Embed Size (px)
Citation preview

Hochschule für Technik und Wirtschaft Dresden
Fakultät Landbau/Umwelt/Chemie
Studiengang Chemieingenieurwesen
Masterarbeit
Untersuchungen zum Einfluss von Isosaccharinsäure auf die
U(VI)- Rückhaltung an Ca-Bentonit unter alkalischen Bedingungen
zur
Erlangung des akademischen Grades
Master of Science (M.Sc.)
eingereicht von: Paul Dullies
geb. am: 01.01.1992 in Gera
Betreuer: Prof. Dr. rer. nat. Jörg Feller (HTW Dresden)
Herr Thimo Philipp (HZDR)
Herr Hannes Brinkmann (HZDR)
Leitung: Prof. Dr. Vinzenz Brendler (HZDR)
Tag der Ausgabe: 01.06.2018
Tag der Einreichung: 30.11.2018

Inhaltsverzeichnis
I
Inhaltsverzeichnis
1 Motivation und Zielstellung .................................................................................. 1
2 Theoretische Grundlagen .................................................................................... 2
2.1 Endlagersituation in Deutschland .................................................................. 2
2.2 Zementkorrosion unter Einfluss von Grundwasser ........................................ 7
2.3 Organische Verbindungen im Endlager ......................................................... 8
2.4 Chemie des Urans ....................................................................................... 13
2.5 U(VI)-ISA-Komplexierung ............................................................................ 16
2.6 U(VI)-Sorption an Ca-Bentonit ..................................................................... 17
2.7 Sorption von U(VI) und anderen RN unter Einfluss von ISA ........................ 19
3 Material und Methoden ...................................................................................... 23
3.1 Material ........................................................................................................ 23
3.1.1 Verbrauchschemikalien ......................................................................... 23
3.1.2 Ca-Bentonit ........................................................................................... 23
3.1.3 Isosaccharinsäure ................................................................................. 25
3.1.4 Uran ...................................................................................................... 26
3.2 Methoden .................................................................................................... 27
3.2.1 pH-Wert Einstellung und Äquilibrierung ................................................ 27
3.2.2 Inductively Coupled Plasma - Mass Spectrometry (ICP-MS) ................ 28
3.2.3 Total Carbon (TC), Total Inorganic Carbon (TIC),
Total Organic Carbon (TOC) ............................................................................. 28
3.2.4 Laugungsverhalten von Ca-Bentonit ..................................................... 29
3.2.5 Batch-Sorptions-Experimente ............................................................... 30
3.2.6 Zeitaufgelöste Laserinduzierte Fluoreszenzspektroskopie (TRLFS) ..... 32
4 Ergebnisse und Diskussion ............................................................................... 37
4.1 Laugung von Ca-Bentonit ............................................................................ 37
4.2 Sorption von ISA an Ca-Bentonit ................................................................. 41
4.3 Einfluss von ISA auf U(VI)-Sorption an Ca-Bentonit .................................... 42
4.4 Löslichkeit von Uran .................................................................................... 45
4.5 Aquatische U(VI) Speziation ........................................................................ 46
5 Zusammenfassung und Ausblick ....................................................................... 56
6 Eidesstaatliche Erklärung über die Eigenständige Erstellung der Arbeit ........... 58
7 Danksagung ...................................................................................................... 59

Abbildungsverzeichnis
II
Abbildungsverzeichnis
Abbildung 1: Prognose für die anfallende Menge an radioaktiven Abfall in
Deutschland bis 2080 Bundesamt für Umweltschutz und
Reaktorsicherheit [3] Grafik Abfallmengen je Kategorie nach
Appunn [4] ......................................................................................... 3
Abbildung 2: Schematische Darstellung eines Einlagerungsbohrloches
Jobmann et al. [2] .............................................................................. 4
Abbildung 3: Bestandteile eines verbrauchten Brennelementes aus einem
durchschnittlichen Leichtwasserreaktor mit einem Abbrand von
33 MWd/kg (HZDR) ........................................................................... 5
Abbildung 4: (a) Anordnung des oktaedrisch geschichteten Netzwerkes unter
Bildung von Sechsringen. (b) Struktur eines 2:1 Schichtsilikates. Die
negativ geladenen Einheiten stapeln sich mit den zwischen den
Schichten gelagerten Ionen. Es kommt zum Ladungsausgleich.
Modifiziert nach Jasmund, K. und Lagaly [7] ..................................... 6
Abbildung 5: Schema der pH-Puffer-Stadien der Zementzersetzung in Endlagern
für LILW (CSH – Calcium-Silikat-Hydrat) Abrahamsen et al. [12] ..... 7
Abbildung 6: Schnitt-Darstellung eines 200 L-Fasses mit komprimierten LILW -
Beispiel der Endlagerung von organischen Abfällen in der
Tschechischen Republik Abrahamsen et al. [12] .............................. 9
Abbildung 7: Peeling-Reaktion von Cellulose Shaw et al. [20] ............................. 10
Abbildung 8: Molekülstrukturen und Umwandlung der ISL-, HISA- und ISA-Form
von Isosaccharinsäure nach H. Brinkmann (HZDR) (Strukturen
optimiert von M. Patzschke) ............................................................ 11
Abbildung 9: Pourbaix-Diagram der komplexen Redoxchemie von Uran, basierend
auf der OECD-NEA-Datenbank Takeno et al. [31] .......................... 14
Abbildung 10: Modellierung (MINTEQ 3.1) der Uranspezies in Abhängigikeit vom pH-
Wert ([U] = 4,2*10-7 M (100 µg/L), I = 0, T = 25 °C) Zhang et al. [32] ..
....................................................................................................... 15
Abbildung 11: Uranyl-Sorption in den Zwischenschichten und an den Kanten von
Montmorillonit Maher et al. [37] ....................................................... 17

Abbildungsverzeichnis
III
Abbildung 12: U(VI)-Sorption an Montmorillonit in Abwesenheit von CO2 (Dreiecke)
und mit atmosphärischem pCO2 (Rauten)
Marques Fernandes et al. [45] ........................................................ 18
Abbildung 13: allgemeines Jablonski-Diagramm für Fluoreszenz und
Phosphoreszenz.............................................................................. 33
Abbildung 14: Relative Energieniveaus des Uranyl-Valenz-Orbitals. 3σu ist HOMO
und 1Φu LUMO Denning [60] ........................................................... 34
Abbildung 15: relativer Anteil der Ca-Bentonit Bestandteile nach dem
Laugungsversuch gemessen an der Gesamteinsatzmenge an Ca-
Bentonit (10 g/L) in Abhängigkeit des pH-Wertes............................ 37
Abbildung 16: Ermittelter organischer Kohlenstoff (TOC) aus Ca-Bentonit-Laugung
in Abhängigkeit vom pH-Wert, (*) Bengtsson et al. [51] ................... 39
Abbildung 17: Ermittelter gesamter (TC) und anorganischer Kohlenstoff (TIC) aus
Ca-Bentonit-Laugung in Abhängigkeit vom pH-Wert, (*) Literaturwerte
für TIC und TOC nach Bengtsson et al. [51] ................................... 40
Abbildung 18: Ermittelter organischer Kohlenstoff (TOC) im Überstand nach dem
Batch-Sorptions-Experiment mit ISA-Zugabe von 0,5 mmol/L in
Abhängigkeit vom pH-Wert mit Angabe des 100 %-Wertes der ISA-
Zugabe ............................................................................................ 41
Abbildung 19: U(VI)-Sorption (5*10-7 mol/L) an Ca-Bentonit (10 g/L) in 0,1 M NaCl in
Abwesenheit von ISA und bei den U-ISA-Verhältnissen 1: 1000,
1: 10.000 und 1: 100.000 ................................................................ 43
Abbildung 20: Vergleich der relative U(VI)-Konzentration in Bezug auf die
Einsatzmenge von 0,5 µmol/L Uran in 0,1 M NaCl bei pH 1-13, ohne
ISA (blau) und mit U(VI): ISA = 1: 1000 (orange) ............................ 46
Abbildung 21: Darstellung der Problematik des stark fluoreszierenden Hintergrundes
am Beispiel des Lumineszenzspektrums von pH 1 ......................... 47
Abbildung 22: Lumineszenzspektren von U(VI) (0,5 µmol/L) in 0,1 M NaCl bei
pH 1- 13 .......................................................................................... 48
Abbildung 23: Referenzspektren für UO22+ von H. Brinkmann (HZDR) .................. 49
Abbildung 24: Lumineszenzspektren zum binärem System U/ISA mit
U: ISA = 1: 1000 im Vergleich mit Uran ohne ISA (mit je
[U] = 0,5 µmol/L in 0,1 M NaCl bei pH 1-13) ................................... 51

Abbildungsverzeichnis
IV
Abbildung 25: Lumineszenzspektren des Überstandes aus dem Sorptionsversuch
Ca-Bentonit/ U/ ISA mit U: ISA = 1: 100.000 im Vergleich zur
aquatischen Speziation ohne ISA ([U] = 0,5 µmol/L in 0,1 M NaCl bei
pH 8-13) .......................................................................................... 54
Abbildung 26: pH-Abhängige Konzentrationen der wichtigsten Ca-Bentonit-
Bestandteile aus dem Laugungsversuch in der überstehenden Lösung
.................................................................................................... XIII
Abbildung 27: Ermittelter organischer Kohlenstoff (TOC) aus Ca-Bentonit-Laugung
in Abhängigkeit vom pH-Wert unter normalen atmosphärischen
Bedingungen (S. Shams (HZDR)) .................................................. XIII

Tabellenverzeichnis
V
Tabellenverzeichnis
Tabelle 1: Übersicht zu den prognostizierten relativen Anteilen der Bestandteile
organischer Abfälle in Endlagern verschiedener europäischer
Länder Abrahamsen et al. [12] .......................................................... 8
Tabelle 2: Zusammenfassung derzeitiger Kenntnisstand zur Radionuklid-ISA-
Komplexierung und deren Einfluss auf Wechselwirkungen mit
Mineraloberflächen .......................................................................... 22
Tabelle 3: Liste der verwendeten Verbrauchschemikalien ............................... 23
Tabelle 4: mineralogische Zusammensetzung von Ca-Bentonit (Calcigel®)
entsprechend der Produktinformationen von Clariant ..................... 23
Tabelle 5: Ergebnisse der RFA-Analyse von Ca-Bentonit (Calcigel®) Reich [50]
........................................................................................................ 24
Tabelle 6: Überblick über Batch-Sorptions-Experimente und die wichtigsten
Versuchsparameter ......................................................................... 30
Tabelle 7: verwendete Geräte- und Software-Parameter für die cryo-TRFLS . 35
Tabelle 8: Überblick über Experimente zur aquatischen Speziation und die
wichtigsten Versuchsparameter ...................................................... 36
Tabelle 9: Peakpositionen und zugeordnete U(VI)-Spezies mit Uran = 0,5 µmol/L
in 0,1 M NaCl bei pH 1- 13 .............................................................. 48
Tabelle 10: Referenzdaten für UO22+ aus cryo-TRLFS von H. Brinkmann (HZDR)
........................................................................................................ 49
Tabelle 11: Referenzdaten für [UO2(ISA)2] und [UO2(ISA)]+ aus cryo-TRLFS von
H. Brinkmann (HZDR) ..................................................................... 53
Tabelle 12: Peakpositionen und Speziationen vom binären System
U : ISA = 1: 1000 mit Uran = 0,5 µmol/L in 0,1 M NaCl bei pH 1-13 ...
........................................................................................................ 53
Tabelle 13: Peakpositionen und Speziationen vom Sorptionsversuch mit Ca-
Bentonit und U: ISA = 1: 100.000 mit Uran = 0,5 µmol/L in 0,1 M NaCl
bei pH 1-13 ...................................................................................... 55

Abkürzungsverzeichnis
VI
Abkürzungsverzeichnis
BGR Bundesanstalt für Geowissenschaften und Rohstoffe
BMUB Bundesamt für Umweltschutz und Reaktorsicherheit
CDP Cellulose-Degradation-Products (Cellulose-Abbauprodukte)
GLU Glukonsäure
GRaZ BMWi-Projekt “Geochemische Radionuklidrückhaltung an
Zementalterationsphasen“
HLW High Level Waste
HOMO highest occupied mulecular orbital - höchstes besetztes Molekülorbital
ICP-MS Inductively Coupled Plasma - Mass Spectrometry
ISA Isosaccharinsäure
LILW Low and Intermediate Level Waste
LUMO lowest unoccupied molecular orbital – niedrigstes unbesetztes
Molekülorbital
MIND Projekt “Microbiology in Nuclear waste Disposal“
OC Organika
PP Polypropylen
RN Radionuklid
TC Total Carbon (Gesamter gebundener Kohlenstoff: TC = TOC + TIC)
TIC Total Inorganic Carbon (gesamter anorganisch gebundener Kohlenstoff)
TOC Total Organic Carbon (gesamter organisch gebundener Kohlenstoff)
TRLFS time-resolved laser-induced fluorescence spectroscopy
XRD Röntgendiffraktometrie

Motivation und Zielstellung
1
1 Motivation und Zielstellung
Radioaktive Abfälle aus Kernkraftwerken, militärischen, medizinischen und
Forschungseinrichtungen stellen aufgrund ihrer Radio- und Chemotoxizität eine große
Gefahr für die Gesundheit und Unversehrtheit von Mensch und Umwelt dar. Deshalb
ist es erforderlich eine sichere und effektive Langzeitverwahrung für diese Art von
Abfällen zu garantieren. Unter Wissenschaftlern und Politikern herrscht international
Konsens darüber, dass radioaktive Abfälle in tiefen geologischen Formationen
gelagert werden sollen. Laut Bundesanstalt für Geowissenschaften und Rohstoffe
(BGR) kommen Steinsalz, Ton und kristalline Gesteine als Wirtsgestein für nukleare
Endlager in Frage [1]. Tongesteine wie Ca-Bentonit sind zudem, unabhängig vom
Wirtsgestein, aufgrund ihrer Verwendung als Verfüllmaterial in Endlagerungssystemen
von entscheidender Bedeutung [2]. In Lagerstätten für radioaktive Abfälle herrschen
aufgrund von Zementzersetzung durch Interaktion mit Grundwasser stark alkalische
Bedingungen vor. Zement und Beton finden sich in Endlagern durch die Verwendung
als Konstruktionsmaterial oder Abfallmatrix. Im worst-case-Szenario eines
Grundwassereinbruchs im Endlager, sollten die meisten Radionuklide (RN) generell
schwer lösliche Hydroxide bilden. Da sich jedoch unter den im Endlager herrschenden
extremen Bedingungen (hoher pH-Wert, hohe Temperatur, Druck und Radiation) aus
organischen Abfall- bzw. Zementbestandteilen gut lösliche organische Liganden bzw.
Abbauprodukte wie Isosaccharinsäure (ISA) bilden bzw. herauslösen können, ist auch
die Bildung von mobileren RN-Spezies mit diesen Liganden möglich. Die
Polyhydroxycarbonsäure ISA ist dabei durch ihre starke Lewis-Basizität für die Bildung
von stabilen und gut löslichen RN-ISA-Komplexen verantwortlich.
Bisher wurden in den beiden Forschungsprojekten GRaZ und MIND ausschließlich die
Sorption von U(VI) an Ca-Bentonit im pH-Bereich 8-13, sowie die Komplexierung von
U(VI) in Gegenwart von ISA bei pH 1-4 untersucht. Aufbauend auf den
Untersuchungen dieser beiden binären Systeme soll diese Arbeit das ternäre System
U(VI)/ Ca-Bentonit/ ISA betrachten. Der Schwerpunkt liegt auf der Untersuchung des
Einflusses von ISA auf die Sorption von U(VI) an Bentonit unter endlagerrelevanten
Bedingungen (pH 8-13, anaerob). Batch-Sorptions-Experimente stehen hierbei im
Vordergrund um zu ermitteln ob und unter welchen Bedingungen ISA zu einer U(VI)-
Mobilisierung führen kann.

Theoretische Grundlagen
2
Dies wurde zuvor in dieser Form noch nicht über einen größeren alkalischen pH-
Bereich untersucht. Mittels zeitaufgelöster laser-induzierte Fluoreszenzspektroskopie
(TRLFS) sollen außerdem die für eine verringerte Sorption verantwortlichen
aquatischen U(VI)-ISA-Spezies identifiziert werden, um Erkenntnisse über die
zugrundeliegenden Rückhaltemechanismen in Anwesenheit von ISA auf der
molekularen Ebene zu gewinnen. Kenntnisse darüber sind für eine
Langzeitsicherheitsanalyse tiefengeologischer Endlager essentiell.
2 Theoretische Grundlagen
2.1 Endlagersituation in Deutschland
In Deutschland werden radioaktive Abfälle in Abfälle mit Wärmeabgabe (HLW) und
Abfälle mit vernachlässigbarer Wärmeabgabe (LILW) unterteilt (siehe Abbildung 1). Im
internationalen Terminus entsprechen die Abfälle mit Wärmeabgabe dem hoch-
radioaktivem Abfall (high level waste - HLW), zu denen beispielsweise verbrauchte
Brennelemente aus Kernkraftwerken zählen. Die Gruppe der Abfälle mit
vernachlässigbarer Wärmeabgabe hingegen werden international noch einmal in
leicht- und mittelradioaktive Abfälle (low and intermediate level waste - LILW) unterteilt
und sind in ihrer Zusammensetzung höchst vielseitig und komplex. In Deutschland
sollen bis 2080 etwa 600.000 m³ schwach-und mittelradioaktive Abfälle, vorrangig aus
dem Rückbau von ehemaligen Kernenergieanlagen eingelagert werden. Die Fraktion
der stark radioaktiven Abfälle fällt dabei mit 28.100 m³ bzw. etwa 5 % des Volumens
der schwach-und mittelradioaktiven Abfälle nicht so groß aus, ist aber für 99 % der
Gesamtaktivität verantwortlich [3].

Theoretische Grundlagen
3
Abbildung 1: Prognose für die anfallende Menge an radioaktiven Abfall in Deutschland bis 2080
Bundesamt für Umweltschutz und Reaktorsicherheit [3] Grafik Abfallmengen je
Kategorie nach Appunn [4]
Da die Endlagersituation in Deutschland noch nicht endgültig geklärt ist, ergeben sich
für die Problemstellung dieser Arbeit zwei Anwendungsfälle, die für die Aussagekraft
der Ergebnisse und deren Bedeutung für künftige Untersuchungen essentiell sind. Die
Anwendungsfälle beziehen sich auf die beiden Arten, in die radioaktiver Abfall in
Deutschland eingeteilt wird. Für beide Anwendungsfälle gilt, dass die Entsorgung
entsprechend einem Multi-Barriere-Konzept erfolgt, bei dem die Radionuklide von
einer technischen (Abfallbehälter), einer geotechnischen (Bohrlochverschluss,
Verfüllmaterial bzw. Buffer) und letztendlich von einer geologischen Barriere
(Wirtsgestein) zurückgehalten werden sollen. In Abbildung 1 ist eben dieses Multi-
Barriere-Konzept beispielhaft für ein Einlagerungsbohrloch basierend auf dem
hypothetischen Endlagermodell NORD dargestellt [2].

