Embed Size (px)
Citation preview

INVESTIGATION OF THE BETA 2 ADRENERGIC RECEPTOR (Β2-AR) PATHWAY IN CANINE HEMANGIOSARCOMA
BY
ROBERTA PORTELA
THESIS
Submitted in partial fulfillment of the requirements
for the degree of Master of Science in VMS – Veterinary Clinical Medicine in the Graduate College of the
University of Illinois at Urbana – Champaign, 2014
Urbana, Illinois
Master’s Committee: Assistant Professor Jackie M. Wypij, Chair
Associate Professor Timothy M. Fan Assistant Professor Stephane Lezmi

ii
ABSTRACT
Canine hemangiosarcoma is a highly metastatic cancer arising from vascular endothelial cells. It
is one of the most aggressive canine cancers and most dogs die from this disease within a few
months of the diagnosis. Despite advancements in veterinary oncology, there has been minimal
improvement in the overall survival time even with standard treatment, which includes surgery
and chemotherapy. Propranolol, an oral drug originally developed for the treatment of
cardiovascular diseases, has been successfully used for the treatment of infantile hemangioma
which is a benign neoplasia of vascular endothelial cells. Propranolol blocks adrenergic
receptors, which would otherwise bind to catecholamines responsible for the “stress signal”
leading to many physiologic changes. Stress has been implicated in many models of
carcinogenesis and tumor progression. Given the relationship between stress and cancer, as well
as similarities between canine hemangiosarcoma and infantile hemangioma, we sought to
investigate the presence of the beta 2 adrenergic receptor and the effects of propranolol in canine
hemangiosarcoma. We demonstrated the presence of the beta 2 adrenergic receptor via
immunohistochemistry in all 18 tissue samples of spontaneous canine splenic hemangiosarcoma
and in canine hemangiosarcoma cell lines Fitz and DEN, cell line receptor expression was also
confirmed with Western blot. Src, a possible intermediary downstream protein involved in
adrenergic signaling was also investigated and both Fitz and DEN exhibited the presence of the
Src protein in Western blot. Activation of this pathway would involve phosphorylation of Src
upon catecholamine binding to the receptor, which was investigated through Western blot. Fitz
and DEN exhibited basal phosphorylation of Src and after treatment with norepinephrine, and
Fitz exhibited greater phosphorylation (23% increase compared to basal control) after 60-minute
exposure to the agonist. Fitz cells were pretreated with a biologically achievable dose of

iii
propranolol followed by the agonist, and a modest decrease in phosphorylation was observed
(11% decrease compared to basal level). Further investigation into the biological effects of
propranolol in Fitz and DEN revealed a decrease in VEGF secretion, increase in proliferation
and decrease in cell migration. Reduction in VEGF secretion was evaluated via ELISA and it
was present at propranolol doses greater than 10 µM for DEN and Fitz, achieving a maximum
reduction of 21% in DEN and 44% in Fitz compared to untreated cells. Cell proliferation was
measured through MTS assay, which revealed an increase in cell proliferation only in Fitz cells
treated with 0.1 µM of NE (25% increase) and cells treated with 0.1 µM of propranolol (30%
increase). Cell migration was evaluated with a scratch assay and was decreased only when cells
were treated with propranolol at a high dose (100 µM). Taken together, the findings of this study
show that beta 2 adrenergic receptors are expressed by canine hemangiosarcoma, Src may be
involved in the downstream signaling from the receptor and blockade of the receptor leads to
mild to moderate effects in cell angiogenesis, proliferation and migration.

iv
For my family,
Luiz, Thais, Rodrigo and Victoria Portela
for never letting me feel alone
and for always believing in me

v
ACKNOWLEDGEMENTS
Thank you to my advisor, Dr. Jackie Wypij, for all her help in this project.
My sincere thank you to Dr. Timothy Fan, for the enormous patience, without whom I
could not have accomplished this.
Thank you to Dr. Stephane Lezmi for the support not only with immunohistochemistry
but also with ideas and encouragement that inspired me to keep going.
A huge thank-you to my friend Evelyn Caporali for her incredible help and support when
I needed it most. Thank you so much.
Thanks to Kevin LeBoedec for taking the time to help me with statistics.
Special thank you to Holly Pondenis, for all the technical knowledge shared and
assistance with experiments. Her expertise is invaluable.
Thank you so much to my resident-mates, Sharon Shor, Zach Neumann and Alycen
Lundberg; technicians Jenny Byrd, Rebecca Kamerer and Tara Bailey, and Dr. Laura Garrett
from the Oncology Service for their words of encouragement throughout the years.
My gratitude to James R. Harkness, Wayne D. and Josephine H Spangler Endowment
fund for the financial assistance in this project.
Lastly, thank you to all my friends everywhere in the world, who at some point have
given me instructions, directions, shelter, a shoulder to cry on, and specially have pushed me to
move forward into success. Their kindness will never be forgotten.

vi
TABLE OF CONTENTS
CHAPTER 1: INTRODUCTION………………………………………………….. ……………. 1
CHAPTER 2: LITERATURE REVIEW………………………………………………………… 3
CHAPTER 3: MATERIALS AND METHODS.………………………………………………. 28
CHAPTER 4: RESULTS……………………………………………………………………….. 38
CHAPTER 5: DISCUSSION AND CONCLUSIONS…………………………………………. 43
CHAPTER 6: FIGURES.………………………………………………………………………. 49
REFERENCES……………………………………………………………………………….….58

1
CHAPTER 1
INTRODUCTION
Canine hemangiosarcoma (cHSA) is a malignant neoplasm of endothelial cells that
affects many species, being more frequent in dogs [1-3]. This disease is highly metastatic and the
prognosis is poor, with a median survival time of about 6 months with standard therapy, which
involves surgery and chemotherapy treatment [4]. Limited advancements have been made to
improve survival time therefore novel therapies are needed. Drug repositioning, which uses
known drugs for alternative purposes (treatment of new diseases), is a strategy that can be
pursued with the advantage of being of lower cost (pharmacokinetics and pharmacodynamics are
already known; drugs already in the market can be selected for their cost) and known tolerability.
Recently, propranolol has received attention for its effects on the treatment of infantile
hemangioma (IH), a benign neoplasm of endothelial cells. It was accidentally discovered while
treating infants with hypertension that propranolol (a β-adrenergic receptor antagonist) has
activity against IH, even in treatment-resistant cases [5]. This prompted many studies, mostly
clinical [6-11], looking at the effects of propranolol as an anti-neoplastic drug. Since both cHSA
and IH are tumors of endothelial cell origin, we developed an interest in investigating the effects
of propranolol in cHSA. Parallel to IH and the recent propranolol discovery, there is also a link
between stress and cancer. Stress is physiologically described as the binding of catecholamines
to the adrenergic receptors, which mediate the “fight-or-flight” response necessary for survival
of an organism. An interesting hypothesis is that this adrenergic signaling may also be
functioning to maintain survival and/or progression of neoplastic cells exposed to the same
catecholamines released during stressful situations, which are common during diagnosis and

2
treatment of cancer. To further support this hypothesis, studies linking breast cancer survival
and β-blockers have been published suggesting that inhibition of the β-adrenergic signaling
pathway can reduce breast cancer progression, recurrence, metastasis and mortality [12, 13].
Many other studies supporting the role of the β-adrenergic pathway in cancer development have
been described and will be discussed in the next section.
We hypothesized that cHSA expresses the receptors necessary for adrenergic signaling
(β2-AR), an intermediary kinase (Src) is involved in the signaling pathway downstream from the
receptor and propranolol treatment would lead to decrease in cell proliferation, angiogenesis and
migration.

3
CHAPTER 2
LITERATURE REVIEW
2.1. Stress and tumorigenesis
The process of tumorigenesis and metastasis has been extensively studied in the scientific
community. Many environmental components have been implicated in cancer development and
new compounds with carcinogenic activity are frequently being discovered. Stress is one
environmental component that may influence the carcinogenic process and potentiate
tumorigenesis. Stress is defined as “A state of mental or emotional strain or tension resulting
from adverse or very demanding circumstances” and “Pressure or tension exerted on a material
object” [14, 15]. In modern life, people are subjected to stress from many different aspects.
Stressful experiences include physical stressors such as pathogens and toxins, and psychological
stressors such as major life events, trauma, abuse, or factors related to the environment in the
home, workplace or family [16].
In 1936, Hans Selye defined stress physiologically as the state in which the
sympathoadrenomedullary system and the limbic-hypothalamic-pituitary-adrenal axis are co-
activated [16, 17]. From a biological standpoint, it is known that stress, whether physical or
mental, culminates in the release of hormones named catecholamines, which mediate the “fight-
or-flight” responses causing activation of the sympathetic nervous system [19]. The most
abundant catecholamines are epinephrine (E), norepinephrine (NE) and dopamine, which are
released from the chromaffin cells of the adrenal medulla and from neuro-muscular junctions
[19, 20]. In humans, acute stress can elevate the levels of norepinephrine and epinephrine by

4
greater than 10-fold in only a few seconds, although chronic stress can lead to fluctuations on the
catecholamine levels as well [19, 20].
Cancer patients are subjected to stress from different aspects, from dealing with the
acceptance of a terminal disease (sometimes associated with anxiety and depression) to the
homeostatic effects of stress on inflammation, immunocompetence, and cancer initiation or
progression [19, 21-28].
Evaluating stress in canine patients is difficult, especially the psychosomatic aspects.
Physiologically there have been attempts at quantifying mediators of stress signaling. Increase in
plasma epinephrine and norepinephrine levels in dogs was documented in a study that compared
levels of these catecholamines in groups subjected to environmental stress of blood collection
immediately after arrival to the laboratory versus 15 minutes after arrival, indicating the rapid
fluctuation of these hormones under different stress conditions [29]. Levels of catecholamines in
dogs subjected to various stressful stimuli were measured and in the literature they ranged
around 65 to 239 pg/mL for epinephrine, 145 to 524 pg/mL for norepinephrine and 20-437
pg/mL for dopamine [14, 29]. The release and degradation of catecholamines is variable and thus
the concentration of these hormones can differ significantly across different tissues and within
the circulating blood [2]. In humans, the difference can be in the order of nanomolar for
circulating NE versus micromolar concentrations in the tissue microenviroment [30].
2.2. Adrenergic pathway
Catecholamines mediate adrenergic signaling via receptors in target cells. Receptors
include α1A,B,D; α2A,B,C; β1,β2 and β3 and are present in a wide variety of cells and tissues,

5
including central nervous system (CNS), lungs, liver, pancreas, kidney, adrenal glands, breast,
ovary, prostate, vasculature, bone marrow and cells of the immune system (macrophages, mast
cells, lymphocytes) [19, 20]. The expression of β1 and β2 adrenergic receptors (AR) has been
extensively studied in the cardiovascular system, with high expression occurring in cardiac
myocytes and vascular smooth muscle cells [31]. These receptors belong to a family called G-
protein coupled receptors (GPCRs) [32], which contain seven transmembrane “serpentine”
segments with the N-terminal facing the extracellular space and the C-terminal immersed in the
cytoplasm. When the ligand binds to the extracellular domain of the receptor, the receptor
undergoes a conformational change, which will then activate the G-protein located in the
intracellular space. The G-protein is composed of a α, β and γ units, with the α and γ subunits
having lipid tails that covalently attach the protein to the cell membrane. In the absence of ligand
binding, the resting G-protein’s alpha subunit is bound to GDP (guanosine diphosphate). When
the signal is initiated by catecholamine binding, the receptor undergoes a conformational change
and GDP is replaced by GTP (guanosine triphosphate) in the alpha subunit of the G-protein. This
protein detaches from the transmembrane receptor and moves on to activate other downstream
proteins (Fig. 2.1). Inactivation of the protein occurs when phosphatase activity removes a
phosphate group from GTP transforming it back to GDP form [33].
Beta 2 adrenergic receptors (β2-AR) are the focus of this project due to its demonstrated
expression in different human vascular tumors [34]. These receptors activate adenylyl cyclase
which in turn will catalyze the conversion of ATP to cyclic AMP (cAMP). Cyclic AMP can act
as a secondary messenger itself or it can bind to other downstream proteins, such as protein
kinase A [33, 35, 36]. Protein kinase A can phosphorylate other target proteins including cAMP-
responsive element binding protein/activating transcription factor (CREB/ATF) and β-adrenergic

6
receptor kinase (BARK) [19]. Upon continuous stimulation, the receptor eventually becomes
inactivated by the inability to bind to G-proteins. That is achieved by phosphorylation of the
intracellular portion of the receptor by BARK, and the phosphorylated protein binds to β-
arrestin, which blocks the connection between the receptor and the G-proteins (Fig. 2.2). β-
arrestin also can function as an adaptor protein, and is this way that it recruits Src to become
bound to the β2-AR [37]. Furthermore, it has also been shown that cAMP/PKA mediates NE-
induced activation of Src [38].
Src is a non-receptor tyrosine kinase ubiquitously expressed in many tissues, and plays
an important role in transmitting signals from the cell surface to the nucleus via phosphorylation
of tyrosine residues of intracellular proteins [39]. Src is involved in the maintenance of normal
cell homeostasis including cell proliferation, survival, maintenance of cytoskeleton, cell adhesion
and motility [38, 39]. Src is overexpressed in many human malignancies, including colorectal,
breast, prostate, pancreas, head and neck carcinoma, lung carcinoma, glioma, melanoma and
sarcomas [38, 40, 41]. A recent study has demonstrated relationship between increased
phosphorylation of Src caused by NE stimulation and tumor growth and progression in ovarian
carcinoma murine models, assessed as number of nodules and tumor weight. Propranolol, a non-
selective β-blocker (described in detail later in this chapter) successfully counteracted the effects
of stress and of adrenergic agonists (isoproterenol and terbutaline) evidenced by decreased tumor
weight and number of nodules in mice, which was similar to control (non-stressed or non-treated
mice) [38]. Src has been also implicated in NE-stimulated vascular endothelial growth factor
(VEGF) production by adipocytes [42, 43]. VEGF is a cytokine known to stimulate angiogenesis
in both normal and neoplastic tissue. In addition, it was demonstrated that Src is present in
canine HSA cell lines and its reduced expression was linked to a decrease in cell viability [44].

7
2.3. Adrenergic pathway and molecular pathogenesis of cancer
2.3.1 Breast cancer
The β2-AR and its signaling pathways have been extensively studied in human oncology.
In breast cancer, preclinical models suggest that this pathway may influence breast cancer
progression through: 1) increased tumor cell survival after exposure to chemotherapeutic agents;
2) increased breast cancer cell proliferation; 3) altering the tumor microenvironment in
angiogenesis and inflammatory response [27].
At therapeutic levels, β-blockers do not seem to be directly cytotoxic; rather the β-AR
blockade may inhibit catecholamine signaling that normally would lead to pro-tumorigenic
effects (cell survival, proliferation, migration, angiogenesis) [45]. Therefore, research efforts are
now focused on the ability of β-blockers to inhibit cancer progression in already established
cancers. In one study using an orthotopic mouse model looking at the effect of chronic stress in
breast cancer progression, mice were injected with luciferase-transfected breast cancer cells into
the 4th mammary pad and physically restrained for 2 hours daily for 20 days, while the control
group was not subjected to stress. The results showed a 37-fold increase in metastasis to the
lungs and 67% increased metastasis to the lymph nodes in the stressed group compared to
control [30]. In a separate experiment, mice were pre-treated with propranolol prior to tumor
inoculation and stress stimuli. There was an increased expression of β2-AR in tumors in
propranolol-treated animals and propranolol significantly decreased the metastatic burden in
stressed animals. There was no change in metastasis for non-stressed animals, and treatment did
not affect primary tumor growth in either group [30]. This study provided some objective
evidence of the role of adrenergic signaling and breast cancer progression.

