Embed Size (px)
Citation preview

Министерство образования и науки Российской Федерации
Л.К. Неудачина, Е.С. Буянова, С.А. Штин, Н.В. Лакиза, М.В. Морозова, Е.Л. Лебедева, А.С. Засухин ХИМИЯ Комплекс методического обеспечения
Учебно-методическое обеспечение модуля «Химия» для студентов, обучающихся по направлению 020400 «Биология»
Подготовлено кафедрой аналитической химии Института естественных наук
Екатеринбург 2011

2
ОГЛАВЛЕНИЕ
ОБЩАЯ ХАРАКТЕРИСТИКА КОМПЛЕКСА .............................................. 3
ПРИЛОЖЕНИЕ А. Спектрофотометрическое определение меди .............. 6
ПРИЛОЖЕНИЕ Б. Определение содержания бензоата натрия и кофеина
в напитках безалкогольных методом высокоэффективной жидкостной
хроматографии..................................................................................................... 32
ПРИЛОЖЕНИЕ В. Определение катионного и анионного состава
природных вод ..................................................................................................... 51

3
ОБЩАЯ ХАРАКТЕРИСТИКА КОМПЛЕКСА
Модуль «Химия» является дисциплиной математического и
естественнонаучного цикла для подготовки студентов, и относится к
вариативной части математического и естественнонаучного цикла Б.2 по
направлению подготовки 020400 «Биология». Это один из фундаментальных
курсов естественных наук, направленных на развитие у студентов
личностных качеств и формирование совокупности компетенций в
соответствии с ФГОС, обеспечивающих их профессиональную мобильность.
Данное учебно-методическое обеспечение направлено на
формирование навыков работы на современной аппаратуре и оборудовании
для выполнения научно-исследовательских и лабораторных работ.
Изучение модуля-дисциплины направлено на освоение студентами
таких результатов обучения, как:
– планирование и проведение химического эксперимента с
использованием современных методов и средств;
– работа на серийной аппаратуре, применяемой для физико-
химических и аналитических исследований;
– выполнение регистрации и обработки результатов химического
эксперимента, метрологического анализа результатов измерений;
и ряда компетенций:
– владение навыками химического эксперимента, основными
синтетическими и аналитическими методами получения и исследования
химических веществ и реакций;
– владение опытом работы на серийной аппаратуре, применяемой в
аналитических и физико-химических исследованиях;
– владение методами регистрации и обработки результатов химических
экспериментов.
Подготовленный в рамках программы развития УрФУ в 2011 комплекс
методического обеспечения по модулю «Химия» направления подготовки
020400 «Биология» включает в себя

4
1) учебно-методические пособия к лабораторным работам:
– спектрофотометрическое определение меди (II) в растворе,
– определение содержания бензоата натрия и кофеина в напитках
безалкогольных методом высокоэффективной жидкостной хроматографии,
– определение катионного и анионного состава природных вод;
2) мультимедийные презентации к каждой из перечисленных работ.
Учебно-методические пособия выполнены в электронном виде в pdf-
формате. Мультимедийные презентации представлены в формате «Microsoft
Power Point».
Основная задача комплекса – привитие навыков практических приемов
проведения различных аналитических операций, овладение процедурой
работы и программным обеспечением современных сложных аналитических
приборов, приемами представления и обработки экспериментальных данных.
Очень важным является формирование у студентов грамотного,
обобщенного, унифицированного подхода к проведению аналитических
операций, навыков сравнения применяемых методов анализа.
Описание каждой работы в учебно-методическом пособии предваряет
краткий теоретический материал об основных характеристиках и
возможностях предлагаемого метода анализа, уделено внимание основным
блокам и узлам современных приборов для данного метода. Затем следует
подробная характеристика прибора, предлагаемого для выполнения данной
работы, с описанием его оптической (электрической) схемы, программного
обеспечения и порядка работы. Для ознакомления с наиболее сложными
приборами и облегчения визуального восприятия методов работы на них (в
данном комплексе, это работы по высокоэффективной жидкостной
хроматографии и капиллярному электрофорезу), студент может использовать
видеолекции и мультимедийные презентации. После ознакомления с
предложенными материалами и закрепив полученные знания с помощью
проведения работы на виртуальном тренажере, студент может быть допущен

5
к проведению реальной лабораторной работы, также подробно описанной в
пособии.
Отчет по выполненной работе должен содержать анализ теоретических
положений метода, подробное описание методики определения и обработку
результатов определения. Для представления и обработки результатов
эксперимента студентам предлагается использовать пакеты современных
компьютерных программ (Excel, Origin).

6
ПРИЛОЖЕНИЕ А. Спектрофотометрическое определение меди

7
Уральский федеральный университет
имени первого Президента России Б.Н.Ельцина
Институт естественных наук
Кафедра аналитической химии
Спектрофотометрическое определение меди
Учебно-методическое пособие
Екатеринбург
2011

8

9
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД
Основные закономерности светопоглощения растворами
Электромагнитное излучение при взаимодействии с веществом может
вызывать в нем процессы разнообразной физической природы, используемые
в методах химического анализа. Общий характер этих процессов зависит от
энергии фотонов. Поэтому для классификации методов анализа весь
диапазон энергий электромагнитных квантов делят на области,
соответствующие тому или иному физическому процессу. Спектроскопия в
УФ и видимой области спектра соответствует спектральному интервалу 200-
400 нм (УФ область) и 400-750 нм (видимая область). Физическим
процессом, ответственным за возникновение спектров поглощения в этой
области, является изменение состояний валентных электронов под
воздействием электромагнитного излучения соответствующей длины волны.
Метод молекулярной абсорбционной спектроскопии в УФ и видимой
областях спектра обычно применяют для исследования растворов, которые
помещают в кювету – сосуд с плоскими параллельными прозрачными
гранями.
Схему прохождения светового потока через светопоглощающий
раствор можно представить следующим образом:
Рисунок 1. Прохождение светового потока через кювету с раствором.
После прохождения светового луча через кювету его интенсивность
уменьшается за счет отражения от стенок кюветы, поглощения образцом и за
счет рассеяния на взвешенных частицах. Для учета отражения и рассеяния
опыт повторяют с аналогичной кюветой, содержащей только растворитель.
Кроме того, рассеяние сводится к минимуму при работе с истинными
поглощениеоще
рассеяние
к детектору
отражение
От источника

10
растворами, а для минимизации отраженного излучения световой поток
должен падать на кювету строго перпендикулярно, а стенки кюветы должны
быть строго параллельны.
Пропускание раствора рассчитывают по следующему уравнению:
0IIT = (1)
где I0 – начальная интенсивность светового потока, а I – интенсивность
светового потока, вышедшего из кюветы. В случае, если используется
измерение относительно кюветы с растворителем или другим раствором
сравнения, то I0 – интенсивность светового потока, прошедшего через кювету
сравнения.
Величину А, рассчитываемую по следующему уравнению:
TIIA lglg 0 −== (2)
называют оптической плотностью.
Пределы возможных изменений величин оптической плотности и
светопропускания можно рассчитать следующим образом:
а) раствор не поглощает световой поток, тогда I = I0 и T=1 (или 100%),
а оптическая плотность А=0;
б) световой поток полностью поглощается раствором, тогда I=0 и Т=0,
а оптическая плотность стремится к бесконечно большой величине. Реально
можно измерять оптическую плотность в интервале от 0 до 2-3 единиц.
Зависимость интенсивности монохроматического светового потока,
прошедшего через слой окрашенного раствора, от интенсивности падающего
потока света, концентрации светопоглощающего вещества и толщины слоя
раствора определяется уравнением
,100kClII −⋅= (3)
где k – коэффициент поглощения, зависящий от природы растворенного
вещества, растворителя, температуры и длины волны падающего на кювету

11
света λ; С – концентрация поглощающего вещества: l – толщина
светопоглощающего слоя (внутренняя длина кюветы).
Если концентрацию С выразить в молях на литр, толщину
светопоглощающего слоя l – в сантиметрах, то коэффициент k обозначают
символом ε и называют молярным коэффициентом поглощения (часто
обозначают еще как εм). Логарифмируя выражение (3) и используя понятие
оптической плотности (уравнение 2), получим
ClA ε= (4)
Это соотношение известно как закон Бугера-Ламберта-Бера,
являющийся основным законом светопоглощения. Этот закон универсален
для всех спектроскопических абсорбционных методов, потому что при его
выводе не делается никаких предположений ни о природе поглощающей
среды, ни о характере поглощаемого излучения.
Молярные коэффициент поглощения численно равен оптической
плотности раствора с концентрацией 1 моль/л, помещенного в кювету с
толщиной поглощающего слоя 1 см. Значения εм даже наиболее интенсивно
светопоглощающих (окрашенных) соединений редко превышают 1·105.
Низкие значения εм характерны для гидратированных ионов и простейших
комплексных соединений. Наиболее высокие значения εм имеют
комплексные соединений ионов металлов с органическими реагентами.
Таблица 1 – Некоторые примеры величин молярных коэффициентов
поглощения
№ п/п Раствор вещества εм
1 Р.з.э. + HCl 1 ÷ 10
2 Комплекс р.з.э. + арсеназо III (30÷90)·103
3 Комплекс титана(IV) с пероксидом водорода 720
4 Комплекс титана(IV) с хромотроповой кислотой 1,7·105
5 Комплекс тория (IV) с арсеназо III 1,27·105

12
Чувствительность и избирательность спектрофотометрических
определений
Графическая интерпретация основного закона светопоглощения
представляет собой прямую, выходящую из начала координат. Тангенс ее
наклона пропорционален величине εм и характеризует чувствительность
спектрофотометрических определений. Чем выше значение молярного
коэффициента поглощения, тем круче идет градуировочный график, тем
выше чувствительность определения.
Большинство веществ неравномерно поглощают отдельные участки
сплошного излучения (например, видимого света). Для характеристики
избирательности поглощения электромагнитного излучения растворами
веществ пользуются кривыми светопоглощения или так называемыми
спектрами поглощения. Это зависимость оптической плотности раствора от
длины волны падающего света. Для раствора, содержащего единственное
достаточно простое по составу светопоглощающее соединение, вид спектра
может быть следующим:
Рисунок 2. Общий вид простого спектра поглощения и схема расчета полуширины полосы
поглощения.
Полоса поглощения характеризуется в первую очередь значением λmax,
т.е. длиной волны, отвечающей максимальной оптической плотности (или
максимальному значению εм). В молекулярном абсорбционном анализе

13
большое значение имеет также ширина полосы поглощения. Чем шире
полоса, тем труднее анализировать смесь нескольких светопоглощающих
соединений, так как при этом более вероятно взаимное наложение полос.
Ширину полосу установить нельзя, так как по обе стороны от λmax
интенсивность поглощения лишь асимптотически приближается к нулю.
Поэтому пользуются такой характеристикой, как полуширина полосы
поглощения. Полушириной полосы поглощения называют расстояние λ”1/2 –
λ’1/2=a в нм, соответствующее половине максимальной оптической плотности
или максимального значения εм. В большинстве случаев полуширина полосы
поглощения простых молекул составляет 80-100 нм. Сложные молекулы
нередко имеют несколько полос поглощения, некоторые из которых могут
накладываться друг на друга.
По спектрам поглощения может быть осуществлен выбор длины
волны, отвечающей максимальной чувствительности
спектрофотометрического определения.
Для рассмотрения этого вопроса сопоставим спектры поглощения
индивидуального соединения при различных концентрациях с
градуировочными графиками, построенными в максимуме светопоглощения
и при любой другой длине волны.

