Embed Size (px)
Citation preview

Núcleo de Telessaúde Técnico-Científico do Rio Grande do Sul Universidade Federal do Rio Grande do Sul – UFRGS
Programa de Pós-Graduação em Epidemiologia - PPGEPI Faculdade de Medicina - FAMED
Módulo 3 de 6 Deficiência Intelectual

Carolina Fischinger Moura de Souza [email protected]
TelessaúdeRS
Encontro 3 de 6 Deficiência Intelectual

Roteiro
• Conceitos;
• Etiologia;
• Classificação;
• Como investigar?
• Como Tratar?

Retardo Mental
A deficiência mental é uma condição complexa. Seu diagnóstico envolve a compreensão da ação combinada de quatro grupos de fatores etiológicos - biomédicos, comportamentais, sociais e educacionais.

Retardo Mental
• Condição freqüente que afeta de 1-10% da população (~ 3%);
• Alto impacto sobre o indivíduo, a família e a sociedade;
• É a terceira condição neurológica mais frequente na infância após a paralisia cerebral e epilepsia;
• A definição do diagnóstico é um grande desafio considerando a grande variabilidade de causas, extensa investigação e custo elevado;

Um grande espectro de causas associadas ao retardo mental
Há cada vez mais evidencias de que fatores genéticos influenciam fortemente a etiologia do RM

Deficiência Mental
• Prejuízo significativo da cognição e funções adaptativas;
• QI: classificação
o leve: 50-55 a 70;
o moderada: 35-40 a 50-55;
o grave: 20-25 a 35-40;
o profunda: < 20-25.

Deficiência Mental
Sexo masculino > sexo feminino
1,5 : 1,0
Genes no cromossomo X

Por que Investigar?
• Procurar respostas para as famílias;
• Somente o reconhecimento da causa pode:
o Ajudar a estabelecer o risco de recorrência;
o Estabelecer o prognóstico;
o Organizar testes laboratoriais adequados, evitar avaliações desnecessárias, de alto custo;
o Iniciar tratamento adequado e referendar a família a grupos de apoio e suporte.

Benefícios da Avaliação Genética
Para os familiares • Qual a causa?
• Como isto aconteceu?
• Quais as complicações médicas?
• Há tratamento?
• Quais são as expectativas?
• Acontecerá em outros filhos?
• Pode ser prevenido?
• Quais recursos de suporte estão disponíveis?
Para os profissionais • Clarificação de etiologia,
prognóstico, mecanismos genéticos, riscos de recorrência, opções de tratamento;
• Evitar exames desnecessários
• Informação sobre manejo e suporte familiar;
• Protocolos de tratamento
• Co-manejo em casos apropriados.

Avaliação Genética
• História: ênfase em dados pré-natais e perinatais. Detalhamento;
• Heredograma de 3 gerações;
• Exame físico dismorfológico: medidas antropométricas;
• Exames complementares:
o Cariótipo, Estudo de microdeleções;
o FISH, análise molecular;
o Imagem do encéfalo;
o Testes metabólicos: Erros Inatos do Metabolismo? Endocrinológicos

Fatores de Risco Associados ao Retardo Mental
• Baixo peso ao nascer;
• Baixo nível educacional familiar;
• Idade materna avançada;
• Múltiplas gestações.

Principais Causas
Fatores ambientais: • Teratógenos;
• Prematuridade;
• Anoxia Perinatal.
Fatores genéticos (2/3): • Hereditário?
• Esporádico?

Diagnóstico Prevalência (%)
Anormalidade cromossômica 4-28
Síndromes reconhecidas 3-7
Condições Monogênicas conhecidas 3-9
Anormalidades estruturais do SNC 7-17
Complicações da prematuridade 2-10
Causas ambientais /Teratogênicas 5-13
Cultural- familiar de RM 3-12
Síndromes monogênicas únicas 1-5
Causas metabólicas 1-5
Desconhecidas 30-50
Curry e cols, 1997

Fatores ambientais:
Teratógenos

Síndrome do álcool Fetal: presente em até 30% dos filhos de gestantes alcóolatras.
Caracterizada por:
• Retardo mental;
• Microcefalia;
• Retardo de crescimento pré e pós-natais;
• Dismorfias faciais.
Apesar da Síndrome do Álcool Fetal estar presente somente em filhos de gestantes etilistas, não existe uma dose considerada segura de álcool na gestação. O preconizado é não ingerir nenhuma dose de bebida alcóolica durante a gestação.
Embriopatia pelo Etanol

• Déficit de crescimento pós natal;
• Baixa estatura;
• Diminuição da mobilidade articular;
• Fosseta sacral;
• Anomalias renais;
• Cardiopatia.