Theoretische Grundlagen
4
Abbildung 2: Schematische Darstellung eines Einlagerungsbohrloches Jobmann et al. [2]
Der erste Anwendungsfall bezieht sich auf die Endlagerung von hochradioaktiven
Abfällen (HLW) bzw. wärmeerzeugende Abfälle. In Deutschland sind etwa 88,3 %
dieser Art von Abfällen auf die Außerdienststellung der Kernkraftwerke
zurückzuführen [5]. Dabei fallen vor allem verbrauchte Brennelemente an, welche je
nach Grad des Abbrandes neben Spaltprodukten und einigen anderen Bestandteilen
etwa 95,76 % Uran beinhalten. Dieses kann wie in Abbildung 3 dargestellt noch einmal
in 238U (94,48 %), 235U (0,86 %) und 236U (0,42 %) unterteilt werden.

Theoretische Grundlagen
5
Abbildung 3: Bestandteile eines verbrauchten Brennelementes aus einem durchschnittlichen
Leichtwasserreaktor mit einem Abbrand von 33 MWd/kg (HZDR)
Der zweite Anwendungsfall nimmt Bezug auf die Endlagerung von schwach- und.
mittel-radioaktiven Abfällen (LILW) bzw. Abfällen mit vernachlässigbarer
Wärmeabgabe. Allgemeinhin sind in dieser Abfallklasse große Volumina an
organischen Substanzen (z.B.: Cellulose, Kunststoffe, Holz und Papier) und geringe
RN-Konzentrationen zu erwarten. Die Situation würde sich jedoch verändern, wenn
die in der Urananreicherungsanlage Gronau (Westfalen) gelagerten 20.870,3 t
(Stand 31. März 2017) an abgereicherten UF6 mit in die Kalkulation einbezogen
werden [6]. Denn aufgrund seiner Aktivität würde das UF6 unter die Kategorie LILW
fallen. Dies würde bedeuten, dass neben den hohen Anteilen an organischen
Substanzen (z.B. ISA) im LILW-Endlager auch signifikante Mengen an Uran vorlägen.
U-23894,48 %
U-2350,86 %
U-2360,42 %
Spaltprodukte3,25 %
Pu0,93 %
Np, Am, Cm0,06 %
Andere Bestandteile4,24 %

Theoretische Grundlagen
6
Tonmineralogie, Montmorillonit und Bentonit
Dieser Abschnitt beschäftigt sich mit der Struktur und den Eigenschaften von
Tonmineralen. Dabei richtet sich der Fokus auf das Mineral Montmorillonit,
Hauptbestandteil von Bentonit. Allgemeinhin sind Tonminerale wasserführende
Aluminium-Silikate mit einer Schichtstruktur. Diese bestehen aus Einheiten von
[SiO4] -Tetraedern und [M (O, OH)6] -Oktaedern (M = Al3+, Fe3+, Fe2+ oder Mg2+). Der
Aufbau der Schichtstruktur ist in Abbildung 4 dargestellt, wobei noch zu erwähnen ist,
dass als Zwischenschicht-Kationen K+ oder Ca+ in Frage kommen. Montmorillonit ist
ein Smectit-Mineral, welches über eine hohe Quellfähigkeit und Bindefähigkeit verfügt.
Damit ist es ihnen möglich sowohl Flüssigkeiten als auch Kationen innerhalb ihres
Schichtsystems effektiv einzubinden [7]. Aufgrund der spezifischen Eigenschaften wie
Quellfähigkeit, Thixotropie, Plastizität, der nano-Poren-Struktur und der extrem
niedrigen Permeabilität, sind Tonminerale speziell für die Anwendung in Endlagern für
radioaktive Abfälle interessant. Denn auf Grund der großen spezifischen Oberfläche,
hohen Kation-Austausch-Fähigkeit und Möglichkeit der Oberflächenkomplexierung,
verfügen Tonminerale über eine ausgeprägte Sorptions-Kapazität in Hinblick auf
Schwermetalle [8]. In der Praxis kommen Tonminerale daher als Wirtsgestein für ein
Endlager und Materialien wie Ca-Bentonit als Verfüllstoff und damit als zusätzliche
geotechnische Barriere in Frage.
Abbildung 4: (a) Anordnung des oktaedrisch geschichteten Netzwerkes unter Bildung von
Sechsringen. (b) Struktur eines 2:1 Schichtsilikates. Die negativ geladenen Einheiten stapeln
sich mit den zwischen den Schichten gelagerten Ionen. Es kommt zum Ladungsausgleich.
Modifiziert nach Jasmund, K. und Lagaly [7]

Theoretische Grundlagen
7
2.2 Zementkorrosion unter Einfluss von Grundwasser
Zement ist aufgrund der Verwendung als Konstruktionsmaterial oder Abfallmatrix in
großen Mengen in einem Endlager zu erwarten. Daraus ergibt sich im worst case
Szenario eines Grundwassereinbruchs folgende Problematik. Tritt der Zement mit dem
Grundwasser in Kontakt, beginnt binnen weniger Wochen dessen Zersetzung [9].
Diese Zersetzung erstreckt sich über vier Phasen(siehe Abbildung 5). Die erste Phase
stellt die Hydratation dar und hat zur Folge, dass sich stark alkalische Porenwässer
bilden, welche von Alkali-Hydroxiden (NaOH, KOH) dominiert werden. Daraus ergeben
sich Bedingungen im Endlager mit pH- Werten >12,5 [9], [10] Da sich Grundwässer
vor allem in Tonhaltigen Schichten nur wenige Zentimeter pro Jahr bewegen [11], ist
davon auszugehen, dass diese stark alkalischen Bedingungen für einen langen
Zeitraum bestehen. In verschiedenen Publikationen wird davon ausgegangen, dass
pH- Werte >12 bis zu 10.000 bis 100.000 Jahre im Endlager vorliegen können [9], [10],
[12]. Der annähernd neutrale pH- Wert des eindringenden Grundwassers wird erst
nach mehr als 100.000 Jahren wieder erreicht (siehe Stufe IV in Abbildung 5) [9], [12].
Abbildung 5: Schema der pH-Puffer-Stadien der Zementzersetzung in Endlagern für LILW
(CSH – Calcium-Silikat-Hydrat) Abrahamsen et al. [12]
Time →
Theoretische Grundlagen
8
2.3 Organische Verbindungen im Endlager
In Endlagern für LILW fallen verschiedenste organische Verbindungen an, die sowohl
über den als Konstruktionsmaterial verwendeten Zement als auch über den Abfall
eingebracht werden. Über den Zement kommt es vorrangig zum Eintrag von
sogenannten "Superplastisizern", welche für den Ausbau der Endlager zur Anwendung
kommen, da diese die rheologischen Eigenschaften des Zements auf verschiedenste
Weise positiv beeinflussen [10], [13]. Bei der Zersetzung der Superplastisizer entsteht
eine Vielzahl an wasserlöslichen organischen Abbauprodukten, von denen
Glukonsäure (C6H12O7) (GLU) den Hauptbestandteil darstellt [13], [14]. Im Abfall selbst
finden sich wiederum organische Substanzen wie Kunststoffe (halogeniert/ nicht
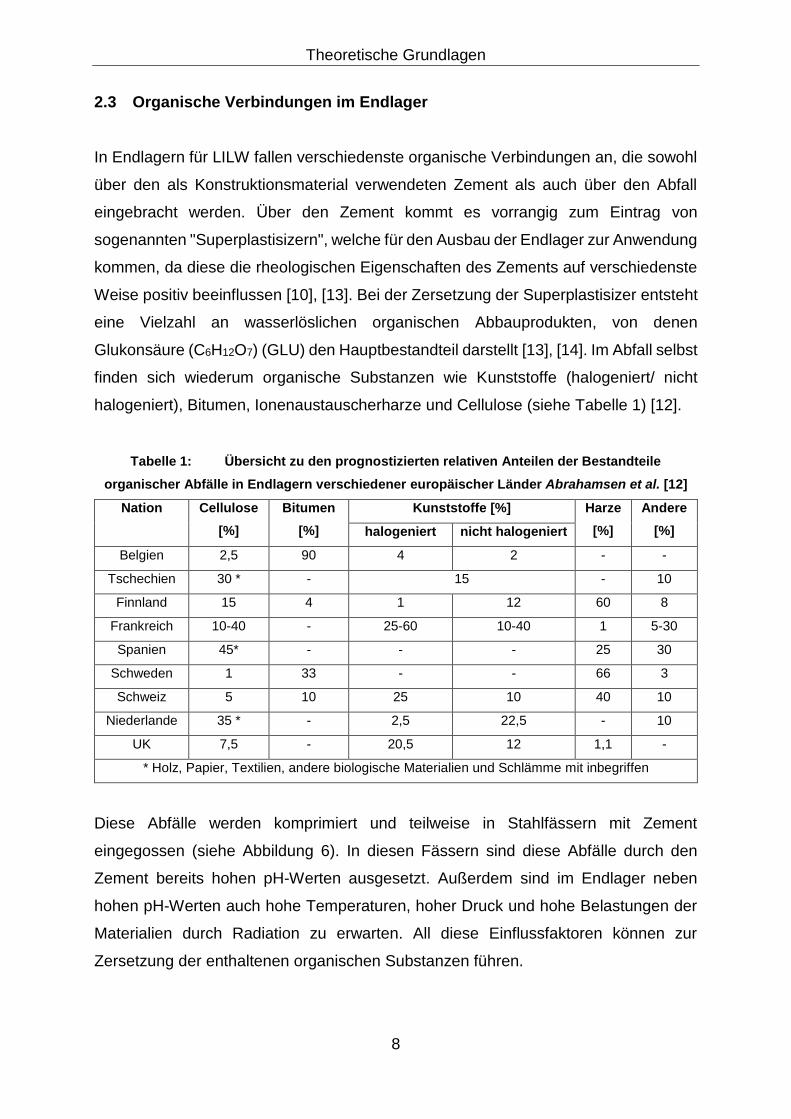
halogeniert), Bitumen, Ionenaustauscherharze und Cellulose (siehe Tabelle 1) [12].
Tabelle 1: Übersicht zu den prognostizierten relativen Anteilen der Bestandteile
organischer Abfälle in Endlagern verschiedener europäischer Länder Abrahamsen et al. [12]
Nation Cellulose
[%]
Bitumen
[%]
Kunststoffe [%] Harze
[%]
Andere
[%] halogeniert nicht halogeniert
Belgien 2,5 90 4 2 - -
Tschechien 30 * - 15 - 10
Finnland 15 4 1 12 60 8
Frankreich 10-40 - 25-60 10-40 1 5-30
Spanien 45* - - - 25 30
Schweden 1 33 - - 66 3
Schweiz 5 10 25 10 40 10
Niederlande 35 * - 2,5 22,5 - 10
UK 7,5 - 20,5 12 1,1 -
* Holz, Papier, Textilien, andere biologische Materialien und Schlämme mit inbegriffen
Diese Abfälle werden komprimiert und teilweise in Stahlfässern mit Zement
eingegossen (siehe Abbildung 6). In diesen Fässern sind diese Abfälle durch den
Zement bereits hohen pH-Werten ausgesetzt. Außerdem sind im Endlager neben
hohen pH-Werten auch hohe Temperaturen, hoher Druck und hohe Belastungen der
Materialien durch Radiation zu erwarten. All diese Einflussfaktoren können zur
Zersetzung der enthaltenen organischen Substanzen führen.

Theoretische Grundlagen
9
Abbildung 6: Schnitt-Darstellung eines 200 L-Fasses mit komprimierten LILW - Beispiel der
Endlagerung von organischen Abfällen in der Tschechischen Republik Abrahamsen et al. [12]
Die Abbauprodukte der organischen Abfälle sind nachgewiesenermaßen dafür
verantwortlich, dass diese durch Komplexierung die Sorption von RN verringern, die
Löslichkeit verbessen und somit deren Mobilität zu erhöhen. Ein wichtiger Vertreter
dieser Abfälle ist Cellulose, welche besonders labil gegenüber den harschen
Bedingungen im Endlager ist [14], [15]. Cellulose und cellulosehaltige Materialien wie
Holz, Papier und Baumwolle sind von großem Interesse, da diese bis zu 40 % des
organischen Abfalls ausmachen [12]. Hauptprodukt des alkalischen Cellulose- Abbaus
ist ISA, neben einer Vielzahl an kleineren aliphatischen Säuren wie Ameisensäure und
Essigsäure [12], [14]–[21].

Theoretische Grundlagen
10
Abbau von Cellulose unter alkalischen Bedingungen
Wie bereits erwähnt, ist ISA das Hauptprodukt des Abbaus von Cellulose, der unter
den stark alkalischen Bedingungen (pH 13,3) abläuft, welche durch das
Zementporenwasser bedingt werden [15]. Unter alkalischen Bedingungen durchläuft
Cellulose einen von den Enden der Cellulose-Kette ausgehende Abbau. Dabei werden
nach und nach einzelne Glukose-Einheiten vom reduzierenden Ende des Cellulose-
Moleküls abgespalten, woraufhin wieder ein neues reduzierendes Ende entsteht, was
eine fortlaufende Reaktion erlaubt. Diese Reaktion wird auch Peeling-Reaktion
genannt (siehe Abbildung 7) [20]. Bei den Untersuchungen zur Identifikation der
Cellulose-Abbauprodukte (CDP) wurde festgestellt, dass die größte Fraktion, mit 70-
80 % des gesamten DOCs, auf die Bildung von α-ISA und β-ISA zurückgeht [15]. Diese
Anteile variieren in Abhängigkeit der untersuchten Cellulosequelle (Wischtücher,
Baumwolle, Recyclingpapier). Des Weiteren entstehen kleinere aliphatische Säuren
wie z. B. Ameisen-, Essig- und Milchsäure mit einem Summenanteil von etwa 10 %
[15].
Abbildung 7: Peeling-Reaktion von Cellulose Shaw et al. [20]

Theoretische Grundlagen
11
Allgemeine Charakterisierung ISA
Isosaccharinsäure (C6H12O6) oder (2S, 4S) - 2,4,5-Trihydroxo-2-hydroxymethyl-
pentansäure (IUPAC) wird zu den Polyhydroxycarbonsäuren (Zuckersäuren) gezählt,
besitzt zwei primäre, eine sekundäre und eine tertiäre Hydroxylgruppe (siehe
Abbildung 8). In Abbildung 8 sind neben der α-D-Isosaccharino-1,4-lacton- (ISL),
Isosaccharinsäure-Form (HISA) und dem Isosacchinat (ISA) auch die pKa-Werte der
Carboxylgruppe dargestellt [22]–[24]. Die Gamma-Lactonform der ISA ist nur bei
niedrigen pH-Werten zu erwarten und die Deprotonierung der OH-Gruppen beginnt
erst bei hohen pH-Werten [23]. Die Wechselwirkungen von ISA mit RN beruhen auf
der starken Lewis-Basizität der ISA.
Abbildung 8: Molekülstrukturen und Umwandlung der ISL-, HISA- und ISA-Form von
Isosaccharinsäure nach H. Brinkmann (HZDR) (Strukturen optimiert von M. Patzschke)

Theoretische Grundlagen
12
Sorption von ISA an Tonmineralen und Zementphasen
Untersuchungen von Van Loon et al. [10] haben ergeben, dass ISA sehr schnell an
Zement-Phasen sorbiert, wobei ein Gleichgewicht bereits nach einem Tag erreicht ist.
Der Sorptionsversuch wurde bei pH 13,3 durchgeführt, was zur Folge hat, dass die
Carboxylgruppe der ISA deprotoniert vorliegt und das Molekül somit negativ geladen
ist. Zementoberflächen sind ebenfalls negativ geladen, allerdings ist die
Zementoberfläche elektrostatisch gesehen nicht gleichmäßig, weshalb es trotz der
großen Abstoßung mit dem ISA-Molekül lokal zu Oberflächen-Interaktionen und
letztendlich doch zur Sorption kommt [25]. Dieser Mechanismus konnte bisher nicht
allumfassend aufgeklärt werden, es wurde aber festgestellt, dass die Sorption
irreversibel ist. In weiteren Untersuchungen von Van Loon et al. [25] wurde beobachtet,
dass ISA an Materialien wie Feldspat praktisch nicht sorbiert. Zur Sorption von ISA
speziell an Ca-Bentonit liegen bisher keinerlei Untersuchungen vor.

Theoretische Grundlagen
13
2.4 Chemie des Urans
Allgemeine Charakterisierung
Uran ist ein chemisches Element mit der Ordnungszahl 92, steht im Periodensystem
der Elemente in der 7. Periode, gehört demzufolge zu den Actiniden und hat die
Elektronenkonfiguration [Rn]5f36d17s2 [26]. Uran ist ein Schwermetall und hat wie alle
Actiniden ausschließlich radioaktive Isotope, von denen drei natürlich vorkommen.
Diese drei Isotope sind 238U (99,274 %), 235U (0,720 %) und 234U (0,005 %), wobei
sowohl 238U als auch 235U primordiale Nuklide darstellen und 234U ein Tochternuklid
von 238U ist. 238U ist das Mutternuklid der Radium-Zerfallsreihe und hat eine
Halbwertszeit von 4,47x109 Jahren, wobei es unter Abgabe eines Alphapartikels zu
234Th zerfällt. 235U hat eine Halbwertszeit von 7,04x108 Jahren, ist Mutternuklid der
Actinium-Zerfallsreihe und durchläuft einen Alpha-Zerfall zu 231Th. 234U zerfällt unter
Abgabe von Alphastrahlung zu 230Th und hat eine Halbwertszeit von 2,46x105 Jahren
[27]. Uran ist ein radio-und chemotoxisches Schwermetall. Die Chemotoxizität des
natürlichen Urans wiegt jedoch wesentlich schwerer als die von ihm ausgehende
radiologische Gefahr. Dies ist neben der geringen spezifischen Aktivität auf die geringe
Reichweite der Alpha-Partikel zurückzuführen, welche erst bei Inkorporation zu einer
ernstzunehmenden Gefahr werden. Die Gefährlichkeit beruht vor allem auf der
Schädigung innerer Organe.
Wie die meisten frühen Actinide verfügt Uran über eine komplexe Redoxchemie. Uran
kommt, abhängig vom Redoxpotential und pH-Wert, in den Oxidationsstufen +3 bis +6
vor [28], [29] (siehe Abbildung 9). Bei höheren Oxidationsstufen (+4, +6) bildet Uran
die sogenannten Uranyl-Ionen (UO2+ und UO2
2+) aus, welche über einen
Bindungswinkel von 180° (O=U=O) verfügen. Die wichtigsten Oxidationsstufen des
Urans sind +4 und +6, da U(III) unter den meisten Umständen instabil ist und somit zu
U(IV) und U(V) oxidiert wird. U(V) wiederum disproportioniert zu U(IV) und U(VI). U(IV)
ist unter reduzierenden Bedingungen stabil und aufgrund seiner schlechten Löslichkeit
weitestgehend immobil. U(VI) ist die bevorzugte Oxidationsstufe des Urans in Ozean-
und Grundwasser, da es unter oxidierenden Bedingungen stabil ist. U(VI) ist gut löslich
und somit auch mobiler als U(IV) [30].