14
Рисунок 3. Зависимости оптической плотности растворов простого соединения от длины
волны (спектры поглощения) и зависимости оптической плотности от концентрации при
двух длинах волн.
Очевидно, что максимальная чувствительность достигается при λmax,
так как при этом наблюдается максимальное значение тангенса угла наклона
градуировочного графика.
Наиболее важной характеристикой, определяющей возможность
анализа достаточно сложных смесей, является селективность определения.
Рассмотрим эту проблему на примере анализа двухкомпонентных смесей.
При рассмотрении этого вопроса следует учесть, что если в растворе
содержится n светопоглощающих компонентов, которые не вступают друг с
другом в химическое взаимодействие, то при условии соблюдения основного
закона светопоглощения оптическая плотность такого раствора будет равна
сумме парциальных оптических плотностей всех содержащихся в растворе
светопоглощающих компонентов. В этом проявляется закон аддитивности
(принцип аддитивности или правило аддитивности) оптических плотностей:
∑=
=+++=n
iiinn CllClClCA
12211 ... εεεε ,(5)
где ε1, ε2, …, εn – молярные коэффициенты поглощения компонентов смеси
по отношению к излучению одной и той же длины волны; С1, С2, …, Сn –
соответствующие молярные концентрации компонентов смеси.
Спектрофотометрический анализ двухкомпонентных систем
Для двухкомпонентных смесей светопоглощающих соединений можно
выделить три типичных ситуации.
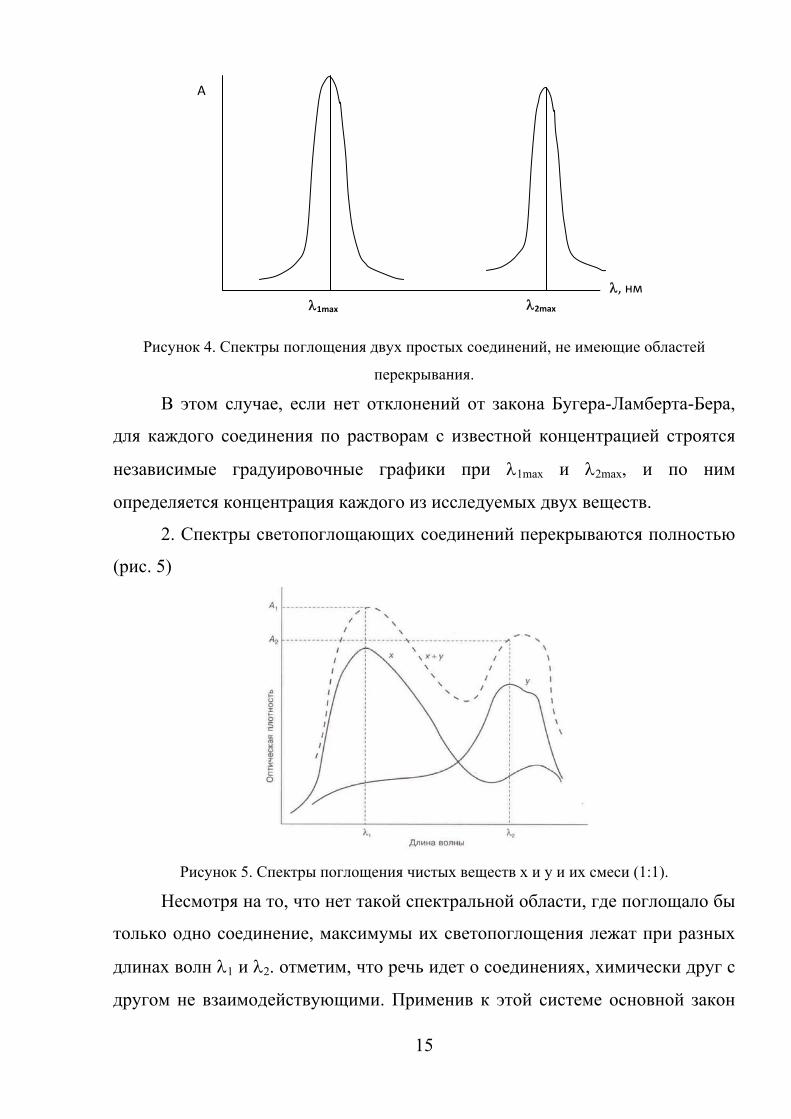
1. Спектры светопоглощающих соединений не перекрываются (рис.4).
15
Рисунок 4. Спектры поглощения двух простых соединений, не имеющие областей
перекрывания.
В этом случае, если нет отклонений от закона Бугера-Ламберта-Бера,
для каждого соединения по растворам с известной концентрацией строятся
независимые градуировочные графики при λ1max и λ2max, и по ним
определяется концентрация каждого из исследуемых двух веществ.
2. Спектры светопоглощающих соединений перекрываются полностью
(рис. 5)
Рисунок 5. Спектры поглощения чистых веществ x и y и их смеси (1:1).
Несмотря на то, что нет такой спектральной области, где поглощало бы
только одно соединение, максимумы их светопоглощения лежат при разных
длинах волн λ1 и λ2. отметим, что речь идет о соединениях, химически друг с
другом не взаимодействующими. Применив к этой системе основной закон
λ2max λ1max
А
λ, нм

16
светопоглощения и закон аддитивности оптических плотностей, получаем
для светопоглощения при λ1 следующее выражение:
lClCA 2211 111 λλλ εε += (6)
При λ2 можно записать аналогичное выражение
lClCA 2211 222 λλλ εε += (7)
Значения молярных коэффициентов светопоглощения либо берут из
таблиц, либо определяют экспериментально, толщина светопоглощающего
слоя указана на кювете, оптические плотности при соответствующих длинах
волн определяются экспериментально. Система двух линейных уравнений с
двумя неизвестными легко решается:
lAA
C)(
2112
1221
2121
221
λλλλ
λλλλ
εεεεεε
−
−= (8)
lAA
C)(
1221
1221
2121
112
λλλλ
λλλλ
εεεεεε
−
−= (9)
3. Спектры двух светопоглощающих соединений перекрываются
частично
В практическом отношении наибольший интерес представляет случай,
когда удается найти такой участок спектра, в котором поглощением одного
из компонентов можно пренебречь (рис. 6).
Рисунок 6. Кривые светопоглощения двух веществ: имеется участок, где поглощает лишь
одно вещество

17
В этом случае при длине волны λ1 по измеренной оптической
плотности А1 обычным способом находят концентрацию первого вещества.
lAС
11
11
λε= (10)
Концентрацию второго вещества находят, подставляя в уравнение (9)
найденное значение С1:
lCA
С22
1122
2
λεε λ−
= (11)
Учет возможных отклонений от основного закона
светопоглощения
Справедливость закона Бугера-Ламберта-Бера доказана и теоретически
и экспериментально и не подвергается сомнениям, однако в реальных
условиях спектрофотометрических измерений имеют место так называемые
кажущиеся отклонения от этого закона. Эти отклонения делят на 2 группы:
инструментальные (физические) и химические.
Физические отклонения. Главной физической причиной кажущихся
отклонений является немонохроматичность поглощаемого светового потока.
Даже в тех случаях, когда в качестве источника света используется лазер,
возможно нарушение монохроматичности светового потока. Схема
прохождения светового потока через кювету с раствором (рис. 1)
предполагает наличие возможности потерь светового потока за счет его
отражения на границе раздела раствор – кювета. Доля этих потерь зависит от
показателей преломления пограничных сред, а показатели преломления
растворов зависят от их концентрации. Поэтому зависимость величины I на
выходе из кюветы, а соответственно, и оптической плотности, от
концентрации раствора будет иметь значительно более сложный характер,
чем это следует из основного закона светопоглощения (уравнение 4).

18
Другой физической причиной нарушения закона Бугера-Ламберта-Бера
является рассеяние света. Это явление может возникнуть в оптической схеме
прибора вследствие отражения света от поверхности зеркал, линз, а, кроме
того, в растворе при возникновении конвективных потоков за счет
неравномерного нагрева. Степень влияния рассеянного света на истинное
значение светопоглощения зависит от диапазона измеряемых оптических
плотностей. Оно увеличивается с возрастанием оптической плотности
анализируемого раствора. Особенно сильное влияние оказывает рассеянный
свет на светопоглощение в УФ области спектра (λ < 220 нм). В современных
высокочувствительных спектрофотометрах уровень рассеяния света не
превышает 0,001%.
Химические причины отклонений от основного закона
светопоглощения.
Главными химическими причинами являются побочные реакции,
которые приводят к различной (и часто недостаточной) степени связанности
аналита в светопоглощающее соединение.
Предположим, что для определения металла М используют реакцию с
органическим реагентом HR:
М+ + HR + H2O MR + H3O+ (12)
В результате этой должна количественно образовываться
аналитическая форма MR, являющаяся фотометрируемым соединением с
молярным коэффициентом поглощения εMR, концентрация которой С(моль/л)
равна исходной концентрации ионов металла М+. Соответственно должно
выполняться соотношение
ClA MRε= ,(13)
где А – истинное значение оптической плотности.
Если ионы металлов под влиянием примесей, присутствующих в
анализируемом растворе, не полностью переходят в аналитическую форму,
концентрация светопоглощающей формы MR уменьшается по сравнению с

19
общей концентрацией иона металла в растворе, и измеренная оптическая
плотность, равная
lCА MRMRизм αε= ,(14)
где αMR – мольная доля светопоглощающей формы MR от общей
концентрации С аналита, будет меньше оптической плотности раствора той
же концентрации иона металла, измеренной при построении
градуировочного графика. Это отрицательное значение отклонения от
основного закона светопоглощения.
Положительные отклонения от закона могут наблюдаться. Когда в
растворе образуются ионные ассоциаты с участием аналита или другие
сложные соединения с молярными коэффициентами поглощения,
превышающими εMR.
Фотометрические формы аналитов и реакции их образования
В фотометрическом анализе используются различные по природе
реагенты, образующие с аналитами светопоглощающие соединения. В
данном пособии рассмотрим только образование хелатных соединений
металлов с органическими реагентами.
Полнота протекания фотометрических реакций. При использовании
фотометрических методов для получения воспроизводимых результатов
необходимо гарантировать, чтобы во всех фотометрируемых растворах
(стандартных и анализируемых) степень перехода аналита в
светопоглощающую форму была одинаковой. Практически это означает, что
с учетом погрешности фотометрического анализа (1-3%) во всех
фотометрируемых растворах аналит должен находиться не менее чем на 99%
в светопоглощающей форме. Также в некоторых случаях из-за невысокой
скорости реакции количественный перевод аналита в светопоглощающее
соединение достигается лишь по истечении определенного времени.
Если используется реакция с образованием комплекса MRn, то
практически полное связывание аналита М в светопоглощающее соединение

20
MRn будет достигаться, когда равновесная концентрация комплекса MRn
составит не менее 99% от исходной концентрации СоМ аналита М. Вводя это
условие в выражение условной константы устойчивости β’MRn комплекса.
получим
nM
n
nM
nM
MM
Mn
RM
nMR nCCn
CCnC
CCC
MRn )(
10)01,0(
99,0)01,0(01,0
99,0)'('
][' 0
)1(2
10
0
00
0 +
+ ====β ,
Откуда
n
nn
MMR nC
n
)1(20 10)('
+
≥β (15)
Если неравенство (15) соблюдается, то фотометрируемое соединение
настолько устойчиво в водном растворе, что даже в отсутствие избытка
реагента R аналит М практически полностью (не менее чем на 99%)
переходит в светопоглощающий комплекс MRn. При невысокой
относительной устойчивости светопоглощающего комплекса MRn, когда
условие (15) не соблюдается, необходимая степень связанности аналита М в
комплекс обеспечивается избытком фотометрического реагента: n
MRR
n
C/1
2
'10' ⎟
⎟⎠
⎞⎜⎜⎝
⎛≥
β (16)
Влияние рН раствора на образование фотометрируемых соединений.
Основные случаи влияния кислотности среды могут быть объединены
в несколько общих вариантов в зависимости от природы аналитического
реагента.
Светопоглощающие комплексы металлов с анионами сильных
кислот. В этом случае наиболее сильное влияние рН наблюдается для
катионов металлов, способных образовывать прочные гидроксокомплексы.
При увеличении рН раствора комплексы с такими металлами могут
разрушаться полностью. Поэтому реакции образования светопоглощающих
соединений ионов металлов с анионами сильных кислот рекомендуется

21
проводить в достаточно кислых средах, где условная константа устойчивости
светопоглощающего комплекса сохраняет наибольшее значение. Анионы
сильных кислот (Cl – , I – , SCN – и др.) даже при высокой концентрации
ионов водорода в растворе практически не протонируются, поэтому
повышение кислотности среды не препятствует полноте образования
светопоглощающих соединений с анионами сильных кислот.
Светопоглощающие комплексы ионов металлов с анионами слабых
кислот. Слабые кислоты и их соли очень широко применяются как
фотометрические реагенты. Большинство органических реагентов относится
к этой группе, например:
S CNH-NH-C6H5
N = N - C6H5дитизон
O
H4NOOC
C
COONH4
OH
OH
COONH4алюминон В таких случаях с определяемым ионом взаимодействует
протонированный анион слабой кислоты. В результате конкуренции между
протоном и ионов металла образуется светопоглощающий комплекс в
соответствии с уравнением реакции:
Xn+ + HR XR(n-1)+ + H+ (17)
Полнота связывания иона в светопоглощающее соединение зависит от
концентрации реагента R- в растворе. Однако эта концентрация в кислых
растворах бывает невелика, так как равновесие реакции (17) сильно сдвинуто
в сторону образования соединения HR. При повышении рН ионы H+
связываются гидроксид-ионами в молекулу воды и равновесие реакции (17)
сдвигается вправо в сторону образования светопоглощающего соединения.
Поэтому комплекс определяемого иона с анионом слабой кислоты следует
всегда получать в возможно менее кислых средах. Однако повышать рН
раствора нужно очень осторожно. При повышении рН растора может

22
происходить образование гидроксокомплексов или гидроксидов
определяемых металлов, может изменяться состав светопоглощающего
соединения вследствие ступенчатого комплексообразования. Следует
считаться также с тем, что при повышении рН определяемый ион может
образовывать иные светопоглощающие соединения с реагентом. Поэтому
максимальный выход светопоглощающего комплекса будет наблюдаться
только в определенном интервале значений рН раствора.
Частным случаем слабых кислот являются органические реагенты,
имеющие свойства кислотно-основных индикаторов, к числу которых
относятся ализарин, диметилглиоксим, эриохромчерный Т и т.д. Они
способны изменять свою окраску в зависимости от величины рН раствора,
так как молекулярная форма реагента-индикатора отличается по структуре и
окраске от солевой формы R-.
В растворе могут иметь место следующие равновесия:
для реагента
HR H+ + R-
окраска 1 окраска 2 (18)
для раствора комплекса
M + HR MR + H+
окраска 3 (19)
Если светопоглощение форм R- и MR характеризуется близкими
значениями λmax (окраски 2 и 3 близки), то даже при отсутствии в растворе
определяемого иона М может быть зафиксировано светопоглощение солевой
формы реагента, что приведет к существенным погрешностям в выполнении
анализа.
Аппаратура и принадлежности для фотометрического анализа
Описание колориметра фотоэлектрического концентрационного
КФК-2

