 ml HNO3 đặc.
Nếu muốn xác định Fe trong mẫu ở các dạng khác nhau thì mỗi lít nước cần phải xử lý bằng 25 ml dung dịch đệm CH3OONa 1 M.
a) Xác định Fe bằng thuốc thử o- phenalthrolin.
Thuốc thử o- phenalthrolin phản ứng với ion Fe2+ tạo thành phức chất có màu tím đỏ. Khoảng pH cho quá trình tạo phức khá rộng, từ 3 - 9. Mn có ảnh hưởng rất lớn đến quá trình xác định nên khi mẫu phân tích có mặt Mn thì không định lượng được Fe bằng phương pháp này. Lượng Cu lớn hơn 10 mg/1 cũng gây ảnh hưởng khi xác định nhưng có thể loại trừ ảnh hưởng đó bằng cách tiến hành phản ứng trong khoảng pH từ 3 đến 4.
Nếu trong mẫu có chứa lượng lớn các chất hữu cơ thì trước đó cần phải vô cơ hoá.
Trình tự phân tích.
Lấy lượng mẫu nước cần phân tích (sao cho lượng Fe trong đó không quá 0,2 mới cho vào bình định mức 50 ml. Thêm 1 ml HCl loãng, cô đến thể tích còn khoảng 40 ml. Làm nguội, thêm 1 ml Hiđroxilamin để khử Fe3+ thành Fe2+, rồi thêm lưu o- phenalthrolin. Nhỏ từng giọt NH3 đến khi giấy chỉ thị Cônggô chuyển sang mầu xanh. Định mức bằng nước cất lắc đều. Sau đó tiến hành so mẫu tại bước sóng λ = 540 nm.
Lập đường chuẩn
Lấy lần lưọt vào mỗi bình 0; 0,5; 1,0; 2,0; 5,0; 10,0; 20,0; 30,0; 40,0; 50,0 ml dung dịch 0,005mg Fe/ml, rồi tiến hành tương tự như đối với mẫu.
Tính kết quả
Dựa vào đường chuẩn xác lập hàm tương quan y = a.x + b
y: hàm lượng Fe trong mẫu;
x: mật độ quang.
Hoá chất
- O- phenalthrolin: Hoà tan 0,28 g o- phenalthrolin trong nước cất thành 100 ml.
- Hiđroxilamin 10%: Hòa tan 10 g Hiđroxilamin trong nước cất thành 100 ml.
- HCl: Dung dịch loãng(1: 9).
- Dung dịch NH3 đặc
- CH3COONa lM: Hoà tan 68g CH3COONa.3H2O trong 500 ml nước, thêm 25 ml CH3COOH 6M sau đó thêm nước cất đến 1 lít.
b. Xác định Fe bằng thuốc thử axit sunfoxalixilic

Ion Fe3+ phản ứng với thuốc thử axit sunfoxalixilic trong môi trường kiềm tạo thành phức có màu vàng. Lượng lớn Al, Cu có trong mẫu sẽ cản trở phép xác định.
Trình tự phân tích.
Cần lấy lượng mẫu nước thích hợp để trong đó có chứa từ 0,01 - 1,00 mg Fe, cho vào cốc có dung dịch 100 ml. Thêm 0,5ml HNO3 đặc, làm bay hơi đến còn khoảng 10 ml. Sau đó pha loãng bằng nước cất, lọc, thu toàn bộ nước lọc và nước rửa vào bình định mức 100 ml, thêm nước cất đến khoảng 90 ml. Thêm 2ml dung dịch NH4Cl, 2ml dung dịch axit sunfoxalixilic và 2ml dung dịch NH3. Thêm nước tới vạch mức, lắc đều, sau 5 phút đo mật độ quang của dung dịch tại bước sóng λ = 430nm.
Lập đường chuẩn
Lần lượt lấy vào mỗi cốc 0; l;2; 3;4; 5; 6; 7; 8; 9; 10 ml dung dịch có nồng độ 0,1 mg Fe/ml. Rồi tiến hành tương tự như với mẫu phân tích.
Tính kết qủa
Dựa vào đường chuẩn lập hàm tương quan y = a.x + b
y: hàm lượng Fe có trong mẫu
x: mật độ quang đo được
Hoá chất
- Axit sunfoxalixilic 20%
- NH3 đặc
- NH4Cl 2N: Hoà tan 1O7 g NH4Cl trong 1 lít nước cất hai lần
- HNO3 đặc.
- Dung dịch chuẩn sắc: Hoà tan 0,864 g NH4Fe(SO4)2.12H2O tinh khiết phân tích trong nước, thêm 2 ml HCl đặc, định mức đến 1 lít, dung dịch này chứa 0,1 mg Fe/ ml.
Lấy 50ml dung dịch chuẩn này pha thành 1 lít được dung dịch chứa 0,005 mg Fe/ ml.
6.4.8. Xác định Mn
Trong nước Mn thường nằm ở hai dạng tan và không tan. Ở dạng tan Mn tồn tại dưới dạng ion Mn2+, còn ở dạng không tan là kết tủa hiđroxit. Hàm lượng Mn có trong nước tuỳ thuộc vào nguồn nước, hàm lượng Mn cao trong nước thải của nhà máy luyện kim và nhà máy hoá chất.
Để xác định tổng lượng Mn có trong nước người ta thường dùng phương pháp trắc quan (so màu). Khi đó Mn sẽ bị ôxy hoá thành Mn7+ bởi chất ôxy hoá mạnh như amonipesunfat (NH4)2S2O8. Chú ý khi lấy mẫu phải thêm vào mỗi lít nước 5ml NH3 đặc.

Trình tự phân tích
Cho 50 đến 100 ml mẫu nước vào cốc 150ml sao cho lượng Mn trong đó là 0,005 - 1 mg, thêm 2ml HNO3 nhỏ từng giọt dung dịch AgNO3 cho đến khi kết tủa hết ion Cl- thêm tiếp 1 - 2 ml dung dịch AgNO3 để lắng rồi lọc. Nếu hàm lượng clorua và các chất hữu cơ lớn thì phải tiến hành vô vơ hoá mẫu, không thể loại trừ Cr bởi AgNO3 khi hàm lượng của chúng quá lớn vì sẽ gây hiện tượng hấp phụ Mn. Thêm 0,5g pesunfat vào nước lọc. Điện dung dịch tới gần sôi khoảng 10 phút. Để nguội rồi chuyển vào bình định mức có đung tích 100 ml, thêm nước tới vạch mức. Tiến hành so màu tại bước sóng λ = 525nm.
Lập đường chuẩn
Cho vào 10 cốc 150 ml chịu nhiệt lần lượt theo thứ tự 0; 0,5; l; 2,5; 5; 10; 20; 30; 40; 50 ml dung dịch chuẩn Mn có nồng độ 0,01 mg/ ml rồi tiến hành như đối với mẫu phân tích
Tính kết quả
Dựa vào thang chuẩn thiết lập mối tương quan hàm số y = a.x + b.
Trong đó: y: hàm lượng Mn
x: mật độ quang đo được
Hoá chất
- HNO3 đặc.
- AgNO3 đặc: Hoà tan 17g AgNO3 trong 500ml nước cất hai lan.
- (NH4)2S2O8 muối rắn.
Dung dịch chuẩn: Hoà tan 0,2748g MnSO4 trong 1 lít nước cất hai lần đã được axit hoá bằng H2SO4, dung dịch này có nồng độ 0,1 mg. Pha loãng dung dịch này 10 lần ta được dung dịch có nồng độ 0,01 mg/ml.
6.4 9. Xác định Cr
Trong môi trường axit, cromat và bicromat phân ứng với điphenycacbazit tạo thành hợp chất tan màu đỏ tím rất thuận lợi cho việc đo màu. Phản ứng này dùng để định lượng Cr có hàm lượng từ 0,005 - 1,00 mg/l.
Các ion Hg2+ và Hg22+ (khi hàm lượng >200 mg/l), Mo
6+ cũng có khả năng tạo phức màu với thuốc thử, tuy nhiên những ion này thường có rất ít trong nước. Sắt với hàm lượng lớn hơn 1 mol cũng phản ứng với thuốc thử tạo thành hợp chất có màu vàng nâu, nhưng có thể loại trừ Fe bằng cách thêm vào lượng nhỏ H3PO4. Nếu trong mẫu có chứa hàm lượng Mn thì phải ôxy hoá bằng pesunfat trong môi trường kiềm hoặc trung tính, nó sẽ tạo thành kết tủa MnO2 và được lọc bỏ bằng bông thuỷ tinh.
Nếu mẫu nước có tính kiềm hay trung tính thì rất khó xác định riêng Cr3+ và Cr6+.

Bởi vì khi axit hoá, nếu trong dung dịch có chất khử như Fe2+, sunfit và các chất hữu cơ thì Cr6+ sẽ bi khử xuống Cr3+. Trong trường hợp này chỉ nên xác định tổng hàm lượng Cr. Muốn xác định riêng ta phải tách riêng Cr3+ bằng cách kết tủa nó bằng MgO (pH = 10,5 - 11). Khi đó Cr(OH)3 kết tủa trên bề mặt MgO, còn Cr6+ vẫn tan trong dung dịch. Lượng lớn chất hữu cơ, các chất khử và ion Cr không cản trở phép xác định
a) Xác định Cr6+
Trình tự phân tích
Lấy lượng mẫu phân tích vào bình định mức sao cho trong đó có khoảng 0,005- 0,1 mg crom. Mẫu nước được cho vào bình nón cỡ 25ml, thêm vài giọt phenotphtalein, nếu dung dịch có màu hồng thì thêm từng giọt H2SO4 1N tới khi mất màu, ghi thể tích dung dịch H2SO4 1N đã dùng: nếu dung dịch không có màu thì thêm từng giọt NaOH lN. Thêm bằng ấy giọt H2SO4 lN hoặc NaOH là vào mẫu để trung hoà. Sau đó thêm 1 ml H2SO4, 0,2ml axitphotphoric và 2 ml dung dịch điphenylcacbant, thêm nước tốt vạch mức, sắc đều. Sau 5 - 10 phút thì đem so màu tại bước sóng λ = 540nm.
Lập đường chuẩn
Chuẩn bị 10 bình định mức có dung tích 100 ml. Lần lượt lấy vào mỗi bình 0; 1;2;5; 10; 15; 30; 40; 50ml dung dịch có nồng độ 0,002mg Cr/ml rồi tiến hành tương tự như đối với mẫu phân tích
Tính kết quả
Dựa vào đường chuẩn thiết lập mối tương quan hàm số y = a.x + b.
Trong đó: y: hàm lượng Cr
x: mật độ quang đo được.
b. Xác định tổng hàm lượng Cr
Lấy lượng mẫu nước cần phân tích cho vào cốc có thể lịch 250ml sao cho lượng Cr trong đó khoảng 0,005 - lmg. Trung hoà bằng H2SO4 hoặc NaOH 1N. Sau đó thêm 0,3ml H2SO4 1N, 5 - 10 ml dung dịch amonipesunfat, đun sôi dung dịch 20 - 25 phút (để phân huỷ hết pesunfat dư). Làm bay hơi bớt để.dung dịch còn khoảng 50ml. Chuyển tất cả vào bình định mức và tiến hành như xác định Cr6+.
Hoá chất dùng để xác định các dạng Cr
- NaOH 1N: Hoà tan 40 g NaOH trong 1 nước cất hai lần.
- H2SO4 1N: Cho 28 ml H2SO4 đặc vào 500 ml nước cất hai lần rồi định mức tới 1 lít.
- H3PO4 đặc 85%.
Điphenylcacbazit 0,5% trong axeton: Hoà tan 0,25g Điphenylcacbazit trong 50ml axeton.

- Amonipesunfat 0,1 %: Hoà tan 0,1 g amonipesunfat trong 100 ml nước cất hai lần.
- Dung dịch chuẩn: Hoà tan 2,8285g K2Cr2O7 trong 1 lít nước cất hai lần được dung dịch có nồng độ 1 mg/ml. Sử dụng dung dịch này để pha thành dung dịch có nồng độ 0,002 mg/ml.
6.4.10. Xác định Ni bằng thuốc thử đimetylglioxim
Trong nước sinh hoạt và nước tự nhiên thường không có Ni hoặc là lượng vết. Ni chỉ có trong nước ở một số hồ hoặc sông mà nguồn nước chảy qua những vùng núi và vùng mỏ có Ni. Ni có trong nước thải của một số nhà máy luyện kim và hoá chất có sử dụng Ni. Trong nước Ni tồn tại dưới dạng Ni2+, dạng phức xianua và dạng ít tan như sunfua, cacbonat, hiđroxit...
Để xác định Ni người ta thường dùng phương pháp so màu khi hàm lượng Ni nằm trong khoảng 1 - 20 mg/l, nếu hàm lượng Ni lớn hơn 5 mg/l thì có thể dùng phương pháp khối lượng, còn khi hàm lượng khoảng 0,02 mg/l dùng phương pháp cực phổ.
Mẫu nước lấy xong chưa phân tích ngay thì phải thêm 2 - 5ml HNO3 đặc vào 1 lít mẫu nước. Nếu cần định lượng riêng Ni ở dạng tan và không tan thì khi lấy mẫu ta phải lọc ngay rồi mới đóng chai bảo quản và xác định Ni ở dạng tan và tổng lượng Ni, từ đó suy ra hàm lượng Ni ở dạng không tan.
Trong môi trường amoniac yếu, khi có mặt chất ôxy hoá mạnh, ion Ni2+ phản ứng với đimetylglioxim tạo thành phức chất có màu đỏ.
Sắt Crom và đồng cản trở phép xác định nên cần loại bỏ trước. Để loại bỏ Fe thêm 2 ml H2O2 3% vào 100 ml mẫu phân tích, đun sôi và kết tủa hiđroxit bằng dung dịch amoniac, lọc bỏ kết tủa.
Loại bỏ Cu bằng cách axit hoá mẫu HCl đến ph = 2, sau đó sục khí H2S, lọc bỏ kết tủa, nước lọc trước khi phân tích phải đun sôi kỹ để đuổi hết H2S dư.
Trình tự phân tích
Cho vào bình định mức 100 ml một thể tích mẫu sao cho lượng Ni trong mẫu là 0,01 - 0,25mg thêm vào đó 10ml dung dịch nước tham bão hoà, lắc hỗn hợp. Sau đó thêm 12ml dung dịch amoniac, 4 ml dung dịch đimetylglioxim, thêm nước cất tới vạch mức. Sau đó tiến hành đo mật độ quang tại bước sóng λ =540nm.
Lập đường chuẩn
Chuẩn bị 8 bình định mức có dung tích 100 ml, lần lượt lấy vào mỗi bình 0; 2,5; 5;10; 20; 30; 40; 50ml dung dịch có nồng độ 0,005 mg/ml. Sau đó pha loãng thành 50 ml bằng nước cất rồi tiến hành như đối với mẫu phân tích
Tính kết quả

Dựa vào đường chuẩn thiết lập mối tương quan hàm số y = ax+b.
Trong đó: y hàm lượng Ni trong mẫu;
x: là mật độ quang đo được.
Hoá chất
- Dung dịch nước brom bão hoà.
- Amoniac đặc.
- Đimetylglioxim 1,2%: Hoà tan 1,2g muối nam đimetylglioxim trong nước cất và pha loãng thành 100 ml.
- Dung dịch chuẩn: Hoà tan là dây Ni và 15ml HNO3 35 %, thêm 5ml H2SO4 (1:3), đun đen bốc khói trắng. Hoà tan phần chất rắn vào thêm nước cất đến 1 lít. Dung dịch này có nồng độ 1mg Ni/ml. Từ dung dịch này pha thành dung dịch có nồng độ 0,005mg/ml.
6.4.11. Xác định Asen(As) bằng phương pháp trắc quang
Trong nước As thường nằm ở dạng asenat. As hầu như không có trong nước ngầm, nó chi có trong các nguồn nước thải của các nhà máy luyện kim, công nghệ hoá học, các nhà máy da và sản xuất chất mầu.
Để xác định Asen người ta thường dùng phương pháp so màu với thuốc thử Ao- đietylđithiocacbamat. Bằng phương pháp này có thể xác định được đến 0,05mg Asen trong 1 lít nước.
Dưới tác dụng của dòng hidro mới sinh, asenat bị khử thành asin (AsH3) và sau đó phản ứng với Ag-đietylđithiocacbamat tạo hợp chất có màu đỏ khi có mặt của piriđi.
Trong điều kiện này, H2S cũng phản ứng với thuốc thử tạo thành hợp chất có màu tương tự. Ta phải loại bỏ ảnh hưởng của H2S bằng cách cho hấp thụ trên ống có đựng giấy lọc có tẩm chì axetat hoặc bằng cách vô cơ hoá nó như một chất hữu cơ SbH3 cũng có khả năng phản ứng tương tự như AsH3 nhưng nó chỉ tạo thành hợp chất màu hồng nhạt khi có hàm lượng cao hơn 0,1 mg/l, cho nên thực tế có thể bỏ qua.
Lượng chất hữu cơ trong nước cũng gây cản trở phép xác định nên cần loại bỏ chúng bằng cách vô cơ hoá mẫu. Lấy lượng mẫu sao cho có 0,002 - 0,4mg As, thêm vào 2 - 3ml H2SO4 đặc 5ml HNO3 đặc Và 0,5ml H2O2 3%, làm bay hơi dung dịch đến khi bốc khói trắng. Để nguội, thêm 5ml nước cất, chuyền toàn bộ vào bình cất, tráng nhiều lần bằng nước cất và thêm nước cất tới thể tích khoảng 50ml.
Nếu lượng chất hữu cơ có trong nước ít thì ta có thể làm giàu asen bằng cách cộng kết nó với hiđroxit. Lấy 100 - 1000m1 mẫu sao cho lượng asen trong đó không nhỏ hơn 0,002mg, cho vào phễu chiết, thêm vào đó 0,5 - 1ml dung dịch FeCl3(hoà tan 115g FeCl3 trong 1 lít nước cất hai lần. Trung hoà bằng NH4OH(1:1) đến khi xuất hiện

kết tủa. Lắc nhẹ phễu chiết khoảng 15 phút, đê yên cho kết tủa lắng xuống, tách lấy phân kết tủa cho vào bình cất của máy cất. Lặp lại kết tủa một lần nữa trong phễu chiết đế tách hết kết tủa. Nếu còn kết tủa thì tách và gộp nó cùng với kết tủa trước. Hoà tan kết tủa bằng một lượng vừa đủ HCl(1:4). Nếu kết tủa không tan hết thì lọc bỏ. Thêm nước cất tới khoảng 50ml.
Trình tự phân tích
Trước khi tiến hành phân tích cần kiểm tra xem hàm lượng asen có trong thuốc thử. Lấy khoảng 50ml mẫu nước cần phân tích cho vào bình tam giác sao cho lượng asen trong bình đó khoảng 0,002 - 0,04mg. Thêm vào đó 15ml dung dịch HCl loãng. Sau đó thêm tiếp 6ml dung dịch KI và 0,5ml dung dịch SnCl2. Lắc đều hỗn hợp và để yên 15 phút. Nhồi giấy lọc đã tẩm chì axetat vào ống thuỷ tinh. Lấy 15ml dung dịch hấp thụ Ag-đietylđithiocacbamat vào bình hấp thụ rồi nối tất cả các bộ phận của bình hấp thụ với nhau. Cho vào bình tam giác 5g kẽm hạt hay 2 - 3g kẽm bột, đậy ngay bình lại. Zn sẽ khó H+ giải phóng ra hiđro. Hiđro mới sinh sẽ khử asen tạo thành asin bay ra và bị dung dịch trong bình hấp thụ. Phản ứng xảy ra trong 60 phút. Nếu thấy phản ứng xảy ra chậm thì cho thêm 0,5ml SnCl2, 5 - 1 5ml HCl đặc qua phễu.
Sau khi phản ứng xong, chuyển dung dịch ở bình hấp thụ vào cuvet và đem đo mật độ quang tại bước sóng 560nm.
Lập đường chuẩn
Pha một dãy dung dịch chuẩn có chứa 0; 2; 5;10; 20; 30; 40; 50ml dung dịch chuẩn có nồng độ 0,001mg/ml. Tiến hành như đối với mẫu phân tích.
Tính kết quả
Dựa vào đường thiết lập mối tương quan hàm số y = ax + b.
Trong đó: y: hàm lượng As trong đất;
x: mật độ quang đo được.
Hoá chất
- Bạc đietylthocacbamat, dung dịch trong pirđin:
+ Dung dịch 1: Hoà tan 2,25g nam đietylthocacbamat trong 100 ml nước cất;
+ Dung dịch 2: Hoà tan 1,7g AgNO3 tinh khiết phân tích trong 100 ml nước cất. Nhỏ dần dung dịch 1 vào dung dịch 2 đến khi kết tủa hết. Lọc, rửa sạch bằng nước cất, sấy khô trong bình hút ẩm ờ áp xuất thấp, kết tủa thu được có màu vàng nhạt. Hoà tan 1 g kết tủa thu được trong 200ml piriđin.
- KI 15%
- SnCl2: dung dịch 40% trong HCl đặc.
- Kẽm hạt tinh khiết phân tích không chứa asen.

- Giấy tẩm dung dịch chì axetat 10% đã sấy khô
- Natri asenit, dung dịch chuẩn:
+ Dung dịch chuẩn gốc: Hoà tan 0.1320g As2O3 tinh khiết phân tích vào 10 ml dung dịch NaOH 1 N tinh khiết phân tích, lắc mạnh, sau khi tan hết thêm vào đó 10ml dung dịch H2SO4 1 N tinh khiết phân tích, rồi thêm nước thành 1 lít, được dung dịch 0,100 mg As/ml;
+ Dung dịch chuẩn sử dụng: Lấy 10 ml dung dịch chuẩn gốc pha loãng bằng nước cất đến 1 lít được dung dịch 0,001 mgAs/ml. Chỉ pha trước khi dùng.
6.4.12. Xác định bạc (Ag)
Bạc thường có trong nước chảy ra từ một số mỏ, và thường có trong nước thải của công nghiệp ảnh. Trong nước bạc tồn tại dưới dạng phức tan hoặc hợp chất không tan, chủ yếu và bạc halogenua. Để xác định bạc trong nước có thể dùng phương pháp trắc quang đithizon hoặc rodanin. Để bảo quản mẫu nước khi phân tích bạc cần thêm 5ml HNO3 đặc vào 1 lít nước
Ion Ag+ phản ứng với rodanin tạo thành kết tủa đỏ, khi lượng bạc nhỏ thì tạo

thành dung dịch keo. Để làm ổn định dung dịch keo đó, thường dùng dung dịch gelatin. Phản ứng này khá đặc trưng và chọn lọc đối với bạc. Trước khi xác định cần phá huỷ các vẩn đục bằng cách vô cơ hoá mẫu nước.
Trình tự phân tích
Lấy 50ml dung dịch mẫu trong suốt (địa được vô cơ hoá), cho vào cốc dung tích 100 ml, thêm vào 2ml dung dịch kalinatritactrat, 2ml NH3 1ml galatin. Sau khi trộn đều hỗn hợp, thêm vào 0,5ml rodanin và sau 5 phút thì tiến hành đo mật độ quang.
Lập đường chuẩn
Đường chuẩn được lập với dãy dung dịch có chứa 0; 0,5; 1; 8; 10 ml dung dịch có chứa 0,01 Ag/ml. Sau đó pha loãng bằng nước cất hai lần tới thể tích 50ml. Tiến hành như đối với mẫu phân tích.
Tính kết quả
Dựa vào đường chuẩn thiết lập mối tương quan hàm số y = a.x + b.
Trong đó: y: hàm lượng Ag có trong mẫu;
x: mật độ quang đo được.
Từ đó sẽ xác định được hàm lượng Ag có trong mẫu
Hoá chất
- Khử natri tactrat 20%.
- Amoniac 10%.
- Gelatin 1 %.
- Rodanin 0,03% trong axeton, đựng trong chai màu nâu, để chỗ mát, dung dịch bền trong 2 tuần
- Dung dịch chuẩn
+ Dung dịch chuẩn gốc: Hoà tan 1,4450g Ag2SO4 tinh khiết phân tích (đã sấy khô ở 1050C) trong nước rồi định mức đến 1 lít trong đó chứa 1 ml HSO4 đặc. Dung dịch này chứa 1,00mgAg/ml.
- Dung dịch chuẩn sử dụng chứa 0,01 mg Ag/ml: Lấy 10 ml dung dịch chuẩn gốc và định mức bằng nước đến 1 lít.
6.4.13. Xác định Beri (Be)
Beri là kim loại màu xám sáng, không gặp trong thiên nhiên ở dạng tinh khiết, tỷ khối l,85; nhiệt độ nóng chảy 12840C, nhiệt độ sôi 24710C, Ben clorua và Beri sunfat ổn định trong nước, nồng độ của chúng giảm đến 30-35% so với nồng độ ban đầu chỉ trong khoảng 5 giây.
Hàm lượng trong nước thiên nhiên của Be dao động trong khoảng 0,00001 -

0,0012mg/l. Hàm lượng Be cao trong nước thiên nhiên thường gặp ở các nơi có vỉa Beri và trong nước có lượng lớn các ion F- và SO4
2-. Beri thường có trong nước thải của các nhà máy luyện kim, chế tạo máy, xử lý dầu mỏ và các xí nghiệp điện tử.
Đối với người và động vật máu nóng, khi hàm lượng Be trong nước cao sẽ gây nhiễm độc cấp tính và tác động tích luỹ. Các muối NO3
-, Cr và SO42- của Be rất độc.
khi hàm lượng Be2+ (mg/kg trọng lượng cơ thể) ở dạng muối sunfat là 80 thì 50% chuột bị chết; ở dạng clorua là 92. Cả hai dạng hợp chất này đều có khả năng tích luỹ.
Với lượng 0,0001 mg/kg thể trọng hay 0,0002 màu nước thì không gây nguy hiểm.
Đối với động vật sống trong nước, khi hàm lượng muối ben clorua là 15mg/l nước cứng thì làm chết cả tuế 0,05mg/1 làm chết rận nước. Khi hàm lượng muối bensunfat là 13mg/l nước mềm thì làm chết cá rô.
Khi nồng độ Be là 10 mg/l thì làm giảm độ trong của nước; 0,5-1,0 mg/l sẽ kìm hãm các quá trình tự làm sạch sinh học trong hồ chứa và quá trình sinh sản của hệ vi thực vật; nồng độ 0,005 mg/l thì không có ảnh hưởng đến quá trình tự làm sạch của hồ chứa. Nồng độ Be từ 15-20ml/l sẽ kìm hãm sự nảy mần của hạt rau diếp và cải canh.
Nồng độ giới hạn cho phép (mg/l).
+ Nước uống: Tuỳ theo tiêu chuẩn từng nước, ở Liên Xô (cũ) là 0,0002;
+ Nước sử dụng nông nghiệp và nước sinh hoạt: Liên Xô (cũ) là 0,0002;
+ Nước tưới cho tất cả các loại đất (tưới lâu dài): 0,5.
Phương pháp xác định
Có thể xác định Be theo phương pháp quang phổ hấp thụ nguyên tử, phương pháp phổ phát xạ plasma ICP (Inductively Coupled Plasma) và phương pháp trắc quang. Tuy nhiên, phương pháp trắc quang cho độ chính xác kém hơn so với hai phương pháp trên.
Phương pháp aluminon
Nguyên lý: Dung dịch thuốc thử aluminon phản ứng với Be tạo thành hợp chất màu đỏ sẫm, hấp thụ cực đại tại bước sóng 515 nm. Thêm một lượng nhỏ chất tạo phức EDTA sẽ ngăn cản ảnh hưởng của Al, Co, Cu, Fe, Mg, Ni, Ti, Zn và Zirconi ở hàm lượng vừa phải đến quá trình xác định Be.
Trong điều kiện như vậy, khi hàm lượng Cu không vượt quá 10 mg trong lượng mẫu phân tích thì không ảnh hưởng đến phép xác định. Nếu lượng cao hơn thì cần tăng lượng EDTA thêm vào. Phức của Cu hấp thụ không đáng kể ở bước sóng 515 nm. Có thể loại trừ ảnh hưởng này bằng cách thêm một lượng Cu tương đương vào thang chuẩn.
Bảo quản mẫu

Axit hoá toàn bộ mẫu phân tích trong khi bảo quản để giữ các kim loại trong dung dịch và ngăn ngừa chúng bám lên thành bình. Nếu nước sạch không có các chất lơ lửng thì mỗi lít mẫu thêm vào l,5ml HNO3 đặc để đưa pH của mẫu đến 2. Nước mặt ở ao hồ và nước thải có thể chứa các trầm tích thì cần phải thêm nhiều axit hơn
Dụng cụ:
Máy so màu quang điện, kính lọc màu xanh lục, bước sóng 515 nm, cuvet dày 5cm.
Thuốc thử
- Dung dịch beri gốc: Hoà tan 0,9837g berisunfat ngậm 4 phân tử nước, tinh khiết 99,9% trong 100ml nước cất, nếu cần thì tốc, sau đó thêm nước cất tới 500ml 1ml dung dịch này chứa 100 μBe.
- Dung dịch chuẩn sử dụng: Hút 5ml dung dịch gốc cho vào bình định mức 100 ml rồi định mức bằng nước cất tới vạch mức, lưu dung dịch này chứa 5 μgBe.
- Dung dịch EDTA: Hòa tan 2,5g EDTA trong 30ml nước cất, thêm vào 1 giọt metyl đỏ 0,05% pha trong rượu. Trung hoà dung dịch bằng NH4OH đặc lạnh và thêm nước cất đến 100 ml.
- Dung dịch đệm aluminon: Cân 500g CH3COONH4 cho vào cốc dung tích 2 lít trong đó đã có sẵn 1 lít nước cất. Thêm vào 80ml axit axetic đặc và khuấy cho đến khi được dung dịch đồng nhất, nếu cần thì lọc. Hoà tan lg aluminon (aurintricacboxilic axit trianmonium) trong 50ml nước cất rồi đô vào cốc chứa dung dịch đệm trên. Hoà tan 3g axit benzoic (C7H6O2) trong 20ml rượu metylic rồi thêm vào cốc đựng dung dịch đệm trên, định mức bằng nước cất tới vạch mức 21ít.
- Gelatin: Cân 10g gelatin cho vào trong cốc có thể tích 400ml đã có sẵn 250ml nước cất, đặt cốc trên bếp cách thuỷ vào khuấy cho đến khi gelatin tan hoàn toàn. Rót gelatin nóng vào trong bình định mức 1 lít đã có sẵn 500ml nước cất. Để nguội đến nhiệt độ phòng, thêm nước cất tới vạch mức và lắc đều. Đổ dung dịch gelatin và dung dịch đệm trên vào lọ thuỷ tinh tối màu có thể tích 41ít. Lắc đều, giữ ở nơi lạnh và tối, dung dịch bền trong khoảng ít nhất 1 tháng.
Trình tự phân tích
Xử lý mẫu: Nếu trong mẫu có chất hữu cơ mà cần xác định hàm lượng Be tổng số thì phải xử lý mẫu bằng dung dịch HNO3 và H2SO4. Còn chỉ xác định Be hoà tan thì lọc qua màng lọc 0,45μm.
Tạo phức với thuốc thử aluminon: Dùng pipet hút 0; 0,1; 0,5; 1; 2; 3; 4 ml dung dịch Be chuẩn sử dụng cho vào các bình định mức 100 ml. Lấy 50ml dung dịch mẫu hoặc lượng mẫu chứa ít hơn 200 μg Be cho vào bình định mức 100 ml. Thêm vào mỗi bình 2ml EDTA rồi thêm nước cất tới thể tích khoảng 75ml, lắc đều. Thêm 15 ml dung dịch đệm aluminon, rồi thêm nước cất tới 100 mlvà lắc đều hỗn hợp. Giữ yên 20 phút

sau khi thêm aluminon, chú ý không đề nơi ánh sáng, lọc nếu thấy cần thiết. Đo mật độ quang trong cuvet có chiều dày 5cm tại bước sóng λ = 515 nm (kính lọc màu xanh lục). Dung dịch so sánh là mẫu trắng.
Tính kết quả
Dựa vào đường chuẩn xây dụng mối tương quan hàm số y = ax + b.
Trong đó: y: hàm lượng Be;
x: mật độ quang
Từ đó thay giá trị mật độ quang đo được của mẫu vào sẽ xác định được hàm lượng Be
Độ chính xác và độ lệch
Kết quả của 32 phòng thí nghiệm khi phân tích mẫu tổng hợp chứa 250 μg Ben; 40 μg As/l; 240μg Se/l; 6μgV/l cho thấy Be đã được phân tích với độ lệch chuẩn tương đối là 7,13% và sai số tương đối là 13%.
6.5. Xác định một số tính chất khác của nước
6.5.1. Xác định hàm lượng ôxy hoà tan trong nước (DO)
Lượng ôxy hoà tan trong nước rất nhỏ, thường khoảng 8- 1 Omg/1. Độ bão hoà của ôxy hoà tan trong nước ngọt sạch ở 00C là 14-15mg/l. Thông thường lượng ôxy hoà tan trong nước cho chiếm 70-85% lượng bão hoà. Lượng ôxy hoà tan trong nước thiên nhiên và nước thai phụ thuộc vào các hoạt tính sinh hoả, hóa học và lý học trong nước. Phân tích DO là một trong những mục tiêu quan trọng để đánh giả mức độ ô nhiễm nước, kiểm tra những hoạt tính và quá trình xử lý chất thải.
Đề xác độ DO có thể sử dụng các phương pháp như: phương pháp cải tiến azid, phương pháp điện cực màng nhạy với ôxy và máy đo.
Phương pháp cải tiến azid được sử dụng cho mẫu nước cống đặc, các mẫu nước sông,...
Nguyên lý của phương pháp này như sau: Trong môi trưởng kiềm Mn2+ bị ôxy hoà tan trong nước ôxy hoá đến Mn4+ dưới dạng MnO2.
Khi có mặt H+, Mn4+ bị I- khử đến Mn2+:
Dùng Na2S2O3 chuẩn lượng I2 giải phóng ra với chỉ thị hồ tinh bột, từ đó xác
định được hàm lượng ôxy hoà tan
Trình tự phân tích

Lấy 300ml mẫu nước cho vào chai có thể tích lớn hơn 300ml. Thêm vào 2ml dung dịch MnSO4 và 2ml dung dịch I-. Đậy nút và dốc ngược chai 15 lần để trộn đều dung dịch. Thêm cần thận 2ml H2SO4 đặc vào thành chai rồi đậy nút và dốc ngược chai vài lần. Lấy 2O4ml dung dịch tương ứng với 200ml mẫu nước cho vào bình tam giác, thêm vào 3-4 giọt tinh bột rồi ( huân độ bằng dung dịch Na2S2S3 0,025N đến khi dung dịch mất màu xanh và trở lên trắng ngà.
Tính kết quả
Lượng ôxy hoà tan được tính theo mẫu. Cứ lưu dung dịch Na2S2O3 0,025N tương đương với 0,2 mg DO
Hoá chất
- Dung dịch MnSO4: Hoà tan 480g MnSO4.4H2O trong nước cất, lọc rồi định mức tới 1 lít
- Dung dịch I- trong kiềm: Hoà tan 500g NaOH (hay 700g KOH) và 135g NaI (hay 150g KI) trong nước cất và định mức tới 1 lít. Thêm vào dung dịch này NaN3 đã hoà tan trong 40ml nước cất.
- H2SO4 đặc
- Hồ tinh bột 1%.
- Dung dịch Na2S2O 0,025N: Hoà tan 6,205g Na2S2O3.5H2o trong nước cất mới sôi rồi làm lạnh bằng nước cất, thêm nước cất tới 1 lít.
Ngoài phương pháp trên người ta còn dùng phương pháp xác định hàm lượng ôxy hoà tan trong nước bằng cách đo trên điện cực
Dưới đây là bảng thể hiện hàm lượng ôxy hoà tan (mg/l) trong nước ở áp suất 760nmHg và độ ẩm tương đối là 100% (theo EAWAF, 1973).
0C 0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 14,60 14,56 14,52 14,48 14,44 14,40 14,36 14,33 14,29 14,25 14,21 14,17 14,13 14,09 14,05 14,02 13,98 13,94 13,90 13,87 13,83 13,79 13,75 13,72 13,68 13,64 13,61 13,57 13,54 13,50 13,46 13,46 13,39 13,36 13,32 13,29 13,25 13,22 13,18 13,15 13,11 13,08 13,O4 13,01 12,98 12,94 12,91 12,88 12,84 12,81 12,78 12,74 12,71 12,68 12,64 12,61 12,58 12,55 12,52 12,48 12,45 12,42 12,39 12,36 12,33 12,29 12,26 12,23 12,20 12,17 12,14 12,11 12,08 12,05 12,02 11,99 11,96 11,93 11,90 11,87 11,84 11,81 11,78 11,76 11,73 11,70 11,67 11,64 11,61 11,58 11,56 11,35 11,50 11,47 11,44 11,42 11,39 11,36 11,34 11,31 11,28 11,25 11,23 11,20 11,17 11,15 11,12 11,10 11,O7 11,O4 11,02 10,99 10,97 10,94 10,91 10,89 10,86 10,84 10,81 10,79 10,76 10,74 10,72 10,69 10,67 10,64 10,62 10,59 10,57 10,55 10,52 10,50 10,47 10,45 10,43 10,40 10,38 10,36 10,34 10,31 10,29 10,27 10,24 10,22 10,20 10,18 10,15 10,13 10,11 10,09 10,07 10,O4 10,02 10,00 9,98 9,96 9,94 9,92 9,98 9,87 9,85 9,83 9,81 9,79 9,77 9,75 9,73 9,71 9,69 9,67 9,65 9,63 9,61 9,59 9,57 9,55 9,53 9,51 9,49 9,47

9,45 9,43 9,41 9,39 9,37 9,36 9,34 9,32 9,30 9,28 9,26 9,24 9,23 9,21 9,19 9,17 9,15 9,13 9,12 9,10 9,08 9,06 9,05 9,03 9,01 8,99 8,98 8,96 8,94 8,92 8,91 8,89 8,87 8,86 8,84 8,82 8,81 8,79 8,77 8,76 8,74 8,72 8,71 8,69 8,67 8,66 8,64 8,63 8,61 8,59 8,58 8,56 8,55 8,53 8,51 8,50 8,48 8,47 8,45 8,44 8,42 8,41 8,39 8,38 8,36 8,35 8,33 8,32 8,30 8,29 8,27 8,26 8,24 8,23 8,21 8,20 8,18 8,17 8,16 8,14 8,13 8,11 8,10 8,08 8,О7 8,06 8,O4 8,03 8,01 8,00 7,99 7,97 7,96 7,94 7,93 7,92 7,90 7,89 7,88 7,86 7,85 7,84 7,82 7,81 7,80 7,78 7,77 7,76 7,74 7,73 7,72 7,70 7,69 7,68 7,66 7,65 7,64 7,63 7,61 7,60 7,59 7,57 7,56 7,55 7,54 7,52 7,51 7,50 7,49 7,47
Nếu gọi γ là hàm lượng O2 hoà tan trong nước ờ một áp suất xác định thì ta có:
Trong đó:
Pm: áp suất tại thời điểm xác định;
S: Hàm lượng O2 hoà tan trong nước tại áp suất 760mm Hg theo bảng trên.
6.5.2. Xác định nhu cầu ôxy hoá (COD)
Chỉ số này dùng để đánh giá hàm lượng chất hữu cơ của nước thải và sự ô nhiễm nước tự nhiên. COD là lượng ôxy cần thiết cho quá trình ôxy hoá hoá học các chất hữu cơ trong nước thành CO2 và nước.
Để xác định con người ta thường sử dụng một chất ôxy hoá mạnh trong môi trường axit, chất thường được sử dụng là K2Cr2O7. Khi đó xảy ra phản ứng:
Lượng dư Cr2O7
2- được chuẩn độ bằng dung dịch muối Fe2+ với chỉ thị axit Phenylanthranilic, màu chỉ thị chuyển từ tím đỏ sang xanh trái cây.
Trình tự phân tích
Lấy 20ml mẫu nước cho vào bình hồi lưu, rồi thêm vào HgSO4 (10 mg Cl- thì cần 0,1 g HgSO4. Thêm vào 10ml dung dịch K2Cr2O7 0,025N và một vài hạt thuỷ tinh. Lắp ống thuỷ sinh hàn thuỷ tinh nhám. Thêm vào từ từ 30ml H2SO4 đặc có chứa Ag2SO4 qua phần cuối ống sinh hàn và lắc đều hỗn hợp trong khi thêm axit. Đun hồi lưu trong hệ giờ. Để nguội và rửa sinh hàn hồi hưu bằng nước cất. Pha loãng hỗn hợp bằng nước cất tới thể tích khoảng 150ml, để nguội. Chuẩn lượng bicronat dư bằng muối Fe2+, với chỉ thị axit Phenylanthranilic. Cũng tiến hành một thí nghiệm mẫu trắng như đối với mẫu phân tích.

Tính kết quả
Trong đó: a: thể tích Fe2+ Chuẩn độ mẫu trắng (ml)
b: thể tích Fe2+ chuẩn độ mẫu (ml)
N: nồng độ đương lượng của Fe2+
Hóa chất
- K2Cr2O7 0,025N: Hoà tan 12,259 g K2Cr2O7 (đã sấy khô 2 giờ ở 1050C) trong nước cất và thêm nước tới vạch mức
- H2SO4 đặc có thêm 22g Ag2SO4 cho một chai 9 lít.
- Dung dịch Fe2+ 0,1 N: Hòa tan 39g Fe(NH4)2SO4.6H2O tinh khiết trong nước cất, thêm 20ml H2SO4 đặc nguội rồi định mức tới 1 lít
- Ag2SO4 tinh khiết hoá học.
- H2SO4 tinh khiết hoá học
Chỉ thị l%: Hoà tan 0,2g axit Phenylanthranilic trong 100 ml Na2SO3 0,2%.
6.5.3. Xác định nhu cầu ôxy sinh hoá (BOD)
Nhu cầu ôxy sinh hoá là lượng ôxy đã sử dụng trong quá trình ôxy hoá các chất hữu cơ bởi các vi sinh vật. Trong nước, khi xảy ra quá trình ôxy hoá sinh học thì các vi sinh vật đã sử dụng ôxy hoà tan. Vì vậy việc xác định tổng lượng ôxy hoà tan cần thiết cho quá trình phân huỷ sinh học là rất quan trọng đề đánh giá ảnh hưởng của dòng chảy đối với nguồn nước.
Trong thực tế, người ta không the xác định được lượng ôxy can thiết để phân huỷ hoàn toàn chất hữu cơ, mà chi cần xác định lượng ôxy cằn thiết cho 5 ngày đầu với nhiệt độ ủ 200C trong phòng tối để tránh quá trình quang hợp, chỉ tiêu này ký hiệu là BOD5.
Phương pháp xác định
Chuẩn bị dung dịch pha loãng: Nước pha loãng được chuẩn bị ở chai to, rộng miệng bằng cách thổi không khí sạch ở 200C vào nước cất và lắc nhiều lần. Cho đến khi bão hoà ôxy sau đó thêm 1 ml dung dịch đệm photphat, 1 ml dung dịch MgSO4, 1 ml CaCl2 và 1 ml FeCl3, rồi định mức tới 1 lít bằng nước cất. Trung hoà mẫu nước phân lịch đến pH = 7 bằng H2SO4 1N hay bằng NaOH 1N. Pha loãng mẫu nước trước khi xác định nước bằng hiếu khí đã chuẩn bị trước theo các bước sau:
+ 0,1 - 1% đối với những mẫu nước có dòng chảy mạnh
+ 15% đối với những mẫu nước cống chưa hoặc đã để lắng

+ 5 - 25% đối với nước đã bị ôxy hoá
+ 25 - 100% đối với nước sông đã bị ô nhiễm
Khi pha loãng cần phải hết sức tránh không để ôxy hoá cuốn theo. Sau khi pha loãng xong cho mẫu vào chai để xác định BOD (thường là chai có thể tích 300ml), đóng kín nút chai, một chai dùng để ủ 5 ngày ở nơi tối tại nhiệt độ 200C, một chai dùng để xác định. Do ban đầu trong mẫu pha loãng.
Tính kết quả
Lượng BOD được tính theo mg O2/l
Trong đó
D1: lượng ôxy hoà tan (màu) của dung dịch mẫu đã pha loãng sau 15 phút;
D2: lượng ôxy hoà tan (máu) trong mẫu sau 5 ngày ủ;
P: hệ số pha loãng được tính như sau
Hoá chất
- Dung dịch đệm photphat: Hoà tan 8,5g K2HPO4, 21,75K2HPO4, 33,4g NaHPO4.7H2O và l,7g NH4Cl trong khoảng 500ml nước cất rồi định mức tới 1 lít.
- Dung dịch MgSO4: Hoà tan 22,5g MgSO4.7H2O trong nước rồi định mức tới 1 lít
- CaCl2: Hoà tan 27,5g trong 1 lít nước cất.
- FeCl3: Hoà tan 0,25g FeCl3.6H2O trong 1 lít nước cất.
- H2SO4 1N và NaOH 1N.
6.5.4. Xác định mầu sắc
Nói chung nước thiên nhiên sạch không có màu. Trong thực tế nước thường có màu do sự có mặt của một số chất màu hữu cơ và các chất của Fe3+. Nước thải có thể có sắc thái khác nhau. Trong nhiều trường hợp màu của nước còn do các vi sinh vật, các hạt bùn, các thực vật sống trong nước, các sunfua, các chất lơ lửng gây nến Trước khi xác định màu cần lọc mẫu nước. Đối với nước có chứa nhiều chất lơ lửng, cần để lắng rồi xác định màu của phần nước trong. Việc xác định chính xác và hoàn toàn khách quan màu của nước là việc tương đối khó, nhiều khi phải mô tả về sắc thái và cường độ màu bằng lời chứ không phải bằng con số định lượng.

Có thể xác định màu sắc của nước bằng mắt dựa trên sự so sánh với một loạt mẫu chuẩn hoặc bằng cách ghi phú hấp thụ của mẫu nước trong miền khả kiến. Cách thứ nhất rõ ràng không được chính xác, chi cho ta biết sắc thái và cường độ gần đúng của màu (độ đậm nhạt). Cách thứ hai chính xác, khách quan, nhưng đòi hỏi phải có thiết bị đắt tiền và không thực hiện được trong điều kiện phân tích tại chỗ.
Dưới đây là phương pháp xác định màu sắc của nước theo cách so sánh với dãy dung dịch chuẩn có màu sắc tương tụ và việc so sánh được thực hiện bằng cách đo trên máy so màu quang điện. Trong thực tế nước thường có màu sắc gần với màu của dung dịch hỗn hợp K2Cr2O7 và CoSO4, nên người ta thường dùng dãy dung dịch của hỗn hợp đó làm dãy chuẩn để so sánh xác định màu của nước.
Trình tự tiến hành
Đường chuẩn: Chuẩn bị 10 ống đo Nessler, dùng pipet lấy lần lượt vào mỗi ống 1, 2, 3, 4, 5, 6, 8, 10, 12, 14 ml dung dịch I và thêm dung dịch II đến thể tích 100ml. Độ màu của dãy dung dịch chuẩn đó được biểu diễn trên thanh màu theo bảng sau:
Thể tích dung dịch I (ml) 0 1 2 3 4 5 6 8 10 12 Thể tích dung dịch II (ml) 100 99 98 97 96 95 94 92 90 84 Độ màu 0 5 10 15 20 25 30 40 50 80
Đo mật độ quang của dung dịch chuẩn trên máy so màu, sử dụng cuvet có chiều dày 1-10mm. Để xác định độ màu của mẫu nước, cho nước vào cuvet và tiến hành đo màu như đã làm đối với mẫu.
Hoá chất và dụng cụ
- Máy so màu quang điện
- Ống đo Nessler, thể tích 100 ml.
- Dung dịch I: Hoà tan 0,0876g K2Cr2O7 2g COSO4.7H2o và lưu dung dịch H2SO4 đặc trong nước cất thành 1 lít.
- Dung dịch II: Dung dịch H2SO4 loãng (1:1000).
6.5.5. Xác minh độ dãn điện riêng
Nước tinh khiết hầu như không dẫn điện, vì nước phân ly rất ít. Nhưng các chất tan trong nước phân ly thành các cation và anion nên các loại nước thiên nhiên cũng như nước thải lại có tính dẫn điện Độ dẫn điện riêng theo định nghĩa là giá trị nghịch đảo của điện trở riêng (là điện trò của dung dịch nằm giữa hai cực có bề mật lcm2 và cách nhau l cm) của dung dịch. Tại một nhiệt độ xác định, độ dẫn điện riêng phụ thuộc vào nồng độ cation và anion, tức là vào thành phẫn của dung dịch.
Để xác định độ dẫn điện riêng của dung dịch người ta dùng máy đo độ dẫn (có thể đo điện trở trong khoảng 50 - 100.000 Ω ) dùng các cực platin và bình đựng dung dịch nghiên cứu kèm theo máy.