Theoretische Grundlagen
14
Abbildung 9: Pourbaix-Diagram der komplexen Redoxchemie von Uran, basierend auf der
OECD-NEA-Datenbank Takeno et al. [31]
Die Mobilität von Uran ist neben der Oxidationsstufe auch von der Speziation
abhängig. Im Fall eines Wassereinbruches in einem Endlager, kann Uran mobilisiert
werden, indem es lösliche Spezies mit den umgebenden Ionen bildet. Die häufigste
Koordinationszahl ist sechs [29]. Außerdem ordnen sich die Liganden aufgrund des
Bindungswinkels (180°) in der äquatorialen Ebene, senkrecht zur Achse der
yl- Bindung an. Uranyl-Ionen sind harte Lewissäuren und bilden bevorzugt Komplexe
mit harten Liganden wie Carbonaten, Hydroxiden, aber auch Halogenen, Phosphaten
und Sulfaten.

Theoretische Grundlagen
15
U(VI)-Speziation in Abhängigkeit vom pH-Wert
Im sauren, neutralen und leicht-alkalischen pH-Bereich ist die aquatische Speziation
von U(VI) bereits gut bekannt. Abhängig vom pH-Wert wechseln die U(VI)- Spezies
von positiven zu negativen Ladungen. In Abbildung 10 ist eine Modellierung von
Zhang et al. [32] zu den zu erwartenden Spezies dargestellt. Diese zeigt, dass bei
niedrigen pH- Werten ausschließlich das positiv geladene Aquo-Ion UO22+ vorliegt. Bei
pH >4 beginnt die Hydrolyse mit geringen (<29 %), aber zunehmenden Anteilen von
UO2(OH)+ bis pH 5. Bei pH 6 stellt UO2(OH)+ die vorherrschende Spezies (≥ 62 %),
während der Anteil der Spezies (UO2)3(OH)5+ ansteigt. Im pH-Bereich 6-8 ist vor allem
eine Mischung der Spezies UO2(OH)+ und (UO2)3(OH)5+ zu erwarten. Die negativ
geladenen Spezies UO2(OH)3- und UO2(OH)4
2- herrschen ab einem pH-Wert von ≥ 9
vor.
Abbildung 10: Modellierung (MINTEQ 3.1) der Uranspezies in Abhängigikeit vom pH-Wert
([U] = 4,2*10-7 M (100 µg/L), I = 0, T = 25 °C) Zhang et al. [32]

Theoretische Grundlagen
16
2.5 U(VI)-ISA-Komplexierung
Die Anzahl von experimentellen Studien zur Interaktion von ISA mit RN ist sehr gering,
dass gilt im Besonderen für U(VI). Die löslichkeitsverbessernden Wechselwirkungen
von ISA (0,04 mol/L) mit U(VI) (10-6 mol/L) wurden von Rai et al. [19] experimentell
beim Versuch der Dekontamination von Stahloberflächen beobachtet. Des Weiteren
experimentierte Warwick et al. [33] mit verschiedenen Methoden, wie etwa der
konduktometrischen Titrationen oder der Schubert Methode (Ionenaustausch-
Methode). Ziel war die Ermittlung von U-ISA-Komplexen und deren
Stabilitätskonstanten. Die Untersuchungen beschränkten sich auf die pH-Werte 7 und
13,5. Die verwendeten U- ISA- Verhältnisse erstreckten sich von 1: 100 bis
1: 10.000.000. Ergebnisse waren die Komplexstabilitätskonstanten der pH-
abhängigen U-ISA-Komplexe [UO2ISA2] (pH 7) und UO2ISA(OH)4 (pH 13,5).
Außerdem wurde ein Löslichkeits-Verstärkungsfaktor für U(VI) gegenüber ISA
ermittelt, welcher sich im pH- Bereich 9- 13,5 als vernachlässigbar gering
herausstellte.
Weitere Untersuchungen beschäftigten sich mit den Wechselwirkungen zwischen RN
und GLU. Dabei untersuchte Colàs et al. [18] den Einfluss von GLU auf die aquatische
Speziation von Th(IV), wobei Th-GLU-Verhältnisse von 1: 100 bis 1: 1000 im
pH- Bereich 9-13 betrachtet wurden. Im Rahmen dieser Untersuchungen wurde die
aquatische Spezies Th(OH)2(GluH)²- identifiziert. In einer anderen Versuchsreihe von
Colàs et al. [18] wurde die Komplexierung von U(VI) mit GLU bei U(VI)-GLU-
Verhältnissen von 1: 1 bis 1: 1000 bei pH 12 untersucht. Dabei wurde die Spezies
UO2(OH)2(GluH3)2- identifiziert.
U(VI)-Komplexe mit ISA und GLU sind sich in Struktur, Bindung,
Koordinationsgeometrie und elektronischem Anregungsverhalten sehr ähnlich,
allerdings gibt es deutliche Unterschiede im Hinblick auf das thermodynamische
Verhalten der beiden Liganden. So unterscheiden sich die Komplexe in Bezug auf die
Gleichgewichtskonstanten um bis zu zwei Größenordnungen [21]. Aufgrund dieser
Erkenntnisse kommen neben Birjkumar et al. [21] auch Guillaumont et al. [34] und
Berry et al. [35] zu dem Schluss, dass der Vergleich der Substanzen zumindest aus
thermodynamischer Sicht nicht sicher ist.

Theoretische Grundlagen
17
2.6 U(VI)-Sorption an Ca-Bentonit
Tonminerale haben aufgrund ihrer Eigenschaften (siehe Kapitel 2.1) ein hohes
Rückhaltevermögen. In natürlichen Gesteinen wie Bentonit, fungieren Tonminerale als
Hauptkomponente für die Actiniden-Sorption [36]. Dabei sind verschiedene
Wechselwirkungs-Mechanismen zwischen den Actiniden in Lösung und den
Tonmineral-Oberflächen möglich. Ionen interagieren mit Tonmineralen sowohl auf den
Grundflächen als auch an den Kanten. Des Weiteren ist ein Ionenaustausch an den
Zwischenschichten möglich (siehe Abbildung 11).
Abbildung 11: Uranyl-Sorption in den Zwischenschichten und an den Kanten von
Montmorillonit Maher et al. [37]

Theoretische Grundlagen
18
Die U(VI)-Sorption an Montmorillonit bei niedriger Ionenstärke und sauren bis leicht
alkalischen pH-Werten ist bereits gut untersucht. Die spezifische Rückhaltung von
U(VI) an Montmorillonit/-Bentonit ist stark abhängig vom pH-Wert, der Ionenstärke und
der An- bzw. Abwesenheit von CO2 [38]–[46]. Allerdings besteht ein Defizit in der
Untersuchung der Mechanismen der U(VI)-Rückhaltung unter starken Ionenstärken
und hyperalkalischen Bedingungen. Schnurr et al. [47] quantifizierte die U(VI)-
Rückhaltung an Illit und Kaolinit bis pH 12 in Abwesenheit von CO2. Für beide Minerale
wurde eine vollständige Sorption bis pH 11 beobachtet. Bei höheren pH-Werten ging
die Sorption dann leicht zurück. Unter Ausschluss von CO2 ist eine maximale Sorption
auch bei höheren pH-Werten möglich (siehe Abbildung 12). Allerdings nimmt die
Sorption in Anwesenheit von CO2 bei pH-Werten größer 7 drastisch ab, was auf die
Bildung von aquatischen Uranyl-Karbonat-Komplexen zurückzuführen ist. Diese
Komplexe sind sehr gut löslich und tendieren stark dazu, nicht an Mineralphasen
anzuknüpfen [34].
Abbildung 12: U(VI)-Sorption an Montmorillonit in Abwesenheit von CO2 (Dreiecke) und mit
atmosphärischem pCO2 (Rauten) Marques Fernandeset al. [45]

Theoretische Grundlagen
19
2.7 Sorption von U(VI) und anderen RN unter Einfluss von ISA
Der derzeitig in der Literatur hinterlegte Kenntnisstand zum Einfluss von organischen
Verbindungen auf die Sorption von Radionukliden erstreckt sich über ein weites Feld
von Versuchsansätzen. So wurden in der Vergangenheit nicht nur verschiedene
Radionuklide, sondern auch verschiedene organische Substanzen verwendet, wobei
wiederum deren Verhältnisse und Konzentrationen über ein breites Spektrum verteilt
sind. Auch der pH-Wert wurde als Einflussgröße erkannt und in einigen Arbeiten
variiert, obgleich die überwiegende Mehrheit der Untersuchungen im stark alkalischen
pH-Bereich (pH ≥ 13) vorgenommen wurde. Im Hinblick auf die Schwerpunklage dieser
Arbeit, wurde der Augenmerk verstärkt auf Untersuchungen mit Uran (VI) als
Radionuklid und Isosaccharinsäure (ISA) bzw. CDP als Ligand im alkalischen
pH- Bereich gelegt. Zur Vervollständigung, wurden aber auch andere Radionuklide
(Th(IV), Eu(III), Ni(II), Am(III)) und GLU mit in die Literatursammlung unter Tabelle 2
einbezogen.
Die Kombination U(VI) und ISA wurde von Baston et al. [16] untersucht, wobei Batch-
Sorption-Versuche bei U(VI)-ISA-Verhältnisse von 1:2,5 bis 1:25.000 durchgeführt
wurden. Die Experimente wurden unter Stickstoffatmosphäre bei pH 7 und pH 12,8
und ein Hintergrundelektrolyt aus synthetischem Grundwasser durchgeführt. Die
Untersuchungen zeigten, dass bei pH 7 (ISA >2*10- 5 mol/L) eine Sorptions-
Verminderung und bei pH 12,8 (ISA = 2*10- 7 mol/L bis 2*10-3 mol/L) eine Sorptions-
Verbesserung um etwa den Faktor 5 zu beobachten war. Als feste Phase für den
Sorptions-Versuch wurde Tuffgestein verwendet.

Theoretische Grundlagen
20
Die folgenden Arbeiten von Felipe-Sotelo et al. [17], [48] und Berry et al. [35] fassen
die Kombination von U(VI) und CDP bzw. GLU als organischen Liganden zusammen
und setzt sich größtenteils mit Batch-Sorptions-Experimenten auseinander.
Berry et al. [35] untersuchte dabei unter N2-Atmosphäre, welchen Einfluss GLU auf die
Sorption von Uran bei den pH-Werten 8 und 11 an Zementplatten und Tongestein hat.
Das verwendeten U(VI)-GLU-Verhältnisse erstreckte sich von 1: 2.000 bis 1: 50.000.
Der beobachtete Effekt ließ darauf schließen, dass GLU im Konzentrationsbereich
2*10-3 mol/L die Sorption von U(VI) stärker beeinflusste als CDP der gleichen
Konzentration, was auf die höhere effektive Liganden-Konzentration im Fall von GLU
zurückzuführen ist. Dieser Effekt war vor allem für niedrige U(VI)-Konzentrationen
(10- 8 mol/L) zu beobachten. Felipe-Sotelo et al. [17], [48] untersuchten die Thematik
in Batch-Sorptions-Experimenten an Zement- Verfüll- Materialien und in Durch-
Diffusions- und Advektions-Versuche mit U(VI) und CDP. Die eingesetzten
U(VI)- CDP-Verhältnisse betrugen etwa 1: 50 bis 1: 10.000.000. Die Experimente
wurden CO2- frei unter N2-Atmosphäre bei den pH-Werten 12,8 bzw. 12,8 und
13,3 durchgeführt. Die Untersuchungen hatten zum Ergebnis, dass die U(VI)-Sorption
am Zement um eine Größenordnung zunahm. Für die Batch- Sorptions- Experimente
ergab sich eine Verbesserung der Löslichkeit von U(VI) um den Faktor zehn, wenn
CDP mit 187 mg/L TOC vorlag. Für die Durch- Diffusions- und Advektions-Versuche
wurde beobachtet, dass nur die aquatische Speziation von U(VI) nicht aber die
Sorptions-Spezies durch CDP beeinflusst wurden und sich die Diffusion nachweislich
verbesserte.
Van Loon et al. [25], Wieland et al. [49] und Tits et al. [14] behandelten Sorptions-
Experimente, wobei andere Radionuklide als Uran betrachtet und deren Verhalten
gegenüber CDP, ISA bzw. GLU untersucht wurden. Van Loon et al. [25] untersuchte
in Batch- Sorptions-Experimenten den Einfluss von CDP bzw. α-ISA auf die Sorption
von Th(IV), Eu(III) und Ni(II) an Feldspat. Dafür wurden alle Arbeiten unter N2-
Atmosphäre, bei einem pH-Wert von 13,3 durchgeführt. Die Experimente befassten
sich mit RN-ISA-Verhältnisse von 1: 10.000 bis 1: 10.000.000. Van Loon et al. [25] gibt
als Grund für die Wahl von Feldspat als Festphase an, dass für diesen eine Sorption
von ISA und somit eine Konkurrenz zur Sorptionen der RN ausgeschlossen werden
kann.

Theoretische Grundlagen
21
Erkenntnis aus den Experimenten war, dass für Th(IV) und Eu(III) α-ISA der effektivste
Ligand der CDPs ist. Für Ni(II) hingegen scheinen noch nicht identifizierte Bestandteile
der CDPs als Liganden wirksam zu sein, welche nicht der reinen Cellulose-Zersetzung
entspringen sondern auf Verunreinigung in dieser zurückzuführen sind. Es wurde
beobachtet, dass erst bei einer α-ISA-Konzentration oberhalb von 10- 4- 10-3 mol/L eine
signifikante Sorptionsminderung von Th(IV) und Eu(III) auftritt. Im Fall von Th(IV)
wurde außerdem beobachtet, dass im alkalischen pH-Bereich ein großer Teil
ohne Komplexierung mit α-ISA irreversibel am Feldspat sorbiert.
Die Batch-Sorptions-Experimente von Wieland et al. [49] behandelten den Einfluss von
ISA auf die Th(IV)-Sorption bei Th(IV)-ISA-Verhältnissen von 1: 10.000 bis
1: 100.000.000. Die Experimente wurden bei einem pH-Wert von 13,3 durchgeführt.
Die Experimente ließen schlussfolgern, dass bei ISA-Konzentrationen größer 10-4
mol/L eine Minderung der Sorption von Th(IV) an der verwendeten Zementpaste
auftritt, was auf die Bildung einer Th(IV)-ISA-Spezies zurückzuführen ist.
Bei den Experimenten von Tits et al. [14] wurde Calcit als Sorptions-Material
verwendet. Die Versuche sollten zum besseren Verständnis des Sorptionsverhalten
der RN Eu(III), Am(III) und Th(IV) gegenüber GLU und ISA in den RN-Ligand-
Verhältnissen 1: 100 bis 1: 10.000.000 beitragen. Dabei wurde beobachtet, dass die
ISA Konzentration erst ab etwa 10-5 mol/L einen signifikanten Einfluss auf die Sorption
der Radionuklide hat und zu einer Verminderung dieser führt. Außerdem gibt
Tits et al. [14] an, dass unter realen Bedingungen im Endlager mit Zementeinbauten,
in der Porenwasser-Zone maximale Konzentrationen von 3*10-6 mol/L für ISA und
10- 7 mol/L für GLU zu erwarten sind, welche den Erkenntnissen zufolge keine
nennenswerten Effekte auf die verwendeten Radionuklide und deren Sorption an
Calcit haben sollten.

Theoretische Grundlagen
22
Tabelle 2: Zusammenfassung derzeitiger Kenntnisstand zur Radionuklid-ISA-
Komplexierung und deren Einfluss auf Wechselwirkungen mit Mineraloberflächen
Radionuklid
Konzentration
Radionuklid
[mol/L]
Konzentration
Organika
[mol/L]
Verhältnis
RN : OC
pH-Wert Effekt Quelle
U(VI)
~8*10-8 ISA:
2*10-7-2*10-3
1:2,5-
1:25.000
7, 12,8 Sorptionsminderun
g Über 2*10-5
mol/L ISA (pH~7)
Sorptionssteigerun
g (pH~12.8) um
Faktor ~5
[16]
1*10-6-4*10-8 GLU: 2*10-3 1:2000-
1:50.000
~8,
~11
Sorptionsminderung
GLU größerer Effekt
als bei CDP bei
[U(VI)] = 10- 8 mol/L
[35]
10-10-10-5 CDP:
~640 mg/L
TOC
ca. 1:100-
1:10.000.000
12,8 Sorptionssteigerung an
Zement um eine
Größenordnung
[17]
10-5 CDP:
187 mg/L TOC
ca. 1: 50
12,8-13,3 U(VI)-Löslichkeit
verbessert um
Faktor 10
Nur aquatische.
Spez. durch CDP
beeinflusst
U(VI)-Diffusion
verbessert
[48]
Th(IV)
6,7 * 10-9 α-ISA:
10-4-10- 3
ca. 1:10.000-
1:10.000.000
13,3 kein Einfluss auf
Sorption
[25]
≤ 10−10 ISA:
10-5-10-2
1:10.000-
1:100.000.000
13,3 Th-Sorption bei
ISA >10- 4 M reduziert
Th(IV)-ISA
[49]
10-11 ISA:
10-4-10-7
1:100-
1:10.000.000
13,3 Kein Einfluss auf
Sorption bei:
[ISA]: 3*10-6 mol/L
[GLU]: 10- 7 mol/L
ISA erst ab 10- 5 mol/L
wirksam
[14]
Eu(III)
6,7*10-10 α-ISA:
10-4-10- 3
ca. 1:10.000-
1:10.000.000
13,3 Sorptionsminderung [25]
10-9 ISA:
10-4-10-7
1:100-
1:10.000.000
13,3 Sorptionsminderung
ab [ISA] 10- 5 mol/L
[14]
Ni(II) 3*10-10 α-ISA:
10-4-10- 3
ca. 1:10.000-
1:10.000.000
13,3 Sorptionsminderung [25]
Am(III) 10-11 ISA:
10-4-10-7
1:100-
1:10.000.000
13,3 Sorptionsminderung
ab [ISA] 10- 5 mol/L
[14]