23
Принцип измерения коэффициента пропускания состоит в том, что на
фотоприемник направляются поочередно различные световые потоки:
полный и прошедший через исследуемую среду, и определяется отношение
этих потоков.
Принципиальная оптическая схема фотометра приведена на рис. 7.
Рисунок 7. Оптическая схема фотоэлектроколориметра КФК-2.
Подготовка к работе
1. Колориметр включить в сеть за 15 минут до начала измерений. Во
время прогрева кюветное отделение должно быть открыто (при этом шторка
перед фотоприемниками перекрывает световой пучок).
2. Ввести необходимый по роду измерения цветной светофильтр.
3. Установить минимальную чувствительность колориметра. Для этого
ручку, «ЧУВСТВИТЕЛЬНОСТЬ» (рис. 8) необходимо установить в
положение 1, ручку «УСТАНОВКА 100 ГРУБО» – в крайнее левое
положение.
12
19
6 81
2 3 4 5 7 9 1014
15
20
11
2118

24
Рисунок 8. Внешний вид колориметра КФК-2.
4. Перед измерениями и при переключении фотоприемников проверить
установку стрелки колориметра на «0» по шкале коэффициентов
пропускания Т при открытом кюветном отделении. При смещении стрелки от
нулевого положения, ее подводят к нулю с помощью потенциометра
«НУЛЬ», выведенного под шлиц.
Порядок работы
Измерение коэффициента пропускания
1. В световой пучок поместить кювету с растворителем или
контрольным раствором, по отношению к которому производятся измерения.
2. Закрыть крышку кюветного отделения.
3. Ручками «ЧУВСТВИТЕЛЬНОСТЬ» и «УСТАНОВКА 100 ГРУБО» и
«ТОЧНО» установить отсчет «100» по шкале колориметра.
4. Затем, поворотом ручки кювету с растворителем или контрольным
раствором заменить кюветой с исследуемым раствором.
5. Снять отсчет по шкале колориметра, соответствующей
коэффициенту пропускания исследуемого раствора в процентах. Для
регистрирующего прибора типа M 907-10 отсчет снимают по шкале
коэффициентов пропускания Т в процентах, или по шкале Д – в единицах
оптической плотности. Абсолютная погрешность измерения коэффициента
пропускания не превышает 1%.
6. Измерение провести 3 – 5 раз и окончательное значение измеренной
величины определить как среднее арифметическое из полученных значений.
1
2
34 5 6
1 2

25
Определение концентрации вещества в растворе
При определении концентрации вещества в растворе следует
соблюдать следующую последовательность в работе:
– выбор светофильтра;
– выбор кюветы;
– построение градуировочной зависимости для данного вещества;
– измерение оптической плотности исследуемого раствора и
определение концентрации вещества в растворе.
Выбор светофильтра
Наличие в колориметре узла светофильтров и набора кювет позволяет
подобрать такое их сочетание, при котором погрешность в определения
концентрации будет наименьшей.
Выбор светофильтра проводят следующим образом. Наливают раствор
в кювету (о выборе размера кювет см. ниже) и определяют оптическую
плотность для всех светофильтров.
По полученным данным строят зависимость, откладывая по
горизонтальной оси длины волн, соответствующие максимуму коэффициента
пропускания светофильтров, указанные в описании колориметра, а по
вертикальной оси – соответствующие значения оптической плотности
раствора. Отмечают тот участок кривой, для которого выполняются
следующие условия:
– оптическая плотность имеет максимальную величину;
– ход кривой примерно параллелен горизонтальной оси,
т. е. оптическая плотность мало зависит от длины волн. Светофильтр
для работы выбирают так, чтобы длина волны, соответствующая максимуму
коэффициента пропускания светофильтра, приходилась на отмеченный выше
участок спектральной кривой испытуемого раствора.
Если эти условия выполняются для нескольких светофильтров, то
выбирают тот из них, для которого чувствительность колориметра выше.
Выбор кюветы

26
Относительная ошибка определения концентрации раствора будет
различной при работе на разных участках шкалы колориметра и достигает
минимума при значении оптической плотности 0.4. Поэтому при работе на
колориметре рекомендуется, путем соответствующего выбора кювет,
работать вблизи указанного значения оптической плотности.
Предварительный выбор кювет проводится визуально, соответственно
интенсивности окраски раствора. Если раствор интенсивно окрашен
(темный), следует пользоваться кюветами с малой рабочей длиной. В случае
слабо окрашенных растворов рекомендуется работать с кюветами с большой
рабочей длиной.
В предварительно подобранную кювету наливают раствор и измеряют
его оптическую плотность, вводя в ход лучей соответствующий для данного
раствора светофильтр.
Второе условие может для некоторых растворов не иметь места, тогда
при выборе светофильтра ограничиваются выполнением первого условия.
При измерении ряда растворов кювету заполняют раствором средней
концентрации. Если полученное значение оптической плотности составляет
примерно 0.3 – 0.5 – выбирают данную кювету для работы с этим раствором.
В том случае, когда это условие не выполняется, следует испробовать другую
кювету. Если величина измеренной оптической плотности больше 0.5 – 0.6,
берут кювету меньшей рабочей длины, если величина оптической плотности
меньше 0.3 – 0.2, следует выбрать кювету с большей рабочей длиной.
Построение градуировочного графика для данного вещества
Построение градуировочного графика проводят следующим образом.
Готовят ряд растворов данного вещества с известными концентрациями,
охватывающими область возможных изменений концентраций этого
вещества в исследуемом растворе.
Измеряют оптические плотности всех растворов и строят
градуировочный график, откладывая по горизонтальной оси известные

27
концентрации, а по вертикальной соответствующие им значения оптической
плотности.
Определение концентрации вещества в растворе
По градуировочному графику в дальнейшем определяют неизвестную
концентрацию вещества в исследуемых растворах. Для этого раствор
наливают в ту же кювету, для которой построена градуировочная
зависимость, и, выбрав тот же светофильтр, определяют оптическую
плотность раствора. Затем по градуировочному графику находят
концентрацию, соответствующую измеренному значению оптической
плотности.
Примечание. Часто в работе бывает удобнее пользоваться
градуировочными таблицами, которые составляются по данным
градуировочной зависимости.
Градуировочный график следует время от времени проверять.
Погрешности измерения оптической плотности
Воспроизводимость (или повторяемость) абсолютных
фотометрических методов анализа, в которых оптическая плотность (или
пропускание) исследуемого или стандартного раствора измеряется
относительно чистого растворителя или раствора «холостого» опыта,
обусловлена погрешностью измерения аналитического сигнала (А или Т).
При заданной абсолютной погрешности ΔТ измерения Т значение
относительной погрешности определения концентрации ΔС/С (sc/C) зависит
от абсолютного значения светопропускания (оптической плотности). Эта
зависимость иллюстрируется следующими рисунками.
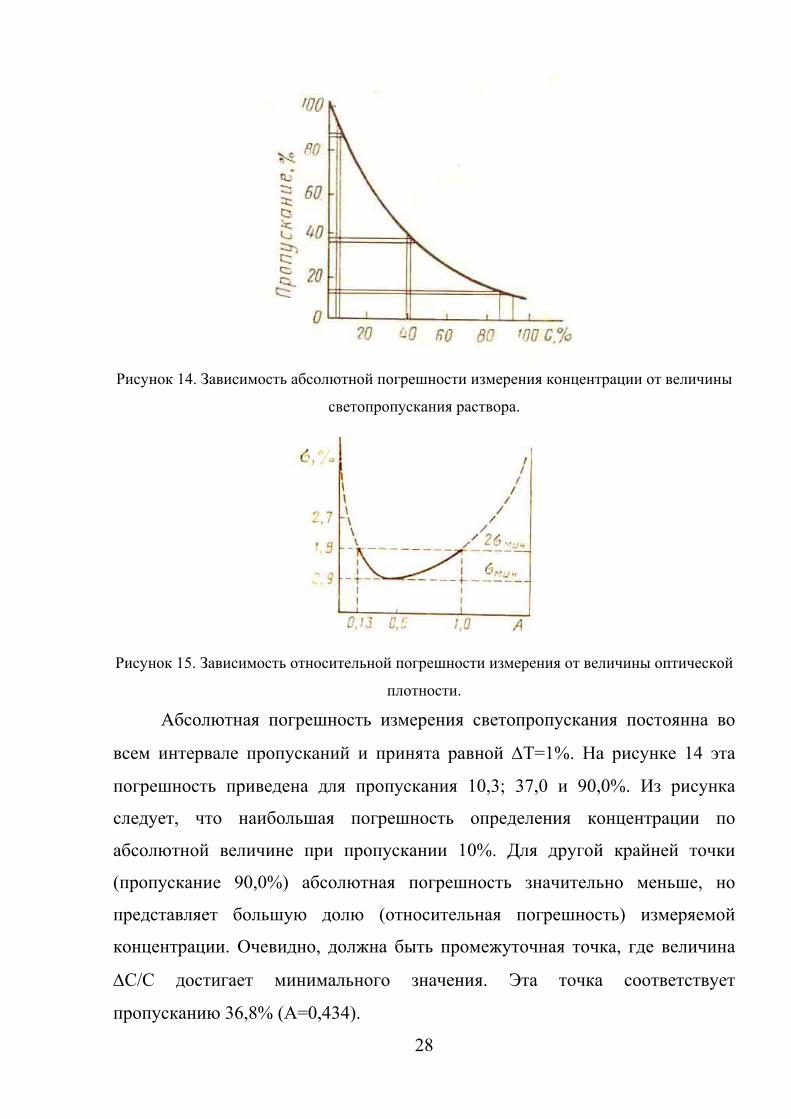
28
Рисунок 14. Зависимость абсолютной погрешности измерения концентрации от величины
светопропускания раствора.
Рисунок 15. Зависимость относительной погрешности измерения от величины оптической
плотности.
Абсолютная погрешность измерения светопропускания постоянна во
всем интервале пропусканий и принята равной ΔТ=1%. На рисунке 14 эта
погрешность приведена для пропускания 10,3; 37,0 и 90,0%. Из рисунка
следует, что наибольшая погрешность определения концентрации по
абсолютной величине при пропускании 10%. Для другой крайней точки
(пропускание 90,0%) абсолютная погрешность значительно меньше, но
представляет большую долю (относительная погрешность) измеряемой
концентрации. Очевидно, должна быть промежуточная точка, где величина
ΔС/С достигает минимального значения. Эта точка соответствует
пропусканию 36,8% (А=0,434).

29
Зависимость относительной погрешности измерения (σмин) от значения
оптической плотности А приведена на рис. 15. Показано, что погрешность
измерения оптической плотности не превышает 2σмин внутри интервала 0,1 –
0,8 единиц оптической плотности. Именно в этом интервале должны
находиться измеряемые оптические плотности. К оптимальной области
измерений анализируемые растворы можно подвести путем подбора кювет
или разбавлением раствора.
Лабораторная работа. Спектрофотометрическое определение меди
(II) в растворе
Раствор соли меди (II) имеет собственную окраску, однако она
недостаточно интенсивна для спектрофотометрического определения. Для
перевода вещества в более интенсивно поглощающее соединение проводят
фотометрическую реакцию. Например, определяя содержание меди, на
раствор действуют избытком аммиака, в результате чего получается
комплексный ион [Cu(NH3)4]2+ интенсивно-синего цвета (λmax=620 нм;
ε=120):
Cu2+ + 4NH3 → [Cu(NH3)4]2+
Оборудование, посуда и реактивы
1. Колориметр фотоэлектрический концентрационный КФК-2.
2. Сульфат меди (II), рабочий раствор с содержанием меди 1.5 мг/мл
2. Аммиак, раствор 1:1
3. Мерные колбы вместимостью 50.0 мл
4. Градуированная пипетка вместимостью 5.0 мл
5. Мерный цилиндр вместимостью 25 мл
Ход работы
1. Приготовление стандартных растворов сульфата меди (II) для
построения градуировочного графика.
В пять мерных колб вместимостью 50.0 мл помещают 1.0; 2.0; 3.0; 4.0 и
5.0 мл рабочего раствора сульфата меди, добавляют 15 мл раствора аммиака,

