Embed Size (px)
Citation preview

Expert Reviews in Molecular Medicinehttp://journals.cambridge.org/ERM
Additional services for Expert Reviews in Molecular Medicine:
Email alerts: Click hereSubscriptions: Click hereCommercial reprints: Click hereTerms of use : Click here
Novel therapeutic strategies for the treatment of proteinmisfolding diseases
JeanChristophe Rochet
Expert Reviews in Molecular Medicine / Volume 9 / Issue 17 / June 2007, pp 1 34DOI: 10.1017/S1462399407000385, Published online: 28 June 2007
Link to this article: http://journals.cambridge.org/abstract_S1462399407000385
How to cite this article:JeanChristophe Rochet (2007). Novel therapeutic strategies for the treatment of proteinmisfolding diseases. Expert Reviews in Molecular Medicine, 9, pp 134 doi:10.1017/S1462399407000385
Request Permissions : Click here
Downloaded from http://journals.cambridge.org/ERM, IP address: 128.143.23.241 on 01 Oct 2012

Novel therapeutic strategies for
the treatment of protein-misfolding
diseases
Jean-Christophe Rochet
Most proteins in the cell adopt a compact, globular fold that determines theirstability and function. Partial protein unfolding under conditions of cellularstress results in the exposure of hydrophobic regions normally buried in theinterior of the native structure. Interactions involving the exposed hydrophobicsurfaces of misfolded protein conformers lead to the formation of toxicaggregates, including oligomers, protofibrils and amyloid fibrils. A significantnumber of human disorders (e.g. Alzheimer disease, Parkinson disease,Huntington disease, amyotrophic lateral sclerosis and type II diabetes) arecharacterised by protein misfolding and aggregation. Over the past five years,outstanding progress has been made in the development of therapeuticstrategies targeting these diseases. Three promising approaches include: (1)inhibiting protein aggregation with peptides or small molecules identified viastructure-based drug design or high-throughput screening; (2) interferingwith post-translational modifications that stimulate protein misfolding andaggregation; and (3) upregulating molecular chaperones or aggregate-clearance mechanisms. Ultimately, drug combinations that capitalise on morethan one therapeutic strategy will constitute the most effective treatment forpatients with these devastating illnesses.
The conformation of a folded protein isstabilised by a network of hydrophobic,electrostatic and hydrogen-bondinginteractions involving residues distributedthroughout the polypeptide chain (Ref. 1).During co- or post-translational proteinfolding, hydrophobic surfaces that are buriedin the final native structure become transientlyexposed and available for incorrectinteractions with neighbouring molecules in
the cell. These interactions promote proteinaggregation, a process that competes withcorrect folding. Similarly, polypeptides thatlack a stable folded structure even in theirnative soluble states (referred to as ‘nativelyunfolded’) expose hydrophobic surfaces and,therefore, have a high propensity to formaggregates at high concentrations. Undernormal conditions, improper interactionsleading to protein aggregation are inhibited by
Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 575 StadiumMall Drive, RHPH 410A, West Lafayette, IN 47907, USA. Tel: +1 765 494 1413; Fax: +1 765 494 1414;E-mail: [email protected]
expert reviewshttp://www.expertreviews.org/ in molecular medicine
1Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

molecular chaperones – cellular factors thatenhance the efficiency of protein folding byshielding exposed hydrophobic surfaces(Fig. 1). However, in the presence of cellularstresses that induce protein unfolding andaggregation, chaperones are no longer able tokeep up with preventing incorrect proteininteractions, and aggregates begin toaccumulate. Examples of cellular stresses thattrigger protein unfolding include heat shock,the introduction of amino acid substitutions asa result of gene mutations, and proteindamage due to post-translational modifications(Fig. 1).
Several diseases are characterised by proteinmisfolding and aggregation. Although theformation of protein aggregates can trigger celldeath by depleting proteins essential for cellsurvival, in many cases cytotoxicity resultsfrom harmful properties of the aggregatesthemselves (Ref. 1). Diseases in which proteinaggregates cause cell death are said tooccur via a toxic gain-of-function mechanism(examples discussed in this review are listed inTable 1). A hallmark feature of these disordersis the presence in diseased tissue ofpathological lesions containing b-sheet-rich,fibrillar protein assemblies. Genetic evidencestrongly suggests that protein aggregationplays a critical role in the pathogenesis of thesediseases. For example, mutations in the geneencoding the amyloid precursor protein (APP)are thought to lower the age of onset ofAlzheimer disease by increasing theproduction of a fibrillogenic variant of theamyloid-beta peptide, Ab1 – 42, relative tothe shorter, less fibrillogenic variant Ab1 – 40.In addition, mutations that expand thepolyglutamine repeat region of huntingtin(by increasing the number of consecutiveglutamine residues) are thought to cause theonset of Huntington disease by increasing thepropensity of huntingtin to form cytotoxicaggregates.
Protein aggregation occurs via a complex self-assembly pathway involving the formation ofsmall oligomers, larger complexes called‘protofibrils’, and mature, b-sheet-rich fibrils.An intensely debated question is whetherprefibrillar aggregates (oligomers, protofibrils)or fibrils are responsible for the underlyingcytotoxicity in protein-misfolding diseases.Multiple lines of evidence suggest that
oligomers or protofibrils play a moreimportant role than fibrils in pathogenesis.Oligomeric forms of Ab are more toxic thanmature fibrils in neuronal cell culture (Ref. 2),and they suppress hippocampal long-termpotentiation and elicit memory deficits in ratsin vivo (Refs 3, 4). Evidence from ground-breaking work in transgenic Caenorhabditiselegans overexpressing Ab1 – 42 suggests thatAb toxicity is suppressed not only viainhibition of protein aggregation by molecularchaperones, but also via a secondarypathway involving conversion of toxicprotofibrils into larger, less harmful aggregates(Ref. 5). Data from another study indicatethat the formation of huntingtin inclusionscorrelates with enhanced neuronal survival ina cell-culture model of Huntington disease(Ref. 6). In addition, oligomeric forms of themammalian prion protein (PrP) have a greaterpropensity to transmit disease than largerfibrils (Ref. 7), and low molecular weightoligomers of transthyretin (TTR) exhibitgreater toxicity than fibrillar species inneuronal cells (Ref. 8). The reason for theenhanced toxicity of oligomeric assembliescompared with fibrils is uncertain, althoughdata from several studies suggest thatoligomers or protofibrils might have a greaterpropensity to trigger harmful membranepermeabilisation (Refs 9, 10, 11).
The purpose of this review is to provide acomprehensive overview of novel therapeuticstrategies for the treatment of protein-misfolding diseases. Three general approachesare considered here: (1) directly targetingproteins that undergo misfolding andaggregation; (2) interfering with post-translational modifications that promoteprotein misfolding and aggregation; and (3)inhibiting a build-up of protein aggregatesby upregulating molecular chaperones orstimulating aggregate clearance. Strategiesthat target cellular pathways modulatedby protein aggregation (e.g. apoptosis,transcription) are not included owing tospace constraints and because theseapproaches have been covered extensively inother reviews (for example, see Refs 12, 13, 14).The discussion in this review is focusedprimarily on research carried out during thepast five years.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
2Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Novobiocinanalogue A4Curcumin
O
O O
O
OOO
O
OH
O
HO
Rapamycin
N
O
HO
HO
OH
O
O O
O
O
HN
O
O
O
OHHO
MeO
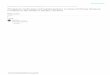
Model illustrating protein misfolding, aggregation and clearance inside the cell, along with opportunities for therapeutic interventionExpert Reviews in Molecular Medicine © 2007 Cambridge University Press
Folded protein
Oxidative damageGene mutationProteolysis
Proteasomaldegradation
ag
h
i
b
Native-statestabilisation
Novobiocinanalogue A4
Curcumin
Hsp90Hsp70Hsp40Hsp27
HSF ↑
Rapamycin
Misfolded species
Prefibrillar oligomers
Aggregates transportedto aggresome
Aggresome
PhosphorylationOxidative damageGene mutationProteolysis
c
d
f
j
eMacroautophagy
Apoptosis
mTOR
O
Hsp90
Figure 1. Model illustrating protein misfolding, aggregation and clearance inside the cell, along withopportunities for therapeutic intervention. (See next page for legend.)
expert reviewshttp://www.expertreviews.org/ in molecular medicine
3Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Therapeutic strategy I: direct targetingof proteins that undergo misfolding
and aggregationOne approach for developing new therapiesto treat protein-misfolding disorders has beento identify agents that bind an aggregatingpolypeptide and keep it from forming toxicassemblies. To target fibrillogenic proteinsor peptides that lack a stable fold undernative conditions, the main strategy has beento identify inhibitors that interfere directlywith aggregation. These inhibitors consist ofsmall molecules or peptides identified viastructure-based design or high-throughputscreening. To target proteins that are foldedunder native conditions, a powerful strategyhas been to identify chemical entities thatstabilise the native conformation and preventunfolding leading to aggregation. Examplesof these different approaches are discussedbelow.
Inhibition of fibril formation: use ofstructure-based approachesOver the past five years there have beenremarkable advances in our understandingof the structures of amyloid fibrils and
protofibrils. In turn, these advances havestimulated the development of therapeuticapproaches targeting structural motifs requiredfor protein self-assembly.
Structure of amyloid fibrilsMany proteins that are susceptible to misfoldingand aggregation form extracellular depositstermed ‘amyloid fibrils’ (Ref. 15). Electronmicroscopy has shown that amyloid fibrilsformed from different proteins are straight,rigid and unbranched, with diameters rangingfrom 5 to 13 nm. Each amyloid fibril consistsof multiple fibrillar subunits, named‘protofilaments’, that wind around each other ina helical fashion (typically, there are two to sixprotofilaments per fibril). Amyloid fibrilsproduce an X-ray diffraction pattern thatsuggests that the amyloid protofilament consistsof a ‘cross-b’ structure [or ‘cross-b spine’(Ref. 15)], with b-strands orthogonal to andbackbone hydrogen bonds parallel to the fibrilaxis. [Note that the terms ‘protofilament’ and‘protofibril’ have different meanings: whereasthe former is a structural unit of the amyloidfibril, the latter is a kinetic intermediate offibrillisation (Ref. 16).]
Figure 1. Model illustrating protein misfolding, aggregation and clearance inside the cell, along withopportunities for therapeutic intervention. (Legend; see previous page for figure.) (a) A folded protein isconverted to a misfolded species (shown enclosed in a red circle) by various stresses, including oxidativedamage, gene mutations and proteolysis. The red asterisk represents a site of modification on the misfoldedprotein as a result of one of these stresses. (b) Under normal conditions, misfolded proteins are degraded bythe ubiquitin–proteasome pathway (shown in the figure) or by chaperone-mediated autophagy (not shown).(c) At high concentrations, a misfolded protein (or a natively unfolded polypeptide such as a-synuclein) canbe induced to form prefibrillar, b-sheet-rich oligomers by various stresses, including phosphorylation,oxidative damage, gene mutations and proteolysis. (d) The oligomers are transported to perinuclearaggresomes, where they may form more mature, fibrillar aggregates such as those present in Lewy bodies.(e) Ultimately, protein aggregates at the aggresome are targeted for degradation via macroautophagy. (f) If b-sheet-rich oligomers accumulate to a point that exceeds the capacity of both the ubiquitin–proteasomepathway and the aggresome–macroautophagy pathway, then they may induce cell death via apoptosis. (g)Misfolding and aggregation of a normally folded protein can be suppressed by stabilising the nativeconformation (an example of Therapeutic Strategy I in the text). (h) Antioxidant compounds (e.g. curcumin)may inhibit protein misfolding and/or aggregation induced by oxidative stress (Therapeutic Strategy II).Curcumin also prevents the formation of toxic aggregates by directly interfering with b-sheet stacking(Therapeutic Strategy I). (i) Protein aggregation can also be blocked by upregulating molecular chaperoneswith an Hsp90 inhibitor, including the novobiocin analogue A4 (Therapeutic Strategy III). Inhibition of Hsp90results in the release of previously bound, inactive HSF1, which undergoes trimerisation andphosphorylation coupled with nuclear transfer and induces the expression of heat-shock proteins includingHsp90, Hsp70, Hsp40 and Hsp27. These heat-shock proteins inhibit protein aggregation, and Hsp27 alsosuppresses oxidative stress. (j) Aggregate clearance can be upregulated via pharmacological inhibition ofmTOR, a kinase that inhibits macroautophagy (Therapeutic Strategy III), for example by the classic mTORinhibitor rapamycin. The chemical structures of curcumin, the novobiocin analogue A4 and rapamycin areshown below the schematic.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
4Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Because amyloid fibrils are insoluble andnoncrystalline, it is generally not possible tosolve their atomic structures using traditionalmethods such as X-ray crystallography orsolution nuclear magnetic resonance (NMR)(Ref. 17). However, recent solid-state NMR (SS-NMR) data have enabled investigators tocharacterise amyloid fibril structure withunprecedented detail. An advantage of SS-NMR is that it provides constraints formodelling fibril secondary structure (foldingof the polypeptide chain into b-strand ornon-b-strand segments), tertiary structure(organisation of b-strands into parallel orantiparallel b-sheets), and quaternary structure(arrangement of b-sheets relative to each other)(Ref. 17). The most intensive SS-NMR studieshave been carried out on fibrils formed fromAb1 – 40. Each Ab1 – 40 fibril consists of a bundleof two to five protofilaments (Refs 17, 18).
Two-dimensional SS-NMR data indicate thateach Ab1 – 40 molecule in a protofilamentconsists of a disordered N-terminal regionspanning residues 1–9, followed by twob-strands encompassing residues 10–22 and30–40 (Fig. 2a). Residues 23–29 form astructured loop enabling the two b-strands tofold over each other and interact via contactsbetween side-chains (Ref. 17). An array ofAb1 – 40 ‘strand–loop–strand’ motifs projectsalong the fibril axis to form a two-layeredstructure (Fig. 2b). Each layer in this structureconsists of an extended b-sheet with individualb-strands aligned parallel and in-register(Ref. 17). Mass per unit length data obtainedvia scanning transmission electron microscopy(STEM) indicate that each Ab1 – 40 protofilamentconsists of four b-sheet layers, equivalent totwo molecular layers (Fig. 2b) (Refs 17, 18). Theprotofilament is stabilised by (1) ‘internal’
Table 1. Proteins involved in misfolding diseases (examples discussed in the text)a
Protein Disease(s) Fibrillar inclusion(s)
a-Synuclein Synucleinopathies, such as Parkinsondisease, dementia with Lewy bodies, andmultiple system atrophy
Lewy bodies, Lewy neurites,andglial cytoplasmic inclusions
Amyloid-beta peptide(Ab1–40, Ab1–42)
Alzheimer disease Extracellular amyloid
Gelsolin Familial amyloidosis, Finnish-type Extracellular amyloid
Islet amyloidpolypeptide
Type II diabetes Extracellular amyloid
Polyglutamineproteins:HuntingtinAtaxin-1Ataxin-3Androgen receptor
Huntington diseaseSpinocerebellar ataxia type 1Machado–Joseph diseaseSpinal and bulbar muscular atrophy
Nuclear and cytoplasmicinclusions
Prion protein (PrP) Transmissible spongiform encephalopathies,such as Creutzfeldt–Jacob disease, bovinespongiform encephalopathy and scrapie
Aggregated PrP (PrPsc)
Superoxidedismutase1 Amyotrophic lateral sclerosis Cytoplasmic inclusions
Tau Tauopathies, such as Alzheimer disease andfrontotemporal dementia
Neurofibrillary tangles
Transthyretin Senile systemic amyloidosisFamilial amyloid polyneuropathy
Extracellular amyloid
aFor more detailed information, see the following review articles: Refs 16, 155, 206, 207, 208, 209, 210, 211.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
5Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

quaternary interactions between two b-strands inthe same molecular layer, and (2) ‘external’quaternary interactions between strands fromdifferent molecular layers (Fig. 2b) (Ref. 17).
The structure of fibrillar Ab1 – 40 describedabove is consistent with constraints determinedusing other methods (Refs 19, 20, 21, 22, 23)and similar to the structure of the Ab1 – 42 fibrildetermined by hydrogen–deuterium exchangeNMR (HX-NMR) and complementation
mutagenesis (Ref. 24). In addition to thesestudies of Ab, biophysical analyses of otherfibrillar assemblies have yielded remarkablestructural insights. For example, a-synuclein,a protein that accumulates in braindisorders referred to as ‘synucleinopathies’ (e.g.Parkinson disease, dementia with Lewy bodies,and multiple system atrophy), forms fibrils witha multilayered structure. Each layer consists ofa parallel, in-register b-sheet similar to the
90°
3533
29
28
3740
17 23
a
b
c
Solid-state nuclear magnetic resonance (SS-NMR) structures of fibrillar amyloid-beta peptide Aβ1–40Expert Reviews in Molecular Medicine 2007 Published by Cambridge University Press
Figure 2. Solid-state nuclear magnetic resonance (SS-NMR) structures of fibrillar amyloid-beta peptideAb1–40. (See next page for legend.)
expert reviewshttp://www.expertreviews.org/ in molecular medicine
6Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