Material und Methoden
23
3 Material und Methoden
3.1 Material
3.1.1 Verbrauchschemikalien
Tabelle 3: Liste der verwendeten Verbrauchschemikalien
Chemikalien Reinheitsgrad Hersteller
NaCl ≥ 99,5 % p.a. Roth
CaCl2 ≥ 99 % p.a. Roth
HCl 37 % p.a. Roth
NaOH ≥ 99 % p.a. Roth
HNO3 ≥ 65 % p.a. Roth
α-Lactose-Monohydrat ≥ 99 % p.a. Sigma
Ca(OH)2 ≥ 96 % p.a. Merck
3.1.2 Ca-Bentonit
Als Mineralphase wurde in allen Batch-Sorptions-Experimenten der Ca-Bentonit vom
Typ Calcigel® verwendet. Laut Hersteller (Clariant) besteht dieser Ca-Bentonit aus
den in Tabelle 4 aufgeführten relativen Bestandteilen.
Tabelle 4: mineralogische Zusammensetzung von Ca-Bentonit (Calcigel®) entsprechend
der Produktinformationen von Clariant
Mineral Anteil in %
Montmorillonit 60 - 70
Quarz 6 - 9
Feldspat 1 - 4
Kaolinit 1 - 2
Glimmer 1 - 6
Andere 5 - 10
Material und Methoden
24
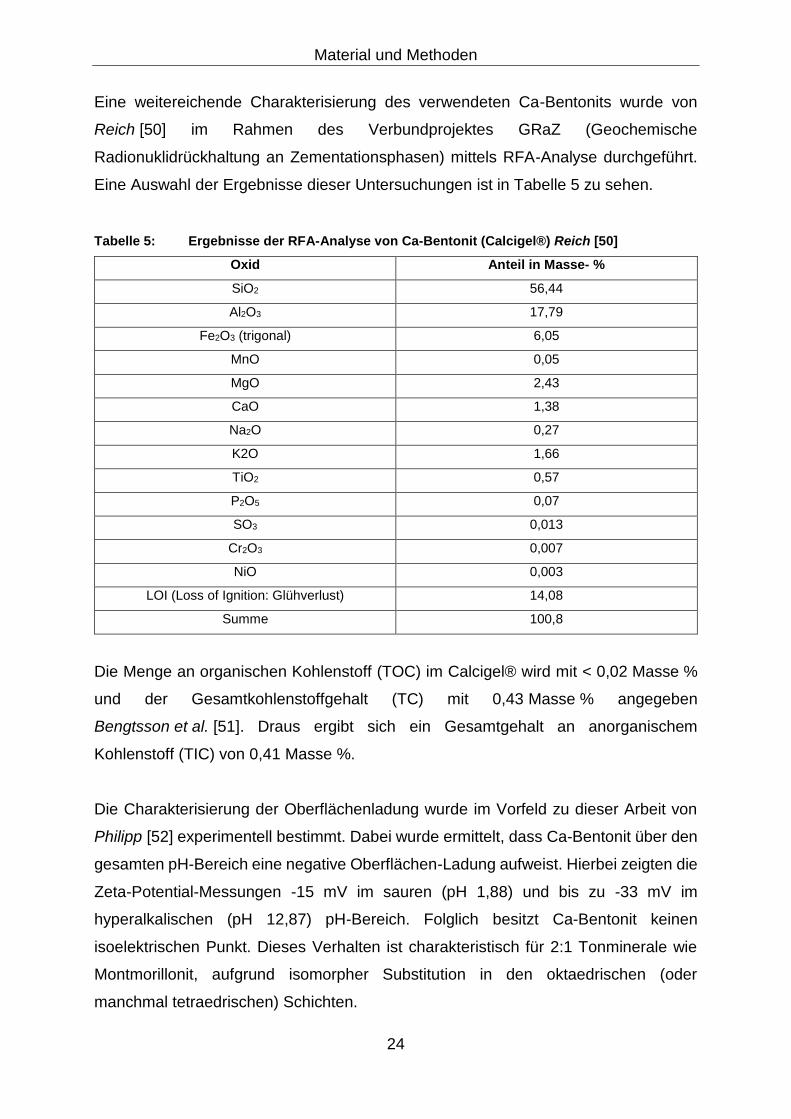
Eine weitereichende Charakterisierung des verwendeten Ca-Bentonits wurde von
Reich [50] im Rahmen des Verbundprojektes GRaZ (Geochemische
Radionuklidrückhaltung an Zementationsphasen) mittels RFA-Analyse durchgeführt.
Eine Auswahl der Ergebnisse dieser Untersuchungen ist in Tabelle 5 zu sehen.
Tabelle 5: Ergebnisse der RFA-Analyse von Ca-Bentonit (Calcigel®) Reich [50]
Oxid Anteil in Masse- %
SiO2 56,44
Al2O3 17,79
Fe2O3 (trigonal) 6,05
MnO 0,05
MgO 2,43
CaO 1,38
Na2O 0,27
K2O 1,66
TiO2 0,57
P2O5 0,07
SO3 0,013
Cr2O3 0,007
NiO 0,003
LOI (Loss of Ignition: Glühverlust) 14,08
Summe 100,8
Die Menge an organischen Kohlenstoff (TOC) im Calcigel® wird mit < 0,02 Masse %
und der Gesamtkohlenstoffgehalt (TC) mit 0,43 Masse % angegeben
Bengtsson et al. [51]. Draus ergibt sich ein Gesamtgehalt an anorganischem
Kohlenstoff (TIC) von 0,41 Masse %.
Die Charakterisierung der Oberflächenladung wurde im Vorfeld zu dieser Arbeit von
Philipp [52] experimentell bestimmt. Dabei wurde ermittelt, dass Ca-Bentonit über den
gesamten pH-Bereich eine negative Oberflächen-Ladung aufweist. Hierbei zeigten die
Zeta-Potential-Messungen -15 mV im sauren (pH 1,88) und bis zu -33 mV im
hyperalkalischen (pH 12,87) pH-Bereich. Folglich besitzt Ca-Bentonit keinen
isoelektrischen Punkt. Dieses Verhalten ist charakteristisch für 2:1 Tonminerale wie
Montmorillonit, aufgrund isomorpher Substitution in den oktaedrischen (oder
manchmal tetraedrischen) Schichten.

Material und Methoden
25
3.1.3 Isosaccharinsäure
Für die durchgeführten Sorptions- und Komplexierungs-Experimente wurde
Isosaccharinsäure als organischer Ligand für das sich in Lösung befindliche 238U
verwendet. Die Synthese von Isosaccharinsäure wurde erstmals von
Whistler und BeMiller [53] beschrieben und entsprechend Bassil et al. [54] und
Vercammen et al. [55] leicht variiert. Dabei wird zuerst das schwer wasserlösliche
Ca(ISA)2 synthetisiert, welches später durch Kationenaustausch in die besser
wasserlösliche Natrium-Form NaISA überführt wird.
Synthese von Ca(ISA)2
Für die Synthese des Calciumsalzes Ca(ISA)2 der Isosaccharinsäure werden 500 ml
entionisiertes Wasser in einem 1 l-Zweihals-Kochrundkolben vorgelegt und unter
Rühren 3 h mit Argon gespült. Daraufhin werden 50 g α-Lactose-monohydrat und
13,6 g Ca(OH)2 zugegeben, der Rundkolben mit einem Septum verschlossen und
unter ständigem Rühren für weitere 30 min mit Argon gespült. Anschließend wird der
Ansatz für drei Tage bei Raumtemperatur gerührt. Dabei ist ein deutlicher Farbwechsel
von weiß über gelb bis hin zu orange und später zu braun zu beobachten. Die
erhaltene Lösung wird für 6 h unter Rückfluss bei 130 °C gekocht, wobei sich die Farbe
ein weiteres Mal zu einem dunklem braun hin verändert. Im Anschluss wird die Lösung
noch heiß über eine Nutsche filtriert. Die Lösung sollte nun eine fast schwarze Färbung
angenommen haben und verströmt einen süßlichen Geruch. Die Lösung wird durch
Kochen und unter ständigem Rühren in einem passenden Becherglas auf 100-200 ml
reduziert. Nach diesem Arbeitsschritt wird die Lösung auf Raumtemperatur abgekühlt,
wobei sich bereits ein beigefarbener Niederschlag bilden kann. Um die Bildung dieses
Niederschlages zu begünstigen, wird die Lösung für mehrere Tage bei 4 °C gelagert.
Der Überstand wird vorsichtig dekantiert und der erhaltene Niederschlag über einen
Whatman-Filter (Nummer 3) vakuumfiltriert und mehrmals mit Wasser und Ethanol
gewaschen. Anschließend wird der vorgereinigte Niederschlag für mehrere Stunden
bei 50 °C im Ofen getrocknet. Der getrocknete Niederschlag wird mit einem Verhältnis
von 1,2 g in 100 ml in kochendem entionisierten Wasser gelöst. Daraufhin erfolgt ein
erneutes Filtern der Lösung über einen Whatman-Filter (Nummer 3).

Material und Methoden
26
Das Volumen der gefilterten Lösung wird auf 10 % des Anfangsvolumens reduziert.
Die erhaltene Lösung wird wiederum bei 4 °C gelagert bis sich ein weißer Niederschlag
bildet. Nach dem dekantieren des Überstandes wird abermals mehrmals mit Wasser
und Ethanol gewaschen. Nachdem der Niederschlag bei 50 °C getrocknet wurde,
erfolgt die Analyse mittels NMR.
Herstellung von NaISA-Stammlösung
Das im ersten Schritt synthetisierte und gereinigte Ca(ISA)2 wird in 500 ml
entionisiertem Wasser mit Chelex-100 (BioRad, in der Na-Form) versetzt und für 3 h
gerührt. Das Harz wird über einen Membranfilter (Millipore 0,2 µm) abgetrennt und das
Volumen des Filtrates durch Kochen auf 50 ml reduziert. Darauf folgt eine
Untersuchung mittels NMR auf Reinheit sowie eine Bestimmung des TOC um die
Konzentration zu erhalten.
3.1.4 Uran
Die verwendete U(VI)-Stammlösung wurde von S. Weiß (HZDR) hergestellt. Dafür
wurde das Ausgangsmaterial Uranylnitrat (UO2(NO3)2 × 6 H2O) über die Zwischenstufe
Uranylperoxid (UO4×4H2O) in Uranoxid (UO3) umgewandelt. UO3 kann dann wiederum
für die Herstellung der Stammlösungen mit den entsprechenden Säuren gelöst
werden. Zu Beginn wurde eine 10 % Uranylnitrat-Lösung unter ständigem Rühren im
Wasserbad auf ca. 70 °C erhitzt. Anschließend bildete sich durch die tropfenweise
Zugabe von 30 % H2O2 ein gelber Niederschlag (UO4×4H2O). Dabei war zu beachten,
dass pro gelöstem Gramm Uranylnitrat 1,5 mL H2O2 zugegeben werden sollten. Der
Niederschlag wurde über spezielle Fritten abgesaugt. Daraufhin wurde der
Niederschlag in der Fritte mit mindestens der 10-fachen Menge heißem Wasser
gewaschen und im Anschluss im Abzug über 12 h getrocknet. Der erhaltene Feststoff
wurde in einen Platintiegel überführt und in den Ofen gestellt, wobei innerhalb von
1,5 h eine Temperatur von 220 – 230 °C erreicht werden sollte, welche dann für
weitere 6 h gehalten werden musste. Nach dem Abkühlen konnte das orangefarbene
Produkt dem Ofen entnommen werden. Das Produkt UO3 konnte daraufhin in
verschiedenen Säuren gelöst werden.

Material und Methoden
27
Es können Ausbeuten von 90 – 95 % erreicht werden. Das Produkt ist röntgenamorph.
Da UO3 je nach Synthesebedingungen 2 - 2,5 M Kristallwasser enthält, muss dieses
für die Einwaage für die Stammlösungen berücksichtigt werden. Für die Experimente
im Rahmen dieser Arbeit wurden 0,1 M U(VI) in 0,5 M HClO4 gelöst und der Uran-
Gehalt mittels ICP-MS untersucht. Die fertige Stammlösung konnte dann problemlos
auf die für die Experimente besser geeignete Ausgangs- Konzentration von
10 mM U(VI) verdünnt werden.
3.2 Methoden
3.2.1 pH-Wert Einstellung und Äquilibrierung
Für die Präparation der Proben zur Durchführung der Batch-Sorptions-Experimente
musste bei jeder Probe vor Beginn der Sorptions-Phase der pH-Wert eingestellt und
äquilibriert werden. Die Bestimmung des pH-Wertes erfolgte dabei mit dem pH-Meter
inoLab pH7110 und der Messkette mit Flüssig-Referenzsystem SenTix® Mic der
Marke WTW der Xylem Analytics Germany GmbH. Vor jeder Messung bzw. an jedem
Messtag musste das Gerät vorerst kalibriert werden. Die Kalibration erfolgte
entsprechend des gewünschten Messbereiches mittels Dreipunkt-Kalibration. Die zur
Kalibration verwendeten Puffer umfassten die pH-Werte: 1,679; 4,006; 6,865; 9,180
und 12.454 und mussten in regelmäßigen Abständen ausgetauscht werden. Das
Einstellen der pH-Werte erfolgt mittels Lösungen von 0,1 M, 1 M und 10 M NaOH bzw.
0,1 M, 1 M und 6 M HCl, welche den Proben mit Hilfe von Transferpipetten hinzudosiert
wurden. Da bei den pH-Werten an den beiden Enden der Skala die Zugabe-Volumina
an NaOH bzw. HCl sehr groß ausfallen können, musste dies bei der Vorbereitung der
Proben berücksichtigt werden. Nach etwa drei Einstellungen sind die Zugabe-
Volumina an NaOH bzw. HCl dann bereits so gering (wenige µl), dass die Proben auf
ihr finales Volumen aufgefüllt werden können. Der pH-Wert der Proben wird so lange
eingestellt und kontrolliert, bis er sich nicht mehr ändert und im Bereich der
Messgenauigkeit des Gerätes bei 0,05 pH-Einheiten um den gewünschten pH-Wert
liegt. Dieser Vorgang lässt sich beschleunigen, indem der pH-Wert in kurzen
Abständen (alle 1-2 Tage) kontrolliert wird. Zumeist ist der pH-Wert aber nach ein bis
maximal zwei Wochen äquilibriert.

Material und Methoden
28
3.2.2 Inductively Coupled Plasma - Mass Spectrometry (ICP-MS)
Die ICP-MS-Messungen wurden von S. Beutner (HZDR) und S. Bachmann (HZDR)
durchgeführt. Im Vorfeld der Analyse wurden die Proben zentrifugiert und zweimal
1 mL aus dem Überstand in Eppendorf-Vials für eine Doppelbestimmung entnommen.
Die Proben in den Vials mussten daraufhin mit destillierter HNO3 angesäuert werden.
Untersucht wurde neben Uran zudem auf die Elemente Si, Al, Ca, Mg und K (NexION
350X, Perkin Elmer). Die Zentrifugation erfolgte bei den Batch-Sorptions-
Experimenten für 30 min bei 6800 g (Avanti J20 XP, Beckman Coulter) und für die
Untersuchungen zur aquatischen Speziation für 60 min bei 77.000 g (Optima XPN-80
Ultracentrifuge, Beckman Coulter).
3.2.3 Total Carbon (TC), Total Inorganic Carbon (TIC),
Total Organic Carbon (TOC)
Der TOC (Total Organic Carbon) ist ein Maß für die in wässrigen Proben enthaltenen
organischen Kohlenstoffverbindungen. Dies schließt sowohl gelöste als auch
ungelöste organische Inhaltsstoffe ein. Bei wässrigen Proben, die keine ungelösten
oder suspendierten Stoffe (Schwebstoffe) enthalten ist der TOC gleichbedeutend mit
dem DOC (Dissolved Organic Carbon), welcher ausschließlich die gelösten
organischen Verbindungen angibt. Enthält eine Probe suspendierte Stoffe, muss diese
vorerst durch Filtration oder Zentrifugation behandelt werden. Die Bestimmung des
TOC erfolgt nach der Differenzmethode, wofür zunächst die beiden Parameter TC
(Total Carbon) und TIC (Total Inorganic Carbon) bestimmt werden müssen. Der TC
wird durch chemischen oder thermischen Aufschluss (T = 800°C) und der TIC durch
Behandlung mit mineralischen Säuren (10 %-ige H3PO4), jeweils durch die Umsetzung
zu CO2 bestimmt. Die Detektion des Kohlendioxids erfolgt mittels nicht dispersive IR-
Detektoren (NDIR-Detektoren). Errechnet wird der TOC analog zu Gleichung 1:
TOC = TC – TIC (1)

Material und Methoden
29
Aufgrund der Differenzbildung ist es zwingend erforderlich, jeden TC- und TIC-Wert
durch mehrere Einzelmessungen statistisch zu sichern. Die Analyse erfolgte am
Multi N/C 2100s von Analytik Jena und die Messzeit betrug je Probe etwa 20 Minuten.
Die Messung von 6 mL des jeweiligen Proben-Überstandes wurde von Frau Carola
Eckardt durchgeführt. Die ISA-Konzentration konnte aus den erhaltenen TOC-Werten
mit Hilfe von Gleichung 2 bestimmt werden.
cSubstanz 𝑖𝑛 [𝑚𝑔
𝐿] =
TOC ∗ MSubstanz
MKohlenstoff ∗ Anzahl C−Atome (Substanz) (2)
3.2.4 Laugungsverhalten von Ca-Bentonit
Beim Laugungs-Experiment, sollte die Stabilität von Ca-Bentonit (10 g/L) im
alkalischen pH- Bereich (pH 8-13) in 0,1 M NaCl-Lösung untersucht werden. Als
Probengefäße kamen 15 mL PP-Zentrifugenröhrchen (Greiner) zum Einsatz. Für den
Laugungsversuch wurde in Doppelbestimmung vorerst der pH-Wert eingestellt (siehe
Kapitel 3.2.1). Danach wurden die Probenröhrchen für eine Laugungsdauer von einer
Woche in einem Überkopfschüttler platziert und im Anschluss daran der Überstand auf
die enthaltenen Ionen (Ca2+, Mg2+, Al3+, Si4+, K+) und den DOC hin untersucht. Dieses
Experiment stellt damit die Voruntersuchungen zu den folgenden Batch-Sorptions-
Experimenten dar.

Material und Methoden
30
3.2.5 Batch-Sorptions-Experimente
Alle Proben wurden in Doppelbestimmung in 15 mL Polypropylen-Zentrifugen-
Röhrchen (Greiner) angesetzt. Zuerst wurden 10 g/L Ca-Bentonit für das jeweilige
Probenvolumen (5 bzw. 10 mL) abgewogen und die Proben mit 0,1 M NaCl-Lösung
voräquilibriert. Dafür wurde der pH-Wert eingestellt und die Proben in einem Überkopf-
oder Horizontal-Schüttler platziert. Für die Experimente wurde in einer Glovebox unter
N2-Atmosphäre bei 25 ± 2 °C gearbeitet und sämtliche verwendete Lösungen mit
entionisiertem Milli-Q-Wasser präpariert. Nach der Äquilibrierung des pH-Wertes
erfolgte die Zugabe der Isosaccharinsäure bzw. des Urans entsprechend den Angaben
in Tabelle 6.
Tabelle 6: Überblick über Batch-Sorptions-Experimente und die wichtigsten
Versuchsparameter
Experiment Elektrolyt S/L
[g/L]
c(U)
[mol/L]
c(ISA)
[mol/L]
pH
ISA-Sorption
0,1 M NaCl
10 g/L
/ 5*10-4
8-13
Uransorption
5*10-7
/
Uransorption
unter
ISA- Einfluss
5*10-4 bis
5*10-2
Aufbauend auf den Voruntersuchungen zur Ca-Bentonit-laugung wurde das
Sorptionsverhalten von ISA an Ca-Bentonit untersucht. Die Proben wurden durch
Einwaage von 10 g/L Ca-Bentonit und Zugabe des Hintergrundelektrolyten (0,1 M
NaCl) hergestellt. Es erfolgte eine wie bereits in Kapitel 3.2.1 beschriebene
Äquilibrierung und pH-Wert-Einstellung. Bei konstantem pH-Wert wurde die
NaISA- Stammlösung (0,636 mol/L) mit Hilfe einer Transferpipette zudosiert, um eine
Zielkonzentration von 5*10-4 mol/L zu erreichen. Danach erfolgt die einwöchige
Sorptions-Phase, während derer die Proben fortwährend geschüttelt wurden.