30
доводят до метки дистиллированной водой и тщательно перемешивают.
Для приготовления раствора сравнения в мерную колбу вместимостью
50.0 мл помещают 15 мл аммиака (1:1), доводят до метки дистиллированной
водой и тщательно перемешивают.
Все работы с аммиаком проводят в вытяжном шкафу!!!
Оптическую плотность растворов измеряют не ранее чем через 10 мин.
2. Выбор светофильтра.
Для выбора светофильтра используют раствор, имеющий наиболее
интенсивную окраску. Измеряют его оптическую плотность относительно
раствора сравнения в диапазоне длин волн 400–750 нм и строят спектр
поглощения в координатах «оптическая плотность – длина волны, нм». По
построенному графику выбирают светофильтр, при котором наблюдается
максимальное светопоглощение раствора. Этот светофильтр используют для
дальнейшей работы.
3. Измерение оптической плотности стандартных растворов и
построение градуировочного графика.
Измерение оптической плотности стандартных растворов,
приготовленных в соответствии с п. 1, относительно раствора сравнения
начинают с раствора с наименьшим содержанием меди (II). С выбранным
светофильтром измеряют оптическую плотность растворов, приготовленных
в соответствии с п.1, начиная с наиболее разбавленного. Измерения проводят
при длине волны 590 нм в стеклянных кюветах с толщиной поглощающего
слоя 2.0 см относительно раствора сравнения. По результатам измерений
строят градуировочный график в координатах «оптическая плотность –
содержание меди, мг/л» (рис. 13).
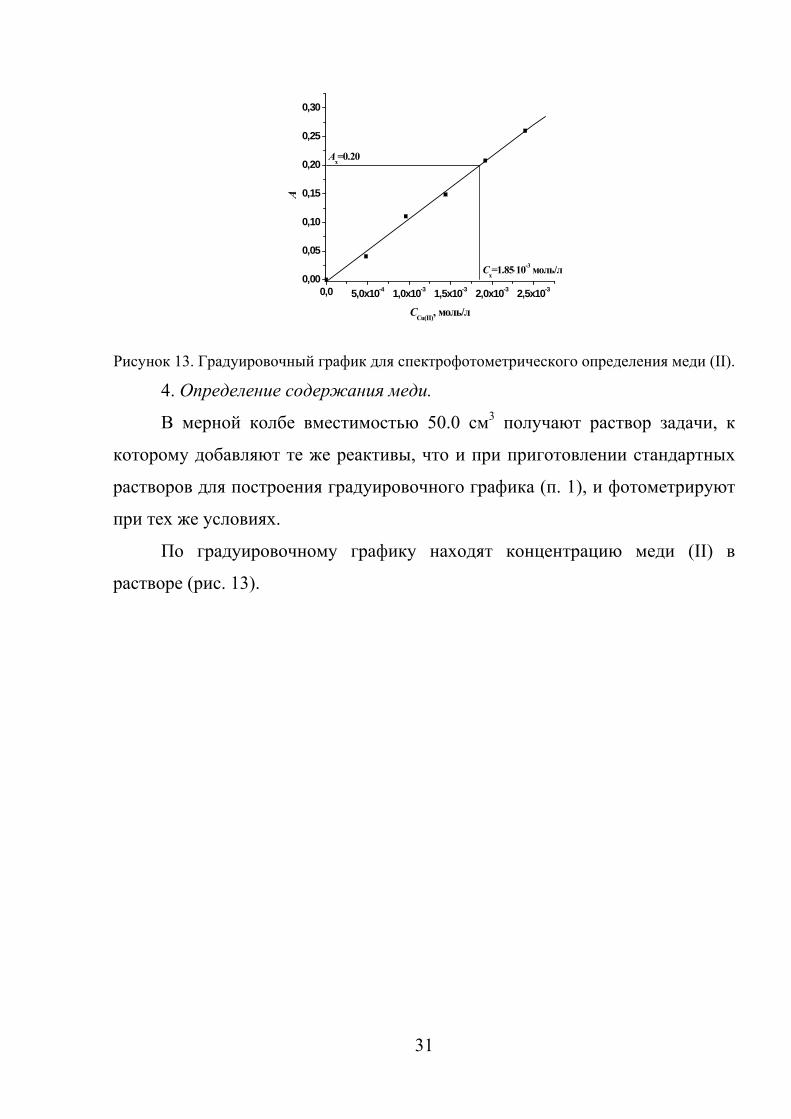
31
0,0 5,0x10-4 1,0x10-3 1,5x10-3 2,0x10-3 2,5x10-30,00
0,05
0,10
0,15
0,20
0,25
0,30
Cx=1.85⋅10-3 моль/л
Ax=0.20
A
CCu(II), моль/л
Рисунок 13. Градуировочный график для спектрофотометрического определения меди (II).
4. Определение содержания меди.
В мерной колбе вместимостью 50.0 см3 получают раствор задачи, к
которому добавляют те же реактивы, что и при приготовлении стандартных
растворов для построения градуировочного графика (п. 1), и фотометрируют
при тех же условиях.
По градуировочному графику находят концентрацию меди (II) в
растворе (рис. 13).

32
ПРИЛОЖЕНИЕ Б. Определение содержания бензоата натрия и кофеина
в напитках безалкогольных методом высокоэффективной жидкостной
хроматографии

33
Уральский федеральный университет
имени первого Президента России Б.Н.Ельцина
Институт естественных наук
Кафедра аналитической химии
Определение содержания бензоата натрия и кофеина в напитках
безалкогольных методом высокоэффективной жидкостной хроматографии
Учебно-методическое пособие
Екатеринбург
2011

34
ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Принцип метода
Жидкостная хроматография (ЖХ) – это метод разделения и анализа
сложных смесей веществ, в котором подвижной фазой служит жидкость.
Метод ЖХ применим для разделения более широкого круга веществ, чем
метод ГХ, поскольку большинство веществ не обладает летучестью, многие
из них неустойчивы при высоких температурах (особенно
высокомолекулярные соединения) и разлагаются при переведении в
газообразное. В ЖХ разделение чаще всего происходит при комнатной
температуре. Особенности всех видов хроматографии обусловлены наличием
жидкой подвижной фазы.
Сорбция компонентов из газа и жидкого элюента осуществляется по-
разному. В отличие от газа, который выполняет только транспортную
функцию и не сорбируется неподвижной фазой, жидкая подвижная фаза –
активный элюент, молекулы которой могут сорбироваться на поверхности.
При прохождении через колонку находящиеся в элюенте молекулы
интересующего нас копонента должны вытеснить молекулы элюента с
поверхностного слоя сорбента, что приводит к уменьшению энергии
взаимодействия молекул вещества с поверхностью сорбента. Поэтому
величины VR, пропорциональны –ΔG (изменению свободной энергии), также
меньше в ЖХ, чем в ГХ; диапазон линейности изотермы сорбции в ЖХ
больше.
Применяя различные элюенты, можно изменять параметры
удерживания и селективность хроматографической системы. Возможно
использование градиентного элюирования. Селективность ЖХ в отличие от
ГХ определяется не одним, а двумя факторами - природой подвижной
(элюент) и неподвижной фаз.
В классическом варианте ЖХ в стеклянную колонку длиной 1–2 м,
заполненную сорбентом (размер частиц ≥100 мкм), вводят анализируемую
пробу и пропускают элюент. Скорость прохождения элюента под действием

35
силы тяжести мала – не более 1 мл/мин, а продолжительность анализа
значительна. Предпринимались попытки увеличить скорость потока путем
использования вакуумных и иных насосов, но это приводило лишь к
ухудшению разделения. Этот результат вполне объясним, поскольку из
хроматографической теории известно, что увеличение линейной скорости
потока подвижной фазы при прочих равных условиях уменьшает число
теоретических тарелок. Довольно скоро стало понятно, что увеличение
эффективности можно достичь только за счет уменьшения размеров зерен
наполнителя. Однако при этом резко возрастает сопротивление потоку
жидкости, преодолеть которое можно лишь путем увеличения давления.
Работа с тонкодисперсными наполнителями требовала столь высоких
давлений, что обычные стеклянные колонки этого не выдерживали.
В конце 1960-х годов появились технические возможности создания
аппаратуры, позволяющей работать с колонками, заполненными
наполнителем с размерами зерен порядка 3–10 мкм. Это способствовало
переходу от классической жидкостной хроматографии низкого давления к
высокоэффективной жидкостной хроматографии (ВЭЖХ) – аналитической
жидкостной хроматографии высокого давления. Быстрый массоперенос при
высокой эффективности позволяет использовать ВЭЖХ для разделения и
определения молекул (адсорбционная и распределительная хроматографии),
для разделения и определения ионов (ионообменная, ионная, ион-парная
хроматографии), для разделения макромолекул (эксклюзионная
хроматография). Методом аффинной и лигандобменной хроматографии
разделяют биологически активные молекулы и оптические изомеры.
Классический вариант до сих пор применяют в лабораторной практике,
поскольку он не требует дорогостоящего оборудования, и применяется,
главным образом, для препаративных целей.
Наряду с сорбентами универсального типа существуют сорбенты
специального назначения, дающие возможность успешно решать
специфические задачи.

36
Наиболее существенными особенностями сорбентов, введенных в
практику ВЭЖХ, являются:
преимущественное использование микрочастиц диаметром 3-10
мкм как нерегулярной, так и сферической формы;
повышенная степень однородности частиц (около 90% основной
фракции);
повышенная степень однородности геометрической внутренней
поверхности частиц и возможность ее регулирования в широких пределах
(диаметр пор 6–10000 нм, удельная площадь поверхности 10–600 м2/г;
удельный объем пор 0.3–1.3 см3/г);
повышенная степень химической однородности широких
пределах емкости сорбционных материалов;
повышенная прочность.
По структуре и характеру протекания диффузионных процессов в
порах сорбенты разделяют на два основных типа: поверхностно-пористые и
объемно-пористые, параметры которых и заполняемых ими колонок
приведены в табл. 1.
Таблица 1 – Параметры сорбентов и колонок
Параметр Поверхностно-
пористые
Объемно-
пористые
ds, мкм 30–40 3–10
Sуд, м2/г 10–15 100–600
Емкость сорбента, мг/г 0.05–0.1 1–5
Типичный диаметр колонки, мм 2–5 1.45
Типичная длина колонки, см 50–100 5–25
Оптимальное значение ВЭТТ, мм 0.2–0.4 0.1–0.01
По химической природе сорбенты для ЖХ можно разделить на три
группы: неорганические (силикагель, оксид алюминия); органические (на
основе полимеров гелевой и макропористой структуры); смешанные

37
(неорганические, капсулированные полимерным слоем, с привитыми
ионогенными группами и неорганические с привитыми органическими
функциональными группами).
Аппаратура
Принципиальная схема жидкостного хроматографа
Основными составляющими оборудования для ВЭЖХ являются
система ввода пробы, насос высокого давления для прокачивания подвижной
фазы, хроматографическая колонка, которая соединена капилляром с
проточным детектором, а также необходимый для качественного и
количественного анализа самописец или электронный интегратор.
Жидкостной хроматограф – более сложный прибор по сравнению с
газовым. Это связано с тем, что система подачи элюента включает ряд
дополнительных узлов: 1 – систему дегазации; 2 – устройство для создания
градиента; 3 – насосы и 4 – измерители давления.
Тщательное обезгаживание всех используемых растворителей
необходимо ввиду того, что появление пузырьков газа в детекторе делает
невозможным его использование.
Жидкостной хроматограф имеет достаточно сложное градентное
устройство, обеспечивающее отбор элюентов из 2–3 емкостей в смеситель,
затем в колонку, а также детекторы, работающие при высоких давлениях.
Система для подачи подвижной фазы
Система для подачи подвижной фазы включает насос высокого
давления и, как правило, устройство для градиентного элюирования. Емкости
для растворителей могут быть заполнены растворителями с различной
полярностью, но при этом хорошо смешивающимися между собой, либо
растворами с различными значениями рН для получения буферного раствора
при смешивании. Растворители должны быть чистыми, дегазированными во
избежание образования пузырей газа и возникновения «воздушных затворов»
в кранах и клапанах насоса. Образование пузырей также может вызвать
возникновение ложных пиков при прохождении через ячейку детектора.

38
Наиболее часто используемые способы дегазации – пропускание гелия или
вакуумная дегазация.
Насосы должны обеспечивать постоянную скорость потока от 0.1 до 10
см3/мин при давлении не менее 15 МПа. В качестве конструкционных
материалов применяют нержавеющую сталь, тефлон, керамику.
Принципиально различают насосы, работающие с постоянным
давлением, и насосы, работающие с постоянным потоком. Современные
ВЭЖХ насосы работают в режиме постоянного тока, это означает, что,
независимо от возникшего падения давления в системе, скорость потока
всегда поддерживается постоянной. Насосы с постоянным потоком можно в
свою очередь разделить на периодические и непрерывные.
Система ввода пробы
Система ввода пробы (дозаторы) должна позволять точно дозировать
объемы от 5 до 500 мкл. При этом в процессе ввода давление в системе
должно оставаться постоянным. Основным типом устройства для
дозирования объема пробы служит петля инжектора (рис. 1). Она соединена с
двумя выходами шестиходового крана. Раствор вводят в петлю при помощи
микрошприца. При этом переключатель шестиходового крана устанавливают
в положение, показанное на рис. 1, а; в колонку в это время непрерывно
(через выходы 2–3) подается поток элюента. Затем для ввода пробы в поток
переключатель крана переводят в положение, показанное на рис. 1, б, после
чего вновь возвращают его в исходное положение.