O Álcool sobre o Sistema Nervoso Fetal
• Interferência do processo de maturação neuronal;
• Interferência na migração das células e na mielinização;
• Interferência na adesão celular;
• Alteração das membranas celulares;
• Alteração da produção ou da resposta aos fatores que regulam o crescimento e divisão celular;
• Interferência na regulação do cálcio intracelular;
• Produção de radicais livres.

Áreas passíveis de lesão secundária à presença álcool durante a gestação

Feniceltonúria Materna
Mae portadora de fenilcetonuria detectada pelo teste do pezinho

Fatores genéticos (2/3):
• Hereditário?
• Esporádico?

Classificação
Monogênicas: OMIM (>19.000), frequência 10/10.000 Rn
• Dominantes;
• Recessivas;
• Ligadas ao X;
• Mitocôndrias.
Cromossômicas
• Numéricas: S. de Down, trissomia do 18 e 13;
• Estruturais: deleção de 4p-, deleção de 9p-
Multifatoriais
• Poligênicas;
• Doenças complexas: autismo, doenças psiquiátricas.

Classificação
Monogênicas: OMIM (>19.000), frequência 10/10.000 Rn
• Dominantes;
• Recessivas;
• Ligadas ao X;
• Mitocôndrias.
Cromossômicas
• Numéricas: S. de Down, trissomia do 18 e 13;
• Estruturais: deleção de 4p-, deleção de 9p-
Multifatoriais
• Poligênicas;
• Doenças complexas: autismo, doenças psiquiátricas.

A História:
• Manchas café com leite;
• Epilepsia;
• Retardo mental;
• Tumor cerebral.
Dominante
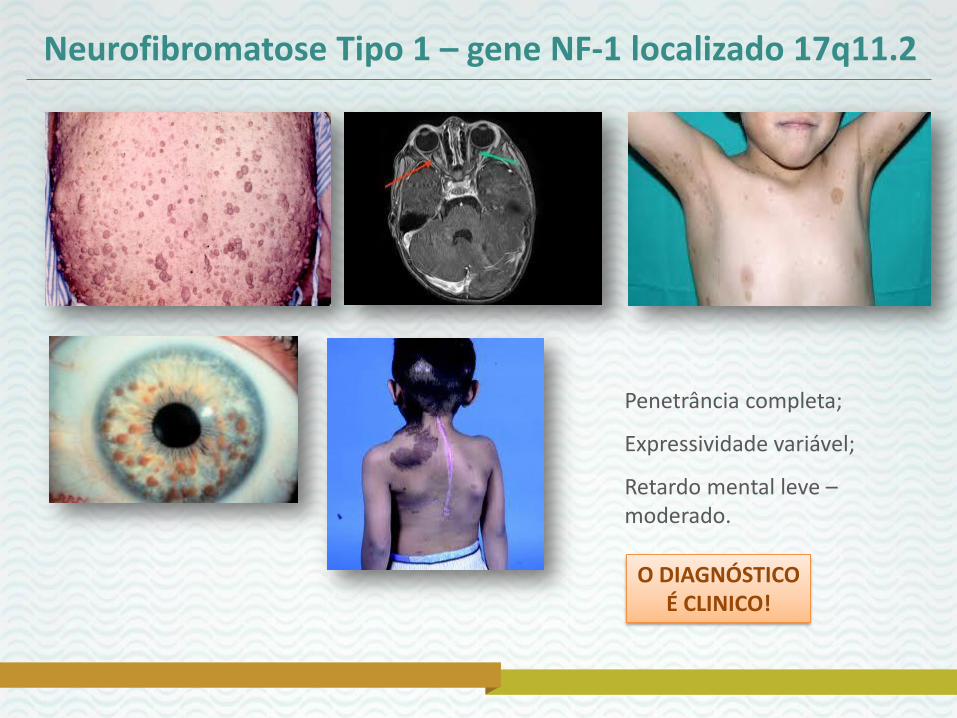
Neurofibromatose Tipo 1 – gene NF-1 localizado 17q11.2
Penetrância completa;
Expressividade variável;
Retardo mental leve – moderado.
O DIAGNÓSTICO
É CLINICO!