Mẫu nước xác định độ dẫn điện thì không được xử lý và nên xác định ngay sau khi lấy mẫu.
Để đo độ dẫn điện của dung dịch, người ta cho dung dịch vào bình đo, nhúng vào đó hai cực platin, nối hai cực với máy đo. Độ dẫn điện riêng được tính theo công thức:
Trong đó: χ : độ dẫn điện riêng;
R: điện trở đo được;
K: hằng số tỷ lệ thường được gọi là hằng số bệnh đo hoặc hằng số hệ cực.
K phụ thuộc vào cấu tạo, kích thước của hệ cực và hình dạng của bình đo. Trước khi xác định độ dẫn điện riêng cửa dung dịch nghiên cứu phải xác định K, hoặc thường nói là chuẩn hoá hệ cực. Để chuẩn hoá hệ cực người ta đo điện trở của dung dịch KCl có nồng độ xác định. K được tính theo công thức:
Trong đó: R: giả trị trong bình của điện trở dung dịch đo được
1278: độ dẫn điện riêng của dung dịch KCl 0,01 M ở 200C, Ωμ -l.cm-1.
Trình tự tiến hành
Chuẩn hoá hệ cực: Tráng bình đo 3 lần, mỗi lần bằng vài ml dung dịch KCl 0,01M, tráng hệ cực cũng bằng dung dịch đó. Ngâm dung dịch và hệ cực trong máy điều nhiệt để có nhiệt độ 200C (kiểm tra bằng nhiệt kế có độ chính xác 0,10C). Đo điện trở của dung dịch trên máy đo độ dẫn và xác định K dựa vào công thức trên. Cho dung dịch chuẩn vào 5 - 10 bình và tiến hành đo như vậy, lấy kết quả trung bình để tính K.
Đổ nước vào hai bình đã được tráng trước 3 lần mỗi lần bằng vài ml nước cần xác định độ dẫn, ngâm các bình vào bề nước của máy điều nhiệt đã điều chỉnh sẵn ở 200C cho đến khô nước trong bình có được nhiệt độ đó. Nhúng hệ cực vào bình thứ nhất nhiều lần để rửa hệ cực. Nhúng hệ cực vào bình thứ hai và đo điện trở. Ghi kết quả R đo được. Nếu phải đo nhiều mẫu có độ dẫn khác nhau thì mỗi mẫu dùng 3 bình, hai bình để rửa hệ cực, bình thứ 3 để đo độ dẫn.
Có thể đo ở nhiệt độ khác nhau, nhưng phải xác định chính xác nhiệt độ đo của nước rồi dùng bảng hiệu chỉnh để đưa về độ dẫn ở 200C. Độ dẫn 200C được tính theo công thức:
Trong đó: K: Hằng số hệ cực
f: Hệ số hiệu chỉnh nhiệt độ

R: Điện trở đo được
Hoá chất và dụng cụ
- Dung dịch KCl 0,01M: Hoà tan 0,7456g KCl tinh khiết hoá học (đã được sấy khô ở 1000C và để nguội trong bình hút ẩm) trong nước cất hai lần thành 1 lít dung dịch ở 200C.
- Máy đo độ dẫn (dùng dòng xoay chiều), có thể đo được điện trở 50 - 100.000 Ω
- Máy điều nhiệt

Chương 7
PHÂN TÍCH ĐẤT
7. 1. Giới thiệu chung
Đất là môi trường hỗ trợ cho sự sinh trưởng và phát triển của thực vật và hệ sinh thảo. Với mục đích như vậy, tính bền vững của đất chủ yếu phụ thuộc vào không gian đối với sự phát triền của bộ rễ thực vật và khả năng trao đổi của nó với nước, không khí, nhiệt độ và các chất dinh dưỡng. Những yếu tố này thê hiện độ phì nhiêu của đất.
Không gian phát triển của rễ thường được xác định bằng sự vươn sâu (solum) của rễ. Tuy nhiên, đối với các loại đất, đá có tính xốp và liên kết không chặt sẽ thúc đẩy khả năng đâm sâu của rễ. Mật độ rễ phụ thuộc lớn với một số tính chất đất như sự thay đổi về dung trọng, thành phần rỗng, mực nước ngầm và sự phân bố của các thành phần như muối, pH, thế khử.
Bảng 7.1. Độ bền của rễ đối với khả năng đâm xuyên
Độ sâu Sự đâm xuyên của rễ Ít hơn 10 cm 10 - 25 cm 25 - 50 cm 50 - 100 cm
Lớn hơn 100 cm
Rất hẹp Hẹp
Trung bình Sâu
Rất sâu
Các nghiên cứu về đất có thể theo một số hướng sau:
Xác định khả năng trao đổi dinh dưỡng
Xác định các vật chất có khả nàng gây ảnh hưởng nguy hại tới nước ngầm.
Xác định các tính chất ăn mòn đối với đường ống
Xác định sự mặn hoá
Kiểm tra hiệu quả lọc
Xác định chất lượng đất
Xác định mức độ nhiễm bẩn môi trường đất
Hàm lượng các chất dinh dưỡng có thể trao đổi từ đất là yếu tố quan trọng quyết định khả năng cung cấp dinh dưỡng cho cây trồng và do vậy việc xác định chúng phải sử dụng phương pháp thống nhất. Từ đó có thể tính toán nhu cầu cung cấp thêm chất dinh dưỡng cho cây (bón phân).
Khả năng trao đổi các chất đinh dưỡng được tổng kết trong bảng 7.2.

Bảng 7.2. Dạng chất dinh dưỡng liên kết trong đất
Dạng liên kết Khả năng trao đổi Xác định bằng
Bị hoà tan nhưng không liên kết trong đất Một phẫn liên kết với các chất trao đổi Không linh động, dễ bị linh động Không linh động, khó bị linh động
Rất dễ Dễ dàng Khó Rất khó
Các chất dinh dưỡng hoà tan trong nước Các chất dinh dưỡng trao đổi (bao gồm phần hoà tan trong nước) Các chất dinh dưỡng trao đổi (bao gồm phẫn hoà tan trong nước) Phần bị dữ lại của các chất dinh dưõng
Tuy nhiên, tỉ lệ trao đổi của các chất dinh dường cơ bản phụ thuộc vào thành phần sét hữu cơ, độ ẩm đất, pH, và thế khử. Hàm lượng các chất bị linh động trong môi trường thực chất phụ thuộc vào các điều kiện khí hậu và tầng đất.
Đánh giá sự nhiễm bẩn các chất vô cơ (kim loại nặng) trong đất là rất khó khăn do nồng độ vốn có của kim loại nặng có thể phụ thuộc rất lớn vào trạng thái khoáng vật học (thành phần đá mẹ). Sử dụng các chất dinh dưỡng (bón phân) cũng là nguyên nhân dẫn đến nhiễm bẩn đất. Đánh giá sự nhiễm bẩn có thể được xem xét từ quan điểm về lợi nhuận hoặc hiệu quả của đất khi xem nó như là một lớp lọc nước tự nhiên. Đối với vấn đề này cần quan tâm ở các điểm sau:
- Vai trò quan trọng của đất như là vật mang đối với cây trồng
- Khả năng lọc của đất đối với việc bảo vệ nước ngầm.
Việc chọn lựa các thí nghiệm đối với việc nghiên cứu khả năng linh động của kim loại nặng đang tồn tại một số vấn đề. Để giải quyết vấn đề này, các kết quả đo đạc tính dễ tan của kim loại nặng đối với thực vật bằng phương pháp chiết cho phép sử dụng đê so sánh với lượng kim loại nặng được hút bởi rễ thực vật. Sử dụng axít mạnh hoặc các chất tạo phức mạnh để chiết kim loại nặng trong đất không cho kết quả ổn định. Sử dụng NH4OH cho kết quả tương đối ổn định đối với việc xác định các dạng linh động của các nguyên tố vết. Ngày này, có rất nhiều phương pháp chiết được sử dụng trong các nghiên cứu liên quan đến kim loại nặng đã được công bố bởi nhiều tác giả. Phương pháp chiết thông dụng nhất thường được thực hiện theo quy trình chiết liên tục đã được công bố bởi Tissier 1979). Khả năng chiết hầu hết các kim loại nặng từ đất lớn hơn hầu hết các nguyên tố thông thường (photpho: 1%, Al 2% đến 3%, Na: 0,1 đến 0,5%). Tuy nhiên, kim loại nặng cũng được biết đến liên kết mạnh với các vật chất humus hơn các nguyên tố đã được đề cập ở trên.
Giới hạn chống chịu của kim loại nặng trong đất là rất quan trọng. Bảng 7.3 cung cấp ngưỡng độc tương ứng với nồng độ của chúng trong cây trồng. Giới hạn này thay

đổi tuỳ thuộc vào loài thực vật.
Bảng 7.3. Khoảng gây độc của mã số kim loại nặng trong đất
Nguyên tố Ngưỡng (mg/kg)
Cu Zn Cd Ni Pb Cr Hg
200-400 500-5000 10-175
200-2000 500-1500 500-1500 10-1000
Nhiễm bẩn đất do các chất hữu cơ như hoá chất bảo vệ thực vật cũng phải được
xem xét đến. Trong quá trình sử dụng hoá chất bảo vệ thực vật, một phần được đưa vào hoặc rơi xuống trực tiếp xuống đất. Lượng hấp phụ vào đất thay đổi theo tuyến và có mối quan hệ không lớn. Sự tồn tại của HCBVTV trong đất được mô phỏng trong hình 7.2.
Khi đi vào đất, các vật chất này thay đổi đặc tính sinh học lệ thuộc vào một số tính chất đất, chúng bị chuyển hoá thậm trí bị khoáng hoá hoàn toàn. Các lớp đất bề mặt có chứa các chất hữu cơ sẽ hấp phụ HCBVTV. Sự thấm sâu của chúng phụ thuộc vào thành phần khoáng sét, thành phần Nitơ, oxít Fe, Al và các lỗ hổng trong đất.
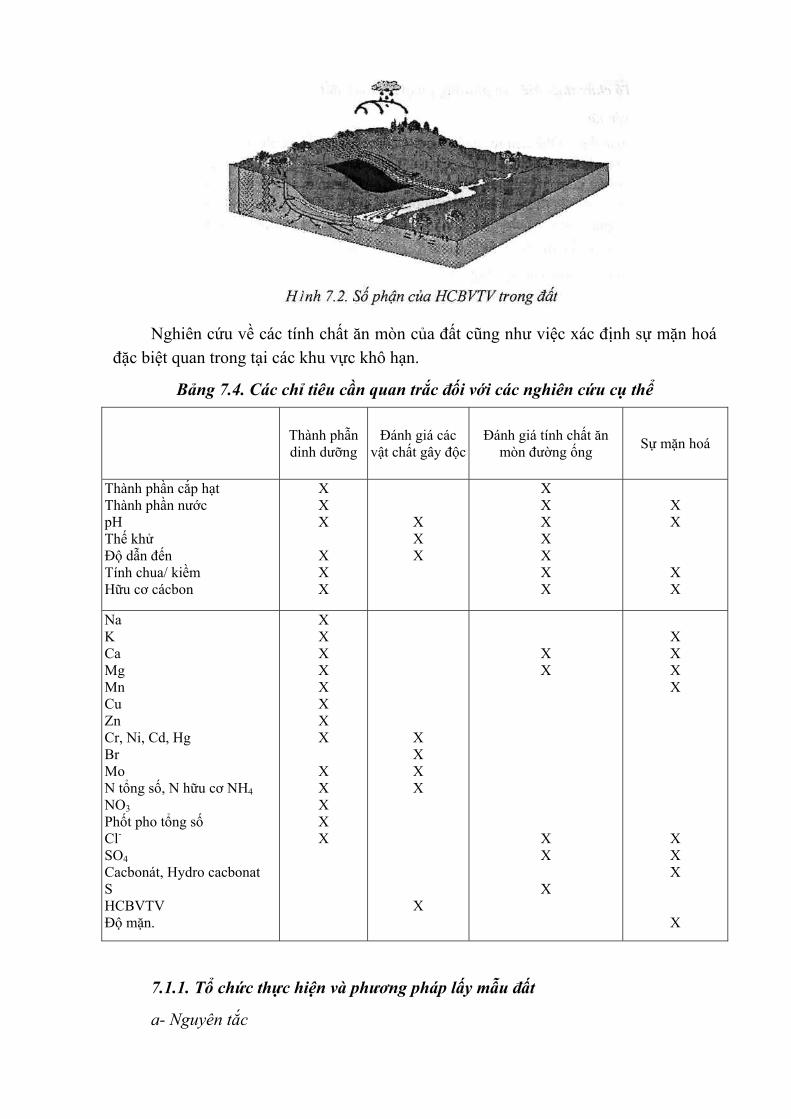
Nghiên cứu về các tính chất ăn mòn của đất cũng như việc xác định sự mặn hoá
đặc biệt quan trong tại các khu vực khô hạn.
Bảng 7.4. Các chỉ tiêu cần quan trắc đối với các nghiên cứu cụ thể
Thành phẫn dinh dưỡng
Đánh giá các vật chất gây độc
Đánh giá tính chất ăn mòn đường ống Sự mặn hoá
Thành phần cắp hạt Thành phần nước pH Thế khử Độ dẫn đến Tính chua/ kiềm Hữu cơ cácbon
X X X
X X X
X X X
X X X X X X X
X X
X X
Na K Ca Mg Mn Cu Zn Cr, Ni, Cd, Hg Br Mo N tổng số, N hữu cơ NH4 NO3 Phốt pho tổng số Cl- SO4 Cacbonát, Hydro cacbonat S HCBVTV Độ mặn.
X X X X X X X X
X X X X X
X X X X
X
X X
X X
X
X X X X
X X X
X
7.1.1. Tổ chức thực hiện và phương pháp lấy mẫu đất
a- Nguyên tắc

Nhiễm bẩn đất có thể đến từ nhiều nguồn, chất nhiễm bẩn có thể tiếp xúc trực tiếp, do chất thải bốc hơi, phát tán do gió, nước mặt và nước ngầm. Khi nghiên cứu một khu vực đã biết trước được các nguồn gây ô nhiễm, vi trí lấy mẫu có thể được lựa trọn trên cơ sở các thông tin xác định về các điểm nóng, tuy nhiên, phương pháp lấy mẫu này cần chú ý về tính chính xác của các thông tin, nếu không sẽ dẫn tới các kết luận sai lệch.
Lớp đất bề mặt có thể bị nhiễm bẩn theo các cách sau:
• Quản lý rác thải không đúng cách
• Tai nạn, như đổ tràn
• Trầm tích không khí
• Hoạt động công nghiệp
• Hoạt động nông nghiệp
• v.v..
Mục đích nghiên cứu cũng cần phải được làm rõ trước khi tiến hành lấy mẫu (mẫu nông hoá, mẫu thổ nhường, mẫu đánh giá nhiễm bẩn. Đối với từng mục đích nghiên cứu, thông tin của địa bàn nghiên cứu cũng cần được làm rõ như: loại đất độ sâu tầng đất, chuỗi địa hình, chuỗi khí hậu, chuỗi sinh học, chuỗi thạch quyển, diện tích...) để phân biệt được tính đồng nhất hay không đồng nhất của khu vục nghiên cứu.
Phương pháp lấy mẫu theo đường kẻ ô có thể sử dụng đối với các khu vực bị nhiễm bẩn trên diện rộng. Để chọn lựa khu vực lấy mẫu đất mặt, kẻ ô vuông trên toàn bộ khu vực dự đinh lấy mẫu. Ô vuông được vẽ dựa trên cơ sở về các thông tin chung của khu vực hoặc sử dụng phương pháp thống kê (số Ô được lấy phải đảm bảo tối thiêu là 25% trên tổng số ô, số mẫu được lấy trên mỗi ô phải đảm bảo tối thiểu là 10 mẫu). Cách làm này có thể dẫn tới việc một số điểm "nóng" bị bỏ qua, nhưng kẻ ô vuông có thể liên kết được toàn bộ các điểm lấy mẫu. Hiện nay, khi tiến hành lấy mẫu các thiết bị định vị toàn cầu thường được sử dụng kèm theo. Công cụ này giúp cho việc xây dụng các bản đồ phân bố chất ô nhiễm tích hợp các thông tin cho cái nhìn tổng thể về sự biến đổi chất lượng đất theo thời gian do ảnh hưởng của các hoạt động sản xuất.

Tuỳ thuộc vào mục đích nghiên cứu trọng lượng mẫu đất được lấy có thể từ 300- 500 gram.
b- Dụng cụ lấy mẫu, túi đựng mẫu
Dụng cụ lấy mẫu có thể dùng bằng cuốc, xẻng, hoặc các dụng cụ chuyên dụng như hình dưới đây.
Túi dụng mẫu có thể dùng túi plastic. Các mẫu đất được lấy yêu cầu phải ghi rõ tên mẫu, địa điểm, thời gian lấy
c. Bảo quản
Cũng giống như đối với mẫu nước, tuỳ thuộc vào đối tượng nghiên cứu sẽ yêu cầu các phương pháp bảo quản mẫu khác nhau. Đối với các mẫu phân tích các chỉ tiêu như NO2
-, NO3-, NH4
+, các chỉ tiêu vi sinh vật... phải được bảo quản trong điều kiện lạnh để tránh sự phân huỷ và hoạt động của các vi sinh vật trong mẫu. Mẫu phân tích các chỉ tiêu này đòi hỏi phải phân tích ngay trong trạng thái mẫu tươi, không qua các
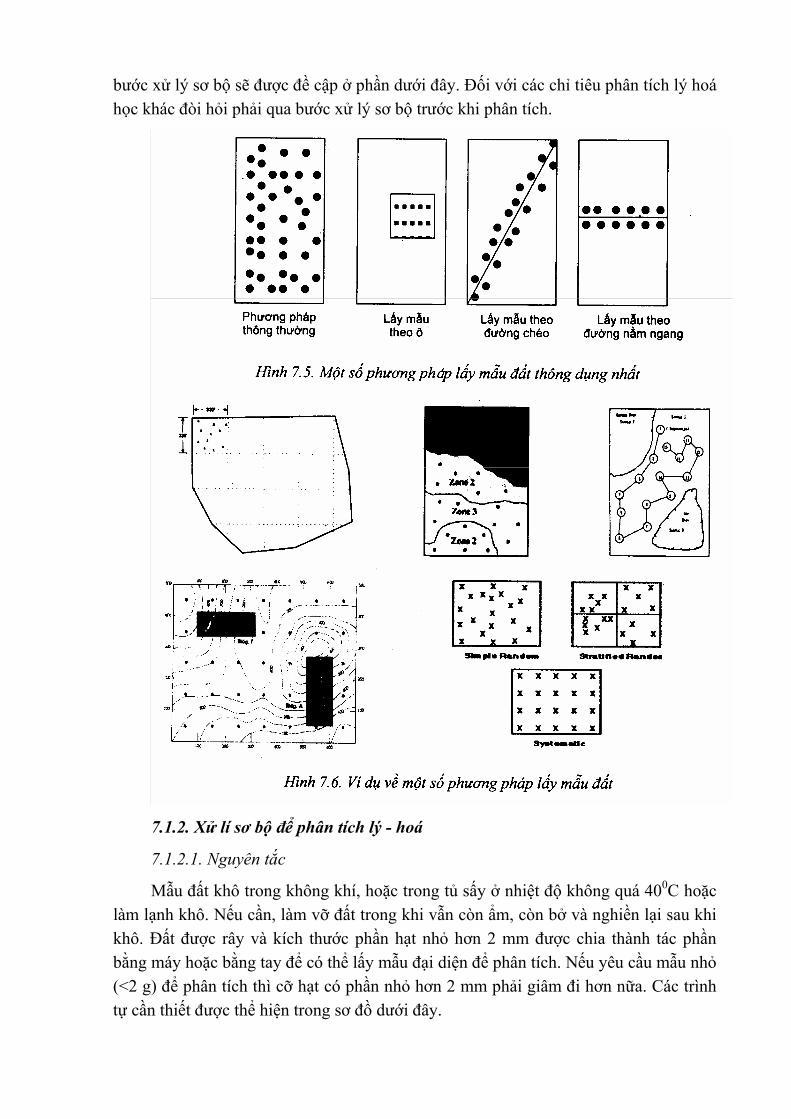
bước xử lý sơ bộ sẽ được đề cập ở phần dưới đây. Đối với các chỉ tiêu phân tích lý hoá học khác đòi hỏi phải qua bước xử lý sơ bộ trước khi phân tích.
7.1.2. Xử lí sơ bộ để phân tích lý - hoá
7.1.2.1. Nguyên tắc
Mẫu đất khô trong không khí, hoặc trong tủ sấy ở nhiệt độ không quá 400C hoặc làm lạnh khô. Nếu cần, làm vỡ đất trong khi vẫn còn ẩm, còn bở và nghiền lại sau khi khô. Đất được rây và kích thước phần hạt nhỏ hơn 2 mm được chia thành tác phần bằng máy hoặc bằng tay để có thể lấy mẫu đại diện để phân tích. Nếu yêu cầu mẫu nhỏ (<2 g) để phân tích thì cỡ hạt có phần nhỏ hơn 2 mm phải giâm đi hơn nữa. Các trình tự cần thiết được thể hiện trong sơ đồ dưới đây.

Chú ý:
1. Làm khô ở nhiệt độ 400C trong tủ sấy thích hợp hơn phơi trong không khỉ ở nhiệt độ phòng, vì tốc độ làm khô nhanh sẽ hạn chế những thay đổi do hoạt động của vi sinh vật.
2. Cần lưu ý rằng mỗi một kiểu xử lý sơ bộ để ảnh hưởng đến một số tính chất đất.
3. Bảo quản các mẫu đất gồm các mẫu đã nhận được, đã phơi khô trong không khí, đã làm lạnh hoặc bảo quản trong môi trường không có ánh sáng, trong một thời gian dài có thể ảnh hưởng đến một số thông số đất đặc biệt là các độ hoà tan của cả phần vô cơ và phần hữu cơ.
4. Đối với các mẫu đất lấy từ nguồn ô nhiễm, nên áp dụng các phép đo đặc biệt, cần tránh tiếp xúc với da và nến có các biện pháp đặc biệt khi làm khô các mẫu đất loại này (thông gió, lưu thông không khí).
5. Khối lượng mẫu yêu cầu phải có sẵn ít nhất 500 g đất vừa mới lấy
7.1.2.2. Thiết bị, dụng cụ
Tủ sấy, tủ lạnh khô, máy làm vỡ, rây phẳng, máy trộn, sàng rung, rây lưới, cân phân tích (độ chính xác tối thiểu tới 1g).
Cách tiến hành
a- Mô tả mẫu
Xem xét mẫu đã nhận được và ghi lại dạng mẫu theo thuật ngữ thích hợp gồm cả các chi tiết về vật ngoại lai, những thực vật còn lại và các đặc điểm đáng lưu ý hoặc có liên quan khác.
b- Làm khô
Làm khô toàn bộ trong không khỉ hoặc trong tủ sấy thông gió đã loại bỏ không khí ẩm hoặc trong tủ làm khô lạnh. Làm khô mẫu cho tới khi khối lượng mẫu đất giảm không quá 5% (m/m) trong 24 h.
Để tăng tốc độ trong quá trình làm khô, đập nhỏ các cục đất có kích thước lớn hơn (lớn hơn 15 mm) trong khi làm khô. Khi mẫu được phơi khô trong không khí, nghiền nhẹ bằng tay bằn cách sử dụng búa gỗ họ một cối giã và chày. Khi mẫu được sấy khô trong tủ sấy, tạm thời sấy mẫu ra khỏi tủ và xử lý như các trên. Các này cũng làm cho việc tác các hạt có kích thước lớn hơn 2 mm dễ dàng hơn.
• Làm khô không khí
Dàn mỏng tất cả các vật liệu thành một lớp không dày quá 15 mm trên một cái khay không hút ẩm từ đất và không nhiễm bẩn.
Chủ yếu là tránh ánh nắng mặt trời trực tiếp ánh sáng mặt trời trực tiếp có thể tạo

ra sự chênh lệch nhiệt độ lớn ở trong mẫu, đặc biệt giữa lớp đất phía trên đã khô một phần hoặc toàn bộ với những lớp đất phía dưới vẫn còn ẩm.
• Làm khô trong tủ sấy
Dàn mỏng tất cả các vật liệu thành một lớp không dày quá 15 mm trên một cái khay không hút ẩm từ đất, không nhiễm bẩn. Đặt khay trong tủ sấy và làm khô ở nhiệt độ không cao quá 400C.
c- Nghiền và loại bỏ các vật liệu thô
• Loại bỏ đá
Trước khi nghiền phải loại bỏ đá. các mảnh thuỷ tinh và rác v.v.. có kích thước lớn hơn 2 mm bằng các rây và nhặt bằng tay. Xác định và ghi lại tổng khối lượng mẫu khô và khối lượng vật liệu nào đó bị loại bỏ ở bước này
• Loại bỏ các vật liệu tự nhiên nhỏ hơn 2 mm
Rây vật liệu nhỏ hơn 2 mm, ghi lại khối lượng của vật liệu lớn và nhỏ hơn 2 mm, nghiền loại lớn hơn 2 mm, dùng máy hoặc bằng tay trộn đều mẫu.
d- Nghiền toàn bộ mẫu
Giảm cỡ hạt lớn hơn 2 mm
Nghiền đất khô thành các hạt không lớn hơn 2 mm bằng các dụng cụ thích hợp. Các dụng cụ cần thiết phải được điều chinh hoặc sử dụng sao cho hạn chế đến mức tối thiểu việc nghiền nhỏ các hạt ban đầu (kết vón và khối liên kết).
e- Rây
Có thể sử dụng rây máy hoặc bằng tay.
f- Lấy mẫu
Để chuẩn bị mẫu thí nghiệm, chứa phần mẫu được làm khô, nghiền nhỏ và rây (kích thước hiện tại < 2 mẫu thành các phần đại diện từ 200 g đến 300 g hoặc theo một trình tự thích hợp khác. Để chuẩn bị một mẫu thử, chia mẫu thí nghiệm thành các phần đại diện cho đến khi đạt được cỡ mẫu yêu cầu. Tránh việc tạo bụi càng nhiều càng tốt
• Lấy mẫu bằng tay (chia tư)
Trộn kỹ mẫu trộn bằng máy hoặc bằng tay và dàn mẫu thành một lớp mỏng trên một cái khay không ảnh hưởng tới thành phần của mẫu. Chia đất thành bốn phần bằng nhau. Gộp hai trong bốn phần theo đường chéo, loại bỏ hai phần kia. Lặp lại trình tự này cho đến khi đạt được khối lượng đất mong muốn.
Sử dụng dụng cụ chia mẫu
Sử dụng loại hộp có nhiều rãnh, chia mẫu thành hai phần bằng nhau.
• Lấy mẫu bằng máy
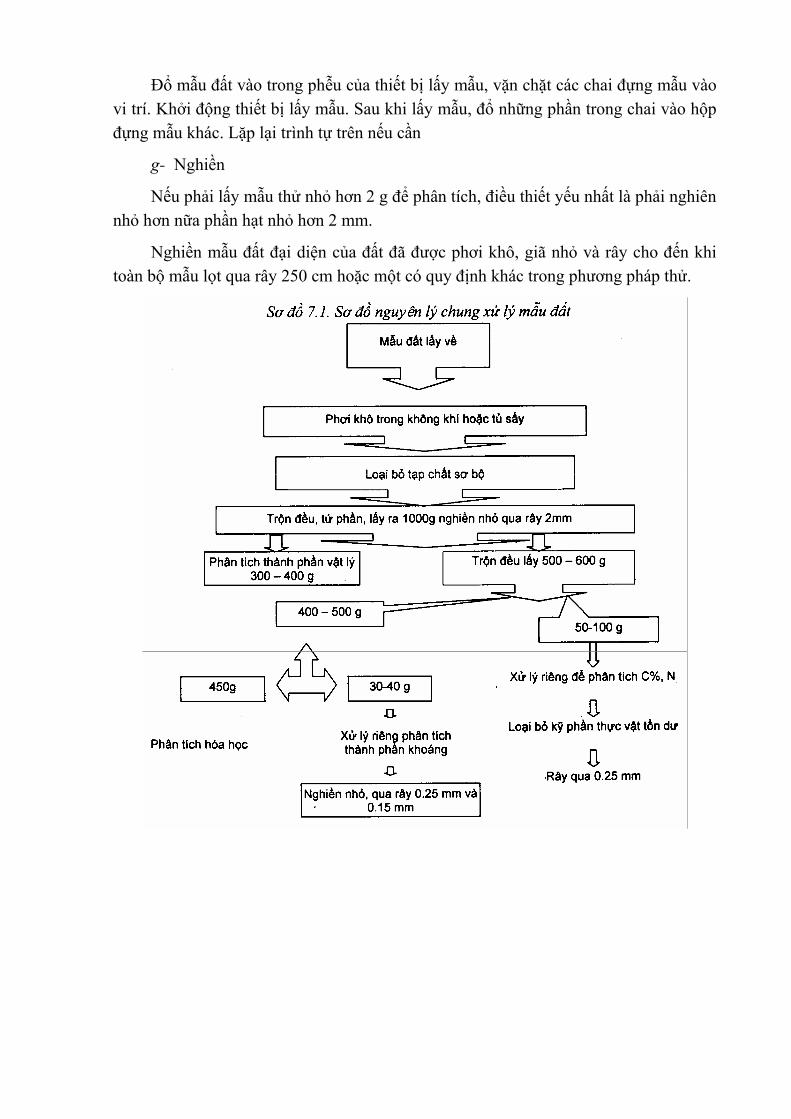
Đổ mẫu đất vào trong phễu của thiết bị lấy mẫu, vặn chặt các chai đựng mẫu vào vi trí. Khởi động thiết bị lấy mẫu. Sau khi lấy mẫu, đổ những phần trong chai vào hộp đựng mẫu khác. Lặp lại trình tự trên nếu cần
g- Nghiền
Nếu phải lấy mẫu thử nhỏ hơn 2 g để phân tích, điều thiết yếu nhất là phải nghiên nhỏ hơn nữa phần hạt nhỏ hơn 2 mm.
Nghiền mẫu đất đại diện của đất đã được phơi khô, giã nhỏ và rây cho đến khi toàn bộ mẫu lọt qua rây 250 cm hoặc một có quy định khác trong phương pháp thử.

7.2. Phân tích một số tính chất lý hoá học cơ bản của đất
7.2.1. Thành phần cơ giới đất
Đất là một hệ thống dị thể gồm những phần tử khoáng, khoáng - hữu cơ và hữu cơ có kích thước khác nhau, từ phân tử đến những nguyên tố cơ học có kích thước lớn như sét, limôn, cát và giăm cuội. Trong nghiên cứu đất, nhiều khi cần phải xác định kích thước của những hợp phần này về mặt số lượng và chất lượng.
Thành phần cơ giới có ý nghĩa quan trọng, nó đặc trưng cho nguồn gốc phát sinh của đất, các tính chất đất và độ phì của đất. Việc phân loại đất về cơ bản cũng dựa vào thành phần cơ giới của đất. Ví dụ, đất cát, cát pha, đất thịt, đất sét... Đối với dinh dưỡng của cây trồng thì thành phần cơ giới đất lại càng có vai trò to lớn, đất có thành phần cơ giới nặng giữ được nhiều chất dinh dưỡng hơn. Nhiều tính chất vật lý của đất như độ xốp, độ trữ ẩm, tính thấm, khả năng giữ khí, giữ nhiệt... đều phụ thuộc vào thành phần cơ giới.
a. Phân loại những nguyên tố cơ học trong đất
Nguyên tố cơ học của đất tồn tại ở 3 dạng: Khoáng, hữu cơ và hữu cơ khoáng (còn gọi là humat). Trong thổ nhưỡng học có nhiều báng phân loại nguyên tố cơ học theo độ lớn của chúng và bảng phân loại của Katrinski được thừa nhận rộng rãi nhất. Trong bảng phân loại này tác giả đã chia thành: đá vụn, sỏi, cuội, cát, limon và sét.
Bảng 7. 5. Phân loại những nguyên tố cơ học của đất (theo Katrinski)
Đường kính (mm) Tên gọi
>3 3-1
1 - 0,5 0,5 - 0,25 0,25 - 0,05 0,05 - 0,01
0,01 - 0,005 0,005 - 0,001
< 0,001 > 0,01 < 0,01
Phần đá vụn của đắt Sỏi cuội Cát thô
Cát trung bình Cát mịn
Limôn thô Limôn trung bình
Limôn mịn Sét
Cát vật lý Sét vật lý.
Tất cả những phân tử có kích thước lớn hơn 1 mm gọi là "phần xương" của đất, những phần tử nhỏ hơn nam gọi là "Phần mịn" của đất. N.M. Xibicxep (1899) phân chia những phân tử của đất thành "cát vật lý" - tức là những cấp hạt lớn hơn 0,01 mm

và "sét vật lý" - những cấp hạt nhỏ hơn 0,01mm. Những khái niệm này hiện nay được sử dụng rộng rãi trong phân loại đất theo thành phần cơ giới.
b. Chuẩn bị đất để phân tích thành phần cơ giới đối với những đất chứa cacbonat
Đối với nhóm đất này việc chuẩn bị phân tích thành phần cơ giới rất phức tạp, người ta thường sử dụng các dung dịch muối khác nhau như: natrioxalat (Na2C2O4); pirophotphat (Na4P2O7) và hecxametaphotphat (Na6P6O18). Tuỳ thuộc vào hàm lượng cacbonat và dung tích hấp phụ của đất mà sử dụng Na2C2O4 0,5N với lượng khác nhau. Việc sử dụng các dung dịch muối nam để xử lý mẫu đất là để đẩy những cacbonat của đất dưới dạng các muối không tan, do đó trong đất sẽ được hình thành những nguyên tố cơ học mới. Những Hợp chất mới này là.(Ca, Mg); C2O4; (Ca2, Mg2) P2O7; (Ca3, Mg3) P6O18; Na2Ca2 (PO3)6. Trong thực tế việc xử lý đất bằng những muối kiềm trên đây thường được kết hợp với những phương pháp cơ học, nghĩa là nghiền, cán đất.
P.F. Menhicốp (1952) đề nghị phương pháp pirophotphat như sau: Cho từ từ dung dịch pirophotphat 5% vào mẫu đất phân tích đến trạng thái sền sệt. Đối với đất mặn và đất cacbonat có thành phần cơ giới nặng thì sử dụng từ 20 - 25ml cho 10 g đất, còn đối với đất cát pha và đất cát thì sử dụng 15ml cho 15g đất. Đối với đất không mặn và không chứa cacbonat có thành phần cơ giới nặng thì sử dụng 5ml; đất thịt, cát pha và cát thì sử dụng 3ml cho lớp đất. Mẫu đất được nhào trộn trong 20 phút, sau đó dùng chày sứ có bịt đầu cao su để nghiền, tán trong 5 phút và đổ vào cốc vừa nghiền tán vừa gạn cho đến khi xuất hiện nước trong khi đất gắng. Cho đất qua rây kích thước 0,1 hoặc 0,25mm vào ống đong có dung tích 1 lít. Thêm nước vào ống đong cho đến 1 lít và phân tích thành phần cơ giới theo phương pháp pipet của Katrmski.
Phương pháp hecxa - metaphotphat để xử lý mẫu đất: lấy khoảng 300g đất, nếu là đất mặn thì trước hết phải loại chỉ bằng cách rửa đất qua giấy lọc trên phễu, dùng nước cất rửa nhiều lần cho đến khi hết lợn Cr (dùng AgNO3 5% trong môi trường được axit hoá bằng HNO3 10% để thử phản ứng clo). Đất sau khi rửa sạch đem phơi ở trong phòng đến trạng thái khô không khí, rây qua rây 1 mm và lấy 20g để phân tích.
Dung dịch dùng để xử lý mẫu đất được chuẩn bị như sau: lấy 3,7 g Na6P6O18 + 7,94g Na2CO3 hoà tan trong 1 lít nước. Natri cacbonat thúc đẩy sự thủy phân của hecxa- metaphotphat thành octophotphat. Lấy 20ml dung dịch này thêm vào mẫu đất đã được rửa (nếu mà đất mặn) và 10 ml đối với đất không mặn, dùng đũa thuỷ tinh đầu có gắn cao su khuấy cẩn thận và sau khoảng 15 - 20 phút dùng nước cất chuyển dần đất sang bình tam giác dung tích 250ml.
Đun dung dịch huyền phù này trong 1 giờ, sau đó chuyển đất vào ống đong có dung tích 1 lít qua rây cỡ 0,1 mm, cần cọ nhẹ và rửa để không cho các phần tử sét bám vào. Dung dịch đất trong ống đong ở trạng thái huyền phù và xảy ra hiện tượng tụ keo.

Nếu quá trình tụ keo xảy ra chậm thì có thể thêm một ít dung dịch xử lý vào dung dịch huyền phù. Trường hợp quá trình tụ keo xảy ra nhanh thì cẩn thận gạn đổ phần nước trong và đổ thêm nước cất cùng với một phần dung dịch xử lý.
c. Nguyên tắc và kỹ thuật phân tích thành phần cơ giới đất
Các phương pháp phân tích thành phần cơ giới đất bao gồm phương pháp ngoài đồng ruộng, phương pháp rây, phương pháp trong môi trường nước (phương pháp gắng gạn, phương pháp tỷ trọng kế, phương pháp pipet).
Phương pháp ngoài đồng ruộng (xác định nhanh, không có dụng cụ
Phương pháp ướt: Tẩm ướt mẫu đất rồi dùng hai lòng bàn tay về thành sợi dài độ 5 - 6cm. Kỹ thuật tẩm ướt đất cần chú ý vừa đủ, không khô quá, không nhau quá, nghĩa là độ ẩm tương ứng với "giới hạn chảy dưới". Sau khi vê thành sợi, tiếp tục uốn thành vòng tròn. Sự thể hiện hình dạng của vòng tròn tương ứng với thành phần cơ giới đất được minh hoạ ở bảng 7.6.
Bảng 7.6. Xác định thành phần cơ giới theo phương pháp ngoài đồng ruộng
Phương pháp này nếu được thực hiện cẩn thận cũng cho những kết quả tốt gần
đúng với kết quả phân tích trong phòng.
- Phương pháp phân tích thành phần cơ giới trong môi trường nước đúng yên tĩnh (phương pháp pipet của Katrinski - Gluskop).
Phương pháp này do Gluskop đề xuất năm 1912. Đến 1922 Robinson đề nghị dùng ống hút với thể tích 25ml để lấy dung dịch huyền phù cho nên còn gọi là ống hút Robinson hay pipet.

Năm 1930, tại Hội nghị Quốc tế Thổ nhưỡng, phương pháp này đã được công nhặn là phương pháp tiêu chuẩn đề xác định thành phần cơ giới đất và được sử dụng rộng rãi ở nhiều nước trên thế giới.
Trình tự phân tích mẫu đất phân tích sau khi phơi trong phòng đến trạng thái "khô không khí", dùng cối chày sứ giã đất và rây qua rây có 1 mm. Trộn đều, lấy 3 mẫu đất để phân tích theo yêu cầu:
Đất thịt và thịt nặng (g) Đất cát và cát pha (g)
- Để xác tính độ hút ẫm không khí (1) 4-5 10
- Để xác tính lượng tiêu hao khi xử lý bằng HCl (2)
10 - 15 20 - 30
- Để chuẩn bị dung dịch huyền phù (3) 10 - 15 20 - 30
Lượng đất để phân tích nhiều hay ít tuỳ thuộc vào đặc tính của đất, đất có thành phần cơ giới càng nhẹ thì lượng đất lấy đê phân tích càng nhiều và ngược lại.
Mẫu đất số 2 và 3 cho vào bát sứ và kiểm tra mức độ chứa cacbonat của đất bằng cách cho nhỏ giọt HCl 10% vào mẫu. Nếu đất có cacbonat thì xuất hiện bọt khí CO2
bay ra và lúc này cần tiếp tục xử lý bằng đồng dịch HCl 0,2N cho đến khi không thấy xuất hiện bọt khí CO2,việc phá huỷ cacbonat khi phân tích thành phần cơ giới là cần thiết vì Ca và Mg trong đất có vai trò chất kết dính, chúng liên kết các nguyên tố cơ học lại với nhau tạo thành những đoàn lạp có kích thước lớn hơn.
Tiếp tục đổ HCl 0,2N vào mẫu đất, dùng đũa thuỷ tinh khuấy đê lắng một thời gian rồi gạn phần nước trong ở bát sứ theo đũa thuỷ tinh qua phễu lọc. Hai mẫu đất được xử lý song song cho đến khi nhỏ HCl 0,2N không thấy xuất hiện bọt khí thì dừng lại.
Sau khi phá huỷ cacbonat, dùng dung dịch HCl 0,05N để chuyển dần mẫu đất ở bát sứ lên giấy lọc đặt trong phễu. Mẫu đất dùng để xác định lượng tiêu hao khi xử lý HCl cần chuyển lên giấy lọc đặt trong phễu nhưng đã cân trước khối lượng của giấy lọc. Đất trên phễu tiếp tục được rửa bằng HCl 0,05N cho đến khi hết phản ứng với Ca2+, Để thử phản ứng, dùng ống nghiệm hứng một ít dịch lọc, thêm hai giọt chỉ thị phenol phtalein và dùng NH4OH 10% để trung hoà theo từng giọt cho đến khi xuất hiện màu hồng, phản ứng lúc này hơi kiềm. Thêm vài giọt CH3COOH 10% để axit hoá dung dịch, thêm vài giọt (NH4)2C2O4 4% và đun đến sôi. Nếu có Ca2+ thì sẽ xuất hiện tinh thể CaC2O4 rất rõ. Nếu hết Ca2+ (không có kết tủa) chuyển hết đất nên phễu bằng dung dịch HCl 0,05N. Tiếp tục rửa mẫu đất trên bằng nước cất cho đến khi hết phản ứng với clo. Để thử phản ứng, dùng ống nghiệm hứng một ít dung dịch lọc, dùng vài giọt HNO3 10% để axit hoá dung dịch, sau đó thêm vài giọt AgNO3 5%, nếu còn do sẽ xuất hiện kết tủa màu trắng AgCl.