39
Рис. Шестиходовый кран с петлей для образца: а – заполнение петли образцом; б –
перевод образца в колонку; 1– элюент; 2 – колонка; 3 – образец.
Колонки для ВЭЖХ
Наилучшие колонки делают на основе прямых трубок правильной
формы из нержавеющей стали. Стандартные насадочные аналитические
колонки в составе жидкостных хроматографов для ВЭЖХ имеют следующие
параметры: длина 150–250 мм, внутренний диаметр 2–4 мм. При заполнении
частицами размером 3 мкм эффективность таких колонок может достигать
100000 теоретических тарелок на метр.
В соответствии с используемыми сорбентами методы разделения
подразделяют на нормальнофазовую, обращеннофазовую и эксклюзионную
хроматографию. За исключением эксклюзионной хроматографии эффект
разделения в жидкостной хроматографии основывается на разной полярности
стационарной и подвижной фазы.
Вид хроматографии, когда полярность неподвижной фазы выше, чем
подвижной, называется нормально-фазовой хроматографии. В противном
случае, когда неподвижная фаза достаточно малополярна (например,
углеводород), а подвижная полярна (вода, метанол) – метод обращенно-
фазовой хроматографией.
В качестве материала для заполнения колонки в нормальнофазовом
варианте используют силикагели или оксид алюминия. Т.к. стационарная
фаза полярная, подвижная фаза должна быть неполярная. В качестве
элюентов используют гексан, гептан, метиленхлорид, уксусный эфир или их
смеси. Из этого сразу вытекает ограничение для анализируемых проб: они
должны быть не только растворимы в этих неполярных растворителях, но и
находится уже растворенными в этих неполярных растворителях. В
неполярных растворителях разделяются преимущественно неполярные
органические вещества. Полярные соединения, даже если они растворимы в
элюентах, могут сильно взаимодействовать с поверхностью силикагеля.

40
Типичным недостатком адсорбционной хроматографии является сильное
влияние следов воды в элюентах. Вода адсорбируется на поверхности
силикагеля и дезактивирует силанольные группы, вследствие чего
удерживание уменьшается. Поэтому используемые колонки нужно часто
регнерировать (сушить).
Неподвижной фазой в жидкостной хроматографии может служить и
химически модифицированная поверхность носителя (химически
закрепленная фаза). Под понятием «полярные привитые стационарные фазы»
понимают, в общем случае, силикагель, поверхность которого
модифицирована полярными группами.
Поверхность полностью гидролизованного силикагеля покрыта
силанольными (гидроксильными) группами. Для создания нормальной фазы
поверхность силикагеля модифицируют такими полярными
функциональными группами, как диольная (–(СН2)3ОСН2СН(ОН)СН2ОН),
нитрильная (–(СН2)3С≡N), аминная (–(СН2)nNH2 (n=3 или 4)),
диметиламинная (–(СН2)3N(CH3)2), диаминная (–(СН2)3NН(CH2)2NH2).
(силанизируют), обрабатывая ее алкилхлорсиланами.
В обращеннофазовой хроматографии работают с противоположным
соотношением полярностей, т.е. стационарная фаза – неполярная, а
подвижная фаза – полярная. В этом случае силикагель модифицируется
химически. Для создания обращенной фазы поверхность силикагеля
гидрофобизируют (силанизируют), обрабатывая ее алкилхлорсиланами. При
этом образуется химически закрепленная фаза силоксана, содержащая Si–O–
Si-связи.
≡Si–ОН + (СН3)3SiСl ↔ ≡Si–О–Si(СН3)3 + НСl
Чаще всего алкильным остатком реагента является н-октадецил (С18),
затем следует н-октил (С8). Чем длиннее алкильный радикал обращенной
фазы, тем больше времена удерживания органических веществ.
В качестве подвижной фазы используется вода, полярные органические
растворители (метанол, ацетонитрил) или их смеси. Эти методом можно

41
разделять широкий круг соединений, т.к. многие вещества растворимы в воде
или подобных растворителях. Развитие техники обращеннофазовой
хроматографии с химически связанными обращенными фазами позволило
значительно расширить область применения ВЭЖХ. Более 70 % анализов
методом ВЭЖХ проводятся сегодня на химически связанных фазах.
В отличие от газовой хроматографии, в которой подвижная фаза
химически инертна и выполняет лишь роль переносчика, в жидкостной
хроматографии между компонентами подвижной фазы и молекулами
разделяемых веществ наблюдаются достаточно сильные физико-химические
взаимодействия. Поэтому выбор подвижной фазы в ЖХ важен, поскольку
она оказывает большое влияние на селективность разделения, эффективность
колонки и скорость движения хроматографической полосы.
Разделения достигают, меняя элюирующую силу подвижной фазы –
растворителя. Элюирующая сила является мерой энергии взаимодействия
молекул растворителя и неподвижной фазы и показывает, во сколько раз
энергия сорбции данного элюента больше, чем энергия сорбции элюента,
выбранного в качестве стандарта, например н-гептана.
По отношению к силикагелю SiO2 элюирующая сила увеличивается в
ряду: пентан (0) < н-гексан (0,01) < СС14 (0,11) < бензол (0,25) < СНСl3 (0,26)
< СН2Сl2 (0,32) < ацетон (0,47) < диоксан (0,49) < ацетонитрил (0,5) < этанол
(0,68) < метанол (0,73) < вода (большая). Расположение растворителей в
соответствии с возрастанием их элюирующей силы называют элюотропным
рядом.
Порядок следования растворителей согласно величинам их
элюирующей силы в общем сохраняется и при переходе от силикагеля к
другой полярной неподвижной фазе, например Al2O3. При использовании
же неполярных фаз порядок изменяется на противоположный. Например,
на обращенной углеводородной фазе сильно полярный растворитель –
вода – обладает гораздо меньшей элюирующей силой, чем неполярный
гексан. Так, для обращенно-фазовой хроматографии на С18 элюотропный ряд

42
имеет вид: метанол (1.0) < ацетонитрил (3.1), этанол (3.1) < изопропанол (8.3)
< н-пропанол (10.1) < диоксан (11.7).
Небольшие колонки длиной от 3 до 10 см, называемые предколонками,
помещают между инжектором и аналитической колонкой. Обычно они
заполняются тем же сорбентом, что и аналитическую колонку. Эти колонки
включаются в хроматографическую систему по двум причинами. Во-первых,
они задерживают мелкие частицы, которые могут попасть из каналов насоса
или из компонентов образца в аналитическую колонку и испортить ее, снизив
эффективность и селективность. Во-вторых, на предколонке удерживаются
сильносорбируемые компоненты пробы, которые в случае их сорбции на
аналитической колонке было бы трудно элюировать. Т.о., предколонка
продлевает срок службы аналитической колонки. Предколонки можно легко
заменять или периодически регенерировать.
ВЭЖХ детекторы
В ВЭЖХ используют следующие два основных принципа
детектирования:
1. Измерение какого-либо общего свойства подвижной фазы,
например, показателя преломления или электропроводности. Этот способ
детектирования является неселективным и поэтому универсальным.
2. Измерение какого-либо специфического свойства разделяемых
веществ – светопоглощение в УФ-области, интенсивность флуоресценции
или сила тока при электролизе вещества на рабочем электроде.
Для ВЭЖХ необходимы детекторы с высокой чувствительностью,
позволяющие определять вещества на уровне мкг–нг. В жидкостной
хроматографии для детектирования используются такие аналитические
параметры, как поглощение света, показатель преломления света, удельная
электрическая проводимость. Очень часто применяют рефрактометрические
детекторы или фотометрические детекторы в УФ-диапазоне.
Рефрактометрический детектор практически невозможно использовать в

43
режиме градиентного элюирования из-за постоянного изменения базовой
линии, а также в тех случаях, когда показатели преломления растворителя и
растворенных веществ имеют достаточно близкие значения. Кроме того,
рефрактометрический детектор чрезвычайно чувствителен к колебаниям
температуры. Этот тип детектора обладает невысокой чувствительностью.
Лучшей чувствительностью обладают фотометрические детекторы.
Наибольшую распространенность (~70 %) фотометрических методов
детектирования определяет то, что большинство органических соединений
(ароматические и гетероциклические соединения; вещества, молекулы
которых содержат функциональные группы С=О, C=S, N=O, N=N и
сопряженные связи) имеют интенсивные полосы поглощения в диапазоне
длин волн 200–800 нм.
Фотометрические детекторы подразделяют следующим образом: 1)
детекторы с фиксированной длиной волны (фотометры); 2) детекторы с
дискретно изменяемой с помощью оптических фильтров длиной волны
(фильтровые фотометры); 3) спектрофотометрические детекторы,
регистрирующие поглощение в определенной области УФ спектра; 4)
спектрофотометрические детекторы на фотодиодных матрицах.
Наиболее простыми и доступными, чаще всего применяемыми в
жидкостных хроматографах являются детекторы с фиксированной длиной
волны (одноволновой детектор), которые в определенном приближении
можно рассматривать как универсальные детекторы, реагирующие на многие
классы органических веществ. Источником УФ света в них служит ртутная
лампа. Детектирование проводят на длине волны, соответствующей наиболее
интенсивной линии ртутной лампы низкого давления, 254 нм. Вблизи этой
длины волны находятся максимумы поглощения всех ароматических
соединений, а также большинства кетонов и альдегидов. УФ-детектор
селективен, позволяет определять 10–9 г, его диапазон линейности ~5
порядков. Шум УФ-детекторов составляет 10–5 ед.поглощения.
Для измерения светопоглощения подвижной фазы на выходе из

44
колонки используют проточные фотометрические кюветы (ячейки) Z-
образной формы (рис. 2). Для предотвращения размывания
хроматографических пиков объем ячейки стараются сделать как можно
меньше – 1–10 мкл. Длина оптического пути ячейки составляет от 2 до 10 мм.
Рис. 2. Проточная фотометрическая ячейка для УФ-детектирования в ВЭЖХ: 1 – корпус; 2
– кварцевое окно; 3, 6 – луч света; 4 – выход элюента; 5 – рабочий объем ячейки; 6 – вход
элюента.
Наряду с одноволновыми применяют и многоволновые детекторы –
сканирующие (с призменными или решеточными монохроматорами) и
многоканальные. Детекторы с фиксированной длиной волны до 20 раз более
чувствительны, чем детекторы с переменной длиной волны.
Спектрофотометрические детекторы позволяют регистрировать
разделяемые вещества по поглощению света при любых длинах волн УФ и
видимого диапазона. В качестве источника света используют дейтериевую
лампу с непрерывным спектром от 190 до 600 нм. Необходимую
спектральную полосу выделяют с помощью дифракционных решеток либо
интерференционных фильтров с заданной шириной спектральной полосы. В
зависимости от выбранной длины волны спектрофотометр может работать
как универсальный или как селективный детектор. Но в любом случае его
универсальность ограничена соединениями определенных классов.

45
В многоканальных детекторах регистрацию осуществляют с помощью
массива фотодиодов – диодной линейки (рис. 3). Информацию представляют
в виде трехмерной диаграммы как зависимость светопоглощения от двух
параметров – времени элюирования и длины волны.
Рис. 3. Оптическая часть детектора с фотодиодной матрицей: 1 – дейтериевая лампа; 2 –
линза; 3 – заслонка; 4 – проточная ячейка; 5 – диодная матрица; 6 – дифракционная
решетка.
В детекторах этого типа непрерывное излучение от источника
проходит через проточную ячейку и попадает на дифракционную решетку.
Луч отклоняется и фокусируется на плоскости, где расположена фотодиодная
матрица. Каждый диод предназначен для измерения узкой полосы спектра.
Традиционные спектрофотометрические детекторы для ВЭЖХ позволяют
осуществлять мониторинг элюируемых компонентов пробы по их
максимумам поглощения. При этом сканирование спектра требует в
большинстве случаев остановки потока и занимает несколько секунд или
даже минут в зависимости от диапазона длин волн. При использовании
фотодиодной матрицы проба сканируется каждые несколько миллисекунд,
т.е. спектральная информация выдается практически непрерывно. При
регистрации подобным детектором светопоглощения элюентом длина волны
используется как третья координата измерения дополнительно к
координатам времени и степени поглощения (трехмерное представление
результатов). В этих условиях можно зафиксировать спектр каждого
компонента и установить максимальное поглощение в конкретной области

46
длин волн. Детекторы на диодной матрице позволяют проводить
количественный анализ, даже если хроматографические пики разрешены
неполностью.
В ВЭЖХ принципиально возможно фотометрическое детектирование и
в ИК-области спектра. Однако поскольку в этом диапазоне большинство
наиболее распространенных растворителей (вода, метанол и др.) интенсивно
поглощает, этот способ детектирования применяют лишь в специальных
случаях.
Лабораторная работа. Определение содержания бензоата натрия и
кофеина в напитках безалкогольных методом высокоэффективной
жидкостной хроматографии
Бензоат натрия используется в качестве консерванта при производстве
безалкогольных напитков, его присутствие позволяет увеличить срок
хранения произведённых напитков без потери вкусовых качеств сроком до
полугода. Кофеин используется в качестве стимулирующей нервную систему
пищевой добавки в газированных напитках типа «Cola» и так называемых
энергетических напитках «Adrenalin rush», «Burn» и т.д. По действующим
нормативам содержание бензоата натрия в напитках не должно превышать
180 мг/л, а кофеина – 300 мг/л для напитков типа «Cola», в энергетических
напитках допускается более высокое содержание. Количество бензоата
натрия также не должно быть очень низким во избежание потери вкусовых
свойств до истечения срока хранения. Контроль содержания бензоата натрия
и кофеина в безалкогольных напитках осуществляется методом
высокоэффективной жидкостной хроматографии (ВЭЖХ) со
спектрофотометрическим детектированием в УФ-области.
Посуда и реактивы
Установка для вакуумной фильтрации и дегазации подвижной фазы.
Фильтры мембранные тефлоновые с размером пор 0.4–0.5 мкм,
диаметром 47 мм.