model described for fibrillar Ab (Refs 25, 26, 27).In addition, X-ray diffraction analysis of crystalsformed by two amyloidogenic peptidesrevealed an extended cross-b spine structure inwhich the b-sheets are held together by highlystabilising ‘zipper-like’ contacts involving side-chains of amino acids (Refs 15, 28, 29). Inprinciple, these advances in our understandingof fibrillar structure make it possible to developstructure-based therapeutics to inhibit proteinfibrillisation. Examples of this type of ‘rational’approach to designing novel inhibitors of Ab
fibril formation are outlined below.
Structure-based design of peptides thatinhibit fibrillisationIn previous work, a peptide spanning residues16–20 of Ab with N-methyl groups at positions17 and 19 was found to interfere with fibrilformation by Ab1 – 40 (Ref. 30). The rationale forthe design of this peptide was that N-methylgroups positioned on the same side of a b-strand should prevent b-sheet formation bydisrupting interstrand hydrogen bonding.Structural evidence that fibrillar Ab1 – 40 consistsof two b-sheet layers (see above) suggested thatfibrillisation should be blocked more effectivelyby targeting both layers simultaneously ratherthan either layer alone. This hypothesis was
addressed by testing the inhibitory effects ofintroducing a pair of alternating N-methylamino acids in each b-strand of Ab1 – 40 (i.e. atpositions 17 and 19 in the N-terminal b-strandand at positions 37 and 39 of the C-terminal b-strand) (Ref. 30). The authors showed thatdisrupting the b-sheet structure in either layerwas insufficient to completely prevent fibrilformation. By contrast, the Ab1 – 40 isoform withN-methyl groups in both b-strands failed toform fibrils altogether, instead remainingmostly soluble and monomeric. These resultsimply that a mixture of N- and C-terminalpeptides targeting both b-sheet layers should beused to inhibit Ab1 – 40 fibrillisation. N-methylpeptides are particularly effective inhibitorsbecause: (1) they are water soluble, andtherefore do not form fibrils themselves; and(2) they adopt a stable, extended conformationthat enables them to ‘cap’ fibril growth viasterically favourable interactions with edgeb-strands (Ref. 30). In a related study, ananalogue of the islet amyloid polypeptide(IAPP) with two N-methyl groups was shownto be a soluble, noncytotoxic, highly potentinhibitor of IAPP fibrillisation (Ref. 31). Giventhat peptides may have limited therapeuticpotential because of their immunogenicity,susceptibility to proteolysis, and weak
Figure 2. Solid-state nuclear magnetic resonance (SS-NMR) structures of fibrillar amyloid-beta peptideAb1–40. (Legend; see previous page for figure.) (a) Structure of the strand–loop–strandmotif spanning residues9–40 (residues1–8arenotshownbecause theyaredisordered).Side-chainspointing inside themotif participatein interactions at the internal interface, whereas side-chains pointing to the exterior from the C-terminal strandparticipate in contacts at the external interface. (b) Structureof anAbprotofilamentwith the longitudinal fibril axisperpendicular or parallel to the page (left or right views, respectively). The mature amyloid fibril consists of abundle of two to five protofilaments. Each Ab molecule contains two b-strands, depicted in red and blue (N-and C-terminal strands, respectively). Strands from neighbouring molecules are organised in separate,parallel b-sheets with 4.8 A displacements along the longitudinal axis and an arbitrarily imposed twist of0.8338/A. The internal interface is located between the red and blue b-sheets, whereas the external interfaceis located between two blue b-sheets. Internal quaternary interactions involve contacts among the side-chains of L17, F19, I32, L34 and V36 in a hydrophobic core (Ref. 17). External quaternary interactionsinvolve contacts between the side-chains of I31 and M35 from one molecular layer and ‘grooves’ formed byG33, G37 and G38 in another (Refs 17, 33). (c) Structure of a complex formed by Ab1–40 and a peptideinhibitor. Gly6 from the inhibitory peptide is in contact with Met35 of Ab, whereas Gly33 and Gly37 of Abpack against the two aromatic side-chains of the inhibitor (Phe4 and Phe8). Panel (a) was reprinted fromRef. 17 [Petkova, A.T., Yau, W.M. and Tycko, R. (2006) Experimental constraints on quaternary structure inAlzheimer’s beta-amyloid fibrils. Biochemistry 45, 498–512 (Published 2006 American Chemical Society)];panel (b) was reprinted, with permission, from Ref. 212 [Tycko, R. (2006) Molecular structure of amyloidfibrils: insights from solid-state NMR. Q Rev Biophys 39, 1–55 (& 2006 Cambridge University Press)]; panel(c) was reprinted, with permission, from Ref. 33 [Sato, T. et al. (2006) Inhibitors of amyloid toxicity based onbeta-sheet packing of Abeta40 and Abeta42. Biochemistry 45, 5503–5516 (& 2006 American ChemicalSociety)].
expert reviewshttp://www.expertreviews.org/ in molecular medicine
7Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

bioavailability, it is uncertain whether theN-methyl peptides described above can be usedto treat Alzheimer disease or other protein-misfolding diseases (Ref. 32). Nevertheless,these peptides are powerful tools to investigatemechanisms of fibrillisation and to test newinhibitory strategies, and they may provide anentry point for developing agents with greaterdrug-like character (e.g. peptidomimetics).
Another way to inhibit Ab fibrillisation involvestargeting interactions between two b-sheet layers.SS-NMR data indicate that fibrils formed byAb1–40 or Ab1–42 are held together by externalquaternary contacts involving the peptidesegment 33GLMVG37 from two stacked molecules(Fig. 2) (Refs 17, 33). Because the b-strands of Ab
are organised in parallel, in-register b-sheets, theglycine residues in the 33GLMVG37 peptide formgrooves on the outside face of the C-terminal layer(Ref. 33). Bulky residues on the surface of theopposite molecular layer form ridges that fit intothe grooves and stabilise the fibrillar structure.Upon examination of these external quaternarycontacts, it was hypothesised that a peptide withalternating small and bulky residues on the sameface of a b-strand should interact in acomplementary manner with grooves and ridgesof fibrillar Ab (Ref. 33). In support of this idea,peptides with the sequence motif RxTzExKz,where x is glycine, alanine or serine and z isleucine, phenylalanine, tyrosine or tryptophan,were found to inhibit Ab1–40 fibrillisation andcause depolymerisation of preformed Ab fibrils.The structure of the complex formed by Ab1–40
and the inhibitory peptide RGTFEGKF(determined by SS-NMR) confirmed that bindingoccurs via complementary interactions (Fig. 2c).These interactions are expected to blockfibrillisation by interfering with b-sheet stacking.This strategy might be widely applicable, giventhat fibrils formed from other proteins such asa-synuclein presumably also contain surfacegrooves and ridges. Although inhibitory peptidesmay not be ideal drug candidates (see above),smaller, more therapeutically suitable moleculeswith appropriately spaced aromatic groups (e.g.the natural product curcumin) may also interferewith fibrillisation by disrupting b-sheet stacking(Fig. 1) (Ref. 33).
Potential pitfalls of fibrillisation inhibitorsAlthough structure-based design of fibrillisationinhibitors may yield new therapies for treating
protein-misfolding disorders, this approach hasan important limitation: namely, one cannot becertain that fibrils produced from synthetic orrecombinant polypeptides have the sameunderlying molecular structure as toxicaggregates involved in disease. One reason forthis uncertainty is that fibril growth conditionsmay differ markedly between in vitro and invivo conditions, such that assemblies withdistinct structures are formed in the twocontexts. This point is highlighted by thedemonstration that synthetic Ab1 – 40 formsfibrils with significantly different conformationsdepending on the incubation conditions(Ref. 18). Additional differences in fibrillarstructure might result from the fact thatfibrillogenic polypeptides undergo distinct post-translational modifications in vitro and in vivo(see below). Given that fibrils generated fromsynthetic or recombinant polypeptides may bestructurally distinct from aggregates involved indisease, fibrillisation inhibitors designed on thebasis of a single high-resolution structure mightnot have the desired mitigating effects ontoxicity. One approach to address this issue maybe to solve the structures of fibrils formed frompeptides with different post-translationalmodifications under various growth conditions.In parallel, fibrils with different molecularstructures should be characterised in terms oftoxicity in cell-culture or animal models, as wasdemonstrated for different structural variants offibrillar Ab1 – 40 (Ref. 18). These analyses willmake it possible to correlate differences infibrillar structure with differences in toxicity,and therefore to identify structural features thatare shared among all toxic assemblies. In turn,the development of fibrillisation inhibitorstargeting these shared structural features wouldbe a sound therapeutic strategy. An alternativeapproach might be to develop ‘cocktails’ ofinhibitors that target multiple structural featuresidentified across various toxic assemblies [forexample, strand–strand inhibitors could bepaired with molecules targeting b-sheet stackinginteractions (Ref. 33)].
Another limitation of inhibiting fibril formationis that this strategy may lead to a build-up of toxicoligomers or protofibrils. One way to address thisproblem is to confirm that soluble oligomers donot accumulate in the presence of a fibrillisationinhibitor (Refs 34, 35). Data from gel filtrationanalyses indicated that Ab1 – 40 with N-methyl
expert reviewshttp://www.expertreviews.org/ in molecular medicine
8Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

groups in both b-strands remained monomericeven upon prolonged incubation (see above),suggesting that a mixture of N- and C-terminalpeptides targeting both b-sheet layers mayprevent a build-up of toxic oligomers (Ref. 30).It is also imperative to verify that fibrillisationinhibitors suppress toxicity in cellular or animalmodels. Two peptides with the sequence motifRxTzExKz suppress cell death induced byAb1 – 42, suggesting that they inhibit Ab
fibrillisation without causing a build-up of toxicoligomers (Ref. 33).
A more challenging approach may be to developtherapies that target protofibril formation directly.This strategy requires a detailed understanding ofhow the structure of protofibrils differs from thatof mature fibrils. A difficulty associated withcharacterising protofibrillar structure using high-resolution methods is that preparations ofprotofibrils tend to be polydisperse, consisting ofmixtures of oligomeric species with potentiallynonidentical structures. In a ground-breakingstudy, the structure of protofibrillar Ab1–40 wascharacterised at the level of individual residuesvia hydrogen-exchange mass spectrometry (HX-MS) combined with proteolysis (Ref. 22). Theprotofibrils were shown to have a similarstructure to Ab1 –40 fibrils, with a b-sheet-richcore comprising residues 20–34 flanked byrelatively unstructured segments spanningresidues 1–19 and 35–40. Importantly, however,the b-sheet-rich core was less ordered in theprotofibrils than in the fibrils. These findingssuggest that a therapeutic strategy aimed atinhibiting the formation of Ab1– 40 protofibrils aswell as fibrils should target residues forming themost highly structured part of the protofibrillarcore. The use of methods such as HX-MS todetermine protofibrillar structure will beessential for the rational design of inhibitors thattarget protofibril formation in various protein-misfolding disorders. As in the case offibrillisation inhibitors, molecules that blockprotofibril formation must be validated withrespect to their therapeutic potential by showingthat they abolish toxicity in cellular or animalmodels.
Inhibition of protein aggregation: use ofhigh-throughput screeningA complementary approach has been to identifysmall molecules that inhibit the formationof cytotoxic protein aggregates via high-
throughput screening. In general, high-throughput screening is carried out using cell-free or cell-based assays. An advantage ofcell-based assays is that they enable theidentification of small molecules that inhibitprotein aggregation indirectly (e.g. by acting oncellular regulatory factors), in addition tocompounds that bind the aggregating proteinand interfere with self-assembly directly. Recenthigh-throughput screening initiatives have ledto the discovery of compounds that inhibit theaggregation of Ab, tau, expanded polyglutamineproteins, or PrP. Importantly, the chemicaldiversity of ‘hit’ molecules identified via high-throughput screening greatly exceeds thediversity that would normally be achieved usingstructure-based methods (this point is wellillustrated by the diversity of compounds inFig. 3). As in the case of structure-based drugdesign, it is critical to ensure that fibrillisationinhibitors identified via high-throughputscreening do not in fact stabilise toxic prefibrillarintermediates.
Ab and tauA library of 3780 biologically active compoundswas screened using a fluorescence-based assayof Ab fibrillisation in a cell-free system(Ref. 36). These efforts led to the identificationof 4,5-dianilinophthalimide (DAPH; Fig. 3a) asa novel inhibitor of Ab fibrillisation that alsodisrupts preformed fibrils. Coincubation of Ab
with DAPH interfered with the conversion ofthe peptide to a form that elicits toxicity in aneuronal cell line (IC50 ¼ �0.7 mM), suggestingthat DAPH inhibits Ab self-assembly withoutstabilising a toxic oligomer. In a second study, alibrary of �1000 compounds was screenedusing an assay that involves monitoringa decrease in the fluorescence of a fusion ofAb1 – 42 and green fluorescent protein (GFP) as areadout of Ab aggregation in an Escherichia coliexpression system (Ref. 37). This screen led tothe discovery of a triazine derivative (Fig. 3a)that suppressed Ab fibrillisation with an IC50 of�30 mM in a secondary cell-free assay. Anadvantage of the E. coli screen is that it isdesigned to identify compounds that inhibitearly oligomer formation, whereas the same isnot necessarily true of cell-free assays of Ab
fibrillisation. Specifically, prefibrillar oligomersformed by the Ab1 – 42–GFP fusion in E. coli areexpected to be nonfluorescent, so that
expert reviewshttp://www.expertreviews.org/ in molecular medicine
9Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

4,5-dianilinophthalimide (DAPH)
NH
NH
HN
O
O
N
NN
NH
HNO
ONH2
HO
O S
N+
S
N
OH
O
3-(2-hydroxyethyl)-2-[2-[[3-(2-hydroxyethyl)-5-methoxy-2-benzothiazolylidene]methyl]-1-butenyl]-5-methoxybenzothiazolium (N744)
OS
N
H2N
2-amino-4,7-dimethyl-benzothiazol-6-ol(PGL-135)
OH
O
O
5-hydroxy-1,4-naphthoquinone(Juglone)
OH O
O
OH
Celastrol
H
CO2H
O
S
Br
Br
2-Phosphono-oxybenzoic acid(Fosfosal)
OHHO
O
OH
O
PO
4-[(1S,2S)-2-amino-1-hydroxy-propyl]benzene-1,2-diol(Levonordefrin)
HO
HO
OH
NH2
Cl
(2R,3S)5-[2-hydroxy-3-(tert-butylamino)-propoxy]tetralin-2,3-diol (Nadolol)
HO
OH
O
HN
HN
HN
OH
5-[4-(4-chlorobenzoyl)-1-piperazinyl]-8-nitroquinoline
–O
N
N+
O
O
N N
Small-molecule modulators of protein aggregation identified via high-throughput screeningExpert Reviews in Molecular Medicine © 2007 Cambridge University Press
a
b
N2-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-N4-butyl-6-decyl-1,3,5-triazine-2,4-diamine
N-(4-bromophenyl)-3-(N-(4-bromophenyl)sulphamoyl)benzamide(C2-8)
Figure 3. Small-molecule modulators of protein aggregation identified via high-throughput screening.Selected examples from the text are shown. (a) Molecules that inhibit the fibrillisation of amyloid-beta peptideAb or tau. (b) Molecules that modulate the aggregation of proteins with expanded polyglutamine repeats.C2-8, fosfosal, levonordefrin and nadolol inhibit aggregation, whereas 5-[4-(4-chlorobenzoyl)-1-piperazinyl]-8-nitroquinoline promotes the formation of large, relatively nontoxic aggregates. The compounds were identifiedby screening chemical libraries in cell-free systems, Escherichia coli, yeast or mammalian cell lines (see text fordetails). The pronounced structural variation among compounds with the same molecular target (e.g. Ab,mutant huntingtin fragment) illustrates the chemical diversity that canbeaccessed via high-throughput screening.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
10Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