Đối với những mẫu đất không có cacbonat thì dùng ngay axit HCl 0,05N để xử lý, sau đó cũng chuyển qua phễu lọc, rửa cho đến khi hết canxi và clo như trình tự trên.
Mẫu đất dùng để tính lượng tiêu hao khi xử lý HCl, sau khi đã rửa đất và giấy lọc được cho vào trong cốc sứ hoặc nhôm đã cân trước khối lượng, sấy khô ở 1050C đến khối lượng không đổi rồi cân. Căn cứ vào hiệu số khối lượng, xác định lượng tiêu hao khi xử lý HCl.
Mẫu đất dùng để phân tích thành phần cơ giới, sau khi đã rửa sạch Ca2+ và do dùng đũa thuỷ tinh chọc thủng giấy lọc, dùng nước cất cọ rửa trực tiếp vào bình có dung tích khoảng 750cm3. Thêm nước cất vào bình đến thể tích khoảng 250cm3 và thêm NaOH là theo dung tích hấp phụ của đất. Cứ 1 ml NaOH là tương đương với 10 mgđl của dung tích hấp phụ. Việc thêm NaOH là với mục địch để phá vỡ hoàn toàn các vi đoàn lạp và chuyển sang các nguyên tố cơ học.
Vì các mẫu đất dùng để phân tích thành phần cơ giới nhiều khi chưa xác định dung tích hấp phụ trước, cho nên để thuận tiện người ta quy định lượng NaOH 1N (ml) thêm vào các loại đất ở bảng sau.
Bảng 7. 7. Lượng NaOH là dùng cho các loại đất khác nhau
Loại đất Lượng NaOH 1N(ml) 1. Đất feralit: Tầng 0 - 50cm Các tầng dưới 2. Đắt feralit trên sản phẩm đá vôi: tầng 0 - 50cm Các tầng dưới 3. Đất macgalit: Tầng 0 - 50cm Các tầng dưới 4. Đất phù sa: Đất phù sa có tầng rửa trôi, bạc màu Các loại phù sa khác
1-2 1 - 1,5 1,5 - 3 2 - 3 2 - 5 2 -3
0,5 - 1,5 0,5 - 1,5
1 - 3
Sau đó đem đun sôi thể huyền phù 1 giờ, thời gian tính từ lúc bắt đầu sôi. Làm nguội dung dịch và chuyển toàn bộ dung dịch vào ống đong có dung tích 1 lít qua rây cỡ 0,25mm. Dùng nước cất rửa phần chứa trên rây và sau đó thêm nước cất đến 1 lít. Như vậy, phần còn lại trên rây gồm những cấp hạt có kích thước từ 0,25 - l,0mm, phần chứa trong ống đong là những cấp hạt có kích thước < 0,25mm. Phần chứa trên rây cho vào chén sứ đã biết khối lượng, sấy ở 1050C trên khối lượng không đổi. Nếu cần tách cấp hạt cát khô 1 - 0,5 mm thì dùng thêm rây 0,5mm và sẽ thu được 2 cấp hạt cất thô: 1 - 0,5mm và cát trung bình 0,5 - 0,25mm.
Tiến hành phân tích các cấp hạt phần trong ống đong dựa vào vận tốc lắng của phương trình Stockes. Tương quan giữa kích thước cấp hạt và độ sâu ống hút (pipet) trong dung dịch như sau:

+ Đối với cấp hạt < 0,050 mm thì độ sâu ống hút là 25cm
+ Đối với cấp hạt < 0,01 mm thì độ sâu ống hút là 10 cm
+ Đối với cấp hạt < 0,005mm thì độ sâu ống hút là 10 cm
+ Đối với cấp hạt < 0,001 mm thì độ sâu ống hút và 7cm.
Thời hạn lấy mẫu từ những độ sâu khác nhau được tính từ khi ngừng khuấy đục dung dịch, thời hạn này thay đổi phụ thuộc vào nhiệt độ và tỷ trọng thể rắn của đất. Trong khi phân tích cần giữ ở điều kiện nhiệt độ cố định. Ví dụ, những ống đong chứa mẫu có đặt trong một bể nước làm bằng miền trong suốt, nước trong bể được điều hoà nhiệt độ nhờ hai vòi nước nóng và lạnh, hoặc ống đong được đậy nắp để giữ nhiệt, hạn chế không cho nhiệt độ dao động gây ảnh hưởng đến vận tốc chìm lắng.
Để theo dõi nhiệt độ, đặt nhiệt kế vào ống đong chứa nước và giữ nhiệt độ như điều kiện của những ống dung dịch đất. Bởi vì khoảng thời gian giữa những lần lắc - hút đối với những cấp hạt < 0,05; < 0,01 và < 0,005 mm không lâu lắm nên nhiệt độ có thể chỉ đo một lần. Khi cần lấy cấp hạt < 0,001 mm nên đo nhiệt độ ba lần: sau khi mắc dụng dịch, trước khi hút và 1 lần vào giữa hai thời điểm trên. Lấy nhiệt độ trung bình sau 3 lần đo, đối chiếu với tốc độ lắng tương ứng chọn để phân tích.
Về tỷ trọng thể rắn, nên lấy giá trị chính xác của từng loại đất nghiên cứu. Tuy nhiên, trong phân tích có thể dùng những tỉ số tương đối gần đúng cho một số trường hợp
Bảng 7.8. Tỷ trọng thể rắn của mã số nhóm đất chính ở Việt Nam
Độ sâu (cm) Nhóm đất feralit (đất đỏ vàng) Nhóm đất phù sa trồng lúa
0 - 20 20-50
50 - 100 100 - 150 150 - 200
2,55 - 2,65 2,60 - 2,65
2,65 2,70 2,70
2,65 (chung cho cả các tầng dưới)
Ở bảng chỉ dẫn thời hạn lấy mẫu dung dịch huyền phù trong ống đong ở các độ sâu khác nhau tương ứng với vận tốc lắng của các cấp hạt theo phương trình Stockes trong điều kiện nhiệt độ và tỉ trọng thể rắn của đất khác nhau.
Vận tốc chìm lắng của các cấp hạt (V) tính theo phương trình Stockes:
Trong đó: r: bánh kính của cấp hạt (mm);
d1: tỷ trọng thể rắn của đất;

d: tỷ trọng chất lỏng khi dùng phân tích;
g: gia tốc trọng trường khi vật rơi tự do (981 cm/s);
η : độ nhớt của chất lỏng (poazơ)
Dụng cụ để phân tích thành phần cơ giới đất bằng pipet theo Katrinski được trình bày ở hình 7.7.
Kỹ thuật lấy mẫu dung dịch huyền phù bằng dụng cụ này như sau khuấy dung dịch huyền phù trong ống đong bằng que khuấy. Que khuấy gồm một ống cao su hình tròn gắn với đũa thuỷ tinh hoặc que kim loại không gỉ. Tầm cao su hình tròn được đục thành nhiều vỗ, đường kính mỗi lỗ khoảng 3mm, que khuấy cho chuyển động từ trên xuống dưới và ngược lại 10 lần trong 20 giây. Theo thời hạn đã tính sẵn (xem phụ lục 8) những ống hút xuống độ sâu quy định hút lấy thành phần cấp hạt cần tìm, dung dịch được hút vào pipet một cách từ từ. Lấy 25ml dung dịch huyền phù khi xác định thành phần cấp hạt nhỏ hơn 0,05mm trong thời hạn 30 giây, đối với cấp hạt < 0,01 mm trong thời hạn 25 giây và cấp hạt nhỏ hơn 0,05 mm trong thời hạn 20 giây. Mẫu lấy xong cho bốc hơi trên bếp cách cát hoặc trên nồi cách thuỷ, sau đó sấy ở nhiệt độ 1050C trên nhiệt độ không đổi. Khối lượng cốc đã được cân sẵn sau khi cân song tính kết quả.
Hàm lượng các cấp hạt được tính ra phần trăm theo công thức:
Trong đó:
x: thành phần cần tìm tính ra %, thành phần nhỏ hơn kích thước cấp hạt nào đó, ví dụ < 0,05mm; < 0,01 mm...
a: khối lượng của thành phần nhỏ hơn kích thước cấp hạt cần tìm (g);
b: thể tích dung dịch huyền phù (ml);
m: khối lượng đất khô tuyệt đối lấy khi phân tích (g)..
Trong thực tế phân tích không nên tính theo đất "khô tuyệt đối" mà tính theo đất

"khô không khí", làm như vậy sẽ có nhiều thuận lợi trong việc tính kết quả. Muốn vậy, nhân công thức trên với hệ số khô kiệt (k). Hệ số k được xác định theo công thức:
Theo công thức này Why là độ hút ẩm không khí (độ ẩm hydroscopic).
Khối lượng thành phần có kích thước xác định (0,05 - 0,01; 0,01 - 0,005mm; 0,005 - 0,001 mm và < 0,001 mm được tính bằng cách trừ khối lượng hoặc hàm lượng phần trăm của thành phần tiếp theo.
Ví dụ cụ thể về cách tính
1) Khối lượng đất khô không khí lấy khí phân tích là 10g;
2) Khối lượng thành phần có kích thước 1 - 0,25mm là 0,81g;
3) Lượng tiêu hao khi xử lý bằng HCl là 0,232g;
4) Khối lượng thành phần trong 25cm3 dung dịch:
Kích thước cấp hạt (mm) Khối lượng thành phần cấp hạt (g)
< 0,05 0,116
< 0,01 0,075
< 0,005 0,015
< 0,001 0,009
5) Thành phần cấp hạt tính ra % so với khối lượng đất khô
Khối lượng cấp hạt(mm) Thành phần (%)
Khi thêm vào dụng dịch huyền phù 2ml NaOH 1N, tương ứng 0,08g, nếu tính với
lượng đất dùng phân tích lớn thì chiếm tỷ lệ 0,8%. Như vậy thực tế hàm lượng cấp hạt

nhỏ hơn 0,001mm chỉ bằng 3,64 - 0,8 = 2,84%.
6) Khối lượng thành phần (%) so với lượng đất
Kích thước cấp hạt (mm) Thành phần (%)
1 - 0,25 = 8,18
0,05 - 0,01 = 46,8 - 30,3 = 16,5
0,01 - 0,005 = 30,3 - 6,06 = 24,24
0,005 - 0,001 = 6,06 - 3,64 = 2,42
< 0,001 = 3,64 - 0,8 = 2,84
7) Lượng tiêu hao khi xử lý bằng HCl:
8) Thành phần cấp hạt có kích thước 0,25 - 0,05mm so với khối lượng đất khô là:
100 - (8,18 +16,5 + 24,24 + 2,42 + 2,84 + 2,34) = 43,48%
Đối với những đấ t có thành phần cơ giới nhẹ, dùng rây khi phân tích có thể phân chia thêm những thành phần chi tiết hơn. Ví dụ, có thể rây cỡ 0,1mm. Như vậy, cùng với cỡ rây có kích thước 0,5 và 0,25mm có thể chia được 1 - 0,5; 0,5 - 0,25 và 0,25 - 0,1mm. Hàm lượng của chúng trong đất được tính bằng %. Tổng lượng của chúng cùng với những thành phần thu được khi hút bằng ống hút cộng gộp lại, lấy 100% trừ cho toàn bộ các thành phần trên sẽ thu được thành phần cấp hạt 0,1 - 0,05 mm, như vậy có thể thu được một đặc trưng chi tiết hơn đối với các thành phần cát của đất nhẹ. Đối với những đất chứa cacbonat, lượng "tiêu hao" khi xử lý bằng HCl nên phân chia thành cột tiếng gọi là "thành phần tổng hợp" cũng gộp vào trong 100% của thành phần cơ giới đất.
d) Các phương pháp thể hiện kết quả phân tích thành phần cơ giới đất
Kết quả phân tích thành phần cơ giới đất thường ghi thành bảng. Trong bảng số liệu này, bên cạnh những thành phần cấp hạt còn ghi thêm cột "độ ẩm không khí" và "lượng tiêu hao" khi xử lý bàng HCl. Trị số về "lượng tiêu hao" đặc trưng cho sự có mặt đất những cacbonat và những muối hoà tan trong đất.
Để hình dung những kết quả phân tích thu được, người ta thường thể hiện dưới dạng đồ thị.
Ví dụ, phương pháp phẫu diện, phương pháp hình tròn (hình 7.8; 7.9)

Việc thể hiện trên đồ thị theo phương pháp phẫu diện hiện nay được sử dụng
rộng rãi trong thổ nhường bởi vì nó có thể biểu thị được những dẫn liệu phân tích theo độ sâu các tầng phát sinh khác nhau.
Trục tung ghi độ sâu, trục hoành ghi hàm lượng % thành cấp hạt. Khi vẽ nhớ tính trị số sau cộng tiếp theo trị số trước ở trục hoành.
e) Ứng dụng thực tiễn của thành phần cơ giới
Có nhiều bảng phân loại thành phần cơ giới đất, nhưng bảng phân loại của Katrinski hiện nay được dùng rộng rãi trong thổ nhường học. Theo bảng phân loại này thì việc xác định tên gọi và phân chia nhóm đất đều dựa vào kết quả phân tích thành phần cơ giới. Đất gọi theo thành phần cơ giới căn cứ vào hàm lượng cát vật lý và sét vật lý và theo ưu thế trội của các thành phần; sỏi, sạn 1 - 3mm, cát 1 - 0,05mm, limon thô 0,01 - 0,05mm, limon 0,01 - 0,001mm và sét < 0,001mm.
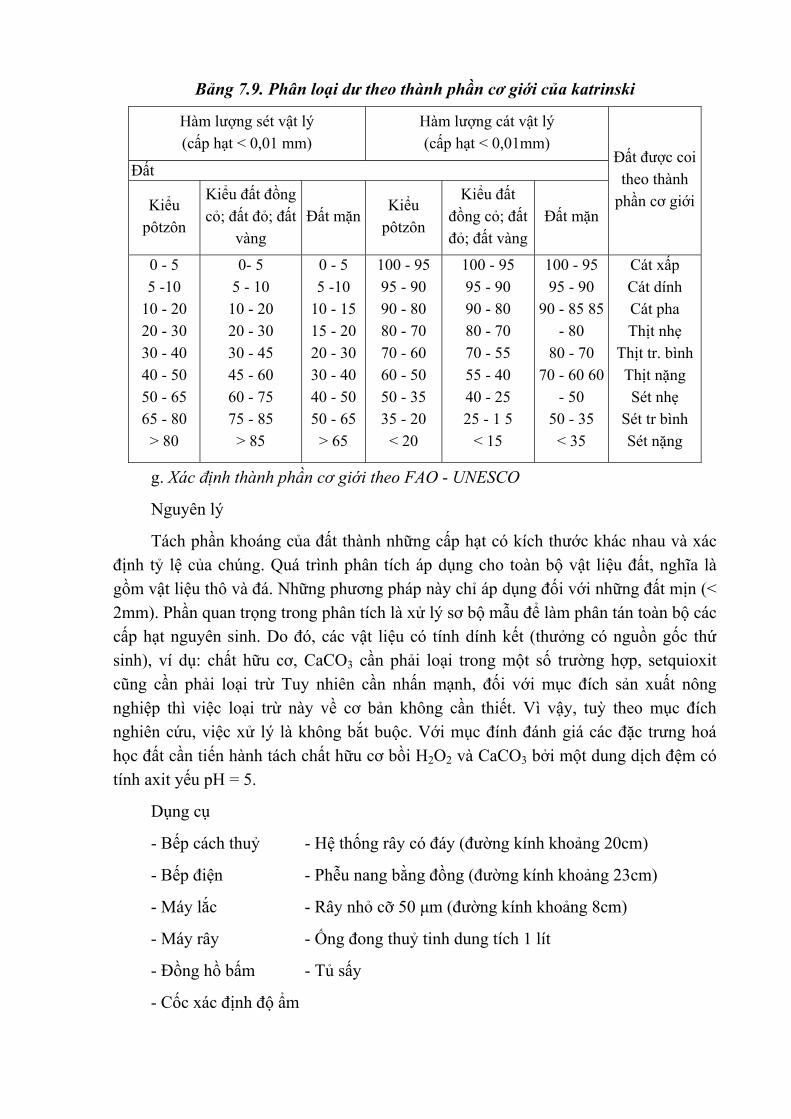
Bảng 7.9. Phân loại dư theo thành phần cơ giới của katrinski
Hàm lượng sét vật lý (cấp hạt < 0,01 mm)
Hàm lượng cát vật lý (cấp hạt < 0,01mm)
Đất
Kiểu pôtzôn
Kiểu đất đồng cỏ; đất đỏ; đất
vàng Đất mặn
Kiểu pôtzôn
Kiểu đất đồng cỏ; đất đỏ; đất vàng
Đất mặn
Đất được coi theo thành
phần cơ giới
0 - 5 5 -10
10 - 20 20 - 30 30 - 40 40 - 50 50 - 65 65 - 80
> 80
0- 5 5 - 10 10 - 20 20 - 30 30 - 45 45 - 60 60 - 75 75 - 85
> 85
0 - 5 5 -10
10 - 15 15 - 20 20 - 30 30 - 40 40 - 50 50 - 65
> 65
100 - 95 95 - 90 90 - 80 80 - 70 70 - 60 60 - 50 50 - 35 35 - 20
< 20
100 - 95 95 - 90 90 - 80 80 - 70 70 - 55 55 - 40 40 - 25 25 - 1 5
< 15
100 - 95 95 - 90
90 - 85 85 - 80
80 - 70 70 - 60 60
- 50 50 - 35
< 35
Cát xấp Cát dính Cát pha Thịt nhẹ
Thịt tr. bình Thịt nặng Sét nhẹ
Sét tr bình Sét nặng
g. Xác định thành phần cơ giới theo FAO - UNESCO
Nguyên lý
Tách phần khoáng của đất thành những cấp hạt có kích thước khác nhau và xác định tỷ lệ của chúng. Quá trình phân tích áp dụng cho toàn bộ vật liệu đất, nghĩa là gồm vật liệu thô và đá. Những phương pháp này chỉ áp dụng đối với những đất mịn (< 2mm). Phần quan trọng trong phân tích là xử lý sơ bộ mẫu để làm phân tán toàn bộ các cấp hạt nguyên sinh. Do đó, các vật liệu có tính dính kết (thưởng có nguồn gốc thứ sinh), ví dụ: chất hữu cơ, CaCO3 cần phải loại trong một số trường hợp, setquioxit cũng cần phải loại trừ Tuy nhiên cần nhấn mạnh, đối với mục đích sản xuất nông nghiệp thì việc loại trừ này về cơ bản không cần thiết. Vì vậy, tuỳ theo mục đích nghiên cứu, việc xử lý là không bắt buộc. Với mục đính đánh giá các đặc trưng hoá học đất cần tiến hành tách chất hữu cơ bồi H2O2 và CaCO3 bởi một dung dịch đệm có tính axit yếu pH = 5.
Dụng cụ
- Bếp cách thuỷ - Hệ thống rây có đáy (đường kính khoảng 20cm)
- Bếp điện - Phễu nang bằng đồng (đường kính khoảng 23cm)
- Máy lắc - Rây nhỏ cỡ 50 μm (đường kính khoảng 8cm)
- Máy rây - Ống đong thuỷ tinh dung tích 1 lít
- Đồng hồ bấm - Tủ sấy
- Cốc xác định độ ẩm

Hoá chất
- H2O2 30%
- Dung dịch đệm axetat nồng độ 1 M: hoà tan 680g CH3COONa.3H2O trong khoảng 4 lít nước, chỉnh tới pH = 5,0 bằng 250ml CH3COOH, thêm nước cất đến 5 lít. Thuốc thử làm phân tán: Dung dịch Pirophotphat 5%: hoà ta 50g Na4P2O7.10H2O trong chai có dung tích 1 lít, sau đó thêm nước cất đến vạch mức.
Dung dịch NaCl bão hoà hoà tan 350g NaCl trong 1 lít nước nóng, làm nguội.
Trình tự phân tích
Đối với đất có chứa cacbonat (pH H2O > 6,8) thì tiến hành loại cacbonat bằng xử lý bởi dung dịch đệm axit yếu. Nếu đất không chứa cacbonat (pHH2O ≤ 6,8) thì tiến hành ôxy hoá chất hữu cơ
1) Loại cacbonat: Cân khoảng 20g đất mịn cho vào cốc dung tích 1 lít (nếu hàm lượng cacbonnat nhiều hơn 10 thì có thể cân nhiều hơn 20g đất). Thêm khoảng 100 ml dung dịch đệm và đun nóng trên bếp cách thuỷ (1000C). Đậy cốc bằng miếng kính. Sau khi ngừng sủi bọt thêm khoảng 25ml dung dịch đệm đến khi sự sủi bọt không tái diễn sau khi thêm xếp dung dịch đệm mới. Trong trường hợp hàm lượng cacbonat rất nhiều có thể thêm 5ml axit axetic băng thay cho dung dịch đệm. Trong trường hợp này có thể dùng giấy quỳ tím đề thử pH.
Li tâm và gạn để qua đêm rồi dùng xi phông hút dung dịch bên trên.
Trường hợp bị tụ keo (Peptit hoá), thêm một vài minilit dung dịch NaCl bão hoà.
Chú ý: Phương pháp rửa này nhằm loại Canxi axetat từ dung dịch lơ lửng vì nó có thể truyền sang dạng canxi oxenat không tan.
2) ôxy hoá chất hữu cơ: Cân khoảng 20g đất mịn, cho vào cốc dung dịch 1 lít thêm 15ml nước vào 15ml H2O2 30% (trong trường hợp xử lý sơ bộ thì không cần thêm H2O2 hoặc thêm ít), đậy cốc bằng miếng kính. Trường hợp sủi bọt mạnh, đặt cốc vào trong bề nước lạnh Cũng có thể ngăn sự sủi bọt bằng cách thêm vài giọt cồn. Để qua đêm, hôm sau đặt lên bếp cách thuỷ (khoảng 800C) và thêm đều đặn 5 - 10 ml H2O2 30% cho đến khi phân huỷ chất hữu cơ xảy ra hoàn toàn (thông thường dung dịch bên trên trở nên trong).
Thêm H2O tiện thể tích khoảng 30ml. Đặt lên bếp nóng và đun sôi cẩn thận trong 1 giờ để loại bỏ toàn bộ H2O2 còn dư. Lấy cốc ra và để nguội. Ly tâm hoặc gạn để các vật liệu lắng trong bình và dùng xi phông hút. Thêm khoảng 300ml H2O và làm phân tán lại kết tủa. Nhắc lại 8 - 9 lần cho đến khi xuất hiện petit hoá. Muốn vậy phải rửa nhiều lần (hơn 4 lần), sau đó thêm một vài ml dung dịch NaCl bão hoà để xúc tiến peptit hoá.
Chú ý: Đối với những đất có chứa thạch cao cần rửa nhiều để hoà tan chúng

3) Tách hợp chất sắt: Thao tác này thường thực hiện sau những xử lý sơ bộ, trước khi làm phân tán đất.
Thuốc thử
Dung dịch đệm Natri xitrat 0,3M và Natri đicacbonat 0,1M: hoà tan 88g Na - xitrat. 2H2O và 8,4g NaHCO3 trong nước và thêm nước tới vạch 1 lít.
Natriđithionit
Trình tự phân tích
Thêm khoảng 200ml dung dịch đệm vào 20g đất mịn chứa trong cốc dung dịch 1 lít. Đun nóng trên bếp cách thuỷ tới khoảng 750C (không được vượt quá 800C vì S nguyên tố sẽ kết tủa). Dùng thìa thêm khoảng lg Nađithionlt và khuấy liên tục khoảng 1 phút, sau đó khuấy ngắt quãng trong 5 phút. Nhắc lại 3 - 4 lần. Ly tâm và gạn lọc cho lắng rồi dùng xiphông hút.
Đối với những mẫu chứa nhiều hơn 5% Fe2O3 có dạng chết dễ rút, cần nhắc lại trình tự 1 - 2 lần màu nâu hoặc màu đỏ của mẫu chứng tỏ chứa loại Fe hoàn toàn. Rửa thêm lần nữa với khoảng 250m hoặc 500ml NaCl 1M. Tiếp tục làm như khâu cuối cùng của phần ôxy hoá chất hữu cơ.
4) Làm phân tán đất: Chuyển hết dung dịch lơ lửng vào chai nhựa dung tích 1 lít (nếu không xứ lý sơ bộ, cân khoảng 20g vào chai). Thêm 25ml thuốc thử phân tán dùng nước đưa thể tích đến khoảng 400ml rồi nút chai, lắc để qua đêm (6 giờ)
5) Tách riêng các cấp hạt: Rây dung dịch lơ lửng qua cỡ 50 μm qua phễu đặt trên ống đong. Dùng nước đưa thể tích trong ống đến 1 lít. Rửa cấp hạt cát trên rây và chuyên vào cốc, cho bay hơi trên bếp cách thuỷ, sấy ở 1050C khoảng 1 giờ.
6) Xác định cấp hạt cát: Chuyển cát vừa sấy khô vào rây ở trên cùng của một chồng rây theo thứ tự 1000μm, 500μm, 250μm, 100μm, 50μm vào đáy
Rây khoảng 10 phút trên máy rây, được thiết đặt biên độ: độ rộng 7,0 và khoảng cách 4 (ở vị trí lắp đặt này các rây sẽ rung với tần số 3000 dao động/phút và biên độ khoảng 2mm). Rửa sạch các cấp hạt ở tùng rây ở các cốc đã cân trước, sấy, cân. Khối lượng A qua E là cấp hạt cát.
Nếu có vật liệu tụ lại ở rây đáy (< 50μm) thì chuyển nó sang dạng lơ lửng trong ống đong để lắng đọng như cách làm ở phần tách riêng các cấp hạt.
7) Xác định limon (slit) và sét (clay):
Cấp hạt < 50μm: Vật liệu < 50 μm có thể đọng lại trong quá trình rây (xem xác định cấp hạt cát), do vậy cần đậy ống đong bằng nút cao su và lắc đều. Đặt ống đong trên bàn, mở nút và dùng pipet hút 20 ml ở giữa tâm ống đong. Chuyển dung dịch vào cốc đã cân trước cho bay hơi trên bếp cách thuỷ và sấy qua đêm ở 1050C. Lấy cốc ra và làm nguội trong bình hút âm rồi cân (khối lượng khô F cho cấp hạt < 50μm).

Cấp hạt < 20μm: Đậy nắp ống đong và lắc đều. Đặt ống đong lên bàn dưới ống hút pipet. Sau đúng 5 phút, dùng pipet hút 20ml ở độ sâu như bảng 7.10. Chuyển dung dịch lơ lửng vừa lấy vào cốc đã cân trước, cho bay hơi trên bếp cách thuỷ, sấy khô ở 1050C, cần.
Khối lượng mẫu G là cấp hạt < 20 cm.
Cấp hạt < 2μm: Sau 5giờ 30 phút dùng pipet hút 20ml ở độ sâu của bảng 7.10.
Chuyển dung dịch mới lấy vào cốc đã cân trước, cho bay hơi trên bếp cách thuỷ, sấy khô ở 1050C và cân.
Khối lượng H+ là cấp hạt < 2μm.
Chú ý: Sau mỗi lần lấy mẫu bằng pipet, đo nhiệt độ của dung dịch lơ lửng.
Bảng 7.10. Độ sâu (cm) lấy mẫu cấp hạt phụ thuộc vào nhiệt độ(0C)
Nhiệt độ (0C)
5 phút (< 0,02mm)
5h30' (0,002 ml
Nhiệt độ (0C)5 phút
(< 0,02mm) 5h30'
(0,002 ml)
19 10,5 6,9 28 13 8,6 20 10,8 7,1 29 13,3 8,8 21 11,0 7,2 30 13,6 9,0 22 11,3 7,4 31 13,9 9,1 23 11,6 7,6 32 14,2 9,3 24 11,9 7,8 33 14,4 9,5 25 12,1 8,0 34 14,8 9,7 26 12,4 8,2 35 15,1 9,9 27 12,7 8,4 36 15,4 10,1
Kết quả theo khối lượng sau khi sấy khô. Đó là tổng của tất cả các cấp hạt riêng biệt.
Cánh tính:
Sét (clay) (< 2μm) = (H x 50) - 0,75g* (khối lượng K)
Limon (silt) (2 -20 μm) = (G x 50) - 0,75 - K (khối lượng L)
Limon (20 -50 μm) = (F x 50) - 0,75 - K - L (khối lượng M)
Cát (> 50 μm) -A +B + C + D +E (khối lượng N)
Khối lượng mẫu = K + L + M + N (tính theo gam)
Khối lượng các cấp hạt theo % tính như sau:

Chú ý: Với cách tính như trên thì các cấp hạt sét, limon và cát tính ra % là của
đất cát mịn (trừ cacbonat và chất hữu cơ đã bị loại trước). Cấp hạt thô > 2mm nếu có, tính ra % của tổng số đất. Nếu tất cả các cấp hạt cần tính ra tổng số đất thì:
Trường hợp tách Fe thì % "sắt tự do" sẽ tính cả vào phần ngoặc đơn của công
thức.
8) Xác định sét mịn (< 0,2 μm):
Nguyên lý

Vì tốc độ lắng chậm của hạt sét nên không thuận tiện cho lắng trong ống đong. Do đó phải dùng máy ly tâm để tăng tốc độ lắng.
Trình tự phân tích
Sau khi dùng pipet lấy mẫu cấp hạt < 2 μm, đậy nút ống đong và lắc đều. Để yên tĩnh khoảng 1 giờ và lấy khoảng 200ml dung dịch lơ lửng và tâm máy ly tâm trong khi quay là khoảng 16cm.
Cho máy quay với vận tốc 1800 vòng/phút theo thời gian chỉ dẫn ở bảng 7.11 (trừ thời gian bắt đầu và kết thúc).
Chú ý: Nếu quay 2500 vòng/phút thì thời gian sẽ ít hơn. Trong trường hợp này sử dụng chai nhựa. Tắt máy ly tâm, lấy các chai ra khỏi máy và đặt dưới pipet. Dùng pipet hút 20ml ở độ sâu 4,5 cm. Đo nhiệt độ của dịch lơ lửng. Chuyển thể tích đã lấy vào cốc đã cân trước, cho bay hơi trên bếp cách thuỷ, sấy ở nhiệt độ 1050C qua đêm. Làm nguội cốc trong bình hút ẩm và cân khối lượng cấp hạt P (g).
Tính kết quả:
Bảng 7.11. Tốc độ ly tâm, thời gian quay và nhiệt độ để
xác định cấp hạt sét mịn < 0,2 μm
Nhiệt độ 0C 1800 vòng/ phút 2500 vòng/ phút 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
32,0 31,0 30,0 29,5 29,0 25,0 27,5 27,0 26,5 26,0 25,0 24,5 24,0 23,5 23,0 22,5 22,0 22,0 21,5 21,0 20,5
16,5 16,1 15,7 15,3 15,0 14,6 14,2 14,0 13,5 13,3 13,0 12,8 12,5 12,3 12,0 11,8 11,5 11,3 11,1 10,9 10,6

9) Sét phân tán trong nước
Nguyên lý
Sét phân tán trong nước là hàm lượng sét xác định được khi không xử lý sơ bộ mẫu và không thêm tác nhân phân tán trước khi lắc với nước.
Trình tự phân tích
Cân khoảng lớp đất mịn cho vào chai dung dịch 1 lít. Thêm khoảng 400ml nước và lắc qua đêm. Chuyển sang ống đong lắc, dung tích 1 lít và thêm nước đến vạch mức. Sau 5h30' dùng pipet hút 20ml ở độ sâu theo bảng 7.10. Chuyển dung dịch lơ lửng vừa lấy vào cốc đã cân nước, cho bay hơi trên bếp cách thuỷ, sấy ở l050C qua tiêm. Làm nguội cốc trong bình hút ẩm rồi cân (khối lượng cấp hạt Q(g).
Cách tính
Trong đó: s: khối lượng máu khô không khí tính theo gam;
mef: hệ số độ ẩm.
10) Một số phương pháp khác trong phân tích thành phần cơ giới đất
Nhìn chung nhiều nhà khoa học trên thế giới đã thừa nhận phương pháp pipet là phương pháp chính xác nhắt, thuận tiện nhất. Tuy nhiên, ở một số nước lại sử dụng những phương pháp riêng.
Ví dụ, ở Mỹ kích thước cấp hạt sét được quy đinh là < 0,002mm; dùng H2O để phá huỷ Cacbonat. Cấp hạt limon được quy định có kích thước từ 0,05 - 0,002mm, trong khi đó cũng cấp hạt li mon này được Hội thổ nhưỡng Quốc tế quy định là 0,02 - 0,002mm. Các nước này đã sử dụng máy lắc như là biện pháp cơ học để phân tán các cấp hạt và khi phân loại người ta sử dụng tam giác đều với thang phân loại như sau:
1. Cát (sand)
2.Cát pha thịt (Loamy - sand)
3.Thit pha cát (Sandy - loam)
4. Thịt (loam)
5. Thịt pha limon (Silty - Loam)
6. Limon (Silt)
7. Thịt pha sét và pha cát (Sandy clay - loam)
8.Thịt pha sét và pha limon (Silty đay - loam)
9. Thịt pha sét (Clay - loam)

10. Sét pha limon (Silty - clay)
11. Sét pha cát (Sandy - clay)
12. Sét (clay)
Cần chú ý rằng khi phân loại các cấp hạt có tác giả dùng tam giác đều hình 7.10,
có tác giả lại dùng tam giác vuông với trục tung biểu thị % của hàm lượng cấp hạt sét; trục hoành (cạnh đáy) biểu thị % của hàm lượng cấp hạt cải còn cạnh huyền để trống.
Thành phần cơ giới theo những phương pháp của Mỹ được trình bày bằng hình tam giác đều (hình 7.8) bao gồm 12 loại; 3 nhóm cấp hạt. sét; li mon và cát được biểu thi ở 3 cạnh. Đỉnh tam giác vuông tương ứng 100%. Từ đáy tam giác đến đỉnh chia thành 10 hàng, mỗi hàng tương ứng 10%. Hàm lượng của 3 nhóm cấp hạt: cát, limon, sét được biểu thị ở 3 đường thẳng song song với đáy tam giác theo điểm giao nhau của ba đường thẳng trong tam giác sẽ biết được loại đất cần tìm.
7.2.2. Xác định dung trọng của đất
Dung trọng (khối lượng của một đơn vị thể tích) của đất là tỷ số giữa khối lượng đất khô tuyệt đối ở trạng thái ty nhìên(kể cà những lỗ hồng của một thể tích xác định với khối lượng của nước có cùng thể tích ở 40C.
Dung trọng đặc trưng cho độ chặt của đất trong thổ nhường học, dung trọng sử đụng để:
- Tính trữ lượng nhiều nguyên tô và chuyên chúng từ % sang thể tích.
- Đánh giả một cách khách quan quá trình rửa trôi theo chiều sâu và việc chuyển rời các nguyên tố từ tầng này sang tầng đất khác.

- Tính độ hồng hay độ xốp của đất.
Có nhiều phương pháp xác định dung trọng nhưng thuận tiện và phổ biến nhất hiện nay và phương pháp dùng ống đóng.
Trình tự phân tích:
Đặt những ống đóng hình trụ có thể tích 100 m3 nên khu vực của thành phẫu diện đất đã được làm bằng phẳng, ít nhất pa xác định 3 lần tập lại và như vậy phải sử dụng 3 ống song song cùng một lúc. Đặt các khuân định hướng nên trên những ống này sau đó dùng choòng (nêm) đặt lên trên cùng dừng búa gỗ đóng các ống vào đất (hình 7.11)
Nhờ hệ thống choòng và ống khuân định hướng nên các ống đóng được đóng thẳng và lún sâu vào đất. Khi ống đóng đã lặn sâu vào đất hết cỡ thì nhắc choòng ra, dùng dao hoặc xẻng đào, gọt, nhắc ống đóng ra dùng tấm gỗ mỏng rồi lật ngược ống đóng dùng dao sắc gọt bằng phẳng đất ờ mặt đáy dưới của ống. Đậy nắp, sau đó gọt phía đầu trên công bằng phẳng như vậy. Lau chùi sạch đất dính bám xung quanh ống. Dùng dao xén cạy đất trong ống vào hộp nhôm lớn, trường hợp không có hộp nhôm thì dùng túi polyetilen hoặc giấy bóng mờ đã gập sẵn thành phong bì. Đem cân hộp nhôm cùng với đất tại độ chính xác 0,01 g.
Sau đó có thể sử dụng mẫu đất này để xác định tỉ trọng thể rắn, độ hút ẩm cực đại...
Biết được khối lượng hộp nhôm (hoặc khối lượng túi pôlyetilen, túi giấy) khối lượng đất và hộp có thể tính được khối lượng đất khô tuyệt đối. Biết khối lượng đất khô tuyệt đối và thể tích của ống đóng ta sẽ tính được dung trọng theo biểu mẫu sau:

Bảng 7. 12. Biểu mẫu ghi và tính kết qủa dung trọng của đất Đất cây
trồng địa điểm
Tầng độ sâu
(cm)
Số hộp
nhôm hoặc túi
Khối lượng
bì (hộp nhôm hoặc túi) (a)
Khối lượng bì
+ đất ướt (b)
Khối lượng đất
tươi (P1 = b -
a)
Độ hút ẩm không
khí (Why%)
Khối lượng đất khô tuyệt đối
hyWPP
+=
100100.1
Thể tích ống đóng -
(V) cm3
Dung trọng
VPDv =
Theo thang đánh giá của Katrinski
Dung trọng nhỏ hơn 1 g/cm3 : đất giàu chất hữu cơ
Từ 1,0 đến 1,1 : điền hình cho đất trồng trọt
1,2 : đất hơi chặt
1,3 đến 1,4 : đất quá chật
1,4 - 1,6 : điển hình cho tầng đế cày.
1,6 - 1,8 : tầng tích tụ quá chặt
7.2.3. Xác định chất hữu cơ trong đất
Sự tích luỹ chất hữu cơ ờ dạng mùn ở trong đất là do hoạt động vi sinh vật, thực vật cũng như bón phân hữu cơ. Hàm lượng, thành phần mùn quyết định hình thái và các tính chất lý, hoá học, độ phì của đất. Trong tầng mùn chứa gần 90% nhơ ở dạng dự trữ và phần lớn các nguyên tố dinh dưỡng như P, S, nguyên tố vi lượng, là kho dự trữ chất dinh dưỡng cho cây trồng.
Hiện có nhiều phương pháp xác định chất hữu cơ của đất phương pháp đốt khô, phương pháp đốt ướt (chiurin, Walkley - Black), phương pháp đốt mùn trong tủ sấy 1500C thời gian 20 phút (Nikitin) và phương pháp ôxy hoá mùn 24 giờ ở nhiệt độ 200C(P. Antanova). Dưới đây là một số phương pháp phổ biến ở Việt Nam.
a) Xác định chất hữu cơ theo phương pháp Chiurin
Nguyên lý
Chất hữu cơ của đất, dưới tác dụng của nhiệt độ, bị dung dịch K2Cr2O7 + H2SO4 (1:1) ôxy hoá:
Lượng K2Cr2O7 còn dư được trung dung dịch muối có tính khử là FeSO4 hay
muối Morh (FeSO4. (NH4+)2SO4.6H2O) để chuẩn:

Chất chỉ thị cho quá trình chuẩn độ này thường dùng là axit phenylanthranilic (C13H11O2N), màu chuyển từ đỏ đậm sang xanh lá cây, hoặc diphenylamin (C12H11N) màu sẽ chuyển từ lam tím sang xanh lá cây.
Trong quá trình chuẩn độ, Fe3+ tạo thành có thể ảnh hưởng tới quá trình chuyển màu chỉ thị, vì vậy trước khi chuẩn độ có thể cho thêm một lượng nhỏ H3PO4 hoặc muối chứa ion F- để tạo phức không màu với Fe3+.
Trình tự phân tích
Đất để phân tích mùn và đạm phải được chuẩn bị cẩn thận lấy 5 - 10g đất đã rây qua rây 1 mm, nhặt hết xác thực vật rồi giã nhỏ, rây qua rây 0,25mm, trộn đều.
Dùng cân phân tích cân 0,2g (đất nghèo mùn - dưới 1 % thì cân 0,4g, còn đất giàu mùn thì cân 0,1 g) cho vào bình tam giác 100 ml.
Dùng buret cho từ từ chính xác loạn K2Cr2O7 0,4N vào bình lắc nhẹ bình, tránh để đất bám nên thành bình. Đậy bình bằng một chiếc phễu nhỏ. Đun trên bếp cách cát cho dung dịch sôi ở nhiệt độ 1800C trong 5 phút.
Lấy ra để nguội dùng nước thêm 10 - 20 ml vào xung quanh thành bình để rửa đicromat bám vào. Cho vào 4 giọt chỉ thị axit phenylanthranilic 0,2% và chuẩn độ bằng dung dịch muối Morh 0,2N đến khi dung dịch chuyên từ màu tím mận sang màu xanh lá cây.
Đồng thời làm một thí nghiệm trắng: cân 0,2g đất đã nung hết chất hữu cơ cho vào bình tam giác, cho vào đúng loạn K2Cr2O7 0,4N và tiến hành các bước như phân tích mẫu.
Tính kết quả:
Trong đó: V0: số mũ muôi Morh dùng chuẩn độ thí nghiệm trắng;
V: số ml muối Morh dùng chuẩn độ mẫu thí nghiệm;
N: nồng độ đương lượng của dung dịch muối Morh;
a: lượng mẫu đất lấy phân tích (g);
K: hệ số chuyển đồi từ mẫu khô không khí sang mẫu khô tuyệt đối
Hoá chất
- K2Cr2O7 0,4N trong H2SO4 (l:1): cân 40g K2Cr2O7 tinh khiết, nghiền bằng chày trong cối sứ, hoà tan trong 500ml nước. Nếu cần, đun nóng nhẹ đề tan hoàn toàn. Để nguội, lọc rồi định mức đến 1 lít. Đổ dung dịch vào bình định mức 2 lít rồi rót từ từ H2SO4 đặc (d = l,84) vào, vừa rót vừa lắc nhẹ, nếu quá nóng thì phải đề nguội rồi mới rót tiếp cho đến thể tích 2 lít. Nồng độ của dung dịch này được kiểm tra bằng dung

dịch FeSO4 (hoặc muối Morh) 0,2N. Có trường hợp sau khi pha xong, để một vài hôm thấy có tinh thể màu đỏ hình kim xuất hiện, trong trường hợp này chỉ cần thêm ít nước, lắc đều tinh thể sẽ mất.
- Dung dịch muôi Morh 0,2N: cân 80g (NH4)2SO4, FeSO4.6H2O hoà tan trong nước, thêm 20ml H2SO4 đặc định mức đến 1 lít. Nồng độ của dung dịch này được xác định bằng cách chuẩn độ với dung dịch KMnO4 chuẩn: lấy lòng muối Morh cho vào bình tam giác 250ml, thêm lưu H2SO4 đặc, 50ml nước cất chuẩn độ bằng dung dịch KMnO4 lần đến khi xuất hiện màu hồng bền trong 1 phút.
- Chỉ thị axit phenylanthranilic 0,2g hoà tan trong 100 ml Na2CO3 0,2%. Sự chuyển màu đen dần về sau này không ảnh hưởng gì đến việc sử dụng chỉ thị.
Chú thích
Trong phương pháp này phải chú ý khống chế nhiệt độ khi đun ôxy hoá mẫu. Nhiệt độ cao quá 1800C sẽ dẫn tới việc phân huỷ Cromic. NikitinB.A. (1972) đưa ra phương pháp ôxy hoá chất hữu cơ bằng cách đun trong tủ sấy tại nhiệt độ 1500C trong 20 phút. Đất chứa nhiều clorua cũng ảnh hưởng đến kết quả phân tích vì có một phần Cr2O7 - tiêu tốn cho ôxy hoá Cr:
Vì vậy khi phân tích đất mặn cần rửa sạch hết Cr trước khi phân tích chất hữu cơ.
b. Xác định chất hữu cơ theo phương pháp Walkley - Black
Nguyên lý
Phương pháp dựa trên nguyên tắc ôxy hoá chất hữu cơ của đất bằng dung dịch K2Cr2O7 sau đó chuẩn lại lượng K2Cr2O7 dư, từ đó tính được hàm lượng chất hữu cơ dư điểm khác nhau ở phương pháp này so với phương pháp Chiurin ở chỗ: nhiệt dùng cho quá trình ôxy hoá được tạo ra do quá trình hoà tan H2SO4 đặc trong trung dịch kalidicromat.
Trình tự phân tích
Cân 1 g đất khô không khí cho vào bình tam giác 500ml thêm 10ml dung dịch K2Cr2O7 lN. Thêm 20ml H2SO4 đặc vào (thêm nhanh theo khả năng có thể thêm). Lắc nhẹ và giữ 30 phút. Thêm 200ml nước cất và lòng H3PO4 85%. Thêm lâu chỉ thị điphenylamin. Chuẩn độ bằng dung dịch FeSO4 0,5N đến khi dung dịch có màu xanh lá cây.
Hoá chất
- Hoà tan 49,039 K2Cr2O7 trong nước cất và thêm thể tích đến 1 lít.
- FeSO4 0,5N: hoà tan 139g FeSO4.7H2O trong 800ml nước, thêm 20ml H2SO4 đặc rồi định mức đến thể tích 1 lít.

- Điphenylamin: hoà tan 0,5g chỉ thị trong 20ml nước rồi thêm tiếp 100 ml H2SO4 đặc.
Tính kết quả
Trong đó:
N: nồng độ đương lượng của muối FeSO4;
V0, V1: thể tích muối FeSO4 dụng đê chuẩn độ thí nghiệm trắng và thuần độ mẫu;
a: lượng mẫu lấy để phân tích (g);
K: hệ số chuyên đôi từ mẫu không khí sang mẫu khô tuyệt đối;
3: đương lượng gam của C;
1,3: hệ số "bù" cho quá trình ôxy hoá chưa hoàn toàn chất hữu cơ trong phương pháp này.
% chất hữu cơ = 2 x C (%)
Trước đây dùng hệ số 1,72 nhưng hiện nay thấy hệ số 2 là thích hợp hơn (ISRIC, 1986).
Chú thích
Trước khi chuẩn độ lượng K2Cr2O7 còn dư (ở cả 2 phương pháp) cần phải để nguội dung dịch ôxy hoá nếu không một phần Fe2+ dùng đề lượng K2Cr2O7 còn dư có thể bị ôxy không khí ôxy hóa.
Với 10 ml K2Cr2O7 1N chỉ có thể ôxy hoá tối đa 25mg C, vì vậy khi áp dụng phương pháp Walkley - Black cần hết sức chú ý lượng mẫu lấy đi phân tích.
Đối với những đất hàm lượng mùn < 2,6% có thể lấy 1 g mẫu đem đi phân tích, nếu hàm lượng mùn cao hơn nên lấy 0,2g đất còn khi hàm lượng mùn >13,5% thì lượng mẫu lấy là 0,1g (Lê Đức - Tạp chí Khoa học Đất 10 - 1998)
Nếu dùng chỉ thị fenoin (0,695g FeSO4.7H2O và 1,485g- phenaltrolinmonohidrat (Cl2H8N2.H2O) trong 100 ml nước thì dùng 4 giọt chỉ thị - khi kết thúc chuẩn độ, màu của dung dịch chuyển từ xanh sang đỏ.
Có thể dùng phương pháp so màu để xác định chất hữu cơ bằng cách đo màu Cr3+ tại bước sóng 625nm. Dùng saccarozơ(C12H22O11) làm trung dịch chuẩn để khử Cr5+ trong K2Cr2O11; làm dung dịch chuẩn đề khử Cr6+ trong K2Cr2O7 thành Cr3+.

7.2.4. Độ chua và cách xác định độ chua của đất
Độ chua là yếu tố độ phí quan trọng của đất, nó ảnh hưởng đến các quá trình lý hoá và sinh học trong đất và có tác động đến cây trồng. Đa số cây trồng thích ứng ở đất trung tính (pH từ 6 đến 7); một số có thể chịu đất chua như chè (pH từ 4,5 đến 5,5), khoai tây pH từ 4,8 đến 5,4.
Đất chua là do có mặt các ion H+ và Al3+ trong dung dịch đất cũng như trong các phức hệ hấp thụ của đất có khả năng trao đổi gây nên.
Khả năng tạo thành H+ và Al3+ càng lớn thì đất càng chua và ngược lại độ chua của đất phụ thuộc vào các phương pháp xác định, trong đó chất chiết rút có ý nghĩa lớn trong trao đổi các ion H+ và Al3+.
Trên cùng một loại đất, sử dụng chất chiết rút NaOH 0,01N (pH = 12) sẽ có độ chua lớn (19mgđl/100g đất); trong khi với NaCH3COO 1N (ph = 8,2) thì độ chua thấp hơn (6,0 mgđl/100g đất); và thấp nhất là khi tác động với đất bằng NaCl 1N (pH = 6,0), độ chua xác định được là 0,2 mgđl/100g đất. Nguyên nhân là do ion OH- có khả năng liên kết mạnh với H+ hằng số phân ly của H2O là 10 - 14) trong khi liên kết của CH3COO- với H+ là nhỏ hơn (hằng số phân ly của CH3COOH là 1,8 x 10-5), Còn Cr hau như không liên kết với H+.
Độ chua của đất thông thường được chia làm 2 loại:
- Độ chua hiện tại (độ chua hoạt tính): là độ chua gây nên do các ion H+ tự do trong dung dịch đất và được xác định khi tác động đất với nước cất và được biểu thị bằng pHH2O.
- Độ chua tiềm tàng: được xác định khi chiết rút đất bằng dung dịch muối. Dựa vào chất triết rút, độ chua tiềm tảng lờ được chia thành độ chua trao đổi và độ chua thuỷ phân
Độ chua trao đổi sử dụng chất chiết rút đất là các dung dịch muối trưng tính như KCl. NaCl, BaCl2 các Cation của các muối này đẩy H+ và một phần Al3+ ra khỏi phức hệ hấp phụ, Al3+ bị thuỷ phân tạo thành độ chua của đất. Độ chua trao đổi là một chỉ số để xác định nhu cầu bón vôi cho đất.
Độ chua thuỷ phân: Độ chua của đất được xác định khi sử dụng chất chiết rút là một muối thuỷ phân (gồm gốc axit yếu và bazơ mạnh như CH3COONa). Thông thường độ chua thuỷ phân có trị số lớn hơn độ chua trao đổi. Vì lúc này gần như toàn bộ H+ và Al3+ trao đổi đã được trao đổi ra ngoài dung dịch đất.
Độ chua thuỷ phân cũng thưởng được sử dụng đề tính lượng vôi bón cải tạo đất chua. Theo nghiên cứu của viên khoa học Kỹ thuật Nông Nghiệp thì đất lúa Việt Nam chỉ nên trung hoà 1/2 độ chua thuỷ phân là tốt nhất.
a) Xác định pH bằng phương pháp cực chọn lọc hiđro

Hiện nay phương pháp đo pa trực tiếp trên máy (pH meter) đã được dùng phổ biến phép đo nhanh, chính xác và phạm vi pH xác định được rộng (pH = 1 ÷ 9).
Nguyên lý
Ion H+ được chiết rút ra bằng chất chiết rút thích hợp (nước cất hoặc muối trung tính) dùng 1 điện cực chỉ thị (điện cực chọn lọc hiđro) và một điện cực so sánh để xác định hiệu thế của dung dịch. Từ đó tính được pH của dung dịch.
Trong các loại máy pH, điện cực chỉ thị thường dùng là điện cực thuỷ tinh, điện cực so sánh là điện cực calomen.
Trình tự phân tích
Lắc 10 g đất (đã qua rây 1 mm) 15 phút trên máy lắc (hoặc sắc tay 30 phút) với 25ml KCl 1N (với pHKCl) hoặc nước cất (pHH2O). Sau đó để yên 2 giờ (không quá 3 giờ), lắc 2 - 3 lần rồi đo pH ngay trong dung dịch huyền phù.
Hiệu chỉnh máy đo phụ: Máy trước khi đo phải hiệu chỉnh bằng cách đo dung dịch đệm pH tiêu chuẩn. Chỉnh cho kim chỉ đúng trị số pH của dung dịch đệm.
Đo mẫu: Giữ cho điện cực cách mặt mẫu đất là 1 cm và ngập nước khoảng 2cm. Khi máy đã ổn định, đọc giá trị pH trên máy.
Ghi chú: Điện cực thủy tinh được ngâm trong nước cất khi không dùng. Tỷ lệ đất và dịch chiết có khác nhau phụ thuộc phương pháp. Vì vậy, trong kết quả phân tích cần ghi rõ tỷ lệ đất: dịch chiết rút và chất chiết rút. Ví dụ: "pH trong KCl 1N - 1: 5W/V". Nghĩa là pH khi chiết rút bằng KCl 1N với tỷ lệ đất dung dịch chiết rút là 1:5 (khối lượng/ thể tích). Nếu không ghi chú gì thì thường được hiệu là pHH2O theo tỷ lệ đất nước là 2:5.
Pha dung dịch đệm tiêu chuẩn
- Dung dịch KHC8H4O4 0,05M: 10,21g KHC8H4O4 pha thành 1000 ml
- Hỗn hợp KH2PO4 + NaHPO4 0,025m: 3,10g KH2PO4 pha thành 1000m1; 3,55g NaHPO4 pha thành 1000ml.
Trộn lẫn 2 dung dịch này thành 2 lít hỗn hợp. Thời hạn sử dụng không quá 2 tháng.
- Dung dịch Na2B4O7 0,01N : 3,81 g Na2B4O8.10H2O pha thành 100 ml
- Dung dịch KHC4H4O6 bão hoà: 6g KHC4H4O6 trong 1 lít nước cất.
- Các dung dịch trên pha song đựng trong bình polyetilen, thời hạn không quá 3 tháng.
- Trị số pH của các dung dịch đệm trên như sau:

Nhiệt độ 0C
KHC8H4O6 bão hoà KHC8H4O4
0,05M KH2PO4 -NaHPO4
0,025M Na2B4O7 0,01M
15 4,00 6,90 9,27 20 4,00 6,88 9,22 25 3,56 4,00 6,86 9,18 30 3,55 4,01 6,85 9.14
Dựa vào độ chua (pHKCl) độ chua của đất được chia ra như sau:
b. Xác định độ chua trao đổi theo phương pháp Daicuhara
Nguyên lý
Sử dụng chất chiết rút KCl, ion K+ sẽ đẩy H+ và Al3+ trao đổi ra khỏi phức hệ hấp thụ (keo đất):
Dùng NaOH chuẩn chuẩn H+ tạo thành với chỉ thị màu phenolphtalein.
Nước chiết này có thể kết hợp đo pHKCl. Xác định nhôm di động (Al - trao đổi) và Ca, Mg trao đổi.
Trình tự phân tích
Cân 40g đất (đã qua rây 1 mm) lắc 1 giờ với 100ml dung dịch KCl 1N (hoặc sắc vài phút rồi đề yên một ngày), sau đó tiến hành lọc.
Lấy 50ml dịch lọc + 3 giọt phenolphtalein rồi chuẩn bằng NaOH 0,02N tiêu chuẩn đến xuất hiện màu hồng không biến mất trong vòng 1 phút
Tính kết quả
Trong đó: Htđ: độ chua trao đổi;
V: số ml NaOH chuẩn độ mẫu;
N: nồng độ đương lượng của NaOH (0,02N)
W: lượng đất đem phân tích (40g)
K: hệ số pha loãng 9100/50 = 2.
Rút gọn ta có:
Htđ(mgđl/100g đất) = V. 0,1
Hoá chất
NaOH 0,02N: lấy 200ml NaOH 0,1N pha thành 1000m1 trong bình định mức

Dùng axit chuẩn (H2SO4 0,02N) kiểm tra lại nồng độ.
- Phenolphtalein: 0,1g phenolphatlein hoà tan trong 60ml rượu etylic rồi pha thành 100 ml bằng nước cất
KCl 1N: 75g KCl pha thành 1 lít.
Ghi chú: Độ chua trao đổi xác định bằng chuẩn độ chỉ áp dụng ở đất chua. Khi đất có PHKCl > 7,5 sẽ không xác định được vì phenolphtalein tạo thành màu hồng ngay trong dung dịch mẫu.
c) Xác định H+ và At3+ trao đổi theo phương pháp Xôlôcôp (1939)
Nguyên lý
Khi tác động với đất bằng dung dịch muối trung tính (KCl chẳng hạn) thì đồng thời cả H+ và Al3+ trao đổi đều được gây ra khỏi tầng hấp thụ trao đối của keo đất
Khi đó Alcl3 lại bị thuỷ phân tạo thành H+
Từ 1 ion Al3+ thuỷ phân sẽ tạo thành 3 ion H+. Như vậy thực chất khí chuẩn độ
xác định độ chua trao đổi đã bao gồm cả H+ trao đổi, H+ tự do trong dung dịch đất, H+ được tạo thành do Al3+ trao đổi được xác định theo công thức:
Al3+ trao đổi = Độ chua trao đổi - H+ trao đổi.
Xôlôcôp sử dụng NaF để liên kết với AI do đó sẽ xác định được riêng H+ trao đổi
Lúc này trong dung dịch chỉ còn H+tự do, dùng phương pháp chuẩn độ để xác
định chúng. Thông thường Al3+ di động tồn tại ở điều kiện pHKCl < 5,5.
Do vậy Al3+ chỉ có ý nghĩa ở các đất chua, và được xác định cùng với khi xác định độ chua trao đổi.
Trình tự phân tích
Cân lòng đất lắc với 250ml KCl là trong 1 giờ rồi lọc
Xác định độ chua trao đổi: Lấy 50ml dịch lọc vào bình tam giác 250ml, đun sôi 5 phút, cho vào 2 giọt phenolphlein, để nguội rồi chuẩn với NaOH 0,02N tiêu chuẩn đến màu hồng bền vững (trong 1 phút).
Xác định H+ trao đổi (gồm H+ trao đổi và H+ có sẵn trong dung dịch đất). Lấy 50ml dịch trên vào bình tam giác 250ml, đun sôi 5 phút, cho vào 5ml NaF 3,5% để nguội, cho vào 2 giọt chỉ thị phenolphtalein, dùng NaOH 0,02N (dung dịch tiêu chuẩn) chuẩn đến màu hồng.