Material und Methoden
31
Für das Experiment zur Uran-Sorption an Ca-Bentonit wurde nach der
pH Äquilibrierung die U(VI)- Stammlösung (10-3 mol/L) mittels Transferpipetten
zugegeben (für U(VI)-Zielkonzentration = 5*10-7 mol/L) und die Proben für den
Zeitraum der Sorption (1 Woche) im Überkopfschüttler platziert. Besondere Bedeutung
dabei hat, dass der pH-Wert nach der Zugabe nicht mehr verändert bzw. kontrolliert
werden sollte. Grund hierfür ist eine mögliche Sorption des Urans an die Glasmembran
der pH-Elektrode, welche spätere Analyseergebnisse verfälschen könnte.
Bei den Experimenten zur Untersuchung des möglichen ISA-Einflusses auf die
U(VI)- Sorption wurde nach der pH-Äquilibrierung zunächst NaISA zugegen und die
Proben für eine Woche im Schüttler platziert, dabei wurden in regelmäßigen
Abständen (2-3 Tage) die pH-Werte der Proben kontrolliert. Daraufhin erfolgte die
Zugabe von U(VI) und erneut eine einwöchige Sorptionsphase im Schüttler.
Nach Abschluss der Batch-Sorptions-Experimente wurden die Proben für die Analysen
vorbereitet, was einschließt, dass der pH-Wert jeder Probe noch einmal überprüft
werden muss. Dafür musste eine Phasentrennung erfolgen, für die die Proben 30 min
bei 6800 g zentrifugiert wurden (Avanti J-20 XP, Beckman Coulter). Anschließend
wurde der Überstand mit einer Transferpipette entnommen um sowohl Volumina für
die Analysen (ICP-MS, TRLFS, TOC) als auch für eine Rückstellprobe zu erhalten.
Um möglichst genaue Sorptions-Daten für die Interaktion zwischen U(VI) und
Ca- Bentonit zu erhalten wurde im Vorfeld zu dieser Arbeit untersucht, ob eine
mögliche Sorption von Uran an die Gefäßwände der Probenröhrchen stattfindet.
Philipp [52] fand diesbezüglich heraus, dass diese U(VI)-Sorption an die Gefäßwände
in signifikantem Maßstab eintritt, dieser Effekt aber nur in Abwesenheit von Ca-
Bentonit auftrat. Diese Erkenntnis lässt vermuten, dass die bisher genutzten
Probenröhrchen sich zwar für die Sorptionsuntersuchungen an Ca-Bentonit eignen,
jedoch für die im folgenden beschriebenen Experimente zur aquatischen Speziation
nicht geeignet sind.

Material und Methoden
32
3.2.6 Zeitaufgelöste Laserinduzierte Fluoreszenzspektroskopie (TRLFS)
Time-resolved laser-induced fluorescence spectroscopy (TRLFS) ist eine Methode, mit
der die Identifizierung verschiedener U(VI)-Komplexe auch bei niedrigen
U(VI) Konzentrationen möglich ist. Grund dafür sind Veränderungen im Spektrum,
welche durch Änderungen im äquatorialen Ligandenfeld hervorgerufen werden [56].
Sie ist eine Weiterentwicklung der konventionellen Lumineszenz-Spektroskopie und
ist eine der wichtigsten spektroskopischen Methoden in der Actiniden-Chemie, da sie
neben der Bestimmung der Peak-Positionen auch eine Bestimmung der
Lumineszenz- Lebensdauer ermöglicht.
Die Methode basiert auf der Lumineszenz der gebildeten U(VI)-Komplexe.
Lumineszenz ist die Emission von Licht, ausgehend von einer Substanz, die zuvor
angeregt wurde. Die Absorption von Licht (z.B.: Laser) regt Elektronen
(Valenzelektronen, Bindungselektronen von Molekülen) an, welche dabei vom
Grundzustand (S0) in einen Zustand höherer Energie (S1, S2… Sn) oder Schwingung
übergehen (siehe Abbildung 13). Zwischen diesen Energiezuständen sind eine
Vielzahl verschiedener Elektronenübergänge möglich [57]. Strahlungsfreie Relaxation
in Verbindung mit einer Veränderung des Energieniveaus (z.B.: S2 → S1) wird auch
als innere Umwandlung (internal conversion) bezeichnet. Außerdem kann
strahlungsfreie Relaxation von höheren Schwingungszuständen des angeregten
Zustandes zum niedrigsten Schwingungszustand eben dieses angeregten Zustandes
auftreten.
Dies ist auf Zusammenstöße mit anderen Atomen und Molekülen zurückzuführen,
welche eine Energieübertragung zur Folge haben. Findet ein Übergang vom
niedrigsten Schwingungszustand des angeregten Zustandes zum Grundzustand
(S1 → S0) statt, wird die überschüssige Energie in Form eines Photons frei und es
kommt zur Fluoreszenz [57]–[59]. Die Energie und Lebensdauer der Lumineszenz sind
charakteristisch für die jeweilige chemische Verbindung.

Material und Methoden
33
Abbildung 13: allgemeines Jablonski-Diagramm für Fluoreszenz und Phosphoreszenz
Bei Lanthanoiden werden die Elektronenübergänge die für die Lumineszenz
verantwortlich sind auf die f-Elektronen zurückgeführt. U(VI) verfügt jedoch formal über
keine f- Elektronen. Die Übergänge finden hier zwischen den Molekülorbitalen des
Uranyls in Verbindung mit Sauerstoff statt [60]. Abbildung 14 zeigt die Molekülorbitale
und relative Energieniveaus des Uranyls, welche aus der Überlappung der
Atomorbitale 6s, 6p, 5f, 6d und 7s des Urans und 2s und 2p des Sauerstoffs
hervorgehen. Die höchsten besetzten Molekülorbitale sind 1πg, 2πu, 3σg und 3σu, was
auf die starke Beteiligung des 2p-Sauerstoff-Orbitals zurückzuführen ist [59], [60]. Das
3σu-Orbital schließt neben der Überlappung des 2p-Sauerstoff-Orbitals mit dem 5f-
Uran-Orbital auch das 6p-Uran-Orbital mit ein. Dies führt zu einem signifikanten
Anstieg des Energieniveaus wodurch das 3σu-Orbital zum höchsten besetzten
Molekülorbital (HOMO) wird. Das niedrigste unbesetzte Molekülorbital (LUMO) ist das
1Φu-Orbital, welches vorwiegend vom 5f-Uran-Orbital beeinflusst wird [60], [61].
Deshalb entspricht die niedrigste benötigte Anregungs-Energie der des Übergangs
3σu → 1Φu. Dieser Übergang ist mit einer Übertragung von elektrischen Ladungen von
Sauerstoff-(2p) zu den dominierenden Uranorbitalen (5f) verbunden (charge transfer).
Bei der Umkehr dieser Ladungsübertragung kommt es zur Lumineszenz.
S0

Material und Methoden
34
Vom niedrigsten Schwingungszustand des angeregten Zustandes sind Übergänge in
verschiedenen Schwingungszustände des Grundzustandes möglich, die zu
unabhängigen Maxima im Uran- Emissions- Spektrum führen (470, 488, 509, 533,
559, 585 nm für das Uran-aquo-Ion [62], [63]. Diese Aufspaltung beruht auf der
symmetrischen Streckschwingungen des Uranyls. Das äquatoriale Ligandenfeld
beeinflusst die Elektronendichte und bestimmt damit die Energiedifferenz zwischen
den Energieniveaus. Daraus resultierend ist die Energie (Wellenlänge) des emittierten
Photons charakteristisch für die jeweiligen Uranyl-Komplexe. Außerdem ist die
Lumineszenz-Lebenszeit spezifisch für den jeweiligen Komplex, da diese mit
möglichen Übergängen korreliert. Diese Lebenszeiten variieren zwischen ns und ms
[59]. Neben der elektronischen Umgebung des Urans hängt die Lebenszeit jedoch von
einer Vielzahl weiterer Faktoren, wie Temperatur oder Quench-Effekten ab. Der
Quench- oder Fluoreszenzlöschungs-Effekt bezeichnet Vorgänge, die eine Abnahme
in der Intensität eines Fluorophors zur Folge haben, ohne dass der Fluorophor zerstört
wird.
Abbildung 14: Relative Energieniveaus des Uranyl-Valenz-Orbitals. 3σu ist HOMO und
1Φu LUMO Denning [60]

Material und Methoden
35
Informationen über die Lumineszenz-Lebensdauer sind vor allem in
Mehrkomponenten-Systemen von großer Bedeutung, da es mit Hilfe der
verschiedenen Zerfallsraten der Lumineszenz-Signale möglich ist, nebeneinander
vorliegende Spezies nachzuweisen. Ein mono-exponentieller Zerfall lässt dabei auf
eine einzelne Spezies, ein bi-exponentieller Zerfall auf zwei Spezies schließen (analog
für multi-exponentielle Zerfälle höherer Ordnung). Zusätzliche Möglichkeiten bietet
eine Messung unter Tieftemperatur (cryo-TRLFS). Bei tiefen Temperaturen ist der
Quench-Effekt für die Lumineszenz-Signale minimal, sodass es möglich ist Spezies zu
detektieren, die bei Raumtemperatur keine Lumineszenz aufweisen.
Die TRLFS-Messungen wurden bei einer Temperatur von 153 K (-120 °C) und unter
den in Tabelle 7 aufgeführten Geräteparametern durchgeführt. Besonderheit der
zeitaufgelösten Messung ist, dass die einzelnen Spektren einer zeitaufgelösten
Messung über 100-120 Einzelmessungen gemittelt und in bestimmten Zeitabständen
aufgenommen werden. Diese Zeitabstände sind aber nicht konstant, sondern folgen
einer bestimmten Funktion, die gegeben Falls für jede Probe angepasst werden muss.
Die Auswertung der Messdaten erfolgte daraufhin mit Hilfe des
Tabellenkalkulationsprogramms Origin.
Tabelle 7: verwendete Geräte- und Software-Parameter für die cryo-TRFLS
Spaltbreite 2000 µm
Impulsbreite 2000 µs
Spannung/ Frequenz 1085-1125 V/ 20 Hz
Energie 0,8-2,8 mJ
Akkumulationen 100-120
Einzelspektren bei zeitaufgelöster Messung 49-51
Gleichung für zeitaufgelöste Messung 0,1 + 0,05*x+x^4/n; n=2000-3000

Material und Methoden
36
Probenpräparation
Aufgrund der Erkenntnisse aus den Voruntersuchungen bei den Batch-Sorptions-
Experimenten, wurden die Proben für die aquatische Speziation in 50 mL
FEP- Röhrchen (Thermo Scientific Nalgene) angesetzt, welche keinerlei
U(VI)- Sorption an die Gefäßwände zulassen. Zu Beginn wurde 0,1 M NaCl-Lösung
vorgelegt der pH-Wert eingestellt, äquilibriert und die Proben in einem Überkopf-
Schüttler platziert. Für die Experimente wurde in einer Glovebox unter N2-Atmosphäre
bei 25 ± 2 °C gearbeitet und sämtliche verwendete Lösungen mit entionisiertem
Milli- Q- Wasser präpariert. Nach der Äquilibrierung des pH-Wertes erfolgte die
Zugabe der Isosaccharinsäure und kurz darauf die des Urans entsprechend den
Angaben in Tabelle 8.
Tabelle 8: Überblick über Experimente zur aquatischen Speziation und die wichtigsten
Versuchsparameter
Experiment
(U-ISA-Verhältnis)
Elektrolyt c(U)
[mol/L]
c(ISA)
[mol/L]
pH
U(VI)
0,1 M NaCl
5*10-7
/
1-13 U/ ISA
1 : 1000
5*10-4
Ca-Bentonit/ U/ ISA
1 : 100.000
5*10-2 8-13

Ergebnisse und Diskussion
37
4 Ergebnisse und Diskussion
4.1 Laugung von Ca-Bentonit
Abbildung 15: relativer Anteil der Ca-Bentonit Bestandteile nach dem Laugungsversuch
gemessen an der Gesamteinsatzmenge an Ca-Bentonit (10 g/L) in Abhängigkeit des pH-Wertes
In Abbildung 15 sind die ermittelten relativen Konzentrationen der
Ca- Bentonit- Bestandteile dargestellt, welche mit Hilfe der ermittelten
Absolutkonzentrationen (siehe Abbildung 26 im Anhang) und den relativen Anteilen
aus der RFA-Analyse von Reich [50] (siehe Tabelle 5) berechnet wurden. Für die
Berechnung wurden nur die Anteile der jeweiligen Elemente, nicht die der Oxide
genutzt. Abbildung 15 zeigt die Konzentrationen von Si, Al, Mg, Ca und K in der
überstehenden Lösung nach Beendigung des Laugungsversuchs in Abhängigkeit des
pH-Wertes. Es ist zu erkennen, dass die Si-Konzentration bis ca. pH 11 konstant
niedrig ist, ab pH 11,5 jedoch zunimmt und bei pH 13 ihr Maximum erreicht. Für die Al-
Konzentration verhält es sich ähnlich, wobei der deutliche Anstieg erst ab pH 12,5 zu
verzeichnen ist. Da Montmorillonit Hauptbestandteil (60-70 %, Clariant) von
Ca- Bentonit ist und Aluminium- Silikat aus verschiedenen oxydischen Aluminium- und
Siliciumverbindngen besteht, könnte an dieser Stelle vermutet werden, dass
Ca- Bentonit ab pH-Werten über 12 die Grenzen seiner Stabilität erreicht.
0
10
20
30
40
50
60
70
80
90
100
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5
% d
er
Besta
nte
ile a
uf
10 g
/L C
a-B
ento
nit
pH-Wert
Silicium Aluminium Magnesium Calcium Kalium

Ergebnisse und Diskussion
38
Für die Elemente Calcium und Magnesium ist eine kontinuierliche Abnahme der
Konzentrationen zu erkennen, was auf eine pH- Wert -abhängige Verringerung der
Löslichkeit zurückzuführen ist. Calcium ist demnach ab pH 12,5 nahezu aus dem
Überstand verschwunden, Magnesium hingegen bei pH 10. Eine Erklärung wäre die
Bildung bzw. Ausfällung der beiden schwer löslichen Erdalkali-Hydroxyde, Portlandit
(Ca(OH)2) und Brucit (Mg(OH)2). Diese Vermutung könnte in zukünftigen
Untersuchungen mittels Festphasen-Analyse (z. B: XRD) aufgeklärt werden. Kalium
ist über den gesamten pH-Bereich nur in sehr geringen Mengen im Überstand zu
finden, wobei auch hier eine leichte Zunahme der Konzentration bei pH 13 zu
erkennen ist. Grund dafür ist womöglich, das Kalium aus den Zwischenschichten des
Montmorillonit-Netzwerkes gelöst wurde, was wiederum eine Austauschreaktion mit
Natrium oder einen weiteren Anhaltspunkt für eine beginnende Instabilität des
Ca- Bentonits, bei pH-Werten über 12 liefern könnte.
Der enthaltene organische Kohlenstoff wurde mittels TOC-Analyse für den durch
Zentrifugation gewonnenen Überstand aus dem Laugungs-Versuch ermittelt. Ziel war
es die organische Grundlast und somit für künftige Experimente einen Korrekturfaktor
für den TOC zu ermitteln. Der Korrekturfaktor ist nötig, da für das Sorptions-Verhalten
von ISA an Ca-Bentonit ebenfalls die TOC-Analyse durchgeführt wird. Die ermittelten
Konzentrationen des TOC in mg/L sind in Abbildung 16 über dem pH-Wert
aufgetragen. Es ist zu beobachten, dass im pH-Bereich 8-11 die gelaugten Mengen
an organischem Kohlenstoff eine gute Übereinstimmung mit dem bereits von
Bengtsson et al. [51] genannten TOC aufweisen. Ab pH 11,5 hingegen ist eine deutlich
höhere Konzentration von organischem Kohlenstoff ermittelt worden. Da die
Konzentration an Ca-Bentonit für jede Probe der Reihe die gleiche war und auch keine
höheren Gehalte an TOC in eben diesem zu vermuten ist, scheint die Quelle für den
zusätzlichen organischen Kohlenstoff eine andere zu sein. Die Vermutung war, dass
im stark alkalischen pH-Bereich bereits eine Laugung von organischem Kohlenstoff
aus den verwendeten PP-Probenröhrchen beginnt. Diese Vermutung wurde parallel
von S. Shams (HZDR) in einer Probenreihe bestätigt (siehe Abbildung 27 im Anhang).
Da der TOC im pH-Bereich 11,5-13 erhöht ist, ist auch der Korrekturfaktor in diesem
pH-Bereich höher.