Esclerose Tuberosa – mutações do gene TSC2
• Sindrome Neurocutanea e Multissistêmica;
• Manchas hipocromicas;
• Fibromas na fronte e ungueais;
• Núdulos periventriculares;
• Calcificações intra cerebrais;
• Mutações no gene TSC2;
• Retardo mental (47%): alta relação com epilepsia antes de 2 anos.

Síndromes de Herança Autossômica Dominante
Síndrome de Cornélia de Lange
Mutações no gene NIPBL (5p)
Síndrome de Noonan:
Mutações nos genes:
TPN11, SOS1, RAF1, KRAS, NRAS and BRAF
herdado ou esporádico

Recessiva
Maioria dos
Erros do
Metabolismo


MPS I
Síndrome de Herança Autossômica Recessiva
Nascimento: peso 3,8 kg, compriment
o 53 cm
1 a 10m: peso: 13
kg (p75), altura 82
cm (p10), fígado
8,5 cm.
Ecocardiograma
normal
5 anos: peso: 16
kg (p10), altura: 94
cm (<p3), fígado
10 cm.
Ecocardiograma:
espessamento
septo, VE, válvula
mitral.
5,5
anos:
óbito

MPS III
Síndrome de Herança Autossômica Recessiva

Ligado ao X

Síndrome de Herança Ligada ao X
Síndrome do X Frágil:

Epidemiologia: 1:1.250 homens; 1:2.500 mulheres é a causa herdada isolada mais comum de RM 40% de todos os casos de RM.
Quadro clínico: face longa
orelhas proeminentes
mandíbulas proeminentes
hipermobilidade articular
macroorquidismo
pés chatos
retardo mental
http://www.fragilex.org/home.htm
Penetrância:
80% homens
30% mulheres
X Frágil

Síndrome do X Frágil

RESOLUÇÃO DO PARADOXO DE SHERMAN
CLONAGEM DO GENE FMR1
Cromossomo X (Xq27.3)
GENE: FMR1 : 38 Kb 17 exons

Síndrome de Herança Ligada ao X Dominante
SINDROME DE RETT Mutações do gene MECP2

Síndrome de Rett Clássica
• Maioria mulheres;
• Meninos: cariótipo 47, XXY;
• Achados neurológicos:
o Um desenvolvimento aparentemente normal até 6 meses;
o Desaceleração do PC a partir de 3 meses;
o Regressão da linguagem;
o Estereotipias de mãos;
o Choro inconsolável;
o Autismo;
o Apnéia ou hiperpnéia;
o Ataxia, apraxia, tremor;
o Convulsões.

Classificação
Monogênicas: OMIM (>19.000), frequência 10/10.000 Rn
• Dominantes;
• Recessivas;
• Ligadas ao X;
• Mitocôndrias.
Cromossômicas
• Numéricas: S. de Down, trissomia do 18 e 13;
• Estruturais: deleção de 4p-, deleção de 9p-
Multifatoriais
• Poligênicas;
• Doenças complexas: autismo, doenças psiquiátricas.