8
Catecholamines alter the cytokine profile of the bone microenvironment and promote the
incidence of metastatic colonization by breast cancer cells. Receptor activator of nuclear factor
kappa-B ligand (RANKL) is a protein involved in bone remodeling as well as dendritic cell
maturation and is secreted by osteoblasts in response to sympathetic activation. RANKL
stimulates breast cancer migration and bone colonization and is recognized as a crucial factor for
cancer cell motility in addition to its well-established role in tumor-induced osteolysis [46]. This
pathway was inhibited by propranolol therapy [47]. Activation of the sympathetic system
promoted bone metastasis in a mouse model of breast cancer. Stimulation of the β2-AR induced
RANKL expression in bone marrow osteoblasts and increased migration of metastatic MDA-
MB-231 mammary carcinoma cells in vitro. Mice models of bone metastasis subjected to chronic
stress had sympathetic activation blocked by propranolol. Sympathetic nerves inhibit osteoblast
proliferation and regulate hematopoietic stem cell proliferation, survival and trafficking [48, 49].
Norepinephrine released from the sympathetic nerves stimulate the formation of osteoclasts [50].
Perhaps one of the most commented serendipitous discovery was that women who
took β-blockers (specifically propranolol) for cardiovascular disease were found to be
significantly less likely to die of their cancer, and less likely to present at higher stages of the
disease, compared to women who did not take propranolol [12]. Another study showed a 57%
reduced risk of metastasis and a 71% reduction in breast cancer mortality after 10 years [13].
Further evidence also showed that β-blocker intake was associated with improved relapse-free
survival but not overall survival in patients with breast cancer, including triple-negative tumors
which are tumors negative for epidermal growth factor receptor (HER2), progesterone receptor
(PR) and estrogen receptor (ER) [51]. Relapse is more frequent among triple-negative breast
cancer patients, especially within the first 3 years of diagnosis, compared to patients with ER

9
positive tumors [52, 53]. High β2-AR expression was found in tumors containing estrogen
receptors that were hormonally positive, and patients carrying these tumors had a worse
prognosis five years later, coincidentally with the discontinuation of tamoxifen therapy. The
prognosis was good within the first five years, hinting at a possible advantage of combination
therapy with propranolol, which could have an impact in breast cancer survival [54].
Despite some encouraging published evidence on the benefits of propranolol in cancer
patients, there is still some controversy. There are studies suggesting an increased risk or no
survival advantage of various cancers including colon, lung, breast and prostate cancer in
patients taking β-blockers [55-57]. However, patients taking β-blockers for various medical
reasons may have already chronically elevated levels of catecholamines, which would promote
cancer progression and worse outcome in cancer-bearing patients. In that sense, the chronically
elevated NE would be a higher risk than the propranolol itself [58]. A large meta-analysis failed
to demonstrate strong evidence that β2-AR blockers promote tumor progression [59]. In
summary, these studies demonstrate that elevated levels of catecholamines as well as blockade of
the β-adrenergic receptor pathway by propranolol may have some impact in breast cancer
progression as well as patient survival.
2.3.2. Pancreatic carcinoma
In pancreatic carcinoma, β2-AR was expressed in cell lines. NE promoted increased
proliferation of these cells in a dose-dependent fashion, also promoted S-phase cell cycle shift
and decreased cells in the G1/G2 phase. Migration was also increased with NE treatment as well
as increase in phosphorylation of p38 (MAPK pathway, important in cell proliferation), which
was blocked by treatment with propranolol [60]. In another study, invasiveness and angiogenic

10
potential of pancreatic carcinoma cell lines were assessed by measurement of matrix
metalloproteinases (MMPs) MMP-2, MMP-9 and VEGF. Norepinephrine promoted the
invasiveness of one of the cell lines in a concentration-dependent manner, and NE increased the
expression of MMP-2, MMP-9, and VEGF. These effects were inhibited by propranolol [61].
Furthermore, propranolol significantly suppressed cell invasion and proliferation in comparison
to β1-adrenergic specific antagonist metoprolol. Treatment with β2-adrenoceptor antagonists
inhibited activation of transcription factors nuclear factor κB (NF-κB), activator protein 1 (AP-1)
and cAMP response element binding protein (CREB). β2-adrenoceptor antagonists also
significantly altered VEGF, cyclooxygenase-2 (COX-2), MMP-2 and MMP-9 expression
confirming previous findings. The β2-adrenergic antagonists suppressed invasion and
proliferation by inhibiting both cAMP/PKA and Ras, which regulate activation of the MAPK
pathway and transcription factors, such as NF-κB, AP-1 and CREB, as well as expression of
their target genes, MMP-9, MMP-2 and VEGF. β1-adrenergic antagonists suppressed invasion
by inhibiting only the cAMP/PKA pathway [62]. Proliferation and migration of human
pancreatic ductal carcinoma is stimulated in vitro by β-AR and subsequent cAMP-dependent
signaling, which in turn leads to cAMP-dependent release of epidermal growth factor, and PKA-
dependent release of VEGF. These effects were also blocked with propranolol treatment [63].
Another study using isoproterenol (a non-selective β-AR agonist) significantly increased cell
proliferation of a pancreatic ductal carcinoma cell line in a dose-dependent manner, with
concomitant activation of ERK/MAPK signal pathway as well as increased levels of
phosphorylated ERK [64, 65]. It has been previously demonstrated that β2-AR causes activation
of the extracellular-signal regulated kinase 1/2 (ERK 1/2).

11
In vivo, mice carrying xenograft tumors had enhanced tumor growth caused by
isoproterenol, which was suppressed by propranolol [64]. Taken together, these studies suggest
that blocking the adrenergic pathway using propranolol may have some impact in pancreatic
carcinoma progression.
2.3.3. Ovarian carcinoma
Immunohistochemistry demonstrated stress-induced increases in levels of basic fibroblast
growth factor (bFGF), MMP-2 and MMP-9 proteins in ovarian carcinoma. Catecholamines have
previously shown to promote VEGF production by ovarian cancer cells in vitro [66, 67].
Chronic behavioral stress resulted in higher levels of tissue catecholamines, greater tumor
burden and more invasive pattern of ovarian cancer growth in a mouse model; effects which
were mediated by β2-AR activation of PKA signaling in the cancer cells [68]. Another
interesting study by Thacker et al used periodic physical restraint to stimulate chronic stress in
nude mice inoculated with human ovarian carcinoma cells in the peritoneal cavity. In mice
receiving 0, 2 or 6 hours of immobilization daily for 21 days, the number of tumor nodules
increased by 259% in the 2 hr stress group and 356% in the 6 hr stress group compared to
unstressed mice. Mean tumor weight increased 242% in the 2 hr stress group and 275% in the 6
hr group compared to unstressed mice. Social isolation as an alternative stressor also showed a
187% increase in tumor weight and a 255% increase in nodule count compared with group-
housed control animals. Stressed animals and non-stressed mice treated with isoproterenol and
terbutaline (β-agonists) had both significantly enhanced mean vessel density (MVD) counts (a
measure of angiogenesis [69]) and that effect was blocked by propranolol. This was
accompanied by significant elevation of VEGF protein and mRNA within tumor tissue.

12
In another model investigating the effects of surgical stress in tumor development, mice
were injected with ovarian carcinoma cells and 4 days later subjected to either a laparotomy
procedure (larger incision, more tissue handling and stress) or a laparoscopy procedure (less
invasive, less stress). Mice in the laparoscopy group had significantly lower tumor weight
compared to mice in the laparotomy group. In the immediate postoperative period, serum levels
of VEGF and MMP-2 were significantly lower in the laparoscopy group as well, further
supporting the mediators of stress-induced tumor progression [70]. Lee et al. had also
demonstrated previously that surgery significantly increased MVD and VEGF expression, which
were blocked by propranolol treatment in mice injected with ovarian carcinoma cell lines [71].
The stress of surgery is suggested to facilitate the post-surgery growth of pre-existing
micrometastases and small residual tumors [72-74]. Taken together, these studies indicate that
psychosomatic and surgical stress, both present in cancer patients, are linked to tumor growth
and angiogenesis and propranolol appears to ameliorate these effects.
2.3.4. Prostatic carcinoma
Many studies have also been performed investigating the role of β2-AR in prostatic
carcinoma. Among the studies investigating possible molecular pathways involved in β blockade
and prostatic carcinoma, there appears to be a relationship between β2-AR and histamine, as
histamine augmented β2-AR-induced cAMP accumulation independently of known histamine
receptors [75]. β2-AR activation also promoted prostate cancer cell proliferation and cell
migration through increasing cellular cAMP and ERK 1/2 activation in another study, where it
also demonstrated the involvement of β-arrestin in this process (β-arrestin participates in agonist-
mediated desensitization of G protein-coupled receptors). The formation of β-arrestin 2/c-Src

13
complex was a key factor in this process [76]. An experiment was performed where PC-3
xenografted mice were implanted with a norepinephrine-releasing micropump which
subsequently led to a 1.6 fold increase in tumor metastasis compared to control [77]. In addition,
behavioral stress activated β2-AR signaling and led to inhibition of apoptosis and accelerated
cancer development in mice. The effects of stress were prevented with treatment using a β2-AR
selective antagonist [78].
β2-AR is a known activator of the androgen receptor, and this receptor is upregulated in
androgen-independent prostatic carcinoma cell lines. Expression of this receptor is greater in
malignant cells compared to benign hyperplasia or normal tissue [79]. One study showed that NE
drives metastasis of PC-3 cells in BALB/C3 nude mice and this process is inhibited by β-
blockers [77]. Loss of BARK-1 in a group of patients with high-grade prostatic carcinoma could
indicate a possible pathway of prostate cancer development involving β2-AR signaling [80].
BARK-1 is a serine/threonine kinase that desensitizes the receptors from catecholamine over-
stimulation (negative feedback via recruitment of beta-arrestin) [81]. In another study, β-blocker
use was not associated with increased risk of prostatic carcinoma development or overall
mortality, however a subgroup of men treated with androgen deprivation therapy and a β-blocker
had a fivefold reduced prostatic carcinoma specific mortality [82].
In summary, these studies indicate that prostatic carcinoma express the β2-AR, β2-AR
activation promote prostate cancer cell proliferation and cell migration, mice exposed to
increased levels of catecholamines have increased metastasis and men treated with β-blockers
may have reduced cancer-related mortality.
2.3.5. Other malignancies

14
Stress has been suggested as a possible factor in human colorectal carcinoma
development. In one study looking at the effects of chronic restraint stress in nude mice bearing
human colorectal carcinoma xenografts, adrenergic signaling-dependent activation of ERK 1/2
promoted cell proliferation, and β-adrenergic antagonism inhibited proliferation and decreased
phosphorylation of ERK 1/2 in vitro and in vivo. Norepinephrine and epinephrine enhanced
colorectal carcinoma cell proliferation and viability in cell culture as well as tumor growth in
mice. These effects were antagonized by propranolol and phentolamine (α-AR antagonist) [83].
The first epidemiological investigation of the effect of post-diagnostic β-blocker usage on
colorectal cancer-specific mortality showed no association. There was some evidence of a weak
reduction in all-cause mortality in β-blocker users which was in part due to the marked effect of
atenolol on cardiovascular mortality [84].
Melanoma cell lines also expressed β1 and β2-ARs. Norepinephrine and E increased
metalloprotease-dependent motility, released interleukin-6 (IL-6), interleukin-8 (IL-8) and
VEGF. The effects of these catecholamines were inhibited by propranolol [85]. Interleukin-6 is
involved in the host immune defense mechanism as well as the modulation of growth and
differentiation in various malignancies [86]. Increased expression of IL-8 and/or its receptors has
been characterized in cancer cells, endothelial cells, infiltrating neutrophils, and tumor-
associated macrophages, suggesting that IL-8 may function as a significant regulatory factor
within the tumor microenvironment [87]. Expression of β-ARs had been previously identified in
melanoma cell lines as well as human melanoma biopsies and NE upregulated production of
VEGF, IL-8, and IL-6 in the cell lines [88]. Overall, the results from preclinical studies support
the suggestion that β-blockers could provide a clinical benefit in melanoma progression however
epidemiological studies are limited by sample size, which is also a limitation in breast, colorectal

15
and prostatic carcinoma studies [89]. In patients with melanoma, the β-blocker-treated group had
an overall improved survival after a median follow-up of four years and for each year of β-
blocker use, the risk of death was reduced by 38% [90]. Another study found that both β1 and
β2-ARs are expressed in tissues from benign melanocytic nevi, atypical nevi and malignant
melanomas and that expression was significantly higher in malignant tumors.
In an in vitro multiple myeloma study, propranolol IC50 values (concentration of
propranolol required for 50% inhibition of proliferation) were decreased over time. There were
also significant increases in caspase 3 activity, in apoptotic cell population, and a decrease in
expression levels of Bcl-2 (anti-apoptotic protein) in response to propranolol treatment [91].
Caspase-3 is an executioner caspase protein, involved in the process of apoptosis (programmed
cell death). Another study showed NE-induced secretion of VEGF in 3 multiple myeloma cell
lines [92].
In conclusion, although the relationship of stress and tumor development/progression has
been more extensively studied in a number of specific carcinomas (breast, ovarian, prostatic,
colorectal, pancreatic), it appears to influence non-carcinoma malignancies as well (melanoma,
myeloma). This illustrates how broad this research field is and indicates that propranolol may
have the potential to be of benefit in the treatment of many other tumors.
2.4. Propranolol
Propranolol is an attractive potential anti-neoplastic therapeutic option since it is
affordable, easily obtainable in the market and the pharmacokinetics have been established for
humans [93] and dogs [94]. It is a non-selective β-blocker, available as propranolol

16
hydrochloride, under the brand name Inderal® in North America by AstraZeneca®. It has been
mainly used for the treatment of hypertension, although other conditions reported include
tachyarrhythmias [95], tachycardia, essential tremor [96, 97], migraine [98, 99], post-traumatic
stress disorder [100-104], glaucoma [105], akathisia (restlessness) [106], schizophrenia (elevated
levels of norepinephrine may play a causative role in the development of this disease) [107],
adrenergic urticaria [108], treatment of burn patients [109-112] and anxiety [113, 114].
In human patients, receiving the diagnosis of cancer creates significant levels of
emotional distress, with intrusive thoughts (emotional memories, flashbacks, nightmares, and
intrusive images) being the most common manifestation among breast cancer survivors. Recently
diagnosed female breast and colorectal cancer patients using β-blockers reported less cancer-
related psychological distress [115]. Although direct psychological stress may not be an
important feature in canine cancer, propranolol could potentially help to control the anxiety
associated with repeated hospital visits required for treatment.
Side effects of propranolol reported in humans most commonly include hypotension,
hypoglycemia, bradycardia, bronchospasm, sleep disturbances (nightmares), acrocyanosis
(cyanotic extremities). Other fewer reported side effects include gastroesophageal reflux, nausea,
vomiting, diarrhea, somnolence, hyperkalemia, tumor lysis syndrome, psoriatic drug rash,
respiratory syncytial virus exacerbation, and dental caries [116-119]. Propranolol is
contraindicated in patients with asthma, heart block and sinus bradycardia. The most serious side
effect of propranolol is hypoglycemia [120, 121]. Propranolol is thought to cause hypoglycemia
by inhibiting glycogenolysis, glyconeogenesis, and lipolysis. Children have lower glycogen
stores and higher glucose consumption rates when fasting and, therefore, are more susceptible to

17
hypoglycemia than adults [122]. Side effect incidence, however, does not appear high. A recent
retrospective study revealed that only 7.6% (10/132) of the children who received propranolol
treatment had to discontinue it due to side effects. No adverse effects requiring hospitalization
were recorded. [123].
In dogs, propranolol is indicated for the treatment of hypertension, atrial fibrillation,
tachyarrhythmias [124, 125], myocarditis [126], ventricular premature contractions and
arrhythmias caused by digitalis toxicity [127]. It is also useful in the treatment of hypertrophic
cardiomyopathy especially when associated with hyperthyroid disease in cats (as it inhibits
conversion of thyroxine to triiodothyronine) [128] and for urinary retention (stimulates bladder
contraction) [129]. It is contraindicated in patients with congestive heart failure unless it is
secondary to a tachyarrhythmia responsive to beta-blockade. The drug is also contraindicated in
patients with 2nd or 3rd degree heart block, sinus bradycardia, asthma and thromboembolic
disease. It should be used with caution in diabetic patients due to risk of hypoglycemia as
described in humans. Side effects are similar to humans and include hypotension, bradycardia,
hypoglycemia, decreased cardiac contractility, bronchoconstriction, peripheral vasoconstriction
and diarrhea. It is recommended that propranolol therapy be gradually withdrawn due to possible
“rebound” effect of catecholamines to the chronically suppressed β2-AR, leading to tachycardia,
arrhythmias and hypertension.
Possible drug interactions with propranolol include [130]:
• Delayed gastrointestinal absorption of propranolol by antacids
• Additive toxic effects when used concomitantly with quinidine, procainamide and
lidocaine (although anti-arrhythmic effects are enhanced)

18
• Hypotensive effects are enhanced by chlorpromazine, cimetidine, furosemide,
phenothiazines and hydralazine
• Increased serum levels of lidocaine
• Increased effects of tubocurarine and succinylcholine
• Increased action of terbutaline
• Antagonizing effects on epinephrine and phenylpropanolamine
• Antagonizing bronchodilatory effects of theophylline
• Bradycardia may be potentiated by concurrent use of digitalis
• Anti-hypertensive effects of propranolol can be inhibited by concurrent use of
salicylates
• Propranolol’s effects can be decreased by concurrent use of thyroid hormone
supplementation and the dose of propranolol may require a decrease in animals
receiving methimazole
A pharmacokinetic study of propranolol in dogs used a racemic mixture as prepared for
pharmaceutical use. A half-life of 1.09 +/- 0.33 hours was observed, which was not different than
what was previously reported for a levo isomer given as an IV bolus, however the distribution
volumes were significantly greater. A mean value of 6.5 L/Kg was observed with a range from
3.44 to 10.47 L/Kg. Total body clearance was calculated to average 68 mL/min/Kg. Both hepatic
and extra-hepatic clearance of propranolol was suggested, and approximately 90% of propranolol
is extracted from the blood as it passes through the liver. This extensive extraction results in poor
bioavailability, with only 2-17% of the dose reaching the systemic circulation unchanged.
Systemic availability increased substantially with multiple dosing (2.1-5.5 mg/Kg every 6 hours),
reaching up to 10.7 times greater than predicted by single dose. Propranolol is rapidly absorbed