Ergebnisse und Diskussion
39
Außerdem hatte der erhöhte TOC bei stark alkalischen pH-Werten zur Folge, dass für
die Batch-Sorptions-Experimente ISA-Konzentrationen im Bereich 0,5-50 mmol/L
verwendet werden mussten. Bei niedrigeren ISA-Konzentrationen wäre es sonst nicht
mehr möglich gewesen den TOC der durch die ISA verursacht wird, von der TOC-
Grundlast des Ca-Bentonits und dem zusätzlichen TOC aus den PP-Probenröhrchen
zu unterscheiden.
Abbildung 16: Ermittelter organischer Kohlenstoff (TOC) aus Ca-Bentonit-Laugung in
Abhängigkeit vom pH-Wert, (*) Bengtsson et al. [51]
Die Bestimmung des anorganischen Kohlenstoffs (TIC) ist von besonderer Bedeutung
um die Menge an im Ca-Bentonit enthaltenem Carbonat zu bestimmen, welches trotz
der Arbeiten unter N2-Atmosphäre dazu führen könnte, dass sich gut lösliche Uran-
Carbonat-Komplexe bilden. In Abbildung 17 sind die ermittelten Konzentrationen für
TC und TIC in Abhängigkeit vom pH-Wert sowie die Literaturwerte von
Bengtsson et al. [51] aufgetragen. Die Werte wurden ausschließlich für den pH-
Bereich 8-11,5 ermittelt, was der Methode, die ein ansäuern und ausblasen der Probe
erfordert und den allgemein geringen Konzentrationen geschuldet ist.
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
5,5
6,0
6,5
7,0
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5
TO
C in m
g/L
pH-Wert
Literaturwert TOC (*)

Ergebnisse und Diskussion
40
Dennoch ist Abbildung 17 zu entnehmen, dass die Konzentrationen an TIC des
verwendeten Ca-Bentonits noch weit unter den erwarteten Literaturwert von
Bengtsson et al. [51] liegt. Ein möglicher Grund für diese Abweichung könnte die
verschiedene Methodik der TC/TIC-Analyse liefern, da Bengtsson et al. [51] ein
thermisches Verfahren nutzte, bei dem sich auch feste Carbonate wie z. B. CaCO3
zersetzen. Der ermittelte Wert für den TIC entspricht in etwa dem Literaturwert für den
TOC und damit einem Gehalt von ~0,02 %, was einer CO32--Konzentration von 143-
612 µmol/L (berechnet aus TIC-Analysedaten) entsprechen würde. Ob diese CO32--
Konzentrationen problematisch sind, muss später anhand des Sorptionsverhaltens
von U(VI) geklärt werden. Der Anstieg von TC und TIC bei pH 11,5 ist abermals auf
den Beginn der Laugung von organischem Kohlenstoff aus den Gefäßwänden
zurückzuführen, welcher sich hier im Summenparameter TC verstärkt niederschlägt.
Aufgrund wiederholt auftretender Komplikationen bei der Analyse, konnten für die
pH- Werte größer 11,5 keine Angaben zum TC und TIC gemacht werden.
Abbildung 17: Ermittelter gesamter (TC) und anorganischer Kohlenstoff (TIC) aus Ca-Bentonit-
Laugung in Abhängigkeit vom pH-Wert, (*) Literaturwerte für TIC und TOC nach
Bengtsson et al. [51]
0
5
10
15
20
25
30
35
40
45
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0
TC
und T
IC in m
g/L
pH-Wert
TC aus Bentonit-Laugung TIC aus Bentonitlaugung
TOC (*)
TIC (*)

Ergebnisse und Diskussion
41
4.2 Sorption von ISA an Ca-Bentonit
Abbildung 18 zeigt die TOC-Daten aus dem Batch-Sorptions-Experiment mit ISA,
wobei die Rohdaten bereits mit den TOC- Werten des Laugungsversuches korrigierten
wurden (siehe Abbildung 16). Der Verlauf der ermittelten Daten weist keinen
besonderen Trend auf. Im Diagramm ist die nach Gleichung 2 (siehe Kapitel 3.2.3)
ermittelte durch ISA zugegebene Menge organischen Kohlenstoffs hervorgehoben.
Beim Vergleich der Daten nach der Sorption mit diesem Ausgangswert und unter
einbeziehen der ebenfalls dargestellten Standardabweichung, kann eine Sorption von
ISA an dem verwendeten Ca-Bentonit unter diesen Bedingungen ausgeschlossen
werden. Auch beim Sorptionsversuch mit U: ISA = 1: 1000 bleibt die ISA-
Konzentration unverändert. Daher kann eine Konkurrenz zwischen Uran- und ISA-
Sorption an Ca-Bentonit, ebenfalls ausgeschlossen werden.
Abbildung 18: Ermittelter organischer Kohlenstoff (TOC) im Überstand nach dem Batch-
Sorptions-Experiment mit ISA-Zugabe von 0,5 mmol/L in Abhängigkeit vom pH-Wert mit
Angabe des 100 %-Wertes der ISA-Zugabe
25
30
35
40
45
50
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5
TO
C in m
g/L
pH-Wert
TOC abzüglich des ermittelten Korrekturwertes aus dem Laugungsversuch
100 % der ISA-Zugabe

Ergebnisse und Diskussion
42
4.3 Einfluss von ISA auf U(VI)-Sorption an Ca-Bentonit
Die folgenden Ergebnisse befassen sich mit dem Sorptionsverhalten von Uran an
Ca- Bentonit unter Einfluss von ISA. Die Sorptionsreihe von Uran an Ca-Bentonit im
pH-Bereich 8-13 soll zum Vergleich mit den U-ISA-Sorptionsreihen dienen, da diese
die "ungestörte" Sorption von Uran an Ca-Bentonit repräsentiert.
Sorptionsverhalten von Uran an Ca-Bentonit
Die Sorption von U(VI) in Abwesenheit von ISA beträgt im pH-Bereich 8-12 etwa 90-
100 % und nimmt erst bei den pH-Werten 12,5 und 13 ab (siehe Abbildung 19). Auf
Grundlage dieser starken Rückhaltung kann ein Einfluss von aus dem Ca-Bentonit
gelöstem Carbonat ausgeschlossen werden, da die U(VI)-Carbonat-Komplexe zu
einer Verbesserung der Löslichkeit und zugleich zu einer verschlechterten Sorption im
betrachteten pH- Bereich geführt hätten [45], [64].

Ergebnisse und Diskussion
43
Sorptionsverhalten von Uran an Ca-Bentonit unter Einfluss von ISA
Alle Daten die aus der ICP-MS-Analyse dieser Sorptionsreihen hervorgingen sind in
Abbildung 19 in direkter Gegenüberstellung mit der unbeeinflussten U(VI)-Sorption
dargestellt.
Abbildung 19: U(VI)-Sorption (5*10-7 mol/L) an Ca-Bentonit (10 g/L) in 0,1 M NaCl in
Abwesenheit von ISA und bei den U-ISA-Verhältnissen 1: 1000, 1: 10.000 und 1: 100.000
Die Gegenüberstellung der Sorptionsreihen zeigt, dass ein 1000- bzw. 10.000-facher
Überschuss an ISA noch keinen nennenswerten Einfluss auf das Sorptionsverhalten
hat. Erst bei einem U(VI)- ISA- Verhältnis von 1: 100.000 ist eine deutliche
Sorptionsminderung zu beobachten, welche sich in ihrer Intensität in Abhängigkeit des
pH-Wertes verändert. Diese Ergebnisse lassen den Schluss zu, dass die
Sorptionsverringerung von U(VI) an Ca-Bentonit ab einem U: ISA-Überschuss
zwischen 1: 10.000 und 1: 100.000 beginnt. Solch ein extremer Überschuss ist jedoch
unter realen Bedingungen in einem Endlager für radioaktive Abfälle nicht zu erwarten.
0
10
20
30
40
50
60
70
80
90
100
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5
% v
on U
O22+
sorb
iert
pH-Wert
U(VI) ohne ISA U : ISA = 1 : 1.000 U : ISA = 1 : 10.000 U : ISA = 1 : 100.000

Ergebnisse und Diskussion
44
Ein möglicher Grund dafür ist, dass der vollständige alkalische Cellulose- Abbau
(99 %), welcher die ISA-Entstehung bedingt, bereits bis zu 5000 Jahre benötigt [65].
Außerdem wird von einer maximalen ISA- Konzentration von 3 µmol/L ausgegangen
[14].
Eine mögliche Erklärung für die Sorptionsverringerung könnte eine pH-Wert abhängige
Konkurrenz zwischen der Bildung des 1:3-U(VI)-OH--Komplexes und U(VI)-ISA-
Komplexen darstellen. Dieser Effekt zeigt sich besonders deutlich bei
U(VI): ISA = 1: 100.000, da der 1:3-U(VI)-OH- -Komplex ohne Einfluss von ISA gut
sorbiert und die Verschlechterung hier nur durch die Bildung von U(VI)-ISA-Komplexen
bedingt sein kann. Entsprechend dieser Hypothese werden zwei
Konkurrenzreaktionen vermutet. Die erste Reaktion (RG 1) besagt, dass mit
steigendem pH-Wert die Bildung von U(VI)-ISA- Komplexen kontinuierlich zurückgeht.
Außerdem ist der ersten Reaktionsgleichung (RG 1) zu entnehmen, dass bei einem
deutlichen ISA-Überschuss auch die Reaktion hin zu den U(VI)-ISA-Komplexen
begünstigt wird. Die Rückreaktion bzw. die Bildung des 1:3-U(VI)-OH- -Komplexes
wiederum wird mit steigendem pH-Wert (steigende OH- -Konzentration) begünstigt. Es
wird vermutet, dass RG 1 für den pH-Bereich 8-11 gültig ist.
[𝑈𝑂2(𝑂𝐻)3]− + 𝑛 𝐼𝑆𝐴 ⇄ 𝑈𝑂2(𝐼𝑆𝐴)𝑛 + 3 𝑂𝐻− (RG 1)
Die zweite Reaktion (RG 2) ist für den pH-Bereich pH 12 gültig. Auch diese Gleichung
besagt, dass mit steigendem pH-Wert die Bildung des 1:4 U(VI)-OH- - Komplexes
begünstigt wird, bei erhöhter ISA-Konzentration jedoch die Bildung von U(VI)-ISA-
Komplexen. Ob der U(VI): ISA-Überschuss von 1: 100.000 ausreichend ist, um die
Bildung des U(VI)-ISA-Komplexes zu begünstigen, muss im Kapitel 4.5 zur
aquatischen Speziation, mittels cryo-TRLFS nachgewiesen werden.
[𝑈𝑂2(𝑂𝐻)4]2− + 𝑛 𝐼𝑆𝐴−𝐻 ⇄ 𝑈𝑂2(𝐼𝑆𝐴−𝐻)𝑛 + 4 𝑂𝐻− (RG 2)

Ergebnisse und Diskussion
45
4.4 Löslichkeit von Uran
Als weiterer Indikator für die Bildung gut löslicher U(VI)-ISA-Komplexe wurde die
U(VI)- Löslichkeit in Abhängigkeit des pH- Wertes untersucht. Die
U(VI)- Konzentrationen in Lösung nach Ultrazentrifugation sind sowohl für die
Probenreihen in Abwesenheit von ISA, als auch für den U/ISA-Überschuss von 1: 1000
in Abbildung 20 dargestellt. Beim Vergleich der beiden Probenreihen ist zu
beobachten, dass die U(VI)-Löslichkeit in Abhängigkeit des pH-Wertes in den pH-
Bereichen 1-4 und 11-13 keine signifikanten Unterschiede aufweist. Im
pH- Bereich 5- 10 fällt jedoch auf, dass die Löslichkeit um 20-50 % erhöht ist, wenn
ISA im 1000-fachen Überschuss zum Uran in Lösung vorliegt. Dies allein lässt die
Vermutung zu, dass im pH- Bereich 5-10 gut lösliche U(VI)-ISA-Komplexe gebildet
werden. Im Vergleich mit den Ergebnissen der Sorptionsversuche, muss jedoch
festgestellt werden, dass selbst bei einem U(VI)-ISA-Überschuss von 1: 10.000 die
Sorption der Komplexbildung mit ISA bei pH 8-10 vorgezogen wird bzw. die gebildeten
U(VI)-ISA-Komplexe gut sorbieren. Allerdings könnten hier auch U(VI)-ISA-Spezies
vorliegen, deren Löslichkeitsgrenze hier ebenfalls nicht überschritten wird. Zudem
besteht die Möglichkeit, dass sowohl U(VI)-OH-- als auch U(VI)-ISA-Komplexe
nebeneinander vorliegen. Aufschluss darüber sollen die TRLFS-Spektren in
Kapitel 4.5 geben. Gleiches gilt für den pH-Bereich 11-13, wobei hier aufgrund der
extrem hohen OH--Konzentration eine Bildung von [UO2(OH)4]2- in beiden
Versuchsreihen wahrscheinlicher ist.
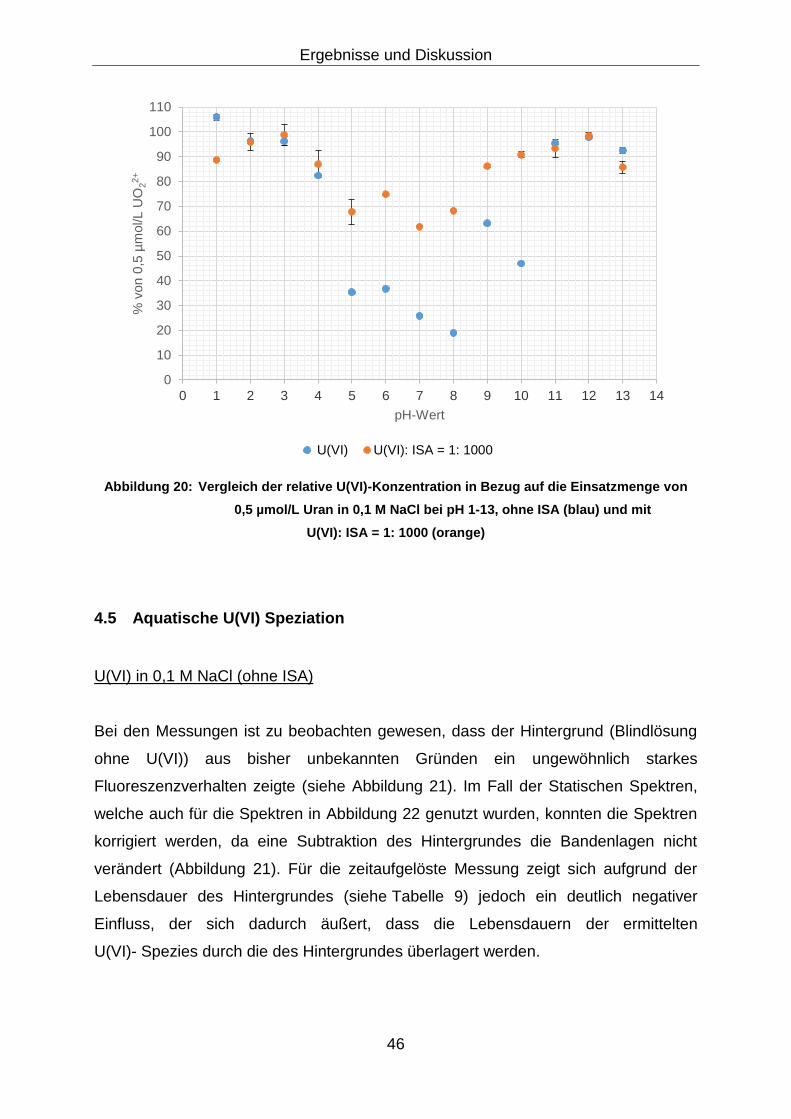
Ergebnisse und Diskussion
46
Abbildung 20: Vergleich der relative U(VI)-Konzentration in Bezug auf die Einsatzmenge von
0,5 µmol/L Uran in 0,1 M NaCl bei pH 1-13, ohne ISA (blau) und mit
U(VI): ISA = 1: 1000 (orange)
4.5 Aquatische U(VI) Speziation
U(VI) in 0,1 M NaCl (ohne ISA)
Bei den Messungen ist zu beobachten gewesen, dass der Hintergrund (Blindlösung
ohne U(VI)) aus bisher unbekannten Gründen ein ungewöhnlich starkes
Fluoreszenzverhalten zeigte (siehe Abbildung 21). Im Fall der Statischen Spektren,
welche auch für die Spektren in Abbildung 22 genutzt wurden, konnten die Spektren
korrigiert werden, da eine Subtraktion des Hintergrundes die Bandenlagen nicht
verändert (Abbildung 21). Für die zeitaufgelöste Messung zeigt sich aufgrund der
Lebensdauer des Hintergrundes (siehe Tabelle 9) jedoch ein deutlich negativer
Einfluss, der sich dadurch äußert, dass die Lebensdauern der ermittelten
U(VI)- Spezies durch die des Hintergrundes überlagert werden.
0
10
20
30
40
50
60
70
80
90
100
110
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
% v
on 0
,5 µ
mol/L U
O22+
pH-Wert
U(VI) U(VI): ISA = 1: 1000

Ergebnisse und Diskussion
47
Damit ist die Vergleichbarkeit mit den Daten aus der Literatur und die Identifikation
einiger der U(VI)- OH- - Spezies auf Grundlage der Lebensdauern nicht möglich,
weshalb von deren Auflistung abgesehen wurde. Nichtsdestotrotz können die
statischen Spektren zum Vergleich herangezogen werden.
Abbildung 21: Darstellung der Problematik des stark fluoreszierenden Hintergrundes am
Beispiel des Lumineszenzspektrums von pH 1
In Abbildung 22 sind die Lumineszenzspektren von 0,5 µmol/L U(VI) in 0,1 M NaCl im
pH- Bereich 1-13 dargestellt. Mit der Gegenüberstellung der Spektren über den
gesamten pH-Bereich sollen pH-Bereiche ermittelt werden, welchen einzelnen
Spezies zugeordnet werden können. Dafür werden die Bandenlage und deren
Verschiebung zueinander betrachtet. Um detailliertere Aussagen die vorliegende
Spezies treffen zu können, wurden die Peakpositionen ermittelt und in Tabelle 9
aufgelisteten. Diese Daten können dann mit bereits bekannten spektralen Daten aus
der Literatur verglichen werden.
380 400 420 440 460 480 500 520 540 560 580 600 620 640 660
0
2000
4000
6000
8000
Flu
ore
szenzin
tensität
[a.u
.]
Wellenlänge [nm]
Hintergrund
pH 1
pH 1 korrigiert

Ergebnisse und Diskussion
48
Abbildung 22: Lumineszenzspektren von U(VI) (0,5 µmol/L) in 0,1 M NaCl bei pH 1-13
Tabelle 9: Peakpositionen und zugeordnete U(VI)-Spezies mit Uran = 0,5 µmol/L in 0,1 M
NaCl bei pH 1- 13
pH-Wert
Peakpositionen
Zugeordnete
Speziation
1 491,8 514,4 539,4
“UO22+“
U-Cl--Komplex
2 492,2 514,9 539,0
3 491,8 514,4 538,5
4 491,8 513,5 537,6
5 492,2 514,0 538,0
U-OH--Komplexe 6 490,4 514,4 538,6
7 494,5 517,2 542,7
8 492,2 513,5 538,6
9 480,6 501,9 521,4 U-OH--Komplexe
10 niedrige Fluoreszenzintensität U-OH--Komplexe
11 489,9 509,8 531,1 U-OH--Komplexe
12 488,5 508,4 531,1
13 490,8 509,8 531,6 [UO2(OH)4]2-
Hintergrund Lebensdauer [µs] = 590 ± 50 /
460 480 500 520 540 560 580 600
Wellenlänge [nm]
pH 13
pH 12
pH 11
pH 10
pH 9
pH 8
pH 7
pH 6
pH 5
pH 4
pH 3
pH 2
pH 1