47
Мембранный вакуумный насос, создающий вакуум от 240 мм рт.ст. и
ниже.
рН-метр.
Микрошприцы для ВЭЖХ вместимостью 10 мм3.
Колбы мерные вместимостью 1000 см3.
Колбы мерные вместимостью 200 см3.
Колба мерная вместимостью 50 см3.
Цилиндр мерный вместимостью 250 см3.
Пипетка градуированная вместимостью 25 см3.
Пипетка градуированная вместимостью 5 см3.
Воронка для фильтрования.
Химический стакан вместимостью 50 см3.
Пузырёк пенициллиновый.
Вода дистиллированная по ГОСТ 6709.
Кислота ортофосфорная квалификации «х.ч.» или «ч.д.а.».
Калий фосфорнокислый однозамещённый (KH2PO4), 3.44 г.
Ацетонитрил квалификации «х.ч.», 350 см3.
Бензоат натрия квалификации «х.ч.» или «ч.д.а.», 0.036 г.
Кофеин квалификации «х.ч.», 0.02 г.
Основные параметры настройки хроматографа
Высокоэффективный жидкостной хроматограф со
спектрофотометрическим детектором с переменной длиной волны или
детектором диодной матрицы, оснащённый системой обработки
хроматографических данных на базе персонального компьютера. Колонка
аналитическая, размером 250 × 4.6 мм (150 × 4.0 мм), заполненная обращено-
фазным сорбентом С18 (С8) с диаметром частиц 5 мкм. Подвижная фаза: 35
% ацетонитрила – 65 % 0.0125 моль/дм3 раствора KH2PO4, доведённого
фосфорной кислотой до значения рН 3.2; скорость потока 1.2–1.5 см3/мин;
детектирование выполняют по поглощению в УФ-области спектра при длине

48
волны 280 нм (основная) и 230 нм (вспомогательная). Инжектируемый объём
образца – 10 мм3.
Порядок выполнения работы
1. Приготовление подвижной фазы
Навеску 1.72 г дигидрофосфата калия KH2PO4 помещают в мерную
колбу вместимостью 1000 см3, растворяют в дистиллированной воде,
перемешивают до полного растворения. Полученный раствор на рН-метре
доводят до значения рН 3.2 ортофосфорной кислотой. Повторно взвешивают
1.72 г KH2PO4, помещают во вторую мерную колбу вместимостью 1000 см3,
растворяют в дистиллированной воде, перемешивают до полного
растворения. Полученный раствор также доводят ортофосфорной кислотой
до значения рН 3.2. Затем из первой колбы в мерный цилиндр вместимостью
250 см3 отливают около 200 см3 полученного буферного раствора,
оставшийся в колбе раствор смешивают со 150 см3 ацетонитрила. Доводят
уровень в мерной колбе буферным раствором из цилиндра до метки,
тщательно перемешивают. Установку для вакуумной фильтрации и
дегазации подвижной фазы подключают к мембранному вакуумному насосу,
на фильтровальную воронку помещают свежий тефлоновый мембранный
фильтр, устанавливают и закрепляют держателем фильтровальную ёмкость,
включают электропитание насоса, отфильтровывают и дегазируют половину
объёма подвижной фазы из первой колбы. По окончанию фильтрации
подготовленную подвижную фазу помещают в специальную ёмкость для
элюента жидкостного хроматографа.
2. Приготовление стандартных растворов и градуировка
На аналитических весах взвешивают навески бензоата натрия 36.0 мг и
кофеина 20.0 мг, переносят их в мерную колбу вместимостью 200 см3, затем
из мерной колбы вместимостью 1000 см3 приливают подвижную фазу до
половины объёма и перемешивают до полного растворения, после чего
доводят до метки подвижной фазой. Получают первый градуировочный
раствор. Далее пипеткой отмеряют 25 см3 градуировочного раствора № 1,

49
переносят во вторую мерную колбу вместимостью 200 см3, доводят
подвижной фазой до метки и тщательно перемешивают. Получают
градуировочный раствор № 2. Рассчитывают концентрацию бензоата натрия
и кофеина в каждом из градуировочных растворов. Включают жидкостной
хроматограф, устанавливают рабочие параметры для данной методики, после
подключения насоса и детектора ожидают выхода хроматографической
системы в режим готовности. Микрошприцем для ВЭЖХ, либо с помощью
автосамплера (если он установлен на данный прибор) вводят в инжектор
хроматографа 10 мм3 градуировочного раствора № 2, дожидаются выхода
пиков обоих компонентов, процедуру выполняют в трёх параллелях. Затем
трижды хроматографируют градуировочный раствор № 1. Полученные
времена удерживания и площади (высоты) хроматографических пиков для
бензоата натрия и кофеина усредняют и с помощью программы обработки
данных хроматографа строят градуировочный график.
3. Подготовка образцов напитков и выполнение хроматографического
анализа
В стакан вместимостью 50 см3 помещают ~25 см3 исследуемого
газированного напитка, оставляют открытым на воздухе для дегазации.
После визуального прекращения выхода пузырьков углекислого газа на
поверхность с помощью пипетки отбирают 5 см3 напитка и переносят в
мерную колбу вместимостью 50 см3, доводят до метки подвижной фазой,
тщательно перемешивают, далее микрошприцем для ВЭЖХ раствор вводят в
хроматограф. Хроматографирование каждого образца выполняют в трёх
параллелях. Определяют средние значения времён удерживания и площадей
(высот) хроматографических пиков бензоата натрия и кофеина. На основании
построенной градуировочной зависимости компьютерная программа
рассчитывает концентрацию бензоата натрия и кофеина в исследуемом
напитке, либо расчёт может быть выполнен по формуле:
aSSCС i
st
sti ××= ,

50
где Сst – концентрация бензоата натрия или кофеина в градуировочном
растворе, мг/дм3; Sst – площадь хроматографического пика бензоата натрия
или кофеина в градуировочном растворе; Si – площадь хроматографического
пика бензоата натрия или кофеина в исследуемом растворе напитка; a –
величина, кратная степени разбавления напитка подвижной фазой.
После выполнения расчёта оценивают результаты анализа на предмет
соответствия содержания бензоата натрия и кофеина в напитке действующим
нормам.

51
ПРИЛОЖЕНИЕ В. Определение катионного и анионного состава
природных вод

52
Уральский федеральный университет
имени первого Президента России Б.Н.Ельцина
Институт естественных наук
Кафедра аналитической химии
Определение катионного и анионного состава природных вод
Учебно-методическое пособие
Екатеринбург
2011

53
КАПИЛЛЯРНЫЙ ЭЛЕКТРОФОРЕЗ
Принцип метода
Капиллярный электрофорез (КЭ) – метод анализа сложных смесей,
реализуемый в капиллярах и основанный на различиях в
электрофоретических подвижностях заряженных частиц.
Впервые электрофорез как метод разделения был предложен в 1930-х
гг. А. Тизелиусом, обнаружившим, что компоненты смеси белков в буферном
растворе движутся под действием электрического поля со скоростями,
зависящими от заряда, размеров и формы их частиц. В 1948 г. эта работа
была удостоена Нобелевской премии. Однако применение нового метода
было ограничено из-за его невысокой эффективности.
В 1967 г. Хиртен предложил проводить электрофоретическое
разделение не на плоскости, а в открытых трубках диаметром 1–5 мм.
Активное развитие метода капиллярного электрофореза началось с 80-х гг.
ХХ века, когда Йоргенсон и Лукас продемонстрировали сепарационные
возможности кварцевого капилляра диаметром 75 мкм с использованием
прямого УФ-детектирования.
Сегодня капиллярный электрофорез находит широкое применение в
анализе объектов окружающей среды, ветеринарии, фармации и клинической
биохимии, криминалистике, химической промышленности, при контроле
качества пищевых продуктов и в других прикладных областях.
Капиллярный электрофорез – универсальный и
высокоавтоматизированный метод анализа. Наиболее близким к нему по
аналитическим возможностям является метод высокоэффективной
жидкостной хроматографии) ВЭЖХ. К преимуществам КЭ перед ВЭЖХ
относятся:
1) сверхэффективность (число теоретических тарелок достигает
сотен тысяч),
2) работа при нормальном давлении (отсутствует необходимость
использования прецизионного микронасоса и крана ввода пробы),

54
3) отсутствие сорбентов, проблем, связанных с их старением и
заменой колонки,
4) повышение скорости анализа, эффективности,
производительности и разрешения путем увеличения напряжения,
5) малый объем анализируемой пробы и буферных растворов (1–2
см3 в день),
6) простая аппаратура, низкая стоимость единичного анализа,
7) возможность проведения разнообразных исследований:
разделения неорганических ионов, радиоактивных изотопов, биополимеров,
энантиомеров, определения подвижности ионов и чисел переноса, состава и
устойчивости комплексов, изучения кинетики реакций и так далее.
Варианты капиллярного электрофореза. Наиболее
распространенным и самым простым вариантом КЭ является капиллярный
зонный электрофорез (КЗЭ), при котором состав буферного раствора,
значение рН и напряженность поля во всем пространстве разделения
остаются постоянными. Проба вводится в виде отдельной зоны на входе в
капилляр, а ее компоненты обнаруживаются в виде дискретных зон на конце
детектора. Полученная электрофореграмма представляет собой
последовательность пиков; время миграции является качественной
характеристикой компонента, площадь (или высота) пика пропорциональна
его концентрации.
Метод КЗЭ позволяет разделять только ионогенные компоненты.
Помимо КЗЭ существуют варианты КЭ, основанные на других принципах
разделения. Некоторые из них перечислены в табл. 1.
Таблица 1
Некоторые варианты метода капиллярного электрофореза
Вариант Условия разделения Принцип разделения
Капиллярный зонный
электрофорез (КЗЭ)
Однородность состава
буферного раствора,
Соотношение зарядов и
размеров ионов

55
Вариант Условия разделения Принцип разделения
постоянство
напряженности в
капилляре
Изоэлектрическое
фокусирование (ИЭФ) Градиент рН в капилляре
Величины
изоэлектрических точек
(pI) амфолитов
Изотахофорез (ИТФ)
Использование ведущего
и замыкающего
электролитов
Соотношение
подвижностей
анализируемого,
ведущего и замыкающего
ионов
Мицеллярная
электрокинетическая
хроматография
(МЭКХ)
Добавление ПАВ,
образующих мицеллы
Распределение частиц
между растворителем и
мицеллой
Микроэмульсионная
электрокинетическая
хроматография
(МЭЭКХ)
Добавление ПАВ для
стабилизации эмульсии
неполярных жидкостей в
воде
Распределение частиц
между водной и
органической фазами
Капиллярный гель-
электрофорез (КГЭ)
Использование
полимеров, образующих
молекулярные сита
Размеры частиц
Капиллярная
электрохроматография
(КЭХ)
Сочетание давления и
напряжения в качестве
движущей силы
разделения
Соотношение зарядов и
размеров частиц,
распределение их между
фазами

56
Физико-химические основы метода. На ион, находящийся в
электрическом поле, действует сила Fм, которая может быть выражена через
напряжённость электрического поля E:
Aм N
zFEF = ,(1)
где F – константа Фарадея (96485 Кл/моль), z – эффективный заряд иона, NА –
число Авогадро (6·1023 моль-1).
При наступлении стационарного скоростного режима сила Fм
уравновешивается силой трения со стороны поверхности кварцевого
капилляра Fтр, которая в соответствии с законом Стокса выражается
следующим образом:
πηurFтр 6= , (2)
здесь η – динамическая вязкость среды, r – стоксовский радиус.
Выражение для скорости заряженной частицы, движущейся под
действием электрического поля, имеет следующий вид:
ANrzFEuπη6
= . (3)
Таким образом, электрофоретическая скорость частиц определяется их
формой и величиной заряда, то есть характеристическими параметрами.
Чаще скорость движения частицы характеризуют подвижностью, т.е.
скоростью в поле единичной напряжённости: uE
μ = . (4)
Подвижности ионов определяют из экспериментальных данных:
UtLL
iм
общэффi =μ , (5)
где Lобщ – общая длина капилляра (от входного до выходного конца), Lэфф –
эффективная длина капилляра (от входного конца до детектора), tm – время
миграции иона от зоны ввода пробы до зоны детектирования (соответствует

57
времени выхода пика на электрофореграмме), U – приложенная разность
потенциалов.
Подвижность иона в КЭ считают положительной, если ион движется
по направлению к детектору (к выходу), и отрицательной, если он движется в
противоположном направлении.
Электроосмотический поток. Чаще всего капилляры для КЗЭ
изготавливают из высокочистого плавленого кварца. Находящиеся на
поверхности кварца силоксановые группы гидролизуются с образованием
удвоенного количества силанольных групп, которые затем гидратируются и
диссоциируют:
SiO2H2O
Si
OH
OH - 2 Н+Si
O
O
Внутренняя поверхность кварцевого капилляра в результате
диссоциации силанольных групп приобретает отрицательный заряд.
Положительно заряженные частицы, противоионы, адсорбируются
поверхностью и выстраиваются вдоль границы раздела, отрицательно
заряженные – отталкиваются электрическими силами от поверхности и
перемещаются вглубь раствора. В результате, соприкасающиеся фазы
приобретают заряды противоположного знака – возникает двойной
электрический слой (рис. 1).
Из-за мощного электростатического взаимодействия с поверхностью
часть катионов, так же как и силанольные группы, частично теряют
гидратирующую воду, в результате чего первый слой катионов,
непосредственно контактирующий с поверхностью, становится весьма мало
подвижным. Остальная часть нейтрализующих отрицательный заряд
катионов распространяется в толщу раствора, образуя так называемую
диффузную часть второй обкладки двойного слоя.