19
with an absorption half-life of 17 minutes. Mean CpMax measured in 5 dogs receiving 40 mg
(2.1-5.5 mg/Kg) in multiple oral doses was 79.54 ng/mL (range 21-162.5), which is equivalent to
a mean of 0.3 µM (range 0.08-0.62 µM) [127]. Another recent study with single administration
of three 40 mg tablets revealed a Cmax of 191.4 +/- 56.52 ng/mL (0.73 +/- 0.21 µM) [131].
Propranolol is available in tablets containing 10, 20, 40, 60, 80, 90 mg and oral solution
containing 4 and 8 mg/m. Injectable propranolol is available in 1 mg/mL concentration. Standard
dosage in dogs: 0.1-0.2 mg/Kg orally every 8 hours (maximum 1.5 mg/Kg), intravenously 0.02
mg/Kg over 5-10 minutes (maximum 1 mg/Kg) [132, 133].
2.5. Propranolol and infantile hemangioma
Fairly recently (2008), propranolol was serendipitously discovered to be effective for the
treatment of infantile hemangioma after two children who were treated with propranolol for
cardiomyopathy were observed to have their hemangioma regress [5]. Based on this observation,
the authors decided to treat nine additional children with propranolol and observed similar
results. Prior to this discovery, hemangiomas were treated with corticosteroids as a first line of
therapy, with other options including interferon alpha, vincristine or surgical removal if the
hemangiomas were progressing despite high dose of corticosteroids [134, 135]. Propranolol thus
came as an effective, inexpensive option for treatment of complicated hemangiomas.
Infantile hemangiomas (IH) are the most common tumors of early childhood, affecting
about 5-10% of infants. It is three times more common in females than males, and most prevalent
in Caucasian children [136]. In general is not considered a severe disease, since most IH will
spontaneously regress necessitating no additional treatment. However, approximately 12% of IH

20
are complicated cases in which the location or the speed of growth can promote significant
morbidity including disfigurement, ulceration, bleeding, visual compromise, airway obstruction,
congestive heart failure and rarely death [137]. Most IH undergo rapid proliferation during the
first months to year of life, reaching an average size of 2-20 cm. These lesions then undergo a
slow involution period over several years and are generally fully regressed by 5-10 years of age
however the duration and rate of growth are variable [136-138].
Histologically these tumors are composed of a mixture of clonal endothelial cells
associated with pericytes, dendritic cells and mast cells [139]. They are densely packed over-
proliferating capillaries with the absence of an open lumen. The origin is still unclear with some
studies suggesting aberrant transplantation of placental endothelial cells [140], predisposing
genetic factors [141, 142] and/or tumor stem cell components [136, 143].
The molecular mechanisms behind the development of infantile hemangioma are still
under investigation. In the early phase of the disease development, angiogenic and growth
factors can contribute for tumor progression; in the late and involuting phase of the disease it is
thought that apoptosis is involved. The proposed mechanisms of propranolol effects include
inhibition of angiogenesis; early vasoconstriction of the tumor (attributed to the blocking of
nitric oxide production) which causes the softening of the tumor as well as change in color from
red to purple and later promotion of tumor apoptosis [5].
During the growth or proliferative phase, two main pro-angiogenic cytokines have been
implicated: VEGF and bFGF [139, 144-146]. Studies have shown that both endothelial and
interstitial cells are actively dividing in that phase. Propranolol has been implicated in
suppression of VEGF protein expression in hemangioma-derived endothelial cells in a dose-

21
dependent manner (25-100 µM) and induced apoptosis by activation of the caspase cascade
(caspase 3 and caspase 9) after 100 µM treatment of propranolol [147]. Serial serum VEGF
decrease was also demonstrated in clinical patients after initiating propranolol treatment. This
same study demonstrated a decrease in serum MMP-9 as well, suggesting another mechanism of
tumor control by propranolol [148]. Another study demonstrated a decrease in the VEGFR
(VEGF receptor) protein expression after 48 and 96 hours of cell treatment with higher
concentrations of propranolol (200-300 µM). Interestingly, there was upregulation of mRNA
expression but downregulation of VEGF by propranolol, indicating an inhibition in mRNA
translation into VEGF protein. This inhibition in VEGF was mediated by hypoxia-inducible
factor 1-alpha (HIF-1α), which is a transcription factor that becomes stabilized in hypoxic
situations. In a normoxic environment, HIF-1α is rapidly degraded; in hypoxia it persists as a
transcription factor for many genes including VEGF, ultimately contributing to angiogenesis.
Another finding was decrease in phosphorylation of PI3/Akt and p38/MAPK in a dose-
dependent fashion [149]. In another study, there was an increase in phosphorylation of p38 in
propranolol-treated IH. Since p38 regulates the production of inflammatory mediators such as
TNFα, IL1β and COX-2 [150], one proposed mechanism of IH remission could be due to
immune-mediated responses [136]. Propranolol treatment also caused decreased levels of
phosphorylated cofilin. Cofilin is a cytoskeletal-binding protein critical for actin microfilament
dynamics and reorganization that severs and depolymerizes actin filaments [151]. Cofilin
phosphorylation could lead to increased cofilin-mediated actin severing, which would disrupt
cell migration and affect cell proliferation because the actin cytoskeleton is intimately related to
regulation of the cell cycle progression [136, 152]. Propranolol also disrupted the cell cycle by
decreasing the expression of key cyclin proteins (cyclin A1, A2, B2, D2 and D3) and increased

22
the expression of important cell cycle inhibitors (p15, p21, p27). No change was noted on
expression of cyclin-dependent kinases Cdk2 and Cdk4 [136]. Propranolol inhibited cell
proliferation at an IC50 of 50 µM and induced an increase in the proportion of cells in the G1
phase while reducing the proportion of cells in S and G2/M phase [136]. Propranolol did not
induce apoptosis of endothelial cells at 50 µM, nor promoted cleavage of caspase 3 and caspase
9, in contrast with pro-apoptotic effects stated by other studies described in this text. Cell
migration (assessed by the scratch/wound healing assay) was significantly inhibited after
endothelial cells were treated with propranolol at 50 µM for 12 hours [136].
HIF-1α serum levels were found elevated in children with proliferative hemangiomas
[153, 154]. HIF-1α protein was decreased in a dose-dependent manner after treatment with
propranolol, which was evaluated both by Western blot and ELISA. Another possible pro-
angiogenic pathway, NF-κB (nuclear factor kappa-β), was analyzed and was upregulated by
treatment with propranolol, indicating a possible compensatory angiogenic mechanism by the
tumor [149]. NF-κB constitutes a non- HIF-1α dependent pathway resulting in VEGF-A
expression and angiogenesis [155]. Interestingly, the same experiments looking at cell viability,
migration, tubule formation, RT-PCR, flow cytometry, western blots and ELISA were performed
in hemangioma endothelial cells and repeated in human umbilical vein endothelial cells
(HUVEC) used as control cells, with the downregulation of VEGF, VEGFR and HIF-1α not
observed in control cells, indicating that the effects of propranolol were confined to the
hemangioma endothelial cells only. Propranolol was also observed to inhibit cell migration and
tubule formation. Apoptosis of hemangioma endothelial cells, but not hemangioma stem cells,
was demonstrated in another study [149]. This could explain the rebound growth of
hemangiomas after propranolol therapy has been discontinued [156-158]. Propranolol also

23
promoted adipogenic differentiation of hemangioma stem cells (involution) [159]. In another
study propranolol did not promote apoptosis of hemangioma stem cells at a concentration of 50
µM; the half maximal inhibition was 133 µM, which is well above the biologically achievable
dose in humans. Inhibition of proliferation was also not achieved in this study. High levels of
VEGF and bFGF were observed in proliferating IH tissue. At low concentrations (0-20 µM),
propranolol was capable of decreasing the VEGF mRNA and VEGF protein levels in
hemangioma-derived stem cells (HemSCs). There was downregulation of bFGF by HemSCs
however it was less pronounced than VEGF. The proportion of VEGF-positive HemSCs was
very low in IH tissue [160]. Zou et al showed that propranolol did not affect proliferation of
endothelial progenitor cells, but it did inhibit migration of these cells in a dose-dependent (0-100
µM) and time-dependent (24-72 hr) manner. This same study investigated the effects of
propranolol in the expression of CXCR4, which was suppressed via Akt and MAPK pathways
[161]. CXCR4 is a receptor for stromal cell derived factor 1α, which promotes mobilization of
the endothelial progenitor cells from the bone marrow to the site of vasculogenesis [154].
In summary, while there is still active research into the molecular pathways involved in
the response of infantile hemangioma to propranolol, most studies suggest that the effects of
propranolol are related to suppression of angiogenesis. Some studies also indicate an effect in
endothelial cell proliferation and survival.
2.6. Canine Hemangiosarcoma
Hemangiosarcoma (HSA) is a malignant neoplasm of vascular endothelial origin. This
cancer occurs more frequently in dogs than in any other species [2, 3]. It accounts for 2.3 to 3.6%
of skin tumors in dogs and 45 to 51% of splenic malignancies [2, 3, 162-164]. Like majority of

24
cancers, it affects mostly middle-aged to older animals [163, 165-168]. Breeds that appear
overrepresented include German shepherds, Golden retrievers and Labrador retrievers [163, 165,
168-170]. The most common primary site for HSA is the spleen, with other frequent sites include
the right atrium, skin, subcutis and liver [1, 2, 165, 166, 168, 169, 171-174].
Being a vascular malignancy, molecular pathways involving angiogenesis have been
explored to further clarify the development of this disease. VEGF expression has been
demonstrated in canine studies [175-177] and plasma levels of VEGF were higher in dogs with
HSA compared to healthy dogs [178]. There was no marked difference between VEGF levels in
effusions associated with malignant versus nonmalignant diseases [179]. Other angiogenic
cytokines important in HSA include bFGF and angiopoietin 1 (Ang-1) [177, 180]. These
cytokines can be secreted by the tumor cells leading to an autocrine growth signaling or can be
secreted by other cells within the microenviroment leading to paracrine stimulation. Another
study showed a significantly higher proportion of platelet derived growth factor (PDGFR-β)
expression in HSAs compared to cutaneous hemangiomas [181].
Mutations in tumor suppressor genes could provide a possible etiology for the disease,
however studies suggested that p53 (tumor suppressor gene) and Ras (oncogene) mutations are
infrequent in canine HSA [182-184]. PTEN (tumor suppressor gene) inactivation was
demonstrated in greater than 50% of evaluated canine HSA samples in one study [185]. Key
growth and apoptosis regulating proteins such as pRB, cyclin D1, Bcl-2 and survivin appear
overexpressed in HSA when compared with hemangiomas or normal tissues [183, 186].
Histopathology is often necessary to establish a diagnosis, since cytology is non-
diagnostic in the majority of time due to excessive bleeding and lack of exfoliation by the tumor

25
[187]. Histologically HSA consists of immature, pleomorphic endothelial cells forming vascular
spaces containing variable amounts of blood and/or thrombi [188]. Immunohistochemistry for
factor VIII (Von Willebrand) or CD31 can be used to confirm endothelial origin and support the
diagnosis [188]. Claudin-5 and CD117 have also been identified as potentially useful markers.
(Kit) [176, 189].
Surgery is the main method of treatment for HSA. Adjuvant chemotherapy is indicated in
most cases (except for dermal hemangiosarcoma) [190]. Protocols involving doxorubicin are
most commonly used, including VAC (vincristine, doxorubicin and cyclophosphamide) [191,
192]; vincristine, cyclophosphamide and methotrexate [168]; doxorubicin and
cyclophosphamide; doxorubicin and minocycline [193] and single-agent doxorubicin [194-197].
Ifosfamide has also been used [198, 199]. Epirubicin and intracavitary pegylated liposomal
encapsulated doxorubicin do not appear to have advantage over conventional doxorubicin [200,
201]. Immunotherapy has been investigated as possible treatment strategy, with one study using
a mixed killed bacterial vaccine following surgery showing some improvement in survival time
[168], and another study looking at doxorubicin/cyclophosphamide combination with liposome-
encapsulated muramyl-tripeptide-phosphatidylethanolamine (L-MTP-PE) which showed a
significant increase in median survival time (5.7 to 9 months) with 40% of the dogs experiencing
long term survival [202]. Radiation therapy is mainly used for non-visceral hemangiosarcoma
and although it can cause reduction in tumor size it does not significantly change the overall
survival [203]. A small study looking at a metronomic chemotherapy protocol using NSAID,
cyclophosphamide and etoposide showed a similar outcome to doxorubicin-based protocols
[204]. A combination of a dose-intensified doxorubicin protocol with deracoxib (COX2
inhibitor) was well tolerated but did not result in overall improvement of survival in dogs with

26
splenic HSA [205]. A study investigating the effects of toceranib phosphate (Palladia®), a
multiple kinase inhibitor (including VEGFR), administered after splenectomy and 5 doses of
doxorubicin given at 2-week intervals has been conducted and the results showed that the
administration of toceranib did not significantly improve disease-free interval or overall survival
time [206].
The prognosis for canine HSA is very poor. Hemangiosarcoma typically has a very
aggressive biologic behavior, with disseminated metastasis occurring very early in the
development of the disease, except for dermal hemangiosarcomas. Metastasis is typically
hematogenous or through transabdominal implantation following rupture. The most frequent
metastatic sites are the liver, omentum, mesentery and lungs. Other reported metastatic sites
include kidney, muscle, peritoneum, lymph nodes, adrenal gland, brain and diaphragm. Dogs
treated with splenectomy alone have a survival time that range from 19-86 days and less than
10% of dogs living one year [3, 164, 168, 207, 208]. Surgery plus adjuvant chemotherapy will
increase median survival times to 141-179 days, however the one year survival rate is still less
than 10% [191, 194, 195, 198, 200]. The average survival time of right atrial HSA undergoing
surgery is 1-4 months [209, 210]. Dermal HSA treated with surgery alone had a median overall
survival time of 987 days and lingual HSA had a median overall survival time of 553 days [211,
212]. One analysis of intramuscular and subcutaneous HSA identified 71 cases, the median time
to tumor progression and overall survival time were 116 and 172 days, respectively and 25%
survived to 1 year [213]. In summary, the literature on canine HSA describes an aggressive
cancer. Unfortunately, there has not been substantial improvement in the outcome of dogs with
HSA and therefore novel therapies are needed.

27
Investigations into the beta adrenergic presence in canine hemangiosarcoma as well as
the effects of its blockade on cell activity are starting to emerge. One study that evaluated canine
hemangiosarcoma cell lines as well as mouse angiosarcoma, hemangioendothelioma and human
dermal microvascular endothelial cell lines revealed that propranolol selectively inhibited
proliferation, survival, and migration of a panel of malignant vascular tumor cells, indicating that
the oncogenic properties of these tumor types are driven, in part, by beta adrenergic signaling.
Propranolol dramatically slowed the proliferation rate of all vascular tumor lines tested, and four
of the five tumor lines exhibited nearly 100% lethality at doses that had been previously reported
to be non-toxic for primary cultures of human endothelial cells. This finding suggested that
malignant endothelial tumors may be more sensitive to beta blockade [214].