Ergebnisse und Diskussion
49
Bei der Auswertung der Fluoreszenzspektren, wurden sechs pH-Bereiche ermittelt,
innerhalb derer sich die spektralen Daten ähneln und daher jeweils eine
vorherrschende U(VI)-Spezies repräsentieren könnten (siehe Tabelle 9). Die erste
Spezies wurde für den pH-Bereich 1-4 identifiziert, wobei es sich um einen U(VI)-Cl--
Komplex handelt. Die Identifizierung erfolgte über den Abgleich mit den cryo-TRLFS-
Daten von H. Brinkmann (HZDR) in Tabelle 10. An dieser Stelle ist es wichtig zu
erwähnen, dass die Untersuchungen von H. Brinkmann (HZDR) den Einfluss von
verschiedenen Hintergrundelektrolyten auf die Bandenlage der Spektren beinhaltet. In
Abbildung 23 ist eine Rotverschiebung der Peakpositionen der Spektren in 1 M NaCl-
Lösung durch den Einfluss von Chlorid-Ionen zu beobachten. Diese Rotverschiebung
ist auch im hier untersuchten System zu beobachten, da die Positionen der ersten
beiden Maxima bei 492 und 514 nm liegen und damit bei eindeutig höheren
Wellenlängen als in der Literatur für das Aquo-Ion hinterlegt [56].
Tabelle 10: Referenzdaten für UO22+ aus cryo-TRLFS von H. Brinkmann (HZDR)
Komplex Peakpositionen [nm] τ [µs] Elektrolyt pH-
Wert
U / ISA
[mmol/L]
UO22+ 491 513 538 564 331,0 ± 4,4 1 M NaCl 3 U: 15 / ISA: 0
485 508 532 558 286,0 ± 3,1 ohne
Abbildung 23: Referenzspektren für UO22+ von H. Brinkmann (HZDR)
460 480 500 520 540 560 580 600 620 640
0
5000
10000
15000
20000
Flu
ore
szenzin
tensität
[a.u
.]
Wellenlänge [nm]
NaCl
ohne Elektrolyt
pH 3 // 0 mmol/L ISA

Ergebnisse und Diskussion
50
Bei der zweiten eindeutig identifizierten Spezies handelt es sich um [UO2(OH)4]2- bei
pH 13. Die Existenz dieser Spezies konnte mit Hilfe der Peakpositionen 491 und
510 nm von Tits et al. [66] bestätigt werden, welcher nahezu identische
cryo- Bedingungen für seine TRLFS-Messungen (153 ± 5 K) verwendete.
Weitere U(VI)-OH-Spezies werden in den pH-Bereichen 5-8, 9,10 und 11-12 vermutet.
Aufgrund des Mangels an geeigneten cryo-TRLFS-Daten aus der Literatur ist es
jedoch nicht im vollen Umfang möglich, diese Spezies in Zusammensetzung und
Struktur aufzuklären. Anhand des Wissens über die U(VI)-Spezies in Abhängigkeit des
pH- Wertes kann jedoch bestimmt werden, dass es sich um U(VI)- OH- - Komplexe
handeln muss. Um detailliertere Aussagen über die Speziation anhand der TRLFS-
Spektren machen zu können, sind an dieser Stelle weitere Untersuchungen
erforderlich. Dies war jedoch im Rahmen dieser Arbeit nicht möglich.
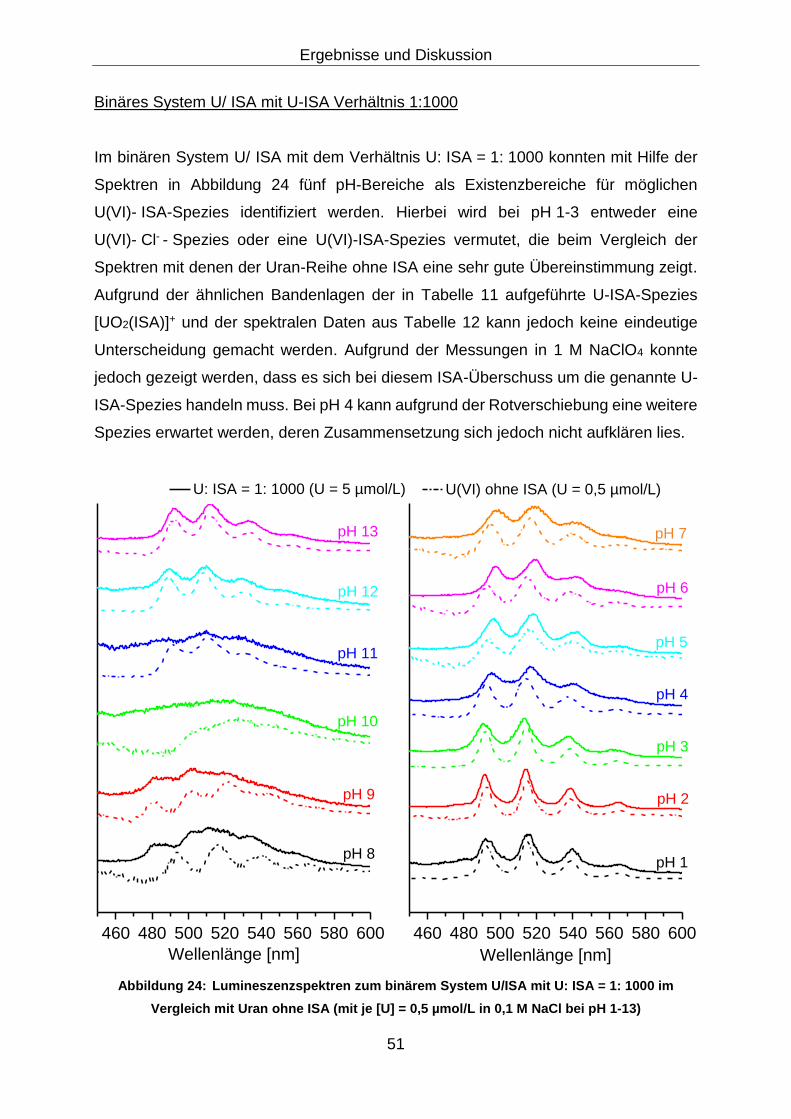
Ergebnisse und Diskussion
51
Binäres System U/ ISA mit U-ISA Verhältnis 1:1000
Im binären System U/ ISA mit dem Verhältnis U: ISA = 1: 1000 konnten mit Hilfe der
Spektren in Abbildung 24 fünf pH-Bereiche als Existenzbereiche für möglichen
U(VI)- ISA-Spezies identifiziert werden. Hierbei wird bei pH 1-3 entweder eine
U(VI)- Cl- - Spezies oder eine U(VI)-ISA-Spezies vermutet, die beim Vergleich der
Spektren mit denen der Uran-Reihe ohne ISA eine sehr gute Übereinstimmung zeigt.
Aufgrund der ähnlichen Bandenlagen der in Tabelle 11 aufgeführte U-ISA-Spezies
[UO2(ISA)]+ und der spektralen Daten aus Tabelle 12 kann jedoch keine eindeutige
Unterscheidung gemacht werden. Aufgrund der Messungen in 1 M NaClO4 konnte
jedoch gezeigt werden, dass es sich bei diesem ISA-Überschuss um die genannte U-
ISA-Spezies handeln muss. Bei pH 4 kann aufgrund der Rotverschiebung eine weitere
Spezies erwartet werden, deren Zusammensetzung sich jedoch nicht aufklären lies.
Abbildung 24: Lumineszenzspektren zum binärem System U/ISA mit U: ISA = 1: 1000 im
Vergleich mit Uran ohne ISA (mit je [U] = 0,5 µmol/L in 0,1 M NaCl bei pH 1-13)
460 480 500 520 540 560 580 600 460 480 500 520 540 560 580 600
Wellenlänge [nm]
pH 13
pH 12
pH 11
pH 10
pH 9
pH 8
Wellenlänge [nm]
pH 1
pH 2
pH 3
pH 4
pH 5
pH 6
pH 7
U(VI) ohne ISA (U = 0,5 µmol/L)U: ISA = 1: 1000 (U = 5 µmol/L)

Ergebnisse und Diskussion
52
Die Untersuchungen von H. Brinkmann (HZDR) haben ergeben, dass in Abhängigkeit
vom ISA-Überschuss bis zu zwei U(VI)-ISA-Spezies im pH- Bereich 1-3 zu erwarten
sind. Die Bandenlagen sind in Tabelle 11 zusammengefasst.
Im pH- Bereich 5-7 ist Im Vergleich zu den Lumineszenzspektren der U(VI)-Reihe
ohne ISA eine deutliche Rotverschiebung der Spektren zu beobachten. Auf Basis der
durchgeführten Löslichkeitsuntersuchungen von U(VI) mit und ohne ISA (siehe
Abbildung 20), lässt sich für diesen pH- Bereich aufgrund der verbesserten Löslichkeit
eindeutig die Bildung einer weiteren U(VI)-ISA-Spezies ableiten. Die nächste
U(VI)- ISA- Spezies wird im pH- Bereich 8-9 beobachtet. Beim Vergleich der hier
bestimmten Peakpositionen 481, 501 und 522 nm zum System ohne ISA mit 492, 513
und 539 nm ist vor allem für pH 8 eine deutliche Veränderung zu beobachten, was
durch eine erhöhte Löslichkeit bestätigt wird. Für die U(VI)-ISA-Komplexe bei pH 8 und
9 kann der 1:2 Komplex [UO2(ISA)2] vermutet werden, was durch den Vergleich mit
den Peakpositionen von H. Brinkmann (HZDR) 482, 502 und 523 nm deutlich wird
(siehe Tabelle 11).
Der pH- Bereich 10-13 wird zusammengefasst, weil die Spektrenqualität keine
belastbaren Aussagen zu U-ISA-Spezies zulassen. Die niedrige Spektrenqualität kann
nicht allein auf die Beteiligung von ISA zurückgeführt werden. Vielmehr sind
wahrscheinlich zwischenzeitlich notwendige Umbauten am Lasersystem für die
veränderte Spektrenqualität verantwortlich. Da sich die Peakpositionen nicht eindeutig
von denen von U(VI) ohne ISA unterscheiden lassen, werden für den stark alkalischen
pH- Bereich bei dem gegebenen U(VI): ISA-Überschuss von 1: 1000 keine U(VI)-ISA-
Spezies vermutet, sondern weiterhin die Bildung anionischer U(VI)-Hydroxo-
Komplexe. Dies bestätigt die bereits beim Sorptionsversuch beobachtete Konkurrenz
zwischen Hydrolyse und Komplexbildung mit ISA.

Ergebnisse und Diskussion
53
Tabelle 11: Referenzdaten für [UO2(ISA)2] und [UO2(ISA)]+ aus cryo-TRLFS von
H. Brinkmann (HZDR)
Komplex Peakpositionen [nm] τ [µs] Elektrolyt pH-
Wert
U / ISA
[mmol/L]
[UO2(ISA)2] 482 502 523 548 606,7 ± 21,5 1 M NaClO4 3 U: 15 / ISA: 100
[UO2(ISA)]+
491 513 537 562 302,4 ± 4,3
1,5
U: 15 / ISA: 240
492 515 539 566 385,3 ± 6,5 1 M NaCl U: 15 / ISA: 200
491 513 538 564 307,6 ± 7,8 ohne
Tabelle 12: Peakpositionen und Speziationen vom binären System U : ISA = 1: 1000 mit
Uran = 0,5 µmol/L in 0,1 M NaCl bei pH 1-13
pH-Wert
Peakpositionen
Speziation
1 491,2 512,5 538,1 “UO22+“
U-Cl--Komplex
[UO2(ISA)]+
2 491,9 513,7 539,3
3 492,5 515,6 539,0
4 494,9 517,1 541,5 U-ISA-Komplexe
5 495,6 515,9 536,5
U-ISA-Komplexe 6 495,6 516,2 537,1
7 498,3 518,1 539,9
8 480,1 501,7 521,8 [UO2(ISA)2]
9 480,8 501,1 522,4
10 482,3 504,5 520,2
U-ISA-Komplexe/
U-OH--Komplexe
11 482,9 505,7 523,6
12 489,1 509,7 521,4
13 491,2 508,8 523,6

Ergebnisse und Diskussion
54
Ternäres System Ca-Bentonit/ U/ ISA mit U-ISA-Verhältnis 1: 100.000
Abbildung 25 zeigt die aufgenommenen Lumineszenzspektren aus dem Überstand
des Sorptionsversuch mit U: ISA = 1: 100.000 im Vergleich mit den aquatischen
Spezies ohne ISA im pH-Bereich 8-13. Ziel dieser Untersuchung ist es die U-ISA-
Spezies zu identifizieren, die für die Sorptionsminderung von U(VI) am Ca-Bentonit bei
einem U-ISA-Verhältnis von 1: 100.000 verantwortlich sind. Der Vergleich der
Spektren in Abbildung 25 lässt auf mindestens zwei pH-Bereiche schließen, bei denen
neue Uran Spezies mit der Beteiligung von ISA vorliegen.
Abbildung 25: Lumineszenzspektren des Überstandes aus dem Sorptionsversuch Ca-Bentonit/
U/ ISA mit U: ISA = 1: 100.000 im Vergleich zur aquatischen Speziation ohne ISA
([U] = 0,5 µmol/L in 0,1 M NaCl bei pH 8-13)
460 480 500 520 540 560 580 600
Wellenlänge [nm]
pH 13
pH 12
pH 11
pH 10
pH 9
pH 8
U: ISA = 1: 100.000 U

Ergebnisse und Diskussion
55
Es wird eine Spezies im pH-Bereich 8-10 und eine zweite im pH- Bereich 11-13
beobachtet. Vergleicht man die Peakpositionen der Spektren bei pH 8-10 mit denen in
Tabelle 11, kann für diesen Bereich wiederum der 1:2 Komplex [UO2(ISA)2]
angenommen werden. Im pH- Bereich 11-13 kann eine weitere deutliche
Blauverschiebung der Peakpositionen beobachtet werden, was auf die Bildung einer
weiteren Spezies schließen lässt. Des Weiteren wird eine deutlich niedrigere
Spektrenqualität beobachtet. Der extreme Unterschied zum vorher beschriebenen
1:4 Hydroxo-Komplex und die deutliche Sorptionsminderung, lässt darauf schließen,
dass bei diesem extremen Überschuss von 1: 100.000 keine U(VI)-OH-Spezies mehr
gebildet werden, sondern U(VI)-ISA-Spezies bzw. möglicherweise eine gemischte
U(VI)- ISA- OH-Spezies wie die von Warwick et al. [33] beschriebene “UO2ISA(OH)4“
im entsprechenden pH-Bereich. Dies steht im Einklang mit der im Kapitel 4.3
angestellten Behauptung, dass der U(VI)-ISA-Überschuss von 1: 100.000 die
Gleichgewichtslage von der Hydrolyse-Spezies hin zum U(VI)-ISA-Komplex
verschiebt.
Tabelle 13: Peakpositionen und Speziationen vom Sorptionsversuch mit Ca-Bentonit und
U: ISA = 1: 100.000 mit Uran = 0,5 µmol/L in 0,1 M NaCl bei pH 1-13
pH-Wert
Peakpositionen
Speziation
8 480,6 501,5 523,4
[UO2(ISA)2] 9 480,9 501,2 523,4
10 481,5 501,8 522,2
11 478,1 499,7 514,5 U-ISA-Komplexe
U-OH/ ISA-Mischkomplexe
UO2ISA(OH)4 [33]
12 477,5 496,3 513,5
13 473,2 499,7 512,9

Zusammenfassung und Ausblick
56
5 Zusammenfassung und Ausblick
Der Laugungsversuch hat gezeigt, dass der verwendete Ca-Bentonit bis zu einem
pH- Wert von 12 stabil ist. Außerdem wurde ermittelt, dass der verwendete
Ca- Bentonit ~0,02 Masse-% organischen Kohlenstoff (~2 mg/L TOC) enthält. Bei
höheren pH-Werten ist zu beobachten gewesen, dass zusätzlich organischer
Kohlenstoff aus den Gefäßwänden der PP- Probenröhrchen gelaugt wurde, welche als
Korrekturwert abgezogen werden musste. Die TOC-Analysen im Sorptionsversuch
ISA/ Ca-Bentonit haben ergeben, dass ISA nicht an Ca-Bentonit sorbiert. Zudem sind
die mit Hilfe der TIC-Analyse ermittelten Carbonat-Konzentrationen (143-612 µmol/L)
aus dem Laugungsversuch von Ca-Bentonit zu gering, um einen Einfluss auf die
Löslichkeit und letztendlich auf die Sorption von U(VI) an Ca-Bentonit zu haben.
Der Löslichkeitsversuch hat ergeben, dass die U(VI)-Löslichkeit bei U: ISA = 1: 1000
im pH-Bereich 5-10 deutlich verbessert ist. Des Weiteren wurde beobachtet, dass die
U(VI)-Sorption an Ca-Bentonit bei U: ISA = 1: 1000 noch nicht beeinflusst ist. Jedoch
zeigt sich im Vergleich mit dem Löslichkeitsversuch, das besonders bei pH 8-10 die
Sorption der Komplexbildung gegenüber bevorzugt wird, bzw. die gebildeten U-ISA-
Komplexe am Ca-Bentonit sorbieren müssen.
Eine deutliche Sorptionsminderung findet dann erst im Bereich zwischen den beiden
U-ISA-Verhältnissen 1: 10.000 und 1: 100.000 statt. Die Sorptionsminderung ist auf
zwei Gleichgewichtsbeziehung zwischen U(VI), ISA und OH- in Abhängigkeit des pH-
Wertes zurückzuführen. Wichtig ist im Zusammenhang der Sorptionsminderung, dass
die verwendeten ISA-Konzentrationen nicht mehr den realen Bedingungen im
Endlager entsprechen, was den Schluss zulässt, dass das Endlager auch im Fall eines
Wassereinbruchs das U(VI) zurückhält.
Im aquatischen System U(VI)-ISA wurden zwei U(VI)-ISA-Spezies, der 1:1- Komplex
[UO2(ISA)]+ und der 1:2-Komplex [UO2(ISA)2] identifiziert. Die Untersuchungen zum
Überstand des Sorptionsversuches mit einem U: ISA-Überschuss von 1: 100.000
lassen den Rückschluss auf die Existenz von zwei U-ISA-Spezies zu, den 1:2-Komplex
[UO2(ISA)2] bei pH 8-10 und eine weitere U-ISA- bzw. U-OH-ISA-Spezies
UO2ISA(OH)4 bei pH 11-13.

Zusammenfassung und Ausblick
57
Zukünftige Untersuchungen könnten den Fokus darauflegen, durch die Verwendung
verschiedener U(VI)-Konzentrationen bei den U(VI)-ISA-Verhältnissen 1: 1000,
1: 10.000 und 1: 100.000 zu ermitteln, ob die Sorptionsminderung an Ca-Bentonit auf
das U(VI)-ISA-Verhältnis oder die Absolutkonzentrationen zurückzuführen ist.
Außerdem interessant erscheinen Wiederholungen der TRLFS-Messungen mit dem
Schwerpunkt vergleichbare Lebensdauern zu ermitteln und diesbezüglich den Einfluss
des Hintergrundes auszuschließen oder gezielt zu untersuchen. Die Wiederholung der
Aufnahme von TRLFS-Spektren ist vor allem für die eindeutige Aufklärung der
Speziation im ternären System U(VI)/ ISA/ Ca-Bentonit erforderlich.