compounds must disrupt these oligomers to bedetected by the E. coli screen (Ref. 37). In a thirdstudy, a small-molecule library was screenedusing a fluorescence-based cell-free assaymonitoring the self-assembly of tau, a proteinthat forms aggregates called ‘neurofibrillarytangles’ (NFTs) in diseases referred to as‘tauopathies’ (Table 1) (Ref. 38). This screenresulted in the identification of a planararomatic compound (N744; Fig. 3a) thatinhibited tau polymerisation with high potency(IC50 ¼ �300 nM) and caused disaggregation ofpreformed filaments.
Expanded polyglutamine proteinsIn a pioneering study, an automated filter-trapassay of huntingtin aggregation was used toscreen a library of �184 000 compounds(Ref. 39). In this assay, fibrils formed by anN-terminal fragment of polyglutamine-expanded (‘mutant’) huntingtin were trappedon a filter membrane in 384-well format andquantified via immunoblotting. The screen ledto the identification of 300 huntingtin-aggregation inhibitors, 25 of which werebenzothiazole derivatives (IC50 ¼ �1–10 mM).Although one of these compounds (PGL-135;Fig. 3b) inhibited huntingtin aggregation in amammalian cell line and in hippocampal slicecultures (IC50 ¼ �40 mM), it was found to bemetabolically unstable and therefore unsuitablefor testing in vivo (Ref. 40). A filter-trap assaywas also used to screen the NINDS CustomCollection of 1040 drugs and bioactivecompounds approved by the US Food andDrug Administration (FDA) (Ref. 41). Thisscreen yielded ten hits that suppressedaggregation with an IC50 , 15 mM. Two of thesecompounds, the natural products juglone andcelastrol (Fig. 3b), prevented the abnormalrelocalisation of full-length mutant huntingtinin a striatal cell line. In another study, a libraryof 16 000 compounds was screened using ayeast-based assay of huntingtin aggregationand toxicity (Ref. 42). This screen resulted inthe discovery of a compound (C2-8; Fig. 3b)that suppressed aggregation by an N-terminalfragment of mutant huntingtin in a neuronalcell line (IC50 ¼ 0.05 mM) and in culturedhippocampal slices from a transgenic mousemodel of Huntington disease. The compoundalso rescued photoreceptor neurodegenerationin huntingtin transgenic flies, suggesting that it
may protect against huntingtin aggregation andtoxicity in vivo. A cell-based screen was alsoused to identify molecules that inhibit theaggregation of two expanded polyglutamineproteins (N-terminal fragments of the androgenreceptor and huntingtin) monitored via asensitive fluorescence resonance energy transfer(FRET) assay (Ref. 43). Of ten hits identifiedby this screen, five partially suppressedphotoreceptor neurodegeneration in huntingtintransgenic flies, and three of these (fosfosal,levonordefrin and nadolol; Fig. 3b) are FDA-approved drugs that could potentially berapidly translated into the clinic. Finally,a library of 37 000 compounds was screenedusing a cell-based assay that monitors thefluorescence of a huntingtin–GFP fusion as areadout of aggregation (Ref. 44). This screen ledto the identification of a compound (5-[4-(4-chlorobenzoyl)-1-piperazinyl]-8-nitroquinoline;Fig. 3b) that enhanced huntingtin aggregationand suppressed huntingtin-induced proteasomedysfunction in mammalian cells. Thiscompound also increased a-synuclein inclusionformation and reduced a-synuclein toxicity in aneuroglioma cell line (Ref. 44). Collectively,these findings suggest that novel therapeuticsin the treatment of protein-misfolding diseasesmay prevent cell death by inhibiting theformation of toxic oligomers or by promotingthe conversion of misfolded proteins to large,relatively benign inclusions.
PrPThe mammalian prion protein PrP exists in twostates: a soluble, nonpathogenic form (PrPc) andan aggregated form (PrPsc) that is thought to beresponsible for prion diseases in the brain. Alibrary of 10 000 drug-like compounds wasscreened using an innovative cell-free assayreferred to as SIFT (‘scanning for intenselyfluorescent targets’) to monitor the association ofPrPc with PrPsc (Ref. 45). This screen led to theidentification of six compounds (four with acommon N’-benzylidene-benzohydrazide corestructure) that suppressed PrPsc formation in aneuronal cell-culture model (IC50¼ �2–6 mM).In another study, a library of 2000 drugs andnatural products was screened using a high-throughput immunoblotting assay (Ref. 46).This screen resulted in the discovery of 15 novel,potent inhibitors of PrPsc formation, includingpolyphenols, antimalarials, antihistamines,
expert reviewshttp://www.expertreviews.org/ in molecular medicine
11Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

phenothiazine analogues, and statins(IC50¼ �0.1–1 mM). Unfortunately, the mostefficacious inhibitor in the cell-free assay(mefloquine, an antimalarial compound) wasshown to be inactive in vivo (Ref. 47). Thisfinding emphasises the importance of testing hitsfrom high-throughput screens in preclinicalanimal models.
Inhibition of protein misfolding andaggregation via native-state stabilisationProteins with a compact, globular structure mustundergo unfolding (or, in the case of oligomericproteins, subunit dissociation followed byunfolding) prior to forming fibrils (Ref. 48). Astrategy to prevent the fibrillisation of theseproteins is to suppress subunit dissociationand/or unfolding using small molecules thatstabilise the native state (Fig. 1). This approachhas two important advantages over interferingwith downstream self-assembly. First, inhibitionof early misfolding precludes a build-up oftoxic oligomers or protofibrils. Second,compounds that stabilise the native statetypically bind in a deep pocket or cavity of afolded protein, and, therefore, they avoidpitfalls faced by molecules that disruptprotein–protein interactions (e.g. weak affinity,poor selectivity) (Refs 49, 50).
Native-state stabilisation of transthyretinThe strategy of inhibiting fibrillisation via native-state stabilisation has been explored extensivelyin a series of elegant papers on TTR (reviewedin Ref. 49). TTR is a 55 kDa homotetramericprotein with two binding sites for the hormonethyroxine (T4) (Fig. 4a). The amyloid diseasessenile systemic amyloidosis and familialamyloid polyneuropathy are characterised bythe fibrillisation of wild-type and mutant TTR,respectively. Amino acid substitutions inpatients with familial amyloid polyneuropathydestabilise the native tetramer (Ref. 49).Biophysical data indicate that TTR fibrillisationoccurs via rate-limiting tetramer dissociation,followed by partial unfolding and aggregationof the dissociated monomer. Previous workshowed that T4 suppresses subunit dissociationby wild-type or mutant TTR (Ref. 49). AlthoughT4 would not be a useful therapeutic inhibitorbecause of its hormonal activity, it washypothesised that the binding of other smallmolecules to the hormone-binding pocket might
stabilise tetrameric TTR and suppresspathogenesis (Ref. 49). Targeting the T4-bindingpockets was a reasonable therapeutic strategybecause TTR is only a minor carrier of T4 in theperiphery, with ,1% of the T4-binding sitesoccupied. Therefore, a high proportion of sitesare available for inhibitor binding (Ref. 51). Anumber of biaryl compounds with structuressimilar to T4, including the nonsteroidal anti-inflammatory drugs (NSAIDs) diclofenac,diflunisal and flufenamic acid (Fig. 4b), werefound to suppress TTR fibrillisation by bindingto the native tetramer and inhibiting subunitdissociation under partially denaturingconditions (Refs 49, 52). Isothermal titrationcalorimetry data indicated that the binding ofinhibitor to one or both T4-binding pocketsincreases the activation energy for tetramerdissociation by stabilising the ground staterelative to the transition state (Fig. 4c) (Refs 49, 52).
Because of its inhibitory activity against TTRfibrillisation and its high oral bioavailability,diflunisal is currently being tested in a clinicaltrial for familial amyloid polyneuropathy(Ref. 49). Nevertheless, diflunisal and otherNSAIDs may not provide a long-termtherapeutic solution because chronic treatmentswith these compounds are likely to producegastrointestinal side effects. Accordingly, recentefforts have focused on identifying inhibitors ofTTR dissociation that lack NSAID activity. Inone study, a library of 55 diflunisal analogueswith various patterns of substitutions byhalogen, hydroxyl or carboxyl groups wasscreened for inhibitory activity against TTRfibrillisation (Ref. 51). The screen yielded threeinhibitors that bound the T4-binding pocket,and two of these analogues bound TTRselectively over other proteins in humanplasma. In other studies, compoundsstructurally unrelated to diflunisal, includingdiethylstilbestrol (Fig. 4b) (Ref. 53),bisaryloxime ethers (Ref. 54) and dibenzofurans(Ref. 55), were found to inhibit TTR subunitdissociation and fibrillisation with high bindingselectivity. The identification of TTRfibrillisation inhibitors from various structuralclasses represents an important advance in thedevelopment of therapeutics that lack NSAIDactivity. Although most inhibitors occupy bothT4-binding sites at sufficiently highconcentrations, recent data indicate that bindingto a single site is sufficient for kinetic
expert reviewshttp://www.expertreviews.org/ in molecular medicine
12Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

stabilisation (Ref. 56). In turn, this findingsuggests that one can minimise toxic sideeffects in vivo by administering a dose of
inhibitor no greater than that required tooccupy a single T4-binding site. Finally,genistein (Fig. 4b), the primary flavone in soy,
Inhibition of transthyretin fibrillisation via native-state stabilisationExpert Reviews in Molecular Medicine © 2007 Cambridge University Press (part b only)
Diethylstilbestrol
HO
OH
2-[2-(2,6-dichlorophenyl)aminophenyl]ethanoic acid(Diclofenac)
Cl
NH
Cl
a c
bOH
O
HO
OH
Genistein
O
O
4′,6′-difluoro-4-hydroxybiphenyl-3-carboxylicacid (Diflunisal)
O
HOFr
ee e
nerg
y
OH
F
F
HN
O OH
FF
F
N-(3-trifluoromethylphenyl)anthranilic acid(Flufenamic acid)
OH
ΔGTI2 > ΔGTI > ΔGT
Foldedmonomer
Amyloidogenicmonomer
Aggregation
TI2
TI
T
TS
ΔGTI2
ΔGTI
ΔGT
Reaction coordinate
Figure 4. Inhibition of transthyretin fibrillisation via native-state stabilisation. (a) The X-ray structure of thecomplex formed by transthyretin (TTR) and thyroxine (T4) (both hormone-binding sites are occupied). Eachmonomer in the TTR tetramer is depicted in a different colour. (b) Small-molecule inhibitors of TTRfibrillisation. (c) Free-energy diagram illustrating how native-state stabilisation inhibits TTR fibrillisation. Whenan inhibitor binds selectively to one or both T4-binding sites (yielding TI or TI2, respectively), the activationenergy for tetramer dissociation (DGT) is increased due to stabilisation of the ground state relative to thetransition state (TS) for subunit dissociation (DGTI22
. DGTI . DGT). As a result, drug binding leads to adecrease in the level of folded monomer with a high propensity to misfold and aggregate. The geometricshapes at the bottom of the graph represent different forms of TTR along the reaction coordinate (square,folded subunit; circle, unfolded subunit; tinted rectangle, aggregate; red dash, drug bound to tetramericTTR). Panel (a) was reprinted, with permission, from Ref. 51 [Adamski-Werner, S.L. et al. (2004) Diflunisalanalogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem 47,355–374 (& 2004 American Chemical Society)]; panel (c) was adapted, with permission, from Ref. 49[Johnson, S.M. et al. (2005) Native state kinetic stabilization as a strategy to ameliorate protein misfoldingdiseases: a focus on the transthyretin amyloidoses. Acc Chem Res 38, 911–921 (& 2005 AmericanChemical Society)].
expert reviewshttp://www.expertreviews.org/ in molecular medicine
13Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

has been shown to inhibit TTR subunitdissociation and fibrillisation, suggesting thatthis natural product may be usedtherapeutically with little risk of toxic sideeffects (Ref. 57).
Native-state stabilisation of SOD1Superoxide dismutase 1 (SOD1), like TTR,undergoes subunit dissociation, partialunfolding, and aggregation as a result ofdisease-linked mutations. SOD1 is ahomodimer whose quaternary structure isdestabilised by mutations linked to familialamyotrophic lateral sclerosis (Ref. 58). Smallmolecules that bind to the dimer interface ofSOD1 were identified by in silico screening(Ref. 59). These compounds were found toinhibit the aggregation of mutant SOD1,suggesting that they may be a new class ofdrugs to treat familial amyotrophic lateralsclerosis (Ref. 59).
A related strategy for the treatment of variousprotein-misfolding disorders is to rescue foldingdefects with ‘pharmacological chaperones’. Apharmacological chaperone is a small molecule(e.g. a ligand or substrate) that binds the folded,functional state of a protein and shifts theequilibrium away from partially unfoldedconformers, thereby enabling folding andtrafficking to the correct subcellularcompartment (reviewed in Ref. 60).
Therapeutic strategy II: targetingpost-translational modifications that
promote protein misfoldingand aggregation
Proteins undergo various types of post-translational modifications in cells under normalor stressed conditions. In some cases, themodified protein has reduced conformationalstability and therefore an increased propensity tomisfold and aggregate. Three post-translationalmodifications that promote the aggregation ofsome proteins are considered below: proteolysis,phosphorylation and oxidation (Fig. 1).Inhibiting these modifications may be a usefultherapeutic strategy in the treatment of someprotein-misfolding disorders.
Inhibition of proteolysisA number of fibrillogenic polypeptides arereleased from larger precursor proteins byproteolysis, and the proteases involved in
catalysing these processing events are potentialtherapeutic targets. The most intensively studiedexample of a fibrillogenic polypeptide generatedby proteolysis is Ab. Other examples discussedbelow include expanded polyglutamineproteins, a-synuclein, tau and gelsolin.
Targeting Ab formation via inhibition ofb- and g-secretaseAb is released from the transmembrane amyloidprecursor protein (APP) via successive cleavagesby two aspartyl proteases: b- and g-secretase(Fig. 5). b-Secretase, also referred to as beta-siteAPP-cleaving enzyme 1 (BACE1) or memapsin 2,is a transmembrane enzyme that cleaves APP toyield a membrane-bound C-terminal fragment(CTFb or C99) (Ref. 61). g-Secretase is atransmembrane multiprotein complex thatcleaves CTFb within the membrane, resulting inthe secretion of Ab1–40 or Ab1–42 (Refs 61, 62,63). Several lines of evidence suggest thatpresenilin (presenilin 1 or 2), a component of themultiprotein complex, acts as the catalyticsubunit of g-secretase (Ref. 62).
In addition to its role in APP proteolysis,g-secretase is responsible for theintramembranous cleavage of Notch, asignalling molecule involved in specifying cellfate during embryogenesis (Ref. 62). Presenilinknock-out mice have an embryonic lethalphenotype similar to that produced by deletingthe Notch gene, and a number of g-secretaseinhibitors have undesired consequences thatresult from disruption of Notch processing(Refs 62, 63). Accordingly, major efforts havefocused on identifying compounds that blockAPP cleavage without perturbing Notchsignalling (Fig. 5). An important breakthroughcame from the discovery that NSAIDsselectively reduce Ab1 – 42 levels in cell cultureand in vivo without targeting Notch, an effectthat is independent of cyclooxygenaseinhibition (Ref. 64). Evidence suggests thatNSAIDs inhibit g-secretase activity by bindingto an allosteric site remote from the active site(Ref. 65). In turn, this binding could trigger aconformational change that specifically disruptsthe enzyme’s cleavage of APP but leaves Notchprocessing unaffected. NSAIDs may alsoblock the formation of Ab1 – 42 by preventingactivation of the small GTP-binding proteinRho, apparently by interfering with Rhoprenylation (Ref. 66). Another compound, EHT
expert reviewshttp://www.expertreviews.org/ in molecular medicine
14Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