28
CHAPTER 3
MATERIALS AND METHODS
3.1. Cells and reagents
Two cHSA cell lines (DEN and Fitz) were provided by Dr. Douglas Thamm (Colorado
State University). Additional cell lines used as controls included HeLa (human cervical
adenocarcinoma), MDA-MB-231 (human metastatic breast carcinoma), and MDCK (Madin-
Darby canine kidney) purchased from American Type Culture Collection (ATCC). Cells were
cultured at 37˚C in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with glutamine
(2 mmol/L), penicillin (100 IU/ml), streptomycin (100 IU/ml), and 10% fetal bovine serum
(FBS) in a humidified atmosphere supplemented with 5% CO2. Cell cultures were maintained in
subconfluent monolayers and passaged as deemed necessary by growth of cell line.
Propranolol hydrochloride (P) was purchased from Sigma-Aldrich (St. Louis, MO -
catalog # P0884). Norepinephrine bitartrate salt monohydrate was purchased from Sigma-
Aldrich (St. Louis, MO - catalog # A9512). Both reagents were diluted with deionized water
based on manufacturer’s instructions and solubility provided.
The anti-β2-AR antibodies (rabbit polyclonal; catalog #AB36956 and #AB135641) used
for Western blot (WB) and immunohistochemistry (IHC) experiments, respectively, were
purchased from Abcam Inc (Cambridge, MA). The anti-Src (mouse monoclonal; catalog #2110)
and anti-phospho-Src (Tyr 416) (rabbit monoclonal; catalog #2113 and rabbit monoclonal;
catalog #6943) were purchased from Cell Signaling Technology (Beverly, MA). Antibody anti-
tyrosine hydroxylase (rabbit polyclonal, Abcam #AB112) and anti-CD31 (mouse monoclonal,

29
Dako, North America Inc., #M0823) were used in splenic cHSA tissue samples. The anti-β actin
antibody used for loading control (rabbit polyclonal; catalog #AB8227) was purchased from
Abcam Inc. Horseradish peroxidase (HRP) conjugated anti-mouse and anti-rabbit secondary
antibodies used for WB were purchased from GE Healthcare, UK. Blocking peptide for the β2-
AR WB (catalog #38102) was purchased from Abcam Inc.
Other reagents used include WB developing solution (SuperSignal West Femto
Chemiluminescent Substrate, Thermo Scientific, Rockford, IL), bovine serum albumin (BSA),
milk, tris-buffered saline and Tween 20 (TBST) and hydrogen peroxide (H2O2).
3.2. Immunohistochemistry
3.2.1 Spontaneous canine HSA samples
Eighteen canine splenic HSA tissue blocks were obtained from the Veterinary Diagnostic
Laboratory of the University of Illinois at Urbana-Champaign. Paraffin-embedded cell pellets
were sectioned every 3 microns, placed on positively charged slides, and dried for one hour at
60°C. Slides were deparaffinized with two sequential xylene washes, and subsequently hydrated
using two ethanol baths for five minutes each; followed by a water rinse. Slides were placed in
3% H2O2 for 15 minutes and rinsed with water. Antigen retrieval was performed with citrate
buffer microwaved for five minutes. Nonspecific staining was minimized using a blocking
solution for 30 minutes (5% BSA in PBST). Slides were then incubated with rabbit polyclonal
anti-β2-AR antibody at a concentration of 1:35 for 30 minutes at 37°C followed by one hour at
room temperature. Slides were rinsed twice with PBST and then incubated with a biotinylated
secondary anti-rabbit antibody for 30 minutes at room temperature. Slides were washed in PBST

30
twice before incubation for 30 minutes with a streptavidin–biotinylated HRP complex, and
developed with 3,3-diaminobenzidine (DAB) (BioGenex, Freemont, CA). Slides were then
counterstained with Mayer’s hematoxylin. The negative control samples underwent the same
process but in the absence of the primary antibody. The isotypic negative control was performed
using tyrosine hydroxylase antibody (1:200), which is very specific for neuronal tissue. This was
performed to rule out non-specific Fc receptor binding or other cellular protein interactions, as
well as confirm the specificity of the primary antibody. The rabbit polyclonal anti-β2-AR
antibody was validated using canine heart tissue as a positive control as well as mouse spleen
and kidney [215-217]. All cHSA tissue samples were reviewed by a single pathologist (S.
Lezmi) and the diagnosis of hemangiosarcoma was confirmed with H&E staining and with IHC
using a CD31 antibody. CD31 antigen is a marker for endothelial cells and is expressed only by
endothelial cells, platelets and macrophages; it is conserved in neoplasia arising from endothelial
cells and validated in canine tissue [218]. The intensity of the β2-AR staining was evaluated
(scored as strong, mild or none) as well as the localization within the cell (nuclear vs.
cytoplasmic).
3.2.2. Cell pellets
Fitz and DEN cell lines and a human positive control cell line, MDA-MB-231 [219],
were used to evaluate β2-AR expression using IHC. Adherent cell cultures were collected and
washed in PBS, then pelleted by centrifugation (10,000 RPM for 10 minutes). Each pellet was
re-suspended in 1 ml 10% formalin for one hour. Formalin was removed and cell pellets were
re-suspended uniformly into 1 ml 4% melted agarose gel by vortexing, then immediately
centrifuged to create an agarose-embedded cell pellet. Cell pellets were then trimmed and

31
processed as for formalin-fixed paraffin-embedded tissue biopsy specimens. Briefly, paraffin-
embedded cell pellets were sectioned at three microns, placed on positively charged slides, and
dried for one hour at 60°C. Slides were deparaffinized with three sequential xylene washes, and
subsequently hydrated using 100%, 95%, and 70% ethanol for two minutes each; followed by a
water rinse. Slides were placed in 3% H2O2 in methanol for 15 minutes, and then treated with
citrate buffer (pH 6.0), placed in a decloaking chamber until the temperature reached 125 °C for
30 seconds with psi between 18-22, followed by 90°C for 10 seconds. Slides were removed
from decloaking chamber and cooled for two minutes followed by a wash in Super Sensitive
Wash Buffer (BioGenex, San Ramon, CA). The rabbit polyclonal anti- β2-AR antibody was used
at a concentration of 1:100 for one hour at room temperature. Slides were rinsed with
SuperSensitive Wash Buffer and treated with Super Enhancer (BioGenex, San Ramon, CA) for
20 minutes at room temperature, and subsequently treated with Polymer-HRP (BioGenex,
Fremont, CA) for 30 minutes at room temperature and incubated with DAB (BioGenex,
Fremont, CA) at room temperature for five minutes. Slides were washed with SuperSensitive
Wash Buffer and counterstained with Mayer’s hematoxylin for one minute. Negative control
samples underwent the same process but in the absence of the primary antibody.
3.3. Cell protein extraction
Cells were grown in culture until confluence and then media was removed and cells were
washed twice with ice-cold PBS. Cells were then placed in trypsin for five minutes to detach
from culture plate, followed by addition of complete media to neutralize the effects of trypsin.
Non-adherent cells were centrifuged at 2,000 rpm at 4°C for five minutes. Supernatant was
removed and the pellet was washed twice in PBS until all trypsin and media was removed. The

32
pellet was then re-suspended in a solution containing Mammalian Protein Extraction Reagent
(M-PER, Pierce, Rockford, IL) and Pierce protease inhibitor cocktail solution (Pierce, Rockford,
IL) diluted 1:100 for final working solution, 10 µL of protease inhibitor to 1000 µL of M-PER.
This was placed on a shaker for 10 minutes, followed by centrifugation at 10,000 RPM, in 4°C
for 10 minutes. The supernatant was placed in separate Eppendorf tubes and placed in -80°C
until analysis.
3.4. Western blot analysis
Cellular protein concentrations were determined using a standard assay kit (Bicinchoninic
Acid Protein Assay, Rockford, IL). For each protein expression analysis, 50 µg samples were
electrophoresed on 12% polyacrylamide gel and then transferred to nitrocellulose membrane.
The membranes were blocked with either TBST with 5% milk (β2-AR and Src) or TBST with
5% BSA (p-Src), followed by addition of their primary antibody at their specific concentrations
and exposure time as described in the next sections. In all WBs, secondary HRP-conjugated
antibody was added at a concentration of 1:1000 for one hour at room temperature after the
membranes were washed three times with TBST for five minutes, and developed using
chemiluminescent substrate followed by detection with enhanced chemiluminescence (ECL)
detection system (Amersham). The blots were imaged using ChemiDoc XRS+ molecular imager
system (Bio-Rad Laboratories, Hercules, CA) with Image Lab software (version 5.1 build 8, Bio-
Rad Laboratories). The evaluation of protein loading within the blots was performed by
incubating the membranes with anti-β-actin antibody (1:2000) without the need for stripping the
membranes since the expected molecular weight of β-actin band was below (46 kDa) the other
bands evaluated. The membranes were subsequently incubated with the secondary antibody and

33
imaged as described previously. The β-actin loading control is important to verify that the
amount of protein loaded into the gel is approximately equal.
3.4.1 β2-AR
Protein expression of β2-AR was performed via Western blotting using cell lysates from
Fitz and DEN, as well as HeLa and MDA-MB-231 cells as positive controls [219, 220]. The
primary antibody was diluted in 5% milk in TBST at a concentration of 1:250. Band specificity
was confirmed with a matching blocking peptide. For evaluation of the blocking peptide, one
membrane containing duplicated samples was separated into two membranes becoming two
exact copies. One portion of the membrane was treated first with the blocking peptide (1:250)
diluted in 5% milk/TBST for one hour at room temperature, rinsed three times with TBST and
then incubated with the primary antibody for one hour at room temperature. The other portion of
the membrane was incubated with only the primary antibody (1:250) for one hour at room
temperature. Subsequent incubation with secondary antibody and analysis was performed as
described above.
3.4.2. Src
Protein expression of Src was performed via Western blotting using cell lysates from Fitz
and DEN, and MDCK as a positive control (per antibody manufacturer). Primary antibody
(1:1000) was diluted in TBST with 5% milk incubated overnight. Basal Src protein expression
was determined in all three cell lines. In addition, protein expression was assessed in Fitz and
DEN exposed to different concentrations of NE and P (0.1 and 10 µM) for 24 hours.
3.4.3. Phospho-Src

34
Protein expression of phosphorylated Src (p-Src) was performed via Western blotting
using cell lysates from Fitz and DEN. The membrane was incubated overnight with the anti-
phospho-Src (1:1000) antibody diluted in TBST with 5% BSA. For the analysis of
phosphorylation status of Src after cell treatment, several experiments were performed under
varying experimental conditions. For positive control of Src phosphorylation in all experiments,
cells were exposed to 3 mM of H2O2 for five minutes per manufacturer’s instructions. First
experiment: DEN and Fitz cells were serum-starved overnight then treated with NE (10 µM) or P
(0.1 µM) for different time exposures (5-30 minutes). Second experiment: To optimize
visualization of the agonism, Fitz cells were serum-starved for 24 hours then treated with NE (10
µM) at increasing time-exposure (5-60 minutes). The goal was to determine if phosphorylation
was positive upon contact with the agonist, if that difference was visible in the WB, and how
much exposure was necessary for optimal visualization. Third experiment: To determine if
propranolol could block NE agonism, Fitz cells were serum-starved for 24 hours, then pre-
treated with propranolol (0-0.1 µM) followed by NE treatment (10 µM) at increased time
exposures (5-60 minutes). Fourth experiment: To determine if propranolol could rescue NE
agonism, DEN and Fitz cells were serum-starved for 24 hours, then pre-treated with NE (0-10
µM) for 45 minutes followed by P (0-100 µM) for 24 hours. Band volume analysis was
performed using Image Lab software and results of the Phospho-Src proteins were adjusted for
β-actin expression levels used as loading control. For the third experiment, results are expressed
as p-Src/ β-actin ratio.
3.5 VEGF ELISA

35
Fitz and DEN cells were plated at a density of 5 x 103 cells/well in a 96-well plate. After
allowing cells to adhere to the plate overnight, the medium was removed and replaced with fresh
medium containing NE and/or P. In the first experiment, optimal agonism was assessed using 0-
10 µM NE for 24 hours. In the second experiment, cells were treated with 0-100 µM P with NE
agonism at 1 µM which was selected as the optimal agonist. Cell culture supernatants were
harvested and soluble VEGF was determined with a commercially available canine VEGF
ELISA immunoassay (Canine VEGF DuoSet, R&D Systems, Minneapolis, MN). Differences in
soluble VEGF secreted by the cHSA cells after each treatment were normalized to cell count
with the use of a colorimetric proliferation assay (CellTiter 96® AQueous One Solution Cell
Proliferation Assay – MTS, Promega, Madison, WI) in which optical density linearly correlates
with viable cell numbers. Specifically, normalized VEGF concentrations were based on the
average of quadruplicate samples for each experimental group expressed as the following ratio:
Normalized VEGF = [Calculated VEGF (pg/mL)]/optical density. Samples were performed with
six replicates per experiment and the results were replicated in three independent experiments.
3.6. Proliferation assay
Fitz and DEN cell proliferation was assessed using a commercial colorimetric assay per
manufacturer’s directions (CellTiter 96® AQueous One Solution Cell Proliferation Assay –
MTS, Promega, Madison, WI). This assay measures the number of viable cells by using a
tetrazolium compound that is bioreduced by metabolically active cells into a colored formazan
product, which is detected via spectrophotometry. Fitz and DEN were seeded into a 96-well plate
at a density of 5 x 103 cells/well. After allowing adherence overnight, the cells were treated with
0, 0.1, 1 and 10 µM of NE or P for 24 hours (first experiment) and with 0, 0.1 or 10 µM NE, 0.1

36
or 10 µM P, 0.1 or 10 µM NE + P (second experiment) for 48 hours. The supernatant was
removed, and 120 µL of fresh media containing 20 µL of the reagent was added to each well.
The plate was then incubated at 37°C in 5% CO2 for 1, 2 and 3 hours. At each hour,
spectrophotometer readings were obtained with a photometric plate reader (Bio-Tek EL 800) at a
490nm absorbance (wavelength correction set to 570 nm). The data collected for treated cells
was compared to untreated cells of the same experiment and the difference was calculated as a
percentage of control cells. The groups were subsequently compared to each other. Samples were
performed with six replicates per experiment and the results were replicated in three independent
experiments.
3.7. Scratch assay
Analysis of cell migration was done using the “scratch assay” method [221]. Fitz and
DEN were grown in culture in 6 well plates until reaching confluence. A scratch was then made
along the center of the cell culture area using a p200 pipette tip. The medium was then replaced
with fresh medium containing P (0.01-100 µM) or no treatment (control). An image of the
scratched cell monolayer was taken (“time 0”) using an inverted microscope (Nikon Eclipse
TS100) with a mounted digital camera (SPOT Insight QE model #4.2, SPOT Imaging Solutions,
Michigan, USA). Cells were then grown in culture for 24 hours and another image was taken at
the same location of the initial image. Captured images were analyzed with Image J software
(National Institutes of Health, Bethesda, MD). A total of 5 measurements of the gap between the
cells were taken for each image and average measurements were compared between time 0 and
24h. Experiment was performed in triplicate with replication.
3.8. Statistical analysis

37
Data from proliferation, VEGF assay and scratch assay were analyzed for normality with
the Kolmogorov-Smirnov test. Because of the small number of samples, histograms were also
performed for visual assessment. Data is reported as mean and standard deviation. One-way
ANOVA was used to evaluate for difference between groups, with Tukey’s multiple comparison
test to detect a difference between groups. GraphPad Instat 3.1 and GraphPad Prism 6.1 software
were used for analysis.

38
CHAPTER 4
RESULTS
4.1. β2-AR is expressed in spontaneous canine splenic hemangiosarcoma
To determine β2-AR protein expression in spontaneous canine hemangiosarcoma, IHC
was performed. First, the 18 selected tissue samples were confirmed cHSA based on H&E
staining and positivity for CD31. All tissue samples demonstrated positive β2-AR expression
(Fig 4.1). Expression of the receptor was identified in the nucleus and cytoplasm, with varying
stain intensity between tissue samples. Five of 18 (27%) samples had strong intensity and were
almost diffusely labeled. Eight of 18 (44%) of the samples had only cytoplasmic staining while
10/18 (56%) had staining of both the nucleus and the cytoplasm. The labeling was frequently
heterogeneous with some cases presenting large unlabeled areas. Isotype control did not reveal
any significant non-specific background staining.
4.2. β2-AR is expressed in canine hemangiosarcoma cell lines
To determine β2-AR protein expression in canine hemangiosarcoma cell lines Fitz and
DEN, WB and IHC were performed. Using WB, we identified a band at the expected molecular
weight of 55 kDa in both human positive control cell lines MDA-MB-231 and HeLa, as well as
the canine cell lines Fitz and DEN (Fig. 4.2b). Qualitatively, Fitz appears to have stronger
protein expression than DEN. Specificity was confirmed with abrogation of the band in the
presence of a blocking peptide. β2-AR protein expression was also confirmed via IHC on Fitz
and DEN cell pellets using the same antibody. Cell pellets demonstrated strong, uniform, diffuse
staining (Fig. 4.2a).