Doenças Cromossômicas
Incidência média de 7 casos em 1000 habitantes. Contribui também com metade dos abortos espontâneos até o 3o. mês de gestação.
Doenças ocasionadas por alterações envolvendo os cromossomos - vários genes.
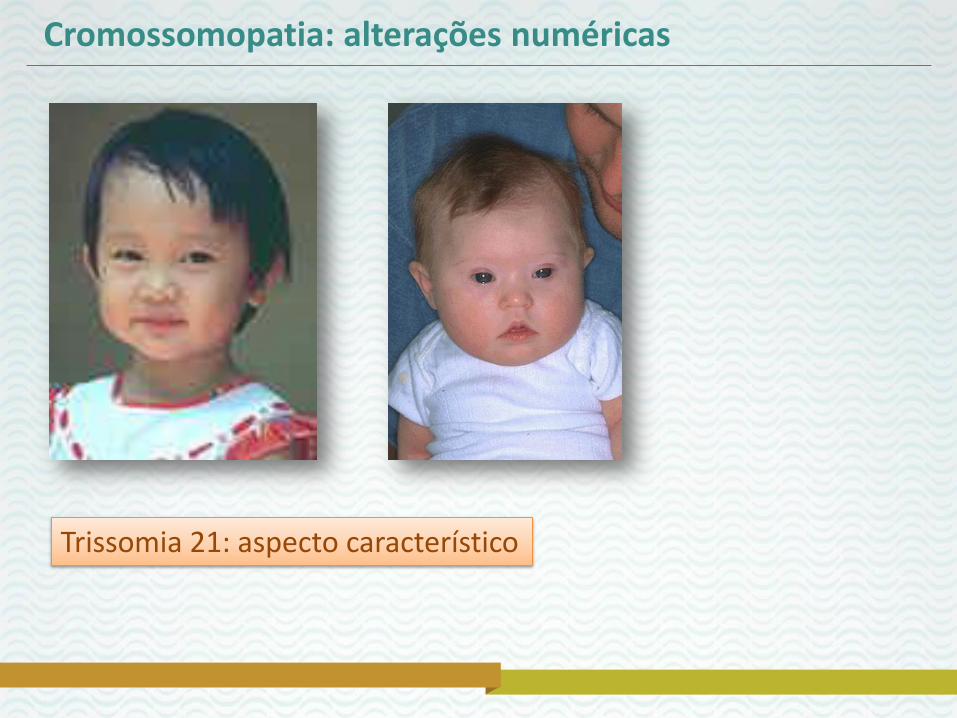
Cromossomopatia: alterações numéricas
Trissomia 21: aspecto característico

Síndrome de Down

Síndrome de Down
Síndrome de Di George/ Velocardio facial – Deleção 22 q 11
Malformação conotruncal:
• Cardiopatia;
• Imunodeficiência: infecções de repetição;
• Hipocalcemia;
• Incompetência velo-faringea;
• Anormalidades renais, esqueléticas, comportamentais e psiquiátricas;
• Hiperatividade.

Classificação
Monogênicas: OMIM (>19.000), frequência 10/10.000 Rn
• Dominantes;
• Recessivas;
• Ligadas ao X;
• Mitocôndrias.
Cromossômicas
• Numéricas: S. de Down, trissomia do 18 e 13;
• Estruturais: deleção de 4p-, deleção de 9p-
Multifatoriais
• Poligênicas;
• Doenças complexas: autismo, doenças psiquiátricas.


Autismo
• Idiopático – 95%
• Secundário – 5% o Agente ambiental;
o Cromossomopatia;
o Gênico.

Como Investigar o Retardo Mental
• Avaliação Clinica!
• Exames laboratoriais Básicos: Hemograma, eletrólitos, função renal, ác úrico, função hepática, lactato, amonia, CPK, Cálcio, Fosforo, glicemia, PH. Urina: cetonas, glicose
• Genéticos:
o Cariótipo;
o Estudo de microdeleção e duplicação: por Fish ou Array;
o Screening para X frágil
o Pesquisa de mutações em genes específicos: MECP2;
• Investigação metabólica: EIM, endócrino;
• Exames de Imagem: RNM de encéfalo (T2, Flair, transversal e coronal);
• Exames de Função: EEG, etc.

Como Investigar o Retardo Mental?
A história: O exame: cariótipo
O Diagnóstico: Sindrome 5p- (Cri du Chat)
• PIG;
• Retardo mental;
• Microcefalia;
• Epilepsia;
• Cardiopatia;
• Dismorfismos.

A história: • Retardo mental leve a moderado;
• Ansiedade;
• Hipermobilidade articular;
• Face característica;
• Alta habilidade de linguagem e musicalização.
O Exame: Citogenética Molecular - Fish
O Diagnóstico: Microdeleção do 7q11.23
Síndrome de Williams

A História:
Menino de 6 anos com diagnóstico do espectro autista.
Aos 4 anos havia sido investigado tendo cariótipo e análise molecular para síndrome do X frágil normal.
O Exame: Estudo de microdeleção e microduplicação por CGH Array do caso e seus pais.
O Diagnóstico: Deleção de 5 Mb de DNA no cromossomo 16. Pais não portadores