58
Рис. 1. Строение двойного электрического слоя на поверхности капилляра.
При наложении электрического поля вдоль капилляра положительно
заряженная диффузная часть ДЭС начинает мигрировать к катоду (-), увлекая
за собой всю массу жидкости в капилляре. Возникает электроосмотический
поток (ЭОП), который осуществляет пассивный перенос раствора в
капилляре.
Скорость ЭОП определяется уравнением:
ηξεε E
vЭОП0= , (6)
где ε0 – диэлектрическая постоянная, ε – диэлектрическая проницаемость
среды, ξ –кинетический (дзета-) потенциал поверхности капилляра, η –
вязкость раствора.
Время, необходимое жидкости для преодоления эффективной длины
капилляра вследствие возникающего ЭОП, называют временем ЭОП (tЭОП);
его определяют экспериментально по времени миграции нейтрального
компонента – маркера ЭОП.
Подвижность ЭОП вычисляют по формуле:
эфф общЭОП
ЭОП
L Lt U
μ = . (7)

59
В условиях КЗЭ наблюдаемая скорость движения частицы является
суммой электрофоретической скорости аналита и скорости
электроосмотического потока электролита в капилляре:
эф ЭОПu u u= + . (8)
Если в капилляр со стороны анода внести небольшой объем раствора
пробы, то ЭОП будет переносить эту зону к катоду. Катионные компоненты
пробы будут обгонять ЭОП, анионные – отставать от него. Если скорость
миграции аниона превышает скорость ЭОП, то он будет выходить в
прианодное пространство.
В сильнокислых растворах ЭОП отсутствует, с увеличением рН его
скорость возрастает за счёт увеличения доли продиссоциировавших
силанольных групп. Увеличение концентрации ведущего электролита
приводит к уменьшению толщины диффузной части ДЭС и
соответствующему уменьшению ЭОП.
Характеристики метода. КЭ относится к группе комбинированных
методов анализа, поскольку объединяет предварительное разделение
компонентов смеси и их определение. Важными характеристиками
разделения являются разрешение, эффективность и селективность. Для
количественного определения компонента наиболее важным параметром
является чувствительность детектора.
Эффективность КЗЭ выражается числом теоретических тарелок N: 2
2/1
54,5 ⎟⎟⎠
⎞⎜⎜⎝
⎛=
wtN м , (9)
где 2/1w – ширина пика на половине высоты. На эффективность разделения
влияют следующие факторы:
1) объём введённой пробы (в идеале должна быть как можно
меньше),
2) температурный градиент в капилляре,
3) адсорбция на стенках капилляра,

60
4) различия в электропроводности пробы и ведущего электролита,
5) гидродинамический поток из-за различия в уровне жидкостей во
входном и выходном сосудах.
Метод КЗЭ характеризуется очень высокой эффективностью.
Обусловлена она уникальным свойство ЭОП – плоским профилем потока (в
отличие от параболического в ВЭЖХ), который практически не вызывает
уширения зон при их движении в капилляре (рис. 2).
Рис. 2. Влияние профиля потока на ширину зоны вещества.
Разрешение в методе КЭ рассчитывают по формуле:
( )21
212wwttRs +
−= , (10)
где t1 и t2 – времена миграции первого и второго компонентов, w1 и w2 –
ширина первого и второго пиков при основании. Разрешение в КЭ, в первую
очередь, управляет эффективностью, а не селективностью. В этом
заключается важное отличие КЭ от ВЭЖХ, где картина прямо
противоположная. Благодаря узким зонам компонентов в капиллярном
электрофорезе даже очень малые различия в электрофоретической
подвижности веществ оказываются достаточны для полного разделения.
Основной вклад в размытие пиков в КЗЭ вносит диффузия. Ширина
зон увеличивается с увеличением времени миграции (эффективной длины
капилляра), поэтому иногда для количественного анализа используют
величину так называемой исправленной площади пика, равную отношению
площади пика ко времени миграции.
Значительную роль в размывании зон играет температурный профиль в
капилляре и соответствующий ему профиль вязкости, а также скорости

61
жидкости в капилляре за счет значительного выделения джоулева тепла Q
при прохождении электрического тока через капилляр:
CEQ элλ2= (11)
где λэл – молярная электропроводность электролита, С – его молярная
концентрация.
Изменение температуры в капилляре сильно влияет на эффективность
разделения и на работоспособность системы в целом. Использование очень
тонких капилляров, разбавленных растворов с низкой электропроводностью
и систем термостатирования (воздушных или жидкостных) помогает свести к
минимуму неблагоприятное влияние теплового фактора.
Разрешение связано с эффективностью:
⎟⎟⎠
⎞⎜⎜⎝
⎛+−
=21
21
2 μμμμNRs , (12)
где 1μ и 2μ – электрофоретические подвижности первого и второго
компонентов.
Фактор селективности в КЗЭ определяется как отношение
электрофоретических подвижностей компонентов:
2
1
μμ
=fS . (13)
Селективность разделения в КЗЭ обычно невелика из-за
осуществления разделения в гомогенной среде. Для повышения
селективности изменяют рН ведущего электролита, вводят в него различные
добавки (ПАВ, макроциклы, органические растворители), проводят
модификацию капилляра.
Чувствительность метода при спектрофотометрическом
детектировании не всегда оказывается достаточной из-за того, что
детектирование происходит в слое малой толщины (внутренний диаметр
капилляра) и вводятся очень малые объемы пробы. Преодолеть этот

62
недостаток можно, используя различные методы концентрирования или
детекторы других типов.
Воспроизводимость результатов анализа в методе КЭ зависит главным
образом от постоянства состояния внутренней стенки капилляра, поэтому
важной задачей является выбор правильной схемы промывки капилляра.
При подготовке к работе капилляр обычно промывают кислотой,
водой, щёлочью, снова водой и раствором ведущего электролита. Кислота
удаляет адсорбированные на поверхности примеси и способствует
первичному гидролизу силоксановых групп. Промывка водой способствует
удалению кислоты и дальнейшему гидролизу поверхности. Щелочная
промывка предназначена для удаления примесей, не реагирующих с
кислотой, и максимальной диссоциации образовавшихся силанольных групп.
Финишная промывка водой имеет целью удалить из капилляра щелочь. Для
того чтобы привести очищенную поверхность в равновесие с раствором
ведущего электролита, капилляр промывают собственно раствором ведущего
электролита. При правильно проведенном кондиционировании времена
миграции веществ остаются постоянными при последовательных вводах.
Схема прибора
Внешний вид системы капиллярного электрофореза «Капель 105М»
показан на рис. 3, блок-схема прибора приведена на рис. 4.
Рис. 3. Внешний вид системы капиллярного электрофореза «Капель 105М».

63
На передней стенке прибора находятся крышка кассетного отделения и
панель индикации с двумя светодиодными индикаторами состояния прибора.
За откидной крышкой находятся кассета с капилляром и карусели входного и
выходного автосемплеров.
На правой стенке расположен выключатель сетевого питания. Левая
стенка имеет закрывающийся отсек, в котором расположены сменный блок
высоковольтного питания и отверстие для ввода воды в систему охлаждения.
На верхней крышке прибора расположен люк, открывающий доступ к
источнику излучения.
Управление работой прибора осуществляется посредством
программного обеспечения «Эльфоран». Последовательность операций (ввод
пробы в капилляр, электрофоретический анализ, промывка капилляра) и их
параметры задаются в виде программы.
Ввод пробы осуществляется гидродинамически: стаканчик с пробой
поднимается, в пространстве над стаканчиком насос создаёт избыточное
давление воздуха, и проба из пробирки поступает в капилляр. Вводимый
объем определяется величиной давления (мбар, 1 мбар = 100 Па) и временем
ввода (с), оба этих параметра задаются оператором. По окончании процедуры
автоматически открывается электромагнитный клапан, давление быстро
падает, а двигатель насоса возвращает поршень в исходное положение.
В системах капиллярного электрофореза «Капель» также реализована
возможность так называемого электрокинетического ввода пробы, когда
проба поступает в капилляр за счет ЭОП, возникающего при подаче
импульса высокого напряжения. Количество введенной пробы зависит от
величины приложенного напряжения (кВ), времени ввода (с) и подвижности
компонентов пробы. В капилляре концентрируются компоненты с высокой
подвижностью, исходный состав пробы при этом изменяется.
Промывка капилляра осуществляется аналогично гидродинамическому
вводу пробы при давлении 1000 мбар.
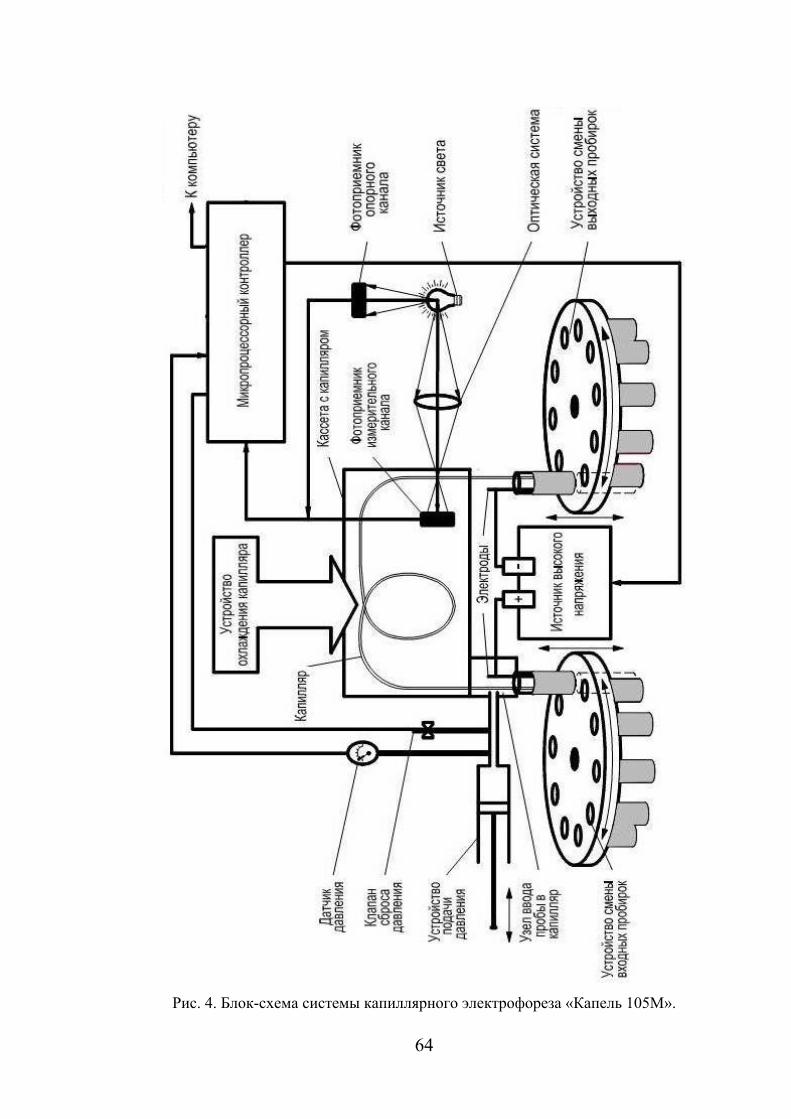
64
Рис. 4. Блок-схема системы капиллярного электрофореза «Капель 105М».

65
Операция «Анализ» осуществляется, когда на входном и выходном
концах капилляра подняты пробирки с ведущим электролитом. На
платиновые электроды, погружённые в раствор, подаётся высокое
напряжение от источника. Электрод выходного узла заземлён, а на электрод
входного узла может быть подано положительное или отрицательное
напряжение путём замены высоковольтного блока. Источник напряжения
имеет блокировки от включения при отсутствии пробирок во входном и
выходном узлах, при попытке открыть крышку кассетного отсека, а также
защиту от перегрузки по току (более 200 мкА).
Под действием электрического поля ионы пробы движутся в капилляре
с определёнными скоростями и образуют отдельные зоны. Эти зоны
детектируются в виде пиков на электрофореграмме. В системе капиллярного
электрофореза «Капель 105М» используется фотометрический детектор.
Источником излучения является дейтериевая лампа. С помощью
диспергирующего элемента – дифракционной решетки – из сплошного
спектра выделяется излучение заданной длины волны в диапазоне 190 – 400
нм. Фотометр проводит непрерывные измерения сигналов в опорном и
измерительном каналах. Сигналы детекторов преобразуются в цифровую
форму и обрабатываются в центральном процессоре. Результат обработки в
виде графика зависимости оптической плотности (в тысячных долях
оптической плотности, mAU) от времени выводится на экран компьютера.
Для отвода тепла, выделяющего в процессе анализа при прохождении
электрического тока, используется система принудительного жидкостного
охлаждения капилляра. Она состоит из резервной ёмкости для теплоносителя
(деионизованная вода), двигателя с центробежным насосом, фильтра,
теплообменника на основе элемента Пельтье, датчиков температуры и
герметичной рубашки вокруг капилляра. При включении насос начинает
прокачивать теплоноситель по замкнутому контуру, а система управлении
поддерживает заданную температуру с точностью до ±0,5 °С.