1864, prevents activation of the small GTP-binding protein Rac1 and suppresses Ab
formation (in neuroblastoma cells and in vivo)without affecting Notch cleavage (Ref. 67).These findings implicate Rho and Rac1 in theselective control of APP processing and suggestthat targeting these small GTP-binding proteinsis a valid strategy to prevent the accumulationof Ab in Alzheimer disease. Consistent withthis idea, Rho and Rac1 are involved inmodulating the assembly or clustering of lipidrafts, and these membrane microdomains are asite for g-secretase cleavage of APP but not ofNotch (Refs 67, 68). Lithium, used in the clinic
to treat bipolar disorder, is another agent thatselectively targets g-secretase cleavage of APP(Ref. 69). Various kinase inhibitors (including ametabolite of Gleevec) inhibit Ab productionwithout affecting Notch processing, apparentlyby interacting with an ATP-binding site onpresenilin 1 (Refs 70, 71). The binding of akinase inhibitor to this site may induce aconformational change in a g-secretase domaininvolved in cleaving APP but not Notch(Ref. 71). Although several tyrosine kinaseinhibitors are known to be safe for use inhumans (Ref. 70), the suitability of thesecompounds to attenuate amyloid plaque
Cytosolicdomain
γ-Secretase
NSAIDsEHT 1864Li+
MRK-560
PeptidomimeticsAcylguanidines
Extracellulardomain
COO–NH3+ APP
711–712713–714671–672
β-Secretase (BACE1)
Inhibition of amyloid-beta peptide (Aβ) release from the amyloid precursorprotein by targeting β- and γ-secretaseExpert Reviews in Molecular Medicine © 2007 Cambridge University Press
Figure 5. Inhibition of amyloid-beta peptide (Ab) release from the amyloid precursor protein by targetingb- and g-secretase. The amyloid precursor protein (APP) is depicted as a transmembrane protein in thephospholipid bilayer (phospholipid headgroups are shown as spheres; acyl chains are drawn as ‘tails’associated with each headgroup). The b- and g-secretase cleavage sites are indicated in the schematicrepresentation of APP: b-secretase (also referred to as beta-site APP-cleaving enzyme 1, BACE1) cleavesbetween residues 671 and 672; and g-secretase cleaves between residues 711 and 712, yielding Ab1–40, or713 and 714, yielding Ab1–42. b- and g-Secretase inhibitors discussed in the text are listed below thecorresponding cleavage sites. The use of a combination of b- and g-secretase inhibitors may be the beststrategy to lower Ab levels while minimising toxic side effects (see text for details). EHT 1864, inhibitor of thesmall GTP-binding protein Rac1; Liþ, lithium ion; MRK-560, N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide (selective g-secretase inhibitor from MerckSharp and Dohme); NSAIDs, nonsteroidal anti-inflammatory drugs.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
15Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

formation may be limited by their weakpenetrance of the blood–brain barrier. Finally,recent data indicate that the potent g-secretaseinhibitor MRK-560 suppresses amyloid plaqueformation and Ab generation in APP transgenicmice without perturbing the Notch signallingpathway (Ref. 72). Because MRK-560 is orallybioavailable and penetrates the blood–brainbarrier, this compound holds promise as a new,selective inhibitor of amyloid formation in thetreatment of Alzheimer disease.
As an alternative to inhibiting g-secretase,several groups have focused on the strategy oflowering Ab levels with small molecules thattarget b-secretase (BACE1) (Fig. 5). The idealBACE1 inhibitor should be both highly potentand selective for BACE1 over other aspartylproteases not involved in APP processing,including BACE2 (memapsin 1), cathepsin D,and pepsin. An advantage of targeting BACE1rather than g-secretase is that a cocrystalstructure of the BACE1 protease domaincomplexed with a transition-state analogue hasbeen solved (Ref. 73). In turn, this informationhas facilitated structure-based design of BACE1inhibitors with optimised potency and selectivity(Refs 74, 75, 76). A series of transition-stateinhibitors containing a nonhydrolysable Leu–Ala dipeptide isostere was described in a recentreport (Ref. 76). One of these compounds washighly potent in an assay of recombinant BACE1activity (Ki ¼ 0.12 nM) and exhibited highselectivity for BACE1 over BACE2 and cathepsinD. Other peptidomimetics (Refs 75, 77) and aseries of acylguanidines (Ref. 74) were alsoreported to inhibit BACE1 in cell-free andcellular assays. A potential limitation of someof these compounds is that they may exhibitweak penetration of the blood–brain barrierbecause of their ‘peptidic character’ (Ref. 78).Various strategies have been reported toimprove the blood–brain barrier permeabilityand Ab-lowering ability of BACE1 inhibitorsin vivo, including covalent attachment of acarrier peptide (Ref. 79) and restriction ofconformational flexibility via macrocyclisation(Ref. 78).
The intense interest in developing BACE1inhibitors has been motivated largely byevidence from early studies that BACE1 nullmice are apparently normal (Refs 62, 63).However, this notion has been challenged bydata from more recent knock-out studies
suggesting that BACE1 is involved inmodulating synaptic plasticity (Ref. 80) andnerve myelination (Refs 81, 82). These findingssuggest that careful dosing of BACE1 inhibitorswill be critical to minimise toxic side effects.One strategy to minimise the doses of BACE1and g-secretase inhibitors while maintainingtherapeutic efficacy might be to treat patientswith both types of drugs simultaneously. Therationale for this type of approach is that thedegree of g-secretase inhibition required tomaintain Ab levels below a toxic threshold maybe reduced if the steady-state levels of CTFbsubstrate are decreased by BACE1 inhibition.Conversely, the extent to which the generationof CTFb must be suppressed by BACE1inhibition may be lower if the downstreamconversion of CTFb to Ab is largely abrogatedby g-secretase inhibition. A combinationtherapy involving BACE1 and g-secretaseinhibitors would also alleviate the accumulationof potentially toxic fragments resulting from asingle cleavage of APP. For example, inhibitionof g-secretase alone might lead to a build-up ofC-terminal fragments of APP, a phenomenonlinked to memory deficits in presenilin knock-out mice (Ref. 83). Dual inhibition of BACE1and g-secretase would be expected to reducethis toxic effect. The results of a recent studyindicate that NSAIDs downregulate expressionof BACE1 by activating the peroxisomeproliferator-activated receptor g (PPARg), arepressor of BACE1 gene transcription (Ref. 84).Therefore, NSAIDs may block APP cleavage byreducing BACE1 and g-secretase activitysimultaneously. Another report showed thatvarious APP-binding molecules inhibit cleavageby both BACE1 and g-secretase (Ref. 85).Because NSAIDs and the APP-binding agentsinterfere with processing by both enzymes, theyare excellent candidates for anti-amyloidtherapies in the treatment of Alzheimer disease.
In addition to targeting BACE1 and g-secretase,another strategy to reduce the amyloid levels inthe brain may be to enhance the destruction ofAb via proteolysis (Refs 86, 87). Interestingly,the activity of one of the major Ab-processingenzymes, neprilysin, is upregulated in corticalneurons by the neuropeptide somatostatin(Ref. 88). These data suggest that the activationof somatostatin receptors might be anothertherapeutic strategy in the treatment ofAlzheimer disease.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
16Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Role of proteolysis in the aggregation ofpolypeptides other than AbEvidence suggests that fibrillogenic polypeptidesother than Ab are generated from larger precursorproteins via proteolysis. Intense research activityhas focused on the role of proteolysis in thepolyglutamine diseases. Recent data indicate thatblocking calpain cleavage reduces huntingtinaggregation and toxicity in a cell-culture model(Ref. 89), and caspase-6 processing is necessaryfor huntingtin-induced neurodegeneration inmice (Ref. 90). In addition, the aggregation ofpolyglutamine-expanded ataxin-3 is triggered bythe removal of an N-terminal segment, possiblyvia caspase-mediated cleavage (Refs 91, 92).The cleavage of huntingtin at some sites on thepolypeptide chain may be associated with thenormal function of the protein (Ref. 93), implyingthat not all forms of huntingtin proteolysis areappropriate targets for therapy. Proteolysis hasalso been implicated in modulating theaggregation and toxicity of the following proteins:(1) a-synuclein, cleaved C-terminally by the 20Sproteasome to yield a truncated, fibrillogenicspecies that seeds aggregation of the full-lengthprotein (Refs 94, 95); (2) tau, processed at itsC-terminus by multiple caspases to yield atruncated species with a high propensity to formfibrils (Refs 96, 97); and (3) gelsolin, cleavedsuccessively by furin and MT1-matrixmetalloproteinase to yield 5 kDa and 8 kDaamyloidogenic peptides (Ref. 98). A reasonabletherapeutic strategy may be to suppress theaggregation of these proteins using compoundsthat inhibit their corresponding proteases.However, as in the case of small moleculestargeting b- and g-secretase, a challenge will be todevelop potent, selective inhibitors with minimaltoxic side effects.
Inhibition of phosphorylationThe aggregation behaviour of some proteins isinfluenced by phosphorylation, and kinases andphosphatases that promote the formation of toxicaggregates are potential therapeutic targets. Theprototypical protein that undergoes aggregationmodulated by phosphorylation is tau. A secondexample discussed below is a-synuclein.
Role of phosphorylation in the aggregationof tauA characteristic feature of tau from NFTs in humantauopathies is the hyperphosphorylation of the
protein at multiple serine and threonine residues.A total of 38 serine/threonine phosphorylationsites have been identified in filamentous tau,compared with only 15 sites in tau from controladult brain (Ref. 99). Hyperphosphorylated tauhas a weakened affinity for microtubulesand readily forms filaments, implying thatphosphorylation promotes NFT formation byreleasing a pool of unbound, fibrillogenic tauin the cytosol (Refs 99, 100, 101). However,evidence from in vivo studies suggests thattau hyperphosphorylation correlates withneurotoxicity but not with NFT assembly per se.Expression of the human tau gene in Drosophilainduced age-dependent neurodegeneration andthe accumulation of phosphorylated tau withoutNFT formation (Ref. 102). Althoughphosphorylated tau and NFTs were detected intransgenic mice expressing human tau, theaccumulation of filamentous tau was inverselycorrelated with the presence of apoptotic nucleiin individual neurons (Ref. 103). In transgenicmice expressing a repressible human tauconstruct, downregulation of transgeneexpression after the onset of NFT formation ledto increased neuronal counts and improvedmemory function without affecting theprogression of tau pathology (Ref. 104).Inhibition of tau hyperphosphorylation with akinase inhibitor suppressed motor dysfunctionwithout reducing NFT levels in transgenic miceexpressing P301L human tau, a mutant form ofthe protein involved in frontotemporal dementiawith parkinsonism-17 (Ref. 105). These findingssuggest that tau hyperphosphorylation triggersneurodegeneration by a mechanism unrelated toNFT formation. Consistent with this idea,hyperphosphorylated tau was found to disruptmicrotubule assembly by a dominant-negativemechanism that involved trapping unmodifiedtau in an inactive complex, whereas thepolymerisation of hyperphosphorylated taueliminated this potentially neurotoxic inhibitoryeffect (Ref. 106).
Regardless of whether NFTs are toxic, benign orprotective, tau hyperphosphorylation generallycorrelates with neurodegeneration in animalmodels or humans (Refs 101, 105, 107).Therefore, the inhibition of tau kinases may bea reasonable therapeutic strategy in thetreatment of tauopathies. Multiple kinaseshave been implicated in tau phosphorylation(Ref. 108). Notable examples include glycogen
expert reviewshttp://www.expertreviews.org/ in molecular medicine
17Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

synthase kinase 3b (GSK3b) (Refs 109, 110),cyclin-dependent kinase 5 (CDK5) (Refs 111,112), protein kinase A (PKA) (Ref. 113) andtau-tubulin kinase 1 (Ref. 114). Conditionaloverexpression of GSK3b induces tauhyperphosphorylation and neurotoxicity in atransgenic mouse model (Ref. 109), andDrosophila co-expressing tau and GSK3b exhibita more severe neurodegenerative phenotypethan flies expressing tau alone (Ref. 110).Oligomeric Ab activates GSK3b viadownregulation of the Akt pathway (Refs 108,115, 116). Accordingly, GSK3b plays a criticalrole in the pathogenesis of Alzheimer disease,linking Ab aggregation with tauhyperphosphorylation. Inhibition of GSK3 withlithium or the small molecule AR-A014418results in decreased tau phosphorylation andaggregation and reduced neurodegeneration intransgenic mice expressing the P301L mutant(Ref. 117). The inhibitor AR-A014418 is apromising lead compound for treatingAlzheimer disease and other tauopathiesbecause it is potent (IC50 ¼ 104+ 27 nM) andhighly selective for GSK3 over other kinases(Ref. 118).
Another kinase considered to be a potentialtherapeutic target in tauopathies is CDK5. Twoactivator proteins, p35 and its more stabletruncated form, p25, modulate CDK5 activity inneurons (Ref. 119). Levels of p25 are increasedin Alzheimer disease brain, resulting inderegulated CDK5 activity. Overexpression ofp25 elicits tau hyperphosphorylation, tauaggregation, and neurodegeneration intransgenic mice (Refs 111, 112). Similarly, co-expression of CDK5 and its activator, p35,enhances tau toxicity in a Drosophila model(Ref. 107). A lentivirally encoded CDK5inhibitory peptide derived from p35 was foundto suppress tau hyperphosphorylation andapoptosis in cortical neurons overexpressingCDK5/p25 or treated with Ab1 – 42 (Ref. 120).These results imply that CDK5/p25 may be auseful therapeutic target in Alzheimer diseaseand other tauopathies. By contrast, data fromanother study indicate that p25-mediated CDK5activation results in the inhibition of GSK3, inturn suggesting that a decrease in CDK5/p25activity could lead to enhanced tauhyperphosphorylation (Ref. 121). Given thesecontradictory findings, it would seem thatGSK3 is a more favourable target than CDK5 to
suppress tau hyperphosphorylation andneurotoxicity.
Role of phosphorylation in the aggregation ofa-synucleinThe most abundant modified form of a-synucleinin Lewy bodies (characteristic inclusions inthe brains of synucleinopathy patients) is aspecies phosphorylated at Ser129 (Refs 122,123). Although this phosphorylated species isalso detected in the soluble portion ofnormal and diseased brain tissue, it is highlyenriched in Lewy bodies. These observationssuggest that the phosphorylation of a-synucleinat Ser129 occurs under normal physiologicalconditions, and this modification convertsthe protein to a form with a high propensityto aggregate. In support of this idea,phosphorylation at Ser129 is necessary forthe formation of a-synuclein-containinginclusions in a neuroblastoma cell line(Ref. 124). By contrast, evidence from studies ofa-synuclein transgenic Drosophila suggests thatphosphorylation at Ser129 prevents theformation of detergent-insoluble aggregates butincreases neurotoxicity, perhaps by eliciting abuild-up of soluble a-synuclein oligomers(Ref. 125). Although it is still unclear howphosphorylation at Ser129 affects a-synucleinaggregation, the fact that this modification isinvolved in neurotoxicity suggests thattargeting serine/threonine kinases may be areasonable therapeutic strategy in thetreatment of Parkinson disease. Kinasesproposed to play a role in phosphorylatinga-synuclein at Ser129 include casein kinases 1and 2 and G-protein-coupled receptor kinase 2(Refs 124, 125, 126). DYRK1A, a member ofthe dual-specificity tyrosine-regulated kinasefamily, has been shown to promotea-synuclein aggregation in a hippocampal cellline and protofibril formation in a cell-freesystem (Ref. 126). Interestingly, DYRK1Aselectively phosphorylates a-synuclein atSer87, suggesting that phosphorylation at sitesother than Ser129 may play a role ina-synuclein aggregation and/or toxicity.Because serine/threonine kinases involved inphosphorylating a-synuclein have manydifferent substrates, the feasibility ofsuppressing a-synuclein toxicity by targetingthese kinases may be limited by harmful off-pathway effects.
expert reviewshttp://www.expertreviews.org/ in molecular medicine
18Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Inhibition of oxidationProtein aggregation and oxidative stress arecoupled phenomena: not only does proteinoxidation generally promote aggregation, butprotein aggregates also tend to elicit a build-upof reactive oxygen species (ROS), therebycreating a vicious cycle (Ref. 127). Thesefindings raise the possibility that antioxidantsmay be beneficial in the treatment of protein-misfolding diseases. Two proteins that exhibitaggregation behaviour and/or toxicitymodulated by oxidative stess are Ab anda-synuclein. Other examples discussed beloware huntingtin, tau and SOD1.
Role of oxidation in the aggregation andtoxicity of AbA large proportion of Ab in the brains ofAlzheimer disease patients is oxidised atMet35 to the corresponding sulphoxide(Refs 128, 129). Evidence from several groupsindicates that oxidation at this positioninhibits the conversion of Ab1 – 40 or Ab1 – 42 tooligomers or fibrils (Refs 130, 131, 132). Thisinhibitory effect can be rationalised byexamining the SS-NMR structure of fibrillarAb1 – 40: methionine sulphoxide is substantiallymore polar than methionine, and an increasein polarity at position 35 would be predictedto disrupt external quaternary interactionsnecessary for self-assembly (Fig. 2a) (Ref. 17).Although the presence of methioninesulphoxide at position 35 interferes with Ab
aggregation, redox reactions involving Met35are thought to contribute to Ab-inducedoxidative stress and neurotoxicity. As onepossibility, one-electron oxidation of Met35 inoligomeric membrane-bound Ab may result inthe formation of a sulphuranyl radical, whichin turn can induce peroxidation ofneighbouring lipids and a build-up of ROS(Ref. 129). Alternatively, the oxidation ofMet35 may be coupled with the reduction ofmetal ions (e.g. Cu2þ, Fe3þ) bound to Ab
(Ref. 128). The reduced forms of these metalions may then react with O2 to generate H2O2
(Refs 127, 128).Recent data indicate that various oxidised
metabolites stimulate Ab aggregation (Ref. 133).A ketoaldehyde derivative of cholesterol and itsaldol product accelerated the conversion ofAb1 – 40 to spherical aggregates and maturefibrils in a cell-free system by lowering the
critical concentration for self-assembly. Thiseffect was attributed to the reaction of Ab1 – 40
with the oxidised cholesterol metabolites,resulting in the formation of covalent adducts.These findings may account for the enhanceddeposition of wild-type Ab in patients withsporadic Alzheimer disease compared withhealthy individuals – namely, Alzheimerdisease patients may have increased levels ofoxidised metabolites that covalently modifywild-type Ab, converting it to a form with ahigh propensity to aggregate. Oxidisedcholesterol metabolites are formed viainflammatory ozonolysis reactions associatedwith atherosclerosis, providing a rationale forobserved links among hypercholesterolaemia,inflammation, atherosclerosis and sporadicAlzheimer disease (Ref. 133).
Given the role of oxidative stress in Ab
aggregation and toxicity, antioxidant therapymay be beneficial in the treatment of Alzheimerdisease. Consistent with this idea, a number ofpolyphenols with the ability to scavenge ROSinhibit Ab fibrillisation and destabilisepreformed Ab fibrils in cell-free systems atconcentrations between 0.1 and 1 mM (Refs 134,135). The polyphenol curcumin (Fig. 1) preventsthe conversion of Ab to potentially toxicoligomers in addition to fibrils (Ref. 136).Curcumin is a promising therapeutic candidatebecause it crosses the blood–brain barrier andreduces amyloid formation in aged APPtransgenic mice (Ref. 136). The pineal hormonemelatonin is another promising candidatebecause it penetrates the blood–brain barrier,attenuates oxidative damage, and prevents Ab
accumulation in the brains of APP transgenicmice, and it has been used safely in the clinicto treat sleep disorders (Refs 137, 138).Antioxidant molecules such as curcumin andmelatonin may inhibit Ab aggregation byscavenging ROS and/or interfering withintermolecular interactions that drive Ab self-assembly (Refs 136, 137). Another antioxidantcompound, acetyl-L-carnitine, suppressesAb1 – 42-induced neurotoxicity and oxidativedamage by increasing cellular levels of theendogenous antioxidant glutathione (Ref. 139).Finally, the metal chelator clioquinol reducesAb neurotoxicity in cell culture, presumably bydisrupting interactions between Ab and metalions (Ref. 128). The results of a pilot Phase IIclinical trial imply that clioquinol may slow
expert reviewshttp://www.expertreviews.org/ in molecular medicine
19Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