39
4.3. Investigating intermediate signaling via Src/p-Src
4.3.1. Src and p-Src are expressed in cHSA cell lines
Src and phosphorylated Src (Tyr416) are intermediaries involved in signal transduction
from β2-AR to the nucleus [19]. To determine their involvement in cHSA cells we performed
WB. We identified bands (Src and p-Src) at the expected molecular weight of 60 kDa in both
canine positive control cell line MDCK as well as the canine cell lines Fitz and DEN (Fig. 4.3a).
4.3.2. Propranolol attenuates norepinephrine-induced Src phosphorylation
Given that Src and phosphorylated Src (Tyr416) are intermediaries involved in signal
transduction from the β2-AR to the nucleus, we sought to determine if the β2-AR agonist (NE)
would lead to phosphorylation of the protein, and if this process could be blocked by the
antagonist (P). To determine the effects of NE and P on Src and p-Src, we performed WB on
cHSA cells. In DEN and Fitz cells treated with different concentrations of NE and P (up to 10
µM), there was no qualitative difference in the expression of total Src (Fig. 4.3b). Both DEN and
Fitz demonstrated significant basal phosphorylation of Src, suspected to be caused by the FBS
used in culture. Overnight serum-starvation in DEN and Fitz cell lines did not result in an
apparent difference in p-Src protein expression with P (low dose 0.1 µM) and increased time-
exposure to NE (Fig. 4.3c). To optimize phosphorylation, Fitz cells were serum-starved for 24
hours, followed by increasing time exposure to NE. Increased protein expression of p-Src was
identified, with phosphorylation more pronounced starting at 15 minutes and persisting through
60 minutes (Fig. 4.3d). Once the positive effects of the agonist NE on phosphorylation were
demonstrated, we proceeded to evaluate if pre-exposure to low-dose propranolol (0.1 µM) was

40
sufficient to block the receptor to the effects of the agonist. In Fitz cells, NE caused a modest
phosphorylation of Src and treatment with low-dose propranolol for 24 hours prior to exposure
led to a modest decrease in phosphorylation. These effects were visible in the WB and therefore
we proceeded to quantify these results by normalizing the adjusted volume of the p-Src band to
the matching β-actin band, to ensure that the increased intensity of the band observed was not
due to increased protein loading (Fig. 4.3e). To determine if propranolol could rescue NE
agonism, DEN and Fitz cells were serum-starved, pre-treated with NE (0-10 µM), then
propranolol (0-100 µM). In Fitz cells, low doses of propranolol reduce p-Src compared to
untreated cells, which is lost at the highest dose in unstimulated cells. In NE-stimulated Fitz
cells, propranolol reduces p-Src compared to basal levels at all doses. Conversely, in DEN cells,
propranolol increases p-Src in untreated and NE stimulated cells (Fig. 4.3f)
4.4. Propranolol affects VEGF secretion of cHSA in vitro
After demonstrating the presence of the receptor as well as a possible intermediate
protein involved in signal transduction, we proceeded to investigate the effects of NE and P on
cell function activities of cHSA cells. Since propranolol’s ability to inhibit tumor angiogenesis
has been largely implicated as a primary anti-neoplastic effect, we sought to evaluate the effects
of NE and P on the ability of cHSA cells to secrete VEGF, a key pro-angiogenic cytokine. In the
first experiment, we used NE at varying doses (0-10 µM) to assess optimal agonism. Normalized
to cell count, basal secretion of VEGF was 1098.83 pg/mL and 464.42 pg/mL for DEN and Fitz
cells, respectively. All doses of NE induced VEGF secretion compared to untreated cells for both
cell lines. Optimal NE agonism was obtained at 1 µM NE, with a significant increase in VEGF
secretion compared to control. At optimal NE agonism, there was an increase in VEGF of 29%

41
for DEN cells (p<0.05 for all treatments compared to control), and 2.6-fold increase for Fitz cells
(p<0.001 for all treatments compared to control) (Fig. 4.4a). Cells were then incubated with the
selected NE dose of 1 µM combined with varying doses of P starting at a biologically achievable
dose (0.1 µM) up to 100 µM. No significant differences were noted at low doses, however there
was a reduction in VEGF secretion in DEN at P ≥ 10 µM (p<0.01) with a maximum decrease in
VEGF of 21% at 100 µM. Similar results were seen with Fitz, with reduced VEGF secretion at P
≥ 10 µM (p<0.001) with maximum 44% reduction at 100 µM; a dose dependence was noted in
the Fitz cell line (p<0.05) (Fig 4.4b). Similar but less profound results were seen without NE
stimulation (results not shown).
4.5. Propranolol affects cell proliferation of cHSA in vitro
The metabolic activity of Fitz and DEN, an indirect measurement of cell viability and
proliferation, was evaluated upon exposure to different doses of NE and P. While there was no
significant difference noted after 24h incubation, when treated for 48h Fitz exhibited significant
difference in proliferation between untreated vs 0.1 µM of NE (25% increase) (p < 0.05) and
untreated vs 0.1 µM of P (30% increase) (p < 0.01). DEN did not exhibit significant difference
between groups (p > 0.05) (Fig. 4.5)
4.6. Propranolol affects cell migration of cHSA in vitro
The ability of cancer cells to move within the microenviroment towards the nearby
vasculature is important in the process of metastasis and therefore we elected to evaluate if P had

42
any effect on cell mobility by evaluating the cells ability to close a gap created between the cells.
Increasing concentrations of P were used (0-100 µM) and an artificially created gap was
measured before and 24 hours after treatment. Data is represented as a percentage of the original
gap (time 0), with being 100% a completely closed gap. The gap remained more noticeably open
at 10 and 100 µM, being statistically significant at 100 µM when compared to untreated control
cells (p<0.05) (Fig. 4.6).

43
CHAPTER 5
DISCUSSION AND CONCLUSIONS
In veterinary medicine, the emotional component of stress may not play an important role
as it does in humans, however the presence of cancer can lead to microenvironmental stressors
such as hypoxia and inflammation which may lead to catecholamine release [222, 223].
Therefore, stress can play a part in canine tumor progression. In this study we were able to
demonstrate the presence of β2-AR in all spontaneous splenic cHSA as well as cell lines
demonstrating the clinical relevance and consistency of expression, and suggesting that this
pathway may be activated by stress-related catecholamines. The Fitz cell line appeared to have
stronger protein expression than the DEN cell line. The expression of β2-AR in spontaneous
tumor samples was heterogeneous in intensity and localization within the cells. The variability of
IHC staining could be explained by a variable expression of the receptor within the neoplastic
cells, as well as integrity and detectability of the antigen depending on the time the tissue has
been stored and the fixative used to initially preserve it as described in a study looking at EGFR
[224, 225]. Since the samples tested were from different cases obtained over several years, the
intensity of the staining obtained could be a reflection of the particular sample rather than
variability within the tumors. However, since tumors undergo frequent mutations, some may
express more of a particular antigen or receptor compared to others and this variability may
account for the difference between cytoplasmic versus nuclear staining. Regarding cellular
localization, upon stimulation β2-AR undergoes internalization, intracellular trafficking and
recycling and thus variability in cytoplasmic localization may reflect variation in stimulation and
internalization [226]. Nuclear membrane localization of β1- and β3-AR as well as downstream

44
signaling partners, but not β2-AR, has been reported in normal cardiac myocytes [227]. Our IHC
results are in concordance with similar studies in human cancer. In a study of β2-AR IHC
expression in hepatocellular carcinoma, 60% of tumors had moderate/strong staining intensity,
31% weak staining intensity and 9% negative staining with nuclear and cytoplasmic distribution
[228]. Another study showed strong β2-AR staining in 41% of angiosarcoma samples that were
analyzed and a 77% total expression indicating heterogeneity in receptor expression, similar to
the results obtained in this project [34]. Further evaluation of localization of β2-AR and signaling
partners could include Western blotting of fractionated cell lysates, confocal microscopy with
immunofluorescence and other methods.
The non-receptor tyrosine kinase Src has been reported as one of the many downstream
proteins involved in β2-AR activation and therefore we elected to investigate if this pathway is
indeed activated and blocked by β2-AR agonism/antagonism [19]. After initially demonstrating
the presence of Src within the cell, we proceeded to verify if this protein is phosphorylated by
NE. The initial challenge was to overcome the strong basal phosphorylation of this protein
within the cHSA, which did not allow for a difference in visualization between untreated and
treated cells. Part of this phosphorylation was probably due to the presence of FBS, which
contains growth factors that can stimulate the Src pathway. After 24h serum-starvation there was
still p-Src activity within the untreated cells, however it was diminished enough that an
appreciable difference was seen compared to NE-treated cells. Norepinephrine agonism of Src
phosphorylation was confirmed, supporting a role for catecholamine-induced tumor stimulation.
The subsequent experiment was performed with pre-treatment of cells with P, showing a
decrease in phosphorylation at low doses of P. With NE agonism prior to P treatment (i.e.
propranolol “rescue”), which may most closely resemble the clinical scenario, P successfully

45
decreased Src phosphorylation in NE pretreated cells, while phosphorylation was variable in
DEN cells. This may be a factor of β2-AR expression, which was qualitatively higher in Fitz
cells. The preliminary results of this experiment warrants further investigation of this pathway,
however due to multiple pathways involved in β2-AR signaling it is possible that alternative
pathways are stimulated or inhibited with propranolol leading to the effects seen. These effects
were related to proliferation, VEGF secretion and cell migration. With respect to VEGF
secretion, cHSA cells secrete basal levels of VEGF with NE causing increased VEGF secretion,
more evident in the Fitz cell line. This supports our hypothesis that NE would increase VEGF
secretion as demonstrated in other studies [229]. The increase in VEGF may be a result of Src
phosphorylation as hypothesized, and the more profound reduction in VEGF in the Fitz cell line
may be related to the increased β2-AR expression as well as the more consistent phosphorylation
effects of P. Taken together, this data supports a role for the β2-AR and the potential role of
propranolol in inhibiting VEGF-mediated HSA angiogenesis.
The effects of NE and P on cell proliferation were also investigated. The initial
hypothesis was that NE would lead to increased proliferation and P would decrease proliferation
[230]. Interestingly, we found that both NE and P caused an increase in proliferation in Fitz cells
at 0.1 µM. There is one report in the literature of P inhibiting proliferation of pulmonary artery
smooth muscle cells at a 10 µM but not at 0.1 µM. The mechanism for this effect on proliferation
remained unexplained [231]. One possible explanation may be that this β-blocker could act as an
agonist to a different receptor or pathway involved in cell proliferation. Additional studies such
as time-dependent effects may elucidate this further. In one rodent study, propranolol caused a
transient increase in markers of cell proliferation (PCNA, mitotic index) that were abrogated
with longer term treatment [232]. The variation observed between the different cell lines in this

46
project was also observed in another study, where beta blockade effectively reduced proliferation
rate among multiple cell lines however some lines were more resistant to these effects than
others [214].
Our last finding was that P at a high dose (100 µM) had an effect on cell migration. This
effect has been described previously in human infantile hemangioma endothelial cells and it also
decreased phosphorylation of cofilin, as a possible mechanism involved (since cofilin is an actin-
severing protein and phosphorylation is an inhibitory event) [136, 233]. The dose necessary for a
significant effect is not biologically achievable, however a time-dependent effect at lower doses
was not investigated in this study and is an avenue for further investigation. Decrease in cell
migration was detected at a 50 µM dose in one study, which was careful to evaluate the effects of
this dose on net cell proliferation to eliminate inhibition of proliferation as a contributing factor
[214]. It is possible that our dose of 100 µM is affecting cell proliferation, which is indirectly
leading to part of the migration effects seen on the scratch assay.
In relating this study to the clinical scenario of canine hemangiosarcoma, the finding of
β2-AR expression in both spontaneous tumors and two cell lines, strong basal phosphorylation of
Src, as well as consistent agonism of downstream signaling by NE supports a role of this
pathway in tumor progression. Variability in expression may suggest a variable response to
pathway blockade. Effects on VEGF secretion and cell migration were most significant at the
highest doses of propranolol, which are not biologically achievable with single-dose treatment at
standard doses. The high doses of propranolol needed to impair tumor cell function is consistent
with that seen in infantile hemangioma – most of these studies show in vitro effect at 100-300
µM, which is not clinically achievable. However, in infantile hemangioma, continuous treatment
is used clinically, which may account for the discordancy between in vitro and in vivo effect. The

47
finding of positive effects on Src phosphorylation at low doses in cHSA may also support a
potential for low-dose propranolol time-dependent effects on VEGF secretion or cell migration,
although this was beyond the scope of this study. Continuous low dose treatment with
propranolol would be reasonable in an adjunctive setting in cHSA due to its low cost and side
effect profile.
Although this study indicated the presence of the β2-ARs in cHSA and that propranolol
exerts some biological effects in cHSA cell lines, it is important to acknowledge some
limitations of this project. There is no direct evidence that Src is the intermediary pathway
connecting β2-AR agonism and cell proliferation, VEGF secretion or cell migration since there
are many other possible pathways involved [19]. We attempted to elicit phosphorylation of Src
by exposing the cells to NE over time in order to detect the optimal moment at which most of the
protein is phosphorylated and thus more easily detectable. We were only able to detect a small
percentage of phosphorylation and even smaller attenuation by propranolol in one experiment.
The necessary amount of phosphorylated Src protein required for downstream activation of
subsequent proteins (such as STAT3 [234]) is unknown and could be as low as 1% and as high
as 100%, therefore the amount of necessary blockade of phosphorylation for biological effect
could also be variable. The use of an agonist with a stronger affinity for β2-AR, such as
isoproterenol [235], could lead to greater phosphorylation of Src and allow for more objective
visualization of the effects of propranolol. The cells were treated with 0.1 µM of propranolol for
one hour, which is a biologically achievable dose; however it does not represent the exposure
time to which an animal is subjected to, being a drug of continuous use. The same critique can be
applied to the other assays in which the cells were treated for either 24 or 48 hours, revealing
only statistically significant results at higher doses, which are not biologically achievable. Most

48
studies looking at the in vitro effects of norepinephrine or propranolol used high doses of
agonist/antagonist, probably because it was necessary to achieve significant findings. Drug
absorption and delivery to the tumor can be variable in each patient depending on tumor blood
flow and microenviroment, which is one limitation of translating in vitro findings to in vivo
effects. Additionally, as noted in one study where propranolol did not completely abolish tumor
cell proliferation in vivo as noted by positive PCNA staining in propranolol-treated tumors,
single-agent propranolol may not be as effective in tumor control however synergistic effects
may be observed with chemotherapeutics warranting further investigation into combination
therapy [214].
In conclusion, our study showed that cHSA express β2-ARs, Src kinase is one pathway
activated by this receptor and agonism/antagonism of this receptor exerts anti-angiogenic effects
in VEGF secretion and minimal effect on cell proliferation. Propranolol at high doses affects cell
migration. Further studies looking at other possible intermediate pathways as well as other
mechanisms involved in cancer development (matrix metalloproteinases or apoptosis) are areas
for further investigation.
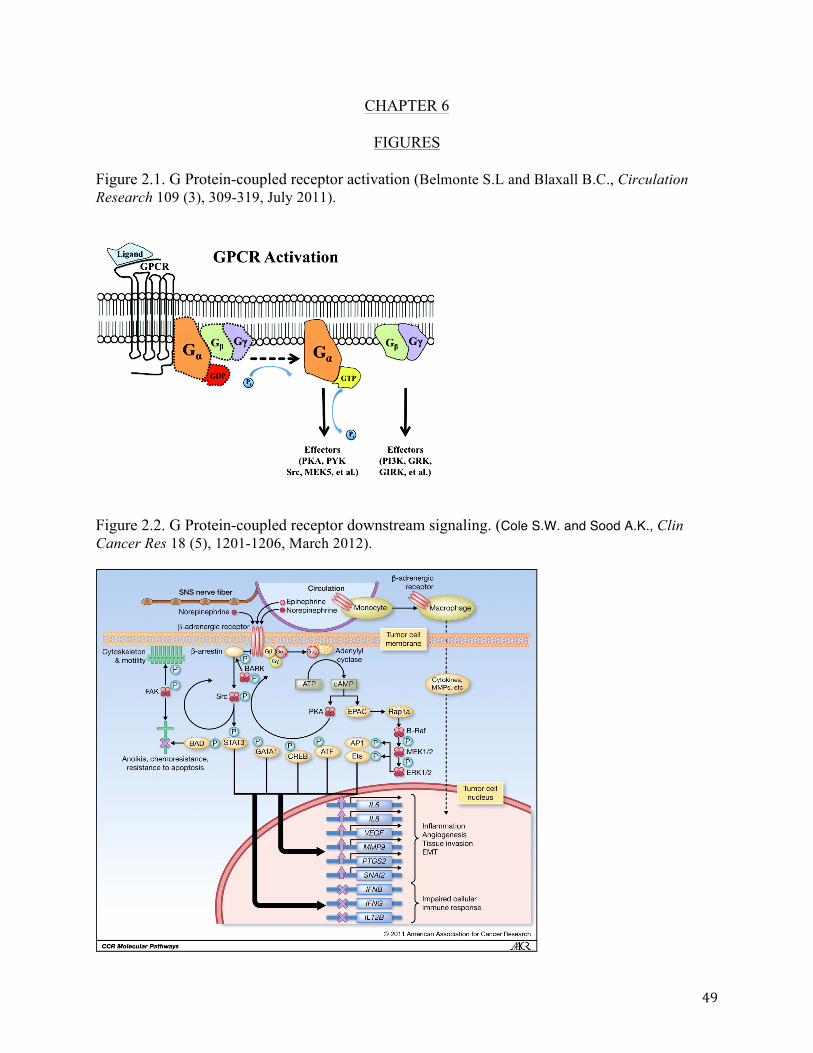
49
CHAPTER 6
FIGURES
Figure 2.1. G Protein-coupled receptor activation (Belmonte S.L and Blaxall B.C., Circulation Research 109 (3), 309-319, July 2011).
Figure 2.2. G Protein-coupled receptor downstream signaling. (Cole S.W. and Sood A.K., Clin Cancer Res 18 (5), 1201-1206, March 2012).