Tính kết quả
hay Al3+ trao đổi = Độ chua trao đổi - H+ trao đổi
Trong đó: V1: số ml NaOH chuẩn độ mẫu không có NaF;
V2: Số mol NaOH chuẩn độ mẫu có NaF;
N: nồng độ đương lượng của NaOH (0,02N);
W: Lượng đất cân (100g);
K: hệ số pha loãng (250/50: 5);
Al3+ thường được biểu thị bằng một mg/100g đất. Lấy kết quả mgđl/100 g đất x 9 hoặc:
Hoá chất
- NaOH 0,02N chuẩn.
- KCl 1N: 75g KCl pha thành 1 lít
Phenolphtalein 0,1% : 0,1g phenolphtalein pha trong rượu etylic 100 ml.
- NaF 3,5%: 3,5g NaF pha trong nước cất 100 ml. NaF pha xong phải chỉnh pH đến trung tính.
Ghi chú
Khi cho đều lượng NaF như nhau vào mẫu, có thể làm đối chứng để khử H+ tự do trong dung dịch NaF.
Thực tế lượng NaF cho vào có thể không cố định, phụ thuộc vào lượng Al3+. Lượng 5ml NaF chỉ đủ tác dụng với 1,80 mgđl Al3+ là tốt nhất. Khi Al3+ lớn hơn 6,3mgđl/100 g đất thì sẽ không đủ. Có thể tính lượng NaF 3,5% cần thiết:
d. Xác định độ chua thuỷ thủy phân theo phương pháp Kappen

Nguyên lý
Dùng một muối kiềm mạnh axit yếu (thường là CH3COONa) để trao đổi H+ và Al3+ từ keo đất:
Ngoài tác dụng trao đổi của Na+, ion CH3COO- có khả năng liên kết với H+ và
Al3+ làm tăng cường quá trinh trao đổi. Do vậy kết quả trao đổi sẽ triệt để hơn dừng muối trung tính (ở độ chua trao đổi). Quá trình thuỷ phân của (CH3COO)3Al làm tăng H+ trong dung dịch:
(CH3COO)3Al + HOH → CH3COOH + Al(OH)3
Chuẩn bị trực tiếp lượng H+ tạo thành bằng dung dịch CH3COONa 1N (pH = 8,2) trong 1 giờ rồi lọc.
Lấy 50 ml dịch lọc + 2 giọt phenolphtalein rồi chuẩn bằng NaOH 0,02 N tới màu hồng (bền trong 1 phút).
Tính kết quả
Trong đó: V: thể tích (ml) NaOH chuẩn độ;
N: nồng độ đương lượng NaOH (0,02N);
K: hệ số pha loãng (100/50 = 2);
W: khối lượng đất (40g);
l,75: dùng CH3COONa trao đổi 1 lần không triệt để. Muốn trao đổi hoàn toàn H+, Al3+ cần lặp lại nhiều lần, như vậy mất thời gian và phức tạp. Qua thực nghiệm đã cho thấy nhân với hệ số 1,75 là thích hợp (hệ số thực nghiệm);
Htf: độ chua thuỷ phân (mgđl/100g đất);
Rút ngọn: Htf (mgđl/100g đất) = V. 0,175
Hoá chất
- CH3COONa 1N (pH = 8,2): 136,1 g CH3COONa.3H2O hoà bằng nước cất đến 1 lít. Điều chỉnh cho pH = 8,2 (vừa làm đổi màu chỉ thị phenolphtalein).
- Hoá chất khác: xem xác đính độ chua trao đổi ở trên.
Ghi chú: Độ chua thuỷ phân thường được sử dụng để tính độ no bazơ của đất và tính lượng vôi bón (theo nghiên cứu của Viện Khoa học Kỹ thuật nông nghiệp, với đất lúa ở nước ta chỉ cần trung hoà 1/2 độ chua thuỷ phân). Nếu lấy độ sâu cần trung hoà 1/2 độ chua thuỷ phân sẽ là:

CAO (tấn/ha) = 0,42.Htf
Trong đó: Htf tính theo mgđl/100 g đất
7.2.5. Xác định dung tích trao đổi cation của đất
Dung tích trao đổi cation của đất hay khả năng trao đổi cation (CEC - cation exchange capacity) là lượng ion lớn nhất được đất hấp thụ lý hoá học được thực hiện nhờ keo đất. Cần phân biệt quá trình trao đổi cation (do keo âm đảm nhận) với trao đổi cation (do keo dương đảm nhận). Dưới đây chỉ đề cập đến việc xác định dung tích trao đổi cation.
Xác định dung tích trao đổi cation theo phương pháp amoni axetat (phương pháp Schachtschabel).
Nguyên lý dùng CH3COONH4 là làm bão hoà dung tích hấp thụ trao đổi cation của đất. Phản ứng trao đổi như sau:
Sau đó cation NH4 đã hấp phụ được trao đổi ra bằng cation K+ (KCl 0,1 N):
Lượng NH4 trau đổi này được xác định bằng chuẩn độ bằng NaOH 0,1 N với sự
có mặt của fomalin (HCHO).
Với 4 NH4 sẽ giải phóng 4H+. Do vậy dựa vào lượng NaOH tiêu tốn mà tính được
lượng NH4 hay lượng cation trao đổi.
Trình tự phân tích
Cân 10g đất (qua rây 1 mua cho vào phễu Mehlich đã chuẩn bi sẵn.
Dùng 100 ml CH3COONH4 1N (pH = 7) chia 10 lần (10 ml x 10 lần) để bão hoà đất bằng NH4. Rửa đất 3 lần bằng rượu etylic (15 ml x 31ần). Dịch trao đổi để xác định thành phần cation trao đổi (Ca2+, Mg2+, Al3+,K+,.. ) nếu cần thiết.
Chuyển toàn bộ phễu và đất sang bình định mức 250ml, rồi dùng 250ml KCl 0,1N trao đổi (25ml x 10 lần) lên thể tích đến 250 ml. Lấy 25ml dịch trao đổi này + 10ml fumalin 20% + 5 giọt phenolphtalein, rồi chuẩn bằng NaOH 0,05N tiêu chuẩn đến màu hồng nhạt (bền trong 1 phút).
Tính kết quả

Trong đó: CEC: dung tích trao đổi cation (mgđl/ 100 g đất);
V: thể tích NaOH chuẩn độ (ml);
N: nồng độ NaOH chuẩn độ (0,05N);
W: lượng đất cân (loa);
K: hệ số pha loãng (250/25 = 10);
Rút gọn: CEC (mgđl/100 đất) = V.5
Hoá chất
CH3COONH4 1N: 77g CH3COONH4 pha thành1 lít bằng nước cất (dùng NH4OH hoặc CH3COOH điều chỉnh cho pH = 7.
- KCl 0,1N; 7,5g KCl pha thành 1 lít.
- Fomalin 20%: pha từ fomalin thông dụng (khoảng 38%). Sau đó trung hòa bằng NaOH 0,05N với chỉ thị phenolphtalein.
- Phenolphtalein 0,1% (pha trong cồn, rượu).
- NaOH 0,05N tiêu chuẩn.
Ghi chú
Phương pháp dùng CH3COONH4 được dùng phổ biến nhưng phù hợp nhất với đất trung tính và không cacbonat. Nó cũng được sử dụng ở các đất chua nhẹ.
NH4 có thể bị giữ chặt bởi các khoáng sét 1:1 (kaolinit) và mùn nên khó thu lại được. Do vậy ở đất nhiều sét, kết quả có thể thấp hơn thực tế.
Lượng đất cân có the thay đôi phụ thuộc đất. Thông thường ở đất thành phần cơ giới nặng có thể cân 5g đất + 5g cát sạch (đã được xử lý bằng H2SO4 đặc) để tăng cường tốc độ trao đổi.
Có thể làm chuẩn độ trắng với focmaLin (đối chứng) để loại trừ H+ chưa được trung hoà. Kết quả xác định mẫu phải trừ đi lượng chuẩn độ này (nếu có).
7.3. Xác định một số kim loại nặng trong đất
Trong dinh dưỡng của thực vật và vi sinh vật, ngoài các nguyên tố nào, photpho và kali, các kim loại nặng như bo, mangan, đồng, kẽm, coban, molipden,.. cũng có ý nghĩa lớn. Một lượng nhỏ của các nguyên tố này cần thiết cho nhiều quá trình sinh học xảy ra trong các cơ thể thực vật và động vật. Nguồn cung cấp chủ yếu các kim loại nặng trong đất là các đá tạo thành đất. Trong quá trình tạo thành đất và hoạt động sống của thực vật và động vật xảy ra qúa trình phân bố lại các kim loại nặng theo phẫu diện đất.

Hàm lượng các kim loại nặng trong các loại đất không vượt quá 10-4 %, trừ mangan, hàm lượng của nó trong một số trường hợp, ví dụ như trong các thành tạo mới của ma ngan - sắt đôi khi được tính đến phần trăm.
Khi nghiên cứu hàm lượng các kim loại nặng trong. đất người ta thường xác định hàm lượng tông số và hàm lượng có thể sử dụng được đối với dinh dưỡng của thực vật như thường được gọi là dạng di động của các kim loại nặng.
Chọn phương pháp xác định, chuẩn bị đất và các thuốc khử
Khi tiến hành nghiên cứu hàm lượng các dạng khác nhau của các kim loại nặng cần lựa chọn các phương pháp xác định chúng, phương pháp phải đảm bảo được độ chính xác, độ nhạy và độ chọn lọc.
Chỉ có thể xác định một lượng nhỏ các chất bằng các phương pháp có độ nhạy cao. Tuy nhiên các phương pháp có độ nhạy cao thường có độ chính xác thấp, ví dụ như phương pháp khối lượng có sai số tương đối tính theo phần trăm < 0,01%, phương pháp trắc quang là 2 - 3%, phương pháp cực phô dòng khuếch tán là 5 - 10 %.
Mẫu đất được nghiền trong cối mà não và rây qua rây làm bằng sợi capron và khung rây làm từ các vật liệu hữu cơ.
Nước dùng trong phân tích là nước cất được cất lại hay cho đi qua nhựa trao đổi lớn, thường sử dụng nước cất hai lần.
Các axit và amoniac cũng được tinh chế bằng cách cất hay sử dụng thuốc thử loại đặc biệt tinh khiết. Các thuốc thử khác được tinh chế theo mô tả trong phương pháp xác định của từng nguyên tố. Các thuốc thử thường bị bẩn nguyên tố kẽm vì thế khi xác định nguyên tố này cần phải tiến hành thí nghiệm kiểm tra sự có mặt của nó trong các thuốc thử sử dụng.
Thành phần của thuỷ tinh để chế tạo dụng cụ thí nghiệm cũng cần phải được lưu tâm, đặc biệt khi xác định bỏ.
Những hoá chất dùng đề pha dung dịch chuẩn nên kết tinh lại. Thường hay pha hai loại dung dịch chuẩn: dung dịch chuẩn gốc và dung dịch sử dụng. Dung dịch chuẩn gốc thường được pha với hàm lượng 0,1 mg nguyên tố trong 1 ml; cân những lượng âm với hàm lượng nguyên tố như vậy cho phép chúng ta có thể tránh được sai số. Dung dịch chuẩn sử dụng được chuẩn bị bằng cách pha loãng dung dịch chuẩn gốc Khi pha loãng 10 lần ta được dung dịch chứa 0,01 mg hay 10μg/ml. Khi pha loãng 100 lần nhận được dung dịch có hàm lượng 0,001 mg hay 1μg/ml. Vì những dung dịch quá loãng không bền nên người ta chỉ chuẩn bị các dung dịch này trong ngày tiến hành phân tích và chỉ giữ trong ngày đó.
Tính kết quả xác định các kim loại nặng
Kết quả xác định các kim loại nặng trong đất được biểu thị bằng mà trong 1 kg đất (mg/kg) hoặc biểu thị bằng ppm (parts pa million) nghĩa là một phần triệu, tương

ứng với 10-4%
7.3.1. Phương pháp phân huỷ mẫu truyền thống
a. Chuẩn bị mẫu
Làm khô các mẫu đất: Những mẫu đất tươi cần phải đưa về trạng thái khô không khí. Làm khô mẫu nên tiến hành ở chỗ sạch, thoáng, trường hợp đặc biệt có thể dùng tủ sấy khống chế nhiệt độ từ 30 - 400C. Dàn mẫu đất (trọng lượng 800 - 1000 g) thành lớp mỏng trên giấy thuộc hoặc giấy bóng mờ, nhặt hết rễ cây, đá, sỏi,... có lẫn trong mẫu. Đập nhỏ mẫu đất đề mẫu có kích thước 5 - 10mm. Có thể dùng máy hay cối sức không có các kim loại nặng để đập nhỏ mẫu trước khi lấy mẫu trung bình.
Việc chọn, nghiền và rây mẫu đất được tiến hành trong phòng riêng có thiết bị thông gió và hút bụi tốt
Lấy mẫu trung bình: Từ mẫu ban đầu lấy ra khoảng 200 g, dùng thiết bị để trộn đều mẫu. Nếu không có thiết bị này có thể dùng phép chia tư để lấy mẫu (hình 7.12). Mẫu đất được trộn rồi rải đều trên giấy sạch thành một lớp mỏng có dạng hình vuông. Chia hình vuông theo hai đường chéo thành 4 phần bằng nhau. Lấy hai phần đối diện (phần 1 và 3 hoặc 2 và 4) gộp lại với nhau, hai phần còn lại có thể bỏ đi hoặc dùng vào việc khác.
Sau khi chọn xong, mẫu được nghiền nhỏ trong cối mã não, rây qua rây có đường kính 1 mm. Nên dùng loại rây làm bằng sợi capron và khung làm bằng vật liệu hữu cơ Mẫu đất sau khi rây được dùng để xác định các kim loại nặng.
b. Phân huỷ đất khi xác định hàm tượng tổng số các kim loại nặng.
Trong đất, các kim loại nặng chủ yếu tham gia trong thành phần các khoáng nguyên sinh thứ sinh và trong các hợp chất vô cơ, hữu cơ khác nhau. Để xác định hàm lượng tổng số các kim loại nặng trong đất cần phải thiêu huỷ các chất hữu cơ và phân huỷ phần khoáng của đất để nhận được những muối dễ hoà tan.
Muốn phân huỷ đất người ta thường sử dụng 2 phương pháp phương pháp nung chảy đất với hỗn hợp cacbonat của các kim loại kiềm và phương pháp phân huỷ bằng các axit vô cơ đặc.
Quá trình nung chảy với hỗn hợp cacbonat của các kim loại kiềm được tiến hành trong các chén phẩm có nắp ờ nhiệt độ 10000C. Sau khi nung chảy, người ta nhận được muối kiềm của axit silicic và các hợp chất dễ hoà tan khác. Chuyển hỗn hợp chảy sang dạng dung dịch, dùng axit clohidric để tách axit silicic, rồi tiến hành xác định các kim loại nặng trong dung dịch. Khi xử lý hỗn hợp bằng axit clohidric (HCl, d = 1,19) sẽ tạo nên một lượng tương đối lớn kết tủa hạt nhỏ SiO2, kết tủa này có thể giữ một

lượng đáng kể các kim loại nặng và như vậy kết quả phân tích sẽ giảm xuống. Axit clohidric dùng để xử lý hỗn hợp có thể chứa các kim loại nặng dưới dạng tạp chất và cũng có thể hoà tan một lượng phẩm ở chén đựng mẫu, nguyên tố này sẽ cản trở việc xác định các kim loại nặng (ví dụ: molipden, vanadi... ).
Phương pháp phân huỷ mẫu bằng hỗn hợp axit HF và H2SO4 là phương pháp được Sử dụng nhiều nhất khi xác định hàm lượng tổng số các kim loại nặng. Phương pháp này đảm bảo phân huỷ hoàn toàn mẫu đất và tách axit silicic dưới dạng SiF4. Tuy nhiên trong trường hợp này có một vài phần khoáng (hay một phần trong đó) như topaz, anđaluzit zirkon, sillimanit sẽ không bị phá huỷ, vì vậy đất chứa nhiều các loại khoáng này không nên phá huỷ mẫu bằng HF.
Việc lựa chọn các phương pháp phân huỷ mẫu đất tuỳ thuộc vào các kim loại nặng cần xác định trong mẫu. Vì HF (loại tinh khiết đề phân tích) có chứa hỗn hợp các kiến loại nặng, cho nên khi xác định các kim loại nặng như kẽm, co ban cần phải cất HF trong các dụng cụ cất đặc biệt chế tạo tù pladi hay platin. Axit clohidric loại đặc biệt tinh khiết không cần phải tinh chế.
Khi sử dụng HF phải hết sức cẩn thận, cần đeo găng tay cao su và chỉ làm ở những nơi có thiết bị thông gió tốt.
Khi xác định kẽm, đồng, co ban trong đất phải dùng axit flohidnc đã cất lại 3 lần để phân huỷ mẫu đất và sau đó để hoà tan kết tủa, axit clohidric phải dùng loại đã cất hai lần.
Đối với các nhóm kim loại nặng khác (molipden, valadi, ma ngan,...) không nhất thiết phải như vậy. Khi xác định các nguyên tố này chỉ cần axit flohidric loại tinh khiết hoá học (không cần cất lại) và axit clohidric đã cất lại một lần. Nhưng theo quá trình phân tích nhóm các nguyên tố này đòi hỏi phải chăng phần còn lại 2 - 3 lần với nước sau khi phân huỷ mẫu (để tách flo triệt để hơn), nung phần còn lại 1 - 2 phút trên đèn khí hay trong lò nung và tiếp theo cần xử lý nước lọc đã cô cạn bằng axit pecloric (HClO4) để tách crom. Khi phân huỷ mẫu bằng HF và H2SO4 còn chú ý đến lượng mẫu lấy để phân huỷ; không nên lấy lượng mẫu lớn hơn 3g vì khi lượng mẫu là 5 - 6g thì khó có thể phân huỷ hoàn toàn phần khoáng của đất và khó tách SiO2.
Dưới đây là một vài cách phân huỷ mẫu bằng hỗn hợp HF và H2SO4 đối với hai nhóm kim loại nặng đã nêu ở phần trên.
Xác định đồng, kẽm, coban trong đất, tiến hành phân huỷ theo phương pháp sau:
Cân mẫu đất xác định hàm lượng tổng số các kim loại nặng trên cân phân tích. Đối với đất sét và đất sét pha lấy 1,5g mẫu, còn đối với đất cát và cát pha lấy 2 -3g mẫu. Sau khi cân xong, cho mẫu vào chén phẩm và nung nóng trong lò nung ờ nhiệt độ 500 - 550oC trong 3 giờ để phân huỷ chất hữu cơ. Tránh nung ở nhiệt độ cao hơn vì có khả năng làm mất các kim loại nặng. Sau khi nung mẫu xong để nguội, cho vào chén 1 -2 ml nước cất 2 lần, 1 ml H2SO4 (d = l,84) và 20 ml axit flohidric, đặt chén lên

bếp điện kín và đun đến khi xuất hiện khí SO2 màu trắng. Không nên đun quá mạnh (không vượt quá 200 - 250oC) để tránh làm bắn các chất ra ngoài.
Sau khi xuất hiện khí SO2, lấy chén ra khỏi bếp điện, để nguội và thêm vào khoảng 10- 15 ml HF rồi lại tiếp tục đun trên bếp điện cho đến khi khô hoàn toàn. Chuyển chén sang bếp điện có khả năng đun nóng hơn tiếp tục đun đến khi hết khí SO3 bay ra. Nhấc chén ra khỏi bếp điện, để nguội rồi cho vào chén 10 ml HCl 22% (loại đặc biệt tinh khiết) và 10 ml nước cất hai lần để hoà tan phần khô còn lại. Tiếp tục đặt lên bếp và đun nóng nhẹ 30 - 40 phút.
Khi phần còn lại đã tan hết, đem lọc dung dịch qua giấy lọc băng trắng (đường kính 9cm) đã được rửa trước bằng HCl 10 % để loại hết vết các kim loại nặng. Sau khi lọc xong rửa giấy lọc bằng nước cất hai lần nóng đã được axit hoá bằng HCl đến khi hết vết sắt. Gộp hai phần nước lọc và nước rửa lại với nhau (khoảng 100 ml) rồi chưng cất dung dịch đến thể tích khoảng 50ml. Dung dịch này dùng để xác định các kim loại nặng (kẽm, coban, đồng) theo các phương pháp khác nhau.
Xác định molipden, valadi, ma gan từ một mẫu đất, dùng phương pháp phá mẫu sau:
Cân 2,5 - 3g đất đã được nghiệm nhỏ đến dạng bột cho vào chén phẩm, đặt chén vào lò nung rồi đốt nóng đến nhiệt độ 500 - 550oC và giữ ở nhiệt độ này ít nhất 3 giờ để phân huỷ chất hữu cơ. Để nguội, cho vào chén 2ml nước cất hai lần, 2ml H2SO4 đặc 20ml HF 30 - 40 %. Đặt chén lên bếp điện và đậy nắp kín, đun đến khi xuất hiện khí SO3 thì thêm vào 0,5ml H2SO4 và 10 ml HF, lại chúng mẫu đến khô. Để tách flo và phần axit còn lại trong chén, ta chẳng phần khô trong chén 2 lần, mỗi lần 5ml nước, đốt nhẹ phần khô còn lại trên đèn khí hay trong lò nung để tách hoàn toàn axit. Làm lạnh chén, xử lý phần còn lại trong chén bằng 20ml HCl 22%, đậy chén lại rồi đun trên bếp điện đến khi tan hoàn toàn phần còn lại. Thêm vào lòng nước cất hai lần lọc dung dịch qua giấy lọc và hứng vào bình 250ml, rửa dung dịch bằng nước nóng đã được axit hoá bằng HCl (98:2) đến khi hết phản ứng của sắt trong nước lọc với KSCN. Chưng dung dịch lọc đến khô, cho vào bình 5ml HCl 22%, 2ml HClO4 và lại chúng dung dịch đến khô để tách crom. Thêm vào phần khô 5ml nước cất 2 lần, lại chóng đến khô để tách hết vết HClO4. Thêm vào phần khô trong bình 2ml HCl để chuyển muối sang dạng clorua, chung khô dung dịch. Thêm 20ml HCl 22% và 15ml và đun nóng nhẹ để hoà tan phần khô. Chuyển dung dịch vào bình định mức 50ml. Thêm nước cất đến vạch mức. Phần dung dịch này được dùng để xác định molipden, valadi, mangan.
7.3.2. Phương pháp phân huỷ mẫu bằng kỹ thuật vi sóng (micyo wave)
a. Giới thiệu về kỹ thuật vi sóng (micro wave)
Vi sóng (micro wave) là sóng cực ngắn hay còn gọi là sóng vi ba có bước sóng từ 1 mm đến 1m. Năng lượng của vi sóng là năng lượng điện từ. Trong phân tích người ta thường sử dụng vi sóng với tan số 2,45 Mhz.

Các phương pháp đốt truyền thống thường dựa trên các hiện tượng dẫn nhiệt, đối lưu và bức xạ. Nhiệt được truyền từ bên ngoài vào thông qua sự tiếp xúc trực tiếp hoặc gián tiếp. Lượng nhiệt này sẽ lan ra toàn bộ vật được đốt nóng nhờ sự chênh lệch nhiệt độ tại hai điểm khác nhau, sự chếch lệch này tỷ lệ nghịch với độ dẫn nhiệt của vật được đốt nóng. Phương pháp này có nhược điềm là chậm và không ổn định.
Sự đốt nóng vi sóng ngược lại với phương pháp đốt nóng truyền thống. Nhiệt
được sinh ra ngay tại trung tâm của vật được đốt nóng và lan theo hướng từ trong ra ngoài. Phương pháp này có ưu điểm:
- Không có quán tính nhiệt (cả khi bắt đầu và kết thúc);
- Đây là năng lượng sạch, dễ tạo và dễ kiểm soát;
- Đốt nóng nhanh;
- Có tác dụng đặc biệt với các phần tủ có liên kết phân cực.
Năng lượng vi sóng được phát ra từ một nguồn phát sóng điện từ (maglletron). Bản chất của vi sóng là sóng điện từ gồm hai yếu tố: yếu tố từ trường và yếu tố điện trường. Quá trình chuyên hoá năng lượng điện từ thành năng lượng nhiệt bao gồm hai cơ chế cơ chế chuyển dẫn ion và cơ chế quay cực phân tử. Nhiệt sinh ra cho sự chuyển dẫn lớn như là kết quả của sự tăng trở kháng của môi trường chống lại sự dịch chuyển của các lớn trong trường điện từ. Còn cơ chế quay cực phân từ là quá trình quay phân tử phân cực theo sự đổi hướng của điện trường. Sự đốt nóng bằng kỹ thuật vi sóng dựa trên sự hấp thu trực tiếp năng lượng vi sóng của mẫu, do vậy các hiện tượng như dẫn nhiệt, đối lưu và bức xạ nhiệt chỉ đóng vai trò thứ yếu trong quá trình cân bằng nhiệt.
Ưu điểm của phương pháp phân huỷ mẫu bằng vi sóng trong phân tích kim loại nặng:
Tiến hành các phản ứng phân huỷ mẫu rất nhanh, tiết kiệm thời gian. Đặc biệt có hiệu quả đối với các mẫu khó phân huỷ bằng các phương pháp thông thường;

- Lượng mẫu sử dụng ít, thường từ 0,5 - 5 g;
- Hiệu suất của phản ứng phân huỷ rất cao do vậy kết quả phân tích thu dược chính xác hơn so với phương pháp khác;
- Tiêu hao hoá chất ít, do vậy chi phí cho quá trình xử lý thấp;
- Thiết bị dễ sử dụng, an toàn và bảo vệ môi trường.
b. Phân huỷ mẫu bằng vi sóng
Các hỗn hợp axit dùng cho phân huỷ mẫu
Các axit thường sử dụng trong quá trình phân huỷ mẫu là H2SO4, HNO3, HCl, HF, HClO4, H2O2.
Việc sử dụng hỗn hợp các axit này trong quá trình phân huỷ mẫu phụ thuộc vào nguyên tố cần xác định ví dụ như xác định các nguyên tố Ba, Si, Pb, Ca, Ao, Hg không nên sử dụng axit sunfumic để công phá mẫu vì muối sunfat của các kim loại này ít tan.
Các axit này có nhiệt độ sôi khác nhau (H2SO4 98%: 3400C; HCl 20,4%: 1100C; HNO3 65%: 1200C; HF 40%: l080C; HClO4 72%: 2030C) do đó tuỳ theo hỗn hợp axit phá huỷ mẫu ta phải sử dụng các bình phá mẫu phù hợp. Đối với các axit có nhiệt độ sôi thấp người ta thường sử dụng loại bình nhựa teflon để phá mẫu. Còn axit có nhiệt độ sôi cao (axit sunfuric) người ta thường sử dụng các loại bình làm từ thuỷ tinh borosilicat hoặc thạch anh (chú ý không sử dụng bình loại này khi có mặt axit flohiđric).
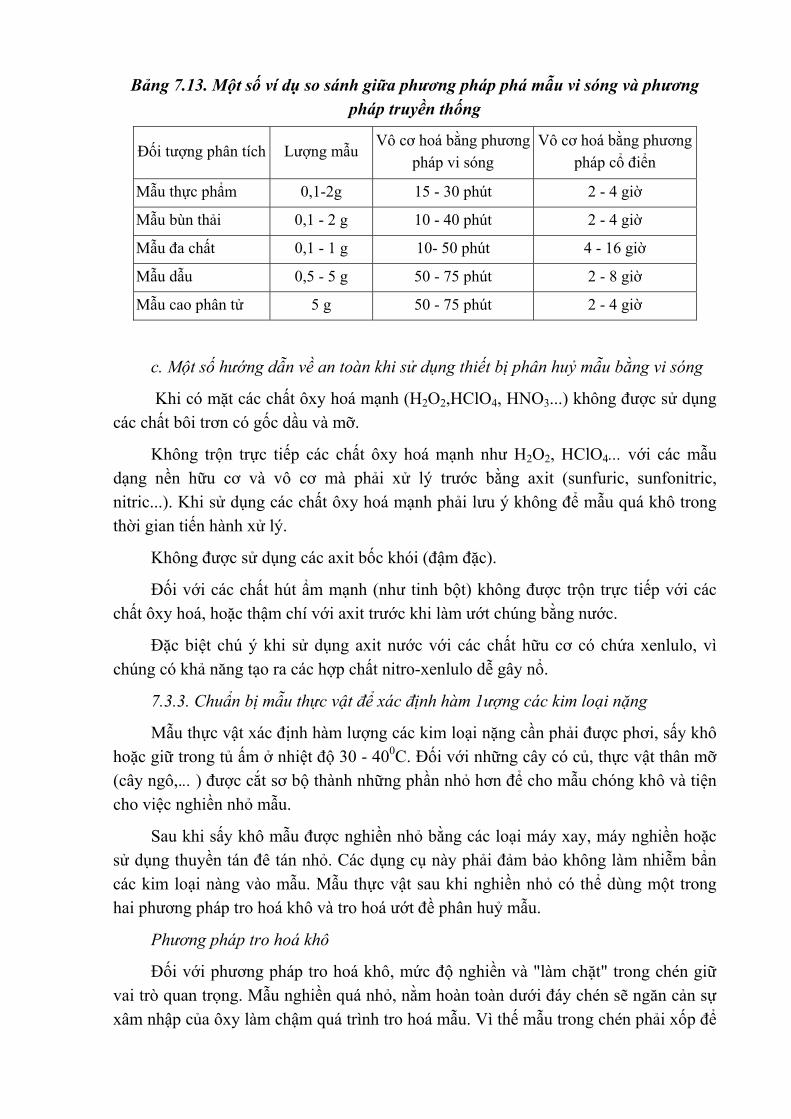
Bảng 7.13. Một số ví dụ so sánh giữa phương pháp phá mẫu vi sóng và phương pháp truyền thống
Đối tượng phân tích Lượng mẫu Vô cơ hoá bằng phương
pháp vi sóng Vô cơ hoá bằng phương
pháp cổ điển
Mẫu thực phẩm 0,1-2g 15 - 30 phút 2 - 4 giờ
Mẫu bùn thải 0,1 - 2 g 10 - 40 phút 2 - 4 giờ
Mẫu đa chất 0,1 - 1 g 10- 50 phút 4 - 16 giờ
Mẫu dẫu 0,5 - 5 g 50 - 75 phút 2 - 8 giờ
Mẫu cao phân tử 5 g 50 - 75 phút 2 - 4 giờ
c. Một số hướng dẫn về an toàn khi sử dụng thiết bị phân huỷ mẫu bằng vi sóng
Khi có mặt các chất ôxy hoá mạnh (H2O2,HClO4, HNO3...) không được sử dụng các chất bôi trơn có gốc dầu và mỡ.
Không trộn trực tiếp các chất ôxy hoá mạnh như H2O2, HClO4... với các mẫu dạng nền hữu cơ và vô cơ mà phải xử lý trước bằng axit (sunfuric, sunfonitric, nitric...). Khi sử dụng các chất ôxy hoá mạnh phải lưu ý không để mẫu quá khô trong thời gian tiến hành xử lý.
Không được sử dụng các axit bốc khói (đậm đặc).
Đối với các chất hút ẩm mạnh (như tinh bột) không được trộn trực tiếp với các chất ôxy hoá, hoặc thậm chí với axit trước khi làm ướt chúng bằng nước.
Đặc biệt chú ý khi sử dụng axit nước với các chất hữu cơ có chứa xenlulo, vì chúng có khả năng tạo ra các hợp chất nitro-xenlulo dễ gây nổ.
7.3.3. Chuẩn bị mẫu thực vật để xác định hàm 1ượng các kim loại nặng
Mẫu thực vật xác định hàm lượng các kim loại nặng cần phải được phơi, sấy khô hoặc giữ trong tủ ấm ở nhiệt độ 30 - 400C. Đối với những cây có củ, thực vật thân mỡ (cây ngô,... ) được cắt sơ bộ thành những phần nhỏ hơn để cho mẫu chóng khô và tiện cho việc nghiền nhỏ mẫu.
Sau khi sấy khô mẫu được nghiền nhỏ bằng các loại máy xay, máy nghiền hoặc sử dụng thuyền tán đê tán nhỏ. Các dụng cụ này phải đảm bảo không làm nhiễm bẩn các kim loại nàng vào mẫu. Mẫu thực vật sau khi nghiền nhỏ có thể dùng một trong hai phương pháp tro hoá khô và tro hoá ướt đề phân huỷ mẫu.
Phương pháp tro hoá khô
Đối với phương pháp tro hoá khô, mức độ nghiền và "làm chặt" trong chén giữ vai trò quan trọng. Mẫu nghiền quá nhỏ, nằm hoàn toàn dưới đáy chén sẽ ngăn cản sự xâm nhập của ôxy làm chậm quá trình tro hoá mẫu. Vì thế mẫu trong chén phải xốp để

tăng khả năng sâm nhập của ôxy làm cho quá trình phân huỷ mẫu diễn ra nhanh hơn. Lượng mẫu lấy để tro hoá tuỳ thuộc vào hàm lượng của kim loại nặng cần xác định ở trong mẫu. Để xác định riêng từng nguyên tố người ta thường lấy lượng mẫu như sau:
Đồng : 0,2 - 0,5 g Kẽm: 0,05 - 0,2 g
Mangan: 1 - 2 g Co ban: 5 - 10 g
Molipden: 1 - 4 g Bo: 0,25 - 1 g
Thực tế người ta thường xác định đồng, kẽm, mangan, molipden trong cùng một mẫu, khi đó lượng mẫu lấy thường là 5 g. Để xác định bỏ và coban người ta lấy các loạn mẫu riêng: Coban: 5 - 10 g; Bo: 0,25 - 1 g.
Tro hoá: Người ta thường tiến hành tro hoá khô trong lò nung ở nhiệt độ 450 - 5000C, thời gian tro hoá là 5 - 8 giờ. Trong quá trình tro hoá chất hữu cơ, những hợp chất của kim loại nặng có nhiệt độ nóng chảy khác nhau được tạo thành. Để tránh mất kim loại nặng, nhiệt độ trong lò nung cần khống chế dưới 5000C (Troitxky, 1957). Để tro hoá, tốt nhất nên dùng chén bằng thạch anh, nếu không có loại này có thể dùng chén sứ hoặc chén thủy tinh chịu nhiệt. Khi dùng chén sứ nếu nung ở nhiệt độ qua 5000C có thể dẫn đến việc nung chảy sản phẩm tro hoá với men của thành chén. Thành phần mẫu tro hoá có ảnh hưởng lớn đến quá trình tro hoá. Đối với mẫu thực vật không những thành phần mẫu có ảnh hưởng đến quá trình tro hoá mà ngay cả nơi thực vật sinh trưởng và phát triển cũng có ảnh hưởng đến quá trình tro hoá mẫu. Người ta đã tiến hành thử nghiệm và thấy rằng những cây thuộc họ hoà thảo trồng ở phía nam tro hoá nhanh hơn những cây trồng ở phía bắc (Gribovxcaia - 1968). Điều đó cho thấy một sự không đồng đều về kiềm và SiO2 trong tế bào thực vật ở những vùng khác nhau.
Một lượng lớn SiO2 sẽ ngăn cản quá trình tro hoá hoàn toàn mẫu thực vật. Phần còn lại không hoà tan của axit silicic, tạo thành do kết quả của quá trình tro hoá mẫu thực vật có thể chứa một lượng đáng kể các kim loại nặng
Để tro hoá ta cân mẫu (mẫu ở trạng thái khô không khí), cho vào chén và đặt vào lò nung. Mẫu thực vật trong chén càng xốp càng tốt, điều đó sẽ làm tăng tốc độ của quá trình tro hoá. Trong quá trình nung mẫu, nên khống chế để nhiệt độ tăng từ từ Nhiệt độ tăng quá nhanh có thể làm mẫu bắn ra ngoài trong quá trình đốt nóng mẫu.
Sau đó, trong quá trình tro hoá nên cần thận mở cửa lò nung để tăng thêm sự tham gia của ôxy vào mẫu tro hoá nhằm nâng cao tốc độ tro hoá và phần tro nhận được sẽ không có hỗn hợp cacbon. Thời gian tro hoá phụ thuộc vào thành phần mẫu tro hoá, mức độ nhiễm mẫu và lượng mẫu đem tro hoá.
Để tăng tốc độ ôxy hoá phần mẫu hữu cơ còn lại, ta thêm vào trong chén 1 - 2 ml HNO3 bậc, chung khô trên bếp điện và nung nóng trong lò nung ở nhiệt độ 450 - 5000C trong 1 giờ. Lặp lại quá trình xử lý mẫu bằng axit rồi chúng và nung trong lò

đến khi ôxy hoá hoàn toàn chất hữu cơ, sau đó hoà tan phần khô bằng axit HCl 10%, lọc dung dịch qua giấy lọc tăng trắng vào bình định mức 100 ml.
Nếu trong chén và trên giấy lọc có một lượng lớn phần không hoà tan (bao gồm SiO2 và hỗn hợp các chất khó tan khác) thì tốt nhất nên phá huỷ phần không tan này đề tránh mất các kim loại nặng can xác định. Để chuyến hợp chất khó tan này sang trạng thái hoà tan, ta chuyển toàn bộ phần này lên trên giấy lọc rồi sấy khô giấy lọc. Cho giấy lọc này vào chén phẩm rồi thiêu huỷ trong lò nung ờ nhiệt độ 450 - 5000C. Qúa trình thiêu huỷ tiến hành đến khi nhận được phần tro không có hợp chất cacbon. Thêm vài giọt nước đế làm ướt phần mẫu đã tro hoá rồi thêm vào O,2m! H2so4' 5ml HF để phá huỷ và tách SiO2 chung mẫu đến khô trên bếp điện. Nếu lượng tro còn lại lớn thì cần phải xử lý bằng HF một lần nữa. Trong trường hợp này, khi xử lý mẫu bằng HF ban đầu ta không cần chung mẫu đến khô mà chỉ chóng đến khi xuất hiện khí SO2.
Hoà tan mẫu bằng HCl 10% rồi lọc qua giấy lọc (đã được rửa sạch trước bằng HCl loãng) sau đó gộp các phần dung dịch lại với nhau.
Phần dịch lọc cuối cùng được dùng để xác định các kim loại nặng trong thực vật theo các phương pháp khác nhau.
Phương pháp tro hoá ướt
Khi tro hóa ướt, các chất hữu cơ được ôxy hoá dưới tác dụng của những chất ôxy hoá mạnh như: H2SO4, HNO3, HClO4. Phương pháp này đơn giản về mặt thực hiện và ưu việt hơn phương pháp khô về mặt tốc độ, đặc biệt trong trường hợp mẫu thực vật có chứa một lượng lớn phần tro không tan. Nếu tuân theo nghiêm ngặt những điều kiện tro hoá ta có thể loại trừ khả năng mất kim loại nặng.
Tro hoá bằng hỗn hợp H2SO4 và HNO3 (Troitxky, 1957): lấy 5g mẫu cho vào bình Kendan dung tích 300ml, cho vào 5ml H2SO4 đặc (d= 1,84) và 5ml hoặc nhiều hơn HNO3 (d = 1,4). Đun nhẹ bình để tránh sủi bọt mạnh (ngoài ra khi đun nóng nhẹ ta có thể duy trì sâu tác dụng của HNO3 vì nó dễ bị phân huỷ và bay hơi).
Sau khi đuổi hết hơi HNO3, làm lạnh bình, nếu như chưa ôxy hoá hoàn toàn chất hữu cơ ở trong mẫu, ta lại thêm HNO3 vào và đun nóng mẫu ở nhiệt độ cao hơn trong khoảng 10 phút rồi làm lạnh. Khi ôxy hoá hoàn toàn chất hữu cơ trong mẫu, dung dịch trở nên trong suốt.
Tro hoá mẫu bằng hỗn hợp các axit H2SO4 và HNO3 và HClO4: lấy 5g mẫu thực vật ở trạng thái khô không khí cho vào bình Ken dan dung tích 300 ml. Thêm vào 5ml H2SO4 đặc (d = 1,84) và 7ml HNO3 (d = 1,4) và cứ mỗi gam mẫu thì thêm vào 4 ml HClO4 (d = 1,54). Để quá trình tro mẫu hoá được tiến hành một cách an toàn, thể tích H2SO4 không được nhỏ hơn 2 ml, đặc biệt trong giai đoạn HNO3 bốc hơi, vì thể tích H2SO4 nhỏ hơn 2ml, có thể đồ bình do sự phân huỷ amoni pecnorat, được tạo thành trong quá trình ôxy hoá chất hữu cơ. Sau khi đã cho hết các axit vào bình, đun nóng dần dần trên bếp điện hoặc đèn khí đến 1000C, khi xuất hiện NO2 màu nâu, lấy bình ra

để nguội. Điều này cần thiết vì nó làm chậm quá trình phân huỷ HNO3, sau đó bình được đốt nóng đến khi xuất hiện khí SO2 màu trắng. Phản ứng cần được tiến hành từ từ.
Sau khi xuất hiện SO2 đun nóng bình từ 5 - 10 phút, rồi tăng nhiệt độ lên và tiếp tục đun nóng 1 - 2 phút. Ở giai đoạn này chất hữu cơ của mẫu được phân huỷ hoàn toàn, dung dịch trở nên không màu.
Nếu mẫu bị tro hoá còn có cacbon thì thêm vào 1 - 2ml HNO3 rồi đun nóng bình đến khi xuất hiện khí SO3. Việc xử lý này tiến hành cho đến khi nào dung dịch trở nên không màu.
Sau khi nguội, đun pha loãng dung dịch trong bình đến 50ml, lọc vào bình định mức dung tích 100 ml. Dung dịch này được dừng để xác định các kim loại nặng. Phương pháp định lượng tương tự như khi xác định chúng ở dạng di động.
7.3.4. Một số ví dụ về giới hạn phát hiện của các phương pháp phần tích công cụ trong phân tích các kim loại nặng
Trong phân tích lượng vết các nguyên tố kim loại nặng, việc lựa chọn phương pháp đóng vai trò hết sức quan trọng. Mỗi phương pháp có khả năng phát hiện và có độ nhạy khác nhau đối với các nguyên tố kim loại nặng.
Giới hạn phát hiện ở đây được tính bằng đơn vị ~g/ml (Bảng 7~14)
7.3.5. Xác định chì trong đất
Hàm lượng chì trung bình trong các đất của Liên Xô (cũ) là 1,2. 10-3% (12mg/kg), trong thực vật là 5.10-5 %, trong đất ở ôxtrâylia: 10 - 130 ppm; ở Trung Quốc là 25ppm, trong các đất ở Mỹ là 17 - 26 ppm. Hàm lượng chì cao hơn ở trong đất thường liên quan đến đất bị ô nhiễm do hoạt động sản xuất của con người.
Bảng 7.14. Giới hạn phát hiện của mã số phương pháp phân tích kim loại nặng
Phương pháp phân tích As Cd Cu Pb Zn Hg Se
Phương pháp quang Phổ - UVNIS - AAS (ngọn lửa) - Phát xạ hồ quang - Phổ phát xạ Plasma - Huỳnh quang tia X - Phổ khối lượng
0,05 0,02 0,004
20 0,05
1 0,006
0,04 0,05 0,001
2 0,005 0,7 0,3
0,03 0,2
0,003 0,05 0,005
1 0,1
0,08 0,6
0,002 2
0,02 2
0,03
0,02 0,02
0,00006 3
0,005 1
0,01
0,08 15 0,2 20
0,02 1
0,06
0,2 0,3
0,006 1
0,002 1
0,1
Phương pháp điện hóa - Cực phổ - Cực phổ xung vi phân - Điện cực chọn lọc ion
0,1 1,004
-
0,2 0,002 0,03
0,2 0,002 0,1
0,4 0,003 0,03
0,5 0,003
-
1 - -
0,5 0,00002
-