cognitive decline in severely affected Alzheimerdisease patients (Ref. 140).
Role of oxidation in the aggregation andtoxicity of a-synucleinVarious covalent oxidative modifications affectthe conversion of a-synuclein to high molecularweight species in cell-free systems. a-Synucleinmodified by nitration (Refs 141, 142), metal-catalysed oxidation (Ref. 143) or dopaminequinone (Refs 34, 35) does not fibrillise, butinstead accumulates as soluble, potentially toxicoligomers. Oligomers also accumulate inmixtures of methionine-oxidised andunoxidised a-synuclein (Refs 34, 144).a-Synuclein species containing nitrotyrosine,methionine sulphoxide, and methioninesulphone were detected in a cellular model ofParkinson disease (Ref. 145), implying thatthese modifications play a role in Parkinsondisease pathogenesis. Oxidised cholesterolmetabolites were shown to accelerate theformation of a-synuclein protofibrils andfibrils in a cell-free system, apparently via anoncovalent mechanism (Ref. 146). Thesemetabolites are elevated in the brains ofsynucleinopathy patients, suggesting that theypromote a-synuclein aggregation and toxicityassociated with Lewy body disorders(Ref. 146). Several compounds with antioxidantactivity inhibit a-synuclein fibrillisation anddestabilise preformed fibrils. Examplesinclude the polyphenols curcumin, myricetin,nordihydroguaiaretic acid, rosmarinic acid andtannic acid, and the flavonoid baicalein(Refs 135, 147, 148). A potential limitation ofthese findings is that the compounds may blockfibrillisation without suppressing the formationof toxic protofibrils. For example, the inhibitionof a-synuclein fibrillisation by baicalein resultsin a build-up of soluble oligomers (Ref. 147).Accordingly, it will be critical to determinewhether antioxidants suppress a-synucleintoxicity in cellular and animal models ofParkinson disease and other synucleinopathies.
Role of oxidation in the aggregation andtoxicity of huntingtin, tau and SOD1Various reports indicate that oxidative stress playsa central role in the aggregation and toxicity ofother proteins involved in protein-misfoldingdisorders, in addition to Ab and a-synuclein.For example, the toxicity of a mutant huntingtin
fragment was shown to be suppressed inyeast lacking the gene encoding kynurenine3-monooxygenase (KMO), an enzyme involvedin the degradation of tryptophan (Ref. 149).Two kynurenine pathway metabolites –3-hydroxykynurenine and quinolinic acid – areknown to induce the formation of ROS.Accordingly, it was hypothesised that mutanthuntingtin causes cell death by triggeringoxidative stress via upregulation of thekynurenine pathway (Ref. 149). In support ofthis hypothesis, the expression of mutanthuntingtin led to a build-up of ROS inSaccharomyces cerevisiae, and this effect wasabolished by genetic or pharmacologicaldisruption of KMO activity. Data from studies inmammalian cell culture suggest that a mutanthuntingtin fragment causes a build-up of ROSvia protein aggregation and iron-dependentredox chemistry, and this oxidative stress issuppressed by antioxidants and heat-shockproteins (Refs 150, 151, 152). In a Drosophilamodel of human tauopathies, tau-inducedneurodegeneration was enhanced or suppressedin flies with reduced or increased expression ofantioxidant enzymes, respectively (Ref. 153).Importantly, neurodegeneration was alsosuppressed in flies treated with the antioxidantcompound a-tocopherol (vitamin E). Finally, theformation of disulphide-linked (i.e. oxidised)SOD1 dimers and oligomers was found to be acritical early step in amyotrophic lateral sclerosispathogenesis (Ref. 154). Together, these findingsunderscore the important role of oxidative stressin various protein-misfolding disorders, andthey suggest that antioxidants are reasonabletherapeutic candidates in the treatment of thesediseases.
Therapeutic strategy III: upregulatingmolecular chaperones or aggregate
clearanceCells have evolved various quality-controlmechanisms to prevent a build-up of toxicprotein aggregates (Ref. 155). An initial line ofdefence is provided by molecular chaperones –cellular factors that inhibit protein misfoldingand/or interfere with non-native interactionsleading to aggregate formation (Fig. 1). In somecases, chaperones promote the correct folding oftheir misfolded protein substrates. Alternatively,chaperones target misfolded polypeptides fordegradation by the ubiquitin–proteasome
expert reviewshttp://www.expertreviews.org/ in molecular medicine
20Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

pathway or by chaperone-mediated autophagy, aclearance mechanism in which a target protein istranslocated into the lysosome with the assistanceof the cytosolic chaperone Hsc70 (Ref. 155).Aggregates formed under conditions wherenormal homeostasis mechanisms areoverwhelmed by a high burden of proteinmisfolding are eliminated by ‘macroautophagy’,a lysosomal clearance pathway in whichcellular material is engulfed in a vesicular bodytermed an ‘autophagosome’ (Fig. 1) (Ref. 156).Enhancing chaperone function and stimulatingaggregate clearance might be useful therapeuticstrategies for the treatment of protein-misfolding disorders.
Enhancing chaperone functionSeveral molecular chaperones are referred toas heat-shock proteins because they areupregulated in response to heat and otherforms of stress that cause a build-up ofmisfolded polypeptides (Ref. 155). Examples ofmolecular chaperones associated with theheat-shock response include Hsp27, Hsp40,Hsp70 and Hsp90 (Fig. 1). Evidence suggeststhat the upregulation of heat-shock proteinssuppresses the aggregation of Ab, a-synucleinand polyglutamine proteins.
Effect of chaperones on the aggregationand toxicity of AbData from studies in a cell-free system indicatethat fibril formation by Ab1 – 42 is inhibited byHsp90 or by Hsp70 in combination with itsco-chaperone, Hsp40 (Ref. 157). The Hsp70–Hsp40 inhibitory effect is dependent on ATPhydrolysis, suggesting that these twochaperones expend energy to convert Ab to aform with a weak propensity to aggregate. Acombination of Hsp90, Hsp70 and Hsp40causes dissociation of preformed oligomers butnot fibrils, implying that the chaperones targetoligomeric intermediates on the Ab aggregationpathway. Other studies have shown that thesmall heat-shock proteins Hsp20, Hsp27 andaB-crystallin bind Ab1 – 40, and this bindingcorrelates with inhibition of Ab aggregationand attenuation of Ab toxicity (Ref. 158). Down-regulation of heat-shock factor 1 (HSF1), atranscription factor that induces the expressionof heat-shock proteins, results in acceleratedparalysis and increased protein aggregation intransgenic worms overexpressing Ab1 – 42 in the
body-wall muscles (Ref. 5). These findingssuggest that molecular chaperones associatedwith the heat-shock response play a critical rolein preventing Ab aggregation and toxicity invivo. Given that they are located primarilyin intracellular compartments, molecularchaperones are likely to protect against Ab
toxicity by binding oligomeric species insidethe cell, as opposed to extracellular oligomersor plaques (Ref. 157).
Effect of chaperones on the aggregationand toxicity of a-synucleinThe overexpression of Hsp70 causes a decrease ina-synuclein toxicity in cell culture and a reductionin high molecular weight and detergent-insolubleforms of a-synuclein in the brains of a-synucleintransgenic mice (Ref. 159). Hsp70 has also beenfound to prevent the accumulation of highmolecular weight a-synuclein species indopaminergic cells treated with the complex Iinhibitor rotenone (Ref. 160). Although Hsp70inhibits dopaminergic cell death in a-synucleintransgenic flies, the chaperone has no effect onthe number, size or morphology of Lewy-likeinclusions in this model (Ref. 161). Hsp70 hasbeen shown to bind protofibrils but not fibrilsformed from recombinant a-synuclein(Refs 162, 163), a result that may explain whyHsp70 has no discernible effect on fibrillarinclusions in a-synuclein transgenic flies. Inother studies, Hsp27 has been shown tointerfere with a-synuclein aggregation andtoxicity in cell-culture models (Refs 164, 165).Together, these findings suggest that Hsp70 andHsp27 may delay neurodegeneration associatedwith synucleinopathies by inhibiting theformation of toxic a-synuclein aggregates in thebrain.
Effect of chaperones on the aggregationand toxicity of polyglutamine proteinsAtomic force microscopy data indicate that Hsp70and Hsp40 block the conversion of a mutanthuntingtin fragment to spherical and ring-likeoligomers by binding and sequestering themonomeric protein (Ref. 166). Evidence fromseveral groups suggests that expandedpolyglutamine proteins bound to Hsp70 aretargeted for degradation via a pathwaymediated by CHIP (‘C-terminus of Hsp70-interacting protein’), an Hsp70 co-chaperonewith E3 ubiquitin ligase activity (Refs 167, 168,
expert reviewshttp://www.expertreviews.org/ in molecular medicine
21Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

169). Other studies have shown that Hsp70 acts incooperation with the cytosolic chaperonin TRiC(CCT) to inhibit the conversion of expandedpolyglutamine proteins to toxic oligomers,perhaps by promoting the formation of benign,off-pathway assemblies (Refs 170, 171, 172).Collectively, these data suggest that Hsp70,Hsp40, CHIP and TRiC interfere with proteinaggregation and neurotoxicity associated withpolyglutamine disorders.
Pharmacological upregulation ofchaperone functionThe observations outlined above suggest thatenhancing the function of molecular chaperonesmight be a useful strategy to inhibit toxicaggregate formation in various protein-misfolding diseases. This type of approach mayhave fewer pitfalls compared with directlytargeting aggregation with small-moleculeinhibitors: whereas chaperones are designed tobind exposed hydrophobic regions of misfoldedpolypeptides, small molecules occupy aninsufficient volume to efficiently disruptaberrant protein–protein interactions involvingthese exposed surfaces (Ref. 173).
Emerging data suggest that it may be possibleto upregulate molecular chaperones using oneof several pharmacological strategies.Notably, the Hsp90 inhibitor geldanamycin(GA) and its derivatives 17-AAG(17-allylamino-demethoxygeldanamycin) and17-DMAG [17-(dimethylaminoethylamino)-17-demethoxygeldanamycin] induce theexpression of Hsp70 and Hsp27 in cancermodels (Refs 174, 175), raising the possibilitythat these agents may be useful for treatingprotein-misfolding diseases (Fig. 1). Consistentwith this idea, GA was shown to inhibit a-synuclein toxicity in a cell-culture model(Ref. 176) and in transgenic Drosophila(Ref. 177). The protective effect of GA occurredin parallel with a decrease in a-synucleinaggregation in cell culture (Ref. 176), whereas itcorrelated with an increase in detergent-resistant, presumably nontoxic a-synucleinspecies in the brains of transgenic flies(Ref. 177). GA and another Hsp90 inhibitor,radicicol, suppressed huntingtin toxicity andaggregation in organotypic slice culturesderived from huntingtin transgenic mice(Ref. 178). The data from these studies supporta model in which GA inhibits the formation of
toxic protein aggregates by upregulating Hsp70(Refs 176, 177, 178). In turn, Hsp70upregulation is thought to involve GA-mediated release of HSF1 from its inactivecomplex with Hsp90, followed by HSF1trimerisation, phosphorylation and nuclearlocalisation (Fig. 1) (Refs 179, 180). 17-AAG and17-DMAG are already being tested in Phase Iand II clinical trials as potential anticanceragents (Ref. 175), suggesting that they may besuitable for administration to patients withprotein-misfolding disorders. A disadvantage ofthese molecules is that they provide a narrowtherapeutic window because the concentrationsat which they induce Hsp70 upregulation aresimilar to the concentrations at which they elicittoxicity as a result of degradation of Hsp90client proteins (Ref. 180). This problem wasaddressed by developing a series of novobiocinanalogues that target a different domain ofHsp90 (the C-terminal ATP-binding domain, incontrast to the N-terminal ATP-binding pockettargeted by GA and its derivatives) (Ref. 180).One analogue (A4; Fig. 1) was found toupregulate Hsp70 and suppress Ab-inducedneurodegeneration in a cellular model ofAlzheimer disease without causing toxicity,suggesting that this compound may provide arelatively broad therapeutic window.
In addition to Hsp90 inhibitors, other smallmolecules have been shown to induce the heat-shock response. Examples include acetyl-L-carnitine, a compound that is naturally presentin mitochondrial membranes and upregulatesHsp72 (Hsp70) (Ref. 139), and celastrol, acomponent of Chinese herbal medicines thatactivates HSF1 (Ref. 181). These smallmolecules may also be useful in the treatmentof protein-misfolding diseases.
Enhancing aggregate clearanceIn cells with excess amounts of misfoldedproteins, the capacity of chaperones and theubiquitin–proteasome pathway to preventprotein aggregation is exceeded (Ref. 155).Under these conditions, misfolded proteinsform aggregates that are transported alongmicrotubules to the perinuclear region, wherethey are recruited into inclusion bodies termed‘aggresomes’ (Fig. 1) (Ref. 182). At theaggresome, it is hypothesised that proteinaggregates are targeted for destruction viamacroautophagy (Refs 183, 184). Examples of
expert reviewshttp://www.expertreviews.org/ in molecular medicine
22Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

aggregated proteins that may be processed viaaggresome formation and macroautophagy area-synuclein and proteins with expandedpolyglutamine repeats.
Clearance of aggregated a-synucleinThe overexpression of a-synuclein in mammaliancells leads to the formation of spherical,prefibrillar oligomers, followed by theappearance of aggresome-like inclusionsenriched with fibrillar a-synuclein (Ref. 185).Prefibrillar a-synuclein oligomers, but not themature fibrils, are eliminated from neuronalcells via lysosomal degradation, and inhibitionof this process augments a-synuclein-inducedcell death (Ref. 186). Data from another studysuggest that macroautophagy plays a role in theclearance of a-synuclein from neuronal cells(Ref. 187). Together, these data support a modelin which toxic a-synuclein oligomers aretransported to the aggresome as part of aprotective mechanism. Once at the aggresome,the oligomers are either converted intorelatively benign fibrils or cleared from the cellvia macroautophagy. Consistent with the ideathat aggresome formation is cytoprotective, thepresence of aggresomes was found to correlatepoorly with the presence of apoptotic markersin a mammalian cell line overexpressinga-synuclein and its binding partner, synphilin-1(Ref. 188). In addition to clearance via theaggresome–macroautophagy pathway, evidencesuggests that a-synuclein is eliminated fromneuronal cells via chaperone-mediatedautophagy (Ref. 156).
Clearance of expanded polyglutamineproteinsAggresome-like inclusion bodies are formed inmammalian cells expressing expandedpolyglutamine proteins, including N-terminalhuntingtin fragments, ataxin-3, and theandrogen receptor (Refs 189, 190, 191).Disruption of macroautophagy by RNAinterference (RNAi) or with small-moleculeinhibitors causes a reduction in the clearance ofcytosolic polyglutamine aggregates (Refs 191,192). The accumulation of autophagic proteinsand lysosomes near the aggresome and theelimination of polyglutamine aggregates requireintact microtubule-dependent transport(Refs 183, 184). Collectively, these findingssuggest that recruitment of the autophagic
machinery to the aggresome is necessary for theefficient removal of protein aggregates from thecytosol (Refs 183, 190, 191).
Pharmacological upregulation ofaggregate clearanceAdvances in understanding aggregate-clearancemechanisms have motivated efforts to activatethese pathways as a strategy to treat protein-misfolding disorders (Ref. 191). Multiple linesof evidence suggest that the accumulation ofcytosolic protein aggregates can be suppresseddramatically via pharmacological upregulationof macroautophagy. This approach may havegreater therapeutic potential than theenhancement of proteasome activity, given thatupregulation of the ubiquitin–proteasomepathway (in contrast to autophagy) is likely toincrease the risk of cancer by causing depletionof short-lived proteins with anti-tumourigenicactivity (e.g. p53) (Ref. 193). The macrolideantibiotic rapamycin (also known as ‘sirolimus’)activates macroautophagy by blocking theactivity of mTOR (‘mammalian target ofrapamycin’), a kinase that inhibitsautophagosome formation (Fig. 1). Rapamycinenhances autophagic clearance and reduces thetoxicity of a-synuclein, tau and polyglutamineproteins in cellular and in vivo models(Refs 187, 194, 195). Rapamycin derivativeswith improved water solubility (CCI-779,RAD001 and AP23576) are already underclinical development as possible anticancerdrugs, and, therefore, they may also be suitablefor use in humans to treat protein-misfoldingdiseases (Refs 193, 196). At the same time, thepotential for toxic side effects associated withlong-term administration of rapamycinderivatives has motivated a search foralternative pharmacological approaches tostimulate macroautophagy. Recent findingssuggest that macroautophagic clearance ofprotein aggregates is stimulated in an mTOR-independent manner by lithium (Ref. 197) andthe disaccharide trehalose (Ref. 198).
Research in progress and outstandingresearch questions
There has been tremendous progress over the pastfive years in the development of novel therapeuticapproaches targeting protein-misfolding diseases.Advances in our understanding of fibrillarstructure have enabled the rational design of
expert reviewshttp://www.expertreviews.org/ in molecular medicine
23Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