66
Лабораторная работа. Определение катионного и анионного
состава природных вод
Для определения катионов щелочных и щелочноземельных металлов в
водных объектах используют источник напряжения положительной
полярности. При этом детектор находится вблизи катода (рис. 5). Ведущий
электролит готовят на основе винной кислоты, анионы которой обладают
малой подвижностью и, следовательно, увеличивают сопротивление
электролита.
Рис. 5. Схема анализа катионов с положительным высоковольтным блоком.
Чтобы зарегистрировать пики компонентов, слабо поглощающих свет,
применяют косвенное детектирование: в состав ведущего электролита вводят
бензимидазол, поглощающий в УФ-области. При разделении катионы пробы
эквивалентно замещают в растворе катион бензимидазолия, что приводит к
снижению оптической плотности в зоне каждого катионного компонента. На
электрофореграмме при этом возникают отрицательные пики. Для удобства
обработки электрофореграмму переворачивают (опция «Инвертировать» в
ПО «Эльфоран»).
При электрофорезе катионы регистрируются в последовательности,
которая определяется их электрофоретической подвижностью. Подвижности
катионов аммония и калия одинаковы, поэтому они выходят одним общим
пиком. Для разделения аммония и калия в состав ведущего электролита
вводят 18-краун-6, являющийся макроциклом с гидрофильной внутренней
полостью, размер которой очень близок размеру ионного радиуса иона калия.
В результате образуются комплексы включения по типу «гость»–«хозяин»,
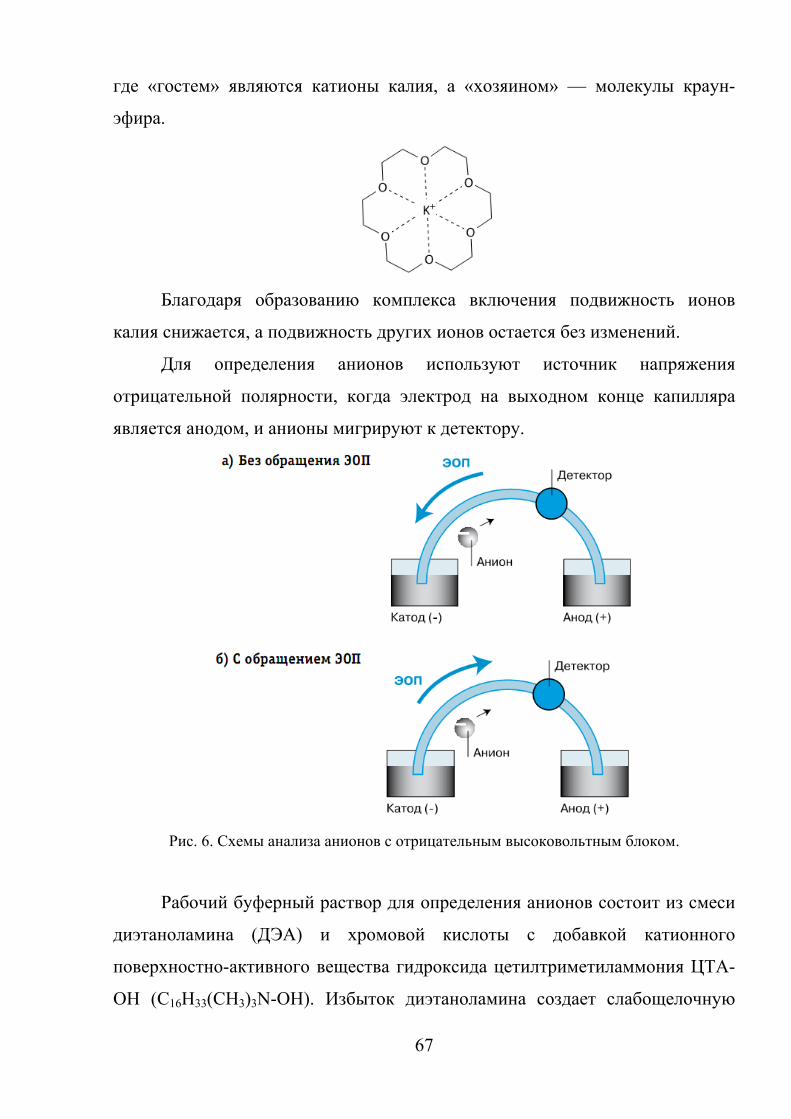
67
где «гостем» являются катионы калия, а «хозяином» — молекулы краун-
эфира.
Благодаря образованию комплекса включения подвижность ионов
калия снижается, а подвижность других ионов остается без изменений.
Для определения анионов используют источник напряжения
отрицательной полярности, когда электрод на выходном конце капилляра
является анодом, и анионы мигрируют к детектору.
Рис. 6. Схемы анализа анионов с отрицательным высоковольтным блоком.
Рабочий буферный раствор для определения анионов состоит из смеси
диэтаноламина (ДЭА) и хромовой кислоты с добавкой катионного
поверхностно-активного вещества гидроксида цетилтриметиламмония ЦТА-
ОН (C16H33(CH3)3N-OH). Избыток диэтаноламина создает слабощелочную

68
среду (рН~9), анион CrO42– обеспечивает необходимое светопоглощение, а
катион ЦТА+, сорбируясь на отрицательно заряженной поверхности
кварцевого капилляра, перезаряжает поверхность на положительную, чем
достигается изменение направления ЭОП (рис. 6).
Приборы и реактивы
1. Система капиллярного электрофореза «Капель 105М» с
источниками высокого напряжения положительной и отрицательной
полярности.
2. Гидроксид натрия, 0,5 моль/дм3 раствор.
3. Соляная кислота, 1 моль/дм3 раствор.
4. Оксид хрома (VI), 0,05 моль/дм3 раствор.
5. Диэтаноламин, 0,10 моль/дм3 раствор.
6. Гидроксид гексадецилтриметиламмония, 0,01 моль/дм3 раствор.
7. Бензимидазол, 0,04 моль/дм3 раствор.
8. Винная кислота, 0,02 моль/дм3 раствор.
9. 18-Краун-6, 0,01 моль/дм3 раствор.
10. Государственные стандартные образцы состава растворов
фосфат-анионов (0,5 мг/см3), хлорид-, нитрат-, фторид-анионов, катионов
калия, аммония, натрия, лития, магния, кальция, стронция, бария (1 мг/см3).
Вспомогательные устройства и материалы
1. Центрифуга.
2. Пробирки одноразовые типа Эппендорф вместимостью 1,5 см3.
3. Бумага индикаторная универсальная.
4. Шприц медицинский одноразовый типа «Луер».
5. Фильтры целлюлозно-ацетатные, размер пор 0,45 мкм, диаметр
25 мм.
6. Насадка для фильтра.
Выполнение работы
1. Приготовление хроматного ведущего электролита для определения
анионов.

69
В чистый сухой химический стакан помещают 0,7 см3 раствора оксида
хрома (VI), 1,0 см3 раствора ДЭА и 2,3 см3 деионизованной воды. Тщательно
перемешивают, а затем добавляют 1,0 см3 раствора ЦТА-ОН. Сразу же после
смешения раствор фильтруют через целлюлозно-ацетатный фильтр в
пластиковую посуду с закрывающейся крышкой (виал). Запрещается
изменять порядок добавления реагентов!
2. Приготовление ведущего электролита на основе бензимидазола для
определения катионов.
В чистый сухой химический стакан помещают 5,0 см3 раствора
бензимидазола, 2,0 см3 раствора винной кислоты, 2,0 см3 раствора 18-крауна-
6 и 1,0 см3 деионизованной воды. Сразу после смешения раствор фильтруют
через целлюлозно-ацетатный фильтр в сухой сосуд с завинчивающейся
крышкой.
3. Приготовление градуировочных растворов для определения анионов.
Смесь 1. В мерную колбу вместимостью 25,0 см3 помещают по 1,25
см3 растворов ГСО состава раствора хлорид- и нитрат-анионов, 2,5 см3 ГСО
состава раствора фосфат-ионов и 0,625 см3 раствора ГСО состава раствора
фторид-ионов, доводят до метки деионизованной водой и перемешивают.
Смесь 2. В чистый сухой сосуд помещают 2,0 см3 смеси 1 и 3,0 см3
деионизованной воды, раствор тщательно перемешивают.
Смесь 3. В чистый сухой сосуд помещают 1,0 см3 смеси 2 и 3,0 см3
деионизованной воды, раствор тщательно перемешивают.
Составы смесей приведены в табл. 2.
Таблица 2– Состав градуировочных смесей для определения анионов

70
Концентрация, мг/дм3 Анионы
Смесь 1 Смесь 2 Смесь 3
Cl- 50 20 5,0
NO3- 50 20 5,0
PO43- 50 20 5,0
F- 25 10 2,5
Смесь 5. В мерную колбу вместимостью 25,0 см3 помещают 2,5 см3
смеси 4, доводят до метки деионизованной водой и тщательно
перемешивают.
Смесь 6. В мерную колбу вместимостью 25,0 см3 помещают 2,5 см3
смеси 5, доводят до метки деионизованной водой и тщательно
перемешивают.
Составы смесей приведены в табл. 3.
Таблица 3
Состав градуировочных смесей для определения катионов
Концентрация, мг/дм3 Катионы
Смесь 4 Смесь 5 Смесь 6
NH4+ 50 5,0 0,5
K+ 50 5,0 0,5
Na+ 50 5,0 0,5
Li+ 5,0 0,5 0,05
Mg2+ 25 2,5 0,25
Sr2+ 50 5,0 0,5
Ba2+ 5,0 0,5 0,05
Ca2+ 50 5,0 0,5
5. Отбор пробы воды.

71
Пробу воды отбирают в пластиковую посуду с завинчивающейся
крышкой, дважды споласкивая её исследуемой водой. Объём отбираемой
пробы должен быть не менее 100 см3, проанализировать её необходимо в
течение суток.
6. Предварительные испытания.
Пробу анализируемой воды фильтруют через целлюлозно-ацетатный
фильтр в виал, отбрасывая первые порции фильтрата. Измеряют рН
фильтрата с помощью универсальной индикаторной бумаги. Если рН пробы
лежит в интервале 5 – 9, то ее можно анализировать непосредственно. Перед
анализом пробу помещают в пробирку Эппендорфа и центрифугируют на
скорости 6000 оборотов в минуту в течение 5 минут.
7. Подготовка прибора к работе.
• Перед работой капилляр промывают кислотой, водой, щелочью и
снова водой по 3 минуты, а затем раствором ведущего электролита 10 минут.
• Между анализами капилляр промывают ведущим электролитом в
течение 3 минут.
• После завершения работы капилляр промывают
дистиллированной водой в течение 10 минут. Концы капилляра оставляют
погруженными в воду.
8. Выполнение измерений и обработка результатов.
Разделение проводят в кварцевом капилляре внутренний диаметром
75 мкм, полной длиной 60 см, эффективной длиной 50 см.
Электрофоретическое определение анионов проводят при следующих
параметрах: ввод пробы гидродинамический при давлении 30 мбар в течение
10 с, напряжение –17 кВ, длина волны детектирования 374 нм, температура
20 °С, время анализа 7 минут. Порядок выхода пиков: хлорид, нитрат,
фторид, фосфат.
Для определения катионов задают следующие параметры анализа: ввод
пробы гидродинамический, давление 30 мбар, время ввода 5 с. Длина волны
детектирования 267 нм, напряжение +13 кВ, температура 20 °С, время

72
анализа 13 минут. Порядок выхода пиков: аммоний, калий, натрий, литий,
магний, стронций, барий, кальций.
С помощью программного обеспечения «Эльфоран» проводят
обработку электрофореграмм: определяют времена выхода пиков и их
площади, задают названия компонентов и их концентрации в
градуировочных растворах. Затем в той же программе строят
градуировочные графики в координатах «площадь пика – концентрация
иона». Размечают электрофореграммы исследуемых проб воды.
Качественный и количественный анализ проводится программой
автоматически на основании построенных градуировочных графиков.
ЛИТЕРАТУРА
1. ОТТО М. Современные методы аналитической химии (в 2-х
томах). М., Техносфера. 2003
2. ТЕРЕК Т. и др. Эмиссионный спектральный анализ. М., "МИР",
1982, (в 2-х томах).
3. ТРОФИМОВА Т.И. Оптика и атомная физика. М., Высшая
школа, 1999.
4. Аналитическая химия. Проблемы и подходы. (в 2-х томах). Под
ред. Р.Кельнера. М., «Мир» «АСТ», 2004.
5. Основы аналитической химии. Практическое руководство: Учеб.
Пособие для вузов/ В.И.Фадеева, Т.Н.Шеховцова, В.М.Иванов и др. Под ред.
Ю.А.Золотова. – М.: Высш. Шк., 2001. – 463 с.
6. Кристиан Г. Аналитическая химия. В двух томах. М.: Бином,
2009
7. Аналитическая химия. В 3 т. Т. 1. Методы идентификации и
определения веществ: Под. Ред. Л.Н.Москвина. – М.: Издательский центр
«Академия». 2008. 576 с.
8. Васильев В.П., Морозова Р.П., Кочергина Л.А. Аналитическая
химия. Лабораторный практикум: учебное пособие для вузов. М.: Дрофа.
2006. – 414 с.