Chỉ có các muối Pb(NO3)2 và Pb(CH3COO)2 tan trong nước. Trong dung dịch axit clohiđric, chì (II) nằm ờ dạng phức với clorua (PbCl3)-. Trong các dung dịch axit yếu, chì bị thuỷ phân và ở pH cao hơn 7, chì ở dạng kết tủa hidroxit màu vàng nâu; trong môi trường kiềm dư bị hoà tan tạo thành ion Pb(OH)4
2-. Vì vậy trong quá trinh phân tích cần lưu ý khả năng thuỷ phân của chì (II).
Chì cũng bị giấy lọc và các dụng cụ thuỷ tinh hấp thụ mạnh, trong các dung dịch axit khi có mặt H2o2 Sự hấp thụ Pb giảm đi đáng kể.
Phương pháp xác định
Phân huỷ đất
- Phân huỷ đất bằng axit: Khi xác định Pb trong đất người ta xử lý mẫu bằng axit HF cùng axit HNO3 hay HClO4. Quá trình phân huỷ tiến hành trong lò nung (để phân huỷ chất hữu cơ), tại nhiệt độ không vượt quá 500oC để tránh mất kim loại nặng do bay hơi. Để phân huỷ mẫu đất và mẫu quặng người ta cũng sử dụng hỗn hợp HNO3 và các chất ôxy hoá như H2O2, HClo4, KClO3. Khi xử lý bằng hỗn hợp này các chất hữu cơ bi khoáng hoá, các hợp chất khó tan của chì chuyển hoàn toàn sang dạng dung dịch. Quá trình phân huỷ tiến hành trong bình Kendan và sinh hàn không khí dưới dạng ống thuỷ tinh.
Phân huỷ bằng phương pháp nung chảy: Để phân huỷ mẫu đất khi xác định chì, có thể sử dụng hỗn hợp Na2CO3 + MgO (1:2) hoặc hỗn hợp Na2CO3 + ZnO (l:4). Quá trình thuỷ phân tiến hành tại nhiệt độ không cao quá 700 - 800oC trong thời gian 1,5 - 2 giờ. Làm lạnh rồi hoà tan trong nước nóng. Chì chuyển vào trong nước dưới dạng Pb(OH)4
2-
- Xác định chì: Để xác định một lượng nhỏ chì, thường sử dụng phương pháp chiết trắc quang với thuốc thử đithizon. Trong môi trường trung tính hoặc kiềm yếu, đithizon tạo với ion Pb (n) thành hợp chất chì đithizonat. Trong các dung môi hữu cơ chì đithizonat có màu đỏ. Độ tan của chì (II) đithizonat trong CHCl3 cao gấp 17 lần so với trong CCl4.
Hệ số hấp thụ phân tử bằng 6,86.1O4 hấp thụ cực đại ở bước sóng 520nm; giới hạn phát hiện của phương pháp là 0,05 μg Pb/ml. Định luật Bia được tuân theo đến khoảng nồng độ 70 μg Pb trong 50ml CCl4.
Điều kiện thích hợp để chiết phức là tại pH = 8 - 10. Tại môi trường kiềm (pH>10,5) và trong môi trường axit (pH < 3,5) nếu tiến hành chiết trong CHCl3 (hay tại pH<4,5 nếu chiết bằng CCl4) chì đithizonat sẽ bị phá huỷ và chì sẽ chuyển sang tướng nước
Trình tự phân tích
Cân 2,5g đất nghiền đến trạng thải bột mịn cho vào bình Kendan, thêm vào đây 1 5ml HNO3 đặc và vài giọt H2O2 đậy bình bằng phễu thuỷ tinh cuống dải dùng làm sinh

hàn. Đun nóng nhẹ đến khi xuất hiện các oxit của não. Để nguội bình rồi thêm tiếp những phần HNO3 và H2O2 mới tiến hành đun cho đến khi mẫu đất trở nên trắng. Xử lý mẫu bằng HCl và lọc vào cốc thuỷ tinh chịu nhiệt. Chưng dung dịch lọc đến khô, thêm vào cốc HCl 1: 1, đun sôi đến 80 - 90oC, lọc và rửa phần không hoà tan của đất. Cho dung dịch lọc vào bình định mức và định mức bằng nước cất đến vạch mức.
Lấy một thể tích dung dịch trên cho vào phễu chiết, thêm 5 - 10ml amoni xitrat 10 % và 1 - 2 giọt chỉ thi thimol xanh. Dùng amoniac trung hoà dung dịch đến pH9-10 (chỉ thị có màu xanh). Sau đó thêm 5ml dung dịch đithizon 0,001 % trong CCl4 rồi lắc đều phễu. Sau khi phân lớp, rót phần hữu cơ vào phễu chiết khác. Tiến hành chiết lại chì trong mẫu đến khi màu của đithizon không thay đổi. Gộp toàn bộ phần chiết. Thêm vào phễu chiết có hỗn hợp các đithizonat của các kim loại 10ml HCl 0,02 N và lắc mạnh để chuyển chì sang tướng nước; chuyển phần tướng nước vào phễu chiết khác. Rửa phần hữu cơ đó bằng nước và nước rửa này cũng gộp vào phễu chiết chứa dung dịch chì. Thêm vào dung dịch chứa chì 5ml amoni xuân 10% và trung hoà bằng amoniac đến pH = 9. Thêm 1ml KCN 5%, 5ml dung dịch đithizon 0,001 % trong CCl4 và lắc mạnh. Sau khi phân lớp, rót phần hữu cơ vào bình định mức dung tích 25 - 50 ml (hoặc ống nghiệm chia độ) và lặp lại quá trình chiết chì đến khi màu của đithizon không thay đổi định mức thê tích đến vạch mức bằng CCl4. Lắc đều và đo màu ở bước sóng 520nm. Dung dịch so sánh là CCl4.
Hoá chất
- Dung dịch chuẩn gốc: 0,1 Pb/ml. Hoà tan 0,1598 g Pb(NO3)2 đã kết tinh lại và sấy khô ở 1100C trong nước, thêm 1 ml HNO3 và thêm nước cất đến vạch mức.
- Dung dịch chuẩn sử dụng: μg Pb/ml.
- Dung dịch đithizon 0,001 % trong CCl4.
- Dung dịch gốc: Hoà tan 0,4g đithizon trong 200ml CCl4, chuyển dung dịch vào phễu chiết 1,5 - 2 lít, thêm vào 500ml NH4OH loãng (1:200), lắc 3 - 5 phút. Loại bỏ phần tướng hữu cơ. Rửa phần nước một vài lần bằng CCl4 mỗi lần 5 - 10ml cho đến khi CCl4 có màu xanh, sau đó thêm vào phễu chiết 12,5ml H2SO4 l:5, lắc đều. Thêm 400ml CCl4 và tắc phễu, rót dung dịch này vài lần bằng nước cất 2 lần để tách hết lương axit dư (thường từ 2 - 3 lần). Lọc dung dịch đithizon qua giấy lọc khô (giấy lọc đã được rửa bằng HCl và nước cất hai lần) vào trong lọ thuỷ tinh màu nâu. Dung dịch phải được bảo quản lạnh.
- Từ dung dịch này pha loãng bằng CCl4 ta được dung dịch đithizon sử dụng có nồng độ 0,01%.
7.3.6. Xác định thuỷ ngân trong đất
Thuỷ ngân từ khí quyền khi xâm nhập vào đất sẽ bị các chất hữu cơ và các hiđroxit của các kim loại hút thu; ma ngan hiđroxit có khả năng hút thu thuỷ ngân

mạnh nhất. Theo Vinagradova A.PA hàm lượng thuỷ ngân trong đất là: 1 - 14.10-6%, theo Glađuseva V.PA và các cộng sự, hàm lượng thuỷ ngân trong đất dao động trong giới hạn 1.10-7 - 1.10-6 %. Hàm lượng trung bình của thuỷ ngân trong thuỷ quyển là 0,03 μg/1, trong thực vật n.10-5 - n.10-7 %. Trong đất ở khoảng pH = 6, thuỷ ngân bị cố định chặt nhất.
Thường hay phân tích hợp chất thuỷ ngân (II). Các hợp chất này tan tốt trong nước nhưng cũng bị thuỷ phân mạnh. Khi pH > 5 sẽ xuất hiện dưới dạng HgO kết tủa màu vàng, khi pH > 11,5 bắt đầu bị hoà tan. Hợp chất của thuỷ ngân với halogen [Hg(hal)]+... [Hg(hal)4]2- là những hợp chất dễ bay hơi. Hg2- còn tham gia tạo phức với các muốn CN-, SCN-, S2O3
2- , NO2 hoặc với NH3.
Hg có thể bị hấp thụ bởi dụng cụ thuỷ tinh, polietylen, teflon cũng hấp thụ một lượng nhỏ thuỷ ngân. Thuỷ tinh thiếc, thạch anh hấp thụ thuỷ ngân ít nhất. Để làm giảm khả năng hấp thụ cần phải xử lý trước thuỷ tinh bằng HNO3 đặc và rửa nước hoặc bằng đung dịch cromic.
Phương pháp xác định
Phân huỷ đất: Có thể dùng phương pháp thiêu kết hoặc xử lý mẫu bằng axit. Trong phương pháp thiêu kết không dùng các muối cacbonat của kim loại kiềm vì khi đó có thể mất hoàn toàn thuỷ ngân. Để xác định thuỷ ngân người ta dùng phương pháp thiêu kết trong các điều kiện khử, khi đó thuỷ ngân sẽ được khử đến kim loại rồi cất trong dụng cụ đặc biệt, thường sử dụng canxi oxit và bột sắt, chì oxit và canxi nitrat làm chất thiêu kết. Hg thu được sẽ được hoà tan trong HNO3.
Trong phương pháp xử lý mẫu bằng axit người ta thường dùng H2SO4 khi có mặt chất ôxy hóa như HNO3, các muối thuật hoặc KMnO4. Quá trình phá mẫu khi đun nóng cho đến khi mẫu trở nên trắng, sau đó loại chất ôxy hoá còn dư bằng cách thêm vào các chất khử như urê, fomalin. Để phân huỷ mẫu thực vật hoặc các đối tượng sinh học khác người ta dùng hỗn hợp H2SO4 và HClO4 khi có mặt nitơ molipdat. Để đẩy nhanh quá trình phân huỷ người ta tẩm ướt mẫu trước bằng dung dịch HNO3 đặc.
Trong môi trường axit khi đã dư thuốc thử, đithizon phản ứng với Hg(II) tạo thành phức Hg(HDZ)2 tan trong các dung môi hữu cơ. Dung dịch thuỷ ngân (li) đithizonat trong CHCl3 và CCl4 có màu vàng da cam; hấp thụ cực đại ở bước sóng 485nm. Hệ số hấp thụ phân tử bằng 7,1.1O4; định luật Bia tuân theo trong khoảng nồng độ đến 50μg Họ trong 50ml dung dịch chiết.
Điều kiện thích hợp để tạo phức và chiết phức là trong dung dịch H2SO4 0,5 N - 1 N hay HNO3 0,5 - 1N.
Trình tự phân tích
Thiêu huỷ 1 g đất đã nghiền đến trạng thái bụi bằng hỗn hợp PbO2 + Ca(NO3)2: lg đất được mải mịn trong cối với 2g PbO2 và 0,15g Ca(NO3)2 đã làm khô trước trong

bình hút ẩm. Cho hỗn hợp vào phần hình cầu của ống 1 của dụng cụ và đun nóng, lúc đầu đun nhẹ ở phần trên ngọn lửa đèn khí và sau đó nung đỏ. Khi thiêu kết dung có được đặt hơi nghiêng. Sau 5 - 6 phút nung nóng, di chuyển ngọn lửa đèn khí đến 3, đốt nóng phần này sau đó lại chuyến xuống dưới. Làm lạnh, hoà tan thuỷ ngân đã thăng hoa gom ở ống 2 trong 1 ml HNO3 1: 1 nóng rồi chuyền dung dịch vào phễu chiết. Rửa ống một vài lần bằng nước cất rồi gộp cả vào phễu chiết trên. Pha loãng dung dịch bằng nước cất đến nồng độ HNO3 1N. Sau đó rót vào phễu chiết dung dịch KMnO4 0,1N đến khi dung dịch có màu hồng rồi lắc đều. Sau 1 - 2 phút khử lượng MnO4
- còn dư bằng 1 - 2 giọt H2O2 30%.
Sau đó thêm vào phễu chiết 1 ml dung dịch complexon III 1 %, 4ml dung dịch Na2SO3 20%, lắc đều. Thêm vào phễu chiết 4 ml dung dịch đithizon 0,001 % trong CHCl3 và tắc phễu chiết 1 phút. Tiến hành chiết đithizonat đến khi đithizon không đổi màu. Sau khi tách lớp, rót phần chiết vào cuvet và đo mật độ quang ở bước sóng 485nm.
Dung dịch chuẩn gốc
Hoà tan 0,3336g Hg(NO3)2 0,5H2O tinh khiết hoá học trong nước cất, thêm lưu HNO3 đặc rồi thêm nước cất đến 1 lít, được dung dịch chuẩn có nồng độ 0,1g Hg/ml. Đường chuẩn được xây dụng từ dung dịch chuẩn có hàm lượng 2μg Hg/ml trong HNO31N.
7.3.7. Xác định dạng di động của một số nguyên tố
Những hợp chất của các kim loại nặng dễ dàng chuyển vào trong các dung dịch chiết rút khác nhau được gọi là dạng di động của các kim loại nặng.
Những hợp chất hoà tan trong nước, những dạng trao đổi và những hợp chất hoà tan trong các axit có nồng độ thấp cũng như trong các dung dịch đệm đều được xếp vào các hợp chất di động. Các dung dịch dùng để chiết rút các hợp chất di động hiện có rất nhiều, thường là các dung dịch axit và các dung dịch đệm. Các dung dịch này khác nhau về lực tác dụng của chứng đối với đất và một số trường hợp dung dịch chiết rút được sử dụng như là một thuốc thử nhóm rối xác định một số kim loại nặng ở trong đó, người ta sử dụng những thuốc thử khác nhau.
Sự lựa chọn một dung dịch chiết rút thích hợp đề chiết rút kim loại nặng di động trong đất được đánh giá bằng cách so sánh những số liệu phân tích nhận được với các

kết quả của các thí nghiệm ngoài đồng có sử dụng các phân vi lượng ở các cây trồng và trong những điều kiện thổ nhưỡng không giống nhau; từ đó giải thích về mức độ đảm bảo của đất về các kim loại nặng đối với dinh dưỡng của thực vật
Đã có nhiều nghiên cứu công phu để chọn lựa, so sánh các dung dịch chiết rút nhóm và các dung dịch chiết rút riêng biệt các kim loại nàng. Các dung dịch chiết rút nhóm thưởng được đề cập đến là:
- Dung dịch axetat-lactat theo phương pháp Egner-Rim-Doming (phương pháp AL).
- Dung dịch HCl là (phương pháp Rinhkis).
- Dung dịch EDTA 0,01 M + dung dịch đệm amoni axetat pH = 4,7 (amoni axetat 0,5N và axit axetic 0,5N).
- Dung dịch đệm oxalat (phương pháp Grigg)
- Dung dịch đệm amoni axetat pH = 4,8 theo phương pháp của Krupski- Alecxandrova (dùng cho đất cacbonat và trung tính).
Dung dịch Baron pH = 4 (gồm amoni axetat, amoni sunfat và axit axetic).
Với từng nguyên tố, sử dụng các dung dịch chiết rút riêng sau đây:
B: nước cất nóng (theo Berger và Trung).
Cu: HNO3 0,43N (theo Vesterhoff); HCl là (theo Rinhkis).
Zn: KCl là (theo Rinhkis); EDTA + (NH4)2CO3 (theo Trierveiler và Lindsay); HCl 0,1N (theo Zommer).
Mo: dung dịch đệm oxalat pH = 3,3 (theo Grigg).
Từ các kết quả phân tích đất, phân tích cây trồng trong các thí nghiệm trong chậu và ngoài đồng, nhiều nhà nghiên cứu đã rút ra nhận xét:
Dung dịch chiết rút có độ axit càng mạnh thì lượng các kim loại nặng được chiết rút ra càng lớn. Lượng các kim loại nàng được chiết rút ra bằng HCl 1N là lớn nhất.
Ba Lan, Cộng hoà Séc sử dụng HCl là làm dung dịch chiết rút nhóm để xác định kẽm, đồng, mangan; Liên Xô (cũ) dùng dung dịch đệm amoni axetat (pH = 4,8) làm dung dịch chiết rút nhóm để xác định mangan, kẽm, đồng và coban trong đất trung tính và đất cacbonat (hàm lượng cacbon không quá 10%).
Ở Liên Xô (cũ) sử dụng phô biến hệ thống dung dịch chiết rút do I.V.Peive và G.I.Rinhkis đưa ra để xác định dạng di động của các kim loại nặng đối với đất không phải cacbonat.
Trong hệ thống này, để chiết rút mỗi kim loại nặng người ta sử dụng những thuốc thử riêng. Phương pháp này đã được phòng thí nghiệm hoá học Đất - Học viện Thổ nhưỡng mang tên V.V. Docutraev cải tiến. Tỷ lệ giữa đất và dung dịch chiết rút là

1:10, lắc trong 1 giờ.
7.3.7.1. Xác định bo di động
Để chiết rút bỏ di động từ đất người ta thường xử lý đất bằng nước sôi. Phương pháp này đã được Berger và Trung đưa ra năm 1939. Hiện nay phương pháp này đã được cải tiến về tỷ lệ giữa đất, nước và thời gian đun sôi.
Chiết bổ di động từ đất Cân 20 - 50g đất khô không khí đã rây qua rây 1 mm cho vào bình làm từ thạch anh hay từ thuỷ tinh không bỏ. Thêm nước cất với lượng gấp hai lượng đất cân theo tỷ lệ 1:2 (đối với đất than bùn 1:10). Đun nóng bình đến sôi và giữ sôi trong 10 phút. Bình đun phải có sinh hàn, nếu không có thể thay bằng một úng thuỷ tinh dài 25 - 30cm làm từ thuỷ tinh không bỏ, cắm vào nút tre. Lọc dung dịch nóng qua giấy lọc không tàn. Lấy 20 - 40 ml (tuỳ thuộc vào hàm lượng bo) dung dịch lọc trong cho vào bát thạch anh hay bát platin, thêm vào lưu dung dịch NaOH hay KOH 1N rồi chúng dung dịch trên bếp cách thuỷ đến khô. Nung phần khô này trong lò nung tại nhiệt độ 450 - 500oC đến khi phá huỷ hoàn toàn chất hữu cơ và thuật (1 - 2 giờ). Sau đó có thể dùngphươngpháp cannin hay phương phápq inazann để xác định bo.
a. Xác định bo di động bằng phương pháp carmin
Hoà tan phần khô sau khi nung trong 5 - 10 ml dung dịch H2SO4 0,5 N, nghiền cẩn thận bằng một que thuỷ tinh khô làm từ thuỷ tinh không bo. Lọc dung dịch qua giấy lọc không tan, trung dung dịch lọc.
Dùng pipet hút 1 ml cho vào ống nghiệm nút nhám, thêm chính xác 9 ml dung dịch carmin 0,005% trong H2SO4 (d = 1,84), nút ống nghiệm, sắc đều và giữ nguyên 1 2 - 18 giờ sau đó đo mật độ quang trong cuvet dày 3mm, đậy cuvet bằng nắp thuỷ tinh. Kết quả phân tích được tính theo đường chuẩn.
Cần chú ý và dung dịch phân tích và dung dịch chuẩn được lấy cùng bằng một phiệt.
Hoá chất
- H2SO4 đặc loại tinh khiết đặc biệt hay tinh khiết hoá học. Nồng độ được xác định bằng tỷ trọng kế.
- Kiểm tra xem có NO3- trong axit này bằng cách rót 9ml nước cất vào trong ống
nghiệm sạch, khô, sau đố rót vào làn H2SO4 đặc, lắc đều. Sau khi nguội, thêm từ từ, dọc theo thành ống 5ml dung dịch diphenylamin trong H2SO4 đặc vào trong ống nghiệm. Nếu có nitrat, trên ranh giới giữa hai chất lỏng xuất hiện một vùng hoặc một vòng màu xanh nước biển.
- Axit bị bẩn HNO3 và những axit có màu nhạt cũng không nên dùng để xác định bo. Để xác định bo tốt nhất là sử dụng loại đặc biệt tinh khiết, có thể sử dụng loại tinh khiết hoá học đã khá trước không có NO3
- và bo.

- Dung dịch cannin 0,005% trong H2SO4 đặc, cần phải thử trước chất lượng carmin; cho dung dịch axit hoặc vào ống nghiệm rồi thêm vào một lượng vừa phải dung dịch can nin trong axit sunfuric đặc. Thêm dung dịch can nin cần phải ở lượng tối thiểu. Nếu dung dịch axit hoặc có màu xanh nước biển thì can nin có thể sử dụng được. Sau khi kiểm tra, cân trên cân phân tích 50mg cannin cho vào bình định mức 1 lít, thêm 500 - 600ml H2SO4 đặc (d=1,84) khuấy đều dung dịch cho đến khi hoà tan hoàn toàn (tốt nhất là để qua đêm), sau đó thêm H2SO4 đặc đến vạch mức. Đậy bình và lật ngược bình một vài lần để dung dịch trở nên đồng nhất.
Dung dịch can nin đựng trong lọ nút nhám để ở chỗ tốt. Dung dịch có màu đỏ, nếu dung dịch có màu khác, không thuận lợi để định lượng bo.
- Axit hoặc H3BO3
Để Xây dụng đường chuẩn cần phải dùng H3BO3 kết tinh lại hai lần. Độ tan của axit hoặc tại 00C là 2g trong 100 mg nước cất, tại 200C là 5 g và tại 1000C là 40g. Cân 10 - 12g axit hoặc, hoà tan khi đun nóng và khuấy liên tục trong 100ml nước cất. Đun dung dịch đến sôi, lọc dung dịch nóng qua giấy lọc băng trắng, hứng dung dịch lọc vào trong bình rộng làm từ thuỷ tinh không bỏ. Làm lạnh bình bằng vòi nước hoặc nước đá. Dùng phễu Busner lọc H3BO3 qua giấy lọc băng trắng, rửa axit này bằng 3 - 4 lần bằng những lượng nhỏ nước cất lạnh. Lấy H3BO3, làm khô ở ngoài không khí, trên tờ giấy lọc, đậy bằng một tờ giấy lọc khác, cùng loại. Chuyển kết tủa vào trong cốc cân rộng đáy, không đậy nắp cốc cân rồi đặt cốc vào trong bình hút ẩm có chứa H2SO4 đặc trên khối lượng không đổi. Axit hoặc kết tinh lại được giữ trong cốc cân đậy nắp và để trong bình hút ẩm.
- Dung dịch chuẩn gốc: Cân 0,2858g axit hoặc đã kết tinh lại trên cân phân tích sau đó chuyển lượng cân vào bình định mức 500ml nút nhám. Hoà tan bằng H2SO4 0,5N. Sau đó định mức đến vạch mức cũng bằng chính dung dịch axit này. Đậy nút và lật ngược bình vài lần để trộn đều dung dịch. Ta được dung dịch chuẩn gốc có hàm lượng bộ là 0,1 mg/ml.
- H2SO4 0,5N: lấy 14ml H2SO4 bắc hoà tan, thêm nước đến 1 lít.
- Dung dịch chuẩn sử dụng: Dùng H2SO4 0,5N pha loãng dung dịch chuẩn gốc lên 10 lần ta được dung dịch chuẩn sử dụng có hàm lượng bo là 0,01mg/ml.
Thang chuẩn: Dùng 2 micro buret có thể tích 1 hoặc 2ml, đựng dung dịch chuẩn sử đụng, một đựng H2SO4 0,5N. Lấy Các thể tích dung dịch chuẩn sử dụng: 0,00; 0,05; 0,10; 0,20; 0,40; 0,60; 0,80; 1.00ml cho vào ống nghiệm sạch, khô làm từ thuỷ tinh không bỏ hoặc làm từ thạch anh; sau đó thêm các thể tích H2SO4 0,5N để thể tích dung dịch trong mỗi ống nghiệm đứng bằng lúa. Lắc đều rồi thêm vào mỗi ống nghiệm chính xác 9ml dung dịch cannin 0,0055. Lắc đều và giữ yên trong 24 giờ để màu ổn định hoàn toàn, ta được thang màu chuẩn.
b. Xác định bộ theo phương pháp quinalizarin

Nguyên lý
Phương pháp dựa trên sự thay đổi màu tím của quinalizanin (1,2,5,8-tetra- oxianthraquinon) trong môi trường H2SO4 đặc sang màu xanh nước biển khi có một bo. Khi có mặt bo, quinaizarin sẽ tạo thành một hợp chất nội phức. Độ nhạy của quinalizann cao hơn so với can nin vì hệ số hấp thụ phân tử cao hơn (ε = 7000 tại bước sóng 615nm).
Vì màu của hợp chất phức boquinalizarin chỉ tuân theo đinh luật Bia khi hàm lượng bộ không vượt quá 1 μg nên trong đường chuẩn cũng chỉ xây dựng với hàm lượng bo từ 0 - 1 μg.
Trình tự phân tích
Lấy dung dịch phân tích cho vào ống nghiệm hay ống trụ làm từ thuỷ tinh không bo hoặc làm từ thạch anh có thể tích là 20 - 25ml.
Tuỳ thuộc vào nồng độ axit sử dụng, dùng buret thêm 19,0-22,0m dung dịch quinalizarin trong axit sunfunc. Đậy nút ống nghiệm, lắc đều dung dịch và giữ yên 12- 18 giờ để cho màu ổn định hoàn toàn sau đó đo màu trên máy trong cuvet 3cm, dùng miếng thuỷ tinh để đậy cuvet.
Hoá chất
- H2SO4 93,56 - 95,60 % loại đặc biệt tinh khiết hoặc tinh khiết hoá học.
- Dung dịch quinalizarin trong axit sunfuric hoà tan 10 - 20mg quinalizarin trong 1 lít H2SO4 đặc. Do số ml dung dịch này thêm vào mẫu phụ thuộc vào hàm lượng phần trăm H2SO4 nên cần phải xác định hàm lượng của nó.
Đĩa cân phân tích bên trái đặt cốc cân hoặc bình tam giác có nút nhám dung tích 100ml; đĩa cân bên phải đặt quả cân 2g. Dùng pipet có quả bóp bằng cao su thêm cần thận H2SO4 đặc vào cốc hoặc bình tam giác cho đến khi cân thăng bằng.
Đậy cốc cân hoặc bình tam giác và xác định chính xác hàm lượng H2SO4 đã lấy. Khi cho axit vào cần chú ý không cho axit rơi ra đĩa cân hay ra ngoài cốc hay bình tam giác. Chuyển toàn bộ lượng axit đã cân vào bình định mức 500ml trong đó đã có sẵn 100 - 200ml nước cất. Rửa cẩn thận cốc cân hay bình tam giác một vài lần bằng nước đề chuyền hết axit sang bình định mức. Thêm nước đến vạch mức và khuấy đều. Dùng pipet lấy 3ml mẫu, mỗi mẫu 25ml cho vào bình tam giác 250ml, thêm vào 2 giọt chỉ thị metyl da cam 0,1 % và dùng dung dịch NaOH 0,1N để chuẩn.
Hàm lượng H2SO4 theo phần trăm được tính theo công thức:
Trong đó: V: thể tích NaOH 0,1 N đã dùng để chuẩn độ (ml);

0,004904: lượng H2SO4 tính ra gam, tương ứng với đúng 1ml dung dịch NaOH 0,1N;
2: lượng H2SO4 tính theo gram.
Tuỳ thuộc vào hàm lượng phần trăm H2SO4 tìm được, gối với 1 ml dung dịch thí nghiệm hoặc dung dịch chuẩn để lấy các lượng quinalizarin khác nhau:
Hàm lượng H2SO4 (tính theo %) Dung dịch quinalizarin (ml) 93 - 94 ≈ 94,5
95 và cao hơn
22,0 20,0 19,0
Khi sử dụng axit có nồng độ cao hơn 94,5 %, thuận lợi hơn cả là nên pha loãng để được dung dịch có nồng độ 93 - 94%.
Thường pha 1 - 2 l hoặc nhiều hơn dung dịch quinalizann trong axit sunfuric. Dung dịch được giữ trong lọ thuỷ tinh nút nhám, đặt ở chỗ tốt.
Thang chuẩn
Dùng nước cất pha loãng dung dịch chuẩn gốc có chứa 0,1 mg Bo/ml ra 100 ml lần ta được dung dịch chuẩn sử dụng. Dùng micro buret cho vào các ống nghiệm nút nhám sạch, khô làm bằng thạch anh hoặc thuỷ tinh không bo. Các thể tích dung dịch chuẩn sử dụng như sau (ml): 0,00;0,20; 0,40; 0,60; 0,80; 1,00, dùng nước cất hai lần đưa thể tích lên đúng 1 ml.
Thêm vào mỗi ống 9ml dung dịch quinalizann trong H2SO4, đậy ống nghiệm, lắc đều rồi giữ yên trong khoảng thời gian từ 12 - 1 8 giờ, sau đó đo mật độ quang và xây dụng đường chuẩn.
7.3.7.2. Xác định đồng di động
a. Xác định đồng di động theo Rinhkis.
Nguyên lý
Phương pháp dựa trên quá trình chiết rút đồng di động từ đất bằng dung dịch HCl 1 N. Tỷ lệ giữa đất và dung dịch là 1:10. Lắc trên máy lắc 1 giờ sau đó đinh lượng Cu dưới dạng phức màu của Cu với dietyldithiocacbaminat trong CCl4.
Trình tự phân tích
Cân trên cân phân tích 5g đất khô không khí đã rây qua rây 1 mm, cho mẫu vào bình tam giác nút nhâm dung tích 100 ml. Thêm chính xác 50ml HCl 1N, lắc trên máy lắc 1 giờ sau đó lọc qua giấy lọc băng trắng.
Dùng pipet hút 10 - 25ml dung dịch lọc (tuỳ thuộc hàm lượng đồng) cho vào phễu chiết 50 - 100 ml, thêm 5ml dung dịch amonixitrat 5%, 1 giọt phenolphtalein rồi thêm từng giọt amoniac đến khi dung dịch có màu hồng nhạt. Cho vào chính xác 15ml

dung dịch chì dietyldithiocacbaminat trong CCl4, lắc trên máy 10 phút. Để dung dịch trong phễu phân lớp rồi rót CCl4 chứa phức của đồng với thuốc thử có màu vàng qua giấy lọc khô vào cuvet 2cm. Đo màu với kính lọc màu 453 mm, dung dịch so sánh là
CCl4. Kết quả phân tích theo công thức: X= ba
Trong đó X: Hàm lượng đồng trong đất mg/kg
a: Lượng đồng trong thể tích lấy đề phân tích, tìm thấy theo đường chuẩn (μg);
b: Khối lượng đất, tương ứng với thể tích lấy đê phân tích (g)
Hoá chất
- Dung dịch chì dietyldithiocacbaminat trong CCl4, lắc 664mg natri dietyldithiocacbaminat với 1 lít CCl4trong phễu chiết; thêm vào phễu dung dịch chì nitrat (489 mg Pb(NO3)2 hoà tan trong 100 ml nước cất hai lần. Lắc phễu 5 phút. Để cho phân lớp rối lọc lớp CCl4có chì dietyldithiocacbaminat tan trong đó qua giấy lọc khô vào trong bình thuỷ tinh màu nâu. Dung dịch được giữ trong tủ lạnh
- Dung dịch chuẩn CuSO4: Hoà tan 3,993g CuSO4.5H2O đã kết tinh lại trong nước cất hai lần, định mức đến 1 lít, được dung dịch chứa leng Cu/ml. Pha loãng dung dịch này 100 lần (10 ml pha loãng đến 1 lít) thu được dung dịch chứa 0,1 mg Cu/ml; lại pha loãng dung dịch đó 10 lần ta được dung dịch chứa 1 μg Cu/ml, sử dụng dung dịch này để pha thang chuẩn.
Bảng 7.15. Thang đánh giá (mg Cu/kg)
Rất nghèo đồng 0,3 Nghèo đồng 0,3 - 1,5 Trung bình 1,5 - 3,3
Cao > 3,3 b. Xác định đồng di động theo Vesterhoff
Hiện nay đã nhiều nước thừa nhận phương pháp xác định đồng di động trong dung dịch HNO3 loãng (để chiết rút đồng) của Vesterhoff là phương pháp chuẩn.
Trình tự phân tích
Lấy lớp đất cho vào bình tam giác 250ml, thêm vào 100 ml HNO3 (30ml HNO3, d = 1,39 định mức đến thể tích 1 lít bằng nước cất hai lần), lắc 2 giờ rồi lọc. Lấy 25ml dung dịch lọc cho vào bình tam giác. Thêm vào 2ml dung dịch KMnO4 0,5% và đun dung đích đến sôi. Cho vào dung dịch nóng 0,5ml axit oxalic 5% để làm mất màu dung dịch. Để dung dịch nguội rồi chuyền dung dịch vào phễu chiết 100 ml (rửa bình tam giác bằng nước cất) và đưa thể tích đến 50ml. Cho vào phễu chiết lưu dung dịch Na3PO4 bão hoà, 4ml dung dịch natri xitrat 50 % và 4ml dung dịch NH4OH đặc (d = 0,92) sau mỗi lần thêm thuốc thử vào đều phải lắc phễu chiết cẩn thận.

Cho vào chính xác 15 ml dung dịch chì đietyldithiocacbaminat trong CCl4 lắc phễu chiết 2 phút. Để cho phân lớp, lọc chì đietyldithiocacbaminat trong CCl4 qua phễu lọc khô vào cuvet chiều dài 2cm. Đo mật độ quang với kính lọc màu xanh là cây (536nm); CCl4 là dung môi so sánh).
Đường chuẩn được xây dụng từ dung dịch có hàm lượng đồng là 10 μg Cu/ml. Để xây dụng đường chuẩn người ta cho vào các phễu chiết 0; 0,2; 0,5; 1,0; 2,0; 3,0; 4,0 ml dung dịch đong, thêm nước cất 2 lần đến thể tích 50 ml rối tiếp tục xử lý như đối với dung dịch phân tích
Bảng 7.16. Thang đánh giá (mg Cu/kg) Đánh giá Đất cát Đất sét và sét pha
Tốt > 3,5 > 2,5 Trung bình 2,1 - 3,5 1,6 - 2,5
Kém < 2,0 < 1,5
7.3.7.3. Xác định mangan di động
a. Phương pháp Dobritxcaia
Nguyên lý
Phương pháp dựa trên quá trình chiết rút ma ngan di động bằng dung dịch H2SO4 0,1N. Tỷ lệ giữa đất và dung dịch là 1: 10. Thời gian tương tác là 1 giờ, ôxy hoá mangan đến trạng thái hoá trị 7 bằng pensunfat khi có mặt bạc nitrat và axit phosphoric.
Trình tự phân tích: Cân trên cân phân tích 5 g đất khô không khí đã rây qua rây 1 mm. Cho lượng cân vào trong bình tam giác nút ni ám dung tích 100 ml; thêm vào 50 ml H2SO4 0,1 N; lắc trên máy lắc 1 giờ, lọc dung dịch lua giấy lọc mịn băng trắng.
Lấy 10 - 15ml dung địch lọc cho vào cốc chịu nhiệt dung tích 50ml. Thêm vào đây 5ml HNO3 đặc và 2ml H2O2 30%; Chưng trên bếp điện đến khô. Lặp lại qúa trình ôxy hoá bằng HNO3 và H2O2 2 - 3 lần sau đó thêm 3ml HNO3 và chưng đến khô. Thêm vào phần khô 25ml H2SO4 10%. Đun trên bếp điện đến khi phần khô tan hoàn toàn, thêm vào cốc 15ml nước, 2ml H3PO4 (d = 1,7) và 2mg AgNO3 1% đun 5 - 10 phút; nếu thấy đục thì cần tiếp tục đun đến sôi rồi lọc dung dịch qua giấy lọc băng xanh. Thêm vào dung dịch nóng trong cốc khoảng 2 g amoni pensunfat (thêm vài lần) khuấy dung dịch cẩn thận bằng que thuỷ tinh, đặt cốc lên trên bếp điện, đun 10 - 15 phút để ôxy hoá nhanh và hoàn toàn ma ngan đến axit manganic, khi đó xảy ra quá trình phân huỷ mãnh liệt amoni pensunfat và có khí ozon thoát ra. Sau khi hết khí thoát ra, lấy cốc ra khỏi bếp, để nguội, chuyển dung dịch vào bình định mức 50ml. Thêm nước cất hai lần đến vạch mức và đo mật độ quang ngay rày lem hay 2cm tại bước sóng 536nm, dung dịch H2SO4 5 % dùng làm dung dịch so sánh.

Đường chuẩn được xây dụng từ dung dịch KMnO4 0,1N (từ ống chuẩn). Lấy 10 ml dung dịch cho vào bình định mức 100 ml, thêm nước cất 2 lần đến vạch mức, khuấy đều. Từ dung dịch mới pha này lấy 10ml cho vào bình định mức 100 ml rồi thêm nước cất hai lần đến vạch mức. Dung dịch này có nồng độ 0,001 N. Trong 1 ml dung dịch chứa 1 μg mangan. Dùng pipet lấy 2; 5; 10; 20; 25ml dung dịch KMnO4 0,001N cho vào bình định mức 50ml. Thêm nước cất đến vạch mức và đo mật độ quang ngay với kính lọc màu xanh lá cây (536nm), dùng cuvet có chiều dài như khi đo với dung dịch phân tích. Kết quả phân tích tính theo công thức:
Trong đó:
X: hàm lượng Mn trong đất (mg/kg)
a: Lượng dung dịch KMnO4 0,001 N nhận được theo đường chuẩn đối với thể tích dung dịch thí nghiệm lấy để phân tích (ml);
b: Khối lượng đất, tương ứng với thể tích dung dịch thí nghiệm lấy để phân tích (g);
11: Hàm lượng Mn tính theo μg trong 1 ml dung dịch KMnO4 0,001 N.
b. Xác định Mn di động theo Schachtschabel
Để đánh giá hàm lượng Mn di động trong đất, Schachtschabel (1957) đề nghị tiến hành xác định ma ngan tan trong sunfit và mangan dễ được khử.
Xác định mangan tan trong dung dịch sunflt: Cân 5g đất đã rây qua rây hàm cho vào bình tam giác 250ml, thêm vào 0,2g tham hoạt tính và 50ml dung dịch chiết rút, lắc trên máy sắc 1 giờ. Sau đó lọc dung dịch qua giấy lọc băng trắng, khô. Để phân tích có thể sử dụng những phần dung dịch lọc đầu tiên. Lấy 15 - 20ml dung dịch lọc cho vào bình định mức 50ml, để che ảnh hưởng khử của chị và để tạo phức với ion sắt, thêm vào bình 2,5ml hỗn hợp gồm thuỷ ngân sunfat, axit nước, bạc thuật và axit photphoric. Sau đó, để ôxy hoá ma ngan lên hoá trị 7 người ta thêm vào 2g amoni pensunfat. Đặt bình với dung dịch vào trong máy ồn nhiệt 20 - 30 phút ở nhiệt độ 90 - 950C đến khi ngừng thoát bọt khí. Nếu dung dịch có màu nâu thì cần phải lấy những phần dịch lọc mới (5 - loạn) rồi lặp lại quá trình xử lý như trên. Dung dịch lọc sau khi nguội được thêm nước đến vạch mức rồi đo màu như đã mô tả ở trên.
Hoá chất
Dung dịch chiết rút: Cân 123,3g MgSO4.7H2O và 2g Na2SO4 hoà tan trong nước rồi định mức đến thể tích 1 lít bằng nước.
Dung dịch thuỷ ngân sunfat: hoà tan 75g Hg2SO4 trong 400ml HNO3 (d=1,4) và 300ml H3PO4 (d=1,7), thêm vào 0,2g AgNO3 và dùng nước cất đưa thể tích đến 1 lít.

Xác định mangan dễ khử (pH = 8,0): Quá trình chiết rút ma ngan tiến hành bằng dung dịch magie sunfat có thểm 2g hiđroquinon trong 1 lít dung dịch. Tất cả quá trình chiết rút và xác định mangan sau này tiến hành như khi xác định mangan tan trong dung dịch sunfit.
Thang đánh giá Mn dễ khử ở pH = 8,0
Nghèo < 20 mg/kg (ppm)
Trung bình 20 - 30 mg/kg
Giàu > 30 mg/kg
7.3.7.4. Xác định kẽm di động
Kẽm di động được chiết rút từ đất bằng dung dịch kim clorua 1N. Phương pháp chỉ sử dụng cho đất axit. Tỷ lệ giữa đất: dung dịch là 1: 10, lắc 1 giờ trên máy. Cuối cùng xác định kẽm bằng phương pháp do màu dưới dạng phức với đithizon theo màu đơn.
Trình tự phân tích
Cân trên cân phân tích 2,5g đất khô không khí đã rây qua rây 1 mm, cho mẫu vào chén thạch anh dung tích 50ml. Thêm vào 25ml dung dịch KCl 1N, lắc trên máy lắc 1 giờ rồi lọc qua phễu lọc thạch anh, dùng giấy lọc báng trắng đã tính chế khỏi các vết kim loại nặng.
Lấy 5ml dung dịch lọc cho vào phễu chiết 50ml, thêm vào 2 giọt chỉ thị metyl da cam, tiến hành trung hòa bằng dung dịch nam axetat 5% đến khi chỉ thị chuyển sang màu vàng, lại thêm vào 0,2ml dung dịch natri axetat 5% và 2ml dung dịch thiosunfat 50%. Chiết kẽm hai lần bằng dung dịch đithizon 0,02% trong CCl4, lần đầu dùng 2ml, lần sau lưu. Sau mỗi lần thêm đithizon, phễu chiết được lắc trên máy 3 phút, giữ yên cho tách tướng rồi chuyển tướng hữu cơ vào phễu chiết sạch khác. Chiết lại tướng nước bằng dung dịch đithizon mới; dung dịch đithizon cũng được rót vào phễu chiết sạch trên. Thêm vào phễu chiết chứa kẽm đithizonat 25ml dung dịch amoniac 0,01 N, lắc 1 - 2 phút. Lớp kẽm đithizonat có màu được chuyển vào bình định mức khô 25ml, rồi thêm CCl4 đến vạch mức. Đo mật độ quang trong cuvet chiều dày 1 cm tại bước sóng 536nm, dung dịch so sánh là CCl4.
Để xây dựng thang chuẩn cho vào các phễu chiết 0; 0,5; 1; 2; 3; 4; 5ml dung dịch kẽm có nồng độ 1 μg Zn/ml thêm nước cất đến 5ml rồi cho vào 2 giọt metyl da cam và tiến hành phân tích như đối với dung dịch thí nghiệm.
Kết quả phân tích theo công thức:
Trong đó:

X: hàm lượng kẽm di động trong đất (mg/kg);
a: lượng kẽm trong thể tích lấy đề phân tích, tìm thấy theo đường chuẩn (μg);
b: khối lượng đất tương ứng với thể tích mẫu lấy để phân tích (g).
Hoá chất
- Tất cả các hoá chất dùng để xây dụng kẽm đều phải tinh chế cẩn thận bằng đithizon.
Dung dịch chuẩn kẽm: Cân 0,2g kẽm kim loại tinh khiết trên cân phân tích cho vào bình định mức 1 lít và hoá tan trong lượng nhỏ nước cất hai lần có lần H2SO4 đặc (d = 1,84). Sau khi tất cả kem đã hoà tan, dùng nước cất hai lần đưa thể tích đến vạch mức. Trong lưu dung dịch này chứa 200 μg kẽm. Pha loãng dung dịch này 200 lần được dung dịch chứa 1 μg Zn/ml.
Bảng 7.17. Thang đánh giá (mg Zn/kg) Rất nghèo < 0,2
Nghèo 0,2 - 1,0 Trung bình 2 - 3
Giàu 4 - 5 Rất giàu > 5
Ghi chú: Đối với đất ngập nước có thể dùng HCl 0,05N làm dung dịch chiết rút: 10g đất và 20ml dung dịch HCl 0,05 N. Lắc 5 phút rồi lọc.
7.3.7.5. Xác định coban di động
Phương pháp dựa trên quá trình chiết coban di động trong đất bằng HNO3 1N. Tỷ lệ giữa đất: dung dịch là 1:10, lắc trên máy lắc 1 giờ. Xác định coban bằng phương pháp đo màu dưới dạng phức màu với muối nitrozo -R.
Trình tự phân tích
Cân lớp đất khô không khí đã rây qua rây hàm trên cân phân tích, cho mẫu vào bình tam giác nút nhám dung tích 250ml. Thêm vào đó chính xác 100ml HNO3 1N lắc trên máy lắc 1 giờ, lọc qua giấy lọc băng trắng.
Lấy 25ml dung dịch lọc cho vào cốc thuỷ tinh chịu nhiệt, thêm vào đó lần HNO3 gác, 1 ml H2O2. Chúng trên bếp điện đến khô. Quá trình xử lý này được lặp lại một lần nữa và chăng dung dịch đến khi bắt đầu kết tinh muốn. Rót vào cốc 6ml nước cất hai lần đã axit hoá bằng HCl (1:10), đun dung dịch đến sôi. Thêm vào 1 ml natrixitrat 40% và đun sôi khoảng 1 phút, pH của dung dịch cần phải lớn hơn 5,5 (kiểm tra theo giấy chỉ thị vạn năng). Nếu chưa đạt giá trị mong muốn, có thể dùng natri xitrat để điều chỉnh thêm vào dung dịch phân tích lưu dung dịch muối nitrozo-R 0,05% rồi đun sôi. Nếu dung dịch phân tích có màu đỏ vàng thì thêm tiếp 1 ml dung dịch nitrozo-R nữa, 5ml nước cất hai lần rồi đun dung dịch đến sôi. Để dung dịch nguội, thêm vào 3ml hỗn

hợp H3PO4 và HNO3 (5 phần H3PO4 85% và 1 phần HNO3 đặc), khuấy dung dịch nhanh và cẩn thận. Chuyển dung dịch vào bình chia độ rối pha loãng đến 10ml. Dung dịch đo phức màu của phức co ban với muối nitrozo-R trong cuvet 2cm, tinh lọc màu xanh lá cây (536nm), dung dịch so sánh là nước cất hai lần.
Thang chuẩn được chuẩn bị như sau: Cân 0,477g CoSO4.7H2O cho vào cốc và hoà tan bằng nước cất hai lần. Chuyển dung dịch vào bình định mức 1 lít, axit hoá bằng 1 ml H2SO4 (d = 1,84) rồi thêm nước cất hai lần đến vạch mức.
Dung dịch nhận được chứa 100 μg Co/ml. Lấy 50ml dung dịch này pha loãng bằng nước cất hai lần đến thể tích 500 ml được dung dịch chứa long Co/ml, được dung dịch chuẩn sử dụng. Lấy các thể tích 0,5; l; 2; 3; 4; 5; 6; 7; 8; 9ml dung dịch chuẩn sử dụng cho vào bình đinh mức 100 ml. Thêm nước cất hai lần đến thể tích 50 ml, thêm 5ml dung dịch natrlxitrat 20% và 5ml dung dịch natriaxetat 40%, thêm loạn dung dịch muối nitrozo-R 0,05% rồi đun đến sôi. Để nguội, thêm vào 5ml hỗn hợp HNO3 và H3PO4, định mức đến vạch mức bằng nước cất hai lần. Tiến hành đo mật độ quang như đối với dung dịch phân tích.
Kết quả phân tích được tính theo công thức
Trong đó: X: hàm lượng co ban trong đất (mg/kg);
N: lượng đất tương ứng với thể tích dung dịch lấy để phân tích (g);
a: Lượng coban tìm thấy theo đồ thị chuẩn μg/ml;
b: thể tích dung dịch so màu (ml).
Hoá chất
Axit nước tinh khiết hoá học: Hút 68,9 ml HNO3 đặc (d = l,40) thêm nước cất hai lần đến thể tích 1 lít được dung dịch HNO3 1N.
- Natrixitrat 3 lần thế (dung dịch 20%).
- Natriaxetat 40%.
- Muối nitrozo-R (dung dịch nước 0,05%).
- Hỗn hợp axit nước và octhophotphoric: Hỗn 2 axit trên theo tỷ lệ 100 ml H3PO4 và 20ml HNO3.
- H2O2
7.3.7.5. Xác định molipden di động
Phương pháp so màu của Grigg (theo Dobritxcala - 1969)
Cân mẫu đất đã rây qua rây mĩm với lượng 15g đối đắt sét, 25g đối với đất cát và

cát pha; cho mẫu vào bình nút nhám, rót vào 150ml hay 250ml (tương ứng với lượng mẫu là 15g hay 25g) dung dịch đệm oxalat của Grigg có pH = 3,3, lắc dung dịch 1 giờ. Lọc dung dịch qua giấy lọc băng trắng, lấy 125ml hay 200ml dung dịch cho vào cốc chịu nhiệt rồi cho dung dịch bay hơi tới khi thề lịch dung dịch còn 1/4. Để phá huỷ các oxalat và các chất hữu cơ trong dịch lọc người ta thêm vào cốc 3ml H2O2 30% và 3 ml HNO3 đặc, tiếp tục chóng đến khô. Thêm vào 5ml nước cất hai lần rối chóng đến khô để tách hết vết axit pecloric. Thêm vào tiếp 5ml HCl 22% để chuyển muối sang dạng clorua rồi lại chung dung dịch đến khô. Hoà tan phân khô bằng cách đun nóng trong 1 1ml HCl 22%, chuyển dung dịch vào bình định mức 50ml, thêm vào 15ml dung dịch NaF 4% (để tạo phức với nhôm, sắt và than), 5ml dung dịch nam thuật 20% (để ngăn ngừa việc khử molipden đến hoá trị thấp hơn +5), thêm nước cất đến vạch mức, khuấy đều dung dịch. Chuyển toàn bộ dung dịch vào phễu chiết 100 ml, rửa thành bình bằng 2 - 3ml nước, rồi rót cả vào phễu chiết trên. Thêm vào phễu chiết 4ml kalithloxianat 20% (để tạo thành phức molipden thioxianat), sắc đều, cho vào phễu 3 - 4ml dung dịch SnCl2 1 0%, lắc phễu và sau khi màu đỏ nâu của phức sắt thioxianat hoàn toàn mất (Fe3+ chuyển thành Fe2+) thì cho vào phễu chiết 20ml dung dịch rượu n-butylic (đã được rửa được bằng nước và dung dịch thiếc clorua bão hoà) và để chiết phức molipden thioxianat cần lắc mạnh phễu chiết 1 phút. Để cho phân lớp rồi tạch lớp nước phía dưới phễu chiết ra nhưng giữ lại khoảng lưu lớp nước. Phần chiết trong phễu có thể lại có màu hồng do một phần Fe2+ đã được ôxy của không khí ôxy hoá chuyền thành Fe3+, vì thế cần phải rửa tiếp phần chiết bằng dung dịch muối thiếc bằng cách cho vào phễu chiết có phần chiết 20ml dung dịch muối thiếc mới pha (2ml SnCl2 10% + 18ml nước), lắc đều phễu chiết. Sau khô phần chiết hoàn toàn mất màu hồng thì tách bỏ lớp nước, còn phần chiết trong tướng hữu cơ cho vào cuvet 3cm. Thêm vào cuvet lậu rượu etylic để loại những giọt nước còn lẫn và 1 - 2 giọt dung dịch muối thiếc, quấy dung dịch trong cuvet cẩn thận bằng que thuỷ tinh nhỏ. Phần chiết lúc này sẽ trở nên trong suốt, không có màu hồng, đem đo mật độ quang của phần chiết với kính lọc màu xanh nước biển (435 nm) (chỉ tiến hành đo mật độ quang ít nhất là sau 20 - 30 phút kể từ khi thêm rượu n-butylic). Dung dịch so sánh là rượu n-butylic, không xử lý bằng thiếc và nước.
Xây dụng đường chuẩn
Cho vào các bình định mức 50ml các thể tích: 0,5; 1.0; 2.0; 3.0; 4.0 và 5.0ml dung dịch molipden chuẩn có hàm lượng μg/ml; thêm vào mỗi bình lúm HCl 6N, 3ml FeCl3 có hàm lượng 5 mgFe3+ 1 ml, khuấy cẩn thận các bình này rồi thêm nước cất đến vạch mức. Sau đó tiến hành như đối với dung dịch phân tích.
Hoá chất
- Dung dịch đệm oxalat: hoà tan 25 g amoni oxalat và 12,6 g axit oxalic trong nước cất hai lần khi đun nóng rồi thêm nước cất đến 1 lít.
- Axit clohidric 22% (6N, d = l,l): Cất hỗn hợp gồm 550ml HCl đặc và 450ml