peptides that inhibit fibrillisation. Chemicallydiverse compounds that block aggregation or,alternatively, that promote the formation oflarge, nontoxic assemblies have been identifiedusing powerful high-throughput screeningtechnologies. In the case of proteins thatnormally have a compact, globular structure,misfolding and aggregation have been inhibitedsuccessfully by compounds that stabilise thenative conformation. A large body of evidencesuggests that the formation of toxic proteinaggregates can be suppressed by interfering withpost-translational modifications (e.g. proteolysis,phosphorylation and oxidation), increasingchaperone function, or upregulating aggregate-clearance mechanisms such as macroautophagy.Several of these highly promising therapeuticstrategies are highlighted in the model shown inFigure 1.
What new developments can be expectedduring the years ahead? One reasonableprediction is that the structures of protofibrillarand fibrillar assemblies will continue to besolved using methods such as SS-NMR, andthis information will enable the structure-baseddesign of many more protein-aggregationinhibitors. In parallel, continued high-throughput screening efforts will lead to theidentification of additional compounds thatinterfere with protein self-assembly. A highpriority will be to assess whether drugcandidates prevent the formation of oligomers,protofibrils or fibrils, and whether they reducetoxicity in animal models of misfoldingdiseases. Future developments will also likelyinclude the implementation of new therapeuticstrategies that have not been discussed in thisreview. For example, emerging data suggestthat the depletion of aggregating proteins usingRNAi methods in vivo may be a powerfulapproach to prevent misfolding diseases(Refs 199, 200, 201).
There are a number of outstanding researchquestions that must be addressed by futureresearch. Which aggregated species (oligomers,protofibrils or fibrils) are responsible for celldeath in protein-misfolding diseases? Why isthe pathology of protein-aggregation disordersgenerally limited to specific types of cells ortissues? Which cytotoxic pathways are triggeredby protein aggregation? Studies carried out ingenetically manipulable model organismsincluding yeast, Drosophila and C. elegans will
provide a powerful approach to answer thethird question by driving the discovery of newpathogenic mechanisms involved in protein-misfolding diseases (see Ref. 202 for a recentexample). The use of proteomic technologies(Refs 160, 203) and genome-wide RNAi(Ref. 204) will also provide important cluesabout disease mechanisms. In turn, theseinsights will make it possible to devisetherapeutic strategies that modulate cellularpathways disrupted by protein misfolding andaggregation, including apoptosis (Ref. 12),signal transduction (Refs 13, 205) andtranscription (Ref. 14). Ultimately, drugcombinations targeting protein aggregation aswell as the resulting perturbation of cellularpathways will provide the most effectivetreatment for patients suffering from protein-misfolding diseases.
Acknowledgements and fundingCurrent funding is provided by the NationalInstitute of Aging, National Institute ofNeurological Disorders and Stroke, AmericanParkinson Disease Association, Parkinson’sDisease Foundation, and National ParkinsonFoundation. I am grateful to the peer reviewersfor their insightful comments and to themembers of my research group for stimulatingdiscussions.
References1 Dobson, C.M. (2003) Protein folding and
misfolding. Nature 426, 884-890
2 Dahlgren, K.N. et al. (2002) Oligomeric and fibrillar
species of amyloid-beta peptides differentially
affect neuronal viability. J Biol Chem 277,
32046-32053
3 Walsh, D.M. et al. (2002) Naturally secreted
oligomers of amyloid beta protein potently inhibit
hippocampal long-term potentiation in vivo.
Nature 416, 535-539
4 Lesne, S. et al. (2006) A specific amyloid-beta
protein assembly in the brain impairs memory.
Nature 440, 352-357
5 Cohen, E. et al. (2006) Opposing activities protect
against age-onset proteotoxicity. Science 313,
1604-1610
6 Arrasate, M. et al. (2004) Inclusion body formation
reduces levels of mutant huntingtin and the risk
of neuronal death. Nature 431, 805-810
7 Silveira, J.R. et al. (2005) The most infectious prion
protein particles. Nature 437, 257-261
expert reviewshttp://www.expertreviews.org/ in molecular medicine
24Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

8 Reixach, N. et al. (2004) Tissue damage in the
amyloidoses: transthyretin monomers and
nonnative oligomers are the major cytotoxic
species in tissue culture. Proc Natl Acad Sci U S A
101, 2817-2822
9 Volles, M.J. et al. (2001) Vesicle permeabilization
by protofibrillar a-synuclein: implications for
the pathogenesis and treatment of Parkinson’s
disease. Biochemistry 40, 7812-7819
10 Lashuel, H.A. et al. (2002) Neurodegenerative
disease: amyloid pores from pathogenic
mutations. Nature 418, 291
11 Kayed, R. et al. (2004) Permeabilization of lipid
bilayers is a common conformation-dependent
activity of soluble amyloid oligomers in protein
misfolding diseases. J Biol Chem 279, 46363-46366
12 Hashimoto, M. et al. (2003) Role of protein
aggregation in mitochondrial dysfunction and
neurodegeneration in Alzheimer’s and
Parkinson’s diseases. Neuromolecular Med 4,
21-36
13 Mattson, M.P. and Sherman, M. (2003) Perturbed
signal transduction in neurodegenerative
disorders involving aberrant protein aggregation.
Neuromolecular Med 4, 109-132
14 Rego, A.C. and de Almeida, L.P. (2005) Molecular
targets and therapeutic strategies in Huntington’s
disease. Curr Drug Targets CNS Neurol Disord 4,
361-381
15 Nelson, R. and Eisenberg, D. (2006) Recent atomic
models of amyloid fibril structure. Curr Opin
Struct Biol 16, 260-265
16 Rochet, J.C. and Lansbury, P.T., Jr. (2000) Amyloid
fibrillogenesis: themes and variations. Curr Opin
Struct Biol 10, 60-68
17 Petkova, A.T., Yau, W.M. and Tycko, R. (2006)
Experimental constraints on quaternary structure
in Alzheimer’s beta-amyloid fibrils. Biochemistry
45, 498-512
18 Petkova, A.T. et al. (2005) Self-propagating,
molecular-level polymorphism in Alzheimer’s
beta-amyloid fibrils. Science 307, 262-265
19 Torok, M. et al. (2002) Structural and dynamic
features of Alzheimer’s Abeta peptide in amyloid
fibrils studied by site-directed spin labeling. J Biol
Chem 277, 40810-40815
20 Williams, A.D. et al. (2004) Mapping abeta
amyloid fibril secondary structure using
scanning proline mutagenesis. J Mol Biol 335,
833-842
21 Whittemore, N.A. et al. (2005) Hydrogen-
deuterium (H/D) exchange mapping of Abeta 1-40
amyloid fibril secondary structure using nuclear
magnetic resonance spectroscopy. Biochemistry
44, 4434-4441
22 Kheterpal, I. et al. (2006) Structural differences in
Abeta amyloid protofibrils and fibrils mapped
by hydrogen exchange–mass spectrometry with
on-line proteolytic fragmentation. J Mol Biol 361,
785-795
23 Shivaprasad, S. and Wetzel, R. (2006) Scanning
cysteine mutagenesis analysis of Abeta-(1–40)
amyloid fibrils. J Biol Chem 281, 993-1000
24 Luhrs, T. et al. (2005) 3D structure of Alzheimer’s
amyloid-beta(1-42) fibrils. Proc Natl Acad Sci U S A
102, 17342-17347
25 Der-Sarkissian, A. et al. (2003) Structural
organization of alpha-synuclein fibrils studied
by site-directed spin labeling. J Biol Chem 278,
37530-37535
26 Del Mar, C. et al. (2005) Structure and properties
of alpha-synuclein and other amyloids determined
at the amino acid level. Proc Natl Acad Sci U S A
102, 15477-15482
27 Heise, H. et al. (2005) Molecular-level secondary
structure, polymorphism, and dynamics of full-
length alpha-synuclein fibrils studied by solid-
state NMR. Proc Natl Acad Sci U S A 102,
15871-15876
28 Makin, O.S. et al. (2005) Molecular basis for
amyloid fibril formation and stability. Proc Natl
Acad Sci U S A 102, 315-320
29 Sambashivan, S. et al. (2005) Amyloid-like fibrils of
ribonuclease A with three-dimensional domain-
swapped and native-like structure. Nature 437,
266-269
30 Sciarretta, K.L. et al. (2006) Spatial separation of
beta-sheet domains of beta-amyloid: disruption of
each beta-sheet by N-methyl amino acids.
Biochemistry 45, 9485-9495
31 Yan, L.M. et al. (2006) Design of a mimic of
nonamyloidogenic and bioactive human islet
amyloid polypeptide (IAPP) as nanomolar affinity
inhibitor of IAPP cytotoxic fibrillogenesis. Proc
Natl Acad Sci U S A 103, 2046-2051
32 Sciarretta, K.L., Gordon, D.J. and Meredith, S.C.
(2006) Peptide-based inhibitors of amyloid
assembly. Methods Enzymol 413, 273-312
33 Sato, T. et al. (2006) Inhibitors of amyloid toxicity
based on beta-sheet packing of Abeta40 and
Abeta42. Biochemistry 45, 5503-5516
34 Conway, K.A. et al. (2001) Kinetic stabilization
of the a-synuclein protofibril by a dopamine-a-
synuclein adduct. Science 294, 1346-1349
35 Rochet, J.C. et al. (2004) Interactions among alpha-
synuclein, dopamine, and biomembranes: some
expert reviewshttp://www.expertreviews.org/ in molecular medicine
25Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

clues for understanding neurodegeneration in
Parkinson’s disease. J Mol Neurosci 23, 23-34
36 Blanchard, B.J. et al. (2004) Efficient reversal of
Alzheimer’s disease fibril formation and
elimination of neurotoxicity by a small molecule.
Proc Natl Acad Sci U S A 101, 14326-14332
37 Kim, W. et al. (2006) A high-throughput screen
for compounds that inhibit aggregation of the
Alzheimer’s peptide. ACS Chem Biol 1,
461-469
38 Chirita, C., Necula, M. and Kuret, J. (2004) Ligand-
dependent inhibition and reversal of tau filament
formation. Biochemistry 43, 2879-2887
39 Heiser, V. et al. (2002) Identification of
benzothiazoles as potential polyglutamine
aggregation inhibitors of Huntington’s disease by
using an automated filter retardation assay. Proc
Natl Acad Sci U S A 99 Suppl 4, 16400-16406
40 Hockly, E. et al. (2006) Evaluation of the
benzothiazole aggregation inhibitors riluzole and
PGL-135 as therapeutics for Huntington’s disease.
Neurobiol Dis 21, 228-236
41 Wang, J. et al. (2005) Reversal of a full-length
mutant huntingtin neuronal cell phenotype by
chemical inhibitors of polyglutamine-mediated
aggregation. BMC Neurosci 6, 1
42 Zhang, X. et al. (2005) A potent small molecule
inhibits polyglutamine aggregation in
Huntington’s disease neurons and suppresses
neurodegeneration in vivo. Proc Natl Acad Sci
U S A 102, 892-897
43 Desai, U.A. et al. (2006) Biologically active
molecules that reduce polyglutamine aggregation
and toxicity. Hum Mol Genet 15, 2114-2124
44 Bodner, R.A. et al. (2006) Pharmacological
promotion of inclusion formation: a therapeutic
approach for Huntington’s and Parkinson’s
diseases. Proc Natl Acad Sci U S A 103, 4246-4251
45 Bertsch, U. et al. (2005) Systematic identification
of antiprion drugs by high-throughput screening
based on scanning for intensely fluorescent targets.
J Virol 79, 7785-7791
46 Kocisko, D.A. et al. (2003) New inhibitors of
scrapie-associated prion protein formation in a
library of 2000 drugs and natural products.
J Virol 77, 10288-10294
47 Kocisko, D.A. and Caughey, B. (2006) Mefloquine,
an antimalaria drug with antiprion activity in vitro,
lacks activity in vivo. J Virol 80, 1044-1046
48 Colon, W. and Kelly, J.W. (1992) Partial
denaturation of transthyretin is sufficient for
amyloid fibril formation in vitro. Biochemistry 31,
8654-8660
49 Johnson, S.M. et al. (2005) Native state kinetic
stabilization as a strategy to ameliorate protein
misfolding diseases: a focus on the transthyretin
amyloidoses. Acc Chem Res 38, 911-921
50 Fry, D.C. (2006) Protein-protein interactions as
targets for small molecule drug discovery.
Biopolymers 84, 535-552
51 Adamski-Werner, S.L. et al. (2004) Diflunisal
analogues stabilize the native state of transthyretin.
Potent inhibition of amyloidogenesis. J Med
Chem 47, 355-374
52 Hammarstrom, P. et al. (2003) Prevention of
transthyretin amyloid disease by changing protein
misfolding energetics. Science 299, 713-716
53 Morais-de-Sa, E. et al. (2004) The crystal structure
of transthyretin in complex with diethylstilbestrol:
a promising template for the design of amyloid
inhibitors. J Biol Chem 279, 53483-53490
54 Johnson, S.M. et al. (2005) Bisaryloxime ethers as
potent inhibitors of transthyretin amyloid fibril
formation. J Med Chem 48, 1576-1587
55 Petrassi, H.M. et al. (2005) Potent and selective
structure-based dibenzofuran inhibitors of
transthyretin amyloidogenesis: kinetic stabilization
of the native state. J Am Chem Soc 127, 6662-6671
56 Wiseman, R.L., Green, N.S. and Kelly, J.W. (2005)
Kinetic stabilization of an oligomeric protein
under physiological conditions demonstrated
by a lack of subunit exchange: implications for
transthyretin amyloidosis. Biochemistry 44,
9265-9274
57 Green, N.S., Foss, T.R. and Kelly, J.W. (2005)
Genistein, a natural product from soy, is a potent
inhibitor of transthyretin amyloidosis. Proc Natl
Acad Sci U S A 102, 14545-14550
58 Hough, M.A. et al. (2004) Dimer destabilization in
superoxide dismutase may result in disease-
causing properties: structures of motor neuron
disease mutants. Proc Natl Acad Sci U S A 101,
5976-5981
59 Ray, S.S. et al. (2005) Small-molecule-mediated
stabilization of familial amyotrophic lateral
sclerosis-linked superoxide dismutase mutants
against unfolding and aggregation. Proc Natl Acad
Sci U S A 102, 3639-3644
60 Loo, T.W. and Clarke, D.M. (2007) Chemical and
pharmacological chaperones as new therapeutic
agents. Expert Rev Mol Med 9, 1-18
61 De Jonghe, C. et al. (2001) Pathogenic APP
mutations near the gamma-secretase cleavage
site differentially affect Abeta secretion and APP
C-terminal fragment stability. Hum Mol Genet 10,
1665-1671
expert reviewshttp://www.expertreviews.org/ in molecular medicine
26Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