50
Figure 4.1. Expression of β2-AR in spontaneous cHSA. Immunohistochemistry was performed on formalin-fixed paraffin-embedded sections containing tissue from splenic cHSA. All samples stained positive for the β2-AR antibody and the degree of staining was variable. Examples of strong β2-AR stain intensity (top row) and mild β2-AR stain intensity (bottom row) are shown. The diagnosis of splenic cHSA was confirmed after review of H&E staining and CD31. Negative control was performed using an isotypic control antibody (1000x).
Figure 4.2a. Expression of β2-AR in cHSA cell lines. Immunohistochemistry was performed on formalin-fixed paraffin-embedded sections of agar-embedded cell pellets containing cHSA cells Fitz and DEN. Human mammary carcinoma cell line MDA-MB-231 was used as a positive control. Negative control was the absence of the primary antibody (10x).

51
Figure 4.2b. Expression of β2-AR protein in cHSA cell lines. Western blot was performed using whole cell lysates of Fitz and DEN. Cell lines HeLa and MDA-MB-231 were used as positive control.
Figure 4.3a. Expression of Src and p-Src in cHSA cell lines. Western blot using an antibody for Src and p-Src was performed using whole cell lysates of Fitz, DEN and MDCK (control). For better visualization of p-Src the cells were serum-starved and stimulated with 3 mM of H2O2 for 5 minutes. All cells demonstrated the presence of basal Src protein as well as phosphorylated Src protein.
Figure 4.3b. Expression of Src in cHSA cell lines exposed to different concentrations of NE and P. Fitz and DEN cells were treated with different concentrations of NE and P for 24 hours as indicated below. Western blot revealed bands of similar size across all treatments, indicating that the presence of total Src protein is independent of the stimulus.
52
Figure 4.3c. Expression of p-Src in cHSA cell lines during increasing exposure to NE and P. Fitz and DEN cells were serum-starved overnight and treated with 10 µM of NE and 0.1 µM of P for different lengths of time as indicated below. Despite 12hr serum-starvation there was still marked baseline phosphorylation of Src, which did not allow for qualitative distinction between control and treated groups.
Figure 4.3d. Expression of p-Src in Fitz cell line during increasing exposure to NE. Fitz was serum-starved for 24 hr and subsequently increasingly exposed to 10 µM of NE. There was a qualitative difference between untreated control and 15-60 minute exposure.

53
Figure 4.3e. Expression of p-Src in Fitz cell line during increasing exposure to NE after pretreatment with P. Fitz was serum-starved for 24 hr and cells received either no pre-treatment or 0.1µM of propranolol for 24 hours. Both groups were subsequently increasingly exposed to 10 µM of NE. There was a modest increase in phosphorylation of Src over time (23% at 60’) and there was modestly decreased phosphorylation when the cells had been pretreated with propranolol (11% at 60’). This difference was quantified as the ratio of chemiluminescence emitted by p-Src and β-actin for each band.
Figure 4.3f. Expression of p-Src in DEN and Fitz cell line pretreated with NE and rescued with propranolol. DEN and Fitz were serum-starved for 24 hr and cells received either no pre-treatment or 10µM of NE for 45 minutes. Cells were subsequently exposed to 0-100µM of P. In non-pretreated Fitz cells, low doses of propranolol reduce p-Src, which is lost at the highest dose. With NE agonism in Fitz cells, propranolol reduces p-Src compared to basal levels at all doses. In DEN cells, propranolol increases p-Src in untreated and NE stimulated cells.

54
Figure 4.4a. VEGF secretion by cHSA cell lines after treatment with NE. The graph represent an average of VEGF secretion by Fitz and DEN quantified by ELISA and normalized to cell count. Cells were treated with increasing doses of NE (0-10 µM) for 24 hours. In both cell lines, compared to untreated cells, there is a significant difference in VEGF secretion (DEN p<0.05, Fitz p<0.001). Error bars represent SD.
Figure 4.4b. VEGF secretion by cHSA cell lines after treatment with NE and P. The graphs represent an average of VEGF secretion by Fitz and DEN quantified by ELISA and normalized to cell count. All cells were treated with 1 µM NE (agonist) and P at 0-100µM for 24 hours. At propranolol doses of ≥10 µM, there was a significant difference of VEGF secretion by both cell lines compared to untreated cells (DEN p<0.01, Fitz p<0.001). For Fitz, there was a significant difference between 10 and 100 µM. p<0.05. Error bars represent SD.

55
Figure 4.5. Proliferation activity of cHSA cell lines after treatment with NE and P. Fitz and DEN were treated with different concentrations of NE and P for 24 hours (top two graphs) and subsequently analyzed by MTS assay. There was no statistical difference between the groups in both cell lines (p > 0.5). When cells were treated for 48 hours (bottom 2 graphs), Fitz exhibited significant difference between untreated vs 0.1 µM of NE (p < 0.05) and untreated vs 0.1 µM of P (p < 0.01). Error bars represent SD.

56
Figure 4.6. Migration of cHSA cells when exposed to increasing doses of propranolol. A scratch assay was performed with Fitz and DEN exposed to increasing concentrations of propranolol for 24 hours, and an average measurement of the residual gap in each treatment group was compared to an untreated control. There was a significant difference between the untreated group and 100 µM (p < 0.05). Error bars represent SD.

57
Figure 4.6 (cont). Migration of cHSA cells when exposed to increasing doses of propranolol. A scratch assay was performed with Fitz and DEN exposed to increasing concentrations of propranolol for 24 hours, and an average measurement of the residual gap in each treatment group was compared to an untreated control. There was a significant difference between the untreated group and 100 µM (p < 0.05). Error bars represent SD.

58
REFERENCES
1. Priester, W.A., Hepatic angiosarcomas in dogs: an excessive frequency as compared with man. J Natl Cancer Inst, 1976. 57(2): p. 451-‐4.
2. Priester, W.A. and F.W. McKay, The occurrence of tumors in domestic animals. Natl Cancer Inst Monogr, 1980(54): p. 1-‐210.
3. Spangler, W.L. and M.R. Culbertson, Prevalence, type, and importance of splenic diseases in dogs: 1,480 cases (1985-‐1989). J Am Vet Med Assoc, 1992. 200(6): p. 829-‐34.
4. Thamm, D.H., Hemangiosarcoma, in Small Animal Clinical Oncology, S.J. Withrow, Vail D.M., Page, R.L., Editor. 2013, Elsevier: St. Louis, MO. p. 679-‐688.
5. Leaute-‐Labreze, C., et al., Propranolol for severe hemangiomas of infancy. N Engl J Med, 2008. 358(24): p. 2649-‐51.
6. Leboulanger, N., et al., Propranolol in the therapeutic strategy of infantile laryngotracheal hemangioma: A preliminary retrospective study of French experience. Int J Pediatr Otorhinolaryngol, 2010. 74(11): p. 1254-‐7.
7. Kurzyna, A., et al., [Propranolol for treatment of subglottic hemangioma]. Otolaryngol Pol, 2010. 64(6): p. 388-‐91.
8. Jadhav, V.M. and S.N. Tolat, Dramatic response of propranolol in hemangioma: report of two cases. Indian J Dermatol Venereol Leprol, 2010. 76(6): p. 691-‐4.
9. Fay, A., et al., Propranolol for isolated orbital infantile hemangioma. Arch Ophthalmol, 2010. 128(2): p. 256-‐8.
10. Baetz, J., et al., [Infantile hemangioma. Successful treatment with propranolol]. Hautarzt, 2010. 61(4): p. 290-‐2.
11. Denoyelle, F., et al., Role of Propranolol in the therapeutic strategy of infantile laryngotracheal hemangioma. Int J Pediatr Otorhinolaryngol, 2009. 73(8): p. 1168-‐72.
12. Barron, T.I., et al., Beta blockers and breast cancer mortality: a population-‐ based study. J Clin Oncol, 2011. 29(19): p. 2635-‐44.
13. Powe, D.G., et al., Beta-‐blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget, 2010. 1(7): p. 628-‐38.
14. Buhler, H.U., et al., Plasma adrenaline, noradrenaline and dopamine in man and different animal species. J Physiol, 1978. 276: p. 311-‐20.
15. http://www.oxforddictionaries.com/us/definition/american_english/stress). . 16. Reiche, E.M., S.O. Nunes, and H.K. Morimoto, Stress, depression, the immune system, and
cancer. Lancet Oncol, 2004. 5(10): p. 617-‐25. 17. Chrousos, G.P., The hypothalamic-‐pituitary-‐adrenal axis and immune-‐mediated inflammation. N
Engl J Med, 1995. 332(20): p. 1351-‐62. 18. Dimsdale, J.M., M, Plasma catecholamines in stress and exercise. JAMA, 1980. 243(2): p. 340-‐
342. 19. Cole, S.W. and A.K. Sood, Molecular pathways: beta-‐adrenergic signaling in cancer. Clin Cancer
Res, 2012. 18(5): p. 1201-‐6. 20. Daly, C.J. and J.C. McGrath, Previously unsuspected widespread cellular and tissue distribution of
beta-‐adrenoceptors and its relevance to drug action. Trends Pharmacol Sci, 2011. 32(4): p. 219-‐26.
21. Carroll, B.T., et al., Screening for depression and anxiety in cancer patients using the Hospital Anxiety and Depression Scale. Gen Hosp Psychiatry, 1993. 15(2): p. 69-‐74.
22. Sellick, S.M. and A.D. Edwardson, Screening new cancer patients for psychological distress using the hospital anxiety and depression scale. Psychooncology, 2007. 16(6): p. 534-‐42.
23. Mantovani, A., et al., Cancer-‐related inflammation. Nature, 2008. 454(7203): p. 436-‐44.

59
24. Candido, J. and T. Hagemann, Cancer-‐related inflammation. J Clin Immunol, 2013. 33 Suppl 1: p. S79-‐84.
25. Pollock, R.E. and J.A. Roth, Cancer-‐induced immunosuppression: implications for therapy? Semin Surg Oncol, 1989. 5(6): p. 414-‐9.
26. Jenkins, F.J., B. Van Houten, and D.H. Bovbjerg, Effects on DNA Damage and/or Repair Processes as Biological Mechanisms Linking Psychological Stress to Cancer Risk. J Appl Biobehav Res, 2014. 19(1): p. 3-‐23.
27. Obeid, E.I. and S.D. Conzen, The role of adrenergic signaling in breast cancer biology. Cancer Biomark, 2013. 13(3): p. 161-‐9.
28. Yang, E.V. and T.D. Eubank, The impact of adrenergic signaling in skin cancer progression: possible repurposing of beta-‐blockers for treatment of skin cancer. Cancer Biomark, 2013. 13(3): p. 155-‐60.
29. Bridle, P.A., et al., Basal levels of plasma epinephrine and norepinephrine in the dog. Hypertension, 1983. 5(6 Pt 3): p. V128-‐33.
30. Sloan, E.K., et al., The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res, 2010. 70(18): p. 7042-‐52.
31. Taylor, M.R., Pharmacogenetics of the human beta-‐adrenergic receptors. Pharmacogenomics J, 2007. 7(1): p. 29-‐37.
32. Audet, M. and M. Bouvier, Insights into signaling from the beta2-‐adrenergic receptor structure. Nat Chem Biol, 2008. 4(7): p. 397-‐403.
33. Rosenbaum, D.M., S.G. Rasmussen, and B.K. Kobilka, The structure and function of G-‐protein-‐coupled receptors. Nature, 2009. 459(7245): p. 356-‐63.
34. Chisholm, K.M., et al., beta-‐Adrenergic receptor expression in vascular tumors. Mod Pathol, 2012. 25(11): p. 1446-‐51.
35. Lipka, E., et al., In vivo non-‐linear intestinal permeability of celiprolol and propranolol in conscious dogs: evidence for intestinal secretion. Eur J Pharm Sci, 1998. 6(1): p. 75-‐81.
36. Vauquelin, G.a.v.M., B. , G-‐protein coupled receptors. G Protein-‐Coupled Receptors: Molecular Pharmacology from Academic Concept to Pharmaceutical Research, 2007: p. 77-‐230.
37. Luttrell, L.M., et al., Beta-‐arrestin-‐dependent formation of beta2 adrenergic receptor-‐Src protein kinase complexes. Science, 1999. 283(5402): p. 655-‐61.
38. Armaiz-‐Pena, G.N., et al., Src activation by beta-‐adrenoreceptors is a key switch for tumour metastasis. Nat Commun, 2013. 4: p. 1403.
39. Guarino, M., Src signaling in cancer invasion. J Cell Physiol, 2010. 223(1): p. 14-‐26. 40. Summy, J.M. and G.E. Gallick, Src family kinases in tumor progression and metastasis. Cancer
Metastasis Rev, 2003. 22(4): p. 337-‐58. 41. Shor, A.C., et al., Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines
and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res, 2007. 67(6): p. 2800-‐8.
42. Fredriksson, J.M., et al., Norepinephrine induces vascular endothelial growth factor gene expression in brown adipocytes through a beta -‐adrenoreceptor/cAMP/protein kinase A pathway involving Src but independently of Erk1/2. J Biol Chem, 2000. 275(18): p. 13802-‐11.
43. Fredriksson, J.M. and J. Nedergaard, Norepinephrine specifically stimulates ribonucleotide reductase subunit R2 gene expression in proliferating brown adipocytes: mediation via a cAMP/PKA pathway involving Src and Erk1/2 kinases. Exp Cell Res, 2002. 274(2): p. 207-‐15.
44. Dickerson, E.B., et al., Imatinib and Dasatinib Inhibit Hemangiosarcoma and Implicate PDGFR-‐beta and Src in Tumor Growth. Transl Oncol, 2013. 6(2): p. 158-‐68.
45. Powe, D.G. and F. Entschladen, Targeted therapies: Using beta-‐blockers to inhibit breast cancer progression. Nat Rev Clin Oncol, 2011. 8(9): p. 511-‐2.

60
46. Guise, T.A., et al., Evidence for a causal role of parathyroid hormone-‐related protein in the pathogenesis of human breast cancer-‐mediated osteolysis. J Clin Invest, 1996. 98(7): p. 1544-‐9.
47. Campbell, J.P., et al., Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice. PLoS Biol, 2012. 10(7): p. e1001363.
48. Katayama, Y., et al., Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell, 2006. 124(2): p. 407-‐21.
49. Calvi, L.M., Osteoblastic activation in the hematopoietic stem cell niche. Ann N Y Acad Sci, 2006. 1068: p. 477-‐88.
50. Elefteriou, F., Neuronal signaling and the regulation of bone remodeling. Cell Mol Life Sci, 2005. 62(19-‐20): p. 2339-‐49.
51. Melhem-‐Bertrandt, A., et al., Beta-‐blocker use is associated with improved relapse-‐free survival in patients with triple-‐negative breast cancer. J Clin Oncol, 2011. 29(19): p. 2645-‐52.
52. Haffty, B.G., et al., Locoregional relapse and distant metastasis in conservatively managed triple negative early-‐stage breast cancer. J Clin Oncol, 2006. 24(36): p. 5652-‐7.
53. Lin, N.U., et al., Sites of distant recurrence and clinical outcomes in patients with metastatic triple-‐negative breast cancer: high incidence of central nervous system metastases. Cancer, 2008. 113(10): p. 2638-‐45.
54. Powe, D.G., et al., Alpha-‐ and beta-‐adrenergic receptor (AR) protein expression is associated with poor clinical outcome in breast cancer: an immunohistochemical study. Breast Cancer Res Treat, 2011. 130(2): p. 457-‐63.
55. Jansen, L., et al., Beta blocker use and colorectal cancer risk: population-‐based case-‐control study. Cancer, 2012. 118(16): p. 3911-‐9.
56. Friedman, G.D., N. Udaltsova, and L.A. Habel, Norepinephrine antagonists and cancer risk. Int J Cancer, 2011. 128(3): p. 737-‐8; author reply 739.
57. Shah, S.M., et al., Does beta-‐adrenoceptor blocker therapy improve cancer survival? Findings from a population-‐based retrospective cohort study. Br J Clin Pharmacol, 2011. 72(1): p. 157-‐61.
58. Fitzgerald, P.J., Beta blockers, norepinephrine, and cancer: an epidemiological viewpoint. Clin Epidemiol, 2012. 4: p. 151-‐6.
59. Bangalore, S., et al., Antihypertensive drugs and risk of cancer: network meta-‐analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol, 2011. 12(1): p. 65-‐82.
60. Huang, X.Y., et al., Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via beta-‐adrenergic receptor-‐dependent activation of P38/MAPK pathway. Hepatogastroenterology, 2012. 59(115): p. 889-‐93.
61. Guo, K., et al., Norepinephrine-‐induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol Rep, 2009. 22(4): p. 825-‐30.
62. Zhang, D., et al., beta2-‐adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFkappaB and AP-‐1. Cancer Biol Ther, 2010. 10(1): p. 19-‐29.
63. Al-‐Wadei, H.A., M.H. Al-‐Wadei, and H.M. Schuller, Prevention of pancreatic cancer by the beta-‐blocker propranolol. Anticancer Drugs, 2009. 20(6): p. 477-‐82.
64. Lin, X., et al., Beta-‐adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology, 2012. 59(114): p. 584-‐8.
65. Wisler, J.W., et al., A unique mechanism of beta-‐blocker action: carvedilol stimulates beta-‐arrestin signaling. Proc Natl Acad Sci U S A, 2007. 104(42): p. 16657-‐62.
66. Thaker, P.H., et al., Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med, 2006. 12(8): p. 939-‐44.
67. Lutgendorf, S.K., et al., Stress-‐related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin Cancer Res, 2003. 9(12): p. 4514-‐21.