Eidesstaatliche Erklärung über die Eigenständige Erstellung der Arbeit
58
6 Eidesstaatliche Erklärung über die Eigenständige Erstellung der Arbeit
Hiermit erkläre ich, Paul Dullies, dass die vorgelegte Arbeit mit dem Titel:
„Untersuchungen zum Einfluss von Isosaccharinsäure auf die U(VI)-Rückhaltung an
Ca-Bentonit unter alkalischen Bedingungen“
selbständig von mir verfasst wurde, keine anderen als die angegebenen Quellen und
Hilfsmittel verwendet sowie alle wörtlich oder sinngemäß übernommenen Stellen in
der Arbeit als solche und durch Angabe der Quelle gekennzeichnet wurden. Dies gilt
auch für bildliche Darstellungen und Quellen aus dem Internet. Mir ist bewusst, dass
die Hochschule für Technik und Wirtschaft Dresden Qualifizierungsarbeiten
stichprobenartig mittels Verwendung von Software zur Erkennung von Plagiaten
überprüft.
……………………… ……………………...
Ort, Datum Unterschrift Student

Danksagung
59
7 Danksagung
An dieser Stelle möchte ich mich bei allen meinen Kolleginnen und Kollegen am Institut
für Ressourcenökologie am HZDR bedanken, die mich während der erlebnis- und
lehrreichen Zeit während meines Masterstudiums begleitet haben. Ein besonderer
Dank gilt dabei meinen Betreuern, T. Philipp, H. Brinkmann und Prof. Dr. V. Brendler
für die großartige Unterstützung und Beratung bei den theoretischen und
experimentellen Arbeiten. Außerdem gilt mein Dank Dr. K. Schmeide
(wissenschaftliche Debatten), S. Weiß (Zentrifugen, Uran-Stammlösung), S. Beutner
und S. Bachmann (ICP-MS), C. Eckardt (TC/TIC/TOC), Dr. R. Steudtner (TRLFS),
A. Rumpel und S. Henke (Labor und Gloveboxen) und H. Lösch (Glovebox, TRLFS).
Des Weiteren möchte ich S. Shams Aldin Azzam danken für seine engagierte Hilfe bei
den täglichen Arbeiten im Labor und im Büro. Ein großer Dank gilt auch dem BMWi für
die Finanzierung im Rahmen des GRaZ-Projektes (Geochemische
Radionuklidrückhaltung an Zementalterationsphasen) (No. 02 E11415B), ohne die
auch diese Arbeit nicht möglich gewesen wäre.

Literaturverzeichnis
VII
Literaturverzeichnis
[1] C. Fahrenholz, B. Förster, U. Noseck, and I. Müller-Lyda,
“Ressortforschungsberichte zur kerntechnischen Sicherheit und zum
Strahlenschutz,” 2013.
[2] M. Jobmann et al., “Sicherheits- und Nachweismethodik für ein Endlager im
Tongestein in Deutschland,” 2017.
[3] Bundesamt für Umweltschutz und Reaktorsicherheit, “Inventory of Radioactive
Waste: Current Inventory as at August 2015,” 2015.
[4] K. Appunn, “What to do with the nuclear waste-the storage question 2017,”
cleanenergywire.org/factsheets, 2017. [Online]. Available:
https://www.cleanenergywire.org/factsheets/what-do-nuclear-waste-storage-
question. [Accessed: 17-Aug-2018].
[5] Nuclear Energy Agency, “Radioactive waste managment programmes in
OECD/NEA member countries: Germany,” 2005.
[6] G. Akbulut, D. Fraktion, and D. I. E. Linke, “Deutscher Bundestag Antwort,” no.
August, 2018.
[7] G. Jasmund, K. and Lagaly, Tonminerale und Tone. Steinkopff Verlag
Darmstadt, 1993.
[8] D. Heim, Tone und Tonminerale : Grundlagen der Sedimentologie und
Mineralogie. Ferdinand Enke Verlag, Stuttgart, 1990.
[9] U. Berner, “A Thermodynamic Description of the Evolution of Pore Water
Chemistry and Uranium Speciation during the Degradation of Cement,” PSI-
Bericht, vol. 62, p. 78, 1990.
[10] L. R. Van Loon, M. A. Glaus, S. Stallone, and A. Laube, “Sorption of
isosaccharinic acid, a cellulose degradation product, on cement,” Environ. Sci.
Technol., vol. 31, no. 4, pp. 1243–1245, 1997.
[11] R. W. U. C. Bannick, B. Engelmann, R. Fendler, J. Frauenstein, H. Ginzky, C.
Hornemann, O. Ilvonen, B. Kirschbaum G. Penn-Bressel, J. Rechenberg, S.
Richter, L. Roy, “Grundwasser in Deutschland,” Ref. Öffentlichkeitsarbeit des
Bundesministerium für Umwelt, Naturschutz und Reakt., vol. 1, p. 72 (8, 9), 2008.
[12] L. Abrahamsen et al., “A review of anthropogenic organic wastes and their
degradation behaviour,” p. 97, 2015.

Literaturverzeichnis
VIII
[13] J. Garcia, D., Grive, M., Duro, L., Brassinnes, S., de Pablo, “The potential role of
the degradation products of cement superplasticizers on the mobility of
radionuclides,” Appl. Geochemistry, vol. 98, pp. 1–9, 2018.
[14] J. Tits, E. Wieland, and M. H. Bradbury, “The effect of isosaccharinic acid and
gluconic acid on the retention of Eu(III), Am(III) and Th(IV) by calcite,” Appl.
Geochemistry, vol. 20, no. 11, pp. 2082–2096, 2005.
[15] L. R. Van Loon, M. a Glaus, a Laube, and S. Stallone, “Degradation of Cellulosic
Materials Under the Alkaline Conditions of a Cementitious Repository for Low-
and Intermediate-Level Radioactive Waste. II. Degradation Kinetics,” J. Polym.
Environ., vol. 7, no. 1, pp. 41–51, 1999.
[16] G. M. N. Baston, J. A. Berry, K. A. Bond, K. A. Boult, M. Brownsword, and C. M.
Linklater, “Effects of cellulosic degradation products on uranium sorption in the
geosphere,” J. Alloys Compd., vol. 213/214, no. C, pp. 475–480, 1994.
[17] M. Felipe-Sotelo, J. Hinchliff, N. Evans, P. Warwick, and D. Read, “Sorption of
radionuclides to a cementitious backfill material under near-field conditions,”
Mineral. Mag., vol. 76, no. 8, pp. 3401–3410, 2012.
[18] I. R. Elisenda Colàs, Mireia Grivé, “Complexation of Uranium(VI) by Gluconate
in Alkaline Solutions,” J. Solut. Chemestry, vol. 42, no. 7, pp. 1545–1557, 2013.
[19] D. Rai, L. Rao, R. C. Moore, R. Bontchev, and K. Holt, “Development of
biodegradable isosaccharinate-containing foams for decontamination of
actinides: thermodynamic and kinetic reactions between isosaccharinate and
actinides on metal and concrete surfaces,” pp. 1–3, 2004.
[20] P. B. Shaw, “Studies of the Alkaline Degradation of Cellulose and the Isolation
of Isosaccharinic Acids,” 2013.
[21] K. H. Birjkumar, N. D. Bryan, and N. Kaltsoyannis, “Is gluconate a good model
for isosaccharinate in uranyl(vi) chemistry? A DFT study,” Dalt. Trans., vol. 41,
no. 18, pp. 5542–5552, 2012.
[22] D. Rai and A. Kitamura, “Evaluation of equilibrium constants for deprotonation
and lactonisation of α-D-isosaccharinic acid,” J. Nucl. Sci. Technol., vol. 53, no.
4, pp. 459–467, 2016.
[23] S. Ekberg, C. Ekberg, and Y. Albinsson, “Characterization of α-isosaccharinic
acid: Lactone and carboxylic conformations,” J. Solution Chem., vol. 33, no. 5,
pp. 465–477, 2004.

Literaturverzeichnis
IX
[24] L. Cho, H., Rai, D., Hess, N. J., Xia, Y., Rao, “Acidity and structure of
isosaccharinate in aqueous solution: a nuclear magnetic resonance study,” J.
Solut. Chem., vol. 32, pp. 691–702, 2003.
[25] L. R. Van Loon, M. a Glaus, A. Laube, and S. Stallone, “Degradation of Cellulosic
Materials under the Alkaline Conditions of a Cementitious Repository for Low-
and Intermediate Level Radioactive Waste. Part III: Effect of Degradation
Products on the Sorption of Radionuclides on Feldspar,” J. Polym. Environ., vol.
86, no. 1, pp. 183–189, 1999.
[26] W. Hollemann, A.F., “Lehrbuch der Anorganischen Chemie,” vol. 102, p. 2181,
2007.
[27] K. Lieser, Einführung in die Kernchemie. VCH Verlagsgesellschaft, Weinheim,
1991.
[28] J. A. N. Choppin, G., Liljenzin, J.-O. and Rydberg, Radiochemistry and Nuclear
Chemistry (Second Edition). 1995.
[29] S. Cotton, Lanthanide and Actinide Chemistry. John Wiley & Sons Ltd,
Chichester, England, 2006.
[30] H. Silva, R.J. and Nitsche, “Actinide environmental Chemistry,” Radiochim. Acta,
vol. 70/71, 1995.
[31] N. Takeno, “Atlas of Eh-pH diagrams Intercomparison of thermodynamic
databases,” AIST Tokyo, no. 419, p. 285, 2005.
[32] Y. Zhang et al., “Adsorption and desorption of uranium(VI) onto humic acids
derived from uranium-enriched lignites,” Water Sci. Technol., vol. 77, no. 4, pp.
920–930, 2018.
[33] P. Warwick, N. Evans, and S. Vines, “Studies on some divalent metal α-
isosaccharinic acid complexes,” Radiochim. Acta, vol. 94, no. 6–7, pp. 363–368,
2006.
[34] V. Guillaumont, R., Fanghänel, T., Fuger, J., Grenthe, I., Neck and M. H. Palmer,
D. A., Rand, “Update on the Chemical Thermodynamics of Uranium, Neptunium,
Plutonium, Americium and Technetium,” Chem. Thermodyn., vol. 5, 2003.
[35] J. A. Berry, K. A. Bond, D. R. Ferguson, and N. J. Pilkington, “Studies of the
effects of organic materials on the sorption of uranium and plutonium.,”
Radiochim. Acta, vol. 52/53, pp. 201–210, 1991.

Literaturverzeichnis
X
[36] T. Hartmann, E., Geckeis, H., Rabung, T., Lützenkirchen, J., Fanghänel,
“Sorption of radionuclides onto natural clay rocks,” Radiochim. Acta, vol. 96,
2008.
[37] J. Maher, K., Bargar, J.R. and Brown, G.E., “Environmental speciation of
actinides,” Inorg Chem, vol. 52, pp. 3510–3532, 2013.
[38] H. Akcay, “Aqueous speciation and pH effect on the sorption behavior of uranium
by montmorillonite,” J. Radioanal. Nucl. Chem., vol. 237, pp. 133–137, 1998.
[39] D. E. Chisholm-Brause, C.J., Berg, J.M., Little, K.M., Matzner, R.A. and Morris,
“Uranyl sorption by smectites: spectroscopic assessment of thermodynamic
modeling,” J. Colloid Interface Sci., vol. 277, pp. 366–382, 2004.
[40] M. H. Bradbury and B. Baeyens, “Modelling the sorption of Mn(II), Co(II), Ni(II),
Zn(II), Cd(II), Eu(III), Am(III), Sn(IV), Th(IV), Np(V) and U(VI) on montmorillonite:
Linear free energy relationships and estimates of surface binding constants for
some selected heavy metals and actinide,” Geochim. Cosmochim. Acta, vol. 69,
no. 4, pp. 875–892, 2005.
[41] G. E. Catalano, J.G. and Brown, “Uranyl adsorption onto montmorillonite:
Evaluation of binding sites and carbonate complexation,” Geochim. Cosmochim.
Acta, vol. 69, pp. 2995–3005, 2005.
[42] B. J. Bachmaf, S., Planer-Friedrich, B. and Merkel, “Effect of sulfate, carbonate,
and phosphate on the uranium(VI) sorption behavior onto bentonite,” Radiochim.
Acta, vol. 96, 2008.
[43] C. Meleshyn, A., Azeroual, M., Reeck, T., Houben, G., Riebe, B. and
Bunnenberg, “Influence of (Calcium-)Uranyl-Carbonate Complexation on U(VI)
Sorption on Ca- and Na-Bentonites,” Environ. Sci. Technol., vol. 43, pp. 4896–
4901, 2009.
[44] S. Ivanov, P., Griffiths, T., Bryan, N.D., Bozhikov, G. and Dmitriev, “The effect of
humic acid on uranyl sorption onto bentonite at trace uranium levels,” J. Environ.
Monit., vol. 14, pp. 2968–2975, 2012.
[45] M. Marques Fernandes, B. Baeyens, R. Dähn, A. C. Scheinost, and M. H.
Bradbury, “U(VI) sorption on montmorillonite in the absence and presence of
carbonate: A macroscopic and microscopic study,” Geochim. Cosmochim. Acta,
vol. 93, pp. 262–277, 2012.

Literaturverzeichnis
XI
[46] P. K. Verma, P.K., Pathak, P., Mohapatra, M., Yadav, A.K., Jha, S.,
Bhattacharyya, D. and Mohapatra, “Spectroscopic investigations on sorption of
uranium onto suspended bentonite: effects of pH, ionic strength and complexing
anions,” Radiochim. Acta, vol. 103, 2015.
[47] A. Schnurr, “Untersuchungen zur Radionuklidsorption an
Tonmineraloberflächen bei hohen Ionenstärken,” Karlsruher Institut für
Technologie (KIT), Karlsruhe, 2015.
[48] M. Felipe-Sotelo, J. Hinchliff, L. P. Field, A. E. Milodowski, O. Preedy, and D.
Read, “Retardation of uranium and thorium by a cementitious backfill developed
for radioactive waste disposal,” Chemosphere, p. 46, 2017.
[49] E. Wieland, J. Tits, J. P. Dobler, and P. Spieler, “The effect of α-isosaccharinic
acid on the stability of and Th(IV) uptake by hardened cement paste,” Radiochim.
Acta, vol. 90, no. 9–11, pp. 683–688, 2002.
[50] T. Reich, “Teilprojekt A : Arbeitsprogramm der Universität Mainz im Rahmen des
Verbundprojektes GRaZ,” 2015.
[51] A. Bengtsson and K. Pedersen, “Microbial sulphide-producing activity in water
saturated Wyoming MX-80, Asha and Calcigel bentonites at wet densities from
1500 to 2000 kg m− 3,” Appl. Clay Sci., vol. 137, pp. 203–212, 2017.
[52] T. Philipp, “Progress Report 1 : Sorption of U ( VI ) by Ca-bentonite under saline
and ( hyper ) alkaline conditions,” 2017.
[53] J. N. Whistler, R. L.; BeMiller, “Methods in carbohydrate chemistry: alpha-D-
Isosaccharino-1,4-Lactone,” vol. 2, p. 477ff, 1963.
[54] J. R. Bassil, N.M., Bryan, N. and Lloyd, “Microbial degradation of isosaccharinic
acid at high pH,” ISME J., vol. 9, no. 2, pp. 310–320, 2015.
[55] K. Vercammen, M. A. Glaus, and L. R. van Loon, “Complexation of calcium by
α-isosaccharinic acid under alkaline conditions,” Acta Chemica Scandinavica,
vol. 53. pp. 241–246, 1999.
[56] C. Moulin, I. Laszak, V. Moulin, and C. Tondre, “Time-resolved laser-induced
fluorescence as a unique tool for low-level uranium speciation,” Appl. Spectrosc.,
vol. 52, no. 4, pp. 528–535, 1998.
[57] J. R. Lakowicz, Principles of fluorescence spectroscopy. New York: Springer,
2006.

Literaturverzeichnis
XII
[58] R. Steudtner, “Zur Wechselwirkung von Uran mit den Bioliganden Citronensäure
und Glucose,” Forschungszentrum Dresden-Rossendorf, TU Dresden, Dresden,
Deutschland, 2010.
[59] B. Drobot et al., “Speciation Studies of Metals in Trace Concentrations: The
Mononuclear Uranyl(VI) Hydroxo Complexes,” Anal. Chem., vol. 88, no. 7, pp.
3548–3555, 2016.
[60] R. G. Denning, “Electronic Structure and Bonding in Actinyl Ions and their
Analogs,” J. Phys. Chem., vol. A 111, pp. 4125–4143, 2007.
[61] E. Pierloot, K. und van Besien, “Electronic structure and spectrum of UO2 2+
and UO2Cl4 2,” J. Chem. Phys., vol. 123, no. 204309, 2005.
[62] R. E. Bell, J.T. und Biggers, “Absorption spectrum of the Uranyl Ion in
Perchlorate Media,” J. Mol. Spectrosc., vol. 25, pp. 312–329, 1968.
[63] X. Tan, X., Fang, M. und Wang, “Sorption speciation of lanthanides/actinides on
minerals by TRLFS, EXAFS and DFT studies: a review,” Molecules, vol. 15, pp.
8431–8468, 2010.
[64] C. Richter, “Sorption of environmentally relevant radionuclides (U(VI), Np(V))
and lanthanides (Nd(III)) on feldspar and mica,” Institut für Ressourcenökologie
des Helmholtz- Zentrums Dresden-Rossendorf, TU Dresden, Dresden,
Germany, 2015.
[65] M. J. Keith-Roach, M. Lindgren, and K. Källström, “R-14-03 Assessment of
complexing agent concentrations in SFR,” no. SKB R-14-03, p. 38, 2014.
[66] J. Tits, G. Geipel, N. Macé, M. Eilzer, and E. Wieland, “Determination of
uranium(VI) sorbed species in calcium silicate hydrate phases: A laser-induced
luminescence spectroscopy and batch sorption study,” J. Colloid Interface Sci.,
vol. 359, no. 1, pp. 248–256, 2011.

Anhang
XIII
Anhang
Abbildung 26: pH-Abhängige Konzentrationen der wichtigsten Ca-Bentonit-Bestandteile aus
dem Laugungsversuch in der überstehenden Lösung
Abbildung 27: Ermittelter organischer Kohlenstoff (TOC) aus Ca-Bentonit-Laugung in
Abhängigkeit vom pH-Wert unter normalen atmosphärischen Bedingungen (S. Shams (HZDR))
7,0
7,5
8,0
8,5
9,0
9,5
10,0
10,5
11,0
11,5
12,0
Ko
nze
ntr
atio
n in m
mo
l/L
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 13,5
Ko
nze
ntr
atio
n in m
mo
l/L
pH-Wert
Silicium Aluminium Magnesium Calcium Kalium
Konze
ntr
atio
n in m
mol/L
3,5
4,0
4,5
5,0
5,5
6,0
6,5
7,5 8 8,5 9 9,5 10 10,5 11 11,5 12 12,5 13 13,5
TO
C in
mg
/L
pH-Wert





