nước cất hai lần trong dụng cụ cắt làm bằng thuỷ tinh, thu được 550ml HCl 22% - Thiếc (II) clorua 10%: Nung chảy thiết kim loại trong chén sứ và nghiền nhanh bằng chày sứ đến khi nhận được bột thiếc mịn. Cân 5,3 g bột này và hoà tan khi đun nóng trong 20ml HCl 6N. Dung dịch nên chuẩn bị trước khi dùng. Có thể chuẩn bị dung dịch này từ muối SnCl2 tinh khiết hoá học bằng cách hoà tan lớn muối này khi đun nóng trong 20ml HCl 6N, sau đó thêm một ít thiếc kim loại.
- Rượu n-butylic: Rửa n-butylic bằng nước cất hai lần để loại hết chất ôxy hoá có trong đó, cho 250ml rượu vào trong phễu chiết lớn, thêm 250ml nước cất hai lần, lắc 1 phút, sau khi tách tưởng, rót lớp nước phía dưới, còn lớp rượu tiến hành bão hoà bằng thiếc (II) clorua (cứ 100 ml rượu butylic thêm vào 10 SnCl2 10%, lắc 3 - 5 phút; thêm vào 5ml lượn etylic, khuấy đều).
- Dung dịch chuẩn molipden: Hoà tan muối amoni molipdat tinh khiết trong NH4OH trặc ích; đun nóng liên tục (ở mọi thời điểm dung dịch đều phải có phàn ứng kiềm), sau đó làm lạnh và kết tủa amoni molipdat bằng rượu etylic, cho kết tủa vào những tờ giấy lọc để làm khô muối.
Lấy 5 - 6g muối đã kết tinh cho vào chén sứ rồi nung trong lò nung ở nhiệt độ 450 - 480oC trong thời gian 40 - 60 phút. Cân 1,5g MoO3 thu được rồi hoà tan trong 10ml NaOH 1N, thêm vào dung dịch 10ml HCl 22%, dùng nước cất hai lần định mức đến 1 lít Ta được dung dịch chứa leng Mo/ml. Pha loãng dung dịch này 1000 lần được dung dịch chuẩn sử dụng.
Chú thích
+ Có thể tách chất hữu cơ ra khỏi dung dịch phân tích bằng cách chung thể tích mẫu đến khô trên bếp cách thuỷ, sau đó đặt chén sứ với phần khô vào trong lò nung ở 4500C trong 4 giờ.
+ Có thể dùng ete (tinh khiết hoá học) thay cho rượu n-butylic để chiết phức của molipden với thioxianat.
7.3.7.7. Xác định dạng di động của đồng, kẽm, coban, mangan khi chiết rút các nguyên tố này trong đất bằng dung dịch đệm amoni axetat
Phương pháp dựa trên sự chiết rút Cu, Co, Zn và Mà bằng dung dịch amoni axetat (pH = 4,8). Tỷ lệ giữa đất: dung dịch chiết rút là 1:5. Thời gian lắc trên máy lắc là 30 phút (Crupxky, 1994). Phương pháp này dùng cho đất cacbonat và không cacbonat.
a. Chuẩn bị dung dịch đệm
Dung dịch được chuẩn bị từ axit axetic và amoniac. Pha loãng 108ml axit axetic đặc (98%) bằng nước cất hai lần đến thể tích 600 - 700ml, thêm vào 75ml NH4OH 25%, khuấy đều; Sau khi nguội đưa thể tích dung dịch đến 1 lít. Kiểm tra pH dung dịch bằng pH mét. Axit axetic và amoniac dùng để chuẩn bị dung dịch đệm cần phải

tinh chế khỏi vết các kim loại nặng. Axit axetic được cất trong bình thuỷ tinh có sinh hàn nhám. Khi cất cần thêm một lượng vừa phải nam axetat khan vào bình cất. Nồng độ axit được kiểm tra theo tỷ trọng.
Để tinh chế amoniac khỏi vết các kim loại, người ta rót NH4OH 25% vào đáy bình hút ẩm lớn, phần trên bình hút âm đặt các cốc thuỷ tinh lớn đã được rót vào nước cất hai lần đến 1/2 thể tích cốc. Đậy nắp bình hút ẩm và giữ yên ít nhất là 48 giờ. Để nhận được amoniac có nồng độ cao hơn cần xử lý nước bão hoà amoniac một lần nữa bằng cách thay dung dịch amoniac trong bình hút ẩm bằng dung dịch amoniac 25% mới. Nếu xử lý nước bão hoà amoniac hai lần có thể nhận được amoniac có nồng độ 20%. Kiểm tra tỷ trọng dung dịch amoniac nhận được, tính lượng amoniac, axit axetic cần thiết đê pha dung dịch đệm theo tỷ lệ đã nêu trên.
b. Chiết rút các kim loại nặng
Cân trên cân phân tích 50g đất đã rây qua rây mỉm, cho lượng cân trên vào bình tam giác nút nhám thể tích 500ml, cho vào chính xác 250ml dung dịch đệm amoni axetat, lắc trên máy lắc 1 giờ. Lọc qua giấy lọc băng trắng. Lấy 150ml dung dịch lọc cho vào bát sứ, cho vào bát 10ml HNO3 (d = 1,4), 10ml H2O2 30%, rồi chưng dung dịch trong bát trên bếp cách thuỷ đến trạng thái sệt. Thêm vào 2ml HNO3, 2ml H2O2 rồi chưng đến khô. Hoà tan phần khô trong 50ml nước cất hai lần (có 3 - 4ml HCl cất lại hai lần) khi đun nóng. Lọc dung dịch thu được vào bình định mức 100 ml, rửa giấy lọc bằng nước cất hai lần đã được axit hoá bằng HCl. Dùng nước cất hai lần đưa thể tích dung dịch đến vạch mức và lấy dung dịch này để xác định các kim loại nặng. Nếu xác định Zn, cần chuẩn bị phần dịch chiết rút riêng trong dụng cụ thạch anh. Sau đó định lượng theo các phương pháp đã nêu.
7.3.7.8. Xác định hàm lượng Cu Pb, Zn di động trong dung dịch chiết rút amoni axetat theo phương pháp Von-Ampe hoà tan
Lấy 10g đất khô không khí cho vào bình tam giác 250ml, thêm vào 100 ml dung dịch đệm anloni axetat có pH = 4,8. Lắc trên máy lắc 1 giờ. Lọc dung dịch qua giấy lọc băng trắng Lấy 80ml dung dịch lọc cho vào cốc hay bát thạch anh, thêm vào 1ml HNO3 đặc và chưng trên bếp điện đến trạng thái sệt. Thêm vào 2ml HNO3 đặc nữa rồi lại chưng dung dịch trong cốc. Quá trình xử lý mẫu bằng HNO3 tiến hành đến khi phần còn lại chứa ít nước có màu trắng hay màu vàng sáng. Sau đó chúng lượng chứa trong cốc đến khô, "xới nhẹ" phần khô bằng HCl đặc rồi chúng dung dịch trong cốc đến trạng thái muối ẩm hoà tan muối khi đun nóng trong 20ml nước cất hai lần đã được axit hoá bằng HCl (1:100). Lọc dung dịch qua giấy lọc đã được rửa được bằng HCl loãng, hứng dung dịch lọc vào trong bình định mức 50ml. Rửa phần còn lại trên giấy lọc bằng nước cất nóng đã được axit hoá rồi đưa thể tích dung dịch đến vạch mức.
Để xác định đồng, kẽm, cờ người ta lấy 8ml dung dịch cho vào cốc dung tích 50 - 100ml, thêm vào 0,5ml dung dịch Cd (4μg/ml) dùng làm dung dịch chuẩn trong, một

lượng nhỏ axit ascorbic. Khuấy đều dung dịch đê hoà tan hoàn toàn axit ascothic. Thêm vào trong cốc l,5ml dung dịch đệm amoni axetat (NH4OH 1N; NH4Cl 2,7M, CH3COOH 2M). Chuyển dung dịch vào bình điện phân tạo giọt thuỷ ngân; cho hùng (hay nuôi qua dung dịch rồi tiến hành điện phân tại thế - 1,2 V trong 3 phút. Sau đó ngừng cho hidro qua; giữ yên dung dịch 30 giây, bắt đầu ghi đường biểu diễn quá trình hoà tan quật các kim loại đồng, chì, kẽm. Sau khi ghi được mỗi một lúc đều cần dừng không cho thế biến thiên trong một khoảng thời gian ngắn. Nồng độ của nguyên tố được xác định theo phương pháp đường chuẩn.
7.4. Sử dụng phương pháp quang phổ hấp thụ nguyên tử để xác định các kim loại nặng
Từ khi quang phổ hấp thụ nguyên từ hidro ra đời, nhiều nhà nghiên cứu đã vận dựng để xác định lượng vết các nguyên tố trong các đối tượng khác nhau.
L.A. Lemer và D.N. Ivanov (1970) đã xác định hàm lượng đồng, kẽm, mangan, coban trong các dung dịch chiết rút đất HCl 1N; HNO3 1N; HCl 0,1 N; HNO3 0,1 N; H2SO4 0,1 N; CH3COOH 0,5N; CH3COONH4 1N; dung dịch đệm amoni axetat (pH = 4,8).
Những số liệu nhận được chỉ ra rằng: có thể xác định trực tiếp Mn trong tất cả các dung dịch chiết rút trên; kẽm trong tất cả các dung dịch chiết rút trên trừ amoni axetat; đồng và co ban chỉ trong hai dung dịch chiết rút HCl 1N và HNO3 1N.
Khi dùng các dung dịch axit mạnh khác nhau về tính chất nhưng có cùng nồng độ đương lượng, để chiết rút các kim loại nặng thì kết quả xác định hàm lượng của chúng rất gần nhau.
Khi sử dụng dung dịch KCl 1N, chế độ chảy của ngọn lửa bị vi phạm nhiều.
Tỷ lệ giữa đất và dung dịch chiết rút là 1: 10, thời gian lắc là 1 giờ.
Dãy các mẫu đầu của các nguyên tố cần xác định được chuẩn bị bằng cách hoà tan các muối tinh khiết của các nguyên tố cần xác định trong các dung dịch chiết rút tương ứng. Đôi khi có thể tiến hành phân tích theo các mẫu đầu trong nước.
Độ nhạy khi xác định các nguyên tố như sau:
Bảng 7.18. Độ nhạy của phương pháp quang phổ hấp phụ nguyên tử khi xác định các kim loại nặng
Nguyên tố Vạch phân
tích (0A )
Dòng của đèn (μA)
Chiều rộng của khe
(μm)
Chiều cao của chùm sáng khi đi qua trên phần trên
của đèn khí (mm)
Độ nhay μg/ml/1%
Cu Co Zn Mn
3247,54 2407,25 2138,56 2794,82
8 20 10 14
50 25
300 100
2 2 2 2
0,05 0,1
0,02 0,07

Khi các điều kiện làm việc của các thiết bị đã ôn định thì qua vòi phun, đưa các dung dịch mẫu đầu và dung dịch phân tích vào trong ngọn lửa rồi xác định các giá trị mật độ quang D tương ứng.
Theo các giá trị mật độ quang thu được và nồng độ các mẫu đất tương ứng, vẽ đồ thị chuẩn trên hai trục; dựa vào giá trị mật độ quang đo được của mẫu phân tích và đường chuẩn người ta xác định được nồng độ của nguyên tố cần phân tích.
Khi xác định các nguyên tố có nồng độ cao thì mật độ quang tương ứng DL của các nguyên tố đó sẽ có giá trị cao hơn đáng kể so với giá trị mật động quang của sự hấp thụ không chọn lọc Di(DL ≈ Di), khi đó cần phải hiệu chinh kết quả giá trị sự hấp thụ không chọn lọc.
Sự hiệu chỉnh kết quả theo sự hấp thụ không chọn lọc được thực hiện như sau: Sau khi đo các giá trị mật độ quang (DL +i) theo các mạch hấp thụ của các nguyên tố tương ứng, tiến hành xác định giá trị mật độ quang DL, khi sử dụng giải phổ liên tục của đèn hiđro, hay là những vạch không hấp thụ của sắt (những vạch gần với các vạch phân tích) được phát ra từ đèn phóng điện kép nhiều nguyên tố Mật độ quang DL của nguyên tố cần xác định sẽ bằng DL = DL +i - Di. Sử dụng giá trị DL và đồ thị chuẩn đã vẽ xác định được nồng độ chưa biết. Sự hiệu chỉnh theo sự hấp thụ không chọn lọc gắn liền với việc thực hiện hai quá trình đo liên tục khi nghiêm ngặt tuân theo tất cả các điều kiện phân tích.

Chương 8
PHÂN TÍCH KHÍ
8.1. Giới thiệu chung
8.1.1. Các chất độc hại gây nhiễm môi trường không khí
Nguồn gốc các chất độc hại, gây ô nhiễm môi trường không khí là do sản xuất công nghiệp, nông nghiệp và do quá trình đốt nhiên liệu. Các chất độc hại đi vào cơ thể người qua đường hô hấp, tiêu hoá và qua da.
Chất độc hại trong không khí đi vào cơ thể người qua đường hô hấp là nguy hiểm nhất và thường gặp nhất. Nó xâm nhập qua phế quản và các tế bào đi vào máu.
Chất độc hại thấm qua da (chủ yếu là các chất có thể hoà tan trong mỡ và trong nước) vào máu như benzen, rượu etylic.
Dựa vào tác dụng chủ yếu của chất độc hại có thể chia ra các nhóm sau
Nhóm 1: Chất gây bỏng, kích thích da, niêm mạc ví dụ như a xít đặc, kiềm đặc và loãng (vôi tôi, amoniac).
Nhóm 2: Chất kích thích đường hô hấp trên như: Clo, NH3, SO3, SO2, NO, HCl, hơi flo.
Nhóm 3: Chất gây ngạt
Gây ngạt đơn thuần như CO2, etan, me tan...
Gây ngạt hoá học: Co hòa hợp với các chất khác làm mất khả năng vận chuyển oxy của hồng cầu làm rối loạn hô hấp.
Nhóm 4: Chất có tác dụng thần kinh trung ương, gây mê, gây tê như các loại rượu, các hợp chất hydro cacbua, H2S, Cs2, Xăng...
Nhóm 5. Chất gây độc
Chất gây độc tổn thương cơ thể: các loại hydro cacbưa, halogen, clorua metin, bromua metin...
Chất gây độc tổn thương cho hệ thống tạo máu như benzen, phenol, chì, asen. Các kim loại và á kim độc như: chì, thuỷ ngân, ma ngan, photpho, fluo, cadimi, hợp chất asen...
Độc tính của các chất ô nhiễm trong không khí có thể tham khảo trong rất nhiều tài liệu liên quan.
8.1.2. Phương pháp lấy mẫu khí
Tầng thấp nhất của khí quyển đến độ cao 11 khi được gọi là tầng đối lưu. Các khí

trộn vẫn trong tầng đổi lưu có thành phần tương đối ổn đinh và có ý nghĩa lớn đối với đời sống sinh vật ờ đây, sự vận chuyên không khí xảy ra mạnh và hình thành các đám mây được xem như là yếu tồ thời tiết có ảnh hường tới các hệ sinh thái của sinh quyển. Bằng việc sử dụng các dụng cự đặc biệt, người ta có thể xác định được các tính chất khác nhau của không khí. Những phương pháp sau đây đã được sử dụng để lấy mẫu không khí. Có hai nhóm phương pháp chính
a. Phương pháp tiếp cận lấy mẫu ướt
Sử dụng chai hoặc ống lấy mẫu được làm đầy bằng chất lỏng có khả năng hoà tan ít các chất khí. Trong nhiều trường hợp hay sử dụng nước đã được a xít hoá. Khi nước tháo đi. Không khí sẽ thâm nhập vào thay thế. Trong phòng thí nghiệm, mẫu không khí có thể chuyển sang dụng cụ phân tích bằng cách cho chất lỏng tương tự vào bình lấy mẫu.
Nhược điểm của phương pháp này là một số chất khí có thể hoà tan vào chất lòng
Ngược lại, chất lỏng cũng có thể bay hơi vào trong mẫu khí. Hơn nữa, khi lấy mẫu khỉ cũng như khi chuyền chất khí vào dụng cụ phân tích cần phải sử dụng lượng khá tìm các chất lỏng.
b. Phương pháp lấy mẫu khô
Trong phương pháp này, khí được lấy vào trong bình lấy mẫu nhờ hệ thống bơm hoặc máy hút cho đến khi thê tích trong bình được trao đổi với không khí ít nhất là 6 lần. Trong phòng thí nghiệm, không khí trong bình lấy mẫu được chuyển trực tiếp tới dụng cụ phân tích bằng chất lỏng (thích hợp nhất là dùng thủy ngân).

Thông thường, để kiểm tra những hợp phần đặc biệt như hơi cồn, hơi chất độc
trong không khí tại vùng công nghiệp, mẫu khí cỏ thể được đưa trực tiếp vào trong các dụng cụ đo tại vị trí cần xác định. Bản chất của phép đo là khác nhau từ đơn giản như ống hấp phụ với các chất đặc biệt để cho vùng màu sắc tương ứng với hàm lượng của chứng, đến dụng cụ đắt tiền để đo Co và SO2. Không khí cần phân tích được bơm qua bình phản ứng nới đã có sẵn các hoá chất cần thiết. Sản phản được hình thành có thể được xác định bằng phương pháp so màu hoặc một số kỹ thuật khác.
Để lấy một lượng nhỏ mẫu khí, có thể sử dụng bơm hút bằng nhựa có nắp đậy rất thuận tiện để lấy mẫu và chuyền mẫu khí vào dụng cụ đo.
Một số dụng cụ khác để lấy mẫu khí là một bình có thể bơm khí vào ở áp suất lớn hơn khí quyển. Do được nén ở áp suất lớn nên chúng dễ dàng chuyển sang dụng cụ đo. Tuy nhiên, do bị nén ở áp suất cao nên các khí có thể khuếch tán qua vỏ bình với các tốc độ khác nhau. Vì vậy, không nên để mẫu quá lau sẽ ảnh hưởng đến thành phần của chúng.
Đối với một chất khí nào đó có khả năng hoà tan tốt trong nước, có thể dùng nước để hấp thụ làm tăng hàm lượng của chứng cho quá trình phân tích. Thường sử dụng nước có thểm những chất thích hợp để tăng khả năng hấp thụ của chúng.
Khí được phân tích được lọc qua màng lọc xác định các phần tử rắn bằng kính hiển vi hay phương pháp hoá học.
Để quan trắc khói xả từ nhà máy hoặc xe cộ trong không khí việc phân tích cần tiến hành ở cả dòng đốt kiệt và tại ống sả của xe.

8.2. Phân tích khí
8.2.1. Xác định mã số tính chất vật lý của không khí
a. Cường độ gió: Gió được xem là yếu tố sinh thái quan trọng vì nó có khả năng làm thay đôi không khí và các yếu tố môi trường như nhiệt độ, ánh sáng, cân bằng nước... Gió cũng là yếu tố ảnh hưởng đến đời sống của thực vật cũng như động vật Bawfort đưa ra 12 cấp đánh giá dựa vào tốc độ gió như sau
Bảng 8.1. Phân loại cấp độ gió Cấp gió Loại gió Tốc độ gió (km/giờ)
0 Lặng gió (Calm) <1 1 Khi chuyển động nhẹ (Light air) 1-5 (3) 2 Gió rất nhẹ (Light breeze) 6-11 (9) 3 Gió nhẹ (Gentle breeze) 12-19 (16) 4 Gió mát (Fresh breeze) 20-28 (24) 5 Gió trung bình (Moderate breeze) 29-38 (34) 6 Gió mạnh (Strong breeze) 39-49 (44) 7 Gần lốc (Near gale) 50-61 (55) 8 Lốc (Gale) 62-74 (68) 9 Lốc mạnh (Strong gale) 75-88 (82) 10 Bão (Storm) 89-102 (96) 11 Bão mạnh (Violent storm) 103-117 (110) 12 Bão rất mạnh (Huricance) > 117
Có nhiều dụng cụ để đo tốc độ gió (được gọi chung là máy đo gió Anemometer).
Loại quay vòng: Gồm ba nửa hình cấu hướng gió và làm quay đồng hồ đo gắn ở trục xung quanh. Loại này được sử dụng phổ biến ở các trạm khí tượng. Tốc độ gió được tính bằng m/s hoặc km/h.
Loại đo gió dạng địa áp lực: Địa áp lực có đường kính khoảng 30cm. Khi gió tác động vào đĩa này sẽ chuyển thành sự chuyển động của tỷ lệ chia bảng chia độ.
b. Hướng gió: Hướng gió được xác định dựa vào địa bàn từ tính. Các hướng được chia làm 16 tỷ lệ theo hướng địa lý.
c. Áp suất khí quyển: Khi lên cao áp suất sẽ giảm, trung bình sẽ giảm đi 1/30 khi lên cao 275 m. Tuy nhiên, tỷ lệ và thành phần của khu quyền thì hầu như không thay đổi so với tầng gần mặt đất. áp suất không chi thay đổi theo độ cao mà còn thay đổi theo nhiệt độ. Tại độ cao 0 m (so với mặt biển trung bình) thì áp suất không khí và 10 13,25 milibar (760 mmHg). Do sự biến động của áp suất khi quyên giữa các vùng khác nhau nên hình thành sự chuyển động không khí trên mặt đất, áp suất khí quyển có liên quan tới nhiều yếu tố vật lý như thời tiết và khí hậu. Áp suất khí quyển cũng là một chỉ tiêu môi trường.

Để đo áp suất, người ta sử dụng máy đo (Barometer) dựa trên nguyên tắc nâng
cao hay hạ thấp của cột thuỷ ngân bên trong ống thuỷ tinh. Kết quả được biểu diễn bằng mmHg hoặc milibars.
d. Nhiệt độ: Nhiệt độ là yếu tố sinh thái được đề cập tới rất nhiều vì tác động quan trọng của nó tới các quá trình tổng họp vật chất, sinh trưởng và phát triền của sinh vật. Các sinh vật có khả năng thích ứng với biên độ dao động rộng của nhiệt độ được gọi là Eurythemlal và dao động hẹp được gọi là Stenothermal.
Nhiệt độ biến động mạnh trong môi trường không khí và nước có ảnh hưởng tới môi trường, nhất là độ ẩm và gió. Tuy nhiên dao động nhiệt theo ngày đêm, mùa có ý nghĩa quan trọng hơn trong sinh thái học so với các chỉ tiêu đơn lẻ.
e. Độ ẩm tương đối: Độ ẩm hiển thị hàm lượng hơi nước có trong không khí thông thường dao động trong khoảng 0,02 - 4% khối lượng. Độ ẩm phụ thuộc rất nhiều vào độ cao và nhiệt độ. Độ ẩm sẽ giảm dần khi lên cao gần như bằng không tại độ cao 8 - 10 km. Độ ẩm tuyệt đối là lượng hơi nước có mặt trong không khí (biểu thị bằng tỷ lệ và áp suất nào đó). Biên độ ẩm có ảnh hưởng đến quá trình bay hơi nước. Nó có ảnh hưởng đến nhiệt độ, ánh sáng và có vai trò quan trọng nhằm điều chính sự phân bố và hoạt động của sinh vật.
Để đo độ ầm tương đối người ta dừng máy đo độ ẩm loại quay số (Dial type hydrometer), loại giữ đồ thị (Hydrographs), hay bộ cảm ứng điện tử (Electrlcal sensing device). Tuy nhiên loại đo độ ẩm (Psychrometer) được sử dụng nhiều nhất.

g. Xác định hàm lượng bụi
1. Phương pháp trọng lượng xác định bụi lắng
Nguyên lý:
Phương pháp dựa trên sự cân dụng cụ hứng mẫu có phản ứng chất bắt dính trước và sau khi lấy mẫu để xác định lượng bụi lắng trong thời gian không mưa. Kết quả được biểu thị bằng g/m2 ngày hoặc mg/m2 ngày.
Dụng cụ:
Khay hứng mẫu: bằng nhôm hoặc thuỷ tinh - khay cũng có chiều dày 1 mm, chiều cao 11 mm, đường kính trong 85 mm, diện tích 57 cm2 được bồi một lớp vazơlin với khối lượng trong khoảng 50 mm - 60 mm; đã sấy trong tủ sấy từ 5 - 10 phút ở nhiệt độ 400C để tạo mặt bằng đều trên khay. Xác định trọng lượng khay trước khi hứng mẫu (m1).
Khay được đậy nắp, cho vào túi PE, xếp trong hộp bào quản
Lấy mẫu:
Khay lấy mẫu bụi lắng khô được đặt trên các giá ở độ cao đồng nhất cách mặt đất 1,5 hoặc 3,5 m.
Điểm lấy mẫu phải bố trí ở nơi thoáng gió từ mọi phía, khoảng cách giữa các điểm lấy mẫu với các vật cân (nhà cao tầng, cây cao,...) phải đảm bảo sao cho góc tạo thành giữa đỉnh của vật cản với điểm đo và mặt nằm ngang không lớn hơn 300.
Số lượng mẫu, sự phân bố các điểm lấy mẫu trong khu vực quan tâm được xác định theo yêu cầu cụ thể nhưng không ít hơn 4 mẫu cho mỗi điểm đo.
Thời gian hứng mỗi mẫu bụi lắng khô ở khu công nghiệp, dân cư tập trung không ít hơn 24 giờ nhưng không quá 7 ngày.

Xử lý mẫu:
Dùng khăn sạch lau cẩn thận bên ngoài khay, sau đó đặt vào tủ sẩy, sấy ở nhiệt độ 400C trong 2 giờ.
Sau khi sấy, cân khay hứng trên cần phân tích với độ chính xác ± 0,1 mg (m2).
Tính toán kết quả:
Trong đó: m1, m2: kết quả cân khay trước và sau khi hứng mẫu (g, mg)
S: diện tích hứng mẫu (m2)
T: thời gian hứng mẫu, ngày (24 giờ)
2. Phương pháp xác định lượng bụi lắng tổng số
Nguyên lý:
Phương pháp dựa trên việc cân trọng lượng bụi thu được trong bệnh hứng mẫu bao gồm dạng hoà tan trong nước. Sử dụng để xác định lượng bởi lắng tổng cộng tháng, kết quả được biểu thị bằng g/m2 hoặc m/km2.
Dụng cụ:
Dụng cụ lấy mẫu: Bình hứng mẫu hình trụ, đáy phẳng, đường kính không nhỏ hơn 12 cm, chiều dày khoảng nhỏ hơn 2 lần đường kính miệng. Chiều dày của thành bình không quá 3 lắm. Có thể sử dụng bình thuỷ tinh hoặc kim loại không gỉ.
Hoá chất:

Hoá chất chống tảo, nấm... clorofom hoặc H2O2 tinh kiết.
- Nước cất hai lần.
Lấy mẫu:
Vị trí đặt thêm hứng mẫu như phương pháp trên
Thời gian lấy mẫu là 10 ngày hoặc 30 ngày.
Trước khi lấy mẫu được rửa sạch, tráng nước cất, đậy nắp.
Trước khi đặt lấy mẫu cho vào bình hứng 250 ml nước cất, 2 - 4 ml hoá chất chống tảo nấm
Trong thời gian lấy mẫu, cần bổ sung lượng nước cất đề giữ mẫu, tránh bình hứng bị khô. Trong quá trình hứng mẫu, nếu lượng mưa hứng được trung bình đạt độ cao 2/3 độ cao bình hứng, thay bình hứng khác đê hứng mẫu tiếp, các mẫu này tại mỗi điểm trong cùng một khoảng thời gian lấy mẫu (10 hoặc 30 ngày) có thể xử lý riêng biệt rồi cộng gộp kết quả.
Cách tiến hành:
Xác định các chất không hoà tan trong nước (ml):
- Loại bỏ các vật ngoại lai không mang tính chất bụi: có thể dùng tranh đề gắp các vật ngoại lai có kích thước lớn hơn 1 mm, trước khi bỏ các vật này cần tráng qua với nước cất, nước cất thu được nhập chung vào một mẫu. Hoặc có thể lọc qua rây 1 mm, dùng nước cất đê rửa bình hứng cho qua rây và nhập nước này và mẫu chung.
- Lọc toàn bộ mẫu qua phễu lọc thuỷ tinh xốp, sạch, khô đã biết trước khối lượng.
- Dùng nước cất rửa cận trên phễu, nước này nhập vào mẫu chung.
- Sấy khô phễu lọc thuỷ tinh xốp với cặn ở 1050C trong 2 giờ làm nguội trong bình hút ẩm, cân với độ chính xác ± 0,1 mg.
Hiệu của hai lần cân phễu thuỷ tinh xốp trước và sau khi lọc và lượng chất không tan trong nước, ml.
Xác định lượng chất tan trong nước m2:
- Xác định toàn bộ thể tích của mẫu sau khi lọc bằng ống đong: V ml
- Lấy đại diện 250 ml dung dịch mẫu lọc cho vào cốc sứ, cho bốc hơi hết trên bếp cách thuỷ, sau đó sấy khô ở 1050C trong 2 giờ, để cho nguội trong bình hút ẩm rồi cân trên cân phân tích với độ chính xác ± 0,1 mg.
- Sấy đến khối lượng không đổi.
- Tổng lượng chất hoà tan trong nước:

- Lượng bụi lắng tổng số tháng tính bằng g/m2 hay mg/m2:
Trong đó: S: là diện tích miệng bình hứng m2
Trong trường hợp lấy mẫu 10 ngày, lượng bụi lắng tháng là tông của 3 kết quả bụi lắng trong 10 ngày trong tháng đó
8.2.2. Xác định một số tính chất hoá học của không khí
a. Xác định NO và NO2
Nitơ oxít và nitơ đioxít được xác định bằng phương pháp gián tiếp như sau. Dùng dung dịch pemanganat xử lý hỗn hợp chứa các khi oặt nitơ. Khi đó nitơ oxít và nitơ đioxít được oxy hoá thành nitrat.
Phản ứng hoá học diễn ra như sau:
Lượng nitrat tạo ra tương ứng với hàm lượng toàn phần của NO và NO2. Lượng
pemanganat đã được dùng để oxy hoá các NO và NO2 thành NO3 được xác định bằng cách thêm vào một lượng dư Fe(II) sunphát, sau đó chuẩn lượng dư đó bằng dung dịch Kali pemanganat, còn hàm lượng thuật thì chuẩn tiếp theo bằng dung dịch Fe(II) sunphat trong môi trường a xít H2SO4 đậm đặc. Xác định điểm tương đương theo phản ứng toà thành sản phần kết hợp Fe2(SO4)3.2NO là cho dung dịch có màu hồng.
Tiến hành xác định
Việc xác định lượng tiến hành trong pipet đã được chuẩn hoá và có cấu trúc đặc biệt. Rút chân không trong pipet trước, rui đưa qua phễu vào đó 25ml dung dịch kali pemanganat 0,5N và tráng phễu bằng một lượng nước đo trước. Sau đó cho hỗn hợp khí khảo sát và pipet qua một khoá 3 nhánh, cân bằng nhiệt độ và áp suất, lắc mạnh pipet cho vào 25 ml dung dịch Fe(n) sunphat 0,5N và một ít nước (để tráng phễu) và lắc pipét trong vài phút. Hút chất lỏng đã bị mất màu ra khỏi pipet qua khoá 3 nhánh vào bình hình nón và tráng kỹ pipet bằng nước cất. Chuẩn độ lượng Fe(II) sunphat dư bằng dung dịch kali pemanganat 0,5N. Như vậy xác định được lượng dung dịch kali pemanganat tiêu tốn để oxy hoá NO và NO2.
Muốn xác định hàm lượng toàn phần của Nitơ đã chuyển thành nitrat người ta chuyển toàn bộ dung dịch cần chuẩn độ vào bình định mức dung tích 500 ml, tráng

bằng một lượng nhỏ nước cất. Rồi vừa cẩn thận thêm lượng nhỏ axít sunphuric đậm đặc, vừa liên tục làm lạnh bình bằng hỗn hợp nước và nước đá, dùng a xít sunphunc đưa thể tích dung dịch lên đến vạch mức. Dùng pipet hút 2 mẫu, mỗi mẫu 100 ml dung dịch thu được vào một hình nón có dung tính 500 ml. Thêm vài tinh thể kim bicromat (để giải phóng ôxy hoà tan) và vừa liên tục xoay bình vừa chuẩn bằng dung dịch Fe(II) sunphat 0,5N. Đầu tiên dung dịch không màu, rồi có màu lục hơi vàng nhạt và tại điểm tương đương có màu hồng. Lượng dư của Fe(II) sunphat tạo cho dung dịch màu đỏ tía đậm.
Tính kết quả
Theo những phương trình phản ứng trình bày ở trên, lượng chống của khí NO và NO2 tương đương với lượng thuật tạo thành. Dựa theo những phản ứng ở trên để xác định thể tích của NO và NO2 ta có phương trình sau:
Trong đó: N: lượng chung của các khí NO và NO2 (ml)
P: lượng dung dịch kim pemanganat 0,5N dùng để oxy hoá (ml).
Trong chuẩn độ lượng thuật bằng phép chuẩn tiếp theo với Fe(II) sunphat, 1 ml dung dịch Fe(n) sunphat 0,5N tương ứng 5,6 ml khí trên.
b. Xác định hàm lượng lưu huỳnh dioxit (SO2)
Xác định lưu huỳnh theo phương pháp trắc quang thorin
Phương pháp này được dùng để xác định nồng độ SO2 trong không khí xung quanh ở các khoảng nồng độ 3,5 - 150 μg/m3. Với thể tích lấy mẫu 2m3 và thể tích dung dịch là 50 ml. Với những nồng độ cao hơn, phải pha loãng trước khi xác định.
Nguyên lý
Cho mẫu không khí đi qua dung dịch H2O2 (đã được axít hoá) trong một thời gian quy định. Lượng SO2 có trong mẫu sẽ được hấp thụ và oxy hoá thành SO4
2- . Kết tủa SO4
2- dưới dạng BaSO4 bằng một lượng Ba(ClO4)2 dư. Bari dư được xác định bằng cách tạo phức màu với thorin và đo ở bước sóng 520 nm bằng máy so màu.
Hoá chất
- H2O2 27 - 30%
- Dung dịch hấp thụ: Pha loãng 10 ml dung dịch H2O2 27% vi nước thành 1 lít.
- Điều chỉnh pH dung dịch về khoảng 4- 4,5 bằng dung dịch HClO4, dùng điện cực pH để kiểm tra.
- Dung dịch Bari peclorat: Hoà tan 0,252 g Ba(ClO4)2 trong một lượng nhỏ HClO4 0,1 M trong bình định mức 250 ml rồi định mức đến vạch bằng axít HClO4

0,01 M.
- Bari peclorat/dioxan: Cho 10 ml dung dịch ban peclorat và 40 ml nước vào bình định mức 1000 ml, thêm dioxan đến vạch mức và trộn đều.
- Dung dịch H2SO4 0,5: Từ ống nghiệm tiêu chuẩn pha thành 1 lít dung dịch.
- Thorin 0,25%: Hoà tan 0,25 g thorin trong 40 ml dung dịch sunphat tiêu chuẩn và thêm nước cất đến thể tích dung dịch 100 ml.
Quá trình xác định
Từ bình hấp thụ lấy ra 5 ml dung dịch, trộn đều với 10 ml đung dịch ban peclorat/dioxan và 0,25 ml thorin. Khi cường độ màu của dung dịch tỷ lệ với lượng ban dư và tỳ lệ nghịch với lượng lớn SO4
2- có trong mẫu. Dựa theo đường chuẩn để tính hàm lượng SO2 có trong mẫu.
Trong khi tiến hành, thorin có thể tạo phức với nhiều ton kim loại làm cản trở phép xác định do đó việc lọc bụi ờ giai đoạn thu mẫu có thể loại bỏ được ảnh hưởng này. Sự có mặt của PO4
3- sẽ tạo thành kết tủa với Ba3(PO4)2 làm cản trở phép xác định. Khi hàm lượng SO2 lớn, đặc biệt ở những nguồn thải tĩnh, có thể dùng phương pháp H2O2/Ba(ClO4)2/thorin thay bằng phương pháp trắc quang thorin. Trong phương pháp này người ta chuẩn độ màu bằng dung dịch Ba(ClO4)2 với chỉ thị thorin. Tại điểm cuối của quá trình chuẩn độ, dung dịch xuất hiện màu hồng.
c. Xác định CO bằng phương pháp sắc ký
Nguyên lý
Cho một thể tích mẫu không khí đi qua cột sắc ký để tách CO ra khỏi các thành phần khí khác. Khử CO vừa tách ra thành me tan bằng dòng khí hydro với xúc tác niken nóng (có thể dùng các chất xúc tác và chất hỗ trợ khác).
Tín hiệu sắc ký nhân được tỷ lệ với lượng Co có trong mẫu.
Hoá chất và thiết bị
- Các khí:
Hydro được sử dụng là khi mang trong tách sắc ký khí, làm chất khử Co thành metan và cũng cần cho hoạt động của detector ngọn lửa ion hoá.
Nitơ, heli: Đối với một số đầu đốt, khí nhở hoặc hai hoặc hỗn hợp cả hai khí này được thêm vào khí đốt để tăng độ nhạy và độ ổn định.
Các khí metan và Co chuẩn
Cột sắc ký: Dùng cột sắc ký để tách Co ra khỏi các thành phần khác có trong mẫu.

Điều quan trọng là nguyên liệu nhồi cột không thưởng xuyên giữ nước vì cột sẽ được rửa ngược giữa các lần phun mẫu
Quy trình tiến hành
Trong khi tiến hành cần chú ý những điểm sau:
- Khoảng những nồng độ dự kiếm
- Tín hiệu sắc ký và nồng độ tối thiểu có thể phát hiện so với tín hiệu nhiễm;
- Số mẫu gián đoạn được đo trong một đơn vị thời gian;
- Độ trôi của "điểm không" và thời gian trôi;
- Độ lặp lại của phép đo;
- Hiệu suất chuyển hoá;
- Khoảng nhiệt độ ở đó các mẫu sẽ được lấy.
1. Chuẩn bị xúc tác: Bão hoà niken nitrat hecxahiđrat (Ni(NO3)2.6H2O) trong 25 ml nước cất, lọc bỏ phần không tan. Ngâm 10 g diatomit có kích cỡ từ 0,125 - 0,15 mm trong dung dịch này, lọc chân không và làm khô qua đêm ở nhiệt độ 1100C. Nung trong không khí 5 giờ ở nhiệt độ 5000C.
2. Chuẩn bị ống chuyển hoá: Nhồi diatomit đã xử lý với niken nitrat vào một ống thép không gỉ.
3. Lò chuyển hoá chất: Lắp ráp hệ thống lấy mẫu, ống chuyền hoá vào máy sắc ký khí. Điều chỉnh hệ thống sao cho nó có thể hoạt động theo cách thức đường vòng. Trong giai đoạn này nút điêu kiện tự động và bộ ghi đều không hoạt động. Bật máy sắc ký và điều chỉnh các chỉ dẫn của nhà sản xuất.
Điêu chỉnh nhiệt độ lò chuyển hoá đến 3500C và vận hành hệ thống trong 10 giờ để khử các niken oxít thành niken kim loại.
4. Xác định: Dùng bơm lấy mẫu hút liên tục khổ qua vòng mẫu và đường vòng của nó. Lưu lượng của dòng khí mẫu (khoảng 100 cm3/giây) đủ để làm sạch và làm đầy vòng mẫu. Vào những thời điểm thích hợp trong chương trình lấy mẫu, khí chứa trong vòng mẫu sẽ được phun tự động vào hệ thống sắc ký đê đo hàm lượng CO.
Kết quả
Nồng độ Co được xác định bằng cách so sánh các diện tích peak thu được từ các hỗn hợp khí chuẩn và mẫu.
Nồng độ Co được tính chuyển tù mg/m3 thành ppm như sau:
- Ở nhiệt độ 250C và áp suất 101,3 kPa:
1 ppm ≥ 1,14 mg/m3
- Ở các nhiệt độ khác với áp suất 101,3 kPa:

Trong đó từ nhiệt độ tính bằng 0C.
d. Phương pháp indophenol xác định hàm lượng amoniac
Nguyên lý
Phương pháp dựa trên cơ sở tác dụng của amoniac với hipoclorit và phenol có sự tham gia của chất ổn định phản ứng là natri nitro pruxit, tạo ra hợp chất màu xanh, hấp phụ cực đại tại bước sóng 625 nm.
Phương pháp này được sử dụng để xác định hàm lượng amoniac trong không khí từng lần vào trung bình ngày đêm trong khoảng từ 0,1 đến 1,0 mg/m3
Các amin thơm và fomadehit gây cản trở phép xác định.
Dụng cụ
- Máy hút khí có lưu lượng kế có vạch chia đến 0,005 lít/phút; nhiệt kế, áp kế, ẩm
- Bình hấp thụ Ricte có vạch đo 6 ml hoặc dụng cụ hấp thụ khác.
- Máy so màu có kính lọc màu 625 mm, hoặc máy quang phổ, cuvet có bề dày khám.
- Ống nghiệm có nút mài dung tích 100 ml.
- Phễu Bucne.
Hoá chất và thuốc thử
- Sử dụng loại tinh khiết hoá học hay loại tinh khiết để phân tích.
- NaOH
- KI - dung dịch 10%
- NH4Cl
- Natri nitro pruxit Na2[Fe(CN)5]NO
- NaCO3
- H2SO4 (d= 1,18) trung dịch 10% theo thể tích
- NaCl
- Axít salixilic [HO(C6H4)COOH]
- Natri thiosunphat: Na2S2O3.5H2O dung dịch 0,05 mol/1
- A xít oxalic H2C2O4
- Phenol C6H5OH
- Corua vôi CaOCl2

- Nước cất hai lần đã khử amoniac
Chuẩn bị hoá chất
- Dụng cụ hấp thụ: pha loãng 0,5 ml H2SO4 (d= 1,84) trong 1 lít nước cất. Bảo quản dung dịch trong bình hình ống trụ đậy kín bằng nút có ống thuỷ tinh chứa đầy tinh thể axít oxalic.
Thuốc thử phenol: Hoà tan 5 g phenol mới chưng cất, 25 mg natri nitro pruxit trong 100 ml nước. Bão hoà thuốc thử không quá 6 tháng ở 40C.
Hồ tinh bột, dung dịch 0,5%: trộn đều 0,25 g tinh bột với 10 ml nước, cho thêm 40 ml nước nóng 60-700C dung đến sôi, để dung dịch sau 1 phút làm lạnh
- Thuốc thử hipoclorit: Hoà tan 10 g NaOH và 11,7 g NaCl trong 100 ml nước đã qua bão hoà với Clo (nồng độ từ 0,6 - 0,8 g Clo). Thuốc thử được bảo quản không quá 6 tháng. Thuốc thử phải chứa đủ lượng chỉ từ 0,6 đến 0,8 g chỉ trong 100 ml.
Tiến hành xác định hàm lương chỉ hoạt tính: Lấy 20 ml dung dịch thuốc thử cho vào bình cầu có nút mài, thêm 10 ml H2SO4 10%, 10 ml KI 10%. Đậy kín bình cầu và để trong bóng tối khoảng 10 phút, sau đó chuẩn độ I2 giải phóng bằng Na2S2O3 0,05 mol/l đến vàng nhạt. Thêm vào vài giọt hồ tinh bột, chuẩn tiếp bằng Na2S2O3 đến khi dung dịch mất mầu; 1 ml dung dịch natri thiosunphat 0,05 mol/l tương ứng với 0,00354 g clo.
Có thể dùng clorua vôi để chuẩn bị thuốc thử
Dung dịch chuẩn amoniac gốc: 100 μg amoniac trong 1 lít; hòa tan 0,3141 g NH4Cl trong 1 lít nước. Bảo quản dung dịch không quá hai tháng.
Dung dịch chuẩn amoniac sử dụng có hàm lượng 1 μ amoniac trong 1 ml. Dung dịch này được chuẩn bị bằng cách pha loãng dung dịch chuẩn gốc.
Chỉ pha loãng dung đình chuẩn trước khi dùng.
Lấy mẫu
Để xác định hàm lượng từng lần amoniac, cho không khí cần nghiên cứu đi qua hai ống hấp thụ mắc nối tiếp nhau, mỗi ống chứa 5 ml dung dịch hấp thụ với lưu lượng 0,51/phút, liên tục trong thời gian từ 10 đến 30 phút
Để xác định hàm lượng amoniac trưng bình ngày đêm có thể lấy mẫu theo hai cách:
- Lấy mẫu như xác định hàm lượng từng lần amoniac với số lan không ít hơn 6, cách nhau đều trong một ngày đêm
- Cho không khí (trong mẫu) cần nghiên cứu đi qua dung dịch hấp thụ có chứa 50 ml dung dịch hấp thụ với lưu lượng 0,2 lít/phút liên tục trong 24 giờ.
Phải theo dõi quá trình lấy mẫu, nếu dung dịch bị cạn cần bổ sung thêm nước cất

Tiến hành phân tích
- Dùng nước cất hai lần thêm vào dung dịch trong bình hấp thụ đến 10 mg.
- Hút 2 ml dung dịch mẫu thử cho vào ống nghiệm nút mài, thêm 3 ml dung dịch hấp thụ, 1 mg thuốc thử phenol. Sau đó vừa lắc.
- Sau 2 giờ tiến hành đo mật độ quang của dung dịch, sử dụng cuvet 1 cm, bước sóng 625 nm, dung dịch so sánh là nước cất.
Thời gian cho thuốc thử cuối cùng đến lúc đo mật độ quang của dung dịch đối với tất cả các mẫu phải như sau.
- Chuẩn bị mẫu trắng từ dung dịch hấp thụ; đo mật độ quang của 5 ml dung dịch thuốc thử đã điều chế thành "mẫu trắng"; giá trị mật độ quang của mẫu trắng không vượt quá 0,02. Nếu quá kiểm tra độ sạch của nước, dụng cụ, cuvet và chất lượng dung dịch.
Lượng NH3 trong mẫu thử được xác định bằng phương pháp đường chuẩn theo hiệu số giữa mật độ quang của mẫu đem đi phân tích và mật độ quang của mẫu trắng. Lấy 5 ml dung dịch từ mỗi bình trên ống nghiệm nút mài rồi tiến hành làm như dung dịch thí nghiệm
Tính kết quả
Hàm lượng amoniac (C) trong không khí được tính bằng mg/m3 theo công thức:
Trong đó: a: lượng NH3 trong dung dịch mẫu lấy đê phân tích(mg);
B: tổng lượng dung dịch mẫu thử (ml);
Ba: thể tích dung dịch mẫu thử lấy để phân tích (ml);
Vo: thể tích mẫu không khí đã đưa về điều kiện tiêu chuẩn (m3)
Khi xác định nồng độ trung bình ngày đêm thì thể tích không khí không cần đưa về điều kiện tiêu chuẩn.