61
68. Jemal, A., et al., Cancer statistics, 2004. CA Cancer J Clin, 2004. 54(1): p. 8-‐29. 69. Hlatky, L., P. Hahnfeldt, and J. Folkman, Clinical application of antiangiogenic therapy:
microvessel density, what it does and doesn't tell us. J Natl Cancer Inst, 2002. 94(12): p. 883-‐93. 70. Lee, J.W., et al., The effect of surgical wound on ovarian carcinoma growth in an animal model.
Anticancer Res, 2013. 33(8): p. 3177-‐84. 71. Lee, J.W., et al., Surgical stress promotes tumor growth in ovarian carcinoma. Clin Cancer Res,
2009. 15(8): p. 2695-‐702. 72. Allendorf, J.D., et al., Increased tumor establishment and growth after open vs laparoscopic
bowel resection in mice. Surg Endosc, 1998. 12(8): p. 1035-‐8. 73. Abramovitch, R., et al., Stimulation of tumour growth by wound-‐derived growth factors. Br J
Cancer, 1999. 79(9-‐10): p. 1392-‐8. 74. Belizon, A., et al., Major abdominal surgery increases plasma levels of vascular endothelial
growth factor: open more so than minimally invasive methods. Ann Surg, 2006. 244(5): p. 792-‐8. 75. Ramos-‐Jimenez, J., et al., Histamine augments beta2-‐adrenoceptor-‐induced cyclic AMP
accumulation in human prostate cancer cells DU-‐145 independently of known histamine receptors. Biochem Pharmacol, 2007. 73(6): p. 814-‐23.
76. Zhang, P., et al., beta-‐arrestin2 mediates beta-‐2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncology reports, 2011. 26(6): p. 1471-‐7.
77. Palm, D., et al., The norepinephrine-‐driven metastasis development of PC-‐3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-‐blockers. Int J Cancer, 2006. 118(11): p. 2744-‐9.
78. Hassan, S., et al., Behavioral stress accelerates prostate cancer development in mice. J Clin Invest, 2013. 123(2): p. 874-‐86.
79. Ramberg, H., et al., Hormonal regulation of beta2-‐adrenergic receptor level in prostate cancer. Prostate, 2008. 68(10): p. 1133-‐42.
80. Prowatke, I., et al., Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br J Cancer, 2007. 96(1): p. 82-‐8.
81. Pippig, S., et al., Overexpression of beta-‐arrestin and beta-‐adrenergic receptor kinase augment desensitization of beta 2-‐adrenergic receptors. J Biol Chem, 1993. 268(5): p. 3201-‐8.
82. Grytli, H.H., et al., Use of beta-‐blockers is associated with prostate cancer-‐specific survival in prostate cancer patients on androgen deprivation therapy. Prostate, 2013. 73(3): p. 250-‐60.
83. Lin, Q., et al., Effect of chronic restraint stress on human colorectal carcinoma growth in mice. PLoS One, 2013. 8(4): p. e61435.
84. Hicks, B.M., et al., beta-‐Blocker usage and colorectal cancer mortality: a nested case-‐control study in the UK Clinical Practice Research Datalink cohort. Ann Oncol, 2013. 24(12): p. 3100-‐6.
85. Moretti, S., et al., beta-‐adrenoceptors are upregulated in human melanoma and their activation releases pro-‐tumorigenic cytokines and metalloproteases in melanoma cell lines. Lab Invest, 2013. 93(3): p. 279-‐90.
86. Guo, Y., et al., Interleukin-‐6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev, 2012. 38(7): p. 904-‐10.
87. Waugh, D.J. and C. Wilson, The interleukin-‐8 pathway in cancer. Clin Cancer Res, 2008. 14(21): p. 6735-‐41.
88. Yang, E.V., et al., Norepinephrine upregulates VEGF, IL-‐8, and IL-‐6 expression in human melanoma tumor cell lines: implications for stress-‐related enhancement of tumor progression. Brain Behav Immun, 2009. 23(2): p. 267-‐75.
89. De Giorgi, V., et al., beta-‐adrenergic-‐blocking drugs and melanoma: current state of the art. Expert Rev Anticancer Ther, 2012. 12(11): p. 1461-‐7.

62
90. De Giorgi, V., et al., Effect of beta-‐blockers and other antihypertensive drugs on the risk of melanoma recurrence and death. Mayo Clin Proc, 2013. 88(11): p. 1196-‐203.
91. Kozanoglu, I., et al., New indication for therapeutic potential of an old well-‐known drug (propranolol) for multiple myeloma. J Cancer Res Clin Oncol, 2013. 139(2): p. 327-‐35.
92. Yang, E.V., et al., VEGF is differentially regulated in multiple myeloma-‐derived cell lines by norepinephrine. Brain Behav Immun, 2008. 22(3): p. 318-‐23.
93. Shand, D.G., Pharmacokinetics of propranolol: a review. Postgrad Med J, 1976. 52 Suppl 4: p. 22-‐25.
94. Mills, P.C., G.A. Siebert, and M.S. Roberts, A model to study intestinal and hepatic metabolism of propranolol in the dog. J Vet Pharmacol Ther, 2004. 27(1): p. 45-‐8.
95. Akin, A., et al., The efficacy of amiodarone-‐propranolol combination for the management of childhood arrhythmias. Pacing Clin Electrophysiol, 2013. 36(6): p. 727-‐31.
96. Hedera, P., F. Cibulcik, and T.L. Davis, Pharmacotherapy of Essential Tremor. J Cent Nerv Syst Dis, 2013. 5: p. 43-‐55.
97. Schneider, S.A. and G. Deuschl, The treatment of tremor. Neurotherapeutics, 2014. 11(1): p. 128-‐38.
98. Hepp, Z., L.M. Bloudek, and S.F. Varon, Systematic review of migraine prophylaxis adherence and persistence. J Manag Care Pharm, 2014. 20(1): p. 22-‐33.
99. Shamliyan, T.A., et al., Preventive pharmacologic treatments for episodic migraine in adults. J Gen Intern Med, 2013. 28(9): p. 1225-‐37.
100. Hruska, B., P.K. Cullen, and D.L. Delahanty, Pharmacological modulation of acute trauma memories to prevent PTSD: Considerations from a developmental perspective. Neurobiol Learn Mem, 2014.
101. Fitzgerald, P.J., J.R. Seemann, and S. Maren, Can fear extinction be enhanced? A review of pharmacological and behavioral findings. Brain Res Bull, 2013.
102. Tawa, J. and S. Murphy, Psychopharmacological treatment for military posttraumatic stress disorder: an integrative review. J Am Assoc Nurse Pract, 2013. 25(8): p. 419-‐23.
103. de Kleine, R.A., B.O. Rothbaum, and A. van Minnen, Pharmacological enhancement of exposure-‐based treatment in PTSD: a qualitative review. Eur J Psychotraumatol, 2013. 4.
104. Searcy, C.P., et al., Pharmacological prevention of combat-‐related PTSD: a literature review. Mil Med, 2012. 177(6): p. 649-‐54.
105. Black, J.W., et al., A NEW ADRENERGIC BETARECEPTOR ANTAGONIST. Lancet, 1964. 1(7342): p. 1080-‐1.
106. Basu, B., et al., A Case of Akathisia induced by Escitalopram: Case Report & Review of Literature. Curr Drug Saf, 2014. 9(1): p. 56-‐9.
107. Fitzgerald, P.J., Is elevated norepinephrine an etiological factor in some cases of schizophrenia? Psychiatry Res, 2014. 215(3): p. 497-‐504.
108. Hogan, S.R., J. Mandrell, and D. Eilers, Adrenergic urticaria: Review of the literature and proposed mechanism. J Am Acad Dermatol, 2013.
109. Finnerty, C.C. and D.N. Herndon, Is propranolol of benefit in pediatric burn patients? Adv Surg, 2013. 47: p. 177-‐97.
110. Herndon, D.N., et al., Long-‐term propranolol use in severely burned pediatric patients: a randomized controlled study. Ann Surg, 2012. 256(3): p. 402-‐11.
111. Olah, G., et al., Increased poly(ADP-‐ribosyl)ation in skeletal muscle tissue of pediatric patients with severe burn injury: prevention by propranolol treatment. Shock, 2011. 36(1): p. 18-‐23.
112. Rojas, Y., et al., Burns: an update on current pharmacotherapy. Expert Opin Pharmacother, 2012. 13(17): p. 2485-‐94.

63
113. Nandhra, H.S., C.L. Murphy, and A. Sule, Novel pharmacological agents targeting memory and cognition in the treatment of anxiety disorders. Hum Psychopharmacol, 2013.
114. Altamura, A.C., et al., Understanding the pharmacokinetics of anxiolytic drugs. Expert Opin Drug Metab Toxicol, 2013. 9(4): p. 423-‐40.
115. Lindgren, M.E., et al., Beta-‐blockers may reduce intrusive thoughts in newly diagnosed cancer patients. Psychooncology, 2013. 22(8): p. 1889-‐94.
116. Lawley, L.P., E. Siegfried, and J.L. Todd, Propranolol treatment for hemangioma of infancy: risks and recommendations. Pediatr Dermatol, 2009. 26(5): p. 610-‐4.
117. Pavlakovic, H., et al., Hyperkalemia complicating propranolol treatment of an infantile hemangioma. Pediatrics, 2010. 126(6): p. e1589-‐93.
118. Abbott, J., et al., Diarrhea associated with propranolol treatment for hemangioma of infancy (HOI). Pediatr Dermatol, 2010. 27(5): p. 558.
119. Giron-‐Vallejo, O., et al., Dental caries as a side effect of infantile hemangioma treatment with propranolol solution. Pediatr Dermatol, 2010. 27(6): p. 672-‐3.
120. Bonifazi, E., et al., Severe hypoglycemia during successful treatment of diffuse hemangiomatosis with propranolol. Pediatr Dermatol, 2010. 27(2): p. 195-‐6.
121. McBride, J.T., M.C. McBride, and P.H. Viles, Hypoglycemia associated with propranolol. Pediatrics, 1973. 51(6): p. 1085-‐7.
122. Holland, K.E., et al., Hypoglycemia in children taking propranolol for the treatment of infantile hemangioma. Arch Dermatol, 2010. 146(7): p. 775-‐8.
123. Kwon, E.K., et al., Retrospective review of adverse effects from propranolol in infants. JAMA Dermatol, 2013. 149(4): p. 484-‐5.
124. Volmer, P.A., Human Drugs of Abuse, in Kirk's Current Veterinary Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co.: Philadelphia. p. 144-‐145.
125. Wright, K.N., Assessment and Treatment of Supraventricular Tachyarrhythmias, in Kirk's Current Veterinary Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co: Philadelphia. p. 722-‐727.
126. Schober, K.E., Myocarditis, in Kirk's Current Veterinary Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co: Philadelphia. p. 804-‐808.
127. Kates, R.E., B.W. Keene, and R.L. Hamlin, Pharmacokinetics of propranolol in the dog. Journal of Veterinary Pharmacology and Therapeutics, 1979. 2(1): p. 21-‐26.
128. Trepanier, L.A., Medical Treatment of Feline Hyperthyroidism, in Kirk's Current Veterinary Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co: Philadelphia. p. 175-‐179.
129. Westropp, I.F.L.J.L., Urinary Incontinence and Micturition Disorders: Pharmacologic Management, in Kirk's Current Veterinary Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co.: Philadelphia. p. 955.
130. Dana G. Allen, J.K.P., Dale Smith, Handbook of Veterinary Drugs. 1993. 131. Feng, X.M., et al., Preparation and evaluation of a novel delayed-‐onset sustained-‐release system
of propranolol hydrochloride. J Pharm Pharmacol, 2008. 60(7): p. 817-‐22. 132. Papich, M.G., Table of Common Drugs: Approximate Dosages, in Kirk's Current Veterinary
Therapy XIV, J.D.B.D.C. Twedt, Editor. 2009, Saunders Co: Philadelphia. p. 1329. 133. Plumb, Plumb's Veterinary Drug Handbook. Vol. 6th. 2008. 134. Fonseca Junior, N.L., et al., [Therapeutical effectiveness of interferon alpha in a child with
craniofacial giant hemangioma: case report]. Arq Bras Oftalmol, 2008. 71(3): p. 423-‐6. 135. Enjolras, O., et al., [Vincristine treatment for function-‐ and life-‐threatening infantile
hemangioma]. Arch Pediatr, 2004. 11(2): p. 99-‐107. 136. Stiles, J., et al., Propranolol treatment of infantile hemangioma endothelial cells: A molecular
analysis. Exp Ther Med, 2012. 4(4): p. 594-‐604.

64
137. Drolet, B.A., et al., Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics, 2013. 131(1): p. 128-‐40.
138. Shah, S. and I.J. Frieden, Treatment of infantile hemangiomas with beta-‐blockers: a review. Skin Therapy Lett, 2013. 18(6): p. 5-‐7.
139. Frieden, I.J., et al., Infantile hemangiomas: current knowledge, future directions. Proceedings of a research workshop on infantile hemangiomas, April 7-‐9, 2005, Bethesda, Maryland, USA. Pediatr Dermatol, 2005. 22(5): p. 383-‐406.
140. Haggstrom, A.N., et al., Measuring the severity of infantile hemangiomas: instrument development and reliability. Arch Dermatol, 2012. 148(2): p. 197-‐202.
141. Boye, E. and B.R. Olsen, Signaling mechanisms in infantile hemangioma. Curr Opin Hematol, 2009. 16(3): p. 202-‐8.
142. Grimmer, J.F., et al., Familial clustering of hemangiomas. Arch Otolaryngol Head Neck Surg, 2011. 137(8): p. 757-‐60.
143. Khan, Z.A., et al., Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest, 2008. 118(7): p. 2592-‐9.
144. Guimaraes, S. and D. Moura, Vascular adrenoceptors: an update. Pharmacol Rev, 2001. 53(2): p. 319-‐56.
145. Wolter, N.E., et al., Propranolol as a novel adjunctive treatment for head and neck squamous cell carcinoma. J Otolaryngol Head Neck Surg, 2012. 41(5): p. 334-‐44.
146. Neufeld, G., et al., Vascular endothelial growth factor (VEGF) and its receptors. Faseb j, 1999. 13(1): p. 9-‐22.
147. Ji, Y., et al., Effects of propranolol on the proliferation and apoptosis of hemangioma-‐derived endothelial cells. J Pediatr Surg, 2012. 47(12): p. 2216-‐23.
148. Zhao, Z.F., et al., [The change of serum vascular endothelial growth factor and matrix metalloproteinases-‐9 in proliferative hemangioma treated with propranolol]. Zhonghua Zheng Xing Wai Ke Za Zhi, 2011. 27(5): p. 359-‐61.
149. Chim, H., et al., Propranolol induces regression of hemangioma cells through HIF-‐1alpha-‐mediated inhibition of VEGF-‐A. Ann Surg, 2012. 256(1): p. 146-‐56.
150. Schieven, G.L., The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem, 2005. 5(10): p. 921-‐8.
151. Bamburg, J.R., Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol, 1999. 15: p. 185-‐230.
152. Street, C.A. and B.A. Bryan, Rho kinase proteins-‐-‐pleiotropic modulators of cell survival and apoptosis. Anticancer Res, 2011. 31(11): p. 3645-‐57.
153. Kelly, B.D., et al., Cell type-‐specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-‐inducible factor 1. Circ Res, 2003. 93(11): p. 1074-‐81.
154. Kleinman, M.E., et al., Hypoxia-‐induced mediators of stem/progenitor cell trafficking are increased in children with hemangioma. Arterioscler Thromb Vasc Biol, 2007. 27(12): p. 2664-‐70.
155. Greenberger, S., et al., Targeting NF-‐kappaB in infantile hemangioma-‐derived stem cells reduces VEGF-‐A expression. Angiogenesis, 2010. 13(4): p. 327-‐35.
156. Bagazgoitia, L., A. Hernandez-‐Martin, and A. Torrelo, Recurrence of infantile hemangiomas treated with propranolol. Pediatr Dermatol, 2011. 28(6): p. 658-‐62.
157. Chang, L., et al., Recurrence of infantile hemangioma after termination of propranolol treatment. Ann Plast Surg, 2014. 72(2): p. 173-‐5.
158. Shehata, N., et al., Late rebound of infantile hemangioma after cessation of oral propranolol. Pediatr Dermatol, 2013. 30(5): p. 587-‐91.