• A técnica molecular Array CGH (array Comparative Genomic Hybridization) permite análise genômica nas mudanças na estrutura e no número de cópias com alta resolução detectando ganhos e perdas de material genético em regiões específicas do genoma;
• O limite de resolução do Array CGH é de 1Mb;
• Aplicado para testar em pacientes com anomalias congênitas múltiplas, retardo do desenvolvimento psicomotor, desordens do espectro autista;
• Tem eliminado a “Odisséia” de exames para o diagnóstico de muitas condições.
Estudos Microdeleção – CGH Array
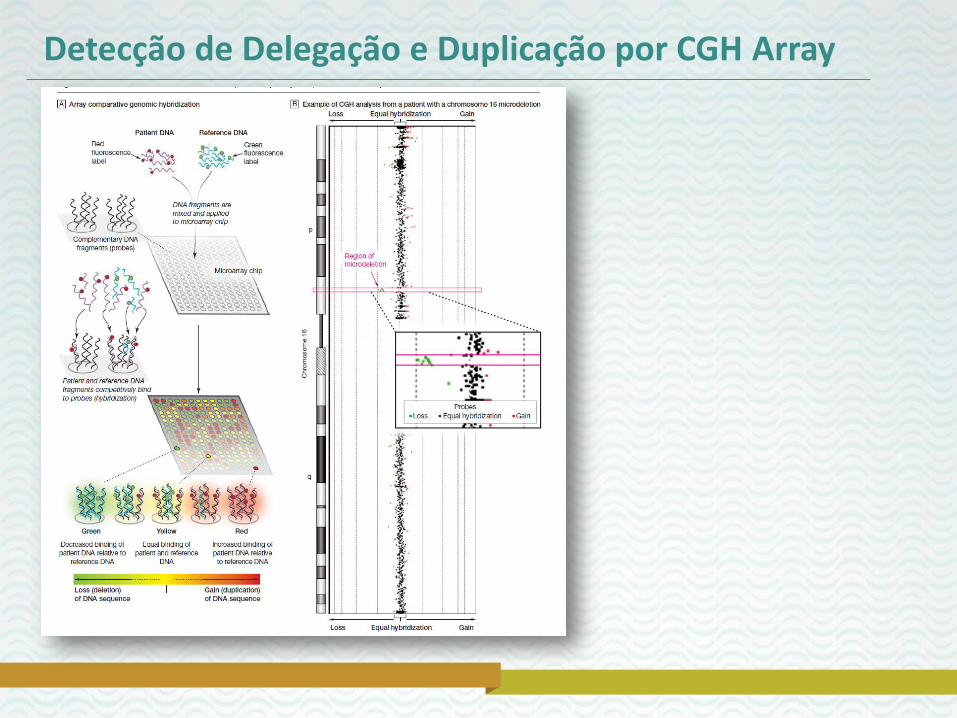
Detecção de Delegação e Duplicação por CGH Array
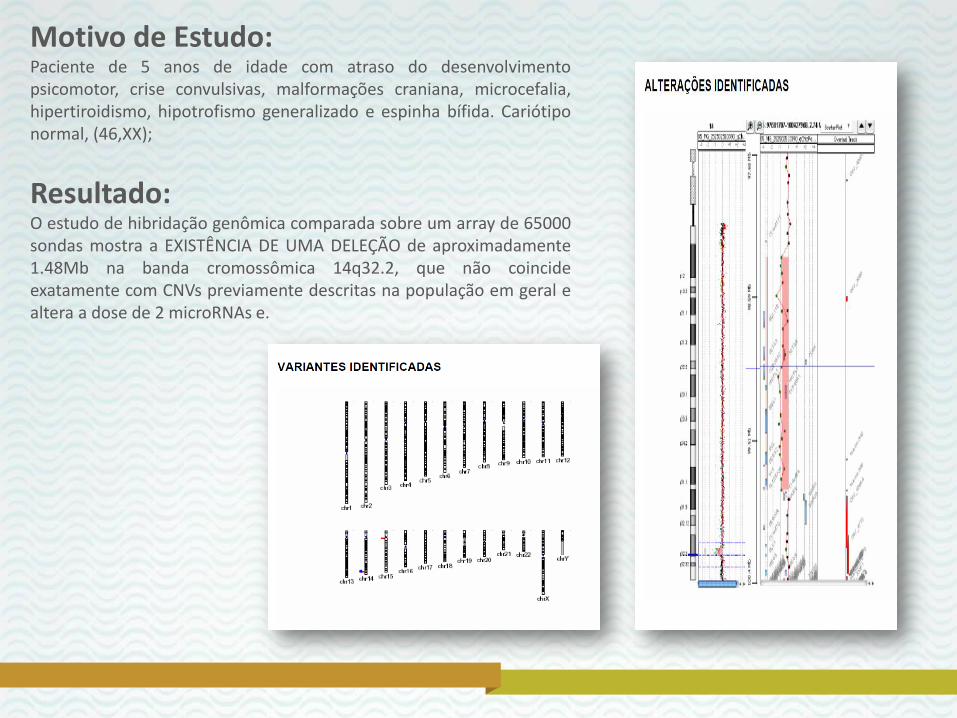
Motivo de Estudo: Paciente de 5 anos de idade com atraso do desenvolvimento psicomotor, crise convulsivas, malformações craniana, microcefalia, hipertiroidismo, hipotrofismo generalizado e espinha bífida. Cariótipo normal, (46,XX);
Resultado: O estudo de hibridação genômica comparada sobre um array de 65000 sondas mostra a EXISTÊNCIA DE UMA DELEÇÃO de aproximadamente 1.48Mb na banda cromossômica 14q32.2, que não coincide exatamente com CNVs previamente descritas na população em geral e altera a dose de 2 microRNAs e.