Chương 9
CÁC THÔNG SỐ Ô NHIẺM CẦN KIỂM SOÁT
9.1. Khái niệm chung
Ô nhiễm môi trường (ONMT)
Ô nhiễm môi trường là sự làm thay đổi trực tiếp hoặc gián tiếp các tính chất vật lý, hoá học, sinh học của bất kỳ thành phần môi trường nào làm cho tiêu chuẩn chất lượng của thành phần môi trường đó bị vi phạm dẫn đến làm nguy hại hoặc có khả năng nguy hại cho môi trường và sức khoẻ con người (Cục Môi trường, 12/2000).
Kiểm soát ô nhiễm môi trường (KSONMT)
Kiểm soát ô nhiễm môi trường (KSONMT) là sự tổng hợp các hoạt động, hành động, biện pháp và công cụ nhằm phòng ngừa, khống chế không cho sự ô nhiễm xảy ra, hoặc khi có sự ô nhiễm xảy ra thì có thể chủ động xử lý, làm giảm thiêu hay loại trừ được nó (Cục Môi trường, 2000).
9.2. Các nguyên tắc chung trong kiểm soát ô nhiễm môi trường:
Kiểm soát ô nhiễm môi trường ở bất kỳ một lĩnh vực, đối tượng nào cũng đều phải tuân thủ những nguyên tắc chính sau:
- Đảm bảo sự phát triển kinh tế - xã hội một cách bền vững.
- Đảm bảo tính lồng ghép: đối với các nhà quản lý, một trong những trọng tâm quan trọng đối với công tác lồng ghép là cách tiếp cận liên ngành đối với công tác kiểm soát ô nhiễm. Ngoài ra, nhu cầu lồng ghép xuyên suốt các trách nhiệm quản lý môi trường nhằm đảm bảo tập trung được nguồn lực về con người, công nghệ và tài chính cho các vấn đề được ưu tiên. Lồng ghép các khu vực, các ngành, các đối tượng kiểm soát ô nhiễm với nhau sẽ tạo cơ hội thuận lợi trong công tác kiềm soát ô nhiễm, nhất là trong khâu tổng hợp, xử lý mang tính khu vực tìm ra các quy luật chung nhất.
Ngăn ngừa ô nhiễm môi trường là nguyên tắc chu đạo trong công tác kiểm soát ô nhiễm. Khắc phục và phục hồi là quan trọng; Các tiêu chuẩn môi trường, chất thải và tiêu chuẩn sử dụng công nghệ là chỗ dựa và căn cứ chính.
- Một số lưu ý cụ thể:
Phải sử dụng các thông số và chỉ thị môi trường để kiểm soát ô nhiễm.
Phải thực hiện đánh giá hiện trạng và tác động môi trường: phạm vi, các bước tiến hành.
Các tiêu chuẩn về môi trường tiêu chuẩn xạ hay phát thái (emission oi emuent standards); tiêu chuẩn chất lượng môi trường bao quanh hay tiêu chuẩn chất lượng của nguồn tiếp nhận chất thải (ambient standards).

Các nguyên tắc giảm thiểu ngay từ nguồn phát sinh, sử dụng lại chất thải: việc giảm thiểu lượng chất thải là một hướng rất quan trọng, có ý nghĩa rất lớn đối với các chất thải, đặc biệt những chất thải bền vững khó phân huỷ và những chất nguy hại. Vì vậy việc giảm thiểu và sử dụng lại các chất thải là những việc làm rất cần thiết nhưng đồng thời là những việc làm rất phức tạp.
Quan trắc chất lượng môi trường (Monitoring Environmental Quality).
Để xác định hiệu quả của các hoạt động cải thiện chất lượng môi trường cần thiết phải lượng hoá chất lượng môi trường theo các thông số môi trường: các số liệu về nguồn thải và lượng thải các chất ô nhiễm; các thông số cần quan trắc, tập trung chủ yếu vào quan trắc các nguồn thải gây ô nhiễm môi trường.
9.3. Các thông số chất lượng hay ô nhiễm môi trường cần kiểm soát
9.3.1. Các chất ô nhiễm môi trường
Các chất gây ô nhiễm là những chất không có trong tự nhiên hoặc vốn có trong tự nhiên nhưng tới một thời điểm nào đó có hàm lượng lớn hơn và gây tác động có hại cho môi trường tự nhiên, cho con người cũng như sinh vật sống. Chất gây ô nhiễm có thể do hoạt động của con người (chất thải công nghiệp và sinh hoạt) hoặc do các hiện tượng tự nhiên gây ra. Trong môi trường, cấu trúc hoá học cũng như nồng độ của nó sẽ quyết định mức độ ô nhiễm của môi trường nước, đất và không khí.
Việc xác định các chất ô nhiễm rất quan trọng. Đó là cơ sở cho việc xác định nguồn gốc và mức độ ô nhiễm môi trường. Theo phương thức xuất hiện, chất ô nhiễm được phân loại thành chất ô nhiễm sơ cấp (xâm nhập trực tiếp tù nguồn phát sinh) và ô nhiễm thứ cấp (là những chất ô nhiễm được hình thành từ ô nhiễm sơ cấp trong điều kiện tự nhiên). Tuy nhiên để tiện lợi cho quá trình kiểm soát ô nhiễm, việc phân loại các chất ô nhiễm được xác định dựa vào đặc tính của chúng theo các nhóm như sau:
9.3.1.1. Đối với môi trường nước và đất
a. Các chất hữu cơ
Đây là những chất tiêu thụ ôxy trong môi trường nước. Do đặc tính không bền, chúng có xu hướng bị ôxy hoá thành các dạng đơn giản hơn Quá trình này ảnh hưởng trực tiếp đến độ hoà tan ôxy trong nước (DO). Ngoài ra các chỉ tiêu khác mưu BOD (nhu cầu ôxy sinh hoá) và COD (nhu cầu ôxy hoá học) được sử dụng để đánh giá mức độ ô nhiễm hữu cơ trong nước.
Nguồn gốc các chất ô nhiễm dạng hữu cơ có thể từ các dạng sau:
- Nước thải sinh hoạt: Hầu hết các chất hữu cơ dạng này đều có khả năng phân huỷ sinh học. Là nguyên nhân chính gây ra hiện tượng thiếu hụt ôxy trong nước.
- Nước cuốn trôi bề mặt: Thành phần hợp chất hữu cơ rất đa dạng, tuỳ thuộc vào đặc tính bề mặt.

- Sinh ra do quá trình phát triển - chết của động thực vật phù du, động thực vật đáy.
Đây là nguồn phát sinh đáng kể trong các lưu vực giàu chất dinh dưỡng.
- Từ hoạt động động sản xuất hoặc sản phẩm của công nghiệp: Thông thường các chất hữu cơ dạng này bền, khả năng phân huỷ sinh học thấp, gây ô nhiễm nặng nề cho các nguồn nước. Đó là các chất nhiên liệu, chất dẻo, chất màu, thuốc trừ sâu, phở gia dược phẩm thực phẩm mà nguồn gốc từ các nhà máy thực phẩm, giấy, thuộc da, đồ hộp, hoá chất, hoặc do tưới tiêu
* Một số chất hữu cơ tổng hợp điển hình tồn tại trong ty nhiên gây ô nhiễm môi trường nước Đó là:
Hoá chất bảo vệ thực vật: Bao gồm thuốc trừ sâu, thuốc diệt cỏ, thuốc diệt nấm mốc, diệt loài gặm nhấm, thuốc trừ côn trùng. Theo quan điểm hoá học các chất bảo vệ thực vật được phân thành các dạng sau:
- Hợp chất hữu cơ halogen
- Hợp chất hữu cơ phối pho
- Các cacbamat
- Các clorophennoxyaxit
Các chất bảo vệ thực vật thâm nhập vào cơ thể con người thông qua quá trình phát tán trong nước hoặc do tồn lưu sinh vật, sau khi sinh vật chết bị cuốn trôi theo nước. Chúng được tích tụ trong chuỗi thức ăn mà mắt xích cuối cùng là con người. Chất bảo vệ thực vật có trong nước sẽ tác động trực tiếp đến quá trình phát triển của sinh vật, thay đổi cấu trúc sinh học, gây ra các các bệnh lý như ung thư, quái thai...
* Xà phòng, các chất tẩy rửa và phụ gia
Là những nguồn tiềm tàng các chất ô nhiễm dạng hữu cơ do có khả năng tạo nhũ tương tạo các chất hữu cơ lơ lừng trong nước. Trong quá trình này các anion tạo ra các mixel xà phòng dạng keo
* Các chất hữu cơ tổng hợp khác.
Tất cả các chất hữu cư có trong nước đều là những chất tiêu thụ ôxy do đặc tính không bền và có xu hưởng ôxy hoá thành chất đơn giải. Trong nước khi chỉ số DO thấp, BOD và COD cao chứng tỏ nước bị ô nhiễm nặng bởi các chất hữu cơ tiêu thụ ôxy. Một số dạng đặc trưng của chất hữu cơ điển hình trong đất, nước cần quan tâm như bảng 9.l:

Bảng 9.1. Một số chất ô nhiễm hữu cơ điển hình trong đất, nước TT Loại hợp chất hữu cơ Ví dụ 1 Hợp chất hyđrocachon Cydohexen, Benzine, Benzen, toluen, Styren.
Naphtalen, Benzopyren2 Hợp chất halogen hyđrocacbon Chloroform, Vinyclorua, tetrachloethen,
Hexachorychohexen. Hexachlobenxen, polydorua, Byphenyl
3 Pôlyclođbenzodioxin 2,3,7,8 tetracl(Hlibenzodioxin 4 Hợp chất phốtpho hữu cơ Tributyphotphat 5 Hợp chất nhơ hữu cơ Acrylamid,Acrylnitnt, O-nitrotoluen 6 Hợp chất hữu cơ kim loại Methyclorua thuỷ ngân 7 Hợp chất hữu cơ lưu huỳnh Methyl-mercaptan 8 Chất hoạt động bề mặt Alkybenzensunfonat 9 Alkohole & Ather Methyl-hexanol, dipphenylether
10 Alđehyt, keton Formaldehyd. axeton, axitbenzoic- 11 Phenol Phenol, Cresol 12 Hợp chất thiên nhiên Mỡ, xít amin, lòng trắng trứng
- Các hợp chất hữu cơ hyđrocacbon mạch thẳng hay mạch vòng thông thường và sản phẩm của dầu mỏ, thâm nhập và làm ô nhiễm nguồn nước thông qua các quá trình khai thác, vận chuyển, gia công, sử dụng. Ảnh hưởng của các hợp chất thơm này gây ra mùi rất khó chịu. Đối với người và thực vật, chúng gây nên các bệnh mãn tính và cấp tính như ung thư, ảnh hưởng đến hệ thần kinh trung ương, mắt, bệnh ngoài da. Các hợp chất hyđrôcacbon đa vòng được các hạt keo hấp thụ hoặc bám dính trên các chất hoạt tính bề mặt, do vậy chúng có khả năng tích tụ lớn và cũng có khả năng gây ung thư.
- Các hợp chất hữu cơ halogen là những chất vô cùng độc hại. Các hợp chất này bao gồm Cacbuahydro clorua, polyclorua byphenyl, thuốc trừ sâu chứa do, các phênol clo, PCDD, PCDF.
- Các hợp chất pôlyclobiphenyl (PCB).
- Đây là những chất ô nhiễm được tìm thấy trong các nguồn trên toàn thế giới, thậm chí trong cả các mô tế bào chim và cá. Các PCB có độ bền hoá học, nhiệt và sinh học rất cao. Nguồn gốc của PCB chủ yếu từ dung dịch lạnh cách điện, làm thẩm thấu bông và sợi amiăng, làm hoá chất dẻo và làm các chất phụ gia cho một số loại sơn.
* Ô nhiễm dầu mỏ
Ô nhiễm nước do dầu mỏ và sản phẩm của chúng (Xăng, mazut, dầu bôi trơn,..) thể hiện như sau:
- Làm giảm tính chất lý hoá của nước (như thay đổi màu, mùi vị): Nước sẽ có mùi đặc trưng khi nồng độ của nó đạt tới 0,5 mg/l, các chỉ tiêu hoá học sẽ thay đổi mạnh khi nồng độ lớn hơn 100 mg/l.

- Tác động đến quần thể sinh vật: Nước bị ô nhiễm gây thiệt hại vô cùng đối với sinh vật có độ nhạy cảm cao, quần thể sinh vật giảm xuống rất nhanh do sự phân huỷ của dầu trong cơ thế sống và dơ lớp vàng dầu ngăn cản quá trinh trao đổi ôxy giữa pha nước và khí.
Hàm lượng dầu trong nước đạt 20-30mg/l sẽ gây rối loạn các hoạt động phản xạ của cá hàm lượng lớn hơn có thể gây chết cá. Khi hàm lượng các hợp chất thơm của đấu đạt tới 0,3 mg/l thì quần thể sinh vật trong nước có thể bị chết.
b. Các chất ô nhiễm dưới dạng vô cơ
Có rất nhiều chất vô cơ gây ô nhiễm nước, đất. Tuy nhiên có một số nhóm điển hình cần lưu ý như sau
* Các loại phân bón vô cơ
Đó là hợp chất vô cơ mà thành phần chủ yếu là cacbon, hyđro và ôxy ngoài ra chúng còn chứa các nguyên tố như N, P, K cùng các nguyên tố vi lượng khác. Các loại hoá chất này sẽ đi vào nước do một phần phân bón bị cuốn trôi khi sử dụng, bốc hơi hoặc chuyển hoá thành các dạng khác và lưu tồn trong môi trường. Việc dư thừa các chất dinh dưỡng (Phốt phát, muối amoni, urê, nitrat, muối kali...) gây nên sự phát triển nhanh của một số loài thực vật bậc thấp như tảo, rong rêu và các thực vật thân mềm, gây nên hiện tượng thực vật chết hàng loạt. Chúng bị phân huỷ và tạo thành lượng lớn hợp chất hữu cơ. Mặt khác do phát triển ồ ạt, một lượng lớn ôxy trong nước sẽ bị tiêu thụ gây nên tình trạng thiếu hụt ôxy một cách trầm trọng (BOD cao), xuất hiện các quá trình khử, các chất có tính khử như H2S, NH3 tăng lên. Các ion kim loại và HPO4
- được chuyển hoá từ các chất lắng cặn, hoà tan vào nước gây độc cho nguồn nước mặt.
Các khoáng axít
Nguồn gốc chính là từ các mỏ than không còn khai thác FeS2 (Có nhiều trong mỏ) là chất bền trong môi trường thiếu ôxy. Khi tiếp xúc với môi trường không khí, có sự tham gia của vi sinh vật sẽ tạo thành H2SO4 và Fe(OH)3 màu đỏ. Sự xuất hiện các chất này là nguyên nhân gây nên tình trạng mất cân bằng sinh thái trong nguồn nước (động thực vật chết).
Cặn
Nguồn phát sinh do xói mòn (chủ yếu) nước thải sản xuất, sinh hoạt.
Các chất cặn đáy thường ờ trong tình trạng yếm khí, tham gia quá trình khử, gây nên tình trạng thiếu hụt ôxy trong nước. Các chất cặn lơ lửng và hạt huyền phù là môi trường hấp thụ rất tốt, chúng như là các kho chứa các kim loại nặng như Cr, Cu, Mo, Ni, Co, Mn... gây độc cho nguồn nước.
* Các nguyên tố vết
Đây là các nguyên tố có rất ít trong nước, tuy nhiên khả năng gây độc rất cao cho
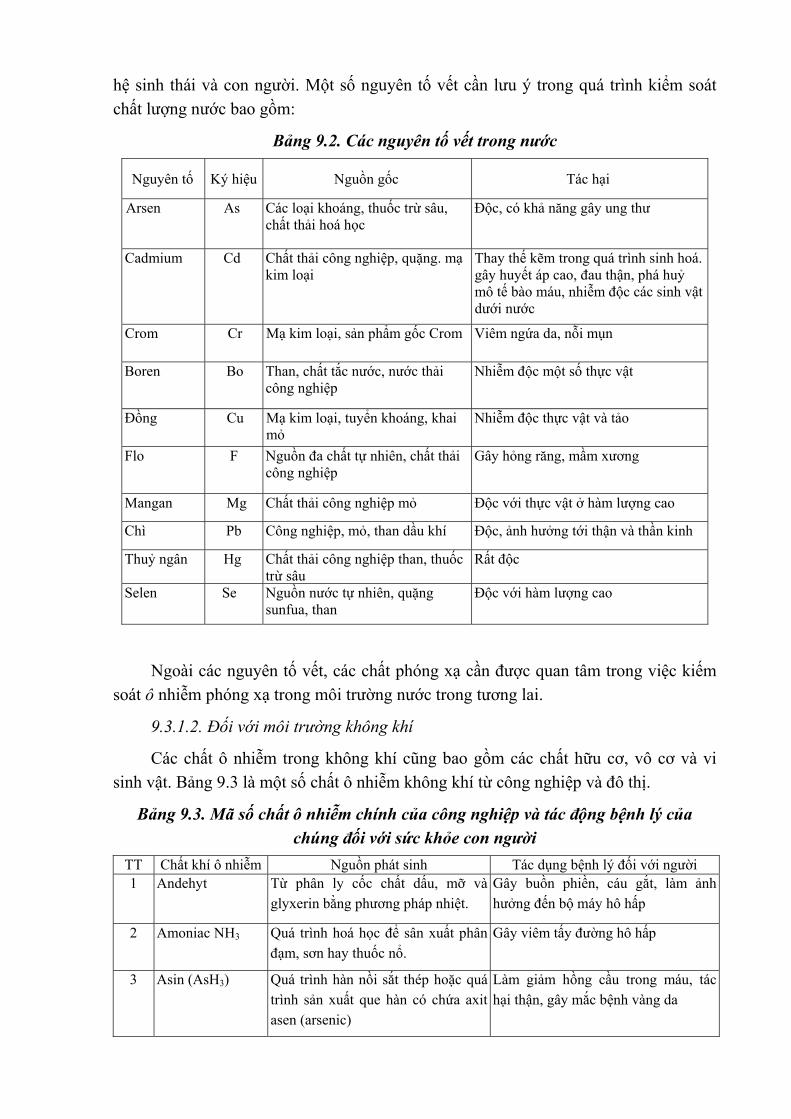
hệ sinh thái và con người. Một số nguyên tố vết cần lưu ý trong quá trình kiểm soát chất lượng nước bao gồm:
Bảng 9.2. Các nguyên tố vết trong nước
Nguyên tố Ký hiệu Nguồn gốc Tác hại
Arsen As Các loại khoáng, thuốc trừ sâu, chất thải hoá học
Độc, có khả năng gây ung thư
Cadmium Cd Chất thải công nghiệp, quặng. mạ kim loại
Thay thế kẽm trong quá trình sinh hoá. gây huyết áp cao, đau thận, phá huỷ mô tế bào máu, nhiễm độc các sinh vật dưới nước
Crom Cr Mạ kim loại, sản phẩm gốc Crom Viêm ngứa da, nỗi mụn
Boren Bo Than, chất tắc nước, nước thải công nghiệp
Nhiễm độc một số thực vật
Đồng Cu Mạ kim loại, tuyển khoáng, khai mỏ
Nhiễm độc thực vật và tảo
Flo F Nguồn đa chất tự nhiên, chất thải công nghiệp
Gây hỏng răng, mầm xương
Mangan Mg Chất thải công nghiệp mỏ Độc với thực vật ở hàm lượng cao
Chì Pb Công nghiệp, mỏ, than dầu khí Độc, ảnh hưởng tới thận và thần kinh
Thuỷ ngân Hg Chất thải công nghiệp than, thuốc trừ sâu
Rất độc
Selen Se Nguồn nước tự nhiên, quặng sunfua, than
Độc với hàm lượng cao
Ngoài các nguyên tố vết, các chất phóng xạ cần được quan tâm trong việc kiếm soát ô nhiễm phóng xạ trong môi trường nước trong tương lai.
9.3.1.2. Đối với môi trường không khí
Các chất ô nhiễm trong không khí cũng bao gồm các chất hữu cơ, vô cơ và vi sinh vật. Bảng 9.3 là một số chất ô nhiễm không khí từ công nghiệp và đô thị.
Bảng 9.3. Mã số chất ô nhiễm chính của công nghiệp và tác động bệnh lý của chúng đối với sức khỏe con người
TT Chất khí ô nhiễm Nguồn phát sinh Tác dụng bệnh lý đối với người 1 Andehyt Từ phân ly cốc chất dấu, mỡ và
glyxerin bằng phương pháp nhiệt. Gây buồn phiền, cáu gắt, làm ảnh hưởng đến bộ máy hô hấp
2 Amoniac NH3 Quá trình hoá học để sân xuất phân đạm, sơn hay thuốc nổ.
Gây viêm tấy đường hô hấp
3 Asin (AsH3) Quá trình hàn nồi sắt thép hoặc quá trình sản xuất que hàn có chứa axit asen (arsenic)
Làm giảm hồng cầu trong máu, tác hại thận, gây mắc bệnh vàng da
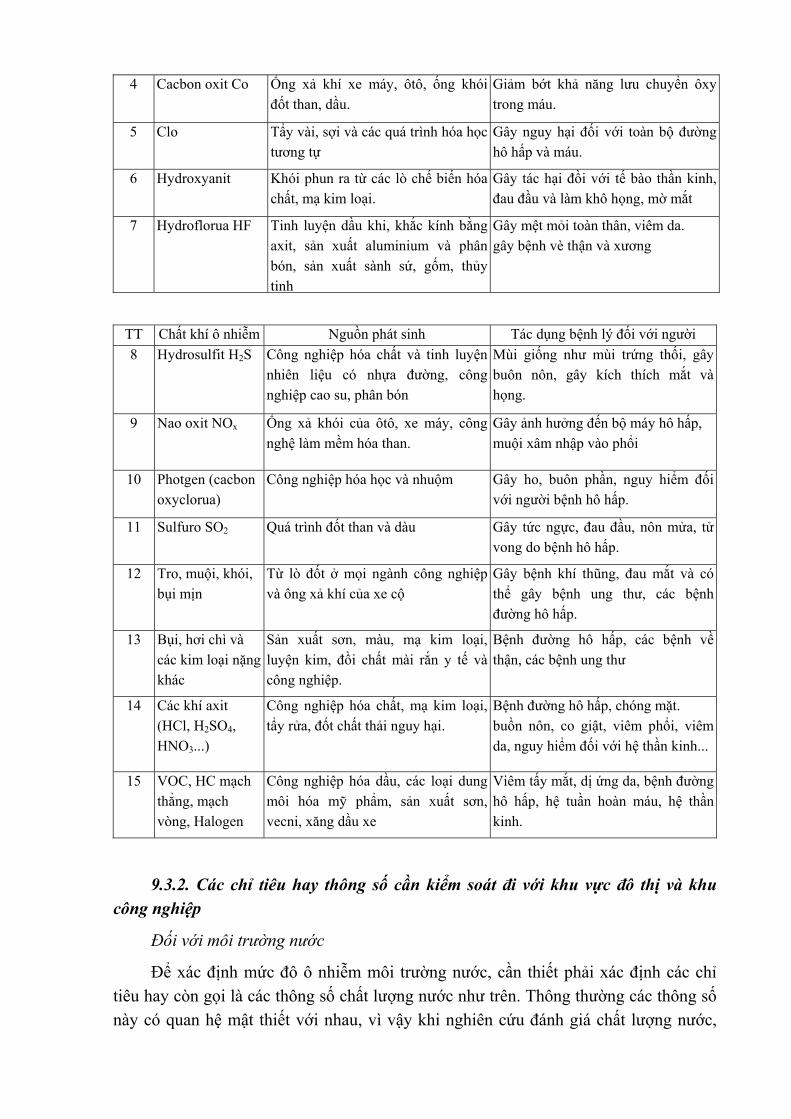
4 Cacbon oxit Co Ống xả khí xe máy, ôtô, ống khói đốt than, dầu.
Giảm bớt khả năng lưu chuyển ôxy trong máu.
5 Clo Tẩy vài, sợi và các quá trình hóa học tương tự
Gây nguy hại đối với toàn bộ đường hô hấp và máu.
6 Hydroxyanit Khói phun ra từ các lò chế biến hóa chất, mạ kim loại.
Gây tác hại đồi với tế bào thần kinh, đau đầu và làm khô họng, mờ mắt
7 Hydroflorua HF Tinh luyện dầu khi, khắc kính bằng axit, sản xuất aluminium và phân bón, sản xuất sành sứ, gốm, thủy tinh
Gây mệt mỏi toàn thân, viêm da. gây bệnh vè thận và xương
TT Chất khí ô nhiễm Nguồn phát sinh Tác dụng bệnh lý đối với người 8 Hydrosulfit H2S Công nghiệp hóa chất và tinh luyện
nhiên liệu có nhựa đường, công nghiệp cao su, phân bón
Mùi giống như mùi trứng thối, gây buôn nôn, gây kích thích mắt và họng.
9 Nao oxit NOx Ống xả khói của ôtô, xe máy, công nghệ làm mềm hóa than.
Gây ảnh hưởng đến bộ máy hô hấp, muội xâm nhập vào phổi
10 Photgen (cacbon oxyclorua)
Công nghiệp hóa học và nhuộm Gây ho, buôn phần, nguy hiểm đối với người bệnh hô hấp.
11 Sulfuro SO2 Quá trình đốt than và dàu Gây tức ngực, đau đầu, nôn mửa, tử vong do bệnh hô hấp.
12 Tro, muội, khói, bụi mịn
Từ lò đốt ở mọi ngành công nghiệp và ông xả khí của xe cộ
Gây bệnh khí thũng, đau mắt và có thể gây bệnh ung thư, các bệnh đường hô hấp.
13 Bụi, hơi chì và các kim loại nặng khác
Sản xuất sơn, màu, mạ kim loại, luyện kim, đồi chất mài rắn y tế và công nghiệp.
Bệnh đường hô hấp, các bệnh vềthận, các bệnh ung thư
14 Các khí axit (HCl, H2SO4, HNO3...)
Công nghiệp hóa chất, mạ kim loại, tẩy rửa, đốt chất thải nguy hại.
Bệnh đường hô hấp, chóng mặt. buồn nôn, co giật, viêm phổi, viêm da, nguy hiểm đối với hệ thần kinh...
15 VOC, HC mạch thẳng, mạch vòng, Halogen
Công nghiệp hóa dầu, các loại dung môi hóa mỹ phẩm, sản xuất sơn, vecni, xăng dầu xe
Viêm tấy mắt, dị ứng da, bệnh đường hô hấp, hệ tuần hoàn máu, hệ thần kinh.
9.3.2. Các chỉ tiêu hay thông số cần kiểm soát đi với khu vực đô thị và khu công nghiệp
Đối với môi trường nước
Để xác định mức đô ô nhiễm môi trường nước, cần thiết phải xác định các chỉ tiêu hay còn gọi là các thông số chất lượng nước như trên. Thông thường các thông số này có quan hệ mật thiết với nhau, vì vậy khi nghiên cứu đánh giá chất lượng nước,

cần thiết phải xem xét một cách cẩn thận, chuẩn xác. Các thông số này được chia thành các nhóm: vật lý, hoá học và sinh học.
Nhóm chỉ tiêu vật lý: Để đánh giá mức độ ô nhiễm hay chất lượng nước theo các chỉ tiêu này người ta xác định nhiệt độ, mầu mùi, độ trong, độ đục hay hàm lượng cặn ràn lơ lửng, tổng hàm lượng các chất rắn, độ dẫn điện,...
- Mùi: Nguyên nhân tạo ra mùi, là các quá trình phân huỷ chất hữu cơ (tạo mùi thối), hoặc mùi tanh của sắt. Như vậy, thông qua mùi có thể đánh giá mức độ phân huỷ chất hữu cơ (hiếu khí, kỵ khí), quá trình phát triển của động thực vật phù du,...
- Mầu: Căn cứ chỉ thị mầu có thể đánh giá sơ bộ chất lượng nước. Ví dụ: căn cứ mầu xanh để xác định mức độ dinh dưỡng trong sông hồ, ảnh hưởng của nước thải công nghiệp có mầu đặc trưng, ảnh hưởng cục bộ bởi nước thải sinh hoạt có mầu nâu xám, nguồn nước trong môi trường kỳ khu H2S tác động với Fe+2 tạo thành FeS có mầu đen,...
- Các chất rắn hay cặn lơ lửng thường là các chất bị cuốn trôi theo dòng nước, do xác động thực vật chết hay do hoạt động của người, súc vật tạo ra. Hàm lượng cặn lớn sẽ làm giảm khả năng tự làm sạch của nguồn nước, cản trở quá trình sinh thái diễn ra trong nước hoặc cản trở quá trình diệt khuẩn trong nước.
Trong quá trình kiểm soát chất lượng nước, tuỳ từng trường hợp cụ thể cũng như đặc tính chất phát thải mà ta đưa ra các chất ô nhiễm cần kiểm soát cũng như các chỉ tiêu đánh giá chất lượng nguồn nước.
Nhóm chỉ tiêu hoá học
Đây là nhóm chỉ thì rất quan trọng. Trạng thái tồn tại của chúng phản ánh rõ nét chất lượng nguồn nước cũng như nguyên nhân chính gây nên.
- pH: Biểu thị nồng độ H+ trong nước, nếu pH bé hơn 7, nước có tính axit; lớn hơn 7 có tính kiềm; bằng 7 có tính trung tính. Chất nước ở trạng thái bình thường pH = 6-8,5 (nguồn A) và 5.5-9 (nguồn loại B).
- Nhóm ô nhiễm hữu cơ:
Đặc trưng cho nhóm này là BOD (nhu cầu ôxy hoá sinh hoá) và DO (ôxy hoà tan).
Các thông số này phản ánh đầy đủ trạng thái ô nhiễm chất hữu cơ trong nguồn nước cũng như trạng thái của hệ sinh thái của nó (quan hệ chặt chẽ với quá trình sinh vật trong nước và bùn đáy).
Đồng thời với BOD còn phản xác định nhu cầu ôxy hoá học (COD).
- Ngoài các hợp chất hữu cơ tác động đến nồng độ ôxy trong nước còn có các hợp chất vô cơ như ammonla, nitrite, Sulphit, Hydrogen sulphit, Fe2+
- Mức dinh dưỡng:
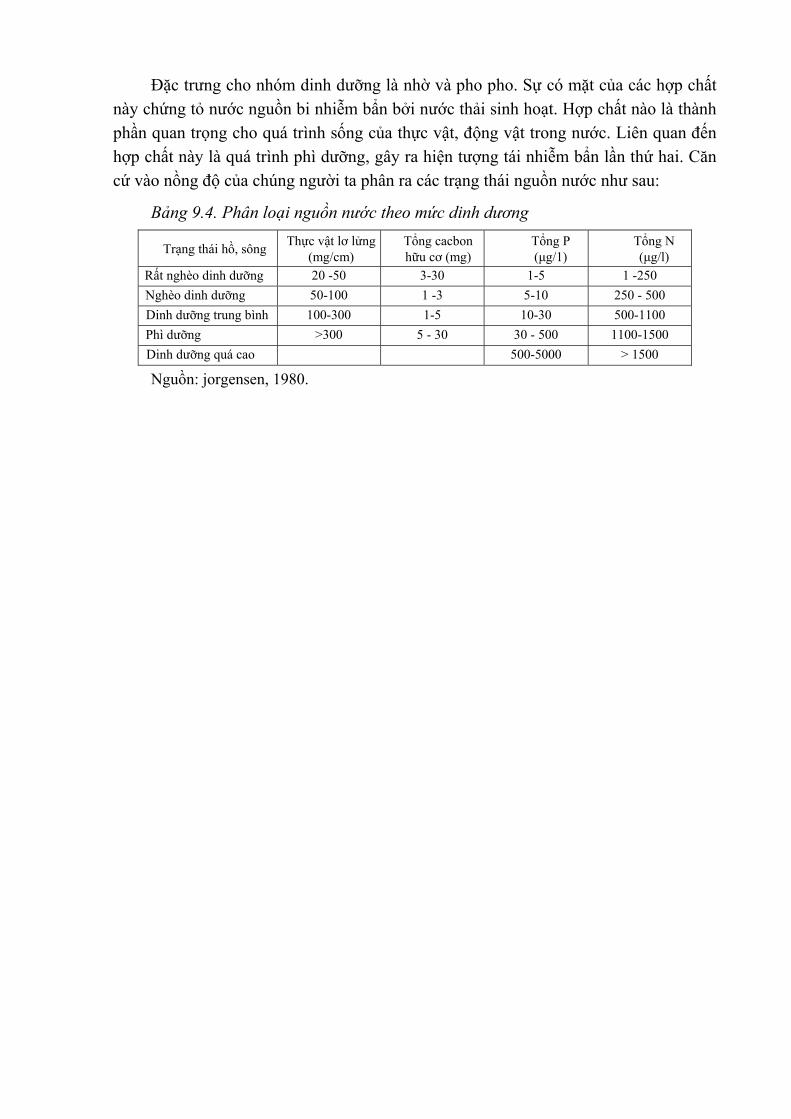
Đặc trưng cho nhóm dinh dưỡng là nhờ và pho pho. Sự có mặt của các hợp chất này chứng tỏ nước nguồn bi nhiễm bẩn bởi nước thải sinh hoạt. Hợp chất nào là thành phần quan trọng cho quá trình sống của thực vật, động vật trong nước. Liên quan đến hợp chất này là quá trình phì dưỡng, gây ra hiện tượng tái nhiễm bẩn lần thứ hai. Căn cứ vào nồng độ của chúng người ta phân ra các trạng thái nguồn nước như sau:
Bảng 9.4. Phân loại nguồn nước theo mức dinh dương
Trạng thái hồ, sông Thực vật lơ lửng (mg/cm)
Tổng cacbon hữu cơ (mg)
Tổng P (μg/1)
Tổng N (μg/l)
Rất nghèo dinh dưỡng 20 -50 3-30 1-5 1 -250 Nghèo dinh dưỡng 50-100 1 -3 5-10 250 - 500 Dinh dưỡng trung bình 100-300 1-5 10-30 500-1100 Phì dưỡng >300 5 - 30 30 - 500 1100-1500 Dinh dưỡng quá cao 500-5000 > 1500
Nguồn: jorgensen, 1980.

Phần 3
THỰC HÀNH
Chương 10
XỬ LÝ SỐ LIỆU THỐNG KÊ TRONG MÔI TRƯỜNG
Mục tiêu:
- Giúp học sinh hiểu được kiến thức thống kê cơ bản ứng dụng trong môi trường. Giúp học sinh tính toán và xử lý được các phép tính thống kê cơ bản, tương quan nhiều chiều, tính sai khác, hồi quy tuyến tính, và xác định các yếu tố hạn chế trong bố trí thí nghiệm.
Yêu cầu:
Máy tính có cấu hình tốt và cài đặt phần mềm xử lý số liệu thống kê
Cài đặt phần mềm SAS
1. Tính thống kê cơ bản kết quả phân tích số liệu của các nguyên tố hoá học trong đất.
Yêu cầu
- Giá trị trung bình
- Giá trị bé nhất
- Giá trị lớn nhất
- Tổng
- Độ lệch chuẩn
- Hệ số biến động
Cách tính toán
Data direct;
input Số liệu X1, X2, X3, X4,... Xn;
cards;
21.08 24.1 1242.0 2074.1
28.9 24.4 1129.0 1232.0
22.18 25.0 1507.0 1659.0
27.31 24.2 12630.0 1861.1

proc corr data-direct;
var X1, X2, X3, X4,...Xn;
run;
2. Phân tích tương quan một chiều và nhiều chiều giữa các yếu tố thí nghiệm
Yêu cầu
- Tính tương quan một nhân tố
- Tính tương quan nhiều nhân tố
- Lập bảng tương quan nhiều nhân tố
Cách tính toán:
Data direct;
input Số liệu X1, X2, X3 X4... Xn;
cards;
21.08 24.1 1242.0 2074.1
28.9 24.41 129.0 1232.0
22.1 825.0 1507.0 1659.0
27.31 24.21 263.0 1861.1
;
proc corr data-direct;
var Xl, X2, X3 X4... Xn;
run;
3. Xác định các yếu tố ảnh hưởng
Yêu cầu
Phân tích sai số thí nghiệm
Cách tính toán
Data direct;
input Số liệu X1, X2, X3, X4...Xn;
cards;
21.08 24.11 242.0 2074.1
28.92 4.41 129.0 1232.0
22.18 25.0 1507.0 1659.0

27.31 24.2 1263.0 1861.1;
proc reg data=direct;
model Y1 = X1, X2, X3, X4,... Xn/method=forward sle=0.05;
run;
4. Phân tích sai khác giữa các công thức thí nghiệm
Yêu cầu
Xảc định các yếu tố hạn chế
Phân tích mức độ ảnh hưởng của các yếu tố thí nghiệm
Cách tính toán:
data lsd;
input treat $ germ @@;
cards;
;
proc anova;
class treat;
model germ treat;
means treat~sd;
means treat/1sd alpha-o~05;
run;

Chương 11
THỰC HÀNH PHÂN TÍCH TRONG PHÒNG THÍ NGHIỆM
l. Xác định pH của nước
Ngày nay, việc xác định pa trở nên đơn giản do sự phát triển của khoa học công nghệ. Giá trị pa được xác định bằng phép đo trên máy đo pa (pa meter). Điểm cần lưu ý khi đo pH là việc bảo quản điện cực do các đặc điểm về khí hậu của vùng nhiệt đới. Trước khi tiến hành đo cần phải kiêm tra máy bằng cách điều chỉnh máy với các dung dịch đệm có giá trị pH = 4,00; 7,00; 10,00. Điện cực phải luôn được bảo quản trong dung dịch bảo quản. Tiến hành đọ bằng cách lắc đều mẫu dung dịch sau đó cho đầu cảm ứng điện cực ngập 1/3 trong dung dịch đo, quan sát đồng hồ báo giá trị pH ở trạng thái cân bằng để ghi giá trị.
2. Xác định độ cứng của nước
Lấy 100 ml nước cần xác định độ cứng, thêm vào 10 ml dung dịch đệm (nếu độ kiềm của mẫu nước lớn thì phải dùng dung dịch HCl 0,1N để điều chỉnh cho pH dung dịch bằng 7- 8 theo giấy chỉ thị tổng hợp trước khi thêm dung dịch đệm), thêm một lượng nhỏ chất chỉ thị (khoảng bằng một hạt đỗ xanh), dung dịch có màu đỏ. Chuẩn dung dịch bằng dung dịch chuẩn Complexon III cho tới khi dung chuyển từ màu đỏ sang màu xanh. Nếu trong dung dịch có mặt các ion cản trở như Fe3+, Cu2+ thì cần chuẩn độ lại với mẫu nước khác và cách tiến hành như sau: Sau khi cho 10ml dung dịch đệm, ta thêm vài giọt KCN 5% để che ion cản trở rồi mới thêm chất chỉ thị và tiến hành chuẩn độ như trên
Tính kết quả: Độ cứng toàn phần của nước được tính như sau:
Trong đó: a: thể tích dung dịch Complexon III tiêu tốn (ml);
V: thể tích mẫu nước lấy để phân tích (ml);
0,025: nồng độ dung dịch Complexon III (mol/1);
2: số đương lượng gam độ cứng tương ứng với 1 mol Complexon III.
Hoá chất
- Dung dịch Complexon III 0,025M: hoà tan 9,306g Complexon III trong 1 lít nước cất hai lần
- KCN 50%: cân 5g KCN hoà tan trong 100 ml nước cất hai lần.
- Enocrom T đen: cân 0,05g chất chỉ thị trộn với 10g NaCl.

- Đệm NH4OH + NH4Cl (pH= 10), hoà tan 25 g NH4Cl trong 100 ml nước thêm 200ml NH4OH 20%, sau đó thêm nước cất đến 1 lít.
3. Xác định Ca và Mg trong nước sinh hoạt
Trình tự phân tích
Cho mẫu nước vào bình tam giác có V = 250ml, thể tích mẫu nước được lấy để không chứa quá 15mg Ca. Dùng nước cất pha loãng thành 100 ml, nếu thấy cần thiết. Nếu mẫu nước có tính axit thì cần trung hoà bằng dung dịch NaOH. Nếu mẫu nước có độ kiềm cao hơn 6 mđlg/1 thì cần thêm vào lượng tương đương dung dịch HCl 0,1N, đun sôi dung dịch và đề nguội. Sau đó thêm vào 2ml NaOH 2M và một ít chất chỉ thị murexit. Chuẩn độ từ từ bằng đung dịch EDTA 0,025M cho đến khi dung dịch chuyển từ màu hồng sang tím.
Tính kết quả:
Trong đó: a: thể tích dung dịch EDTA 0,025M (ml);
V: thể tích mẫu nước chuẩn độ (ml);
1000: 2 x 0,025 x 20 x 1000.
Hóa chất
- EDTA 0,025M: Xem phần trên.
- NaOH 2M: hoà tan 8g NaOH trong 100 ml nước cất hai lần.
HCl 0,1N: pha 8,2ml HCl đặc trong 1 lít nước cất hai lần
(Cách xác định Mg tính tương tự như Ca)
Trình tự phân tích
Lấy vào bình tam giác có V: 250ml, lượng thể tích như trong xác định Ca. Xử lý mẫu như đã làm đối với Ca. Thêm vào 5ml Hiđroxilamin 10%, vài giọt KCN 10% và một lượng nhỏ chỉ thị Eriocrom T đen rồi tiến hành chuẩn độ bằng EDTA 0,025M.
Tính kết quả
Trong đó: V1: thể tích EDTA chuẩn Ca + Mg (ml); V2: thể tích Ca (ml);
V: thể tích mẫu nước (ml).
Hoá chất
- Hidroximalin 10% hoà tan 10g NH2OH.HCl trong 100 ml nước cất hai lần

4. Xác định nhu cẩu ôxy hóa học (COD)
Trình tự phân tích
Lấy 20ml mẫu nước cho vào bình hồi lưu, rồi thêm vào HgSO4 (trung Cr thì cần 0,1 g HgSO4). Thêm vào 10ml dung dịch K2Cr2O7 0,025N và một vài hạt thuỷ tinh. Lắp ống thuỳ sinh hàn thuỷ tinh nhảm. Thêm vào từ từ 30ml H2SO4 đặc có chứa Ag2SO4 qua phần cuối ống sinh hàn và lắc đều hỗn hợp trong khi thêm axit. Đun hồi lưu trong hệ giờ. Để nguội và rửa sinh hàn hồi lưu bằng nước cất. Pha loãng hỗn hợp bằng nước cất tới thể tích khoảng 150ml, để nguội. Chuẩn lượng bicronat dư bằng muối Fe2+, với chỉ thị axit Phenylanthranilic. Cũng tiến hành một thí nghiệm mẫu trắng như đối với mẫu phân tích.
Tính kết quả
Trong đó:
a: thể tích Fe2+ chuẩn độ mẫu ứng (ml)
b: thể tích Fe2+ chuẩn độ mẫu (ml)
N: nồng độ đương lượng của Fe2+
Hóa chất:
- K2Cr2O7 0,025N: Hoà tan 12,259 g K2Cr2O7 (đã sấy khô 2 giờ ở 1050C) trong nước cất và thêm nước tới vạch mức.
- H2SO4 đặc có thểm 22g Ag2SO4 cho một chai 9 lít.
- Dung dịch Fe2+ 0,1 N: Hòa tan 39g Fe(NH4)2SO4.6H2O tinh khiết trong nước cất, thêm 20ml H2SO4 đặc nguội rồi định mức tới 1 lít
- Ag2SO4 tinh khiết hoá học.
- H2SO4 tinh khiết hoá học.
Chỉ thị 1%: Hoà tan 0,2g axit Phenylanthranilic trong 100 ml Na2SO3 0,2%.
5. Xác định pH trong đất
Trình tự phân tích
Lắc lớp đất (đã qua rây 1 mm) 15 phút trên máy lắc (hoặc lắc tay 30 phút) với 25ml KCl là (với pHKCl) hoặc nước cất (pHH2O) sau đó để yên 2 giờ (không quá 3 giờ), lắc 2 - 3 lần rồi đo pH ngay trong dung dịch huyền phù.

Hiệu chỉnh máy đo phụ: Máy trước khi đo phải hiệu chỉnh bằng cách đo dung dịch đệm pH tiêu chuẩn. Chỉnh cho kim chỉ đúng tri số pH của dung dịch đệm.
Đo mẫu: Giữ cho điện cực cách mặt mẫu đất là lem và ngập nước khoảng 2cm. Khi máy đã ổn định, đọc giá trị pH trên máy
Ghi chú: Điện cực thủy tinh được ngâm trong nước cất khi không dùng. Tỷ lệ đất và dịch chiết có khác nhau phụ thuộc phương pháp. Vì vậy, trong kết quả phân tích cần ghi rõ tỷ lệ đất dịch chiết rút và chất chiết rút. Ví dụ: "pH trong KCl 1N - 1:5W/V". Nghĩa là pH khi chiết rút bằng KCl 1N với tỷ lệ đất dung dịch chiết rút là 1:5 (khối lượng/ thể tích). Nếu không ghi chú gì thì thường được hiệu là pHH2O theo tỷ lệ đất nước là 2:5.
Pha dung dịch đệm tiêu chuẩn
- Dung dịch KHC8H4O4 0,05M: 10,21g KHC8H4O4 pha thành 1000 ml.
- Hỗn hợp KH2PO4 + NaHPO4 0,025m: 3,10g KH2PO4 pha thành 1000 ml; 3,55g NaHPO4 pha thành 1000 m1.
- Trộn lẫn 2 dung dịch này thành 2 lít hỗn hợp. Thời hạn sử dụng không quá 2 tháng.
- Dung dịch Na2B4O7 0,01N: 3,81g Na2B4O8.10H2O pha thành 100ml
- Dung dịch KHC4H4O6 bão hoà: 6g KHC4H4O6 trong 1 lít nước cất.
- Các dung dịch trên pha song đựng trong bình polietilen, thời hạn không quá 3 tháng.
- Trị số pH của các dung dịch đệm trên như sau:
6. Xác định chất hữu cơ trong đất (Walkley-Black)
Trình tự phân tích
Cân là đất khô không khí cho vào bình tam giác 500ml thêm 10 ml dung dịch K2Cr2O7 1N. Thêm 20ml H2SO4 đặc vào (thêm nhanh theo khả năng có thể thêm). Lắc nhẹ và giữ 30 phút. Thêm 200ml nước cất và 10ml H3PO4 85%. Thêm 1 ml chỉ thị điphenylamin. Chuẩn độ bằng dung dịch FeSO4 0,5N đến khi dung dịch có màu xanh lá cây.
Hoá chất
Hoà tan 49,039 K2Cr2O7 trong nước cất và thêm thể tích đến 1 lít
- FeSO4 0,5N: hoà tan 139g FeSO4.7H2O trong 800ml nước, thêm 20ml H2SO4 đặc rồi định mức đến thể tích 1 lít.
- Điphenylamin: hoà tan 0,5g chỉ thị trong 20ml nước rồi thêm tiếp 100 ml H2SO4 đặc.

Tính kết quả
Trong đó:
N: nồng độ đương lượng của muối FeSO4
V0, V1: thể tích muối FeSO4 dùng để chuẩn độ thí nghiệm trắng và chuẩn độ mẫu;
a: lượng mẫu lấy để phân tích (g);
K: hệ số chuyển đổi từ mẫu không khí sang mẫu khô tuyệt đối;
0,39 = 3x 10-3 x 100% x 1,3;
3: đương lượng gam của C;
13: hệ số "bù" cho quá trình oxy hoá chưa hoàn toàn chất hữu cơ trong phương pháp này.
% chất hữu cơ = 2xC (%)
Trước đây dùng hệ số 1,72 nhưng hiện nay thấy hệ số 2 là thích hợp hơn (ISRIC, 1986).
Chú thích
Trước khi chuẩn độ lượng K2Cr2O7 còn dư (ở cả 2 phương pháp) cần phải đề nguội dung dịch ôxy hoá, nếu không một phần Fe2+ dùng để lượng K2Cr2O7 còn dư có thể bị ôxy không khí ôxy hoá.
Với 10ml K2Cr2O7 là chỉ có thể ôxy hoá tối đa 25mg C, vì vậy khi áp dụng phương pháp Walkley - Black cần hết sức chú ý lượng mẫu lấy đi phân tích.
Đối với những đất hàm lượng mùn < 2,6% có thể lẫy 1 g mẫu đem đi phân tích, nếu hàm lượng mùn cao hơn nên lấy 0,2g đất còn khi hàm lượng mùn >13,5% thì lượng mẫu lấy là 0,1 g (Lê Đức - Tạp chí Khoa học Đất, 10 - 1998).
Nếu dùng chỉ thị ferroin (0,695g FeSO4.7H2O và 1,485go - phenaltrolininonohidrat (Cl2H8N2.H2O) trong 100 ml nước thì dùng 4 giọt chỉ thị - khi kết thúc chuẩn độ, màu của dung dịch chuyển từ xanh sang đỏ.
Có thể dùng phương pháp so màu để xác định chất hữu cơ bằng cách đo màu Cr3+ tại bước sóng 625nm. Dùng saccarozơ(C12H22O11) làm trung dịch chuẩn để khử Cr6+ trong K2Cr2O11; làm dung dịch chuẩn để khử Cr6+ trong K2Cr2O7 thành Cr3+.