65
159. Wong, A., et al., Propranolol accelerates adipogenesis in hemangioma stem cells and causes apoptosis of hemangioma endothelial cells. Plast Reconstr Surg, 2012. 130(5): p. 1012-‐21.
160. Zhang, L., et al., Propranolol inhibits angiogenesis via down-‐regulating the expression of vascular endothelial growth factor in hemangioma derived stem cell. Int J Clin Exp Pathol, 2013. 7(1): p. 48-‐55.
161. Zou, H.X., et al., Propranolol inhibits endothelial progenitor cell homing: a possible treatment mechanism of infantile hemangioma. Cardiovasc Pathol, 2013. 22(3): p. 203-‐10.
162. Rostami, M., et al., Tumors in domestic animals examined during a ten-‐year period (1980 to 1989) at Miyazaki University. J Vet Med Sci, 1994. 56(2): p. 403-‐5.
163. Day, M.J., V.M. Lucke, and H. Pearson, A review of pathological diagnoses made from 87 canine splenic biopsies. J Small Anim Pract, 1995. 36(10): p. 426-‐33.
164. Spangler, W.L. and P.H. Kass, Pathologic factors affecting postsplenectomy survival in dogs. J Vet Intern Med, 1997. 11(3): p. 166-‐71.
165. Schultheiss, P.C., A retrospective study of visceral and nonvisceral hemangiosarcoma and hemangiomas in domestic animals. J Vet Diagn Invest, 2004. 16(6): p. 522-‐6.
166. Oksanen, A., Haemangiosarcoma in dogs. J Comp Pathol, 1978. 88(4): p. 585-‐95. 167. Arp, L.H. and R.L. Grier, Disseminated cutaneous hemangiosarcoma in a young dog. J Am Vet
Med Assoc, 1984. 185(6): p. 671-‐3. 168. Brown, N.O., A.K. Patnaik, and E.G. MacEwen, Canine hemangiosarcoma: retrospective analysis
of 104 cases. J Am Vet Med Assoc, 1985. 186(1): p. 56-‐8. 169. Srebernik, N. and E.C. Appleby, Breed prevalence and sites of haemangioma and
haemangiosarcoma in dogs. Vet Rec, 1991. 129(18): p. 408-‐9. 170. Moe, L., et al., Canine neoplasia-‐-‐population-‐based incidence of vascular tumours. APMIS Suppl,
2008(125): p. 63-‐8. 171. Hargis, A.M., et al., A retrospective clinicopathologic study of 212 dogs with cutaneous
hemangiomas and hemangiosarcomas. Vet Pathol, 1992. 29(4): p. 316-‐28. 172. Ward, H., et al., Cutaneous hemangiosarcoma in 25 dogs: a retrospective study. J Vet Intern
Med, 1994. 8(5): p. 345-‐8. 173. Ware, W.A. and D.L. Hopper, Cardiac tumors in dogs: 1982-‐1995. J Vet Intern Med, 1999. 13(2):
p. 95-‐103. 174. Aupperle, H., et al., Primary and secondary heart tumours in dogs and cats. J Comp Pathol, 2007.
136(1): p. 18-‐26. 175. Goritz, M., et al., Canine splenic haemangiosarcoma: influence of metastases, chemotherapy and
growth pattern on post-‐splenectomy survival and expression of angiogenic factors. J Comp Pathol, 2013. 149(1): p. 30-‐9.
176. Sabattini, S. and G. Bettini, An immunohistochemical analysis of canine haemangioma and haemangiosarcoma. J Comp Pathol, 2009. 140(2-‐3): p. 158-‐68.
177. Yonemaru, K., et al., Expression of vascular endothelial growth factor, basic fibroblast growth factor, and their receptors (flt-‐1, flk-‐1, and flg-‐1) in canine vascular tumors. Vet Pathol, 2006. 43(6): p. 971-‐80.
178. Clifford, C.A., et al., Plasma vascular endothelial growth factor concentrations in healthy dogs and dogs with hemangiosarcoma. J Vet Intern Med, 2001. 15(2): p. 131-‐5.
179. Clifford, C.A., et al., Vascular endothelial growth factor concentrations in body cavity effusions in dogs. J Vet Intern Med, 2002. 16(2): p. 164-‐8.
180. Kato, Y., et al., Gene expressions of canine angiopoietin-‐1 and -‐2 in normal tissues and spontaneous tumours. Res Vet Sci, 2006. 81(2): p. 280-‐6.
181. Asa, S.A., et al., Expression of platelet-‐derived growth factor and its receptors in spontaneous canine hemangiosarcoma and cutaneous hemangioma. Histol Histopathol, 2012. 27(5): p. 601-‐7.

66
182. Mayr, B., et al., Tumour suppressor gene p53 mutation in a case of haemangiosarcoma of a dog. Acta Vet Hung, 2002. 50(2): p. 157-‐60.
183. Yonemaru, K., et al., The significance of p53 and retinoblastoma pathways in canine hemangiosarcoma. J Vet Med Sci, 2007. 69(3): p. 271-‐8.
184. Tamburini, B.A., et al., Gene expression profiling identifies inflammation and angiogenesis as distinguishing features of canine hemangiosarcoma. BMC Cancer, 2010. 10: p. 619.
185. Dickerson, E.B., et al., Mutations of phosphatase and tensin homolog deleted from chromosome 10 in canine hemangiosarcoma. Vet Pathol, 2005. 42(5): p. 618-‐32.
186. Murakami, M., et al., Expression of the anti-‐apoptotic factors Bcl-‐2 and survivin in canine vascular tumours. J Comp Pathol, 2008. 139(1): p. 1-‐7.
187. Bertazzolo, W., et al., Canine angiosarcoma: cytologic, histologic, and immunohistochemical correlations. Vet Clin Pathol, 2005. 34(1): p. 28-‐34.
188. Gamlem, H. and K. Nordstoga, Canine vascular neoplasia-‐-‐histologic classification and inmunohistochemical analysis of 221 tumours and tumour-‐like lesions. APMIS Suppl, 2008(125): p. 19-‐40.
189. Jakab, C., et al., Claudin-‐5 protein is a new differential marker for histopathological differential diagnosis of canine hemangiosarcoma. Histol Histopathol, 2009. 24(7): p. 801-‐13.
190. Thamm, D.H., Miscellaneous tumors (hemangiosarcoma), in Withrow and MacEwen's Small Animal Clinical Oncology, S.J.W.D.M.V.R.L. Page, Editor. 2013, Saunders Co. p. 679-‐688.
191. Hammer, A.S., et al., Efficacy and toxicity of VAC chemotherapy (vincristine, doxorubicin, and cyclophosphamide) in dogs with hemangiosarcoma. J Vet Intern Med, 1991. 5(3): p. 160-‐6.
192. Alvarez, F.J., et al., VAC protocol for treatment of dogs with stage III hemangiosarcoma. J Am Anim Hosp Assoc, 2013. 49(6): p. 370-‐7.
193. Sorenmo, K., et al., Canine hemangiosarcoma treated with standard chemotherapy and minocycline. J Vet Intern Med, 2000. 14(4): p. 395-‐8.
194. Sorenmo, K.U., et al., Efficacy and toxicity of a dose-‐intensified doxorubicin protocol in canine hemangiosarcoma. J Vet Intern Med, 2004. 18(2): p. 209-‐13.
195. Ogilvie, G.K., et al., Surgery and doxorubicin in dogs with hemangiosarcoma. J Vet Intern Med, 1996. 10(6): p. 379-‐84.
196. Ogilvie, G.K., et al., Phase II evaluation of doxorubicin for treatment of various canine neoplasms. J Am Vet Med Assoc, 1989. 195(11): p. 1580-‐3.
197. Wiley, J.L., et al., Efficacy of doxorubicin-‐based chemotherapy for non-‐resectable canine subcutaneous haemangiosarcoma. Vet Comp Oncol, 2010. 8(3): p. 221-‐33.
198. Payne, S.E., et al., Treatment of vascular and soft-‐tissue sarcomas in dogs using an alternating protocol of ifosfamide and doxorubicin. Vet Comp Oncol, 2003. 1(4): p. 171-‐9.
199. Rassnick, K.M., et al., Evaluation of ifosfamide for treatment of various canine neoplasms. J Vet Intern Med, 2000. 14(3): p. 271-‐6.
200. Kim, S.E., et al., Epirubicin in the adjuvant treatment of splenic hemangiosarcoma in dogs: 59 cases (1997-‐2004). J Am Vet Med Assoc, 2007. 231(10): p. 1550-‐7.
201. Sorenmo, K., et al., Clinical and pharmacokinetic characteristics of intracavitary administration of pegylated liposomal encapsulated doxorubicin in dogs with splenic hemangiosarcoma. J Vet Intern Med, 2007. 21(6): p. 1347-‐54.
202. Vail, D.M., et al., Liposome-‐encapsulated muramyl tripeptide phosphatidylethanolamine adjuvant immunotherapy for splenic hemangiosarcoma in the dog: a randomized multi-‐institutional clinical trial. Clin Cancer Res, 1995. 1(10): p. 1165-‐70.
203. Hillers, K.R., et al., Effects of palliative radiation therapy on nonsplenic hemangiosarcoma in dogs. J Am Anim Hosp Assoc, 2007. 43(4): p. 187-‐92.

67
204. Lana, S., et al., Continuous low-‐dose oral chemotherapy for adjuvant therapy of splenic hemangiosarcoma in dogs. J Vet Intern Med, 2007. 21(4): p. 764-‐9.
205. Kahn, S.A., et al., Doxorubicin and deracoxib adjuvant therapy for canine splenic hemangiosarcoma: a pilot study. Can Vet J, 2013. 54(3): p. 237-‐42.
206. Gardner, H.L.L., C.A; Portela, R.F.; Nguyen, S.; Rosenberg, M.P.; Klein, M.K.; Clifford, C.; Thamm, D.H.; Vail, D.M; Bergman, P.J.; Crawford-‐Jakubiak, M.; Henry, C.; Locke, J.; Garrett, L.D.; Cronin, K.L., Maintenance therapy with toceranib following doxorubicin-‐based chemotherapy for canine splenic hemangiosarcoma. Manuscript submitted for publication., 2014.
207. Prymak, C., et al., Epidemiologic, clinical, pathologic, and prognostic characteristics of splenic hemangiosarcoma and splenic hematoma in dogs: 217 cases (1985). J Am Vet Med Assoc, 1988. 193(6): p. 706-‐12.
208. Wood, C.A., et al., Prognosis for dogs with stage I or II splenic hemangiosarcoma treated by splenectomy alone: 32 cases (1991-‐1993). J Am Anim Hosp Assoc, 1998. 34(5): p. 417-‐21.
209. Dunning, D., et al., Analysis of prognostic indicators for dogs with pericardial effusion: 46 cases (1985-‐1996). J Am Vet Med Assoc, 1998. 212(8): p. 1276-‐80.
210. Aronsohn, M., Cardiac hemangiosarcoma in the dog: a review of 38 cases. J Am Vet Med Assoc, 1985. 187(9): p. 922-‐6.
211. Szivek, A., et al., Clinical outcome in 94 cases of dermal haemangiosarcoma in dogs treated with surgical excision: 1993-‐2007*. Vet Comp Oncol, 2012. 10(1): p. 65-‐73.
212. Burton, J.H., B.E. Powers, and B.J. Biller, Clinical outcome in 20 cases of lingual hemangiosarcoma in dogs: 1996-‐2011. Vet Comp Oncol, 2012.
213. Shiu, K.B., et al., Predictors of outcome in dogs with subcutaneous or intramuscular hemangiosarcoma. J Am Vet Med Assoc, 2011. 238(4): p. 472-‐9.
214. Stiles, J.M., et al., Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PLoS One, 2013. 8(3): p. e60021.
215. Rada, T., L. Okruhlicova, and J. Slezak, Immunohistochemical localization of beta-‐adrenergic receptors in the heart. Bratisl Lek Listy, 1991. 92(3-‐4): p. 138-‐41.
216. Vroon, A., et al., Taxol normalizes the impaired agonist-‐induced beta2-‐adrenoceptor internalization in splenocytes from GRK2+/-‐ mice. Eur J Pharmacol, 2007. 560(1): p. 9-‐16.
217. Neubauer, B., et al., Renin expression in large renal vessels during fetal development depends on functional beta1/beta2-‐adrenergic receptors. Am J Physiol Renal Physiol, 2011. 301(1): p. F71-‐7.
218. Ferrer, L., et al., Immunohistochemical detection of CD31 antigen in normal and neoplastic canine endothelial cells. J Comp Pathol, 1995. 112(4): p. 319-‐26.
219. Slotkin, T.A., et al., Beta-‐adrenoceptor signaling and its control of cell replication in MDA-‐MB-‐231 human breast cancer cells. Breast Cancer Res Treat, 2000. 60(2): p. 153-‐66.
220. Tallman, J.F., C.C. Smith, and R.C. Henneberry, Induction of functional beta-‐adrenergic receptors in HeLa cells. Proc Natl Acad Sci U S A, 1977. 74(3): p. 873-‐7.
221. Liang, C.C., A.Y. Park, and J.L. Guan, In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc, 2007. 2(2): p. 329-‐33.
222. Avni, R., B. Cohen, and M. Neeman, Hypoxic stress and cancer: imaging the axis of evil in tumor metastasis. NMR Biomed, 2011. 24(6): p. 569-‐81.
223. Coussens, L.M. and Z. Werb, Inflammation and cancer. Nature, 2002. 420(6917): p. 860-‐7. 224. Ramos-‐Vara, J.A., Technical aspects of immunohistochemistry. Vet Pathol, 2005. 42(4): p. 405-‐
26. 225. Atkins, D., et al., Immunohistochemical detection of EGFR in paraffin-‐embedded tumor tissues:
variation in staining intensity due to choice of fixative and storage time of tissue sections. J Histochem Cytochem, 2004. 52(7): p. 893-‐901.

68
226. Xiao, K. and S.K. Shenoy, Beta2-‐adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J Biol Chem, 2011. 286(14): p. 12785-‐95.
227. Boivin, B., et al., Functional beta-‐adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res, 2006. 71(1): p. 69-‐78.
228. Chen, D., et al., The beta2-‐adrenergic receptor is a potential prognostic biomarker for human hepatocellular carcinoma after curative resection. Ann Surg Oncol, 2012. 19(11): p. 3556-‐65.
229. Yang, E.V., et al., Norepinephrine up-‐regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-‐2, and MMP-‐9 in nasopharyngeal carcinoma tumor cells. Cancer Res, 2006. 66(21): p. 10357-‐64.
230. Hajighasemi F. , M.A., Propranolol effect on proliferation and vascular endothelial growth factor secretion in human immunocompetent cells. Journal of Clinical Immunology and Immunopathology Research, 2010. 2(2): p. 22-‐27.
231. Fujio, H., et al., Carvedilol inhibits proliferation of cultured pulmonary artery smooth muscle cells of patients with idiopathic pulmonary arterial hypertension. J Cardiovasc Pharmacol, 2006. 47(2): p. 250-‐5.
232. Cardani, R., et al., Influence of beta-‐adrenergic antagonists on cell proliferation rates in the kidney of untreated and diethylnitrosamine-‐treated male F344 rats. Chem Biol Interact, 1999. 118(3): p. 217-‐31.
233. Arber, S., et al., Regulation of actin dynamics through phosphorylation of cofilin by LIM-‐kinase. Nature, 1998. 393(6687): p. 805-‐9.
234. Schreiner, S.J., A.P. Schiavone, and T.E. Smithgall, Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem, 2002. 277(47): p. 45680-‐7.
235. Lefkowitz, R.J., E. Haber, and D. O'Hara, Identification of the cardiac beta-‐adrenergic receptor protein: solubilization and purification by affinity chromatography. Proc Natl Acad Sci U S A, 1972. 69(10): p. 2828-‐32.





