62 Wolfe, M.S. (2002) APP, Notch, and presenilin:
molecular pieces in the puzzle of Alzheimer’s
disease. Int Immunopharmacol 2, 1919-1929
63 Hamaguchi, T., Ono, K. and Yamada, M. (2006)
Anti-amyloidogenic therapies: strategies for
prevention and treatment of Alzheimer’s disease.
Cell Mol Life Sci 63, 1538-1552
64 Weggen, S. et al. (2001) A subset of NSAIDs lower
amyloidogenic Abeta42 independently of
cyclooxygenase activity. Nature 414, 212-216
65 Beher, D. et al. (2004) Selected non-steroidal anti-
inflammatory drugs and their derivatives target
gamma-secretase at a novel site. Evidence for an
allosteric mechanism. J Biol Chem 279,
43419-43426
66 Zhou, Y. et al. (2003) Nonsteroidal anti-
inflammatory drugs can lower amyloidogenic
Abeta42 by inhibiting Rho. Science 302, 1215-1217
67 Desire, L. et al. (2005) RAC1 inhibition targets
amyloid precursor protein processing by gamma-
secretase and decreases Abeta production in vitro
and in vivo. J Biol Chem 280, 37516-37525
68 Manes, S. et al. (2003) From rafts to crafts:
membrane asymmetry in moving cells. Trends
Immunol 24, 320-326
69 Phiel, C.J. et al. (2003) GSK-3alpha regulates
production of Alzheimer’s disease amyloid-beta
peptides. Nature 423, 435-439
70 Netzer, W.J. et al. (2003) Gleevec inhibits beta-
amyloid production but not Notch cleavage.
Proc Natl Acad Sci U S A 100, 12444-12449
71 Fraering, P.C. et al. (2005) gamma-Secretase
substrate selectivity can be modulated directly
via interaction with a nucleotide-binding site.
J Biol Chem 280, 41987-41996
72 Best, J.D. et al. (2007) The novel gamma secretase
inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-
(2,5-difluorophenyl)cyclohexyl]-1,1,1-
trifluoromethanesulfonamide (MRK-560) reduces
amyloid plaque deposition without evidence of
notch-related pathology in the Tg2576 mouse.
J Pharmacol Exp Ther 320, 552-558
73 Hong, L. et al. (2000) Structure of the protease
domain of memapsin 2 (beta-secretase) complexed
with inhibitor. Science 290, 150-153
74 Cole, D.C. et al. (2006) Acylguanidines as small-
molecule beta-secretase inhibitors. J Med Chem 49,
6158-6161
75 Freskos, J.N. et al. (2007) Design of potent
inhibitors of human beta-secretase. Part 2.
Bioorg Med Chem Lett 17, 78-81
76 Ghosh, A.K. et al. (2006) Design, synthesis
and X-ray structure of protein-ligand
complexes: important insight into selectivity of
memapsin 2 (beta-secretase) inhibitors. J Am
Chem Soc 128, 5310-5311
77 Hanessian, S. et al. (2005) Structure-based design,
synthesis, and memapsin 2 (BACE) inhibitory
activity of carbocyclic and heterocyclic
peptidomimetics. J Med Chem 48, 5175-5190
78 Stachel, S.J. et al. (2006) Macrocyclic inhibitors of
beta-secretase: functional activity in an animal
model. J Med Chem 49, 6147-6150
79 Chang, W.P. et al. (2004) In vivo inhibition of Abeta
production by memapsin 2 (beta-secretase)
inhibitors. J Neurochem 89, 1409-1416
80 Laird, F.M. et al. (2005) BACE1, a major
determinant of selective vulnerability of the brain
to amyloid-beta amyloidogenesis, is essential for
cognitive, emotional, and synaptic functions.
J Neurosci 25, 11693-11709
81 Hu, X. et al. (2006) Bace1 modulates myelination in
the central and peripheral nervous system. Nat
Neurosci 9, 1520-1525
82 Willem, M. et al. (2006) Control of peripheral nerve
myelination by the beta-secretase BACE1. Science
314, 664-666
83 Saura, C.A. et al. (2005) Conditional inactivation of
presenilin 1 prevents amyloid accumulation and
temporarily rescues contextual and spatial
working memory impairments in amyloid
precursor protein transgenic mice. J Neurosci 25,
6755-6764
84 Sastre, M. et al. (2006) Nonsteroidal anti-
inflammatory drugs repress beta-secretase
gene promoter activity by the activation of
PPARgamma. Proc Natl Acad Sci U S A 103,
443-448
85 Espeseth, A.S. et al. (2005) Compounds that bind
APP and inhibit Abeta processing in vitro suggest a
novel approach to Alzheimer disease therapeutics.
J Biol Chem 280, 17792-17797
86 Leissring, M.A. et al. (2003) Enhanced proteolysis
of beta-amyloid in APP transgenic mice prevents
plaque formation, secondary pathology, and
premature death. Neuron 40, 1087-1093
87 Choi, D.S. et al. (2006) PKCepsilon increases
endothelin converting enzyme activity and
reduces amyloid plaque pathology in transgenic
mice. Proc Natl Acad Sci U S A 103, 8215-8220
88 Saito, T. et al. (2005) Somatostatin regulates brain
amyloid beta peptide Abeta42 through
modulation of proteolytic degradation. Nat
Med 11, 434-439
89 Gafni, J. et al. (2004) Inhibition of calpain cleavage
of huntingtin reduces toxicity: accumulation of
expert reviewshttp://www.expertreviews.org/ in molecular medicine
27Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

calpain/caspase fragments in the nucleus. J Biol
Chem 279, 20211-20220
90 Graham, R.K. et al. (2006) Cleavage at the caspase-6
site is required for neuronal dysfunction and
degeneration due to mutant huntingtin. Cell 125,
1179-1191
91 Berke, S.J. et al. (2004) Caspase-mediated
proteolysis of the polyglutamine disease protein
ataxin-3. J Neurochem 89, 908-918
92 Haacke, A. et al. (2006) Proteolytic cleavage of
polyglutamine-expanded ataxin-3 is critical for
aggregation and sequestration of non-expanded
ataxin-3. Hum Mol Genet 15, 555-568
93 Cattaneo, E., Zuccato, C. and Tartari, M. (2005)
Normal huntingtin function: an alternative
approach to Huntington’s disease. Nat Rev
Neurosci 6, 919-930
94 Li, W. et al. (2005) Aggregation promoting
C-terminal truncation of alpha-synuclein is a
normal cellular process and is enhanced by the
familial Parkinson’s disease-linked mutations.
Proc Natl Acad Sci U S A 102, 2162-2167
95 Liu, C.W. et al. (2005) A precipitating role for
truncated alpha-synuclein and the proteasome in
alpha-synuclein aggregation: implications for
pathogenesis of Parkinson disease. J Biol Chem
280, 22670-22678
96 Gamblin, T.C. et al. (2003) Caspase cleavage of
tau: linking amyloid and neurofibrillary tangles
in Alzheimer’s disease. Proc Natl Acad Sci U S A
100, 10032-10037
97 Yin, H. and Kuret, J. (2006) C-terminal truncation
modulates both nucleation and extension phases
of tau fibrillization. FEBS Lett 580, 211-215
98 Page, L.J. et al. (2005) Metalloendoprotease
cleavage triggers gelsolin amyloidogenesis. Embo
J 24, 4124-4132
99 Chen, F. et al. (2004) Posttranslational
modifications of tau–role in human tauopathies
and modeling in transgenic animals. Curr Drug
Targets 5, 503-515
100 Alonso, A. et al. (2001) Hyperphosphorylation
induces self-assembly of tau into tangles of paired
helical filaments/straight filaments. Proc Natl
Acad Sci U S A 98, 6923-6928
101 Duff, K. and Planel, E. (2005) Untangling memory
deficits. Nat Med 11, 826-827
102 Wittmann, C.W. et al. (2001) Tauopathy in
Drosophila: neurodegeneration without
neurofibrillary tangles. Science 293, 711-714
103 Andorfer, C. et al. (2005) Cell-cycle reentry and cell
death in transgenic mice expressing nonmutant
human tau isoforms. J Neurosci 25, 5446-5454
104 Santacruz, K. et al. (2005) Tau suppression in a
neurodegenerative mouse model improves
memory function. Science 309, 476-481
105 Le Corre, S. et al. (2006) An inhibitor of tau
hyperphosphorylation prevents severe motor
impairments in tau transgenic mice. Proc Natl
Acad Sci U S A 103, 9673-9678
106 Alonso Adel, C. et al. (2006) Polymerization of
hyperphosphorylated tau into filaments
eliminates its inhibitory activity. Proc Natl Acad Sci
U S A 103, 8864-8869
107 Shulman, J.M. and Feany, M.B. (2003) Genetic
modifiers of tauopathy in Drosophila. Genetics
165, 1233-1242
108 Avila, J. (2006) Tau phosphorylation and
aggregation in Alzheimer’s disease pathology.
FEBS Lett 580, 2922-2927
109 Lucas, J.J. et al. (2001) Decreased nuclear beta-
catenin, tau hyperphosphorylation and
neurodegeneration in GSK-3beta conditional
transgenic mice. Embo J 20, 27-39
110 Jackson, G.R. et al. (2002) Human wild-type tau
interacts with wingless pathway components and
produces neurofibrillary pathology in Drosophila.
Neuron 34, 509-519
111 Cruz, J.C. et al. (2003) Aberrant Cdk5 activation
by p25 triggers pathological events leading to
neurodegeneration and neurofibrillary tangles.
Neuron 40, 471-483
112 Noble, W. et al. (2003) Cdk5 is a key factor in tau
aggregation and tangle formation in vivo. Neuron
38, 555-565
113 Liu, S.J. et al. (2004) Tau becomes a more favorable
substrate for GSK-3 when it is prephosphorylated
by PKA in rat brain. J Biol Chem 279, 50078-50088
114 Sato, S. et al. (2006) Tau-tubulin kinase 1 (TTBK1),
a neuron-specific tau kinase candidate, is involved
in tau phosphorylation and aggregation.
J Neurochem 98, 1573-1584
115 Hoshi, M. et al. (2003) Spherical aggregates of beta-
amyloid (amylospheroid) show high neurotoxicity
and activate tau protein kinase I/glycogen
synthase kinase-3beta. Proc Natl Acad Sci U S A
100, 6370-6375
116 Takashima, A. (2006) GSK-3 is essential in the
pathogenesis of Alzheimer’s disease. J Alzheimers
Dis 9, 309-317
117 Noble, W. et al. (2005) Inhibition of glycogen
synthase kinase-3 by lithium correlates with
reduced tauopathy and degeneration in vivo.
Proc Natl Acad Sci U S A 102, 6990-6995
118 Bhat, R. et al. (2003) Structural insights and
biological effects of glycogen synthase kinase
expert reviewshttp://www.expertreviews.org/ in molecular medicine
28Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

3-specific inhibitor AR-A014418. J Biol Chem 278,
45937-45945
119 Patrick, G.N. et al. (1999) Conversion of p35 to
p25 deregulates Cdk5 activity and promotes
neurodegeneration. Nature 402, 615-622
120 Zheng, Y.L. et al. (2005) A Cdk5 inhibitory peptide
reduces tau hyperphosphorylation and apoptosis
in neurons. Embo J 24, 209-220
121 Plattner, F., Angelo, M. and Giese, K.P. (2006) The
roles of cyclin-dependent kinase 5 and glycogen
synthase kinase 3 in tau hyperphosphorylation.
J Biol Chem 281, 25457-25465
122 Fujiwara, H. et al. (2002) alpha-Synuclein is
phosphorylated in synucleinopathy lesions. Nat
Cell Biol 4, 160-164
123 Anderson, J.P. et al. (2006) Phosphorylation of
Ser-129 is the dominant pathological modification
of alpha-synuclein in familial and sporadic Lewy
body disease. J Biol Chem 281, 29739-29752
124 Smith, W.W. et al. (2005) alpha-Synuclein
phosphorylation enhances eosinophilic
cytoplasmic inclusion formation in SH-SY5Y cells.
J Neurosci 25, 5544-5552
125 Chen, L. and Feany, M.B. (2005) alpha-Synuclein
phosphorylation controls neurotoxicity and
inclusion formation in a Drosophila model
of Parkinson disease. Nat Neurosci 8,
657-663
126 Kim, E.J. et al. (2006) Dyrk1A phosphorylates
alpha-synuclein and enhances intracellular
inclusion formation. J Biol Chem 281, 33250-33257
127 Tabner, B.J. et al. (2005) Hydrogen peroxide is
generated during the very early stages of
aggregation of the amyloid peptides implicated in
Alzheimer disease and familial British dementia.
J Biol Chem 280, 35789-35792
128 Barnham, K.J. et al. (2003) Neurotoxic, redox-
competent Alzheimer’s beta-amyloid is released
from lipid membrane by methionine oxidation.
J Biol Chem 278, 42959-42965
129 Butterfield, D.A. and Boyd-Kimball, D. (2005) The
critical role of methionine 35 in Alzheimer’s
amyloid beta-peptide (1-42)-induced oxidative
stress and neurotoxicity. Biochim Biophys Acta
1703, 149-156
130 Hou, L. et al. (2002) Methionine 35 oxidation
reduces fibril assembly of the amyloid abeta-(1–
42) peptide of Alzheimer’s disease. J Biol Chem
277, 40173-40176
131 Palmblad, M., Westlind-Danielsson, A. and
Bergquist, J. (2002) Oxidation of methionine 35
attenuates formation of amyloid beta -peptide 1-40
oligomers. J Biol Chem 277, 19506-19510
132 Bitan, G. et al. (2003) A molecular switch in
amyloid assembly: Met35 and amyloid beta-
protein oligomerization. J Am Chem Soc 125,
15359-15365
133 Zhang, Q. et al. (2004) Metabolite-initiated protein
misfolding may trigger Alzheimer’s disease. Proc
Natl Acad Sci U S A 101, 4752-4757
134 Ono, K. et al. (2003) Potent anti-amyloidogenic
and fibril-destabilizing effects of polyphenols in
vitro: implications for the prevention and
therapeutics of Alzheimer’s disease. J Neurochem
87, 172-181
135 Masuda, M. et al. (2006) Small molecule inhibitors
of alpha-synuclein filament assembly.
Biochemistry 45, 6085-6094
136 Yang, F. et al. (2005) Curcumin inhibits formation of
amyloid beta oligomers and fibrils, binds plaques,
and reduces amyloid in vivo. J Biol Chem 280,
5892-5901
137 Matsubara, E. et al. (2003) Melatonin increases
survival and inhibits oxidative and amyloid
pathology in a transgenic model of Alzheimer’s
disease. J Neurochem 85, 1101-1108
138 Feng, Z. et al. (2006) Early melatonin
supplementation alleviates oxidative stress in a
transgenic mouse model of Alzheimer’s disease.
Free Radic Biol Med 40, 101-109
139 Abdul, H.M. et al. (2006) Acetyl-L-carnitine-
induced up-regulation of heat shock proteins
protects cortical neurons against amyloid-beta
peptide 1-42-mediated oxidative stress and
neurotoxicity: implications for Alzheimer’s
disease. J Neurosci Res 84, 398-408
140 Ritchie, C.W. et al. (2003) Metal-protein attenuation
with iodochlorhydroxyquin (clioquinol) targeting
Abeta amyloid deposition and toxicity in
Alzheimer disease: a pilot phase 2 clinical trial.
Arch Neurol 60, 1685-1691
141 Norris, E.H. et al. (2003) Effects of oxidative and
nitrative challenges on a-synuclein fibrillogenesis
involve distinct mechanisms of protein
modifications. J Biol Chem 278, 27230-27240
142 Yamin, G., Uversky, V.N. and Fink, A.L. (2003)
Nitration inhibits fibrillation of human
a-synuclein in vitro by formation of soluble
oligomers. FEBS Lett 542, 147-152
143 Cole, N.B. et al. (2005) Metal-catalyzed oxidation of
alpha synuclein: helping to define the relationship
between oligomers, protofilaments and filaments.
J Biol Chem 280, 9678-9690
144 Glaser, C.B. et al. (2005) Methionine oxidation,
alpha-synuclein and Parkinson’s disease. Biochim
Biophys Acta 1703, 157-169
expert reviewshttp://www.expertreviews.org/ in molecular medicine
29Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