7. Xác định đạm dễ tiêu (NH4+) trong đất
Trình tự phân tích
Cân 5 gam đất và đổ vào bình tam giác, sau đó cho 50ml dung dịch 2M.KCL, đậy nút lại và lắc cơ học trong vòng 1 giờ đồng hồ. Để lặng đất và lọc và tách đất và dung dịch cho phân tích. Nếu mẫu phân tích mà chưa phân tích ngay thì phải giữ trong tủ lạnh giữ. Khi phân tích, lắc đều ông nghiệm đựng mẫu và lấy 10 ml dung dịch mẫu, bỏ vào ông nghiệm chuyên dùng để phân tích, sau đó phân tích bằng máy Spectrophotometter.
Hoá chất
Dung dịch KCl 2 M: Hoà 149 gam KCl trong 800 ml nước cất và đổ vào bình tam giác 1l, và sau đó đổ thêm nước cất vào bình cho đến khi chạm vạch đánh dấu.
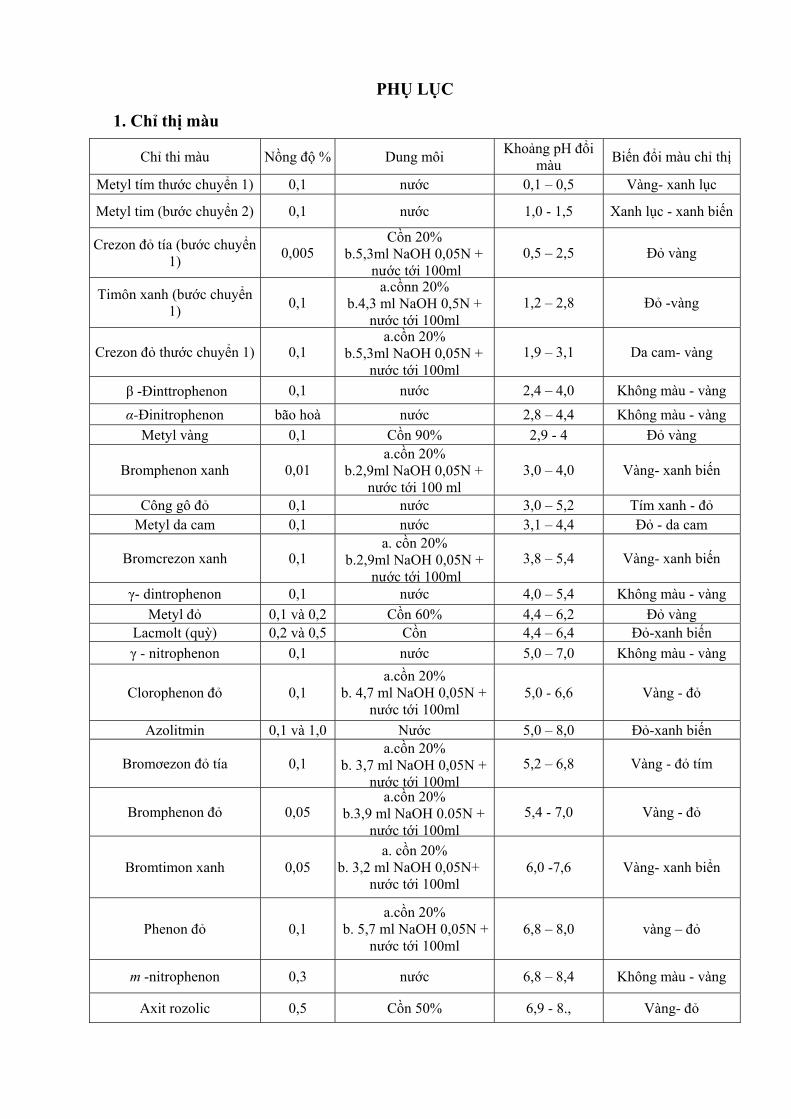
PHỤ LỤC
1. Chỉ thị màu
Chỉ thi màu Nồng độ % Dung môi Khoảng pH đổi màu Biến đổi màu chỉ thị
Metyl tím thước chuyển 1) 0,1 nước 0,1 – 0,5 Vàng- xanh lục
Metyl tim (bước chuyển 2) 0,1 nước 1,0 - 1,5 Xanh lục - xanh biến
Crezon đỏ tía (bước chuyển 1) 0,005
Cồn 20% b.5,3ml NaOH 0,05N +
nước tới 100ml0,5 – 2,5 Đỏ vàng
Timôn xanh (bước chuyển 1) 0,1
a.cồnn 20% b.4,3 ml NaOH 0,5N +
nước tới 100ml1,2 – 2,8 Đỏ -vàng
Crezon đỏ thước chuyển 1) 0,1 a.cồn 20%
b.5,3ml NaOH 0,05N + nước tới 100ml
1,9 – 3,1 Da cam- vàng
β -Đinttrophenon 0,1 nước 2,4 – 4,0 Không màu - vàng
α-Đinitrophenon bão hoà nước 2,8 – 4,4 Không màu - vàng Metyl vàng 0,1 Cồn 90% 2,9 - 4 Đỏ vàng
Bromphenon xanh 0,01 a.cồn 20%
b.2,9ml NaOH 0,05N + nước tới 100 ml
3,0 – 4,0 Vàng- xanh biến
Công gô đỏ 0,1 nước 3,0 – 5,2 Tím xanh - đỏ Metyl da cam 0,1 nước 3,1 – 4,4 Đỏ - da cam
Bromcrezon xanh 0,1 a. cồn 20%
b.2,9ml NaOH 0,05N + nước tới 100ml
3,8 – 5,4 Vàng- xanh biến
γ- dintrophenon 0,1 nước 4,0 – 5,4 Không màu - vàng Metyl đỏ 0,1 và 0,2 Cồn 60% 4,4 – 6,2 Đỏ vàng
Lacmolt (quỳ) 0,2 và 0,5 Cồn 4,4 – 6,4 Đỏ-xanh biến γ - nitrophenon 0,1 nước 5,0 – 7,0 Không màu - vàng
Clorophenon đỏ 0,1 a.cồn 20%
b. 4,7 ml NaOH 0,05N + nước tới 100ml
5,0 - 6,6 Vàng - đỏ
Azolitmin 0,1 và 1,0 Nước 5,0 – 8,0 Đỏ-xanh biến
Bromơezon đỏ tía 0,1 a.cồn 20%
b. 3,7 ml NaOH 0,05N + nước tới 100ml
5,2 – 6,8 Vàng - đỏ tím
Bromphenon đỏ 0,05 a.cồn 20%
b.3,9 ml NaOH 0.05N + nước tới 100ml
5,4 - 7,0 Vàng - đỏ
Bromtimon xanh 0,05 a. cồn 20%
b. 3,2 ml NaOH 0,05N+ nước tới 100ml
6,0 -7,6 Vàng- xanh biển
Phenon đỏ 0,1 a.cồn 20%
b. 5,7 ml NaOH 0,05N + nước tới 100ml
6,8 – 8,0 vàng – đỏ
m -nitrophenon 0,3 nước 6,8 – 8,4 Không màu - vàng
Axit rozolic 0,5 Cồn 50% 6,9 - 8., Vàng- đỏ
Crezon đỏ (bước chuyển 2) 0,1 a.cồn 20%
b. 5,3 ml NaOH 0,05N + nước tới 100ml
7,2 – 8,8 Vàng hỗ phách - đỏ hơi tía
α -naphtophtalein 1,0 và 0,1 Cồn 50% 7,3 – 8,7 Hồng vàng - lục xanh
Crezon đỏ tía 0,5 a.cồn 20%
b. 5,2 ml NaOH 0,05N + nước tới 100ml
7,6 – 9,2 Xanh vàng - đỏ tía
Timôn xanh (bước chuyền 2) 0,1
a.cồn 20% b. 4,3 ml NaOH 0,05N +
Nước tới 100ml 8,0 – 9,6 Vàng - xanh biển
γ-Crezonphtalein 0,2 Cồn 90% 8,2 – 9,8 Không màu – đỏ
Phenonphtalein 0,1 và 1,0 Cồn 60% 8,2 - 10,0 Không màu – hồng
Thimonphtalein 0,1 Cồn 90% 9,3 – 10,5 Không màu- xanh biển
Xanh Nil B 0,1 nước 10,0 - 11,0 Xanh biến - đỏ
Alizarinddor (bước chuyển 2) 0,1 nước 10,0 – 12,0 Tím - vàng nhạt
Nitroamin 0,1 Cồn 60% 10,8 - 13,0 Không màu -đỏ nâu
Indigocacmin 0,25 Cồn 50% 11,6 - 14,0 Xanh biển - vàng
Tassiro (chỉ thị hỗn hợp) -
0,15 g metyl đỏ trong 100ml còn và 0,05 g
metilen xanh trong 5ml nước cất và trộn đều
54 Tím - xanh lục
2. Dung dịch đệm
Hỗn hợp đệm CH3COOH +CH3COO Na pH CH3COOH 1N pH CH3COOH 1N pH CH3COOH 1N
3,8 3,9 4,0 4,1 4,2 4,3 4,4 4,5 4,6
421,5 345,1 284,4 236,2 197,9 167,4 143,3 124,1 108,9
4,67 4,7 4,8 4,9 5,0 5,1 5,2 5,3 5,4
100,0 96,8 87,2 79,5 73,4 68,6 64,8 61,7 59,3
5,5 5,6 5,7 5,8 5,9 6,0 6,1 6,2 6,3
57,4 55,9 54,7 52,7 53,0 52,3 51,9 51,5 51,2
Để hỗn hợp đệm với giá trị pH cần thiết, lấy những thể tích dung dịch CH3COOH là ở bảng trên, thêm 50ml NaOH 1N, thêm nước đến 500ml rồi lắc đều.
Hỗn hợp đệm CH3COOH + CH3COONH4 pH CH3COOH 0,2N NH4OH 3,0 3,4 3,8 4,2 4,6 5,0 5,4 5,8 6,2 6,6 7,0 7,4 7,8 8,2 8,6 9,0
99,24 93,40 88,20 80,20 66,00 58,60 54,60 50,90 50,40 50,30 50,00 48,40 47,80 46,40 44,70 41,00
0,76 6,60 11,80 19,80 34,00 41,40 45,40 49,10 49,60 49,70 50,00 51,60 52,20 63,60 55,30 59,00
9,4 9,8 10,2 10,6 11,0
34,00 28,80 16,00 9,80 2,48
66,00 76,20 84,00 90,20 97,52
Hỗn hợp những thể tích CH3COOH 0,2N và NH4OH 0,2N lấy theo bảng trên rồi thêm nước đến 200ml và lắc đều.
3. Pha dung dịch % axít và amoniac (ml dung dịch đặc để pha thành 1 lít)
Hoá chất để pha
Tỉ trọng của hoá chất ở
150C
Nồng độ phần %
Các nồng độ phần trăm muối pha
HCl 1,19 37,23 634,8 496,8 236,4 115,2 45,5 22,6 H2SO4 1,84 95,6 167,7 129,9 60,6 29,3 11,5 5,60 HNO3 1,40 65,6 313,6 243,6 115,0 56,0 22,0 10,80
CH3COOH 1,05 99,5 217,8 196,7 97,1 48,2 19,2 9,00
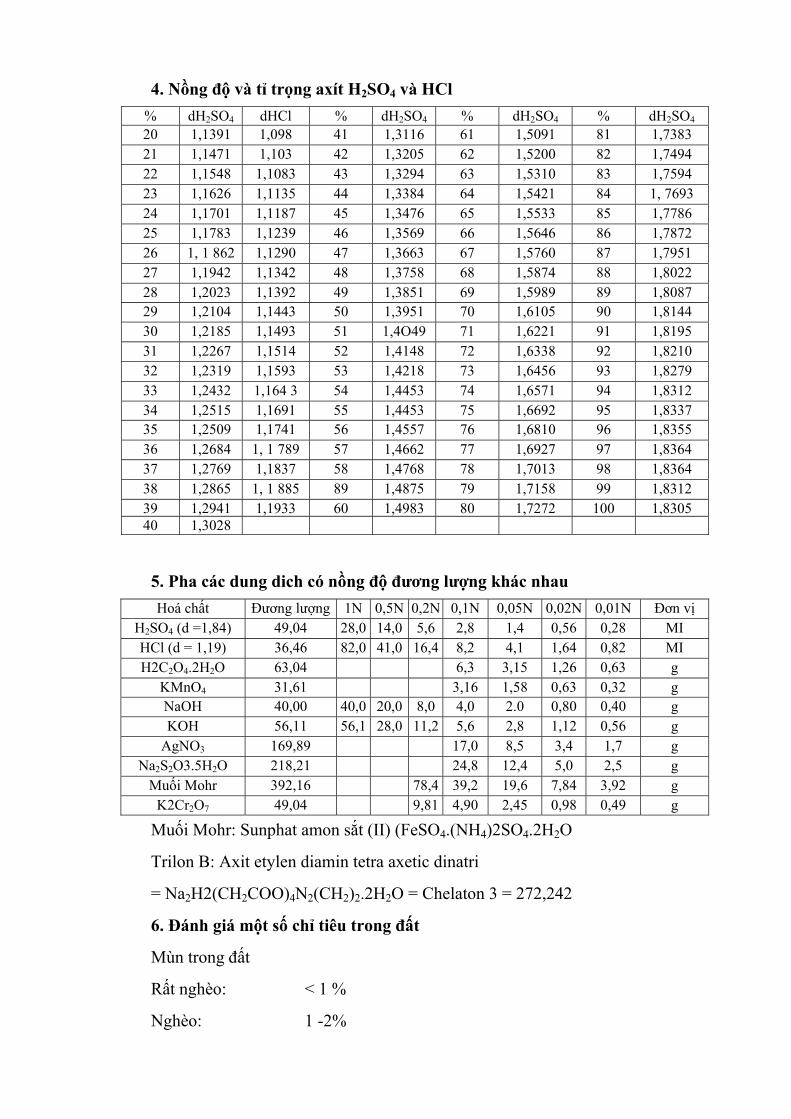
4. Nồng độ và tỉ trọng axít H2SO4 và HCl % dH2SO4 dHCl % dH2SO4 % dH2SO4 % dH2SO4 20 1,1391 1,098 41 1,3116 61 1,5091 81 1,7383 21 1,1471 1,103 42 1,3205 62 1,5200 82 1,7494 22 1,1548 1,1083 43 1,3294 63 1,5310 83 1,7594 23 1,1626 1,1135 44 1,3384 64 1,5421 84 1, 7693 24 1,1701 1,1187 45 1,3476 65 1,5533 85 1,7786 25 1,1783 1,1239 46 1,3569 66 1,5646 86 1,7872 26 1, 1 862 1,1290 47 1,3663 67 1,5760 87 1,7951 27 1,1942 1,1342 48 1,3758 68 1,5874 88 1,8022 28 1,2023 1,1392 49 1,3851 69 1,5989 89 1,8087 29 1,2104 1,1443 50 1,3951 70 1,6105 90 1,8144 30 1,2185 1,1493 51 1,4O49 71 1,6221 91 1,8195 31 1,2267 1,1514 52 1,4148 72 1,6338 92 1,8210 32 1,2319 1,1593 53 1,4218 73 1,6456 93 1,8279 33 1,2432 1,164 3 54 1,4453 74 1,6571 94 1,8312 34 1,2515 1,1691 55 1,4453 75 1,6692 95 1,8337 35 1,2509 1,1741 56 1,4557 76 1,6810 96 1,8355 36 1,2684 1, 1 789 57 1,4662 77 1,6927 97 1,8364 37 1,2769 1,1837 58 1,4768 78 1,7013 98 1,8364 38 1,2865 1, 1 885 89 1,4875 79 1,7158 99 1,8312 39 1,2941 1,1933 60 1,4983 80 1,7272 100 1,830540 1,3028
5. Pha các dung dich có nồng độ đương lượng khác nhau Hoá chất Đương lượng 1N 0,5N 0,2N 0,1N 0,05N 0,02N 0,01N Đơn vị
H2SO4 (d =1,84) 49,04 28,0 14,0 5,6 2,8 1,4 0,56 0,28 MI HCl (d = 1,19) 36,46 82,0 41,0 16,4 8,2 4,1 1,64 0,82 MI H2C2O4.2H2O 63,04 6,3 3,15 1,26 0,63 g
KMnO4 31,61 3,16 1,58 0,63 0,32 g NaOH 40,00 40,0 20,0 8,0 4,0 2.0 0,80 0,40 g KOH 56,11 56,1 28,0 11,2 5,6 2,8 1,12 0,56 g
AgNO3 169,89 17,0 8,5 3,4 1,7 g Na2S2O3.5H2O 218,21 24,8 12,4 5,0 2,5 g
Muối Mohr 392,16 78,4 39,2 19,6 7,84 3,92 g K2Cr2O7 49,04 9,81 4,90 2,45 0,98 0,49 g
Muối Mohr: Sunphat amon sắt (II) (FeSO4.(NH4)2SO4.2H2O
Trilon B: Axit etylen diamin tetra axetic dinatri
= Na2H2(CH2COO)4N2(CH2)2.2H2O = Chelaton 3 = 272,242
6. Đánh giá một số chỉ tiêu trong đất
Mùn trong đất
Rất nghèo: < 1 %
Nghèo: 1 -2%
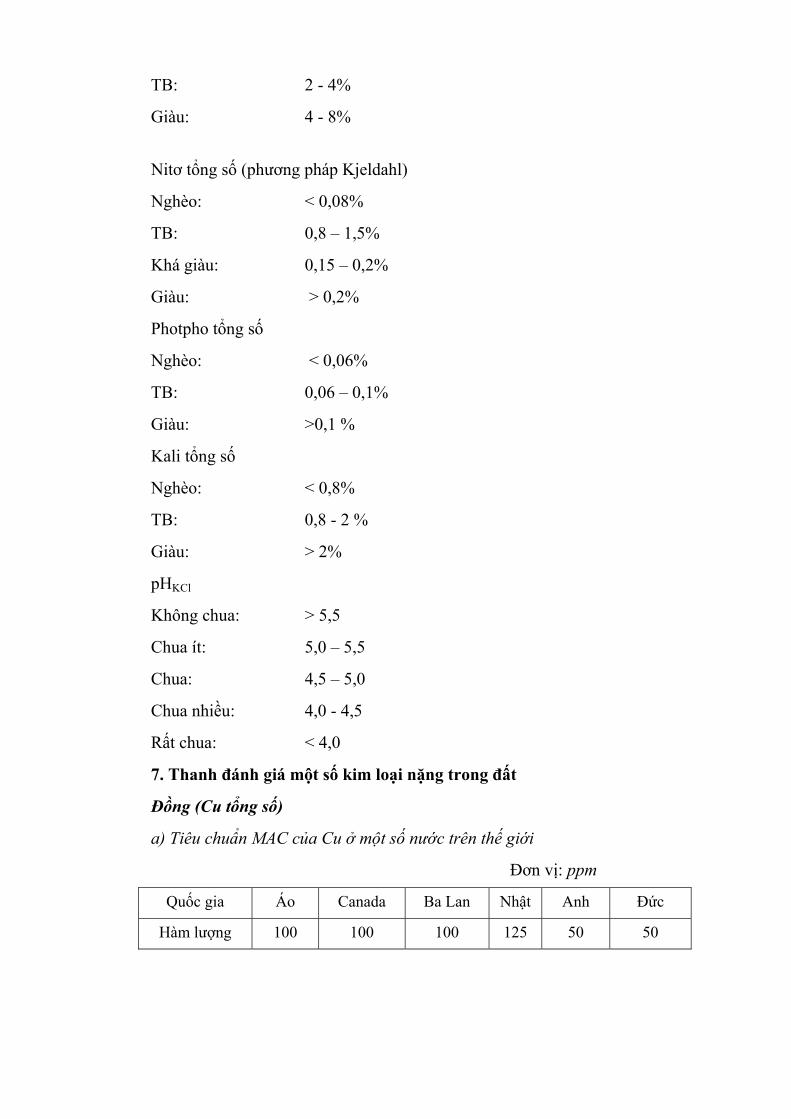
TB: 2 - 4%
Giàu: 4 - 8%
Nitơ tổng số (phương pháp Kjeldahl)
Nghèo: < 0,08%
TB: 0,8 – 1,5%
Khá giàu: 0,15 – 0,2%
Giàu: > 0,2%
Photpho tổng số
Nghèo: < 0,06%
TB: 0,06 – 0,1%
Giàu: >0,1 %
Kali tổng số
Nghèo: < 0,8%
TB: 0,8 - 2 %
Giàu: > 2%
pHKCl
Không chua: > 5,5
Chua ít: 5,0 – 5,5
Chua: 4,5 – 5,0
Chua nhiều: 4,0 - 4,5
Rất chua: < 4,0
7. Thanh đánh giá một số kim loại nặng trong đất
Đồng (Cu tổng số)
a) Tiêu chuẩn MAC của Cu ở một số nước trên thế giới
Đơn vị: ppm
Quốc gia Áo Canada Ba Lan Nhật Anh Đức
Hàm lượng 100 100 100 125 50 50

b) Đánh giá mức độ ô nhiễm đắt một bởi đồng của Ba Lan
Đơn vị: ppm Nhóm đất 0 I II II IV V
A B C
< 15 < 25 < 40
1 5 - 30 25 - 50 40 - 70
30 - 50 50 - 80 70 - 100
50 - 80 80 - 100 100 - 150
80 - 300 100 - 500 150 - 750
> 300 > 500 > 750
(Nguồn: Kabata & Pendias, 1992)
Nhóm đất A: đất có thành phần cơ giới nhẹ và trung bình, pH < 5.5
Nhóm đất B: đất có thành phần cơ giới nặng và trung bình, pH < 5.5
Nhóm đất C: đất có thành phần cơ giới nặng, giàu chất hữu cơ, pH: 5.5 – 6.5 Hướng dẫn sử dụng các loại đất trên:
* Đất ô nhiễm loại I: đất bi nhiễm đồng nhẹ, có thể canh tác với tất cả các cấy trồng, không nên trồng rau cho trẻ nhỏ
* Đất ô nhiễm loại II: đất bị ô nhiễm trung bình, có thể trồng các loại ngũ cốc, tránh ra diếp và rau Bina.
* Đất ô nhiễm loại III: đất bị ô nhiễm tương đối nặng, loại đất này chỉ nên sử dụng cho việc trồng cây công nghiệp
* Đất ô nhiễm loại IV: đất bị ô nhiễm nặng, tuyệt đối không nên trồng các cây làm thức ăn, nên trồng cây công nghiệp đặc biệt là các loại cây lấy cồn, lấy dầu dùng làm chất đốt.
Đất ô nhiễm V: đất bị ô nhiễm rất nặng, phải được làm sạch trước khi sử dụng.
Kẽm
Tiêu chuẩn MA C của Zn ở mã số nước trên thế giới
Đơn vị: ppm
Quốc gia Áo Canada Ba Lan Nhật Anh Đức
Hàm lượng 300 400 300 250 150 300
Chì
Tiêu chuẩn MA C của Pb ở mã số nước trên thế giới
Đơn vị: ppm
Quốc gia Áo Canada Ba Lan Nhật Anh Đức
Hàm lượng 100 200 100 400 50 500
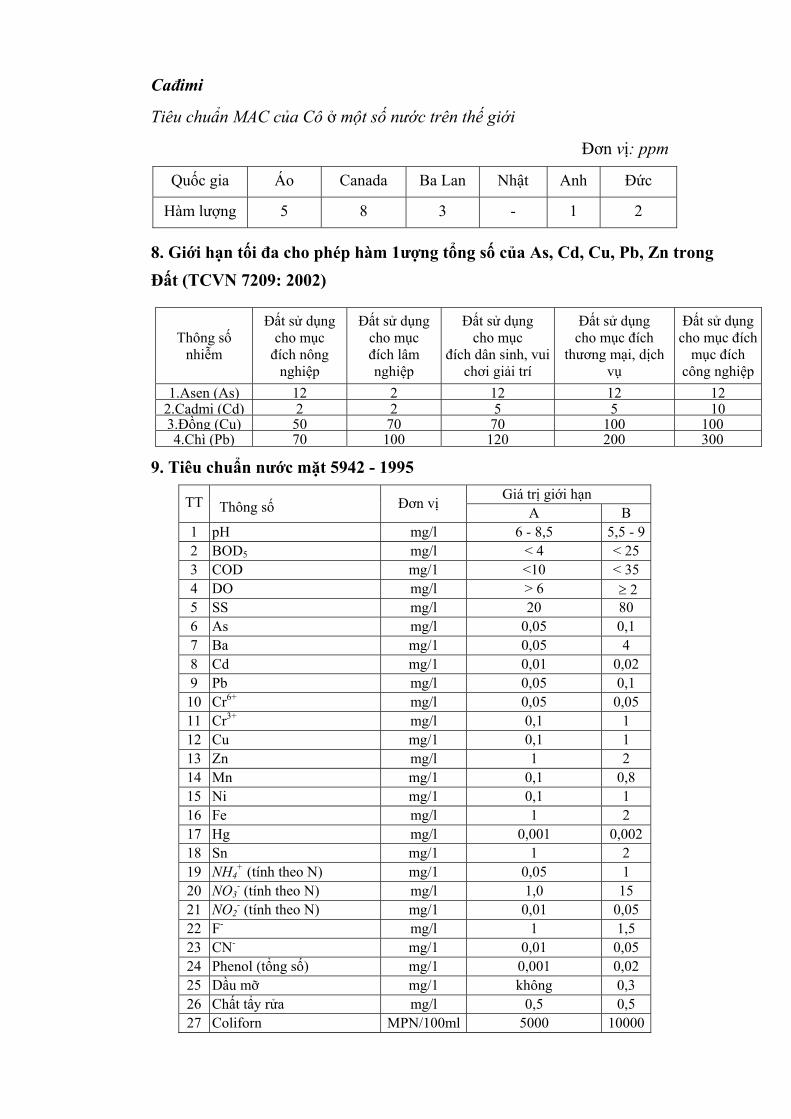
Cađimi
Tiêu chuẩn MAC của Cô ở một số nước trên thế giới
Đơn vị: ppm
Quốc gia Áo Canada Ba Lan Nhật Anh Đức
Hàm lượng 5 8 3 - 1 2
8. Giới hạn tối đa cho phép hàm 1ượng tổng số của As, Cd, Cu, Pb, Zn trong Đất (TCVN 7209: 2002)
9. Tiêu chuẩn nước mặt 5942 - 1995 Giá trị giới hạn TT
Thông số
Đơn vị A B
1 pH mg/l 6 - 8,5 5,5 - 9 2 BOD5 mg/l < 4 < 25 3 COD mg/1 <10 < 35 4 DO mg/l > 6 ≥ 2 5 SS mg/l 20 80 6 As mg/l 0,05 0,1 7 Ba mg/1 0,05 4 8 Cd mg/1 0,01 0,02 9 Pb mg/l 0,05 0,1
10 Cr6+ mg/l 0,05 0,05 11 Cr3+ mg/l 0,1 1 12 Cu mg/1 0,1 1 13 Zn mg/l 1 2 14 Mn mg/1 0,1 0,8 15 Ni mg/1 0,1 1 16 Fe mg/l 1 2 17 Hg mg/l 0,001 0,002 18 Sn mg/1 1 2 19 NH4
+ (tính theo N) mg/1 0,05 1 20 NO3
- (tính theo N) mg/l 1,0 15 21 NO2
- (tính theo N) mg/1 0,01 0,05 22 F- mg/l 1 1,5 23 CN- mg/1 0,01 0,05 24 Phenol (tổng số) mg/1 0,001 0,02 25 Dầu mỡ mg/1 không 0,3 26 Chất tẩy rửa mg/l 0,5 0,5 27 Coliforn MPN/100ml 5000 10000
Thông số nhiễm
Đất sử dụng cho mục đích nông
nghiệp
Đất sử dụng cho mục đích lâm nghiệp
Đất sử dụng cho mục
đích dân sinh, vui chơi giải trí
Đất sử dụng cho mục đích
thương mại, dịch vụ
Đất sử dụng cho mục đích
mục đích công nghiệp
1.Asen (As) 12 2 12 12 122.Cadmi (Cd) 2 2 5 5 103.Đồng (Cu) 50 70 70 100 100 4.Chì (Pb) 70 100 120 200 300
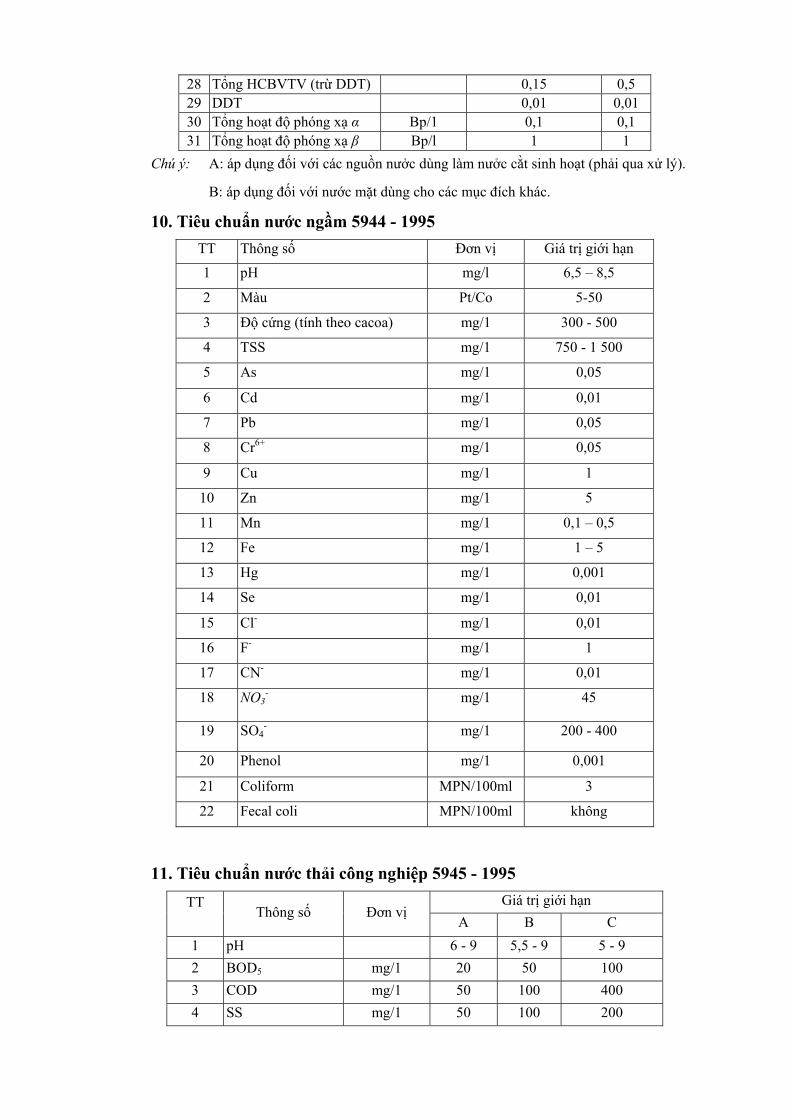
28 Tổng HCBVTV (trừ DDT) 0,15 0,5 29 DDT 0,01 0,01 30 Tổng hoạt độ phóng xạ α Bp/1 0,1 0,1 31 Tổng hoạt độ phóng xạ β Bp/l 1 1
Chú ý: A: áp dụng đối với các nguồn nưởc dùng làm nưởc cằt sinh hoạt (phải qua xử lý).
B: áp dụng đối với nước mặt dùng cho các mục đích khác.
10. Tiêu chuẩn nước ngầm 5944 - 1995 TT Thông số Đơn vị Giá trị giới hạn
1 pH mg/l 6,5 – 8,5
2 Màu Pt/Co 5-50
3 Độ cứng (tính theo cacoa) mg/1 300 - 500
4 TSS mg/1 750 - 1 500
5 As mg/1 0,05
6 Cd mg/1 0,01
7 Pb mg/1 0,05
8 Cr6+ mg/1 0,05
9 Cu mg/1 1
10 Zn mg/1 5
11 Mn mg/1 0,1 – 0,5
12 Fe mg/1 1 – 5
13 Hg mg/1 0,001
14 Se mg/1 0,01
15 Cl- mg/1 0,01
16 F- mg/1 1
17 CN- mg/1 0,01
18 NO3- mg/1 45
19 SO4- mg/1 200 - 400
20 Phenol mg/1 0,001
21 Coliform MPN/100ml 3
22 Fecal coli MPN/100ml không
11. Tiêu chuẩn nước thải công nghiệp 5945 - 1995 Giá trị giới hạn TT
Thông số Đơn vị
A B C 1 pH 6 - 9 5,5 - 9 5 - 9 2 BOD5 mg/1 20 50 100 3 COD mg/1 50 100 400 4 SS mg/1 50 100 200
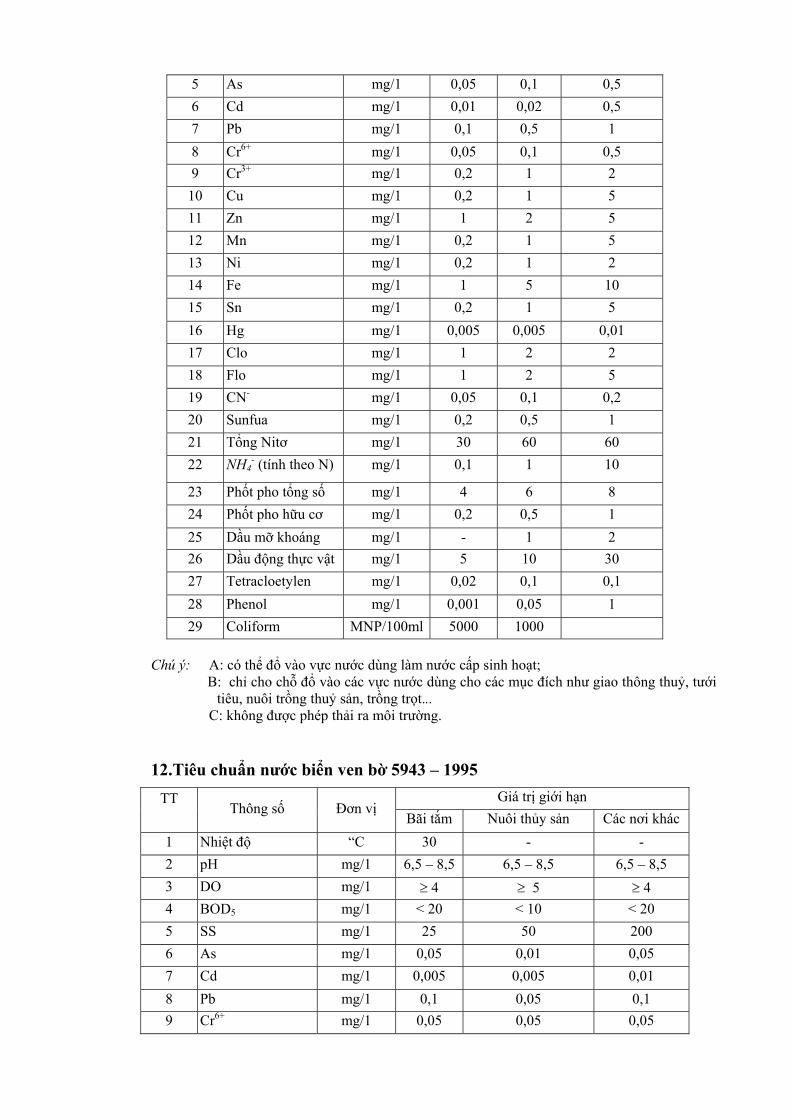
5 As mg/1 0,05 0,1 0,5 6 Cd mg/1 0,01 0,02 0,5 7 Pb mg/1 0,1 0,5 1 8 Cr6+ mg/1 0,05 0,1 0,5 9 Cr3+ mg/1 0,2 1 2
10 Cu mg/1 0,2 1 5 11 Zn mg/1 1 2 5 12 Mn mg/1 0,2 1 5 13 Ni mg/1 0,2 1 2 14 Fe mg/1 1 5 10 15 Sn mg/1 0,2 1 5 16 Hg mg/1 0,005 0,005 0,01 17 Clo mg/1 1 2 2 18 Flo mg/1 1 2 5 19 CN- mg/1 0,05 0,1 0,2 20 Sunfua mg/1 0,2 0,5 1 21 Tổng Nitơ mg/1 30 60 60 22 NH4
- (tính theo N) mg/1 0,1 1 10
23 Phốt pho tổng số mg/1 4 6 8 24 Phốt pho hữu cơ mg/1 0,2 0,5 1 25 Dầu mỡ khoáng mg/1 - 1 2 26 Dầu động thực vật mg/1 5 10 30 27 Tetracloetylen mg/1 0,02 0,1 0,1 28 Phenol mg/1 0,001 0,05 1 29 Coliform MNP/100ml 5000 1000
Chú ý: A: có thể đổ vào vực nước dùng làm nước cấp sinh hoạt;
B: chỉ cho chỗ đổ vào các vực nước dùng cho các mục đích như giao thông thuỷ, tưới tiêu, nuôi trồng thuỷ sản, trồng trọt...
C: không được phép thải ra môi trường.
12.Tiêu chuẩn nước biển ven bờ 5943 – 1995 Giá trị giới hạn TT
Thông số Đơn vị
Bãi tắm Nuôi thủy sản Các nơi khác 1 Nhiệt độ “C 30 - - 2 pH mg/1 6,5 – 8,5 6,5 – 8,5 6,5 – 8,5 3 DO mg/1 ≥ 4 ≥ 5 ≥ 4 4 BOD5 mg/1 < 20 < 10 < 20 5 SS mg/1 25 50 200 6 As mg/1 0,05 0,01 0,05 7 Cd mg/1 0,005 0,005 0,01 8 Pb mg/1 0,1 0,05 0,1 9 Cr6+ mg/1 0,05 0,05 0,05
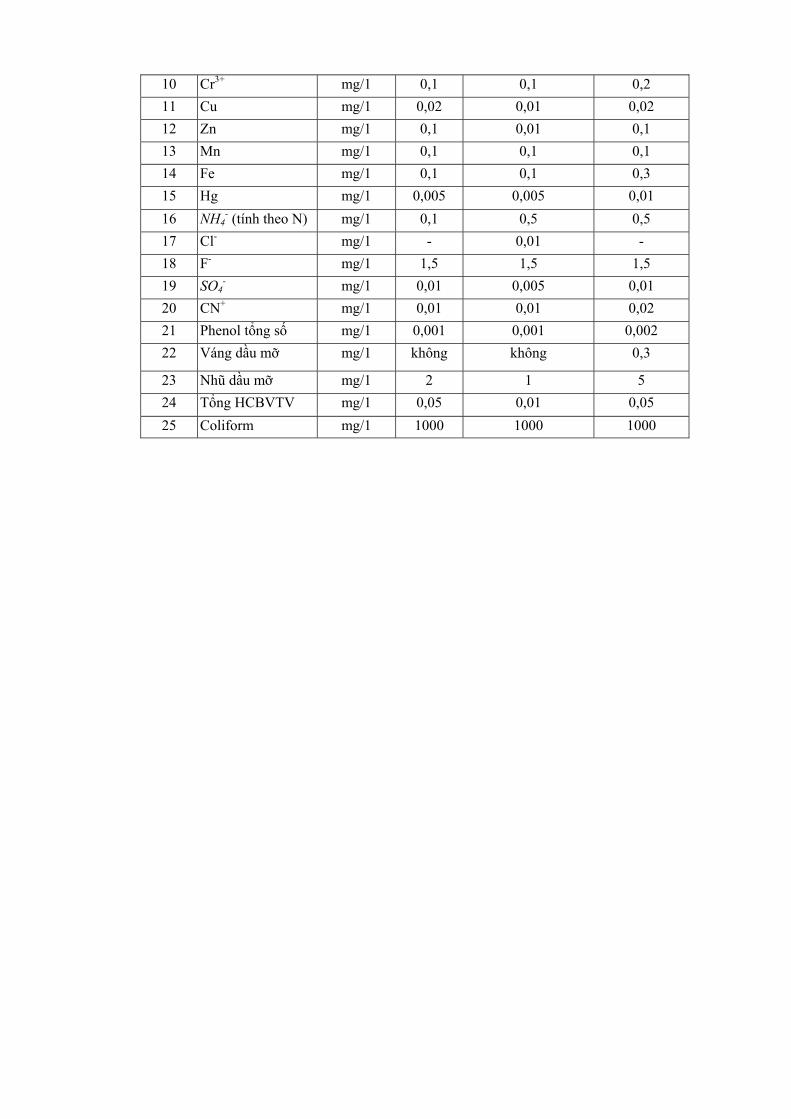
10 Cr3+ mg/1 0,1 0,1 0,2 11 Cu mg/1 0,02 0,01 0,02 12 Zn mg/1 0,1 0,01 0,1 13 Mn mg/1 0,1 0,1 0,1 14 Fe mg/1 0,1 0,1 0,3 15 Hg mg/1 0,005 0,005 0,01 16 NH4
- (tính theo N) mg/1 0,1 0,5 0,5 17 Cl- mg/1 - 0,01 - 18 F- mg/1 1,5 1,5 1,5 19 SO4
- mg/1 0,01 0,005 0,01 20 CN+ mg/1 0,01 0,01 0,02 21 Phenol tổng số mg/1 0,001 0,001 0,002 22 Váng dầu mỡ mg/1 không không 0,3
23 Nhũ dầu mỡ mg/1 2 1 5 24 Tổng HCBVTV mg/1 0,05 0,01 0,05 25 Coliform mg/1 1000 1000 1000

TÀI LIỆU THAM KHẢO CHÍNH
1. A.P.Kreskov, người dịch: Tử Trọng Nghi, Trần Tứ Hiếu (1989): Cơ sở hoá học phân tích: Cơ sở lý thuyết phân tích định tính, Nxb Đại học và Giáo dục chuyên nghiệp.
2. A.P.Kreskov, người dịch: Tử Trọng Nghi, Trần Tứ Hiếu (1989): Cơ sở lý thuyết phân tích định lượng, Nxb Đại học và Giáo dục chuyên nghiệp
3. Các tiêu chuẩn nhà nước Việt Nam và môi trường(1995), Hà Nội.
4. Lê Đức và các tác giả (2004): Một số phương pháp phân tích môi trường, Nhà Xuất Bản Đại học Quốc gia Hà Nội
5. F.W.Fifield and P.J. Haines (2000): Environmental Anlytical Chemistry, Blackle Academic & Professional
6. Lê Văn Khoa, Nguyễn Xuân Cự, Bùi Thị Ngọc Dung, Lê Đức, Trần Khắc Hiệp, Cái Văn Tranh (2000): Phương pháp phân tích, nước, phân bón và cây trồng, NXB Giáo dục, Hà Nội.
7. Từ Văn Mặc(1995): Phân tích hoá lý, NXB KHKT, Hà Nội.
8. Từ Vọng Nghi, Huỳnh Văn Trung, Trần Tứ Hiếu(1986): Phân tích nước, NXB KHKT, Hà Nội.
9. AmoldE. Greenberg et al.(1992): Sandard Methods for the Examination of Water and Waste Water, 18h - Enition, Amencan Public Health Association
10. G.R. Chhatwal et al.(1989): Environmental Analysis, Anmol Publications, NewDelhi.
11. Pradyot Patnaik (1997): Handbook of EnvironmentaIAnaIysis, Lewis Publishers.
12. Stewart E.Allen (1989): Chemical analysls of Ecologlcal materials, Blackwell Scientific Publications.
13. Lâm Minh Triết, Diệp Ngọc Sương (2000): Các phương pháp phân tích kim loại trong nước và nước thải, Nhà xuất băn khoa học và kỹ thuật.
14. W.Schneider (1988): Water Analysls, Springer - Verlag Berlin Heidelberg

MỤC LỤC
Trang
LỜI MỞ ĐẦU ............................................................................................................................2 Phần 1: NHỮNG VẤN ĐỀ CHUNG .........................................................................................3 Chương 1: MỞ ĐẦU..................................................................................................................3
1.1. Môi trường...................................................................................................................3 1.2. Phân tích môi trường ...................................................................................................3 1.3. Sự lựa chọn phương pháp để phân tích môi trường ....................................................3 l.4. Giá trị của các số liệu trong phân tích môi trường .......................................................4 1.5. Ảnh hưởng của cân bằng .............................................................................................5
Chương 2: ĐỘ CHÍNH XÁC VÀ ĐỘ TIN CẬY CỦA PHÉP PHÂN TÍCH ............................8 2.1. Bảo đảm và kiểm soát chất lượng trong phân tích môi trường ...................................8 2.2. Sai số và độ chính xác .................................................................................................8 2.3. Đồ thị kiểm tra...........................................................................................................15
Phần 2: MỘT SỐ PHƯƠNG PHÁP DÙNG TRONG PHÂN TÍCH MÔI TRƯỜNG.............17 Chương 3: PHƯƠNG PHÁP TRẮC QUANG.........................................................................17
3.1. Phương pháp so màu quang điện...............................................................................17 3.2. Phương pháp quang kế ngọn lửa (Flamephotomet)...................................................20 3.3. Phương pháp quang phổ hấp thụ nguyên tử (AAS) ..................................................23
Chương 4: PHƯƠNG PHÁP ĐIỆN HOÁ................................................................................29 4.1. Cực chọn lọc ion........................................................................................................29 4.2. Phương pháp cực phổ ................................................................................................39
Chương 5: CÁC PHƯƠNG PHÁP PHÂN TÍCH SẮC KÝ.....................................................49 5.1. Mở đầu.......................................................................................................................49 5.2. Một số khái niệm .......................................................................................................49 5.3. Sắc ký lỏng hiệu năng cao .........................................................................................54 5.4. Sắc ký khí (GC - Gas chromatography) ....................................................................56
Chương 6: CÁC LOẠI NƯỚC VÀ CÁC PHƯƠNG PHÁP PHÂN TÍCH NƯỚC.................60 6.1. Đại cương về các loại nước .......................................................................................60 6.2. Phân tích nước ...........................................................................................................62 6.3. Lấy và bảo quản mẫu nước........................................................................................63 6.4. Xác định thành phần hoá học của nước.....................................................................67 6.5. Xác định một số tính chất khác của nước..................................................................87
Chương 7: PHÂN TÍCH ĐẤT..................................................................................................95 7. 1. Giới thiệu chung .......................................................................................................95 7.2. Phân tích một số tính chất lý hoá học cơ bản của đất..............................................105 7.3. Xác định một số kim loại nặng trong đất.................................................................135 7.4. Sử dụng phương pháp quang phổ hấp thụ nguyên tử để xác định các kim loại nặng........................................................................................................................................164
Chương 8: PHÂN TÍCH KHÍ.................................................................................................166 8.1. Giới thiệu chung ......................................................................................................166 8.2. Phân tích khí ............................................................................................................169
Chương 9: CÁC THÔNG SỐ Ô NHIẺM CẦN KIỂM SOÁT...............................................181 9.1. Khái niệm chung......................................................................................................181 9.2. Các nguyên tắc chung trong kiểm soát ô nhiễm môi trường:..................................181 9.3. Các thông số chất lượng hay ô nhiễm môi trường cần kiểm soát............................182
Phần 3: THỰC HÀNH ........................................................................................................... 190 Chương 10: XỬ LÝ SỐ LIỆU THỐNG KÊ TRONG MÔI TRƯỜNG.................................190 Chương 11: THỰC HÀNH PHÂN TÍCH TRONG PHÒNG THÍ NGHIỆM........................193

l. Xác định pH của nước .................................................................................................193 2. Xác định độ cứng của nước ........................................................................................193 3. Xác định Ca và Mg trong nước sinh hoạt...................................................................194 4. Xác định nhu cẩu ôxy hóa học (COD) .......................................................................195 5. Xác định pH trong đất ................................................................................................195 6. Xác định chất hữu cơ trong đất (Walkley-Black).......................................................196 7. Xác định đạm dễ tiêu (NH4
+) trong đất ......................................................................198 PHỤ LỤC ...............................................................................................................................199 TÀI LIỆU THAM KHẢO CHÍNH ........................................................................................209


![A · Web viewVận dụng giải đề thi Đại học – Cao đẳng: [ĐH-Mỏ địa chất Hà Nội-1995] [ĐH-Mỹ thuật CN Hà Nội-1996] [ĐH-GTVT Hà Nội-2000] [ĐH](https://img.pdfslide.tips/doc/110x75/5e48f321f0e9fc64755f8060/a-web-view-vn-dng-gii-thi-i-hc-a-cao-ng-h-m.jpg)


