Microdeleção 15q11.3

Microdeleção 15q11.3

4 mutações mais freqüentes:
R168X, R255X, R270X e R294X
MLPA
O Exame:
Pesquisa de mutações do Gene MECP2
A história: • Menina com hipotonia mais
marcada a partir de 6 meses;
• Epilepsia;
• Microcefalia pós natal;
• Estereotipias.

Estudos sobre Etiologia no Brasil
• Llerena Jr JC, Santa-Rosa AA, Correia P, Horovitz D, Silva EJC, Mascarenhas EF, Silva R, Camacho L, Raggio R
Investigação do retardo mental e doenças genéticas a partir de um estudo transversal em escolas do estado do Rio de Janeiro.
Informe Epidemiol SUS 2000; 9(4):251-262.

Félix TM, Leite JCL, Maluf SW, Coelho JC
A genetic diagnostic survey in a population of 202 mentally retarded institutionalized patients in the south of Brazil.
Clinical Genetics 1998; 54: 219-223.

Suspeita
Clínica
Diagnóstico Clínico
Diagnóstico Laboratorial específico:
Bioquímico e Molecular
Decisões terapêuticas:
Tratamento farmacológico
Tratamento não farmacológico
Seguimento multidisciplinar
Aconselhamento genético

Aconselhamento Genético
É um processo de comunicação ou informação acerca da ocorrência de uma situação de causa ou predisposição genética, seus possíveis mecanismos etiológicos, riscos de recorrência, implicações e possibilidades atuais e futuras de prevenção ou tratamento.
O AG tem como base a verdade, a imparcialidade e a confidencialidade.
O AG deve preceder qualquer teste de diagnóstico pré natal e a testagem de indivíduos em risco de ter ou desenvolver uma doença.

Tratamento
Modelo da Medicina Preventiva:
• Prevenção primária – redução dos fatores e condições que levam ao desenvolvimento das perturbações associadas ao RM. Ex Vacinação para Rubéola;
• Prevenção secundária – tratar a condição associada ao RM, de modo a abreviar o curso da doença. Ex Dieta para EIM ;
• Prevenção terciária – minimizar seqüelas e comprometimentos conseqüentes. EX uso de anticonvulsivantes.

Tratamento
Atenção multidisciplinar: • Fisioterapia, Fonoterapia, Terapia Ocupacional, Educador Especial,
Psicomotricidade, Psicologia, etc;
• Educação familiar ;
• Inclusão, escola normal, escola especial.
Medicamentoso: minimizar as co- morbidades (epilepsia, agitação, etc)
Dietético: doenças metabólicas hereditárias

Doença do Xarope do Bordo







![PROTOCOLO DE TRATAMENTO DE INFLUENZA 2015 · Protocolo de tratamento de Influenza: 2015 [recurso eletrônico] ... (disfunção cognitiva, lesão medular, epilepsia, paralisia cerebral,](https://img.pdfslide.tips/doc/110x75/5c4b9be793f3c34c5509777d/protocolo-de-tratamento-de-influenza-protocolo-de-tratamento-de-influenza-2015.jpg)










![M.O [7] - Paralisia Facial](https://img.pdfslide.tips/doc/110x75/5438ce15afaf9fbe2e8b49ea/mo-7-paralisia-facial.jpg)