145 Mirzaei, H. et al. (2006) Identification of rotenone-
induced modifications in alpha-synuclein using
affinity pull-down and tandem mass spectrometry.
Anal Chem 78, 2422-2431
146 Bosco, D.A. et al. (2006) Elevated levels of oxidized
cholesterol metabolites in Lewy body disease
brains accelerate alpha-synuclein fibrilization. Nat
Chem Biol 2, 249-253
147 Zhu, M. et al. (2004) The flavonoid baicalein
inhibits fibrillation of alpha-synuclein and
disaggregates existing fibrils. J Biol Chem 279,
26846-26857
148 Ono, K. and Yamada, M. (2006) Antioxidant
compounds have potent anti-fibrillogenic and
fibril-destabilizing effects for alpha-synuclein
fibrils in vitro. J Neurochem 97, 105-115
149 Giorgini, F. et al. (2005) A genomic screen in yeast
implicates kynurenine 3-monooxygenase as a
therapeutic target for Huntington disease. Nat
Genet 37, 526-531
150 Wyttenbach, A. et al. (2002) Heat shock protein 27
prevents cellular polyglutamine toxicity and
suppresses the increase of reactive oxygen species
caused by huntingtin. Hum Mol Genet 11,
1137-1151
151 Firdaus, W.J. et al. (2006) Analysis of oxidative
events induced by expanded polyglutamine
huntingtin exon 1 that are differentially restored
by expression of heat shock proteins or treatment
with an antioxidant. Febs J 273, 3076-3093
152 Firdaus, W.J. et al. (2006) Huntingtin inclusion
bodies are iron-dependent centers of oxidative
events. Febs J 273, 5428-5441
153 Dias-Santagata, D. et al. (2007) Oxidative stress
mediates tau-induced neurodegeneration in
Drosophila. J Clin Invest 117, 236-245
154 Deng, H.X. et al. (2006) Conversion to the
amyotrophic lateral sclerosis phenotype is
associated with intermolecular linked insoluble
aggregates of SOD1 in mitochondria. Proc Natl
Acad Sci U S A 103, 7142-7147
155 Muchowski, P.J. and Wacker, J.L. (2005)
Modulation of neurodegeneration by molecular
chaperones. Nat Rev Neurosci 6, 11-22
156 Cuervo, A.M. et al. (2004) Impaired degradation of
mutant alpha-synuclein by chaperone-mediated
autophagy. Science 305, 1292-1295
157 Evans, C.G., Wisen, S. and Gestwicki, J.E. (2006)
Heat shock proteins 70 and 90 inhibit early stages
of amyloid beta-(1-42) aggregation in vitro. J Biol
Chem 281, 33182-33191
158 Wilhelmus, M.M. et al. (2006) Small heat shock
proteins inhibit amyloid-beta protein aggregation
and cerebrovascular amyloid-beta protein toxicity.
Brain Res 1089, 67-78
159 Klucken, J. et al. (2004) Hsp70 reduces alpha-
synuclein aggregation and toxicity. J Biol Chem
279, 25497-25502
160 Zhou, Y. et al. (2004) Analysis of alpha-synuclein-
associated proteins by quantitative proteomics.
J Biol Chem 279, 39155-39164
161 Auluck, P.K. et al. (2002) Chaperone suppression of
alpha-synuclein toxicity in a Drosophila model for
Parkinson’s disease. Science 295, 865-868
162 Dedmon, M.M. et al. (2005) Heat shock protein 70
inhibits alpha-synuclein fibril formation via
preferential binding to prefibrillar species. J Biol
Chem 280, 14733-14740
163 Huang, C. et al. (2006) Heat shock protein 70
inhibits alpha-synuclein fibril formation via
interactions with diverse intermediates. J Mol Biol
364, 323-336
164 Zourlidou, A., Payne Smith, M.D. and Latchman,
D.S. (2004) HSP27 but not HSP70 has a potent
protective effect against alpha-synuclein-induced
cell death in mammalian neuronal cells.
J Neurochem 88, 1439-1448
165 Outeiro, T.F. et al. (2006) Small heat shock proteins
protect against alpha-synuclein-induced toxicity
and aggregation. Biochem Biophys Res Commun
351, 631-638
166 Wacker, J.L. et al. (2004) Hsp70 and Hsp40
attenuate formation of spherical and annular
polyglutamine oligomers by partitioning
monomer. Nat Struct Mol Biol 11, 1215-1222
167 Miller, V.M. et al. (2005) CHIP suppresses
polyglutamine aggregation and toxicity in vitro
and in vivo. J Neurosci 25, 9152-9161
168 Jana, N.R. et al. (2005) Co-chaperone CHIP
associates with expanded polyglutamine protein
and promotes their degradation by proteasomes.
J Biol Chem 280, 11635-11640
169 Al-Ramahi, I. et al. (2006) CHIP protects from the
neurotoxicity of expanded and wild-type ataxin-1
and promotes their ubiquitination and
degradation. J Biol Chem 281, 26714-26724
170 Behrends, C. et al. (2006) Chaperonin TRiC
promotes the assembly of polyQ expansion
proteins into nontoxic oligomers. Mol Cell 23,
887-897
171 Kitamura, A. et al. (2006) Cytosolic
chaperonin prevents polyglutamine toxicity with
altering the aggregation state. Nat Cell Biol 8,
1163-1170
172 Tam, S. et al. (2006) The chaperonin TRiC controls
polyglutamine aggregation and toxicity through
expert reviewshttp://www.expertreviews.org/ in molecular medicine
30Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

subunit-specific interactions. Nat Cell Biol 8,
1155-1162
173 Gestwicki, J.E., Crabtree, G.R. and Graef, I.A.
(2004) Harnessing chaperones to generate small-
molecule inhibitors of amyloid beta aggregation.
Science 306, 865-869
174 Drysdale, M.J. et al. (2006) Targeting Hsp90 for
the treatment of cancer. Curr Opin Drug Discov
Devel 9, 483-495
175 McCollum, A.K. et al. (2006) Up-regulation of heat
shock protein 27 induces resistance to 17-
allylamino-demethoxygeldanamycin through a
glutathione-mediated mechanism. Cancer Res
66, 10967-10975
176 McLean, P.J. et al. (2004) Geldanamycin induces
Hsp70 and prevents alpha-synuclein aggregation
and toxicity in vitro. Biochem Biophys Res
Commun 321, 665-669
177 Auluck, P.K., Meulener, M.C. and Bonini, N.M.
(2005) Mechanisms of suppression of {alpha}-
synuclein neurotoxicity by geldanamycin in
Drosophila. J Biol Chem 280, 2873-2878
178 Hay, D.G. et al. (2004) Progressive decrease in
chaperone protein levels in a mouse model of
Huntington’s disease and induction of stress
proteins as a therapeutic approach. Hum Mol
Genet 13, 1389-1405
179 Shen, H.Y. et al. (2005) Geldanamycin induces
heat shock protein 70 and protects against MPTP-
induced dopaminergic neurotoxicity in mice.
J Biol Chem 280, 39962-39969
180 Ansar, S. et al. (2007) A non-toxic Hsp90 inhibitor
protects neurons from Abeta-induced toxicity.
Bioorg Med Chem Lett 17, 1984-1990
181 Westerheide, S.D. et al. (2004) Celastrols as
inducers of the heat shock response and
cytoprotection. J Biol Chem 279, 56053-56060
182 Johnston, J.A., Ward, C.L. and Kopito, R.R. (1998)
Aggresomes: a cellular response to misfolded
proteins. J Cell Biol 143, 1883-1898
183 Iwata, A. et al. (2005) HDAC6 and microtubules are
required for autophagic degradation of aggregated
huntingtin. J Biol Chem 280, 40282-40292
184 Ravikumar, B. et al. (2005) Dynein mutations
impair autophagic clearance of aggregate-prone
proteins. Nat Genet 37, 771-776
185 Lee, H.J. and Lee, S.J. (2002) Characterization of
cytoplasmic alpha-synuclein aggregates. Fibril
formation is tightly linked to the inclusion-forming
process in cells. J Biol Chem 277, 48976-48983
186 Lee, H.J. et al. (2004) Clearance of alpha-synuclein
oligomeric intermediates via the lysosomal
degradation pathway. J Neurosci 24, 1888-1896
187 Webb, J.L. et al. (2003) alpha-Synuclein is degraded
by both autophagy and the proteasome. J Biol
Chem 278, 25009-25013
188 Tanaka, M. et al. (2004) Aggresomes formed by
alpha-synuclein and synphilin-1 are
cytoprotective. J Biol Chem 279, 4625-4631
189 Waelter, S. et al. (2001) Accumulation of mutant
huntingtin fragments in aggresome-like inclusion
bodies as a result of insufficient protein
degradation. Mol Biol Cell 12, 1393-1407
190 Taylor, J.P. et al. (2003) Aggresomes protect cells by
enhancing the degradation of toxic polyglutamine-
containing protein. Hum Mol Genet 12, 749-757
191 Iwata, A. et al. (2005) Increased susceptibility of
cytoplasmic over nuclear polyglutamine
aggregates to autophagic degradation. Proc Natl
Acad Sci U S A 102, 13135-13140
192 Ravikumar, B., Duden, R. and Rubinsztein, D.C.
(2002) Aggregate-prone proteins with
polyglutamine and polyalanine expansions are
degraded by autophagy. Hum Mol Genet 11,
1107-1117
193 Rubinsztein, D.C. (2006) The roles of intracellular
protein-degradation pathways in
neurodegeneration. Nature 443, 780-786
194 Ravikumar, B. et al. (2004) Inhibition of mTOR
induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse
models of Huntington disease. Nat Genet 36,
585-595
195 Berger, Z. et al. (2006) Rapamycin alleviates toxicity
of different aggregate-prone proteins. Hum Mol
Genet 15, 433-442
196 Vignot, S. et al. (2005) mTOR-targeted therapy of
cancer with rapamycin derivatives. Ann Oncol 16,
525-537
197 Sarkar, S. et al. (2005) Lithium induces autophagy
by inhibiting inositol monophosphatase. J Cell Biol
170, 1101-1111
198 Sarkar, S. et al. (2007) Trehalose, a novel mTOR-
independent autophagy enhancer, accelerates the
clearance of mutant huntingtin and {alpha}-
synuclein. J Biol Chem 282, 5641-5652
199 Miller, V.M. et al. (2004) Targeting Alzheimer’s
disease genes with RNA interference: an efficient
strategy for silencing mutant alleles. Nucleic Acids
Res 32, 661-668
200 Saito, Y. et al. (2005) Transgenic small interfering
RNA halts amyotrophic lateral sclerosis in a mouse
model. J Biol Chem 280, 42826-42830
201 Sapru, M.K. et al. (2006) Silencing of human alpha-
synuclein in vitro and in rat brain using lentiviral-
mediated RNAi. Exp Neurol 198, 382-390
expert reviewshttp://www.expertreviews.org/ in molecular medicine
31Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

202 Cooper, A.A. et al. (2006) alpha-Synuclein blocks
ER-Golgi traffic and Rab1 rescues neuron loss in
Parkinson’s models. Science 313, 324-328
203 Poon, H.F. et al. (2005) Redox proteomics
analysis of oxidatively modified proteins in G93A-
SOD1 transgenic mice–a model of familial
amyotrophic lateral sclerosis. Free Radic Biol Med
39, 453-462
204 Nollen, E.A. et al. (2004) Genome-wide RNA
interference screen identifies previously
undescribed regulators of polyglutamine
aggregation. Proc Natl Acad Sci U S A 101,
6403-6408
205 Morfini, G. et al. (2006) JNK mediates pathogenic
effects of polyglutamine-expanded androgen
receptor on fast axonal transport. Nat Neurosci 9,
907-916
206 Kelly, J.W. (1996) Alternative conformations of
amyloidogenic proteins govern their behavior.
Curr Opin Struct Biol 6, 11-17
207 Zoghbi, H.Y. and Orr, H.T. (2000) Glutamine
repeats and neurodegeneration. Annu Rev
Neurosci 23, 217-247
208 Taylor, J.P., Hardy, J. and Fischbeck, K.H. (2002)
Toxic proteins in neurodegenerative disease.
Science 296, 1991-1995
209 Caughey, B. and Lansbury, P.T. (2003) Protofibrils,
pores, fibrils, and neurodegeneration: separating
the responsible protein aggregates from the
innocent bystanders. Annu Rev Neurosci 26,
267-298
210 Ross, C.A. and Poirier, M.A. (2004) Protein
aggregation and neurodegenerative disease. Nat
Med 10 Suppl, S10-17
211 Chaudhuri, T.K. and Paul, S. (2006) Protein-
misfolding diseases and chaperone-based
therapeutic approaches. Febs J 273, 1331-1349
212 Tycko, R. (2006) Molecular structure of amyloid
fibrils: insights from solid-state NMR. Q Rev
Biophys 39, 1-55
Further reading, resources and contacts
TextbooksUversky, V.N. and Fink, A.L. (2006) Protein Misfolding, Aggregation and Conformational Diseases: Part A:
Protein Aggregation and Conformational Diseases, SpringerThis textbook provides in-depth analysis of the molecular underpinnings of protein-misfolding diseases. The
major topics include biophysical properties of proteins resulting in misfolding and aggregation, toxicity ofoligomers versus amyloid fibrils, effects of post-translational modifications, the role of molecularchaperones and protein-clearance mechanisms, methods to visualise protein aggregates, and the use ofanimal models to elucidate disease pathways.
Sipe, J.D. (2005) Amyloid Proteins: The Beta Sheet Conformation and Disease, John Wiley & SonsThis is a comprehensive volume that discusses the biochemical properties and pathobiology of a broad range of
disease-linked fibrillogenic proteins. The book covers a number of important topics including structuralproperties of amyloid fibrils, thermodynamics of fibrillisation, the role of post-translational modificationsand cellular factors in modulating fibril formation, the relation between oligomeric intermediates andcytotoxicity, and current and emerging therapeutic strategies.
Smith, H.J., Simons, C. and Sewell, R.D.E. (2007) Protein Misfolding in Neurodegenerative Diseases:Mechanisms and Therapeutic Strategies, CRC Press
This textbook is a valuable resource with detailed information about the role of protein misfolding inneurodegenerative disorders such as Alzheimer disease, Parkinson disease, Huntington disease, and theprion diseases. Key topics include protein targets associated with each disease, reasons for the limitedsuccess of current therapeutic strategies, and ground-breaking research aimed at generating more-effective therapies.
(continued on next page)
expert reviewshttp://www.expertreviews.org/ in molecular medicine
32Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses

Further reading, resources and contacts (continued)
Review articlesEstrada, L.D. and Soto, C. (2006) Inhibition of protein misfolding and aggregation by small rationally-designed
peptides. Curr Pharm Des 12, 2557-2567This is an informative review article that discusses the use of peptide inhibitors to suppress protein aggregation
associated with various misfolding diseases.
Herbst, M. and Wanker, E.E. (2006) Therapeutic approaches to polyglutamine diseases: combating proteinmisfolding and aggregation. Curr Pharm Des 12, 2543-2555
This insightful review presents strategies to treat polyglutamine diseases with compounds identified viachemical screening. Emphasis is placed on small molecules that inhibit protein aggregation, upregulatemolecular chaperones, or interfere with downstream toxic pathways. The therapeutic approaches coveredby this article are applicable to other protein-misfolding disorders.
May, B.C., Govaerts, C. and Cohen, F.E. (2006) Developing therapeutics for the diseases of protein misfolding.Neurology 66, S118-122
This review article is a useful resource that discusses therapeutic strategies to treat prion diseases, withemphasis on reducing PrP expression, stabilising the native state, enhancing cellular clearancemechanisms, and screening for chemical inhibitors of PrP misfolding. The topics covered by this articleare relevant to other protein-aggregation diseases.
WebsitesThe PubChem website is a useful link with information relating to the biological activities of small molecules.
It includes a database of chemical structures and the results of NIH-funded high-throughput screens:
http://pubchem.ncbi.nlm.nih.gov/2
The ClinicalTrials.gov website is an important resourcewith up-to-date information about federally and privatelysponsored clinical trials in humans. The link includes details about the goals of each trial, criteria forparticipation, and contact information:
http://clinicaltrials.gov/2
The disease-foundation websites listed below provide useful information about protein-misfolding disorders,including disease mechanisms, ongoing research and clinical trials, current available therapies, and patientresources:
http://www.alz.org/ (Alzheimer’s Association)
http://www.alsa.org/ (ALS Association)
http://www.hdfoundation.org/ (Hereditary Disease Foundation)
http://www.apdaparkinson.org (American Parkinson Disease Association)
http://www.michaeljfox.org/ (Michael J. Fox Foundation)
http://www.parkinson.org (National Parkinson Foundation)
http://www.pdf.org/ (Parkinson’s Disease Foundation)
Features associated with this article
FiguresFigure 1. Model illustrating protein misfolding, aggregation and clearance inside the cell, along with
opportunities for therapeutic intervention.Figure 2. Solid-state nuclear magnetic resonance (SS-NMR) structures of fibrillar amyloid-beta peptide Ab1–40.
(continued on next page)
expert reviewshttp://www.expertreviews.org/ in molecular medicine
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses
33Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press

Features associated with this article (continued)
Figure 3. Small-molecule modulators of protein aggregation identified via high-throughput screening.Figure 4. Inhibition of transthyretin fibrillisation via native-state stabilisation.Figure 5. Inhibition of amyloid-beta peptide (Ab) release from the amyloid precursor protein by targeting b- and
g-secretase.
TableTable 1. Proteins involved in misfolding diseases (examples discussed in the text)a.
Citation details for this article
Jean-Christophe Rochet (2007) Novel therapeutic strategies for the treatment of protein-misfolding diseases.Expert Rev. Mol. Med. Vol. 9, Issue 17, June 2007, DOI: 10.1017/S1462399407000385
expert reviewshttp://www.expertreviews.org/ in molecular medicine
34Accession information: DOI: 10.1017/S1462399407000385; Vol. 9; Issue 17; June 2007
&2007 Cambridge University Press
Nove
lthe
rapeu
ticstrategiesforthetrea
tmen
tofp
rotein-m
isfoldingdisea
ses